
Biblografia Endocrino BQ
64
ENDOCRINOLOGY CONCEPTS FOR MEDICAL STUDENTS H. Maurice Goodman Department of Physiology, University of Massachusetts Medical School, Worcester, Massachusetts 01655 ADV PHYSIOL EDUC 25: 213–224, 2001. WHAT IS ENDOCRINOLOGY? The explosive growth of information in endocrinol- ogy made possible by unprecedented advances in technology and by expansion of the number of inves- tigators engaged in endocrinological research pre- sents a difficult and growing challenge to those of us who teach endocrine physiology to medical students. The scope of research has so extended the bound- aries of endocrinology and blurred the distinctions among disciplines that even defining endocrinology is problematic. Additionally, it has become increasingly difficult to decide what should be taught to first-year medical students and in what context. Regulation of cellular functions by hormones represents only a sub- set of the larger field of chemical communication that includes aspects of neurobiology, cell biology, immu- nology, and developmental biology. From the per- spective of the target cell, what we call a hormone is quite arbitrary. Cellular and molecular processes as- sociated with production, secretion, and actions of hormones are no different from actions of hundreds of other paracrine and autocrine factors, immuno- modulators, neurotransmitters, growth factors, and so forth. The exquisite sensitivity of molecular bi- ological tools has uncovered hormone production and hormone receptors in the most unexpected places. A host of nonendocrine tissues produce some of the same molecules secreted by the classic endocrine glands and use them to serve as local paracrine modulators or neurotransmitters. It is now apparent that hormones act on many more cells than their classically defined targets and that virtually every tissue in the body participates in some endocrine function. Despite all of the above, the endocrine system re- mains a vital component of any course in medical physiology, and mastery of the principles and phe- nomena it encompasses is essential for later study of clinical medicine. The role of the endocrine system is to coordinate and integrate cellular activity within the whole body, regulating cellular and organ function from a distance, with factors produced locally, often in response to hormones, governing local fine tuning. From this perspective, it becomes more logical to focus on the physiological processes that are gov- erned by the endocrine system rather than the classi- cal morphologically based gland by gland survey, al- though students need to master basic facts connected with each gland and hormone. We can regard the endocrine system as having the following physiolog- ical missions ● Regulation of sodium and water balance: preserva- tion of the volume/pressure reservoir required for tissue perfusion ● Regulation of calcium balance: preservation of ex- tracellular fluid concentrations required for mem- brane integrity, intracellular signaling, hemostasis, etc., and preservation of skeletal integrity ● Regulation of energy balance: preserving, access- ing, and interconverting metabolic fuels to meet cellular energy demands ● Coordination of processes for coping with a hostile environment ● Coordination of growth and development A P S R E F R E S H E R C O U R S E R E P O R T 1043 - 4046 / 01 – $5.00 – COPYRIGHT © 2001 THE AMERICAN PHYSIOLOGICAL SOCIETY VOLUME 25 : NUMBER 4 – ADVANCES IN PHYSIOLOGY EDUCATION – DECEMBER 2001 213
-
Upload
muriel-nunez -
Category
Documents
-
view
34 -
download
2
Transcript of Biblografia Endocrino BQ

ENDOCRINOLOGY CONCEPTS
FOR MEDICAL STUDENTS
H. Maurice Goodman
Department of Physiology, University of Massachusetts Medical School, Worcester, Massachusetts 01655
ADV PHYSIOL EDUC 25: 213–224, 2001.
WHAT IS ENDOCRINOLOGY?
The explosive growth of information in endocrinol-
ogy made possible by unprecedented advances in
technology and by expansion of the number of inves-
tigators engaged in endocrinological research pre-
sents a difficult and growing challenge to those of us
who teach endocrine physiology to medical students.
The scope of research has so extended the bound-
aries of endocrinology and blurred the distinctions
among disciplines that even defining endocrinology is
problematic. Additionally, it has become increasingly
difficult to decide what should be taught to first-year
medical students and in what context. Regulation of
cellular functions by hormones represents only a sub-
set of the larger field of chemical communication that
includes aspects of neurobiology, cell biology, immu-
nology, and developmental biology. From the per-
spective of the target cell, what we call a hormone is
quite arbitrary. Cellular and molecular processes as-
sociated with production, secretion, and actions of
hormones are no different from actions of hundreds
of other paracrine and autocrine factors, immuno-
modulators, neurotransmitters, growth factors, and
so forth. The exquisite sensitivity of molecular bi-
ological tools has uncovered hormone production
and hormone receptors in the most unexpected
places. A host of nonendocrine tissues produce
some of the same molecules secreted by the classic
endocrine glands and use them to serve as local
paracrine modulators or neurotransmitters. It is
now apparent that hormones act on many more
cells than their classically defined targets and that
virtually every tissue in the body participates in
some endocrine function.
Despite all of the above, the endocrine system re-mains a vital component of any course in medicalphysiology, and mastery of the principles and phe-nomena it encompasses is essential for later study ofclinical medicine. The role of the endocrine system isto coordinate and integrate cellular activity within thewhole body, regulating cellular and organ functionfrom a distance, with factors produced locally, oftenin response to hormones, governing local fine tuning.From this perspective, it becomes more logical tofocus on the physiological processes that are gov-erned by the endocrine system rather than the classi-cal morphologically based gland by gland survey, al-though students need to master basic facts connectedwith each gland and hormone. We can regard theendocrine system as having the following physiolog-ical missions
● Regulation of sodium and water balance: preserva-tion of the volume/pressure reservoir required fortissue perfusion
● Regulation of calcium balance: preservation of ex-tracellular fluid concentrations required for mem-brane integrity, intracellular signaling, hemostasis,etc., and preservation of skeletal integrity
● Regulation of energy balance: preserving, access-ing, and interconverting metabolic fuels to meetcellular energy demands
● Coordination of processes for coping with a hostileenvironment
● Coordination of growth and development
A P S R E F R E S H E R C O U R S E R E P O R T
1043 - 4046 / 01 – $5.00 – COPYRIGHT © 2001 THE AMERICAN PHYSIOLOGICAL SOCIETY
VOLUME 25 : NUMBER 4 – ADVANCES IN PHYSIOLOGY EDUCATION – DECEMBER 2001
213

● Coordination of processes associated with repro-duction and lactation
From this perspective, it is clear that at least someaspect of virtually every physiological system lieswithin the realm of endocrine control. No single hor-mone or endocrine gland can accomplish any of thesemissions alone, and virtually every hormone partici-pates in fulfilling multiple missions. Consequently,students need to understand not only how hormonesact but also how they interact. Some basic conceptstranscend the wide range of physiological actions ofhormones and may provide a foundation for under-standing hormonal regulation. Many of these con-cepts are also the bases for diagnosis and treatment ofendocrine disorders.
CONCEPTS RELATED TO CONTROL OFHORMONE SECRETION
Negative feedback control. Understanding negativefeedback lies at the heart of understanding endocrinecontrol systems.
The essence of negative feedback control of hormonesecretion is that some consequence of secretionblocks or dampens further secretion (Fig. 1). Themodel depicted would ensure constancy of a regu-lated parameter at some set point except for thetransient negative deviations that initiate the cycle
and the positive overshoots that stop it. This model is
inflexible and permits no opportunity for adaptation
to changing environmental demands. The added ele-
ments of changing the set point or overriding the set
point, shown in Fig. 2, are more likely to meet phys-
iological requirements.
Many biological examples appear more complex but
are simply superimposition of the same principles.
The pituitary and adrenal glands are in a negative
feedback relationship (Fig. 3), with cortisol acting as
an inhibitor of both pituitary secretion of ACTH and
hypothalamic secretion of corticotropin-releasing
hormone and arginine vasopressin. Hypothalamic in-
put to the negative feedback system allows for epi-
sodic override and adjustment of the set point in
response to environmental inputs. Input to the hypo-
thalamus comes also from circadian elements that
impose periodic adjustments of the set point. Positive
drive to the system is imposed by stress, in this case
hypoglycemia. Another aspect of negative feedback
illustrated here is negation of the positive drive im-FIG. 1.
Simple negative feedback.
FIG. 2.
Negative feedback including features that allow for
adaptive changes: adjustment of the set point or addi-
tional input to override the set point.
A P S R E F R E S H E R C O U R S E R E P O R T
VOLUME 25 : NUMBER 4 – ADVANCES IN PHYSIOLOGY EDUCATION – DECEMBER 2001
214

posed by hypoglycemia when glucose production isincreased as a consequence of cortisol secretion.
Negative feedback systems operating in opposite di-rections combine to maintain blood glucose concen-trations within narrow limits (Fig. 4). This illustrationalso incorporates feed-forward elements. Minimizingupward deviations is facilitated by the feed-forwardeffects of the intestinal hormone, glucagon-like pep-tide 1, which stimulates insulin secretion in anticipa-tion of absorption of a dietary glucose load. A similarbut perhaps more ambiguous role is played by para-sympathetic stimulation of both the a- and b-cellsduring the cephalic phase of eating. Override of neg-ative feedback to permit blood glucose to increase isprovided by the sympathetic innervation of the isletsand circulating catecholamines from the adrenal me-
dulla. Such a transient override ensures adequacy ofhigh energy fuels to meet the needs of episodic mus-cular activity or other responses to environmentallyimposed emergencies.
Positive feedback. In positive feedback systems, theconsequences of hormone secretion feed back to re-inforce the drive for secretion rather than dampen it.Rather than maintaining matters stable and unchang-ing, positive feedback creates instability and leads toexplosive changes (Fig. 5). Consequently, positivefeedback is rare in biology. The best example is oxy-tocin secretion by the posterior pituitary lobe duringthe birthing process. Stretch exerted on the uterinecervix is a powerful stimulus for oxytocin secretionand is transmitted to the oxytocin-secreting magno-cellular elements in the paraventricular and supraop-
FIG. 3.
Feedback relationships governing ACTH and cortisol secretion. Note that
the simple negative feedback relationship of the pituitary and adrenal
cortex is extended to include the paraventricular nucleus (PVN) of the
hypothalamus and corticotropin-releasing hormone (CRH) and arginine
vasopressin (AVP). Adjustment of the set point throughout the day ac-
counts for circadian rhythm, and overriding of the set point, at least
transiently, is imposed by the stress of hypoglycemia. A broader negative
feedback relationship between hypoglycemia and the adrenal cortex is
also present as glucocorticoid hormones contribute to alleviation of the
hypoglycemia.
A P S R E F R E S H E R C O U R S E R E P O R T
VOLUME 25 : NUMBER 4 – ADVANCES IN PHYSIOLOGY EDUCATION – DECEMBER 2001
215

tic hypothalamic nuclei by sensory neural inputs.With progressively greater stretch sensed by cervicalnerve endings by the contractions of uterine muscle,more and more oxytocin is released, stimulating esca-lating contractile force and stretch on the cervix.Ultimately, the cycle is broken by the explosive evac-uation of the uterus with the birth of the baby (Fig. 6).
CONCEPTS RELATED TO SPECIFICITY
Because of “internal secretion” of hormones into theblood, hormones are widely disseminated throughoutthe body and have access to virtually all cells. How-ever, only certain cells respond to any particular hor-mone. These “target” cells differ from all other cells inthe respect that they express receptors for that hor-mone (Fig. 7). In this example, only the cells thatexpress receptors change color in response to hor-mones.
Receptors are proteins located within a cell or on itssurface and contain a hormone recognition site that
binds its hormone with high affinity and selectivity
and a signal-transducing domain. Typical cells express
thousands to tens of thousands of copies of receptors
for a particular hormone. Binding the hormone pro-
duces some intramolecular change that activates the
signal-transducing domain. Some receptors initiate
signaling through their intrinsic enzymatic activity,
but most do so by the change in their interactions
with other proteins that may or may not have enzy-
matic activity.
The information delivered to the target cell is present
in the structure and three-dimensional conformation
of the hormone and is sufficient only to activate the
receptor. It appears that activation of the receptor is
an all-or-none phenomenon, with gradations in re-
sponse resulting from gradations in the numbers of
receptors that are activated in each cell. The receptor,
by virtue of the biochemical changes it triggers in
transducing the signal, initiates a particular biochem-
ical change or group of changes (Fig. 8). Once the
FIG. 4.
Feedback regulation of pancreatic islets. Note that glucose has opposite
effects on insulin and glucagon secretion, and override of negative feed-
back is achieved by parasympathetic and sympathetic input as well as by
the feed-forward input of glucagon-like peptide 1.
A P S R E F R E S H E R C O U R S E R E P O R T
VOLUME 25 : NUMBER 4 – ADVANCES IN PHYSIOLOGY EDUCATION – DECEMBER 2001
216

hormone has delivered its message by activating thereceptor, except in very rare cases, it plays no role inshaping the response. Rather, the signals generated inthe target cell are determined by the signal-transduc-ing component of the receptor. In many cases, thereis more than a single class of receptors for a particularhormone, and each class usually activates a differentbiochemical pathway. Only in rare instances can areceptor bind more than a single entity of hormonewith high affinity, and when this occurs (e.g., para-thyroid hormone and the parathyroid hormone-re-lated peptide), both agents produce identical cellularresponses. It is possible to express chimeric receptorsthat contain the hormone recognition component ofone hormone with the signal-transducing componentof a second hormone. The biochemical changes set inmotion are invariably characteristic of the signalingdomain of the receptor (Fig. 8).
The nature of the final response elicited in a target cellis not determined by the intracellular signal generatedby the receptor but, rather, by the effective machin-ery expressed in the cell as a consequence of its
differentiated state. For example, receptors for theparathyroid hormone are present in the basal mem-branes of cells of both the proximal and distal por-tions of the nephron (Fig. 9). Binding of the hormoneinitiates the same signaling cascade in both cell types,but the proximal tubules respond by decreasing phos-phate reabsorption from the glomerular filtrate andincreasing hydroxylation of vitamin D, while the distalcells respond by increasing reabsorption of calcium.
CONCEPTS RELATED TO TARGET CELLRESPONSIVENESS
Responsiveness of target cells to stimulation by theirhormones is not constant but may vary widely indifferent physiological states and is often adjusted bythe actions of other hormones or local paracrine orautocrine agents as well as the primary hormone.
Factors that govern the magnitude of the re-sponse to a hormone. 1) The most obvious deter-minant of the magnitude of the response is the con-centration of hormone that is available to bind to
FIG. 5.
Positive feedback is initiated by some perturbation and culminates in
some explosive event.
A P S R E F R E S H E R C O U R S E R E P O R T
VOLUME 25 : NUMBER 4 – ADVANCES IN PHYSIOLOGY EDUCATION – DECEMBER 2001
217

receptors. That concentration, in turn, is determinedby
● The rate of hormone secretion
● The rate of delivery by the circulation to the targetcell surface, which is slower if the hormone circu-lates bound to plasma proteins than if it is unbound
● The rate at which the hormone is degraded orexcreted
2) Of equal importance to the hormone concentra-tion is the number of competent target cells thatexpress functional receptors.
3) The sensitivity of target cells to hormonal stimula-tion is not constant and depends on
● The number of functional receptors that are ex-pressed
● The affinity of the receptor for the hormone
● The status of postreceptor amplification mecha-nisms
● The status and abundance of effector molecules
The relationship between the magnitude of response toa hormone and the concentration of hormone produc-ing that response is described by a sigmoidal curve (Fig.10). The sensitivity to a hormone is often defined as theconcentration needed to produce a half-maximal re-sponse. Target organ sensitivity is not constant and isoften adjusted in accordance with physiological circum-stances. In the example shown in Fig. 10, we mayassume that curve B is the basal sensitivity that may be
FIG. 6.
Positive feedback of oxytocin secretion during childbirth.
A P S R E F R E S H E R C O U R S E R E P O R T
VOLUME 25 : NUMBER 4 – ADVANCES IN PHYSIOLOGY EDUCATION – DECEMBER 2001
218
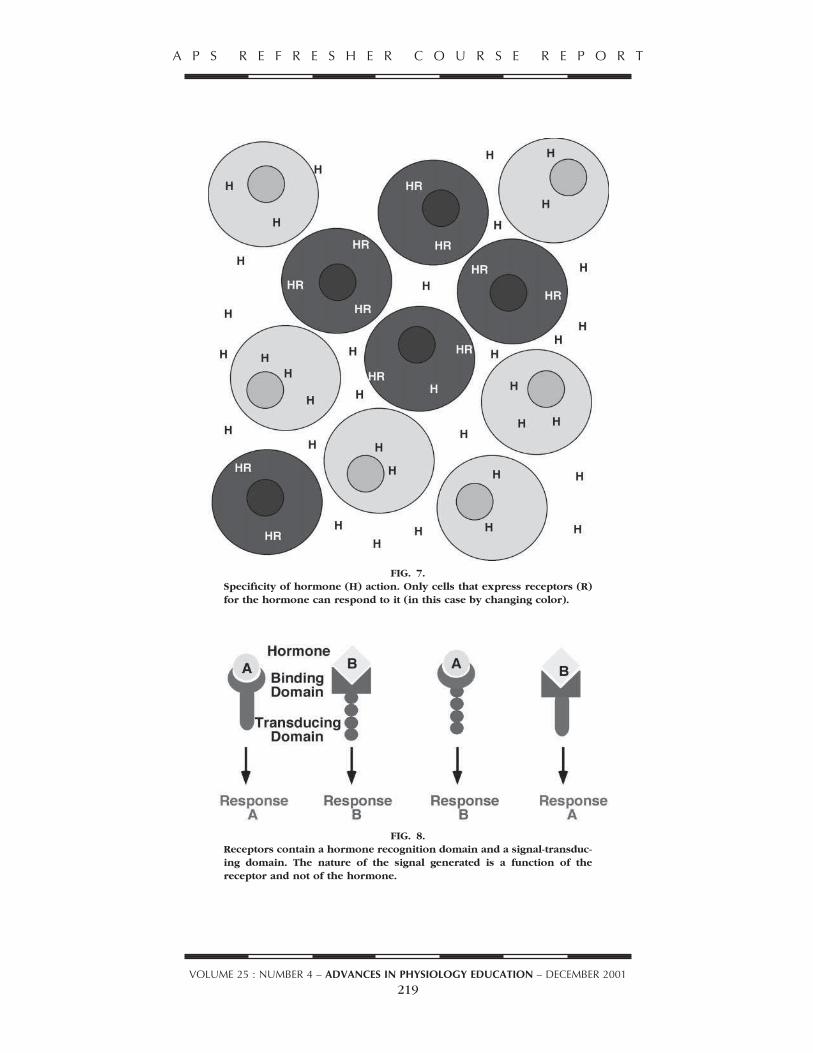
FIG. 7.
Specificity of hormone (H) action. Only cells that express receptors (R)
for the hormone can respond to it (in this case by changing color).
FIG. 8.
Receptors contain a hormone recognition domain and a signal-transduc-
ing domain. The nature of the signal generated is a function of the
receptor and not of the hormone.
A P S R E F R E S H E R C O U R S E R E P O R T
VOLUME 25 : NUMBER 4 – ADVANCES IN PHYSIOLOGY EDUCATION – DECEMBER 2001
219

increased (curve A) or decreased (curve C). With in-creased sensitivity, a lower concentration of hormone isrequired to produce a half-maximal response.
Sensitivity does not necessarily parallel hormone
binding by the receptor and, therefore, is not neces-
sarily a function of the affinity of the receptor for the
hormone. Because it depends on many postreceptor
events, the response to a hormone may be at a max-
imum at a hormone concentration that does not sat-
urate all of the receptors (Fig. 11). When ,100% of
the receptors need to be occupied to obtain a maxi-
mum response, cells are said to express “spare recep-
tors.” For example, glucose uptake by the fat cell is
stimulated in a dose-dependent manner by insulin,
but the response reaches a maximum when only a
few percent of the available and functional insulin
receptors are occupied by insulin. The affinity of the
receptor for the hormone is defined as the concen-
tration at which half of the receptors are occupied by
hormone. Because the response is related to the num-
ber of receptors that are activated and can therefore
produce a biochemical response, the consequence
for cells that express spare receptors is that they are
more sensitive to the hormone than would be pre-
dicted from their binding affinity. In the example
shown in Fig. 11, A twofold excess of receptors re-
duced by half the concentration of hormone needed
to produce a half-maximal response.
FIG. 9.
Proximal and distal renal tubular cells respond to parathyroid hormone (PTH) by
increasing cAMP production, but cAMP initiates different events in each cell in
accordance with the capabilities programmed during cellular differentiation.
FIG. 10.
Concentration-response curves showing 3 different lev-els (curves A–C) of sensitivity as defined by the concen-tration of hormone required to produce a half-maximalresponse (ED50).
A P S R E F R E S H E R C O U R S E R E P O R T
VOLUME 25 : NUMBER 4 – ADVANCES IN PHYSIOLOGY EDUCATION – DECEMBER 2001
220

Under a variety of circumstances, cells may increase(upregulate) or decrease (downregulate) the numberof functional receptors they express. Upregulation ordownregulation can be achieved by adjusting the rel-ative rates of receptor synthesis and degradation, re-ceptor endocytosis and sequestration, or covalentmodification through phosphorylation or dephos-phorylation. The consequences of upregulation ordownregulation for sensitivity are shown in Fig. 12.Small changes in receptor abundance are of littleconsequence in the presence of a large number ofspare receptors but can be quite profound for cellsthat express no spare receptors. It is more commonfor cells to adjust the number of receptors rather thanfor receptor affinity to regulate their sensitivity to ahormone.
The apparent sensitivity to hormonal stimulation isnot a function only of receptor number. Down-stream events also contribute to the concentration-response relationship. On the cellular level, upregu-lation or downregulation of effector moleculessuch as enzymes, ion channels, and contractile pro-teins, etc., may increase or reduce the maximumcapacity for a response even though the sensitivity,as defined earlier, is unchanged (Fig. 13). On atissue or organ level, the measured response is the
FIG. 11.
The presence of “spare receptors” lowers the concen-
tration of hormone needed to produce a half-maximal
response below that needed to produce half-satura-
tion of receptor binding sites (i.e., the affinity of the
receptor for the hormone). Kd, dissociation constant.
FIG. 12.
Regulation of receptor number changes the concen-
tration of hormone needed for a half-maximal re-
sponse (i.e., the sensitivity).
FIG. 13.
A change in the capacity of effector elements down-
stream from the receptor or in the number of compe-
tent cells changes the magnitude of the response with-
out necessarily changing the concentration of hormone
needed to produce a half-maximal response.
A P S R E F R E S H E R C O U R S E R E P O R T
VOLUME 25 : NUMBER 4 – ADVANCES IN PHYSIOLOGY EDUCATION – DECEMBER 2001
221

aggregate of the contributions of all of the respond-ing cells, so that the magnitude of the response toa particular concentration of hormone is a functionof the number of available cells as well as thecompetence of each cell. Thus a change in re-sponse capacity will result in the need for a greateror lesser concentration of hormone to produce agiven level of response and, therefore, appears as achange in sensitivity, even though the concentra-tion of hormone required for the half-maximal re-sponse may remain unchanged.
Factors that govern the duration of the responseto a hormone. Of equal concern to the magnitude ofresponse is its duration. Factors that govern the dura-tion of the response to a hormone include
1) the duration of hormone availability, which is de-termined by
● The duration of secretion
FIG. 14.
Responses to multiple hormonal inputs may be addi-
tive (glucagon 1 epinephrine) or synergistic (gluca-
gon 1 epinephrine 1 cortisol).
FIG. 15.
Reinforcement. Different effects of cortisol in different tissues reinforce
hepatic actions to increase glucose production.
A P S R E F R E S H E R C O U R S E R E P O R T
VOLUME 25 : NUMBER 4 – ADVANCES IN PHYSIOLOGY EDUCATION – DECEMBER 2001
222

● The rate of hormone clearance from the blood,usually described as its half-life
2) whether the response results from
● A rapidly reversible covalent change, i.e., phosphor-ylation or dephosphorylation of key enzymes
● Or genomic events involving synthesis of proteinsand the half-lives of the proteins
CONCEPTS RELATED TO INTEGRATION
Hormones seldom act alone or on a single tissue incarrying out their missions of regulation and coordi-nation. The following examples illustrate some con-cepts of integration.
Additivity and synergy. Multiple hormones oftenwork in concert, and in some instances, they mayappear to be redundant. Figure 14 illustrates theinteractions of glucagon, epinephrine, and cortisolto increase blood glucose concentrations. The datashown are redrawn from Eigler et al. (1). Bothepinephrine and glucagon, when administeredalone to dogs, produced an increase in blood glu-cose, and when given together, their effects wereadditive. Cortisol alone had little effect on theblood glucose concentration, but when epineph-rine plus glucagon were given to cortisol-treateddogs, the rise in glucose was far greater than thesum of the increases produced by the individualhormones alone. These data illustrate the conceptof synergism, wherein the response to a combina-tion of hormones is greater than the sums of theirindividual actions.
Reinforcement. As shown in Fig. 14, the actions ofseveral hormones may converge to regulate the pro-cess of glucose production. Figure 15 shows an ex-ample of how the diverse actions of a single hormoneexerted on several different tissues may converge toreinforce a critical action. One of the primary effectsof glucocorticoids is to stimulate gluconeogenesis inthe liver. Cortisol increases the enzymatic capacity ofthe liver for gluconeogenesis and renders the hepato-cyte sensitive to gluconeogenic stimulation by gluca-gon and catecholamines. Efficient gluconeogenesis,however, requires a supply of substrate. Cortisol pro-
motes the breakdown of protein in skeletal muscleand the release of amino acids into the circulation. Italso facilitates the breakdown of triglycerides in adi-pose tissue and the release of glycerol and fatty acidsinto the blood. Amino acids and glycerol (along withlactate) are the principal substrates for gluconeogen-esis.
Push-pull. Secretion of glucose by the liver is underboth positive and negative control. In emergency sit-uations or during exercise, there is increased demandfor glucose. Just increasing the secretion of glucagonand epinephrine would increase the rate of glycogenbreakdown and would, therefore, increase glucoseproduction. Stimulation by these hormones becomesmuch more effective if the restraining effect of insulinis simultaneously relieved. This push-pull mechanismallows for rapid, unhindered glucose mobilization(Fig. 16).
Dozens of other examples of patterns of hormoneinteractions at the molecular, cellular, organ, and
FIG. 16.
Push-pull mechanism for producing a rapid increase
in glucose production.
A P S R E F R E S H E R C O U R S E R E P O R T
VOLUME 25 : NUMBER 4 – ADVANCES IN PHYSIOLOGY EDUCATION – DECEMBER 2001
223

organismal levels can be cited to underscore con-cepts of integration. The concept of homeostasis isa fundamental tenet of physiology, and the endo-crine system is its principal defender. The ever-changing challenges to maintaining the constancyof the internal environment and ensuring survivalof the species demand that endocrine control bedynamic, adaptable, and precise. The concepts pre-sented here will hopefully provide students withsome insight into the workings of the endocrinesystem. The concepts presented here are notunique to endocrinology, however, and perhaps
will also contribute to their understanding of phys-iology as a whole.
Address for reprint requests and other correspondence: H. M. Good-
man, Dept. of Physiology, Univ. of Massachusetts Medical School,
Worcester, MA 01655 (E-mail: [email protected]).
REFERENCES
1. Eigler N, Sacca L, and Sherwin RS. Synergistic increments of
glucagon, epinephrine, and cortisol in the dog. J Clin Invest
63: 114, 1979.
A P S R E F R E S H E R C O U R S E R E P O R T
VOLUME 25 : NUMBER 4 – ADVANCES IN PHYSIOLOGY EDUCATION – DECEMBER 2001
224

Review
Mechanisms of nongenomic actions of thyroid hormone
Paul J. Davis a,b,*, Jack L. Leonard c, Faith B. Davis a
a Ordway Research Institute, Inc., 150 New Scotland Avenue, Albany, NY 12208, USAb Albany Medical College and Stratton Veterans Affairs Medical Center, Albany, NY, USA
c Department of Physiology, University of Massachusetts Medical School, Worcester, MA, USA
Available online 5 October 2007
Abstract
The nongenomic actions of thyroid hormone require a plasma membrane receptor or nuclear receptors located in cytoplasm. Theplasma membrane receptor is located on integrin aVb3 at the Arg-Gly-Asp recognition site important to the binding by the integrinof extracellular matrix proteins. L-Thyroxine (T4) is bound with greater affinity at this site than 3,5,3 0-triiodo-L-thyronine (T3). Mito-gen-activated protein kinase (MAPK; ERK1/2) transduces the hormone signal into complex cellular/nuclear events including angiogen-esis and tumor cell proliferation. Acting at the integrin receptor and without cell entry, thyroid hormone can foster ERK1/2-dependentserine phosphorylation of nuclear thyroid hormone receptor-b1 (TRb1) and de-repress the latter. The integrin receptor also mediatesactions of the hormone on intracellular protein trafficking and on plasma membrane ion pumps, including the sodium/protein antiporter.Tetraiodothyroacetic (tetrac) is a T4 analog that inhibits binding of iodothyronines to the integrin receptor and is a probe for the par-ticipation of this receptor in cellular actions of the hormone. Tetrac blocks thyroid hormone effects on angiogenesis and cancer cell pro-liferation. Acting on a truncated form of nuclear TRa1 (TRDa1) located in cytoplasm, T4 and 3,3 0,5 0-triiodothyronine (reverse T3), butnot T3, cause conversion of soluble actin to fibrous (F) actin that is important to cell motility, e.g., in cells such as glia and neurons.Normal development of the central nervous system requires such motility. TRb1 in cytoplasm mediates action of T3 on expression ofcertain genes via phosphatidylinositol 3-kinase (PI 3-K) and the protein kinase B/Akt pathway. PI 3-K and, possibly, cytoplasmicTRb1 are involved in stimulation by T3 of insertion of Na,K-ATPase in the plasma membrane and of increase in activity of this pump.Because ambient thyroid hormone levels are constant in the euthyroid intact organism, these nongenomic hormone actions are likely tobe contributors to basal rate-setting of transcription of certain genes and of complex cellular events such as angiogenesis and cancer cellproliferation.Ó 2007 Elsevier Inc. All rights reserved.
Keywords: Thyroxine; 3,5,3 0-Triiodo-L-thyronine; Integrin aVb3; Nuclear thyroid hormone receptor-b1 (TRb1); TRDa1; Actin; Protein kinase B/Akt;
Mitogen-activated protein kinase (MAPK); ERK1/2
1. Introduction
Themechanism of action of thyroid hormone on or in cellshas been assumed to begin in the cell nucleus and to requireparticipation of receptor proteins for thyroid hormone in thenuclear compartment. These receptors are transcriptionallyactive proteins that cause expression of thyroid hormone-responsive genes. This classical or genomic mechanism (seebelow) has been complimented in the past decade by descrip-
tions of thyroid hormone action that are now understood toinvolve novel extranuclear (nongenomic) mechanisms in avariety of cells, including those of the central nervous system(CNS). Such nongenomic mechanisms appear to be relevantto proliferation and motility of endothelial cells and certaintumor cells, including glioma cells that are models for humanbrain tumors. Themechanisms are also important to the stateof the actin cytoskeleton and motility of normal nerve andglial cells. The current review is focused on recent insightsinto such nongenomic actions in a variety of cells and oninterfaces of nongenomic and genomic actions of the hor-mone that are now known to exist.
0091-3022/$ - see front matter Ó 2007 Elsevier Inc. All rights reserved.
doi:10.1016/j.yfrne.2007.09.003
* Corresponding author.E-mail address: [email protected] (P.J. Davis).
www.elsevier.com/locate/yfrne
Frontiers inNeuroendocrinology
Available online at www.sciencedirect.com
Frontiers in Neuroendocrinology 29 (2008) 211–218

2. Genomic mechanisms of thyroid hormone action
The classical molecular mechanism of thyroid hormoneaction involves uptake of L-thyroxine (T4) or 3,5,3
0-triiodo-L-thyronine (T3) by target cells, access of T3 to the cellnucleus and complexing of the hormone with nuclear thy-roid hormone receptor (TR) protein [61–63]. TR is foundin the nucleus as a heterodimer with retinoic acid X recep-tor (RXR). The heterodimeric complex sheds corepressorproteins when T3 is bound and recruits coactivators thatfacilitate binding of the heterodimer-T3 complex to thyroidhormone response elements (TREs) of hormone-responsivegenes and consequent gene transcription [63]. Activation ofTR may involve phosphorylation of the receptor [12,58].This genomic mechanism of hormone action has been dem-onstrated in a variety of thyroid hormone-responsive cellsand leads to modulation of transcription of a hundred ormore genes [24,43]. Characteristics of genomic actions ofthe hormone include the requirement for access of the hor-mone to the cell interior, translocation of the hormone tothe nucleus, altered rates of gene transcription, generationof specific mRNAs, translation and changes in cell contentor secretion of specific gene products. Several or morehours are usually required for genomic mechanisms to bemanifest.
L-Thyroxine (T4) can act via nuclear TR, but the affinityof the receptor for T4 is much lower than that for T3. Thus,T3 is the natural ligand of TR [61–63]. In the genomic con-cept of hormone action, T4 is viewed as a prohormone thatyields the more metabolically active T3 via action of tissuedeiodinase activities. That T4 acts as a hormone as well as aprohormone will be considered below and has beenreviewed elsewhere [16].
3. Nongenomic mechanisms of thyroid hormone action
For more than two decades, actions of thyroid hormonein a variety of cells have been described that do not primar-ily involve nuclear TR (reviewed in [2,14]) and thus are‘nongenomic.’ The mechanisms of several of these nonge-nomic actions of thyroid hormone are now understood,at least in part, and depend upon cellular signal transduc-tion systems and either novel cell surface receptors for thy-roid hormone [3,14] or extranuclear TRb [33,45] orderivatives of TRa ([28]; see section on actin, below). It isimportant to point out that actions of thyroid hormonethat begin nongenomically at a cell surface receptor mayculminate in complex nuclear and cellular events. Oneexample is phosphorylation of Ser-142 of the TRb1 iso-form [12] that results in altered transcriptional activity(de-repression) of the receptor due to shedding of corepres-sor proteins and recruitment of coactivators [12]. Estrogenreceptor-a (ERa) in the nucleus may also be specificallyphosphorylated under the direction of thyroid hormonethat is acting exclusively at the cell surface [60]. These areexamples of interfaces between nongenomic and genomicmechanisms of action of iodothyronines. Other complex
cellular events directed by thyroid hormone from theplasma membrane include cell proliferation in endothelialcells and specific tumor cells ([13,15]; see below).
Acting exclusively at the cell surface, thyroid hormonecan also modulate intracellular protein trafficking, suchas translocation from cytoplasm to the nucleus of nuclearhormone receptor superfamily members—including TRb1and ERa—and moieties such as signal transducer and acti-vator of transcription-3 (STAT3) [39], STAT1a [39], andTrip230 [7], a coactivator for the nuclear receptor for thy-roid hormone. Another action of the hormone that is initi-ated at the cell surface is modulation of the activity of theNa+/H+-antiporter or exchanger (sodium/hydrogen ionexchanger, NHE) [10,29]. This effect does not appear toinvolve the cell nucleus.
When actions of thyroid hormone are initiated withinthe cell, but outside the nucleus, nongenomic mechanismsmay involve a derivative of cytoplasmicTRa1 that regu-lates the state of actin, converting soluble into fibrous (F)actin, and regulates cell motility. These effects have beenwell-characterized in CNS cells [21,22] and are reviewedin detail below. TRb1 also exists in cytoplasm and remark-ably is involved in transduction of the thyroid hormone sig-nal through a specific signal transduction cascade intospecific gene expression, e.g., the immodulatory protein,ZAKI-4 [44,45].
From the foregoing, it is apparent that nongenomicactions of thyroid hormone result in changes in cell func-tion. These actions have usually been demonstrated in thy-roprival cells or tissues in vitro that are acutely re-exposedto thyroid hormone. This paradigm has resulted in charac-terization of nongenomic actions of the hormone as ‘acute’or ‘rapid onset’ (seconds or minutes) when compared topurely genomic mechanisms that require gene transcriptionand consequent translation of mRNA. This characteriza-tion is not accurate in the context of the intact organism,where ambient concentrations of thyroid hormone nor-mally are stable. In this setting we assume that the hor-mone contributes to basal activity rates of the functionsreported, e.g., proton efflux of NHE-1 [10], rates of proteintrafficking [14] or background levels of phosphorylation ofspecific nucleoproteins [12,60]. The hormone is assumed tocondition the state of actin in the cell and the rate of migra-tion of cells toward or away from trophic factors. Theterm, acute actions, is also inaccurate when interfacesbetween nongenomic and genomic mechanisms are studied.Nongenomic mechanisms thus are best characterized asthose which at initiation do not primarily depend uponintranuclear complexing of TR and thyroid hormone.
Another feature of nongenomic mechanisms of thyroidhormone action is the plurality of hormone analogues thatmay initiate specific actions. Genomic actions, as notedabove, are T3-dependent. In contrast, the nongenomicmechanism of action of the hormone on the state of actinin the cell requires T4 or reverse-T3 (rT3, 3,3
0,5 0-triiodothy-ronine) and is insensitive to T3. On the other hand, nonge-nomic actions that affect the NHE are conditioned by T3
212 P.J. Davis et al. / Frontiers in Neuroendocrinology 29 (2008) 211–218

[10,29], whereas intracellular protein trafficking and thephosphorylation of nucleoproteins that is initiated at thecell surface are primarily T4-responsive effects [14]. As willbe noted below, tetraiodothyroacetic acid (tetrac), a T4
derivative, is purely inhibitory of the actions of T4 andT3 that are initiated at the cell surface [3]; inside the cell,however, tetrac has low potency thyromimetic actions,e.g., suppressing TSH release by pituitary gland thyro-tropes [4]. Although the nongenomic actions of thyroidhormone on mitochondria are not included in this review,it should be noted that 3,5-diiodothyronine (T2) is an effec-tive modulator of cellular respiration [41] and perhapsmore potent than T3 in this regard.
4. Receptors for nongenomic actions of thyroid hormone
Studies of thyroid hormone action on cell surface events,such as calcium efflux [11,49] or glucose uptake [54,55], sev-eral decades ago implied the existence of one or moreplasma membrane receptors for T3 or T4. This was partic-ularly the case when experiments were conducted in mem-brane vesicles from enucleate cells, such as the humanmature erythrocyte (RBC) [49] or when rates of glucosetransport in intact cells were increased in a time framein vitro that precluded transcription and translation. Bind-ing sites for the hormone were also described on RBCs thatinferred the saturation of binding sites on human thyrox-ine-binding globulin (TBG) (‘T3 uptake’) by a partitionof labeled iodothyronine between erythrocytes and plasma[26]. The latter sites, however, were not seen to have a spe-cific function and thus could not be characterized as‘receptors.’
Recently, a cell surface receptor for iodothyronines hasbeen described on a structural protein of the plasma mem-brane of virtually all cells [3,14]. This protein is integrinaVb3, a heterodimer that interacts with a substantial num-ber of proteins of the extracellular matrix (ECM) [1,50].The integrin is highly plastic and is capable of transducinga number of discrete signals. These include ECM proteinsignals that are converted into cellular responses (outside-in conduction) and signals that originate within the cellto the external milieu (inside-out conduction). The thyroidhormone receptor domain is at or near the Arg-Gly-Asp(RGD) recognition site on the integrin [3,8]. ECM proteinssuch as vitronectin, fibronectin and osteopontin that arebound at discrete sites on the integrin must also containan RGD sequence to achieve the bound state [50]. Thebinding of T4, T3 and tetrac at this site has been modeled[8]. The Kd for T4 at this site is subnanomolar and the affin-ity of the site for T4 is higher than for T3 [3]. T4 and T3 areagonists at this site (see below), whereas tetrac is an antag-onist and inhibits T4- and T3-binding at the receptor [3].
The classification of this site on integrin aVb3 as areceptor required definition downstream of functional con-sequences of binding site occupancy. This definition camewhen T4 and—less potently, T3—at this site were shownto activate from the cell surface a serine–threonine kinase,
mitogen-activated protein kinase (MAPK, ERK1/2). Thisactivation (tyrosine phosphorylation of the enzyme) resultsimmediately in nuclear translocation of phosphoMAPK(pMAPK). In the nucleus, the activated kinase is foundin a complex with TRb1 and serine phosphorylates thisnuclear receptor [12]. As noted above, this phosphorylationstep de-represses TR and induces a basal rate of transcrip-tion in the receptor. Full activation of transcription appar-ently requires the presence of T3 in the nucleus. We alsopointed out above that T4, acting via the integrin receptor,can also cause MAPK to phosphorylate Ser-118 of nuclearERa [60]. When this occurs in ERa-positive human breastcancer (MCF-7) cells in vitro in the absence of estrogen, cellproliferation is enhanced [60]. In this context, T4 acts likean estrogen. These observations re-initiated concern thatthyroid hormone may have a proliferative effect on certaintumor cells in the clinical setting. This issue is discussedbelow in a consideration of actions of the hormone on glialtumor cells and other cancer cell lines. The mechanisms ofthese actions of thyroid hormone are shown in Fig. 1.
Interestingly, thyroid hormone-activated MAPKappears to be a nidation factor for a complex in the nucleusof transcription factors. Immunoprecipitation of nucleo-proteins with anti-pMAPK in thyroid hormone-treatedcells results in the recovery of TRb1, ERa, STAT1a, p53and retinoic acid X receptor (RXR) (H.-Y. Lin, F.B.Davis, P.J. Davis: unpublished observations). As notedabove, each of these proteins is subject to serine phosphor-ylation by activated MAPK (ERK1/2). The function ofsuch complexes is not known. Complexes of transcriptionfactors and co-factors, such as corepressor proteins, havebeen termed ‘enhanceosomes’ [6]. We speculate thatpMAPK-associated nucleoproteins serve to organize theunliganded superfamily of nuclear hormone receptors(transcription factors TRb1, ERa, RXR) in anticipationof interactions with nuclear nonpeptide ligands [14].
The collaborating Refetoff and Seo laboratories andIngbar and Mariash have reported that nongenomic mech-anisms of action of thyroid hormone can involve a signaltransduction pathway other than MAPK (ERK1/2). Forexample, X Cao, Kambe, Moeller, Refetoff and Seo haveimplicated protein kinase B (PKB)/Akt in transduction ofthe thyroid hormone signal that culminates in the tran-scription of the ZAKI-4 gene [5]. The two ZAKI-4 geneproduct isoforms (a and b) are calcineurin inhibitors. Thisgroup of investigators has shown the transduction pathwayin this human skin fibroblast system to include phosphati-dylinositol 3-kinase (PI 3-K)-Akt/PKB, then nuclear mam-malian target of rapamycin (mTOR) and finallyphosphorylation of a nuclear mTOR substrate, p70S6kinase
[5]. The first step in this interesting cascade is an interactionin cytoplasm of TRb1 and the p85a regulatory subunit ofPI 3-K [5]. A number of laboratories have reported theexistence of TRb1 in cytoplasm [42,64], but whether thispool of ‘nuclear’ receptor was nascent or functional hasnot been clear. In addition, L.C. Moeller et al. from thesame laboratory have shown that the same signal transduc-
P.J. Davis et al. / Frontiers in Neuroendocrinology 29 (2008) 211–218 213

tion mechanism induces the expression of several othergenes [45]. The latter include hypoxia-inducible factor-1a(HIF1a). HIF1a protein targets several genes relevant tocarbohydrate handling by cells and causes their expression.These include a glucose transporter (GLUT1), platelet-typephosphofructokinase (PFKAP) and monocarboxylate
transporter-4 (MCT4) [45]. These nongenomic actions ofthyroid hormone are depicted in Fig. 1.
In interesting studies of the actions of T3 on rat lungalveolar cells, Lei, Nowbar, Mariash and Ingbar haveshown that the hormone nongenomically increases (a) theinsertion of the sodium pump (Na,K-ATPase) in theplasma membrane and (b) the activity of the membrane-bound enzyme [33]. These effects of thyroid hormone weresubsequently shown by the same group to depend uponhormonal activation of PI 3-K activity via Src kinase[34]. Lei and co-workers have also described the acquisitionduring rat embryonic lung development of T3-sensitivity ofNa,K-ATPase [35]. It is not yet clear what the first step is,upsteam of Src kinase, in this action of T3 in lung cells. Thepossibilities exist that the initial step involves TRb1 in res-idence in the cytoplasm, as in the case of modulation oftranscription of ZAKI-4, or an extranuclear TRa1 deriva-tive, as described below in the regulation by T4 of the actincytoskeleton and mobility in nerve and glial cells. An addi-tional consideration is that plasma membrane receptors inaddition to integrin aVb3 may exist that primarily trans-duce the T3 signal.
As noted above, we have shown that the MAPK path-way downstream of the integrin thyroid hormone receptortransduces the hormonal signal into angiogenesis andtumor cell growth. We have recently examined the possibil-ity in one of our tumor cell models that PI 3-K might alsomediate cell proliferation. T4 activates MAPK in humanglioblastoma (U87) cells. We found that PI 3-K was acti-vated by T3, but not by T4. T3 and T4 both stimulatedtumor cell proliferation, measured by proliferating cellnuclear antigen (PCNA), but a pharmacologic inhibitorof PI 3-K, LY294002 [18], did not block the proliferativeeffects of either hormone analogue (H.-Y. Lin, F.B. Davis,P.J. Davis: unpublished observations). Thus, PI 3-K didnot mediate the action on proliferation in this human cellline of either T3 or T4 and these two analogues differ intheir abilities to activate PI 3-K.
5. Complex tissue responses induced nongenomically by
thyroid hormone
In addition to the effects on individual nuclear proteins[12,29,39,60] that are dictated from the cell surface receptorby thyroid hormone analogues, the latter can induce fromits integrin receptor certain complex cellular or tissueresponses. Among these are tumor cell proliferation [15],angiogenesis [3,13] and cellular migration (S.A. Mousa,L. O’Connor, P.J. Davis: unpublished observations).Tumor cell proliferation is discussed in the next sectionof this review. That thyroid hormone promotes new bloodvessel formation has been extensively documented in mod-els of the chick chorioallantoic membrane (CAM) [3,13,47]and tubule formation by human dermal microvascularendothelial cells (HDMEC) [46]. These actions can be mim-icked by agarose-T4 that does not gain access to the cellinterior. Tetrac inhibits thyroid hormone-induced angio-
T4 or rT3
T4 TR∆α1
Soluble actin F actin
CYTOPLASM
NUCLEUS
T4
T3
MAPK
PLCPKCα
Na/H
ERαTRβ1
STATsERα
TRβ1Serine
phosphorylation
and change in
transcriptional
activity
ZAKI-4
GLUT1
HIF1αSpecific
gene
transcription
T4
T3
MAPK
NaK-ATPase activity
and membrane insertion
PI 3-KTRβ1
T3
CYTOPLASM
NaK-ATPase
T3
TRβ1PI 3-K
Tumor cell
proliferation;
angiogenesis
αvβ3
exchanger
Trafficking
NHE
1
3
1
2
3
↑
αvβ3
Fig. 1. Schematic representation of nongenomic cellular actions of
thyroid hormone analogues T4, T3 and rT3. T4 can act via a plasma
membrane receptor on integrin aVb3 to activate the mitogen-activated
protein kinase (MAPK; ERK1/2) signal transduction cascade via phos-
pholipase C (PLC) and protein kinase Ca (PKCa). Activated MAPK can
translocate to the nucleus to phosphorylate nuclear thyroid hormone
receptor TRb1 (Ser-142) or nuclear estrogen receptor ERa (Ser-118). T4-
activated MAPK also modulates intracellular protein trafficking of ER
and TR from cytoplasm to nucleus and can act locally at the plasma
membrane to activate the sodium proton exchanger (NHE). Via the
integrin receptor and MAPK, T4 also is pro-angiogenic and causes
proliferation of certain tumor cells. T3 may also act via the integrin
receptor, but the affinity of the receptor for T3 is lower than for T4.
Nongenomic mechanisms of action initiated at the integrin receptor are
designated ‘1’ in the figure. T4 and reverse T3 (rT3), but not T3, may
interact with cytoplasmic TRa-derived polypeptide (TRDa1) to cause a
change in the state of intracellular actin that supports cell migration
(conversion of soluble actin to F actin). Nongenomic mechanisms that are
initiated at cytoplasmic TRDa1 are designated ‘2’ in the figure. T3, but
apparently not T4, may interact with cytoplasmic TRb1 to activate the
phosphatidylinositol 3-kinase (PI 3-K) signal transduction pathway. The
end results of this process include change in numbers of pumps inserted in
the membrane and increased activity of the sodium pump (Na,K-ATPase)
in the plasma membrane and the transcription of specific genes, such as
ZAKI-4, an anti-calcineurin, and hypoxia-inducible factor-1a (HIF1a). A
number of genes are targets of HIF1a protein, including a glucose
transporter (GLUT1). Nongenomic actions that are initiated at cytoplas-
mic TRb1 are designated ‘3’ in the figure.
214 P.J. Davis et al. / Frontiers in Neuroendocrinology 29 (2008) 211–218

genesis. Induction of angiogenesis by thyroid hormone is acomplex process that is dependent, at least in part, on tran-scription of the basic fibroblast growth factor (bFGF) geneand release of the gene product into the medium of theCAM assay [13]. Addition of anti-bFGF to the mediumof thyroid hormone-exposed CAM blocks the pro-angio-genic activity of iodothyronines (T4 and T3). Tetrac alsoinhibits the hormone-induced process. Because tetrac is aprobe for thyroid hormone-related events that begin atthe integrin aVb3, the proangiogenic activity of T4 is inte-grin-requiring. Anti-integrin aVb3 was also inhibitory [13].We also know that thyroid hormone enhances mobility ofHDMEC in a Boyden chamber (S.A. Mousa, L. O’Con-nor, P.J. Davis: unpublished observations). This factor isalso likely to contribute to the angiogenic process and vas-cular tubule formation.
While it is clear that tetrac inhibits the pro-angiogeniceffect of iodothyronines, it has also been shown that inthe absence of thyroid hormone tetrac is anti-angiogenic[48]. This is a remarkable finding that we attribute to theproximity of the integrin thyroid hormone receptor site atwhich tetrac binds and the RGD recognition site on thesame protein. As pointed out earlier, this recognition siteis required for angiogenesis induced by a variety of vascu-lar endothelial growth factors, including VEGF andbFGF. Thus, tetrac is a small molecule with potentiallyimportant usefulness as an anti-angiogenic agent.
6. Nongenomic actions of thyroid hormone on tumoral glial
cells
In 2003, Hercbergs and co-workers reported that thepharmacological induction with propylthiouracil (PTU)of mild biochemical hypothyroidism significantly improvedsurvival time in patients with advanced, recurrent glioblas-toma multiforme (GBM) [27]. Control and treated patientsalso received high-dose tamoxifen that appeared to havelittle effect on survival. We subsequently showed that phys-iological concentrations of T4 and T3 were proliferationfactors for several mouse glioma cell lines [15] that aremodels for GBM. Tetrac blocked glioma cell proliferation,as did anti-integrin aVb3. In separate studies, tetrac hasbeen shown to inhibit C6 glioma cell migration in a Boydenchamber (S.A. Mousa, L. O’Connor, P.J. Davis: unpub-lished observations). Because GBM is a highly vasculartumor, it is desirable to include an anti-angiogenic strategyin its management and tetrac has the desirable feature ofbeing an anti-angiogenic small molecule. Thus, this anti-thyroid hormone that acts at the cell membrane receptorfor thyroid hormone on glioma cells acts by at least 3 path-ways to oppose glioma cell growth. Tetrac is anti-prolifer-ative [15] and anti-angiogenic [3,46,47], as well as aninhibitor of cell migration. Because thyroid hormone tendsto alkalinize tumor cells by enhancing transport activity ofthe NHE [10,29], endogenous hormone levels may stimu-late the activity of multi-drug resistance (MDR) pumpsin the plasma membrane [17]. By opposing this action of
the hormone, tetrac may support acidification of the cells,a state that decreases activity of the MDR pumps andincreases retention by tumor cells of chemotherapeuticagents.
Among the other cancer cell types on which iodothyro-nines have proliferative activity are ERa-positive humanbreast cancer cells [60], as mentioned above, and humanpapillary and follicular thyroid cancer [40], human head-and-neck cancer cell lines (H.-Y. Lin, F.B. Davis, H.J.Cao et al.: unpublished observations) and lung cancer celllines [51].
7. Mechanisms of thyroid hormone action mediated through
the actin cytoskeleton
Twenty years ago, Faivre-Sarrailh and Rabie [19] foundthat the quantity of filamentous actin in the hypothyroidneonatal cerebellum was markedly reduced compared tonormal and that acute treatment with T4 normalized thisdefect. This was the first hint of the ability of thyroid hor-mone to regulate the dynamics of cytoskeleton remodeling,a key structural component required for cells to migrateand to interact with their environment. Coincident withRabie’s work, we were examining the molecular events thatparticipate in the nongenomic regulation of thyroid hor-mone metabolism in the brain [21,56]. Interestingly, theability of astrocytes to adhere to the culture dish was foundto depend on the presence of thyroid hormone and subse-quent analysis of the actin cytoskeleton revealed that theprominent actin cables observed in normal astrocytes werelost when T4 was removed from the culture medium [56].Replacement with either T4 or reverse T3 restored theseactin cables to normal within 20 min, while the T3 hadno effect on the actin cytoskeleton. Since astrocytes lackchromatin-bound TRs [38], but express both the truncateddelta versions derived from the TRa gene, TRDa1 andTRDa2, [36], it was clear that these dynamic, structuralchanges in the cytoarchitecture were not mediated bydirect, regulated gene expression.
Subsequent work showed that the laminin receptor, amember of the integrin family of transmembrane adhesionmolecules, was largely responsible for the initial binding ofastrocytes to the culture dish [20,23]. In both astrocytes andneuronal growth cones, bundled actin cables anchor thistransmembrane integrin receptor in place by interactionsbetween their cytoplasmic tail and filamentous actin cyto-skeleton [9,25,59]. This allows the cell to take a footholdon the extracellular matrix and for the integrin receptorto initiate intracellular signaling. Importantly, astrocytessecrete most, if not all, of laminin in brain, and subsequentwork revealed that loss of the actin cables in hypothyroidastrocytes eliminated their ability to anchor this guidancemolecule on their cell surface, leading to a loss of thiskey molecule both in vitro and in the developing cerebellum[21].
While the ability of thyroid hormone to regulate thedynamics of actin fiber remodeling is a convenient biolog-
P.J. Davis et al. / Frontiers in Neuroendocrinology 29 (2008) 211–218 215

ical event readily studied in cell culture, the biological con-sequences of such a molecular pathway are more far reach-ing. In astrocytes and neuronal growth cones, bundledactin cables anchor transmembrane integrin receptor(s),one of several classes of adhesion molecules, in place byinteractions between their cytoplasmic tail and filamentousactin cytoskeleton [25,53,57]. Both neuron pathfinding andguided neurite elongation rely on integrins that projectfrom the leading edge of the neuronal growth cone andbind to guidance cues in the extracellular matrix (ECM).Thus, cell migration depends, in part, on interactionsbetween the ECM and the extracellular domain(s) of inte-grins that are stabilized by anchoring to the intracellular fil-amentous actin cytoskeleton.
One of the most obvious developmental events regulatedby thyroid hormone is the programmed migration of gran-ule cells from the external granular layer to the inner gran-ular layer in the neonatal cerebellum [30–32]. Here, just asin cultured astrocytes, hypothyroidism delays and dimin-ishes the timing of appearance and quantity of astrocyte-derived laminin found in the molecular layer of the devel-oping rat cerebellum as compared to that in the euthyroidcerebellum [20]. Thus, the inability of the hypothyroidastrocyte to anchor the laminin receptor leads to disruptionof the deposition and patterning of secreted laminin andultimately the loss of a key guidance cue for the migratinggranule cell.
Analysis of the ligand activation properties of this noveleffector molecule responsible for the T4-dependent modula-tion of actin polymerization revealed that the alanine sidechain played an essential role [52]. A net negative chargeon the alanine side chain of T4 destroyed its ability to mod-ulate the dynamics of microfilament remodeling, whereasan uncharged or positively charged alanine side chain onT4 retained functional control. Importantly, these ligandrequirements differ markedly from those determined forthe TR and for other T4-binding proteins [52], indicatingthat the mediator of T4-dependent actin polymerizationpossesses a unique ligand specificity. Importantly, the fail-ure of tetrac to initiate actin polymerization [52] differsfrom its ability to block glioma cell proliferation, or inhibitmigration of C6 cells (see above) suggesting that the anti-cancer properties of this thyroid hormone analog do notutilize regulated actin polymerization to exert its biologicaleffects.
Recent studies done in astrocytes from the TR0/0 mousethat lack all TRa-derived gene products revealed a disorga-nized actin cytoskeleton and marginal quantities of lamininbound to the cell surface; thyroid hormone has no effect onthese two processes [37]. Unlike the full length TRa geneproducts, both the TRDa1- and TRDa2-encoded polypep-tides bind T4 and rT3 with high affinity, but do not specif-ically bind T3. Expression of transfactor TRa1 did notrepair this defect in laminin deposition, while expressionof TRDa1, but not TRDa2 in TRa-null astrocytes restoredT4-dependent actin polymerization and led to the T4-dependent deposition of laminin on the cell surface [37].
These data imply that the delta forms derived from theTRa gene are biologically active and provide a clear molec-ular pathway by which thyroid hormone can regulate a keyelement of the developmental program of the brain.
References
[1] M.A. Arnaout, B. Mahalingam, J.P. Xiang, Integrin structure,
allostery, and bidirectional signaling, Annu. Rev. Cell Dev. Biol. 21
(2005) 381–410.
[2] J.H. Bassett, C.B. Harvey, G.R. Williams, Mechanisms of thyroid
hormone receptor-specific nuclear and extranuclear actions, Mol.
Cell. Endocrinol. 213 (2003) 1–11.
[3] J.J. Bergh, H.Y. Lin, L. Lansing, S.N. Mohamed, F.B. Davis, S.
Mousa, P.J. Davis, Integrin avb3 contains a cell surface receptor site
for thyroid hormone that is linked to activation of mitogen-activated
protein kinase and induction of angiogenesis, Endocrinology 146
(2005) 2864–2871.
[4] A.G. Burger, D. Engler, C. Sakaloff, V. Staeheli, The effects of
tetraiodothyroacetic and triiodothyroacetic acids on thyroid function
in euthyroid and hyperthyroid subjects, Acta Endocrinol. 92 (1979)
455–467.
[5] X. Cao, F. Kambe, L.C. Moeller, S. Refetoff, H. Seo, Thyroid
hormone induces rapid activation of Akt/protein kinase B-mamma-
lian target of rapamycin-p70S6K cascade through phosphatidylino-
sitol 3-kinase in human fibroblasts, Mol. Endocrinol. 19 (2005) 102–
112.
[6] M. Carey, The enhanceosome and transcriptional activity, Cell 92
(1998) 5–8.
[7] Y. Chen, P.L. Chen, C.F. Chen, Z.D. Sharp, W.H. Lee, Thyroid
hormone, T3-dependent phosphorylation and translocation of
Trip230 from the Golgi apparatus to the nucleus, Proc. Natl. Acad.
Sci. USA 96 (1999) 4443–4448.
[8] V. Cody, P.J. Davis, F.B. Davis, Molecular modeling of the
thyroid hormone interactions with avb3 integrin, Steroids 72 (2007)
165–170.
[9] H. Colognato, D.A. Winkelmann, P.D. Yurchenko, Laminin poly-
merization induces a receptor-cytoskeleton network, J. Cell Biol. 145
(1999) 619–631.
[10] S. D’Arezzo, S. Incerpi, F.B. Davis, F. Acconcia, M. Marino, R.N.
Farias, P.J. Davis, Rapid nongenomic effects of 3,5,3 0-triiodo-L-
thyronine on the intracellular pH of L-6 myoblasts are mediated by
intracellular calcium mobilization and kinase pathways, Endocrinol-
ogy 145 (2004) 5694–5703.
[11] F.B. Davis, V. Cody, P.J. Davis, L.J. Borzynski, S.D. Blas,
Stimulation by thyroid hormone analogues of red blood cell Ca2+-
ATPase activity in vitro. Correlation between hormone structure and
biological activity in a human cell system, J. Biol. Chem. 258 (1983)
12373–12377.
[12] P.J. Davis, A. Shih, H.Y. Lin, L.J. Martino, F.B. Davis, Thyroxine
promotes association of mitogen-activated protein kinase and nuclear
thyroid hormone receptor (TR) and causes serine phosphorylation of
TR, J. Biol. Chem. 275 (2000) 38032–38039.
[13] F.B. Davis, S.A. Mousa, L. O’Connor, S. Mohamed, H.Y. Lin, H.J.
Cao, P.J. Davis, Proangiogenic action of thyroid hormone is
fibroblast growth factor-dependent and is initiated at the cell surface,
Circ. Res. 94 (2004) 1500–1506.
[14] P.J. Davis, F.B. Davis, V. Cody, Membrane receptors mediating
thyroid hormone action, Trends Endocrinol. Metab. 16 (2005)
429–435.
[15] F.B. Davis, H.Y. Tang, A. Shih, T. Keating, L. Lansing, A.
Hercbergs, R.A. Fenstermaker, A. Mousa, S.A. Mousa, P.J. Davis,
H.Y. Lin, Acting via a cell surface receptor, thyroid hormone is a
growth factor for glioma cells, Cancer Res. 66 (2006) 72707275.
[16] P.J. Davis, F.B. Davis, H.Y. Lin, L-Thyroxine acts as a hormone as
well as a prohormone at the cell membrane, Immun. Endocr. Metab.
Agents Med. Chem. 6 (2006) 235–240.
216 P.J. Davis et al. / Frontiers in Neuroendocrinology 29 (2008) 211–218

[17] A. De Milito, S. Fais, Proton pump inhibitors may reduce tumour
resistance, Expert Opin. Pharmacother. 6 (2005) 1049–1054.
[18] Z. Dong, C. Huang, W.Y. Ma, PI-3 kinase in signal transduction, cell
transformation, and as a target for chemoprevention of cancer,
Anticancer Res. 19 (1999) 3743–3747.
[19] C. Faivre-Sarrailh, A. Rabie, A lower proportion of filamentous to
monomeric actin in the developing cerebellum of thyroid-deficient
rats, Brain Res. 469 (1988) 293–297.
[20] A.P. Farwell, S.A. Dubord-Tomasetti, Thyroid hormone regulates
the extracellular organization of laminin on astrocytes, Endocrinol-
ogy 140 (1999) 5014–5021.
[21] A.P. Farwell, S.A. Dubord-Tomasetti, A.Z. Pietrzykowski, J.L.
Leonard, Regulation of cerebellar neuronal migration and neurite
outgrowth by thyroxine and 3,30,5 0-triiodothyronine, Brain Res. Dev.
Brain Res. 154 (2005) 121–135.
[22] A.P. Farwell, R.M. Lynch, W.C. Okulicz, A.M. Comi, J.L. Leonard,
The actin cytoskeleton mediates the hormonally regulated translation
of type II iodothyronine 50-deiodinase in astocytes, J. Biol. Chem. 265
(1990) 18546–18553.
[23] A.P. Farwell, M.P. Tranter, J.L. Leonard, Thyroxine-dependent
regulation of integrin-laminin interactions in astrocytes, Endocrinol-
ogy 136 (1995) 3909–3915.
[24] X. Feng, Y. Jiang, P. Meltzer, P.M. Yen, Thyroid hormone
regulation of hepatic genes in vivo detected by complimentary
DNA microarray, Mol. Endocrinol. 14 (2000) 947–955.
[25] R.B. Fishman, M.E. Hatten, Multiple receptor systems promote CNS
neural migration, J. Neurosci. 13 (1993) 3485–3495.
[26] M.W. Hamolsky, A. Golodetz, A.S. Freedberg, The plasma protein-
thyroid hormone complex in man. III. Further studies on the use of
the in vitro red blood cell uptake of I131-L-triiodothyronine as a
diagnostic test of thyroid function, J. Clin. Endocrinol. Metab. 19
(1959) 103–116.
[27] A.A. Hercbergs, L.K. Goyal, J.H. Suh, S. Lee, C.A. Reddy, B.H.
Cohen, G.H. Stevens, S.K. Reddy, D.M. Peereboom, P.J. Elson,
M.K. Gupta, G.H. Barnett, Propylthiouracil-induced chemical hypo-
thyroidism with high-dose tamoxifen prolongs survival in recurrent
high grade glioma: a Phase I/II study, Anticancer Res. 23 (2003)
617–626.
[28] Y. Hiroi, H.H. Kim, H. Ying, F. Furuya, Z. Huang, T. Simoncini, K.
Noma, K. Ueki, N.H. Nguyen, T.S. Scanlan, M.A. Moskowitz, S.Y.
Cheng, J.K. Liao, Rapid nongenomic actions of thyroid hormone,
Proc. Natl. Acad. Sci. USA 103 (2006) 14104–14109.
[29] S. Incerpi, P. Luly, P. De Vito, R.N. Farias, Short-term effects of
thyroid hormones on the Na/H antiporter in L-6 myoblasts: high
molecular specificity for 3,305-triiodo-L-thyronine, Endocrinology 140
(1999) 683–689.
[30] J.M. Lauder, Hormonal and humoral influences on brain develop-
ment, Psychoneuroendocrin 8 (1983) 121–155.
[31] J. Legrand, [Analysis of the morphogenetic action of thyroid
hormones on the cerebellum of young rats], Arch Anat. Microsc.
Morphol. Exp. 56 (1967) 205–244.
[32] J. Legrand, Thyroid hormones and maturation of the nervous system,
J. Physiol. 78 (1982) 603–652.
[33] J. Lei, S. Nowbar, C.N. Mariash, D.H. Ingbar, Thyroid hormone
stimulates Na–K-ATPase activity and its plasma membrane insertion
in rat alveolar epithelial cells, Am. J. Physiol. 285 (2003) L762–L772.
[34] J. Lei, C.N. Mariash, S.H. Ingbar, 3,3 05-Triiodo-L-thyronine up-
regulation of Na,K-ATPase activity and cell surface expression in
alveolar epithelial cells is Src kinase- and phosphoinositide 3-kinase-
dependent, J. Biol. Chem. 279 (2004) 47589–47600.
[35] J. Lei, C.H. Wendt, D. Fan, C.N. Mariash, D.H. Ingbar, Develop-
mental acquisition of T3-sensitive Na–K-ATPase stimulation by rat
alveolar epithelial cells, Am. J. Physiol. 292 (2007) L6–L-14.
[36] J.L. Leonard, A.P. Farwell, Cytoskeletal actions of iodothyronines,
Hot Thyroidology, December (<www.hotthyroidology.com>) 2006.
[37] J.L. Leonard, A.P. Farwell, Cytoskeletal actions of iodothyronines,
in: Proceedings of the 88th Annual Meeting of The Endocrine
Society, Boston, MA, 2006.
[38] J.L. Leonard, A.P. Farwell, P.M. Yen, W.W. Chin, M. Stula,
Differential expression of thyroid hormone receptor isoforms in
neurons and astroglial cells, Endocrinology 135 (1994) 548–555.
[39] H.Y. Lin, A. Shih, F.B. Davis, P.J. Davis, Thyroid hormone
promotes the phosphorylation of STAT3 and potentiates the action
of epidermal growth factor in cultured cells, Biochem. J. 338 (1999)
427–432.
[40] H.Y. Lin, H.Y. Tang, A. Shih, T. Keating, G. Cao, P.J. Davis, F.B.
Davis, Thyroid hormone is a MAPK-dependent growth factor for
thyroid cancer cells and is anti-apoptotic, Steroids 72 (2007) 180–187.
[41] A. Lombardi, A. Lanni, M. Moreno, M.D. Brand, F. Goglia, Effect
of 3,5-di-iodo-L-thyronine on the mitochondrial energy-transduction
apparatus, Biochem. J. 330 (1998) 521–526.
[42] P. Maruvada, C.T. Baumann, G.L. Hager, P.M. Yen, Dynamic
shuttling and intranuclear mobility of nuclear hormone receptors, J.
Biol. Chem. 278 (2003) 12425–12432.
[43] L.D. Miller, P. McPhie, H. Suzuki, Y. Kato, E.T. Liu, S.Y. Cheng,
Multi-tissue gene-expression analysis in a mouse model of thyroid
hormone resistance, Genome Biol. 5 (2004) R31.
[44] Y. Mizuno, Y. Kanou, M. Rogatcheva, T. Imai, S. Refetoff, H. Seo,
Y. Murata, Genomic organization of mouse ZAKI-4 gene that
encodes ZAKI-4, alpha and beta isoforms, endogenous calcineurin
inhibitors, and changes in the expression of these isoforms by thyroid
hormone in adult mouse brain and heart, Eur. J. Endocrinol. 150
(2004) 371–380.
[45] L.C. Moeller, X. Cao, A.M. Dumitrescu, H. Seo, S. Refetoff, Thyroid
hormone mediated changes in gene expression can be initiated by
cytosolic action of the thyroid hormone receptor beta through the
phosphatidylinositol 3-kinase pathway, Nucl. Recept. Signal. 4 (2006)
e020.
[46] S.A. Mousa, L. O’Connor, J.J. Bergh, F.B. Davis, T.S. Scanlan, P.J.
Davis, The proangiogenic action of thyroid hormone analogue GC-1
is initiated at an integrin, J. Cardiovasc. Pharmacol. 46 (2005)
356–360.
[47] S.A. Mousa, L. O’Connor, F.B. Davis, P.J. Davis, Proangiogenesis
action of the thyroid hormone analog 3,5-diiodothyropropionic acid
(DITPA) is initiated at the cell surface and is integrin-mediated,
Endocrinology 147 (2006) 1602–1607.
[48] S.A. Mousa, L. O’Connor, F.B. Davis, J.J. Bergh, P.J. Davis,
Tetraiodothyroacetic acid inhibits angiogenesis. Annual Meeting of
the American Thyroid Association, Abstract 107, Thyroid (2006) 16,
p. 893.
[49] L.K. Nieman, F.B. Davis, P.J. Davis, E.E. Cunningham, S. Gutman,
S.D. Blas, M. Schoenl, Effect of end-stage renal disease on respon-
siveness to calmodulin and thyroid hormone of calcium-ATPase in
human red blood cells, Kidney Int. 16 (1983) S167–S170.
[50] E.F. Plow, T.A. Haas, L. Zhang, J. Loftus, J.W. Smith, Ligand
binding to integrins, J. Biol. Chem. 275 (2000) 21785–21788.
[51] C. Rho, Tzirogiannis, T. Keating, A. Hercbergs, F.B. Davis, P.J.
Davis, H.Y. Tang, J. Bergh, S. Mousa, H.Y. Lin, Enhanced
proliferation of human lung adenocarcinoma and small cell lung
carcinoma cells directed from the cell surface by thyroid hormone, in:
Proceedings of the 89th Annual Meeting of The Endocrine Society,
Toronto, Ont., Canada, Abstract P1-602, 2007.
[52] M. Safran, A. Farwell, H. Rokos, J. Leonard, Structural require-
ments of iodothyronines for the rapid inactivation and internalization
of type II iodothyronine 5 0-deiodinase in glial cells, J. Biol. Chem. 268
(1993) 14224–14229.
[53] S.M. Schoenwaelder, K. Burridge, Bidirectional signaling between the
cytoskeleton and integrins, Curr. Opin. Cell Biol. 11 (1999) 274–286.
[54] J. Segal, S.H. Ingbar, Evidence that an increase in cytoplasmic
calcium is the initiating event in certain plasma membrane-mediated
responses to 3,5,30-triiodothyronine in rat thymocytes, Endocrinology
124 (1989) 1949–1955.
[55] J. Segal, S.H. Ingbar, 3,5,3 0-Triiodothyronine enhances sugar trans-
port in rat thymocytes by increasing the intrinsic activity of the
plasma membrane sugar transporter, J. Endocrinol. 124 (1990)
133–140.
P.J. Davis et al. / Frontiers in Neuroendocrinology 29 (2008) 211–218 217

[56] C.A. Siegrist-Kaiser, C. Juge-Aubry, M.P. Tranter, D.M. Ekenbar-
ger, J.L. Leonard, Thyroxine-dependent modulation of actin poly-
merization in cultured astrocytes. A novel extranuclear action of
thyroid hormone, J. Biol. Chem. 265 (1990) 5296–5302.
[57] T.N. Stitt, U.E. Gasser, M.E. Hatten, Molecular mechanisms of glial-
guided neuronal migration, Ann. N. Y. Acad. Sci. 633 (1991)
113–121.
[58] A. Sugawara, P.M. Yen, J.W. Apriletti, R.C. Ribeiro, D.B. Sacks,
J.D. Baxter, W.W. Chin, Phosphorylation selectively increases
triiodothyronine receptor homodimer binding to DNA, J. Biol.
Chem. 269 (1994) 433–437.
[59] J.W. Tamkun, D.W. DeSimone, D. Fonda, R.S. Patel, C. Buck, A.F.
Horwitz, R.O. Hynes, Structure of integrin, a glycoprotein involved
in the transmembrane linkage between fibronectin and actin, Cell 46
(1986) 271–282.
[60] H.Y. Tang, H.Y. Lin, S. Zhang, F.B. Davis, P.J. Davis, Thyroid
hormone causes mitogen-activated protein kinase-dependent phos-
phorylation of the nuclear estrogen receptor, Endocrinology 145
(2004) 3265–3272.
[61] Y. Wu, R.J. Koenig, Gene regulation by thyroid hormone, Trends
Endocrinol. Metab. 11 (2000) 207–211.
[62] P.M. Yen, S. Ando, X. Feng, Y. Liu, P. Maruvada, X. Xia, Thyroid
hormone action at the cellular, genomic and target gene level, Mol.
Cell. Endocrinol. 246 (2006) 121–127.
[63] J. Zhang, M.A. Lazar, The mechanism of action of thyroid
hormones, Annu. Rev. Physiol. 62 (2000) 439–466.
[64] X.G. Zhu, J.A. Hanover, G.L. Hager, S.Y. Cheng, Hormone-induced
translocation of thyroid hormone receptors in living cells visualized
using a receptor green fluorescent protein chimera, J. Biol. Chem. 273
(1998) 27058–27063.
218 P.J. Davis et al. / Frontiers in Neuroendocrinology 29 (2008) 211–218

Minireview: The Sodium-Iodide Symporter NIS
and Pendrin in Iodide Homeostasis of the Thyroid
Aigerim Bizhanova and Peter Kopp
Division of Endocrinology, Metabolism, and Molecular Medicine, Feinberg School of Medicine, Northwestern
University, Chicago, Illinois 60611
Thyroid hormones are essential for normal development and metabolism. Thyroid hormone bio-
synthesis requires iodide uptake into the thyrocytes and efflux into the follicular lumen, where it
is organified on selected tyrosyls of thyroglobulin. Uptake of iodide into the thyrocytes is mediated
by an intrinsic membrane glycoprotein, the sodium-iodide symporter (NIS), which actively cotrans-
ports two sodium cations per each iodide anion. NIS-mediated transport of iodide is driven by the
electrochemical sodium gradient generated by the Na1/K1-ATPase. NIS is expressed in the thyroid,
the salivary glands, gastric mucosa, and the lactating mammary gland. TSH and iodide regulate
iodide accumulation by modulating NIS activity via transcriptional and posttranscriptional mech-
anisms. Biallelic mutations in the NIS gene lead to a congenital iodide transport defect, an auto-
somal recessive condition characterized by hypothyroidism, goiter, low thyroid iodide uptake, and
a low saliva/plasma iodide ratio. Pendrin is an anion transporter that is predominantly expressed
in the inner ear, the thyroid, and the kidney. Biallelic mutations in the SLC26A4 gene lead to
Pendred syndrome, an autosomal recessive disorder characterized by sensorineural deafness, goi-
ter, and impaired iodide organification. In thyroid follicular cells, pendrin is expressed at the apical
membrane. Functional in vitro data and the impaired iodide organification observed in patients
with Pendred syndrome support a role of pendrin as an apical iodide transporter. (Endocrinology
150: 1084–1090, 2009)
The iodide-containing thyroid hormones T3 and its precursor
T4 are crucial for normal development, growth, and regu-
lation of numerous metabolic pathways. The main function of
the thyroid gland is to concentrate iodide and to make it available
for biosynthesis of thyroid hormones. The significance of this
mechanism is evident in light of the scarcity of iodide in most of
the environment and the fact that insufficient dietary supply of
iodide remains a major public health issue in many parts of the
world (1).
The synthesis of thyroid hormones requires a normally de-
veloped thyroid gland, an adequate nutritional intake of iodide,
and a series of sequential biochemical steps. Thyroid hormone
synthesis takes place in the follicles, the functional units of the
gland (2). Each follicle consists of a single layer of thyroid epi-
thelial cells surrounding the follicular lumen. The follicular lu-
men is filled with colloid, which is predominantly composed of
thyroglobulin, a large glycoprotein that serves as the scaffold for
thyroid hormone synthesis (3).
The synthesis of thyroid hormones requires uptake of iodide
across the basolateral membrane into the thyrocytes, transport
across the cell, and efflux through the apical membrane into the
follicular lumen. Uptake of iodide is mediated by the sodium-
iodide symporter (NIS), which cotransports two sodium ions
along with one iodide ion, with the sodium gradient serving as
the driving force (Fig. 1) (1). The energy required to produce the
sodium gradient is provided by the ouabain-sensitive Na1/K1-
ATPase (4). The efflux of iodide across the apical membrane is
mediated, at least in part, by pendrin (5). Once iodide reaches the
cell-colloid interface, it is oxidized and rapidly organified by
incorporation into selected tyrosyl residues of thyroglobulin.
This reaction, referred to as organification, is catalyzed by thy-
roid peroxidase (TPO) in the presence of hydrogen peroxide
and results in the formation of mono- and diiodotyrosines
(MIT and DIT). The generation of hydrogen peroxide is me-
diated by the calcium-dependent reduced nicotinamide ade-
nine dinucleotide phosphate (NADPH) dual oxidase type 2
ISSN Print 0013-7227 ISSN Online 1945-7170
Printed in U.S.A.
Copyright © 2009 by The Endocrine Society
doi: 10.1210/en.2008-1437 Received October 14, 2008. Accepted January 22, 2009.
First Published Online February 5, 2009
Abbreviations: CICn5, Chloride channel 5; DIT, diiodotyrosine; DUOX2, dual oxidase type
2; ITD, iodide transport defect; MDCK, Madin-Darby canine kidney; MIT, monoiodoty-
rosine; NIS, sodium-iodide symporter; PKA, protein kinase A; SLC5A, solute carrier 5A; TPO,
thyroperoxidase.
M I N I R E V I E W
1084 endo.endojournals.org Endocrinology, March 2009, 150(3):1084–1090

(DUOX2). TPO also catalyzes the coupling of two iodoty-
rosines to form either T3 or T4. To release thyroid hormones,
thyroglobulin is engulfed by pinocytosis, digested in lyso-
somes, and then secreted into the bloodstream at the baso-
lateral membrane (2). The mechanisms of hormone secretion
at the basolateral membrane and the involved channel(s) have
not been characterized.
NIS
NIS is an integral plasma membrane glycoprotein localized at the
basolateral plasma membrane of thyrocytes (6). This protein
plays an essential role in thyroid physiology by mediating uptake
of iodide into the thyrocytes, a key step in thyroid hormone
synthesis. The ability of the thyroid gland to accumulate radio-
iodine is also a cornerstone in the diagnosis and treatment of
thyroid disorders (7).
The molecular characterization of NIS began in 1996 when
the cDNA encoding rat NIS was isolated by expression-cloning
in Xenopus laevis oocytes (6). Subsequently, the human cDNA
has been cloned by a RT-PCR approach taking advantage of the
homology to rat NIS (8). Rat NIS is predicted to have 618 amino
acids with a relative molecular mass of 65,196 Daltons (6), and
human NIS contains 643 amino acids and exhibits an 84%
amino acid identity and 93% similarity to rat NIS (9). The hu-
man NIS gene is located on chromosome 19p12-13.2 and con-
tains 14 introns and 15 exons (9).
NIS (SLC5A5) belongs to the solute carrier family 5A
(SLC5A). All members of this protein family depend on an elec-
trochemical sodium gradient as the driving force for transport of
anions across the plasma membrane (10). The current secondary
structure model predicts that NIS contains 13 transmembrane
domains with the amino terminus located extracellularly and the
carboxy terminus facing the cytosol (1). The mature NIS protein
is approximately 87 kDa in size and has three asparagine-linked
glycosylation sites (1). Glycosylation does not seem to be re-
quired for stability, activity, or targeting of the NIS molecule to
the plasma membrane (11). The expression of NIS is differen-
tially regulated and is subjected to various posttranslational
modifications in each tissue in which it is expressed (1, 12).
In addition to thyroid follicular cells, NIS is expressed in sev-
eral other tissues, including the salivary glands, gastric mucosa,
and the lactating mammary gland, where it mediates active trans-
port of iodide (1). In the lactating mammary gland, NIS plays an
important role by concentrating iodide in the milk, thereby sup-
plying newborns with iodide for thyroid hormone synthesis.
Although NIS has a high affinity for iodide, it is able to me-
diate transport of other ions (13). Large anions such as thiocy-
anate and perchlorate can inhibit accumulation of iodide in the
thyroid by competing with iodide (14, 15). Perchlorate is 10–100
times more potent than thiocyanate in inhibiting iodide accu-
mulation in the thyroid (1). The ability of perchlorate to block
iodide transport has been used in the therapy of hyperthyroid-
ism, and it is used in the perchlorate discharge test, which serves
to detect defects in iodide organification (16). In normal indi-
viduals, administration of perchlorate blocks subsequent accu-
mulation of iodide in the thyrocytes but does not cause any dis-
charge of previously accumulated radioiodine because of its
rapid organification. In contrast, in individuals with a total or
partial iodide organification defect, administration of perchlor-
ate results in the rapid release of the unorganified fraction of the
tracer from the thyrocytes (16). Until recently, it has been con-
troversial whether perchlorate acts as a blocker or as a substrate
that is transported by NIS (13, 17, 18). Two recent studies pro-
vide evidence that perchlorate is actively transported by NIS (19,
20). Perchlorate transport could be demonstrated in a polarized
cell system (19) as well as by direct measurement with mass
spectrometry (20). Moreover, it has been shown that perchlor-
ate, a widely found pollutant, is transported into the milk (19).
Remarkably, the stoichiometry of the NIS-mediated Na1/ClO42
transport is electroneutral, which contrasts with the electrogenic
transport of iodide (two Na1 and one I2) (19). These findings
indicate that NIS is able to transport different substrates with
distinct stoichiometries (19).
Apical membrane
Basolateral membraneNa/K-ATPase NIS
TG
MIT
DIT
Hydrolysis
NADP+ NADPH
Ca2+
Na+
Na+ 2 Na+ I-K+
DEHAL1
DUOX2
DUOXA2
TPO
H2O2
PDS
I-
I- I-
?
T3
T4
Io
dinated
TG
T4O CH2-HO
I
I
I
I
TG
T3O CH2-HO
I
II
TG
DIT
MIT
-HO CH2
-HO CH2
I
I
I
TG
DIT
DIT
-HO CH2
-HO CH2
I
I
I
I
TG
-HO CH2
-HO CH2
TG
Organification
TPO
H2O2
I-
Coupling
TPO
H2O2
I-
Follicular lumen
FIG. 1. Main steps in thyroid hormone synthesis. At the basolateral membrane
of thyroid follicular cells, which form the follicles, iodide is transported into
thyrocytes by the NIS. NIS is dependent on the sodium gradient created by the
Na/K-ATPase. At the apical membrane, iodide efflux is, in part, mediated by
pendrin (PDS/SLC26A4). At the cell-colloid interface, iodide is oxidized by TPO in
the presence of H2O2. H2O2 is produced by the calcium- and reduced
nicotinamide adenine dinucleotide phosphate-dependent (NADPH) enzyme
DUOX2. DUOX2 requires a specific maturation factor, DUOXA2. Thyroglobulin
(TG), which is secreted into the follicular lumen, serves as matrix for synthesis of
T4 and T3. First, TPO catalyzes iodination of selected tyrosyl residues
(organification), which results in the formation of MIT and DIT. Subsequently,
two iodotyrosines are coupled to form either T4 or T3 in a reaction that is also
catalyzed by TPO. Iodinated thyroglobulin is stored as colloid in the follicular
lumen. Upon a demand for thyroid hormone secretion, thyroglobulin is
internalized into the follicular cell by pinocytosis and digested in lysosomes, which
generates T4 and T3 that are released into the bloodstream through unknown
mechanisms. The unused MIT and DIT are retained in the cell and deiodinated by the
iodotyrosine dehalogenase 1 (DEHAL1). The released iodide is recycled for thyroid
hormone synthesis.
Endocrinology, March 2009, 150(3):1084–1090 endo.endojournals.org 1085

Regulation of NIS protein expression
TSH is the major regulator of thyroid cell proliferation, differ-
entiation, and function, including iodide uptake (21). The effects
of TSH are primarily mediated through the activation of the
cAMP cascade via the GTP-binding protein Gsa (22). TSH stim-
ulates iodide accumulation by positively regulating NIS expres-
sion at the protein and mRNA level via the cAMP pathway (23).
Hypophysectomized rats with low circulating levels of TSH have
a decreased protein expression of NIS, whereas a single injection
of TSH leads to a prompt increase in NIS expression (24). Rats
maintained on an iodide-deficient diet or treated with propyl-
thiouracil, an agent blocking iodide organification, have high
concentrations of TSH, which correlates with an increase in NIS
protein expression. These findings are in agreement with the
results obtained in human thyroid primary cultures (25, 26) and
the rat thyroid FRTL-5 cell line (23). In FRTL-5 cells, withdrawal
of TSH results in a decrease in intracellular cAMP concentration
and iodide uptake activity (23). Re-addition of TSH increases
NIS mRNA and protein expression and subsequently restores
iodide uptake activity.
Recent studies have shown that TSH not only regulates NIS
transcription and biosynthesis but also mediates NIS activity by
posttranscriptional mechanisms (27). In the presence of TSH,
NIS is active and inserted in the basolateral membrane of thy-
rocytes (27). Upon TSH withdrawal, NIS protein half-life de-
creases from 5 to 3 d, and it translocates from the plasma mem-
brane to intracellular compartments (27). As of yet, the
mechanisms regulating the subcellular distribution of NIS are
only partially elucidated. It is known that NIS has several con-
sensus sites for kinases, including protein kinase A (PKA) and
protein kinase C (1). Although it has been shown that NIS is
phosphorylated in vivo (27), the functional significance of this
modification needs further characterization. A more recent study
identified five amino acid residues in NIS that are phosphory-
lated in vivo, but the phosphorylation status of these amino acid
residues does not affect targeting of NIS to the plasma membrane
(28). NIS contains several sorting sequences that are known to
play a role in targeting, retention, and endocytosis of other mem-
brane proteins (29). For example, the PDZ motif (T/S-X-V/L)
located at the carboxy terminus of NIS is one of the sequences
involved in protein-protein interactions (1). The PDZ motif is
recognized by PDZ-binding proteins that have been implicated in
internalization of other transporters (29). NIS also has a
dileucine motif, L557L558, which is known to interact with the
clathrin-coated system (30). This interaction leads to incorpo-
ration of integral membrane proteins into coated vesicles that are
then carried to different destinations within the cell (31).
Iodide is another factor that can regulate iodide accumulation
in the thyroid. In 1948, Wolff and Chaikoff (32) reported that
high doses of iodide block iodide organification in the rat thyroid
in vivo. This phenomenon, known as the acute Wolff-Chaikoff
effect, is a reversible process, because iodide organification re-
sumes when the iodide concentration in the serum decreases. The
mechanisms underlying the Wolff-Chaikoff effect are complex
and involve acute regulation of several key genes and proteins
within the thyrocytes. Several studies have examined the effect of
iodide on NIS mRNA and protein expression in vivo and in vitro
(33–35). In vivo data suggest that high concentrations of iodide
lead to reduction in both NIS mRNA and protein levels, partially
by a transcriptional mechanism. In vitro results suggest that ex-
posure to high doses of iodide results in a decrease in NIS protein
levels that is, at least in part, due to an increase in NIS protein
turnover (33–35).
Congenital iodide transport defect (ITD)
Biallelic mutations in the NIS gene cause a congenital ITD. ITD
is an autosomal recessive condition characterized by hypothy-
roidism, goiter, reduced or absent thyroid uptake of radioiodide,
and a low saliva/plasma iodide ratio (1, 36). Currently, at least
12 ITD-causing mutations of NIS have been identified (37). Six
of these mutations, namely 226delH, T354P, G395R, Q267E,
G543E, and V59E have been characterized more thoroughly
(38–41). The G543E substitution leads to retention of NIS in
intracellular compartments as a result of improper maturation
and trafficking of the protein, the other mutants are still targeted
to the membrane but result in a loss of function (38–41). Struc-
tural and functional analysis of the T354P mutant protein dem-
onstrated that a hydroxyl group at the b-carbon of the residue at
position 354 is crucial for proper NIS function (38). Substitu-
tions of the glycine residue at position 395 residue with several
amino acids indicated that the presence of a small and an un-
charged amino acid residue at this position is required for NIS
function (39). Lastly, a recent study revealed that the histidine
residue at position 226 is important for the iodide transport
activity of NIS (37).
Pendrin
Pendrin is a highly hydrophobic membrane protein located at the
apical membrane of thyrocytes (2, 4). In addition to the thyroid,
pendrin is also expressed in the kidney and in the inner ear (42,
43). In the kidney, pendrin plays an important role in acid-base
metabolism as an exchanger of chloride and bicarbonate in b-in-
tercalated cells (44). In the inner ear, pendrin is important for
generation of the endocochlear potential (45).
Pendrin belongs to the SLC26A family, which includes several
anion transporters, as well as the motor protein prestin that is
expressed in outer hair cells (46, 47). Pendrin is encoded by the
SLC26A4 gene, which was cloned in 1997 (48). The SLC26A4
gene is located on chromosome 7q21-31 and contains 21 exons
with an open reading frame of 2343 bp (48).
Pendrin is a glycoprotein composed of 780 amino acids (2). It
contains three putative extracellular asparagine-glycosylation
sites (4, 49). Pendrin usually appears as a single protein band
with a molecular mass of 110–115 kDa when isolated from
human thyroid membranes (49). Pendrin is proposed to have 12
transmembrane domains with both amino and carboxy termini
located inside the cytosol (4, 50). Like other members of the
SLC26A family, pendrin contains a so-called STAS (sulfate
transporter and antisigma factor antagonist) domain (51). The
1086 Bizhanova and Kopp Minireview Endocrinology, March 2009, 150(3):1084–1090

exact function of this domain has not been elucidated. Recent
studies, however, suggest that the STAS domain can interact with
the regulatory domain of CFTR (cystic fibrosis transmembrane
conductance regulator) in certain epithelial cells (52–54).
Pendred syndrome
Mutations in the SLC26A4 gene lead to Pendred syndrome (2).
Pendred syndrome is an autosomal recessive disorder character-
ized by sensorineural deafness, goiter, and a partial defect in
iodide organification (55, 56). Deafness or hearing impairment
is the leading clinical sign of Pendred syndrome (56). In many
patients, hearing loss is prelingual; in some individuals, however,
the hearing loss develops later in childhood (57). Patients with
Pendred syndrome display an enlarged endolymphatic sac and
duct (58–60). A subset of patients presents with a so-called
Mondini defect, which is characterized by replacement of the
cochlear turns by a single cavity or a rudimentary cochlea (57,
58). Variability in the hearing loss observed in patients with
SLC26A4 mutations suggests that the phenotype is influenced by
environmental factors and/or genetic modifiers (61).
Goiter usually develops during childhood. There is, however,
a substantial variation within and between families and different
geographic regions (62–64). Nutritional iodide intake appears
to play an important role as a modifier of the thyroidal pheno-
type (65, 66). Under conditions of high iodide intake, most in-
dividuals have no or only a mild enlargement of the thyroid (60,
67). If the nutritional iodide is scarce, patients with Pendred
syndrome not only develop goiter but may also present with mild
or overt hypothyroidism (68, 69).
Mutations in the SLC26A4 gene, found in patients with Pen-
dred syndrome, are highly heterogeneous (56). Currently, more
than 150 mutations of the SLC26A4 gene have been reported
(56). Most of the mutations are missense mutations, and a
smaller number of mutations consists mainly of nonsense and
intronic mutations (56). Loss of function of some of the mutants
results from the retention of the mutated and misfolded protein
in intracellular compartments, most likely the endoplasmic re-
ticulum (70, 71).
Role of pendrin in the thyroid
Initial functional studies of pendrin in Xenopus oocytes have
demonstrated that pendrin is able to mediate transport of chlo-
ride and iodide (5) and that it can act as a chloride/formate
exchanger (72). The ability of pendrin to mediate iodide efflux
(5), the localization of pendrin at the apical membrane of thy-
rocytes (4, 73), as well as the defect in iodide organification
observed in patients with Pendred syndrome (62, 74), suggested
that pendrin could function as an apical iodide transporter in
thyroid cells (46). The results obtained from a number of inde-
pendent studies performed in heterologous systems support the
role of pendrin in mediating, at least in part, apical iodide efflux.
Yoshida et al. (75) have demonstrated that iodide efflux is much
higher in nonpolarized Chinese hamster ovary cells expressing
NIS and pendrin than in cells expressing NIS alone. Electrophys-
iological studies with transfected COS-7 cells also indicate that
pendrin mediates iodide transport and that it is more efficient at
high extracellular concentrations of chloride (76). In addition,
iodide efflux/chloride influx appears to be more efficient than
chloride efflux/iodide influx (76). These findings are consistent
with results obtained in polarized Madin-Darby canine kidney
(MDCK) cells (50). MDCK cells expressing NIS and pendrin
independently or simultaneously were cultured in a bicameral
system, which allowed measuring iodide uptake at the basolat-
eral membrane and iodide efflux at the apical membrane. Cells
transfected with NIS alone have a significant increase in intra-
cellular iodide uptake compared with untransfected control
cells. In contrast, cells expressing NIS and pendrin show a sig-
nificant increase in iodide transport into the apical chamber and
consequently a significant decrease in the intracellular iodide
content. These findings support the notion that pendrin may
have a role in facilitating vectorial iodide transport at the apical
membrane (50). The partial organification defect found in pa-
tients with Pendred syndrome suggests, however, that iodide can
reach the follicular lumen independently of the presence of
pendrin.
Questions concerning the role of pendrin asan apical iodide transporter
The traditional concept held that iodide simply crosses the apical
membrane due to the electrochemical gradient that is present
between the cytosol and the follicular lumen (77). However,
autoradiography studies revealed that iodide first accumulates in
the cytosol and subsequently moves to the follicular lumen (78).
This transport of iodide across the apical membrane is rapidly
stimulated by TSH (64, 79). Hence, it has been proposed that
apical iodide efflux is mediated through a specific transporter or
channel. This concept has risen based on several findings. Elec-
trophysiological studies performed with thyroid membrane ves-
icles suggested the presence of two apical iodide transporters
(80). Functional studies performed in heterologous cells, includ-
ing polarized cells (50, 75, 76, 81), along with the iodide organi-
fication defects found in patients with Pendred syndrome (62,
74), suggested that pendrin mediates apical iodide efflux in thy-
rocytes. The physiological role of pendrin has, however, been
questioned for several reasons (77). Patients with biallelic mu-
tations in the SLC26A4 gene display a mild or no thyroidal phe-
notype under conditions of sufficient iodide intake (65). The
pendrin knockout mice, studied under normal iodide intake con-
ditions, do not develop a goiter or abnormal thyroid hormone
levels (45). In addition, it is intriguing that pendrin may have
distinct roles in the thyroid, the inner ear, and the kidney (77).
This led to the proposal that pendrin may be a part of multipro-
tein complex, the composition of which may vary among differ-
ent cell types, thereby potentially explaining a variability in an-
ion selectivity of pendrin (77). Other proteins (SLC5A8 and
chloride channel 5 ClCn5) have been proposed to mediate apical
iodide efflux (82, 83). Functional studies performed in Xenopus
oocytes and polarized MDCK cells clearly demonstrate that
Endocrinology, March 2009, 150(3):1084–1090 endo.endojournals.org 1087

SLC5A8, originally designated as human apical iodide trans-
porter (hAIT) (82), does not mediate iodide uptake or efflux (84).
Localization of the ClCn5 protein at the apical membrane of
thyrocytes and a thyroidal phenotype of the ClCn5-deficient
mice that is reminiscent of Pendred syndrome suggest that ClCn5
could be, possibly in conjunction with other chloride channels,
involved in mediating apical iodide efflux or iodide/chloride ex-
change (83). This possibility has, as of yet, not been corroborated
by further experimental data.
Regulation of pendrin expression
TSH stimulates iodide efflux across the apical membrane of thy-
rocytes (79, 85, 86). After exposure to TSH, iodide efflux is
rapidly stimulated in FRTL-5 cells (85) and in polarized porcine
thyrocytes (79, 86). In polarized porcine thyrocytes grown in a
bicameral system, measurement of iodide transport in both di-
rections demonstrates that TSH up-regulates iodide efflux at the
apical membrane, whereas efflux across the basolateral mem-
brane does not change (79). Treatment of rat thyroid PCCl3 cells
with TSH for a short time results in the rapid translocation of
pendrin from intracellular compartments, specifically endo-
somes, to the plasma membrane, thus suggesting a role of pen-
drin in rapid regulation of apical iodide efflux (87). Insertion of
pendrin in the apical membrane correlates with the phosphory-
lation of pendrin, but it remains unknown whether phosphory-
lation is necessary or sufficient for this translocation. These
events occur through the PKA pathway and can be inhibited by
H89, a specific PKA inhibitor (87). Interestingly, translocation of
pendrin from the cytosol to the plasma membrane through a
PKC-dependent pathway has been demonstrated after exposure
of cultured rat thyroid cells to insulin for 10, 20, and 40 min (88).
At least in rat FRTL-5 cells, TSH does not significantly modify
SLC26A4 gene expression (4). Interestingly, thyroglobulin has
been shown to up-regulate SLC26A4 mRNA levels in FRTL-5
cells while suppressing expression of several thyroid-specific
genes, including the TSH receptor, NIS, TPO, and TG genes (4).
Treatment with iodide does not affect expression of the
SLC26A4 gene (89). Exposure to thyroglobulin leads to a de-
creased NIS gene and protein expression and subsequently re-
sults in a reduced iodide uptake in vitro (90). In contrast, accu-
mulation of thyroglobulin in the follicular lumen suppresses
iodide uptake in vivo (90). It has been suggested that the inverse
relationship between the concentration of thyroglobulin in the
follicular lumen and iodide uptake in vivo may be important in
the regulation of thyroid function under constant TSH levels (91)
and promote iodide efflux into the follicular lumen (89).
Conclusions
• NIS mediates the active transport of iodide at the basolateral
membrane of thyrocytes.
• Biallelic mutations in NIS cause a congenital iodide transport
defect, an autosomal recessive condition, characterized by hy-
pothyroidism, goiter, low thyroid iodide uptake, and low sa-
liva/plasma iodide ratio.
• Pendrin is involved in the apical iodide efflux in thyroid cells.
It can also exchange chloride and bicarbonate.
• Pendrin is encoded by the SLC26A4 gene. Biallelic mutations
in the SLC26A4 gene cause Pendred syndrome, an autosomal
recessive disorder, characterized by deafness, goiter, and im-
paired iodide organification.
• In the inner ear, pendrin is important for anion and fluid
transport and for maintenance of the endocochlear potential.
• In the kidney, pendrin plays a role in acid-base metabolism as
a chloride/bicarbonate exchanger.
• In addition to pendrin, other apical iodide channels or trans-
porters may be involved in regulation of apical iodide efflux
in thyrocytes.
Acknowledgments
Address all correspondence and requests for reprints to: Peter Kopp,
M.D., Associate Professor, Director ad interim Center of Genetic Med-
icine, Division of Endocrinology, Metabolism, and Molecular Medicine,
Northwestern University, Tarry 15, 303 East Chicago Avenue, Chicago,
Illinois 60611. E-mail: [email protected].
Part of this work has been supported by Grant 1R01DK63024-01
from the National Institute of Diabetes and Digestive and Kidney Dis-
eases, National Institutes of Health (to P.K.).
References
1. Dohan O, De la Vieja A, Paroder V, Riedel C, Artani M, Reed M, Ginter CS,
Carrasco N 2003 The sodium/iodide symporter (NIS): characterization, reg-
ulation, and medical significance. Endocr Rev 24:48–77
2. Kopp P 2005 Thyroid hormone synthesis: thyroid iodine metabolism. In:
Braverman L, Utiger R, eds. Werner and Ingbar’s the thyroid: a fundamental
and clinical text. 9th ed. New York: Lippincott Williams Wilkins; 52–76
3. Arvan P, Di Jeso B 2005 Thyroglobulin structure, function, and biosynthesis.
In: Braverman L, Utiger R, eds. Werner and Ingbar’s the thyroid: a fundamental
and clinical text. New York: Lippincott Williams Wilkins; 77–95
4. Royaux IE, Suzuki K, Mori A, Katoh R, Everett LA, Kohn LD, Green ED 2000
Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical
porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5
cells. Endocrinology 141:839–845
5. Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP 1999 The Pendred
syndrome gene encodes a chloride-iodide transport protein. Nat Genet 21:
440–443
6. Dai G, Levy O, Carrasco N 1996 Cloning and characterization of the thyroid
iodide transporter. Nature 379:458–460
7. Mazaferri EL 2000 Carcinoma of the follicular epithelium. In: Braverman L,
Utiger R, eds. Thyroid: a fundamental and clinical text. 8th ed. New York:
Lippincott Williams & Wilkins; 904–930
8. Smanik PA, Liu Q, Furminger TL, Ryu K, Xing S, Mazzaferri EL, Jhiang SM
1996 Cloning of the human sodium iodide symporter. Biochem Biophys Res
Commun 226:339–345
9. Smanik PA, Ryu KY, Theil KS, Mazzaferri EL, Jhiang SM 1997 Expression,
exon-intron organization, and chromosome mapping of the human sodium
iodide symporter. Endocrinology 138:3555–3558
10. Reizer J, Reizer A, Saier Jr MH 1994 A functional superfamily of sodium/solute
symporters. Biochim Biophys Acta 1197:133–166
11. Levy O, De la Vieja A, Ginter CS, Riedel C, Dai G, Carrasco N 1998 N-linked
glycosylation of the thyroid Na1/I2 symporter (NIS). Implications for its sec-
ondary structure model. J Biol Chem 273:22657–22663
12. De La Vieja A, Dohan O, Levy O, Carrasco N 2000 Molecular analysis of the
sodium/iodide symporter: impact on thyroid and extrathyroid pathophysiol-
ogy. Physiol Rev 80:1083–1105
1088 Bizhanova and Kopp Minireview Endocrinology, March 2009, 150(3):1084–1090

13. Eskandari S, Loo DD, Dai G, Levy O, Wright EM, Carrasco N 1997 Thyroid
Na1/I2 symporter. Mechanism, stoichiometry, and specificity. J Biol Chem
272:27230–27238
14. Carrasco N 1993 Iodide transport in the thyroid gland. Biochim Biophys Acta
1154:65–82
15. Wolff J 1964 Transport of iodide and other anions in the thyroid gland. Physiol
Rev 44:45–90
16. Baschieri L, Benedetti G, Deluca F, Negri M 1963 Evaluation and limitations
of the perchlorate test in the study of thyroid function. J Clin Endocrinol Metab
23:786–791
17. Yoshida A, Sasaki N, Mori A, Taniguchi S, Mitani Y, Ueta Y, Hattori K, Sato
R, Hisatome I, Mori T, Shigemasa C, Kosugi S 1997 Different electrophysi-
ological character of I2, ClO42, and SCN2 in the transport by Na1/I2 sym-
porter. Biochem Biophys Res Commun 231:731–734
18. Yoshida A, Sasaki N, Mori A, Taniguchi S, Ueta Y, Hattori K, Tanaka Y,
Igawa O, Tsuboi M, Sugawa H, Sato R, Hisatome I, Shigemasa C, Grollman
EF, Kosugi S 1998 Differences in the electrophysiological response to I2 and
the inhibitory anions SCN2 and ClO42, studied in FRTL-5 cells. Biochim
Biophys Acta 1414:231–237
19. Dohan O, Portulano C, Basquin C, Reyna-Neyra A, Amzel LM, Carrasco N
2007 The Na1/I2 symporter (NIS) mediates electroneutral active transport of
the environmental pollutant perchlorate. Proc Natl Acad Sci USA 104:20250–
20255
20. Tran N, Valentin-Blasini L, Blount BC, McCuistion CG, Fenton MS, Gin E,
Salem A, Hershman JM 2008 Thyroid-stimulating hormone increases active
transport of perchlorate into thyroid cells. Am J Physiol Endocrinol Metab
294:E802–E806
21. Vassart G, Dumont JE 1992 The thyrotropin receptor and the regulation of
thyrocyte function and growth. Endocr Rev 13:596–611
22. Laglia G, Zeiger MA, Leipricht A, Caturegli P, Levine MA, Kohn LD, Saji M
1996 Increased cyclic adenosine 39,59-monophosphate inhibits G protein-cou-
pled activation of phospholipase C in rat FRTL-5 thyroid cells. Endocrinology
137:3170–3176
23. Weiss SJ, Philp NJ, Ambesi-Impiombato FS, Grollman EF 1984 Thyrotropin-
stimulated iodide transport mediated by adenosine 39,59-monophosphate and
dependent on protein synthesis. Endocrinology 114:1099–1107
24. Levy O, Dai G, Riedel C, Ginter CS, Paul EM, Lebowitz AN, Carrasco N 1997
Characterization of the thyroid Na1/I2 symporter with an anti-COOH ter-
minus antibody. Proc Natl Acad Sci USA 94:5568–5573
25. Saito T, Endo T, Kawaguchi A, Ikeda M, Nakazato M, Kogai T, Onaya T 1997
Increased expression of the Na1/I2 symporter in cultured human thyroid cells
exposed to thyrotropin and in Graves’ thyroid tissue. J Clin Endocrinol Metab
82:3331–3336
26. Kogai T, Curcio F, Hyman S, Cornford EM, Brent GA, Hershman JM 2000
Induction of follicle formation in long-term cultured normal human thyroid
cells treated with thyrotropin stimulates iodide uptake but not sodium/iodide
symporter messenger RNA and protein expression. J Endocrinol 167:125–135
27. Riedel C, Levy O, Carrasco N 2001 Post-transcriptional regulation of the
sodium/iodide symporter by thyrotropin. J Biol Chem 276:21458–21463
28. Vadysirisack DD, Chen ES, Zhang Z, Tsai MD, Chang GD, Jhiang SM 2007
Identification of in vivo phosphorylation sites and their functional significance
in the sodium iodide symporter. J Biol Chem 282:36820–36828
29. Fanning AS, Anderson JM 1999 PDZ domains: fundamental building blocks
in the organization of protein complexes at the plasma membrane. J Clin Invest
103:767–772
30. Tan PK, Waites C, Liu Y, Krantz DE, Edwards RH 1998 A leucine-based motif
mediates the endocytosis of vesicular monoamine and acetylcholine transport-
ers. J Biol Chem 273:17351–17360
31. Marks MS, Ohno H, Kirchnausen T, Bonracino JS 1997 Protein sorting by
tyrosine-based signals: adapting to the Ys and wherefores. Trends Cell Biol
7:124–128
32. Wolff J, Chaikoff IL 1948 Plasma inorganic iodide as a homeostatic regulator
of thyroid function. J Biol Chem 174:555–564
33. Eng PH, Cardona GR, Fang SL, Previti M, Alex S, Carrasco N, Chin WW,
Braverman LE 1999 Escape from the acute Wolff-Chaikoff effect is associated
with a decrease in thyroid sodium/iodide symporter messenger ribonucleic acid
and protein. Endocrinology 140:3404–3410
34. Eng PH, Cardona GR, Previti MC, Chin WW, Braverman LE 2001 Regulation
of the sodium iodide symporter by iodide in FRTL-5 cells. Eur J Endocrinol
144:139–144
35. Spitzweg C, Joba W, Morris JC, Heufelder AE 1999 Regulation of sodium iodide
symporter gene expression in FRTL-5 rat thyroid cells. Thyroid 9:821–830
36. Wolff J 1983 Congenital goiter with defective iodide transport. Endocr Rev
4:240–254
37. Wu SL, Ho TY, Liang JA, Hsiang CY 2008 Histidine residue at position 226
is critical for iodide uptake activity of human sodium/iodide symporter. J
Endocrinol 199:213–219
38. Levy O, Ginter CS, De la Vieja A, Levy D, Carrasco N 1998 Identification of
a structural requirement for thyroid Na1/I2 symporter (NIS) function from
analysis of a mutation that causes human congenital hypothyroidism. FEBS
Lett 429:36–40
39. Dohan O, Gavrielides MV, Ginter C, Amzel LM, Carrasco N 2002 Na1/I2
symporter activity requires a small and uncharged amino acid residue at po-
sition 395. Mol Endocrinol 16:1893–1902
40. De La Vieja A, Ginter CS, Carrasco N 2004 The Q267E mutation in the
sodium/iodide symporter (NIS) causes congenital iodide transport defect (ITD)
by decreasing the NIS turnover number. J Cell Sci 117:677–687
41. De la Vieja A, Ginter CS, Carrasco N 2005 Molecular analysis of a congenital
iodide transport defect: G543E impairs maturation and trafficking of the
Na1/I2 symporter. Mol Endocrinol 19:2847–2858
42. Soleimani M, Greeley T, Petrovic S, Wang Z, Amlal H, Kopp P, Burnham CE
2001 Pendrin: an apical Cl2/OH2/HCO32 exchanger in the kidney cortex.
Am J Physiol Renal Physiol 280:F356–F364
43. Everett LA, Morsli H, Wu DK, Green ED 1999 Expression pattern of the
mouse ortholog of the Pendred’s syndrome gene (Pds) suggests a key role for
pendrin in the inner ear. Proc Natl Acad Sci USA 96:9727–9732
44. Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green
ED 2001 Pendrin, encoded by the Pendred syndrome gene, resides in the apical
region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl
Acad Sci USA 98:4221–4226
45. Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI,
Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED 2001 Targeted disrup-
tion of mouse Pds provides insight about the inner-ear defects encountered in
Pendred syndrome. Hum Mol Genet 10:153–161
46. Everett LA, Green ED 1999 A family of mammalian anion transporters and
their involvement in human genetic diseases. Hum Mol Genet 8:1883–1891
47. Zheng J, Shen W, He DZ, Long KB, Madison LD, Dallos P 2000 Prestin is the
motor protein of cochlear outer hair cells. Nature 405:149–155
48. Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani
E, Nassir E, Baxevanis AD, Sheffield VC, Green ED 1997 Pendred syndrome
is causedbymutations inaputative sulphate transporter gene (PDS).NatGenet
17:411–422
49. Porra V, Bernier-Valentin F, Trouttet-Masson S, Berger-Dutrieux N, Peix JL,
Perrin A, Selmi-Ruby S, Rousset B 2002 Characterization and semiquantita-
tive analyses of pendrin expressed in normal and tumoral human thyroid
tissues. J Clin Endocrinol Metab 87:1700–1707
50. Gillam MP, Sidhaye AR, Lee EJ, Rutishauser J, Stephan CW, Kopp P 2004
Functional characterization of pendrin in a polarized cell system. Evidence for
pendrin-mediated apical iodide efflux. J Biol Chem 279:13004–13010
51. Aravind L, Koonin EV 2000 The STAS domain: a link between anion trans-
porters and antisigma-factor antagonists. Curr Biol 10:R53–R55
52. Ko SB, Shcheynikov N, Choi JY, Luo X, Ishibashi K, Thomas PJ, Kim JY, Kim
KH, Lee MG, Naruse S, Muallem S 2002 A molecular mechanism for aberrant
CFTR-dependent HCO32 transport in cystic fibrosis. EMBO J 21:5662–5672
53. Ko SB, Zeng W, Dorwart MR, Luo X, Kim KH, Millen L, Goto H, Naruse S,
Soyombo A, Thomas PJ, Muallem S 2004 Gating of CFTR by the STAS domain
of SLC26 transporters. Nat Cell Biol 6:343–350
54. Shcheynikov N, Ko SB, Zeng W, Choi JY, Dorwart MR, Thomas PJ, Muallem
S 2006 Regulatory interaction between CFTR and the SLC26 transporters.
Novartis Found Symp 273:177–186; discussion 186–192, 261–264
55. Morgans ME, Trotter WR 1958 Association of congenital deafness with goi-
tre; the nature of the thyroid defect. Lancet 1:607–609
56. Kopp P, Pesce L, Solis SJ 2008 Pendred syndrome and iodide transport in the
thyroid. Trends Endocrinol Metab 19:260–268
57. Reardon W, Coffey R, Phelps PD, Luxon LM, Stephens D, Kendall-Taylor P,
Britton KE, Grossman A, Trembath R 1997 Pendred syndrome: 100 years of
underascertainment? QJM 90:443–447
58. Phelps PD, Coffey RA, Trembath RC, Luxon LM, Grossman AB, Britton KE,
Kendall-Taylor P, Graham JM, Cadge BC, Stephens SG, Pembrey ME, Reardon
W 1998 Radiological malformations of the ear in Pendred syndrome. Clin Radiol
53:268–273
59. Fugazzola L, Mannavola D, Cerutti N, Maghnie M, Pagella F, Bianchi P,
Weber G, Persani L, Beck-Peccoz P 2000 Molecular analysis of the Pendred’s
syndrome gene and magnetic resonance imaging studies of the inner ear are
essential for the diagnosis of true Pendred’s syndrome. J Clin Endocrinol
Metab 85:2469–2475
60. Pryor SP, Madeo AC, Reynolds JC, Sarlis NJ, Arnos KS, Nance WE, Yang Y,
Zalewski CK, Brewer CC, Butman JA, Griffith AJ 2005 SLC26A4/PDS gen-
otype-phenotype correlation in hearing loss with enlargement of the vestibular
Endocrinology, March 2009, 150(3):1084–1090 endo.endojournals.org 1089

aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are
distinct clinical and genetic entities. J Med Genet 42:159–165
61. Azaiez H, Yang T, Prasad S, Sorensen JL, Nishimura CJ, Kimberling WJ, Smith
RJ 2007 Genotype-phenotype correlations for SLC26A4-related deafness.
Hum Genet 122:451–457
62. Fraser GR, Morgans ME, Trotter WR 1960 The syndrome of sporadic goitre
and congenital deafness. Q J Med 29:279–295
63. Fraser GR 1965 Association of congenital deafness with goitre (Pendred’s
syndrome): a study of 207 families. Ann Hum Genet 28:201–249
64. Nilsson LR, Borgfors N, Gamstorp I, Holst HE, Liden G 1964 Nonendemic
goitre and deafness. Acta Paediatr 53:117–131
65. Sato E, Nakashima T, Miura Y, Furuhashi A, Nakayama A, Mori N,
Murakami H, Naganawa S, Tadokoro M 2001 Phenotypes associated with
replacement of His by Arg in the Pendred syndrome gene. Eur J Endocrinol
145:697–703
66. Gausden E, Coyle B, Armour JA, Coffey R, Grossman A, Fraser GR, Winter
RM, Pembrey ME, Kendall-Taylor P, Stephens D, Luxon LM, Phelps PD,
Reardon W, Trembath R 1997 Pendred syndrome: evidence for genetic ho-
mogeneity and further refinement of linkage. J Med Genet 34:126–129
67. Usami S, Abe S, Weston MD, Shinkawa H, Van Camp G, Kimberling WJ 1999
Non-syndromic hearing loss associated with enlarged vestibular aqueduct is
caused by PDS mutations. Hum Genet 104:188–192
68. Kopp P, Arseven OK, Sabacan L, Kotlar T, Dupuis J, Cavaliere H, Santos CL,
Jameson JL, Medeiros-Neto G 1999 Phenocopies for deafness and goiter de-
velopment in a large inbred Brazilian kindred with Pendred’s syndrome asso-
ciated with a novel mutation in the PDS gene. J Clin Endocrinol Metab 84:
336–341
69. Gonzalez Trevino O, Karamanoglu Arseven O, Ceballos CJ, Vives VI, Ramirez
RC, Gomez VV, Medeiros-Neto G, Kopp P 2001 Clinical and molecular anal-
ysis of three Mexican families with Pendred’s syndrome. Eur J Endocrinol
144:585–593
70. Rotman-Pikielny P, Hirschberg K, Maruvada P, Suzuki K, Royaux IE, Green
ED, Kohn LD, Lippincott-Schwartz J, Yen PM 2002 Retention of pendrin in
the endoplasmic reticulum is a major mechanism for Pendred syndrome. Hum
Mol Genet 11:2625–2633
71. Schnyder S, Chen L, Chan L, Turk A, Gillam MP, Kopp P 2005 Pendrin
mutations that are retained in intracellular compartments induce the IRE1/
XBP1 and the ATF6 unfolded protein response pathways. Thyroid 15(Suppl
1):S-4–S-5 (Abstract O11)
72. Scott DA, Karniski LP 2000 Human pendrin expressed in Xenopus laevis
oocytes mediates chloride/formate exchange. Am J Physiol Cell Physiol 278:
C207–C211
73. Bidart JM, Mian C, Lazar V, Russo D, Filetti S, Caillou B, Schlumberger M
2000 Expression of pendrin and the Pendred syndrome (PDS) gene in human
thyroid tissues. J Clin Endocrinol Metab 85:2028–2033
74. Kopp P 1999 Pendred’s syndrome: clinical characteristics and molecular basis.
Current Opin Endocrinol Diabetes 6:261–269
75. Yoshida A, Taniguchi S, Hisatome I, Royaux IE, Green ED, Kohn LD, Suzuki
K 2002 Pendrin is an iodide-specific apical porter responsible for iodide efflux
from thyroid cells. J Clin Endocrinol Metab 87:3356–3361
76. Yoshida A, Hisatome I, Taniguchi S, Sasaki N, Yamamoto Y, Miake J, Fukui
H, Shimizu H, Okamura T, Okura T, Igawa O, Shigemasa C, Green ED, Kohn
LD, Suzuki K 2004 Mechanism of iodide/chloride exchange by pendrin. En-
docrinology 145:4301–4308
77. Wolff J 2005 What is the role of pendrin? Thyroid 15:346–348
78. Andros G, Wollman SH 1967 Autoradiographic localization of radioiodide in
the thyroid gland of the mouse. Am J Physiol 213:198–208
79. Nilsson M, Bjorkman U, Ekholm R, Ericson LE 1990 Iodide transport in
primary cultured thyroid follicle cells: evidence of a TSH-regulated channel
mediating iodide efflux selectively across the apical domain of the plasma
membrane. Eur J Cell Biol 52:270–281
80. Golstein P, Abramow M, Dumont JE, Beauwens R 1992 The iodide channel
of the thyroid: a plasma membrane vesicle study. Am J Physiol 263:C590–
C597
81. Taylor JP, Metcalfe RA, Watson PF, Weetman AP, Trembath RC 2002 Mu-
tations of the PDS gene, encoding pendrin, are associated with protein mislo-
calization and loss of iodide efflux: implications for thyroid dysfunction in
Pendred syndrome. J Clin Endocrinol Metab 87:1778–1784
82. Rodriguez AM, Perron B, Lacroix L, Caillou B, Leblanc G, Schlumberger M,
Bidart JM, Pourcher T 2002 Identification and characterization of a putative
human iodide transporter located at the apical membrane of thyrocytes. J Clin
Endocrinol Metab 87:3500–3503
83. van den Hove MF, Croizet-Berger K, Jouret F, Guggino SE, Guggino WB,
Devuyst O, Courtoy PJ 2006 The loss of the chloride channel, ClC-5, delays
apical iodide efflux and induces a euthyroid goiter in the mouse thyroid gland.
Endocrinology 147:1287–1296
84. Paroder V, Spencer SR, Paroder M, Arango D, Schwartz Jr S, Mariadason JM,
Augenlicht LH, Eskandari S, Carrasco N 2006 Na1/monocarboxylate trans-
port (SMCT) protein expression correlates with survival in colon cancer: mo-
lecular characterization of SMCT. Proc Natl Acad Sci USA 103:7270–7275
85. Weiss SJ, Philp NJ, Grollman EF 1984 Effect of thyrotropin on iodide efflux
in FRTL-5 cells mediated by Ca21. Endocrinology 114:1108–1113
86. Nilsson M, Bjorkman U, Ekholm R, Ericson LE 1992 Polarized efflux of iodide
in porcine thyrocytes occurs via a cAMP-regulated iodide channel in the apical
plasma membrane. Acta Endocrinol 126:67–74
87. Pesce L, Kopp P 2007 Thyrotropin rapidly regulates pendrin membrane abun-
dance via PKA dependent and PKC dependent pathways in rat thyroid cells.
Thyroid 17(Suppl 1):S-136 (Abstract 282)
88. Muscella A, Marsigliante S, Verri T, Urso L, Dimitri C, Botta G, Paulmichl M,
Beck-Peccoz P, Fugazzola L, Storelli C 2008 PKC-«-dependent cytosol-to-
membrane translocation of pendrin in rat thyroid PC Cl3 cells. J Cell Physiol
217:103–112
89. Suzuki K, Kohn LD 2006 Differential regulation of apical and basal iodide
transporters in the thyroid by thyroglobulin. J Endocrinol 189:247–255
90. Suzuki K, Mori A, Saito J, Moriyama E, Ullianich L, Kohn LD 1999 Follicular
thyroglobulin suppresses iodide uptake by suppressing expression of the so-
dium/iodide symporter gene. Endocrinology 140:5422–5430
91. Kohn LD, Suzuki K, Nakazato M, Royaux I, Green ED 2001 Effects of thy-
roglobulin and pendrin on iodide flux through the thyrocyte. Trends Endo-
crinol Metab 12:10–16
1090 Bizhanova and Kopp Minireview Endocrinology, March 2009, 150(3):1084–1090

GLUCOCORTICOIDS AND MOOD
Glucocorticoid Signaling in the Cell
Expanding Clinical Implications to Complex HumanBehavioral and Somatic Disorders
George P. Chrousosa,b and Tomoshige Kinob
aFirst Department of Pediatrics, Athens University Medical School, Athens, Greece
bPediatric Endocrinology Section, Program in Reproductive and Adult Endocrinology,
Eunice Kennedy Shriver National Institute of Child Health and Human Development,
National Institutes of Health, Bethesda, Maryland, USA
Glucocorticoids contribute to the maintenance of basal and stress-related homeosta-
sis in all higher organisms, and influence a large proportion of the expressed human
genome, and their effects spare almost no organs or tissues. Glucocorticoids regulate
many functions of the central nervous system, such as arousal, cognition, mood, sleep,
the activity and direction of intermediary metabolism, the maintenance of a proper
cardiovascular tone, the activity and quality of the immune and inflammatory reaction,
including the manifestations of the sickness syndrome, and growth and reproduction.
The numerous actions of glucocorticoids are mediated by a set of at least 16 gluco-
corticoid receptor (GR) isoforms forming homo- or hetero-dimers. The GRs consist of
multifunctional domain proteins operating as ligand-dependent transcription factors
that interact with many other cell signaling systems, including large and small G pro-
teins. The presence of multiple GR monomers and homo- or hetero-dimers expressed
in a cell-specific fashion at different quantities with quantitatively and qualitatively
different transcriptional activities suggest that the glucocorticoid signaling system
is highly stochastic. Glucocorticoids are heavily involved in human pathophysiology
and influence life expectancy. Common behavioral and/or somatic complex disorders,
such as anxiety, depression, insomnia, chronic pain and fatigue syndromes, obesity, the
metabolic syndrome, essential hypertension, diabetes type 2, atherosclerosis with its
cardiovascular sequelae, and osteoporosis, as well as autoimmune inflammatory and
allergic disorders, all appear to have a glucocorticoid-regulated component.
Key words: metabolic syndrome; osteoporosis; CDK5; GR phosphorylation; stress sys-
tem; glucocorticoid resistance; glucocorticoid hypersensitivity
Introduction
Glucocorticoids are among the most per-
vasive hormones in mammalian organisms.1,2
These steroid molecules reach all tissues,
including the brain, readily penetrate the
cell membrane, and interact with ubiquitous
cytoplasmic/nuclear glucocorticoid receptors
Address for correspondence: George P. Chrousos, MD, MACP, MACE,
FRCP (London), Professor and Chairman, First Department of Pediatrics,
Athens University Medical School, Aghia Sophia Children’s Hospital,
115 27 Athens, Greece. Voice: +30-210-7794023; fax: +30-210-7759167.
(GRs), through which they exert markedly
diverse actions.1,2 Using DNA microarray tech-
nology, we found that about 20% of the ex-
pressed human leukocyte genome was posi-
tively or negatively affected by glucocorticoids.3
This is many-fold higher than the proportion
of genes that change in the transformation of
a normal cell to a tumor cell and involves a
broad array of functions, affecting every as-
pect of resting and stress-related homeostasis,
including a large number of genes expressed
by the immune system.3,4 The pervasive
nature of glucocorticoids, the rapid advances
Glucocorticoids and Mood: Ann. N.Y. Acad. Sci. 1179: 153–166 (2009).doi: 10.1111/j.1749-6632.2009.04988.x c© 2009 New York Academy of Sciences.
153

154 Annals of the New York Academy of Sciences
in our general knowledge of the human and
other mammalian genomes, and the massive
amount of information that increasingly accu-
mulates, dictate a new model of thinking and
testing of hypotheses regarding the actions of
these hormones and their involvement in hu-
man physiology and pathophysiology.
GR Gene Polymorphisms andComplex Human Pathophysiology
Wust et al. reported a convincing asso-
ciation between the hypothalamic-pituitary-
adrenal (HPA) axis response to a standardized
socio-emotional stimulus (Trier test) and poly-
morphisms of the GR gene.5 This study fol-
lowed others that used a similar rationale and
examined HPA axis indices and other end-
points, such as arterial blood pressure, body
mass index and markers of the metabolic syn-
drome, and bone mineral density.6–13 These
studies have had some overlap and have pro-
duced mostly concordant results, but have also
shown inconsistencies. This should have been
expected because these studies were performed
in a limited number of subjects in different eth-
nic populations, and because the altered GR
would have been expected to function differ-
ently in the context of different genetic back-
grounds characterized by different panels of
genes with differing epistatic effects upon the
ability of the GR to exert its actions.14
Glucocorticoid Effects: Physiologyand Pathophysiology
Empirically, experimentally, and intuitively,
physicians and scientists have made major ad-
vances in the general understanding of glu-
cocorticoids and their involvement in human
physiology and pathophysiology, and in using
these hormones extensively and effectively in
the treatment of a wide spectrum of human
diseases.1,2 As the end product of the HPA
axis, glucocorticoids are literally present in ev-
ery organ system of the human organism, in
almost all physiologic, cellular and molecu-
lar networks, and in many crucial modules of
these networks.2,3,14 Glucocorticoids, further-
more, participate in a pivotal fashion in the
unfolding of vital biologic programs employing
synchronously or in tandem several networks,
including the behavioral and physical response
to stress, the inflammatory reaction and the
consequent “sickness syndrome,” i.e., the col-
lection of “nonspecific symptoms” caused by
excessive inflammatory cytokines during infec-
tious or inflammatory illness, as well as the pro-
cess of sleep, and long-term functions, such as
growth and reproduction.4
As is true with many other homeostatic sys-
tems, too much or too little HPA axis and/or
glucocorticoid activity may be associated with
pathology—for instance, Cushing syndrome
versus Addison disease, respectively.15–17 Since
the responsiveness of the target tissues to glu-
cocorticoids is crucial for the end-effect of
these hormones, similar pathology may re-
sult from hypersensitivity or resistance of these
target tissues to these hormones, respectively
(Table 1).16,18 However, because the brain and
the pituitary are also the targets of glucocor-
ticoids, and because the organism strives for
homeostasis in time-integrated free cortisol ex-
posure, any generalized change in the gluco-
corticoid signaling system would be expected
to be followed by corrective, “compensatory”
changes in the activity of the HPA axis. In-
deed, in the rare GR-mediated genetic dis-
order Primary Glucocorticoid Resistance, the
majority of the clinical manifestations are not
Addisonian, but are rather due to the compen-
satory hyperfunction of the HPA axis leading
to adrenal androgen and mineralocorticoid hy-
persecretion while the opposite would be ex-
pected in primary generalized glucocorticoid
hypersensitivity (Fig. 1A and B).19,20
However, an absence of complete concor-
dance between HPA axis activity and the
target tissues outside those responsible for
feedback regulation, be it slightly excessive
or deficient, could result in a state called

Chrousos & Kino: Glucocorticoids and Pathophysiology 155
TABLE 1. Expected Clinical Manifestations in Target Tissue Hypersensitivity or Resistance to Gluco-corticoids
Glucocorticoid excess = Glucocorticoid deficiency =
Target area Glucocorticoid hypersensitivity Glucocorticoid resistance
Central nervous system Insomnia, anxiety, depression,
defective cognition
Fatigue, somnolence, malaise,
defective cognition
Liver +Gluconeogenesis, +lipogenesis Hypoglycemia, resistance to
diabetes mellitus
Fat Accumulation of visceral fat
(metabolic syndrome)
Loss of weight, resistance to weight
gain
Blood vessels Hypertension Hypotension
Bone Stunted growth, osteoporosis
Inflammation/immunity Immune suppression,
anti-inflammation, vulnerability
to certain infection and tumors
+Inflammation, +autoimmunity,
+allergy
Modified from Refs. 22 & 61.
allostasis, more accurately termed cacostasis,
leading to target tissue pathology, as occurs
in chronically stressed or depressed individu-
als in whom there is frequently mild chronic
hypercortisolism.21,22 This has been known for
several decades. The key question is whether
it is possible to have discordance between the
feedback regulation of the HPA axis by glu-
cocorticoids and peripheral target tissue sen-
sitivity to these hormones in totally “normal”
individuals.
Indeed it appears to be possible. The glu-
cocorticoid signaling system of the suprahy-
pothalamic, hypothalamic, and pituitary
glucocorticoid-sensing network is different
from the signaling systems of the reward,
arousal, associative, cardiovascular, metabolic
and immune systems, which are influenced by
glucocorticoids. The feedback centers of the
HPA axis sense and thus determine the cir-
culating glucocorticoid levels, while other tis-
sues passively accept the actions of circulating
glucocorticoids. Indeed, any change in one or
more molecules or processes that participate in
the glucocorticoid signaling system could po-
tentially have a different impact on the HPA
feedback system and other target tissues. Such
discrepancies in the glucocorticoid sensing net-
work between the HPA axis and peripheral tis-
sues could therefore produce peripheral tissue
“hypercortisolism” or “hypocorticosolism,” de-
pending on their combinations (Fig. 1C and
Table 1).16 As an example, in a recent study by
Alevizaki et al., both high HPA axis reactivity
to stress and increased peripheral tissue sensi-
tivity to glucocorticoids were associated with
increased severity of coronary artery disease.23
Naturally, GR is not alone in defining the sen-
sitivity of the feedback system and other tis-
sues to glucocorticoids. Numerous GR isoforms
with different activities, other molecules or pro-
cesses with considerable input into the activity
of the cellular glucocorticoid signaling system
have been described (Table 2).18,24,25
In our studies of the glucocorticoid signal-
ing system, we have identified several molecules
from a variety of different signaling systems that
interact in many ways with and influence the
activity of the GR and vice versa, and are de-
picted in Figure 2 and Table 2.18,24–33 These
include the β subunit of the G protein trimeric
complex, small G proteins, components of the
tumor necrosis factor-α/Fas ligand signaling
systems, such as FLASH, the chaperone protein
14-3-3, the HIV accessory proteins Vpr and
Tat, the adenoviral protein E1A, and molec-
ular components of brain cyclin-dependent
kinase 5 (CDK5).26–31,33 In light of the ma-
jor physiologic homeostatic influence of glu-
cocorticoids on many brain functions, as
156 Annals of the New York Academy of Sciences
Figure 1. (A) Feedback-regulated compensatory changes in the activity of the HPA axis and their ef-fects in peripheral tissues, such as the liver, fat, and blood vessels. Note that glucocorticoid sensitivity inthe HPA axis and the peripheral tissues can be independently regulated, and the former determines theserum-free cortisol levels; thus, a combination of their directions influences net peripheral action of thishormone. ACTH: adrenocorticotropic hormone; AVP: arginine vasopressin; CRH: corticotropin-releasing hor-mone; DOC: deoxycorticosterone; B: corticosterone. Modified from Refs. 68, 73 & 74. (B) Known GRmutations that cause familial/sporadic glucocorticoid resistance syndrome. Localization of GR mutations thatcause familial/sporadic glucocorticoid resistance syndrome are shown in the human GR gene (top) and in thehuman GR protein (bottom). DBD: DNA-binding domain; LBD: ligand-binding domain. From Refs. 19 & 20.(C) Alteration of net glucocorticoid effects in target tissues by activity of the central HPA axis and peripheraltissue sensitivity to glucocorticoids. Net glucocorticoid action in peripheral tissues, such as the CNS, liver, fatand vasculature, is determined by two components: the central HPA axis and sensitivity of peripheral tissuesto glucocorticoids. (From Ref. 68.)

Chrousos & Kino: Glucocorticoids and Pathophysiology 157
Figure 1. Continued.
well as the major pathologic effects of these
hormones on the central nervous system
(CNS), we elected to summarize below our
work on the interactions of CDK5 and the
GR.34
Glucocorticoids and the Brain: CNSCDK5 Regulates Glucocorticoid
Actions in the Brain byPhosphorylating GR
The transcriptional activity of GR is reg-
ulated by direct phosphorylation of this re-
ceptor by serine/threonine kinases.35 GR has
several phosphorylation sites, all of which are
located in the AF-1 domain of its N-terminal
domain (NTD),24,36 suggesting that AF-1 acts
as an interface for phosphorylation-dependent
intracellular molecular signals (Fig. 3A). For
example, yeast CDK p34CDC28 phosphory-
lates rat GR at serines 224 and 232, which
are orthologous to serines 203 and 211 of hu-
man GR, with the resultant phosphorylation
enhancing rat GR transcriptional activity in
the yeast.35 These residues are also phospho-
rylated after activation of GR with agonists
or antagonists, and the phosphorylated recep-
tor shows reduced translocation to the nucleus
and/or altered subcellular localization in mam-
malian cells.36,37 The p38 mitogen-activated
protein kinase (MAPK) phosphorylates serine
211 of human GR and enhances its transcrip-
tional activity.38 p38 MAPK and JNK also
phosphorylate serine 226 of human GR and
suppress its transcriptional activity by enhanc-
ing nuclear export of the receptor.39 Threo-
nine 171 of rat GR is phosphorylated by p38
MAPK and glycogen synthase kinase-3. Threo-
nine 171-phosphorylated GR demonstrates re-
duced transcriptional activity in yeast and hu-
man cells; however, human GR does not have
a threonine residue equivalent to that of the rat
GR.40
In addition to these kinases that phospho-
rylate GR and regulate its transcriptional ac-
tivity, we recently found that CDK5, which
is a member of the CDK family,41 phospho-
rylates GR and modulates its transcriptional
activity.33 In contrast to other CDKs, which
function in the control of the cell cycle, CDK5
has no activity during mitosis but is essential
for brain development and neuronal morpho-
genesis and survival.42,43 CDK5 is expressed

158 Annals of the New York Academy of Sciences
TABLE 2. Factors Influencing GR Functions
Ligands Membrane transporters of
glucocorticoids
11β-hydroxysteroid dehydrogenases
Agonists, antagonists (ex. RU 486)
Chemical
compounds
Ursodeoxycholic acid, cortivazol,
thioredoxin, carnitine
Chemical
modifications
Phosphorylation, nitrosylation,
acetylation, methylation,
sumoylation
Chaperones, co-
chaperones
Heat shock proteins, RAP46,
FK506-binding proteins
Receptor
isoforms
Glucocorticoid receptor α and β
isoforms (16 or more, 256 or more
combinations of dimers)
Transcriptional
co-regulators
Coactivators/corepressors,
SWI/SNF, TRAP/DRIP
complex, SMAD6
Viral proteins: adenoviral E1A,
Human Immunodeficiency virus
type-1 Vpr and Tat
Transcription
factors
Nuclear factor-κB, activator
protein-1, CREB-binding protein,
p53, chicken ovalbumin upstream
promoter-transcription factor-II,
GATA-1, SP-1, Nuclear factor-1,
CLOCK/BMAL1 PPARα
Other proteins 14-3-3, FLASH, Rho-type guanine
nucleotide exchange factors (Brx
and c-Lbc), autoimmune
regulator gene (AIRE), guanine
nucleotide-binding protein β
RNAs SRA, Gas5
Modified from Refs. 18, 24, 26–29, 32, 68–72 & 75.
ubiquitously in many tissues, but its activity
is restricted primarily to the nervous system
due to neuron-specific expression of its activa-
tor molecules p35 and p39.44,45 In addition to
these physiologic roles of CDK5, recent evi-
dence suggests that aberrant CDK5 activation
caused by proteolytic conversion of p35 to p25
may play a role in the pathogenesis of neu-
rodegenerative disorders, such as Alzheimer’s
disease and amyotrophic lateral sclerosis.43,46
In these conditions, calpain-directed proteoly-
sis of p35 deprives the membrane-associated
p35 of its N-terminal myristoylated membrane
tether, releasing p25 into the cytoplasm and
activating CDK5.47,48
We found that the CDK5/p35 complex
physically interacts with the GR ligand-binding
domain (LBD) and modulates GR-induced
transcriptional activity by phosphorylating ser-
ines 203, 211, and 226 of human GR.33 The
C-terminal part of p35 binds the GR LBD in a
ligand-dependent fashion, while both the GR
LBD and NTD are necessary for CDK5/p35
to modulate GR transcriptional activity, indi-
cating that these subdomains of GR respec-
tively act as interactor and effector surfaces
for CDK5/p35.33 We found that CDK5 posi-
tively or negatively regulates the transcriptional
activity of GR on endogenous glucocorticoid-
responsive genes in rat cortical neuronal cells in
microarray analyses, indicating that the effect
of CDK5 on GR-induced transcriptional ac-
tivity is gene promoter–specific.33 Since phos-
phorylation of GR at serines 203, 211, and 226
alters the attraction of cofactor molecules to
the promoter-bound GR,33 it is possible that
phosphorylation of the receptor may respec-
tively facilitate or inhibit the attraction of tran-
scription cofactors on promoter-bound GR,
through as yet undetermined mechanisms that
appear to be gene-specific (Fig. 3B). The tu-
mor susceptibility gene 101 (TSG101), which
interacts with the co-integulator molecules
p300/CBP and inhibits the transcriptional
activity of GR by modulating GR-induced
attraction of coactivators, preferentially in-
teracts with a nonphosphorylated form of
GR.36,40,49 Thus, TSG101 and/or similar
molecules that specifically bind nonphospho-
rylated or phosphorylated forms of GR40 may
mediate phosphorylation-dependent transcrip-
tional modulation by changing accumulation of
cofactors on promoter-bound GR.
Glucocorticoids play an essential role in the
homeostasis of the CNS50,51; these hormones
indeed regulate cognition, memory, mood, and
sleep, and influence the anatomic structure
of the brain, causing reduction of hippocam-
pal volume, ventricular enlargement, and re-
versible cortical atrophy.51 Alteration of CDK5
activity in neuronal cells can potentially in-
fluence any of these glucocorticoid actions in

Chrousos & Kino: Glucocorticoids and Pathophysiology 159
Figure 2. Regulation of tissue GR activity by distinct signaling pathways. Transcriptionalactivity of GR in glucocorticoid target tissues is regulated by numerous signaling pathwaysthrough distinct mechanisms. (From Refs. 18, 24, 26–29, 32, 69, 71 & 72.) (In color inAnnals online.)
the CNS. It is also particularly interesting
whether the regulatory actions of CDK5 on
GR transcriptional activity contribute to the
development of neurodegenerative disorders,
since glucocorticoids have strong activity on
consolidation of memory and neuronal cell sur-
vival.51,52 Proteolytically produced cytoplasmic
p25, whose excessive production has been as-
sociated with Alzheimer’s disease,53,54 demon-
strated a much stronger effect than p35 in reg-
ulating GR transcriptional activity.33 Thus, it
is possible that CDK5 may exert pathologic
effects by altering GR transcriptional activity
through aberrant conversion of p35 to p25.
Glucocorticoids, the MetabolicSyndrome and Other Somatic
Sequelae of Stress
Endogenous or exogenous Cushing syn-
drome is associated with the full metabolic
profile of the metabolic syndrome and with
substantially increased cardiovascular mor-
bidity and mortality.55,56 Glucocorticoids di-
rectly cause insulin resistance of peripheral
target tissues in proportion to their levels
and to the particular target tissue’s sensitiv-
ity to these hormones. Over time, glucocorti-
coids also cause progressive accumulation of
visceral fat, leading to worsening manifesta-
tions of the metabolic syndrome. Thus, when
polymorphisms of the GR gene lead to an
unfavorable discordance between the activity
of the HPA axis and the sensitivity of mus-
cle, fat and/or liver to glucocorticoids, the
increased glucocorticoid effect in these tis-
sues could influence the metabolic profile and
the longevity of humans in a negative fash-
ion, similar to that which occurs in Cushing
syndrome.7,15,57–60
Genetic and developmental factors, nutri-
tion, lifestyle, and cumulative chronic or in-
termittent stress may lead to the development

160 Annals of the New York Academy of Sciences
Figure 3. Regulation of GR transcriptional activity through phosphorylation. (A) HumanGR has three major phosphorylation sites at serines 203, 211, and 226, and two minorphosphorylation sites at serines 113 and 141. The former sites are phosphorylated by severalkinases as indicated. From Refs. 24 & 34–39. (B) Schematic regulation of GR-induced tran-scriptional activity by CDK5. CDK5 regulates GR-induced transcriptional activity by changingattraction of transcriptional cofactors to responsive promoters in the CNS, possibly throughmolecules that specifically interact with phosphorylated or nonphosphorylated ligand-boundGR. The direction and size of the effect depends also on the presence of other gene promoter–,tissue- or brain region–specific transcriptional cofactors. (Modified from Refs. 33 & 34.)
of obesity, primarily of the visceral type with
concurrent loss of lean body mass, and, hence,
the metabolic syndrome with its components of
insulin resistance, dyslipidemia, chronic smol-
dering inflammation, blood hypercoagulation,
arterial hypertension, and/or diabetes mellitus
type 2 (Fig. 4).61 These changes lead to endothe-
lial inflammation, atherosclerosis, and cardio-
vascular disease, ultimately resulting in pre-
mature cardiovascular morbidity and death.61
Similarly, through decreased bone formation
and/or increased bone resorption, osteopenia
or osteoporosis may ensue.
Glucocorticoids contribute to the pathogen-
esis of obesity and the metabolic syndrome,
not only through unfavorable genetic varia-
tions that increase both the activity of the HPA
axis and the sensitivity of tissues to glucocorti-
coids, but also because of fetal programming of
the HPA axis by an adverse intrauterine envi-
ronment, which may lead to a postnatally hy-
peractive axis, and because of chronic cortisol

Chrousos & Kino: Glucocorticoids and Pathophysiology 161
Figure 4. Endogenous/exogenous inputs to the stress system and their effects on themetabolic and cardiovascular systems and bone. ABP: arterial blood pressure; APR: acutephase reactants; AVP: arginine vasopressin; CRH: corticotropin-releasing hormone; E2: estra-diol; EDS: excessive daytime sleepiness; GH: growth hormone; HDL: high-density lipopro-tein; HPA axis: hypothalamic-pituitary-adrenal axis; IGF-1: insulin-like growth factor-1; IL-6:interleukin-6; LC: locus cæruleus; LDL: low-density lipoprotein; LH: luteinizing hormone; NE:norepinephrine; T: testosterone; T3: triiodothyronine; TG: triglyceride; TSH: thyroid-stimulatinghormone. (From Ref. 68.) (In color in Annals online.)
hypersecretion owing to concurrent real or per-
ceived stress.15,21,62
The opposite result is possible as well. Pa-
tients may be protected from obesity, the
metabolic syndrome, and premature death be-
cause of favorable genetic variations causing
a decreased activity of their HPA axis and
their tissue sensitivity to glucocorticoids, as
well as because of normal or opposite fetal
programming and decreased exposure to real
or perceived stress.15,21,62 In the studies by
Van Rossum et al.62a,62b Syed et al. and oth-
ers,58,62a,62b,63–66 favorable genetic variations in
the GR gene, in which carriers of a particu-
lar polymorphism had peripheral target tissues
with decreased sensitivity to glucocorticoids, re-
sulted in increased sensitivity of the same tis-
sues to insulin and hence a healthier metabolic
profile.
Beyond Glucocorticoids and theMetabolic Syndrome
Despite their obvious importance, glucocor-
ticoids and their signaling system are only one
of several physiologic and molecular networks
that participate in the development of obesity
and the metabolic syndrome, with a resultant
adverse effect on longevity.61 Other major hor-
mones of the stress and other systems and their
receptors also participate in these phenomena
(Fig. 4).4,17
The stress system includes brain nuclei,
such as the paraventricular nucleus of the
hypothalamus, the brainstem locus cæruleus–
norepinephrine/autonomic nervous system
nuclei, and two powerful peripheral neuroen-
docrine limbs—the HPA axis and the systemic
sympathetic and adrenomedullary systems.4,50

162 Annals of the New York Academy of Sciences
Figure 5. Central regulation of the stress system in normal (left panel) and chronically stressed andstress-hyperresponsive individuals (right panel). The stress system (PVN CRH/AVP and LC/NE) activates theamygdala and the MCLS and receives activating signals from the amygdala and suppressive signals fromthe MCLS and the hippocampus. Chronic stress has behavioral and somatic consequences summarized inthe bottom of right panel. MCLS = mesocorticolimbic (reward) system; PVN = paraventricular nucleus; CRH= corticotropin-releasing hormone; AVP = arginine vasopressin. (Modified from Ref. 4.)
The stress system normally receives positive
regulatory input from the amygdala (fear), neg-
ative tonic input from the hippocampus and
negative regulation from the mesocorticolimbic
dopaminergic system (MCLS, reward), while
itself regulates these systems by providing posi-
tive influences on all three (Fig. 5, left panel).4
Abnormally increased chronic activity and/or
reactivity of the stress system can be primary
or secondary to excessive input from the amyg-
dala or defective input from the hippocam-
pus and/or MCLS (Fig. 5, right panel).4 The
normally positive input of the stress system to
the MCLS becomes negative in response to
chronic hyperactivity of the former, perhaps as
a result of the characteristic tolerance of the
latter.4
The main central molecular mediators of
the stress system are corticotropin-releasing
hormone, arginine vasopressin, and nore-
pinephrine. The key peripheral molecular me-
diators are corticotropin, cortisol, arginine va-
sopressin, norepinephrine, epinephrine and,
interestingly, interleukin-6 (IL-6).4,15,50 The
genes that code for the synthesis, regulation,
actions, and metabolism of these mediators
and their receptors are major participants
in the adaptation to stress.4,15,50 The stress
system is activated in a coordinated fashion
during acute, time-limited stress, influencing
central and peripheral functions that are im-
portant for adaptation and survival.4 Chronic
activation of the stress system, however, is asso-
ciated with many negative manifestations and

Chrousos & Kino: Glucocorticoids and Pathophysiology 163
TABLE 3. Gene Networks Subserving Functions Important for Human Survival and Species Preservation,Which May Produce Pathology in Contemporary Western Societies due to Changes in Lifestyle
Response to survival threat Selective advantage Contemporary diseases
Combat starvation Energy conservation Obesity
Combat dehydration Fluid and electrolyte conservation Hypertension
Combat infectious diseases Potent immune reaction Autoimmunity/allergy
Anticipate adversaries Arousal/fear Anxiety/insomnia
Minimize exposure to danger Withdrawal from danger Depression
Prevent tissue strain and injury Retain tissue integrity and reserve Pain and fatigue syndromes
Modified from Refs. 61 & 68.
sequelae beyond obesity/metabolic syndrome,
atherosclerosis and loss of bone mineral density,
which include a long list of behavioral disorders
(Fig. 5, right panel, bottom).4
In addition to noninflammatory stress,
even very mild, asymptomatic inflammation
stimulates secretion of IL-6 and other inflam-
matory cytokines, while adipose tissue is a ma-
jor source of circulating tumor necrosis factor-
α and IL-6.14,50,67 Both glucocorticoids and
IL-6 synergistically stimulate the acute phase
response, including C-reactive protein, fibrino-
gen, and plasminogen activator inhibitor 1, all
of which increase the ability of blood to coag-
ulate and through their pro-atherosclerosis ac-
tion have a negative effect on longevity.14,50,67
Thus, chronic stress, an indolent infection,
an active autoimmune process, and visceral
obesity are all associated with mild hypercy-
tokinemia and low-grade inflammation, which
ultimately results in blood hypercoagulability,
endothelial dysfunction, atherosclerosis, and
cardiovascular disease. Finally, it is evident that
the metabolic syndrome, regardless of its cause,
is a major risk factor for the development of dia-
betes type 2 and the polycystic ovary syndrome
in patients with a genetic propensity to develop
these very common disorders.61,68
As a species, we have survived because we
have been able to adapt to potentially lethal
evolutionary stressors during our life on Earth.
Thus, selective pressures on our genome have
allowed adaptive changes that, at this time
in our evolutionary history and with our cur-
rent lifestyle, have become somewhat maladap-
tive in a large proportion of the population
(Table 3).4,22,50,67 Thus, gene networks dedi-
cated to adaptation and survival, with a finite
number of members, are probably responsi-
ble for much of the contemporary nosology of
Western societies presented in Table 3. Even
though cancer is not included in this table, it is
likely that modification of the immune system
and the inflammatory reaction by stress could
increase the susceptibility of the organism to
certain neoplasias.
Conclusions
To understand the roles of polymorphisms
of multiple genes related to the HPA axis and
the glucocorticoid signaling system in human
physiology and pathophysiology, one will have
to study large populations of normal subjects,
including adequate numbers of representative
racial and ethnic subpopulations, as well as
populations of patients afflicted by states and
diseases that may result from dysfunction of
this system, which are summarized in Tables 1
and 3. Once crucial genes and their polymor-
phisms have been defined, new, existing and
constantly improving methods could be em-
ployed to screen for changes in the entire gene
networks of choice, which, in the appropriate
context, could predict the relative risk for devel-
oping these common disorders. Also, granted
that a large subgroup of this gene network plays
a major role in regulating immune function,
this information could be useful in predicting
vulnerability to certain infections and tumors.
Finally, this knowledge might help individual-
ize medications and doses for subjects with the

164 Annals of the New York Academy of Sciences
above conditions, depending on their genetics
in a rational way—an effort that is developing
into the field of pharmacogenomics.
Acknowledgments
This is a synoptic review of work supported
by the University of Athens, Athens, Greece,
and the Intramural Research Program of the
Eunice Kennedy Shriver National Institute of Child
Health and Human Development, National In-
stitutes of Health, Bethesda, MD.
Conflicts of Interest
The authors declare no conflicts of interest.
References
1. Chrousos, G.P. 2001. Glucocorticoid therapy. In En-
docrinology and Metabolism. P. Flig & L. Frohman, Eds.:
609–632. McGraw-Hill. New York, NY.
2. Franchimont, D. et al. 2003. Glucocorticoids and in-
flammation revisited: the state of the art. NIH Clini-
cal Staff Conference. Neuroimmunomodulation 10: 247–
260.
3. Galon, J. et al. 2002. Gene profiling reveals unknown
enhancing and suppressive actions of glucocorticoids
on immune cells. FASEB J. 16: 61–71.
4. Chrousos, G.P. 1998. Stressors, stress, and neuroen-
docrine integration of the adaptive response. The
1997 Hans Selye Memorial Lecture. Ann. N. Y. Acad.
Sci. 851: 311–335.
5. Wust, S. et al. 2004. Common polymorphisms in
the glucocorticoid receptor gene are associated with
adrenocortical responses to psychosocial stress. J.
Clin. Endocrinol. Metab. 89: 565–573.
6. Buemann, B. et al. 1997. Abdominal visceral fat is as-
sociated with a BclI restriction fragment length poly-
morphism at the glucocorticoid receptor gene locus.
Obes. Res. 5: 186–192.
7. Dobson, M.G. et al. 2001. The N363S polymorphism
of the glucocorticoid receptor: potential contribution
to central obesity in men and lack of association with
other risk factors for coronary heart disease and di-
abetes mellitus. J. Clin. Endocrinol. Metab. 86: 2270–
2274.
8. Huizenga, N.A. et al. 1998. A polymorphism in the
glucocorticoid receptor gene may be associated with
and increased sensitivity to glucocorticoids in vivo. J.
Clin. Endocrinol. Metab. 83: 144–151.
9. Lin, R.C., W.Y. Wang & B.J. Morris. 1999. High
penetrance, overweight, and glucocorticoid receptor
variant: case-control study. BMJ 319: 1337–1338.
10. Panarelli, M. et al. 1998. Glucocorticoid recep-
tor polymorphism, skin vasoconstriction, and other
metabolic intermediate phenotypes in normal hu-
man subjects. J. Clin. Endocrinol. Metab. 83: 1846–
1852.
11. Rosmond, R. et al. 2000. A glucocorticoid recep-
tor gene marker is associated with abdominal obe-
sity, leptin, and dysregulation of the hypothalamic-
pituitary-adrenal axis. Obes. Res. 8: 211–218.
12. Ukkola, O. et al. 2001. Glucocorticoid receptor Bcl
I variant is associated with an increased athero-
genic profile in response to long-term overfeeding.
Atherosclerosis 157: 221–224.
13. Weaver, J.U., G.A. Hitman & P.G. Kopelman. 1992.
An association between a Bc1I restriction fragment
length polymorphism of the glucocorticoid receptor
locus and hyperinsulinaemia in obese women. J. Mol.
Endocrinol. 9: 295–300.
14. Chrousos, G.P. 2000. The stress response and im-
mune function: clinical implications. The 1999
Novera H. Spector Lecture. Ann. N. Y. Acad. Sci. 917:
38–67.
15. Chrousos, G.P. 2000. The role of stress and the
hypothalamic-pituitary-adrenal axis in the pathogen-
esis of the metabolic syndrome: neuro-endocrine and
target tissue-related causes. Int. J. Obes. Relat. Metab.
Disord. 24: S50–S55.
16. Chrousos, G.P., S.D. Detera-Wadleigh & M. Karl.
1993. Syndromes of glucocorticoid resistance. Ann.
Intern. Med. 119: 1113–1124.
17. McEwen, B.S. 1998. Protective and damaging ef-
fects of stress mediators. N. Engl. J. Med. 338: 171–
179.
18. Kino, T. et al. 2003. Tissue glucocorticoid resis-
tance/hypersensitivity syndromes. J. Steroid Biochem.
Mol. Biol. 85: 457–467.
19. Charmandari, E. et al. 2008. Generalized glucocor-
ticoid resistance: clinical aspects, molecular mecha-
nisms, and implications of a rare genetic disorder. J.
Clin. Endocrinol. Metab. 93: 1563–1572.
20. Charmandari, E. et al. 2008. A novel point mutation
in the DNA-binding domain of the human glucocor-
ticoid receptor gene causing generalized glucocorti-
coid resistance. Endocrine 90: 280.
21. Chrousos, G.P. & P.W. Gold. 1992. The concepts of
stress and stress system disorders. Overview of phys-
ical and behavioral homeostasis. JAMA 267: 1244–
1252.
22. Gold, P.W. & G.P. Chrousos. 2002. Organization of
the stress system and its dysregulation in melancholic
and atypical depression: high vs low CRH/NE states.
Mol. Psychiatry 7: 254–275.

Chrousos & Kino: Glucocorticoids and Pathophysiology 165
23. Alevizaki, M. et al. 2007. High anticipatory stress
plasma cortisol levels and sensitivity to glucocor-
ticoids predict severity of coronary artery dis-
ease in subjects undergoing coronary angiography.
Metabolism 56: 222–226.
24. Chrousos, G.P. & T. Kino. 2005. Intracellular glu-
cocorticoid signaling: a formerly simple system turns
stochastic. Sci. STKE 2005: pe48.
25. Kino, T., E. Charmandari & G.P. Chrousos. 2003.
Basic and clinical implications of glucocorticoid
action—focus on development. National Institutes
of Health, Bethesda, Maryland, USA. June 17–18,
2003. Introduction and abstracts. Horm. Metab. Res.
35: 628–648.
26. Kino, T. et al. 2005. G protein β interacts with the
glucocorticoid receptor and suppresses its transcrip-
tional activity in the nucleus. J. Cell Biol. 169: 885–
896.
27. Kino, T., et al. 2006. Rho family guanine nucleotide
exchange factor Brx couples extracellular signals to
the glucocorticoid signaling system. J. Biol. Chem.
281: 9118–9126.
28. Kino, T. & G.P. Chrousos. 2003. Tumor necrosis fac-
tor α receptor- and Fas-associated FLASH inhibit
transcriptional activity of the glucocorticoid receptor
by binding to and interfering with its interaction with
p160 type nuclear receptor coactivators. J. Biol. Chem.
278: 3023–3029.
29. Kino, T. et al. 2003. Protein 14-3-3σ interacts with
and favors cytoplasmic subcellular localization of the
glucocorticoid receptor, acting as a negative regulator
of the glucocorticoid signaling pathway. J. Biol. Chem.
278: 25651–25656.
30. Kino, T. et al. 1999. The HIV-1 virion-associated pro-
tein vpr is a coactivator of the human glucocorticoid
receptor. J. Exp. Med. 189: 51–62.
31. Kino, T. et al. 2002. Human immunodeficiency virus
type-1 accessory protein Vpr induces transcription
of the HIV-1 and glucocorticoid-responsive promot-
ers by binding directly to p300/CBP coactivators. J.
Virol. 76: 9724–9734.
32. Ichijo, T. et al. 2005. The Smad6-histone deacety-
lase 3 complex silences the transcriptional activity of
the glucocorticoid receptor: potential clinical impli-
cations. J. Biol. Chem. 280: 42067–42077.
33. Kino, T. et al. 2007. Cyclin-dependent kinase 5 dif-
ferentially regulates the transcriptional activity of
the glucocorticoid receptor through phosphoryla-
tion: clinical implications for the nervous system re-
sponse to glucocorticoids and stress. Mol. Endocrinol.
21: 1552–1568.
34. Kino, T. 2007. Tissue glucocorticoid sensitivity: be-
yond stochastic regulation on the diverse actions of
glucocorticoids. Horm. Metab. Res. 39: 420–424.
35. Krstic, M.D. et al. 1997. Mitogen-activated and
cyclin-dependent protein kinases selectively and dif-
ferentially modulate transcriptional enhancement by
the glucocorticoid receptor. Mol. Cell Biol. 17: 3947–
3954.
36. Ismaili, N. & M.J. Garabedian. 2004. Modulation
of glucocorticoid receptor function via phosphoryla-
tion. Ann. N. Y. Acad. Sci. 1024: 86–101.
37. Wang, Z., J. Frederick & M.J. Garabedian. 2002. De-
ciphering the phosphorylation “code” of the gluco-
corticoid receptor in vivo. J. Biol. Chem. 277: 26573–
26580.
38. Miller, A.L. et al. 2005. p38 Mitogen-activated
protein kinase (MAPK) is a key mediator in
glucocorticoid-induced apoptosis of lymphoid cells:
correlation between p38 MAPK activation and site-
specific phosphorylation of the human glucocorticoid
receptor at serine 211. Mol. Endocrinol. 19: 1569–
1583.
39. Itoh, M. et al. 2002. Nuclear export of glucocorticoid
receptor is enhanced by c-Jun N-terminal kinase-
mediated phosphorylation. Mol. Endocrinol. 16: 2382–
2392.
40. Ismaili, N., R. Blind & M.J. Garabedian. 2005. Sta-
bilization of the unliganded glucocorticoid receptor
by TSG101. J. Biol. Chem. 280: 11120–11126.
41. Kesavapany, S., B.S. Li & H.C. Pant. 2003. Cyclin-
dependent kinase 5 in neurofilament function and
regulation. Neurosignals 12: 252–264.
42. Ohshima, T. et al. 1996. Targeted disruption of the
cyclin-dependent kinase 5 gene results in abnor-
mal corticogenesis, neuronal pathology and peri-
natal death. Proc. Natl. Acad. Sci. USA 93: 11173–
11178.
43. Dhavan, R. & L.H. Tsai. 2001. A decade of CDK5.
Nat. Rev. Mol. Cell Biol. 2: 749–759.
44. Tsai, L.H. et al. 1994. p35 is a neural-specific reg-
ulatory subunit of cyclin-dependent kinase 5. Nature
371: 419–423.
45. Tang, D. & Wang, J.H. 1996. Cyclin-dependent ki-
nase 5 (Cdk5) and neuron-specific Cdk5 activators.
Prog. Cell Cycle Res. 2: 205–216.
46. Lau, L.F. et al. 2002. Cdk5 as a drug target for the
treatment of Alzheimer’s disease. J. Mol. Neurosci. 19:
267–273.
47. Lee, M.S. et al. 2000. Neurotoxicity induces cleavage
of p35 to p25 by calpain. Nature 405: 360–364.
48. Kusakawa, G. et al. 2000. Calpain-dependent prote-
olytic cleavage of the p35 cyclin-dependent kinase 5
activator to p25. J. Biol. Chem. 275: 17166–17172.
49. Sun, Z. et al. 1999. Tumor susceptibility gene 101
protein represses androgen receptor transactivation
and interacts with p300. Cancer 86: 689–696.
50. Chrousos, G.P. 1995. The hypothalamic-pituitary-
adrenal axis and immune-mediated inflammation.
N. Engl. J. Med. 332: 1351–1362.

166 Annals of the New York Academy of Sciences
51. Kino, T. & G.P. Chrousos. 2005. Glucocorticoid ef-
fects on gene expression. In Handbook of Stress and the
Brain. T. Steckler, N.H. Kalin & J.M.H.M. Reul, Eds.:
295–311. Elsevier B.V. Amsterdam, Netherlands.
52. Holsboer, F. & N. Barden. 1996. Antidepressants
and hypothalamic-pituitary-adrenocortical regula-
tion. Endocr. Rev. 17: 187–205.
53. Ahlijanian, M.K. et al. 2000. Hyperphosphorylated
tau and neurofilament and cytoskeletal disruptions
in mice overexpressing human p25, an activator of
cdk5. Proc. Natl. Acad. Sci. USA 97: 2910–2915.
54. Patrick, G.N. et al. 1999. Conversion of p35 to p25
deregulates Cdk5 activity and promotes neurodegen-
eration. Nature 402: 615–622.
55. Friedman, T.C. et al. 1996. Carbohydrate and lipid
metabolism in endogenous hypercortisolism: shared
features with metabolic syndrome X and NIDDM.
Endocr. J. 43: 645–655.
56. Miller, W.L. & G.P. Chrousos. 2001. The adrenal
cortex. In Endocrinology & Metabolism. P. Felig & L.A.
Frohman, Eds.: 387–524. McGraw-Hill. New York,
NY.
57. Buemann, B. et al. 2005. The N363S polymorphism
of the glucocorticoid receptor and metabolic syn-
drome factors in men. Obes. Res. 13: 862–867.
58. DeRijk, R. & E.R. de Kloet. 2005. Corticosteroid re-
ceptor genetic polymorphisms and stress responsivity.
Endocrine 28: 263–270.
59. Marti, A. et al. 2006. Meta-analysis on the effect of the
N363S polymorphism of the glucocorticoid receptor
gene (GRL) on human obesity. BMC Med. Genet. 7:
50–60.
60. Stevens, A. et al. 2004. Glucocorticoid sensitivity is
determined by a specific glucocorticoid receptor hap-
lotype. J. Clin. Endocrinol. Metab. 89: 892–897.
61. Chrousos, G.P. 2004. The glucocorticoid receptor
gene, longevity, and the complex disorders of Western
societies. Am. J. Med. 117: 204–207.
62. Phillips, D.I. et al. 1998. Elevated plasma cortisol con-
centrations: a link between low birth weight and the
insulin resistance syndrome? J. Clin. Endocrinol. Metab.
83: 757–760.
62a. Manenschijn, L., E. L. T. van den Akker, S. W. J.
Lamberts & E. F. C. Van Rossum. 2009. Clinical fea-
tures associated with glucocorticoid receptor poly-
morphisms: an overview. Ann. N. Y. Acad. Sci. Gluco-
corticoids and Mood. Clinical Manifestations, Risk
Factors, and Molecular Mechanisms. In Press.
62b. Spijker, A. T. & E. F. C. Van Rossum. 2009. Glucocor-
ticoid receptor polymorphisms in major depression:
focus on glucocorticoid sensitivity and neurocogni-
tive functioning. Ann. N. Y. Acad. Sci. Glucocorticoids
and Mood: Clinical Manifestations, Risk Factors, and
Molecular Mechanisms. In press.
63. Syed, A.A. et al. 2006. Association of glucocorticoid
receptor polymorphism A3669G in exon 9β with
reduced central adiposity in women. Obesity (Silver
Spring) 14: 759–764.
64. van Rossum, E.F. et al. 2002. A polymorphism in the
glucocorticoid receptor gene, which decreases sen-
sitivity to glucocorticoids in vivo, is associated with
low insulin and cholesterol levels. Diabetes 51: 3128–
3134.
65. van Rossum, E.F. & S.W. Lamberts. 2004. Polymor-
phisms in the glucocorticoid receptor gene and their
associations with metabolic parameters and body
composition. Recent Prog. Horm. Res. 59: 333–357.
66. van Rossum, E.F. et al. 2004. The ER22/23EK poly-
morphism in the glucocorticoid receptor gene is asso-
ciated with a beneficial body composition and muscle
strength in young adults. J. Clin. Endocrinol. Metab. 89:
4004–4009.
67. Papanicolaou, D.A. et al. 1998. The pathophysiologic
roles of interleukin-6 in human disease. Ann. Intern.
Med. 128: 127–137.
68. Chrousos, G.P. & T. Kino. 2007. Glucocorticoid ac-
tion networks and complex psychiatric and/or so-
matic disorders. Stress 10: 213–219.
69. Kino, T., T. Ichijo & G.P. Chrousos. 2004. FLASH
interacts with p160 coactivator subtypes and differ-
entially suppresses transcriptional activity of steroid
hormone receptors. J. Steroid Biochem. Mol. Biol. 92:
357–363.
70. Lanz, R.B. et al. 1999. A steroid receptor coactivator,
SRA, functions as an RNA and is present in an SRC-
1 complex. Cell 97: 17–27.
71. Kino, T. & G.P. Chrousos. 2004. Glucocorticoid and
mineralocorticoid receptors and associated diseases.
Essays Biochem. 40: 137–155.
72. De Martino, M.U. et al. 2004. The glucocorticoid
receptor and the orphan nuclear receptor chicken
ovalbumin upstream promoter-transcription factor II
interact with and mutually affect each other’s tran-
scriptional activities: implications for intermediary
metabolism. Mol. Endocrinol. 18: 820–833.
73. Charmandari, E., T. Kino & G.P. Chrousos. 2004.
Glucocorticoids and their actions: an introduction.
Ann. N. Y. Acad. Sci. 1024: 1–8.
74. Kino, T., A. Vottero & G.P. Chrousos. 2001. Min-
eralocorticoid and glucocorticoid receptors. In Nu-
clear Receptor and Genetic Disease. T.P. Burris & E.R.B.
McCabe, Eds.: 297–307. Academic Press. London,
England.
75. Nader, N., G.P. Chrousos & T. Kino. 2009. Circadian
rhythm transcription factor CLOCK regulates the
transcriptional activity of the glucocorticoid receptor
by acetylating its hinge region lysine cluster: potential
physiologic implications. FASEB J. 23: 1572–1583.

Review
Corticosteroid hormones in the central stress response: Quick-and-slow
E. Ronald de Kloet a,*, Henk Karst b, Marian Joels b
a Department of Medical Pharmacology, LACDR, Leiden University Medical Center, PO Box 9502, 2300 RA Leiden, The Netherlandsb SILS-CNS, University of Amsterdam, The Netherlands
Available online 24 October 2007
Abstract
Recent evidence shows that corticosteroid hormones exert rapid non-genomic effects on neurons in the hypothalamus and the hippo-campal CA1 region. The latter depend on classical mineralocorticoid receptors which are accessible from the outside of the plasma mem-brane and display a 10-fold lower affinity for corticosterone than the nuclear version involved in neuroprotection. Consequently, this‘membrane’ receptor could play an important role while corticosteroid levels are high, i.e. during the initial phase of the stress response.We propose that during this phase corticosterone promotes hippocampal excitability and amplifies the effect of other stress hormones.These permissive non-genomic effects may contribute to fast behavioral effects and encoding of stress-related information. The fast effectsare complemented by slower glucocorticoid receptor-mediated effects which facilitate suppression of temporary raised excitability, recov-ery from the stressful experience and storage of information for future use.Ó 2007 Elsevier Inc. All rights reserved.
Keywords: Stress; Brain; Behavior; Electrophysiology; Feedback; Glucocorticoid receptors; Mineralocorticoid receptors; Non-genomic steroid action;
Genomic steroid action; CRH
1. Introduction
Glucocorticoids are secreted from the adrenal in hourlypulses which are thought to synchronize and coordinatesleep related and daily events [42]. At any time a glucocor-ticoid response can be triggered by a stressor. In concertwith other stress mediators, the stress-induced rise in gluco-corticoid concentration facilitates adaptation to stress andrestores homeostasis, (among other things) by enhancingemotional arousal and promoting motivational and cogni-tive processes [6,17,26]. A glucocorticoid response that isexcessive, prolonged or inadequate impairs adaptation tostress and is considered a risk factor for stress-related dis-eases. The hormones have profound effects on brain devel-opment and are a significant factor in the aging process.
The key towards understanding these fundamental pro-cesses underlying homeostasis and health is in the receptorsthat mediate the action of the corticosteroids. These are the
classical glucocorticoid receptors (GR), selective for natu-rally occurring and synthetic glucocorticoids; and the min-eralocorticoid receptors (MR), which retain corticosteroneand aldosterone with a very high affinity, i.e. about 10-foldhigher than to the GR [6]. The MR also binds progesteroneand deoxycorticosterone with relatively high affinity [3] andhence this receptor is considered promiscuous in non-epi-thelial cells. In epithelial cells the MR is aldosterone-selec-tive because the naturally occurring glucocorticoids aremetabolized by the 11b-steroid-dehydrogenase type 1[13]. The MR and GR are nuclear receptors that mediategenomic actions of the naturally occurring glucocorticoidscorticosterone in rodents and cortisol in man [32,43].
Our contribution to this special issue of Frontiers willfocus on the MR and GR. These receptors are abundantlyexpressed in the limbic brain where they mediate distinctand complementary actions. While most emphasis in thepast decades was on their genomic action there obviouslywas a problem. Thus, over the years findings were reportedthat showed fast effects of corticosteroids on feedbackoperation in the HPA axis [4]. Fast effects within minuteswere also described for violent behavior propagated by a
0091-3022/$ - see front matter Ó 2007 Elsevier Inc. All rights reserved.
doi:10.1016/j.yfrne.2007.10.002
* Corresponding author. Fax: +31 71 5274715.E-mail address: [email protected] (E.R. de Kloet).
www.elsevier.com/locate/yfrne
Available online at www.sciencedirect.com
Frontiers in Neuroendocrinology 29 (2008) 268–272
Frontiers inNeuroendocrinology

fast feedforward mechanism [21] which were not genomic,but obeyed the pharmacology dictated by the classicalnuclear receptors. Moreover, corticosteroid effects wereobserved on cognitive operations in appraisal of novel sit-uations [29,30], extinction processes [2] and more recentlyon retrieval processes [7,35]. Recent discoveries using anelectrophysiological approach have supplied a mechanisticbasis to these fast behavioral and neuroendocrine effects.We first will review these fast cellular effects, and nextexamine the fast effects in the chain of events that takeplace from the first encounter with a real or imagined stres-sor to its long-term outcome on cognitive performance.
2. Fast effects of corticosteroid hormones on neuronal
function
Parvocellular neurons in the PVN are critically involvedin the production and release of CRH. These processes areunder fast and delayed negative control of corticosterone[4]. Insight in the putative neurobiological substrate ofthe fast negative feedback was recently provided by Taskerand co-workers [38]. They showed that glucocorticoids rap-idly and non-genomically suppress the frequency (but notamplitude) of miniature excitatory postsynaptic currents(mEPSCs), which reflect the postsynaptic response to thespontaneous release of a single glutamate-containing vesi-cle [8]; inhibitory events were changed in magnocellularbut parvocellular neurons [9,40]. It was argued that corti-costerone attenuates the release probability of glutamate-containing vesicles, a presynaptic phenomenon. Interest-ingly, this effect was found to depend on a postsynapticG-protein linked pathway, a process blocked by leptin[24]. This supports the involvement of retrograde messen-gers. In accordance, corticosteroids stimulate endocannab-inoid synthesis and release from the postsynapticcompartment which subsequently via a presynaptic CB1receptor leads to suppression of glutamate release [8,24].
The nature of the receptors mediating these fast effectsof corticosteroid hormones on PVN parvocellular neuronsis still unclear. Corticosterone was only active when admin-istered on the outside of the plasma membrane. This doesnot congrue with a nuclear MR or GR localization. More-over, antagonists for the classical nuclear MR and GR didnot interfere with the suppression of mEPSC frequency.Possibly a so far unknown G-protein coupled receptor isresponsible for these actions.
Rapid effects of corticosteroid hormones have also beenseen in the hippocampus, but the characteristics differ fromthose seen in the hypothalamus. Thus, it was observed thatrapidly after corticosterone application the frequency ofmEPSCs in hippocampal CA1 neurons is enhanced [18].No effect was observed on mEPSC amplitude or kineticproperties. As paired pulse facilitation was decreased, itwas concluded that corticosterone rapidly increases therelease probability of glutamate-containing vesicles.Enhanced release of glutamate from the hippocampusin vivo, shortly after administration of corticosterone, was
indeed reported [39]. Preliminary evidence indicates thatthe rapid effect on mEPSC frequency involves a presynap-tic ERK1/2 pathway, independent of retrograde messen-gers [31].
The corticosterone-induced enhancement of mEPSC fre-quency was quickly reversed upon removal of the steroidhormone [18], which points to a non-genomic mechanism.This was confirmed in follow-up experiments where theincrease in mEPSC frequency with corticosterone was alsodemonstrated in the presence of a protein synthesis inhibitor.Moreover, nuclear localization of the hormone-receptorcomplex appeared not necessary, since a corticosterone-BSAconjugate evoked effects comparable to the natural hor-mone itself. The effects of corticosterone were seen with�10 nM of the hormone, which is a concentration that canbe reached in the hippocampus under stressful conditions.
Because the hormone concentration necessary to seechanges in mEPSC frequency is closer to the Kd of theGR than of the MR it was assumed that the rapid effectsinvolve the GR. However, application of a selective GR-agonist was ineffective. The effect of corticosterone couldnot be blocked by a GR-antagonist and was still presentin forebrain specific GR knockouts [18]. By contrast, themineralocorticoid aldosterone strongly increased mEPSCfrequency, an effect that was fully suppressed by the MR-antagonist spironolactone. No change in mEPSC fre-quency was induced by corticosterone in forebrain specificMR knockouts. Collectively, these data indicate that in thehippocampus corticosterone can increase the release prob-ability of glutamate-containing vesicles in a manner requir-ing the ‘classical’ MR, which for some reason is insertedinto the membrane and has a 10-fold lower apparent affin-ity than the nuclear MR [18].
More recently, other rapid effects were also reported forCA1 hippocampal neurons. For instance, it has been foundthat corticosterone suppresses the K+-conductance IA, viaa postsynaptic membrane MR coupled to a G-proteindependent pathway [31]. Effectively, this could mean thatincreased presynaptic release of glutamate is accompaniedby an enhanced likelihood that action potentials are gener-ated postsynaptically. This could contribute to the fact thatlong-term potentiation in the CA1 area is facilitated whencorticosterone is present during high-frequency stimulationof the Schaffer collaterals, although it should be noted thatthis phenomenon is not sensitive to GR- as well as MR-blockers [41]. Interestingly, corticosterone was also foundto rapidly facilitate the excitatory effect of b-adrenergicagonists on LTP in the dentate gyrus [34]. By contrast, ifcorticosterone was applied several hours before the b-ago-nist, so that genomic effects were allowed to develop, thesteroid suppressed excitatory effects via the b-agonist onLTP [34].
3. Corticosteroid hormones in the stress response
How can these rapid cellular effects of corticosteronecontribute to the central stress response?
E.R. de Kloet et al. / Frontiers in Neuroendocrinology 29 (2008) 268–272 269

With respect to the HPA regulation, it has been knownalready for a long time that rising levels of corticosteronecan exert a fast negative control over the release of CRHand ACTH [4]. This effect may be short-lived, because assoon as corticosteroid levels plateau the negative influenceis lost, only to re-appear some time later when gene-medi-ated effects kick in. This transient character of fast cortico-steroid effects does not seem to hold for limbic actions.Thus, in vitro studies show that the enhancement ofmEPSC frequency lasts as long as corticosterone concen-trations are high (Karst, unpublished observation). Thiscould signify that during the initial stage of the centralstress response—i.e. when levels of corticosterone, peptideslike CRH and vasopressin, and of catecholamines are high-stress-induced levels of corticosterone promote hippocam-pal excitability and amplify the effect of other stress hor-mones (Fig. 1). These permissive effects of corticosterone,which at least partly involve membrane MRs, may contrib-ute to fast encoding of stress-related information, toappraisal processes and the selection of behavioralresponses to cope with the stressor [27,29,30].
By the time that hormone levels subside, gene-mediatedeffects by corticosterone have developed. These slow effectsare mostly mediated via nuclear GRs (reviewed in [15,17]).In the hippocampal CA1 area, activation of nuclear GRsraises the threshold for induction of LTP, so that any infor-mation reaching this area several hours after the encodingof stress-related information has started must be very sali-
ent in order to surpass the threshold for LTP induction.This may help to preserve the earlier information. Gene-mediated GR actions further slowly attenuate the transferof excitatory information, suppress excitatory b-adrenergicactions and enhance inhibitory effects of serotonin(reviewed in [15,17]). This fits with the notion that at least1 h after stress exposure the temporary raised excitability isreversed and normalized to pre-stress levels [16], thus pre-venting the hippocampal response from overshooting orpersisting when no longer necessary. This can be regardedas the limbic correlate of the slow negative feedback in thehypothalamus. Gene patterns contributing to these delayedgenomic effects of corticosteroids have been identified[5,28].
At last: what about the nuclear form of the MR? It hasbeen demonstrated that nuclear MR-mediated actions arecrucial for the stability of neuronal networks, survival ofneurons in the hippocampus under adverse conditions aswell as the threshold/sensitivity of the stress system andbehavioral responses [20,23,37]. Recent studies usingmouse lines where the MR was knocked out [1,12] or over-expressed [11,22,36] generally support this point of view.
4. Perspectives
While electrophysiological studies over the past yearshave firmly established the existence of rapid non-genomiccorticosteroid effects in brain, numerous questions are still
Stress Reaction
Rapid nongenomic
corticosteroid effects:
-Enhanced Glu release
probability
-Suppression IA-Synergy with NA
-Facilitation LTP
Excitability up→ →
Recovery & Adaptation
Delayed genomic
corticosteroid effects:
-Attenuated transfer of
excitatory information
-Increased 5-HT inhibition
-Suppression of NA action
-Impaired LTP
Excitability normalized
Neuroendocrine
feedback in
hypothalamus
Behavioral
feedback in
hippocampus
[cort
icoste
rone]
0 hrs1 2
threshold to activate
‘membrane’ MR
stressor
Fig. 1. Shortly after exposure to a stressor (vertical arrow), corticosteroid levels rise. Rapid as well as delayed negative feedback has been described (grey
bars), the latter causing normalization of corticosteroid levels some hours after stress exposure. The onset of the delayed negative feedback varies
(indicated here by horizontal arrow), depending e.g. on modulatory input to the PVN. Recently, rapid non-genomic cellular effects were described in the
hypothalamus, which may provide a neurobiological substrate for the fast feedback. In the hippocampus, corticosterone can also exert a number of rapid
non-genomic actions which require relatively high levels of the hormone. Hence, these will take place only when a certain threshold concentration of the
hormone is surpassed (striped line). We propose that overall these rapid effects in CA1 neurons promote excitability and amplify the effects of other stress
hormones. These rapid effects contribute to the overall ‘central’ stress reaction, comprising facets of appraisal, attention and alertness. At the same time,
however, gene-mediated pathways are started [28] which some hours later (i.e. by the time that hormone levels are declining) result in attenuated
neurotransmission, thus normalizing the earlier raised activity. This phase is important for recovery and adaptation.
270 E.R. de Kloet et al. / Frontiers in Neuroendocrinology 29 (2008) 268–272

unanswered. First and foremost it will be important to linkthe presently described cellular effects to the sets of behav-ioral and neuroendocrine observations: the cellular non-genomic effects are likely to underlie fast changes in mem-ory formation and HPA activity, but causative evidencehas not yet been provided. In addition to the rapid hor-monal influences on encoding of information, there is alsoample evidence for non-genomic modulation by stress hor-mones of retrieval of information stored in limbic regions[35]. As yet, a neurobiological substrate for this phenome-non is not available at the cellular level.
Clearly, a host of mechanistic questions are still open.The fast non-genomic effects in part require ‘classical’nuclear corticosteroid receptors [18]. Are these membranereceptors identical to or variants of the ones mediatinggenomic effects? If they are identical, then why do theynot translocate to the nucleus but instead travel towardsthe plasma membrane? And is the proportion of receptorsin (or close to) the membrane dependent on external fac-tors, like the availability of corticosteroid hormones overa longer period of time? In part, however, the receptorsmediating fast corticosteroid receptors do not display thepharmacological profile of nuclear receptors [8,41]. There-fore, the existence of a so far unknown G-protein coupledreceptor to which corticosterone binds with relatively highaffinity cannot be excluded, as was described for estrogens[33].
The comparison between hypothalamus and hippocam-pus shows that rapid non-genomic effects of corticosteroidsare regionally differentiated. It will therefore be very impor-tant to also examine rapid effects in other limbic areasimportant for the central stress response, like amygdalarnuclei or subareas of the prefrontal cortex [10,19,25].Extracellular recording studies in fact support rapid effectsof stress on firing patterns in the prefrontal cortex [14].Extension of the current studies to these areas will be nec-essary to get a full comprehension of the role of rapid non-genomic corticosteroid effects during the initial and latephases of the central stress response.
Acknowledgments
The support by the Netherlands Organisation for Scien-tific Research and the Royal Netherlands Academy of Artsand Sciences is gratefully acknowledged.
References
[1] S. Berger, D.P. Wolfer, O. Selbach, H. Alter, G. Erdmann, H.M.
Reichardt, A.N. Chepkova, H. Welzl, H.L. Haas, H.P. Lipp, G.
Schutz, Loss of the limbic mineralocorticoid receptor impairs
behavioral plasticity, Proc. Natl. Acad. Sci. USA 103 (2006) 195–200.
[2] B. Bohus, E.R. de Kloet, Adrenal steroids and extinction behavior:
antagonism by progesterone, deoxycorticosterone and dexametha-
sone of a specific effect of corticosterone, Life Sci. 28 (1981) 433–440.
[3] M.P. Carey, E.R. de Kloet, Interaction of progesterone with the
hippocampal mineralocorticoid receptor, Ann. N. Y. Acad. Sci. 746
(1994) 434–437.
[4] M.F. Dallman, Fast glucocorticoid actions on brain: back to the
future, Front. Neuroendocrinol. 26 (2005) 103–108.
[5] N.A. Datson, J. van der Perk, E.R. de Kloet, E. Vreugdenhil,
Identification of corticosteroid-responsive genes in rat hippocampus
using serial analysis of gene expression, Eur. J. Neurosci. 14 (2001)
675–689.
[6] E.R. De Kloet, M. Joels, F. Holsboer, Stress and the brain: from
adaptation to disease, Nat. Rev. Neurosci. 6 (2005) 463–475.
[7] D.J. De Quervain, B. Roozendaal, J.L. McGaugh, Stress and
glucocorticoids impair retrieval of long-term spatial memory, Nature
394 (1998) 787–790.
[8] S. Di, R. Malcher-Lopes, K.C. Halmos, J.G. Tasker, Nongenomic
glucocorticoid inhibition via endocannabinoid release in the hypo-
thalamus: a fast feedback mechanism, J. Neurosci. 23 (2003) 4850–
4857.
[9] S. Di, R. Malcher-Lopes, V.L. Marcheselli, N.G. Bazan, J.G. Tasker,
Rapid glucocorticoid-mediated endocannabinoid release and oppos-
ing regulation of glutamate and gamma-aminobutyric acid inputs to
hypothalamic magnocellular neurons, Endocrinology 146 (2005)
4292–4301.
[10] D.M. Diamond, A.M. Campbell, C.R. Park, J. Halonen, P.R.
Zoladz, The temporal dynamics model of emotional memory
processing: a synthesis on the neurobiological basis of stress-induced
amnesia, flashbulb and traumatic memories, and the Yerkes–Dodson
law, Neural Plast. (2007) 60803.
[11] D. Ferguson, R. Sapolsky, Mineralocorticoid receptor overexpression
differentially modulates specific phases of spatial and nonspatial
memory, J. Neurosci. 27 (2007) 8046–8052.
[12] P. Gass, O. Kretzer, D.P. Wolfer, S. Berger, F. Tronche, H.M.
Reichardt, C. Kellendonk, H.P. Lipp, W. Schmid, G. Schutz, Genetic
disruption of mineralocorticoid receptor leads to impaired neurogen-
esis and granule cell degeneration in the hippocampus of adult mice,
EMBO Rep. 1 (2000) 447–451.
[13] M.C. Holmes, J.R. Seckl, The role of 11beta-hydroxysteroid
dehydrogenases in the brain, Mol. Cell. Endocrinol. 248 (2006) 9–
14.
[14] M.E. Jackson, B. Moghaddam, Distinct patterns of plasticity in
prefrontal cortex neurons that encode slow and fast responses to
stress, Eur. J. Neurosci. 6 (2006) 1702–1710.
[15] M. Joels, Steroid hormones and excitability in the mammalian brain,
Front. Neuroendocrinol. 18 (1997) 2–48.
[16] M. Joels, Z. Pu, O. Wiegert, M.S. Oitzl, H.J. Krugers, Learning under
stress: how does it work? Trends Cogn. Sci. 10 (2006) 152–158.
[17] M. Joels, H. Karst, H.J. Krugers, P.J. Lucassen, Chronic stress:
implications for neuronal morphology, function and neurogenesis,
Front. Neuroendocrinol. 28 (2007) 72–96.
[18] H. Karst, S. Berger, M. Turiault, F. Tronche, G. Schutz, M. Joels,
Mineralocorticoid receptors are indispensable for nongenomic mod-
ulation of hippocampal glutamate transmission by corticosterone,
Proc. Natl. Acad. Sci. USA 102 (2005) 19204–19207.
[19] A. Kavushansky, R.M. Vouimba, H. Cohen, G. Richter-Levin,
Activity and plasticity in the CA1, the dentate gyrus, and the
amygdala following controllable vs. uncontrollable water stress,
Hippocampus 16 (2006) 35–42.
[20] H.J. Krugers, S. Maslam, S.M. van Vuuren, J. Korf, M. Joels,
Postischemic steroid modulation: effects on hippocampal neuronal
integrity and synaptic plasticity, J. Cereb. Blood Flow Metab. 19
(1999) 1072–1082.
[21] M.R. Kruk, J. Halasz, W. Mellis, J. Haller, Fast positive feedback
between the adrenocortical stress response and a brain mechanism
involved in aggressive behavior, Behav. Neurosci. 118 (2004) 1062–
1070.
[22] M. Lai, K. Horsburgh, S.E. Bae, R.N. Carter, D.J. Stenvers, J.H.
Fowler, J.L. Yau, C.E. Gomez-Sanchez, M.C. Holmes, C.J. Kenyon,
J.R. Seckl, M.R. Macleod, Forebrain mineralocorticoid receptor
overexpression enhances memory, reduces anxiety and attenuates
neuronal loss in cerebral ischaemia, Eur. J. Neurosci. 25 (2007) 1832–
1842.
E.R. de Kloet et al. / Frontiers in Neuroendocrinology 29 (2008) 268–272 271

[23] M.R. MacLeod, I.M. Johansson, I. Soderstrom, M. Lai, G. Gido, T.
Wieloch, J.R. Seckl, T. Olsson, Mineralocorticoid receptor expression
and increased survival following neuronal injury, Eur. J. Neurosci. 17
(2003) 1549–1555.
[24] R. Malcher-Lopes, S. Di, V.S. Marcheselli, F.J. Weng, C.T. Stuart,
N.G. Bazan, J.G. Tasker, Opposing crosstalk between leptin and
glucocorticoids rapidly modulates synaptic excitation via endocan-
nabinoid release, J. Neurosci. 24 (2006) 6643–6650.
[25] M. Maroun, G. Richter-Levin, Exposure to acute stress blocks
the induction of long-term potentiation of the amygdala-
prefrontal cortex pathway in vivo, J. Neurosci. 23 (2003) 4406–
4409.
[26] B.S. McEwen, Physiology and neurobiology of stress and adaptation:
central role of the brain, Physiol. Rev. 87 (2007) 873–904.
[27] J.L. McGaugh, B. Roozendaal, Role of adrenal stress hormones in
forming lasting memories in the brain, Curr. Opin. Neurobiol. 12
(2002) 205–210.
[28] M.C. Morsink, P.J. Steenbergen, J.B. Vos, H. Karst, M. Joels, E.R.
de Kloet, N.A. Datson, Acute activation of hippocampal glucocor-
ticoid receptors results in different waves of gene expression
throughout time, J. Neuroendocrinol. 18 (2006) 239–252.
[29] M.S. Oitzl, E.R. de Kloet, Selective corticosteroid antagonists
modulate specific aspects of spatial orientation learning, Behav.
Neurosci. 106 (1992) 62–71.
[30] M.S. Oitzl, M. Fluttert, E.R. de Kloet, The effect of corticosterone on
reactivity to spatial novelty is mediated by central mineralocorticos-
teroid receptors, Eur. J. Neurosci. 6 (1994) 1072–1079.
[31] J.E. Olijslagers, E.R. de Kloet, M. Joels, H. Karst, Rapid enhance-
ment of hippocampal glutamate transmission by corticosterone is
mediated via the MEK/ERK pathway. FENS Forum Meeting,
abstract 152.17 (2006).
[32] L. Pascual-Le Tallec, M. Lombes, The mineralocorticoid receptor: a
journey exploring its diversity and specificity of action, Mol.
Endocrinol. 19 (2005) 2211–2221.
[33] E.R. Prossnitz, J.B. Arterburn, L.A. Sklar, GPR30: a G protein-
coupled receptor for estrogen, Mol. Cell. Endocrinol. 265–266 (2007)
138–142.
[34] Z. Pu, H.J. Krugers, M. Joels, Corticosterone time-dependently
modulates beta-adrenergic effects on long-term potentiation in the
hippocampal dentate gyrus, Learn Mem. 14 (2007) 359–367.
[35] B. Roozendaal, Stress and memory: opposing effects of glucocorti-
coids on memory consolidation and memory retrieval, Neurobiol.
Learn Mem. 78 (2002) 578–595.
[36] A.M. Rozeboom, H. Akil, A.F. Seasholtz, Mineralocorticoid recep-
tor overexpression in forebrain decreases anxiety-like behavior and
alters the stress response in mice, Proc. Natl. Acad. Sci. USA 104
(2007) 4688–4693.
[37] R.S. Sloviter, G. Valiquette, G.M. Abrams, E.C. Ronk, A.l. Sollas,
L.A. Paul, S. Neubort, Selective loss of hippocampal granule cells in
the mature rat brain after adrenalectomy, Science 243 (1989) 535–538.
[38] J.G. Tasker, S. Di, R. Malcher-Lopes, Minireview: rapid glucocor-
ticoid signaling via membrane-associated receptors, Endocrinology
147 (2006) 5549–5556.
[39] C. Venero, J. Borrell, Rapid glucocorticoid effects on excitatory
amino acid levels in the hippocampus: a microdialysis study in freely
moving rats, Eur. J. Neurosci. 11 (1999) 2465–2473.
[40] J.M. Verkuyl, H. Karst, M. Joels, GABAergic transmission in the rat
paraventricular nucleus of the hypothalamus is suppressed by
corticosterone and stress, Eur. J. Neurosci. 21 (2005) 113–121.
[41] O. Wiegert, M. Joels, H. Krugers, Timing is essential for rapid effects
of corticosterone on synaptic potentiation in the mouse hippocampus,
Learn Mem. 13 (2006) 110–113.
[42] E.A. Young, J. Abelson, S.L. Lightman, Cortisol pulsatility and its
role in stress regulation and health, Front. Neuroendocrinol. 25
(2004) 69–76.
[43] J. Zhou, J.A. Cidlowski, The human glucocorticoid receptor: one
gene, multiple proteins and diverse responses, Steroids 70 (5–7) (2005)
407–417.
272 E.R. de Kloet et al. / Frontiers in Neuroendocrinology 29 (2008) 268–272

The central role of ions in the control of cellular excit-ability has been recognized for more than a centuryand dates back to the pioneering work of Ringer [1].For a long time, electrical excitability was a propertybelieved to be confined to a small group of highly so-phisticated cells such as nerve and muscle cells inwhich the role and need of electrical signalling wasobvious. However, during the 1960s and 1970s it be-came obvious that a number of endocrine cells sharethis capacity and that they utilize changes in theirmembrane potential to transduce changes in their
environment to acceleration of hormone secretion[2, 3].
The pancreatic beta cell is electrically excitableIn 1968 Dean and Matthews [3] provided the first evi-dence for glucose-stimulated electrical activity in thepancreatic beta cell. The salient features of this elec-trical activity are summarized in Figure 1. In the ab-sence of glucose, or at substimulatory glucose con-centrations (< 7–8 mmol/l), the membrane potentialof the beta cell is negative (–60 mV). Following eleva-tion of glucose to insulin-releasing concentrations,the beta cell depolarizes and once the cell becomessufficiently depolarized (i.e. exceeds the “thresholdpotential”), electrical activity is generated. This elec-trical activity consists of slow oscillations in mem-brane potential between a depolarized plateau, onwhich action potentials are superimposed (active
Diabetologia (1997) 40: 487–495
ReviewThe pancreatic beta-cell as a fuel sensor:an electrophysiologist’s viewpoint*P. RorsmanDepartment of Islet Cell Physiology, Novo Nordisk A/S, Copenhagen, Denmark
Springer-Verlag 1997
Summary The pancreatic beta cell serves as the fuelsensor of the entire body and controls, via secretionof the hypoglycaemic hormone insulin, the blood glu-cose concentrations within narrow limits by regula-tion of glucose uptake and release. During the last30 years, a combination of biochemical and ultra-structural approaches has resulted in dramatic pro-gress in the understanding of the processes by whichglucose and other nutrients modulate the release ofinsulin. The beta cells have also been investigatedusing electrophysiological techniques and were thusfound to be electrically excitable and to undergocomplex changes in their membrane potential whenexposed to glucose and other stimulators of secretion.The application of the patch-clamp technique to the
pancreatic islet preparations has revolutionized theunderstanding of how bioelectrical processes partici-pate in the fuel-sensing of the beta cell. An importantachievement was the identification of an ATP-sensi-tive K +-channel as the resting and glucose-sensitivemembrane conductance of the beta cell. This channelalso constitutes the target of the hypoglycaemic sul-phonylureas: a group of compounds which havebeen used successfully in the treatment of insulin-de-pendent diabetes mellitus for several decades. [Dia-betologia (1997) 40: 487–495]
Keywords Ion channels, ATP, insulin, exocytosis,pancreas.
* The 31st Minkowski lecture given in the Austria Centre,Vienna, Austria, 4 September, 1996.
Corresponding author: Dr. P. Rorsman, Department of IsletCell Physiology, Novo Nordisk A/S, Fruebjergvej 3, DK-2100Copenhagen, DenmarkAbbreviations: K-ATP channel, ATP-sensitive potassiumchannel; PKA, protein kinase A.

phase), and repolarized electrically silent intervals.The beta cell responds to glucose in a graded fashion:the fraction active phase increases as the glucose con-centration is raised until electrical activity becomescontinuous at concentrations above 20 mmol/l. Theinduction of electrical activity is a key event in the se-quence of events that culminates in the release of in-sulin and it has been possible to demonstrate thatthe periods of electrical activity coincide with pulsa-tile insulin secretion [4]. More extensive accounts ofthe beta cell electrical activity have been publishedand interested readers are referred to these for a de-tailed description [5, 6].
ATP-sensitive K + -channels: the biophysical basis forthe fuel-sensing of the beta cellRecordings using intracellular electrodes impaledinto the beta cell in intact pancreatic islets havebeen invaluable in determining the effects of insu-lin-releasing agents on the membrane potential andelectrical activity of the beta cell (reviewed in [5]).However, because it was not possible to control themembrane potential of the preparation using thistechnique, the identity and characteristics of the ionchannels underlying the electrical activity could notbe determined. Such analyses had to await the appli-cation of the patch-clamp technique to pancreatic is-let cells [7]. This technique, the features of which re-levant to the study of pancreatic beta cells havebeen described at length elsewhere [6], permits therecording under voltage-clamp conditions (i. e. themembrane potential is kept constant irrespective ofany activation of membrane currents) of both theminute single-channel and the whole-cell currents;the latter reflecting the summed activity of all theion channels in the entire plasma membrane. The in-troduction of the patch-clamp technique revolutio-nized electrophysiology and in just a few years trans-formed it from an “art”, practised in only a few la-boratories, into a standard technique of cell physiolo-gy. Using the various recording modes of the patch-clamp technique it became possible to demonstrate
that the glucose-sensitive resting conductance of thebeta cell is due to the activity of K + -channels whichare inhibited by intracellular ATP (the K-ATP chan-nel) [7–11]. The regulation of the K-ATP channel isextremely complex but there is now agreement thatchanges in the cytoplasmic ATP/ADP-ratio repre-sents an important determinant of channel activity[11, 12]. In parallel with the work on the K-ATPchannel, the voltage-dependent membrane currentsparticipating in the generation of the beta cell actionpotential were characterized. In mouse beta cells,which represent the “classic” preparation for electro-physiological experiments, the depolarizing phase ofthe action potential is attributable to the activationof voltage-gated Ca2 + -channels which are sensitiveto dihydropyridines such as nifedipine (L-typeCa2 + -channels) [13, 14]. The repolarization of the ac-tion potential principally results from the opening ofvoltage-dependent K + -channel with a time courseof activation which is delayed relative that of theCa2 + -channels (hence delayed rectifying K + -cur-rent) [13, 15, 16].
The K-ATP channel: a “target” of the hypoglycaemicsulphonylureasFollowing the identification of the K-ATP channel asthe glucose-sensitive membrane conductance of thebeta cell it was proposed that this channel also repre-sents the target of the hypoglycaemic sulphonylureas,compounds which have been used in the treatment ofnon-insulin-dependent diabetes for several decades.With the aid of the patch-clamp technique it was pos-sible to show that this was indeed the case and thattherapeutic concentrations of the sulphonylureasproduce a concentration-dependent inhibition of theK-ATP channel [17–19]. The exact nature of the in-teractions between the sulphonylureas and the K-ATP channel could not be resolved in the firstpatch-clamp experiments, but the observation thatthey remained inhibitory in isolated membrane pat-ches enabled the conclusion that their effect is notsecondary to interference with beta cell metabolism.Some of the sulphonylureas were found to be verypotent inhibitors of the K-ATP channel. For exam-ple, glibenclamide was effective at nanomolar con-centrations [19]. By using the sulphonylureas as li-gands it was thereby possible to purify and eventuallyto clone the sulphonylurea receptor. Thanks to theseefforts we now know that the K-ATP channel is acomplex of a 145 kDa sulphonylurea receptor(SUR; [20]) and an inward rectifier K + -channel pro-tein (KIR6.2; [21, 22]). Hopefully this novel molecu-lar information can be exploited in current and fu-ture endeavours to develop new and more tissue-se-lective antidiabetic compounds. In this context it ispertinent that K-ATP channels in different tissues
P.Rorsman: The pancreatic beta-cell as a fuel sensor488
Fig. 1. The membrane potential (V) of a single beta cell withinan intact pancreatic islet recorded in the presence of 6.5 and10 mM glucose as indicated by the staircase

have distinct molecular composition. For example,the K-ATP channels in cardiac and beta cells containdistinct isoforms of SUR (denoted SUR2 and SUR1,respectively) [23]. This accounts for the differentpharmacological properties of cardiac and beta cellK-ATP channels. It is not known which SUR is pre-sent in the K-ATP channels in smooth muscle cells(e.g. those in the blood vessels) but it is likely to re-present yet another isoform of SUR as neitherSUR1 nor SUR2 is expressed in smooth muscle pre-parations.
A model for glucose-stimulated insulin secretionA simple model for the stimulus-secretion couplingof the pancreatic beta cell is shown in Figure 2. Inthe absence of glucose, the cytoplasmic ATP/ADPratio is low and the K-ATP channels are active.Each beta cell is equipped with thousands of K-ATPchannels and their summed activity effectivelyclamps the beta-cell membrane potential at the K +
equilibrium potential which (with the K + -gradientsexisting over the beta-cell membrane) is around–70 mV. Ion channels other than the K-ATP channelsare present, and perhaps active even in the absence ofglucose, but they are too few to influence the mem-brane potential as long as the K-ATP channels re-main active. When the beta cell is exposed to glucose,the associated acceleration of glucose metabolismleads to a rise in the cytoplasmic ATP/ADP-ratioand the K-ATP channels close. Once they are almost
completely inhibited ( > 90%), the remaining K-ATP conductance is unable to balance the depolariz-ing influence of (tonically active?) background con-ductance(s) and the beta cell depolarizes. When thisdepolarization is large enough, voltage-gated Ca2 + -channels become activated in a feed-forward manner(i. e. the initial depolarization up to the threshold po-tential causes the opening of a few voltage-gatedCa2 + -channels which in turn causes a bigger depolar-ization and the opening of additional Ca2 + -channelswith resultant further depolarization, etc), thus ac-counting for the upstroke of the beta-cell action po-tential. The associated Ca2 + -influx causes a transientelevation of the cytoplasmic Ca2 + -concentration([Ca2 + ]i) [24] which, via a series of poorly defined re-actions, culminates in the exocytosis of the insulin-containing granules.
The unique feature of the pancreatic beta cell, es-sential for its ability to serve as the body’s fuel sensor,is the presence of the K-ATP channels. As discussedabove, the activity of these channels sets the mem-brane potential of the beta cell and thus determinesits electrical and secretory activities. If the beta cellhad not been equipped with K-ATP channels, itwould have been tonically active and constantly re-leasing its insulin into the circulation regardless ofthe glucose concentration. Indeed, these are preciselythe characteristics of beta-cells isolated from patientswith persistent hyperinsulinaemic hypoglycaemia ofinfancy [25]. This rare hereditary disease is linked tomutations in SUR [26], which is part of the K-ATPchannel complex (see above), and thus results in theformation of non-functional K-ATP channels. Thefact that K-ATP channels play such an importantrole in the stimulus-secretion coupling of the betacell does not exclude, however, that metabolic regu-lation of more distal processes also contributes tothe overall fuel-sensing of the beta cell. For example,there is evidence suggesting that glucose metabolismmay control both the functional state of the voltage-gated Ca2 + -channels [27] (thus determining theamount of Ca2 + -entry and the extent of Ca2 + -in-duced exocytosis) as well as the insulin secretory pro-cess itself [28]. The objective of beta-cell electrical ac-tivity is to generate the intracellular signal that initi-ates the exocytosis of the insulin-containing granules.In the remainder of this review I shall therefore dis-cuss the control of exocytosis in the insulin-secretingbeta cell and consider various modulatory mechan-isms.
Capacitance measurements of insulin secretionElucidation of the fundamental properties of exocy-tosis requires a means to record secretion in singlecells with high temporal (millisecond) resolution.Unfortunately, none of the traditional biochemical
P.Rorsman: The pancreatic beta-cell as a fuel sensor 489
Fig. 2. Schematic model for the stimulus-secretion coupling ofthe pancreatic beta cell. Glucose produces, via its metabolism,an increased cytoplasmic ATP/ADP-ratio which inhibits theK-ATP channels. This results in membrane depolarizationand the opening of voltage-dependent Ca2 + -channels, an in-crease in [Ca2 + ]i and the initiation of exocytosis of the insu-lin-containing secretory granules. Also indicated in the modelis the site of action of the hypoglycaemic sulphonylureas whichinhibit the K-ATP channels by a direct effect which is not de-pendent on glucose metabolism

approaches for measuring insulin release haveachieved this resolution. We have therefore been ob-liged to monitor changes in cell capacitance as an in-dicator of exocytosis. The membrane capacitance(C) is an electrical property of the cell which is pro-portionally related to the cell surface area. When theinsulin-containing granules undergo exocytosis, theirmembranes are incorporated into the plasma mem-brane (Fig. 3). The resulting increase in cell surfacearea can be monitored as an increase in cell capaci-tance using electrophysiological techniques [29]. Themajor advantages of capacitance measurements overmore traditional approaches to study secretion are:1) the measurements can be carried out in single cellswith millisecond resolution; 2) the experiments arecarried out in a voltage-clamped preparation: i. e.any effects of a compound on membrane conductan-ces and membrane potential will not influence the re-sults; and 3) large proteins such as antibodies can beapplied intracellularly by including them into the pip-ette solution which replaces the cytoplasm when uti-lizing the whole-cell recording mode of the patch-clamp technique. The specific capacitance of biologi-cal membranes (including those of the plasma mem-brane and the secretory granules) is ≈ 10 fF/ m2.With a diameter of 250 nm [30], each beta-cell gran-ule can be estimated to add 0.2 m2 of membranearea which corresponds to 2 fF of capacitance. Capa-citance increases with this unitary amplitude havebeen recorded [31] and may accordingly reflect singleexocytotic events (i. e. the fusion of an individualgranule with the plasma membrane). It must be keptin mind that recordings of cell capacitance do not
involve measurements of insulin secretion as suchbut changes in a physical property of the cell whichhopefully reflect exocytosis. To ascertain that this isindeed the case we have combined capacitance mea-surements with fluorimetric and/or electrochemicaldetection of insulin secretion [32]. So far we have en-countered no discrepancies between the capacitancemeasurements and secretion detected by the alterna-tive methods making it reasonable to conclude thatan increase in cell capacitance can be equated to exo-cytosis. The conclusion is reinforced by the observa-tions that insulin secretion and exocytosis as reportedby the capacitance measurements exhibit the sameCa2 + -dependence [32], are equally affected by cool-ing [33] and similarly modulated by hormones andneurotransmitters [34, 35]. Capacitance measure-ments finally offer the unique possibility of studyingthe retrieval of the secreted membranes by endocyto-sis. In this case, membrane retrieval results in a de-creased membrane surface area and thus a reductionof the cell capacitance [36].
Exocytosis in the beta cell is rapidUsing capacitance measurements it was possible todemonstrate that the latency between the opening ofthe Ca2 + -channels and the onset of exocytosis is lessthan 50 ms. This is shorter than the time required for[Ca2 + ]i to equilibrate within the beta cell which sug-gests that the Ca2 + -channels, the secretory granulesand the release sites are situated in the near vicinityof each other. During the first 50 ms the rate of capa-citance increase approached 1 pF/s. Using the conver-sion factor of 2 fF/granule this can be converted to anexocytotic rate of 500 granules/s. Since a beta cellcontains 13000 granules [30], this value correspondsto a release rate of 4% of the total granule numberbeing released per second! Clearly, exocytosis canonly continue at this high rate for a very limited peri-od and during protracted stimulations exocytosis pro-ceeds at much slower average rates. This decline inthe exocytotic rate reflects the gradual depletion ofthe pool of granules which is immediately availablefor release (“the readily releasable pool”). Once thispool has been depleted other processes, such as therefilling of the readily releasable pool (see below),become rate-limiting to exocytosis.
It should be made clear that the short latency(< 50 ms) observed in the capacitance measurementsdoes not imply that the release process (including thedissolution of the Zn2 + -insulin crystal) is completedwithin this time span: the latency is simply a reflectionof the time required for the granular membrane tofuse with the plasma membrane and thus be detectedby the capacitance measurements. Even so the aboveconsiderations illustrate the high speed and capacityof the exocytotic machinery in the beta cell.
P.Rorsman: The pancreatic beta-cell as a fuel sensor490
Fig. 3. Principle for capacitance measurements of exocytosis.The exocytosis of a secretory granule (left and centre) resultsin the incorporation of the granular membrane with the plasmamembrane. The added membrane causes an increase in cellsurface area which can be detected as an increase in cell capa-citance (Cm), an electrical parameter of the cell which is pro-portionally related to the surface area (A; i. e. Cm ∝ A). Thespecific membrane capacitance is 10 fF/ m2. The surface areaof a secretory granule can be estimated as 0.2 m2 assumingspherical geometry and a granule diameter of 0.25 m [30].The fusion of a single granule accordingly results in a capaci-tance increase of 2 fF. When a granule is subsequently re-trieved by endocytosis, a corresponding decrease in cell capaci-tance can be recorded (right)

Modulation of Ca2 + -induced exocytosisExocytosis in the beta cell is clearly Ca2 + -dependentbut Ca2 + should perhaps be regarded as an initiatorrather than a determinant of exocytosis. This is sug-gested by the observation that the amplitude of theexocytotic responses depends to a greater extent onthe activity of protein kinases and phosphatases thanthe actual [Ca2 + ]i. For example, agents which in-crease cytoplasmic cyclic AMP levels, such as gluca-gon and glucagon-like peptide-1, potentiate glucose-stimulated insulin secretion by a protein kinase A(PKA)-dependent mechanism almost 10-fold withoutmuch affecting Ca2 + -influx or [Ca2 + ] [34, unpub-lished data]. In the case of glucagon-like peptide-1(GLP-1), no stimulation of exocytosis was observedin the absence of glucose suggesting that ATP de-rived by glucose metabolism is required for thePKA-dependent phosphorylation. Our capacitancemeasurements indicate that this stimulation princi-pally results from PKA accelerating granule mobili-zation from the reserve pool into the readily releasa-ble pool thus increasing the size of the latter overfivefold. When exocytosis is subsequently initiatedby elevation of [Ca2 + ]i, a greater number of granulesare available for release. We have previouslypostulated that cAMP acts by “sensitizing” the secre-tory machinery [34]. However, the fact that the rela-tionship between [Ca2 + ]i and exocytosis remains thesame in the absence and presence of cAMP is hardto reconcile with such a concept and an increasedsize of the readily releasable pool may produce ef-fects on exocytosis which at first glance are difficultto distinguish from a sensitizing mechanism.
The molecular mechanism by which PKA acceler-ates granule mobilization remains obscure. In nerveendings, PKA is known to exert a similar action as inthe beta cell by phosphorylation of the protein synap-sin-1 which controls the interactions between the vesi-cles and the cytoskeleton. Synapsin-1 is not expressedin pancreatic beta cells but a synapsin-1-like protein,which may fulfill the function of synapsin-I,has recent-ly been characterized in insulin-secreting cells [37].
Ca2 + -induced exocytosis is also enhanced byagents which activate protein kinase C, such as AChand the phorbol ester 4- -phorbol-12- -myristate-13- -acetate (PMA) [35]. In general, conditionswhich promote protein phosphorylation lead to en-hancement of secretion. Conversely one would ex-pect that agents which produce the activation of pro-tein phosphatases inhibit exocytosis. Indeed, thisseems to be the mechanism by which the inhibitoryhormones and neurotransmitters somatostatin, gala-nin and adrenaline suppress glucose-stimulated insu-lin secretion. The action of these compounds is medi-ated by activation of an inhibitory (pertussis toxin-sensitive) G-protein and culminates in the activationof the protein phosphatase calcineurin [38].
Hypoglycaemic sulphonylureas stimulate insulinsecretion by interaction with exocytotic machineryPerhaps the most surprising finding that has emanat-ed from the capacitance measurements is that thesulphonylureas, in addition to closing the K-ATPchannels, also stimulate insulin secretion by direct in-teraction with the exocytotic machinery [39] (but see[40] for conflicting data). Such an effect is not easilydetected in ordinary assays of insulin secretion as thestimulation of secretion resulting from the closure ofthe K-ATP effectively obscures any contribution ofa late mechanism. Using capacitance measurementsit became possible to separate the two effects as themembrane potential was voltage-clamped and thusheld constant irrespective of K-ATP channel activity.An effect of the sulphonylureas on exocytosis wouldalso be consistent with the ultrastructural and bio-chemical evidence indicating that as much as 90 % ofthe sulphonylurea-binding in the beta cell is intracel-lular and localized to the secretory granules [41, 42].
The observations that the sulphonylureas inter-fere with exocytosis, possibly by binding to granularsulphonylurea receptors, raises several interestingquestions: 1) Is the granular sulphonylurea receptorthe same as the 145 kDa SUR which is part of theK-ATP channel? 2) Do the granular sulphonylureareceptors couple to ion channels in the granulemembrane. Sulphonylurea-sensitive membrane cur-rents have been demonstrated in pancreatic zymo-gen granules and have been proposed to control thefusion process [43]. It is attractive to speculate thatthe sulphonylureas modulate exocytosis in the betacell by interference with similar conductances in theinsulin-containing granules. In this context it is of in-terest that the 145 kDa sulphonylurea receptor hasbeen reported to promiscuously couple to K + -chan-nel proteins other than KIR6.2 and it is thereforepossible that it may also associate with other channelproteins [44]; 3) What is the physiological role of thegranular sulphonylurea receptors and do they parti-cipate in “normal” Ca2 + -induced exocytosis? Theanswers to these questions are clearly central to theunderstanding of how the sulphonylureas stimulateexocytosis in the beta cell. However the significanceof these results may not be limited to the under-standing of the control of insulin secretion. The sul-phonylureas have been postulated to enhance glu-cose uptake in fat cells [45, 46]; an effect whichseems as controversial [47] as the effect on exocyto-sis in the beta cell [40]. Glucose uptake in fat cellsinvolves the insertion of the glucose transportersinto the plasma membrane by exocytosis of their in-tracellular storage vesicles [48, 49]. Since exocytosisin various cell types appears to involve the same mo-lecular processes it is attractive to speculate that themechanism we have described in the pancreaticbeta cell is also operational in other cells (such as
P.Rorsman: The pancreatic beta-cell as a fuel sensor 491

adipocytes) and accounts for some of the reportedextrapancreatic actions of the sulphonylureas.
Metabolic regulation of exocytosisIn this review I emphasize the metabolic regulationof the beta cell. I have already described how ATP,via regulation of the K-ATP channels, controls themembrane potential and thereby Ca2 + -influx andCa2 + -induced exocytosis. However, there is evidencesuggesting that ATP also controls insulin secretion ina more direct way. Experiments on permeabilized in-sulin-secreting cells have indicated that withdrawalof ATP from the cytoplasm results in 90% inhibitionof exocytosis [50]. Moreover, glucose exerts a strongstimulatory action (particularly at late times) in cellswhich are already maximally depolarized by high ex-tracellular K +, i. e. under conditions where the sugaris unable to act via depolarization and elevation of[Ca2 + ]i [28]. Collectively these observations indicatethat access to cytoplasmic ATP (or another glucosemetabolite) is somehow rate-limiting to exocytosis.We have applied capacitance measurements in con-junction with photorelease of caged Ca2 + from itscaged “precursor” Ca2 + /NP-EGTA to test this hy-pothesis. A representative experiment is shown inFigure 4. Here [Ca2 + ]i was elevated in two differentcells which were dialysed with an ATP-containing oran ATP-free solution. Whereas elevation of [Ca2 + ]i
in the presence of ATP produced a biphasic stimula-tion of exocytosis (seen as rapid initial increase incell capacitance followed by a sustained second slow-er phase), no change in cell capacitance was observedin the absence of ATP. If anything, the response inthe latter cell consisted of a transient decrease in cellcapacitance which may reflect endocytosis of gran-ules that had been inserted in the plasma membraneduring the period required for the wash-in of theCa2 + /NP-EGTA complex and the concomitantwash-out of the endogenous ATP. Ca2 + -induced exo-cytosis in the beta cell is clearly highly dependent onaccess to cytoplasmic ATP. In this respect the pan-creatic beta cell differs from other neuroendocrinecells. In both chromaffin and pituitary cells, largeexocytotic responses can be obtained long after com-plete wash-out of ATP [51]. As I shall try to explainbelow, this does not necessarily imply that the bio-chemical regulation of exocytosis in the beta cell dif-fers from that in the other cell types in any funda-mental way.
As alluded to above, both ultrastructural and func-tional studies have suggested that the granules in en-docrine cells exist in pools of different “releasability”[52]. A small fraction of the total granule populationis immediately available for release and accordinglydesignated as “the readily releasable pool”. Thesegranules are probably located just beneath the
membrane and are the first to undergo exocytosiswhen [Ca2 + ]i is elevated. The vast majority of gran-ules are not immediately available for release andpresumably located further away from the plasmamembrane. These granules need to be “mobilized”into the readily releasable pool before they can be re-leased and are referred to as the “reserve pool”. The“mobilization“ of the granules may involve eithertheir physical translocation within the cell, a chemicalmodification of the granules (such that the releaseprobability is increased) or both.
According to current biochemical models of exo-cytosis, hydrolysis of ATP is required in the chemicalmodification of the granules which precedes exocyto-sis (“priming”). When [Ca2 + ]i subsequently rises toexocytotic levels, the primed granules (and onlythose) can be released in a process which does not re-quire any further consumption of ATP. However, thepool of primed/energized granules is of limited sizeand once these granules have been released the poolneeds to be replenished in an ATP-dependent wayby mobilization of granules from the reserve pool. Asimple explanation to the apparent greater ATP-de-pendence of exocytosis in the beta cell than in theother neuroendocrine cells is therefore that the insu-lin-secreting cell contains fewer primed granules. Inthe pituitary cell, the size of this pool has been esti-mated both functionally and by electron microscopy.Both methods suggest that the primed pool compri-ses ≈ 4000 granules. This is considerably higher thanthe corresponding number in the pancreatic beta
P.Rorsman: The pancreatic beta-cell as a fuel sensor492
Fig. 4. Exocytosis is ATP-dependent. Exocytosis (monitored asan increase in cell capacitance; Cm) was elicited by photore-lease of Ca2 + from its caged precursor Ca2 + /NP-EGTA whichhad been preloaded into the cell. Upon irradiation with ultra-violet light, the affinity of NP-EGTA for Ca2 + is dramaticallyreduced resulting in the “release” of Ca2 + . Photolysis was ef-fected as indicated by the dotted vertical lines in the presence(right) and absence of ATP (left). Note the failure of Ca2 + toelicit exocytosis in the absence of ATP. The capacitance in-crease in the presence of ATP ( ≈ 1000 fF) corresponds to therelease of 500 secretory granules

cell. Based on our capacitance measurements, we es-timate that only 15–50 granules (Fig. 5), or 0.1–0.3%of the total granule population ( ≈ 13000 [30]), existin the “primed” state and are capable of being re-leased in an ATP-independent fashion. Since thenumber of energized granules in the beta cell isonly ≈ 1% of that in the pituitary cell, it is not surpris-ing that insulin secretion exhibits a higher ATP de-pendence. It seems possible that this represents animportant functional adaptation as keeping the num-ber of energized granules low provides the beta cellwith the means of rapidly adjusting its secretory capa-city to the metabolic state.
Comparison with insulin secretionA rapid elevation of [Ca2 + ]i produces a biphasic sti-mulation of exocytosis consisting of an initial rapidincrease in cell capacitance followed by a secondslower phase (Fig. 4). This biphasic response can beinterpreted as the release of different pools of gran-ules. Whereas the first rapid phase is likely to corre-spond to the release of granules situated immediatelybeneath the membrane (the readily releasable and/orprimed pool), the second slower phase reflects themobilization of the granules from the reserve pool.The biphasic increase in cell capacitance is clearly re-miniscent of the biphasic nature of glucose-stimulat-ed insulin secretion [53] and it is attractive to specu-late that it can be explained in similar terms (Fig. 6).In fact, the size of the readily releasable pool whichcan be released in an ATP-independent fashion com-pares favourably with the number of granules thatcan be estimated to undergo exocytosis during thefirst phase of glucose-stimulated insulin secretion (L.Eliasson, P. Rorsman, unpublished data). Using pho-torelease of ATP from a caged precursor we believeit has been possible to estimate the time required fora granule to pass from the reserve pool into the read-ily releasable pool and to be released. We consistent-ly observed a delay of ≈ 10 s between the applicationof ATP and the onset of secretion under experimen-tal conditions that ensure that the primed/readily re-leasable pool was previously depleted. The long la-tency argues that the process of mobilization involvesphysical translocation, and not just chemical modifi-cation, of the granules.
Glucose metabolism, ATP and diabetesIn this review I have attempted to illustrate how glu-cose metabolism, via changes in the cytoplasmicATP concentration, exerts its control of insulin secre-tion. The action of ATP is exerted at several levels.First, it controls the size of the readily releasablepool of granules by regulating the rate of granule mo-bilization/priming. Secondly, ATP generated by glu-cose metabolism determines the amplitude of Ca2 + -evoked secretion via protein kinase A and C-depen-dent phosphorylation of exocytosis-regulating pro-teins. Thirdly, glucose metabolism modulates the ac-tivity of the voltage-dependent Ca2 + -channels andthus Ca2 + -entry and Ca2 + -induced secretion. Finally,by controlling the activity of the ATP-sensitive K + -channel, ATP regulates the membrane potential ofthe beta cell and thereby the electrical and secretoryactivities. Because ATP exerts so many regulatoryfunctions it is not surprising that conditions which in-terfere with the ability of the beta cell to generateATP have marked effects on its secretory capacity.For example, it has been reported that increased
P.Rorsman: The pancreatic beta-cell as a fuel sensor 493
Fig. 5. Comparison of the number of primed granules in thebeta cell and in a pituitary cell. The pituitary cell contains amuch larger number pool of primed (energized) granules,which are located just beneath the plasma membrane and thatcan be rapidly released in a seemingly ATP-independent fash-ion when [Ca2 + ]i is elevated, than the beta cell
Fig. 6. Biphasic glucose-stimulated insulin secretion can be ex-plained as the release of distinct pools of granules. The firstphase of insulin secretion can be accounted for by the releaseof readily releasable (primed/docked) granules which arelocated immediately below the plasma membrane. The secondslower phase results from the time- and ATP-dependent mobi-lization of granules situated further away from the plasmamembrane

activity of ATP-consuming substrate cycles of glu-cose metabolism (e.g. glucose → glucose 6-phosphate→ glucose; reactions catalysed by glucokinase andglucose 6-phosphatase, respectively) is an early signof human non-insulin-dependent diabetes [54, 55].Such a defect will not only interfere with the abilityof glucose to depolarize the beta cell but also, as out-lined above, reduce the refilling of the readily relea-sable pool thus possibly accounting for absence of afirst phase of glucose-stimulated insulin secretion inthese patients [56]. Finally, it deserves pointing outthat currently available pharmacological principlesfor the treatment of non-insulin-dependent diabetes,such as the sulphonylureas, only rectify the inabilityof glucose to close the K-ATP channel but fail to cor-rect the other ATP-dependent steps and these ac-cordingly require alternative therapeutic approaches.
Acknowledgements. I thank the present and past members ofmy group (Dr. K. Bokvist, Dr. P.A. Smith, Dr. O. Larsson, Dr.C. Ammala, Dr. L. Eliasson, Dr. E. Renstrom, Dr. J. Gromada,Dr. L. Best and Dr. W.-G. Ding) for their hard work, loyaltyand enthusiasm. During the last 10 years I have had a fruitfuland productive collaboration with Professor F.M. Ashcroft(Oxford) and Professor P.O. Berggren (Stockholm). Finally Iwish to express my gratitude to Dr. G. Trube for first introduc-ing me to the patch-clamp technique. Supported in part byThe Juvenile Diabetes Foundation, The Swedish Medical Re-search Council, The Nordic Insulin Foundation, The SwedishDiabetes Association and the Novo Nordisk A/S.
Note added in proof. Recently Isomoto et al. (J Biol Chem 271:24321-24324, 1996) describe an isoform of the cardiac sulpho-nylurea receptor (SUR2B). This isoform is present in smoothmuscle cells and, when it is coexpressed with KIR6.2, formsK-ATP channels with the pharmacological properties of thesmooth muscle type of the channel.
References1. Ringer S (1883) A further contribution regarding the influ-
ence of the different constituents of blood on the contrac-tion of the heart. J Physiol 4: 29–42
2. Dean PM, Matthews EK (1968) Electrical activity in pan-creatic islet cells. Nature 219: 389–390
3. Taraskevich PS, Douglas WW (1977) Action potentials oc-cur in cells of the normal anterior pituitary gland and arestimulated by the hypophysiotropic peptide thyrotropin re-leasing hormone. Proc Natl Acad Sci USA 74: 4064–4067
4. Barbosa RM, Silva AM, Tome AR, Stamford JA, SantosRM, Rosario LM (1996) Real time electrochemical detec-tion of 5-HT/insulin secretion from single pancreatic islets:effects of glucose and K + depolarization. Biochem Bio-phys Res Commun 228: 100–104
5. Henquin JC, Meissner HP (1984) Significance of ionicfluxes and changes in membrane potential for stimulus-se-cretion coupling in pancreatic B-cells. Experientia 40:1043–1052
6. Ashcroft FM, Rorsman P (1989) Electrophysiology of thepancreatic -cell. Prog Biophys Molec Biol 54: 87–143
7. Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ(1981) Improved patch-clamp techniques for the recording
from cells and cell-free membrane patches. Pflugers Arch291: 85–100
8. Cook DL, Hales CN (1984) Intracellular ATP directlyblocks K + -channels in pancreatic B-cells. Nature 312:271–273
9. Ashcroft FM, Harrison DE, Ashcroft SJH (1984) Glucoseinduces closure of single potassium channels in isolated ratpancreatic -cells. Nature 312: 446–448
10. Rorsman P, Trube G (1985) Glucose-dependent K + -chan-nels in pancreatic -cells are regulated by intracellularATP. Pflugers Arch 405: 305–309
11. Misler S, Falke LC, Gillis K, McDaniel ML (1986) A meta-bolite-regulated potassium channel in rat pancreatic B-cells. Proc Natl Acad Sci USA 83: 7119–7123
12. Dunne MJ, Petersen OH (1986) Intracellular ADP acti-vates K + -channels that are inhibited by ATP in an insu-lin-secreting cell line. FEBS Lett 208: 59–62
13. Rorsman P, Trube G (1986) Calcium and potassium cur-rents in mouse pancreatic -cells under voltage-clamp con-ditions. J Physiol 374: 531–550
14. Rorsman P, Ashcroft FM, Trube G (1988) Single Ca chan-nel currents in mouse pancreatic B-cells. Pflugers Arch412: 597–603
15. Smith PA, Bokvist K, Arkhammar P, Berggren PO, Rors-man P (1990) Delayed rectifying and calcium activatedK + -channels and their significance for action potential re-polarization in mouse pancreatic -cells. J Gen Physiol 95:1041–1059
16. Bokvist K, Rorsman P, Smith PA (1990) Effects of externaltetraethylammonium and quinine on delayed rectifyingK + -channels in mouse pancreatic -cells. J Physiol 423:311–325
17. Sturgess NC, Ashford MLJ, Cook DL, Hales CN (1985)The sulfonylurea receptor may be an ATP-sensitive potas-sium channel. Lancet II: 474–475
18. Trube G, Rorsman P, Ohno-Shosaku T (1986) Opposite ef-fects of tolbutamide and diazoxide on the ATP-dependentK + -channel in mouse pancreatic -cells. Pflugers Arch407: 493–499
19. Zunkler BJ, Lenzen S, Manner K, Panten U, Trube G(1988) Concentration-dependent effects of tolbutamide,meglitinide, glipizide, glibenclamide and diazoxide onATP-regulated K + -currents in pancreatic B-cells. ArchPharmacol 337: 225–230
20. Aguilar-Bryan L, Nichols CG, Herrera-Sosa H, Nguy K,Bryan J, Nelson DA (1995) Cloning of the -cell high-affi-nity sulfonylurea receptor: a regulator of insulin secretion.Science 268: 423–426
21. Inagaki N, Gonoi T, Clement JP IV et al. (1995) Reconsti-tution of IKATP: an inward rectifier subunit plus thesulfonylurea receptor. Science 270: 1166–1170
22. Sakura H, Ammala C, Smith PA, Gribble FM, AshcroftFM (1996) Cloning and the functional expression of thecDNA encoding a novel ATP-sensitive potassium channelsubunit expressed in pancreatic -cells, brain, heart andskeletal muscle. FEBS Lett 377: 338–344
23. Nichols CG, Shyng SL, Nestorowicz A et al. (1996) Adeno-sine diphosphate as an intracellular regulator of insulin se-cretion. Science 272: 1785–1787
24. Rorsman P, Ammala C, Berggren PO, Bokvist K, LarssonO (1992) Cytoplasmic calcium transients due to single ac-tion potentials and voltage-clamp depolarizations in mousepancreatic B-cells. EMBO J 11: 2877–2884
25. Kane C, Shepherd RM, Squires PE et al. (1996) Loss offunctional KATP channels in pancreatic -cells causes per-sistent hyperinsulinemic hypoglycemia of infancy. NatureMed 2: 1344–1347
P.Rorsman: The pancreatic beta-cell as a fuel sensor494

26. Thomas PM, Cote GJ, Wohlik N et al. (1995) Mutationsof the sulfonylurea receptor gene in familial persistenthyperinsulinemic hypoglycemia of infancy. Science 268:426–429
27. Smith PA, Rorsman P, Ashcroft FM (1989) Modulation ofdihydropyridine-sensitive Ca2 + -channels by glucose meta-bolism in mouse pancreatic B-cells. Nature 342: 550–553
28. Detimary P, Gilon P, Nenquin M, Henquin JC (1994) Twosites of glucose control of insulin release with distinct de-pendence on the energy state in pancreatic B-cells. Bio-chem J 297: 455–461
29. Neher E, Marty A (1982) Discrete changes of cell mem-brane capacitance observed under conditions of enhancedsecretion in bovine chromaffin cells. Proc Natl Acad SciUSA 79: 6712–6716
30. Dean PM (1973) Ultrastructural morphometry of the pan-creatic beta-cell. Diabetologia 9: 115–119
31. Ammala C, Eliasson L, Bokvist K, Larsson O, AshcroftFM, Rorsman P (1993) Exocytosis elicited by action poten-tials and voltage-clamp calcium currents in individualmouse pancreatic B-cells. J Physiol 472: 665–688
32. Bokvist K, Eliasson L, Ammala C, Renstrom E, Rorsman P(1995) Co-localization of L-type Ca2 + -channels and insu-lin-containing granules and its significance for the initiationof exocytosis in mouse pancreatic B-cells. EMBO J 14: 50–57
33. Renstrom E, Eliasson L, Bokvist K, Rorsman P (1996)Cooling inhibits exocytosis in single mouse B-cells by sup-pression of granule mobilization. J Physiol 494: 41–52
34. Ammala C, Ashcroft FM, Rorsman P (1993) Ca2 + -inde-pendent potentiation of insulin release by cyclic AMP inpancreatic -cells. Nature 363: 556–558
35. Ammala C, Eliasson L, Bokvist K et al. (1994) Activationof protein kinases and inhibition of protein phosphatasesplay a central role in the regulation of exocytosis in insu-lin-secreting mouse pancreatic B-cells. Proc Natl Acad SciUSA 91: 4343–4347
36. Eliasson L, Proks L, Ammala C et al. (1996) Endocytosis ofsecretory granules in pancreatic -cells evoked by transientelevation of cytosolic calcium. J Physiol 493: 755–767
37. Matsumoto K, Fukunaga K, Miyazaki J, Schichiri M, Miya-moto E (1995) Ca2 + /calmodulin-dependent protein kinaseII and synapsin 1-like protein in mouse insulinoma MIN-6cells. Endocrinology 136: 3784–3793
38. Renstrom E, Ding WG, Bokvist K, Rorsman P (1996) Neu-rotransmitter-induced inhibition of exocytosis in insulin-se-creting -cells by activation of calcineurin. Neuron 17: 513–522
39. Eliasson L, Renstrom E, Ammala C et al. (1996) PKC-de-pendent stimulation of exocytosis by sulfonylureas in pan-creatic cells. Science 271: 813–815
40. Garciabarrado MJ, Jonas JC, Gilon P, Henquin JC (1996)Sulfonylureas do not increase insulin secretion by a me-chanism other than a rise in cytoplasmic Ca2 + in pancreaticB-cells. Eur J Pharmacol 298: 279–286
41. Carpentier JL, Sawano F, Ravazzola M, Malaisse WJ(1986) Internalization of 3H-glibenclamide in pancreatic is-let cells. Diabetologia 29: 259–26
42. Ozanne SE, Guest PC, Hutton JC, Hales CN (1995) Intra-cellular localization and molecular heterogeneity of thesulphonylurea receptor in insulin-secreting cells. Diabeto-logia 38: 277–282
43. Thevenod F, Chathadi KV, Jiang B, Hopfer U (1992) ATP-sensitive K + conductance in pancreatic zymogen granules:block by glyburide and activation by diazoxide. J MembrBiol 129: 253–256
44. Ammala C, Moorhouse A, Gribble F et al. (1996) Promis-cuous coupling between the sulphonylurea receptor and in-wardly rectifying potassium channels. Nature 379: 545–548
45. Tsiani E, Ramlal T, Leiter LA, Klip A, Fantus IG (1995)Stimulation of glucose uptake and increased plasma mem-brane content of glucose transporters in L6 skeletal musclecells by the sulfonylureas gliclazide and glyburide. Endocri-nology 136: 2505–2512
46. Muller G, Wied S (1993) The sulfonylurea drug, glimepir-ide, stimulates glucose transport, glucose transporter trans-location, and dephosphorylation in insulin-resistant rat adi-pocytes in vitro. Diabetes 42: 1852–1867
47. Panten U, Schwanstecher M, Schwanstecher C (1992) Pan-creatic and extrapancreatic sulfonylurea receptors. HormMetabol Res 24: 549–554
48. Simpson IA, Cushman SW (1986) Hormonal regulation ofmammalian glucose transport. Ann Rev Biochem 55:1055–1089
49. Lienhard GE (1983) Regulation of cellular membranetransport by the exocytotic insertion and endocytotic re-trieval of transporters. Trends Biochem Sci 8: 125–127
50. Regazzi R, Wollheim CB, Lang J et al. (1995) Vamp-2 andcellubrevin are expressed in pancreatic -cells and are es-sential for Ca2 + but not for GTP!S induced insulin secre-tion. EMBO J 14: 2723–2730
51. Parsons TD, Coorssen JR, Horstmann H, Almers W (1995)Docked granules, the exocytotic burst and the need forATP hydrolysis in endocrine cells. Neuron 15: 1085–1096
52. Neher E, Zucker RS (1993) Multiple calcium dependentprocesses related to secretion in bovine chromaffin cells.Neuron 10: 21–30
53. Grodsky GM (1994) An update on implications of phasicinsulin secretion. In: Pickup J, Williams G (eds) Textbookof diabetes. Blackwell Scientific Publications, Oxford, Vol.1, pp. 421–430
54. Efendic S, Wajngot A, Vranic M (1985) Increased activityof the glucose cycle in the liver: early characteristic of type2 diabetes. Proc Natl Acad Sci USA 82: 2965–2969
55. Khan A, Ostenson CG, Efendic S (1994) Glucose cycling inpancreatic islets. In: Flatt P, Lenzen S (eds) Frontiers of in-sulin secretion and pancreatic B-cell research. Smith-Gor-don, London pp. 103–111
56. Ramirez LC, Raskin P (1991) Pancreatic abnormalities innon-insulin-dependent diabetes mellitus. In: Pickup J, Wil-liams G (eds) Textbook of diabetes. Blackwell ScientificPublications, Oxford, Vol. 1, pp. 198–204
P.Rorsman: The pancreatic beta-cell as a fuel sensor 495

NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | DECEMBER 2010 | 689
Department of Medicine, University of Cincinnati, Metabolic Diseases Institute, Office E-217, 2170 East Galbraith Road, Cincinnati, OH 45237, USA (K. M. Habegger, K. M. Heppner, M. H. Tschöp). Institute of Food, Nutrition and Health, Swiss Federal Institute of Technology Zürich, Schorenstraβe 16, 8603 Schwerzenbach, Switzerland (N. Geary). Department of Biology, Georgia State University, 33 Gilmer Street Southeast, Atlanta, GA 30303-3044, USA (T. J. Bartness). Department of Chemistry, Indiana University, 107 South Indiana Avenue, Bloomington, IN 47405-7000, USA (R. DiMarchi).
Correspondence to: M. H. Tschöp tschoemh@ ucmail.uc.edu
The metabolic actions of glucagon revisitedKirk M. Habegger, Kristy M. Heppner, Nori Geary, Timothy J. Bartness, Richard DiMarchi
and Matthias H. Tschöp
Abstract | The initial identification of glucagon as a counter-regulatory hormone to insulin revealed this
hormone to be of largely singular physiological and pharmacological purpose. Glucagon agonism, however,
has also been shown to exert effects on lipid metabolism, energy balance, body adipose tissue mass and
food intake. The ability of glucagon to stimulate energy expenditure, along with its hypolipidemic and satiating
effects, in particular, make this hormone an attractive pharmaceutical agent for the treatment of dyslipidemia
and obesity. Studies that describe novel preclinical applications of glucagon, alone and in concert with
glucagon-like peptide 1 agonism, have revealed potential benefits of glucagon agonism in the treatment of the
metabolic syndrome. Collectively, these observations challenge us to thoroughly investigate the physiology and
therapeutic potential of insulin’s long-known opponent.
Habegger, K. M. et al. Nat. Rev. Endocrinol. 6, 689–697 (2010); published online 19 October 2010; doi:10.1038/nrendo.2010.187
Introduction
The isolation of insulin in 1921 by Banting, Best, Collip and Macleod transformed our understanding of the hor-monal regulation of glucose metabolism and has provided a life-saving treatment for millions of patients with dia-betes mellitus. Of far lesser prominence was the simulta-neous description of a pancreatic contaminant observed to elicit a temporary increase in blood glucose levels.1 This contaminant was later studied by Murlin et al.2 and found to oppose insulin in the control of glucose homeo-stasis, both in healthy animals and in those whose pan-creas had been removed. Murlin and colleagues named this new hormone glucagon, as they thought it was a glucose agonist. Later work by Sutherland, Park and Exton described the counter- regulatory actions of gluca-gon relative to insulin pharma cology. Specifically, the researchers determined that glucagon stimulates hepatic glyco genolysis and gluconeogenesis in hypo glycemic states to restore glucose homeostasis.3,4 The patho-physiological role of hyper glucagonemia and unopposed glucagon action in diabetes mellitus has been empha-sized.5–9 By contrast, the possibility that glucagon may also have benefits in the treatment of metabolic disease has received little or no attention. However, new studies that describe novel preclinical applications of glucagon, either alone or in concert with glucagon-like peptide 1 (GLP-1) agonism, have revealed the prospect of harness-ing the bene fits of glucagon action for the treatment of the metabolic syndrome.10,11
Glucagon
Expression
Glucagon is a hormone produced in the α cells of the pan creatic islets. Encoded by the proglucagon gene, the pro glucagon peptide is processed by neuro endocrine convertase-2 (NEC2) to produce the 29-amino acid native glucagon peptide. In addition, the proglucagon gene contains sequences that encode GLP-1, GLP-2, oxyntomodulin and glicentin. These hormones are pro-cessed from the proglucagon peptide via NEC1-mediated cleavage in the brain and in L cells of the intestine.12 Studies on the mechanisms of proglucagon transcription have elucidated the minimal promoter region, as well as four enhancer elements.13 Regulation of transcription has been shown to occur through homeodomain transcrip-tion factors, influenced by amino acids and cyclic AMP (cAMP) in the pancreas and intestinal cells,14 as well as Wnt signaling in the intestine.15 Insulin is also known to exert inhibitory effects on proglucagon expression in α cells.16,17 Interestingly, this effect is reversed in the intes-tine, where insulin at pathological concentrations stimu-lates proglucagon expression, which results in increased GLP-1 production.18
Secretion
Glucagon secretion, similar to that of insulin, is tightly regulated and intimately tied to blood glucose levels (Figure 1). Converse to the inhibition of insulin secre-tion by hypoglycemia, low levels of blood glucose directly stimulate the pancreatic α cells to secrete glu-cagon.19 The secretion of glucagon is promoted via the action of voltage-dependent sodium (Na+) and calcium (Ca2+) channels, which maintain action potentials during times of low glucose. Depolarization increases the Ca2+
influx and subsequent glucagon secretion,20 which is sup-ported by the activity of ATP-sensitive potassium (K
ATP)
Competing interests
N. Geary declares an association with the following company: Novo Nordisk. R. DiMarchi declares an association with the following company: Marcadia Biotech. See the article online for full details of the relationships. The other authors declare no competing interests.
REVIEWS
© 20 Macmillan Publishers Limited. All rights reserved10

690 | DECEMBER 2010 | VOLUME 6 www.nature.com/nrendo
channels.21 As glucose levels rise, secretion of glucagon is inhibited through the elevation of cytosolic ATP, block-ade of K
ATP channels and termination of the Na+-induced
and Ca2+-induced action potentials. This process inhibits Ca2+ influx and ends glucagon secretion. Although the cellular signals regulating glucagon secretion are fairly well-established, the role of glucose, whether directly, or indirectly via β-cell activation, is still a matter of debate. Studies in rats suggest that mediatory, paracrine signaling from the β cell is essential for the inhibition of glucagon secretion by elevated glucose levels.22,23 Investigations in mice and humans, however, suggest that glucose directly inhibits glucagon secretion at concentrations which are too low to stimulate insulin secretion.21 Furthermore, this inhibition has been demonstrated in vitro in both isolated α cells and in intact pancreatic islets.24 However, a study in individuals with type 1 diabetes mellitus sug-gests that insulin is the primary signal that inhibits gluca-gon secretion in humans.25 In addition to glucose, several other physiological parameters are known regulators of glucagon secretion, including GLP-1,26 GLP-2,27 fatty acids,28 the autonomic nervous system29 and circulating amino acids.30
Signaling
Glucagon receptor
The effects of glucagon are mediated by the binding of glucagon to its membrane-bound receptor—a seven-transmembrane protein and a member of the class II guanine nucleotide-binding protein (G protein) coupled receptor superfamily.31 Glucagon receptors (encoded by the GCGR gene) are expressed abundantly in the liver and kidney and to a lesser extent in heart, adipocytes, lymphoblasts, spleen, endocrine pancreas, brain, retina,
Key points
In addition to its well-known effects on glycemia, increased glucagon ■
signaling directly regulates triglyceride, free fatty acid, apolipoprotein and bile
acid metabolism
Glucagon action can be inhibited via receptor desensitization by excess dietary ■
fat intake
Energy expenditure and thermogenesis are increased by glucagon agonism ■
Glucagon administration stimulates satiety and decreases food intake ■
Glucagon action, in combination with incretins such as glucagon-like peptide 1, ■
may be a crucial tool in the treatment of the metabolic syndrome
Fatty acids
Metabolites
Paracrine and
endocrine input
Neural inputs
Lipolysis
Fatty acid oxidation
Ketogenesis
Satiation
Thermogenesis
Energy expenditure
Bile acid synthesis
Glucose production
Glucagonsecretion
Cell
Food intake
Figure 1 | Physiological stimuli and outcomes of glucagon secretion.
adrenal gland and the gastrointestinal tract.32 Previous studies suggest that glucagon receptors are also expressed in islet α cells.33 In the liver, glucagon receptors are located mainly in hepatocytes, but can also be found on the surface of Kupffer cells.34 Interestingly, glucagon recep-tors in the pancreas are predominately located on β cells, which, together with the data described above, suggests a bidirectional feedback mechanism.35 Further evidence supporting this hypothesis is the fact that gluca gon, at physiological concentrations, was found to stimulate insulin release via these receptors.36
Much knowledge concerning glucagon action has been gained from genetically altered animal models. Glucose homeostasis and pancreatic function has been elaborately studied in mice with a null mutation of the glucagon receptor (Gcgr−/−). As expected, blood glucose levels are markedly lower and glucose tolerance is improved in Gcgr−/− mice compared with wild-type con-trols.37,38 The improvement in glucose tolerance is not owing to an increase in insulin secretion, but rather to an improvement in insulin sensitivity, as demonstrated by hyperinsulinemic euglycemic clamp studies.39 When fed a high-fat diet (HFD), Gcgr−/− mice do not display diet-induced insulin resistance, which is potentially a result of enhanced insulin sensitivity in these mice. However, after prolonged fasting, Gcgr−/− mice experience severe hypoglycemia,37 which illustrates the essential role of glucagon for the maintenance of blood glucose levels.38 Gcgr−/− mice were observed to display an enlargement of the pancreas primarily attributed to α-cell hyperplasia, which indicates glucagon receptor signaling is essential for normal endocrine-cell proliferation.37
Further indication of glucagon’s role in endocrine-cell function was demonstrated in transgenic mice that overexpress the glucagon receptor specifically on pan-creatic β cells. These mice display enhanced glucagon-stimulated and glucose-stimulated insulin secretion, as well as improved glucose tolerance.40 A reduction in both fasting hyperglycemia and impaired glucose tolerance is observed when these transgenic mice are exposed to a HFD,40 suggesting that enhanced glucagon signaling on β cells improves the function of these cells. The studies that show improvements in glucose homeostasis in mice overexpressing the glucagon receptor on β cells are in contrast to studies in mice treated with streptozotocin, which showed that ablation of glucagon signal ing has protective effects. Streptozotocin treatment destroys β cells, impairs insulin secretion and induces hyper-glycemia in wild-type mice; however, when strepto-zotocin is admini stered to Gcgr−/− mice, euglycemia is maintained even after HFD exposure.38 This finding indicates that lack of glucagon receptor signaling results in resistance to streptozotocin-mediated β-cell destruc-tion and hyper glycemia in vivo. The mechanism behind the streptozotocin resistance in Gcgr−/− mice remains unknown; however, given that other reports demonstrate that increased concentration of circulating GLP-1 results in resistance to streptozotocin-induced β-cell destruc-tion, the increased levels of circulating GLP-1 determined in Gcgr−/− mice have been speculated to be an important
REVIEWS
© 20 Macmillan Publishers Limited. All rights reserved10

NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | DECEMBER 2010 | 691
factor.41,42 These data illustrate that both enhanced, as well as lack of, glucagon receptor signaling have positive effects on glucose homeostasis and pan creatic function and, therefore, further studies are needed to understand the cause of these results.
Signaling pathway and targets
Binding of glucagon to its receptor elicits activation of a heterotrimeric, stimulatory G protein (G
s) in a process
dependent on GTP and magnesium.43 In liver cells, the glucagon receptor and G
s proteins have been postulated
to be linked in a multimeric configuration, which dis-engages following activation.44 The activated G
s protein
undergoes a conformational change, upon which the GTP-bound subunit G
sα is released from the G-protein
complex that comprises two additional subunits, Gsβ and
Gsγ. This activation leads to interaction and subsequent
stimulation of adenylate cyclase, elevated cAMP levels and enhanced intracellular signaling via Rap guanine nucleotide exchange factor 3 (RAPGEF3; also known as EPAC1), cAMP response element-binding protein (CREB)-regulated transcription coactivator 2 (CRTC2; also known as TORC2) and protein kinase A (PKA). The activation of PKA results in the phosphorylation and nuclear localization of CREB.45,46 Once phosphorylated in the liver, CREB binds to the cAMP response element of target genes, resulting in the recruitment of coactiva-tors, such as hepatic nuclear factor 4α (HNF-4α),47 per-oxisome proliferator-activated receptor γ co activator 1-α (PGC-1α)48 and the gluco corticoid receptor.47 In addi-tion to this well-described pathway, gluca gon has also been implicated in signaling via 5'-AMP-activated protein kinase (AMPK),49 mitogen-activated pro-tein kinase (MAPK) and in a c-Jun N-terminal kinase (JNK)-dependent manner.50
Beyond glucose homeostasis
The role of glucagon in glucose homeostasis has been well-studied and previously reviewed elsewhere.51 How-ever, this pancreatic peptide has additional metabolic effects of notable importance. Glucagon agonism has been shown to regulate lipid metabolism and energy expenditure, as well as reduce histamine-induced cardiac injury during reperfusion.52
Glucagon and lipid metabolism
Plasma lipid homeostasis
Studies in the early 1960s investigated glucagon’s action independent of glucose homeostasis. These studies described a lipid-mobilizing effect for glucagon in a range of species.53–57 The regulation of plasma lipids by glucagon was first highlighted in studies which suggested that gluca gon acts to decrease plasma cholesterol, total esterified fatty acids and arachidonic acid levels.53,58 As epinephrine, insulin and exogenous glucose all failed to lower levels of plasma cholesterol, the investigators concluded that these effects were not an indirect result of altered carbo hydrate metabolism.59 In both canines and humans, the researchers observed a total decrease in levels of plasma cholesterol, as well as plasma total lipid
concentrations, within 30 min of intravenous gluca-gon administra tion. Intriguingly, this depression was not found for whole-blood total lipid levels, suggesting a repartitioning of lipids from serum to platelet-rich fractions. Incubation of the blood at 37 °C led to partial or full restoration of plasma cholesterol and total lipid levels, further supporting the hypothesis of repartition-ing.58 Reports from other groups provided additional support for a role of glucagon in the regulation of plasma lipids.55,56
In the context of triglyceride metabolism, several reports indicated that triglyceride production is decreased in perfused livers treated with glucagon.60–62 Although these studies suggest hepatic lipoprotein metabolism as the site of such regulation, the mol ecular mechanism by which glucagon achieved this result remained elusive. Building upon these observations, the role of glucagon on lipid suppression was investigated in a rat model of hyper lipidemia.63 This study demonstrated that glucagon sig nificantly decreased levels of serum triglycerides, cho-lesterol, and VLDL cholesterol in hyperlipidemic rats, as well as in control eulipidemic rats. Furthermore, a decrease in synthesis of hepatic lipo protein apo proteins was observed,63 complementing earlier studies that described depression of triglyceride synthesis in livers treated with glucagon. A caveat of this study is that it involved supra-physiological concentrations of glucagon administered over a 4-day period. Thus, the relevance of acute signal-ing by endo genous glucagon remained uncertain. Guettet et al.64–66 continued the investigation of chronic effects of glucagon signaling in the Wistar rat. Their initial studies described a decrease in concentrations of plasma cho-lesterol, phospholipids and tri glycerides. Interestingly, these reductions were not seen in the liver or in erythro-cytes of these rats, which suggested another target tissue as the site of action. Continued investigation of cholesterol turnover revealed increased urinary secretion of cho-lesterol, as well as elevated transformation to bile acids. Of special importance was the reported observation that the chronic treatment of Wistar rats with twice-daily gluca gon (20 μg per day) for 3 weeks reduced blood glucose and insulin concentrations. This report thus indicated that chronic glucagon pharmacology was neither transien t nor detrimental to glucose homeostasis.64
Further studies by Guettet et al.65,66 evaluated the effect of chronic glucagon treatment on lipoprotein composition in fed and fasted rats, as well as rats fed a cholesterol-rich diet. Cholesterol, phospholipids and total protein levels were proportionally decreased in chylomicrons, VLDL cholesterol, LDL cholesterol and HDL cholesterol, which suggested a reduction in the number of lipo protein par-ticles. Triglycerides were reported to be decreased only in the chylomicron and VLDL fractions. Evaluation of the lipoproteins indicated a decline in levels of apolipo-protein E (ApoE) and an increase in ApoB.65 Collectively, these findings suggested glucagon- stimulated targeting of the ApoE-rich lipoproteins to limit the accumulation that typically occurs with high-cholesterol feeding.66 A follow-up study showed that glucagon treatment had no effect on secretion rates of triglyceride-rich particles in either
REVIEWS
© 20 Macmillan Publishers Limited. All rights reserved10

692 | DECEMBER 2010 | VOLUME 6 www.nature.com/nrendo
fed or fasted conditions. Conversely, glucagon treatment accelerated the rate of triglyceride removal (fractional catabolic rate constant) from the plasma compart ment.67 Taken together, the work of Guettet et al.64–66 revealed that increased glucagon signaling may directly regulate lipid catabolism.
Rudling and Angelin68 extended these findings by the direct study of glucagon’s effects on LDL recep-tors (LDLRs). The investigators found that glucagon administra tion to rats resulted in a dose-dependent increase in binding of LDL cholesterol to the receptor, with no apparent effect on receptor mRNA expression. Concomitant with this increased binding was a decrease in levels of cholesterol, ApoB, and ApoE. Furthermore, these effects were completely antagonized by insulin administration. Rudling and Angelin hypothesized a mechanism such as post-translational receptor modifica-tion, whereby glucagon increases LDLR activity without affecting expression levels.68 The findings in rodents were complemented by studies in dairy cattle which showed that subcutaneous injections of glucagon led to vari-able decreases, some of sizable magnitude, in circulating plasma concentrations of VLDL-triglycerides, HDL
1-
phospholipids and HDL2-free cholesterol.69 Improved
glucose status with decreased nonesterified fatty acids and β-hydoxybutyrate was also reported.70
Genetic manipulation of the glucagon receptor has led to inconclusive results regarding glucagon’s role in lipid metabolism. Conarello et al.38 found that Gcgr−/− mice are resistant to HFD-induced liver steatosis; however, Longuet et al.71 showed that Gcgr−/− mice have enhanced suscepti-bility to hepatic steatosis. Interestingly, these studies used genetically modified null mice generated in an identical manner and of the same background (C57B6). Further-more, the effect was observed in mice fed a similar diet (45% fat). The length of diet differed between the two studies (8 versus 12 weeks); however, mice fed a HFD for the longer duration did not exhibit steatosis. Taken together, the contribution of glucagon receptor agonism to liver steatosis has yet to be conclusively described.
Longuet et al.71 also demonstrated the importance of glucagon signaling in the adaptive response to fasting. Following exposure to a prolonged fasting period, Gcgr−/− mice showed increased levels of plasma triglycerides and free fatty acids.71 During either glucagon treatment or a prolonged fast, wild-type mice had an increased expres-sion of genes involved in fatty acid oxidation, including Decr2, carnitine O-palmitoyltransferase (Cpt) 1a, Cpt2 and Acadm. These changes in expression were correlated with an increased capacity of the liver to oxidize free fatty acids. By contrast, this increased gene expression was not observed in Gcgr−/− mice, which displayed a reduced oxi-dation of free fatty acids in both the fed and the fasted state. These data highlight the essential physiological role of glucagon-receptor signaling in the regulation of gene expression during prolonged fasting.
Glucagon-mediated lipolysis and ketogenesis
Glucagon-mediated regulation of lipid metabolism is not limited to plasma triglycerides and cholesterol. Glucagon
treatments as low as 10–8 mol/l have been implicated in promoting lipolysis in white adipose tissue.72 This lipo-lytic effect appears to be independent of sympathetic nervous system innervation,73 as denervation of white adipose tissue does not block glucagon-induced glycerol release, whereas it decreases the release of nonesterified fatty acids. The latter, however, is reported to be the result of re-esterification,73 not a blockade of glucagon’s lipolytic effects. Mechanistically, glucagon is known to stimulate the activity of hormone-sensitive lipase (HSL) in adipo-cytes, resulting in an increase of nonesterified fatty acids in the circulation.74 These fatty acids, usually bound to albumin, are transported to the heart, skeletal muscle, kidney and liver, where they are catabolyzed or, in the case of the liver, alternatively converted to ketone bodies.
Ketone bodies provide up to two-thirds of the energy for the brain during times of glucose deficiency, thus sparing glucose utilization and reducing proteo lysis.75 Supporting a role for glucagon in the regulation of keto-genesis, suppression of glucagon secretion via somato-statin prevented the development of keto acidosis in patients with type 1 diabetes mellitus.76 Pegorier et al.77 elucidated the regulation of fatty acid oxidation and keto-genesis by glucagon in rabbit hepatocytes. The re searchers reported increased ketone-body production in the pres-ence of either glucagon or dibutyryl cAMP. Oxidation of exogenous oleate was increased by both glucagon and cAMP, effects which were reduced in the presence of insulin. Furthermore, exposure to glucagon or cAMP completely inhibited lipogenesis and decreased malonyl-CoA concentration and stimulated fatty acid oxidation. These effects were suggested to be driven by a fall in the sensitivity of CPT1 to malonyl-CoA, releasing the inhibi-tion of this rate-limiting enzyme in the transport of fatty acids across the mitochondrial membranes.77,78 The rele-vance of these studies was confirmed by the observation that glucagon directly controls ketone-body production in primary human hepatocytes.79 Specifically, glucagon increased ketone-body production and fatty acid oxida-tion, whereas it decreased fatty acid esterification, similar to the effects of cAMP.79
Lipid-induced glucagon resistance
In addition to glucagon’s regulation of lipid metabolism, previous studies suggest that lipids may also regulate glucagon signaling. Studies by Charbonneau et al.80 show that rats fed a HFD display hepatic steatosis that is associated with glucagon resistance. Specifically, exercise training in rats was used to increase plasma glucagon levels and improve fatty liver. In trained rats, a nega-tive correlation between liver adiposity and density of the glucagon receptors in the plasma membrane was observed. Additional studies described a reduction in both hepatic glucagon-receptor density and G
sα protein
content at the plasma membrane.81,82 The mechanism for this decrease in receptor density was explored in a study which proposed that HFD promoted glucagon receptor proteolysis.81 A reduction in glucagon recep-tors at the plasma membrane was notable, accompanied by a marked increase in the number of endosomal and
REVIEWS
© 20 Macmillan Publishers Limited. All rights reserved10

NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | DECEMBER 2010 | 693
lysosomal compartments. These effects were correlated with an increase of protein kinase Cα (PKCα) on the plasma membrane, which is known to phosphorylate G-protein-related kinases. Such action, in turn, is known to inhibit receptor internalization which leads to receptor desensitiza tion.83 The same association seems to be con-served in humans, as glucagon resistance is associated with pathological hyperlipidemia in humans.84
Glucagon and bile acid metabolism
Although the canonical role of bile acids is associated with the absorption of dietary lipids and cholesterol homeostasis, new studies suggest that these acids have additional signaling roles in glucose homeostasis. This emerging function was highlighted in a study by Song and Chiang,85 who investigated cholesterol 7-alpha- monooxygenase (CYP7A1) as a possible target of gluca-gon signaling. Their study demonstrated that gluca gon, as well as cAMP, represses CYP7A1 expression in human primary hepatocytes. CYP7A1 is the rate- limiting enzyme in bile acid synthesis, and its expression is tightly regu-lated as a means to control flux through the pathway.86 Song and Chiang86 further showed that gluca gon’s regula-tion of CYP7A1 expression is mediated via PKA signaling to HNF-4α. Given that no direct measure of bile acid syn-thesis or secretion was available, the direct result on bile acid metabolism remains unknown. The findings of their earlier study,85 however, were complemented by work from Hylemon et al.,87 who showed similar decreases in CYP7A1 expression in response to glucagon or cAMP exposure in rat hepatocytes. These in vitro studies support initial observations that suggest a role for glucagon in the regulation of bile acid metabolism and provide a plausible mechanism for the glucagon-induced decrease in plasma cholesterol levels.
Glucagon and energy expenditure
In addition to regulating glucose and lipid metabolism, glucagon participates in the control of energy expen-diture and thermogenesis. Early studies by Davidson
et al.88,89 showed that pharmacological infusion of gluca-gon increased oxygen consumption in rats. This effect was mirrored by a report in human study participants, in whom infusion of a pharmacological dose of gluca-gon increased resting energy expenditure during acute insulin deficiency produced by the additional infusion of somatostatin.90 Moreover, a physiological dose of gluca-gon increased energy expenditure in humans during euinsulinemia, and hyperinsulinemia blunted gluca-gon’s thermogenic activity.91 Studies conducted in vitro showed that hyperglucagonemia may increase energy expenditure via stimulation of oxygen consumption and heat production in brown adipose tissue.92,93 Studies in rats confirmed that glucagon administration increased whole-body oxygen consumption, core body tempera-ture, blood flow,94 as well as temperature and mass of brown adipose tissue.95,96 Furthermore, cold exposure was found to increase plasma glucagon levels,97 which implicates glucagon in nonshivering thermogenesis. The stimulation of thermogenic activity in brown adipose
tissue by glucagon98 has become even more relevant, as novel findings suggest a renewed importance for brown adipose tissue in human energy metabolism.99
The mechanism by which glucagon-induced thermo-genesis is regulated is probably complex and may involve the sympathetic nervous system. Supporting this hypo-thesis, glucagon-induced increases in oxygen consump-tion, blood flow and thermogenesis in brown adipose tissue were blocked by the nonselective β adrenergic receptor blocker, propranolol,100,101 whereas denervation of brown adipose tissue blunted the thermogenic action of glucagon.98 Furthermore, although glucagon injec tion in ducklings was associated with an increase in norepi-nephrine, blockade of catecholamines via guanethidine sympathectomy (the interruption of the transmission of sympathetic nerve impulses by a chemical agent that blocks the secretion of epinephrine and norepinephrine at postganglionic nerve endings) decreased metabolic rate compared to nonsympathectomized birds treated with glucagon or saline.102 Glucagon might also have a direct effect on thermogenesis, as glucagon incuba-tion of brown adipocytes of rat and mice, but not those of Syrian hamsters, markedly increased oxygen con-sumption.100 However, the concentration of glucagon required for such in vitro effects has been speculated to be supraphysiological.100
The role of glucagon in mediating cold-induced thermo genic responses has been further questioned by a report that denervated interscapular brown adipose tissue decreased the number of glucagon receptors as compared to the contralateral adipose tissue pad that served as an internal (within animal) control.103 Given that cold exposure is a well-known stimulator of the sym-pathetic nervous system in brown adipose tissue,104,105 a decrease in glucagon receptor protein or gene expres-sion mitigates the role for glucagon in cold-triggered brown adipose tissue thermogenesis via innervation of the sympathetic nervous system. Taken together, these data suggest a pivotal role for the sympathetic nervous system in glucagon-induced thermogenesis and suggest a thermogenic basis for anti-obesity effects of glucagon.
Glucagon and regulation of food intake
Early studies indicated that glucagon administration diminishes the sense of hunger and decreases food intake in humans106–108 and rats.109 Additional studies in rats confirmed that glucagon specifically decreases meal size owing to increased satiation rather than illness or aversion. Intravenous infusions similarly produced a dose-related decrease in eating.109–113 Evidence for an endogenous role of glucagon in satiation was provided by demonstrations that glucagon concentration increases physiologically during meals, in many situations,114–116 and that antago-nism of glucagon by pre prandial administra tion of gluca-gon antibodies increases the amount of food consumed in one meal (meal size).117,118 Finally, intra venous infusion of a physiological dose of glucagon during meals was shown to reduce meal size in humans.119
A study in rats with both hepatic portal vein and vena
cava infusion catheters indicated that both exo genous
REVIEWS
© 20 Macmillan Publishers Limited. All rights reserved10

694 | DECEMBER 2010 | VOLUME 6 www.nature.com/nrendo
and endogenous glucagon act in the liver to limit meal size.120 Furthermore, as for several other peripheral signals that control eating, vagal afferents relay the signal to the brain.121 In the case of glucagon, the hepatic branch of the abdominal vagus is critical,122 consistent with the hepatic site of action. Although the inhibitory effect of glucagon on feeding probably arises, at least in part, from a hepatic metabolic action of the hormone, novel studies in sheep suggest that glucagon acts directly in the central nervous system to inhibit food intake.123
Genetically modified animal models also demon-strate that glucagon signaling is involved in regulating food intake and body composition. Gcgr−/− mice display a decreased adipose tissue mass and an increased lean mass despite similar food intake and body weight compared to their wild-type littermates.37 Furthermore, Gcgr−/−
mice on a HFD are resistant to diet-induced obesity,38 which can be primarily attributed to a lower food intake compar ed with controls.
Glucagon-based pharmacological therapy
Glucagon-based drug therapy has largely been restricted to acute emergency use to treat episodes of hypo-glycemia in patients with type 1 diabetes mellitus and as an esopha geal muscle relaxant to prepare patients for radiological procedures. Nevertheless, glucagon’s hypo-lipidemic, energy expenditure-stimulatory and satiat-ing effects make it an attractive pharmaceutical agent for the treatment of dyslipidemia and obesity. Such chronic uses, however, must be considered carefully in light of gluca gon’s ability to accelerate the development of glucose intolerance and insulin resistance. Studies in cattle suggest that glucagon may be an effective treat-ment for fatty liver and dys lipidemia124 without deleteri-ous effects on glucose homestasis, as both single and multiple injections of 5 mg glucagon over 14 days con-sistently improve the carbo hydrate status of dairy cows and decreased concentrations of plasma nonesterified fatty acids.70 Of course, these actions must be verified in nonruminant species.
Glucagon and GLP-1 coagonism
Two prominent studies published in July 2009, have put forth the hypothesis that glucagon agonism may be beneficial in the pharmacological treatment of obesity and, possibly, obesity-associated glucose intolerance.10,11 Interestingly, both studies combined agonists of two proglucagon-derived peptides to elicit these effects in mouse models. Day et al.10 combined the antidiabetic properties of GLP-1 receptor (GLP-1R) agonism with the hypo lipidemic properties of glucagon receptor agonism to create a dual agonist, which they conjugated to a poly-ethylene glycol polymer to prolong pharmacokinetic action. The researchers then compared 1-week adminis-tration of this dual agonist with an equimolar amount of a structurally comparable, but selective, GLP-1R agonist in diet-induced obese mice. Both peptides significantly decreased food intake, body weight and adipose tissue mass when compared with saline-injected controls. Significant decreases in blood glucose and insulin con-centrations and a profound increase in glucose tolerance were also reported. Intriguingly, the peptide with balanced glucagon and GLP-1 coagonism exhibited significantly greater efficacy, as measured by changes in body weight, adipose tissue mass and glucose homeo stasis, compared with the GLP-1R agonist monotherapy.10 Studies con-ducted over a 1-month period revealed similar effects on body weight, adiposity and blood glucose levels, although cumulative total food intake in this study was not impres-sively altered. Interestingly, energy expenditure in the 1-month study was significantly increased in the animals treated with the coagonist, with a trend for a decreased respiratory quotient. Lipid metabolism was markedly improved with decreases in levels of total cholesterol, LDL cholesterol, HDL cholesterol and tri gylcerides, as well as reversal of liver steato sis and activation of HSL in white adipose tissue.
Similar to the study by Day et al.,10 Pocai et al.11 tested a dual agonist of GLP-1R and the glucagon receptor in diet-induced obese mice. The investigators injected the coagonist daily for 2 weeks and compared these mice with those treated with either a GLP-1R agonist with the same degree of GLP-1R agonism as the coagonist or vehicle. Significant enhancements in measures of body weight and glucose tolerance were reported in mice treated with the coagonist. Results of animals treated only with a GLP-1R agonist were intermediate between those of mice treated with either the coagonist peptide or vehicle, for all measured parameters.11 Also, similar to the study by Day and colleagues,10 Pocai et al.11 showed decreases in liver steatosis, total cholesterol and tri glyceride levels in the coagonist-treated animals, with the single- agonist-treated mice again displaying intermediate values in comparison with the other two groups.
Together, these two studies exemplify a novel and potentially important new direction for therapy of obesity and metabolic disease (Figure 2). An advantage of the described approach is the use of single-agent pep-tides that represent two full hormone-receptor agonists that elicit additive therapeutic effects, while at the same time adverse effects can be minimized. These studies
Glucagonsecretion
Cell
GLP-1secretion
L Cell
Food intake
Body weight
Pharmacological synergism
Insulin secretion
Gastric emptying
GLP-1-speci"c effects
Glucose production
Thermogenesis
Glugagon-speci"c effects
Obesity
Figure 2 | Individual and synergystic effects of glucagon-like peptide 1 and glucagon in obesity and pharmacotherapy.
REVIEWS
© 20 Macmillan Publishers Limited. All rights reserved10

NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | DECEMBER 2010 | 695
come with the caveat that the glucagon agonism caused by the coagonist peptide far exceeds the actions elicited by endogenous gluca gon. Furthermore, these studies did not compare coagonism with glucagon monotherapy; any effect observed is, therefore, a possible, and probable, interaction between the two receptor agonists. Taken together, interpretation of glucagon ph ysiology on the basis of these findings must be tempered. Possibly of greater importance, these studies challenge our deeply rooted understanding of the full physiology of a pan creatic hormone that partners with insulin in the manage ment of glucose and lipid homeostas is and body weight.
The fact that excessive glucagon receptor agonism leads to glucose intolerance and insulin dysregulation is well-established. Observations of elevated glucagon concen-tration in insulin-resistant individuals, as well as animal models of insulin resistance, have supported this view. The studies of Day et al.10 and Pocai et al.11 build upon a foundation of less evident metabolic actions of glucagon reported in this Review. They provide a rationale to recon-sider whether glucagon administered at modest concentra-tions and frequency of exposure constructively contributes to proper glucose and lipid metabolism directly by acting at specific target tissues and indirectly by modulating body weight. The prospect of using glucagon in combination with other agents to address obesity-associated glucose intolerance and insulin resistance, as well as dyslipidemia, is something worthy of scholarly consideration.
Conclusions
The limited data available on the pharmacology of chronically administered glucagon is the result of a focus on acute glucagon action and its disadvantageous
diabetogenic properties that render it poorly suited for the treatment of obesity and metabolic diseases. Development of stable, soluble glucagon agonists of varying pharmaco-kinetics and of glucagon-like co agonists in conjunction with GLP-1, which are suitable for pharmacological study, however, provide an unprecedented opportunity to explore the full physiological character and pharmaco logical potential of this fascinating hormone. When viewed in combination with historical observations, the first reports resulting from studies of these agonistic peptides suggest that glucagon may potently regulate glucose metabolism and lipid homeostasis, as well as energy balance, body adipose tissue mass and food intake. Clearly, excessive and un opposed glucagon action is catabolic and must be avoided if chronic use is considered. Nevertheless, col-lectively, novel observations challenge us to thoroughly investigate and broadly take into considera tion the physiology and thera peutic potential of this long-known counter-regulatory hormone to insulin.
Review criteria
Articles referenced in this Review were selected on the
basis of relevance to the less appreciated effects of
glucagon on lipid metabolism, food intake and energy
expenditure. Articles were searched for in the PubMed
database. Search terms used included “glucagon”,
“glucagon AND food intake”, “glucagon AND cholesterol”,
“glucagon AND triglycerides”, “glucagon AND energy
expenditure”. Full-text articles published in English
were chosen, and referenced papers were utilized for
additional leads. Articles from all years were used;
however, whenever possible, the most recent references
were used.
1. Banting, F. G. & Best, C. H. The internal
secretion of the pancreas. J. Lab. Clin. Med. 7,
251–266 (1922).
2. Murlin, J. R., Clough, H. D., Gibbs, C. B. F. &
Stokes, A. M. Aqueous extracts of the pancreas.
I. Influence on the carbohydrate metabolism of
depancreatized animals. J. Biol. Chem. 56,
253–296 (1923).
3. Exton, J. H. & Park, C. R. The role of cyclic AMP
in the control of liver metabolism. Adv. Enzyme
Regul. 6, 391–407 (1968).
4. Robison, G. A., Butcher, R. W. & Sutherland, E. W.
Cyclic AMP. Annu. Rev. Biochem. 37, 149–174
(1968).
5. Gu, W. et al. Long-term inhibition of the glucagon
receptor with a monoclonal antibody in mice
causes sustained improvement in glycemic
control, with reversible alpha-cell hyperplasia
and hyperglucagonemia. J. Pharmacol. Exp. Ther.
331, 871–881 (2009).
6. Wang, M. Y. et al. Leptin therapy in insulin-
deficient type I diabetes. Proc. Natl Acad. Sci.
USA 107, 4813–4819 (2010).
7. Brown, R. J., Sinaii, N. & Rother, K. I. Too much
glucagon, too little insulin: time course of
pancreatic islet dysfunction in new-onset type 1
diabetes. Diabetes Care 31, 1403–1404 (2008).
8. Dunning, B. E. & Gerich, J. E. The role of alpha-
cell dysregulation in fasting and postprandial
hyperglycemia in type 2 diabetes and
therapeutic implications. Endocr. Rev. 28,
253–283 (2007).
9. Gromada, J., Franklin, I. & Wollheim, C. B. Alpha-
cells of the endocrine pancreas: 35 years of
research but the enigma remains. Endocr. Rev.
28, 84–116 (2007).
10. Day, J. W. et al. A new glucagon and GLP-1
co-agonist eliminates obesity in rodents. Nat.
Chem. Biol. 5, 749–757 (2009).
11. Pocai, A. et al. Glucagon-like peptide 1/glucagon
receptor dual agonism reverses obesity in mice.
Diabetes 58, 2258–2266 (2009).
12. Baggio, L. L. & Drucker, D. J. Biology of
incretins: GLP-1 and GIP. Gastroenterology 132,
2131–2157 (2007).
13. Herzig, S., Fuzesi, L. & Knepel, W. Heterodimeric
Pbx-Prep1 homeodomain protein binding to the
glucagon gene restricting transcription in a cell
type-dependent manner. J. Biol. Chem. 275,
27989–27999 (2000).
14. Gevrey, J. C. et al. Protein hydrolysates stimulate
proglucagon gene transcription in intestinal
endocrine cells via two elements related to
cyclic AMP response element. Diabetologia 47,
926–936 (2004).
15. Yi, F., Brubaker, P. L. & Jin, T. TCF-4 mediates cell
type-specific regulation of proglucagon gene
expression by beta-catenin and glycogen
synthase kinase-3beta. J. Biol. Chem. 280,
1457–1464 (2005).
16. Philippe, J. Insulin regulation of the glucagon
gene is mediated by an insulin-responsive DNA
element. Proc. Natl Acad. Sci. USA 88,
7224–7227 (1991).
17. Artner, I. et al. MafB: an activator of the
glucagon gene expressed in developing islet
alpha- and beta-cells. Diabetes 55, 297–304
(2006).
18. Yi, F. et al. Cross talk between the insulin and
Wnt signaling pathways: evidence from
intestinal endocrine L cells. Endocrinology 149,
2341–2351 (2008).
19. Quesada, I., Todorova, M. G. & Soria, B. Different
metabolic responses in alpha-, beta-, and delta-
cells of the islet of Langerhans monitored by
redox confocal microscopy. Biophys. J. 90,
2641–2650 (2006).
20. Gromada, J. et al. Adrenaline stimulates
glucagon secretion in pancreatic A-cells by
increasing the Ca2+ current and the number of
granules close to the L-type Ca2+ channels.
J. Gen. Physiol. 110, 217–228 (1997).
21. MacDonald, P. E. et al. A K ATP channel-
dependent pathway within alpha cells regulates
glucagon release from both rodent and human
islets of Langerhans. PLoS Biol. 5, e143 (2007).
22. Franklin, I., Gromada, J., Gjinovci, A., Theander, S.
& Wollheim, C. B. Beta-cell secretory products
activate alpha-cell ATP-dependent potassium
channels to inhibit glucagon release. Diabetes
54, 1808–1815 (2005).
23. Olsen, H. L. et al. Glucose stimulates glucagon
release in single rat alpha-cells by mechanisms
that mirror the stimulus-secretion coupling in
beta-cells. Endocrinology 146, 4861–4870
(2005).
REVIEWS
© 20 Macmillan Publishers Limited. All rights reserved10

696 | DECEMBER 2010 | VOLUME 6 www.nature.com/nrendo
24. Ravier, M. A. & Rutter, G. A. Glucose or insulin,
but not zinc ions, inhibit glucagon secretion from
mouse pancreatic alpha-cells. Diabetes 54,
1789–1797 (2005).
25. Cooperberg, B. A. & Cryer, P. E. Beta-cell-
mediated signaling predominates over direct
alpha-cell signaling in the regulation of glucagon
secretion in humans. Diabetes Care 32,
2275–2280 (2009).
26. Dunning, B. E., Foley, J. E. & Ahrén, B. Alpha cell
function in health and disease: influence of
glucagon-like peptide-1. Diabetologia 48,
1700–1713 (2005).
27. Meier, J. J., Kjems, L. L., Veldhuis, J. D.,
Lefèbvre, P. & Butler, P. C. Postprandial
suppression of glucagon secretion depends on
intact pulsatile insulin secretion: further
evidence for the intraislet insulin hypothesis.
Diabetes 55, 1051–1056 (2006).
28. Bollheimer, L. C. et al. Stimulatory short-term
effects of free fatty acids on glucagon secretion
at low to normal glucose concentrations.
Metabolism 53, 1443–1448 (2004).
29. Ahrén, B. Autonomic regulation of islet hormone
secretion—implications for health and disease.
Diabetologia 43, 393–410 (2000).
30. Dumonteil, E. et al. Glucose regulates proinsulin
and prosomatostatin but not proglucagon
messenger ribonucleic acid levels in rat pancreatic
islets. Endocrinology 141, 174–180 (2000).
31. Mayo, K. E. et al. International Union of
Pharmacology. XXXV. The glucagon receptor
family. Pharmacol. Rev. 55, 167–194 (2003).
32. Svoboda, M., Tastenoy, M., Vertongen, P. &
Robberecht, P. Relative quantitative analysis of
glucagon receptor mRNA in rat tissues. Mol. Cell
Endocrinol. 105, 131–137 (1994).
33. Kedees, M. H., Grigoryan, M., Guz, Y. &
Teitelman, G. Differential expression of glucagon
and glucagon-like peptide 1 receptors in mouse
pancreatic alpha and beta cells in two models of
alpha cell hyperplasia. Mol. Cell Endocrinol. 311,
69–76 (2009).
34. Watanabe, J., Kanai, K. & Kanamura, S.
Glucagon receptors in endothelial and Kupffer
cells of mouse liver. J. Histochem. Cytochem. 36,
1081–1089 (1988).
35. Kieffer, T. J., Heller, R. S., Unson, C. G.,
Weir, G. C. & Habener, J. F. Distribution of
glucagon receptors on hormone-specific
endocrine cells of rat pancreatic islets.
Endocrinology 137, 5119–5125 (1996).
36. Huypens, P., Ling, Z., Pipeleers, D. & Schuit, F.
Glucagon receptors on human islet cells
contribute to glucose competence of insulin
release. Diabetologia 43, 1012–1019 (2000).
37. Gelling, R. W. et al. Lower blood glucose,
hyperglucagonemia, and pancreatic alpha cell
hyperplasia in glucagon receptor knockout mice.
Proc. Natl Acad. Sci. USA 100, 1438–1443
(2003).
38. Conarello, S. L. et al. Glucagon receptor knockout
mice are resistant to diet-induced obesity and
streptozotocin-mediated beta cell loss and
hyperglycaemia. Diabetologia 50, 142–150
(2007).
39. Sørensen, H. et al. Glucagon receptor knockout
mice display increased insulin sensitivity and
impaired beta-cell function. Diabetes 55,
3463–3469 (2006).
40. Gelling, R. W. et al. Pancreatic beta-cell
overexpression of the glucagon receptor gene
results in enhanced beta-cell function and mass.
Am. J. Physiol. Endocrinol. Metab. 297,
E695–E707 (2009).
41. Conarello, S. L. et al. Mice lacking dipeptidyl
peptidase IV are protected against obesity and
insulin resistance. Proc. Natl Acad. Sci. USA 100,
6825–6830 (2003).
42. Pospisilik, J. A. et al. Dipeptidyl peptidase IV
inhibitor treatment stimulates beta-cell survival
and islet neogenesis in streptozotocin-induced
diabetic rats. Diabetes 52, 741–750 (2003).
43. Rodbell, M., Birnbaumer, L., Pohl, S. L. &
Krans, H. M. The glucagon-sensitive adenyl
cyclase system in plasma membranes of rat
liver. V. An obligatory role of guanylnucleotides in
glucagon action. J. Biol. Chem. 246, 1877–1882
(1971).
44. Rodbell, M. The complex regulation of receptor-
coupled G-proteins. Adv. Enzyme Regul. 37,
427–435 (1997).
45. Jelinek, L. J. et al. Expression cloning and
signaling properties of the rat glucagon receptor.
Science 259, 1614–1616 (1993).
46. Koo, S. H. et al. The CREB coactivator TORC2 is
a key regulator of fasting glucose metabolism.
Nature 437, 1109–1111 (2005).
47. Yoon, J. C. et al. Control of hepatic
gluconeogenesis through the transcriptional
coactivator PGC-1. Nature 413, 131–138 (2001).
48. Herzig, S. et al. CREB regulates hepatic
gluconeogenesis through the coactivator PGC-1.
Nature 413, 179–183 (2001).
49. Kimball, S. R., Siegfried, B. A. & Jefferson, L. S.
Glucagon represses signaling through the
mammalian target of rapamycin in rat liver by
activating AMP-activated protein kinase. J. Biol.
Chem. 279, 54103–54109 (2004).
50. Chen, J., Ishac, E. J., Dent, P., Kunos, G. &
Gao, B. Effects of ethanol on mitogen-activated
protein kinase and stress-activated protein
kinase cascades in normal and regenerating
liver. Biochem. J. 334 (Pt 3), 669–676 (1998).
51. Jiang, G. & Zhang, B. B. Glucagon and regulation
of glucose metabolism. Am. J. Physiol.
Endocrinol. Metab. 284, E671–E678 (2003).
52. Rosic, M. et al. Glucagon effects on ischemic
vasodilatation in the isolated rat heart.
J. Biomed. Biotechnol. 2010, 231832 (2010).
53. Caren, R. & Corbo, L. Glucagon and cholesterol
metabolism. Metabolism 9, 938–945 (1960).
54. Salter, J. M., Ezrin, C., Laidlaw, J. C. &
Gornall, A. G. Metabolic effects of glucagon in
human subjects. Metabolism 9, 753–768
(1960).
55. Paloyan, E. & Harper, P. V. Jr. Glucagon as a
regulating factor of plasma lipids. Metabolism
10, 315–323 (1961).
56. Amatuzio, D. S., Grande, F. & Wada, S. Effect of
glucagon on the serum lipids in essential
hyperlipemia and in hypercholesterolemia.
Metabolism 11, 1240–1249 (1962).
57. De Oya, M., Prigge, W. F., Swenson, D. E. &
Grande, F. Role of glucagon on fatty liver
production in birds. Am. J. Physiol. 221, 25–30
(1971).
58. Caren, R. & Corbo, L. Transfer of plasma lipid to
platelets by action of glucagon. Metabolism 19,
598–607 (1970).
59. Caren, R. & Corbo, L. Glucagon and plasma
arachidonic acid. Metabolism 14, 684–692
(1965).
60. De Oya, M., Prigge, W. F. & Grande, F.
Suppression by hepatectomy of glucagon-
induced hypertriglyceridemia in geese. Proc. Soc.
Exp. Biol. Med. 136, 107–110 (1971).
61. Heimberg, M., Weinstein, I. & Kohout, M. The
effects of glucagon, dibutyryl cyclic adenosine
3',5'-monophosphate, and concentration of free
fatty acid on hepatic lipid metabolism. J. Biol.
Chem. 244, 5131–5139 (1969).
62. Penhos, J. C., Wu, C. H., Daunas, J., Reitman, M.
& Levine, R. Effect of glucagon on the
metabolism of lipids and on urea formation by
the perfused rat liver. Diabetes 15, 740–748
(1966).
63. Eaton, R. P. Hypolipemic action of glucagon in
experimental endogenous lipemia in the rat.
J. Lipid Res. 14, 312–318 (1973).
64. Guettet, C., Mathé, D., Riottot, M. & Lutton, C.
Effects of chronic glucagon administration on
cholesterol and bile acid metabolism. Biochim.
Biophys. Acta 963, 215–223 (1988).
65. Guettet, C., Mathé, D., Navarro, N. & Lecuyer, B.
Effects of chronic glucagon administration on rat
lipoprotein composition. Biochim. Biophys. Acta
1005, 233–238 (1989).
66. Guettet, C. et al. Effect of chronic glucagon
administration on lipoprotein composition in
normally fed, fasted and cholesterol-fed rats.
Lipids 26, 451–458 (1991).
67. Guettet, C., Rostaqui, N., Navarro, N., Lecuyer, B.
& Mathe, D. Effect of chronic glucagon
administration on the metabolism of
triacylglycerol-rich lipoproteins in rats fed a high
sucrose diet. J. Nutr. 121, 24–30 (1991).
68. Rudling, M. & Angelin, B. Stimulation of rat
hepatic low density lipoprotein receptors by
glucagon. Evidence of a novel regulatory
mechanism in vivo. J. Clin. Invest. 91,
2796–2805 (1993).
69. Bobe, G., Ametaj, B. N., Young, J. W. &
Beitz, D. C. Effects of exogenous glucagon on
lipids in lipoproteins and liver of lactating dairy
cows. J. Dairy Sci. 86, 2895–2903 (2003).
70. Bobe, G., Sonon, R. N., Ametaj, B. N.,
Young, J. W. & Beitz, D. C. Metabolic responses
of lactating dairy cows to single and multiple
subcutaneous injections of glucagon. J. Dairy
Sci. 86, 2072–2081 (2003).
71. Longuet, C. et al. The glucagon receptor is
required for the adaptive metabolic response to
fasting. Cell Metab. 8, 359–371 (2008).
72. Richter, W. O., Robl, H. & Schwandt, P. Human
glucagon and vasoactive intestinal polypeptide
(VIP) stimulate free fatty acid release from
human adipose tissue in vitro. Peptides 10,
333–335 (1989).
73. Lefebvre, P., Luyckx, A. & Bacq, Z. M. Effects of
denervation on the metabolism and the
response to glucagon of white adipose tissue of
rats. Horm. Metab. Res. 5, 245–250 (1973).
74. Perea, A., Clemente, F., Martinell, J., Villanueva-
Peñacarrillo, M. L. & Valverde, I. Physiological
effect of glucagon in human isolated adipocytes.
Horm. Metab. Res. 27, 372–375 (1995).
75. Nair, K. S., Welle, S. L., Halliday, D. &
Campbell, R. G. Effect of beta-hydroxybutyrate on
whole-body leucine kinetics and fractional mixed
skeletal muscle protein synthesis in humans.
J. Clin. Invest. 82, 198–205 (1988).
76. Gerich, J. E. et al. Prevention of human diabetic
ketoacidosis by somatostatin. Evidence for an
essential role of glucagon. N. Engl. J. Med. 292,
985–989 (1975).
77. Pegorier, J. P. et al. Induction of ketogenesis and
fatty acid oxidation by glucagon and cyclic AMP
in cultured hepatocytes from rabbit fetuses.
Evidence for a decreased sensitivity of carnitine
palmitoyltransferase I to malonyl-CoA inhibition
after glucagon or cyclic AMP treatment.
Biochem. J. 264, 93–100 (1989).
78. Prip-Buus, C., Pegorier, J. P., Duee, P. H., Kohl, C.
& Girard, J. Evidence that the sensitivity of
carnitine palmitoyltransferase I to inhibition by
malonyl-CoA is an important site of regulation
of hepatic fatty acid oxidation in the fetal
and newborn rabbit. Perinatal development and
effects of pancreatic hormones in cultured rabbit
hepatocytes. Biochem. J. 269, 409–415 (1990).
REVIEWS
© 20 Macmillan Publishers Limited. All rights reserved10

NATURE REVIEWS | ENDOCRINOLOGY VOLUME 6 | DECEMBER 2010 | 697
79. Vons, C. et al. Regulation of fatty-acid
metabolism by pancreatic hormones in cultured
human hepatocytes. Hepatology 13, 1126–1130
(1991).
80. Charbonneau, A., Couturier, K., Gauthier, M. S. &
Lavoie, J. M. Evidence of hepatic glucagon
resistance associated with hepatic steatosis:
reversal effect of training. Int. J. Sports Med. 26,
432–441 (2005).
81. Charbonneau, A., Unson, C. G. & Lavoie, J. M.
High-fat diet-induced hepatic steatosis reduces
glucagon receptor content in rat hepatocytes:
potential interaction with acute exercise.
J. Physiol. 579 (Pt 1), 255–267 (2007).
82. Charbonneau, A., Melancon, A., Lavoie, C. &
Lavoie, J. M. Alterations in hepatic glucagon
receptor density and in Gsalpha and Gialpha2
protein content with diet-induced hepatic
steatosis: effects of acute exercise. Am. J.
Physiol. Endocrinol. Metab. 289, E8–E14 (2005).
83. Savage, A., Zeng, L. & Houslay, M. D. A role for
protein kinase C-mediated phosphorylation in
eliciting glucagon desensitization in rat
hepatocytes. Biochem. J. 307 (Pt 1), 281–285
(1995).
84. Eaton, R. P. & Schade, D. S. Glucagon resistance
as a hormonal basis for endogenous
hyperlipaemia. Lancet 1, 973–974 (1973).
85. Song, K. H. & Chiang, J. Y. Glucagon and cAMP
inhibit cholesterol 7alpha-hydroxylase (CYP7A1)
gene expression in human hepatocytes:
discordant regulation of bile acid synthesis and
gluconeogenesis. Hepatology 43, 117–125
(2006).
86. Chiang, J. Y. Bile acid regulation of gene
expression: roles of nuclear hormone receptors.
Endocr. Rev. 23, 443–463 (2002).
87. Hylemon, P. B. et al. Hormonal regulation of
cholesterol 7 alpha-hydroxylase mRNA levels
and transcriptional activity in primary rat
hepatocyte cultures. J. Biol. Chem. 267,
16866–16871 (1992).
88. Davidson, I. W., Salter, J. M. & Best, C. H. The
effect of glucagon on the metabolic rate of rats.
Am. J. Clin. Nutr. 8, 540–546 (1960).
89. Davidson, I. W., Salter, J. M. & Best, C. H.
Calorigenic action of glucagon. Nature 180,
1124 (1957).
90. Nair, K. S. Hyperglucagonemia increases resting
metabolic rate in man during insulin deficiency.
J. Clin. Endocrinol. Metab. 64, 896–901 (1987).
91. Calles-Escandón, J. Insulin dissociates
hepatic glucose cycling and glucagon-induced
thermogenesis in man. Metabolism 43,
1000–1005 (1994).
92. Joel, C. D. Stimulation of metabolism of rat
brown adipose tissue by addition of lipolytic
hormones in vitro. J. Biol. Chem. 241, 814–821
(1966).
93. Kuroshima, A. & Yahata, T. Thermogenic
responses of brown adipocytes to noradrenaline
and glucagon in heat-acclimated and cold-
acclimated rats. Jpn. J. Physiol. 29, 683–690
(1979).
94. Yahata, T., Habara, Y. & Kuroshima, A. Effects of
glucagon and noradrenaline on the blood flow
through brown adipose tissue in temperature-
acclimated rats. Jpn. J. Physiol. 33, 367–376
(1983).
95. Doi, K. & Kuroshima, A. Modified metabolic
responsiveness to glucagon in cold-acclimated
and heat-acclimated rats. Life Sci. 30, 785–791
(1982).
96. Billington, C. J., Briggs, J. E., Link, J. G. &
Levine, A. S. Glucagon in physiological
concentrations stimulates brown fat
thermogenesis in vivo. Am. J. Physiol. 261 (Pt 2),
R501–R507 (1991).
97. Edwards, C. I. & Howland, R. J. Adaptive changes
in insulin and glucagon secretion during cold
acclimation in the rat. Am. J. Physiol. 250 (Pt 1),
E669–E676 (1986).
98. Billington, C. J., Bartness, T. J., Briggs, J.,
Levine, A. S. & Morley, J. E. Glucagon stimulation
of brown adipose tissue growth and
thermogenesis. Am. J. Physiol. 252 (Pt 2),
R160–R165 (1987).
99. Cypess, A. M. et al. Identification and
importance of brown adipose tissue in adult
humans. N. Engl. J. Med. 360, 1509–1517
(2009).
100. Dicker, A., Zhao, J., Cannon, B. & Nedergaard, J.
Apparent thermogenic effect of injected glucagon
is not due to a direct effect on brown fat cells.
Am. J. Physiol. 275 (Pt 2), R1674–R1682
(1998).
101. Heim, T. & Hull, D. The effect of propranalol on
the calorigenic response in brown adipose
tissue of new-born rabbits to catecholamines,
glucagon, corticotrophin and cold exposure.
J. Physiol. 187, 271–283 (1966).
102. Filali-Zegzouti, Y. et al. Role of catecholamines in
glucagon-induced thermogenesis. J. Neural
Transm. 112, 481–489 (2005).
103. Morales, A. et al. Sympathetic control of
glucagon receptor mRNA levels in brown adipose
tissue of cold-exposed rats. Mol. Cell Biochem.
208, 139–142 (2000).
104. Brito, N. A., Brito, M. N. & Bartness, T. J.
Differential sympathetic drive to adipose tissues
after food deprivation, cold exposure or
glucoprivation. Am. J. Physiol. Regul. Integr.
Comp. Physiol. 294, R1445–R1452 (2008).
105. Young, J. B., Saville, E., Rothwell, N. J.,
Stock, M. J. & Landsberg, L. Effect of diet and
cold exposure on norepinephrine turnover in
brown adipose tissue of the rat. J. Clin. Invest.
69, 1061–1071 (1982).
106. Stunkard, A. J., Van Itallie, T. B. & Reis, B. B.
The mechanism of satiety: effect of glucagon on
gastric hunger contractions in man. Proc. Soc.
Exp. Biol. Med. 89, 258–261 (1955).
107. Schulman, J. L., Carleton, J. L., Whitney, G. &
Whitehorn, J. C. Effect of glucagon on food
intake and body weight in man. J. Appl. Physiol.
11, 419–421 (1957).
108. Penick, S. B. & Hinkle, L. E. Jr. Depression of
food intake induced in healthy subjects by
glucagon. N. Engl. J. Med. 264, 893–897 (1961).
109. Martin, J. R. & Novin, D. Decreased feeding in
rats following hepatic-portal infusion of
glucagon. Physiol. Behav. 19, 461–466 (1977).
110. Weick, B. G. & Ritter, S. Dose-related
suppression of feeding by intraportal glucagon
infusion in the rat. Am. J. Physiol. 250 (Pt 2),
R676–R681 (1986).
111. Salter, J. M. Metabolic effects of glucagon in
the Wistar rat. Am. J. Clin. Nutr. 8, 535–539
(1960).
112. Holloway, S. A. & Stevenson, J. A. Effect of
glucagon on food intake and weight gain in the
young rat. Can. J. Physiol. Pharmacol. 42,
867–869 (1964).
113. Le Sauter, J. & Geary, N. Hepatic portal glucagon
infusion decreases spontaneous meal size in
rats. Am. J. Physiol. 261 (Pt 2), R154–R161
(1991).
114. de Jong, A., Strubbe, J. H. & Steffens, A. B.
Hypothalamic influence on insulin and glucagon
release in the rat. Am. J. Physiol. 233,
E380–E388 (1977).
115. Langhans, W., Pantel, K., Müller-Schell, W.,
Eggenberger, E. & Scharrer, E. Hepatic handling
of pancreatic glucagon and glucose during
meals in rats. Am. J. Physiol. 247 (Pt 2),
R827–R832 (1984).
116. Unger, R. H. & Orci, L. Physiology and
pathophysiology of glucagon. Physiol. Rev. 56,
778–826 (1976).
117. Le Sauter, J., Noh, U. & Geary, N. Hepatic portal
infusion of glucagon antibodies increases
spontaneous meal size in rats. Am. J. Physiol.
261 (Pt 2), R162–R165 (1991).
118. Langhans, W., Zeiger, U., Scharrer, E. & Geary, N.
Stimulation of feeding in rats by intraperitoneal
injection of antibodies to glucagon. Science 218,
894–896 (1982).
119. Geary, N., Kissileff, H. R., Pi-Sunyer, F. X. &
Hinton, V. Individual, but not simultaneous,
glucagon and cholecystokinin infusions inhibit
feeding in men. Am. J. Physiol. 262 (Pt 2),
R975–R980 (1992).
120. Geary, N., Le Sauter, J. & Noh, U. Glucagon acts
in the liver to control spontaneous meal size in
rats. Am. J. Physiol. 264 (Pt 2), R116–R122
(1993).
121. Martin, J. R., Novin, D. & Vanderweele, D. A.
Loss of glucagon suppression of feeding after
vagotomy in rats. Am. J. Physiol. 234,
E314–E318 (1978).
122. Geary, N. & Smith, G. P. Selective hepatic
vagotomy blocks pancreatic glucagon’s satiety
effect. Physiol. Behav. 31, 391–394 (1983).
123. Kurose, Y. et al. Effects of central administration
of glucagon on feed intake and endocrine
responses in sheep. Anim. Sci. J. 80, 686–690
(2009).
124. Bobe, G., Ametaj, B. N., Young, J. W. &
Beitz, D. C. Potential treatment of fatty liver with
14-day subcutaneous injections of glucagon.
J. Dairy Sci. 86, 3138–3147 (2003).
Author contributions
All authors researched the data for the article,
provided a substantial contribution to discussions of
the content, contributed equally to writing the article
and reviewed and/or edited the manuscript before
submission.
REVIEWS
© 20 Macmillan Publishers Limited. All rights reserved10





![[XLS] · Web viewSummary DAYWORK 1 BQ-10 BQ-9 BQ-8 BQ-7 BQ-6 BQ-5 BQ-4 BQ-3 BQ-2 BQ-1 Preamble Contents Example Notes Multiple post sign support assemblies (each type and size) Multiple](https://static.fdocuments.in/doc/165x107/5aff741f7f8b9aa34d906f7c/xls-viewsummary-daywork-1-bq-10-bq-9-bq-8-bq-7-bq-6-bq-5-bq-4-bq-3-bq-2-bq-1-preamble.jpg)
![SISTEMA ENDOCRINO[1].pptx](https://static.fdocuments.in/doc/165x107/577c852f1a28abe054bc0da0/sistema-endocrino1pptx.jpg)











