
Inhibition of Mammalian Target of Rapamycin Reverses Alveolar Epithelial Neoplasia...
11
Inhibition of Mammalian Target of Rapamycin Reverses Alveolar Epithelial Neoplasia Induced by Oncogenic K-ras Marie Wislez, 1 M. Loreto Spencer, 2 Julie G. Izzo, 3 Denise M. Juroske, 1 Kamna Balhara, 1 Dianna D. Cody, 4 Roger E. Price, 4 Walter N. Hittelman, 3 Ignacio I. Wistuba, 1,2 and Jonathan M. Kurie 1 Departments of 1 Thoracic/Head and Neck Medical Oncology, 2 Pathology, 3 Experimental Therapeutics, and 4 Imaging Physics, University of Texas M.D. Anderson Cancer Center, Houston, Texas Abstract The serine/threonine kinase AKT and its downstream mediator mammalian target of rapamycin (mTOR) are activated in lung adenocarcinoma, and clinical trials are under way to test whether inhibition of mTOR is useful in treating lung cancer. Here, we report that mTOR inhibition blocked malignant progression in K-ras LA1 mice, which undergo somatic activation of the K-ras oncogene and display morphologic changes in alveolar epithelial cells that recapit- ulate those of precursors of human lung adenocarcinoma. Levels of phospho-S6 Ser236/235 , a downstream mediator of mTOR, increased with malignant progression (normal alve- olar epithelial cells to adenocarcinoma) in K-ras LA1 mice and in patients with lung adenocarcinoma. Atypical alveolar hyperplasia, an early neoplastic change, was prominently associated with macrophages and expressed high levels of phospho-S6 Ser236/235 . mTOR inhibition in K-ras LA1 mice by treatment with the rapamycin analogue CCI-779 reduced the size and number of early epithelial neoplastic lesions (atypical alveolar hyperplasia and adenomas) and induced apoptosis of intraepithelial macrophages. LKR-13, a lung adenocarcinoma cell line derived from K-ras LA1 mice, was resistant to treatment with CCI-779 in vitro . However, LKR-13 cells grown as syngeneic tumors recruited macrophages, and those tumors regressed in response to treatment with CCI- 779. Lastly, conditioned medium from primary cultures of alveolar macrophages stimulated the proliferation of LKR-13 cells. These findings provide evidence that the expansion of lung adenocarcinoma precursors induced by oncogenic K-ras requires mTOR-dependent signaling and that host factors derived from macrophages play a critical role in adenocar- cinoma progression. (Cancer Res 2005; 65(8): 3226-35) Introduction Lung cancer is the most common cause of cancer-related death in the United States, and its incidence is increasing worldwide (1). The high mortality of lung cancer reflects its invasive nature and its resistance to current treatment modalities. Because the prognosis is grim once lung cancer has reached advanced stages, inves- tigators have attempted to intervene earlier, before the develop- ment of overt disease (2). Efforts to develop effective lung cancer prevention strategies have stimulated interest in understanding the biology of lung neoplasia. Lung adenocarcinoma, the most common subtype of non–small cell lung cancer (NSCLC), often occurs near sites of atypical alveolar hyperplasia (AAH). A subgroup of AAH cells contains activating mutations in K-ras , a genetic event found in 30% to 50% of lung adenocarcinomas (3). Because of their proximity and shared genetic changes, AAH is considered a precursor of lung adenocarcinoma. Another type of lung cancer, bronchioloalveolar cell carcinoma (BAC), is a noninvasive, slowly progressive tumor of alveolar epithelial cells that may evolve into invasive adenocarci- noma (4). Activating K-ras mutations are also common in BAC. Based on these observations, two models for lung adenocarcinoma progression have been proposed, one in which AAH evolves directly to adenocarcinoma and another in which AAH evolves to BAC, which can subsequently develop into adenocarcinoma. The biochemical events required for malignant progression of the bronchial epithelium have been the focus of recent investi- gation. The serine/threonine kinase AKT is activated in NSCLC and in bronchial dysplasia, an early event in squamous bronchial neoplasia (5). NSCLC cells with activated AKT undergo prolifera- tive arrest after treatment with inhibitors of phosphatidylinositol 3-kinase (PI3K), an upstream activator of AKT (6–8). Activation of AKT-dependent signaling contributes to the development of glio- blastoma multiforme, endometrial cancer, prostate cancer, breast cancer, etc. (9–13). Several biochemical events have been identified downstream of AKT that enhance cell survival, increase cell proliferation, and alter cell metabolism. AKT phosphorylates and inactivates downstream substrates, including BAD, FOXO proteins, GSK3, and tuberin, the protein product of Tsc2 (14). Phosphorylation of tuberin leads to activation of mammalian target of rapamycin (mTOR, encoded by the gene frap1 in mice), a critical mediator of pro- tein translation (15–19). mTOR substrates required for protein translation include the serine/threonine kinase S6K1 (p70 S6K / p85 S6K ) and the 4E binding protein 1 (20–22). Phosphorylation of 4E binding protein 1 causes it to dissociate from eukaryotic initiation factor (eIF) 4E, which then binds to the eIF4G scaffold protein, promoting the assembly of the eIF4F initiation complex. Of note, eIF4E is overexpressed in BAC and lung adenocarcinoma (23–25). These cumulative observations have led to derivatives of the mTOR inhibitor rapamycin being tested in clinical trials of lung cancer. In this study, we hypothesized that mTOR signaling is activated in precursors of lung adenocarcinoma and that activation contributes to lung tumor progression. We addressed this question Note: M. Wislez is a postdoctoral fellow of La Fondation pour la Recherche Medicale and La Formation Continue du Corps Me ´dical des Ho ˆpitaux de Paris. Supplementary data for this article are available at Cancer Research Online (http:// cancerres.aacrjournals.org). Requests for reprints: Jonathan M. Kurie, Department of Thoracic/Head and Neck Medical Oncology, University of Texas M.D. Anderson Cancer Center, Box 432, 1515 Holcombe Boulevard, Houston, TX 77030. Phone: 713-792-6363; E-mail: [email protected]. I2005 American Association for Cancer Research. Cancer Res 2005; 65: (8). April 15, 2005 3226 www.aacrjournals.org Research Article Research. on June 4, 2018. © 2005 American Association for Cancer cancerres.aacrjournals.org Downloaded from
Transcript of Inhibition of Mammalian Target of Rapamycin Reverses Alveolar Epithelial Neoplasia...

Inhibition of Mammalian Target of Rapamycin Reverses Alveolar
Epithelial Neoplasia Induced by Oncogenic K-ras
Marie Wislez,1M. Loreto Spencer,
2Julie G. Izzo,
3Denise M. Juroske,
1Kamna Balhara,
1
Dianna D. Cody,4Roger E. Price,
4Walter N. Hittelman,
3Ignacio I. Wistuba,
1,2
and Jonathan M. Kurie1
Departments of 1Thoracic/Head and Neck Medical Oncology, 2Pathology, 3Experimental Therapeutics, and 4Imaging Physics, University ofTexas M.D. Anderson Cancer Center, Houston, Texas
Abstract
The serine/threonine kinase AKT and its downstreammediator mammalian target of rapamycin (mTOR) areactivated in lung adenocarcinoma, and clinical trials areunder way to test whether inhibition of mTOR is useful intreating lung cancer. Here, we report that mTOR inhibitionblocked malignant progression in K-rasLA1 mice, whichundergo somatic activation of the K-ras oncogene and displaymorphologic changes in alveolar epithelial cells that recapit-ulate those of precursors of human lung adenocarcinoma.Levels of phospho-S6Ser236/235, a downstream mediator ofmTOR, increased with malignant progression (normal alve-olar epithelial cells to adenocarcinoma) in K-rasLA1 mice andin patients with lung adenocarcinoma. Atypical alveolarhyperplasia, an early neoplastic change, was prominentlyassociated with macrophages and expressed high levels ofphospho-S6Ser236/235. mTOR inhibition in K-rasLA1 mice bytreatment with the rapamycin analogue CCI-779 reduced thesize and number of early epithelial neoplastic lesions(atypical alveolar hyperplasia and adenomas) and inducedapoptosis of intraepithelial macrophages. LKR-13, a lungadenocarcinoma cell line derived from K-rasLA1 mice, wasresistant to treatment with CCI-779 in vitro . However, LKR-13cells grown as syngeneic tumors recruited macrophages, andthose tumors regressed in response to treatment with CCI-779. Lastly, conditioned medium from primary cultures ofalveolar macrophages stimulated the proliferation of LKR-13cells. These findings provide evidence that the expansion oflung adenocarcinoma precursors induced by oncogenic K-rasrequires mTOR-dependent signaling and that host factorsderived from macrophages play a critical role in adenocar-cinoma progression. (Cancer Res 2005; 65(8): 3226-35)
Introduction
Lung cancer is the most common cause of cancer-related deathin the United States, and its incidence is increasing worldwide (1).The high mortality of lung cancer reflects its invasive nature and itsresistance to current treatment modalities. Because the prognosis
is grim once lung cancer has reached advanced stages, inves-tigators have attempted to intervene earlier, before the develop-ment of overt disease (2). Efforts to develop effective lung cancerprevention strategies have stimulated interest in understanding thebiology of lung neoplasia.Lung adenocarcinoma, the most common subtype of non–small
cell lung cancer (NSCLC), often occurs near sites of atypicalalveolar hyperplasia (AAH). A subgroup of AAH cells containsactivating mutations in K-ras , a genetic event found in 30% to 50%of lung adenocarcinomas (3). Because of their proximity andshared genetic changes, AAH is considered a precursor of lungadenocarcinoma. Another type of lung cancer, bronchioloalveolarcell carcinoma (BAC), is a noninvasive, slowly progressive tumor ofalveolar epithelial cells that may evolve into invasive adenocarci-noma (4). Activating K-ras mutations are also common in BAC.Based on these observations, two models for lung adenocarcinomaprogression have been proposed, one in which AAH evolves directlyto adenocarcinoma and another in which AAH evolves to BAC,which can subsequently develop into adenocarcinoma.The biochemical events required for malignant progression of
the bronchial epithelium have been the focus of recent investi-gation. The serine/threonine kinase AKT is activated in NSCLCand in bronchial dysplasia, an early event in squamous bronchialneoplasia (5). NSCLC cells with activated AKT undergo prolifera-tive arrest after treatment with inhibitors of phosphatidylinositol3-kinase (PI3K), an upstream activator of AKT (6–8). Activation ofAKT-dependent signaling contributes to the development of glio-blastoma multiforme, endometrial cancer, prostate cancer, breastcancer, etc. (9–13).Several biochemical events have been identified downstream of
AKT that enhance cell survival, increase cell proliferation, and altercell metabolism. AKT phosphorylates and inactivates downstreamsubstrates, including BAD, FOXO proteins, GSK3, and tuberin,the protein product of Tsc2 (14). Phosphorylation of tuberinleads to activation of mammalian target of rapamycin (mTOR,encoded by the gene frap1 in mice), a critical mediator of pro-tein translation (15–19). mTOR substrates required for proteintranslation include the serine/threonine kinase S6K1 (p70S6K/p85S6K) and the 4E binding protein 1 (20–22). Phosphorylation of4E binding protein 1 causes it to dissociate from eukaryoticinitiation factor (eIF) 4E, which then binds to the eIF4G scaffoldprotein, promoting the assembly of the eIF4F initiation complex.Of note, eIF4E is overexpressed in BAC and lung adenocarcinoma(23–25). These cumulative observations have led to derivatives ofthe mTOR inhibitor rapamycin being tested in clinical trials oflung cancer.In this study, we hypothesized that mTOR signaling is activated
in precursors of lung adenocarcinoma and that activationcontributes to lung tumor progression. We addressed this question
Note: M. Wislez is a postdoctoral fellow of La Fondation pour la RechercheMedicale and La Formation Continue du Corps Medical des Hopitaux de Paris.
Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org).
Requests for reprints: Jonathan M. Kurie, Department of Thoracic/Head andNeck Medical Oncology, University of Texas M.D. Anderson Cancer Center, Box432, 1515 Holcombe Boulevard, Houston, TX 77030. Phone: 713-792-6363; E-mail:[email protected].
I2005 American Association for Cancer Research.
Cancer Res 2005; 65: (8). April 15, 2005 3226 www.aacrjournals.org
Research Article
Research. on June 4, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

by testing human tissues and K-rasLA1 mice, which develop lungadenocarcinoma through somatic activation of a K-ras allelecarrying an activating mutation in codon 12 (G12D; ref. 26). Wechose K-rasLA1 as a model because K-ras is the most commonlymutated proto-oncogene in lung adenocarcinoma, because mutantK-ras activates PI3K/AKT-dependent signaling (27), and becausetumors in K-rasLA1 mice recapitulate certain morphologic changesin alveolar epithelial cells that precede human lung adenocarcino-ma, evolving through a series of morphologic stages from AAH toadenocarcinoma. The predominant epithelial changes observed in4- to 8-week-old mice are AAH-like lesions and small adenomasfollowed by the appearance of adenomas with papillary or atypicalfeatures and, at 6 to 8 months, well-differentiated adenocarcino-mas that invade regional lymph nodes or, less often, metastasize todistant sites. The K-ras mutations, the growth patterns alongalveolar surfaces (lepidic growth), and the infiltration of inflam-matory cells (macrophages) observed in this model are allcharacteristic of human adenocarcinoma that evolves throughBAC. Whereas the earliest precursor of lung adenocarcinoma inhumans and K-rasLA1 mice is a hyperplastic phenotype (AAH), thesubsequent morphologic changes that precede the appearance ofadenocarcinoma are different in humans (BAC) and in K-rasLA1
mice (adenoma).
Materials and Methods
Animals, cells, and reagents. We studied K-rasLA1 mice, which carry a
latent K-ras allele with two copies of exon 1, one wild-type and the other theG12D mutant (26). The latent allele is stochastically activated in cells
through homologous recombination, which results in deletion of the wild-
type copy of exon 1 and expression of an oncogenic form of the K-ras gene.Multifocal lung adenocarcinomas develop spontaneously in 100% of these
mice.
The LKR-13 and LKR-10 cell lines were derived by serial passage of
minced lung adenocarcinoma tissues from two tumors isolated fromseparate lobes of the same K-rasLA1 mouse. The cells were passaged in RPMI
1640 supplemented with 10% fetal bovine serum on standard plasticware
(Falcon, Becton Dickinson, Bedford, MA) at 37jC and in a 5% CO2
atmosphere. Alveolar macrophages were recovered from bronchoalveolarlavage (BAL) fluid from wild-type (129/Sv) littermates of K-rasLA1 mice as
follows. Briefly, after the mice were killed by cervical dislocation, three 2 mL
aliquots of PBS were injected directly into the trachea. The liquid wasrecovered by gentle aspiration and centrifuged. The resultant cell pellet was
suspended in serum-free RPMI 1640 containing antibiotics [100 IU/mL
penicillin G, 100 Ag/mL streptomycin, and 0.25 Ag/mL amphotericin B (Life
Technologies, Gaithersburg, MD)] and plated in 24-well tissue culture platesfor 24 hours at 37jC and 5% CO2. Macrophage conditioned medium was
then recovered and frozen at �80jC until use. Macrophages (f1 � 104 �8 � 104) were present per milliliter of BAL and viability was z98% as
assessed by trypan blue exclusion. Morphologic analysis of cytospinpreparations stained with H&E revealed the cells to be 97.2 F 0.5%
macrophages, 1.9 F 0.4% lymphocytes, and 0.8 F 0.07% neutrophils.
We purchased rabbit polyclonal antibodies against phospho-S6Ser236/235
(p-S6), p-S6K (Thr289), S6K, p-mTOR (Ser2448), mTOR, and cleaved caspase-3(Cell Signaling Technology, Beverly, MA), rat monoclonal anti-mouse F4/80
(Serotec, Oxford, United Kingdom), murine monoclonal anti-human CD68
(DAKOCytomation, Carpinteria, CA), and murine monoclonal antibodiesagainst proliferating cell nuclear antigen (PCNA; BioGenex Laboratories,
San Ramon, CA). Blocking peptides for p-S6 antibodies were purchased
from Cell Signaling Technology and mouse immunoglobulin was from
DAKOCytomation. For fluorescence staining, we used rabbit secondaryTRITC and rat secondary FITC (Immunology Consultants Laboratory, Inc.,
Newburg, OR) and 4V,6-diamidino-2-phenylindole (DAPI) II (Vysis, Inc.,
Downers Grove, IL) for nuclear counterstaining. The rapamycin analogue
CCI-779 (Temsirolimus) was provided by Wyeth-Ayerst (Philadelphia, PA).
Tween 80 and trypsin-EDTA were from Life Technologies, andglutaraldehyde, paraformaldehyde, formalin, 3-(4,5-dimethylthiazol-2-yl)-
2,5-diphenyltetrazolium bromide (MTT), bisbenzimide (Hoescht 33342),
and polyethylene glycol-400 were from Sigma (St. Louis, MO).
K-rasLA1 mice experiments. Sixteen-week-old K-rasLA1 mice were giveni.p. injections of CCI-779 at either 20 mg/kg/d (high-dose group, n = 12) or
0.1 mg/kg/d (low-dose group, n = 14) for 5 days/wk for 4 weeks; another 12
control mice were given vehicle (5% Tween 80/5% polyethylene glycol-400)
on the same schedule. Mice were subjected to respiratory-gated micro–
computed tomography (micro-CT) scans under general anesthesia as
described elsewhere (28) at the beginning and at the end of the treatment;they were killed by cervical dislocation within 6 hours of the final micro-CT
scan. During the autopsy procedure, visible lesions were counted on the
surfaces of both lungs by investigators blinded as to treatment group (M.W.
and D.J.). The lungs were then perfused with PBS and removed from the
body. One lung was kept at �80jC for protein extraction and the other was
fixed in 4% glutaraldehyde/paraformaldehyde for 30 minutes followed by10% formalin overnight before being embedded in paraffin as described
previously (29).
Micro-CT scan analysis. Mice were anesthetized and intubated by
experienced veterinary personnel and connected to a SAR-830/P smallanimal ventilator (CWE, Ardmore, PA). A single respiratory-gated three-
dimensional micro-CT image set was acquired for each mouse using 80 kVp,
405 AA, 100 ms per frame, 5 frames per view, 360 views, and 1jincrementation per view. This acquisition resulted in a set of contiguousaxial DICOM formatted images through each mouse thorax with voxels of
dimensions 91 � 91 � 91 Am. Lesions were characterized on the initial and
final micro-CT scans by one investigator (M.W.) who was blinded as totreatment group and autopsy results. Lesions visualized on the CT images
included solid or ‘‘ground-glass’’ opacities or areas of consolidation
resembling adenocarcinoma with bronchioloaveolar features in humans
(30). Lesion volume was measured with ‘‘Analyze’’ software (AnalyzeDirect,Inc., Lenexa, KS) as follows. The volumes of three to five lesions were
measured in each mouse (10-30 or more axial images per lesion) on the final
micro-CT scan, after which anatomic landmarks were used to locate the
same lesions on the initial micro-CT scan and the volumes were measuredagain. Lesion growth was defined as the difference between the final and the
initial volumes. If a lesion from the final micro-CT scan could not be located
on the initial micro-CT image set, it was considered a new lesion arisingduring the treatment.
Tissue microarrays and immunohistochemical analyses. Three types
of tissue microarrays were constructed with cores from formalin-fixed,
paraffin-embedded blocks (microarrays are summarized in Supplementary
Table S1). The first array was made with specimens of human normal lung,
AAH, BAC, and invasive adenocarcinoma. The first array was made with
specimens of human normal lung (n = 30), AAH (n = 27), BAC (n = 41), and
invasive adenocarcinoma (n = 97) obtained from surgical resections
performed between 1994 and 2004 at the University of Texas M.D. Anderson
Cancer Center (Houston, TX). The histologic groups (AAH, BAC, and
adenocarcinoma) were balanced with respect to age gender and smoking
history (data not shown). The adenocarcinomas included pathologic stages
I, II, and IIIA. None of the patients received neoadjuvant therapy. The BAC
lesions were not pure BAC but rather were considered ‘‘mixed adenocar-
cinoma with predominant BAC features’’ according to the 1999 WHO
definitions (31). Three cores were obtained from each specimen, with core
diameters of 1 mm for adenocarcinoma and 1.5 mm for BAC and AAH. The
second array consisted of specimens of murine normal lung (n = 30), AAH
(n = 40), adenoma (n = 206), and adenocarcinoma [n = 11; according to the
histologic criteria established by Johnson et al. (26)]. The third array
comprised all lesions (identified by histologic analysis) from the mice in the
current study treated with CCI-779 or vehicle (47 AAH, 153 adenomas,
and 36 normal lung). Each murine lesion was sampled with a single core of
1 mm diameter; the lesions were too small to permit multiple cores to be
obtained.
For immunohistochemical analyses, 4 Am sections were deparaffinized,
rehydrated, and washed with PBS as described previously (29). Antigenswere retrieved with 0.01 mol/L citrate buffer (pH 6, DAKOCytomation) for
mTOR, Macrophage Survival, and Lung Adenocarcinoma
www.aacrjournals.org 3227 Cancer Res 2005; 65: (8). April 15, 2005
Research. on June 4, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

30 minutes in a steamer. To detect the macrophage antigen F4/80, slideswere exposed to 0.025% trypsin-EDTA for 10 minutes. Samples were blocked
for endogenous activity in 3% hydrogen peroxide/PBS, avidin/biotin
solution (Zymed, South San Francisco, CA), and DAKO serum-free protein
block (DAKOCytomation) before incubation with the primary antibodyovernight at 4jC. Standard avidin-biotin immunoperoxidase methods with
diaminobenzine as the chromogen were used for detection. The Animal
Research Kit (DAKOCytomation) was used to reduce nonspecific binding of
the murine PCNA antibody to murine tissues. For dual-fluorescencestaining, we followed the same protocol, except for the detection step for
which we used a secondary antibody coupled with a fluorochrome.
Immunofluorescence-generated signals were visualized with a Zeiss
Axioplan epifluorescence microscope (Nikon, Inc., Melville, NY) equippedwith oil immersion objective and single band pass filters for FITC, Texas
red, and DAPI. Digitized images of each fluorochrome were captured
individually with a high-resolution image analysis system (MetaMorph,Universal Imaging Corp., Downington, PA) with a cooled charge-coupled
device camera (Hamamatsu C4742-95-12, Hamamatsu Photonocis K.K.,
Hamamatsu City, Japan).
As negative controls for determining the specificity of the immunostain-ing results, we omitted the primary antibody (cleaved caspase-3), pretreated
the samples with blocking peptides (p-S6) or isotype immunoglobulins
(PCNA), and stained a paraffin-embedded pellet of H1607 human lung
adenocarcinoma cells for the macrophage antigens F4/80 and CD68. Aspositive controls, we used paraffin-embedded pellets of H1607 cells (which
are PTEN�/�; p-S6), normal lung tissue from mouse (F4/80) and human
(CD68), murine lymphoma tissue (cleaved caspase-3), and normal humantonsil tissue (PCNA).
Staining in normal lung and lung lesions was quantified by one
investigator (L.S.), blinded as to treatment group, using two approaches:
frequency of staining (defined as the presence of any positive intralesional
cells in a single tissue section) and degree of staining [a combined score
based on staining intensity and extension in a single tissue section
(intensity � extension)]. Staining intensity for p-S6 was recorded as
undetectable (0), weak (1), medium (2), or strong (3). For F4/80, CD68, and
cleaved caspase-3, staining intensity was defined as undetectable (0) or
detectable (1). Staining extension was evaluated for the whole of the tumor
and defined as the percentage of positive cells per 20� square ( for p-S6,
F4/80, and CD68), per 40� square ( for PCNA), or per 100� square ( for
cleaved caspase-3) magnification. The degree of staining was compared
between each histologic category and treatment group and expressed as
the mean F SE.
Western blot analysis. Lysates from cell lines or mouse tissue
containing the same amounts of protein were separated by SDS-PAGEand transferred onto a polyvinylidene fluoride nitrocellulose membrane
(Bio-Rad Laboratories, Hercules, CA). Membranes were immunoblotted
overnight at 4jC with primary antibodies in TBS containing 5% nonfat drymilk. Antibody binding was detected with an enhanced chemiluminescence
kit according to the manufacturer’s directions (Amersham, Inc., Arlington
Heights, IL).
Apoptosis analysis. Nuclei from alveolar macrophages obtained from
BAL fluid were stained with bisbenzimide (Hoechst 33342) as follows. Cells
were fixed in 4% formaldehyde, washed with PBS, incubated with Hoechst
33342 (10 Ag/mL) for 15 minutes at room temperature, and counted with a
fluorescence microscope (with a UV filter) at a magnification of �63 with
oil immersion objective. Results were expressed as the percentage of 200
counted cells with the morphologic characteristics of apoptosis (chromatin
condensation and nuclear fragmentation).
Proliferation assay. To measure sensitivity to the rapamycin analogueCCI-779 in vitro , LKR-13 cells were plated (1,000 cells/well) in 96-well tissue
culture plates and incubated for 24 hours before being exposed to different
concentrations of CCI-779 or conditioned medium from macrophages.
Proliferation was quantified after 3 days by MTT assays.Syngeneic tumors. To measure the sensitivity of LKR-13 cells to CCI-779
in vivo , 107 LKR-13 cells (in 100 AL PBS) were injected s.c. into 129/Sv mice.
Five days later, when tumor volume had reached 50 mm3, mice were
randomly assigned to one of two experimental groups [CCI-779 20 mg/kg/d
(n = 10) or vehicle (n = 10) given i.p.]. Tumor diameters were measured dailyby an investigator (M.W.) who was blinded to treatment group. Tumor
volumes were calculated as follows: 0.5 (greatest diameter) � (shortest
diameter)2. Treatment was continued until one tumor reached 1.5 cm3
(8 days), at which time all mice were killed. Tumors (n = 36) were thenremoved and either frozen (�80jC) for protein extraction or fixed in 10%
formalin overnight before being embedded in paraffin.
Statistical analysis. For statistical analysis, the immunostaining scores,
tumor numbers, and tumor volumes were considered continuous variables,whereas new tumor formation was considered a categorical variable. The
Mann-Whitney nonparametric test was used to compare the continuous
variables with Bonferroni correction. P = 0.05 was considered significant
for two pair-wise comparisons (experiments with syngenic tumors), 0.017for three pair-wise comparisons (mouse experiments with three treatment
arms), and 0.012 for four pair-wise comparisons (comparison of expression
in lung lesions). Fisher’s exact test was used to compare categoricalvariables. Spearman’s r coefficient was used for correlative studies between
quantitative variables and P = 0.05 was considered significant. Data were
processed with StatView and Survival Tools 5.0 (Abacus Concepts,
Berkeley, CA).
Results
Activation of AKT-dependent signaling with malignantprogression. We examined p-S6, which increases with mTORactivation, in lung tissues from K-rasLA1 mice (relevant signalingpathways are summarized in Supplementary Fig. S1). We assessed
Figure 1. Typical histologic features of normal lung (A), AAH (B ), adenoma (C ),and adenocarcinoma (D ) from 16- to 20-week-old K-rasLA1 mice stained withH&E and visualized at �20 magnification. Columns, mean PCNA score ofpositive cells per square at �40 magnification for each histologic subtype; bars,SE (E).
Cancer Research
Cancer Res 2005; 65: (8). April 15, 2005 3228 www.aacrjournals.org
Research. on June 4, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

p-S6 by immunohistochemical analysis of a tissue microarraycontaining samples of murine normal lung, AAH, adenoma, andadenocarcinoma to determine whether p-S6 levels changed duringmalignant progression. To compare the K-rasLA1 mice to patientswith lung adenocarcinoma, we also examined p-S6 in a tissuemicroarray containing human normal lung, AAH, BAC and adeno-carcinoma. Typical histologic features of precursor lesions andadenocarcinoma from the K-rasLA1 mice are illustrated in Fig. 1Ato D . Phenotypically, the precursors were low-grade, noninvasiveepithelial cells, demonstrating no evidence of invasion through thebasement membrane and low proliferative activity, as shown byPCNA staining (Fig. 1E).Although the frequency of p-S6 staining was not different in cells
along the progression to lung adenocarcinoma in humans or inK-rasLA1 mice (p-S6 was detected in 60-100% of the lesions in eachcategory), the degree of staining (intensity � extension) increasedwith progression from normal lung to adenocarcinoma in K-rasLA1
mice (Fig. 2A-D) and from normal lung to AAH in human tissues(Fig. 2E -H ). Thus, mTOR signaling was activated in earlyprecursors of lung adenocarcinoma in K-rasLA1 mice and humans.Macrophages infiltrate AAH and express p-S6. A subset of the
cells within AAH and adenomas that expressed p-S6 had typicalepithelial morphology. In addition, a morphologically distinctpopulation of cells in K-rasLA1 mice also stained positively for p-S6.This population exhibited more intense cytoplasmic staining andmorphologic characteristics of macrophages. To better characterizethis population, we did immunohistochemical analysis of F4/80,a murine macrophage marker, and observed a population ofintralesional cells that stained positively (Fig. 3A-D). Immunoflu-orescence staining for F4/80 and p-S6 revealed a population of cellsin which p-S6 colocalized with F4/80 (Fig. 3E-H), supporting the
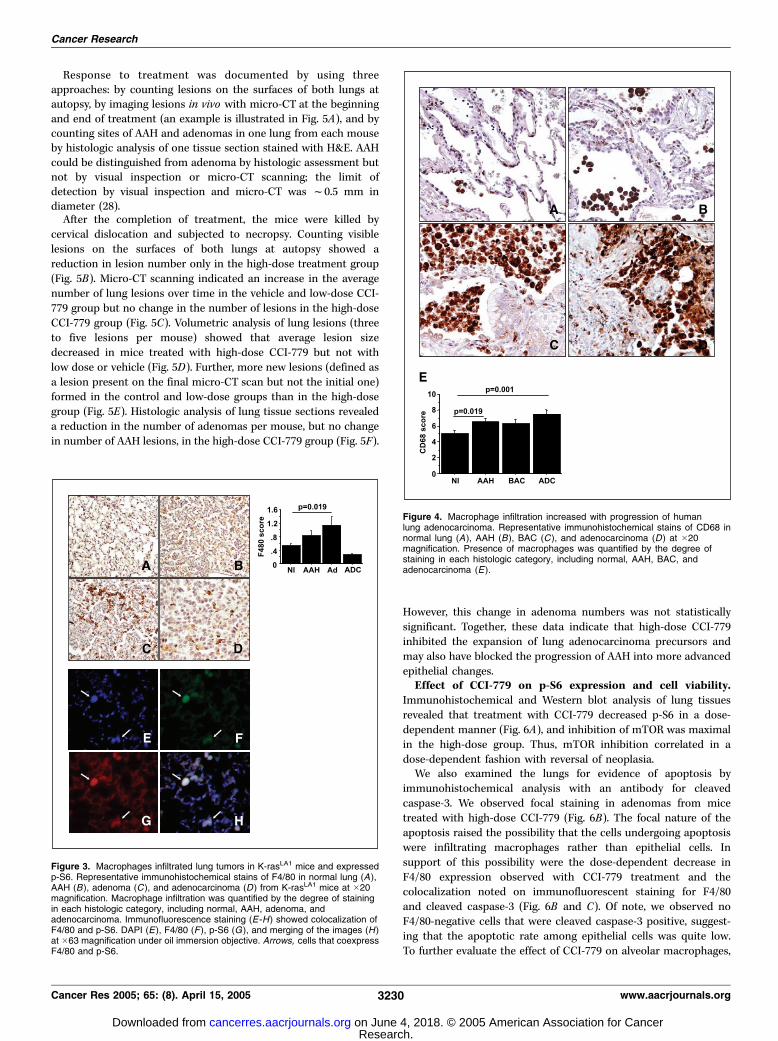
immunohistochemical evidence that macrophages were expressingp-S6. We next examined whether the prevalence of macrophageschanged with malignant progression. In samples from bothhumans and K-rasLA1 mice, the numbers of macrophages increasedwith malignant progression ( from normal lung to adenocarcinomain human and from normal lung to adenoma in mouse; Fig. 4).The prevalence of macrophages in K-rasLA1 mice dropped withthe progression of adenoma to adenocarcinoma, indicating thatthe association of macrophages with epithelial precursors wasstage specific (Fig. 3).CCI-779 inhibited malignant progression in K-rasLA1 mice.
Based on these findings, we hypothesized that S6 phosphorylationis required for malignant progression. Because S6Ser236/235 isphosphorylated by S6K through mTOR-dependent mechanisms,we investigated the effect of treatment with CCI-779, a mTORinhibitor, in K-rasLA1 mice, hypothesizing that loss of p-S6 wouldblock progression of AAH to adenoma. CCI-779 treatment wasbegun at 16 weeks of age, at which time AAH and adenomas wereidentifiable but adenocarcinomas were not yet evident. The dosesof CCI-779 (0.1 or 20 mg/kg daily for 4 weeks) were chosen basedon a previous study that showed dose-dependent growthinhibition of phosphatase and tensin homologue on chromosome10 (PTEN) wild-type xenografts in severe-combined immunodefi-cient mice (13). The only appreciable toxicity we observed withCCI-779 treatment was weight loss, which occurred in the high-dose group. Mean changes in body weight (expressed as thepercentage of total body weight F SD) were 2.64 F 3.27% in thecontrol group, 1.80 F 3.03% in the low-dose group, and �3.99 F4.74% in the high-dose group. Weight loss in the high-dose groupdid not correlate with treatment-induced changes in meannumber of tumors (P = 0.44, q = 0.21).
Figure 2. p-S6 increased with malignant progression in lungtissue from K-rasLA1 mice and patients with adenocarcinoma.Murine (A -D ) and human (E-H ) lung tissues illustrated includerepresentative normal lung (A and E), AAH (B and F ), adenoma(C ), BAC (G ), and adenocarcinoma (D and H ) at �20magnification. Degree of p-S6 staining for each histologiccategory, including normal (Nl), AAH, adenoma (Ad ), BAC, andadenocarcinoma (ADC ) in K-rasLA1 mice (top ) and patients withlung adenocarcinoma (bottom ).
mTOR, Macrophage Survival, and Lung Adenocarcinoma
www.aacrjournals.org 3229 Cancer Res 2005; 65: (8). April 15, 2005
Research. on June 4, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from
Response to treatment was documented by using threeapproaches: by counting lesions on the surfaces of both lungs atautopsy, by imaging lesions in vivo with micro-CT at the beginningand end of treatment (an example is illustrated in Fig. 5A), and bycounting sites of AAH and adenomas in one lung from each mouseby histologic analysis of one tissue section stained with H&E. AAHcould be distinguished from adenoma by histologic assessment butnot by visual inspection or micro-CT scanning; the limit ofdetection by visual inspection and micro-CT was f0.5 mm indiameter (28).After the completion of treatment, the mice were killed by
cervical dislocation and subjected to necropsy. Counting visiblelesions on the surfaces of both lungs at autopsy showed areduction in lesion number only in the high-dose treatment group(Fig. 5B). Micro-CT scanning indicated an increase in the averagenumber of lung lesions over time in the vehicle and low-dose CCI-779 group but no change in the number of lesions in the high-doseCCI-779 group (Fig. 5C). Volumetric analysis of lung lesions (threeto five lesions per mouse) showed that average lesion sizedecreased in mice treated with high-dose CCI-779 but not withlow dose or vehicle (Fig. 5D). Further, more new lesions (defined asa lesion present on the final micro-CT scan but not the initial one)formed in the control and low-dose groups than in the high-dosegroup (Fig. 5E). Histologic analysis of lung tissue sections revealeda reduction in the number of adenomas per mouse, but no changein number of AAH lesions, in the high-dose CCI-779 group (Fig. 5F).
However, this change in adenoma numbers was not statisticallysignificant. Together, these data indicate that high-dose CCI-779inhibited the expansion of lung adenocarcinoma precursors andmay also have blocked the progression of AAH into more advancedepithelial changes.Effect of CCI-779 on p-S6 expression and cell viability.
Immunohistochemical and Western blot analysis of lung tissuesrevealed that treatment with CCI-779 decreased p-S6 in a dose-dependent manner (Fig. 6A), and inhibition of mTOR was maximalin the high-dose group. Thus, mTOR inhibition correlated in adose-dependent fashion with reversal of neoplasia.We also examined the lungs for evidence of apoptosis by
immunohistochemical analysis with an antibody for cleavedcaspase-3. We observed focal staining in adenomas from micetreated with high-dose CCI-779 (Fig. 6B). The focal nature of theapoptosis raised the possibility that the cells undergoing apoptosiswere infiltrating macrophages rather than epithelial cells. Insupport of this possibility were the dose-dependent decrease inF4/80 expression observed with CCI-779 treatment and thecolocalization noted on immunofluorescent staining for F4/80and cleaved caspase-3 (Fig. 6B and C). Of note, we observed noF4/80-negative cells that were cleaved caspase-3 positive, suggest-ing that the apoptotic rate among epithelial cells was quite low.To further evaluate the effect of CCI-779 on alveolar macrophages,
Figure 3. Macrophages infiltrated lung tumors in K-rasLA1 mice and expressedp-S6. Representative immunohistochemical stains of F4/80 in normal lung (A),AAH (B ), adenoma (C ), and adenocarcinoma (D ) from K-rasLA1 mice at �20magnification. Macrophage infiltration was quantified by the degree of stainingin each histologic category, including normal, AAH, adenoma, andadenocarcinoma. Immunofluorescence staining (E-H) showed colocalization ofF4/80 and p-S6. DAPI (E), F4/80 (F ), p-S6 (G ), and merging of the images (H )at �63 magnification under oil immersion objective. Arrows, cells that coexpressF4/80 and p-S6.
Figure 4. Macrophage infiltration increased with progression of humanlung adenocarcinoma. Representative immunohistochemical stains of CD68 innormal lung (A ), AAH (B), BAC (C ), and adenocarcinoma (D) at �20magnification. Presence of macrophages was quantified by the degree ofstaining in each histologic category, including normal, AAH, BAC, andadenocarcinoma (E ).
Cancer Research
Cancer Res 2005; 65: (8). April 15, 2005 3230 www.aacrjournals.org
Research. on June 4, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

we treated primary cultures of alveolar macrophages derived fromK-rasLA1 mice (n = 4) with CCI-779 using doses comparable withthe 15 Amol/L serum levels achieved in mice treated with a singledose of CCI-779 at 20 mg/kg.5 Staining with bisbenzimide(Hoechst 33342) revealed that treatment with CCI-779 inducedchromatin condensation and nuclear fragmentation, morphologicchanges consistent with apoptosis (Fig. 6D).Sensitivity of K-rasLA1-derived tumor cells to CCI-779
differed in vitro and in vivo . Several host factors are importantin lung tumor progression, including the intratumoral migration ofendothelial cell precursors and inflammatory cells (32–34). Giventhe prominence of macrophages in this model and their sensitivityto CCI-779, we hypothesized that macrophages promote lungtumorigenesis and that macrophage loss is required for theantitumor effect of CCI-779 in K-rasLA1 mice. We examined theeffect of CCI-779 treatment on LKR-13 cells, an adenocarcinomacell line derived from K-rasLA1 mice. Treatment of LKR-13 cellswith 20 Amol/L CCI-779 in vitro inhibited p-S6 levels (Fig. 7A) but
had no detectable effect on LKR-13 cell proliferation (data notshown), indicating that p-S6 inhibition was not sufficient to inhibitgrowth. To examine LKR-13 cells in an in vivo context, weestablished LKR-13 cells as syngeneic tumors in K-rasLA1 wild-typelittermates. LKR-13 cells were highly tumorigenic; tumors grew inall 20 of the injected mice and showed central necrosis at necropsy.Daily treatment of the mice with CCI-779 (20 mg/kg) decreasedtumor volume relative to that of controls (P < 0.05; Fig. 7B).Immunofluorescence staining revealed a population of intra-tumoral cells that coexpressed F4/80 and p-S6, indicating thatmacrophages infiltrated the tumor and expressed p-S6 (Fig. 7C).Treatment with CCI-779 decreased intratumoral p-S6 levels (Fig.7D), demonstrating mTOR inhibition, and F4/80 levels (Fig. 7E),indicating that CCI-779 decreased intratumoral macrophages.Taken together, treatment with CCI-779 inhibited the growth ofLKR-13 cells in vivo but not in vitro , raising the possibility that CCI-779 mediated its antitumor effect by inhibiting host factorsrequired for the growth of LKR-13 cells in the in vivo setting.Finally, we examined whether factors secreted from alveolar
macrophages have a direct effect on tumor cells from K-rasLA1
mice. We isolated alveolar macrophages from wild-type littermates(as opposed to K-rasLA1 mice) to avoid contaminating neutrophils
Figure 5. Treatment with high-dose CCI-779 decreased the size and number of precursor lesions. A, representative micro-CT scan axial images of one mouse atthe beginning (left ) and end (right ) of treatment with CCI-779. Lesions that appear as consolidations in the right lower lobe and left lower lobe (arrows ) decreasedmarkedly with treatment. B, lesion numbers were determined for the three treatment groups by autopsy. Total lesions (C ) and new lesions (E ) were quantified forthe three treatment groups by micro-CT scanning done at the beginning and end of treatment. Mean changes in lesion volume (D ) were determined for the threetreatment groups by volumetric analysis of three to five lesions per mouse at the beginning and end of treatment. F, numbers of AAH and adenomas were counted ina single tissue section from each mouse in the three treatment groups [mean (SE)].
5 J. Gibbons, personal communication.
mTOR, Macrophage Survival, and Lung Adenocarcinoma
www.aacrjournals.org 3231 Cancer Res 2005; 65: (8). April 15, 2005
Research. on June 4, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

and tumor cells. F4/80 staining of cytospin preparations confirmedthat >97% of the cells were macrophages (data not shown).We collected conditioned medium from alveolar macrophagesamples, transferred conditioned medium (or control medium) to
LKR-13 cells or a second cell line derived from a K-rasLA1 lungadenocarcinoma (LKR-10) for 24 hours, and examined changesin proliferation by MTT assay. We did seven experiments withLKR-13 cells and seven experiments with LKR-10 cells, each with
Figure 6. CCI-779 decreased p-S6 in a dose-dependent manner and induced apoptosis of intralesional macrophages. Western blot analysis of p-S6 and total S6 (A)and CD68 and actin (C ) in whole lung extracts from 12 mice and representative immunohistochemical stains of p-S6 (A ), cleaved caspase-3 (CC-3 ; B), and F4/80 (C)in K-rasLA1 mice treated with vehicle, low-dose CCI-779 (Low ), or high-dose CCI-779 (High). Columns, mean degree of immunohistochemical staining in thethree treatment groups; bars, SE. B, representative immunofluorescent staining (�63 magnification under oil immersion objective) for DAPI (a ), F4/80 (b), cleavedcaspase-3 (c), and merging of the images (d) of a lung section from a mouse treated with high-dose CCI-779. Arrowheads, cells that coexpress F4/80 and cleavedcaspase-3. D, alveolar macrophages isolated from 129/Sv mice were subjected to treatment with different doses of CCI-779 for 24 hours and stained with Hoechst33342 dye. Typical morphologic characteristics of cells examined by fluorescence microscopy (�63 under oil immersion objective) treated with CCI-779 or mediumalone. Columns, mean percentages of cells with morphologic characteristics of apoptotic cells (chromatin condensation and nuclear fragmentation); bars, SE.
Cancer Research
Cancer Res 2005; 65: (8). April 15, 2005 3232 www.aacrjournals.org
Research. on June 4, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

a separate conditioned medium sample. Relative to control,conditioned medium increased cell proliferation by a factor of 1.26F 0.15 (mean F SD for the 14 experiments; P = 0.01; results ofrepresentative experiments are in Supplementary Fig. S2).
Discussion
Genetic mouse models of cancer are tools for investigating thesufficiency of specific genetic events in tumorigenesis. In K-rasLA1
mice, the rapidity with which AAH develops and progresses toadenoma supports the concept that K-ras mutation is an initiatingevent in lung adenocarcinoma. Here, we report that mTOR wasactivated in AAH, and mTOR inhibition was sufficient to decrease the
size and number of AAH and adenomas and may reverse progressionof AAH to adenoma, suggesting that the expansion of lungadenocarcinoma precursors induced by oncogenic K-ras requiresmTOR-dependent signaling. Human AAH and invasive lung adeno-carcinoma share a variety of genetic and biochemical features otherthan K-ras mutations (35–40). Certain genetic changes described inNSCLC are sufficient to induce AAH when modeled in mice (41).Thus, AAH is a histologic change caused by a variety of genetic eventsthat can initiate the development of lung adenocarcinoma.Several genetic events described previously in human NSCLC can
activate mTOR, including deletion of the tumor suppressors PTENand LKB1/STK11 , amplification of the p110 catalytic subunit of PI3K(PI3KCA), and activating mutations in PI3KCA , epidermal growth
Figure 7. Sensitivity of K-rasLA1-derivedtumor cells to CCI-779 differed in vitro andin vivo . A, Western analysis was done onLKR-13 cells (40 Ag/sample) treated for24 hours with 20 Amol/L CCI-779 (+) ormedium alone (- ). B-E, LKR-13 cells wereestablished as syngenic tumors in 129/Svmice, and the mice were treated withCCI-779 or vehicle alone. B, tumors weremeasured daily. Points, mean tumorvolumes; bars, SE. C, the mice were killed,and the tumors were subjected toimmunofluorescence staining with DAPI(a), F4/80 (b), and p-S6R (c ), and mergingof the images (d ). Magnification, �63.Arrowheads, cells that coexpress p-S6 andF4/80. D and E, effect of treatment withCCI-779 on intratumoral p-S6 (D ) wasexamined by Western blotting (40 Ag/sample) and immunohistochemicalanalysis, and changes in F4/80 (E) wereexamined by immunohistochemicalanalysis. Columns, mean degree ofimmunohistochemical staining in thetreatment groups; bars, SE.
mTOR, Macrophage Survival, and Lung Adenocarcinoma
www.aacrjournals.org 3233 Cancer Res 2005; 65: (8). April 15, 2005
Research. on June 4, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

factor receptor, or K-ras (42–46). The increase in p-S6 we observedwith progression from normal lung to AAH in K-rasLA1 micesupports a role for oncogenic K-ras in S6 activation. However, twoother models of lung tumorigenesis that conditionally expressmutant K-ras (K-rasG12V-IRES-BGeo and Lox-K-rasG12D ; refs. 47, 48)differ with respect to activation of AKT-dependent signaling; AKTis activated in K-rasG12V-IRES-BGeo but not Lox-K-rasG12D mice.Given the presence of mutant K-ras in all three models, it is not clearwhy they differ with respect to evidence for AKT activation. Onepotential contributing factor may be ongoing genetic damage,which was evident in K-rasG12V-IRES-BGeo but not Lox-K-rasG12D . Infact, stochastic genetic events contribute to the phenotypes of othermouse models of human cancer (49, 50). Interestingly, we observedthat p-S6 increased with the progression of AAH to adenocarcino-ma, supporting the possibility that genetic events other than K-rasmutations contributed to S6 activation in K-rasLA1 mice.In the setting of PTEN gene loss or AKT transgene expression,
inhibition of mTOR with low doses of CCI-779 (1 nmol/L) orrapamycin induces proliferative arrest or apoptosis (51, 52). In cellswithout these genetic changes, treatment with low doses of theseagents inhibits mTOR but has no effect on proliferation or survival,indicating that mTOR inhibition is not sufficient to inhibit growth;high doses, on the other hand, are cytostatic (13). Our findings in K-rasLA1 mice are partly consistent with the latter study in that a highdose of CCI-779 was required to inhibit tumor progression but,unlike the latter group, low-dose treatment was not sufficient tocompletely suppress p-S6, and the cytostatic effect correlated ina dose-dependent manner with complete suppression of p-S6.Although we do not know the minimal dose of CCI-779 required forcomplete suppression of p-S6 in K-rasLA1 mice, this minimal dosemay have targets other than mTOR.Cells derived from genetic mouse models of cancer are tools for
investigating survival pathways in the setting of specific oncogenicevents. For example, in PTEN heterozygous cells, mTOR inhibition isprimarily cytostatic, whereas selective activation of AKT sensitizescells to apoptosis in response to mTOR inhibition, suggesting thatcells transformed by PTEN loss may have survival pathways notavailable to cells transformed by AKT activation (11, 51–53). In thisstudy, treatment of LKR-13 cells with CCI-779 in vitro inhibited p-S6but had no effect on LKR-13 cell proliferation, suggesting that cellstransformed by mutant K-ras have survival pathways other thanthose activated by mTOR. In contrast to the resistance we observedin LKR-13 cells, CCI-779 induced apoptosis of macrophages,indicating a cell type–specific role of mTOR in cell survival.Intraepithelial macrophages in K-rasLA1 mice expressed p-S6 atlevels higher than those of adjacent epithelial cells, raising thepossibility that high levels of mTOR sensitized alveolar macrophagesto mTOR inhibition. The mechanisms responsible for S6 phosphor-ylation in macrophages are unclear. Macrophages from both wild-type and K-rasLA1 mice showed high levels of p-S6 in tissue section(Figs. 3E-F and 7C) and after isolation by BAL (data not shown),indicating that the presence of K-ras mutations is not required. Onepossibility is S6 phosphorylation by cytokines released from Ras-transformed epithelial cells.
Interactions between lung cancer cells and inflammatorycells can affect lung tumor growth positively or negatively.
However, a growing body of experimental and clinical data has
led to the proposal of a model in which lung cancer cells secrete
CC chemokines, recruiting intratumoral macrophages, whichinteract with tumor cells to enhance tumor cell proliferation
and invasion through a variety of mediators (54–58). Several lines
of evidence presented here suggest that alveolar macrophagesmay also contribute to the transformation of alveolar epithelial
cells. In human lung tissues, numbers of intraepithelial and
airspace macrophages increased with malignant progression. InK-rasLA1 mice, we observed intraepithelial macrophage infiltration
in AAH, indicating that alveolar epithelial cells recruited macro-
phages in the early stages of neoplasia induced by oncogenicK-ras . The inhibition of AAH progression to adenoma induced by
treatment with CCI-779 was accompanied by macrophage loss.
LKR-13 cells were resistant to CCI-779 in vitro , but when they
were established as syngeneic tumors LKR-13 cells recruitedintratumoral macrophages and were sensitive to treatment with
CCI-779. Lastly, conditioned medium from primary cultures of
alveolar macrophages stimulated the proliferation of LKR-13 cells,which is consistent with previous reports demonstrating a
stimulatory effect of alveolar macrophages on the proliferation
of normal proximal and distal bronchial epithelial cells in rat andbovine models (59, 60). Together, these findings provide
substantial evidence that macrophages may be important in the
expansion of early adenocarcinoma precursors and in maintain-
ing the survival of established lung cancer cells induced byoncogenic K-ras .Finally, the similarities we observed between lung tissues from
K-rasLA1 mice and patients with lung adenocarcinoma suggest that
findings from this study may have clinical applications. Findings
presented here suggest that host factors are required for theantitumor effect of CCI-779 in K-rasLA1 mice. We have shown that
one of these host factors may be intraepithelial macrophages,
which secrete several cytokines that stimulate epithelial cellproliferation and tumor angiogenesis (54–58). However, our
findings have not excluded the possibility that CCI-779 also had
direct effects on intratumoral epithelial cells or vascularendothelial cells, inhibiting tumor growth directly or indirectly.
Although the current clinical application of mTOR inhibition is
targeted at late-stage cancers, the response of this and other early
neoplastic disease models to mTOR inhibition (53) raises thepossibility that these agents may find application in treating early
lung cancers in humans.
Acknowledgments
Received 12/10/2004; revised 1/28/2005; accepted 2/4/2005.Grant support: National Cancer Institute grants R01 CA105155, P50 CA070907
(Lung Cancer Specialized Program of Research Excellence), and P30 CA016672.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
We thank Diane Liu for assistance in performing statistical analysis.
References
1. Greenlee RT, Murray T, Bolden S, Wingo PA. Cancerstatistics, 2000. CA Cancer J Clin 2000;50:7–33.
2. Tsao AS, Kim ES, Hong WK. Chemoprevention ofcancer. CA Cancer J Clin 2004;54:150–80.
3. Westra WH. Early glandular neoplasia of the lung.Respir Res 2000;1:163–9.
4. Lee KS, Kim Y, Han J, Ko EJ, Park CK, Primack SL.Bronchioloalveolar carcinoma: clinical, histopatholo-gic, and radiologic findings. Radiographics 1997;17:1345–57.
Cancer Research
Cancer Res 2005; 65: (8). April 15, 2005 3234 www.aacrjournals.org
Research. on June 4, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

5. Tsao AS, McDonnell T, Lam S, et al. Increasedphospho-AKT (Ser(473)) expression in bronchialdysplasia: implications for lung cancer preventionstudies. Cancer Epidemiol Biomarkers Prev 2003;12:660–4.
6. West KA, Brognard J, Clark AS, et al. Rapid Aktactivation by nicotine and a tobacco carcinogenmodulates the phenotype of normal human airwayepithelial cells. J Clin Invest 2003;111:81–90.
7. Brognard J, Clark AS, Ni Y, Dennis PA. Akt/proteinkinase B is constitutively active in non-small cell lungcancer cells and promotes cellular survival andresistance to chemotherapy and radiation. Cancer Res2001;61:3986–97.
8. Lee HY, Srinivas H, Xia D, et al. Evidence thatphosphatidylinositol 3-kinase- and mitogen-activatedprotein kinase kinase-4/c-Jun NH2-terminal kinase-dependent pathways cooperate to maintain lung cancercell survival. J Biol Chem 2003;278:23630–8.
9. Avdulov S, Li S, Michalek V, et al. Activation oftranslation complex eIF4F is essential for the genesisand maintenance of the malignant phenotype inhuman mammary epithelial cells. Cancer Cell 2004;5:553–63.
10. Wendel HG, De Stanchina E, Fridman JS, et al.Survival signalling by Akt and eIF4E in oncogenesis andcancer therapy. Nature 2004;428:332–7.
11. Majumder PK, Yeh JJ, George DJ, et al. Prostateintraepithelial neoplasia induced by prostate restrictedAkt activation: the MPAKT model. Proc Natl Acad SciU S A 2003;100:7841–6.
12. Ruggero D, Montanaro L, Ma L, et al. The translationfactor eIF-4E promotes tumor formation and cooper-ates with c-Myc in lymphomagenesis. Nat Med 2004;10:484–6.
13. Neshat MS, Mellinghoff IK, Tran C, et al. Enhancedsensitivity of PTEN-deficient tumors to inhibition ofFRAP/mTOR. Proc Natl Acad Sci U S A 2001;98:10314–9.
14. Downward J. PI 3-kinase, Akt and cell survival. SeminCell Dev Biol 2004;15:177–82.
15. Manning BD, Tee AR, Logsdon MN, Blenis J, CantleyLC. Identification of the tuberous sclerosis complex-2tumor suppressor gene product tuberin as a target ofthe phosphoinositide 3-kinase/akt pathway. Mol Cell2002;10:151–62.
16. Gao X, Zhang Y, Arrazola P, et al. Tsc tumoursuppressor proteins antagonize amino-acid-TOR sig-nalling. Nat Cell Biol 2002;4:699–704.
17. Gao X, Pan D. TSC1 and TSC2 tumor suppressorsantagonize insulin signaling in cell growth. Genes Dev2001;15:1383–92.
18. Potter CJ, Huang H, Xu T. Drosophila Tsc1 functionswith Tsc2 to antagonize insulin signaling in regulatingcell growth, cell proliferation, and organ size. Cell2001;105:357–68.
19. Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J.Tuberous sclerosis complex gene products, Tuberin andHamartin, control mTOR signaling by acting as aGTPase-activating protein complex toward Rheb. CurrBiol 2003;13:1259–68.
20. Burnett PE, Barrow RK, Cohen NA, Snyder SH,Sabatini DM. RAFT1 phosphorylation of the translationalregulators p70 S6 kinase and 4E-BP1. Proc Natl Acad SciU S A 1998;95:1432–7.
21. Dennis PB, Jaeschke A, Saitoh M, Fowler B, KozmaSC, Thomas G. Mammalian TOR: a homeostatic ATPsensor. Science 2001;294:1102–5.
22. Isotani S, Hara K, Tokunaga C, Inoue H, Avruch J,Yonezawa K. Immunopurified mammalian target ofrapamycin phosphorylates and activates p70 S6 kinasea in vitro . J Biol Chem 1999;274:34493–8.
23. Seki N, Takasu T, Mandai K, et al. Expression ofeukaryotic initiation factor 4E in atypical adenomatoushyperplasia and adenocarcinoma of the human periph-eral lung. Clin Cancer Res 2002;8:3046–53.
24. Rosenwald IB, Hutzler MJ, Wang S, Savas L, FraireAE. Expression of eukaryotic translation initiationfactors 4E and 2a is increased frequently in bronchio-loalveolar but not in squamous cell carcinomas of thelung. Cancer 2001;92:2164–71.
25. Boffa DJ, Luan F, Thomas D, et al. Rapamycin inhibitsthe growth and metastatic progression of non-small celllung cancer. Clin Cancer Res 2004;10:293–300.
26. Johnson L, Mercer K, Greenbaum D, et al. Somaticactivation of the K-ras oncogene causes early onsetlung cancer in mice. Nature 2001;410:1111–6.
27. Rodriguez-Viciana P, Warne PH, Khwaja A, et al. Roleof phosphoinositide 3-OH kinase in cell transformationand control of the actin cytoskeleton by Ras. Cell1997;89:457–67.
28. Cavanaugh D, Johnson E, Price RE, Kurie J, Travis EL,Cody DD. In vivo respiratory-gated micro-CT imaging insmall-animal oncology models. Mol Imaging 2004;3:55–62.
29. Lee HY, Suh YA, Lee JI, et al. Inhibition of oncogenicK-ras signaling by aerosolized gene delivery in a mousemodel of human lung cancer. Clin Cancer Res 2002;8:2970–5.
30. Mirtcheva RM, Vazquez M, Yankelevitz DF, HenschkeCI. Bronchioloalveolar carcinoma and adenocarcinomawith bronchioloalveolar features presenting as ground-glass opacities on CT. Clin Imaging 2002;26:95–100.
31. Travis WD, Colby TV, Corrin B, Shimosato Y,Branhamera, editors. Histologic typing of lung andpleural tumors. WHO international histological classi-fication of tumours. Berlin: Springer; 1999.
32. Balkwill F, Mantovani A. Inflammation and cancer:back to Virchow? Lancet 2001;357:539–45.
33. Wislez M, Rabbe N, Marchal J, et al. Hepatocytegrowth factor production by neutrophils infiltratingbronchioloalveolar subtype pulmonary adenocarcino-ma: role in tumor progression and death. Cancer Res2003;63:1405–12.
34. Kataki A, Scheid P, Piet M, et al. Tumor infiltratinglymphocytes and macrophages have a potential dualrole in lung cancer by supporting both host-defense andtumor progression. J Lab Clin Med 2002;140:320–8.
35. Nakanishi K, Kawai T, Kumaki F, Hiroi S, Mukai M,Ikeda E. Survivin expression in atypical adenomatoushyperplasia of the lung. Am J Clin Pathol 2003;120:712–9.
36. Tominaga M, Sueoka N, Irie K, et al. Detection anddiscrimination of preneoplastic and early stages of lungadenocarcinoma using hnRNP B1 combined with thecell cycle-related markers p16, cyclin D1, and Ki-67.Lung Cancer 2003;40:45–53.
37. Yamasaki M, Takeshima Y, Fujii S, et al. Correlationbetween genetic alterations and histopathologicalsubtypes in bronchiolo-alveolar carcinoma and atypicaladenomatous hyperplasia of the lung. Pathol Int 2000;50:778–85.
38. Hosomi Y, Yokose T, Hirose Y, et al. Increasedcyclooxygenase 2 (COX-2) expression occurs frequentlyin precursor lesions of human adenocarcinoma of thelung. Lung Cancer 2000;30:73–81.
39. Hayashi H, Ito T, Yazawa T, et al. Reducedexpression of p27/Kip1 is associated with the devel-opment of pulmonary adenocarcinoma. J Pathol 2000;192:26–31.
40. Awaya H, Takeshima Y, Yamasaki M, Inai K.Expression of MUC1, MUC2, MUC5AC, and MUC6 inatypical adenomatous hyperplasia, bronchioloalveolarcarcinoma, adenocarcinoma with mixed subtypes, andmucinous bronchioloalveolar carcinoma of the lung.Am J Clin Pathol 2004;121:644–53.
41. Nikitin AY, Alcaraz A, Anver MR, et al. Classificationof proliferative pulmonary lesions of the mouse:recommendations of the mouse models of humancancers consortium. Cancer Res 2004;64:2307–16.
42. Ohshima S, Shimizu Y, Takahama M. Detection ofc-Ki-ras gene mutation in paraffin sections of
adenocarcinoma and atypical bronchioloalveolar cellhyperplasia of human lung. Virchows Arch 1994;424:129–34.
43. Samuels Y, Wang Z, Bardelli A, et al. High frequencyof mutations of the PIK3CA gene in human cancers.Science 2004;304:554.
44. Massion PP, Kuo WL, Stokoe D, et al. Genomic copynumber analysis of non-small cell lung cancer usingarray comparative genomic hybridization: implicationsof the phosphatidylinositol 3-kinase pathway. CancerRes 2002;62:3636–40.
45. Forgacs E, Biesterveld EJ, Sekido Y, et al. Mutationanalysis of the PTEN/MMAC1 gene in lung cancer.Oncogene 1998;17:1557–65.
46. Sordella R, Bell DW, Haber DA, Settleman J.Gefitinib-sensitizing EGFR mutations in lung canceractivate anti-apoptotic pathways. Science 2004;305:1163–7.
47. Guerra C, Mijimolle N, Dhawahir A, et al. Tumorinduction by an endogenous K-ras oncogene is highlydependent on cellular context. Cancer Cell 2003;4:111–20.
48. Tuveson DA, Shaw AT, Willis NA, et al. Endogenousoncogenic K-ras(G12D) stimulates proliferation andwidespread neoplastic and developmental defects.Cancer Cell 2004;5:375–87.
49. Moody SE, Sarkisian CJ, Hahn KT, et al. Conditionalactivation of Neu in the mammary epithelium oftransgenic mice results in reversible pulmonary metas-tasis. Cancer Cell 2002;2:451–61.
50. D’Cruz CM, Gunther EJ, Boxer RB, et al. c-MYCinduces mammary tumorigenesis by means of apreferred pathway involving spontaneous Kras2 muta-tions. Nat Med 2001;7:235–9.
51. Podsypanina K, Lee RT, Politis C, et al. An inhibitor ofmTOR reduces neoplasia and normalizes p70/S6 kinaseactivity in Pten+/� mice. Proc Natl Acad Sci U S A2001;98:10320–5.
52. Aoki M, Blazek E, Vogt PK. A role of the kinasemTOR in cellular transformation induced by the onco-proteins P3k and Akt. Proc Natl Acad Sci U S A 2001;98:136–41.
53. Majumder PK, Febbo PG, Bikoff R, et al. mTORinhibition reverses Akt-dependent prostate intraepi-thelial neoplasia through regulation of apoptotic andHIF-1-dependent pathways. Nat Med 2004;10:594–601.
54. Arenberg DA, Keane MP, DiGiovine B, et al.Macrophage infiltration in human non-small-cell lungcancer: the role of CC chemokines. Cancer ImmunolImmunother 2000;49:63–70.
55. Meyer AM, Dwyer-Nield LD, Hurteau GJ, et al.Decreased lung tumorigenesis in mice geneticallydeficient in cytosolic phospholipase A2. Carcinogenesis2004;25:1517–24.
56. Barbera-Guillem E, Nyhus JK, Wolford CC, FrieceCR, Sampsel JW. Vascular endothelial growth factorsecretion by tumor-infiltrating macrophages essentiallysupports tumor angiogenesis, and IgG immune com-plexes potentiate the process. Cancer Res 2002;62:7042–9.
57. Chen JJ, Yao PL, Yuan A, et al. Up-regulation of tumorinterleukin-8 expression by infiltrating macrophages: itscorrelation with tumor angiogenesis and patientsurvival in non-small cell lung cancer. Clin Cancer Res2003;9:729–37.
58. Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9supplied by bone marrow-derived cells contributes toskin carcinogenesis. Cell 2000;103:481–90.
59. Leslie CC, McCormick-Shannon K, Cook JL, Mason RJ.Macrophages stimulate DNA synthesis in rat alveolartype II cells. Am Rev Respir Dis 1985;132:1246–52.
60. Takizawa H, Beckmann JD, Shoji S, et al. Pulmonarymacrophages can stimulate cell growth of bovinebronchial epithelial cells. Am J Respir Cell Mol Biol1990;2:245–55.
mTOR, Macrophage Survival, and Lung Adenocarcinoma
www.aacrjournals.org 3235 Cancer Res 2005; 65: (8). April 15, 2005
Research. on June 4, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

2005;65:3226-3235. Cancer Res Marie Wislez, M. Loreto Spencer, Julie G. Izzo, et al.
K-rasAlveolar Epithelial Neoplasia Induced by Oncogenic Inhibition of Mammalian Target of Rapamycin Reverses
Updated version
http://cancerres.aacrjournals.org/content/65/8/3226
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2005/04/18/65.8.3226.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/65/8/3226.full#ref-list-1
This article cites 58 articles, 21 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/65/8/3226.full#related-urls
This article has been cited by 33 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/65/8/3226To request permission to re-use all or part of this article, use this link
Research. on June 4, 2018. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from


















