
Transcriptome analysis methods for RNA-Seq...
39
Transcriptome analysis methods for RNA-Seq data Colin Dewey Biostatistics & Medical Informatics and Computer Sciences Bo Li Computer Sciences University of Wisconsin-Madison
Transcript of Transcriptome analysis methods for RNA-Seq...

Transcriptome analysismethods for RNA-Seq data
Colin DeweyBiostatistics & Medical Informatics and Computer
Sciences
Bo LiComputer Sciences
University of Wisconsin-Madison

RNA-Seq Revolution
“RNA-Seq [...] is expected torevolutionize the manner in whicheukaryotic transcriptomes are analysed”
-Z. Wang, M. Gerstein, M. Snyder(Nature Genetics 2009)

How do we analyze RNA-Seqdata?
• RNA-Seq protocol• Gene expression analysis methods• Novel transcript discovery methods• Future challenges for RNA-Seq
analysis
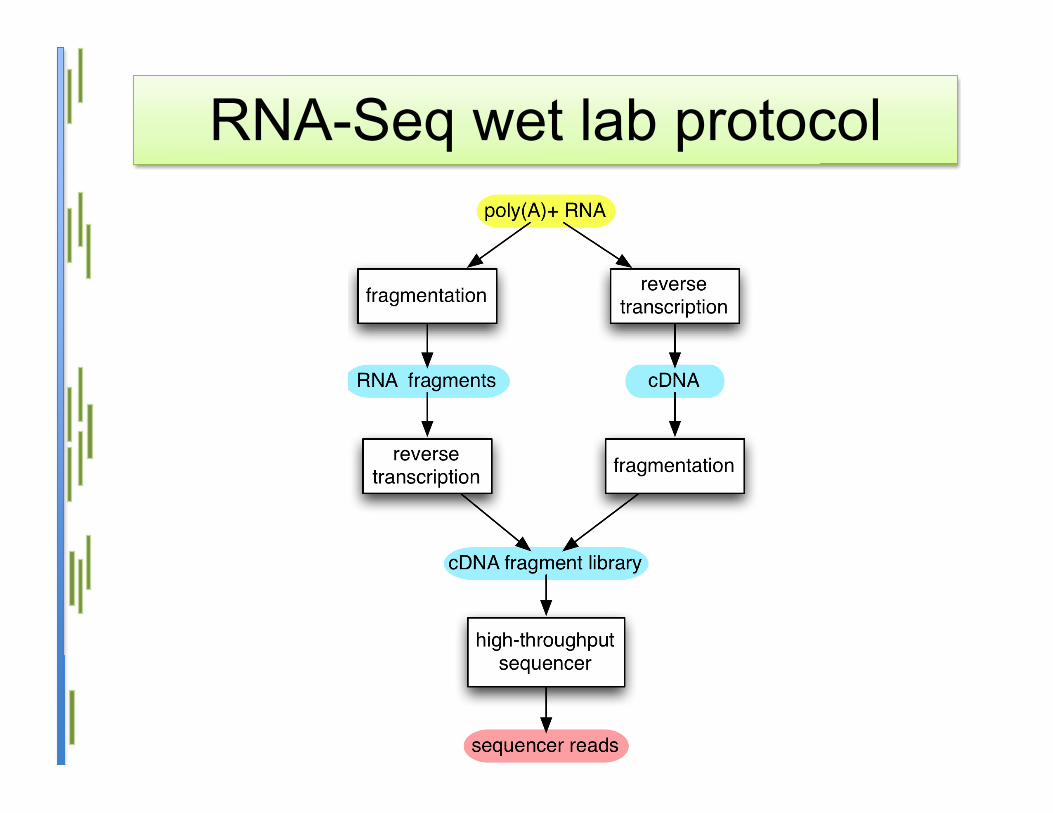
RNA-Seq wet lab protocol

RNA-Seq computationalprotocol

What has been done with RNA-Seq?
• Applied to transcriptome analysis in– Yeast (Nagalakshmi et al., 2008)– Human (Cloonan et al., Morin et al., Marioni et
al. 2008)– Mouse (Mortazavi et al., 2008)– Arabidopsis (Lister et al., 2008)– Butterfly (reference free!) (Vera et al., 2008)
• What has been discovered?– High accuracy and reproducibility– Low-level expressors– Novel splice junctions– Transcript chimeras
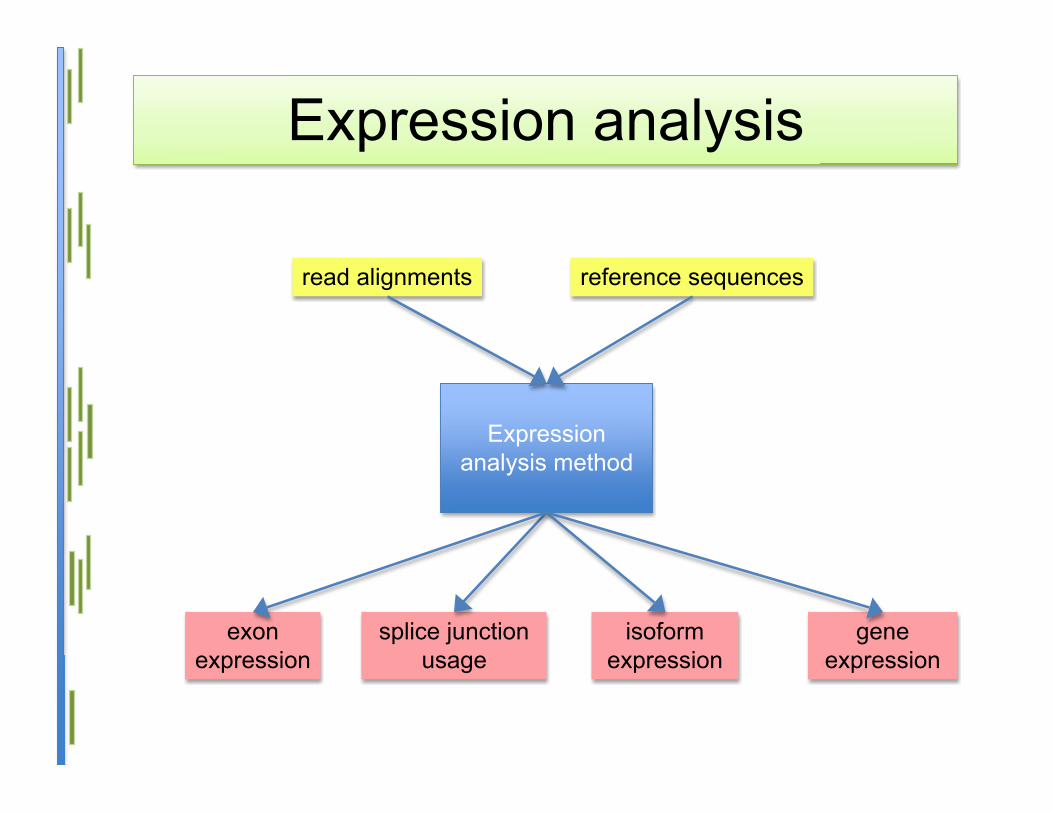
Expression analysis
Expressionanalysis method
read alignments reference sequences
geneexpression
splice junctionusage
isoformexpression
exonexpression

From counts to expressionlevels
• Primary assumption: reads are I.I.D.uniformly across the transcriptome
• Number of reads mapping to each gene measure of fraction of nucleotides intranscriptome made up by that gene
• Relative expression estimates

In symbols• ci: count of reads mapping to transcript i• νi: fraction of nucleotides in transcriptome
– νi × 106 is in units of “nucleotides per million”NPM
• N: number of (mapped) reads• ci / N is maximum likelihood estimator for νi
• Assuming reads can start at any positionalong a transcript– mostly true for a poly(A)+ sample

Normalizing for length• τi: fraction of transcripts in transcriptome
made up by transcript i– τi × 106 is in units of “transcripts per million”
TPM
• Allows comparison between expression ofdifferent transcripts

The RPKM measure• Introduced by Mortazavi et al. 2008 and used in
a number of studies• RPKM: Reads Per Kilobase per Million mapped
reads
• Normalization factor includes mean transcriptlength

Why TPM instead of RPKM?• TPM values are comparable across
experiments even when the meantranscript length changes
• Mean transcript length can change evenwhen the transcript fraction stays the same
• Examples:– τi is the same but νi differs across experiments– τi and νi both the same but li differs across
experiments• Warning: using νi or raw counts has same
problem

How do we get read counts?

The origins of multireads• Two classes
– Gene multireads• Paralogous genes• Low complexity or repetitive sequence
– Isoform multireads• Sharing of exonic sequence between isoforms
• Read mapping must allow for mismatches– Sequencing error– Polymorphism– Reference sequence error

How significant aremultireads?
Data set
%unmappe
d
%uniqu
e
%multiread
s
%filtere
dMouseliver
(Mortazaviet al.2008)
46.2 44.4 9.2 0.2
Maizesimulation 47.5 25.0 27.1 0.4
25 base reads, 2 mismatches allowed

Classes of expressionmethods
• Unique reads only• Mappability methods• Rescue methods• Statistical models• Isoform expression estimation

Unique reads only• Most straightforward approach• Used by some of the first RNA-Seq studies
– Nagalakshmi et al. 2008, Marioni et al. 2008,etc.
• Discard multireads• Advantages
– Fast!• Disadvantages:
– Throws away data– Underestimates expression of repetitive genes– Overestimates expression of relatively unique
genes

Mappability• Morin et al., 2008• Uses unique reads only• Adjusts read count for each exon by its
mappability
• Essentially fraction of positions in exon that giverise to uniquely mapping reads
• Advantages:– Fast, after preprocessing to determine
mappabilities– Corrects for repetitive sequence bias
• Disadvantages:– Also throws away data
1 if read starting at jis unique
Possible readsequencesoverlapping i

Rescue method 1: ERANGE• Mortazavi et al., 2008• Calculate initial expression levels from unique
reads• Allocate (fractions of) multireads based on using
initial expression levels
• Advantages:– Uses all data– Shown to improve correlation with microarray
• Disadvantages:– Gene-level expression only– Heuristic allocation of multireads
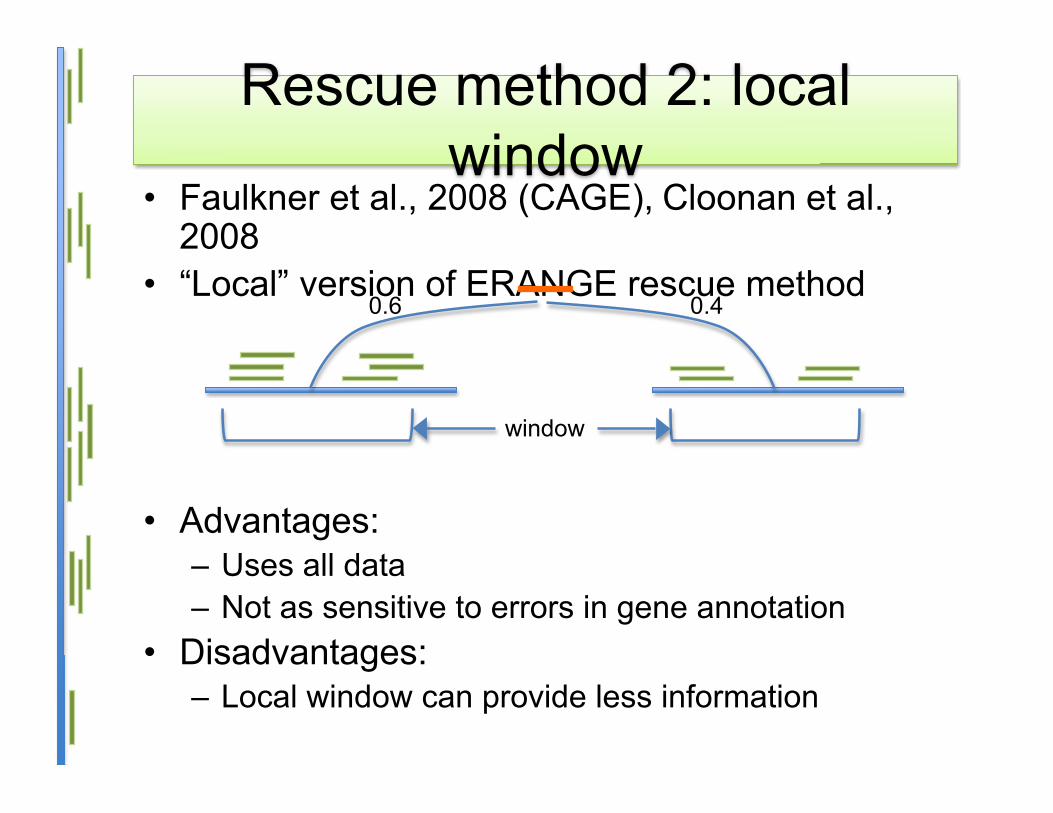
Rescue method 2: localwindow
• Faulkner et al., 2008 (CAGE), Cloonan et al.,2008
• “Local” version of ERANGE rescue method
• Advantages:– Uses all data– Not as sensitive to errors in gene annotation
• Disadvantages:– Local window can provide less information
0.6 0.4
window
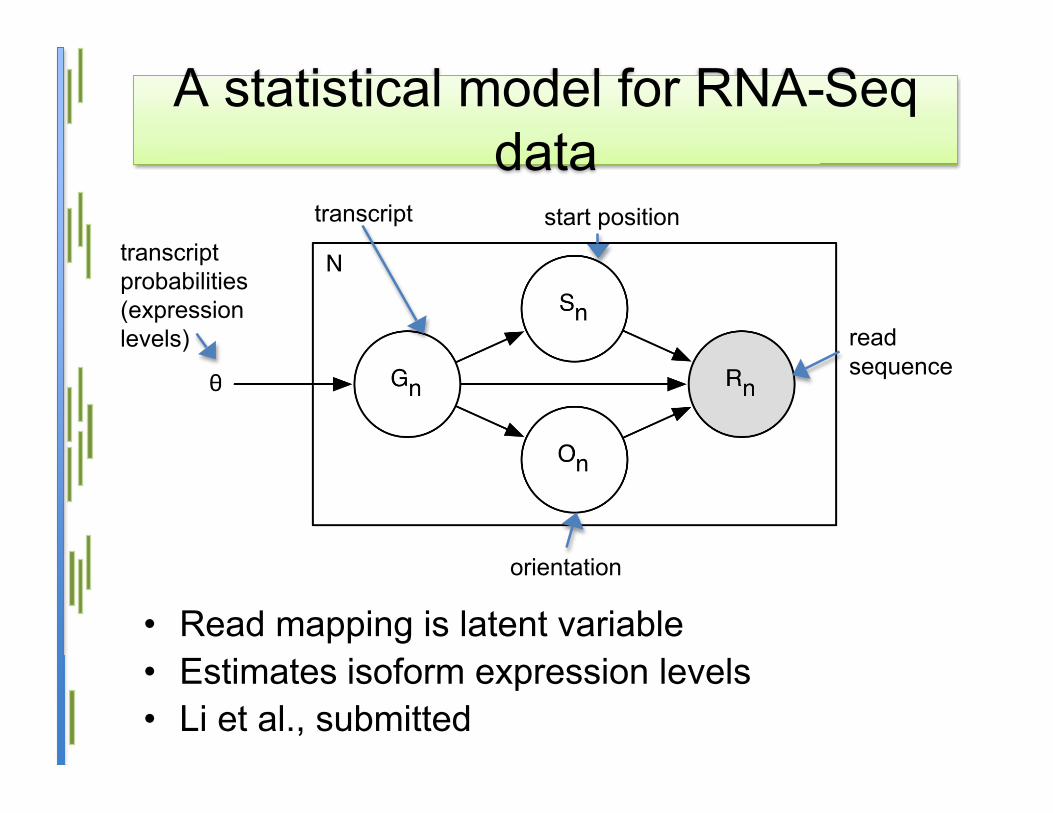
A statistical model for RNA-Seqdata
• Read mapping is latent variable• Estimates isoform expression levels• Li et al., submitted
start position
orientation
readsequence
transcripttranscriptprobabilities(expressionlevels)

EM Algorithm• Expectation-Maximization for RNA-Seq
– E-step: Compute expected read counts– M-step: Compute expression values
maximizing likelihood given expected readcounts
– Repeat E & M steps until convergence• E-step:
• Rescue methods are essentially oneiteration!
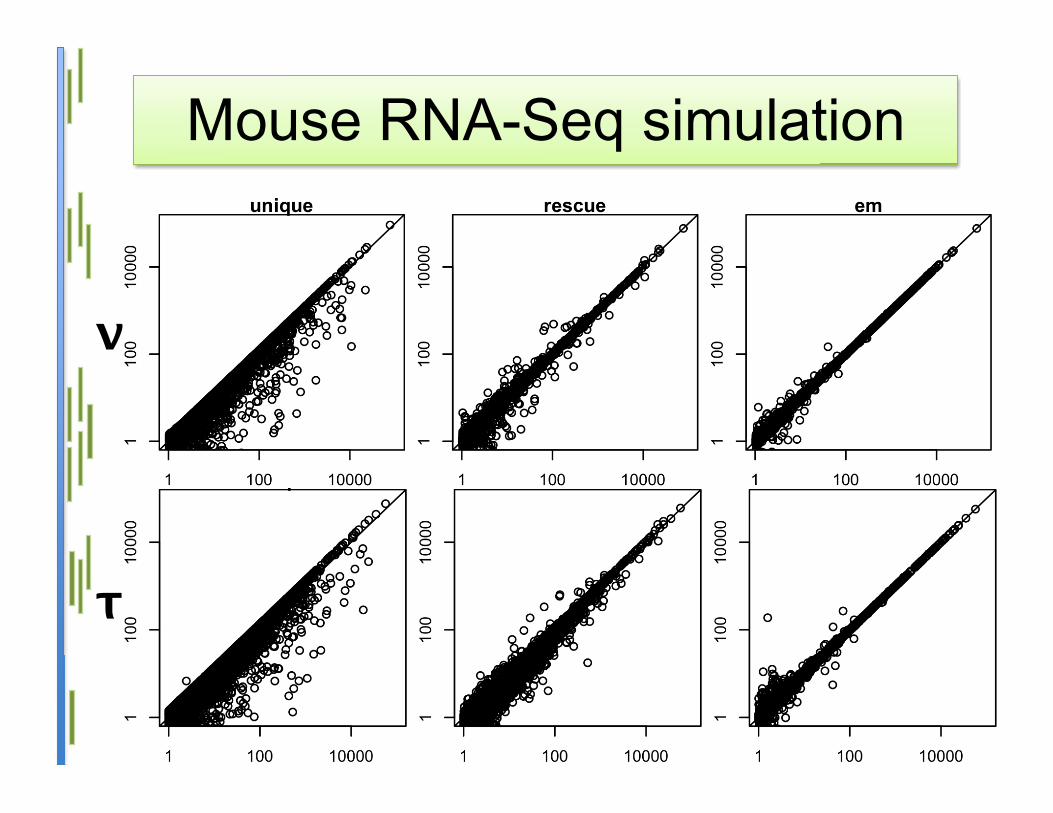
Mouse RNA-Seq simulation
τ
ν
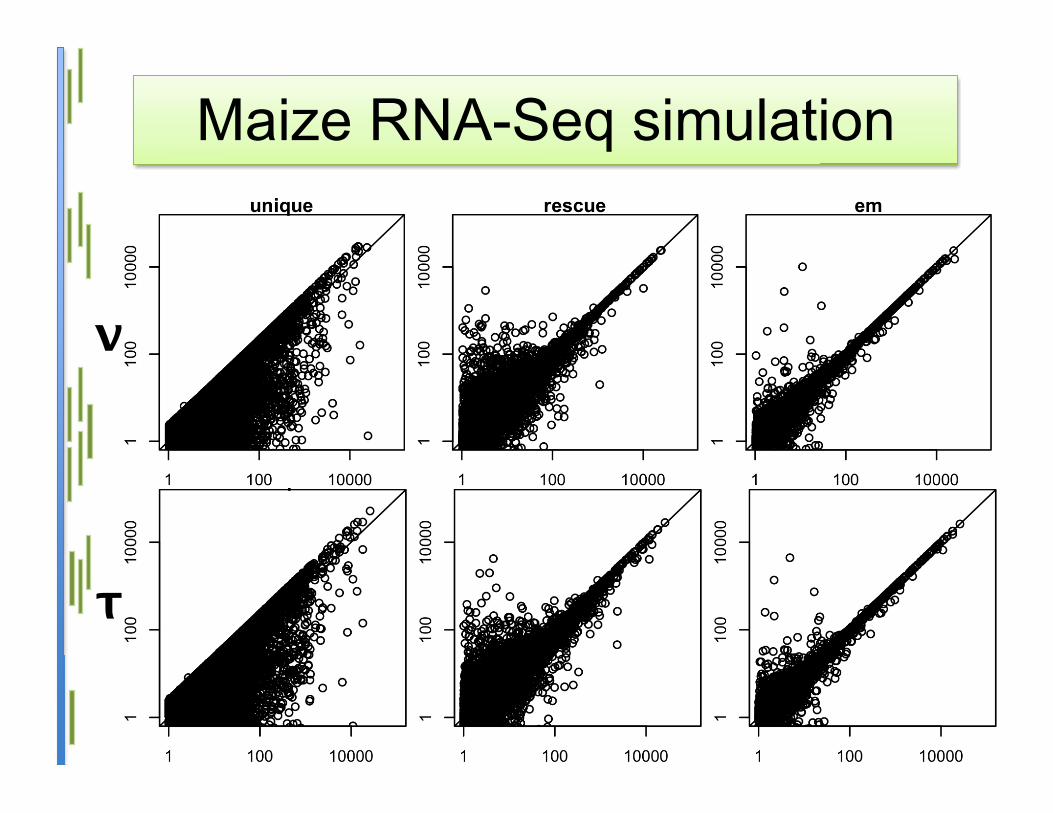
Maize RNA-Seq simulation
τ
ν
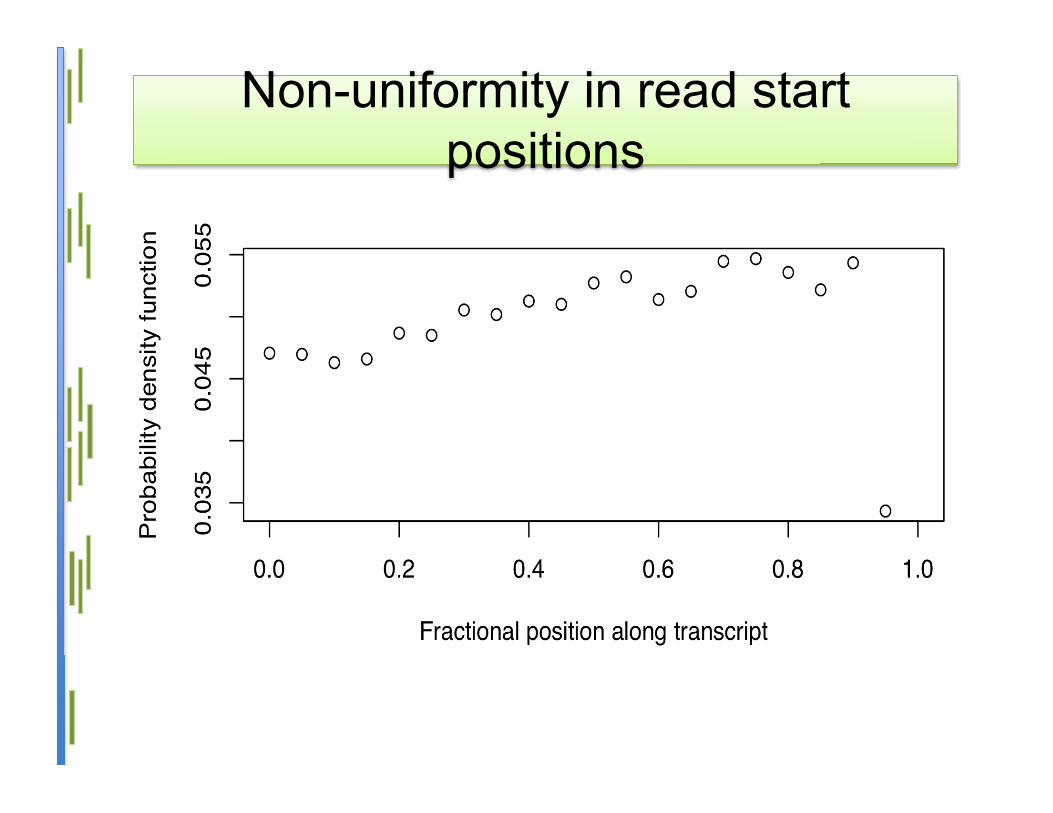
Non-uniformity in read startpositions

Statistical model• Advantages:
– Statistically-grounded method– Most accurate on simulated data– Isoform expression level estimation
• Disadvantages:– More computationally intensive (1-2 hours)– Requires accurate gene models

Isoform expression estimation• Jiang & Wong, 2009• Handles isoform multireads• Each “atomic” genomic interval generates
reads according to a Poisson distribution• ML estimates via coordinate-wise hill
climbing• Confidence intervals via importance
sampling• Advantages:
– Confidence intervals– Potentially faster
• Disadvantages:– Does not handle gene multireads– Requires accurate gene models
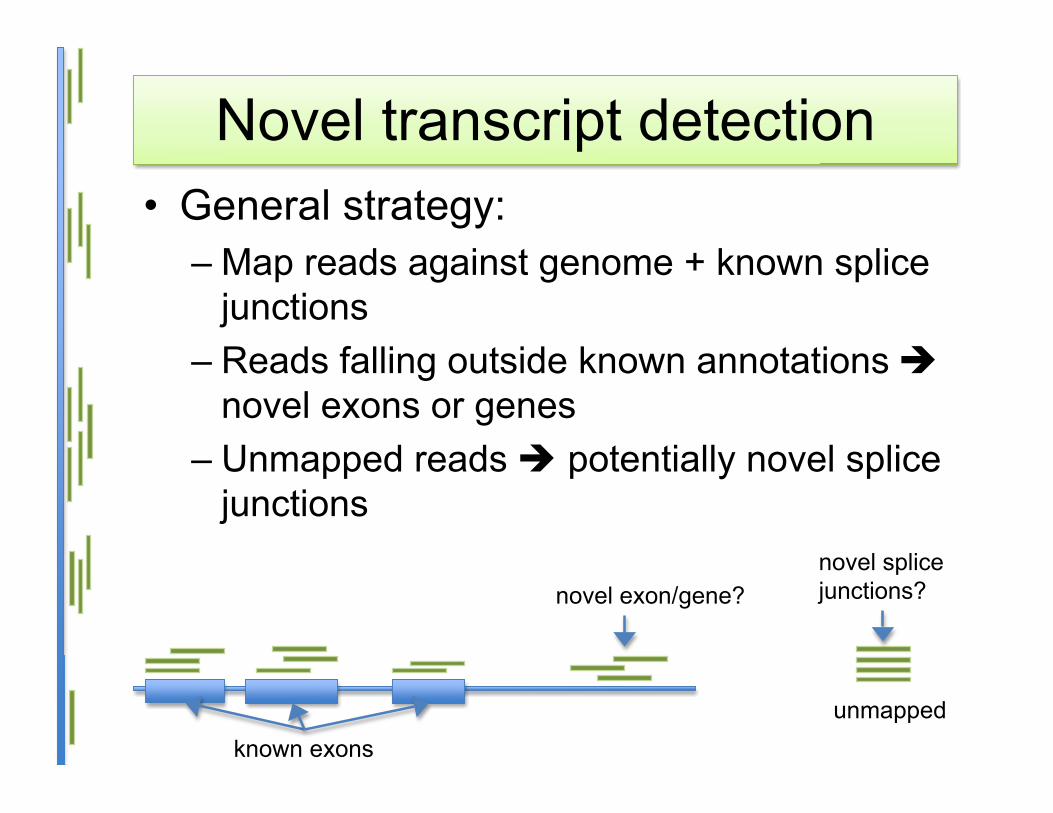
Novel transcript detection• General strategy:
– Map reads against genome + known splicejunctions
– Reads falling outside known annotations novel exons or genes
– Unmapped reads potentially novel splicejunctions
unmapped
novel exon/gene?novel splicejunctions?
known exons

QPALMA• De Bona et al., 2008• Halves of unmappable reads mapped to
genome• Alignments seed splice junction search• Splice-site-aware Smith-Waterman• Parameters learned via large margin
approach
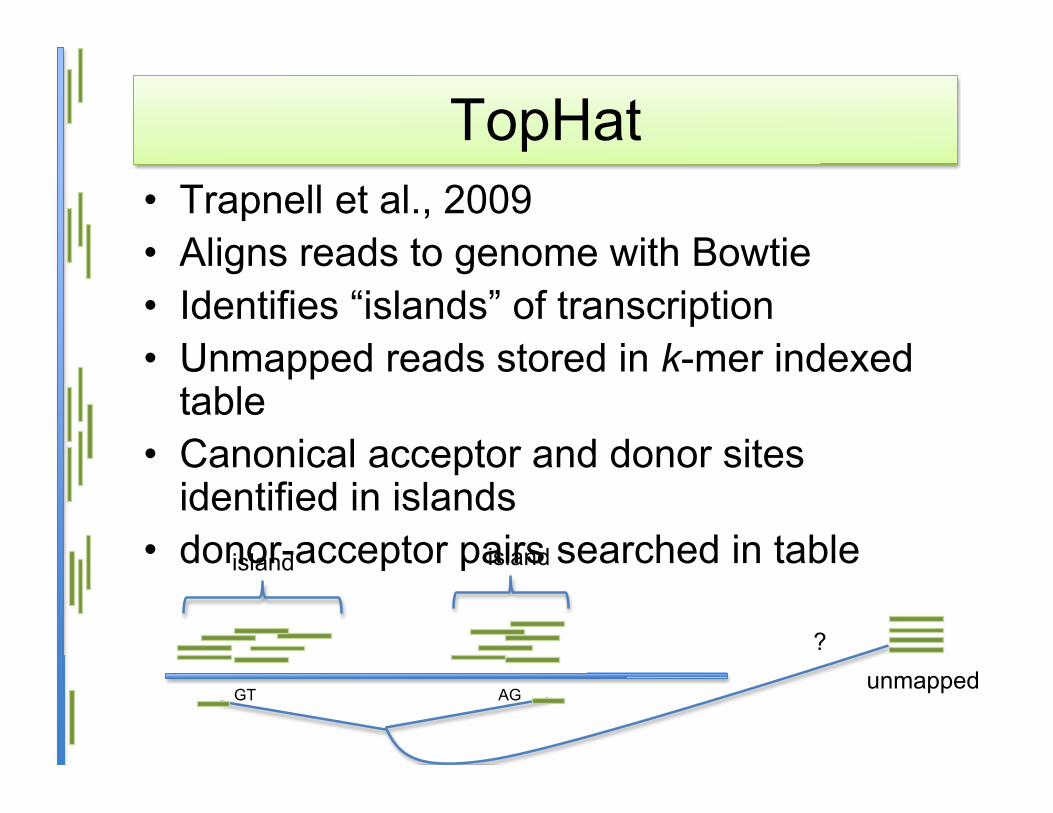
TopHat• Trapnell et al., 2009• Aligns reads to genome with Bowtie• Identifies “islands” of transcription• Unmapped reads stored in k-mer indexed
table• Canonical acceptor and donor sites
identified in islands• donor-acceptor pairs searched in table
unmappedGT AG
?
islandisland

Future challenges• Changing technology• Sequencing biases• Alternative splicing• Reference-free RNA-Seq

Moving technological target• Sequencing technology is rapidly
advancing– Read length– Number of reads
• Methods often designed with specifictechnologies in mind
• How much time should be invested inmethods for specific technologicalparameters?

Do multireads go away?• Not as quickly as you might think• For mouse:
– 25 base reads: 17% multireads– 75 base reads: 10% multireads– 75 base reads (paired-end): 8% multireads

Optimal read length
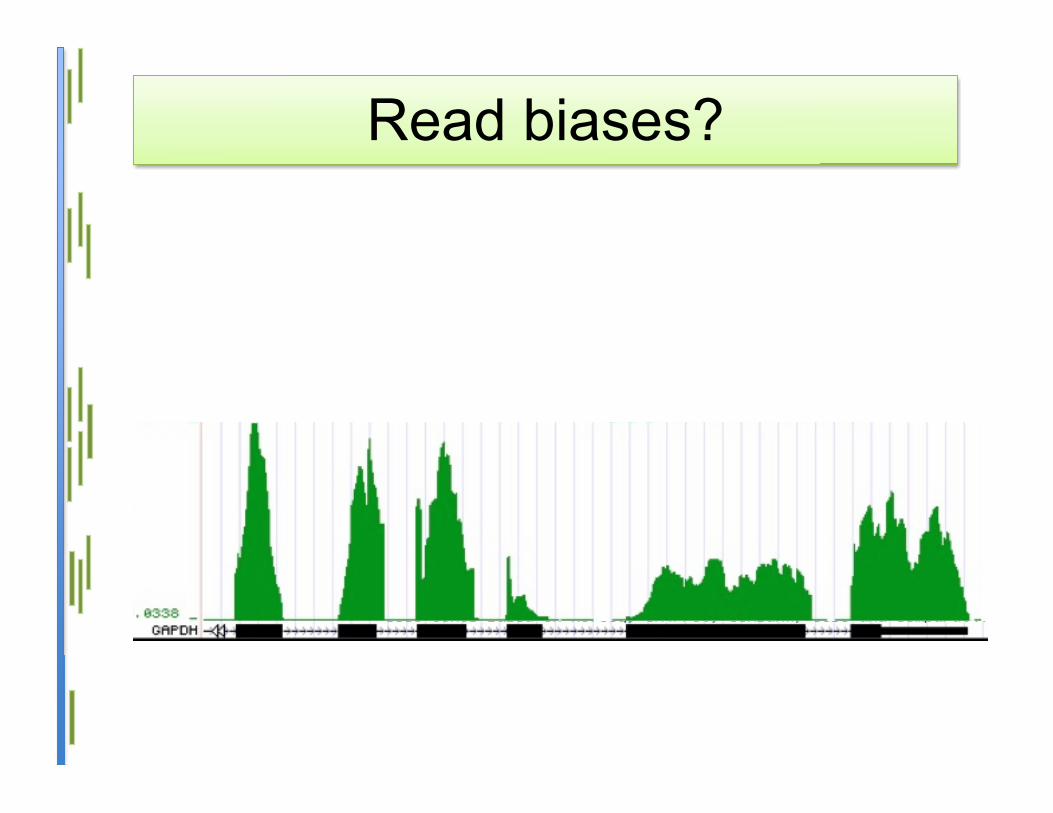
Read biases?

The mystery of the unmapped• RNA-Seq data sets often contain a large
fraction of unmapped reads– 40-50% in some cases
• Where do these come from?– Problems with the protocol?– Higher sequencer error than advertised?– Unknown splice junctions?– Chimeric transcripts?– Unknown biological phenomena?

Measuring alternative splicing
7 known isoforms18 possible given known splice junctions and start/stop sites
Few reads may be unique to any single isoform

Reference-free transcriptomics• Most methods rely heavily on a known
reference genome and/or transcript set• What can we do without a reference?
– Sequence assembly• Birol et al., 2009 (ABySS)• Complicated by alternative splicing
– Comparative approach• Collins et al., 2008 (Pachycladon enysii)• Large dependence on divergence between
species

Acknowledgments• Thomson Lab, UW-Madison
– Victor Ruotti– Ron Stewart– James Thomson
• Ali Mortazavi, Cal Tech
• CAMDA 2009 Organizers & Reviewers


















