
RESEARCH ARTICLE Open Access De novo root transcriptome of
9
RESEARCH ARTICLE Open Access De novo assembly and characterization of the root transcriptome of Aegilops variabilis during an interaction with the cereal cyst nematode De-Lin Xu 1,2 , Hai Long 1* , Jun-Jun Liang 1,2 , Jie Zhang 1,2 , Xin Chen 1,2 , Jing-Liang Li 1,2 , Zhi-Fen Pan 1 , Guang-Bing Deng 1 and Mao-Qun Yu 1* Abstract Background: Aegilops variabilis No.1 is highly resistant to cereal cyst nematode (CCN). However, a lack of genomic information has restricted studies on CCN resistance genes in Ae. variabilis and has limited genetic applications in wheat breeding. Results: Using RNA-Seq technology, we generated a root transcriptome at a sequencing depth of 4.69 gigabases of Ae. variabilis No. 1 from a pooled RNA sample. The sample contained equal amounts of RNA extracted from CCN-infected and untreated control plants at three time-points. Using the Trinity method, nearly 52,081,238 high-quality trimmed reads were assembled into a non-redundant set of 118,064 unigenes with an average length of 500 bp and an N50 of 599 bp. The total assembly was 59.09 Mb of unique transcriptome sequences with average read-depth coverage of 33.25×. In BLAST searches of our database against public databases, 66.46% (78,467) of the unigenes were annotated with gene descriptions, conserved protein domains, or gene ontology terms. Functional categorization further revealed 7,408 individual unigenes and three pathways related to plant stress resistance. Conclusions: We conducted high-resolution transcriptome profiling related to root development and the response to CCN infection in Ae. variabilis No.1. This research facilitates further studies on gene discovery and on the molecular mechanisms related to CCN resistance. Background Cereal cyst nematode (causal agent Heterodeta avenae) causes cereal disease in many regions of the world [1-5], and results in economic losses of billions of dollars annu- ally [6]. Although CCNs have caused serious economic losses over the last 40 years [1], only a few CCN resistance genes have been genetically mapped on the genomes of wheat (Cre1 and Cre8) and its relatives, such as those in the genus Aegilops (Cre2-7), Secale cereale (CreR) (reviewed in Smiley and Nicol (2009) [7]) and Hordeum vulgare (Ha1-4) (reviewed in Bakker et al. (2006) [8]). The molecu- lar mechanism of CCN resistance remains unknown. Members of the genus Aegilops readily hybridize with bread wheat as the male parent [9]. Aegilops species are valuable genetic resources for breeding for disease resistance in wheat; for example, for resistance to Cochlio- bolus sativus (spot blotch), Tilletia indica (Karnal bunt), and powdery mildew [10,11]. Ae. variabilis accession No.1 (2n = 4x = 28, UUS v S v ) (syn. Triticum peregrinum (Hack In J. Fraser) Marie & Hackel) was reported to harbor resistance genes to both CCN and root knot nematode (Meloidogyne naasi) [12,13]. A greater understanding of the mechanism of CCN resistance in Ae. variabilis is necessary for wheat breeding. However, the major barrier against using genomic approaches to improve Ae. variabilis is that the genome sequence, cDNA libraries, EST databases, and microarray platform information are not available [14]. Recent developments in RNA-Seq technology have enabled very efficient probing of transcriptomic data [15-19]. This method not only detects transcripts that correspond to existing genomic sequences, but it can also be used for de novo assembly of short reads for gene discov- ery and expression profiling in organisms for which there is no reference genome [17,20-26]. * Correspondence: [email protected]; [email protected] 1 Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu, Sichuan, China Full list of author information is available at the end of the article © 2012 Xu et al.; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Xu et al. BMC Genomics 2012, 13:133 http://www.biomedcentral.com/1471-2164/13/133
Transcript of RESEARCH ARTICLE Open Access De novo root transcriptome of

Xu et al. BMC Genomics 2012, 13:133http://www.biomedcentral.com/1471-2164/13/133
RESEARCH ARTICLE Open Access
De novo assembly and characterization of theroot transcriptome of Aegilops variabilis during aninteraction with the cereal cyst nematodeDe-Lin Xu1,2, Hai Long1*, Jun-Jun Liang1,2, Jie Zhang1,2, Xin Chen1,2, Jing-Liang Li1,2, Zhi-Fen Pan1,Guang-Bing Deng1 and Mao-Qun Yu1*
Abstract
Background: Aegilops variabilis No.1 is highly resistant to cereal cyst nematode (CCN). However, a lack of genomicinformation has restricted studies on CCN resistance genes in Ae. variabilis and has limited genetic applications inwheat breeding.
Results: Using RNA-Seq technology, we generated a root transcriptome at a sequencing depth of 4.69 gigabases ofAe. variabilis No. 1 from a pooled RNA sample. The sample contained equal amounts of RNA extracted fromCCN-infected and untreated control plants at three time-points. Using the Trinity method, nearly 52,081,238high-quality trimmed reads were assembled into a non-redundant set of 118,064 unigenes with an average lengthof 500 bp and an N50 of 599 bp. The total assembly was 59.09 Mb of unique transcriptome sequences with averageread-depth coverage of 33.25×. In BLAST searches of our database against public databases, 66.46% (78,467) of theunigenes were annotated with gene descriptions, conserved protein domains, or gene ontology terms. Functionalcategorization further revealed 7,408 individual unigenes and three pathways related to plant stress resistance.
Conclusions: We conducted high-resolution transcriptome profiling related to root development and the responseto CCN infection in Ae. variabilis No.1. This research facilitates further studies on gene discovery and on themolecular mechanisms related to CCN resistance.
BackgroundCereal cyst nematode (causal agent Heterodeta avenae)causes cereal disease in many regions of the world [1-5],and results in economic losses of billions of dollars annu-ally [6]. Although CCNs have caused serious economiclosses over the last 40 years [1], only a few CCN resistancegenes have been genetically mapped on the genomes ofwheat (Cre1 and Cre8) and its relatives, such as those inthe genus Aegilops (Cre2-7), Secale cereale (CreR) (reviewedin Smiley and Nicol (2009) [7]) and Hordeum vulgare(Ha1-4) (reviewed in Bakker et al. (2006) [8]). The molecu-lar mechanism of CCN resistance remains unknown.Members of the genus Aegilops readily hybridize with
bread wheat as the male parent [9]. Aegilops species arevaluable genetic resources for breeding for disease
* Correspondence: [email protected]; [email protected] Institute of Biology, Chinese Academy of Sciences, Chengdu,Sichuan, ChinaFull list of author information is available at the end of the article
© 2012 Xu et al.; licensee BioMed Central Ltd.Commons Attribution License (http://creativecreproduction in any medium, provided the or
resistance in wheat; for example, for resistance to Cochlio-bolus sativus (spot blotch), Tilletia indica (Karnal bunt),and powdery mildew [10,11]. Ae. variabilis accession No.1(2n=4x=28, UUSvSv) (syn. Triticum peregrinum (Hack InJ. Fraser) Marie & Hackel) was reported to harbor resistancegenes to both CCN and root knot nematode (Meloidogynenaasi) [12,13]. A greater understanding of the mechanismof CCN resistance in Ae. variabilis is necessary for wheatbreeding. However, the major barrier against using genomicapproaches to improve Ae. variabilis is that the genomesequence, cDNA libraries, EST databases, and microarrayplatform information are not available [14].Recent developments in RNA-Seq technology have
enabled very efficient probing of transcriptomic data[15-19]. This method not only detects transcripts thatcorrespond to existing genomic sequences, but it can alsobe used for de novo assembly of short reads for gene discov-ery and expression profiling in organisms for which there isno reference genome [17,20-26].
This is an Open Access article distributed under the terms of the Creativeommons.org/licenses/by/2.0), which permits unrestricted use, distribution, andiginal work is properly cited.

Xu et al. BMC Genomics 2012, 13:133 Page 2 of 9http://www.biomedcentral.com/1471-2164/13/133
In the present study, we analyzed the root transcriptomeof Ae. variabilis using RNA-Seq technology. We usedtwo methods, SOAPdenovo and Trinity, for de novo assem-bly of the transcriptome, and compared their results.Characterization of the transcriptome data assembled byTrinity give a high-resolution insight into the genesinvolved in several major metabolic pathways associatedwith root development and plant defense. This research willserve as a public information platform for further studieson the evolution and function of genes in Ae. variabilis, andprovides a thorough insight into the gene expressionprofiles associated with the response to CCN infection inAe. variabilis.
MethodsPlant material and pathogen infectionAe. variabilis accession No.1 was used for transcrip-tomic profiling of genes expressed in roots. Grains ofAe. variabilis No.1 were surface-sterilized in a solutioncontaining 3% (v/v) hypochlorite and 0.01% (v/v) Tween20 for 5 min and rinsed three times with sterile water[27]. The seeds were germinated in Petri dishes (5-cmdiameter) on wet paper at 20 C under a 16-h light/8-hdark photoperiod. After 10 days, seedlings were dividedinto two groups. One group was inoculated with 1,000second-stage juveniles (J2) of CCN per plant, and theother group (negative control) was not inoculated withCCN [28,29]. Thirty hours after inoculation, the rootswere thoroughly washed three times with sterile water(each 10 min) to remove CCNs adhering to roots. Then,plants were transplanted into 500-ml glass containersfilled with sterilized perlite, and were grown at 20 Cunder a 16-h light/8-h dark photoperiod. These condi-tions prevented further CCN penetration and ensuredsynchronized development of syncytia [27,30].
RNA isolationSuccessful CCN inoculation was confirmed by observingroots under a microscope (Additional file 1). Roots ofCCN-infected and non-infected plants were sampled at30 hpi (hours post inoculation), 3 dpi (days post inocu-lation) and 9 dpi for RNA extraction [27,31,32]. Eachsample consisted of 15 individuals. Total RNA wasextracted with a Biomiga RNA kit according to the man-ufacturer’s protocol (Biomiga, San Diego, CA, USA).The concentration and quality of each RNA sample wasdetermined using a NanoDrop 2000™ micro-volumespectrophotometer (Thermo Scientific, Waltham, MA,USA). Equal amounts of total RNA from each samplewere pooled to construct the cDNA library. Pooling is acost-effective strategy when the primary research goalis to identify gene expression profiles. This strategywas well-justified based on statistical and practicalconsiderations [33-35].
Construction of cDNA library and Illumina deep-sequencingThe cDNA library was constructed using an mRNA-Seqassay for paired-end transcriptome sequencing. Thelibrary construction and sequencing were performed bythe Beijing Genomics Institute (BGI)-Shenzhen, Shenzhen,China. Briefly, mRNA was enriched from 20 μg total RNAusing oligo dT magnetic beads, and was then cleaved into200–700 nt fragments by incubation with RNA Fragmen-tation Reagent. The fragmented mRNA was converted intodouble-stranded cDNA by priming with random hexamer-primers, purified with a QiaQuick PCR extraction kit(QIAGEN Inc., Valencia, CA, USA), and then washed withEB buffer for end repairing and single nucleotide adenineaddition. Finally, sequencing adaptors were ligated ontothe fragments, and the required fragments were purifiedby agarose gel electrophoresis and enriched by PCR ampli-fication to construct the cDNA library. The library wasloaded onto the channels of an Illumina HiSeq™ 2000instrument for 4 gigabase in-depth sequencing, which wasused to obtain more detailed information about geneexpression. Each paired-end library had an insert size of200–700 bp. The average read length of 90 bp was gener-ated as raw data. The data sets are available at the NCBISRA database with the accession number of SRA050454.
De novo assembly and sequence clusteringThe clean reads were obtained from raw data by filteringout adaptor-only reads, reads containing more than 5%unknown nucleotides, and low-quality reads (reads con-taining more than 50% bases with Q-value ≤ 20). Then denovo assembly of the clean reads was performed to gen-erate non-redundant unigenes. We used two methodsfor de novo assembly; SOAPdenovo 63mer-V1.05 [36]with optimized k-mer length of 41, and the Trinitymethod [19] with optimized k-mer length of 25.Sequence directions of the resulting unigenes were
determined by performing BLASTX searches againstprotein databases, with the priority order of NR (non-redundant protein sequences in NCBI), Swiss-Prot, KyotoEncyclopedia of Genes and Genomes database (KEGG),and COG (E-value≤1e-5) if conflicting results wereobtained. ESTScan software [37] was also used to determinethe directions of sequences that were not aligned to thosein any of the databases mentioned above.The expression levels of unigenes were measured as the
number of clean reads mapped to its sequence. The num-ber of clean reads mapped to each annotated unigene wascalculated and then normalized to RPKM (reads per Kbper million reads) with ERANGE3.1 software [18] andadjusted by a normalized factor [38].
Functional categorization of unigenesThe unigenes assembled by the Trinity method that werelonger than 200 bp were annotated according to their

Table 2 Length and number distribution of the unigenesand contigs
Lengthrange (bp)
SOAPdenovo Trinity
Contig No. Unigene No. Contig No. Unigene No.
Xu et al. BMC Genomics 2012, 13:133 Page 3 of 9http://www.biomedcentral.com/1471-2164/13/133
sequence similarity to previously annotated genes. Weused sequence-based and domain alignments to comparesequences. Sequence-based alignments were performedagainst three public databases (NR, Swiss-Prot, andKEGG; significant thresholds of E-value ≤ 1e-5). Domain-based alignments were carried out against the COGdatabase at NCBI with a cut-off E-value of ≤1e-5.The resulting BLAST hits were processed by Blast2GO
software [39] to retrieve associated Gene Ontology (GO)terms describing biological processes, molecular functions,and cellular components [40]. By using specific gene iden-tifiers and accession numbers, Blast2GO produces GOannotations as well as corresponding enzyme commissionnumbers (EC) for sequences with an E-value ≤1e-5.KEGG mapping was used to determine the metabolic
pathways [41,42]. The sequences with corresponding ECsobtained from Blast2GO were mapped to the KEGG meta-bolic pathway database. To further enrich the pathway an-notation and to identify the BRITE functional hierarchies,sequences were also submitted to the KEGG AutomaticAnnotation Server (KAAS) [43], and the single-directionalbest hit information method was selected. KAAS annotatesevery submitted sequence with KEGG orthology (KO) iden-tifiers, which represents an orthologous group of genes dir-ectly linked to an object in the KEGG pathways and BRITEfunctional hierarchy [43,44]. Therefore, these methods in-corporate different types of relationships that exist in bio-logical systems (i.e. genetic and environmental informationprocessing, cellular processes, and organism systems).
ResultsTranscriptome sequencing, de novo assembly, andsequence analysisWe constructed a cDNA library of pooled RNA samplesto generate a transcriptomic view of genes expressed inthe root of uninfected and CCN-infected Ae. variabilis.Approximately 4,687,311,420 base pairs of raw data weregenerated, yielding a total of 54,267,786 clean reads thatwere 90 bp in length (Table 1). Of the clean reads,91.63% had a Phred quality score of≤Q20 level (errorprobability of 0.01).All trimmed reads were de novo assembled by SOAP
denovo and Trinity programs (Table 1). SOAPdenovo
Table 1 Summary of de novo sequence assembly
Sequences(n)
Base pairs(Mbp)
Lengthrange (bp)
Meanlength (bp)
N50(bp)
Clean reads 52,081,238 4,687.31 90-90 90 90
SOAP contigs 336,641 60.21 60-3911 200 229
Trinity contigs 481,672 92.50 75-3696 192 250
SOAP unigenes 130,487 45.86 150-4113 351 392
Trinity unigenes 118,064 59.09 200-4214 500 599
produced 336,641 contigs of 60 to 3,911 bp with an aver-age length of 200 bp and an N50 of 229 bp (i.e., 50% ofthe assembled bases were incorporated into contigs of229 bp or longer). The majority of the contigs wereshorter than 200 bp (71.97%), and 2,722 contigs (0.81%)were longer than 1,000 bp. Trinity generated 481,672contigs ranging from 75 to 3696 bp with an averagelength of 192 bp and an N50 of 250 bp. Similar to theSOAPdenovo assembly, most contigs were shorter than200 bp (79.65%) but there was a greater number oflonger contigs—11,394 contigs (2.37%) were longer than1000 bp. The size distribution of these contigs is shownin Table 2. A total of 130,487 unigenes were further gen-erated by SOAPdenovo. The unigenes had an averagelength of 351 bp and an N50 of 392 bp. Among the uni-genes, 37,828 (28.99%) were shorter than 200 bp and4,702 (3.60%) were longer than 1,000 bp. The Trinitymethod generated fewer unigenes (118,064). These uni-genes had an average length of 500 bp and an N50 of599 bp. Among the unigenes, 64,330 unigenes (54.49%)were 200 to 400 bp in length. There were no unigenesshorter than 200 bp and 9.00% (10,622) of all generatedunigenes were longer than 1,000 bp (Table 2).To assess the quality of the data set, we evaluated the
assembled unigenes to determine the presence and lengthof gaps in the sequences. The analysis showed that 1.56%of the unigenes assembled by SOAPdenovo containedgaps, whereas those assembled by Trinity contained nogaps (Figure 1a).Because there is no transcriptome profile of Ae. varia-
bilis available for comparison, we used a web-based tool,ESTcal [45], to evaluate the depth and breadth of ourdata set. The read-depth coverage for 35.29% of SOAP-denovo-generated unigenes and for 22.79% of Trinity-generated unigenes was greater than 20 fold (Figure 1b),with an average read-depth coverage of 33.54-fold and33.25-fold, respectively.
<200 242,275 37,828 383,651 0
200-299 51,635 42,043 41,094 41,031
300-399 19,366 19,062 18,886 23,299
400-499 9,163 10,457 9,454 13,917
500-999 11,480 16,395 17,193 29,195
1000-1999 2,497 4,120 8,913 10,027
2000-3000 208 505 1969 572
>3000 17 77 512 23
Total 336,641 130,487 481,672 118,064
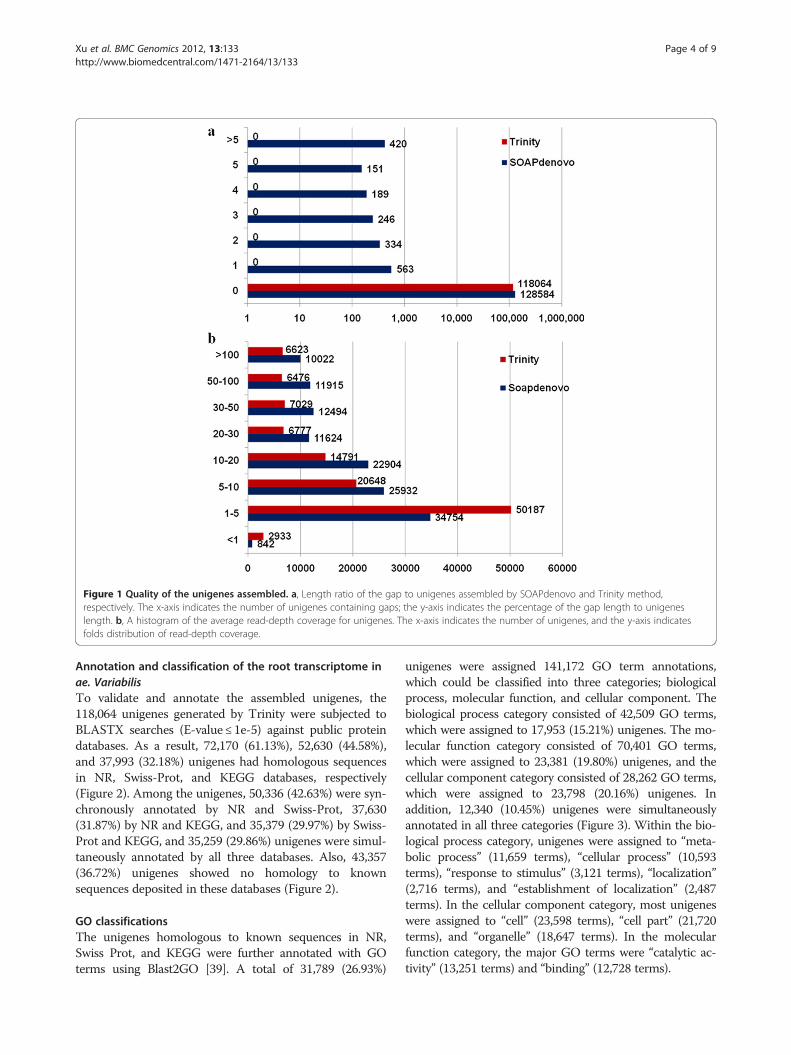
Figure 1 Quality of the unigenes assembled. a, Length ratio of the gap to unigenes assembled by SOAPdenovo and Trinity method,respectively. The x-axis indicates the number of unigenes containing gaps; the y-axis indicates the percentage of the gap length to unigeneslength. b, A histogram of the average read-depth coverage for unigenes. The x-axis indicates the number of unigenes, and the y-axis indicatesfolds distribution of read-depth coverage.
Xu et al. BMC Genomics 2012, 13:133 Page 4 of 9http://www.biomedcentral.com/1471-2164/13/133
Annotation and classification of the root transcriptome inae. VariabilisTo validate and annotate the assembled unigenes, the118,064 unigenes generated by Trinity were subjected toBLASTX searches (E-value≤ 1e-5) against public proteindatabases. As a result, 72,170 (61.13%), 52,630 (44.58%),and 37,993 (32.18%) unigenes had homologous sequencesin NR, Swiss-Prot, and KEGG databases, respectively(Figure 2). Among the unigenes, 50,336 (42.63%) were syn-chronously annotated by NR and Swiss-Prot, 37,630(31.87%) by NR and KEGG, and 35,379 (29.97%) by Swiss-Prot and KEGG, and 35,259 (29.86%) unigenes were simul-taneously annotated by all three databases. Also, 43,357(36.72%) unigenes showed no homology to knownsequences deposited in these databases (Figure 2).
GO classificationsThe unigenes homologous to known sequences in NR,Swiss Prot, and KEGG were further annotated with GOterms using Blast2GO [39]. A total of 31,789 (26.93%)
unigenes were assigned 141,172 GO term annotations,which could be classified into three categories; biologicalprocess, molecular function, and cellular component. Thebiological process category consisted of 42,509 GO terms,which were assigned to 17,953 (15.21%) unigenes. The mo-lecular function category consisted of 70,401 GO terms,which were assigned to 23,381 (19.80%) unigenes, and thecellular component category consisted of 28,262 GO terms,which were assigned to 23,798 (20.16%) unigenes. Inaddition, 12,340 (10.45%) unigenes were simultaneouslyannotated in all three categories (Figure 3). Within the bio-logical process category, unigenes were assigned to “meta-bolic process” (11,659 terms), “cellular process” (10,593terms), “response to stimulus” (3,121 terms), “localization”(2,716 terms), and “establishment of localization” (2,487terms). In the cellular component category, most unigeneswere assigned to “cell” (23,598 terms), “cell part” (21,720terms), and “organelle” (18,647 terms). In the molecularfunction category, the major GO terms were “catalytic ac-tivity” (13,251 terms) and “binding” (12,728 terms).

Figure 2 Detection of Homologous genes in public databases.The numbers of annotated and unmapped unigenes were indicatedin the ellipses, respectively.
Xu et al. BMC Genomics 2012, 13:133 Page 5 of 9http://www.biomedcentral.com/1471-2164/13/133
The five subcategories, “response to stimulus”, “death”,“immune system process”, “cell killing” and “antioxidant ac-tivity”, are all involved in resistance-related biological pro-cesses in the responses to abiotic and biotic stimulus/stress,based on their function explanations (Additional file 2).
Figure 3 GO annotations of unigenes by Blast2GO. The x-axis indicatedand the unigene numbers assigned with same GO terms were indicated on
KEGG pathway mappingTo identify biological pathways activated in the root of Ae.variabilis, the assembled unigenes were annotated withEnzyme Commission (EC) numbers from BLASTX align-ments against the KEGG database (E-value≤ 1e-5). Theassigned EC numbers were subsequently mapped to thereference canonical pathways. As a result, 37,993 unigenes(32.18% of 118,064) matched 57,975 members involved in119 KEGG pathways (Additional file 3). Of the 37,993unigenes, 9,596 were related to metabolic pathways, 4,815to biosynthesis of secondary metabolites, 3,355 to spliceo-some, 3,216 to plant-pathogen interaction, and 2,275 toribosome.Furthermore, 3,798 (including 3,712 individual unigenes)
of the 57,975 members were sorted into the plant immuneresponse pathways category, which includes plant-pathogeninteraction, phosphatidylinositol signaling system, and ABCtransporters (Additional file 3). These pathways are closelyrelated to plant defense against biotic/abiotic stress.
COG classificationAll assembled unigenes were further annotated based onCOG category [46]. A total of 28,126 unigenes wereassigned 64,441 functional annotations, which could begrouped into 25 functional categories (Figure 4). The lar-gest category was “General function prediction only”(7,888 COG annotations, 12.24% of 64,441). Approxi-mately 36.5% of the COG categories were associated with
the sub-categories and the y-axis indicates the number of unigenes,the top of the rectangle bars.
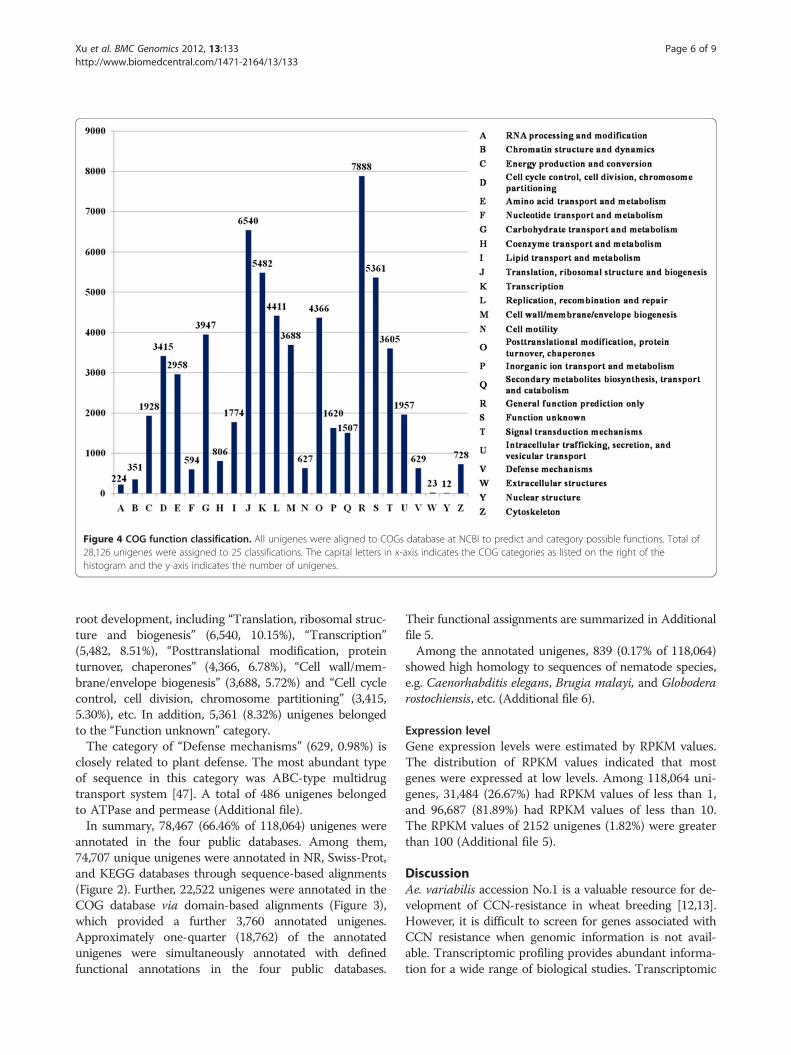
Figure 4 COG function classification. All unigenes were aligned to COGs database at NCBI to predict and category possible functions. Total of28,126 unigenes were assigned to 25 classifications. The capital letters in x-axis indicates the COG categories as listed on the right of thehistogram and the y-axis indicates the number of unigenes.
Xu et al. BMC Genomics 2012, 13:133 Page 6 of 9http://www.biomedcentral.com/1471-2164/13/133
root development, including “Translation, ribosomal struc-ture and biogenesis” (6,540, 10.15%), “Transcription”(5,482, 8.51%), “Posttranslational modification, proteinturnover, chaperones” (4,366, 6.78%), “Cell wall/mem-brane/envelope biogenesis” (3,688, 5.72%) and “Cell cyclecontrol, cell division, chromosome partitioning” (3,415,5.30%), etc. In addition, 5,361 (8.32%) unigenes belongedto the “Function unknown” category.The category of “Defense mechanisms” (629, 0.98%) is
closely related to plant defense. The most abundant typeof sequence in this category was ABC-type multidrugtransport system [47]. A total of 486 unigenes belongedto ATPase and permease (Additional file).In summary, 78,467 (66.46% of 118,064) unigenes were
annotated in the four public databases. Among them,74,707 unique unigenes were annotated in NR, Swiss-Prot,and KEGG databases through sequence-based alignments(Figure 2). Further, 22,522 unigenes were annotated in theCOG database via domain-based alignments (Figure 3),which provided a further 3,760 annotated unigenes.Approximately one-quarter (18,762) of the annotatedunigenes were simultaneously annotated with definedfunctional annotations in the four public databases.
Their functional assignments are summarized in Additionalfile 5.Among the annotated unigenes, 839 (0.17% of 118,064)
showed high homology to sequences of nematode species,e.g. Caenorhabditis elegans, Brugia malayi, and Globoderarostochiensis, etc. (Additional file 6).
Expression levelGene expression levels were estimated by RPKM values.The distribution of RPKM values indicated that mostgenes were expressed at low levels. Among 118,064 uni-genes, 31,484 (26.67%) had RPKM values of less than 1,and 96,687 (81.89%) had RPKM values of less than 10.The RPKM values of 2152 unigenes (1.82%) were greaterthan 100 (Additional file 5).
DiscussionAe. variabilis accession No.1 is a valuable resource for de-velopment of CCN-resistance in wheat breeding [12,13].However, it is difficult to screen for genes associated withCCN resistance when genomic information is not avail-able. Transcriptomic profiling provides abundant informa-tion for a wide range of biological studies. Transcriptomic

Xu et al. BMC Genomics 2012, 13:133 Page 7 of 9http://www.biomedcentral.com/1471-2164/13/133
data gives fundamental insights into biological processes.It can reveal gene expression profiles after experimentaltreatments or infection, and analyses of conserved ortholo-gous genes can be used for phylogenomic purposes, etc.[48]. Here, we used high-throughput deep sequencingtechnology to profile the root transcriptome of Ae. variabi-lis using the Illumina HiSeq™ 2000 platform. To the bestof our knowledge, this is the first report on this subject forAe. variabilis. The cDNA library was constructed usingpooled RNA samples from CCN-infected and non-infectedplants at three time points. This maximized the number ofexpressed transcripts included in the analysis, especiallythose related to CCN resistance.Accurate sequencing and reliable read assembly are
essential for downstream applications of transcriptomedata [49]. In this study, we used two popular assemblers,SOAPdenovo and Trinity, for de novo assembly of thetranscriptomic data of Ae. variabilis. The SOAPdenovoprogram has been widely used in many studies [25,50],while the Trinity method is a newly developed tool. Trin-ity was reported to recover more full-length transcriptsacross a broad range of expression levels, and to providea unified, sensitive solution for transcriptome recon-struction in species without a reference genome, similarto methods that rely on genome alignments [19]. The twomethods showed similar average read-depth coveragevalues. SOAPdenovo produced more unigenes than Trin-ity; however, many of the sequences assembled by SOAP-denovo were shorter than 200 bp (37,828 out of 130,487).On the other hand, Trinity generated 118,064 unigenes,the unigenes did not contain gaps, and the average unigenelength was nearly twice that of those produced by SOAP-denovo (mean length of 599 bp using Trinity, 351 bp usingSOAPdenovo). Therefore, Trinity was a better approachthan SOAPdenovo for assembly in this research.The Roche 454 GS FLX platform produces long reads
(>400 bp), whereas the Illumina sequencer generates morereads with a shorter length (90 bp). In this study, however,most of the assembled unigenes (130,487 from SOAPdenovo (≥150 bp) or 118,064 from Trinity (≥200 bp))achieved a higher coverage of ~33×. This indicates thatshort-read sequencing combined with an in-depth sequen-cing strategy and an effective assembly tool is an appropri-ate strategy to analyze transcriptome profiles.Compared with other transcriptome studies, the length
distribution of the 130,487 and 118,064 unigenes gener-ated in this work tended towards shorter-length reads.There are several possible explanations for this. First, Ae.variabilis (2n = 4x = 28, UUSvSv) is an allotetraploid spe-cies of the tribe Titiceae and it has an enormouslyexpanded repeated genome. This may present a substan-tial barrier to assembling short unigenes into long onesusing current and upcoming sequencing technology[51,52]. Second, the total RNA for sequencing in our
work was pooled from six samples, which may negativelyaffect read assembly [53]. The high dynamic range ofmRNA expression is a problem for comprehensive denovo mRNA sequencing and assembly [50]. Third, highfrequencies of alternative splicing and fusion events mayhave restricted the assembly of short sequences intolonger ones [54,55]. Another important reason is thatmore than 80% of unigenes in this study were expressedat low levels. Therefore, there would be fewer reads cor-responding to these unigenes for sequencing and for usein sequence assembly. Even so, the de novo transcrip-tome of Ae. variabilis provided abundant unigene infor-mation without gaps in sequences. This genetic dataenriches the genomic resources for the tribe Titiceae.A total of 7,408 individual unigenes (6.27% of 118,064)
were associated with plant defense and resistance(Additional file 7). These unigenes could be classifiedinto five GO sub-categories, three pathways, and a COGfunction group. More attention should be paid to thethree pathways related to plant defense, which included3712 unigenes. In the “plant-pathogen interaction” path-way, unigenes were mainly involved with the hypersensi-tive response, cell wall reinforcement, stomatal closure,and defense-related gene induction (Additional file 8).In the “phosphatidylinositol signaling system” pathway,unigenes were mainly related to reactions involving phos-phatidylinositol and its derivatives (Additional file 9). Inthe “ABC transporters” pathway, unigenes were related toeukaryotic-type transporters only, such as the ABCA sub-family, ABCB subfamily, ABCC subfamily, ABCG subfam-ily, and other putative ABC transporters (Additional file 10).These pathways provide a starting point to explore thegenes related to CCN resistance and to understand its mo-lecular mechanism.Interestingly, 839 unigenes showed high homology to
genes from nematode species (Additional file 6), probablybecause the root had been invaded by CCNs. As there is nogenomic information available for CCN, we cannot thor-oughly filter sequences of H. avenae genes from the tran-scriptome database. However, the detection of CCNunigenes confirmed that the method used for CCN inocula-tion was successful. More importantly, these unigenes rep-resent those expressed during the interaction with aresistant host. Therefore, this experimental system and theunigene dataset obtained from it build a platform for com-bining genetic, genomic, and expression information on theinteraction between CCN and its host in future studies [56].
ConclusionsThis is the first report of transcriptome profiling of Ae. var-iabilis using high-throughput deep sequencing technology.The sequencing was at a depth of 4.69 gigabase pairs. Atotal of 118,064 unigenes were assembled and 78,467 uni-genes were functionally annotated. By including RNA

Xu et al. BMC Genomics 2012, 13:133 Page 8 of 9http://www.biomedcentral.com/1471-2164/13/133
samples from CCN-infected plants, the dataset shown heremay reveal important information about gene expressionrelated to the plant response to, and defense against, CCNinvasion. Consequently, the large number of transcriptomicsequences and their functional annotations will provide suf-ficient information to discover novel genes and to explorethe molecular mechanism of CCN resistance in Ae. varia-bilis. Therefore, the results of this study will be useful forimproving CCN resistance in wheat breeding programs.
Additional files
Additional file 1: Roots invaded by CCN. A prep-experimentconfirmed the CCN J2 could parasitize plant root effectively before RNAextraction. Few hours later after the CCN inoculation, one nematode wasdetected being piercing root epidermis (Figure S1a). Utilizing themicroscope, one CCN J2 was found invading into a root tip of plant,already (Figure S1b; part of the CCN cover was removed).
Additional file 2: Resistance related unigenes from GO classificationCCN.
Additional file 3: KEGG pathway mapping.
Additional file 4: Defending unigenes from COG alignment.
Additional file 5: Unigene annotations in public databases.
Additional file 6: Nematode-like unigenes list in the transcriptomedatabase.Additional file 7: Resistance candidate unigenes in this study.
Additional file 8: Unigenes involved in plant-pathogen interactionpathway.
Additional file 9: Unigenes involved in phosphatidy linositolsignaling system pathway.
Additional file 10: Unigenes involved in ABC transporters pathway.
Competing interestsThe authors declare that they have no competing interests.
AcknowledgementsWe thank Associate Prof. Feng Liu of ShanDong Agricultural University forCCN supply, Associate Prof. Wen-Kun Huang of Chinese Academy ofAgricultural Sciences for technique aiding in CCN inoculation, Prof. Xiang-Zhen Li, CIB of CAS, for kindly suggestions on the manuscript. We also thankfor the patience of Editor Board and constructive comments of theanonymous reviewers to improve our manuscript. This work was supportedby the National Natural Science Foundation of China (30971903) and theNational S&T Key Project of China on GMO Cultivation for New Varieties(2008ZX08009-003).
Author details1Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu,Sichuan, China. 2Graduate University of the Chinese Academy of Sciences,Beijing, China.
Authors’ contributionsMQY and HL conceived this study. DLX and HL designed the experimentalplan, drafted and revised the manuscript. DLX, HL, JJL, JZ, XC, JLL, ZFP andGBD participated in sample collection, RNA preparation, performedexperiments, analyzed and interpreted the sequence data. All authors readand approved the final manuscript.
Received: 15 August 2011 Accepted: 11 April 2012Published: 11 April 2012
References1. Holgado R, Andersson S, Magnusson C: Management of cereal cyst
nematodes, Heterodera spp., in norway. Comm Appl Biol Sci GhentUniversity 2006, 71(3a):7.
2. Williamson V, Kumar A: Nematode resistance in plants: the battleunderground. Trends in Genet 2006, 22(7):396–403.
3. Sharma SN, Sain RS, Mathur BN, Sharma GL, Bhatnagar VK, Singh H, MidhaRL: Development and validation of the first cereal cyst nematode(Heterodera avenae) resistant wheat CCNRV 1 for northern India. SABRAOJ Breeding and Genet 2007, 39(1):1–16.
4. Bonfil DJ, Dolgin B, Mufradi I, Asido S: Bioassay to Forecast Cereal CystNematode Damage to Wheat in Fields. Precis Agric 2004, 5(4):329–344.
5. Nicol JM, Rivoal R: Global Knowledge And Its Application For TheIntegrated Control And Management Of Nematodes On Wheat. InIntegrated Management and Biocontrol of Vegetable and Grain CropsNematodes. vol. 2nd edition. Edited by Ciancio A, Mukerji KG.: SpringerNetherlands; 2007:251–294.
6. McCarter J: Molecular Approaches Toward Resistance to Plant-ParasiticNematodes. In Cell Biology of Plant Nematode Parasitism. vol. 15th edition.Edited by Berg R, Taylor C.: Springer Berlin/Heidelberg; 2009:239–267.
7. Smiley RW, Nicol JM: Nematodes which Challenge Global WheatProduction. In: Wheat Science and Trade. Wiley-Blackwell; 2009:171–187.
8. Bakker E, Dees R, Bakker J, Goverse A: Mechanisms Involved in PlantResistance to Nematodes. In: Multigenic and Induced Systemic Resistance inPlants. Edited by Tuzun S, Bent E: Springer US; 2006: 314–334.
9. Coriton O, Barloy D, Huteau V, Lemoine J, Tanguy A-M, Jahier J: Assignmentof Aegilops variabilis Eig chromosomes and translocations carryingresistance to nematodes in wheat. Genome 2009, 52(4):338–346.
10. Spetsov P, Mingeot D, Jacquemin JM, Samardjieva K, Marinova E: Transfer ofpowdery mildew resistance from Aegilops variabilis into bread wheat.Euphytica 1997, 93(1):49–54.
11. Mujeeb-Kaz A, Gul A, Farooq M, Rizwan S, Mirza JI: Genetic diversity ofAegilops variabilis (2n = 4x = 28; UUSS) for wheat improvement: morpho-cytogenetic characterization of some derived amphiploids and theirpractical significance. Pak J Bot 2007, 39(1):57–66.
12. Person-Dedryver F, Jahier J: Cereals as hosts of Meloidogyne naasiFranklin. III. Investigations into the level of resistance of wheat relatives.Agronomie 1985, 5(7):6.
13. Barloy D, Lemoine J, Abelard P, Tanguy A, Rivoal R, Jahier J: Marker-assistedpyramiding of two cereal cyst nematode resistance genes from Aegilopsvariabilis in wheat. Mol Breed 2007, 20(1):31–40.
14. Kilian B, Mammen K, Millet E, Sharma R, Graner A, Salamini F, Hammer K,Özkan H: Aegilops. In: Wild Crop Relatives: Genomic and Breeding Resources.Edited by Kole C: Springer Berlin Heidelberg; 2011: 1–76.
15. González-Ballester D, Casero D, Cokus S, Pellegrini M, Merchant SS,Grossman AR: RNA-Seq Analysis of Sulfur-Deprived Chlamydomonas CellsReveals Aspects of Acclimation Critical for Cell Survival. Plant Cell 2010, 22(6):2058-2084
16. Lee S, Seo CH, Lim B, Yang JO, Oh J, Kim M, Lee S, Lee B, Kang C, Lee S:Accurate quantification of transcriptome from RNA-Seq data by effectivelength normalization. Nucleic Acids Res 2011, 39(2):e9.
17. Wang Z, Gerstein M, Snyder M: RNA-Seq: a revolutionary tool fortranscriptomics. Nat Rev Genet 2009, 10(1):57–63.
18. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B: Mapping andquantifying mammalian transcriptomes by RNA-Seq. Nat Meth 2008, 5(7):621–628.
19. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, AdiconisX, Fan L, Raychowdhury R, Zeng Q et al: Full-length transcriptomeassembly from RNA-Seq data without a reference genome. Nat Biotechnol2011, 29:644-652.
20. Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, Hanski I,Marden JH: Rapid transcriptome characterization for a nonmodelorganism using 454 pyrosequencing. Mol Ecol 2008,17(7):1636–1647.
21. Meyer E, Aglyamova G, Wang S, Buchanan-Carter J, Abrego D, Colbourne J,Willis B, Matz M: Sequencing and de novo analysis of a coral larvaltranscriptome using 454 GSFlx. BMC Genomics 2009, 10(1):219.
22. Kristiansson E, Asker N, Forlin L, Larsson DJ: Characterization of the Zoarcesviviparus liver transcriptome using massively parallel pyrosequencing.BMC Genomics 2009, 10(1):345.
23. Graham IA, Besser K, Blumer S, Branigan CA, Czechowski T, Elias L,Guterman I, Harvey D, Isaac PG, Khan AM, et al: The Genetic Map ofArtemisia annua L. Identifies Loci Affecting Yield of the AntimalarialDrug Artemisinin. Science 2010, 327(5963):328–331.
24. Wang Z, Fang B, Chen J, Zhang X, Luo Z, Huang L, Chen X, Li Y: De novoassembly and characterization of root transcriptome using Illumina

Xu et al. BMC Genomics 2012, 13:133 Page 9 of 9http://www.biomedcentral.com/1471-2164/13/133
paired-end sequencing and development of cSSR markers insweetpotato (Ipomoea batatas). BMC Genomics 2010, 11(1):726.
25. Shi C-Y, Yang H, Wei C-L, Yu O, Zhang Z-Z, Jiang C-J, Sun J, Li Y-Y, Chen Q,Xia T, et al: Deep sequencing of the Camellia sinensis transcriptomerevealed candidate genes for major metabolic pathways of tea-specificcompounds. BMC Genomics 2011, 12(1):131.
26. Natarajan P, Parani M: De novo assembly and transcriptome analysis offive major tissues of Jatropha curcas L using GS FLX titanium platform of454 pyrosequencing. BMC Genomics 2011, 12(1):191.
27. Seah S, Miller C, Sivasithamparam K, Lagudah ES: Root responses to cerealcyst nematode (Heterodera avenae) in hosts with different resistancegenes. New Phytol 2000, 146(3):527–533.
28. Simonetti E, Alba E, Montes M, Delibes Á, López-Braña I: Analysis ofascorbate peroxidase genes expressed in resistant and susceptiblewheat lines infected by the cereal cyst nematode. Heterodera avenae PlantCell Rep 2010, 29(10):1169–1178.
29. Andres MF, Melillo MT, Delibes A, Romero MD, Bleve-Zacheo T: Changes inwheat root enzymes correlated with resistance to cereal cyst nematodes.New Phytol 2001, 152(2):343–354.
30. Mazarei M, Liu W, Al-Ahmad H, Arelli P, Pantalone V, Stewart C: Geneexpression profiling of resistant and susceptible soybean lines infectedwith soybean cyst nematode. TAG Theoretical and Applied Genetics 2011,123(7):1193–1206.
31. Williamson VM, Hussey RS: Nematode pathogenesis and resistance inplants. The Plant Cell 1996, 8:11.
32. Das S, Ehlers JD, Close TJ, Roberts PA: Transcriptional profiling of root-knotnematode induced feeding sites in cowpea (Vigna unguiculata L. Walp)using a soybean genome array. BMC Genomics 2010, 11(1):480.
33. Peng X, Wood C, Blalock E, Chen K, Landfield P, Stromberg A: Statisticalimplications of pooling RNA samples for microarray experiments. BMCBioinforma 2003, 4(1):26.
34. Liu S, Lin L, Jiang P, Wang D, Xing Y: A comparison of RNA-Seq andhigh-density exon array for detecting differential gene expressionbetween closely related species. Nucleic Acids Res 2010, 39(2):578–588.
35. Everett MV, Grau ED, Seeb JE: Short reads and nonmodel species:exploring the complexities of next-generation sequence assembly andSNP discovery in the absence of a reference genome. Mol Ecol Resour2011, 11:93–108.
36. Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, etal: De novo assembly of human genomes with massively parallel shortread sequencing. Genome Res 2009, 20(2):265–272.
37. Iseli C, Jongeneel CV, Bucher P: ESTScan: a program for detecting,evaluating, and reconstructing potential coding regions in ESTsequences. Proceedings/International Conference on Intelligent Systems forMolecular Biology; ISMB International Conference on Intelligent Systems forMolecular Biology 1999:138–148.
38. Robinson M, Oshlack A: A scaling normalization method for differentialexpression analysis of RNA-seq data. Genome Biol 2010, 11(3):R25.
39. Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M: Blast2GO: auniversal tool for annotation, visualization and analysis in functionalgenomics research. Bioinformatics 2005, 21(18):3674–3676.
40. Botstein D, Cherry JM, Ashburner M, Ball CA, Blake JA, Butler H, Davis AP,Dolinski K, Dwight SS, Eppig JT, et al: Gene Ontology: tool for theunification of biology. Nat Genet 2000, 25(1):25–29.
41. Kanehisa M, Goto S: KEGG: Kyoto Encyclopedia of Genes and Genomes.Nucleic Acids Res 2000, 28(1):27–30.
42. Rismani-Yazdi H, Haznedaroglu B, Bibby K, Peccia J: Transcriptomesequencing and annotation of the microalgae Dunaliella tertiolecta:Pathway description and gene discovery for production of next-generation biofuels. BMC Genomics 2011, 12(1):148.
43. Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M: KAAS: an automaticgenome annotation and pathway reconstruction server. Nucleic Acids Res2007, 35(suppl 2):W182–W185.
44. Mao X, Cai T, Olyarchuk JG, Wei L: Automated genome annotation andpathway identification using the KEGG Orthology (KO) as a controlledvocabulary. Bioinformatics 2005, 21(19):3787–3793.
45. Wall PK, Leebens-Mack J, Chanderbali A, Barakat A, Wolcott E, Liang H,Landherr L, Tomsho L, Hu Y, Carlson J, et al: Comparison of nextgeneration sequencing technologies for transcriptome characterization.BMC Genomics 2009, 10(1):347.
46. Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS,Kiryutin B, Galperin MY, Fedorova ND, Koonin EV: Genetic diversity of
Aegilops variabilis for wheat improvement. Nucleic Acids Res 2001,29(1):7.
47. Peng Y, Abercrombie LLG, Yuan JS, Riggins CW, Sammons RD, Tranel PJ,Stewart CN: Characterization of the horseweed (Conyza canadensis)transcriptome using GS-FLX 454 pyrosequencing and its application forexpression analysis of candidate non-target herbicide resistance genes.Pest Manage Sci 2010, 66(10):1053–1062.
48. Surget-Groba Y, Montoya-Burgos JI: Optimization of de novotranscriptome assembly from next-generation sequencing data. GenomeRes 2010, 20(10):1432–1440.
49. Feldmeyer B, Wheat C, Krezdorn N, Rotter B, Pfenninger M: Short readIllumina data for the de novo assembly of a non-model snail speciestranscriptome (Radix balthica, Basommatophora, Pulmonata), and acomparison of assembler performance. BMC Genomics 2011, 12(1):317.
50. Adamidi C, Wang Y, Gruen D, Mastrobuoni G, You X, Tolle D, Dodt M,Mackowiak SD, Gogol-Doering A, Oenal P, et al: De novo assembly andvalidation of planaria transcriptome by massive parallel sequencing andshotgun proteomics. Genome Res 2011, 21(7):1193–1200.
51. Ness R, Siol M, Barrett S: De novo sequence assembly and characterizationof the floral transcriptome in cross- and self-fertilizing plants. BMCGenomics 2011, 12(1):298.
52. Mach J: On the Habits of Transposons: Dissociation Mapping in Maizeand Megabase Sequencing in Wheat Reveal Site Preferences,Distribution, and Evolutionary History. The Plant Cell Online 2010, 22(6):1650–1652.
53. Chen S, Yang P, Jiang F, Wei Y, Ma Z, Kang L: De novo analysis oftranscriptome dynamics in the migratory locust during the developmentof phase traits. PLoS One 2010, 5(12):e15633.
54. Asmann YW, Hossain A, Necela BM, Middha S, Kalari KR, Sun Z, Chai H-S,Williamson DW, Radisky D, Schroth GP et al: A novel bioinformaticspipeline for identification and characterization of fusion transcripts inbreast cancer and normal cell lines. Nucleic Acids Research 2011.
55. Maher CA, Palanisamy N, Brenner JC, Cao X, Kalyana-Sundaram S, Luo S,Khrebtukova I, Barrette TR, Grasso C, Yu J, et al: Chimeric transcriptdiscovery by paired-end transcriptome sequencing. Proc Natl Acad Sci2009, 106(30):12353–12358.
56. Jacob J, Mitreva M: Transcriptomes of Plant-Parasitic Nematodes. In:Genomics and Molecular Genetics of Plant-Nematode Interactions. Edited byJohn Jones GG, Carmen Fenoll: Springer Netherlands; 2011: 119–138.
doi:10.1186/1471-2164-13-133Cite this article as: Xu et al.: De novo assembly and characterization ofthe root transcriptome of Aegilops variabilis during an interaction withthe cereal cyst nematode. BMC Genomics 2012 13:133.
Submit your next manuscript to BioMed Centraland take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution
Submit your manuscript at www.biomedcentral.com/submit











![Comparative Transcriptome Profiling of Maize Coleoptilar · Comparative Transcriptome Profiling of Maize Coleoptilar Nodes during Shoot-Borne Root Initiation1[C][W][OPEN] Nils Muthreich2,](https://static.fdocuments.in/doc/165x107/5e4d3e829c5ec8732734cd8d/comparative-transcriptome-proiling-of-maize-comparative-transcriptome-proiling.jpg)






