
Computational Chemical Dynamics of Complex Systems
45
Final Report For EMSL Computational Grand Challenge Grant Period covered: October 1, 2006 – September 30, 2009 Date of report: November 2, 2009 Computational Chemical Dynamics of Complex Systems Abstract The goal of this project is to apply powerful new simulation techniques to tackle computationally challenging problems in chemical dynamics, with special emphasis on electrochemistry, heterogeneous catalysis, nanoparticles, solid-state dynamics, and photochemistry. These calculations are carried out with new high-throughput integrated software that we are developing. New research capabilities in computational chemical dynamics are expected to play a significant role in enabling environmental scientists worldwide to address environmental challenges facing DOE and the nation. Recent advances in computer power and algorithms have made possible accurate calculations of many chemical properties for both equilibria and kinetics. Nonetheless, applications to complex chemical systems, such as reactive processes in the condensed phase, remain problematic due to the lack of a seamless integration of computational methods that allow modern quantum electronic structure calculations to be combined with state- of-the-art methods for chemical thermodynamics and reactive dynamics. These problems are often exacerbated by unvalidated methods and limited software reliability. Our consortium is developing an integrated software suite that combines electronic structure packages with dynamics codes and efficient sampling algorithms for the following kinds of condensed-phase modeling problems: thermochemical kinetics and rate constants, photochemistry and spectroscopy, chemical and phase equilibria, electrochemistry, and heterogeneous catalysis. These fundamental areas of research are important for solar energy, fuel-cell technology, environmental remediation, weather modeling, pollution modeling, and atmospheric chemistry. Photochemical creation of excited states offers a means to control chemical transformations because different wavelengths of light can be used to create different vibronic states, thereby directing chemical reactions along different pathways. It is crucial to understand how energy deposited into the system is used; this is particularly complicated in condensed phase systems where there are many ways to dissipate excess energy. Similar opportunities and challenges present themselves in the areas of electrochemistry and catalysis. We carry out prototype large- scale applications on environmental problems as well as other applications to complex chemical dynamics processes, focusing on three high-impact areas. In the computational electrochemistry area, we are especially concerned with processes that enhance the design of fuel cell technology and with the calculation of in situ reduction potentials. For heterogeneous, nanoparticle, and solid-state dynamics, we develop an array of methods for multi-time-scale simulation of nucleation of crystals in solution, reactions of radicals at solution-phase interfaces and in ice, zeolite catalysis, structure and dynamics of gallazane precursors to gallium nitride nanocrystals, the regulatory role of metal ions in the reactivity of inorganic phosphates, nanoparticles structure and dynamics, and ice dynamics. In the computational photochemistry area, we construct 1
Transcript of Computational Chemical Dynamics of Complex Systems

Final Report For EMSL Computational Grand Challenge Grant
Period covered: October 1, 2006 – September 30, 2009 Date of report: November 2, 2009
Computational Chemical Dynamics of Complex Systems Abstract The goal of this project is to apply powerful new simulation techniques to tackle computationally challenging problems in chemical dynamics, with special emphasis on electrochemistry, heterogeneous catalysis, nanoparticles, solid-state dynamics, and photochemistry. These calculations are carried out with new high-throughput integrated software that we are developing. New research capabilities in computational chemical dynamics are expected to play a significant role in enabling environmental scientists worldwide to address environmental challenges facing DOE and the nation. Recent advances in computer power and algorithms have made possible accurate calculations of many chemical properties for both equilibria and kinetics. Nonetheless, applications to complex chemical systems, such as reactive processes in the condensed phase, remain problematic due to the lack of a seamless integration of computational methods that allow modern quantum electronic structure calculations to be combined with state-of-the-art methods for chemical thermodynamics and reactive dynamics. These problems are often exacerbated by unvalidated methods and limited software reliability. Our consortium is developing an integrated software suite that combines electronic structure packages with dynamics codes and efficient sampling algorithms for the following kinds of condensed-phase modeling problems: thermochemical kinetics and rate constants, photochemistry and spectroscopy, chemical and phase equilibria, electrochemistry, and heterogeneous catalysis. These fundamental areas of research are important for solar energy, fuel-cell technology, environmental remediation, weather modeling, pollution modeling, and atmospheric chemistry. Photochemical creation of excited states offers a means to control chemical transformations because different wavelengths of light can be used to create different vibronic states, thereby directing chemical reactions along different pathways. It is crucial to understand how energy deposited into the system is used; this is particularly complicated in condensed phase systems where there are many ways to dissipate excess energy. Similar opportunities and challenges present themselves in the areas of electrochemistry and catalysis. We carry out prototype large-scale applications on environmental problems as well as other applications to complex chemical dynamics processes, focusing on three high-impact areas. In the computational electrochemistry area, we are especially concerned with processes that enhance the design of fuel cell technology and with the calculation of in situ reduction potentials. For heterogeneous, nanoparticle, and solid-state dynamics, we develop an array of methods for multi-time-scale simulation of nucleation of crystals in solution, reactions of radicals at solution-phase interfaces and in ice, zeolite catalysis, structure and dynamics of gallazane precursors to gallium nitride nanocrystals, the regulatory role of metal ions in the reactivity of inorganic phosphates, nanoparticles structure and dynamics, and ice dynamics. In the computational photochemistry area, we construct
1

potential energy surfaces for a number of photochemical reactions and employ them for dynamics calculations based on the new decay of mixing with coherent switches algorithm. We also consider solvatochromic shifts on conical intersections that govern selected photo chemical processes. Participants for the past three years: University of Minnesota
Donald Truhlar, Principal Investigator Aleksandr Marenich, Project Manager for UMN
John Alecu Elizabeth Amin Kelly Anderson Boris Averkiev Alessandro Cembran Christopher Cramer Luke Fiedler Jiali Gao Andreas Heyden (collaborator, University of South Carolina) Mark Iron Hannah Leverentz Hai Lin (collaborator, University of Colorado - Denver) Steven Mielke Ewa Papajak Jake Rafferty J. Ilja Siepmann Anastassia Sorkin Oksana Tishchenko Rosendo Valero Brian White Ke Yang Darrin York Yan Zhao Jingjing Zheng
Pacific Northwest National Laboratory
Marat Valiev, Project Manager for PNNL Peng-Dong Fan Bruce Garrett Shawn Kathmann Sriram Krishnamoorthy Karol Kowalski Chris Mundy
2

Financial support for the past three years (University of Minnesota): Agency: National Science Foundation, Chemistry Division Project title: Quantum Mechanical Structure and Dynamics of Complex Systems Principal investigator: Donald G. Truhlar Period: 5/1/07 to 4/30/10 Agency: U.S. Department of Energy, Office of Basic Energy Sciences Project title: Variational Transition State Theory Principal investigator: Donald G. Truhlar Period: 12/1/07 to 11/30/10 Agency: Office of Naval Research Project title: Integrated Tools for Computational Chemical Dynamics Principal investigator: Donald G. Truhlar Period: 3/1/05 to 2/1/10 Agency: Air Force Office of Scientific Research Project title: Orbital-Dependent Density Functionals for Chemical Catalysis Principal investigator: Donald G. Truhlar Period: 3/1/08 to 2/1/11 Agency: Army Research Office Project title: Enabling Computational Technologies for the Accurate Prediction/Description of Molecular Interactions in Condensed Phases Principal investigator: Christopher J. Cramer Co-principal investigator: Donald G. Truhlar Period: 7/14/09 to 7/13/12 Agency: NIH (Trans-NIH Recovery Act Research Support) Project title: A New Paradigm for Biomolecular Simulations Principal investigator: Jiali Gao Co-principal investigator: Donald G. Truhlar Period: 9/30/09 to 9/29/11
3

Number of hours allocated for the past three years: 1,825,000 node-hours Number of hours actually used in the past three years: 1,301,622 node-hours
4

Overview of the past three year’s accomplishments and activities: Minnesota density functionals: application to supramolecules One of the most vigorously developing technological research areas is the field of supramolecular chemistry, which involves the use of noncovalent interactions to assemble molecules into stable, well-defined structures called supramolecules. Most of the experimental studies of supramolecular self-assembly are carried out in the condensed phase since the gas-phase supramolecular experiments are technically too demanding. An alternative is theoretical and computational studies that can be carried out for gas-phase supramolecular systems quite reliably nowadays, and such theoretical studies can shed light on intrinsic supramolecular structures and energetics, thus complementing the experimental investigations. One promising approach is density functional theory, which has an excellent performance-to-cost ratio. However, the popular B3LYP density functional and most other older functionals are inaccurate for noncovalent (for instance, aromatic-aromatic) interactions. One of the ways to improve the performance of DFT is to optimize the exchange-correlation functionals for a broad range of properties including rare-gas dimers, aromatic-aromatic interactions, and other data sensitive to medium-range correlation energy, such as barrier heights. This approach has been used for the development of the M06-2X density functional which has been validated for π...π stacking and has also been employed to study supramolecular assembly involving a nanoring and to study the dimerization of coronene as a model for interacting graphene sheets in multiwalled carbon-based assemblies. The M06 suite of Minnesota density functionals includes M06-2X, M06, M06-HF, and M06-L, all of which have been incorporated in the NWChem computational package developed at PNNL and they are now available for all NWChem users. Minnesota solvation models We have developed a new charge-density based solvation model called SMD. The SMD algorithm involves an integration of the nonhomogeneous Poisson equation for electrostatics. The SMD model has been parametrized using a large training set of neutral and ionic solvation free energies for various solutes in water and organic solvents (nearly 3000 data). The non-bulk-electrostatic part of the SMD model utilizes the cavity–dispersion–solvent-structure formalism that was worked out for previous solvation models developed at the University of Minnesota. The SMD model parameters are recommended to be used for the calculation of solvation free energies within the frameworks of the Polarizable Continuum Models implemented in Gaussian 03 and GAMESS, and it can be used in conjunction with the Generalized Conductor-like Screening Model (GCOSMO) implemented in NWChem. (Note that NWChem calculations were performed at the EMSL computing facilities whereas Gaussian and GAMESS calculations were carried out at the Minnesota Supercomputing Institute facilities.) The new SMD model complements the recent SM8 and SM8AD models based on the generalized Born equation for electrostatics.
5

Calculation of reaction rates for barrierless association reactions using variational transition theory This project was focused on calculations of quantitatively accurate reaction rate for barrierless association reactions by means of variational transition state theory. One of the challenges for predicting accurate reaction rates of barrierless association reactions is to calculate accurate potential energy surface in the transition state regions which are often accompanied with a strong multireference character of the corresponding electronic wavefunction. The investigated association reactions are CN + NH3 and OH + SO2. High level ab inito methods, namely the coupled cluster CCSDT and CCSDTQ methods with augmented triple-zeta basis sets, were used to generate an accurate minimum reaction path of the association reactions. Note that the inclusion of explicit triple (T) and quadruple (Q) electronic excitations is often critical for accurate treatment of bond breaking/forming processes. However, density functional theory (DFT) is obviously much more affordable for the use in direct dynamics calculations in terms of variational transition state theory. Thus, we employed the CCSDT and CCSDTQ calculations for validation of the DFT results in order to identify those combinations of density functionals and basis sets that provide most accurate potential energy surfaces. The NWChem computational package developed at PNNL was intensively used in this project. We found the implementation of NWChem at the Chinook supercomputer very effective in such expensive calculations as those employing CCSDTQ. QM/MM free energy simulations of reactions of ions and radicals in aqueous systems The focus of this project is to gain a theoretical understanding of factors that control ground and excited-state properties of chemical systems in condensed phase and other complex environments. This is a challenging problem requiring simultaneous consideration of the electronic structure of reactive species, collective degrees of freedom associated with the environment, reaction dynamics, and thermal fluctuations. To address these issues, we are developing multi-scale, multi-physics approaches that recognize the advantages of using different theoretical models (multi-physics) to address the natural decomposition of the chemical system into distinct regions (multi-scale). These theoretical models can be associated with different parts of the overall chemical system and/or can coexist in a layered fashion. This flexibility in the system description significantly extends the scope, accuracy, and reliability of ab initio modeling of ground and excited-state processes in complex systems. Based on this methodology, we are pursuing novel approaches for efficient utilization of high-level electronic structure methods such as coupled cluster theory in real condensed phase applications. We recently presented a method for calculating free energy profiles for chemical reactions in solution utilizing high-level ab initio methods. We begin with the standard separation of the condensed phase system into a reactive part, which is treated quantum mechanically (QM), and the rest of the system, which is treated by a classical molecular mechanics (MM) model. We use a thermodynamic cycle to take advantage of the state function property of the potential of mean force and combine simulations of free energy differences with pure MM models that allow sampling to be performed over ensembles that are sufficiently large to converge statistical
6

averages with high-level electronic structure calculations for the QM region to accurately predict energetics. Large-scale parallel calculations with combined coupled cluster and molecular mechanics formalism: excitation energies of zinc-porphyrin in aqueous solution Zinc-porphyrin (ZnP) is important in understanding photochemical properties of π-conjugated systems. Although vertical excitation energies of ZnP monomer were studied previously, due to the size of the system, the role of high order correlation effects (e.g. triply excited configurations) still remained unknown. The solvent effect on the excited states of ZnP have not been studied either. To address the above issues and demonstrate the performance of our parallel codes, we carried out cutting edge large scale benchmark excited state calculations of the ZnP monomer in aqueous solution using combined coupled cluster molecular mechanics formalism, in which the coupled cluster (CC) methods are equation of motion approach with singles and doubles (EOMCCSD) and completely renormalized non-iterative triples corrections to EOMCCSD method, CR-EOMCCSD(T). The calculations show that the non-iterative triples corrections ranges from 0.24 eV to 0.29 eV, and the CC approaches provide consistent energetics of low-lying excited states for all environments, while the TD-DFT approach can not adequately describe prototype charge transfer states dominated by excitations from central zinc and nitrogen atoms to carbon atoms. Such breakdown of TD-DFT is best observed in aqueous environment. The parallel performance of our code scales cross 1000 CPUs. The scalability is 1.92 and 1.94 for the CCSD and EOMCCSD methods, respectively, when going from 256 CPUs to 512 CPUs, and 1.66 and 1.74, respectively, from 512 CPUs to 1024 CPUs. Investigation of hydrous silica melts Understanding the structure of hydrous silica (SiO2) melts is important for gaining insight into the structure of the Earth’s upper mantle. Pressures within the mantle range from 1 GPa to 135 GPa at the core-mantle boundary and temperatures range from several hundred to a few thousand Kelvin degrees. This wide range of combined high temperatures and pressures makes experimental studies of these systems challenging. Simulation provides an alternative means to examine structural properties. Previously, our group used an empirical force field to examine hydrous silica melts at high temperatures and moderate pressures (0.25-10 GPa) using Monte Carlo (MC) simulations in the isobaric-isothermal ensemble. We have sought to probe these systems via first-principles molecular dynamics (FPMD) simulations using Kohn-Sham density functional theory to represent the interactions. These simulations were performed under isobaric-isothermal conditions using the CP2K simulation package and the Gaussian plane wave method implemented therein to calculate energies and forces. It was found that in the absence of water, the silica melt forms a highly networked liquid. The addition of even relatively small amounts of water (3.2 wt.%) is sufficient to substantially change the structure of the liquid. Owing to PNNL’s support, one of the graduate students involved in the project (Katie Maerzke) participated in the 2008 Summer Research Institute program at PNNL.
7

Quantum mechanical/molecular mechanical simulation methods for catalysis The molecular mechanism of hairpin ribozyme catalysis is studied with molecular dynamics simulations using a combined quantum mechanical and molecular mechanical (QM/MM) potential with a recently developed semiempirical AM1/d-PhoT model for phosphoryl transfer reactions. Simulations are used to derive one- and two-dimensional potentials of mean force to examine specific reaction paths and assess the feasibility of proposed general acid and base mechanisms. Density-functional calculations of truncated active site models provide complementary insight to the simulation results. The computational methods are general and may also be used for heterogeneous catalysis. Part of these calculations was performed at PNNL’s computational facility, and with the PNNL resources, we could carried these simulations out to longer, more meaningful time scales using more reliable QM/MM models. For example, the CHARMM simulation package ported to the MPP2 supercomputer (this supercomputer is retired now) could generate simulations of 1-1.5 ps per CPU-hour on single CPU. The maximum speedup for CHARMM running on MPP2 was 20 using 32 CPUs. For comparison, another simulation package (NAMD) installed on MPP2 achieved 2-3 ps per CPU-hour on single CPU, and its maximum speedup was around 10 ps using 32 CPUs. We substantially improved the performance of NAMD so that calculations on large-scale parallel machines could be run much more efficiently with our implementation of NAMD rather than with its original implementation. Orotidine monophosphate decarboxylase: anatomy of the catalytic mechanism by nature’s most proficient enzyme Orotidine 5'-monophosphate decarboxylase (OMPDC) catalyzes the exchange of CO2 for a proton at the C6 position to form uridine 5'-monophosphate (UMP), providing a rate acceleration of 17 orders of magnitude over that of the uncatalyzed reaction. The remarkable catalytic proficiency of this enzyme has attracted numerous experimental and computational studies and a number of mechanisms have been proposed to account for its activity. In this project, by means of combined quantum mechanical and molecular mechanical (QM/MM) simulations, we provided some new insights into the factors that contribute to the OMPDC catalysis. In particular, we computed the free energy of activation for the decarboxylation reaction in water solution (34.4 kcal/mol) and in the enzyme (16.9 kcal/mol), which compare well with the experimental data (38.5 and 16.5 kcal/mol, respectively). We identified Lys72 hydrogen bond with the ribose 2'-OH group in the Michaelis complex structure as a key point for understanding the OMPDC catalysis. There is a subtle, but significant, conformational transition involving Lys72 that migrates more than 1 Å towards the ribose ring relative to the crystal structure. As the recent literature suggests that the presence of a specific water molecule in the active site is not compatible with catalysis, we tested for the effects of removing such a water molecule: our calculations showed only a marginal (3 kcal/mol) energy barrier reduction, which cannot account for the energy barrier reduction performed by the enzyme (about 22 kcal/mol).
8

In addition, to determine the amount of catalysis performed by the enzyme through transition-state stabilization, we computed the potential of mean force in the enzyme and in water for the proton transfer between Lys72 and the uracil C6 carbanion, where the latter closely resembles the transition-state for the decarboxylation reaction. By these means, we found that at most 11 kcal/mol can be accounted for by the transition-state stabilization, and that about 7 kcal/mol for the computed barrier reduction must derive from other factors. By analyses of the structures of reactant and transition state, it was possible to identify a distortion operated by binding of negatively charged OMP upon the protein backbone through repulsion with Asp70. We proposed that part of the catalytic power of the enzyme is achieved by the relieving of this strain upon reaching the transition-state. Molecular dynamics simulation for the transport of ions through the phospholamban membranes Phospholamban (PLN) is a single pass membrane protein that reversibly binds and regulates the sarcoplasmic reticulum (SR) Ca2+-ATPase (SERCA). The SERCA/PLN complex is responsible for Ca2+ ions transport from the cytoplasm into the SR lumen, causing muscle relaxation. PLN exists in equilibrium between the pentameric and monomeric forms, and it is the monomeric form that is the active, inhibitory species, regulating Ca2+ transport. However, the function of the oligomeric configuration has puzzled biochemists and biophysicists for many years, and the literature suggests that it might act as a Cl- or Ca2+ ion channel. We computed through molecular dynamics (MD) simulations the potential of mean force (PMF) for the Cl- and Ca2+ ion conduction through the PLN pentamer pore. We determined the barriers for the passage of Cl- and Ca2+ to be 19 and 41 kcal/mol, respectively, values that are too high for the translocation to take place. This result, together with electrochemical measurements performed by collaborators, reinforced the hypothesis of PLN pentamer to be a storage form and not an ion channel.
The structure and dynamics of the monomeric and pentameric forms of PLN have also been investigated with molecular dynamics simulations. Specifically, our research focused on the structural and dynamical effects due to the protein interaction with the lipid bilayer. We compared our results to NMR data. We highlighted the different dynamics of the cytoplasmic loop when embedded at the membrane/water interface or when fully exposed to the solution. Multiscale model development, implementation, and application The objective of this project has been to develop, test, and implement a highly efficient conservative multiscale modeling algorithm for simulating large systems that require an extensive sampling of a complex phase space to determine the system properties, but that requires only locally, i.e., close to an active site, a high level of resolution and accuracy. The method is called adaptive partitioning (AP) of the Lagrangian (APL) and it can often permit a molecular dynamics (MD) simulation of a complex system with an accuracy of an equivalent high level of theory / resolution simulation at the computational cost of a low level of theory /
9

resolution simulation. Using our methodology, we have recently begun studying the oxygen mobility in perovskite and double perovskite oxide anode materials such as doped LaGaO3 and Sr2MgMoO6 for solid oxide fuel cells. Our objective is to identify practical descriptors that allow for rational design of novel anode materials in solid oxide fuel cells with excellent catalytic activity, electronic conductivity, and oxygen mobility. Within this project, we have also developed the electrostatically embedded many-body (EE-MB) method for calculating the energetics of large molecular clusters. The EE-MB method is a cost-effective way to accurately approximate the ab initio energies of large systems. We believe this method will ultimately be usable in Monte Carlo simulations of atmospheric nucleation.
10

List of all publications resulting from this work: “Thermochemical kinetics of hydrogen-atom transfers between methyl, methane, ethynyl, ethyne, and hydrogen,” J. Zheng, Y. Zhao, and D. G. Truhlar, J. Phys. Chem. A 2007, 111, 4632. “Attractive noncovalent interactions in Grubbs second-generation Ru catalysts for olefin metathesis,” Y. Zhao and D. G. Truhlar, Org. Lett. 2007, 9, 1967. “Size-selective supramolecular chemistry in a hydrocarbon nanoring,” Y. Zhao and D. G. Truhlar, J. Am. Chem. Soc. 2007, 129, 8440. “Hybrid approach for free energy calculations with high-level methods: Application to the SN2 reaction of CHCl3 and OH- in water,” M. Valiev, B. C. Garrett, M.-K. Tsai, K. Kowalski, S. M. Kathmann, G. K. Schenter, and M. Dupuis, J. Chem. Phys. 2007, 127, 051102. “Molecular computational investigation of electron-transfer kinetics across cytochrome-iron oxide interfaces,” S. Kerisit, K. M. Rosso, M. Dupuis, and M. Valiev, J. Phys. Chem. C 2007, 111, 11363. “Density functionals with broad applicability in chemistry,” Y. Zhao and D. G. Truhlar, Acc. Chem. Res. 2008, 41, 157. “A prototype for graphene material simulation: Structures and interaction potentials of coronene dimers,” Y. Zhao and D. G. Truhlar, J. Phys. Chem. C 2008, 112, 4061. “Density functional theory in transition-metal chemistry: relative energies of low-lying states of iron compounds and the effect of spatial symmetry breaking,” A. Sorkin, M. A. Iron, and D. G. Truhlar, J. Chem. Theory Comput. 2008, 4, 307. “How well can new-generation density functionals describe the energetics of bond dissociation reactions producing radicals?” Y. Zhao and D. G. Truhlar, J. Phys. Chem. A 2008, 112, 1095. “Benchmark data for interactions in zeolite model complexes and their use for assessment and validation of electronic structure methods,” Y. Zhao and D. G. Truhlar, J. Phys. Chem. C 2008, 112, 6860. “Computational characterization and design of buckyball tweezers: density functional study of concave-convex pi...pi interactions,” Y. Zhao and D. G. Truhlar, Phys. Chem. Chem. Phys. 2008, 10, 2813. “Perspective on foundations of solvation modeling: the electrostatic contribution to the free energy of solvation,” A. V. Marenich, C. J. Cramer, and D. G. Truhlar, J. Chem. Theory Comput. 2008, 4, 877. “Construction of a generalized gradient approximation by restoring the density-gradient
11

expansion and enforcing a tight Lieb-Oxford bound,” Y. Zhao and D. G. Truhlar, J. Chem. Phys. 2008, 128, 184109. “Large-scale parallel calculations with combined coupled cluster and molecular mechanics formalism: excitation energies of zinc-porphyrin in aqueous solution,” P. D. Fan, M. Valiev, and K. Kowalski, Chem. Phys. Lett. 2008, 458, 205. “Exploring the limit of accuracy of the global hybrid density functional for main-group thermochemistry, kinetics, and noncovalent interactions,” Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput. 2008, 4, 1849. “Benchmark energetic data in a model system for Grubbs II metathesis catalysis and their use for assessment and validation of electronic structure methods,” Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput. 2009, 5, 324. “On the function of pentameric phospholamban: ion channel or storage form?” L. Becucci, A. Cembran, C. B. Karim, D. D. Thomas, R. Guidelli, J. Gao, and G. Veglia, Biophys. J. 2009, 96, L60. “Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions,” A. V. Marenich, C. J. Cramer, and D. G. Truhlar, J. Phys. Chem. B 2009, 113, 6378. “Interactions of Cl- and OH radical in aqueous solution,” M. Valiev, R. D'Auria, D. J. Tobias, and B. C. Garrett, J. Phys. Chem. A 2009, 113, 8823. “Thermochemical kinetics for multireference systems: Addition reactions of ozone,” Y. Zhao, O. Tishchenko, J. R. Gour, W. Li, J. J. Lutz, P. Piecuch, and D. G. Truhlar, J. Phys. Chem. A 2009, 113, 5786.
12

List of presentations resulting from this work: “Interfacing electronic structure theory with dynamics,” D. G. Truhlar, H. Lin, O. Tischenko, Y. Zhao, and R. Valero, 232nd ACS National Meeting, San Francisco, CA, September 10-14, 2006. “New semiempirical density functionals,” D. G. Truhlar, Y. Zhao, N. E. Schultz, E. E. Dahlke, and M. Iron, 232nd ACS National Meeting, San Francisco, CA, September 10-14, 2006. “Benchmark study of noncovalent interactions in the zeolite model complexes and their use for validation of density functionals and force fields,” Y. Zhao and D. G. Truhlar, 232nd ACS National Meeting, San Francisco, CA, September 10-14, 2006. “Density functional theory: The M06 suite,” Y. Zhao and D. G. Truhlar, Symposium on Practicing Chemistry with Theoretical Tools on the Occasion of Professor Mark S. Gordon's 65th Birthday: MSG-65, Maui, HI, January 15-18, 2007. “DFT study of the interaction potentials of coronene dimers,” Y. Zhao and D. G. Truhlar, NWChem Meeting on Science Driven Petascale Computing and Capability Development at Environmental Molecular Science Laboratory, Richland Washington, January 23-26, 2007. “Toward more realistic simulations for large-size reactive systems by combining quantum mechanics and molecular mechanics in new ways,” H. Lin and G. G. Truhlar, NWChem Meeting on Science Driven Petascale Computing and Capability Development at Environmental Molecular Science Laboratory, Richland Washington, January 23-26, 2007. “Adaptive partitioning in multilevel/multiscale simulations,” A. Heyden and D. G. Truhlar, 233rd ACS National Meeting, Chicago, IL, March 25-29, 2007. “Density functionals for noncovalent interaction energies of biological importance,” Y. Zhao and D. G. Truhlar, 233rd ACS National Meeting, Chicago, IL, March 25-29, 2007. “Universal solvation models and their applications,” D. G. Truhlar, Barry Honig 65th Birthday Symposium: Biological Applications of Implicit Solvation Models, National Meeting of the American Chemical Society, Chicago, IL, March 25-29, 2007. “Modeling free energies of solvation, vapor pressure, partitioning, solubility, and condensed-phase properties and reaction mechanisms,” C. J. Cramer, D. G. Truhlar, C. P. Kelly, A. C. Chamberlin, and N. Elmasry, 90th Canadian Chemistry Conference, Winnipeg Manitoba, May 26-30, 2007. “Advances in modeling and simulation,” D. G. Truhlar, Workshop on Multi-Scale and Large-Scale Simulations in DoD Materials Science, McLean, VA, June 14-15, 2007. “Density functionals for dynamics,” Y. Zhao and D. G. Truhlar, Conference on the Dynamics of Molecular Collisions, Santa Fe, NM, July 8-13, 2007.
13

“A new density functional for spectroscopy,” Y. Zhao and D. G. Truhlar, Gordon Research Conference on Time-Dependent Density Functional Theory, Waterville, ME, July 15-20, 2007. “Density functional theory,” D. G. Truhlar, MURI Software Transition Meeting, Army Research Office, Aberdeen, MD, July 19, 2007. “New density functionals: a meta GGA and three hybrid meta GGAs with good performance for thermochemistry, thermochemical kinetics, noncovalent interactions, and spectroscopy,” IMA Summer Program on Quantum Approaches in Molecular Modeling, Minneapolis, MN, August 1-3, 2007. “New density functionals for making bold predictions,” Y. Zhao and D. G. Truhlar, 234th ACS National Meeting, Boston, MA, August 19-23, 2007. “Development and use of a small representative benchmark suite for thermochemical kinetics,” J. Zheng, Y. Zhao, B. J. Lynch, and D. G. Truhlar, 234th ACS National Meeting, Boston, MA, August 19-23, 2007. “Predicting accurate oxidation and reduction potentials in condensed phases,” C. J. Cramer, P. Jaque, C. P. Kelly, A. V. Marenich, and D. G. Truhlar, 234th ACS National Meeting, Boston, MA, United States, August 19-23, 2007 (2007). “Using statistical mechanics with quantum mechanics for enzymes and nanoparticles,” D. G. Truhlar, D. Bhatt, Divesh, A. Dybala-Defratyka, J. Gao, M. Garcia-Viloca, Z. H. Li, P. Paneth, J. Pu, J. I. Siepmann, 234th ACS National Meeting, Boston, MA, August 19-23, 2007. “A conservative algorithm for an on-the-fly change of resolution in mixed atomistic / coarse-grained multiscale simulations,” A. Heyden and D. G. Truhlar, 2007 Annual Meeting of the American Institute for Chemical Engineers, Salt Lake City, UT, November 4-9, 2007. “Density functionals with broad applicability for main group and transition metal chemistry, spectroscopy, kinetics, and noncovalent interactions,” D. G. Truhlar, Max T. Rogers Lecture Series, Michigan State University, April 22, 2008. “Quantum photochemistry,” D. G. Truhlar, Michigan State University, April 23, 2008. “New density functionals with broad applicability for thermochemistry, thermochemical kinetics, noncovalent interactions, transition metals, and spectroscopy,” D. G. Truhlar, Houk65 Symposium: Computational Chemistry: Orgo, Bio, Nano, in honor of Kendall N. Houk, Los Angeles, CA, June 27-28, 2008. “New density functionals for a broad range of applications,” D. G. Truhlar, plenary lecture, 6th Congress of the International Society for Theoretical and Chemical Physics, Vancouver, Canada, July 19-24, 2008.
14

“Open boundary approaches for combined quantum mechanical and molecular mechanical calculations,” D. G. Truhlar, Gordon Research Conference On Computational Chemistry, Mt. Holyoke, MA, July 27-31, 2008. “Density functional study of concave-convex Pi–Pi interactions in supramolecular assemblies,” Y. Zhao and D. G. Truhlar, 236th ACS National Meeting, Philadelphia, PA, United States, August 17-21, 2008. “New density functionals with broad applicability for thermochemistry, thermochemical kinetics, noncovalent interactions, transition metals, and spectroscopy,” D. G. Truhlar, plenary lecture, WATOC 2008: Congress of the World Association of Theoretical and Computational Chemists, Sydney, Australia, September 14-19, 2008. “New density functionals with broad applicability for thermchemistry, thermochemical kinetics, noncovalent interactions, transition metals, and spectroscopy,” D. G. Truhlar, invited lecture at Workshop on Mathematical and Algorithmic Challenges in Electronic Structure Theory, Institute for Mathematics and Its Applications, Minneapolis, MN, October 1, 2008. “Orotidine monophosphate decarboxylase: A simple reaction in a complex environment,” A. Cembran and J. Gao, XXVIII Midwest Enzyme Chemistry Conference, Chicago, IL, October 4, 2008. “Massively parallel variational transition state theory calculations for association reactions with POLYRATE,” S. Zhang, J. Zheng, and D. G. Truhlar, paper presented at SC08: 21st International Conference for High-Performance Computing, Networking, Storage, and Analysis, Austin, TX, November 18, 2008. “New approaches to including charge polarization in simulations,” D. G. Truhlar, invited lecture at symposium on Progress in Polarizable Force Fields and Simulation: New Polarizable Models and Methods, National ACS Meeting, Salt Lake City, UT, April 22-26, 2009. “Inclusion of correlation and polarization effects in molecular modeling,” D. G. Truhlar, 13th Internaitonal Congress on Quantum Chemistry, Helsinki, Finland, June 22-27, 2009. “New approaches to the calculation of reaction rates,” D. G. Truhlar, Conference on the Dynamics of Molecular Collisions, Snowbird, Utah, July 5-9, 2009. “Orotidine monophosphate decarboxylase: A simple reaction in a complex environment,” A. Cembran and J. Gao, Thermodynamics of Biomolecular Machinery, Beijing, China, July 8, 2009. “Orotidine monophosphate decarboxylase: A simple reaction in a complex environment,” A. Cembran and J. Gao, The 23rd Annual Symposium of The Protein Society, Boston, MA, July 25-29, 2009.
15

“Orotidine monophosphate decarboxylase: A simple reaction in a complex environment,” A. Cembran and J. Gao, Molecular Modeling 2009 Meeting, Surfers Paradise, Qeensland, Australia, July 26-29, 2009.
16

List of significant methods/routines or codes developed with a brief description of each: MN-NWCHEMFM (Yan Zhao and Donald G. Truhlar) MN-NWCHEMFM (Minnesota NWChem Functional Module) is a module written in FORTRAN 90 to incorporate the M06 suite of density functionals into NWChem–v. 5.0. This program adds the M06-L, M06-HF, M06, M06-2X, and VS98 (also called VSXC) density functionals to NWChem. See http://comp.chem.umn.edu/mn-nwchemfm for more detail. NWCHEMRATE 2007 (Jingjing Zheng, Mark A. Iron, Benjamin A. Ellingson, José C. Corchado, Yao-Yuan Chuang, and Donald G. Truhlar) NWCHEMRATE is a set of FORTRAN subroutines and Unix scripts for interfacing the POLYRATE and NWChem computer programs for the purpose of carrying out direct dynamics calculations of gas-phase chemical reaction rates of polyatomic species (and also atoms and diatoms as special cases) using the electronic structure methods available in NWChem to calculate the potential energy surface and POLYRATE for the dynamics. The interface is based on the POLYRATE hooks protocol. The dynamical methods used are variational or conventional transition state theory and multidimensional semiclassical approximations for tunneling and nonclassical reflection. Rate constants may be calculated by any of the methods available in POLYRATE for canonical or microcanonical ensembles or for specific vibrational states of selected modes with translational, rotational, and other vibrational modes treated thermally. Bimolecular and unimolecular reactions are included. Both single-level and dual-level calculations may be carried out. In single-level mode, optimized geometries, potential energies, gradients, and Hessians can be calculated by any of the density functionals in the NWChem package or with the MP2 ab initio method. In dual-level mode, the lower-level data is calculated by NWChem, and the higher-level data is read in from an external file. See http://comp.chem.umn.edu/nwchemrate for more detail.
17

Report Appendices
18

Appendix A Full report of first year activities and accomplishments (FY07) 1. Density functionals with broad applicability in chemistry Yan Zhao and Donald G. Truhlar
Although density functional theory is widely used in the computational chemistry community, the most popular density functional, B3LYP, has some serious shortcomings: (i) it is less accurate for transition metal compounds than for main-group chemistry; (ii) it systematically underestimates reaction barrier heights; (iii) it is inaccurate for interactions dominated by medium-range correlation energy, such as van der Waals attraction, aromatic–aromatic stacking, and alkane isomerization energies. We have developed a variety of databases for testing and designing new density functionals, including databases for main-group thermochemistry, thermochemical kinetics, noncovalent interactions, transition metal chemistry, and spectroscopy. We used these data to design new density functionals, called M06-class (and, earlier, M05-class) functionals, for which we enforced some fundamental exact constraints such as the uniform-electron-gas limit and the absence of self-correlation energy. Our M06-class functionals depend on spin-up and spin-down electron densities (i.e., spin densities), spin density gradients, spin kinetic energy densities, and, for nonlocal functionals, Hartree-Fock exchange. We have developed four new functionals that overcome the above mentioned difficulties: (a) M06, a hybrid meta functional, is a functional with good accuracy “across-the-board” for transition metals, main group thermochemistry, medium-range correlation energy, and barrier heights. (b) M06-2X, another hybrid meta functional, is not good for transition metals but has excellent performance for main group chemistry, predicts accurate valence and Rydberg electronic excitation energies, and is an excellent functional for aromatic–aromatic stacking interactions. (c) M06-L is not as accurate as M06 for barrier heights but is the most accurate functional for transition metals and is the only local functional (no Hartree-Fock exchange) with better across-the-board average performance than B3LYP; this is very important because only local functionals are affordable for many demanding applications on very large systems. (d) M06-HF has good performance for valence, Rydberg, and charge transfer excited states with the minimal sacrifice of accuracy for ground-state calculations. Based on our analysis of the performance of the new Minnesota density functionals versus the other popular density functionals on diverse databases, we recommend 1) the M06-2X, BMK, and M05-2X functionals for main-group thermochemistry and kinetics, 2) M06-2X, M05-2X, and M06 for systems where main-group thermochemistry, kinetics, and noncovalent interactions are all equally important, 3) M06-L and M06 for transition metal thermochemistry, 4) M06 for problems involving multireference rearrangements or reactions where both organic and transition-metal bonds are formed or broken, 5) M06-2X, M05-2X, M06-HF, M06, and M06-L for the study of noncovalent interactions, 6) M06-HF when the use of full Hartree-Fock exchange is important, for example to avoid the error of self-interaction at long-range, 7) M06-L when a local functional is required, because a local functional has much lower cost for large systems. The availability of these functionals is described at http://comp.chem.umn.edu/info/DFT.htm.
19

2. Insight into the role of Mg2+ in hammerhead ribozyme catalysis from X-ray crystallography and molecular dynamics simulation Tai-Sung Lee, Carlos Silva López, Monika Martick, William G. Scott, and Darrin M. York The hammerhead ribozyme is an archetypal system to study RNA catalysis. A detailed understanding of the hammerhead mechanism provides insight into the inner workings of more complex cellular catalytic RNA machinery such as the ribosome, and ultimately may aid the rational design of new medical therapies and biotechnology. Despite a tremendous amount of experimental and theoretical efforts, the details of the hammerhead ribozyme mechanism have been elusive. In particular, one of the main puzzles involves the apparent inconsistency between the interpretation of thio effect experiments and mutational data with available crystallographic structural information of the minimal hammerhead sequence. Results from the biochemical experiments suggest that a pH-dependent conformational change, inconsistent with crystallographic data, must precede or be concomitant with the catalytic chemical step. This includes a possible metal ion bridge between the A9 and scissile phosphates that in previous crystal structures were ~20 Å apart. Moreover, the function of the 2’OH group of G8 remains unclear.
Figure 1. The full length hammerhead ribozyme. Recently, the research group of Professor Darrin York in the Department of Chemistry, U of MN, in collaboration with Professor Bill Scott of the Department of Chemistry, UCSC, have performed molecular simulations that probe the conformational and chemical events that leads to ribozyme catalysis. A series of 12 ns molecular dynamics (MD) simulations of the reactant state (with and without a Mg(II) ion), early and late transition states are presented based on a recent crystal structure of a full-length hammerhead RNA reported by Martick and Scott (Figure 1) using new molecular mechanical force field models and combined quantum mechanical/ molecular mechanical potentials. Their simulation results support a catalytically active conformation with a Mg(II) ion bridging the A9 and scissile phosphates, preliminary data generated prior to the PNNL Computational Grand Challenge allocation for which has been published in a letter in J. Chem. Theory Comput. 3, 325-327, (2007). With the PNNL resources, these simulations are now being carried out to longer, more meaningful time scales and with a more realistic QM/MM models.
In the reactant state, the Mg(II) ion most probably occupies a position near the 2’OH group of G8, but remains fairly distant from the leaving group O5’ position. In the early TS mimic simulation, where the nucleophilic O2’ and leaving group O5’ are equidistant from the
20

phosphorus, the Mg(II) ion remains tightly coordinated to the 2’OH of G8, but is positioned closer to the O5’ leaving group, stabilizing the accumulating charge. In the late TS mimic simulation, the coordination around the bridging Mg(II) ion undergoes a transition whereby the coordination with the 2’OH of G8 is replace by the leaving group O5’ that has developed significant charge. At the same time, the 2’OH of G8 forms a hydrogen bond with the leaving group O5’ and is positioned to act as a general acid catalyst. This work represents the first simulations of the full-length hammerhead structure and TS mimics, and provides direct evidence for the possible role of a bridging Mg(II) ion in catalysis that is consistent with both crystallographic and biochemical data. The different relative positions of the Mg(II) to key oxygen atoms are shown in Figure 2.
Figure 2. The Radial distribution of the Mg(II)-O pairs for the proposed catalytically active Mg(II). Further studying of the full-length hammerhead ribozyme system requires much longer simulation time and hence more advanced hardware and software advances. The simulation package CHARMM has been ported to PNNL’s MPP2 supercomputer and it produces simulations of 1-1.5 ps per CPU-hour on single CPU. The maximum speedup for CHARMM running on MPP2 is 20 using 32 CPU’s thus the maximum possible speed of simulation is 500ps per day. Another simulation package, NAMD, already installed on MPP2, produces 2-3 ps per CPU-hour on a single CPU, and its maximum speedup is ~10 using 32 CPUs. Hence the maximum possible simulation speed for this Hammerhead ribozyme is ~500 ps per day on MPP2. We have optimized the performance of NAMD so that calculations can be run efficiently on large-scale parallel machines. The original version of NAMD at PNNL exhibited very poor
21

scaling with the number of processors. Various compilation and inter-node network options have been tested and benchmarked. For single CPU, the performance has been improved by over 100%. For the parallel performance, the maximum speedup has been improved substantially as well. The maximum possible simulation speed for this ribozyme system now reaches ~12,000 ps per day on MPP2 due to the optimization. The performance improvement is shown in Figure 3.
0
2
4
6
8
10
12
14
0 50 100 150 200 250 300Number of CPU
Sim
ulat
ion
Rat
e (n
s pe
r day
)
PNL
PNL Old
Figure 3. The simulation performance of the original NAMD code (PNNL old) and the optimized PNNL code (PNNL). The y-axis is the simulation time that can be obtained in one day. The x-axis is the number of CPU's used to perform the simulation. Our optimization has made much longer simulation on the hammerhead ribozyme possible. This allows us to examine the role of large-scale conformational events, including relaxation of the solvent and ion atmosphere, on ribozyme catalysis. The methods developed here can be broadly applied to other systems that involve bio-catalysis.
22

3. QM/MM study of the H/D kinetic isotope effect in the reduction reaction catalyzed by dihydrofoloate reductase (DHFR) Jingzhi Pu, Shuhua Ma, Mireia Garcia-Viloca, Jiali Gao, and Donald G. Truhlar Dihydrofoloate reductase (DHFR) is a small, monomeric protein that has been extensively studied experimentally and computationally. DHFR catalyzes the reduction of 7,8-dihydrofolate (DHF) to 5,6,7,8-tetrahydrofolate (THF) with the key chemical step being the transfer of a hydride ion from the nicotinamide ring of the cofactor nicotinamide adenine dinucleotide phosphate (NADPH). At pH = 7, product release is partly rate-limiting and the H/D kinetic isotope effect is about 1.1-1.3, but the intrinsic KIE on the hydride transfer step is >3. Previously, we have investigated the catalytic mechanism and the factors that affect the reaction rate (“Reaction-path energetics and kinetics of the hydride transfer reaction catalyzed by dihydrofolate reductase,” M. Garcia-Viloca, D. G. Truhlar, J. Gao, Biochem., 2003, 42, 13558) and have computed the temperature dependence of the kinetic isotope effects for the hydride transfer reaction (“Small temperature dependence of the kinetic isotope effect for the hydride transfer reaction catalyzed by Escherichia coli dihydrofolate reductase,” J. Pu, S. Ma, J. Gao, D. G. Truhlar, J. Phys. Chem. B, 2005, 109, 8551). Recently, Kohen and coworkers (PNAS, 2006, 103, 15753) described a study of the wild-type, single mutant (G121V) and double mutant (DM) (G121V and M42W) of the Escherichia coli DHFR (ecDHFR) enzyme at different temperatures and reported KIE and kinetic data. Analysis of Arrehnius plots revealed the follow kinetic data: WT: Ea = 5.6 kcal/mol, TΔS = -11.8 kcal/mol; G121V: Ea = 5.3 kcal/mol, TΔS = -12.2 kcal/mol, and G121V/M42W: Ea = 3.9 kcal/mol, TΔS = -15.7 kcal/mol. This work raises interesting questions, especially concerning the contributions of enzyme dynamics to enzyme catalysis. The availability of Arrhenius parameters both for the wild-type and mutant enzymes provided an opportunity to address these questions by computation. The work has been carried out for ecDHFR at three different temperatures both for the WT and DM, by computing the potentials of mean force (PMF) and analysis of structural and kinetic parameters. The work involves multiple dynamics simulations consisting of at least 15 free energy calculations for each PMF, each lasting 200 to 500 ps.
23

4. Thermochemical kinetics of hydrogen-atom transfers between methyl, methane, ethynyl, ethyne, and hydrogen Jingjing Zheng, Yan Zhao, and Donald G. Truhlar Saddle point properties of three symmetric and one asymmetric hydrogen–transfer reaction and the energies of reaction of the asymmetric reactions were investigated. These reactions,
H + H2 → H2 + H (R1) CH3 + CH4 → CH4 + CH3 (R2) HCC + HCCH → HCCH + CCH (R3) HCC + H2 → HCCH + H (R4)
were calculated by various density functionals, many of which were developed in recent years, by coupled cluster theory, and by multicoefficient correlation methods based on wave function theory. Instead of comparing calculated results to “semi-experimental” values, we compared them to very accurate theoretical values (e.g., to values obtained by the Weizmann-1 method). Coupled cluster theory and the multicoefficient correlation methods MC–QCISD/3 and MCQCISD–MPW are found to be very accurate for these reactions with mean unsigned errors below 0.94 kcal/mol. Diagnostics for multireference character were calculated to aid in the interpretation and help assess the reliability of the calculations. The newly developed hybrid density functional M06-2X shows very good performance for these reactions with a mean unsigned error of only 0.77 kcal/mol. The MPW1K and BB1K density functionals, can also predict these reactions well with mean unsigned errors less than 1.42 kcal/mol.
24

5. Attractive noncovalent interactions in the mechanism of Grubbs second-generation Ru catalysts for olefin metathesis Yan Zhao and Donald G. Truhlar Grubbs’ second-generation Ru metathesis catalysts are a hundred to a thousand times more active than first-generation Ru metathesis catalysts, and they also exhibit greater thermal and chemical stability. The difference is the substitution of one of the phosphine ligands, usually tricyclohexylphospine, PCy3, of the bisphosphine first-generation precatalyst by a N-heterocyclic carbene (NHC), usually 1,3-dimesityl-4,5-dihydro-2-ylidene, henceforth denoted H2IMes. Liquid-phase mechanistic studies established that olefin metathesis with these 16-electron (five-coordinate) Ru precatalysts proceeds by phosphine dissociation to generate the 14-electron (four-coordinate) active species. A surprising discovery (later confirmed in the gas phase) was that phosphine dissociation in (PCy3)2Cl2Ru=CHPh (1) is faster than in (H2IMes)(PCy3)Cl2Ru=CHPh (2), so that the rate of generation of active species does not correlate with catalytic activity. Since the reverse association reaction is believed to be barrierless, the relative dissociation rates are attributed to a smaller Ru-P bond dissociation energy (BDE) in 1 than in 2 (Scheme 1). Understanding the factors that control catalyst initiation is critical to rational ligand-design strategies for new catalysts. Scheme 1. First step: generation of the catalyst from the precatalysts.
Ru
PCy3
PCy3Cl
Cl Ph
Ru
PCy3
Cl
ClPh
NN
BDE (1)
BDE (2)
(1)
(2)
Ru
PCy3
Cl
Cl Ph+ PCy3
+ PCy3Ru
Cl
ClPh
NN
We have applied the recently developed M06-L density functional and the popular B3LYP, BP86, and PW91 functionals along with two more recent (but older than M06-L) high-quality functionals, PBEh and TPSSh, to the Ru benzylidene compounds 1 and 2 employing a double-zeta-quality (DZQ) basis set and a triple-zeta-quality (TZQ) basis set explained elsewhere. In some cases we also included a counterpoise correction (CP) for basis set superposition error. The results are listed in Table 1. At the M06-L/TZQ-CP level of theory, we obtain ΔBDE = –4.0 kcal/mol, in good agreement with experiment. This shows the importance of medium-range correlation energy for the description of large transition metal complexes. Table 1 shows that M06-L gives larger BDEs for both 1 and 2 with the DZQ basis set than the TZQ basis set, but the CP correction brings the results closer. Furthermore, all four M06-L calculations, with and without the CP correction, give very similar values for ΔBDE, in the range -3.4 to -4.1 kcal/mol, and the negative sign is the right trend. The BP86, PW91, PBE0 and TPSSh functionals are better than the most popular functional, B3LYP, but they share the same flaw as B3LYP for the sign of ΔBDE.
25

Table 1. Bond dissociation energies (kcal/mol) for the Grubbs catalysts method BDE(1) BDE(2) ΔBDE Exp. -3.4 ± 2aM06-L/TZQ 36.1 40.2 -4.1 M06-L/TZQ-CP b 34.2 38.2 -4.0 M06-L/DZQ 41.7 45.2 -3.5 M06-L/DZQ-CP 38.3 41.7 -3.4 B3LYP/DZQ 19.0 17.4 1.6 B3LYP/DZQ-CP 15.6 14.0 1.7 BP86/DZQ 20.0 18.8 1.2 PW91/DZQ 26.1 25.7 0.4 PBEh/DZQ 28.9 28.1 0.8 TPSSh/DZQ 24.6 23.5 1.1
Removed RuCl2 c M06-L/DZQ 9.9 14.4 -4.5 B3LYP/DZQ -4.5 -8.0 3.5
aAs inferred from experiment. b Results marked CP are corrected for the basis set superposition error by the counterpoise approach. c We removed the Ru2+ cation and the two Cl anions from the complexes, without reoptimizing the geometries of either the original complexes or their dissociation products The success of the M06-L density functional in explaining the difference between first- and second-generation Grubbs catalysts allows us to use it to obtain insight into the origin of the substituent effect on bond energy and hence on the kinetics of the key step by which catalyst is generated from precatalyst. In particular we can assess the role of noncovalent interactions. To do this we removed the Ru2+ cation and the Cl anions from the complexes, without reoptimizing the geometries of either the original complexes or their dissociation products. The change in energy upon dissociation of the remaining moieties may now be fairly ascribed to noncovalent interactions. With the M06-L functional, we obtain dissociation energies of 14.4 and 9.9 kcal/mol, respectively, for the demetalized 1 and 2, with a difference of -4.5 kcal/mol. This explains the sign of the ΔBDE value of the original complexes. The positive BDEs, even in the absence of Ru, shows that the noncovalent interactions between the large ligands are attractive. However, the B3LYP functional give a negative BDE and the sign of ΔBDE for demetalized 1 and 2. This is consistent with results for test suites on which we have found that M06-L is more accurate than the other functionals considered above for both noncovalent interaction energies and metal-ligand bond energies. We conclude that noncovalent attractive interactions between the large ligands in the precatalysts play a decisive role in the effect of ligand variation on the mechanism and activity of ruthenium-based olefin metathesis. The roles of covalent and dative electronic effects (electron-donating effects) and noncovalent repulsive interactions (steric effects) have been widely discussed, but the noncovalent attractive interactions have been ignored in previous work. The ability of new density functionals to analyze and accurately model such attractive interactions due to medium-range correlation energy opens new possibilities for computer-aided catalyst design.
26

6. Size-selective supramolecular chemistry in a hydrocarbon nanoring Yan Zhao and Donald G. Truhlar New-generation density functionals (M06-L and M06-2X) include an accurate treatment of medium-range correlation energy and have been applied to investigate host-guest interactions in supramolecular complexes in which a carbon nanoring, [6]paraphenyleneacetlene ([6]CPPA), acts as the host molecule. Guests include fullerenes and carbon nanotubes. The nature of the interactions has been discussed and analyzed. The size-selective supramolecular chemistry in the nanoring has been investigated by varying the size of the guest molecules and optimizing inclusion structures as large as C128H44. We found that the (5,5) armchair-type nanotube fits in the [6]CPPA hydrocarbon nanoring better than the (3,3) or (4,4) ones, and C70 is bound more strongly than C60. The predicted host-guest binding energies of the (4,4), (5,5), C60, and C70 structures are 24, 43, 25, and 28 kcal/mol, respectively.
(4,4) (4,4)@[6]CPPA
+ →
HMB HMB@[6]CPPA
C60 C60@[6]CPPA
(3,3) (3,3)@[6]CPPA
C70 C70@[6]CPPA
(5,5) (5,5)@[6]CPPA
27

7. Free energies of formation of metal clusters and nanoparticles from molecular simulations Zhen Hua Li, Divesh Bhatt, Nathan E. Schultz, J. Ilja Siepmann, and Donald G. Truhlar Metal nanoparticles are important in several emerging technologies, but their size-selected thermodynamic properties are hard to obtain from experiment. We have calculated the global and local minima of unsupported neutral Aln (2 ≤ n ≤ 65) particles (clusters and nanoparticles) by combining big-bang random searches with molecular dynamics quenching and using a recently validated many-body analytic potential. The distribution of the local energy minima and the finite-temperature thermodynamics of the Aln particles have been calculated, the latter by evaluating the rovibrational partition functions of the low-energy isomers. This analysis indicates that structural stability depends on temperature, and that one must consider statistical mechanics as well as electronic structure in determining the properties of nanoparticles. Some particles with electronic magic numbers are shown to become unstable at elevated temperatures, when one considers thermal and entropy effects. The electronic magic numbers are n = 13, 19, 23, 38, and 55, but n = 28 becomes unstable at elevated temperatures. Furthermore, for several n, the global minimum of potential energy need not be the dominant structure at moderate temperatures, and the second- or third-lowest-energy isomer may be dominant at temperatures as low as room temperature. The isomeric energy (EIso), which is the difference between the thermal average energy of the particle in the ground electronic state and thermal average energy of the structure corresponding to the global minimum in the ground electronic state, is an indicator of how well the thermochemical properties of a multi-isomer particle can be represented by those of the global minimum, and we calculate EIso for n = 2–65. We also characterize the energy landscapes by determining the probability of finding the second, third, and fourth lowest-energy structures as a function of particle size, temperature and isomer energy. An efficient simulation method was developed for determining the standard Gibbs free energy changes for the reactions, M + Mn-1 ↔ Mn (R1), involved in the formation of atomic clusters and nanoparticles (also called particles) in the vapor phase. The standard Gibbs free energy of formation ( ) of particles is obtained from these Gibbs free energy changes ( ) by a
recursion relationship using the experimental of the monomer. This method has been
applied to reactions involving Al
oGfΔ oGΔ
oGfΔ
n particles with n = 2 – 60. This method has been validated for n = 2, where the experimental thermodynamic properties of Al2 have been recompiled using the latest available experimental or accurate theoretical data. For n = 2– 4, two completely different approaches, a Monte Carlo configuration integral (MCCI) integration of partition functions and an aggregation-volume-bias Monte Carlo (AVBMC) direct simulation of the equilibrium constants, employing four well validated potential energy functions have been used to calculate
of R1. Excellent agreement is observed for these two methods. Although different potential energy functions give different stage-1 results for n ≤ 10, three high-level correction (HLC) terms, namely a correction for the potential energy difference of the global minima, another for the electronic excitation contribution, and a third based on calculating isomeric-rovibrational contributions have been applied to mitigate deficiencies in the potential energy functions. For n =
oGΔ
28

2, good agreement has been found between the corrected simulation results and experimental data. For larger n, the more efficient AVBMC method has been used. Finally, accurate of R1 and thus of Al
oGΔoGfΔ n particles with n = 2 – 60 have been determined. This is the first
example of the determination of nanoparticle free energies of formation. The association reactions of Al atom with Aln particles, and the unimolecular dissociation of Aln particles have been studied using classical molecular dynamics trajectory simulations. Thermal reaction rate constants of both reactions have been determined; these rate constants are the key parameters of clustering mechanisms for particle growth. It was found that the association rate constants depend weakly on temperature, and the reaction is barrierless. For the unimolecular dissociation reactions, the rate constant depends strongly on temperature and is fitted using the Arrhenius equation. The results indicate that the unimolecular dissociation reaction has a high activation barrier that tends to increase with particle size. The results indicate that monomer emission is always the dissocation process with the largest rate constant. With both the forward (monomer association) and backward (monomer emission) rate constants, the standard Gibbs free energy changes of the Al + Aln-1 ↔ Aln reactions with n = 2 – 60 have been determined. The standard Gibbs free energy changes determined by the molecular dynamics simulations agree fairly well with those determined by our previous Monte Carlo equilibrium simulations.
29

8. Hybrid approach for excited states and for free energy calculations with high-level methods Marat Valiev, Bruce C. Garrett, Ming-Kang Tsai, Karol Kowalski, Shawn M. Kathmann, Gregory K. Schenter, and Michel Dupuis Evolution of the excited state energies of cytosine base in the native DNA environment was investigated using a hybrid coupled cluster and classical molecular dynamics approach. The time averaged excitation energies obtained with the variant of the completely renormalized equation-of-motion with singles, doubles, and non-iterative triples approach that includes a bulk of the correlation effects for excited states, are compared with the analogous calculations in the gas phase. Significant blue shifts for the two lowest singlet excitation energies can be observed as a result of the interaction of the quantum system with the surrounding environment. We also presented an approach to calculate the free energy profile along a condensed-phase reaction path based on high-level electronic structure methods for the reactive region. The bulk of statistical averaging is shifted toward less expensive descriptions by using a hierarchy of representations that includes molecular mechanics, density functional theory, and coupled cluster theories. As an application of this approach we study the reaction of CHCl3 with OH− in aqueous solution.
30

Appendix B Full report of second year activities and accomplishments (FY08) 1. Use of Minnesota density functionals in studying supramolecular compounds Yan Zhao and Donald G. Truhlar One of the most vigorously developing technological research areas is the field of supramolecular chemistry, which involves the use of noncovalent interactions to assemble molecules into stable, well-defined structures called supramolecules. Most of the experimental studies of supramolecular selfassembly are carried out in the condensed phase since the gas-phase supramolecular experiments are technically too demanding. An alternative is theoretical and computational studies that can be carried out nowadays for gas-phase supramolecular systems quite reliably. Such theoretical studies can shed light on intrinsic supramolecular structures and energetics, thus complementing the experimental investigations. One promising approach is density functional theory, which has an excellent performance-to-cost ratio. However, the popular B3LYP density functional and most other older functionals are inaccurate for noncovalent (for instance, aromatic-aromatic) interactions. One of the ways to improve the performance of DFT is to optimize the exchange-correlation functionals for a broad range of properties including rare-gas dimers, aromatic-aromatic interactions, and other data sensitive to medium-range correlation energy, such as barrier heights. This approach has been used for the development of the M06-2X density functional which has been validated for π...π stacking and has also been employed to study supramolecular assembly involving a nanoring and to study the dimerization of coronene as a model for interacting graphene sheets in multiwalled carbon-based assemblies.
One of the most exciting topics of the project is an application of the Minnesota density functionals (M06-L and M06-2X) study the geometries and binding energies of a recently synthesized tweezers-like buckyball catcher (C60H28) and its supramolecular complexes with buckminsterfullerene C60 (see the figure). The C60H28 buckycatcher has two corannulene pincers and a tetrabenzo-cyclooctatetraene tether. The C60@C60H28 supramolecule is bound by the strong attractive concave-convex aromatic-aromatic interactions between the corannulene pincers and the C60 molecule. However, due to the
entropy penalty, the calculated gas-phase free energy of association of the C60@corannulene supramolecule is positive 3.5 kcal/mol; and this entropy penalty explains why it is difficult to observe such C60@corannulene supramolecules experimentally.
to
We have also been using coronene dimers as prototypes for graphene material simulation. Graphene sheets are the building blocks of carbon nanotubes and a variety of functionalized nanomaterials. Methods to be used for computer-aided design of such materials or for the study of aromatic-aromatic interactions in biopolymers and other soft materials should be validated for smaller systems where reliable estimates of interaction energies are available. In our work, we first validated the M06-2X functional against the S22 database of noncovalent interaction
31

energies of biological importance. We then applied the M06-2X functional to study aromatic-aromatic interactions in coronene dimers. We located six stationary points on the potential energy surface of coronene dimer, we calculated the potential energy curves for the sandwich, T-shaped, and parallel-displaced configurations of this prototype of aromatic-aromatic interactions, and we found that a parallel displaced configuration is the global minimum. The potential curves for the coronene dimers will aid the development of new force fields and potential energy functions that are computationally efficient and capable of modeling large graphene or aromatic clusters.
32

2. Benchmark data for interactions in zeolite model complexes and their use for assessment and validation of electronic structure methods Yan Zhao and Donald G. Truhlar We have obtained benchmark binding energies for five zeolite model complexes, with four of the adsorbates bound noncovalently and one covalently. The binding energies were determined as the sum of the infinite-basis-set limit of Møller-Plesset second-order perturbation theory (MP2) energies and a CCSD(T) correction term evaluated with the aug-cc-pVDZ basis set. The basis set limit of MP2 energies was determined by two-point extrapolation using the aug-cc-pVXZ basis sets for X = D and T and separate extrapolation of the Hartree-Fock and correlation energies. We found that correlation contributions beyond MP2 to the final binding energies are small; their magnitude is in the range of 0.02-1.0 kcal/mol. For the MP2 method to describe the interactions in these zeolite model systems accurately, one needs to use a basis set at least the size of aug-cc-pVTZ in conjunction with counterpoise corrections. Final binding energies of the model complexes are in the range of 3.5-19.5 kcal/mol, and they were used as reference data to test 6 wave function methods and 41 density functionals. Among the tested density functional methods, M06-L/6-31+G(d,p) gives a mean unsigned error (MUE) without counterpoise correction of 0.87 kcal/mol. With counterpoise corrections, the M06-2X and M05-2X functionals give the best performance. The MUE with counterpoise corrections for the M06-2X/6-311+G(2df,2p)//MP2/6-311+G(2df,2p) level of theory is 0.39 kcal/mol. With the DFT/6-31+G(d,p) geometries and the 6-311+G(2df,2p) basis set, M05-2X and M06-2X give MUEs with counterpoise corrections of 0.40 and 0.52 kcal/mol, respectively. Tests against the binding energies of four complexes (two noncovalent and two covalent) of the adsoption of isobutene on a large 16T zeolite model cluster confirmed that M06-L, M06, M05-2X, and M06-2X are very promising quantum mechanical methods for hybrid quantum mechanical/molecular mechanical (QM/MM) simulations of zeolites. In fact, the performance of these four Minnesota functionals, as compared to other high-quality functionals, is relatively even better for the larger 16T clusters than for the smaller 3T ones.
33
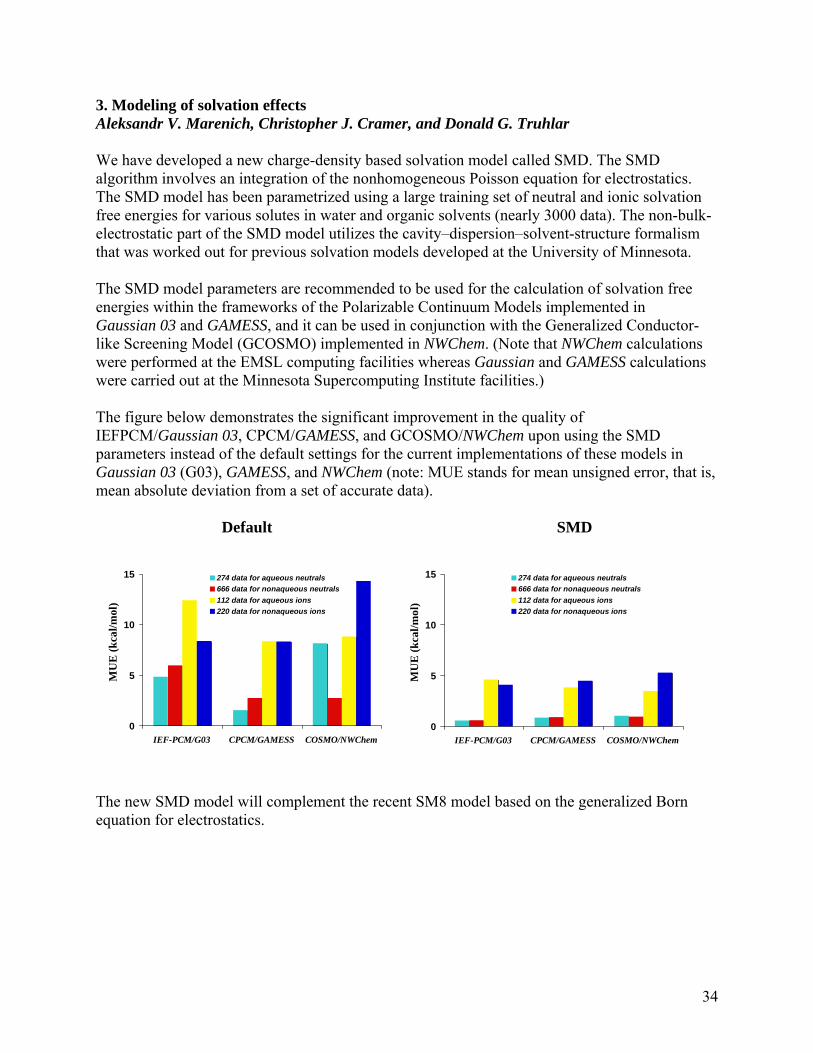
3. Modeling of solvation effects Aleksandr V. Marenich, Christopher J. Cramer, and Donald G. Truhlar We have developed a new charge-density based solvation model called SMD. The SMD algorithm involves an integration of the nonhomogeneous Poisson equation for electrostatics. The SMD model has been parametrized using a large training set of neutral and ionic solvation free energies for various solutes in water and organic solvents (nearly 3000 data). The non-bulk-electrostatic part of the SMD model utilizes the cavity–dispersion–solvent-structure formalism that was worked out for previous solvation models developed at the University of Minnesota. The SMD model parameters are recommended to be used for the calculation of solvation free energies within the frameworks of the Polarizable Continuum Models implemented in Gaussian 03 and GAMESS, and it can be used in conjunction with the Generalized Conductor-like Screening Model (GCOSMO) implemented in NWChem. (Note that NWChem calculations were performed at the EMSL computing facilities whereas Gaussian and GAMESS calculations were carried out at the Minnesota Supercomputing Institute facilities.) The figure below demonstrates the significant improvement in the quality of IEFPCM/Gaussian 03, CPCM/GAMESS, and GCOSMO/NWChem upon using the SMD parameters instead of the default settings for the current implementations of these models in Gaussian 03 (G03), GAMESS, and NWChem (note: MUE stands for mean unsigned error, that is, mean absolute deviation from a set of accurate data).
Default SMD
The new SMD model will complement the recent SM8 model based on the generalized Born equation for electrostatics.
0
5
10
15
IEF-PCM/G03 CPCM/GAMESS COSMO/NWChem
MU
E (k
cal/m
ol)
274 data for aqueous neutrals
0
5
10
15
IEF-PCM/G03 CPCM/GAMESS COSMO/NWChem
MU
E (k
cal/m
ol)
274 data for aqueous neutrals666 data for nonaqueous neutrals666 data for nonaqueous neutrals112 data for aqueous ions112 data for aqueous ions220 data for nonaqueous ions220 data for nonaqueous ions
34

4. Large-scale parallel calculations with combined coupled cluster and molecular mechanics formalism: excitation energies of zinc-porphyrin in aqueous solution Peng-Dong Fan, Marat Valiev, and Karol Kowalski Zinc-porphyrin (ZnP) is important in understanding photochemical properties of π-conjugated systems. Vertical excitation energies of ZnP monomer was previously studied by SAC-CI method and TD-DFT method. However, due to the size of the system (consisting 37 atoms and 190 electrons), the high order correlation effects (e.g. triply excited configurations) still remain unknown. The environment effect on the excited states of ZnP have not been studied either. To address the above issues and demonstrate the performance of our parallel codes, we performed cutting edge large scale benchmark excited state calculations of ZnP monomer in aqueous solution using combined coupled cluster molecular mechanics formalism, in which the coupled cluster (CC) methods are equation of motion approach with singles and doubles (EOMCCSD) and completely renormalized non-iterative triples corrections to EOMCCSD method (CR-EOMCCSD(T)).
The calculations show that the non-iterative triples corrections ranges from 0.24 eV to 0.29 eV, and the CC approaches provide consistent energetics of low-lying excited states for all environments, while the TD-DFT approach cannot adequately describe prototype charge transfer states dominated by excitations from central zinc and nitrogen atoms to carbon atoms. Such breakdown of TD-DFT is best observed in aqueous environment. The parallel performance of our code scales across 1000 CPUs. The scalability is 1.92 and 1.94 for the CCSD and
EOMCCSD methods, respectively, when going from 256 CPUs to 512 CPUs, and 1.66 and 1.74, respectively, from 512 CPUs to 1024 CPUs.
35

5. Calculation of reaction rates for barrierless association reactions using variational transition theory Jingjing Zheng and Donald G. Truhlar This project was focused on calculations of quantitatively accurate reaction rate for barrierless association reactions by means of variational transition state theory. One of the challenges for predicting accurate reaction rates of barrierless association reactions is to calculate accurate potential energy surface in the transition state regions which are often accompanied with a strong multireference character of the corresponding electronic wavefunction. The investigated association reactions are CN + NH3 and OH + SO2. High level ab inito methods, namely the coupled cluster CCSDT and CCSDTQ methods with augmented triple-zeta basis sets, were used to generate an accurate minimum reaction path of the association reactions. Note that the inclusion of explicit triple (T) and quadruple (Q) electronic excitations is often critical for accurate treatment of bond breaking/forming processes. However, density functional theory (DFT) is obviously much more affordable for the use in direct dynamics calculations in terms of variational transition state theory. Thus, we employed the CCSDT and CCSDTQ calculations for validation of the DFT results in order to identify those combinations of density functionals and basis sets that provide most accurate potential energy surfaces. The NWChem computational package developed at PNNL was intensively used in this project. We found the implementation of NWChem at the Chinook supercomputer very effective in such expensive calculations as those employing CCSDTQ.
36

6. Investigation of hydrous silica melts Kelly E. Anderson, Katie A. Maerzke, J. Ilja Siepmann, and Christopher J. Mundy Understanding the structure of hydrous silica (SiO2) melts is important for gaining insight into the structure of the Earth’s upper mantle. Pressures within the mantle range from 1 GPa to 135 GPa at the core-mantle boundary and temperatures range from several hundred to a few thousand Kelvin. This wide range of combined high temperatures and pressures makes experimental studies of these systems challenging. Simulation provides an alternative means to examine structural properties. Previously, our group used an empirical force field to examine hydrous silica melts at high temperatures and moderate pressures (0.25-10 GPa) using Monte Carlo (MC) simulations in the isobaric-isothermal ensemble (see Anderson, K. E.; Siepmann, J. I.; Hirschmann, M. M. J. Phys. Chem. B 2008, 112, 13005; Anderson, K. E.; Grauvilardell, L. C.; Siepmann, J. I.; Hirschmann, M. M. J. Phys. Chem. B 2008, 112, 13015). We have sought to probe these systems via first-principles molecular dynamics (FPMD) simulations using Kohn-Sham density functional theory to represent the interactions. These simulations were performed under isobaric-isothermal conditions using the CP2K simulation package and the Gaussian plane wave method implemented therein (VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter J. Comp. Phys. Comm. 2005, 167, 103) to calculate energies and forces. The BLYP functional was used in conjunction with the Goedecker-Teter-Hutter pseudo-potentials and a Gaussian double-ζ valence basis set and a charge density cutoff at 280 Ry. The lengths of the trajectories were 50 ps with a time step of 0.5 fs. The last 80% of the trajectory was analyzed. Analysis of both the MC and FPMD simulations shows that more than 95% of Si atoms have at least three O nearest neighbors (rSiO ≤ 2.35 Å; taken from the minimum of the radial distribution function). In the absence of water, the silica melt forms a highly networked liquid. The addition of even relatively small amounts of water (3.2 wt.%) is sufficient to substantially change the structure of the liquid. A more detailed analysis of the first principles simulations is being prepared for publication. Katie Maerzke participated in PNNL’s 2008 Summer Research Institute.
37

7. Molecular dynamics simulation for the transport of ions through the phospholamban membranes Alessandro Cembran and Jiali Gao Phospholamban (PLN) is a single pass membrane protein that reversibly binds and regulates the sarcoplasmic reticulum (SR) Ca2+-ATPase (SERCA). The SERCA/PLN complex is responsible for Ca2+ ions transport from the cytoplasm into the SR lumen, causing muscle relaxation. PLN exists in equilibrium between the pentameric and monomeric forms, and it is the monomeric form that is the active, inhibitory species, regulating Ca2+ transport. However, the function of the oligomeric configuration has puzzled biochemists and biophysicists for many years, and the literature suggests that it might act as a Cl- or Ca2+ ion channel. We computed through molecular dynamics (MD) simulations the potential of mean force (PMF) for the Cl- and Ca2+ ion conduction through the PLN pentamer pore. We determined the barriers for the passage of Cl- and Ca2+ to be 19 and 41 kcal/mol, respectively, values that are too high for the translocation to take place. This result, together with electrochemical measurements performed by collaborators, reinforced the hypothesis of PLN pentamer to be a storage form and not an ion channel. The structure and dynamics of the monomeric and pentameric forms of PLN have also been investigated with molecular dynamics simulations. Specifically, our research focused on the structural and dynamical effects due to the protein interaction with the lipid bilayer. We compared our results to NMR data. We highlighted the different dynamics of the cytoplasmic loop when embedded at the membrane/water interface or when fully exposed to the solution.
38
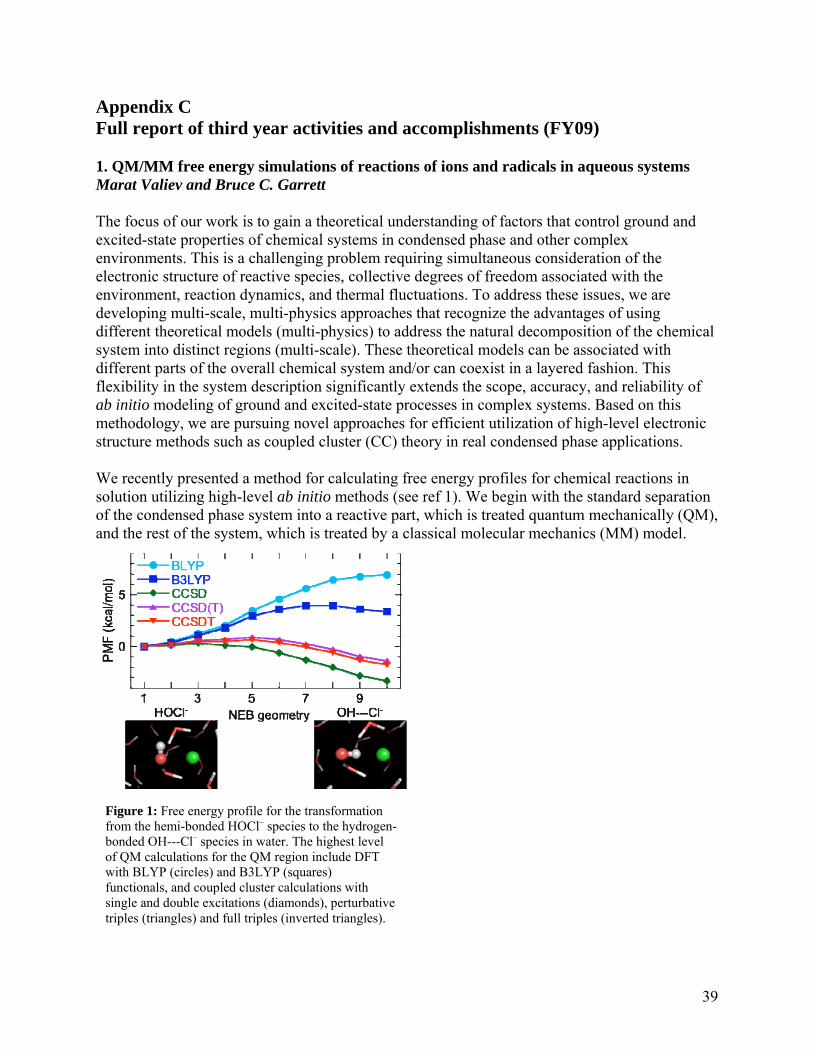
Appendix C Full report of third year activities and accomplishments (FY09) 1. QM/MM free energy simulations of reactions of ions and radicals in aqueous systems Marat Valiev and Bruce C. Garrett The focus of our work is to gain a theoretical understanding of factors that control ground and excited-state properties of chemical systems in condensed phase and other complex environments. This is a challenging problem requiring simultaneous consideration of the electronic structure of reactive species, collective degrees of freedom associated with the environment, reaction dynamics, and thermal fluctuations. To address these issues, we are developing multi-scale, multi-physics approaches that recognize the advantages of using different theoretical models (multi-physics) to address the natural decomposition of the chemical system into distinct regions (multi-scale). These theoretical models can be associated with different parts of the overall chemical system and/or can coexist in a layered fashion. This flexibility in the system description significantly extends the scope, accuracy, and reliability of ab initio modeling of ground and excited-state processes in complex systems. Based on this methodology, we are pursuing novel approaches for efficient utilization of high-level electronic structure methods such as coupled cluster (CC) theory in real condensed phase applications. We recently presented a method for calculating free energy profiles for chemical reactions in solution utilizing high-level ab initio methods (see ref 1). We begin with the standard separation of the condensed phase system into a reactive part, which is treated quantum mechanically (QM), and the rest of the system, which is treated by a classical molecular mechanics (MM) model.
Figure 1: Free energy profile for the transformation from the hemi-bonded HOCl– species to the hydrogen-bonded OH---Cl– species in water. The highest level of QM calculations for the QM region include DFT with BLYP (circles) and B3LYP (squares) functionals, and coupled cluster calculations with single and double excitations (diamonds), perturbative triples (triangles) and full triples (inverted triangles).
39

We use a thermodynamic cycle to take advantage of the state function property of the potential of mean force (PMF), W, and combine simulations of free energy differences with pure MM models that allow sampling to be performed over ensembles that are sufficiently large to converge statistical averages with high-level electronic structure calculations for the QM region to accurately predict energetics. The potential of mean force (PMF) was calculated for transformation from a hydrogen bonded OH---Cl– species to a hemi-bonded HOCl– species. In the PMF calculation, OH and Cl– represent the QM region and waters are treated with a MM model. The nudged elastic band (NEB) method was used to calculate a sequence of geometries along a reaction path at T = 0K. The approach mentioned above using a thermodynamic cycle takes advance MM, DFT, and coupled cluster (CC) methods in the QM regions. The results displayed in Figure 1 show that a subtle interplay of electronic structure and solvation determines the most stable structure. DFT methods commonly used for condensed phase simulations are inadequate for predicting the most stable species. The best estimates of the energies show that the hemi-bonded structure is only about 1 kcal/mol higher than the hydrogen-bonded species and therefore could play an important role in reactions that lead to the formation of Cl2 (see ref 2).
(1) “Hybrid Approach for Free Energy Calculations with High-Level Methods: Application to the SN2 Reaction of CHCl3 and OH- in Water,” M. Valiev, B. C. Garrett, M.-K. Tsai, K. Kowalski, S. M. Kathmann, G. K. Schenter, and M. Dupuis, J. Chem. Phys. 2007, 127, 051102.
(2) “Interactions of Cl- and OH radical in aqueous solution,” M. Valiev, R. D'Auria, D. J. Tobias, and B. C. Garrett, J. Phys. Chem. A 2009, 113, 8823-8825.
40

2. Orotidine monophosphate decarboxylase: Anatomy of the catalytic mechanism by nature’s most proficient enzyme Alessandro Cembran and Jiali Gao Orotidine 5'-monophosphate decarboxylase (OMPDC) catalyzes the exchange of CO2 for a proton at the C6 position to form uridine 5'-monophosphate (UMP), providing a rate acceleration of 17 orders of magnitude over that of the uncatalyzed reaction. The remarkable catalytic proficiency of this enzyme has attracted numerous experimental and computational studies and a number of mechanisms have been proposed to account for its activity. In this work, by means of combined quantum mechanical and molecular mechanical (QM/MM) simulations, we provide some new insights into the factors that contribute to the OMPDC catalysis. In particular, we computed the free energy of activation for the decarboxylation reaction in water solution (34.4 kcal/mol) and in the enzyme (16.9 kcal/mol), which compare well with the experimental data (38.5 and 16.5 kcal/mol, respectively). The main results can be summarized in the following points:
1) We identified Lys72 hydrogen bond with the ribose 2'-OH group in the Michaelis complex structure as a key point for understanding OMPDC catalysis. There is a subtle, but significant, conformational transition involving Lys72 that migrates more than 1 Å towards the ribose ring relative to the crystal structure. In test studies where this hydrogen bond is broken, the computed free energy barriers are 5 kcal/mol or more higher than the experimental value. Noticeably, experiments using a 2'-deoxyOMP substrate showed a comparable increase in energy barrier of 4.6 kcal/mol.
2) As recent literature suggests that the presence of a specific water molecule in the active site is not compatible with catalysis, we tested for the effects of removing such water: our calculations showed only a marginal (3 kcal/mol) energy barrier reduction, which cannot account for the energy barrier reduction performed by the enzyme (about 22 kcal/mol).
3) To determine the amount of catalysis performed by the enzyme through transition-state stabilization, we computed the potential of mean force in the enzyme and in water for the proton transfer between Lys72 and the uracil C6 carbanion, where the latter closely resembles the transition-state for the decarboxylation reaction. By these means, we found that at most 11 kcal/mol can be accounted for by transition-state stabilization, and that about 7 kcal/mol for the computed barrier reduction must derive from other factors.
4) By analyses of the structures of reactant and transition state, it was possible to identify a distortion operated by binding of negatively charged OMP upon the protein backbone through repulsion with Asp70. We proposed that part of the catalytic power of the enzyme is achieved by the relieving of this strain upon reaching the transition-state. Recent experiments on the D70G mutant show that the mutation increases the free energy of activation for the decarboxylation reaction by about 5 kcal/mol, but does not affect the pKa of UMP in the enzyme (in other words, this effect is not due to a change in transition-state stabilization). Our computed result for the mutant decarboxylation reaction (22 kcal/mol) is in agreement with the experiment (21 kcal/mol), and strengthens
41

the view of Asp70 as a handle to store strain energy as protein backbone distortion.
5) Through principal component analysis of the computed trajectories, we identified a particular mode involving a hinge-bending motion of helices 1, 2, and 8 that is coupled with the reaction coordinate. We identified Asp70 as a key residue to couple the reaction coordinate motion with the equilibrium transition between different enzyme conformational substates.
Two papers that show the results of this work are in preparation. This research has been also orally presented at the following venues: − XXVIII Midwest Enzyme Chemistry Conference, Chicago, IL (Oct. 2008). − Molecular Modeling 2009 Meeting, Surfers Paradise, Qeensland, Australia (Jul. 2009). − Protein Society Meeting, Boston, MA (Jul. 2009). − Thermodynamics of Biomolecular Machinery, Beijing, China (Jul. 2009).
Figure 1: Decarboxylation free energy profile in water (red) and in the enzyme (black).
Figure 2: Mode associated with the reaction coordinate motion: active site with Asp70 and Lys72 (left) and hinge-bending motion (right).
42

3. Multiscale model development, implementation, and application Jong Hyuk Park and Andreas Heyden The objective of this project has been to develop, test, and implement a highly efficient conservative multiscale modeling algorithm for simulating large systems that require an extensive sampling of a complex phase space to determine the system properties, but that requires only locally, i.e., close to an active site, a high level of resolution and accuracy. The method is called adaptive partitioning (AP) of the Lagrangian (APL) (see refs 1-3) and it can often permit a molecular dynamics (MD) simulation of a complex system with an accuracy of an equivalent high level of theory / resolution simulation at the computational cost of a low level of theory / resolution simulation. In the last couple of months we started to apply our methodology to study the oxygen mobility in perovskite and double perovskite oxide anode materials such as doped LaGaO3 and Sr2MgMoO6 for solid oxide fuel cells. Our objective is to identify practical descriptors that allow for the rational design of novel anode materials for solid oxide fuel cells with excellent catalytic activity, electronic conductivity, and oxygen mobility.
Nanoparticle
High level of theory / high resolution
Solid surface
Low level of theory / low resolution
Figure 1. Schematic representation of the AP-MD method for a nanoparticle (blue spheres) or an oxide surface (grey, and red spheres) in contact with a fluid phase (fluid molecules not shown). Fluid molecules close to the surface / nanoparticle (within the light blue spheres) are treated during a molecular dynamics simulation at a high level of theory / high level of resolution while fluid molecules away from the surface / nanoparticle (outside the light blue spheres) are treated at a low level of theory / low level of resolution.
(1) “Adaptive partitioning in combined quantum mechanical and molecular mechanical calculations of potential energy functions for multiscale simulations,” A. Heyden, H. Lin, D. G. Truhlar, J. Phys. Chem. B 2007, 111, 2231-2241.
(2) “Conservative algorithm for an adaptive change of resolution in mixed atomistic/coarse-grained multiscale simulations,” A. Heyden, D. G. Truhlar, J. Chem. Theory Comput. 2008, 4, 217-221.
(3) “Solving the equations of motion for mixed atomistic and coarse-grained systems,” J. H. Park, A. Heyden, Mol. Sim. 2009, 35, 962-973.
43

4. The electrostatically-embedded many-body approximation for large systems Hannah R. Leverentz and Donald G. Truhlar The electrostatically-embedded many-body (EE-MB) approximation is a fragment-based method for the calculation of the energies, gradients, and Hessians of moderately-sized to large systems molecules. In order to calculate, for example, the electrostatically-embedded three-body (EE-3B) approximation of the total energy of a given system, one must divide the larger system into smaller fragments, or monomers and then embed each monomer in a field of point charges that roughly represents the electrostatic influence of the rest of the larger system on the given monomer. Every possible pair of monomers (i.e., dimer), group of three monomers (i.e., trimer) is electrostatically-embedded in a similar manner, and a linear combination of the calculated energies of the embedded monomers, dimers, and trimers is taken to approximate the energy of the entire system. At a given level of electronic structure theory, individual monomer, dimer, trimer, etc. calculations may be performed completely independently of one another, making the EE-MB approximation particularly well suited for computational parallelization.
44

5. The hydrogen-transfer isomerization of the butoxyl radical: barrier heights and reaction energies Jingjing Zheng and Donald G. Truhlar We studied five hydrogen-transfer isomerization reactions involving the butoxyl radical (see Scheme 1) to obtain the best-estimated barrier heights and reaction energies for these five reactions. The corresponding single-point energies at the M06-2X/MG3S optimized molecular geometries were calculated at the coupled-cluster singles and doubles level augmented by a perturbative correction for connected triple excitations CCSD(T) using the augmented correlation-consistent polarized basis sets of double-, triple-, and quadruple-ζ quality. The CCSD(T) total energies were extrapolated then to the complete basis set level. The relativistic and core−valence corrections were taken into consideration. The calculations were carried out at the EMSL’s Chinook supercomputer using the 2008.1 version of Molpro. Scheme 1
45


















