
Proptosis por Celulitis Orbitaria en paciente Pediátrico ...
Upload
hospital-pediatrico-de-sinaloaCategory
view
209download
4
UNIVERSIDAD AUTÓNOMA DE SINALOAHOSPITAL PEDIATRICO DE SINALOA
“DR. RIGOBERTO AGUILAR PICO”
"INCIDENCIA DE CROMOSOMOPATIAS EN EL LABORATORIO DECITOGENETICA DEL HOSPITAL PEDIATRICO DE SINALOA”
TESISDE POSTGRADO PARA OBTENER EL TITULO
DE LA ESPECIALIDAD DEPEDIATRIA MEDICA
PRESENTA:MIGUEL ANGEL SANTOS LEON
TUTOR DE TESIS:
JESUS ERNERSTO DUEÑAS ARIAS
CULIACAN, SINALOA; NOVIEMBRE DE 2013

AGRADECIMIENTOS
A mis padres cuyo apoyo ha resultado indispensable para tolerar la distancia y el arduo camino que mi especialidad en pediatria me ha llevado a recorrer, a mi madre que con su amor y cariño me ha mostrado que es posible superar cualquier obstaculo en la vida y en especial a mis niños, mis pacientes donde cada uno ha aportado una enseñanza para cada dia ser no tan solo mejor medico, sino por igual, mejor persona y ser humano.

ÍNDICE
CAPITULO I: Introducción
a) Marco teórico..........................................................................................Pag.1b) Antecedentes Científicos........................................................................Pag 1c) Planteamiento del Problema...................................................................Pag 9d) Justificación............................................................................................Pag.14e) Objetivo General y específico................................................................Pag.16
CAPITULO II.- Material y Métodos
a) tipo de estudio........................................................................................Pag.17b) Población objetivo con su ubicación espaciotemporal..........................Pag.17c) criterios de selección:.............................................................................Pag.17
• Criterios de inclusión.........................................................................Pag.17• Criterios de exclusión........................................................................Pag.17• Criterios de eliminación.....................................................................Pag.17
d) Metodología: Técnicas y procedimientos realizados.............................Pag.18
CAPITULO III.- Resultados
Incidencia por total de casos analizados................................................Pag. 19Incidencia por total de casos analizados por sexo.................................Pag. 19Frecuencia de muestras analizadas por año..........................................Pag. 20Frecuencia de muestras analizadas por grupos de edad.......................Pag. 21Incidencia de cromosomopatias encontradas .......................................Pag. 22Diagnosticos cromosomicos mas frecuentemente encontrados y representacion percentual..............................,.......................................Pag. 24Cromosomopatias por grupos de edad...................................................Pag. 24
Cromosomopatias por sexo....................................................................Pag. 25

CAPITULO IV.- Discusión
Discusion.................................................................................................... Pag. 24
CAPITULO VConclusiones........................................................................................... Pag. 25
BIBLIOGRAFIA.......................................................................................... Pag. 29
1

Capítulo I: Introducción.
Alteraciones Cromosómicas: Cariotipo como herramienta fundamental.
El termino cariotipo alude a al número y tipo de los cromosomas presentes en un
individuo, y cariograma se utiliza a menudo para designar la imagen impresa de
los cromosomas. El Cariotipado es el proceso de empatar y ordenar todos los
cromosomas de un organismo, con lo que se obtendría una foto instantánea lo
suficientemente amplia de los cromosomas de cualquier individuo. El cariotipo se
preparan usando tinciones estandarizadas que revelaran características
estructurales de cada cromosoma, con los que con ayuda de citogenetistas
clínicos se pueden detectar cambios genéticos gruesos. En el cariotipo se pueden
identificar cambios en el número de cromosomas lo que se como alteraciones
aneuploides o bien con un análisis más amplio se pueden identificar cambios
sutiles en su conformación pudiendo ser: Deleciones, duplicaciones y
translocaciones o inversiones. (1)
Preparación de un Cariotipo.
A partir de células de células en fase de mitosis, las cuales son estacionadas en
metafase o prometafase del ciclo celular, cuando los cromosomas muestran una
conformación mas condensada. Pudiendo ser una gran variedad de tejido los
pueden utilizarse como donadores de estas células, así hoy en día para el
diagnósticos oncológicos los especímenes típicos suelen ser biopsias del tumor o
bien muestras de un aspirado de medula ósea; Así, para otros diagnósticos, el
cariotipo es generado a partir de especímenes de sangre periférica o bien la piel,
además el diagnostico prenatal utiliza muestra de liquido amniótico o
vellosidades corionicas como tejidos donadores de células.
Antecedentes científicos: Detección de alteraciones.
Sin tratamiento alguno, los detalles estructurales de los cromosomas son difíciles
de detectar bajo un microscopio de luz. Por lo tanto, para realizar un análisis más
1

efectivo y eficiente, se han desarrollado tinciones que se unen al DNA y generan
patrones de unión característicos para diferentes cromosomas. Previo a estas
nuevas técnicas, el distinguir un cromosoma de otro era una labor muy difícil y por
lo tanto los cromosomas eran simplemente agrupados de acuerdo a su tamaño y
colocación de sus centrómeros.
Los métodos de tinción utilizados en la historia donde en 1970 Tobjom
Caspersson y sus colegas describieron la primer técnica del análisis de patrones
de unión, conocida como “Q-Banding” que involucraba el uso de la tinción
fluorescente con quinacrina y posteriormente con los avances de Caspersson y
colaboradores quienes demostraron que la quinacrina produce patrones de unión
característicos y reproducibles entre los cromosomas individuales. Hoy en dia la
mayoría de los cariotipos son teñidos con Giemsa, lo cual ofrece una mejor
resolución de las bandas individuales y produce una preparación más estable y
puede ser analizada por microscopia de campo brillante.
Las causas moleculares en las diferencias entre una tinción a lo largo de todo un
cromosoma son complejas y abarcan la composición de las bases de DNA y las
diferencias locales en la estructura de cromatina. En general, las regiones
heterocromaticas, las cuales tienden a ser regiones ricas en DNA-AT (por las
bases Adenina/Tiamina) y consideradas relativamente pobres genéticamente
hablando, tenden a teñirse mas obscuras en el G-Banding. En contraste, una
cromatina menos condensada la cual tiende a ser mas rica en DNA-GC (de las
bases Guanina/Citocina) consideradas mas activas transcripcionalmente,
incorpora una menor tinción de Giemsa y por lo tanto estas regiones aparecen
como bandas de luz en el G-Banding. Mas importantemente, el G-Banding
produce patrones reprodubles para cada cromosoma, y estos patrones son
compartidos de entre los individuos de una misma especia.
2

Figura 1: Bandeo cromosomatico revelado por diferentes tecnicas de tincion.
Diferentes tecnicas de tincion cromosomica revelan variaciones en sus
cromosomas. Citogenetistas usan estos patrones para reconocer las diferencias
de entre los cromosomas y asi ligarlos a diferentes fenotipos patologicos. Bandeo
con Giemsa (a), Bando-Q (b), Bandeo-R (c) y Bandeo-C (d).
El acomodar los cromosomas dentro de un cariográma puede simplificar la
identificación de cualquier anormalidad. Ya que los patrones de unión de entre dos
copias de cromosoma o bien “homólogos”, de cualquier autosoma (O cromosoma
no sexual) son casi idénticas. Algunas diferencias sutiles de entre los homólogos
de cualquier cromosoma puede ser atribuible a una variabilidad estructural natural
entre los individuos.
3

Usando el Cariotipo para detectar anormalidades cromosómicas.
Hoy en día los cariográmas basados en la tinción “G-Banded” es la herramienta
rutinaria para el diagnostico de un amplia variedad de anormalidades
cromosómicas en los individuos. Aunque la resolución de los cambios
cromosómicos detectables por un cariotipo convencional es solo de algunas
megabases, estas pueden ser suficientes para diagnosticas ciertas categorías de
anormalidades. Por ejemplo, aneuploidías, las cuales frecuentemente son
causadas por la ausencia o presencia de cromosomas, estas son detectadas por
un simple análisis de un cariotipo. Así también, los citogenetístas pueden también
frecuentemente detectar deleciónes o inserciones más sutiles como una
desviación de los patrones de unión normales. Por lo tanto, las translocaciónes
son rápidamente aparentes en un cariotipo.
El bandeo cromosómico es de gran utilidad en la detección y facilita la
dentificacion correcta de los cromosoms individuales. Las bandas principales de
cada cromosoma se numeran sistemáticamente asi por ejemplo, 14,q32 alude a la
segunda banda de la tercera región del brazo largo del cromosoma 14. Las sub-
bandas se designan por puntos decimales detrás del numero de la banda (p. ej.
14,q32.3 es la tercera sub-banda de la banda 2). Así por la colocación de los
centromeros establece a los cromosomas como metacéntricos (con su colocación
en medio) acrocentricos (colocaciona cerca a la punta) y submetacentricos (por su
colocación en algún lugar entre el centro y la punta). Asi también la clasificación
“q” “p” nos sirva para determinar si se trata de un brazo “corto” (de “Petite”,
francés: pequeño.) yo de un brazo “largo” (de “Queue”, Ingles: dejado atrás o al
final), los brazos “p” y “q” se designan por convección.
4

Alteraciones Cromosómicas: Métodos extendidos. Hibridación fluorescente “In Situ”.
Técnica en la cual un segmento de DNA monocatenario marcado (sonda) el cual
se expone a cromosomas desnaturalizados en metafase, profase o interfase. La
sonda experimenta emparejamiento de bases complementarias (hibridación) solo
con la secuencia complementaria de DNA en una ubicación específica en uno de
los cromosomas desnaturalizados. Al estar marcada la sonda con colorante
fluorescente, es posible visualizar la ubicación donde se hibrida con los
cromosomas del paciente al microscopio de fluorescencia. Un uso común de la
FISH es determinar si una parte de un cromosoma esta suprimida en un paciente.
Así en una persona normal, una sonda se hibrida en dos lugares, en reflejo de la
presencia de dos cromosomas homólogos en un núcleo celular somático. Si una
sonda del segmento cromosómico en cuestión se hibrida solo en uno de los
cromosomas del paciente, probablemente este tiene la delecion en la copia del
cromosoma en el que la sonda no se hibrida. La FISH ofrece una resolución
considerablemente superior a la de los métodos de bandeo de alta resolución,
normalmente puede detectar deleciones de apenas un millo de pares de bases (1
Mb) de tamaño.
El FISH también puede detectar copias adicionales de una región cromosómica.
En este caso, la sonda se hibrida en tres o más lugares en lugar de dos.
Asimismo, puede emplearse combinaciones de sonda de FISH para detectar
reordenamientos cromosómicos tales como translocaciones.
5

Planteamiento de Problema: Alteraciones Cromosómicas: Aspectos Generales.
Las anomalías cromosómicas son responsables de un porcentaje significativo de
las enfermedades genéticas y están presentes en 1 de cada 150 nacimientos
vivos aproximadamente. Constituyen la causa principal conocida de retraso mental
y pérdida fetal. Se observan anormalidades cromosómicas en el 50% de los
abortos espontáneos en el primer trimestre y 20% en el segundo. Por tanto,
representan una importante causa de co-morbilidad aunado a la relevancia de la
incidencia de fenotipos dismorficos en los recién nacidos así como fenotipos que
alertan al médico a la detección de anormalidades genéticas como el síndrome de
Down, trisomía del cromosoma 21 bien conocida y que tiene tiene repercusiones
globales en los sistemas del paciente afectado, relacionándose tempranamente
con la presencia de cardiopatías que pueden poner en riesgo la vida de estos
pacientes, por igual en pacientes con monosomías del cromosoma X como lo es el
Síndrome de Turner (Monosomía X0), en donde como en el síndrome de Down
hasta un 50% de las pacientes afectadas presentaran defectos cardiacos, estas
pacientes tendrán definitivamente problemas de talla, infertilidad y ausencia en el
desarrollo de caracteres sexuales secundarios, así es la relevancia de la
citogenética clínica en el campo medico y para el médico pediatra, ya que la
detección oportuna o bien la simple sospecha de estos trastornos podrán ser
suficientes para activar sistemas de alarma que llevaran a la monitorización de
estos pacientes, así como por igual es importante el conocimiento de todo clínico
de estas herramientas y que el citogenetísta debe de ser parte del manejo
complementario de todos los pacientes bajo sospecha de una alteración genética
ya que esto llevara a la detección oportuna de alteraciones que pongan en riesgo
la vida del paciente a su vez que establecerá factores pronostico para el paciente.
Poliploidía Se dice que una célula que contiene un múltiplo de 23 cromosomas en el núcleo
es euploide (griego, eu = “bueno”, ploid = “conjunto”). Asi, los gametos haploides y
6

las células somaticas diploides son euploides. La poliploidia – presencia de un
conjunto completo de cromosomas adicionales en una celula – se da en humanos,
aunque con mucha menos frecuencia. Los transtornos que se han observado en
humanos son la triploidía (69 cromosomas en el nucleo de cada celula) y la
tetraploidía (92 cromosomas en cada nucleo celular). Los cariotipos de estos dos
transtornos son 69, XXX y 92, XXXX, respectivamente (asumiendo que todos los
cromosomas sexuales fueran X, pueden darse otras combinaciones de
cromosomas X e Y). Dado que el numero de cromosomas presentes en cada uno
de estos transtornos es múltiplo de tres, las células son euploides en todos los
casos. No obstante, los cromosomas adicionales codifican una gran cantidad de
producto génico sobrante, causando multiples anomalías tales como defectos del
corazón y el sistema nervioso central.
La Triploidia solo esta presente en 1 de cada 10.000 nacidos vivos
aproximadamente, pero representa un 15% estimado de las anomalías
cromosómicas que se producen en la concepción. Asi, la gran mayoría de las
concepciones triploides se abortan de manera espontanea y este trnstorno
constituye una de las causas más frecuentes de perdida fetal en los dos primeros
trimestres del embarazo. Normalmente los fetos triploides que sobreviven hast el
nacimiento muere poco después. La tetraploidia es mucho mas infrecuente que la
triploidia, tanto en la concepción como en los nacimientos vivos. Solo se han
observado en pocos nacimientos vivos y los niños solo sobrevivieron un periodo
breve.
Las anomalías cromosómicas son responsables de un porcentaje significativo de
las enfermedades genéticas y están presentes en 1 de cada 150 nacimientos
vivos aproximadamente. Constituyen la causa principal conocida de retraso mental
y pérdida fetal. Se observan anormalidades cromosómicas en el 50% de los
abortos espontáneos en el primer trimestre y 20% en el segundo. Por tanto,
representan una importante causa de co-morbilidad aunado a la relevancia de la
incidencia de fenotipos dismorficos en los recién nacidos así como fenotipos que
7

alertan al médico a la detección de anormalidades genéticas como el síndrome de
Down, trisomía del cromosoma 21 bien conocida y que tiene tiene repercusiones
globales en los sistemas del paciente afectado, relacionándose tempranamente
con la presencia de cardiopatías que pueden poner en riesgo la vida de estos
pacientes, por igual en pacientes con monosomías del cromosoma X como lo es el
Síndrome de Turner (Monosomía X0), en donde por igual que el síndrome de
Down hasta un 50% de las pacientes afectadas presentaran defectos cardiacos,
estas pacientes tendrán definitivamente problemas de talla, infertilidad y ausencia
en el desarrollo de caracteres sexuales secundarios, así es la relevancia de la
citogenética clínica en el campo medico y para el médico pediatra, ya que la
detección oportuna o bien la simple sospecha de estos trastornos podrán ser
suficientes para activar sistemas de alarma que llevaran a la monitorización de
estos pacientes, asi como por igual es importante el conocimiento de todo clínico
de estas herramientas y que el citogenetísta debe de ser parte del manejo
complementario de todos los pacientes bajo sospecha de una alteración genética.
Aneuploidía autosómica Las células que contienen cromosomas individuales ausentes o adiciones se
denominan aneuplóides. Normalmente solo está afectado un cromosoma, pero es
posible que haya más de un cromosoma ausente o duplicado. Las Aneuploidías
de los autosomas se cuentan entre las anomalías cromosómicas clínicamente más
importantes. Consisten principalmente en monosomía (la presencia de solo una
copia de un cromosoma en una célula diploide por lo demás normal) y trisomía
(tres copias de un cromosoma). Las monosomías autosómicas son casi siempre
incompatibles con la supervivencia hasta el nacimiento y solo se han observado
pocos casos en individuos nacidos con vida. En cambio, algunas trisomías se dan
con frecuencia apreciables en los nacimientos con vida. El hecho de que las
trisomías produzcan consecuencias menos graves que las monosomías ilustra un
principio importante en la citogenética clínica, así se dice “el cuerpo puede tolerar
un exceso de material genético con mayor facilidad que su falta”.
8

La causa más frecuente de aneuploidía es la no disyunción, el fallo de los
cromosomas al dividirse normalmente durante la meiosis (Figura), la cual puede
producirse durante la meiosis I o la meiosis II. En el gameto resultante falta un
cromosoma o hay dos copias del mismo, lo que dará lugar a un cigoto
monosomico o trisomico, respectivamente.
Anomalías de la estructura cromosómica. Además de la pérdida o ganancia de cromosomas enteros, es posible que se
pierdan o duplique partes de cromosomas cuando se forman los gametos y se
altere la disposición de los cromosomas. Las anomalías cromosómicas
estructurales pueden presentarse como desequilibradas (el reordenamiento causa
una ganancia o una pérdida de material cromosómico) o equilibradas (el
reordenamiento no produce ni ganancia ni perdida del material cromosómico). Las
anomalías estructurales, sobre todo las desequilibradas, pueden provocar
enfermedad grave en los individuos o sus hijos.
9

Translocaciónes.
Una translocación es el intercambio de material genético entre cromosomas no
homólogos. Las translocaciónes equilibradas representan una de las aberraciones
cromosómicas más frecuentes en los humanos y están presentes en 1 de cada
500 a 1.000 Individuos, existen dos tipos básicos de translocaciónes: reciprocas y
Robertsonianas.
Translocaciónes recíprocas: Tienen lugar cuando se producen roturas en dos
cromosomas diferentes y estos intercambian material. Los cromosomas
resultantes se denominan cromosomas derivativos.
Translocaciónes Robertsonianas: En estas, se pierden los brazos cortos de dos
cromosomas no homólogos y los brazos largos se fusionan en el centrómero para
formar un único cromosoma. Este tipo de translocación está limitado a los
cromosomas acrocéntricos (13,14,15,21 y 22), porque sus brazos cortos son muy
pequeños y no contienen material genético esencial. Dado que los portadores de
translocaciónes Robertsonianas no pierden material genético esencial, son
fenotípicamente normales pero solo tienen 45 cromosomas en cada célula. Sus
hijos, sin embargo, pueden heredar un brazo largo extra o ausente en un
cromosoma acrocéntrico.
Así también otras alteraciones de los cromosomas abarcan las deleciónes en
donde una ruptura cromosómica y la pérdida de material genético el cual al unirse
con un gameto normal para formar un cigoto, concluirá con el carácter portador de
un cromosoma normal y un homologo con la deleción. Tras las aneuploidías
autosómicas antes descritas, los síndromes de deleciónes autosómicas
representan el grupo más frecuente de anomalías cromosómicas clínicamente
significativas, por igual, con los avances tecnológicos actuales se han podido
identificar microdeleciónes, donde con la aparición del bandeo de alta resolución,
es posible identificar microscópicamente un gran número de deleciónes que antes
resultaban demasiado pequeñas para su detección. Además están los avances en
genética molecular, especialmente la técnica de FISH y a aparición de grandes
10

cantidades de polimorfismos que se identifican con facilidad han permitido la
detección de deleciónes que muchas veces son demasiado pequeñas para ser
observadas al microscopio.
Justificacion: Relevancia Clínica.
En México hasta la fecha las alteraciones cromosómicas e indicaciones para su
estudio se encuentran establecidas por la Federación Mexicana de Colegios
de Obstetricia y Ginecología (FEMECOG) donde se especifica los factores de
riesgo y condiciones que tendrán que presentar las pacientes para realizar estudio
con muestras obtenías del liquido amniótico asi como de las vellosidades
coriónicas, los cuales son lineamientos de manera inicial estandarizados por el
Consejo Americano de Ginecólogos y Obstetras (ACOG)(4). En el caso de la
pediatría, en Mexico no existe como tal regulación normativa para las indicaciones
de un cariotipo y bajo que perspectiva clínica deben de ser evaluados estos
pacientes, sin embargo, la Asociacion Española de Pediatria establece una Guía
Clínica para la indicación del estudio genético (5) donde se establece Las
anomalías cromosómicas pueden ser las responsables de rasgos dismórficos y
diversas malformaciones. Por este motivo debemos realizar un cariotipo si estas
anomalías están presentes, sobre todo si se acompañan de retraso mental. Las
indicaciones de cariotipo quedan reflejadas en la tabla 1 . Es importante recordar
que nuestra capacidad para identificar alteraciones cromosómicas estructurales
depende del desarrollo tecnológico. Así, hace unos años solo disponíamos del
bandeo cromosómico con un nivel inferior a 500 bandas. Más recientemente, con
la posibilidad de realizar estudios cromosómicos prometafásicos, aumentó el nivel
de bandas, pudiendo realizarse cariotipos con más de 800 bandas. En la
actualidad las técnicas de citogenética molecular nos permiten reconocer
microdeleciones cromosómicas que no se pueden realizar con las técnicas de
bandeo de alta resolución. Por todo ello, ante un niño con defectos congénitos,
hay que realizar siempre un cariotipo con bandas de alta resolución (cromosomas
11

prometafásicos). Si un paciente tuviera un cariotipo previo normal pero con menos
de 500 bandas, habría que repetírselo. De igual forma, si la clínica lo indica
(pacientes con rasgos dismórficos, defectos congénitos, retraso de crecimiento y
retraso mental), habría que realizar estudios de citogenética molecular para
descartar microdeleciones cromosómicas.
TABLA 1: INDICACIONES PARA REALIZAR
UN CARIOTIPO 5) Periodo de adolescencia:
1) Periodo Prenatal: - Ginecomastia.
- Falta de desarrollo puberal.
- Amenorrea primaria o secundaria
- Retraso mental.
- Rasgos dismórficos.
- Edad mayor de 35 años. - Ansiedad materna.
- Triple screening alterado. - Oligoamnios-polihidramnios.
- Retraso de crecimiento intrauterino (CIR) - Arteria umbilical única.
- Sospecha ecográfica de cromosomopatía - Antecedentes de cromosomopatía balanceada
en un progenitor
6) Periodo del adulto:
- Padres de niños con anomalías cromosómicas
estructurales.
- Abortos de repetición.
- Infertilidad inexplicable.
- Diagnostico prenatal (liquido amniótico y
biopsia de corion).
- Rasgos dismórficos.
2) Periodo neonatal:
- Malformaciones mayores aisladas. - Presencia de 3 o más defectos congénitos menores.
- Recién nacido con rasgos dismórficos. - Recién nacido con genitales ambiguos.
- Parto con producto muerto de causa inexplicable. - Muerte neonatal de causa inexplicada.
7) En todas las edades:
- Procesos malignos (cariotipo constitucional y
tumoral).
- Control de transplantes de medula ósea.
3) Periodo de lactancia:
- Niños con dificultades para el aprendizaje. - Niños con rasgos dismórficos.
- Niños con retraso psicomotor.
4) Periodos Preescolar-Escolar:
- Trastornos del crecimiento.
- Retraso psicomotor. - Rasgos dismórficos.
12

Objetivo Generales
a) Determinar el numero de alteraciones cromosomicas detectadas por el
Laboratorio de Citogenetica del Hospital Pediatrico de Sinaloa de Enero del 2006 a
Diciembre del 2012.
Objetivos Especificos
a) Determinar parametros estadisticos por año, edad y sexo de las
cromosomopatias detectadas.
c) Determinar la cromosomopatia mas frecuentemente encontrada por parte del
laboratorio de citogenetica del Hospital Pediatrico de Sinaloa.
13

Capítulo II: material y metodos.
Tipo de estudio Se realizara un estudio de Incidencia, retrospectivo donde se evaluaran el numero
de casos nuevos para diversas cromosomopatias detectadas por el laboratorio de
Citogenetica del Hospital Pediatrico de Sinaloa de Enero del 2006 al mes de
Diciembre del 2012.
Poblacion objetivo Pacientes pediatricos desde el periodo Neonatal hasta la adultes, marcada como
los 18 años de edad para nuestro pais; y que hayan acudido para determinacion
de cariotipo al laboratorio de citogenetica del Hospital Pediatrico de Sinaloa de
Enero del 2006 al mes de Diciembre del 2012.
Criterios de selección a) Criterios de Inclusion:
Todo aquel paciente que haya acudido en la fecha antes mencionada cuya
informacion se encuentre completa (fecha de ingreso, numero de
expediente, edad, sexo y diagnostico cromosomico).
b) Criterios de exclusion:
Pacientes que no cuenten con datos completos o congruentes por fecha,
edad, sexo o diagnostico cromosomico, pacientes previos al 2006 o bien
posteriores a diciembre del 2012.
14

Metodologia:
Se recabaron los pacientes analizados y registrados en las libretas del
departamento de citogenetica del hospital pediatrico de sinaloa de enero del 2006
a diciembre del 2012 mediante tablas de incidencia por año de estudio,
diagnosticos cromosomicos, sexo y grupos de edad considerados como recien
nacido a 2 años de edad, de 3 a 5 años, de 6 a 13 años y de 13 a 18 años de
edad.
Por igual, se determinaran el numero de cromosomopatias independientes
detectadas en dichas fechas y se dermaran las que tengan mayor incidencia en la
poblacion dada.
15

Capitulo III: Resultados.
Durante el periodo comprendido de Enero del 2006 al mes de Diciembre del 2013
se analizaron un total de 774 cariotipos correspondientes al mismo numero de
pacientes divididos por sexo como se muestra en el Grafico 1.
De el total de pacientes, la relacion
y porcentual por sexo se establecen
en el grafico 2, donde encontramos
un 51.6% de las muestras
correspondientes al sexo masculino
y 48.4% correspondientes al sexo
femenino.
0
20
40
60
80
100
120
140
160
180
200
220
2006 2007 2008 2009 2010 2011 2012
Grafico 1: Pacientes Analizados por Año y Sexo.
Pacientes
Masculino
Femenino
16

Del total de muestras analizadas, los porcentajes correspondientes a la frecuencia
de los cariotipos analizados se tabulan en la tabla 2.
Representando en el
Grafico 3 la tasa
porcentual de los
resultados encontrado
por año, encontrando
un repunte en el año
2011 con 26.3% del
total de la muestra.
Tabla 2: Frecuencia y porcentajes de la muestra
Los resultados y el numero total de cariotipos analizados se distribuyo por grupos
de edad de entre los cuales se establecio desde la edad neonatal hasta los 2 años
0.0
5.0
10.0
15.0
20.0
25.0
30.0
2006 2007 2008 2009 2010 2011 2012
Grafico 3: Porcentajes por año de analisis
Porcentaje
Año Frecuencia Porcentaje Porcentaje
válido
Porcentaje
acumulado
2006 36 4.7 4.7 4.7
2007 76 9.9 9.9 14.6
2008 72 9.4 9.4 24.0
2009 135 17.6 17.6 41.6
2010 121 15.8 15.8 57.4
2011 202 26.3 26.3 83.7
2012 125 16.3 16.3 100.0
Total 767 100.0 100.0
17

de edad, de los 3 a los 6 años de edad, de los 7 a los 12 años de edad y de los 13
a los 18 años de edad, tabulandose los resultados en la tabla 4.
Edad Frecuencia
Porcentaje Porcentaje
válido
Porcentaje acumulado
0-2 años 539 70.3 70.3 70.3
3 - 6 años 108 14.1 14.1 84.4
7- 12 años 87 11.3 11.3 95.7
13 - 18 años 33 4.3 4.3 100.0
Tabla 3: Frecuencia y porcentajes de las muestras por grupos de edad.
En el grafico 4 se ilustra de manera comparativa los tamaños de las muestras por
grupos de edad y su representacion percentual encontrandose un numero mas
importante para el grupo de edad desde el periodo neonatal hasta los 2 años de
edad.
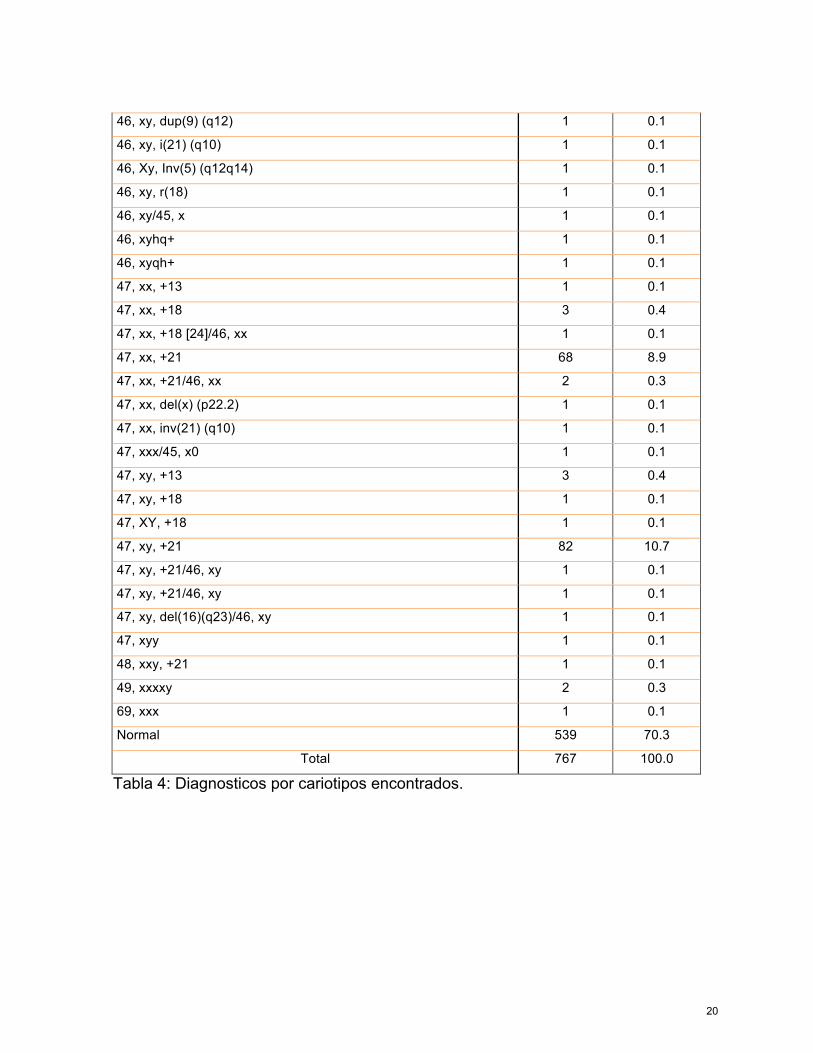
De las muestras reportadas, se tabularon los diagnosticos encontrados (tabla 4)
donde se puede observar como de los diagnosticos mas frecuentes y con
relevancia porcentual, es el Sindrome de Down, tipificado como 47, xx, +21 o bien
47, xy, +21 esto reportandose con una incidencia de hasta en un 8.9% para
mujeres y 10.7% para varones con dicho sindrome, en segundo lugar de
incidencia estaria el sindrome de Turner, encontrado hasta en 1.3% de la muestra,
esto se representa en la Tabla 5.
70.3 14.1 11.3 4.3 0
200
400
600
0-‐2 años 3 -‐ 6 años 7-‐ 12 años 13 -‐ 18 años
Grafico 4: Distribucion percentual por grupos de edad.
Frecuencia
Porcentaje
18

Diagnostico Frecuencia Porcentaje
45, x0 10 1.3
45, x0/46, x, r(x) 1 0.1
45, x0/46, xx 1 0.1
45, x0/46, xx 1 0.1
45, x0[32]/46, x0, indicó(x)(q22) 1 0.1
45, xy -22/46, xy 1 0.1
45, xy, der(14;15) (q10; q10) 1 0.1
46, x, del (x)(p11) 1 0.1
46, x0, +mar 1 0.1
46, x0, delX, (q24-qter) 1 0.1
46, xx add(5)(p15.1) 1 0.1
46, xx, +21 1 0.1
46, xx, del(16), (q22) 1 0.1
46, xx, del(2), (q34) 1 0.1
46, xx, del(9) (q11q13) 1 0.1
46, xx, der(14,21) (q10;q10), +21 1 0.1
46, xx, dup(9) (q12) 1 0.1
46, xx, dup(9) (q12q13) 1 0.1
46, xx, inv(2,3) (q37) 1 0.1
46, xx, inv(21) (q10) 3 0.4
46, xx, inv(9) (p11q12) 1 0.1
46, xx, inv(9) (p12q13) 3 0.4
46, xx, inv(9) (p12q13), add(16)(q24) 1 0.1
46, xx, r(10)/45, xx, -10/47, xx, r(10), +r(10) 1 0.1
46, xx, r(10)/45, xx -10/47, xx, r(10), +r(10) 1 0.1
46, xx/47, xx, +mar 1 0.1
46, xy/45, x0 1 0.1
46, xy, +21 1 0.1
46, xy, del(18)(q21) 1 0.1
46, xy, del(3)(q22q25) 1 0.1
46, xy, del(5) (p15.2) 1 0.1
46, xy, der(14,21) (q10,q10) 2 0.3
46, xy, der(14,21) (q10,q10) +21 1 0.1
46, xy, dup(6) (p12) 1 0.1
19
46, xy, dup(9) (q12) 1 0.1
46, xy, i(21) (q10) 1 0.1
46, Xy, Inv(5) (q12q14) 1 0.1
46, xy, r(18) 1 0.1
46, xy/45, x 1 0.1
46, xyhq+ 1 0.1
46, xyqh+ 1 0.1
47, xx, +13 1 0.1
47, xx, +18 3 0.4
47, xx, +18 [24]/46, xx 1 0.1
47, xx, +21 68 8.9
47, xx, +21/46, xx 2 0.3
47, xx, del(x) (p22.2) 1 0.1
47, xx, inv(21) (q10) 1 0.1
47, xxx/45, x0 1 0.1
47, xy, +13 3 0.4
47, xy, +18 1 0.1
47, XY, +18 1 0.1
47, xy, +21 82 10.7
47, xy, +21/46, xy 1 0.1
47, xy, +21/46, xy 1 0.1
47, xy, del(16)(q23)/46, xy 1 0.1
47, xyy 1 0.1
48, xxy, +21 1 0.1
49, xxxxy 2 0.3
69, xxx 1 0.1
Normal 539 70.3
Total 767 100.0
Tabla 4: Diagnosticos por cariotipos encontrados.
20

Diagnósticos mas frecuentes Numero Porcentaje
Varones con Síndrome de Down 82 10.7
Mujeres con Síndrome de Down 68 8.9
Síndrome de Turner 10 1.3
Tabla 5: Diagnosticos mas frecuentemente encontrados y representacion
percentual.
10.7 8.9 1.3 0
50
100
Varones con Sindrome de
Down
Mujeres con Sindrome de
Down
Sindrome de Turner
Grafico 5: DiagnosBcos mas frecuentes encontrados y representacion percentual
Numero
Porcentaje
0
100
200
300
400
500
600
0-‐2 años 3 -‐ 6 años 7-‐ 12 años 13 -‐ 18 años
Grafico 6: CromosomopaBas detectadas por grupos de edad.
Frecuencia
21

Capitulo IV: Discusion.
Desde el 2006 el laboratorio de citogenetica del Hospital Pediatrico de Sinaloa se
ha postulado como el unico centro medico con capacidad tecnica y humana para
el estudio citogenetico, recibiendo de un gran numero de muestras de diversas
localidades del estado, tanto medios privados como publicos. Lo que permite
tener un mapeo poblacional lo suficientemente representativo para determinar
posteriormente la prevalencia de cromosomopatias dentro de nuestra poblacion,
asi tambien, el seguimiento de ciertas mutaciones con relevancia dentro de
nuestra muestra como los mosaicos, la inversion del cromosoma 9, del
cromosoma 21, la delecion del 14, pudiento rastrear los pacientes para determinar
si existe algun rasgo fenotipico identico entre ellos y llevar a cabo una deteccion
mas oportuna; realizar estudios comparativos con otras poblaciones para valorar si
existe un factor de riesgo no antes considerado asi para nuestra poblacion, al
haber una gran gama de diferencias poblacionales, culturales, ambientales y
demograficas de entre la republica mexicana.
0
100
200
300
400
500
1 2
Grafico 7: CromosomopaBas detectadas por sexo.
Masculino Femenino
22

Asi tambien es importante como las variabilidades geneticas encontradas son
idiferentes del sexo al no haber diferencia estadisticamente significativa, como se
observa en la muestra y se comprueba en la literatura, la cromosomopatia con
mayor incidencia encontrada en nuestro medio es el sindrome de Down, trisomia
del cromosoma 21, seguida por el sindrome de Turner, sin embargo este rastreo
de incidencia permitio identificar como otras alteraciones tanto estructurales como
numericas se encuentran dentro de nuestra poblacion, asi tambien las inversiones
tanto del 9 como del 21 presentan una frecuencia significativa dentro de nuestra
muestra. La determinacion en por sexo demuestra que no existe una importante
diferencia entre las alteraciones cromosomicas presentes pero si es casi un 2%
mas frecuente para sindrome de down en hombres que en mujeres.
Capitulo V: Conclusiones.
Hasta el momento la mayoria de las muestran que llegan a nuestro laboratorio de
citogenetica corresponden a pacientes dentro del rango del recien nacido a los 2
años de edad casi con un 70% de las muestras, sin embargo debemos considerar
ampliar el muestreo de pacientes dentro de los demas grupos de edad que tal vez,
al no contar con el recurso diagnostico antes de la formacion de nuestro
laboratorio, les resultaba imposible la confirmacion diagnostico, esto con afanes
del cribado poblacional.
A su vez, en pediatria, aunque el numero de muestras dentro del grupo de los 0 a
los 2 años de edad es el mas estadisticamente representativo, en este estudio
queda señalado como en mexico no existe una normativa oficial para el cribado
poblacional o bien el analisis de pacientes bajo sospecha de una alteracion
cromosomica y como podemos mediante estudios complementarios determinar la
prevalencia de estas en nuestra poblacion. Asi como en el caso del sindrome de
down, cromosomopatia frecuentemente relacionada con la presencia de
cardiopatias congenitas, la corroboracion clinica y bioquimica de un diagnostico de
sospecha nos llevara a determinar la morbi-mortalidad especifica que tengan
23

nuestros pacientes para ciertas patologias comunmente relacionadas con su
cromosomopatia, repercutiendo en una atencion medica mas preventiva,
delimitando las complicaciones subsecuentes que puedan tener estos y mejorando
la calidad de vida de nuestros pacientes.
24

Bibliografia:
1. Karyotyping for Chromosomal Abnormalities, By: Clare O'Connor,
Ph.D. (Biology Dapartment, Boston College) © 2008 Nature
Education Citation: O'Connor, C. (2008) Karyotyping for chromosomal
abnormalities. Nature Education 1(1) “Biblioteca científica libre del Grupo
editorial Nature” .
2. L.B. Jorde, M.J. Bamshad, J.C. Carey, Genética medica, 4ta Ed. Cap. 6,
Citogenética Clínica: La base cromosómica de la enfermedad humana, Pg.
100 – 127, Ed. Elservier, 2011.
3. Alteraciones genéticas y estrategias diagnósticas en muerte fetal
Medina Castro D, Castro Llamas J, Grether González P, Aguinaga Ríos M.
Ginecol Obstet Mex. 2012 May;80(5):313-9
4. American College of Obstetricians and Gynecologists (ACOG). Invasive
prenatal testing for aneuploidy. Washington (DC): American College of
Obstetricians and Gynecologists (ACOG); 2007 Dec. 9 p. (ACOG practice
bulletin; no. 88)
5. Asociacion Española de Pediatria, AEP, Guia Clinica de Indicaciones del
Estudio Genetico
25


















