
Gas1 cooperates with Cdo and promotes myogenic...
9
Gas1 cooperates with Cdo and promotes myogenic differentiation via activation of p38MAPK Young-Eun Leem a , Ji-Won Han a , Hye-Jin Lee a , Hye-Lim Ha a , Yu-Lim Kwon a , Seok-Man Ho a , Bok-Geon Kim a , Phong Tran a , Gyu-Un Bae a, b, ⁎, Jong-Sun Kang a, ⁎⁎ a Department of Molecular Cell Biology, Center for Molecular Medicine, Samsung Biomedical Research Institute, Sungkyunkwan University School of Medicine, Suwon 440-746, Republic of Korea b College of Pharmacy, Sookmyung Women's University, Seoul 140-742, Republic of Korea abstract article info Article history: Received 5 July 2011 Accepted 18 July 2011 Available online 24 July 2011 Keywords: Myogenic differentiation Gas1 Cdo p38MAPK Cell cycle Skeletal myogenesis is a multistep process that involves cell cycle exit, expression of muscle-specific genes and formation of multinucleated myotubes. Growth arrest specific gene 1 (Gas1) is a GPI-linked membrane protein and originally identified as a growth arrest-linked gene in fibroblasts. Promyogenic cell surface protein, Cdo functions as a component of multiprotein complexes that include other cell adhesion molecules, like Cadherins to mediate cell contact signaling. Here we report that Gas1 and Cdo are coexpressed in muscle cells and form a complex in differentiating myoblasts. Interestingly, Cdo -/- myoblasts display defects in Gas1 induction during differentiation. Overexpression or depletion of Gas1 enhances or decreases myogenic differentiation, respectively. During myoblast differentiation, Gas1 depletion causes defects in downregulation of Cdk2 and Cyclin D1 and up-regulation of miR-322, a negative regulator of Cdk2 activities. Furthermore overexpression or knockdown of Gas1 either enhances or decreases activation of p38MAPK that functions downstream of Cdo. Additionally, Gas1 overexpression in Cdo-depleted C2C12 cells restores p38MAPK activities and differentiation abilities. These data suggest that Gas1 promotes myogenic differentiation through regulation of cell cycle arrest and is critical to activate p38MAPK, most likely via association with Cdo/Cadherin multiprotein complexes. © 2011 Elsevier Inc. All rights reserved. 1. Introduction Cell proliferation and differentiation are usually in an inverse relationship and often mutually exclusive. The inverse relationship is apparent during embryonic development as well as in cancers. Skeletal myogenesis is one of the best models for studying this inverse relationship between cell proliferation and transformation. Skeletal myogenesis is a well orchestrated multistep process that involves cell cycle withdrawal, expression of muscle-specific genes and formation of multinucleated myotubes by cell fusion [1]. The arrest in the G1 phase of the cell cycle is critical for initiation of myogenic differen- tiation. The passage through G1 into S phase of the cell cycle is regulated by cyclin-dependent kinases (Cdks) 4, 6 and 2 which in turn are regulated by Cyclin D (for Cdk4 and Cdk6), Cyclin E, and Cyclin A (for Cdk2). The key event for myogenic differentiation is the downregulation of Cdks and Cyclins expressed during G1 as well as the up-regulation of Cdk inhibitors such as p21 or p27 [2] thereby inducing cell cycle withdrawal [3]. Recently two microRNAs, miR-322 and miR-503 have been shown to be induced upon induction of differentiation, inhibiting Cdk2 via downregulation of Cdc25A and promoting myogenic differentiation [4]. Growth arrest-specific gene 1 (Gas1), a glycosylphosphatidylinositol (GPI)-linked protein, was originally identified as a protein involved in the contact inhibition of fibroblasts [5]. In addition, Gas1 has been shown to play a dual role in the regulation of apoptosis depending on the cellular context [6,7]. Furthermore the expression pattern of Gas1 correlates well with characteristics of a tumor suppressor gene. Gas1 is upregulated in fibroblasts arrested in G0 phase of the cell cycle by serum starvation and high cell density while it is downregulated in proliferating, oncogene-transformed fibroblasts [5,8] and often in cancers [9]. However its role as a true tumor suppressor has not been proven and Gas1 knockout mice do not exhibit an increased incidence in cancers [10]. It appears that Gas1 can induce cell cycle arrest without the GPI anchor in fibroblasts, suggesting that Gas1 may act through another membrane partner to exert its function [11]. Recent studies have shown that Gas1 interacts with a limited number of proteins and functions as a coreceptor to exert its different functions depending on the cellular context [12,13]. Gas1 shares a structural homology with the glial cell- derived neurotrophic factor (GDNF) α receptor (GFRα) and it can negatively regulate GDNF signaling by binding to the membrane receptor tyrosine kinase RET [12,14]. In addition, Gas1 has also been implicated in the modulation of Sonic hedgehog (Shh) signaling. Gas1 Cellular Signalling 23 (2011) 2021–2029 ⁎ Corresponding author. Tel.: + 82 2 2077 7629; fax: + 82 31 299 6157. ⁎⁎ Corresponding author. Tel.: +82 31 299 6135; fax: +82 31 299 6157. E-mail addresses: [email protected], [email protected] (G.-U. Bae), [email protected] (J.-S. Kang). 0898-6568/$ – see front matter © 2011 Elsevier Inc. All rights reserved. doi:10.1016/j.cellsig.2011.07.016 Contents lists available at ScienceDirect Cellular Signalling journal homepage: www.elsevier.com/locate/cellsig
Transcript of Gas1 cooperates with Cdo and promotes myogenic...

Cellular Signalling 23 (2011) 2021–2029
Contents lists available at ScienceDirect
Cellular Signalling
j ourna l homepage: www.e lsev ie r.com/ locate /ce l l s ig
Gas1 cooperates with Cdo and promotes myogenic differentiation viaactivation of p38MAPK
Young-Eun Leem a, Ji-Won Han a, Hye-Jin Lee a, Hye-Lim Ha a, Yu-Lim Kwon a, Seok-Man Ho a,Bok-Geon Kim a, Phong Tran a, Gyu-Un Bae a,b,⁎, Jong-Sun Kang a,⁎⁎a Department of Molecular Cell Biology, Center for Molecular Medicine, Samsung Biomedical Research Institute, Sungkyunkwan University School of Medicine, Suwon 440-746, Republic of Koreab College of Pharmacy, Sookmyung Women's University, Seoul 140-742, Republic of Korea
⁎ Corresponding author. Tel.: +82 2 2077 7629; fax:⁎⁎ Corresponding author. Tel.: +82 31 299 6135; fax:
E-mail addresses: [email protected], [email protected]@skku.edu (J.-S. Kang).
0898-6568/$ – see front matter © 2011 Elsevier Inc. Aldoi:10.1016/j.cellsig.2011.07.016
a b s t r a c t
a r t i c l e i n f oArticle history:Received 5 July 2011Accepted 18 July 2011Available online 24 July 2011
Keywords:Myogenic differentiationGas1Cdop38MAPKCell cycle
Skeletal myogenesis is a multistep process that involves cell cycle exit, expression of muscle-specific genes andformation of multinucleated myotubes. Growth arrest specific gene 1 (Gas1) is a GPI-linked membrane proteinand originally identified as a growth arrest-linked gene in fibroblasts. Promyogenic cell surface protein, Cdofunctions as a component ofmultiprotein complexes that include other cell adhesionmolecules, like Cadherins tomediate cell contact signaling. Here we report that Gas1 and Cdo are coexpressed in muscle cells and form acomplex in differentiating myoblasts. Interestingly, Cdo−/− myoblasts display defects in Gas1 induction duringdifferentiation. Overexpression or depletion of Gas1 enhances or decreases myogenic differentiation,respectively. During myoblast differentiation, Gas1 depletion causes defects in downregulation of Cdk2 andCyclin D1 and up-regulation of miR-322, a negative regulator of Cdk2 activities. Furthermore overexpression orknockdown of Gas1 either enhances or decreases activation of p38MAPK that functions downstream of Cdo.Additionally, Gas1 overexpression in Cdo-depleted C2C12 cells restores p38MAPK activities and differentiationabilities. These data suggest that Gas1 promotes myogenic differentiation through regulation of cell cycle arrestand is critical to activate p38MAPK, most likely via association with Cdo/Cadherin multiprotein complexes.
+82 31 299 6157.+82 31 299 6157.u (G.-U. Bae),
l rights reserved.
© 2011 Elsevier Inc. All rights reserved.
1. Introduction
Cell proliferation and differentiation are usually in an inverserelationship and often mutually exclusive. The inverse relationship isapparent during embryonic development aswell as in cancers. Skeletalmyogenesis is one of the best models for studying this inverserelationship between cell proliferation and transformation. Skeletalmyogenesis is a well orchestrated multistep process that involves cellcycle withdrawal, expression of muscle-specific genes and formationof multinucleated myotubes by cell fusion [1]. The arrest in the G1phase of the cell cycle is critical for initiation of myogenic differen-tiation. The passage through G1 into S phase of the cell cycle isregulated by cyclin-dependent kinases (Cdks) 4, 6 and 2which in turnare regulated by Cyclin D (for Cdk4 and Cdk6), Cyclin E, and CyclinA (for Cdk2). The key event for myogenic differentiation is thedownregulation of Cdks and Cyclins expressed duringG1 aswell as theup-regulation of Cdk inhibitors such as p21 or p27 [2] thereby inducingcell cycle withdrawal [3]. Recently two microRNAs, miR-322 and
miR-503 have been shown to be induced upon induction ofdifferentiation, inhibiting Cdk2 via downregulation of Cdc25A andpromoting myogenic differentiation [4].
Growth arrest-specific gene 1 (Gas1), a glycosylphosphatidylinositol(GPI)-linked protein, was originally identified as a protein involved inthe contact inhibition of fibroblasts [5]. In addition, Gas1 has beenshown to play a dual role in the regulation of apoptosis depending onthe cellular context [6,7]. Furthermore the expression pattern of Gas1correlates well with characteristics of a tumor suppressor gene. Gas1 isupregulated infibroblasts arrested inG0phase of the cell cycle by serumstarvation and high cell density while it is downregulated inproliferating, oncogene-transformed fibroblasts [5,8] and often incancers [9]. However its role as a true tumor suppressor has not beenproven andGas1 knockoutmicedonot exhibit an increased incidence incancers [10]. It appears thatGas1 can induce cell cycle arrestwithout theGPI anchor in fibroblasts, suggesting that Gas1may act through anothermembrane partner to exert its function [11]. Recent studies have shownthat Gas1 interacts with a limited number of proteins and functions as acoreceptor to exert its different functions depending on the cellularcontext [12,13]. Gas1 shares a structural homology with the glial cell-derived neurotrophic factor (GDNF) α receptor (GFRα) and it cannegatively regulate GDNF signaling by binding to the membranereceptor tyrosine kinase RET [12,14]. In addition, Gas1 has also beenimplicated in the modulation of Sonic hedgehog (Shh) signaling. Gas1

2022 Y.-E. Leem et al. / Cellular Signalling 23 (2011) 2021–2029
functions as a positive regulator for Shh signaling by binding directly toShh and its receptor Patched1. Consistently Gas1 knockout mice showsimilar phenotypes to that caused by reduction of Shh signaling [11,15].Gas1 deficient mice are smaller than the control littermates withoutabnormal outgrowth of any tissue, suggesting that Gas1 plays a positiverole in the regulation of cell proliferation and survival [10], whichmightbe due to its function in Shh signaling.
Recently two immunoglobulin (Ig)/fibronectin type III (FNIII)superfamily members, Cdo and Boc have been identified as positiveregulators for Shhsignaling that interact directlywithShhandPatched1,similar to Gas1. Interestingly, these genes are structurally unrelated toGas1 however share striking similarities in the expression patternduring development and are downregulated in response to Shhsignaling. In a recent study employing double mutant mice for CdoandGas1, it has been suggested that Cdo andGas1mayplay a redundantrole in the regulation of Shh signaling in neural tubepatterning [15]. Cdoplays a promyogenic role in vivo and in vitro that appears to beindependent of its role in Shh signaling. Mice lacking Cdo exhibitdelayed skeletal myogenesis, and Cdo−/− primary myoblasts havedefects in myoblast differentiation [16]. Cdo forms multiproteincomplexes that include other cell adhesion molecules, Boc, Cadherinsand Neogenin [17–19] and controls in part the cell contact mediatedpromyogenic signaling via activation of the key kinases, p38MAPK.Similar to Gas1, Cdo is also upregulated in fibroblasts and myoblasts byhigh cell density or serum removal [20], suggesting that Gas1 may beinvolved in cell density-mediated signaling like Cdo. Like Cdo, Gas1 isalso expressed in thedevelopingmuscle compartment dermomyotome,suggestive of a potential role in myogenesis. However what role Gas1plays in myogenesis and whether Gas1 and Cdo interact functionally inmyogenic differentiation is not yet understood.
We report here that Gas1 is expressed in developing hindlimbmuscles and upregulated in differentiating myoblasts at high celldensities. Furthermore, Gas1 promotes myoblast differentiation anddepletion of Gas1 attenuates myoblast differentiation. Interestingly,Gas1 induction during myoblast differentiation is impaired in Cdo-deficient myoblasts, correlating with their defective differentiation.Gas1 depletion causes an increase in BrdU incorporation, failure torepress Cdk2 and Cyclin D1 expressions, and induction of miR-322, anegative regulator for Cdk2. In addition, overexpression or knockdownof Gas1 either enhances or decreases the activity of a key promyogenicregulator, p38MAPK that acts downstream of Cdo-mediated signaling.Gas1 overexpression in Cdo knockdown cells restored p38MAPKactivation and myotube formation. Taken together, these data suggestthat Gas1 promotes myoblast differentiation most likely via cell cyclearrest and its inductionmaybe critical for activation of p38MAPKduringmyoblast differentiation.
2. Material and methods
2.1. Cell culture and expression vectors
C2C12, 293T, andprimarymyoblastsderived fromCdo+/+andCdo−/−
mice were cultured as previously described [21]. Induction of muscledifferentiation was reported previously [21]. In brief, growth medium(DMEM with 15% FBS) of C2C12 or primary myoblasts at nearconfluence was replaced by differentiation medium (DM, DMEMwith 2% horse serum) to induce myogenesis, and myotube formationwas generally observed at about 2–3 days of differentiation. C2C12myoblasts stably expressing Gas1, Gas1 RNAi or Cdo RNAi wereprepared by transfection with lipofectamine 2000 (Invitrogen) andselected for resistance against puromycin. The mouse Gas1 gene wasamplified by RT-PCR. The resulting fragment was cloned into pcDNA.Initially we tested five different Gas1 shRNAs from Thermo Scientificand one of them effectively depleted Gas1 level. The followingsequence was inserted into pSuper expression vector. The sequenceof the effective RNAi is followed below; 5′-CCGGGCGCGGAACTCGGA-
CAAACTTCTCGAGAAGTTTGTCCGAGTTCCGCGCT TTTTG-3′. Cdc25AsiRNA (sc-350357) was purchased from Santa Cruz Biotechnology.Cdo siRNA sequence #1 [22]was subcloned into pSuper vector and usedin similar fashion.
2.2. Western blot analysis and co-immunoprecipitation
Western blot analyses were performed as previously described[21]. Briefly, cells were lysed in lysis buffer (50 mM Tris–HCl, pH7.4,150 mM NaCl, 50 mM NaF, 5 mM sodium pyrophosphate, 0.1% TritonX-100, 1 mM EDTA) with proteinase inhibitor (Roche) and separatedby SDS-PAGE. Antibodies used in this study were as followed: anti-Gas1 (R&D systems), anti-Cdo (Zymed or R&D systems), anti-pan-Cadherin, anti-p38MAPK (Sigma-Aldrich), anti-pp38MAPK (Cellsignaling), anti-MHC (MF20; Developmental Studies HybridomaBank), anti-Troponin T (Sigma-Aldrich), anti-MyoD, anti-myogenin,anti-Cdk2, anti-Cyclin D1, anti-Cdk4, anti-Cyclin E, anti-p21 (SantaCruz), anti-p27 (Abcam), anti-Cdc25A (Santa Cruz), anti-β-tubulinand anti-GFP (Zymed). To study the interaction between Gas1 andCdo, co-immunoprecipitation was performed as described previously[21].
2.3. Immunocytochemisty and microscopy
Cells were fixed with 4% paraformaldehyde (PFA) for 15 min,permeabilized with 0.5% Triton X-100 for 20 min, blocked with 5% calfserum for 30 min and probed with anti-MHC antibody (MF20). Afterincubationwith primary antibodies cells were labeledwith appropriatesecondary antibodies (Alexa Fluor 488 goat anti-rabbit or Alexa Fluor568 goat anti-mouse antibody (1:300; Molecular Probe)). Cells werecounterstained with 4′,6-diamidine-2-phenylindole dihydrochloride(DAPI, Roche) to show the nuclei. A Nikon Eclipse Ti–U microscopeand the NIS-Elements BR version 3.10 (Nikon Corp.) were employed todetectfluorescence and tomeasure thenuclei numbers. For the confocalmicroscopy, C2C12/pSuper or C2C12/Cdo RNAi cells in 12-well plateswere cotransfected with 100 ng of GFP expression vector and 900 ng ofthe indicated DNA constructs and two days later, cells were induced todifferentiate by switching to DM. Immunostaining for pp38 wasperformed as described previously [23]. Images were obtained on aZeiss LSM-510 Meta confocal microscope. Quantification of thefluorescent signal for pp38MAPK was performed with Image-Gaugesoftware (Fuji Film).
2.4. BrdU incorporation and TUNEL assay
To monitor cell proliferation, cells were grown with bromodeoxy-uridine (BrdU; Sigma-Aldrich Corp.) for 10 min. Fixed cells by 4% PFAwere incubated with 2 N HCl for 30 min at 37 °C followed byneutralization with 0.1 M borate buffer (pH8.5). After blocking with5% calf serum cells were probed with anti-BrdU antibody (1:200;Chemicon international Inc.). Fluorescence was detected by Alexa Fluor488 goat anti-mouse antibody (1:100;Molecular Probe). Tomonitor celldeath, C2C12 myoblasts were cultured in growth medium or DM andwere fixed with 4% PFA. Terminal deoxynucleotidyl transferase-dUTPnick end labeling (TUNEL) was performed with a kit according to themanufacturer's instructions (Invitrogen). Nuclei were visualized byDAPI staining.
2.5. RNA extraction and quantitative RT-PCR (qRT-PCR)
Total RNAs from C2C12 cells at different differentiation time pointswere isolated by using easy-Blue reagent (iNtRON Biotechnology,Sungnam, Korea) according to the manufacturer's instructions. ThePCR primers for miR-322 were designed as previously described [24].cDNA synthesis was carried out by using SuperScript III first-strandsynthesis system for RT-PCR (Invitrogen). Real-timePCRwasperformed

2023Y.-E. Leem et al. / Cellular Signalling 23 (2011) 2021–2029
using SYBR Green PCR master mix in an ABI cycler and quantified withABI 7000 software (Applied Biosystems, Foster City, CA). Briefly, 1 μg oftotal RNAs was reverse-transcribed for 5 min at 72 °C, incubated for5 min on ice followed by incubation for 60 min at 42 °C and 5 min at95 °C. 100 ngcDNAand0.2 μl universal reverse (Invitrogen) and specificforward primer were used for the 20 μl PCR reaction. All PCR reactionswere analyzed as triplicates.
3. Results
3.1. Gas1 is expressed in muscle cells and is implicated in myogenicdifferentiation
To investigate the role of Gas1 in myogenesis, we analyzed theexpression pattern of Gas1 in developing muscles. Extracts of hindlimbmuscles obtained from mice at embryonic day 15.5 (E15.5) and E18.5embryos as well as postnatal day 1 (P1) and P5 were analyzed usingantibodies recognizing Gas1, Cdo, and β-tubulin as a loading control.Gas1 and Cdo were expressed robustly in hindlimb muscles at E15.5,which is a critical stage of morphogenetic events of muscle formation,and decreased as muscle development progresses (Fig. 1A). Since cellcontact plays a positive role inmyoblast differentiationwe analyzed theeffect of the high cell density on Gas1 and Cdo expression. C2C12 cellswere plated at low and high cell density in high serum containingmediumso that theywere able to reach50% and 100% confluency in oneday, respectively and analyzed by immunoblotting. The levels of bothGas1 andCdowere substantially increased in C2C12 cells at high density(Fig. 1B).
To investigate the role of Gas1 in myoblast differentiation, C2C12cells were stably transfected with control or Gas1 expression vectorsand induced to differentiate. C2C12 cells at near confluence (D0) wereinduced to differentiate by switching to low serum culture medium(DM). Lysates of C2C12 cells that were proliferating at low density ingrowthmedium(G),D0or inDMfor the indicated timeswere employedto analyze myogenesis. The extent of myogenesis was analyzed byexamining the level of muscle-specific markers, MHC, Myogenin orTroponin-T, and Cadherin serving as a loading control. Overexpressionof Gas1 in C2C12 cells generally resulted in a 2–3 fold increase of Gas1protein (Fig. 1C) and expressions of the muscle-specific genes weresignificantly enhanced in Gas1 overexpressing C2C12 cells, compared tocontrol cells. Next we examined the effect of Gas1 overexpression onmyotube formation. Control and Gas1 overexpressing C2C12 cells wereinduced to differentiate for 2 days, fixed and immunostained withanti-MHC antibodies followed by DAPI staining. Photomicrographs ofthese cells revealed that Gas1 overexpressing C2C12 cells formed largermyotubes with more than six nuclei per myotube than the control cells(Fig. 1D and E). MHC-positive cells were scored as mononucleate,containing two to five nuclei, or containing six or more nuclei. Gas1overexpressing cells showed a significant reduction of mononucleatecells from 66% to 42% and a substantial increase of myotubes containingsix or more nuclei from 11% to 30% (Fig. 1E).
To assess whether Gas1 is rate-limiting for myoblast differentiation,C2C12 cells were stably transfected with Gas1 RNAi, induced todifferentiate fully for 3 days and analyzed for their differentiationability by Western blot analysis as well as immunostaining with anti-MHCantibodies.Wehave tested5differentRNAi expressionvectors andout of five, one RNAi construct reproducibly resulted in a significantknockdown of Gas1. Gas1 protein levels were reduced to 30% in C2C12/Gas1 RNAi cells, relative to that of the control cells (Fig. 1F). Gas1--depleted cells exhibited a stark reduction in the expression of MHC,Myogenin and MyoD, compared to the control cells (Fig. 1F). Further-more these cells formed fewer myotubes, compared to control vector-transfected cells (Fig. 1G). The quantification of MHC-positive cellsshowed that the formation of largermyotubeswithmore than six nucleiper myotube was specifically affected by Gas1 depletion with areduction of myotube formation from 31% to 5%. Conversely mononu-
cleate cells were strongly increased from 40% to 69%, compared to thecontrol cells (Fig. 1H). Thesedata suggest thatGas1 is critical for both thebiochemical as well as the morphological differentiation of C2C12myoblasts.
3.2. Gas1 is critical for the withdrawal from the cell cycle during myoblastdifferentiation most likely via inhibition of Cdk2
Because the cell cycle withdrawal is prerequisite for myoblastdifferentiation, we investigated whether Gas1 promotes myoblastdifferentiation via the regulation of the cell cycle withdrawal. Toanalyze their abilities to cell cycle withdrawal in the initiation ofdifferentiation, C2C12/pSuper and C2C12/Gas1 RNAi cells were inducedto differentiate by switching to the nutrient-poor medium for one dayand cell proliferation was analyzed by BrdU incorporation. Control cellsexhibited only ~8% of BrdU-positive cells whereas ~25% of C2C12/Gas1RNAi cellswerepositive for BrdU(Fig. 2A andB). Thesedata suggest thatGas1 may be important for the cell cycle arrest upon induction ofmyogenic differentiation. In addition, Gas1 has been implicated in theregulation of cell death in a cell context-dependent manner [25].We tested the possibility that the defective differentiation of Gas1-depleted cellswas causedby changes in cell death byusing TUNEL assay.Both the control andC2C12/Gas1RNAi cells showed fewTUNEL-positivecells with Gas1-depeleted cells even displaying a slightly reducednumber of TUNEL-positive cells, compared to the control cells (Fig. 2Cand D). However this small difference in cell death cannot account forthe inefficient differentiation seen in Gas1-depleted C2C12 cells.
To further analyze changes in the cell cycle control, lysates of C2C12/pSuper and C2C12/Gas1 RNAi cells at D0 (the confluent culture in highserum containing medium) and D1 (induced to differentiate by serumremoval) were analyzed for expression of the positive cell cycleregulators (Cdk4, Cyclin D1, Cdk2, Cyclin E), the cell cycle inhibitors(p21 and p27), and β-tubulin as a loading control. Among the positivecell cycle regulators, Cdk4, Cdk2 and Cyclin D1 proteins weremaintained on relatively high levels at D1 in Gas1-depleted cells,compared to the control cells. p21 and p27 levels were only slightlyincreased in Gas1-depleted cells, compared to the control cells. Thesedata suggest that Gas1 depletion results in failure to downregulate thepositive regulators of theG1 cell cycle phase such as Cdk2 andCyclinD1,most likely leading to the failure of the cell cycle withdrawal andinefficientmyogenic differentiation (Fig. 2E). Recently itwas shown thatseveral microRNAs (miRs) are induced during myoblast differentiationand play important roles in regulation of the various steps of thedifferentiation process [26–28]. Two microRNAs, miR-322/424 andmiR-503 have been shown to be induced during myogenesis andinhibiting Cdk2 activities by downregulation of Cdc25A [4]. Hence wetested the possibility that Gas1 may regulate the cell cycle exit viamodulation of the level of miRs during myogenesis. First, we analyzedthe effect of Gas1 depletion on miR-322 expression. RNAs from C2C12/pSuper and C2C12/Gas1 RNAi cells at indicated differentiation timepoints were isolated and analyzed for miR-322 expression byquantitative RT-PCR. As previously reported, miR-322 was increasedduring myoblast differentiation however Gas1 depleted cells displayeda blunted induction of miR-322, compared to the control cells (Fig. 2F).Furthermore cells overexpressing Gas1 resulted in a dramatic increaseof miR-322 mRNA levels during the entire differentiation time course,compared to the control cells (Fig. 2G). Taken together, Gas1 mayregulate the cell cycle arrest via inhibition of Cdk2 expression andactivity by induction of miR-322.
Next we tested whether the defective differentiation capacity ofC2C12/Gas1 RNAi cells are due to failure of Cdk2 inhibition. C2C12 cellswere transiently transfectedwith the control or Cdc25ARNAi oligos andanalyzed for Cdc25A expression. As expected, the level of Cdc25Aprotein was reduced in C2C12/Cdc25A RNAi cells, compared to thecontrol transfected cells (Fig. 2H). C2C12/pSuper and C2C12/Gas1 RNAiwere transiently transfectedwith the control or Cdc25ARNAi oligos and

Fig. 1. Gas1 and Cdo are coexpressed in developing muscles, and overexpression of Gas1 enhances myoblast differentiation while knockdown of Gas1 impairs myogenic differentiation.(A) Extracts of hindlimbmuscles fromembryos at the embryonic stage (E) 15.5 and 18.5 andmice at postnatal days (P) 1 and5wereprobedwith antibodies toGas1, Cdo andβ-tubulin as aloading control. (B) C2C12 cells were cultured either to 50% confluency (LD) or 100% confluency (HD) in growth medium, and lysates were immunoblotted with antibodies recognizingCdo, Gas or Akt. (C) C2C12 cells were stably transfectedwith control or Gas1 expression vectors. Cells were then induced to differentiate for 2 days and lysateswere analyzed byWesternblottingwith antibodies recognizing Gas1, MHC,Myogenin, or Troponin-T, and Cadherin as a loading control. (D) C2C12 cells shown in panel C were cultured in DM for 2 days, fixed, andstained with an antibody to MHC (red). Cellular nuclei were visualized by DAPI staining (blue). (E) Quantification of myotube formation shown in panel D. Values represent means oftriplicate determinations±1 SD. The experimentwas repeated three times with similar results. Significant difference from control, *pb0.01. (F) C2C12 cells were stably transfectedwithGas1 RNAi or control (pSuper) expression vectors, cultured in DM for 3 days and lysates were subjected toWestern blotting with antibodies recognizing Gas1, MHC,Myogenin orMyoD,and Cadherin as a loading control. (G) C2C12 cells shown in F were immunostained with an antibody to MHC (red). Cellular nuclei were visualized by DAPI staining (blue).(H) Quantification of myotube formation shown in panel G. Values represent means of triplicate determinations ±1 SD. The experiment was repeated three times with similar results.Significant difference from control, *pb0.01.
2024 Y.-E. Leem et al. / Cellular Signalling 23 (2011) 2021–2029
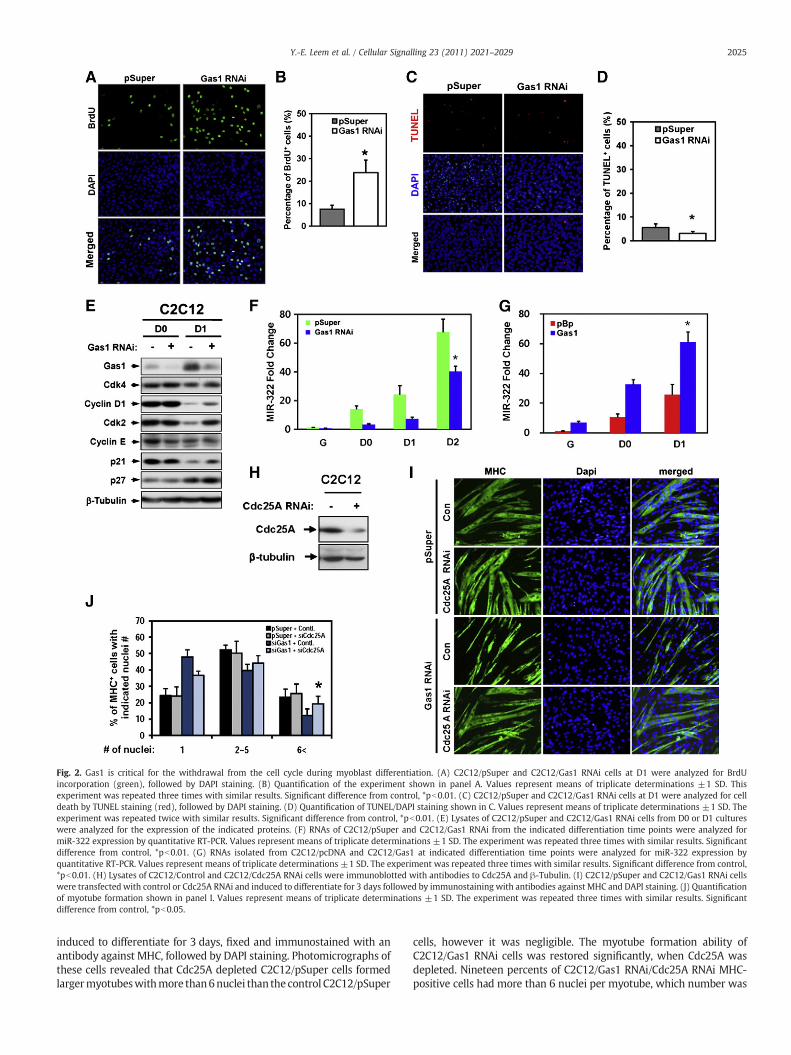
Fig. 2. Gas1 is critical for the withdrawal from the cell cycle during myoblast differentiation. (A) C2C12/pSuper and C2C12/Gas1 RNAi cells at D1 were analyzed for BrdUincorporation (green), followed by DAPI staining. (B) Quantification of the experiment shown in panel A. Values represent means of triplicate determinations ±1 SD. Thisexperiment was repeated three times with similar results. Significant difference from control, *pb0.01. (C) C2C12/pSuper and C2C12/Gas1 RNAi cells at D1 were analyzed for celldeath by TUNEL staining (red), followed by DAPI staining. (D) Quantification of TUNEL/DAPI staining shown in C. Values represent means of triplicate determinations ±1 SD. Theexperiment was repeated twice with similar results. Significant difference from control, *pb0.01. (E) Lysates of C2C12/pSuper and C2C12/Gas1 RNAi cells from D0 or D1 cultureswere analyzed for the expression of the indicated proteins. (F) RNAs of C2C12/pSuper and C2C12/Gas1 RNAi from the indicated differentiation time points were analyzed formiR-322 expression by quantitative RT-PCR. Values represent means of triplicate determinations ±1 SD. The experiment was repeated three times with similar results. Significantdifference from control, *pb0.01. (G) RNAs isolated from C2C12/pcDNA and C2C12/Gas1 at indicated differentiation time points were analyzed for miR-322 expression byquantitative RT-PCR. Values represent means of triplicate determinations ±1 SD. The experiment was repeated three times with similar results. Significant difference from control,*pb0.01. (H) Lysates of C2C12/Control and C2C12/Cdc25A RNAi cells were immunoblotted with antibodies to Cdc25A and β-Tubulin. (I) C2C12/pSuper and C2C12/Gas1 RNAi cellswere transfected with control or Cdc25A RNAi and induced to differentiate for 3 days followed by immunostaining with antibodies against MHC and DAPI staining. (J) Quantificationof myotube formation shown in panel I. Values represent means of triplicate determinations ±1 SD. The experiment was repeated three times with similar results. Significantdifference from control, *pb0.05.
2025Y.-E. Leem et al. / Cellular Signalling 23 (2011) 2021–2029
induced to differentiate for 3 days, fixed and immunostained with anantibody against MHC, followed by DAPI staining. Photomicrographs ofthese cells revealed that Cdc25A depleted C2C12/pSuper cells formedlargermyotubeswithmore than6nuclei than the control C2C12/pSuper
cells, however it was negligible. The myotube formation ability ofC2C12/Gas1 RNAi cells was restored significantly, when Cdc25A wasdepleted. Nineteen percents of C2C12/Gas1 RNAi/Cdc25A RNAi MHC-positive cells had more than 6 nuclei per myotube, which number was
2026 Y.-E. Leem et al. / Cellular Signalling 23 (2011) 2021–2029
close to 25% of myotubes with more than 6 nuclei shown in CdC12/pSuper cells (Fig. 2J). Taken together, these data suggest that inhibitionof Cdk2 activities by Gas1-mediated signaling may require suppressionof Cdc25A as well as Cdk2 itself.
3.3. Gas1 associates with Cdo during myoblast differentiation
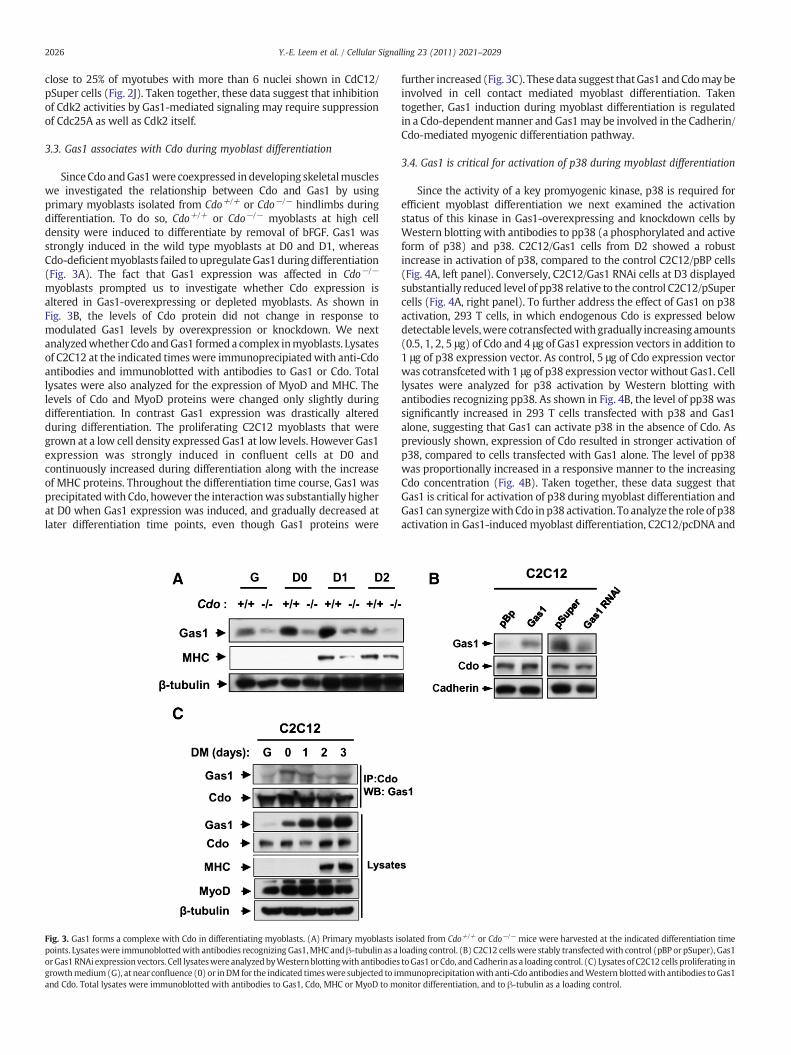
SinceCdo andGas1were coexpressed indeveloping skeletalmuscleswe investigated the relationship between Cdo and Gas1 by usingprimary myoblasts isolated from Cdo+/+ or Cdo−/− hindlimbs duringdifferentiation. To do so, Cdo+/+ or Cdo−/− myoblasts at high celldensity were induced to differentiate by removal of bFGF. Gas1 wasstrongly induced in the wild type myoblasts at D0 and D1, whereasCdo-deficientmyoblasts failed to upregulateGas1 duringdifferentiation(Fig. 3A). The fact that Gas1 expression was affected in Cdo−/−
myoblasts prompted us to investigate whether Cdo expression isaltered in Gas1-overexpressing or depleted myoblasts. As shown inFig. 3B, the levels of Cdo protein did not change in response tomodulated Gas1 levels by overexpression or knockdown. We nextanalyzedwhether Cdo andGas1 formeda complex inmyoblasts. Lysatesof C2C12 at the indicated timeswere immunoprecipiatedwith anti-Cdoantibodies and immunoblotted with antibodies to Gas1 or Cdo. Totallysates were also analyzed for the expression of MyoD and MHC. Thelevels of Cdo and MyoD proteins were changed only slightly duringdifferentiation. In contrast Gas1 expression was drastically alteredduring differentiation. The proliferating C2C12 myoblasts that weregrown at a low cell density expressed Gas1 at low levels. However Gas1expression was strongly induced in confluent cells at D0 andcontinuously increased during differentiation along with the increaseof MHC proteins. Throughout the differentiation time course, Gas1 wasprecipitatedwith Cdo, however the interactionwas substantially higherat D0 when Gas1 expression was induced, and gradually decreased atlater differentiation time points, even though Gas1 proteins were
Fig. 3. Gas1 forms a complexe with Cdo in differentiating myoblasts. (A) Primary myoblasts ipoints. Lysateswere immunoblottedwith antibodies recognizing Gas1,MHC andβ-tubulin as aorGas1 RNAi expressionvectors. Cell lysateswere analyzedbyWesternblottingwith antibodiesgrowthmedium (G), at near confluence (0) or inDMfor the indicated timeswere subjected to imand Cdo. Total lysates were immunoblotted with antibodies to Gas1, Cdo, MHC or MyoD to m
further increased (Fig. 3C). Thesedata suggest thatGas1 andCdomaybeinvolved in cell contact mediated myoblast differentiation. Takentogether, Gas1 induction during myoblast differentiation is regulatedin a Cdo-dependentmanner and Gas1may be involved in the Cadherin/Cdo-mediated myogenic differentiation pathway.
3.4. Gas1 is critical for activation of p38 during myoblast differentiation
Since the activity of a key promyogenic kinase, p38 is required forefficient myoblast differentiation we next examined the activationstatus of this kinase in Gas1-overexpressing and knockdown cells byWestern blotting with antibodies to pp38 (a phosphorylated and activeform of p38) and p38. C2C12/Gas1 cells from D2 showed a robustincrease in activation of p38, compared to the control C2C12/pBP cells(Fig. 4A, left panel). Conversely, C2C12/Gas1 RNAi cells at D3 displayedsubstantially reduced level of pp38 relative to the control C2C12/pSupercells (Fig. 4A, right panel). To further address the effect of Gas1 on p38activation, 293 T cells, in which endogenous Cdo is expressed belowdetectable levels,were cotransfectedwithgradually increasingamounts(0.5, 1, 2, 5 μg) of Cdo and 4 μg of Gas1 expression vectors in addition to1 μg of p38 expression vector. As control, 5 μg of Cdo expression vectorwas cotransfcetedwith 1 μg of p38 expression vector without Gas1. Celllysates were analyzed for p38 activation by Western blotting withantibodies recognizing pp38. As shown in Fig. 4B, the level of pp38 wassignificantly increased in 293 T cells transfected with p38 and Gas1alone, suggesting that Gas1 can activate p38 in the absence of Cdo. Aspreviously shown, expression of Cdo resulted in stronger activation ofp38, compared to cells transfected with Gas1 alone. The level of pp38was proportionally increased in a responsive manner to the increasingCdo concentration (Fig. 4B). Taken together, these data suggest thatGas1 is critical for activation of p38 duringmyoblast differentiation andGas1 can synergizewithCdo in p38 activation. To analyze the role of p38activation in Gas1-inducedmyoblast differentiation, C2C12/pcDNA and
solated from Cdo+/+ or Cdo−/− mice were harvested at the indicated differentiation timeloading control. (B) C2C12 cellswere stably transfectedwith control (pBP or pSuper), Gas1toGas1or Cdo, andCadherin as a loading control. (C) Lysates of C2C12 cells proliferating inmunoprecipitationwith anti-Cdo antibodies andWestern blottedwith antibodies toGas1
onitor differentiation, and to β-tubulin as a loading control.

Fig. 4. Gas1 is required for the activation of p38 during myoblast differentiation. (A) C2C12 cells were stably transfected with the expression vectors for control (pBP or pSuper),Gas1 or Gas1 RNAi. Cell lysates were immunoblotted with antibodies to pp38 and p38. (B) 293T cells were transiently transfected with 4 μg of Gas1, and the various amounts of Cdoexpression vectors plus p38 expression vectors. The control, pcDNA (−) vector-transfected cells serve as a negative control. Lysates were immunoblotted with antibodies to pp38,p38, Cdo or Gas1. (C) C2C12/pcDNA and C2C12/Gas1 cells were induced to differentiate with either the vehicle (DMSO) or SB203580 (a p38 inhibitor) for 3 days and immunostainedwith antibodies to MHC, followed by DAPI staining. (D) Quantification of myotube formation shown in panel C. Values represent means of triplicate determinations ±1 SD. Theexperiment was repeated three times with similar results. Significant difference from control, *pb0.01. (E) Lysates of C2C12/pSuper and C2C12/Cdo RNAi cells were immunoblottedwith antibodies to Cdo and Cadherin. (F) C2C12/pSuper and C2C12/Cdo RNAi cells were transiently transfectedwith either the control or Gas1expression vectors plus GFP expressionvectors and induced to differentiate for 3 days, followed by imunostaining with antibodies to MHC. The transfected cells were visualized by GFP expression. (G) Quantification ofmyotube formation shown in panel F. Values represent means of triplicate determinations ±1 SD. The experiment was repeated three times with similar results. Significantdifference from control, *pb0.01. (H) C2C12/pSuper and C2C12/Cdo RNAi cells were transiently transfected with the control or Gas1 expression vectors plus a GFP vector. Confluentcultures that had not fed with fresh medium for 48 h were then fixed and stained with antibodies to pp38 (red) and visualized for GFP expression (green). Cellular nuclei werevisualized by DAPI (blue). Red arrow indicates untransfected cells and white arrows indicate transfected cells. (I) Quantification of cultures shown in panel H. GFP+ cells were scoredas positive or negative for pp38 staining. Values represent means of triplicate determinations ±1 SD. Asterisk indicates difference from the control at pb0.01. (J) C2C12/pSuper andC2C12/Cdo RNAi cells were transiently transfected with pcDNA or Gas1 expression vectors plus a GFP vector. Cells were incubated in DM for 2 days and then fixed and stained withantibodies to pp38 (red) and visualized for GFP expression (green), followed by DAPI staining (blue). Red arrows indicate untransfected cells and white arrows indicate transfectedcells. (K) Quantification of cultures shown in panel J. The intensity of the immunofluorescent pp38 signal in GFP+ and GFP- cells were quantified, with the values obtained from theuntransfected C2C12/pSuper cultures set to 1.0. Values represent means of triplicate determination ±1 SD. The experiment was repeated with similar results. Asterisk indicatesdifference from the control at pb0.01.
2027Y.-E. Leem et al. / Cellular Signalling 23 (2011) 2021–2029

2028 Y.-E. Leem et al. / Cellular Signalling 23 (2011) 2021–2029
C2C12/Gas1 cells were induced to differentiate with or without 1 μMSB203580, a specific inhibitor of p38 for 3 days. Cells were then fixedand immunostained with anti-MHC antibodies, followed by DAPIstaining. As shown in Fig. 4C and D, SB203580 treated control cellsexhibited a substantial reduction in the formation of larger myotubescontaining more than 6 nuclei per myotube. The enhanced differenti-ation seen in C2C12/Gas1 cells was abrogated by treatment withSB203580 suggesting p38 activation is required for the Gas1-mediatedmyotube formation.
3.5. Gas1 overexpression restores activation of p38 and differentiation ofCdo-depleted myoblasts
Since Cdo and Gas1 activated p38 cooperatively, we next askedwhether the defective differentiation of Cdo-depleted cells can berescued by Gas1 overexpression. To do so, C2C12 cells stablyexpressing pSuper or Cdo RNAi were transiently transfected withpcDNAorGas1 expression vectors aswell as a GFP expression vector tomark the transfected cells. The cells were induced to differentiate for3 days followed by immunostaining with anti-MHC and GFP anti-bodies. Nuclei were visualized by DAPI staining. C2C12/Cdo RNAi cellsdisplayed a reduction in Cdo expression up to 30% of the control cells(Fig. 4E). As previously shown, C2C12/Cdo RNAi cells transfected withpcDNA formed smaller myotubes, compared to the control C2C12/pSuper/pcDNA cells. Interestingly C2C12/Cdo RNAi/Gas1 cells restoredthe differentiation capability almost to the level of the control C2C12/pSuper/pcDNA cells (Fig. 4, F and G).
We next analyzedwhether the rescue in differentiation capacities ofC2C12/CdoRNAi/Gas1 cells is due to reactivation of p38 by immunos-taining. To do so, C2C12/pSuper and C2C12/Cdo RNAi cells weretransiently cotransfected with the control pcDNA or Gas1 expressionvectors and theGFPexpressionvector tomark the transfected cells. Cellsfrom D0 or D2 were fixed and immunostained with anti-pp38antibodies. As shown in Fig. 4H and I, ~54% of GFP-positive, pcDNA-transfceted C2C12/pSuper cells at D0 showed immunoreactivity forpp38 in contrast to the ~82% of Gas1 transfected C2C12/pSuper cells. Aspreviously shown Cdo knockdown cells exhibited a stark reduction inpp38-positive cells and only ~10% of control vector-transfected cellsshowed immunorecativity for pp38. Gas1 overexpression in such cellsincreased pp38 positivity to ~47%, similar to the control vector-transfected C2C12/pSuper cells. Using the D2 conditions most of thecontrol vector-transfected cells formedmultinucleatedmyotubeswith astrong pp38 signal which was increased in C2C12/pSuper cellstransfected with the Gas1 expression vector (Fig. 4J). C2C12/Cdo RNAicells transfected with pcDNA were mainly mononucleate and had avisibly weaker pp38 signal, compared to the control cells. Gas1overexpressing C2C12/Cdo RNAi cells showed a robust pp38 signal(Fig. 4J). To quantify this, we scored the intensity of the immunofluo-rescent pp38 signal in untransfected (GFP−) versus transfected (GFP+)cells on the same cover slips, with the average pp38 signal ofuntransfected C2C12/pSuper cells set to 1.0. Control vector-transfectedC2C12/pSuper cells also had a pp38 signal of ~1.0,whereas the signal foruntransfected C2C12 cells was ~1.1 and for Gas1 transfected C2C12/pSuper cells was ~1.6 (Fig. 4K). In addition, both untransfected andcontrol vector-transfected C2C12/Cdo RNAi cells had a pp38 signal of~0.2, while the signal for Gas1 expressing C2C12/Cdo RNAi cellswas ~0.6 (Fig. 4K). Taken together, these data suggest that Gas1promotesmyogenic differentiationmost likely via inductionof cell cyclearrest and activation of p38. In addition, Cdo and Gas1 appear tocooperate in activation of p38 and myogenic differentiation.
4. Discussion
Tissuehomeostasis requires a precise control of cell proliferation anddifferentiation in response to extracellular cues, including solublefactors or factors involved in cell to cell contact and adhesion. The
mechanisms whereby cell contact regulates cell proliferation anddifferentiation are of fundamental interest, andmyoblast differentiationis an excellent system to study this event. Cadherins and Ig superfamilymembers of cell adhesion molecules are proposed to mediate manypromyogenic signaling events initiated by cell contact [29]. The Igsuperfamily member Cdo forms a multiprotein complex containingother cell adhesion molecules, N-Cadherin, Boc and Neogenin and thiscomplex mediates some of the cell contact-mediated promyogenicsignaling including p38MAPK [17–19]. Gas1 and Cdo are structurallyunrelated however they share a strong correlation in the expressionpattern during embryonic development as well as in their responsive-ness to cell contact and serum starvation. Interestingly both Gas1 andCdo have been linked to growth arrest in fibroblasts and suggested to betumor suppressor genes [20,30]. In recent studies, they have beenidentified as positive regulators for Shh signaling by binding directly toShh and their expression is negatively regulated by Shh signaling [15].Consistently, mice lacking these proteins display a similar, though notidentical Shh-related phenotype [15,22,31].
Cdo's promyogenic effect requires its intracellular domain, whichinitiates signaling through p38MAPK by a direct binding to a scaffoldprotein JLP [32]. In contrast, Cdo's intracellular region is dispensable forits ability to promote Shh signaling. In this study, we demonstrated thatGas1 was expressed in developing skeletal muscles and inducedstrongly myoblast differentiation. It is noteworthy that expressionand interaction of Gas1 with Cdo increased in confluent cultures,regardless of high serum content in the culture medium. It is likely thatGas1 may function in promyogenic signaling initiated by N-Cadherin/Cdo-mediated cell contact. In consistent with this expression pattern,Gas1 overexpression enhanced both biochemical and morphologicaldifferentiation, whereas Gas1-depleted C2C12 cells displayed impairedbiochemical and morphological myoblast differentiation. As previouslyshown for fibroblasts, Gas1 appeared to regulate also cell cyclewithdrawal of myoblasts. Gas1 depleted cells failed to fully downregulate two positive regulators of G1 cell cycle phase, Cyclin D1 andCdk2 and displayed a delay in miR-322 induction during myoblastdifferentiation. miR-322 has been shown to down regulate Cdc25A,thereby negatively regulating Cdk2 activities. In addition, Gas1overexpression resulted in an increased expression of miR-322. Thesedata suggest that Gas1 may control cell cycle withdrawal via inhibitionof Cdk2 activities by regulating Cdk2 negative regulators and Cdk2expression itself. This notion is further supported by the partial rescueof defective differentiation of C2C12/Gas1 RNAi cells by Cdc25Adepletion. The molecular mechanism whereby Gas1 regulates expres-sion of these cell cycle regulators is unclear.
p38MAPK has been proposed to play a key role in the cell contact-mediated cell cycle arrest via modulation of expression and activities ofcell cycle regulators for G1-S transition, such as Cyclin D1 [33]. It is wellestablished that the p38MAPK signaling pathway plays an essential rolein skeletal myogenesis for both biochemical and morphologicaldifferentiation of myoblasts [34,35]. The fact that overexpression ordepletion of Gas1 in C2C12 cells resulted in either increased ordecreased levels of pp38, respectively suggests that Gas1 may controlthe cell cycle arrest and myoblast differentiation via activation ofp38MAPK. Consistently, inhibition of p38MAPK by the treatment withSB203580 blocked the enhanced differentiation induced by Gas1overexpression. We have previously shown that Cdo depletion inC2C12 cells causes impaired p38MAPK activation and myoblastdifferentiation. Interestingly Gas1 rescued defective myoblast differen-tiation causedbyCdodepletionmost likely via reactivation of p38MAPK.It appeared that Gas1 enhanced Cdo-mediated p38MAPK activation in aCdo concentration-dependent manner. Since Cdo-depleted C2C12 cellsexhibited a reduction in Cdo protein to 20–30% of the control cells, therestoration of defective p38 activation may be the cooperative effect ofGas1 with the residual amount of Cdo in Cdo-depleted C2C12 cells.However it appears that Gas1 can activate p38MAPK withoutcotransfection of Cdo in 293 T cells that do not express detectable levels

2029Y.-E. Leem et al. / Cellular Signalling 23 (2011) 2021–2029
of Cdo, suggesting Cdo is not the only interacting protein to beinvolved in Gas1-mediated p38MAPK activation. The fact that Gas1 is aGPI-linked protein suggests it may exert its function by binding withother signaling regulators including the GDNF receptor Ret, Cdo and yetto be identified proteins. Our recent work to define the mechanisms ofcoreceptors for the Shh signaling pathway has revealed that Gas1 caninteract with Cdo and Boc in 293 T cells and Boc's interactionwith Gas1is independent of Cdo [38]. Since Boc is expressed in 293 T and C2C12myoblasts Gas1 may interact with Boc to activate p38MAPK. Similar toCdo−/− myoblasts, Boc−/− myoblasts displayed, with lesser extent,defects in myotube formation and Gas1 induction (Lee et al.,unpublished data), suggesting a potential link between Gas1 and Bocin myoblast differentiation. However whether Boc activates p38MAPKin a Cdo-independent manner during myoblast differentiation isunknown. Further studies will address this aspect of regulatorypathways of myogenesis. Both Gas1 and Cdo interact directly withShh and promote the Shh signaling pathway [15]. Shh signaling hasbeen shown to activate p38MAPK in primary astrocyte cultures [36],however Shh does not activate p38MAPK in myoblasts [37]. Inagreement with this notion, Cyclin D1, a downstream target gene ofthe Shh signaling pathway, was downregulated in Gas1 overexpressingcells or upregulated by Gas1-depletion, suggesting Gas1's promyogenicfunction most likely is independent of its positive role in Shh signaling.These data suggest that Cdo and Gas1 coordinate the actions ofmultiplesignaling pathways, including Shh signaling and cell contact-initiatedprodifferentiation signaling in a cell context-dependent fashion.
Acknowledgements
We thank Dr Ruth Simon for critical reading of the manuscript.Thisworkwas supportedby thepromotion program for the new faculty,Sungkyunkwan University (2009) to JSK and the grant from theNational Research Foundation of Korea Grant funded by the KoreanGovernment (2010-0022490) to YEL.
References
[1] J.D. Molkentin, E.N. Olson, Curr. Opin. Genet. Dev. 6 (1996) 445–453.[2] I. Nilsson, I. Hoffmann, Prog. Cell Cycle Res. 4 (2000) 107–114.[3] T.J. Hawke, A.P. Meeson, N. Jiang, S. Graham, K. Hutcheson, J.M. DiMaio, D.J. Garry,
Am. J. Physiol. Cell Physiol. 285 (2003) 1019–1027.[4] S. Sarkar, B.K. Dey, A. Dutta, Mol. Biol. Cell 21 (2010) 2138–2149.[5] A. Evdokiou, P.A. Cowled, Exp. Cell Res. 240 (1998) 359–367.[6] S. Gobeil, X. Zhu, C.J. Doillon, M.R. Green, Genes Dev. 22 (2008) 2932–2940.
[7] R. Spagnuolo,M. Corada, F. Orsenigo, L. Zanetta, U. Deuschle, P. Sandy, C. Schneider,C.J. Drake, F. Breviario, E. Dejana, Blood 103 (2004) 3005–3012.
[8] D.Kletsas, D. Stathakos, V. Sorrentino, L. Philipson, Exp. Cell Res. 217 (1995)477–483.[9] A.L. Gartel, K. Shchors, Exp. Cell Res. 283 (2003) 17–21.
[10] M. Seppala, M.J. Depew, D.C. Martinelli, C.M. Fan, P.T. Sharpe, M.T. Cobourne,J. Clin. Invest. 117 (2007) 1575–1584.
[11] D.C. Martinelli, C.M. Fan, Cell Cycle 6 (2007) 2650–2655.[12] J.R. Cabrera, L. Sanchez-Pulido, A.M. Rojas, A. Valencia, S. Mañes, J.R. Naranjo, B.
Mellström, J. Biol. Chem. 281 (2006) 14330–14339.[13] J.S. Kang, W. Zhang, R.S. Krauss, Sci. STKE 403 (2007) e50.[14] M.A. López-Ramírez, G. Domínguez-Monzón, P. Vergara, J. Segovia, Int. J. Dev.
Neurosci. 26 (2008) 497–503.[15] B.L. Allen, T. Tenzen, A.P. McMahon, Genes Dev. 21 (2007) 1244–1257.[16] F. Cole, W. Zhang, A. Geyra, J.S. Kang, R.S. Krauss, Dev. Cell 7 (2004) 843–854.[17] J.S. Kang, P.J. Mulieri, Y. Hu, L. Taliana, R.S. Krauss, EMBO J. 21 (2002) 114–124.[18] J.S. Kang, J.L. Feinleib, S. Knox, M.A. Ketteringham, R.S. Krauss, Proc. Natl. Acad. Sci.
U. S. A. 100 (2003) 3989–3994.[19] J.S. Kang, M.J. Yi, W. Zhang, J.L. Feinleib, F. Cole, R.S. Krauss, J. Cell Biol. 167 (2004)
493–504.[20] J.S. Kang, M. Gao, J.L. Feinleib, P.D. Cotter, S.N. Guadagno, R.S. Krauss, J. Cell Biol.
138 (1997) 203–213.[21] G.U. Bae, J.R. Lee, B.G. Kim, J.W. Han, Y.E. Leem, H.J. Lee, S.M. Ho, M.J. Hahn, J.S.
Kang, Mol. Biol. Cell 21 (2010) 2399–2411.[22] W. Zhang, J.S. Kang, F. Cole, M.J. Yi, R.S. Krauss, Dev. Cell 10 (2006) 657–665.[23] G.U. Bae, B.G. Kim, H.J. Lee, J.E. Oh, S.J. Lee, W. Zhang, R.S. Krauss, J.S. Kang, Mol.
Cell. Biol. 29 (2009) 4130–4143.[24] C. Chen, D.A. Ridzon, A.J. Broomer, Z. Zhou, D.H. Lee, J.T. Nguyen, M. Barbisin, N.L.
Xu, V.R. Mahuvakar, M.R. Andersen, K.Q. Lao, K.J. Livak, K.J. Guegler, Nucleic AcidsRes. 33 (2005) e179.
[25] E.M. Ruaro, L. Collavin, G. Del Sal, R. Haffner, M. Oren, A.J. Levine, C.A. Schneider,Proc. Natl. Acad. Sci. U. S .A. 94 (1997) 4675–4680.
[26] P.L. Boutz, G. Chawla, P. Stoilov, D.L. Black, Genes Dev. 21 (2007) 71–84.[27] X. Chen, K. Wang, J. Chen, J. Guo, Y. Yin, X. Cai, X. Guo, G. Wang, R. Yang, L. Zhu, Y.
Zhang, J. Wang, Y. Xiang, C. Weng, K. Zen, J. Zhang, C.Y. Zhang, J. Biol. Chem. 284(2009) 5362–5369.
[28] C.G. Crist, D. Montarras, G. Pallafacchina, D. Rocancourt, A. Cumano, S.J. Conway,M. Buckingham, Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 13383–13387.
[29] R.S. Krauss, F. Cole, U. Gaio, G. Takaesu, W. Zhang, J.S. Kang, J. Cell Sci. 118 (2005)2355–2362.
[30] G. Del Sal, M.E. Ruaro, L. Philipson, C. Schneider, Cell 70 (1992) 595–607.[31] T. Tenzen, B.L. Allen, F. Cole, J.S. Kang, R.S. Krauss, A.P. McMahon, Dev. Cell 10 (2006)
647–656.[32] G. Takaesu, J.S. Kang, G.U. Bae, M.J. Yi, C.M. Lee, E.P. Reddy, R.S. Krauss, J. Cell. Biol.
175 (2006) 383–388.[33] D.E. Todd, R.M. Densham, S.A. Molton, K. Balmanno, C. Newson, C.R. Weston, A.P.
Garner, L. Scott, S.J. Cook, Oncogene 23 (2004) 3284–3295.[34] C. Simone, S.V. Forcales, D.A. Hill, A.N. Imbalzano, L. Latella, P.L. Puri, Nat. Genet. 36
(2004) 738–743.[35] C. Serra, D. Palacios, C. Mozzetta, S.V. Forcales, I. Morantte, M. Ripani, D.R. Jones, K.
Du, U.S. Jhala, C. Simone, P.L. Puri, Mol. Cell 28 (2007) 200–213.[36] P.J. Atkinson, T. Dellovade, D. Albers, D. Von Schack, K. Saraf, E. Needle, P.H.
Reinhart, W.D. Hirst, J. Neurochem. 108 (2009) 1539–1549.[37] M. Lu, R.S. Krauss, Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 4212–4217.[38] G.U. Bae, S. Domene, E. Roessler, K. Schachter, J.S. Kang, M. Muenke, R.S. Krauss,
Am. J. Hum. Genet. (July 27, 2011).


















