







Languages
Pages
Legal


Revista médica de Chile versión impresa ISSN 0034-9887
Rev. méd. Chile v.130 n.3 Santiago mar. 2002
doi: 10.4067/S0034-98872002000300003
Diagnóstico y seguimiento de 23 niños con acidurias orgánicas
Verónica Cornejo E1, Marta Colombo C, Gloria Durán S, Paulina Mabe S, Mónica Jiménez M2, Alicia De la Parra C3, Alf Valiente G4, Erna Raimann B.
Diagnosis and follow up of 23 children with organic acidurias
Correspondencia a: M.Sc. Verónica Cornejo E. Macul 5540 Macul. Fax: 56-2-2941254. Fono: 56-2-6781491. E mail: [email protected]
Background: Propionic aciduria (PA) and Methymalonic aciduria (MMA) result from an inherited abnormality of the enzymes propionyl CoA carboxylase and methylmalonyl CoA mutase respectively. This produces marked increases in the amino acids methionine, threonine, valine and isoleucine (MTVI). Their clinical presentation can be neonatal or late onset forms. Aim: To report 23 children with organic acidurias. Material and methods: Twenty three cases of organic acidurias diagnosed since 1980 (17 PA and 6 MMA) and followed at the Institute of Nutrition and Food Technology, are reported. Results: The average age of diagnosis was 3.9 days for the neonatal form and 8.3 months for the late onset form. The most frequent symptoms were hypotonia, lethargy and vomiting. Neonatal PA had mean ammonemias of 1089±678.3 µg/dl. The figure for MMA was 933±801.9 µg/dl. Seven children were dialyzed and 30% died. 16 children are followed and 81.2% have normal weight for age. Seven children required gastrostomy because of anorexia and failure to thrive. The nutritional treatment is based on natural and artificial proteins without MTVI, with periodical controls, amino acid and ammonia quantification. Some patients were submitted to enzyme assays and molecular studies. Conclusions: An early diagnosis and a very strict follow up allows a normal development of children with organic aciduras. There is a relationship between prognosis and the presentation form, the nutritional status and the emergency treatment during acute episodes. The importance of the enzymatic and molecular studies is emphasized because they facilitate treatment, accurate diagnosis and allow an adequate genetic counseling (Rev Méd Chile 2002; 130: 259-66).

(Key-words: Acidurias, organic; Enzymes; Genetics, medical, Kidney failure)
Recibido el 23 de julio, 2001. Aceptado el 22 de octubre, 2001. Unidad de Genética y Enfermedades Metabólicas, Instituto de Nutrición y Tecnología de los Alimentos (INTA), Universidad de Chile. Laboratorio Clínico, Hospital Van Buren, Valparaíso 1 Nutricionista, Magister Nutrición Humana 2 Nutricionista, Magister en Planificación 3 Psicóloga 4 Bioquímico
Las acidurias orgánicas (AO) son errores innatos del metabolismo de los aminoácidos: metionina, treonina, valina, isoleucina, leucina, de ácido grasos de cadena impar y otros metabolitos. En general se constituyen como patologías poco comunes, siendo las más frecuentes la acidemia propiónica (AP) y la acidemia metilmalónica (AMM).
La AP se produce por deficiencia de la enzima propionil CoA carboxilasa (PCC) (Figura 1), compuesta por 2 subunidades α y ß, codificadas en el gen PCCA y PCCB respectivamente, ambas son dependientes de biotina. El gen de la subunidad α está localizado en el cromosoma 13 y el de la subunidad ß en el brazo largo del cromosoma 3. Cualquier alteración en el gen PCCA o PCCB produce AP1. Se estima una incidencia de 1:100.000 recién nacidos1,25. La AMM se debe al déficit o ausencia de la enzima metilmalonil CoA mutasa (formas mut(°) y mut(-
)) o por defectos de cobalaminas (cblA y cblB) o vitamina B12 (Figura 1). Se ha demostrado que la presencia de vitamina B12 en las formas mut(-), cblA y cblB aumenta la actividad de la enzima mejorando su pronóstico2. El gen que codifica la enzima se ubica en el cromosoma 6p21. La incidencia es de 1:50.000 recién nacidos en la población caucásica3.
Figura 1. Metabolismo del propionato y metilmalonato

1. Propionil CoA carboxilasa (unidad ∂ y ß). 2. Metilmalonil CoA mutasa y cofactor de cobalaminas.
La sintomatología clínica de las AO puede manifestarse en el período neonatal o ser de presentación tardía. Los síntomas son rechazo a la alimentación, vómitos, deshidratación, compromiso de conciencia progresivo hasta el coma, hipotonía, convulsiones, hipoglicemia, quetoacidosis con aumento del anion gap, hiperamonemia, hiperglicinemia, hiperlactatemia, trombopenia, leucopenia y ocasionalmente anemia. La presencia de pancitopenia puede llevar a confundir el cuadro con una sepsis4. Si estas patologías no son diagnosticadas precozmente y tratadas adecuadamente durante los cuadros agudos, se pueden producir graves secuelas neurológicas e incluso la muerte5,6.
El tratamiento consiste en restringir la ingesta de los aminoácidos metionina, treonina, valina e isoleucina (MTVI), evitar el catabolismo proteico endógeno y la lipólisis. Durante la crisis aguda se recomienda realizar diálisis peritoneal, suspender las proteínas e iniciar el manejo nutricional de emergencia con aporte de calorías, biotina, vitamina B12 y l-carnitina7,8.
El Instituto de Nutrición y Tecnología de los Alimentos (INTA) de la Universidad de Chile, Centro de Referencia Nacional para el diagnóstico de estas patologías tiene un programa de seguimiento compuesto por médicos, nutricionistas, bioquímico, neurólogo, psicólogo y asistente social, encargados de realizar evaluaciones periódicas, orientadas a cubrir todas las áreas vulnerables del niño y lograr un crecimiento y desarrollo en rangos de normalidad.
El objetivo del presente estudio es analizar los resultados obtenidos del seguimiento médico, nutricional y bioquímico de los niños con AP y AMM tratados en el policlínico de enfermedades metabólicas del INTA de la Universidad de Chile.
MATERIAL Y MÉTODO
La muestra está conformada por 23 niños con AP y AMM diagnosticados desde 1980 a la fecha por la Unidad de Genética y Enfermedades Metabólicas del INTA.
Durante la primera crisis se evaluaron gases venosos, electrolitos plasmáticos, amonio, y cromatografía de capa fina para aminoácidos en sangre y orina. El diagnóstico se estableció a través del análisis de ácidos orgánicos en orina por cromatografía de gas con espectrometría de masa. La presencia de los ácidos orgánicos 3-hidroxipropiónico, 3-hidroxibutírico y metilcitrato confirmaron una AP y el hallazgo de los ácidos metilmalónico y metilcítrico una AMM.
Protocolo de tratamiento: durante la etapa aguda se suspendió el aporte de proteínas, proporcionando glucosa (10 mg/kg/min) y lípidos intravenosos al 20% (2 gr/kg/día), se corrigió el balance hidroelectrolítico y ácido base. Una amonemia sobre 600 ug/dl fue señal para efectuar una diálisis peritoneal. Se suplementó en forma

paralela 200 a 400 mg de l-carnitina/kg/día, 10 mg biotina/día en una AP y 1,0 mg de B12/día en una AMM, vía oral.
El período de seguimiento crónico incluyó evaluaciones médicas, nutricionales y bioquímicas cada 15 días durante los dos primeros meses y posteriormente cada mes. La evaluación nutricional consistió en establecer los requerimientos individuales de macro y micronutrientes y de los aminoácidos MTVI, basándose en las recomendaciones de la Recommended Dietary Allowance (RDA), 19977. La dieta restringida en los aminoácidos MTVI, es hipercalórica (>120 cals/kg/día), hipoproteica (1,0-1,5 grs/kg/día de origen natural), siendo necesaria la suplementación con leche especial sin MTVI, para completar requerimientos protéicos. Se mantuvo la suplementación de biotina y vitamina B12 y l-carnitina (100 mg/kg/día)7.
Los parámetros antropométricos: peso, talla, circunferencia craneana se compararon con patrones estándares del National Center for Health Statistic (NCHS)9.
Una vez al mes y durante los primeros 6 meses de tratamiento se evaluó el amonio y se cuantificaron los aminoácidos con un analizador de aminoácidos (Biotronik 2000).
Como estudio adicional, en 3 casos con AMM se midió la actividad de la enzima metilmalonil CoA mutasa in vitro para evaluar la respuesta a megadosis de vitamina B12. En 8 casos con AP se determinó actividad de la enzima propionil CoA carboxilasa en cultivo de fibroblasto y se realizó además el estudio molecular para identificar mutaciones en el gen PCCB. Los análisis enzimáticos y moleculares fueron realizados en el Centro de Estudios Moleculares de la Universidad Autónoma de Madrid, España.
Este estudio es del tipo descriptivo, retrospectivo y transversal. Para el análisis estadístico se utilizó el programa de computación Epi Info 6,0 y se calculó el promedio de los datos ± 1 desviación estándar.
RESULTADOS
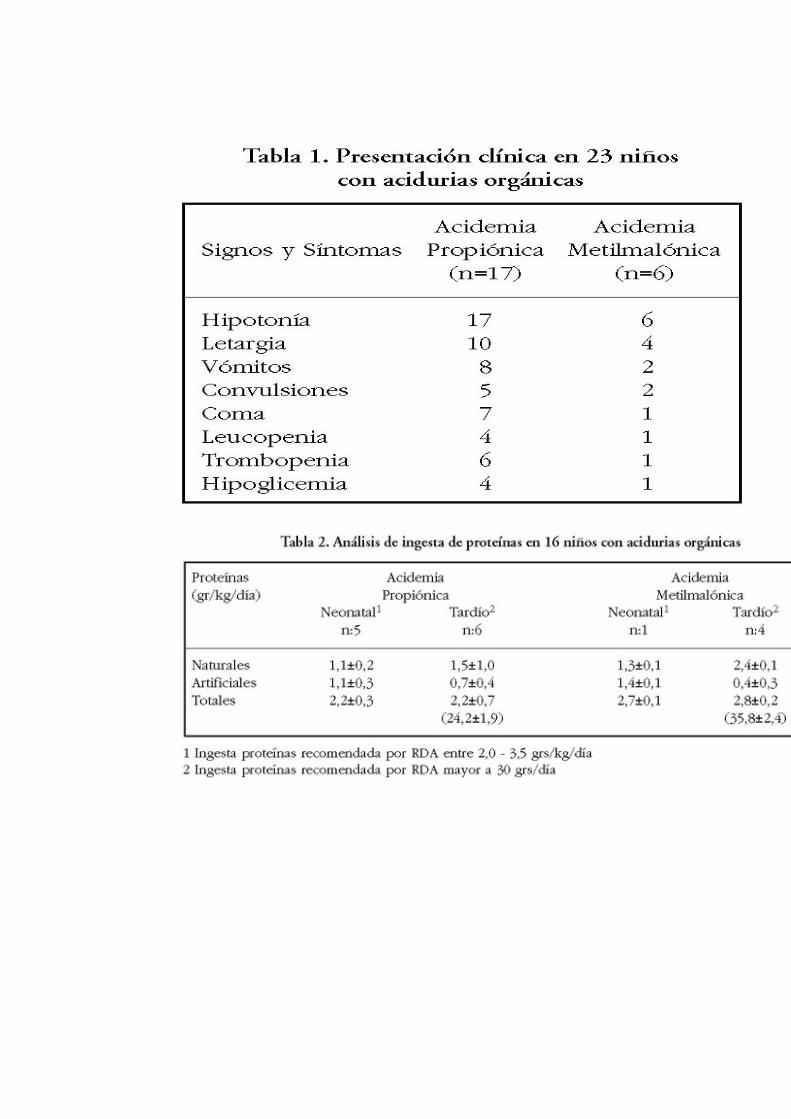
La muestra estuvo compuesta por 23 niños, de los cuales 14 eran de sexo masculino y 9 de sexo femenino. Del total, 17 tenían AP y 6 AMM. En 10 de ellos los síntomas clínicos se presentaron durante el período neonatal (8 AP y 2 AMM) y en los otros 13 pacientes la sintomatología clínica apareció después del 5° mes de vida (9 AP y 4 AMM).
Las AO de presentación neonatal se diagnosticaron en promedio a los 3,9 días de edad (rango de 36 h a 9 días) y los de manifestacion tardía a los 8,3 meses de edad (rango de 5 a 22 meses). Siete de los diez pacientes con la forma neonatal requirieron diálisis peritoneal, correspondiendo a 6 AP y a 1 AMM.
Al momento del diagnóstico los síntomas clínicos de mayor ocurrencia fueron hipotonía, letargia y vómitos (Tabla 1). Dentro de los signos bioquímicos destacaba el elevado nivel de amonio encontrado en las AO de presentación neonatal, siendo en las AP en promedio de 1089

±678,3 µg/dl y en las AMM de 933±801,9 µg/dl. En la forma tardía el amonio estaba bajo, 180 µg/dl para ambas patologías, considerándose valores normales entre 20 y 80 µg/dl. La glicina sérica se encontró en rango de 413 - 1218 µM/l; sólo un niño tuvo glicina de 236 µM/l (valores de referencia 127 - 341 µM/l). La quetoacidosis estuvo presente en 15 de los 23 niños, siendo 8 de presentación neonatal. A la fecha 4 casos de presentación neonatal y 3 con la forma tardía han muerto (30%: 6 AP y 1 AMM); 3 de ellos fallecieron durante un episodio de descompensación grave, realizándose el diagnóstico post mortem; dos pacientes que se encontraban en seguimiento por dos años, fallecieron por una infección grave, ambos tenían AO de presentación neonatal y 2 niños fallecieron por descompensaciones a repetición causadas por desnutrición secundaria a un tratamiento inadecuado.
Actualmente 16 niños con AO (11 AP y 5 AMM) se encuentran en seguimiento, con un promedio de 43,7 meses en las de presentación neonatal y de 80,5 meses en la forma tardía.
Tanto en las AP como en las AMM de presentación neonatal, la ingesta de proteínas de origen natural (leche maternizadas, cereales, frutas y verduras) fue 1,1±0,2 y 1,3±0,1 gr/kg/día y de proteínas artificiales 1,1±0,3 y 1,4±0,1 gr/kg/día respectivamente. En los de presentación tardía la ingesta de proteínas naturales fue en promedio 1,5±1,0 y 2,4±0,1 grs/kg/día en AP y AMM (Tabla 2) respectivamente.
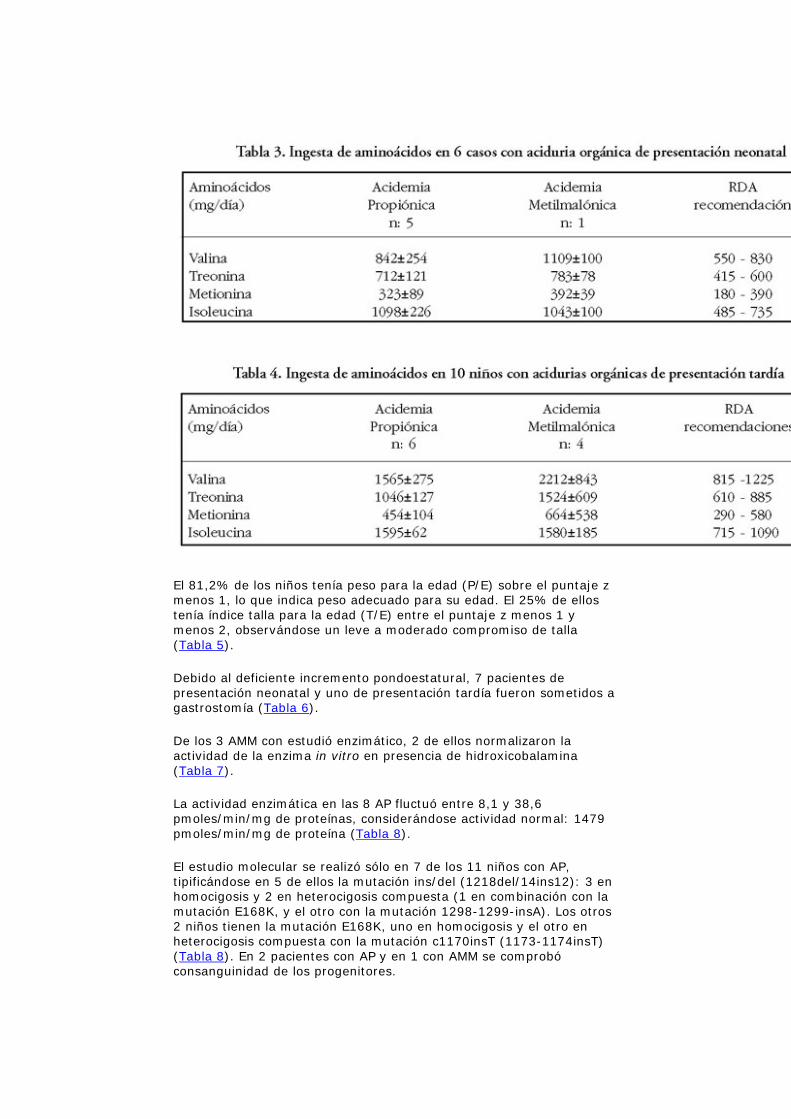
Se observó además que la ingesta de los aminoácidos metionina, treonina y valina estaba en el límite máximo de las recomendaciones de la RDA y la isoleucina por sobre ellas (Tabla 3 y 4).
Doce casos presentaron valina sérica inferior a 95 µM/l (valores recomendados 95-300 µM/l) y 8 niños tuvieron isoleucina inferior a 35 µM/l (valores recomendados 35-105 µM/l). En ambas situaciones se suplementó farmacológicamente de acuerdo a las necesidades individuales.
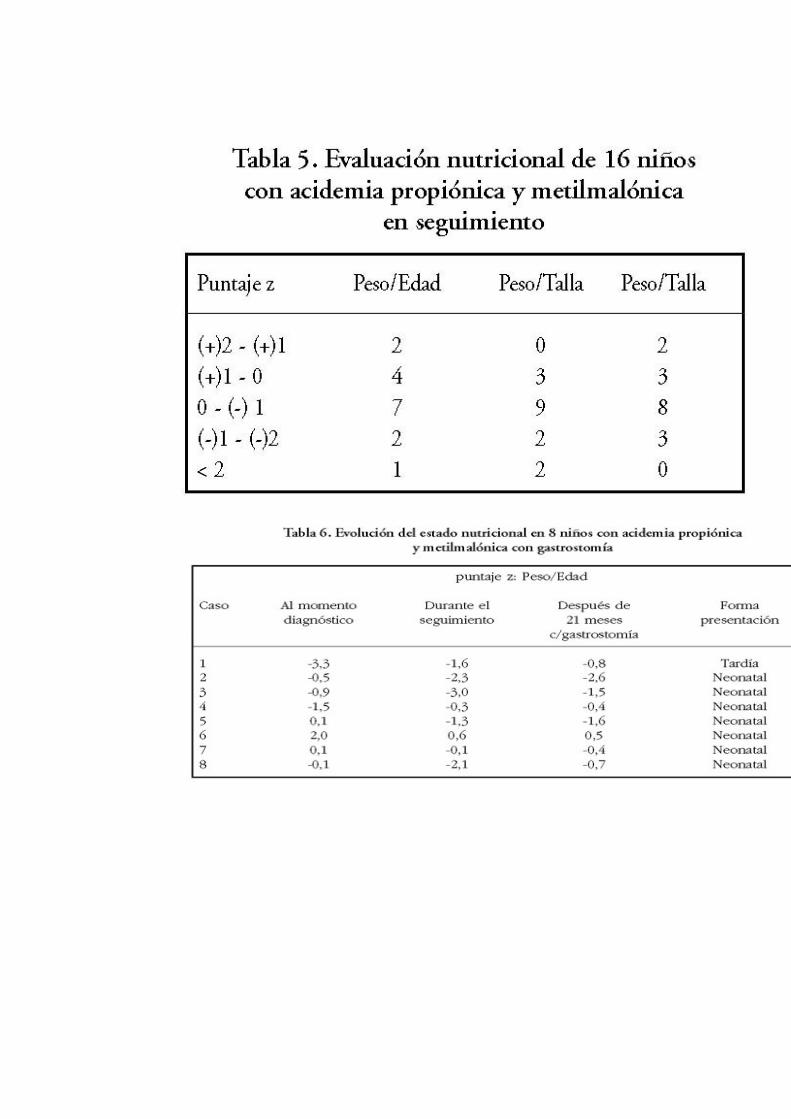
El 81,2% de los niños tenía peso para la edad (P/E) sobre el puntaje z menos 1, lo que indica peso adecuado para su edad. El 25% de ellos tenía índice talla para la edad (T/E) entre el puntaje z menos 1 y menos 2, observándose un leve a moderado compromiso de talla (Tabla 5).
Debido al deficiente incremento pondoestatural, 7 pacientes de presentación neonatal y uno de presentación tardía fueron sometidos a gastrostomía (Tabla 6).
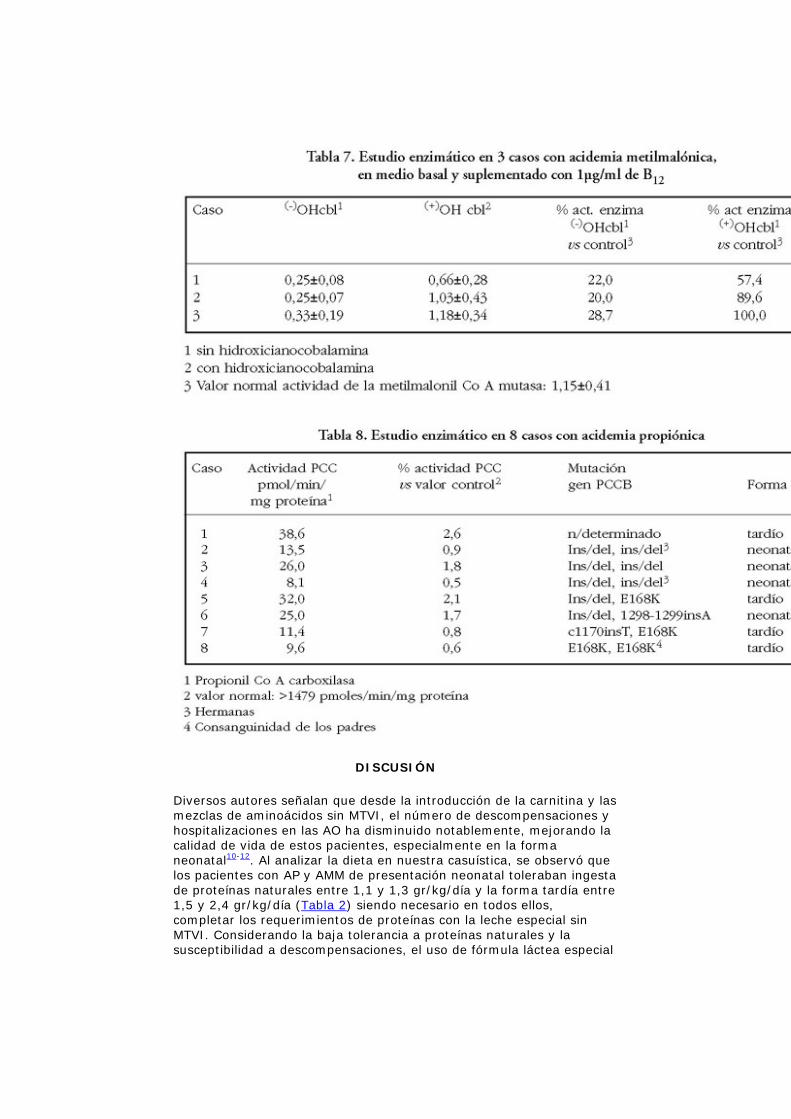
De los 3 AMM con estudió enzimático, 2 de ellos normalizaron la actividad de la enzima in vitro en presencia de hidroxicobalamina (Tabla 7).
La actividad enzimática en las 8 AP fluctuó entre 8,1 y 38,6 pmoles/min/mg de proteínas, considerándose actividad normal: 1479 pmoles/min/mg de proteína (Tabla 8).
El estudio molecular se realizó sólo en 7 de los 11 niños con AP, tipificándose en 5 de ellos la mutación ins/del (1218del/14ins12): 3 en homocigosis y 2 en heterocigosis compuesta (1 en combinación con la mutación E168K, y el otro con la mutación 1298-1299-insA). Los otros 2 niños tienen la mutación E168K, uno en homocigosis y el otro en heterocigosis compuesta con la mutación c1170insT (1173-1174insT) (Tabla 8). En 2 pacientes con AP y en 1 con AMM se comprobó consanguinidad de los progenitores.
DISCUSIÓN
Diversos autores señalan que desde la introducción de la carnitina y las mezclas de aminoácidos sin MTVI, el número de descompensaciones y hospitalizaciones en las AO ha disminuido notablemente, mejorando la calidad de vida de estos pacientes, especialmente en la forma neonatal10-12. Al analizar la dieta en nuestra casuística, se observó que los pacientes con AP y AMM de presentación neonatal toleraban ingesta de proteínas naturales entre 1,1 y 1,3 gr/kg/día y la forma tardía entre 1,5 y 2,4 gr/kg/día (Tabla 2) siendo necesario en todos ellos, completar los requerimientos de proteínas con la leche especial sin MTVI. Considerando la baja tolerancia a proteínas naturales y la susceptibilidad a descompensaciones, el uso de fórmula láctea especial

resulta imprescindible en el tratamiento de las AO, para mantener balance metabólico y fomentar anabolismo13-15.
Se ha descrito que niveles de isoleucina inferiores a 20 µM/l producen pérdida de peso, decoloración de la mucosa bucal, desbalance entre los aminoácidos esenciales y no esenciales, disminución del colesterol plasmático y si esta deficiencia es mantenida en el tiempo afectará el crecimiento ponderal del niño15,16. Todos los niños con AO en este estudio tuvieron ingesta de isoleucina por sobre las recomendaciones de la RDA (Tabla 3). Sin embargo, al evaluar el nivel plasmático de este aminoácido, se detectó que 8 niños tenían la isoleucina bajo los 35 µM/l, siendo necesario la suplementación farmacológica. Ninguno de estos niños manifestó signos clínicos de deficiencia ya que la suplementación fue instaurada precozmente.
Los niños con AO, especialmente las formas neonatales, evolucionan frecuentemente con mal incremento ponderal por anorexia. El uso de gastrostomía previene el deterioro del estado nutricional y disminuye la frecuencia de descompensaciones12,16,17 . En este estudio, 8 niños requirieron gastrostomía porque presentaron descenso en el canal de crecimiento. Después de 21 meses, solamente 4 niños incrementaron su peso (Tabla 6), pero en todos hubo disminución del número de descompensaciones graves.
Todas las formas de AMM presentan la misma sintomatología clínica y bioquímica, pero existen diferencias entre ellas frente a una sobrecarga de B12. El 90% de la forma cblA y un menor porcentaje los tipos mut(-) y cblB responden efectivamente a megadosis de B12, aumentando la actividad de la enzima, siendo mejor su pronóstico12,14,18,26. De las 3 AMM estudiadas, 2 de ellos normalizaron in vitro la actividad enzimática, en presencia de hidroxicianocobalamina. Este hallazgo concuerda con la clínica, ya que al aumentar la dosis oral de B12, de 1,0 mg a 10 mg/día, disminuyó la excreción de ácido metilmalónico en orina de 3094 y 1763 mg/mg de creatinina a 220 y 695 mg/mg/creatinina respectivamente.
En este estudio todas las AP tuvieron actividad enzimática inferior al 3% con respecto a lo normal y no se detectaron diferencias porcentuales de actividad entre la forma neonatal y la tardía (Tabla 8). Esta observación sugiere que la heterogeneidad clínica entre las AP de presentación neonatal y de presentación tardía no sería explicable a través de la actividad enzimática residual19,20. Con el objetivo de interpretar las diferencias fenotípicas, se han buscado las mutaciones que producen una AP, describiéndose a la fecha más de 60 mutaciones21-23. La mutaciones más frecuentes en la población española son: ins/del (31%) y la mutación c1170insT (16,7%)24. En Chile son del 47% y del 6,2% respectivamente. La mutación E168K tiene una frecuencia del 25%.
Al correlacionar el genotipo con el fenotipo, se han demostrado que los niños homocigotos para las mutaciones ins/del y c1170insT o heterocigotos compuestos con la mutación 1298-1299insA, tienen ausencia total de actividad de la enzima propionil Co A carboxilasa24. En nuestro estudio, de las 7 AP estudiadas, los 3 pacientes que poseían la mutación ins/del en homocigosis, tenían AO de presentación

neonatal, al igual que el niño con la mutación ins/del en combinación con la mutación 1298-1299insA, resultados concordantes con lo descrito en la literatura24. La actividad enzimática para estas mutaciones fue mayor a lo descrito (1,2 y 2,4% con respecto a valores normales), deduciéndose que el estudio enzimático por sí sólo, no permite predecir el pronóstico de la enfermedad, siendo necesario el estudio molecular.
Podemos concluir que el diagnóstico oportuno de las AO, seguido de tratamiento estricto y sistemático, permite a los pacientes un desarrollo pondoestatural dentro de parámetros normales. Sin embargo, su evolución está relacionada estrechamente con el tipo de presentación clínica y con el manejo de emergencia que se realice durante cuadros agudos.
REFERENCIAS
1. Fenton W, Rosenberg L. Disorders of Propionate and Methylmalonate Metabolism. En: Scriver C, Beaudet A, Sly W, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. Nueva York: Editorial Mc Graw-Hill, Inc Health Professions Division, 1995; 1423-9. [ Links ]
2. Leonard J. The management and outcome of propionic and methylmalonic acidemia. J Inher Metab Dis 1995; 18: 430-4. [ Links ]
3. Rosemblatt DS, Ledley FD. A molecular study of methylmalonic aciduria: structure-function correlations. Bull Acad Natl Med 1996; 180: 1553-63. [ Links ]
4. Saudubray JM, Ogier H, Charpentier C, Dehondt E, Coude FX et al. Neonatal management of organic acidurias. Clinical update. J Inher Metab Dis 1984; 7: 2-9. [ Links ]
5. Baumgartner R, Leopold D. Propionic (PA) and methylmalonic (MMA) acidemias. En: 25th conference of the European metabolic group, Milupa, european metabolic group. 1992; 4 [ Links ]
6. Baulny O, Wendel U, Saudubray J. Branched-Chain Organic Acidurias. En: Fernández J, Saudubray JM, Van Den Berghe G, eds. Inborn metabolic disease. Diagnosis and treatment. Part VI Organic Acids. New York. Editorial: Springer-Verlag, 1995; 206-21. [ Links ]
7. Acosta P. Nutrition support of infants with propionic acidemia and metilmalonic acidemia. Protocols 10. Nutrition support protocols (A6224), Ohio, USA. Editorial Ross Laboratories, 1993; 253-74. [ Links ]
8. Roe CH, Millington D, Maltby D, Bohan T. L-Carnitine enhances excretion of propionyl coenzyme. A as Propionylcarnitine in propionic acidemia. J Clin Invest 1984; 73: 1785-8. [ Links ]

9. Barrera G. Estándares antropométricos para evaluación del estado nutritivo. Instituto de Nutrición y Tecnología de los Alimentos, Universidad de Chile, 1999. [ Links ]
10. Levy H. Nutritional therapy in inborn errors of metabolism. En: Desnick R. Treatment of genetics diseases. New York, London. Editorial: Churchill Livingstone Inc., 1991; 1-22. [ Links ]
11. Del Valle JA, Merinero B, Jimenez A, Garcia M, Ugarte M. Dietary treatment and biochemical studies on a neonatal case of propionyl-CoA carboxylase deficiency. J Inher Metab Dis 1982; 5: 121-4. [ Links ]
12. Van Der Meer SB, Poggi F, Spada M, Bonnefont JP, Ogier H, et al. Clinical outcome of long-term management of patients with vitamin B12-unresponsive methylmalonic acidemia. J Pediatr 1994; 125: 903-8. [ Links ]
13. Cornejo V. Tratamiento nutricional en algunos errores congénitos del metabolismo. En: Meneghello J, ed. Diálogos en pediatría XII. Santiago, Chile. Editorial: Mediterraneo, 1997; 197-210. [ Links ]
14. Baumgarten E, Viardot C. Long-term follow-up of 77 patients with isolated methylmalonic acidaemia. J Inher Metab Dis 1995; 18: 138-42. [ Links ]
15. Cornejo V, Raimann E. Errores innatos del metabolismo de los aminoácidos En: Colombo M, Cornejo V, Raimann E. eds. Errores innatos del metabolismo del niño. Santiago Chile. Editorial: Universitaria, 1999; 59-112. [ Links ]
16. Thomas E. Dietary management of inborn errors of aminoacid metabolism with protein-modified diets. J Child Neurol 1992; 7: S92-111. [ Links ]
17. Thompson GN, Walter JH, Breson JL, Ford GC, Lyonnet JV et al. Sources of propionate in inborn errors of propionate metabolism. Metabolism 1990; 39: 1133-7. [ Links ]
18. Ninan TK, Thom H, Rusell G. Oral vitamin B12 treatment of cobalamin-responsive methylmalonic aciduria. J Inher Metab Dis 1992; 15: 939-40. [ Links ]
19. Chloupkova M, Ravn K, Schwartz M, Kraus JP. Changes in the carboxyl terminus of the beta subunit of human propionyl-CoA carboxylase affect the oligomer assembly and catalysis: expression and characterization of seven patient-derived mutant forms of PCC in Escherichia coli. Mol Genet Metab 2000; 71: 623-32. [ Links ]
20. Ravn K, Chloupkova M, Christensen E, Jacob N, Simonense H et al. High incidence of propionic acidemia in Greenland is due to a prevalent mutation, 1540insCCC, in the gene for the ß-subunit of propionyl CoA carboxylase. Am J Hum Genet 2000; 67: 203-6. [ Links ]

21. Richards E, Desviat LR, Perez B, Perez-Cerda C, Ugarte M. Three novel splice mutations in the PCCA gene causing identical exon skipping in propionic acidemia patients. Hum Genet 1997; 101: 93-6. [ Links ]
22. Ohura T, Narisawa K, Tada K. Propionic Acidemia: sequence analysis of mutant mRNAs from japanese ß subunit-deficient patients. J Inher Metab Dis 1993; 16: 863-7. [ Links ]
23. Lamhonwah AM, Troxel CE, Scuster S, Grael RA. Two distinct mutations at the same site in the PCCB gene in propionic acidemia. Genomics 1990; 8: 249-54. [ Links ]
24. Rodriguez P, Hoenicka J, Muros, Perez B, Perez-Cerda C, Richard E, et al. Human propionyl -Co A carboxylase ß subunit gene: exon-intron definition and mutation spectrum in spanish and latin american propionic acidemia patients. Am J Hum Genet 1998; 63: 360-9. [ Links ]
25. Naylor EW, Chace DH. Automated tandem mass spectrometry for mass newborn screening for disorders in fatty acid, organic acid and amino acid metabolism. J Child Neurol 1999;14: S4-8. [ Links ]
26. Matsui SM, Mahoney MJ, Rosemberg LE. The natural history of the inherited methylmalonic acidemias. N Engl J Med 1983; 14: 857-61. [ Links ]
© 2010 Sociedad Médica de Santiago
Bernarda Morín 488, Providencia,
Casilla 168, Correo 55 Santiago - 9 - Chile
Teléfono: 56-2-7535520 Fono/Fax:56-2-7535524