
О цен ка качеств а модели пространственной структуры
93
Оценка качества модели пространственной структуры
-
Upload
louis-albert -
Category
Documents
-
view
72 -
download
2
description
О цен ка качеств а модели пространственной структуры. Тернистый путь создания модели. Ян Вермеер Дельфтский Аллегория живописи 1666. Структура белка - это созданная человеком на основании экспериментальных данных модель. - PowerPoint PPT Presentation
Transcript of О цен ка качеств а модели пространственной структуры

Оценка качества модели пространственной структуры

Тернистый путь создания модели
Ян Вермеер ДельфтскийАллегория живописи1666

The structures in the PDB are based on a subjective interpretation of experimental data, which may itself be of variable quality, a process that can lead to errors with varying degrees of impact (Bra¨nde´n & Jones, 1990; Morris et al., 1992; Kleywegt & Jones, 1995, 1996, 1997, 2002; Hooft et al., 1996; Kleywegt, 2000, 2007, 2009; Chen et al., 2010).
For this reason, it is crucial to assess the quality and reliability of the resulting models, a process known as validation (Kleywegt, 2000, 2009).
Gore, Velankar and Kleywegt, Implementing an X-ray validation pipeline for the Protein Data Bank, 2012
Структура белка - это созданная человеком на основании экспериментальных данных модель

1. Примеры ошибок
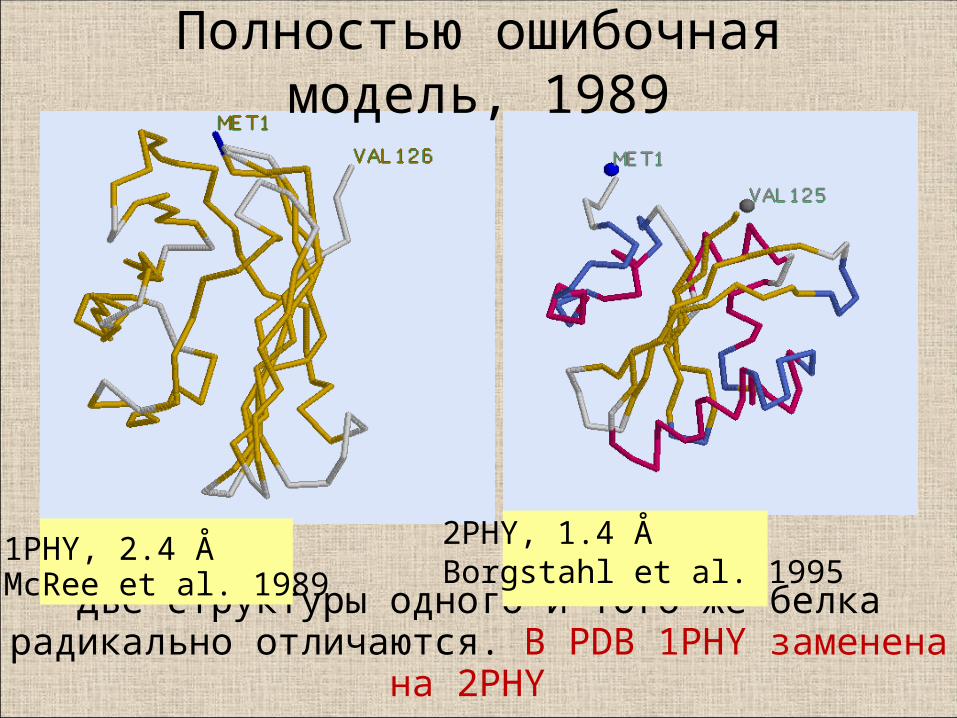
Две структуры одного и того же белка радикально отличаются. В PDB 1PHY заменена на 2PHY
1PHY, 2.4 Å McRee et al. 1989
2PHY, 1.4 ÅBorgstahl et al. 1995
Полностью ошибочная модель, 1989

SCIENCE VOL 314 22 DECEMBER 2006
Полностью ошибочная модель, 2006

Experimental Data forStructure PapersWe are writing to address the retraction of fivepapers on structural studies of ATP-bindingcassette (ABC) transporters—three in Science(G. Chang et al., “Retraction,” Letters, 22 Dec.2006, p. 1875), one in the Proceedings of theNational Academy of Sciences (1), and one inthe Journal of Molecular Biology (2). We havemuch sympathy for your readers but very littlefor the magazine. This is not the first timeincorrect structures have been published inScience (3), and it will not be the last time. Weand all of your readers make mistakes; crystallographyis fortunate that by careful treatmentof the experimental and derived data, most seriousmistakes are caught and corrected beforepublication. The necessary tools and techniquesare well described [for example, (4), andreferences therein] and widely used by ourcommunity. Inherent in structural analysis is adegree of subjectivity (3), which is particularlyrelevant in low-resolution studies such as thosemade by Chang and co-workers. Essentiallycorrect structures have been built at 4.5 Å resolution,but it is not surprising that some of themturn out to be wrong upon further scrutiny.
We are writing to address the retraction of fivepapers on structural studies of ATP-bindingcassette (ABC) transporters—three in Science(G. Chang et al., “Retraction,” Letters, 22 Dec.2006, p. 1875), one in the Proceedings of theNational Academy of Sciences (1), and one inthe Journal of Molecular Biology (2). We havemuch sympathy for your readers but very littlefor the magazine. This is not the first timeincorrect structures have been published inScience (3), and it will not be the last time.
ROBBIE P. JOOSTEN AND GERT VRIEND
13 JULY 2007 VOL 317 SCIENCE

2009 год… Университет Алабамы обращается к руководителям PDB c запросом на удаление следующих структур, депонированных сотрудниками университета. Также просит журналы отозвать 9 соответствующих публикаций
1BEF, 1CMW, 1DF92QID, 1G40, 1G44, 1L6L, 2OU1, 1RID, 1Y8E, 2A01, 2HR0
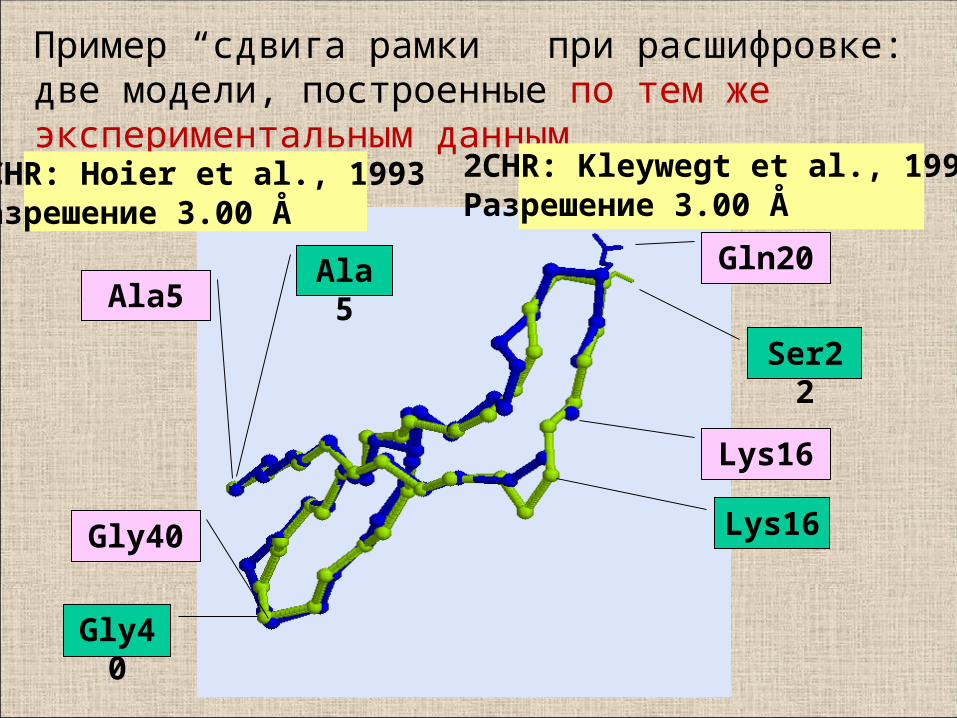
Gln20
Ser22
Ala5Ala5
Lys16
Lys16
Gly40
Gly40
Пример “сдвига рамки” при расшифровке: две модели, построенные по тем же экспериментальным данным
1CHR: Hoier et al., 1993Разрешение 3.00 Å
2CHR: Kleywegt et al., 1996Разрешение 3.00 Å

Выравнивание последовательностей 1CHR и 2CHR по близости C_alpha атомов при
наложении структур
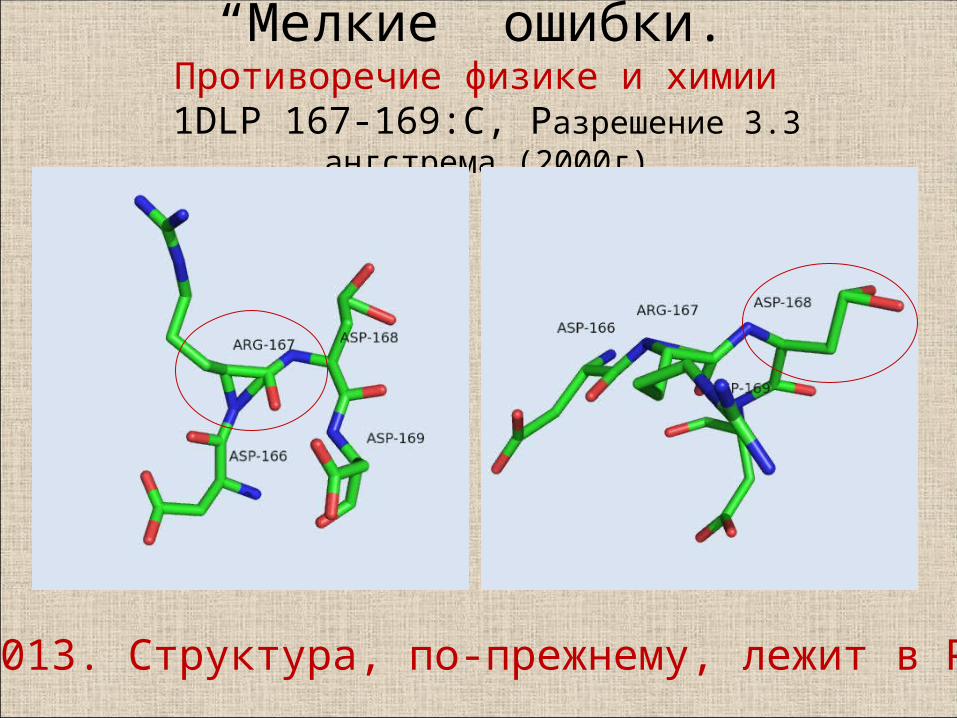
“Мелкие” ошибки. Противоречие физике и химии
1DLP 167-169:C, Разрешение 3.3 ангстрема (2000г)
2013. Структура, по-прежнему, лежит в PDB

Примеры плохого соответствия модели “экспериментальной” ЭП
Это явная неточность расшифровки.… сталкиваемся с полным произволом авторов структуры. То есть если на уровне подрезки 1 на месте бокового радикала этого остатка ещё есть какая-то электронная плотность, то на этом уровне от неё не остаётся и следа.Наибольшая электронная плотность (3 sigma) наблюдается вокруг Сα-атома, наименьшая (около 0.1 sigma!) – у Сδ – атома.
Из протокола Алисы Гараевой
Из протокола Льва Шагама, 2006Уровень подрезки2.5 sigma

2. Источники ошибок

Этапы РСАЭтап
1. Кристаллизация
2. Рентгеновский эксперимент
3. Решение фазовой проблемы
4. Построение черновой модели
5. Оптимизация модели
6. Проверка качества
Результат
1. Кристалл
2. Структурные факторы= амплитуды
гармоник Фурье, кристаллографическая ячейка, группа симметрий, разрешение
3. Фазы гармоник Фурье, функция ЭП
4. Модель (“черновой” pdb файл)
5. Модель для PDB

Ошибки, влияющие на качество модели
• Ошибки измерения структурных факторов
• Ошибки в размерах и симметриях кристаллографической ячейки
• Плохие фазы
• Ошибки вписывания атомов в черновую ЭП
• Неудачная оптимизация модели

3. Разрешение
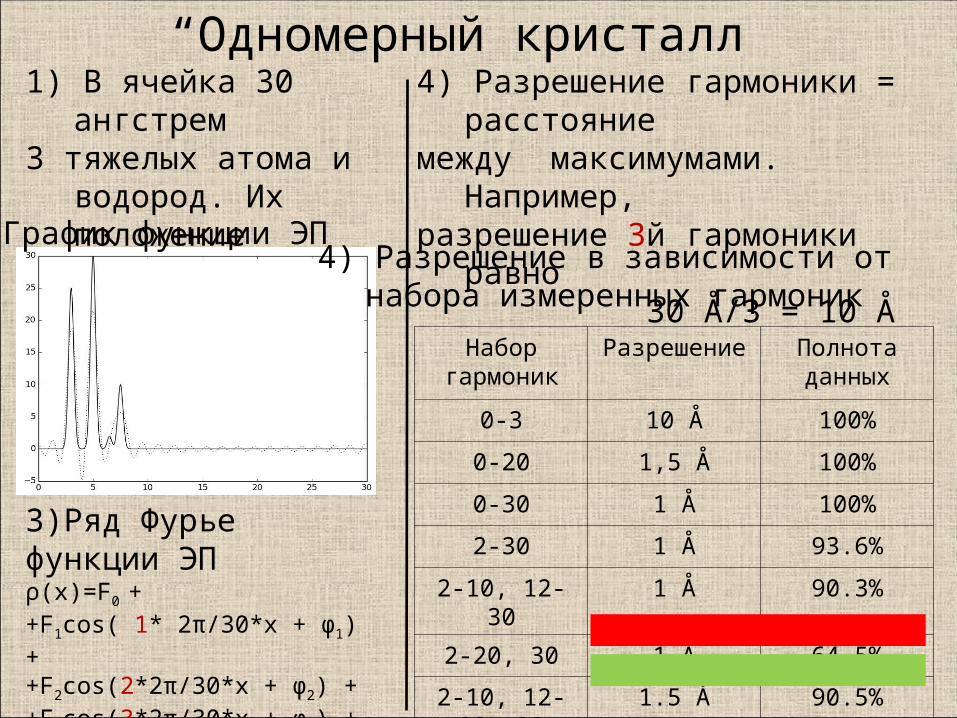
“Одномерный кристалл”1) В ячейка 30 ангстрем3 тяжелых атома и
водород. Их положение известно.
Набор гармоник
Разрешение Полнота данных
0-3 10 Å 100%
0-20 1,5 Å 100%
0-30 1 Å 100%
2-30 1 Å 93.6%
2-10, 12-30 1 Å 90.3%
2-20, 30 1 Å 64.5%
2-10, 12-20, 30 1.5 Å 90.5%
3)Ряд Фурье функции ЭПρ(x)=F0 + +F1cos( 1* 2π/30*x + φ1) ++F2cos(2*2π/30*x + φ2) ++F3cos(3*2π/30*x + φ3) ++ …
2) График функции ЭП4) Разрешение в зависимости от
набора измеренных гармоник
4) Разрешение гармоники = расстояние между максимумами. Например,разрешение 3й гармоники равно 30 Å/3 = 10 Å

Разрешение структуры• Проведен РСА эксперимент: найдены параметры кристаллической
ячейки и получен файл структурных факторов : CRYST1 77.553 192.966 93.740 90.00 90.00 90.00 0 0 4 211.0
0 0 6 1642.7 ………………. 1 1 3 160.9
• Для каждой гармоники (h, k, l) рассчитываем разрешение dhkl (параметры кристаллической решетки известны!)
• Имеем множество измеренных рефлексов (h, k, l) (см. рис)
• Если измерены все рефлексы с разрешением d и больше, и d – минимальное с таким свойством, то говорят, что разрешение структуры d (ангстрем)
• Слово “все” следует заменить на слова “почти все” (добавив, для честности, параметр “полнота данных”) потому, что это эксперимент, а не теория.
• Так, например, рефлексы, отвечающие самым маленьким тройкам чисел (h,k,l): (0,0,0), (1,0,0) …. не могут быть измерены [почему?]
• Кроме того, некоторые измеренные амплитуды не используют из-за их плохого качества

Разрешение как показатель качества модели структуры
• Характеризует экспериментальные данные, а не модель!
• Остается небольшая доля субъективности в определении разрешения структуры
• По данным РСА с плохим разрешением можно построить хорошую модель; при хорошем разрешении в модели могут быть ошибки

Условные градации разрешения
• Высокое разрешение: <1.5 Å• Хорошее разрешение: 1.5 – 2.5 Å
(в моделях с разрешением < 2.5 Å обычно моделируют и молекулы воды)
• Удовлетворительное: 2.5 – 3.5 Å• Низкое: > 3.5 Å
Но и модели с разрешением 5 Å и более могут быть очень важными!Например, первые расшифровки рибосомы

Менее 10% структур, полученных с помощью рентгеноструктурного анализа, имеют разрешение менее, чем 1,5 ангстрем

4. R-фактор и R-free
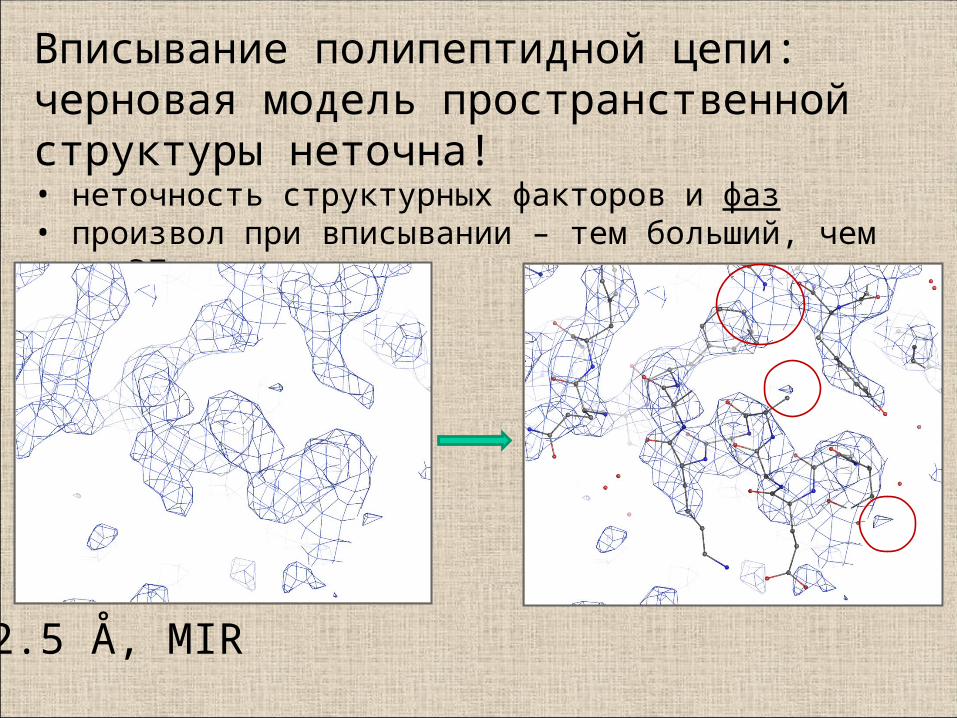
2.5 Å, MIR
Вписывание полипептидной цепи: черновая модель пространственной структуры неточна!• неточность структурных факторов и фаз• произвол при вписывании – тем больший, чем хуже ЭП
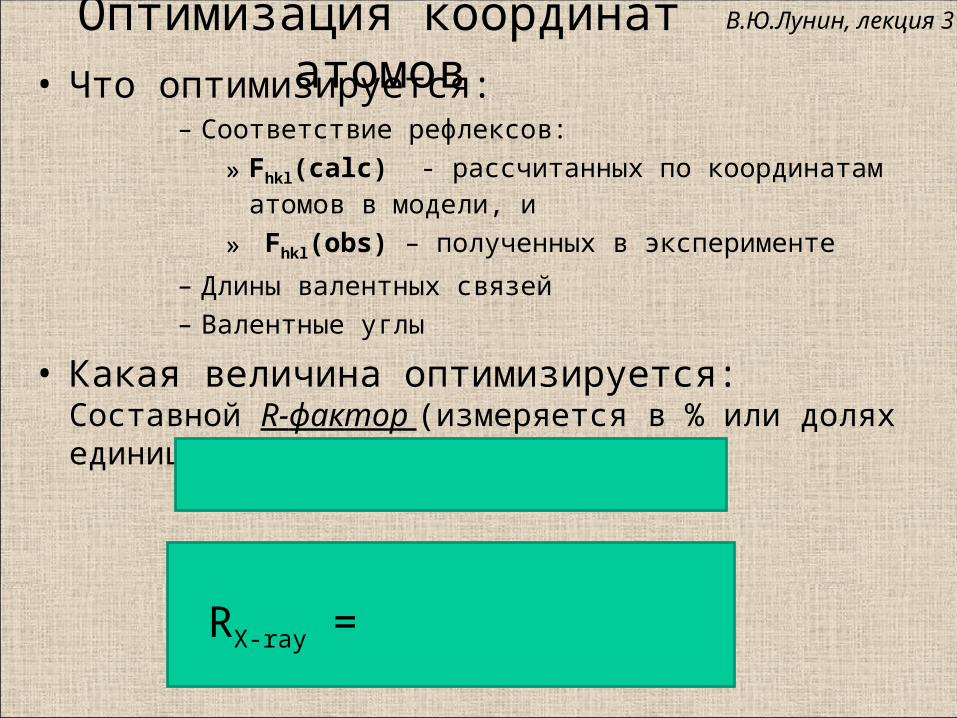
Оптимизация координат атомов• Что оптимизируется:
– Соответствие рефлексов:
» Fhkl(calc) - рассчитанных по координатам атомов в модели, и
» Fhkl(obs) – полученных в эксперименте
– Длины валентных связей
– Валентные углы
• Какая величина оптимизируется:Составной R-фактор (измеряется в % или долях единицы):
angleangledistdistrayXrayX RwRwRwR
%100
hkl
obshkl
hkl
obshkl
calchkl
F
FFR
В.Ю.Лунин, лекция 3
RX-ray =

Оптимизация координат атомов• Как оптимизируется
– Немножко меняются координаты всех атомов в текущей n-й модели , получаем новую, (n+1)-ю модель
– Рассчитывается Rn+1 для новой модели
– Если Rn > Rn+1 , то берем (n+1)-ю модель– Поступаем так до тех пор, пока R-фактор не
перестанет уменьшаться
• Существуют алгоритмы как выбирать смещения атомов для очередной модели
В.Ю.Лунин, лекция 3

R-фактор отражает соответствие модели и эксперимента:
чем меньше R-фактор, тем модель лучше!
Но…
Хорошие значения: R<25%

Борис КустодиевСапожник1924
Подгонка под ответ!
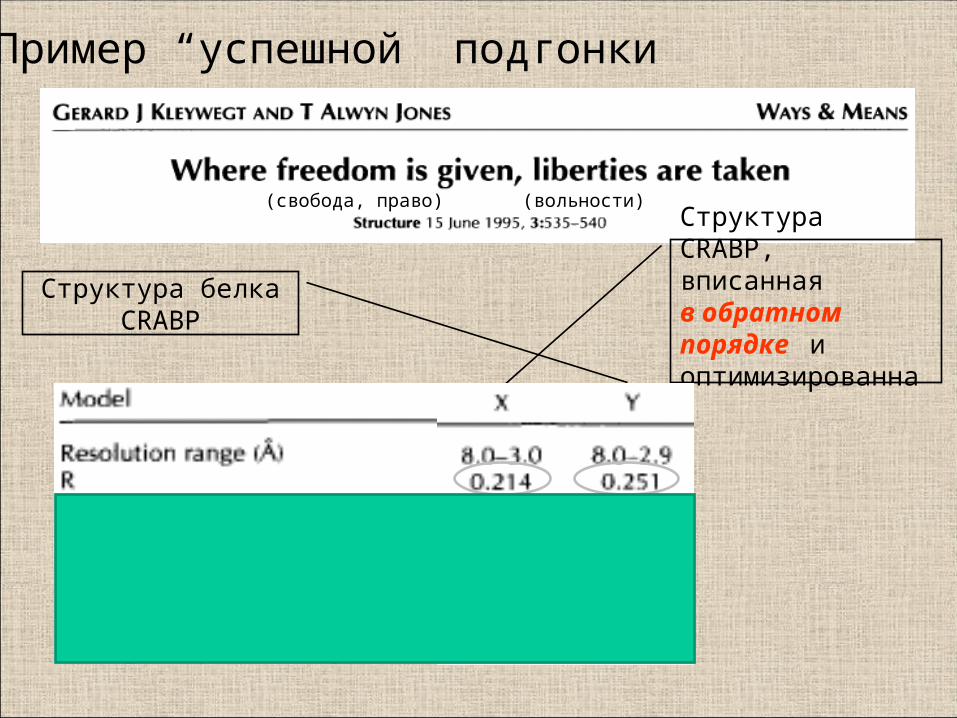
Структура белка CRABP
Структура CRABP, вписанная в обратном порядке и оптимизированная
Пример “успешной” подгонки
(свобода, право) (вольности)

• R-фактор является тем параметром, который минимизируется в процессе уточнения модели
• При минимизации уточняются координаты всех атомов => “подкручиваются много тысяч ручек” (параметров)
• С помощью тысяч “ручек” один параметр можно минимизировать почти до нуля по случайным причинам!
ЧТО ПОЛУЧАЕТСЯ:

R-free
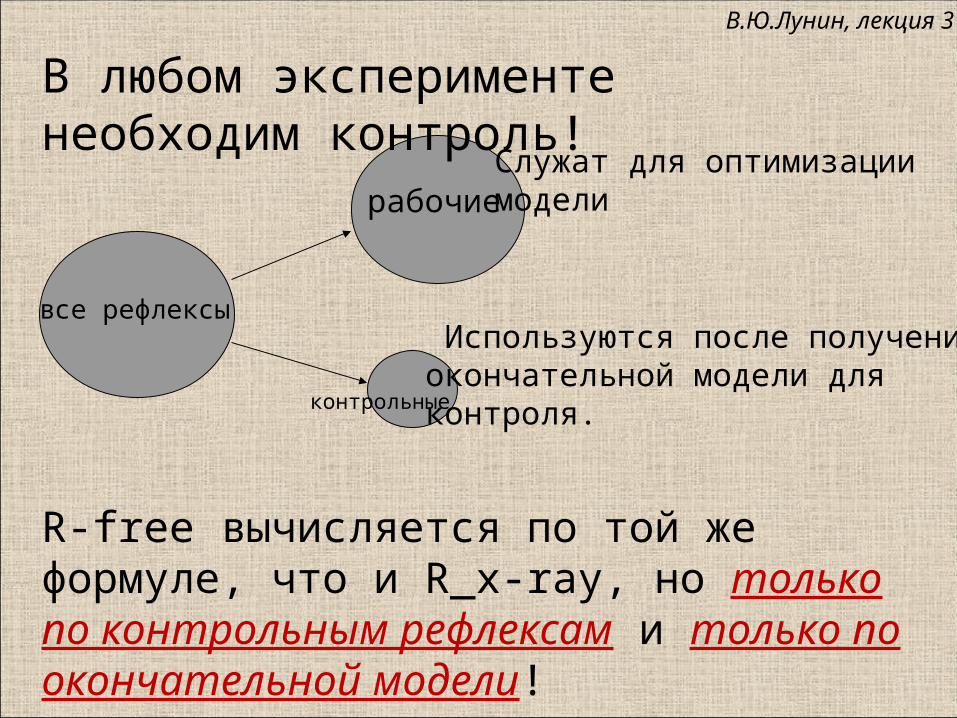
все рефлексы
рабочие
контрольные
Служат для оптимизации модели
Используются после полученияокончательной модели для контроля.
R-free вычисляется по той же формуле, что и R_x-ray, но только по контрольным рефлексам и только по окончательной модели!
В.Ю.Лунин, лекция 3
В любом эксперименте необходим контроль!

• Если модель правильная, то R-free окажется примерно равным R-X-ray или немногим больше!
• Может ли быть так, что R-free < R?
• Если модель подогнана под рабочие рефлексы – “переоптимизирована”, - то
R_free окажется большим!

Интерпретация R_free
• Хорошие значения: R_free<20%• Плохие значения: R_free>40%• Значения (R_free – R)>10%
настораживают в отношении переоптимизации (ovefitting)

REMARK 3 REMARK 3 FIT TO DATA USED IN REFINEMENT. REMARK 3 CROSS-VALIDATION METHOD : THROUGHOUT REMARK 3 FREE R VALUE TEST SET SELECTION : RANDOM REMARK 3 R VALUE (WORKING + TEST SET) : 0.15621 REMARK 3 R VALUE (WORKING SET) : 0.15185 REMARK 3 FREE R VALUE : 0.19471 REMARK 3 FREE R VALUE TEST SET SIZE (%) : 10.1 REMARK 3 FREE R VALUE TEST SET COUNT : 5989 REMARK 3
• Авторы получили в эксперименте 59 297 рефлексов• Они утверждают, что 5 989 (10%) рефлексов спрятали в сейф, опечатали и никому не показывали • По оставшимся 53 308 рефлексам оптимизировали модель и получили R=15%• После этого достали тайные рефлексы из сейфа и рассчитали R-фактор по ним. Это и есть Rfree . Авторы получили Rfree = 19%.• Все общественно признанные критерии удовлетворены:
• Rfree <20% - приемлемое значение для хорошей модели• Rfree- R <10% - нет доводов в пользу переоптимизации
• Значит, можно отправлять в PDB и публиковать статью!

Структура белка CRABP
Структура CRABP, вписанная в обратном порядке и оптимизированная
Польза R_free демонстрируется в работе

5. Карта РамачандранаСтепени свободы полипептидной цепи
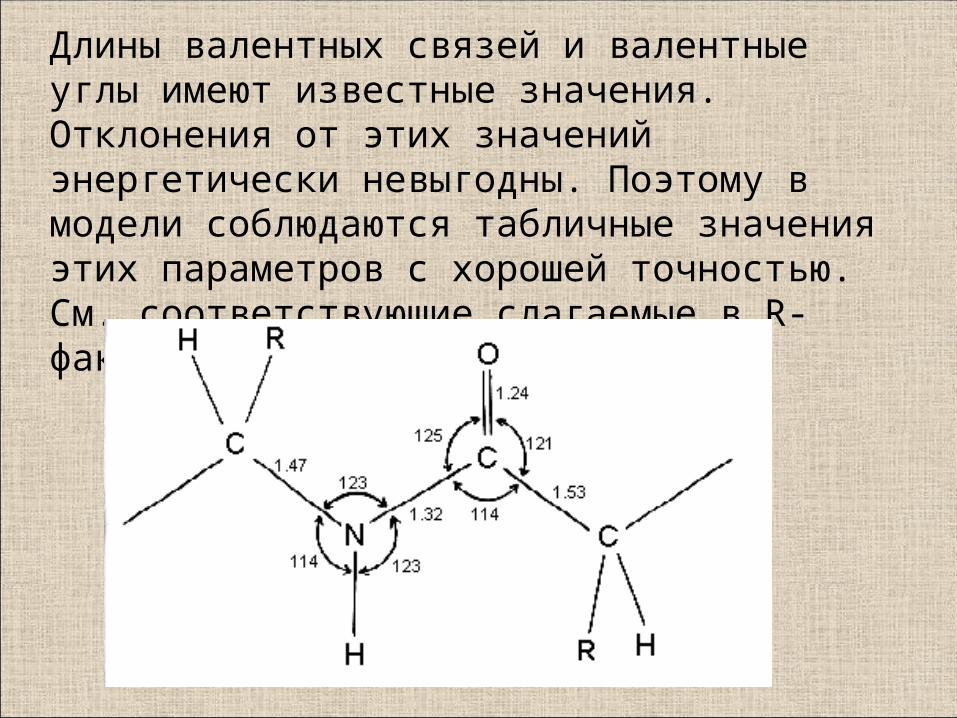
Длины валентных связей и валентные углы имеют известные значения. Отклонения от этих значений энергетически невыгодны. Поэтому в модели соблюдаются табличные значения этих параметров с хорошей точностью.См. соответствующие слагаемые в R-факторе.
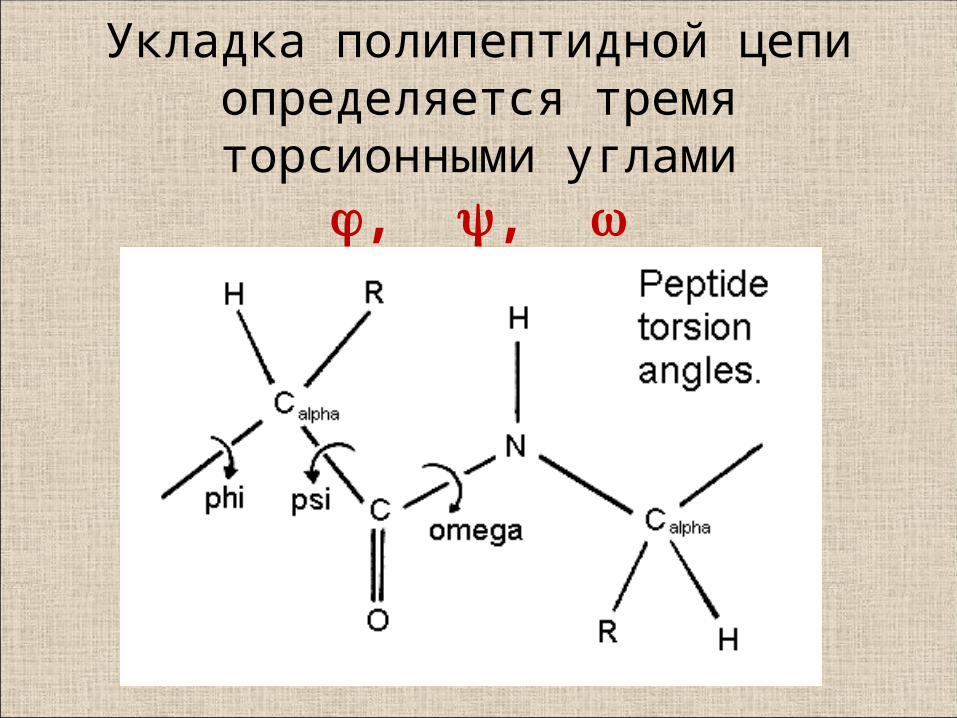
Укладка полипептидной цепи определяется тремя торсионными углами
, ,
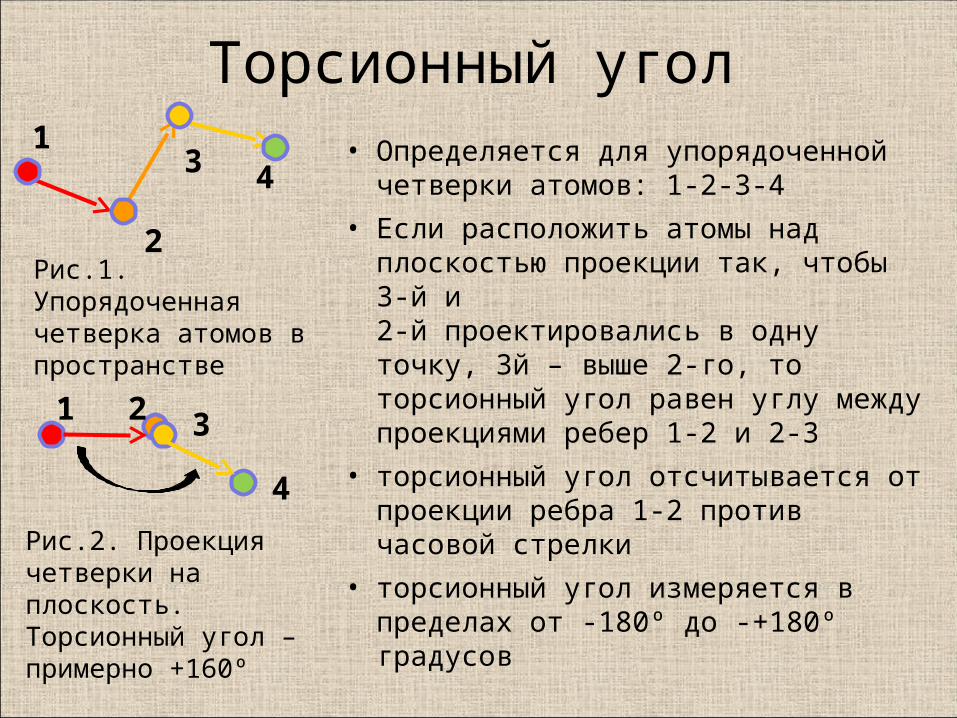
Торсионный угол• Определяется для упорядоченной
четверки атомов: 1-2-3-4
• Если расположить атомы над плоскостью проекции так, чтобы 3-й и 2-й проектировались в одну точку, 3й – выше 2-го, то торсионный угол равен углу между проекциями ребер 1-2 и 2-3
• торсионный угол отсчитывается от проекции ребра 1-2 против часовой стрелки
• торсионный угол измеряется в пределах от -180º до -+180º градусов
2
143
4
321
Рис.1. Упорядоченная четверка атомов в пространстве
Рис.2. Проекция четверки на плоскость.Торсионный угол – примерно +160º

Угол принимает определенные значения:180° почти всегда (trans - конформация) 0° крайне редко (cis - конформация), бывает, в основном, у пролина
Cα(i+1)
C
Cα(i) N
NCα(i)
C
Cα(i+1)
Рис.1 Cis и trans конформации полипептидной цепи
Рис.2 Trans конформация: “кочерга”
Рис.3 Cis конформация: “чашка”

Существенно разные (но не любые) значения могут принимать углы , .
Пара чисел от -180° до +180 ° соответствует точке в квадрате на координатной плоскости
Квадрат -180° до +180 °, предназначенный для отображения торсионных углов (φ,ψ) называется картой Рамачандрана
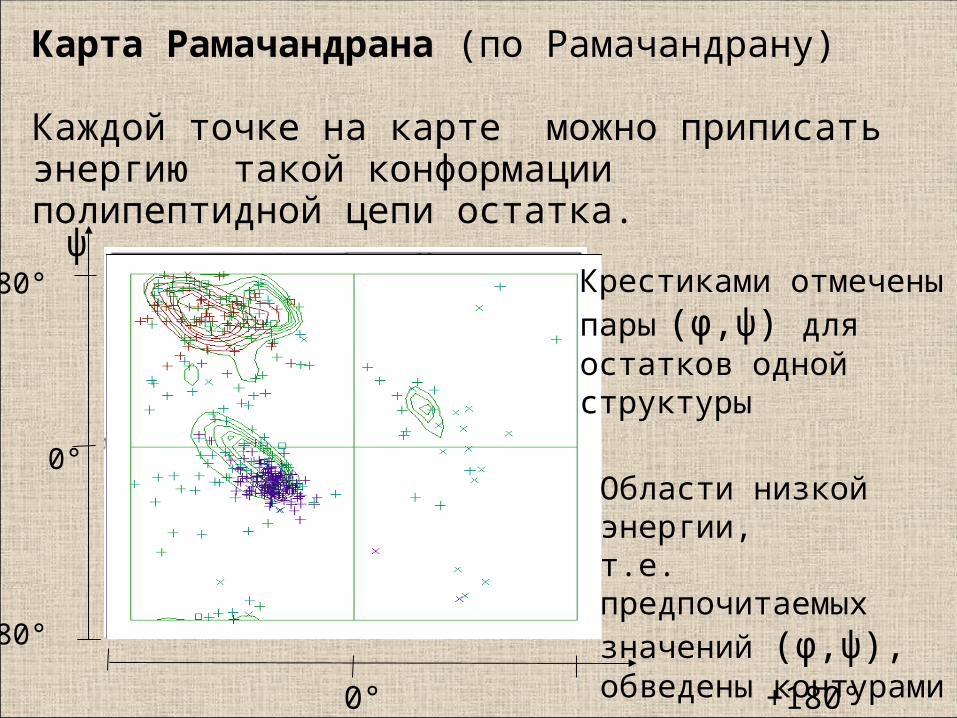
Карта Рамачандрана (по Рамачандрану)
Каждой точке на карте можно приписать энергию такой конформации полипептидной цепи остатка.
-180° 0° +180° φ
ψ+180°
0°
-180°
Области низкой энергии,т.е. предпочитаемых значений (φ,ψ),обведены контурами
Крестиками отмеченыпары (φ,ψ) для остатков одной структуры
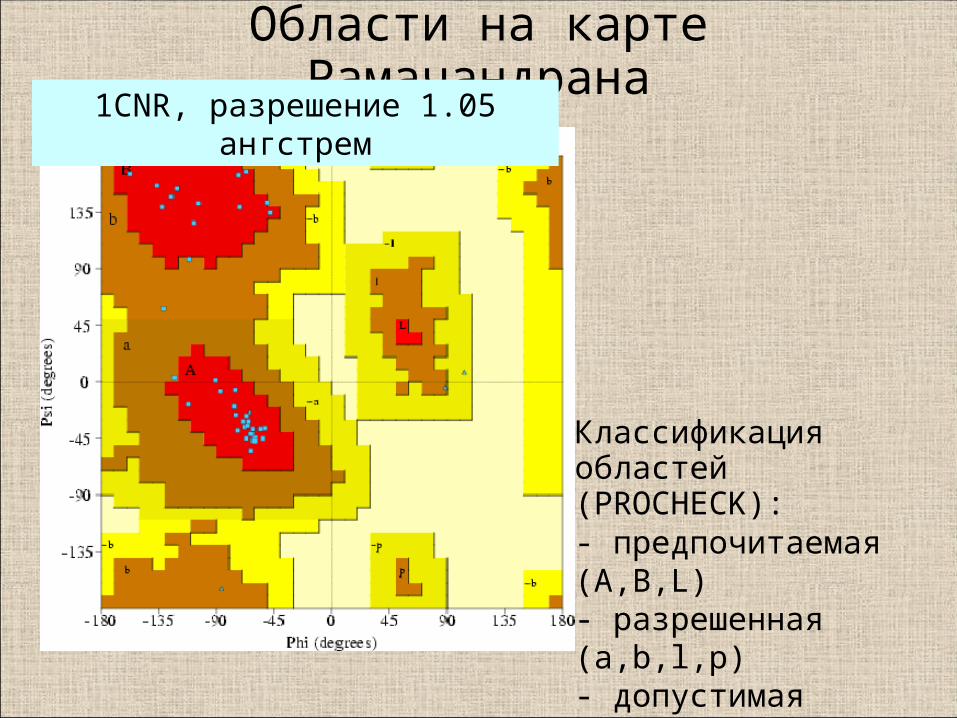
Области на карте Рамачандрана
Классификация областей (PROCHECK):- предпочитаемая (A,B,L) - разрешенная (a,b,l,p)- допустимая (~a,~b,~l,~p)- запрещенная
1CNR, разрешение 1.05 ангстрем

Карта Рамачандрана по последним данным (Lovell et al., 2003; Davis et al., 2004)
Внутренний контур окружает 98% остатковВнешний - 99.95 % остатков (!)

Области на карте Рамачандрана, используемые в программах,
определены на основании статистики по PDB
Рекомендуемая VTF программа - MolProbity

Выводы
• ВСЕ остатки, не попадающие в обведенные области на карте Рамачандрана, следует считать маргинальными:– либо координаты остовных атомов
существенно неточны– либо это крайне редкие особенности
(встречаются реже 1 на 2000 случаев), требующие объяснения

Карта Рамачандрана модели белка как индикатор её качества
• Этот индикатор хорош потому, что независим от процедуры оптимизации модели (как правило)
• В хорошей модели >90% остатков, не считая Gly, Pro, находятся в предпочитаемой области
• Этот критерий нынче известен всем, поэтому авторы стараются подогнать модель!

Сравните две карты Рамачандрана

Инверсия пептидной цепи(pep-flip)
Индикатором возможности инверсии служат два идущих подряд остатка на карте Рамачандрана попавшие в неблагоприятныеобласти

6. Торсионные углы боковых цепей. Ротамеры

Конформации боковых цепей
• Боковые цепи имеют от 0 (Gly, Ala) до 4х (Lys, Arg) степеней свободы
• Эти степени свободы - вращения вокруг ковалентных связей – торсионные углы
• Соответствующие торсионные углы обозначаются χ
1, ..., χ
4, отсчёт идет от связи
C_alpha – C_beta
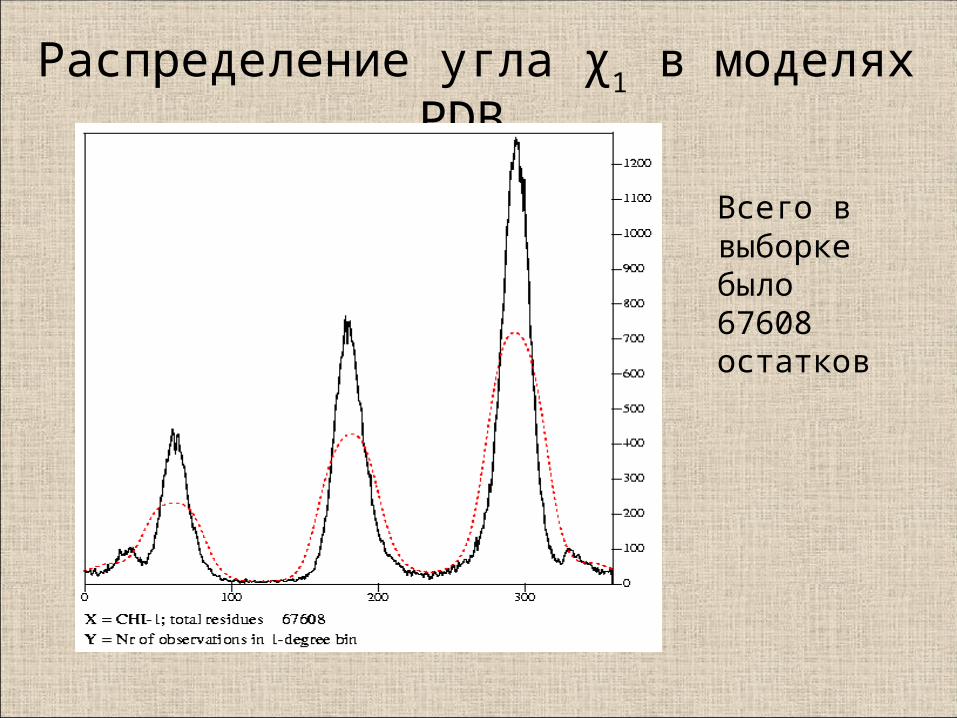
Распределение угла χ1 в моделях PDB
Всего в выборкебыло 67608 остатков
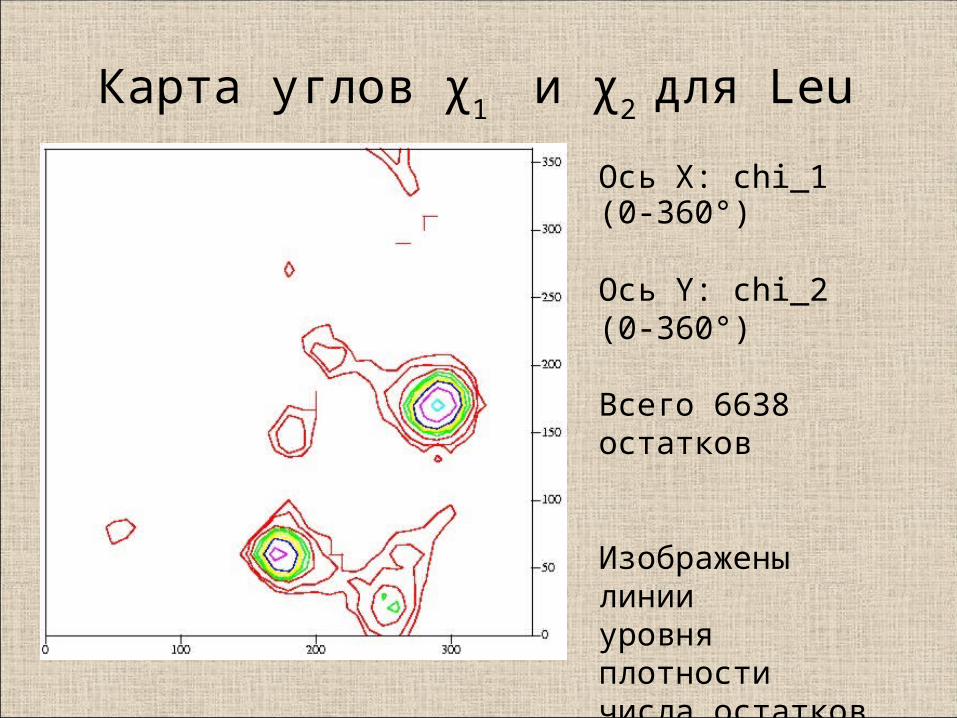
Карта углов χ1 и χ
2 для Leu
Ось X: chi_1 (0-360°)
Ось Y: chi_2 (0-360°)
Всего 6638 остатков
Изображены линии уровня плотности числа остатков

Ротамеры - это боковые цепи в типичных для данного типа остатка конформациях
У каждого типа остатков (Leu, Trp, Arg и т.п.) свое число ротамеров
Имеются базы данных ротамеров, для каждого остатка указаны средние и доверительные интервалы
Программы “знают” ротамеры боковых цепей

Боковые цепи, конформация которых существенно отличается от
одной из предпочитаемых, считаются маргиналами.
Рекомендуемая VTF программа - MolProbity

7. Пространственный R-фактор (RSR)

Простой метод найти ошибку в положении атомов – посмотреть как согласуются экспериментальная электронная плотность и построенная по модели.
Проблема в том, что … эксперимент не даёт функцию электронной плотности!
Эл.пл-ть, полученая в результате решения фазовой проблемы, служит только для построения черновой модели структуры!
RSR оценивает соответствие модели экспериментальным данным

Как рассчитать “экспериментальную” электронную
плотность?
• Фазы - по модели (больше неоткуда взять!)
• Рефлексы – из эксперимента
• Используют трюк “2F_эксп – F_модель” для контрастирования ошибок. В результате трюка получается лучшее приближение к правильной электронной плотности

Продолжение
• Карты электронной плотности моделей, для которых в PDB есть файл структурных факторов, доступны на сайте Electron Density Server (EDS)

Как сравнить “экспериментальную” электронную плотность с электронной плотностью, построенной по модели?
• Real Space R (RSR) характеризует насколько модель атомов (или даже отдельного атома) соответствует “экспериментальной” электронной плотности
Сумма берется по узлам пространственной решетки в окружении всех атомов (или группы атомов)
Хорошие значения: RSR<10%Плохие: >20%
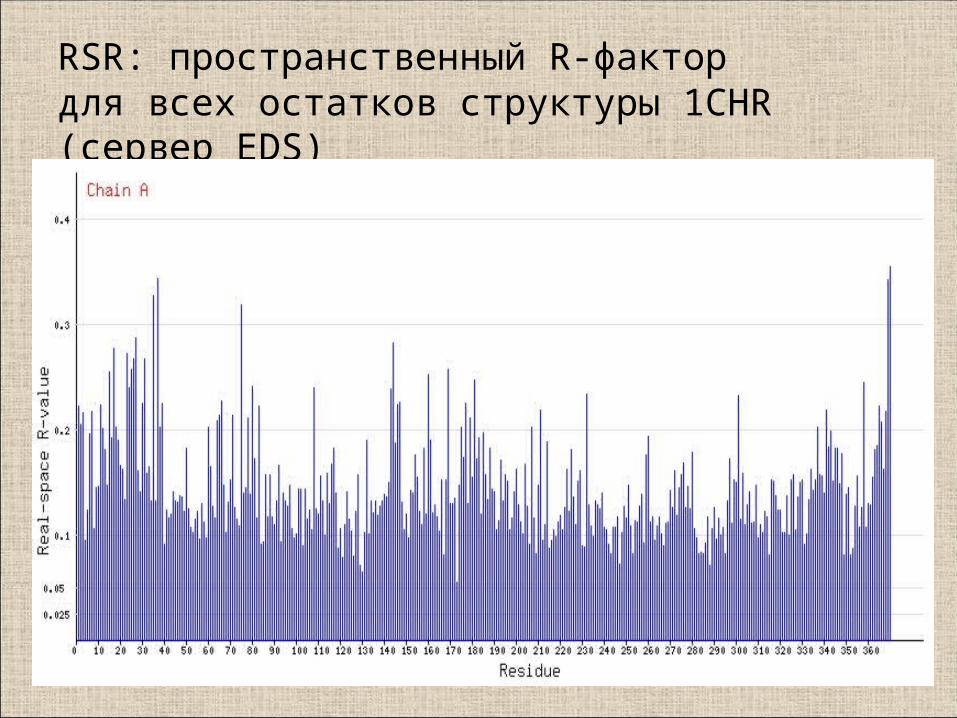
RSR: пространственный R-фактор для всех остатков структуры 1CHR (сервер EDS)

Для маргиналов с RSR>20% имеет смысл посмотреть как остаток
вписан в электронную плотность

Относительная оценка RSR: RSR – Z-score
• Для вычисления Z остатка (напр. Ala57) его RSR сравнивается со средним RSR для того же типа остатков (Ala) по выборке из PDB с примерно таким же разрешением (напр. 1.5-1.8 Å)
• Если RSR плохой, а RSR-Z – хороший, то значит координаты атомов расшифрованы плохо, но не хуже, чем в других подобных структурах.
Z = (RSR - <RSRresolution>) / Sigmaresolution
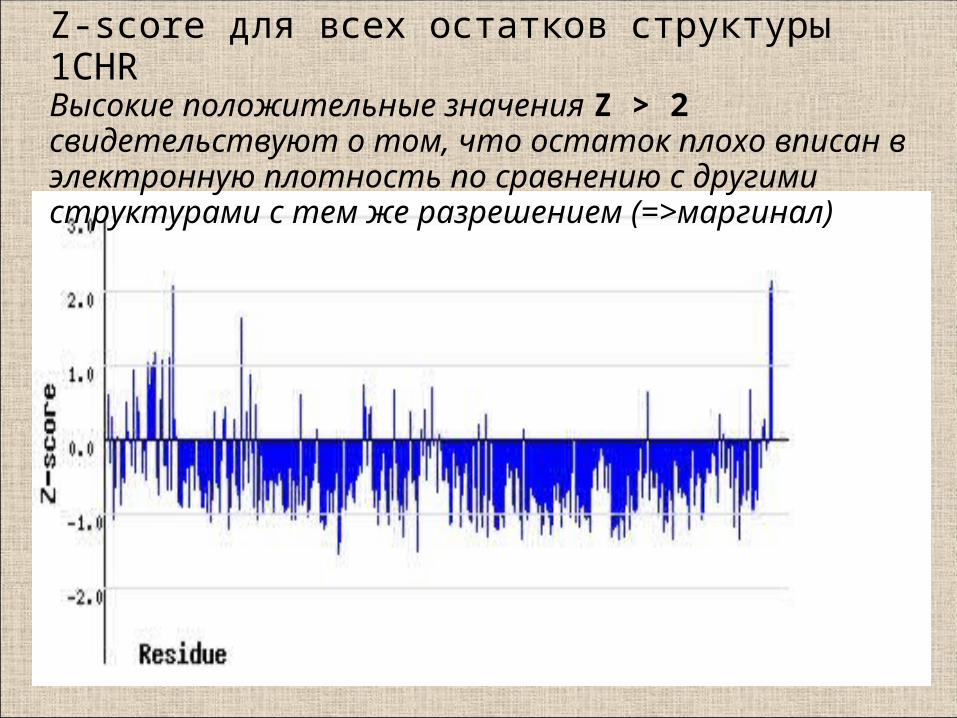
Z-score для всех остатков структуры 1CHRВысокие положительные значения Z > 2 свидетельствуют о том, что остаток плохо вписан в электронную плотность по сравнению с другими структурами с тем же разрешением (=>маргинал)

8. “Комфортность” окружения атома

• Пересечения ван-дер-Ваальсовых радиусов, включая водород, (более, чем на 0.4 Å), физически невозможны (clash)
• Заряд остатка должен компенсироваться взаимодействием с зарядом противоположного знака
• Донорам/акцепторам протона выгодно образовывать водородную связь; если таковой нет, то остаток - маргинал
• Неполярным атомам предпочтительно находиться в гидрофобном окружении
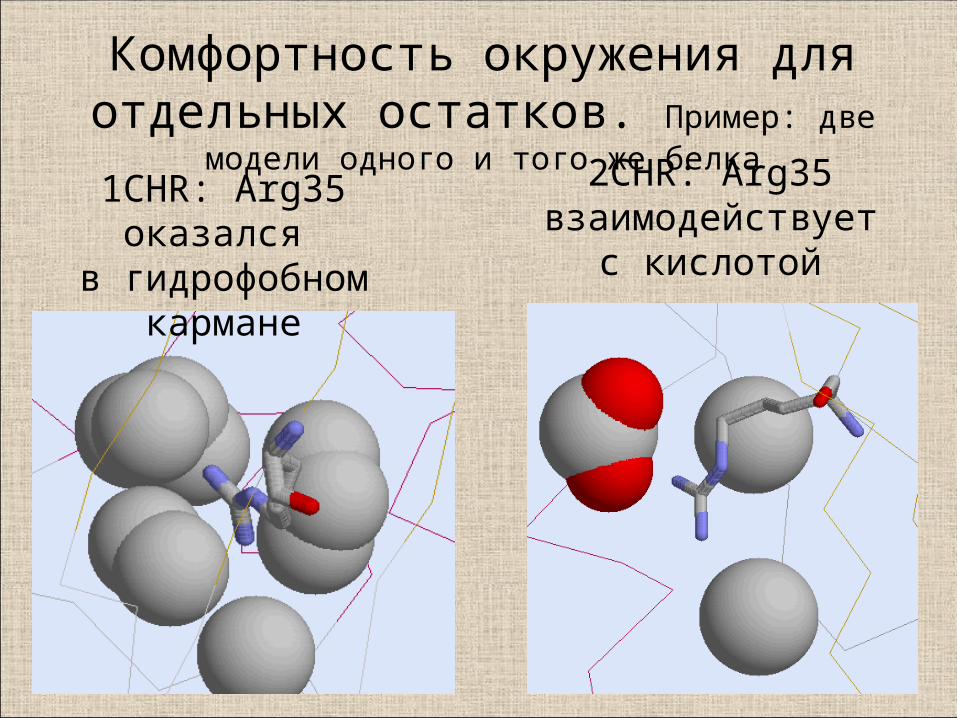
1CHR: Arg35 оказался в гидрофобном
кармане
2CHR: Arg35 взаимодействует с
кислотой
Комфортность окружения для отдельных остатков. Пример: две модели одного и того же белка
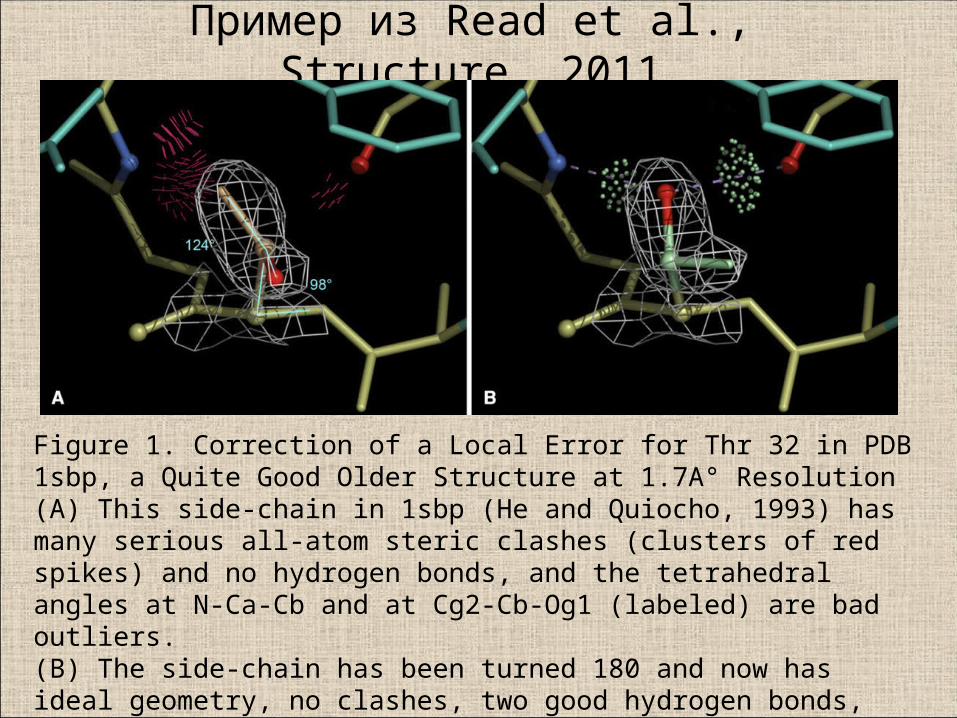
Пример из Read et al., Structure, 2011
Figure 1. Correction of a Local Error for Thr 32 in PDB 1sbp, a Quite Good Older Structure at 1.7A° Resolution(A) This side-chain in 1sbp (He and Quiocho, 1993) has many serious all-atom steric clashes (clusters of red spikes) and no hydrogen bonds, and the tetrahedralangles at N-Ca-Cb and at Cg2-Cb-Og1 (labeled) are bad outliers.(B) The side-chain has been turned 180 and now has ideal geometry, no clashes, two good hydrogen bonds, and a slightly better fit to the density.

Существуют несколько интегральных критериев комфортности окружения.
Можно использовать те, которые приведены в протоколах PDBReport, создаваемых программой WhatCheck

Интегральная оценка комфортности
окружения остатка• В программе WhatCheck рассчитывается Z-
score для комфортности окружения каждой боковой цепи
• Маргиналы – Z-score < -5• Более показательны участки цепи с низким Z,
для их обнаружения строится сглаженный график зависимости Z от номера остатка
• Маргиналов по окружению стоит проверять визуально: часто маргинальность объясняется выходом на поверхность глобулы, контактом с белком из соседней ячейки и др.

Некоторые специальные ситуации
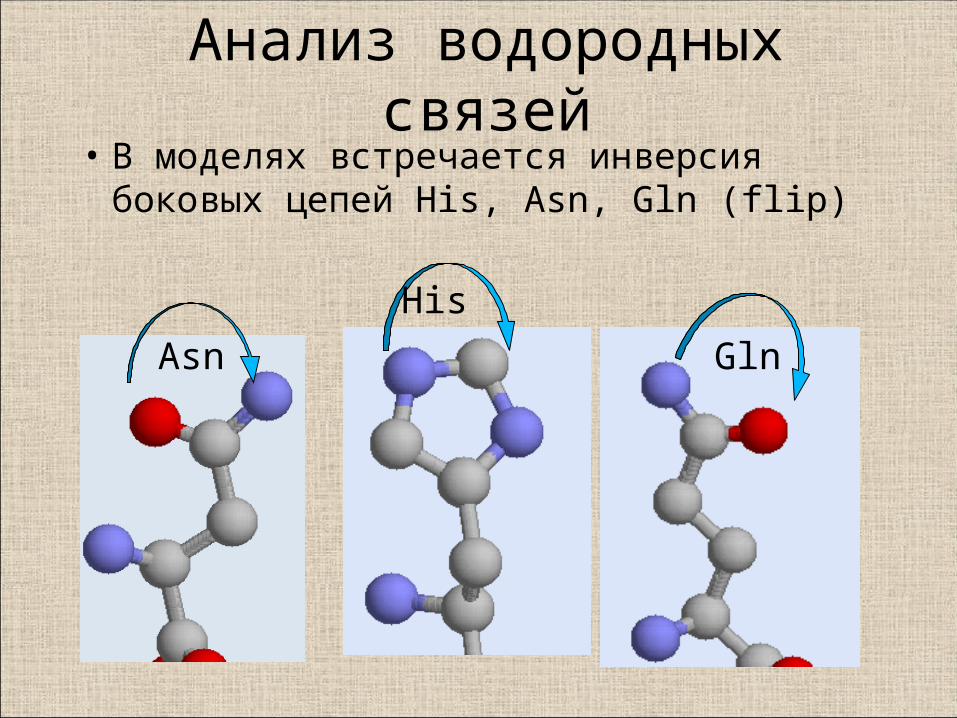
Анализ водородных связей• В моделях встречается инверсия
боковых цепей His, Asn, Gln (flip)
Asn
His
Gln
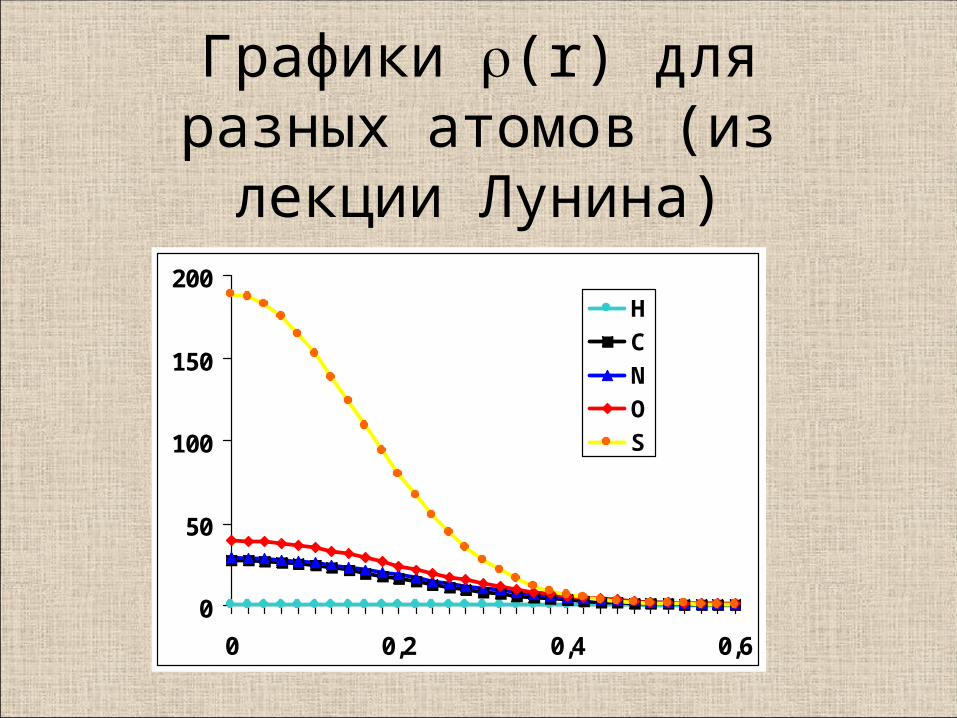
Графики (r) для разных атомов (из лекции Лунина)
0
50
100
150
200
0 0,2 0,4 0,6
H
C
N
O
S

Инверсия (?) в Asn51 гомеодомена №2
Атом ND2
Атом OD1
Атом N6 –донор H
Атом N7 –акцептор H
??
№1
№2
A103
Asn51
Еще в 36 структурах гомеодоменов – так же, как в 1й;еще в 2х – как во второй

9. Молекулы воды и ионы
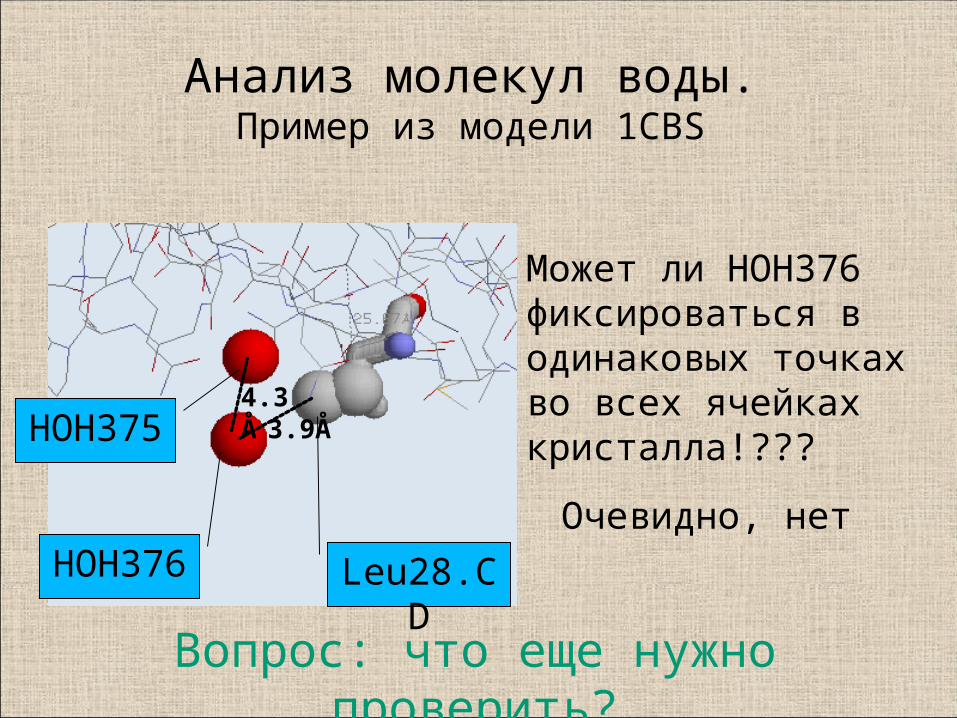
Анализ молекул воды.Пример из модели 1CBS
HOH376
HOH375
Leu28.CD
3.9Å4.3Å
Может ли HOH376фиксироваться в одинаковых точках во всех ячейкахкристалла!???
Очевидно, нет
Вопрос: что еще нужно проверить?

10. Две одинаковых молекулы в асимметрической ячейке

Два мономера в асимметрической ячейкеПример. Хлормуконат циклоизомераза из Alcaligenes eutrophus:
структуры 1chr и 2chr
1CHR: Hoier et al., 1993Разрешение 3.00 Å
2CHR: Kleywegt et al., 1996Разрешение 3.00 Å
Использованы те же экспериментальные данные!

Совмещение остовов 1CHR_A и 2СHR

11. Фильтрация экспериментальных данных
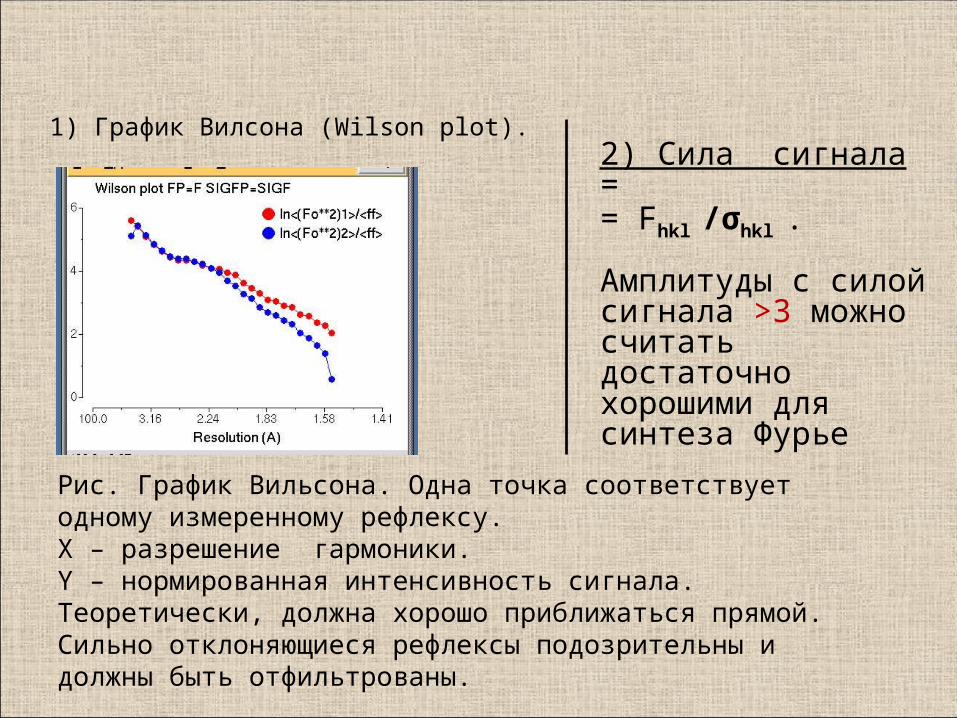
1) График Вилсона (Wilson plot).
Рис. График Вильсона. Одна точка соответствует одному измеренному рефлексу. X – разрешение гармоники.Y – нормированная интенсивность сигнала.Теоретически, должна хорошо приближаться прямой.Сильно отклоняющиеся рефлексы подозрительны и должны быть отфильтрованы.
2) Сила сигнала = = Fhkl /σhkl .
Амплитуды с силой сигнала >3 можно считать достаточно хорошими для синтеза Фурье

12. Повторная оптимизация pdb файла (re-refinment)

Science 2007: Vol. 317. no. 5835, pp. 195 - 196
PDB Improvement Starts with Data DepositionRobbie P. Joosten, Gert Vriend
In 1996, Hooft et al. (4) reported one million anomalies in the PDB, and we recently detected 10 times as many anomalies in a PDB that is 10 times as large. Most of these anomalies are of minor importance, and a small fraction are genuine discoveries that warrant further studies. However, a substantial number are serious errors. Using today's tools, we can correct many of the erroneous structures, provided that the original experimental x-ray data are available.
We re-refined all 1195 PDB files that had a reported resolution of 2.0 Å and that were deposited after 1992 with the use of an experimental data file that included an Rfree set.

http://swift.cmbi.ru.nl/pdb_redo/

13. Две задачи:- Выбор лучшей PDB модели из нескольких
(интегральная оценка)- Выявление маргинальных групп атомов
(локальная оценка)
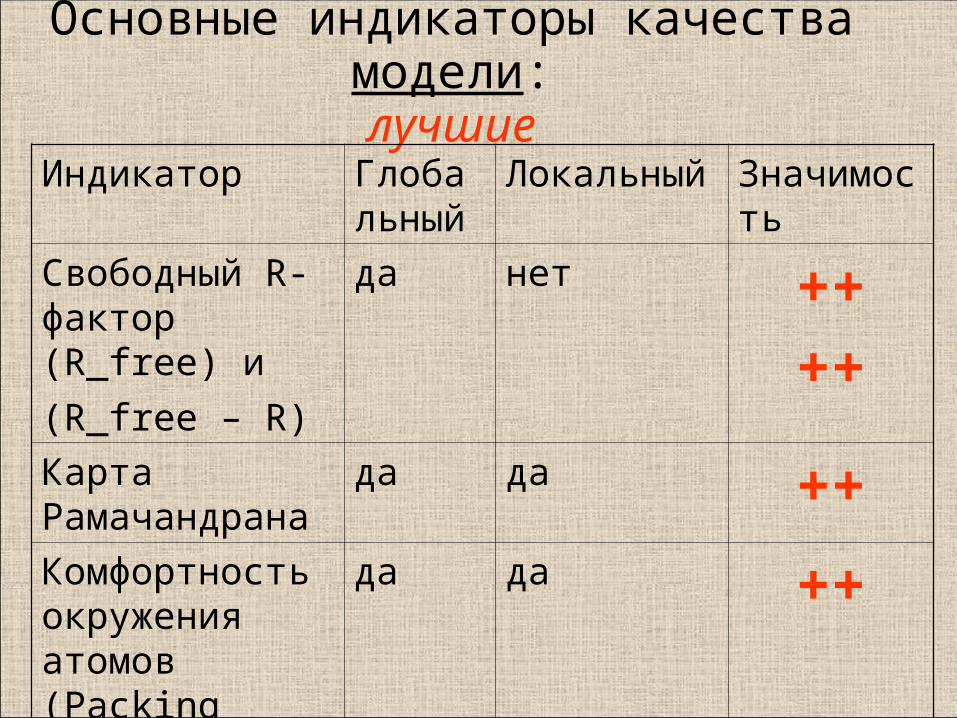
Основные индикаторы качества модели:лучшие
Индикатор Глобальный
Локальный Значимость
Свободный R-фактор (R_free) и
(R_free – R)
да нет ++++
Карта Рамачандрана
да да ++Комфортность окружения атомов (Packing score)
да да ++

Основные индикаторы:хорошие
Индикатор Глобальный
Локальный Значимость
Разрешение да нет +Ротамеры да да +Пространственный R-фактор (RSR)
да да +
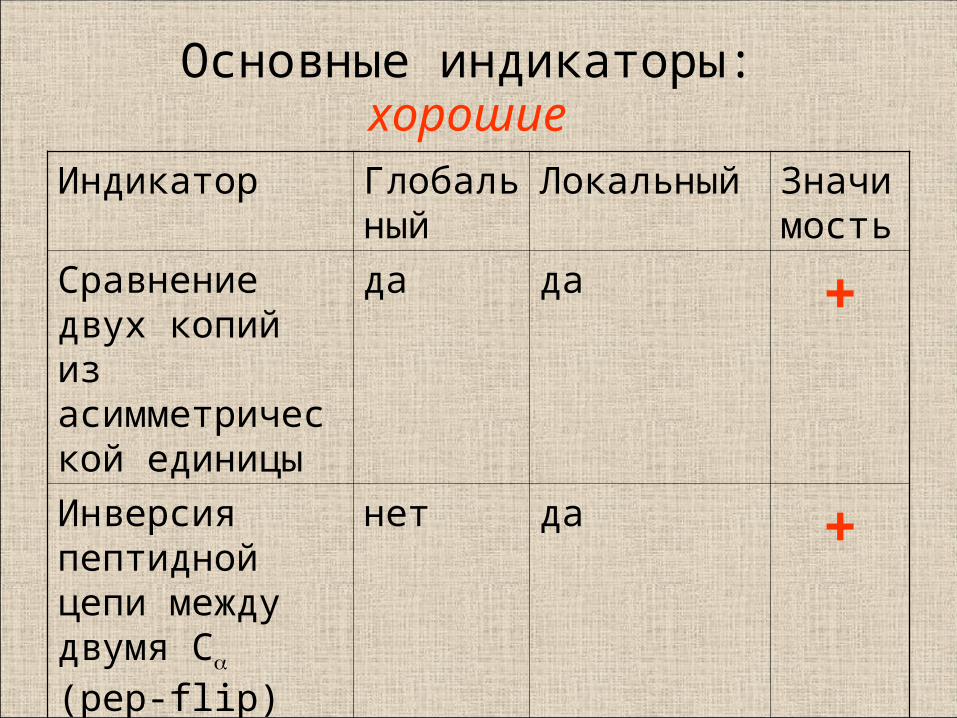
Основные индикаторы:хорошие
Индикатор Глобальный
Локальный Значимость
Сравнение двух копий из асимметрической единицы
да да +
Инверсия пептидной цепи между двумя C
(pep-flip)
нет да +
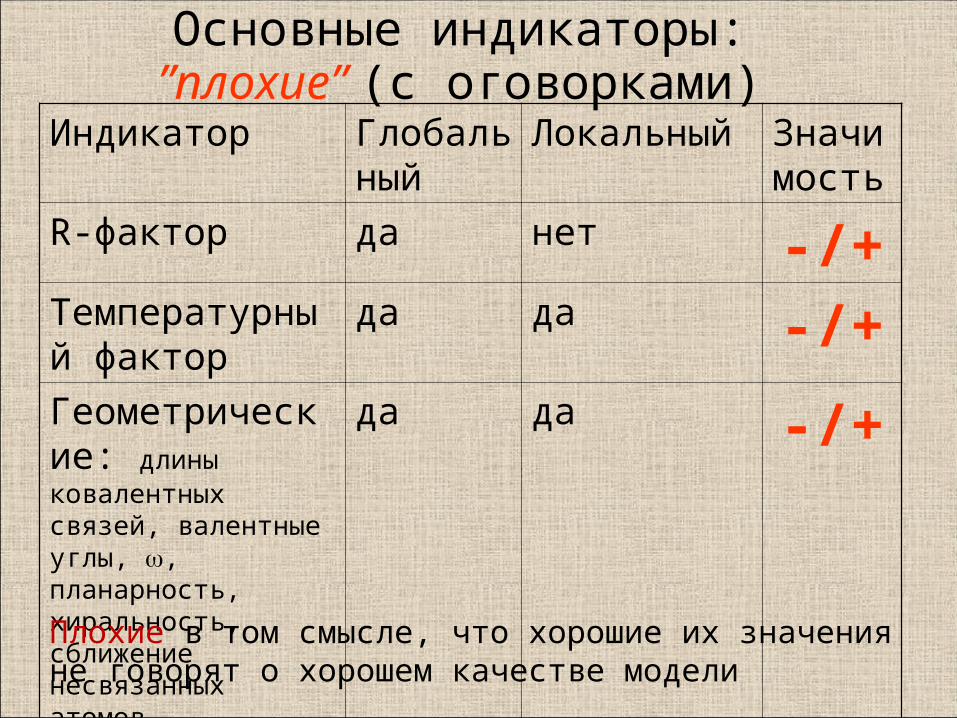
Основные индикаторы:”плохие” (с оговорками)
Индикатор Глобальный
Локальный Значимость
R-фактор да нет -/+Температурный фактор
да да -/+Геометрические: длины ковалентных связей, валентные углы, , планарность, хиральность, сближение несвязанных атомов
да да -/+
Плохие в том смысле, что хорошие их значения не говорят о хорошем качестве модели

Сервисы и программы
• PDBsum
• PDB
• PDBCheck (программа WhatCheck из пакета WhatIf)
• MolProbity
• PROCHECK
• PDBReport
• EDS (RSR во всех видах, файлы с электронными плотностями)

Продолжение
• Ramachandran plots for all types of residues (http://xray.bmc.uu.se/gerard/supmat/ramarev.html)
• Rotamers for all types of side chains (http://xray.bmc.uu.se/gerard/supmat/chi.html)

Validation Task Forses (VTF),2008
• Организация экспертов, созванная PDB
• Вырабатывает рекомендаций по – построению моделей структур– методам проверки; создает поток (pipline) по
автоматической проверке PDB-файлов и выявлению ошибок

Конец







![e g g h k l b 2016 ± 2017 m q [ g u c ] hds390mr.ru/wp-content/uploads/2016/09/Rabochaya... · качеств в соответствии с возрастными и индивидуальными](https://static.fdocuments.in/doc/165x107/604033ccfe964a68615187c6/e-g-g-h-k-l-b-2016-2017-m-q-g-u-c-.jpg)










