
Next-generation transcriptome assembly · transcriptome by deep RNA sequencing (RNA-seq), even...
12
Identifying the full set of transcripts — including large and small RNAs, novel transcripts from unannotated genes, splicing isoforms and gene-fusion transcripts — serves as the foundation for a comprehensive study of the transcriptome. For a long time, our knowledge of the transcriptome was largely derived from gene predictions and limited EST evidence and has there- fore been partial and biased. Recently, however, whole- transcriptome sequencing using next-generation sequencing (NGS) technologies, or RNA sequencing (RNA-seq), has started to reveal the complex landscape and dynamics of the transcriptome from yeast to human at an unprecedented level of sensitivity and accuracy 1–4 . Compared with traditional low-throughput EST sequencing by Sanger technology, which only detects the more abundant transcripts, the enormous sequencing depth (100–1,000 reads per base pair of a transcript) of a typical RNA-seq experiment offers a near-complete snapshot of a transcriptome, including the rare tran- scripts that have regulatory roles. In contrast to alterna- tive high-throughput technologies, such as microarrays, RNA-seq achieves base-pair-level resolution and a much higher dynamic range of expression levels, and it is also capable of de novo annotation 1,2 . Despite these advantages, sequence reads obtained from the common NGS platforms, including Illumina, SOLiD and 454, are often very short (35–500 bp) 5 . As a result, it is neces- sary to reconstruct the full-length transcripts by tran- scriptome assembly, except in the case of small classes of RNA — such as microRNAs, PIWI-interacting RNAs (piRNAs), small nucleolar (snoRNAs) and small inter- fering (siRNAs) — which are shorter than the sequencing length and do not require assembly. Reconstructing a comprehensive transcriptome from short reads has many informatics challenges. Similar to short-read genome assembly, transcriptome assembly involves piecing together short, low-quality reads. Typical NGS data sets are very large (several gigabases to tera- bases), which requires computing systems to have large memories and/or many cores to run parallel algorithms. Several short-read assemblers have been developed to tackle these challenges 6–9 , including Velvet 6 , ABYSS 7 and ALLPATHS 8 . Although these tools have achieved reason- able success in the assembly of genomes 9,10 , they cannot directly be applied to transcriptome assembly, mainly because of three considerations. First, whereas DNA sequencing depth is expected to be the same across a genome, the sequencing depth of transcripts can vary by several orders of magnitude. Many short-read genome assemblers use sequencing depth to distinguish repetitive regions of the genome, a feature that would mark abun- dant transcripts as repetitive. Sequencing depth is also used by assemblers to calculate an optimal set of parame- ters for genome assembly, which would probably result in only a small set of transcripts being favoured in the tran- scriptome assembly. Second, unlike genomic sequencing, in which both strands are sequenced, RNA-seq experi- ments can be strand-specific. Transcriptome assemblers will need to take advantage of strand information to resolve overlapping sense and antisense transcripts 11–14 . Finally, transcriptome assembly is challenging, because transcript variants from the same gene can share exons and are difficult to resolve unambiguously. Given the complexity of most transcriptomes and the above chal- lenges, exclusively reconstructing all of the transcripts and their variants from short reads has been difficult. Lawrence Berkeley National Laboratory, DOE Joint Genome Institute, 2800 Mitchell Drive, MS100 Walnut Creek, California 94598, USA. e‑mails: [email protected]; [email protected] doi:10.1038/nrg3068 Published online 7 September 2011 RNA sequencing (RNA-seq). An experimental protocol that uses next- generation sequencing technologies to sequence the RNA molecules within a biological sample in an effort to determine the primary sequence and relative abundance of each RNA. Sequencing depth The average number of reads representing a given nucleotide in the reconstructed sequence. A 10× sequence depth means that each nucleotide of the transcript was sequenced, on average, ten times. Next-generation transcriptome assembly Jeffrey A. Martin and Zhong Wang Abstract | Transcriptomics studies often rely on partial reference transcriptomes that fail to capture the full catalogue of transcripts and their variations. Recent advances in sequencing technologies and assembly algorithms have facilitated the reconstruction of the entire transcriptome by deep RNA sequencing (RNA-seq), even without a reference genome. However, transcriptome assembly from billions of RNA-seq reads, which are often very short, poses a significant informatics challenge. This Review summarizes the recent developments in transcriptome assembly approaches — reference-based, de novo and combined strategies — along with some perspectives on transcriptome assembly in the near future. STUDY DESIGNS REVIEWS NATURE REVIEWS | GENETICS VOLUME 12 | OCTOBER 2011 | 671 © 2011 Macmillan Publishers Limited. All rights reserved
Transcript of Next-generation transcriptome assembly · transcriptome by deep RNA sequencing (RNA-seq), even...

Identifying the full set of transcripts — including large and small RNAs, novel transcripts from unannotated genes, splicing isoforms and gene-fusion transcripts — serves as the foundation for a comprehensive study of the transcriptome. For a long time, our knowledge of the transcriptome was largely derived from gene predictions and limited EST evidence and has there-fore been partial and biased. Recently, however, whole- transcriptome sequencing using next-generation sequencing (NGS) technologies, or RNA sequencing (RNA-seq), has started to reveal the complex landscape and dynamics of the transcriptome from yeast to human at an unprecedented level of sensitivity and accuracy1–4. Compared with traditional low-throughput EST sequencing by Sanger technology, which only detects the more abundant transcripts, the enormous sequencing depth (100–1,000 reads per base pair of a transcript) of a typical RNA-seq experiment offers a near-complete snapshot of a transcriptome, including the rare tran-scripts that have regulatory roles. In contrast to alterna-tive high-throughput technologies, such as microarrays, RNA-seq achieves base-pair-level resolution and a much higher dynamic range of expression levels, and it is also capable of de novo annotation1,2. Despite these advantages, sequence reads obtained from the common NGS platforms, including Illumina, SOLiD and 454, are often very short (35–500 bp)5. As a result, it is neces-sary to reconstruct the full-length transcripts by tran-scriptome assembly, except in the case of small classes of RNA — such as microRNAs, PIWI-interacting RNAs (piRNAs), small nucleolar (snoRNAs) and small inter-fering (siRNAs) — which are shorter than the sequencing length and do not require assembly.
Reconstructing a comprehensive transcriptome from short reads has many informatics challenges. Similar to short-read genome assembly, transcriptome assembly involves piecing together short, low-quality reads. Typical NGS data sets are very large (several gigabases to tera-bases), which requires computing systems to have large memories and/or many cores to run parallel algorithms. Several short-read assemblers have been developed to tackle these challenges6–9, including Velvet6, ABYSS7 and ALLPATHS8. Although these tools have achieved reason-able success in the assembly of genomes9,10, they cannot directly be applied to transcriptome assembly, mainly because of three considerations. First, whereas DNA sequencing depth is expected to be the same across a genome, the sequencing depth of transcripts can vary by several orders of magnitude. Many short-read genome assemblers use sequencing depth to distinguish repetitive regions of the genome, a feature that would mark abun-dant transcripts as repetitive. Sequencing depth is also used by assemblers to calculate an optimal set of parame-ters for genome assembly, which would probably result in only a small set of transcripts being favoured in the tran-scriptome assembly. Second, unlike genomic sequencing, in which both strands are sequenced, RNA-seq experi-ments can be strand-specific. Transcriptome assemblers will need to take advantage of strand information to resolve overlapping sense and antisense transcripts11–14. Finally, transcriptome assembly is challenging, because transcript variants from the same gene can share exons and are difficult to resolve unambiguously. Given the complexity of most transcriptomes and the above chal-lenges, exclusively reconstructing all of the transcripts and their variants from short reads has been difficult.
Lawrence Berkeley National Laboratory, DOE Joint Genome Institute, 2800 Mitchell Drive, MS100 Walnut Creek, California 94598, USA.e‑mails: [email protected]; [email protected]:10.1038/nrg3068Published online 7 September 2011
RNA sequencing(RNA-seq). An experimental protocol that uses next- generation sequencing technologies to sequence the RNA molecules within a biological sample in an effort to determine the primary sequence and relative abundance of each RNA.
Sequencing depthThe average number of reads representing a given nucleotide in the reconstructed sequence. A 10× sequence depth means that each nucleotide of the transcript was sequenced, on average, ten times.
Next-generation transcriptome assemblyJeffrey A. Martin and Zhong Wang
Abstract | Transcriptomics studies often rely on partial reference transcriptomes that fail to capture the full catalogue of transcripts and their variations. Recent advances in sequencing technologies and assembly algorithms have facilitated the reconstruction of the entire transcriptome by deep RNA sequencing (RNA-seq), even without a reference genome. However, transcriptome assembly from billions of RNA-seq reads, which are often very short, poses a significant informatics challenge. This Review summarizes the recent developments in transcriptome assembly approaches — reference-based, de novo and combined strategies — along with some perspectives on transcriptome assembly in the near future.
S T U DY D E S I G N S
REVIEWS
NATURE REVIEWS | GENETICS VOLUME 12 | OCTOBER 2011 | 671
© 2011 Macmillan Publishers Limited. All rights reserved

Paired-end protocolA library construction and sequencing strategy in which both ends of a DNA fragment are sequenced to produce pairs of reads (mate pairs).
ContigsAn abbreviation for contiguous sequences that is used to indicate a contiguous piece of DNA assembled from shorter overlapping sequence reads.
In the past 3 years, improvements in data quality and the rapid evolution of assembly algorithms have made it possible to address the above challenges. In this Review, we show how these exciting advances have led to a wealth of assembled transcriptomes from short reads15–25, and we provide practical guidelines for imple-menting a transcriptome assembly experiment. After describing the experimental and informatics considera-tions that need to be made before assembly, we discuss three assembly strategies: assembly based on a reference genome, de novo assembly and a combined approach that merges the two strategies. We focus on the strengths and weaknesses of the three strategies in the context of both gene-dense transcriptomes and large transcrip-tomes with pervasive alternative splicing. Finally, we give some perspectives on the future of transcriptome assembly in light of the rapid evolution of sequencing technologies and high-performance computing.
Considerations prior to assemblyTo ensure a high-quality transcriptome assembly, par-ticular care should be taken in designing the RNA-seq experiment. The steps of a typical transcriptome assem-bly experiment are shown in FIG. 1. In the data generation phase (FIG. 1a), total RNAs or mRNAs are fragmented and converted into a library of cDNAs containing sequencing adaptors. The cDNA library is then sequenced by next-generation sequencers to produce millions to billions of short reads from one end or both ends of the cDNA fragments. In the data analysis phase (FIG. 1b), these short reads are pre-processed to remove sequencing errors and other artefacts. The reads are subsequently assembled to reconstruct the original RNAs and to assess their abun-dance (‘expression counting’). Expression counting is not trivial for transcriptomes with extensive alternative splicing26: transcripts often share some exons, causing uncertainty as to which transcript each read belongs to. The accuracy and precision of gene expression counting are influenced by cDNA library construction methods, sequencing technologies and data pre-treatment tech-niques27. Similarly, these factors can influence the quality of assembled transcriptomes, as discussed below.
Library construction. To increase the number of assem-bled transcripts, especially the less abundant ones, riboso-mal RNA (rRNA) and abundant transcripts are removed during the first steps of library construction. Poly(A) selection is very effective at enriching mRNAs in eukary-otes, but this selection approach will miss non-coding RNAs (ncRNAs) and mRNAs that lack a poly(A) tail. In order to retain RNAs without a poly(A) tail in the assem-bled transcriptome, rRNA contamination can instead be removed by hybridization-based depletion methods28,29. These methods increase the opportunity for the detec-tion and assembly of rare transcripts by reducing the representation of rRNAs and other highly abundant tran-scripts30, which often constitute most of the reads in RNA-seq data sets. Note that these depletion methods may bias the quantification of highly abundant transcripts, and so if quantification is a goal of the study, then the sequencing of non-depleted libraries will be required.
Another consideration to make during library con-struction, provided the starting RNA quantities are not limiting, is whether to eliminate the PCR amplifica-tion step from the protocol. PCR amplification results in a low sequencing coverage for transcripts or regions within a transcript that have a high GC content31. This can, in turn, cause gaps in the assembled transcripts and can cause other transcripts to be missing from the assembly altogether. Amplification-free protocols have been developed to overcome this problem31,32. The latest single-molecule sequencing technologies from Helicos and Pacific Biosciences do not require PCR amplifica-tion before sequencing33. The Helicos system can even directly sequence RNAs without cDNA library construc-tion1,34, which should greatly reduce biases in sequencing coverage. However, these single-molecule technologies suffer from high error rates. Overall, sequencing cover-age of the transcriptome from amplification-free pro-tocols is more even and contiguous across transcripts, making it easier for assemblers to construct full-length transcripts across GC-rich regions of the transcriptome.
Last, the use of strand-specific RNA-seq protocols27 aids in the assembly and quantification of overlapping transcripts that are derived from opposite strands of the genome. This consideration is especially important for gene-dense genomes, such as those of bacteria, archaea and lower eukaryotes, but it is also important for detect-ing antisense transcription, which is common in higher eukaryotes.
Sequencing. The major factors to consider before sequencing a sample are: the choice of sequencing plat-form, the sequencing read length and whether to use a paired-end protocol. All of the current NGS technolo-gies have successfully been used to assemble transcrip-tomes35–37, and they differ mostly in their throughput and cost.
The choice of sequencing technology largely depends on the technology to which a user has access and the budget constraints for sequencing. In general, the assembly of large and complex transcriptomes (plants and mammals) requires extensive sequencing and is fre-quently done on Illumina or SOLiD platforms. The 454 technology offers longer reads, and it can be used alone for small transcriptomes. Illumina, SOLiD and 454 technol-ogy can also be combined in a ‘hybrid assembly’ strategy: short reads that are sequenced at a greater depth are assembled into contigs, and long reads are subsequently used to scaffold the contigs and resolve variants38,39.
For transcriptome assembly, longer reads are gener-ally preferred, as they greatly reduce the complexity of the assembly. It is worth noting, however, that the prob-lem posed by short reads can be alleviated by using a paired-end protocol, in which 75–150 bp are sequenced from both ends of short DNA fragments (100–250 bp), and the overlapping reads are computationally joined together to form a longer read40. Paired reads from long inserts (500–1,000 bp) offer long-range exon con-nectivity, in a similar way to reads obtained using 454 technology. For this reason, some assemblers, such as ALLPATHS, require at least two libraries with different
R E V I E W S
672 | OCTOBER 2011 | VOLUME 12 www.nature.com/reviews/genetics
© 2011 Macmillan Publishers Limited. All rights reserved
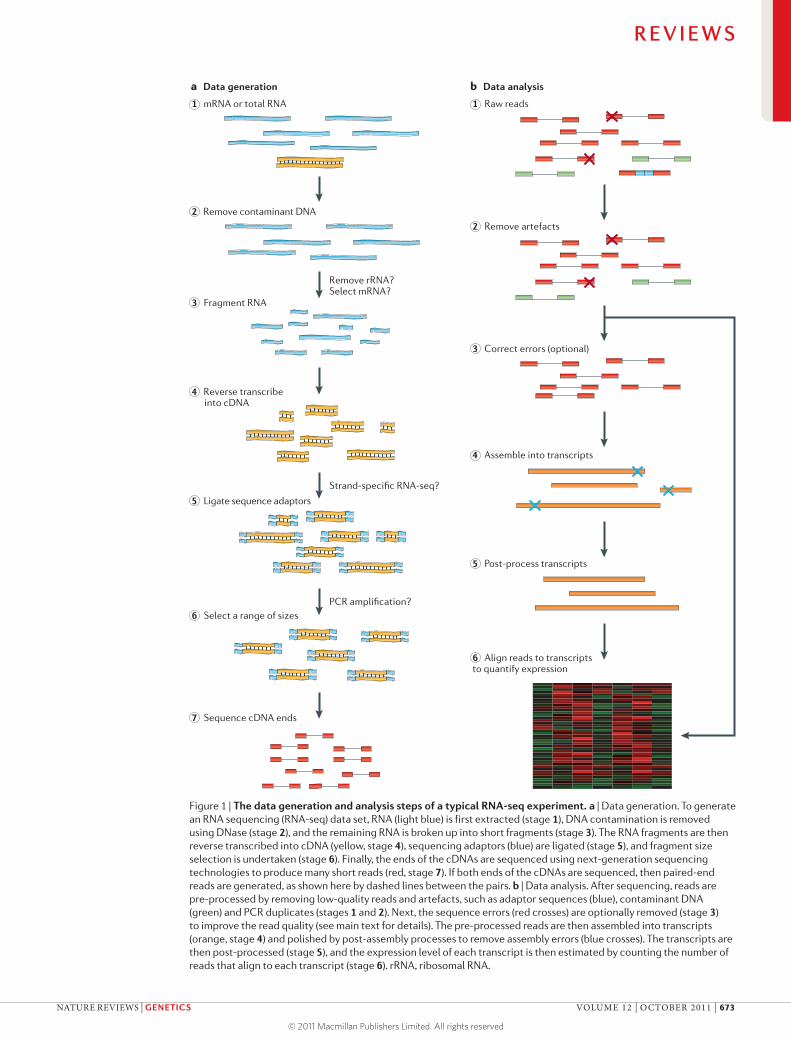
Figure 1 | The data generation and analysis steps of a typical RNA-seq experiment. a | Data generation. To generate an RNA sequencing (RNA-seq) data set, RNA (light blue) is first extracted (stage 1), DNA contamination is removed using DNase (stage 2), and the remaining RNA is broken up into short fragments (stage 3). The RNA fragments are then reverse transcribed into cDNA (yellow, stage 4), sequencing adaptors (blue) are ligated (stage 5), and fragment size selection is undertaken (stage 6). Finally, the ends of the cDNAs are sequenced using next-generation sequencing technologies to produce many short reads (red, stage 7). If both ends of the cDNAs are sequenced, then paired-end reads are generated, as shown here by dashed lines between the pairs. b | Data analysis. After sequencing, reads are pre-processed by removing low-quality reads and artefacts, such as adaptor sequences (blue), contaminant DNA (green) and PCR duplicates (stages 1 and 2). Next, the sequence errors (red crosses) are optionally removed (stage 3) to improve the read quality (see main text for details). The pre-processed reads are then assembled into transcripts (orange, stage 4) and polished by post-assembly processes to remove assembly errors (blue crosses). The transcripts are then post-processed (stage 5), and the expression level of each transcript is then estimated by counting the number of reads that align to each transcript (stage 6). rRNA, ribosomal RNA.
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 12 | OCTOBER 2011 | 673
© 2011 Macmillan Publishers Limited. All rights reserved

Low-complexity readsShort DNA sequences composed of stretches of homopolymer nucleotides or simple sequence repeats.
Quality scoresAn integer representing the probability that a given base in a nucleic acid sequence is correct.
k-mer frequencyThe number of times that each k-mer (that is, a short oligonucleotide of length k) appears in a set of DNA sequences.
Splice-aware alignerA program that is designed to align cDNA reads to a genome.
TraversingA method for systematically visiting all nodes in a mathematical graph.
Seed-and-extend alignersAn alignment strategy that first builds a hash table containing the location of each k-mer (seed) within the reference genome. These algorithms then extend these seeds in both directions to find the best alignment (or alignments) for each read.
Burrows–Wheeler transform(BWT). This reorders the characters within a sequence, which allows for better data compression. Many short-read aligners implement this transform in order to use less memory when aligning reads to a genome.
Parallel computingA computer programming model for distributing data processing across multiple processors, so that multiple tasks can be carried out simultaneously.
insert sizes8. The combination of short and long insert libraries should be helpful in capturing transcripts of various sizes while also helping to resolve alternatively spliced isoforms.
Data pre-processing. Removing artefacts from RNA-seq data sets before assembly improves the read quality, which, in turn, improves the accuracy and computa-tional efficiency of the assembly. This step is straight-forward and can be executed using several tools41–44. In general, three types of artefacts should be removed from raw RNA-seq data: sequencing adaptors43,44, which origi-nate from failed or short DNA insertions during library preparation; low-complexity reads43; and near-identical reads that are derived from PCR amplification15. Adaptor and low-complexity sequences can lead to misassem-blies. PCR duplicates are more common in long-insert libraries, and their presence can skew mate-pair sta-tistics that are used by many assemblers for scaffold-ing. When their identities are known, rRNA and other RNA contaminants should also be removed to improve assembly speed.
Sequencing errors in NGS reads can be removed or corrected by analysing the quality score and/or the k-mer frequency. For most NGS data sets, low quality scores indicate possible sequencing errors. Sequencing errors can also be empirically inferred by looking at the fre-quencies of each k-mer in the data set. As the same RNA molecule is sequenced many times, k-mers without errors in them will occur multiple times. By contrast, k-mers that occur in the data set at very low frequencies are probably sequencing errors or are from transcripts with a low abundance. Reads containing these errors can be removed, trimmed or corrected to improve the assembly quality and to decrease the amount of random access memory (RAM) required10,15,42. However, k-mer-based error removal carries a side effect, in that reads derived from rare transcripts may also be removed. This should not be a large problem, as the shallow sequenc-ing depth for these transcripts would not be sufficient to assemble them, even if these reads were retained.
Transcriptome assembly strategiesDepending on whether a reference genome assembly is available, current transcriptome assembly strategies gen-erally fall into one of three categories: a reference-based strategy, a de novo strategy or a combined strategy that merges the two (FIGS 2–4). In the following sections, we discuss each of these three strategies in detail, includ-ing their pros and cons for the assembly of simple and complex transcriptomes.
Reference-based strategyWhen a reference genome for the target transcriptome is available, the transcriptome assembly can be built upon it. In general, this strategy — which is known as ‘reference-based’ or ‘ab initio’ assembly — involves three steps. First, RNA-seq reads are aligned to a refer-ence genome using a splice-aware aligner, such as Blat45, TopHat46, SpliceMap47, MapSplice48 or GSNAP49 (TABLE 1; FIG. 2a). Second, overlapping reads from each locus are
clustered to build a graph representing all possible iso-forms (FIG. 2b). The final step involves traversing the graph to resolve individual isoforms (FIG. 2c,d). Examples of methods that use the reference-based strategy include Cufflinks20, Scripture16 and others17,50 (TABLE 2).
Splice-aware aligners generally fall into two classes: seed-and-extend aligners and Burrows–Wheeler transform (BWT) aligners, each of which has clear trade-offs. The seed-and-extend algorithms, such as BLAT and GSNAP, start by quickly finding a ‘seed’ — a substring of the read — that exactly matches the genome and then locally extending the match using Smith–Waterman alignment algorithms. BWT aligners are optimized to align reads with few errors in them and are therefore generally faster than seed-and-extend aligners. Each aligner differs in its implementation for aligning reads across introns. In gen-eral, seed-and-extend aligners shift the gaps in the local alignment to match known splice sites, whereas BWT aligners, such as TopHat, create a database of all possible combinations of splicing junctions within a locus and then align to this database the reads that failed to align to the genome.
After the reads are aligned to the genome, two meth-ods are typically used for graph construction and tra-versal16,20. Cufflinks20 creates an overlap graph from all of the reads that align to a single locus and then traverses this graph to assemble isoforms by finding the minimum set of transcripts that ‘explain’ the intron junctions within the reads (that is, a minimum path cover of the graph). Scripture16, by contrast, constructs a splice graph contain-ing each base of a chromosome and adds edges (connec-tions) between bases if there is a read that joins the two bases. Scripture then finds all paths through the graph that have a statistically significant read coverage. These differences in graph construction and traversal methods suggest that Cufflinks is more conservative in its choice of which transcripts to re-construct, whereas Scripture may produce a larger set of transcripts from a locus.
Advantages. The reference-based transcriptome assembly strategy has several advantages. Because this approach transforms a large assembly problem (mil-lions of reads) into many smaller assembly problems (for example, independent assemblies of each locus that contain thousands of reads or less), assembly can be solved using parallel computing and can run effi-ciently on machines with only a few gigabytes of RAM. Contamination or sequencing artefacts are not a major concern for this strategy, because they are not expected to align to the reference genome. More importantly, the reference-based strategy is very sensitive and can assem-ble transcripts of low abundance. Because the underlying genome sequence is already known20, small gaps within the transcript that have been caused by a lack of read coverage can be filled in using the reference sequence17. Similarly, this strategy tends to generate longer UTRs, which usually have a lower sequencing coverage16. Owing to the high sensitivity of this approach, it allows users to discover novel transcripts that are not present in the current annotation, as in general such transcripts have lower expression levels.
R E V I E W S
674 | OCTOBER 2011 | VOLUME 12 www.nature.com/reviews/genetics
© 2011 Macmillan Publishers Limited. All rights reserved
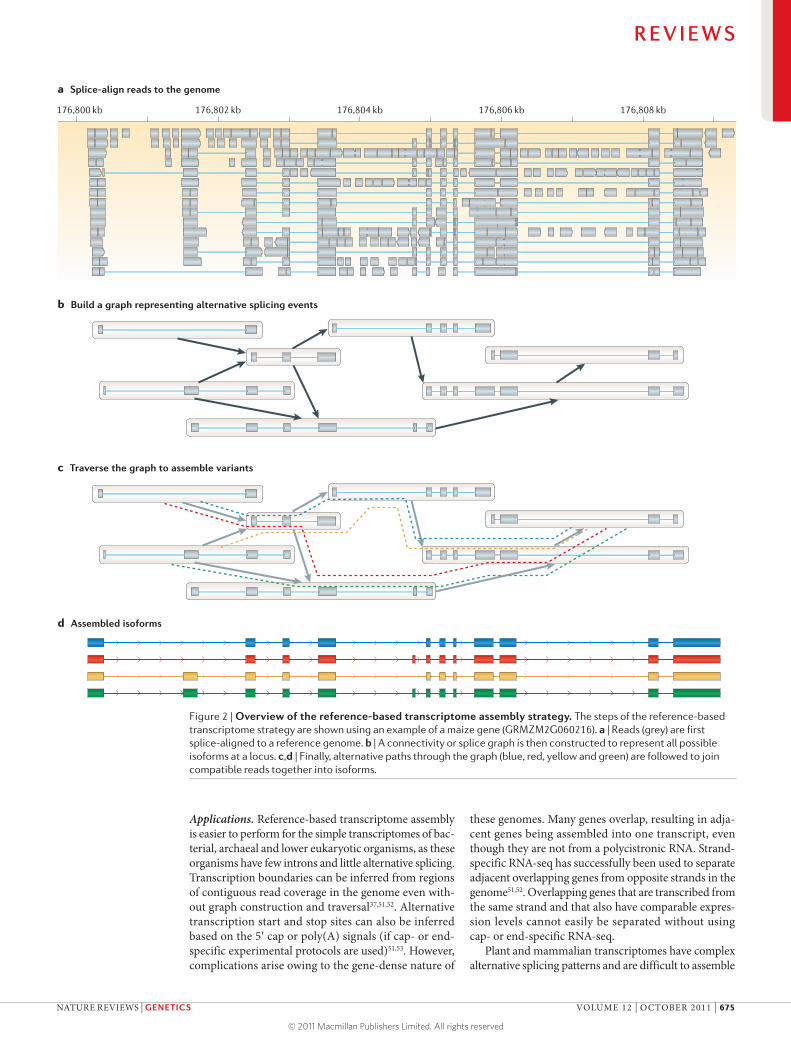
Figure 2 | Overview of the reference-based transcriptome assembly strategy. The steps of the reference-based transcriptome strategy are shown using an example of a maize gene (GRMZM2G060216). a | Reads (grey) are first splice-aligned to a reference genome. b | A connectivity or splice graph is then constructed to represent all possible isoforms at a locus. c,d | Finally, alternative paths through the graph (blue, red, yellow and green) are followed to join compatible reads together into isoforms.
Applications. Reference-based transcriptome assembly is easier to perform for the simple transcriptomes of bac-terial, archaeal and lower eukaryotic organisms, as these organisms have few introns and little alternative splicing. Transcription boundaries can be inferred from regions of contiguous read coverage in the genome even with-out graph construction and traversal37,51,52. Alternative transcription start and stop sites can also be inferred based on the 5′ cap or poly(A) signals (if cap- or end-specific experimental protocols are used)51,53. However, complications arise owing to the gene-dense nature of
these genomes. Many genes overlap, resulting in adja-cent genes being assembled into one transcript, even though they are not from a polycistronic RNA. Strand-specific RNA-seq has successfully been used to separate adjacent overlapping genes from opposite strands in the genome51,52. Overlapping genes that are transcribed from the same strand and that also have comparable expres-sion levels cannot easily be separated without using cap- or end-specific RNA-seq.
Plant and mammalian transcriptomes have complex alternative splicing patterns and are difficult to assemble
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 12 | OCTOBER 2011 | 675
© 2011 Macmillan Publishers Limited. All rights reserved
ACCGC CCGCC CGCCC
GCCCA CCCAC
CTTCC TTCCT
TCCTG CCTGC CTGCT TGCTG CTGGT TGGTC GGTCT GTCTC TCTCT
TCCTC CCTCT
GCCCACAGC
GCCCTCAGC
CAGCGCTTCCT
TCCTGCTGGTCTCT
CTCTTGTTGGTCGTAG
ACCGCCCACAGCGCTTCCTGCTGGTCTCTTGTTG
ACCGCCC
ACCGC
CCGCC
CGCCC
CCCAC
CCACA
CACAG
GCCCA
ACAGC
CAGCG
AGCGC
GCGCT
CGCTT
GCTTC
CTTCC
TTCCT
TCCTG
CCTGC
CTGCT
TGCTG
GCTGG
CTGGT
TGGTC
GGTCT
GTCTC
TCTCT
CTCTT
TCTTG
CTTGT
CGCCCTCAGCGCTTCCTCTTGTTGGTCGTAG
TCCTCT
TTGTT
TGTTG
CGCCC
GCCCT
CCCTC
CCTCA
CTCAG
TCAGC
CAGCG
AGCGC
GCGCT
CGCTT
GCTTC
CTTCC
TTCCT
TCCTC
CCTCT
CTCTT
TCTTG
CTTGT
TTGTT
TGTTG
GTTGG
TTGGT
TGGTC
GGTCG
GTCGT
TCGTA
CGTAG
CCACA CACAG ACAGC
GCTGG
GCCCT CCCTC CCTCA CTCAG TCAGC
CAGCG AGCGC GCGCT CGCTT GCTTC
CTCTT TCTTG CTTGT TTGTT TGTTG GTTGG TTGGT TGGTC GGTCG GTCGT TCGTA CGTAG
GCCCACAGC
GCCCTCAGC
CAGCGCTTCCT
TCCTGCTGGTCTCT
CTCTTGTTGGTCGTAGACCGCCC
TCCTCT
ACCGCCCACAGCGCTTCCT--------CTTGTTGGTCGTAGACCGCCCACAGCGCTTCCTGCTGGTCTCTTGTTGGTCGTAG
ACCGCCCTCAGCGCTTCCTGCTGGTCTCTTGTTGGTCGTAGACCGCCCTCAGCGCTTCCT--------CTTGTTGGTCGTAG
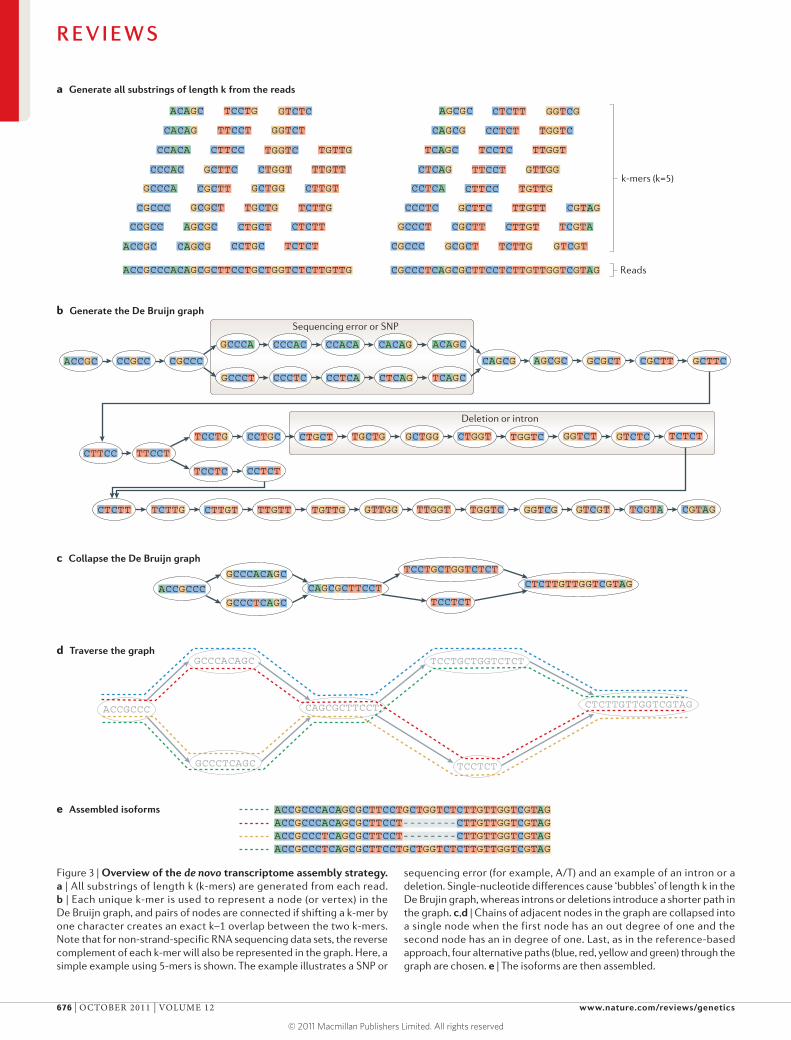
Figure 3 | Overview of the de novo transcriptome assembly strategy. a | All substrings of length k (k-mers) are generated from each read. b | Each unique k-mer is used to represent a node (or vertex) in the De Bruijn graph, and pairs of nodes are connected if shifting a k-mer by one character creates an exact k–1 overlap between the two k-mers. Note that for non-strand-specific RNA sequencing data sets, the reverse complement of each k-mer will also be represented in the graph. Here, a simple example using 5-mers is shown. The example illustrates a SNP or
sequencing error (for example, A/T) and an example of an intron or a deletion. Single-nucleotide differences cause ‘bubbles’ of length k in the De Brujin graph, whereas introns or deletions introduce a shorter path in the graph. c,d | Chains of adjacent nodes in the graph are collapsed into a single node when the first node has an out degree of one and the second node has an in degree of one. Last, as in the reference-based approach, four alternative paths (blue, red, yellow and green) through the graph are chosen. e | The isoforms are then assembled.
R E V I E W S
676 | OCTOBER 2011 | VOLUME 12 www.nature.com/reviews/genetics
© 2011 Macmillan Publishers Limited. All rights reserved
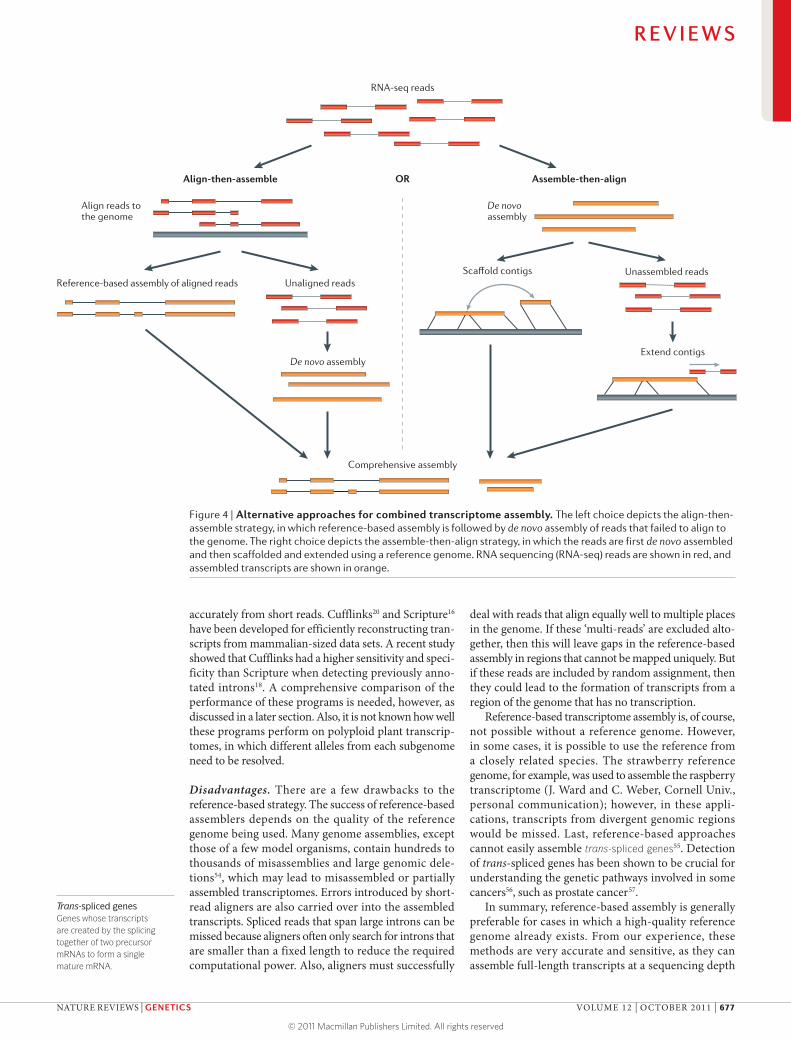
Figure 4 | Alternative approaches for combined transcriptome assembly. The left choice depicts the align-then-assemble strategy, in which reference-based assembly is followed by de novo assembly of reads that failed to align to the genome. The right choice depicts the assemble-then-align strategy, in which the reads are first de novo assembled and then scaffolded and extended using a reference genome. RNA sequencing (RNA-seq) reads are shown in red, and assembled transcripts are shown in orange.
Trans-spliced genesGenes whose transcripts are created by the splicing together of two precursor mRNAs to form a single mature mRNA.
accurately from short reads. Cufflinks20 and Scripture16 have been developed for efficiently reconstructing tran-scripts from mammalian-sized data sets. A recent study showed that Cufflinks had a higher sensitivity and speci-ficity than Scripture when detecting previously anno-tated introns18. A comprehensive comparison of the performance of these programs is needed, however, as discussed in a later section. Also, it is not known how well these programs perform on polyploid plant transcrip-tomes, in which different alleles from each subgenome need to be resolved.
Disadvantages. There are a few drawbacks to the reference-based strategy. The success of reference-based assemblers depends on the quality of the reference genome being used. Many genome assemblies, except those of a few model organisms, contain hundreds to thousands of misassemblies and large genomic dele-tions54, which may lead to misassembled or partially assembled transcriptomes. Errors introduced by short-read aligners are also carried over into the assembled transcripts. Spliced reads that span large introns can be missed because aligners often only search for introns that are smaller than a fixed length to reduce the required computational power. Also, aligners must successfully
deal with reads that align equally well to multiple places in the genome. If these ‘multi-reads’ are excluded alto-gether, then this will leave gaps in the reference-based assembly in regions that cannot be mapped uniquely. But if these reads are included by random assignment, then they could lead to the formation of transcripts from a region of the genome that has no transcription.
Reference-based transcriptome assembly is, of course, not possible without a reference genome. However, in some cases, it is possible to use the reference from a closely related species. The strawberry reference genome, for example, was used to assemble the raspberry transcriptome (J. Ward and C. Weber, Cornell Univ., personal communication); however, in these appli-cations, transcripts from divergent genomic regions would be missed. Last, reference-based approaches cannot easily assemble trans-spliced genes55. Detection of trans-spliced genes has been shown to be crucial for understanding the genetic pathways involved in some cancers56, such as prostate cancer57.
In summary, reference-based assembly is generally preferable for cases in which a high-quality reference genome already exists. From our experience, these methods are very accurate and sensitive, as they can assemble full-length transcripts at a sequencing depth
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 12 | OCTOBER 2011 | 677
© 2011 Macmillan Publishers Limited. All rights reserved
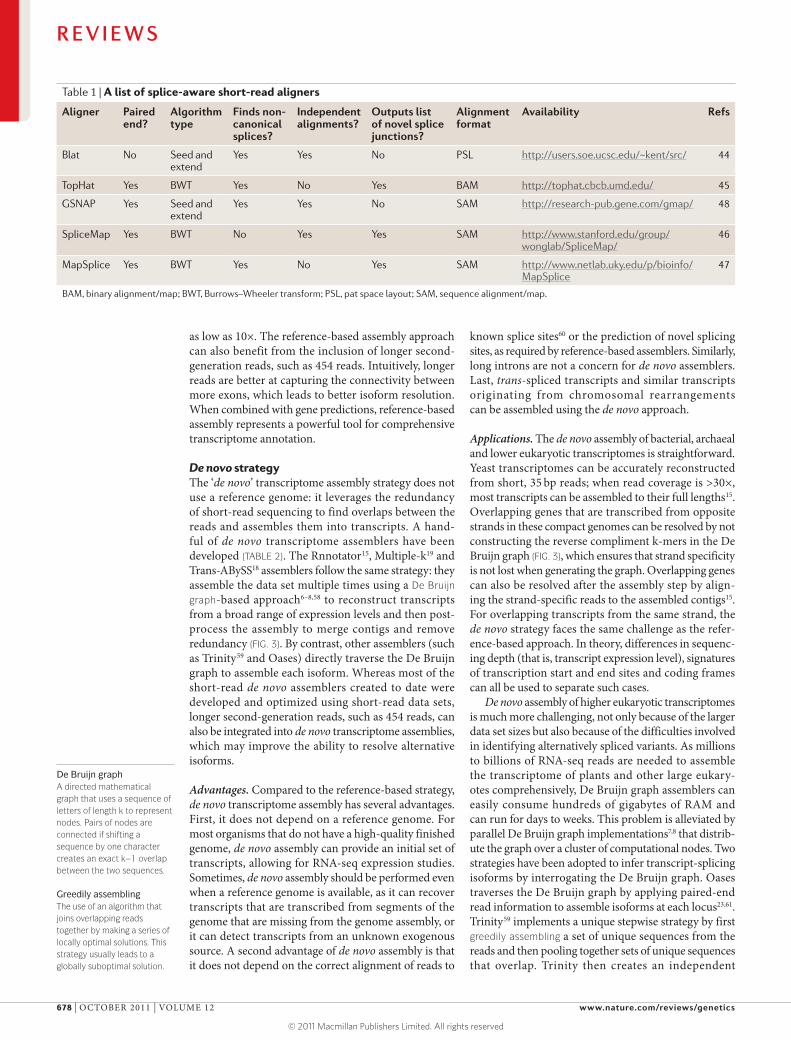
Table 1 | A list of splice-aware short-read aligners
Aligner Paired end?
Algorithm type
Finds non-canonical splices?
Independent alignments?
Outputs list of novel splice junctions?
Alignment format
Availability Refs
Blat No Seed and extend
Yes Yes No PSL http://users.soe.ucsc.edu/~kent/src/ 44
TopHat Yes BWT Yes No Yes BAM http://tophat.cbcb.umd.edu/ 45
GSNAP Yes Seed and extend
Yes Yes No SAM http://research-pub.gene.com/gmap/ 48
SpliceMap Yes BWT No Yes Yes SAM http://www.stanford.edu/group/wonglab/SpliceMap/
46
MapSplice Yes BWT Yes No Yes SAM http://www.netlab.uky.edu/p/bioinfo/MapSplice
47
BAM, binary alignment/map; BWT, Burrows–Wheeler transform; PSL, pat space layout; SAM, sequence alignment/map.
De Bruijn graphA directed mathematical graph that uses a sequence of letters of length k to represent nodes. Pairs of nodes are connected if shifting a sequence by one character creates an exact k–1 overlap between the two sequences.
Greedily assemblingThe use of an algorithm that joins overlapping reads together by making a series of locally optimal solutions. This strategy usually leads to a globally suboptimal solution.
as low as 10×. The reference-based assembly approach can also benefit from the inclusion of longer second-generation reads, such as 454 reads. Intuitively, longer reads are better at capturing the connectivity between more exons, which leads to better isoform resolution. When combined with gene predictions, reference-based assembly represents a powerful tool for comprehensive transcriptome annotation.
De novo strategyThe ‘de novo’ transcriptome assembly strategy does not use a reference genome: it leverages the redundancy of short-read sequencing to find overlaps between the reads and assembles them into transcripts. A hand-ful of de novo transcriptome assemblers have been developed (TABLE 2). The Rnnotator15, Multiple-k19 and Trans-ABySS18 assemblers follow the same strategy: they assemble the data set multiple times using a De Bruijn graph-based approach6–8,58 to reconstruct transcripts from a broad range of expression levels and then post-process the assembly to merge contigs and remove redundancy (FIG. 3). By contrast, other assemblers (such as Trinity59 and Oases) directly traverse the De Bruijn graph to assemble each isoform. Whereas most of the short-read de novo assemblers created to date were developed and optimized using short-read data sets, longer second-generation reads, such as 454 reads, can also be integrated into de novo transcriptome assemblies, which may improve the ability to resolve alternative isoforms.
Advantages. Compared to the reference-based strategy, de novo transcriptome assembly has several advantages. First, it does not depend on a reference genome. For most organisms that do not have a high-quality finished genome, de novo assembly can provide an initial set of transcripts, allowing for RNA-seq expression studies. Sometimes, de novo assembly should be performed even when a reference genome is available, as it can recover transcripts that are transcribed from segments of the genome that are missing from the genome assembly, or it can detect transcripts from an unknown exogenous source. A second advantage of de novo assembly is that it does not depend on the correct alignment of reads to
known splice sites60 or the prediction of novel splicing sites, as required by reference-based assemblers. Similarly, long introns are not a concern for de novo assemblers. Last, trans-spliced transcripts and similar transcripts originating from chromosomal rearrangements can be assembled using the de novo approach.
Applications. The de novo assembly of bacterial, archaeal and lower eukaryotic transcriptomes is straightforward. Yeast transcriptomes can be accurately reconstructed from short, 35 bp reads; when read coverage is >30×, most transcripts can be assembled to their full lengths15. Overlapping genes that are transcribed from opposite strands in these compact genomes can be resolved by not constructing the reverse compliment k-mers in the De Bruijn graph (FIG. 3), which ensures that strand specificity is not lost when generating the graph. Overlapping genes can also be resolved after the assembly step by align-ing the strand-specific reads to the assembled contigs15. For overlapping transcripts from the same strand, the de novo strategy faces the same challenge as the refer-ence-based approach. In theory, differences in sequenc-ing depth (that is, transcript expression level), signatures of transcription start and end sites and coding frames can all be used to separate such cases.
De novo assembly of higher eukaryotic transcriptomes is much more challenging, not only because of the larger data set sizes but also because of the difficulties involved in identifying alternatively spliced variants. As millions to billions of RNA-seq reads are needed to assemble the transcriptome of plants and other large eukary-otes comprehensively, De Bruijn graph assemblers can easily consume hundreds of gigabytes of RAM and can run for days to weeks. This problem is alleviated by parallel De Bruijn graph implementations7,8 that distrib-ute the graph over a cluster of computational nodes. Two strategies have been adopted to infer transcript-splicing isoforms by interrogating the De Bruijn graph. Oases traverses the De Bruijn graph by applying paired-end read information to assemble isoforms at each locus23,61. Trinity59 implements a unique stepwise strategy by first greedily assembling a set of unique sequences from the reads and then pooling together sets of unique sequences that overlap. Trinity then creates an independent
R E V I E W S
678 | OCTOBER 2011 | VOLUME 12 www.nature.com/reviews/genetics
© 2011 Macmillan Publishers Limited. All rights reserved
De Bruijn graph for each group of sequences and assembles isoforms within the group, which can run in parallel across a computational cluster to speed up the assembly process.
Disadvantages. Beside the fact that the computing resources needed to assemble large transcriptomes de novo can be overwhelming, there are several aspects of the de novo assembly strategy that need to be further improved. In general, de novo transcriptome assem-bly requires a much higher sequencing depth for full-length transcript assembly than does the reference-based assembly strategy. Whereas a reference-based assem-bler can reconstruct full-length transcripts with <10× sequencing coverage18, a de novo assembler usually requires more than 30× coverage for the same task15. Furthermore, de novo transcriptome assemblers are very sensitive to sequencing errors and to the presence of chimeric molecules in the data set62. Although algo-rithms have been developed to correct error-containing reads from abundant transcripts, this distinction is more difficult to make for reads that are sequenced from low-abundance transcripts, as discussed earlier. There is currently no effective way to discriminate chimeric reads that are artefacts of library preparation from true trans-spliced reads. Highly similar transcripts (for exam-ple, from different alleles or paralogues) are likely to be assembled into a single transcript and will require addi-tional post-assembly steps to resolve. Short repeats pose less of a challenge for transcriptome assembly, as repeats most often occur within intergenic regions. These intergenic regions are not present in the transcriptome, and repeats that are present can often be resolved by paired-end reads that span the repeated segment.
Combined strategyReference-based and de novo strategies can be combined to create a more comprehensive transcriptome. By bring-ing together these two complementary strategies, one
can take advantage of the high sensitivity of reference-based assemblers while leveraging the ability of de novo assemblers to detect novel and trans-spliced transcripts. Generally, the combined assembly strategy can be carried out by first either aligning the reads to the refer-ence genome or by de novo assembling the reads63 (FIG. 4). There has been no systematic evaluation to determine which strategy is better, and the choice is likely to be dependent on several factors, which are discussed below.
Align-then-assemble. Intuitively, when a reference genome is available, the combined approach should start by assembling the data set using the reference genome, followed by de novo assembling the reads that failed to align to the genome (FIG. 4). Alternatively, the transcripts that result from the reference-based assembly could also serve as input to the de novo assembly if the de novo assembler supports both long and short reads, as do Trans-ABySS and Oases. As mentioned earlier, de novo assembly requires more computing resources, particularly memory, compared with the alignment-based reference strategy. With a nearly complete refer-ence, most of the reads will be assembled, leaving only a small fraction of the reads to be de novo assembled. This align-then-assemble approach is also the preferred method for quickly filtering out unwanted sequences: for example, for pathogen detection64, in which reads of human origin are filtered out before assembly.
Assemble-then-align. If the quality of the reference genome is called into question or if the reference genome is from a different but closely related species, de novo assembly should be performed first, followed by align-ment of the contigs to the reference to extend and scaffold contigs (FIG. 4). The main advantage of this approach is that errors in the genome assembly do not get propagated into the assembled transcripts. As mentioned earlier, de novo assembly generates more fragmented transcripts than reference-based assembly. By aligning both the
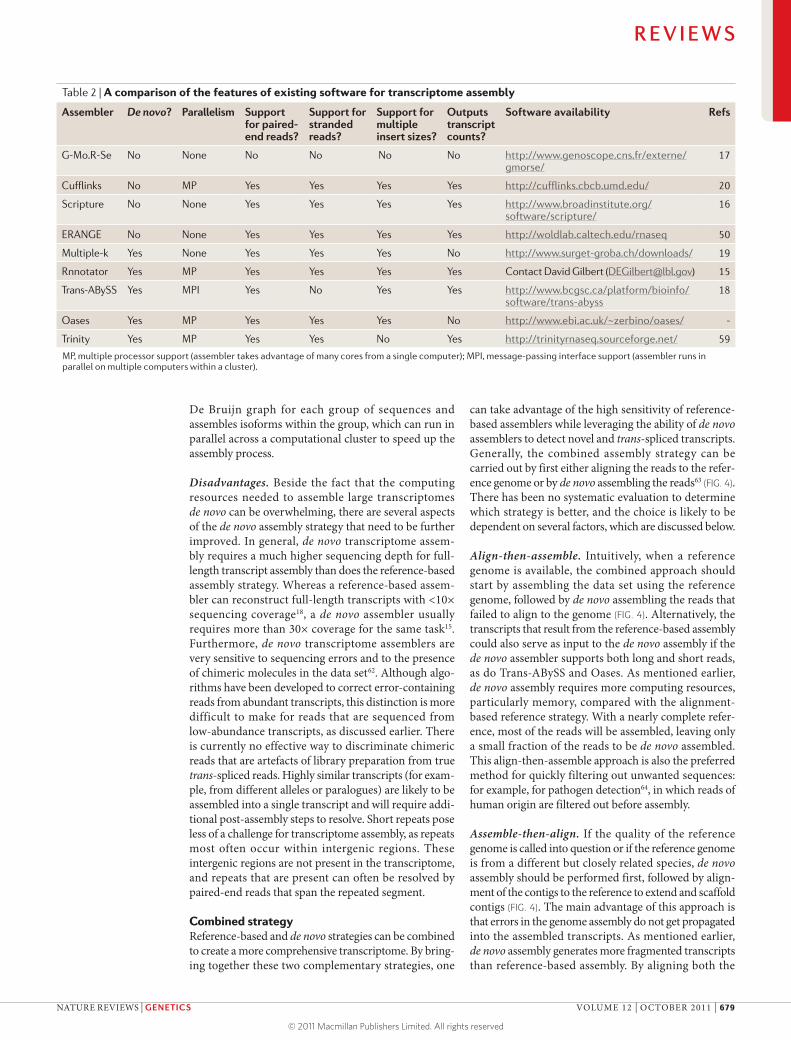
Table 2 | A comparison of the features of existing software for transcriptome assembly
Assembler De novo? Parallelism Support for paired-end reads?
Support for stranded reads?
Support for multiple insert sizes?
Outputs transcript counts?
Software availability Refs
G-Mo.R-Se No None No No No No http://www.genoscope.cns.fr/externe/gmorse/
17
Cufflinks No MP Yes Yes Yes Yes http://cufflinks.cbcb.umd.edu/ 20
Scripture No None Yes Yes Yes Yes http://www.broadinstitute.org/ software/scripture/
16
ERANGE No None Yes Yes Yes Yes http://woldlab.caltech.edu/rnaseq 50
Multiple-k Yes None Yes Yes Yes No http://www.surget-groba.ch/downloads/ 19
Rnnotator Yes MP Yes Yes Yes Yes Contact David Gilbert ([email protected]) 15
Trans-ABySS Yes MPI Yes No Yes Yes http://www.bcgsc.ca/platform/bioinfo/software/trans-abyss
18
Oases Yes MP Yes Yes Yes No http://www.ebi.ac.uk/~zerbino/oases/ -
Trinity Yes MP Yes Yes No Yes http://trinityrnaseq.sourceforge.net/ 59
MP, multiple processor support (assembler takes advantage of many cores from a single computer); MPI, message-passing interface support (assembler runs in parallel on multiple computers within a cluster).
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 12 | OCTOBER 2011 | 679
© 2011 Macmillan Publishers Limited. All rights reserved

N50 sizeThe size at which half of all assembled bases reside in contigs of this size or longer.
assembled transcripts and the unassembled reads to the reference genome, or to a closely related genome, incom-plete transcripts can be merged or extended to form longer, possibly full-length transcripts. Gaps between fragments of the same transcript can also be joined and filled in using the reference genomic sequence. Note that protein sequences can also be used for the align-ment step if the sequence similarity at the RNA level is not sufficient. For example, in a recent study, catfish transcripts were aligned to the stickleback proteome
to achieve substantially longer transcripts (the N50 size increased by 27%)19. The mosquito transcriptome was also scaffolded using this technique22.
No automated software pipelines exist that can carry out the combined assembly strategy. A systematic study is needed to explore which errors are introduced by com-bining assembly approaches. In the align-then-assemble approach, methods need to be developed to detect the errors in the reference-based assemblies to prevent them from being incorporated into the final assembly. In the assemble-then-align approach, measures must be taken to avoid incorrectly joining segments from different genes, thus, in turn, avoiding chimaeras.
Choosing a strategyThe choice of transcriptome assembly strategy depends on many factors, including the existence or complete-ness of a reference genome, the availability of sequencing and computing resources, the type of data set gener-ated and, most importantly, the overarching goal of the sequencing project. For comprehensive annotation of the transcriptome with a reference genome, one should make multiple pair-end libraries, sequence the transcrip-tome at a great depth and then use a combined strat-egy of reference-based and de novo assembly. Because the information provided by the RNA-seq data set is so rich, even a partial analysis can quickly lead to impor-tant discoveries. For example, in a recent study of the rice genome65, the use of Cufflinks led to the discovery of 649 genes that were missing from the rice annota-tion but that were found to be differently expressed in response to salinity stress. Sometimes, only one aspect of the transcriptome needs to be examined. In a study of Alzheimer’s disease66, it was hypothesized that alterna-tive splicing was involved in disease pathogenesis. The authors assembled the transcriptome using a reference-based assembler and discovered two genes with alter-native start sites and splicing patterns that may help to explain the progression of Alzheimer’s disease. As good-quality reference genomes are increasingly becoming available, the reference-based approach is well suited for many projects. If no reference genome exists, then a de novo assembly approach is the logical choice.
Choosing an assembly program. After an assembly strat-egy has been chosen, it can still be challenging to decide which assembly program to use. In general, most assem-blers were developed using a particular organism and NGS platform and, consequently, the tool is likely to per-form better on a similar data set. The NGS platform used for sequencing may also greatly limit the number of tools that can be used on that data type. The SOLiD sequenc-ing platform, for example, produces reads in colour space, which is not explicitly handled by most assembly algo-rithms. Other platforms have a unique error model that is best handled by the assembler from the sequencing ven-dor. Reads from the 454 platform, for example, are usually assembled using Newbler, the software distributed with 454 sequencing machines. Newbler can correct for long stretches of homopolymers of an unknown length, which are caused by ambiguities in the signal intensity.
Box 1 | Proposed quality metrics for assessing transcriptome assemblies
We suggest five metrics for evaluating the quality of an assembled transcriptome, given a set of reference transcripts that are expressed in the sample and are derived from the same transcriptome.
AccuracyThe accuracy metric is defined as the percentage of the correctly assembled bases estimated using the set of expressed reference transcripts (N). If reference transcripts are not available, then the reference genome can be used as an alternative. Accuracy can be formally written as:
×ΣΣ
where Li is the length of alignment between a reference transcript and an assembled
transcript Ti, A
i is the correct bases in transcript T
i, and M represents the number of best
alignments between assembled transcripts and reference.
CompletenessThe completeness metric is defined as the percentage of expressed reference transcripts covered by all the assembled transcripts and is written as:
×δΣ
where the indicator function, I, represents whether (1) or not (0) Ci (the percentage of
a reference transcript, i, that is covered by assembled transcripts) is greater than some arbitrary threshold, δ: for example, 80%.
ContiguityThe contiguity metric is defined as the percentage of expressed reference transcripts covered by a single, longest-assembled transcript and is similarly written as:
×δΣ
where the indicator function, I, represents whether (1) or not (0) Ci (the percentage of
a reference transcript, i, that is covered by a single, longest-assembled transcript) is greater than some arbitrary threshold, δ: for example, 80%.
ChimerismThe percentage of chimaeras that occur owing to misassemblies among all of the assembled transcripts. A chimeric transcript is one that contains non-repetitive parts from two or more different reference genes. They can arise from biological sources (gene fusions or trans-splicing), experimental sources (intermolecular ligation) or informatics sources (misassemblies). Misassembled chimeric transcripts can be distinguished from true chimaeras by determining whether the number of reads spanning the chimeric junction is significant when compared to the number of reads spanning other segments of the transcript.
Variant resolutionThe percentage of transcript variants assembled. This can be calculated by the average of the percentage of assembled variants within the reference set as:
×Σ
where Ci and E
i are the number of correctly and incorrectly assembled variants for
reference gene i, respectively, and Vi is the total number of variants for i.
R E V I E W S
680 | OCTOBER 2011 | VOLUME 12 www.nature.com/reviews/genetics
© 2011 Macmillan Publishers Limited. All rights reserved

RACEAn experimental protocol termed Rapid Amplification of cDNA Ends, which is used to determine the start and end points of gene transcription.
Cloud computingThe abstraction of underlying hardware architectures (for example, servers, storage and networking) to a shared pool of computing resources that can be readily provisioned and released.
Still, for a given sequencing project, there are sev-eral choices of assemblers. The most recent compari-son comes from the authors of Trinity. They compared six assembly algorithms and found that the number of full-length transcripts assembled in mice was higher for reference-based strategies; in yeast, however, two de novo assemblers outperformed all of the reference- based assemblers. Perhaps more importantly, the study found that, in general, reference-based assemblers dis-covered a greater number of unique splicing patterns than did de novo approaches, highlighting the greater sensitivity of reference-based assembly. An unbiased comparison of the performance of the current tran-scriptome assemblers is still needed to help users decide which assembler to use.
Assessing assembly qualityAlthough criteria to assess genome assemblies are under development54,67, standards for systematically assessing the quality of transcriptome assemblies have not been established. In a recent study15, such standards were pro-posed for a simple transcriptome in which alternative splicing is rare. Here we propose to extend these metrics for both simple and complex transcriptomes. These met-rics include accuracy, completeness, contiguity and chi-maera and variant resolution. They allow for the direct comparison of different assemblies with each other and the optimization of assembly parameters (BOX 1).
All of these metrics require a set of well-established expressed transcripts as a reference. Ideally, the refer-ence set should include both short and long transcripts, as long transcripts are particularly useful for estimat-ing the contiguity and chimerism metrics. It should also include transcripts with different expression lev-els, as weakly expressed transcripts can provide a good estimate for completeness, as well as pinpoint novel transcripts. Such a reference set can be difficult to find. For example, if possible, the reference would contain a set of known variants of different expression levels for estimating the variant resolution metric. This kind of data set is often not available, as the ability to detect different expression levels is one of the problems that transcriptome assembly is trying to address. A reference set of transcripts can also be derived from complemen-tary experimental methods. For example, the degree to which full-length protein-coding genes are assembled can be evaluated by checking whether the alternative isoforms encode full-length ORFs and by validating
the isoforms using proteomics assays25. UTRs can be evaluated through other experimental approaches, such as RACE68.
It is worth noting that optimizing some of these metrics may negatively affect others. For example, an assembler that creates many spurious overlaps would yield a high contiguity metric; however, the number of chimeric transcripts due to misassemblies would also be high. Exactly which metrics to optimize largely depends on the underlying scientific goals.
Conclusions and future perspectivesAdvances in both reference-based and de novo tran-scriptome assembly have expanded RNA-seq applica-tions to practically any genome. This is particularly important, because only a small number of species cur-rently have a high-quality reference genome available. Most species, especially polyploid plants, lack a refer-ence genome owing to the size and complexity of their genomes. Another area that is expected to be substan-tially improved by the advances in de novo transcriptome assembly is metatranscriptomics, in which thousands of transcriptomes from an entire microbial community are studied simultaneously.
Advances in high-performance computing (HPC) will greatly reduce the time required to assemble a large transcriptome or metatranscriptome data set. Almost all of the currently available transcriptome assemblers have some level of built-in parallelism that takes advan-tage of HPC clusters with thousands of computing cores (TABLE 2). Alternatively, cloud computing69 is an attrac-tive framework for parallel computing, as computing resources can be rented as a service on an as-needed basis. A cloud-based genome assembler called Contrail has already been developed, and hopefully cloud-based tran-scriptome assemblers will emerge as scalable solutions to the large transcriptome assembly problem.
Meanwhile, experimental RNA-seq and sequencing protocols are continually improving and should greatly reduce the informatics challenges. RNA-seq reads from third-generation sequencers, such as PacBio70, are longer (up to several kilobases). PacBio sequencers are capable of sequencing a single transcript to its full length in a single read. If this technology reaches a throughput that is comparable to the second-generation technologies, then the need for transcriptome assembly will probably be eliminated. Hopefully, the future of transcriptome assembly will be ‘no assembly required’.
1. Ozsolak, F. & Milos, P. M. RNA sequencing: advances, challenges and opportunities. Nature Rev. Genet. 12, 87–98 (2011).This Review provides a good, up‑to‑date summary of the RNA‑seq experimental protocol and its usefulness in addressing important biological questions.
2. Wang, Z., Gerstein, M. & Snyder, M. RNA-seq: a revolutionary tool for transcriptomics. Nature Rev. Genet. 10, 57–63 (2009).
3. Marguerat, S. & Bahler, J. RNA-seq: from technology to biology. Cell. Mol. Life Sci. 67, 569–579 (2010).
4. Wilhelm, B. T. & Landry, J. R. RNA-seq-quantitative measurement of expression through massively parallel RNA-sequencing. Methods 48, 249–257 (2009).
5. Metzker, M. L. Sequencing technologies — the next generation. Nature Rev. Genet. 11, 31–46 (2010).This Review provides a good introduction to NGS technologies and the analysis challenges that they pose.
6. Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
7. Simpson, J. T. et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123 (2009).
8. Butler, J. et al. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 18, 810–820 (2008).
9. Paszkiewicz, K. & Studholme, D. J. De novo assembly of short sequence reads. Brief. Bioinform. 11, 457–472 (2010).
10. Miller, J. R., Koren, S. & Sutton, G. Assembly algorithms for next-generation sequencing data. Genomics 95, 315–327 (2010).This paper provides a good introduction to the current algorithms used in next‑generation genome assembly and the challenges posed by these approaches.
11. Makalowska, I., Lin, C. F. & Makalowski, W. Overlapping genes in vertebrate genomes. Comput. Biol. Chem. 29, 1–12 (2005).
12. Normark, S. et al. Overlapping genes. Annu. Rev. Genet. 17, 499–525 (1983).
13. Johnson, Z. I. & Chisholm, S. W. Properties of overlapping genes are conserved across microbial genomes. Genome Res. 14, 2268–2272 (2004).
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 12 | OCTOBER 2011 | 681
© 2011 Macmillan Publishers Limited. All rights reserved

14. Fukuda, Y., Washio, T. & Tomita, M. Comparative study of overlapping genes in the genomes of Mycoplasma genitalium and Mycoplasma pneumoniae. Nucleic Acids Res. 27, 1847–1853 (1999).
15. Martin, J. et al. Rnnotator: an automated de novo transcriptome assembly pipeline from stranded RNA-seq reads. BMC Genomics 11, 663 (2010).This paper describes the first de novo transcriptome assembler to automate the use of several k‑mers for assembly. It also provides a good overview of methods used for the pre‑ and post‑processing of de novo transcriptome assemblies.
16. Guttman, M. et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nature Biotech. 28, 503–510 (2010).This paper introduces the Scripture algorithm, which was one of the first reference‑based assemblers that effectively tackled the assembly of alternative isoforms using NGS data.
17. Denoeud, F. et al. Annotating genomes with massive-scale RNA sequencing. Genome Biol. 9, R175 (2008).
18. Robertson, G. et al. De novo assembly and analysis of RNA-seq data. Nature Methods 7, 909–912 (2010).
19. Surget-Groba, Y. & Montoya-Burgos, J. I. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 20, 1432–1440 (2010).
20. Trapnell, C. et al. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotech. 28, 511–515 (2010).The Cufflinks algorithm is introduced in this paper, which, like the Scripture algorithm described in reference 16, was one of the first reference‑based assemblers that effectively tackled the assembly of alternative isoforms using NGS data.
21. Birol, I. et al. De novo transcriptome assembly with ABySS. Bioinformatics 25, 2872–2877 (2009).
22. Crawford, J. E. et al. De novo transcriptome sequencing in Anopheles funestus using Illumina RNA-seq technology. PLoS ONE 5, e14202 (2010).
23. Garg, R., Patel, R. K., Tyagi, A. K. & Jain, M. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 18, 53–63 (2011).
24. Yassour, M. et al. Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing. Proc. Natl Acad. Sci. USA 106, 3264–3269 (2009).
25. Adamidi, C. et al. De novo assembly and validation of planaria transcriptome by massive parallel sequencing and shotgun proteomics. Genome Res. 21, 1193–1200 (2011).
26. Katz, Y., Wang, E. T., Airoldi, E. M. & Burge, C. B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nature Methods 7, 1009–1015 (2010).
27. Levin, J. Z. et al. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nature Methods 7, 709–715 (2010).This paper provides an excellent comparison of different RNA‑seq protocols and how they affect the quantification of expression levels.
28. He, S. et al. Validation of two ribosomal RNA removal methods for microbial metatranscriptomics. Nature Methods 7, 807–812 (2010).
29. Chen, Z. & Duan, X. Ribosomal RNA depletion for massively parallel bacterial RNA-sequencing applications. Methods Mol. Biol. 733, 93–103 (2011).
30. Christodoulou, D. C., Gorham, J. M., Herman, D. S. & Seidman, J. G. Construction of normalized RNA-seq libraries for next-generation sequencing using the crab duplex-specific nuclease. Curr. Protoc. Mol. Biol. 1 Apr 2011 (doi:10.1002/0471142727.mb0412s94).
31. Kozarewa, I. et al. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nature Methods 6, 291–295 (2009).
32. Mamanova, L. et al. FRT-seq: amplification-free, strand-specific transcriptome sequencing. Nature Methods 7, 130–132 (2010).
33. Sam, L. T. et al. A comparison of single molecule and amplification based sequencing of cancer transcriptomes. PLoS ONE 6, e17305 (2011).
34. Ozsolak, F. et al. Amplification-free digital gene expression profiling from minute cell quantities. Nature Methods 7, 619–621 (2010).
35. Chen, S. et al. De novo analysis of transcriptome dynamics in the migratory locust during the development of phase traits. PLoS ONE 5, e15633 (2010).
36. Schwartz, T. S. et al. A garter snake transcriptome: pyrosequencing, de novo assembly, and sex-specific differences. BMC Genomics 11, 694 (2010).
37. Passalacqua, K. D. et al. Structure and complexity of a bacterial transcriptome. J. Bacteriol. 191, 3203–3211 (2009).
38. Dalloul, R. A. et al. Multi-platform next-generation sequencing of the domestic turkey (Meleagris gallopavo): genome assembly and analysis. PLoS Biol. 8, e1000475 (2010).
39. Jackman, S. D. & Birol, I. Assembling genomes using short-read sequencing technology. Genome Biol. 11, 202 (2010).
40. Rodrigue, S. et al. Unlocking short read sequencing for metagenomics. PLoS ONE 5, e11840 (2010).
41. Shi, H., Schmidt, B., Liu, W. & Muller-Wittig, W. A parallel algorithm for error correction in high-throughput short-read data on CUDA-enabled graphics hardware. J. Comput. Biol. 17, 603–615 (2010).
42. Kelley, D. R., Schatz, M. C. & Salzberg, S. L. Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 11, R116 (2010).
43. Falgueras, J. et al. SeqTrim: a high-throughput pipeline for pre-processing any type of sequence read. BMC Bioinformatics 11, 38 (2010).
44. Lassmann, T., Hayashizaki, Y. & Daub, C. O. TagDust—a program to eliminate artifacts from next generation sequencing data. Bioinformatics 25, 2839–2840 (2009).
45. Kent, W. J. BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
46. Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-seq. Bioinformatics 25, 1105–1111 (2009).
47. Au, K. F., Jiang, H., Lin, L., Xing, Y. & Wong, W. H. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 38, 4570–4578 (2010).
48. Wang, K. et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 38, e178 (2010).
49. Wu, T. D. & Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881 (2010).
50. Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nature Methods 5, 621–628 (2008).
51. Nagalakshmi, U. et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349 (2008).
52. Perkins, T. T. et al. A strand-specific RNA-seq analysis of the transcriptome of the typhoid bacillus Salmonella typhi. PLoS Genet. 5, e1000569 (2009).
53. Ozsolak, F. et al. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell 143, 1018–1029 (2010).
54. Salzberg, S. L. & Yorke, J. A. Beware of mis-assembled genomes. Bioinformatics 21, 4320–4321 (2005).This study highlights the importance of having standardized metrics to assess the quality of NGS assemblies.
55. Kinsella, M., Harismendy, O., Nakano, M., Frazer, K. A. & Bafna, V. Sensitive gene fusion detection using ambiguously mapping RNA-seq read pairs. Bioinformatics 27, 1068–1075 (2011).
56. McPherson, A. et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-seq data. PLoS Comput. Biol. 7, e1001138 (2011).
57. Tomlins, S. A. et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 448, 595–599 (2007).
58. Pevzner, P. A., Tang, H. & Waterman, M. S. An Eulerian path approach to DNA fragment assembly. Proc. Natl Acad. Sci. USA 98, 9748–9753 (2001).This paper introduces the idea of using a De Bruijn graph for the purposes of assembly.
59. Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nature Biotech. 29, 644–652 (2011).The Trinity de novo assembly program is introduced in this paper. This was the first NGS transcriptome assembly strategy not to rely on a genome assembler while also addressing the assembly of alternative isoforms.
60. Burset, M., Seledtsov, I. A. & Solovyev, V. V. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 28, 4364–4375 (2000).
61. Jager, M. et al. Composite transcriptome assembly of RNA-seq data in a sheep model for delayed bone healing. BMC Genomics 12, 158 (2011).
62. Cocquet, J., Chong, A., Zhang, G. & Veitia, R. A. Reverse transcriptase template switching and false alternative transcripts. Genomics 88, 127–131 (2006).
63. Haas, B. J. & Zody, M. C. Advancing RNA-seq analysis. Nature Biotech. 28, 421–423 (2010).
64. Greninger, A. L. et al. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS ONE 5, e13381 (2010).
65. Mizuno, H. et al. Massive parallel sequencing of mRNA in identification of unannotated salinity stress-inducible transcripts in rice (Oryza sativa L.). BMC Genomics 11, 683 (2010).
66. Twine, N. A., Janitz, K., Wilkins, M. R. & Janitz, M. Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer’s disease. PLoS ONE 6, e16266 (2011).
67. Meader, S., Hillier, L. W., Locke, D., Ponting, C. P. & Lunter, G. Genome assembly quality: assessment and improvement using the neutral indel model. Genome Res. 20, 675–84 (2010).
68. Schaefer, B. C. Revolutions in rapid amplification of cDNA ends: new strategies for polymerase chain reaction cloning of full-length cDNA ends. Anal. Biochem. 227, 255–273 (1995).
69. Taylor, R. C. An overview of the Hadoop/MapReduce/HBase framework and its current applications in bioinformatics. BMC Bioinformatics 11 (Suppl. 12), S1 (2010).
70. Eid, J. et al. Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138 (2009).
AcknowledgementsThe work conducted by the US Department of Energy (DOE) Joint Genome Institute is supported by the Office of Science of the DOE under contract number DE-AC02-05CH11231. The views and opinions of the authors expressed herein do not necessarily state or reflect those of the United States government, or any agency thereof, or the Regents of the University of California.
Competing interests statementThe author declares no competing interests.
FURTHER INFORMATIONZhong Wang’s homepage: http://www.epernicus.com/people/zhongwangNature Reviews Genetics series on Applications of Next-Generation Sequencing: http://www.nature.com/nrg/series/nextgeneration/index.htmlNature Reviews Genetics series on Study Designs: http://www.nature.com/nrg/series/studydesigns/index.htmlUS Department of Energy Joint Genome Institute: http://jgi.doe.gov/
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
R E V I E W S
682 | OCTOBER 2011 | VOLUME 12 www.nature.com/reviews/genetics
© 2011 Macmillan Publishers Limited. All rights reserved


















