
Hiperplasia Suprarrenal Congenita
32
UNIVERSIDAD AUTÓNOMA DE ZACATECAS ¨FRANCISCO GARCÍA SALINAS¨ AREA: CIENCIAS DE LA SALUD PROGRAMA: MEDICINA HUMANA UDI: PATOLOGIA CLINICA V TEMA: DIAGNOSTICO POR LABORATORIO DE HIPERPLASIA SUPRARRENAL CONGENITA DR. MARIA CALIXTA MARTINEZ VAZQUEZ ALUMNO: JHONNATAN SANDOVAL SANDOVAL ALAN BENJAMIN VILLA REVELES GRUPO: 9° F
-
Upload
jhonnatan-sandoval-sandoval -
Category
Documents
-
view
31 -
download
2
Transcript of Hiperplasia Suprarrenal Congenita

UNIVERSIDAD AUTÓNOMA DE ZACATECAS¨FRANCISCO GARCÍA SALINAS¨
AREA: CIENCIAS DE LA SALUD
PROGRAMA: MEDICINA HUMANA
UDI: PATOLOGIA CLINICA V
TEMA: DIAGNOSTICO POR LABORATORIO DE HIPERPLASIA SUPRARRENAL CONGENITA
DR. MARIA CALIXTA MARTINEZ VAZQUEZ
ALUMNO: JHONNATAN SANDOVAL SANDOVALALAN BENJAMIN VILLA REVELES
GRUPO: 9° F



Introducción
• Autosómica Recesiva• Deficiencia 21 Hidroxilasa 90%
• Forma Clásica - Perdedora de sal 75% - Virilizante simple 25% No Clásica 1-2:1.000 RN vivos Portación 1:60-80
1:15.000 RN vivos
Hh Hh
HH Hh Hh hh

Hiperplasia suprarrenal congénita
PATOGENIA
• Es una familia de trastornos autosómicos recesivos de la esteroidogénesis suprarrenal, en la que está alterada la actividad de las enzimas necesarias para la síntesis de cortisol.

Hiperplasia suprarrenal congénita: PATOGENIA

Clasificacion
HIPERPLASIA SUPRARRENAL
CONGÉNITA LIPOIDEA.
DEFICIENCIA DE 3β-HIDROXIESTEROIDE
DESHIDROGENASA/4,5 ISOMERASA.
DEFICIENCIA DE 17 α-HIDROXILASA/17,20
LIASA.
DEFICIENCIA DE 21-HIDROXILASA.
DEFICIENCIA DE 11β-HIDROXILASA

Deficiencia de 21-hidroxilasa.
• Es la causa más frecuente de HSC.• La incapacidad de 21-hidroxilar adecuadamente 17-
hidroxiprogesterona en 11-desoxicortisol, provocan: una deficiencia de cortisol y aldosterona.↑ de ACTHHiperplasia suprarrenal.↑de la secreción de andrógenos suprarrenales:
17-hidroxiprogesteronaAndrostendiona DHEA Testosterona

Deficiencia de 21-hidroxilasa

Etiologia genetica: cromosoma 6p21.3
CYP21P CYP21
CYP21P CYP21
P: brazo largopesudogen (CYP21P): enzima truncada gen activo (CYP21)

FormaClásica
FormaNO Clásica
FormaVirilizante
Simple
FormaPerdedora de Sal
Pubarquiaprematura
PubertadPrecozCentral
Acné
Clínicas de Presentación de deficiencia de 21 OHasa
Hirsutismo
Oligomenorrea
Neonato Lactante Escolar Adolescente Adulto
Expresión clínica
SudorApocrino
Talla Alta
INFERTILIDAD
Hipertrofia clitoris Fusion pliegues labioescrotales
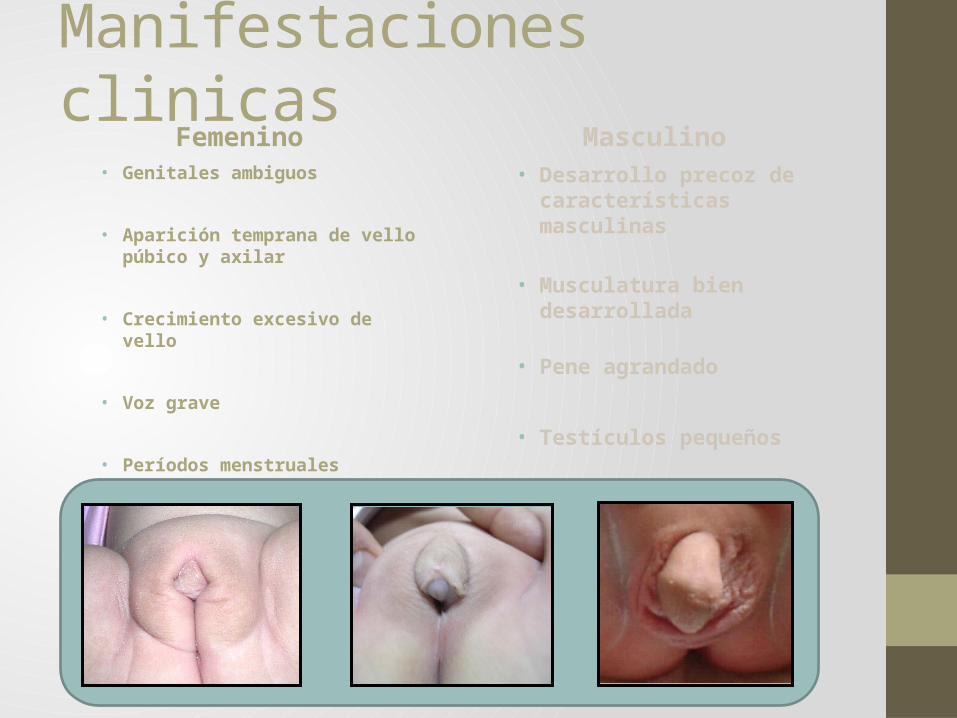
Manifestaciones clinicasFemenino Masculino
• Genitales ambiguos
• Aparición temprana de vello púbico y axilar
• Crecimiento excesivo de vello
• Voz grave
• Períodos menstruales anormales
• Ausencia de la menstruación
• Desarrollo precoz de características masculinas
• Musculatura bien desarrollada
• Pene agrandado
• Testículos pequeños
• Aparición temprana de vello púbico y axilar

Diagnóstico:17-OH Progesterona serica
• <35ng/dl• <20ng/dlValores Normales
• Hasta 100 ng/dlHSC clasica virilizante simple:
• hasta 1000ng/dlHSC clasica pierde sal:
Muestra: Sangre por Venopuncion

Test de ACTH
1000
100
10
1
Stimulated
17-OHP
ng/mL
1 10 100 1000
basal 17-OHP, ng/mL
Determinación basal de 17hidroxiprogesterona,
Administración de 250 μg/m2 de ACTH,
Determinación de 17-hidroxiprogesterona a
los 60 minutos.
• Diagnostico forma no clasica
• se recomienda valoresbasales superiores a 5ng/mL,
>15 ng/mL= HSC
10-15 ng/mL= repetir prueba

Estudio Genetico: Dx Definitivo

Electrolitos Sericos: (forma perdedora sal)
Hiponatremia: Na=135-145 mEq/l
Hiperkalemia: K= 3.5-5.1mEq/l
Acidosis metabolica: HCO3: 22-26mEq/l; PH:
7.35-7.45
Hipoglucemia
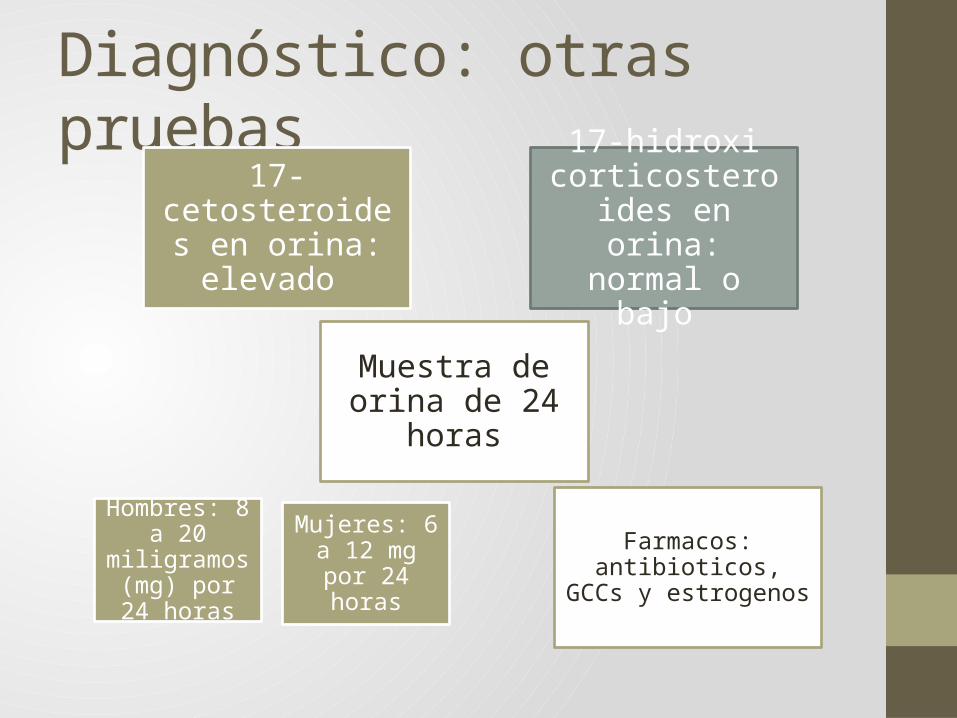
Diagnóstico: otras pruebas
17-cetosteroides en orina: elevado
17-hidroxi corticosteroides en
orina: normal o bajo
Muestra de orina de 24 horas
Hombres: 8 a 20 miligramos
(mg) por 24 horas
Mujeres: 6 a 12 mg por 24
horasFarmacos: antibioticos,
GCCs y estrogenos

Tamiz neonatal basico
Fenilcetonuria Hipotiroidismo congénito
Enfermedad de orina de jarabe de
arce o «maple»Homocistinuria
Galactosemia Hiperplasia suprarrenal congénita
Fibrosis quística.
Sangre capilar de
talon
Tarjeta de guthrie

Tamiz neonatal
17 OHP de sangre capilar
17 OHP > p95th
Para el peso de RN
17 OHP > p95th
Para el peso de RNNo más investigación
normal
normal
Re-llamado
Limites 17-hidroxiprogesterona totalBasada en el peso al nacer: > 3,000 g < de 17.3 ng/ml 2,500-3,000 g < de 22.71,500-2,500 g < de 27.3< 1,500 g < de 45.5

17 OHP > p95th
Para el peso de RN
TEST de ACTH
17 OHP< 15 ng/mL
17 OHP15-100 ng/mL
17 OHP>100 ng/mL
<3 ng/mLNo afectado
3-15 ng/mLHeterocigoto
HSR-NC
HSR-C


Diagnostico prenatal• Última fase del primer trimestre mediante el análisis del ADN
obtenido por biopsia de vellosidades coriónicas.
• Durante el segundo trimestre mediante amniocentesis.
• Analisis del gen CYP21P del brazo corto del cromosoma 6.PCR

CASO CLINICOHiperplasia Suprarrenal Congenita

• Se presenta el caso de un recién nacido con fecha de nacimiento e ingreso, al Servicio de Neonatología, del 3 de octubre del 2012 por presentar genitales ambiguos.
• Madre de 18 años de edad con escolaridad preparatoria, toxicomanías negadas y sin antecedentes heredofamiliares o personales patológicos de importancia. En unión libre, con menarca a los 14 años. Producto de la primera gesta, resuelto por cesárea debida a presentación pélvica en la semana 36 de gestación. Peso al nacer de 2,550 g, talla 46 cm, Apgar 99 en tiempos convencionales.
• A la exploración física se encontraron genitales ambiguos caracterizados por hipertrofia de clítoris, fusión labioescrotal y seno urogenital abajo del falo o clítoris. No se palparon testículos y el ano estaba permeable. El resto de la exploración se encontró sin anormalidades.
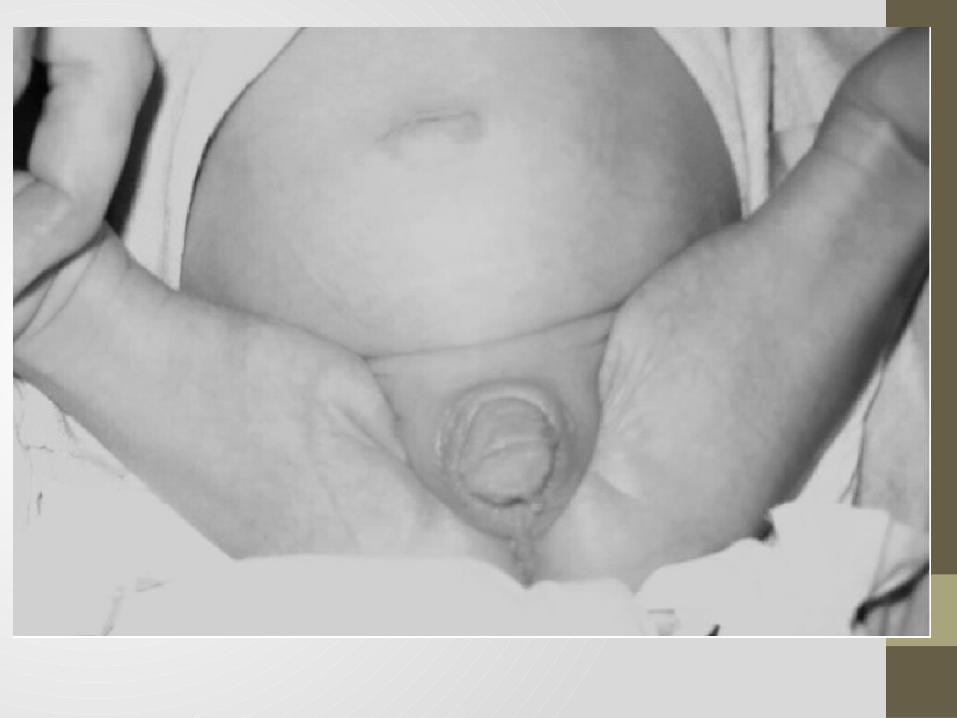

• Los estudiosde laboratorio dentro de valores de referencia . El ecosonograma pélvico no visualizó órganos sexuales internos femeninos. Cariotipo 46 XX. Egresó del servicio para continuar estudio en la consulta externa.
• El paciente reingresó a los 26 días de vida, al ser enviado del poblado Miguel Alemán, Hermosillo, con el diagnóstico de choque hipovolémico secundario a vómito y rechazo al alimento. A su llegada se le encontró con coloración pálida, hipoactivo, hiporreactivo, con presencia de signos universales de deshidratación, respiración de kussmaull y llenado capilar de 3 segundos. Recibió hidratación endovenosa con solución salina a razón de 50 ml/kg/hora más reanimación con presión positiva intermitente, con lo que mejoraron sus condiciones clínicas. El sodio sérico se reportó en 109 mEq/L, potasio 8.9 mEq/L, cloro 86 mEql/L, urea 154 mg/dL y creatinina 1.4 mg/dL. La biometría hemática con hemoglobinade 12.8 g/dL, hematocrito de 37 5%, leucocitos en 15,300 mm3, con 71% de neutrófilos y plaquetas de 580,000 mm3. La gasometría arterial con acidosis metabólica.

• El manejo posterior incluyó la administración endovenosa de solución glucoelectrolítica, corrección de acidosis con bicarbonato de sodio y asociación antimicrobiana de ampicilina/cefotaxima. El cuadro clínico se consideró como característico de crisis adrenal por lo que se agregó al manejo hidrocortisona endovenosa. El sodio se elevó a 129 mmol/L veinticuatro horas después y se inició mineralocorticoide (fludrocortisona) al tratamiento.
• En el servicio de Neonatología se confirmó el diagnóstico de hiperplasia suprarrenal con la determinación de 17OHP en cifras de 339 ng/ml. Fue dado de alta con normalización de electrolitos séricos y tratamiento por vía oral a base de hidrocortisona (1.2 mg cada 8 horas) y fludrocortisona (0.1 mg cada 24 horas).

ARTICULOSHiperplasia Suprarrenal Congenita

Confirmation of congenital adrenal hyperplasia by adrenalsteroid profiling of filter paper dried blood samples usingultra-performance liquid chromatography-tandem massspectrometry• Claudia Rossi1,2,*, Lisa Calton3, Heather A. Brown3, Scott Gillingwater3, A. Michael Wallace4, Francesca Petrucci1,2,
Domenico Ciavardelli1,5, Andrea Urbani6,7, Paolo Sacchetta1,2 and Michael Morris3• 1 Centre of Study on Aging (Ce.S.I.), ‘‘G. d’Annunzio’’ University Foundation, Chieti, Italy• 2 Department of Biomedical Science, ‘‘G. d’Annunzio University’’ Chieti-Pescara, Italy• 3 Clinical Operations Group, Waters Corporation, Atlas Park, Manchester, UK• 4 Department of Clinical Biochemistry, Glasgow Royal Infirmary, Glasgow, Scotland, UK• 5 Faculty of Motor and Health Sciences, Kore University of Enna, Enna, Italy• 6 Department of Internal Medicine,‘‘Tor Vergata’’ University, Rome, Italy• 7 IRCCS-Santa Lucia Foundation, Rome, Italy
Clin Chem Lab Med 2011;49(4):677–684
• AbstractBackground: The specificity of screening for congenital adrenal hyperplasia by direct measurement of 17-hydroxyprogesterone in filter paper dried blood spot samples by immunoassay is low and has a high false-positive rate. In order to reduce the false-positive rate of this test, we developed a rapid, robust, specific confirmatory procedure in which cortisol, 4-androstene-3,17-dione and 17-hydroxyprogesterone were measured simultaneously by ultra-performance liquid chromatography-tandem mass spectrometry.Methods: After extraction, samples were analysed by ultraperformance liquid chromatography-tandem mass spectrometry and 17-hydroxyprogesterone was quantified accurately. Other steroids were determined using stable deuterated internal standards. In total, 25 patient blood spot samples and 92 control samples were analysed.Results: The assay was linear for 17-hydroxyprogesterone, with a coefficient of determination )0.997 and imprecision F6.5%. An upper limit of normal for 17-hydroxyprogesterone of 4.45 nmol/L was established by analysing a cohort of samples from unaffected newborns. In addition, a cut-off of3.5 for the peak areas ratio (17-hydroxyprogesteroneq4- androstene-3,17-dione)/cortisol, allows confirmation of the affected steroidogenic enzyme.Conclusions: A high throughput method for the detection of steroids related to congenital adrenal hyperplasia has been developed, allowing the false-positive rate associated with screening for 17-hydroxyprogesterone by immunoassay to be determined.

Molecular testing in congenital adrenalhyperplasia due to 21 -α hydroxylase deficiencyin the era of newborn screening
Sarafoglou K, Lorentz CP, Otten N, Oetting WS, Grebe SKG. aDepartment of Pediatrics, and bCollege of Pharmacology, Institute of Human Genetics, University of Minnesota, Minneapolis, MN, USA, and cDepartment of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN, USA
Clin Genet 2012: 82: 64–70. © John Wiley & Sons A/S, 2011
AbstractNewborn screening (NBS) identifies the majority of classical [salt-wasting (SW) and simple-virilizing (SV)] cases of congenital adrenal hyperplasia (CAH) due to 21α-hydroxylase (21α-OHase) during the first days of life.Diagnosis of classical CAH is confirmed by follow-up serum 17-hydroxyprogesterone and/or the adrenocorticotropin stimulation test; however, neither test definitively distinguishes between the classical subtypes. After confirmation, all newborns are started on hydrocortisone (glucocorticoid) and fludrocortisone (mineralocorticoid) treatment. While initiating fludrocortisone treatment in classical CAH patients, independent of subtype and before SW signs or symptoms occur, prevents a life-threatening SW crisis, it may later complicate distinguishing between the classical subtypes. Genotype–phenotype correlations in 21α-Ohase deficiency are excellent; however, molecular testing is not a regular part of the diagnostic workup. Molecular testing on 39 patients (25 identified by NBS) with an already established diagnosis of CAH identified 11 SW patients (8 identified by NBS) whose mutations suggested further biochemical and clinical reassessment of their subtype. Overall, SW accounted for 57.6% of our classical CAH patients, below the generally accepted figure that >75% of classical CAH are comprised of the SW form. In the era of NBS, molecular testing is a valuable supplemental tool identifying patients who may benefit from reassessment of their salt-retaining ability.

Referencias bibliograficas
• Confirmation of congenital adrenal hyperplasia by adrenal steroid profiling of filter paper dried blood samples using ultra-performance liquid chromatography-tandem mass spectrometry Claudia Rossi1,2,*, Lisa Calton3, Heather A. Brown3 Clin Chem Lab Med 2011;49(4):677–684
• Hiperplasia suprarrenal congenita.L. Soriano Guillén, M. Velázquez de Cuéllar Paracchi. Unidad de Endocrinología Infantil. Servicio de Pediatría. Fundación Jiménez Díaz. Madrid. Pediatr Integral 2007;XI(7):601-610.
• Resultados del tamiz neonatal ampliado, como nueva estrategia para la prevención de los defectos al nacimiento.Antonio Velázquez,*,** Marcela Vela-Amieva,*,*** Edwin W Naylor,***Donald H Chace**** Velázquez A. y cols: Tamiz neonatal ampliado.• Rev Mex Pediatr 2000; 67(5); 206-213
• Hiperplasia Suprarrenal Congénita: Reporte de un caso clínico. Miguel A. MartínezMedina* Jorge I. HernándezBlanquel** Bol Clin Hosp Infant Edo Son 2007; 24(1): 3841

GRACIAS


















