
Xavier F. Figueroa, Chien-Chang Chen, Kevin P. Campbell ... · 1Departamento de Ciencias...
14
doi:10.1152/ajpheart.01368.2006 293:1371-1383, 2007. First published May 18, 2007; Am J Physiol Heart Circ Physiol Kathleen H. Day, Susan Ramos and Brian R. Duling Xavier F. Figueroa, Chien-Chang Chen, Kevin P. Campbell, David N. Damon, You might find this additional information useful... 70 articles, 39 of which you can access free at: This article cites http://ajpheart.physiology.org/cgi/content/full/293/3/H1371#BIBL including high-resolution figures, can be found at: Updated information and services http://ajpheart.physiology.org/cgi/content/full/293/3/H1371 can be found at: AJP - Heart and Circulatory Physiology about Additional material and information http://www.the-aps.org/publications/ajpheart This information is current as of July 30, 2008 . http://www.the-aps.org/. ISSN: 0363-6135, ESSN: 1522-1539. Visit our website at Physiological Society, 9650 Rockville Pike, Bethesda MD 20814-3991. Copyright © 2005 by the American Physiological Society. intact animal to the cellular, subcellular, and molecular levels. It is published 12 times a year (monthly) by the American lymphatics, including experimental and theoretical studies of cardiovascular function at all levels of organization ranging from the publishes original investigations on the physiology of the heart, blood vessels, and AJP - Heart and Circulatory Physiology on July 30, 2008 ajpheart.physiology.org Downloaded from
Transcript of Xavier F. Figueroa, Chien-Chang Chen, Kevin P. Campbell ... · 1Departamento de Ciencias...
doi:10.1152/ajpheart.01368.2006 293:1371-1383, 2007. First published May 18, 2007;Am J Physiol Heart Circ Physiol
Kathleen H. Day, Susan Ramos and Brian R. Duling Xavier F. Figueroa, Chien-Chang Chen, Kevin P. Campbell, David N. Damon,
You might find this additional information useful...
70 articles, 39 of which you can access free at: This article cites http://ajpheart.physiology.org/cgi/content/full/293/3/H1371#BIBL
including high-resolution figures, can be found at: Updated information and services http://ajpheart.physiology.org/cgi/content/full/293/3/H1371
can be found at: AJP - Heart and Circulatory Physiologyabout Additional material and information http://www.the-aps.org/publications/ajpheart
This information is current as of July 30, 2008 .
http://www.the-aps.org/.ISSN: 0363-6135, ESSN: 1522-1539. Visit our website at Physiological Society, 9650 Rockville Pike, Bethesda MD 20814-3991. Copyright © 2005 by the American Physiological Society. intact animal to the cellular, subcellular, and molecular levels. It is published 12 times a year (monthly) by the Americanlymphatics, including experimental and theoretical studies of cardiovascular function at all levels of organization ranging from the
publishes original investigations on the physiology of the heart, blood vessels, andAJP - Heart and Circulatory Physiology
on July 30, 2008 ajpheart.physiology.org
Dow
nloaded from

Are voltage-dependent ion channels involved in the endothelial cell controlof vasomotor tone?
Xavier F. Figueroa,1 Chien-Chang Chen,2 Kevin P. Campbell,3,4,5 David N. Damon,6 Kathleen H. Day,6
Susan Ramos,6 and Brian R. Duling6
1Departamento de Ciencias Fisiologicas, Facultad de Ciencias Biologicas, Pontificia Universidad Catolica de Chile,Santiago, Chile; 2Institute of Biomedical Science, Academia Sinica, Nankang, Taipei, Taiwan; 3Howard Hughes MedicalInstitute and Departments of 4Physiology and Biophysics and 5Neurology, University of Iowa, Iowa City, Iowa;and 6Department of Molecular Physiology and Biological Physics, University of Virginia, Charlottesville, Virginia
Submitted 14 December 2006; accepted in final form 18 May 2007
Figueroa XF, Chen CC, Campbell KP, Damon DN, Day KH,Ramos S, Duling BR. Are voltage-dependent ion channels involvedin the endothelial cell control of vasomotor tone? Am J Physiol HeartCirc Physiol 293: H1371–H1383, 2007. First published May 18,2007; doi:10.1152/ajpheart.01368.2006.—In the microcirculation,longitudinal conduction of vasomotor responses provides an essentialmeans of coordinating flow distribution among vessels in a complexnetwork. Spread of current along the vessel axis can display aregenerative component, which leads to propagation of vasomotorsignals over many millimeters; the ionic basis for the regenerativeresponse is unknown. We examined the responses to 10 s of focalelectrical stimulation (30 Hz, 2 ms, 30 V) of mouse cremasterarterioles to test the hypothesis that voltage-dependent Na� (Nav) andCa2� channels might be activated in long-distance signaling in mi-crovessels. Electrical stimulation evoked a vasoconstriction at the siteof stimulation and a spreading, nondecremental conducted dilation.Endothelial damage (air bubble) blocked conduction of the vasodila-tion, indicating an involvement of the endothelium. The Nav channelblocker bupivacaine also blocked conduction, and TTX attenuated it.The Nav channel activator veratridine induced an endothelium-depen-dent dilation. The Nav channel isoforms Nav1.2, Nav1.6, and Nav1.9were detected in the endothelial cells of cremaster arterioles byimmunocytochemistry. These findings are consistent with the involve-ment of Nav channels in the conducted response. BAPTA buffering ofendothelial cell Ca2� delayed and reduced the conducted dilation,which was almost eliminated by Ni2�, amiloride, or deletion of �1H
T-type Ca2� (Cav3.2) channels. Blockade of endothelial nitric oxidesynthase or Ca2�-activated K� channels also inhibited the conductedvasodilation. Our findings indicate that an electrically induced signalcan propagate along the vessel axis via the endothelium and caninduce sequential activation of Nav and Cav3.2 channels. The resultantCa2� influx activates endothelial nitric oxide synthase and Ca2�-activated K� channels, triggering vasodilation.
conducted vasodilation; T-type calcium channels; hypertension; gapjunction; voltage-gated sodium channels
CONTROL OF BLOOD PRESSURE and blood flow depends on theregulation of vessel diameter and on the integrated responses ofdifferent segments of the arteriolar resistance network (12, 47,48). Thus, vascular function depends on precise, well-inte-grated activation of adjacent smooth muscle and endothelialcells (27, 48, 49), processes that are accomplished, in largepart, by cell-cell conduction of electrical signals via gapjunctions (12, 16, 20, 27, 50, 62, 65). Vasodilator signals
generated in the endothelium can extend many millimetersalong the vessel axis without noticeable decay (12, 14, 16, 17),and such nondecremental conduction suggests the involvementof a regenerative mechanism that would likely require one ormore voltage-dependent ion channels. Consistent with thishypothesis, focal electrical stimulation of arterioles with amicropipette activates a vasoconstriction that is restricted to ashort vessel segment at the stimulation site and, in addition,causes a nondecremental, conducted vasodilation that is non-neurally mediated and dependent on the presence of connexin40 (Cx40) (20), a gap junctional protein confined to theendothelium (12, 20). These findings raise two questions: 1)How is a regenerative conduction initiated and/or sustained? 2)How is a conducted electrical signal linked to vasodilation?
Voltage-dependent ion channels are not generally thought tobe present in the endothelium, and endothelial cells are typi-cally assumed to be electrically unexcitable (38), but a numberof observations suggest otherwise. Electrical field stimulationinduces a nonneuronal, endothelium-dependent vasodilationin vitro, and such stimulation can cause nitric oxide (NO)release from cultured endothelial cells (7, 17, 23, 24, 53).Moreover, although not widely appreciated, voltage-dependentNa� (Nav) and Ca2� (Cav) channels have been detected inendothelial cells (2, 3, 21, 25, 26, 55, 57, 58, 61, 63, 66, 70),and these ion channels have been reported to be functional.T-type, voltage-dependent Ca2� (Cav3) channels of the sub-type �1H (Cav3.2) are essential for normal relaxation of themurine coronary arteries (10), and the Nav channels are in-volved in the endothelial response to shear stress (55).
On the basis of the above-described findings, we hypothe-sized that Nav and Cav channels might be activated in long-distance signaling in microvessels and, thus, could play a rolein the regulation of vasomotor tone. Our experiments supportthe proposition that electrical stimulation induces regenerativeactivation of endothelial cell Nav channels. Furthermore, ourfindings are consistent with the idea that a depolarizationinduced by opening of Nav channels leads to activation ofendothelial cell Cav3.2 channels, providing a key trigger forlong-distance, endothelium-dependent activation of NO pro-duction and Ca2�-activated K� (KCa) channels, which leads tovasodilation.
Address for reprint requests and other correspondence: B. R. Duling, Dept.of Molecular Physiology and Biological Physics, Univ. of Virginia HealthSciences Center, PO Box 800736, Charlottesville, VA 22908-0736 (e-mail:[email protected]).
The costs of publication of this article were defrayed in part by the paymentof page charges. The article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Am J Physiol Heart Circ Physiol 293: H1371–H1383, 2007.First published May 18, 2007; doi:10.1152/ajpheart.01368.2006.
0363-6135/07 $8.00 Copyright © 2007 the American Physiological Societyhttp://www.ajpheart.org H1371
on July 30, 2008 ajpheart.physiology.org
Dow
nloaded from

MATERIALS AND METHODS
Male C57Bl/6 (wild-type), endothelial NO synthase (eNOS)-knockout (eNOS�/�) (44), �13.2-knockout (�13.2�/�) (10), and �1-subunit of KCa channel-knockout (�1
�/�) mice (4) (22–30 g body wt)were used. Experiments were conducted according to the HelsinkiDeclaration and the “Guiding Principles in the Care and Use ofLaboratory Animals” endorsed by the American Physiological Soci-ety. All animal protocols were approved by the Animal Care and UseCommittee of the University of Virginia. All animals were genotypedafter the experiment.
Mouse Cremaster Preparation
Mice were anesthetized with pentobarbital sodium (Nembutal; 40mg/kg ip, diluted in isotonic saline to 5 mg/ml) and placed on aPlexiglas board. Body temperature was maintained at 35–36°C with aheating pad throughout the surgery and the experiment. The rightcremaster muscle was exposed and opened by a longitudinal incisionon its ventral surface. The testis and epididymis were excised afterligation of the supply vessels, and the cremaster muscle was pinnedout on a silicone rubber pedestal. The mouse was placed on the stageof an Olympus microscope (BX 50 WI, Gibraltar Platform), and thecremaster muscle was continuously superfused at 3 ml/min with abicarbonate-buffered saline solution (mM: 131.9 NaCl, 4.7 KCl, 2.0CaCl2, 1.2 MgSO4, and 20.0 NaHCO3) kept at 35°C and equilibratedwith 95% N2-5% CO2. The preparation was allowed to stabilize for45–60 min before initiation of the experiment. Supplemental doses ofdilute pentobarbital sodium anesthetic in isotonic saline (10 mg/kg ip)were administered as appropriate. At the end of the experiment, theanimals were killed by an anesthetic overdose.
Vessel Diameters
The cremaster muscle was transilluminated, the microscope imagewas projected to a video camera (series 65, Dage-MTI), and the videooutput was displayed on a monitor (model HR1000, Dage-MTI). Theinner diameter of the arterioles (27–67 �m maximum diameter) wascontinuously measured using Diamtrak software (65).
Electrical Stimulation
A Ag-AgCl reference electrode immersed in the superfusate waspositioned symmetrically around the cremaster muscle, and selectedarterioles were stimulated (30 Hz, 2 ms, 30 V) for 10 s withtriple-beveled micropipettes (3–4 �m ID) filled with 1 M NaCl andconnected to the cathode of a stimulator (model SD9, Grass Instru-ment). The location of the stimulating pipette was critical: it wasinserted under the cremaster mesothelium and positioned directlyabove an arteriole at a distance selected to evoke a local constrictionof �50%.
Stimulation With Activators of Voltage-Dependent Na� Channels
Arterioles were stimulated focally with a pressure-pulse ejection(500 ms, 10–15 psi) of the neurotoxins veratridine (100 �M), �-pom-pilidotoxin (�-PMTX, 30 �M), and ATX-II (10 �M) using a micropi-pette (3–4 �m ID), and the vasomotor response was evaluated at theapplication site. Activation of Nav channels of the cremaster striatedmuscle causes constriction of these muscle fibers, which may interferewith the vasomotor response of the vessels. Therefore, the neurotoxinswere applied to a selected arteriolar segment that was not closelysurrounded (�20 �m) by striated muscle.
Blood Pressure Measurement
Ten- to 14-wk-old mice were placed in a dark environment, andblood pressure and heart rate were determined in “blinded” fashionusing a computerized tail-cuff system (Visitech Systems, Cary, NC).The animals were exposed to the measurement procedure for 7 days
before initiation of data recording, and typically three replicate bloodpressure measurement sets were averaged over the course of severaldays. Reported means are the averages for multiples days.
Immunocytochemical Analysis
Anesthetized mice were perfused through the left ventricle with awarmed MOPS-buffered physiological saline solution (PSS) contain-ing 1% fetal calf serum, 10 U/�l heparin, 10 �M ACh, and 10 �Msodium nitroprusside and then with 2% paraformaldehyde in MOPS-buffered PSS to fix the vasculature. The cremaster muscles wereremoved and postfixed overnight. The tissues were dehydrated, em-bedded in paraffin, sectioned (5 �m), placed on charge-coated slides,and deparaffinized using standard procedures. For antigen retrieval,the slides were heated in a microwave oven in a citrate buffer. Thesections were blocked with 0.5% BSA in PBS and incubated over-night at 4°C with a rabbit polyclonal primary antibody (1:250 dilu-tion). Primary antibodies were directed against one of the sevenspecific isoforms of the Na� channel (Nav1.1, Nav1.2, Nav1.3,Nav1.5, Nav1.6, Nav1.7, and Nav1.9) or against all isoforms of theNa� channel, i.e., pan-sodium antibodies (Upstate Biotechnology andAlomone Laboratories). Antibodies against the individual isoforms ofNav channels were purchased from Alomone Laboratories; the Nav1.9channel-specific rabbit antibody obtained from Chemicon Interna-tional, which yielded results very similar to those obtained with theantibody purchased from Alomone Laboratories, was also used. Bind-ing of the primary antibody was analyzed by immunofluorescence andimmunohistochemistry. For immunofluorescence, the sections wereincubated with an Alexa 568-labeled goat anti-rabbit secondary anti-body (Molecular Probes) for 1 h at room temperature, and thefluorescent signal was examined with a confocal microscope (Fluo-view, Olympus, New Hyde Park, NY). For immunohistochemistry,the primary antibody was labeled with a biotinylated secondaryantibody (1 h at room temperature), and the endogenous peroxidaseactivity was blocked with 0.45% H2O2 in methanol (20 min). Thesections were then incubated with Vectastain ABC reagent (VectorLaboratories) for 30 min and developed with 0.1% 3,3�-diaminoben-zidine (Dakocytomation) and 0.023% H2O2 in the dark (5 min). Theresultant immunostaining was observed with an Olympus BX 51microscope and DP 70 camera. As controls, the primary antibodieswere preabsorbed with their respective antigen or omitted entirely(secondary alone).
In some experiments, after the vasculature was fixed by perfusionwith 2% paraformaldehyde, immunofluorescence was used to studyintact cremaster arterioles and aortas. The cremaster muscle wasspread and pinned on a Sylgard pedestal, and selected arterioles wereperfused via micropipette with the primary and secondary antibodiesto target the endothelial cells directly. Aortas were removed and cutinto 3- to 4-mm segments, which were opened longitudinally for enface observation. Silicone rubber adhesive was used to secure thesegment edges to a glass slide, and the tissue was permeabilized with1% Triton X-100 in MOPS-buffered PSS. Incubation with the primaryand secondary antibodies was the same as that described for arterioles.
Isolation of Aortic Endothelial Cells for PCR
The chest cavity of anesthetized mice was opened, and the thoracicaortas were removed, cleared of connective tissue, and filled withMOPS-buffered PSS containing 1 mg/ml collagenase VIII (Sigma-Aldrich) and 3.36 U/ml elastase (Sigma-Aldrich). The vessels werethen incubated for 4 min at 37°C and pulled gently over a blunted,silicone rubber-filled 23-gauge hypodermic needle to dislodge theendothelial cells, which were collected in a microcentrifuge tube onice and immediately pelleted at 10,000 g for 5 min for use in RT-PCR.
RT-PCR
RNA was isolated using the RNaqueous-4 PCR kit (Ambion) andreverse transcribed using Superscript RT-PCR reagents (Invitrogen).
H1372 VOLTAGE-DEPENDENT ION CHANNELS AND CONDUCTED DILATION
AJP-Heart Circ Physiol • VOL 293 • SEPTEMBER 2007 • www.ajpheart.org
on July 30, 2008 ajpheart.physiology.org
Dow
nloaded from

Platelet endothelial cell adhesion molecule (PECAM) and desminwere used to confirm the purity of the endothelial cell samples asspecific markers for endothelial cell and smooth muscle, respectively.Nav1.6 channels, PECAM, and desmin were detected using previouslypublished primer sets (15, 35). For the Nav1.6 channel, the sequencesof the primer pair were 5�-CAT CTT TGA CTT TGT GGT GGTCAT-3� (sense) and 5�-CGG ATA ACT CGG AAT AGG GTT G-3�(antisense), and the thermal cycling conditions were 50°C for 2 min,60°C for 20 min, 95°C for 5 min, and 60 cycles at 94°C for 15 s and59°C for 60 s with an extension at 72°C for 7 min (15, 35). Thesequences of the primer pair were 5�-AGG GGA CCA GCA GCACAT TAG G-3� (sense) and 5�-AGG CCG CTT CTC TTG ACC ACTT-3� (antisense) for PECAM (15, 35) and 5�-GCT CTC CCG TGTTCC CTC GAG CAG G-3� (sense) and 5�-GGC GAA GCG GTCATT GAG CTC TTG C-3� (antisense) for desmin (15, 35), and thecycling conditions were 45°C for 30 min, 95°C for 2 min, and 35cycles of 94°C for 15 s, 55°C for 30 s, and 72°C for 30 s with anextension at 72°C for 5 min.
Experimental Protocols
The changes in diameter were measured first at the stimulation site(local) and then 500, 1,000, and 2,000 �m upstream (Fig. 1A) inresponse to four separate stimuli. Maximal diameter was estimatedduring superfusion of 1 mM adenosine. Variations in diameter areexpressed as percentage of the maximum possible constriction ordilation.
Damage of the endothelium. Selective endothelial cell damagewas produced by injection of an air bubble via a side branch of thestimulated arteriole. A side branch was selected �1,000 �mupstream from the site of electrical stimulation. The side branchwas cannulated with a micropipette (3– 4 �m ID), and a bolus of airwas injected. After 45– 60 min of recovery, the endothelial damagewas evaluated by focal application via pressure-pulse ejection of 10�M ACh (700 ms, 15–20 psi; Fig. 1A) from a micropipette (3–4 �mID) at locations previously examined during responses to electricalstimulation (i.e., local and 500, 1,000, 1,500, and 2,000 �m up-stream), and the change in diameter was analyzed only at the AChapplication site. The localized reduction in the response to ACh (see
RESULTS) provided evidence that the endothelial cell damage caused bythe air bubble was restricted to a short vessel segment that typicallywas closely surrounded by striated muscle. The close associationbetween vessel and striated muscle in many cases precluded selectivestimulation of the vessel with activators of Nav channels because ofsimultaneous activation of striated muscle cells (see Stimulation WithActivators of Nav). Therefore, to assess participation of the endothelialcells in the response to Nav channel activators in an arteriolar segmentnot closely surrounded by striated muscle, we disrupted the endothe-lium along the entire vessel length by perfusing 0.01% saponin (�20s) via micropipette (�10 �m ID) through a side branch. In theseexperiments, the endothelial cell function was also analyzed by focalstimulation with a pulse of ACh.
Buffering of endothelial cell Ca2�. A side branch of the selectedarteriole 2,000 �m upstream from the local site (Fig. 1A) wascannulated with a micropipette (�10 �m ID) and perfused with 10�M BAPTA-AM for �10 min to buffer the endothelial cell Ca2�
(67). Subsequent vasomotor responses to electrical stimulation wereanalyzed 30–40 min after perfusion and restoration of blood flow.BAPTA-AM was prepared in MOPS-buffered saline solution contain-ing 1% low-endotoxin BSA. Chelation of endothelial cell Ca2� wasconfirmed by demonstration of blockade of the vascular response toACh delivered from a micropipette filled with a 10 �M ACh solution.
Focal application of Ni2�, charybdotoxin, or nifedipine. The tip(�10 �m ID) of a micropipette filled with MOPS-buffered salinesolution containing 100 �M Ni2�, 1 �M charybdotoxin (ChTX), or10 �M nifedipine was positioned above the arteriole, and the blockerswere ejected by pressure over a 5- to 10-min period for Ni2� or a 10-to 15-min period for ChTX or nifedipine. Nifedipine was superfusedat the electrical stimulation site (local), and Ni2� or ChTX wasapplied 1,000 �m upstream (Fig. 1A).
Topical application of drugs. The responses were assessed undercontrol conditions and during topical application of the selected drug.A K� channel blocker, 1 �M TTX, 0.5 mM bupivacaine, 10 �MNi2�, 100 �M amiloride, or 100 nM prazosin, was applied for 15 minand 100 �M NG-nitro-L-arginine (L-NNA) for 45 min. K� channelblockers included glibenclamide (1 �M), BaCl2 (50 �M), and tetra-ethylammonium (TEA, 1–10 mM), which were used to block ATP-sensitive K�, inward rectifier K�, and KCa channels, respectively.
Fig. 1. A: schematic representation of experimental proce-dures. Local, 500 �m, 1,000 �m, and 2,000 �m denote sites atwhich vasomotor responses were assessed. References to otherfigures indicate locations of the suffusion pipette when theindicated data were obtained. B: local vasoconstriction at thestimulation site is not required for induction of electricallyinduced conducted vasodilation. The �-adrenoceptor antago-nist prazosin (Pz, 100 nM) was applied topically in combina-tion with focal superfusion via micropipette of 10 �M nifed-ipine (Nif) to block the neural component of response toelectrical stimulation and direct activation of L-type, voltage-dependent Ca2� channels, respectively. Response to electricalstimulation was evaluated at the stimulation site (local) and1,000 �m upstream from the stimulation site. Values aremeans SE. Horizontal bars indicate stimulation period.
H1373VOLTAGE-DEPENDENT ION CHANNELS AND CONDUCTED DILATION
AJP-Heart Circ Physiol • VOL 293 • SEPTEMBER 2007 • www.ajpheart.org
on July 30, 2008 ajpheart.physiology.org
Dow
nloaded from

Chemicals
TTX, glibenclamide, BaCl2, TEA, veratridine, prazosin, nifedipine,and chemicals of analytic grade were purchased from Sigma Chemical(St. Louis, MO). L-NNA and ChTX were obtained from ResearchBiochemicals International (Natick, MA), BAPTA-AM from Molec-ular Probes (Eugene, OR), bupivacaine and pentobarbital sodiumfrom Abbott Laboratories (North Chicago, IL), and BSA from UnitedStates Biochemical (Cleveland, OH). The toxins �-PMTX andATX-II were purchased from Alomone Laboratories.
Statistical Analysis
Values are means SE. Comparisons between groups were madeusing paired or unpaired Student’s t-test, one-way ANOVA plusNewman-Keuls post hoc test, or two-way ANOVA as appropriate.P � 0.05 was considered significant.
RESULTS
Manipulation of tissue associated with the preparation ofin vitro specimens can produce fundamental alterations in thepatterns of cell-cell communication in the vessel wall (62);therefore, the present work was confined to the study ofconduction of vasodilation in arterioles of the intact mousecremaster muscle preparation (20). The cremaster arteriolesmanifest conducted vasomotor stimuli that propagate 2 mmwithout decay in response to ACh (data not shown) or electri-cal stimulation (20). For the experiments reported here, weselected electrical stimulation, because the response to thisstimulus does not depend on activation of a receptor and, thus,could be defined more specifically in terms of the mechanismsinvolved in the conduction of the vasomotor responses. Asdescribed previously (20), electrical stimulation evoked a localvasoconstriction, presumably due to electrical activation of thesmooth muscle cells. At the same time, a conducted vasodila-tion originates in the vessel on either side of the constrictionand spreads along the entire arteriole without decay, suggestingthat a regenerative mechanism of some type was triggered bythe electrical stimulation (see Figs. 1, 2, 5, 7, and 8).
Vasodilator Signal Does Not Depend on SmoothMuscle Stimulation
It could be hypothesized that electrical stimulation openssmooth muscle L-type Ca2� channels, leading to diffusion ofCa2� from the activated smooth muscle cells to endothelialcells via myoendothelial gap junctions (6, 13, 67), with sec-ondary triggering of a delayed, endothelium-dependent vaso-dilator response (67). Two observations indicate that this wasnot the case, however: 1) the local and conducted responses toelectrical stimulation occurred with no detectable delay be-tween them (see Figs. 1, 2, 5, 7, and 8), and 2) the eliminationof the local vasoconstriction and, presumably, the rise insmooth muscle Ca2� with a combination of the �-adrenoceptorantagonist prazosin (100 nmol/l) and focal superfusion viamicropipette of 10 �M nifedipine (Fig. 1A) did not affect theelectrically induced conducted vasodilation (Fig. 1B). Thisobservation also excludes mechanical distortion of the vesselwall or a hemodynamic effect at the site of constriction astriggers for the propagation and supports the hypothesis thatelectrical stimulation directly activates the endothelial cells, ashas been recently proposed (20).
Endothelium Is Required for Propagation of theVasodilator Signal
To assess the cellular pathway followed by the electricallyinduced vasodilator signal, we damaged the endothelium byinjecting an air bubble via a side branch �1,000 �m upstreamfrom the stimulus (Fig. 1A). After injection of the air bubble,endothelial cell function was reevaluated at locations surround-ing the injection site by focal application of 10 �M ACh, anendothelium-dependent vasodilator. The air bubble caused adecrease in baseline diameter (Table 1), and a selective reduc-tion in reactivity to ACh in the region surrounding the site ofinjection (Fig. 2A). Although the disruption of the endotheliumwas restricted to the air bubble injection area (Fig. 2A), the airbubble-induced damage eliminated the regenerative compo-nent of the conducted dilation, leaving in its place a decremen-tal conduction with a length constant of slightly more than 1mm (Fig. 2B), a finding consistent with the previously dem-onstrated Cx40 sensitivity of the electrically induced con-ducted response and with the endothelial cell localization ofCx40 (20). These data indicate that integrity of the endothe-lium is essential for the nondecremental propagation of theelectrically induced vasodilator signal.
Nav Channel Is Involved in Generation of theRegenerative Response
We hypothesized that the conducted vasodilator signal re-flected activation of a regenerative electrical signal at thestimulation site, which would involve one or more voltage-sensitive ion channels, such as Nav and Cav. Although notwidely appreciated, expression of the Nav channel has beenrepeatedly detected in endothelial cells (25, 26, 55, 58), andTTX attenuates the propagation of the vasodilator response(Fig. 2C). However, the effect of TTX was modest, perhapsindicating the involvement of a TTX-resistant Nav channel.We, therefore, applied the local anesthetic bupivacaine, whichblocks TTX-sensitive and TTX-resistant Nav channels (46) andfound that it abolished the regenerative component of the
Table 1. Effect of experimental maneuvers on restingarteriolar diameter
Treatment n
Arteriolar Diameter, �m
Before After
TTX (1 �M) 6 26.82.1 24.52.9Bupivacaine (500 �M) 6 29.53.9 26.44.0*Saponin (0.01%) 4 28.74.6 27.64.2Endothelial damage 4 35.14.4 29.53.9*BAPTA-AM perfusion 5 21.51.5 23.72.1Amiloride (100 �M) 4 27.74.4 22.03.1*L-NNA (100 �M) 4 28.12.5 24.62.5*Ba2� (50 �M) 3 29.93.0 24.13.6*Glibenclamide (1 �M) 3 22.32.8 15.33.1*TEA
1 mM 4 27.63.4 24.12.2*10 mM 5 29.03.0 25.32.3*
Values are means SE. Endothelium was damaged by perfusion of saponinor injection of an air bubble through a side-branch �1,000 �m upstream fromthe local site. BAPTA-AM (10 �M) was perfused for �10 min via a sidebranch. TTX, bupivacaine, amiloride, NG-nitro-L-arginine (L-NNA), Ba2�,glibenclamide, and tetraethylammonium (TEA) were applied topically. *P �0.05 vs. Before (by paired Student’s t-test).
H1374 VOLTAGE-DEPENDENT ION CHANNELS AND CONDUCTED DILATION
AJP-Heart Circ Physiol • VOL 293 • SEPTEMBER 2007 • www.ajpheart.org
on July 30, 2008 ajpheart.physiology.org
Dow
nloaded from
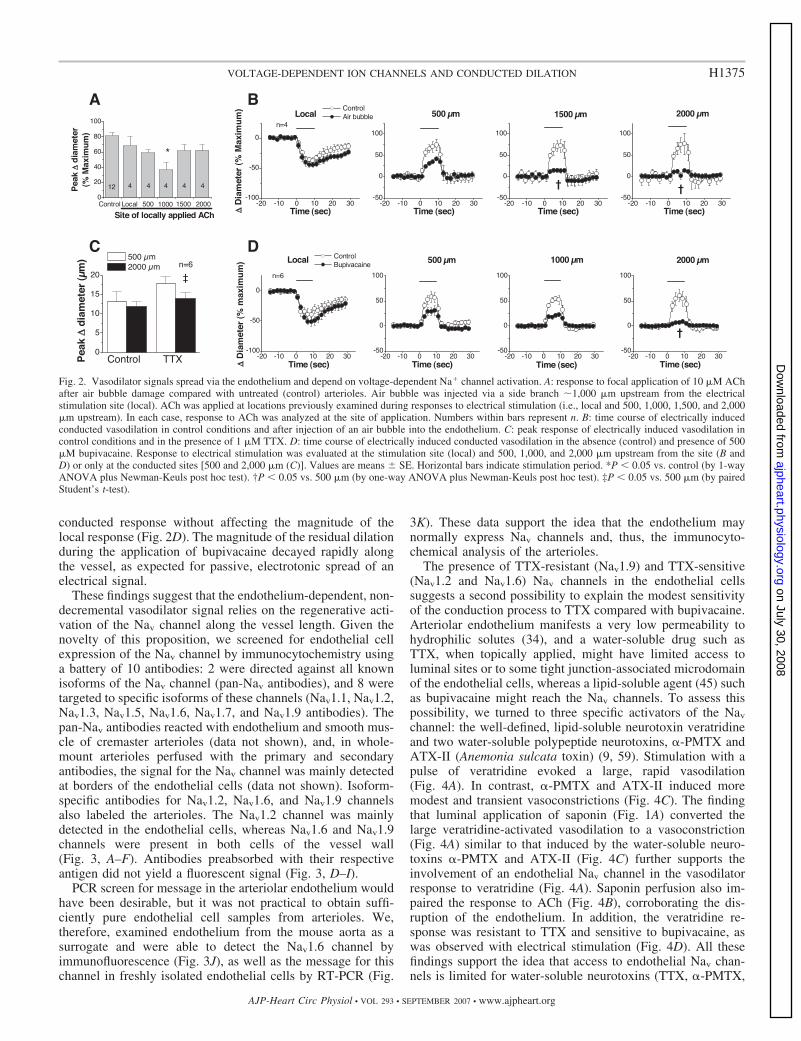
conducted response without affecting the magnitude of thelocal response (Fig. 2D). The magnitude of the residual dilationduring the application of bupivacaine decayed rapidly alongthe vessel, as expected for passive, electrotonic spread of anelectrical signal.
These findings suggest that the endothelium-dependent, non-decremental vasodilator signal relies on the regenerative acti-vation of the Nav channel along the vessel length. Given thenovelty of this proposition, we screened for endothelial cellexpression of the Nav channel by immunocytochemistry usinga battery of 10 antibodies: 2 were directed against all knownisoforms of the Nav channel (pan-Nav antibodies), and 8 weretargeted to specific isoforms of these channels (Nav1.1, Nav1.2,Nav1.3, Nav1.5, Nav1.6, Nav1.7, and Nav1.9 antibodies). Thepan-Nav antibodies reacted with endothelium and smooth mus-cle of cremaster arterioles (data not shown), and, in whole-mount arterioles perfused with the primary and secondaryantibodies, the signal for the Nav channel was mainly detectedat borders of the endothelial cells (data not shown). Isoform-specific antibodies for Nav1.2, Nav1.6, and Nav1.9 channelsalso labeled the arterioles. The Nav1.2 channel was mainlydetected in the endothelial cells, whereas Nav1.6 and Nav1.9channels were present in both cells of the vessel wall(Fig. 3, A–F). Antibodies preabsorbed with their respectiveantigen did not yield a fluorescent signal (Fig. 3, D–I).
PCR screen for message in the arteriolar endothelium wouldhave been desirable, but it was not practical to obtain suffi-ciently pure endothelial cell samples from arterioles. We,therefore, examined endothelium from the mouse aorta as asurrogate and were able to detect the Nav1.6 channel byimmunofluorescence (Fig. 3J), as well as the message for thischannel in freshly isolated endothelial cells by RT-PCR (Fig.
3K). These data support the idea that the endothelium maynormally express Nav channels and, thus, the immunocyto-chemical analysis of the arterioles.
The presence of TTX-resistant (Nav1.9) and TTX-sensitive(Nav1.2 and Nav1.6) Nav channels in the endothelial cellssuggests a second possibility to explain the modest sensitivityof the conduction process to TTX compared with bupivacaine.Arteriolar endothelium manifests a very low permeability tohydrophilic solutes (34), and a water-soluble drug such asTTX, when topically applied, might have limited access toluminal sites or to some tight junction-associated microdomainof the endothelial cells, whereas a lipid-soluble agent (45) suchas bupivacaine might reach the Nav channels. To assess thispossibility, we turned to three specific activators of the Nav
channel: the well-defined, lipid-soluble neurotoxin veratridineand two water-soluble polypeptide neurotoxins, �-PMTX andATX-II (Anemonia sulcata toxin) (9, 59). Stimulation with apulse of veratridine evoked a large, rapid vasodilation(Fig. 4A). In contrast, �-PMTX and ATX-II induced moremodest and transient vasoconstrictions (Fig. 4C). The findingthat luminal application of saponin (Fig. 1A) converted thelarge veratridine-activated vasodilation to a vasoconstriction(Fig. 4A) similar to that induced by the water-soluble neuro-toxins �-PMTX and ATX-II (Fig. 4C) further supports theinvolvement of an endothelial Nav channel in the vasodilatorresponse to veratridine (Fig. 4A). Saponin perfusion also im-paired the response to ACh (Fig. 4B), corroborating the dis-ruption of the endothelium. In addition, the veratridine re-sponse was resistant to TTX and sensitive to bupivacaine, aswas observed with electrical stimulation (Fig. 4D). All thesefindings support the idea that access to endothelial Nav chan-nels is limited for water-soluble neurotoxins (TTX, �-PMTX,
Fig. 2. Vasodilator signals spread via the endothelium and depend on voltage-dependent Na� channel activation. A: response to focal application of 10 �M AChafter air bubble damage compared with untreated (control) arterioles. Air bubble was injected via a side branch �1,000 �m upstream from the electricalstimulation site (local). ACh was applied at locations previously examined during responses to electrical stimulation (i.e., local and 500, 1,000, 1,500, and 2,000�m upstream). In each case, response to ACh was analyzed at the site of application. Numbers within bars represent n. B: time course of electrically inducedconducted vasodilation in control conditions and after injection of an air bubble into the endothelium. C: peak response of electrically induced vasodilation incontrol conditions and in the presence of 1 �M TTX. D: time course of electrically induced conducted vasodilation in the absence (control) and presence of 500�M bupivacaine. Response to electrical stimulation was evaluated at the stimulation site (local) and 500, 1,000, and 2,000 �m upstream from the site (B andD) or only at the conducted sites [500 and 2,000 �m (C)]. Values are means SE. Horizontal bars indicate stimulation period. *P � 0.05 vs. control (by 1-wayANOVA plus Newman-Keuls post hoc test). †P � 0.05 vs. 500 �m (by one-way ANOVA plus Newman-Keuls post hoc test). ‡P � 0.05 vs. 500 �m (by pairedStudent’s t-test).
H1375VOLTAGE-DEPENDENT ION CHANNELS AND CONDUCTED DILATION
AJP-Heart Circ Physiol • VOL 293 • SEPTEMBER 2007 • www.ajpheart.org
on July 30, 2008 ajpheart.physiology.org
Dow
nloaded from

and ATX-II) and that only lipid-soluble drugs (veratridine andbupivacaine) can reach these channels.
Endothelial Cell Ca2� Mediates Vasodilation butnot Conduction
How might an Nav channel-dependent propagating depo-larization lead to dilation? Endothelial cell-mediated dila-
tion typically involves Ca2� as a second messenger; accord-ingly, to test for a role of endothelial cell cytoplasmic Ca2�
in the conducted vasodilator response, we buffered endo-thelial cell Ca2� by perfusing the arterioles for 10 min with10 �M BAPTA-AM via a side branch located upstreamfrom the 1,000 �m-site (Fig. 1A). BAPTA-AM perfusiondid not alter baseline diameter (Table 1) but markedly
Fig. 3. Voltage-dependent Na� (Nav) channel expression in mouse endothelial cells. A–C: immunohistochemical detection of expression of Nav channel-specificisoforms Nav1.2, Nav1.6, and Nav1.9 in endothelial cells of cremaster arterioles. D–I: immunofluorescence of Nav1.2, Nav1.6, and Nav1.9 channels in serialsections of cremaster muscle incubated with primary antibodies in control conditions (D–F) or preabsorbed with their appropriate peptide antigen (G–I).J: immunocytochemistry for Nav1.6 channel in aortic endothelium. Scale bars, 20 �m. K: RT-PCR for desmin, platelet endothelial cell adhesion molecule(PECAM), and Nav1.6 channel in freshly isolated aortic endothelial cells. In different gels prepared with the same endothelial cell sample, mRNA for endothelialcell (EC) and smooth muscle cell (SMC) markers PECAM and desmin was assayed. Positive reaction for PECAM and negative reaction for desmin corroborateselectivity of endothelial cell isolation. RT-PCR confirms Nav1.6 channel mRNA expression in these cells. �RT, omission of reverse transcriptase to rule outdirect amplification of DNA. Lane M, DNA size standards; white boxes, position of 500 bp.
H1376 VOLTAGE-DEPENDENT ION CHANNELS AND CONDUCTED DILATION
AJP-Heart Circ Physiol • VOL 293 • SEPTEMBER 2007 • www.ajpheart.org
on July 30, 2008 ajpheart.physiology.org
Dow
nloaded from
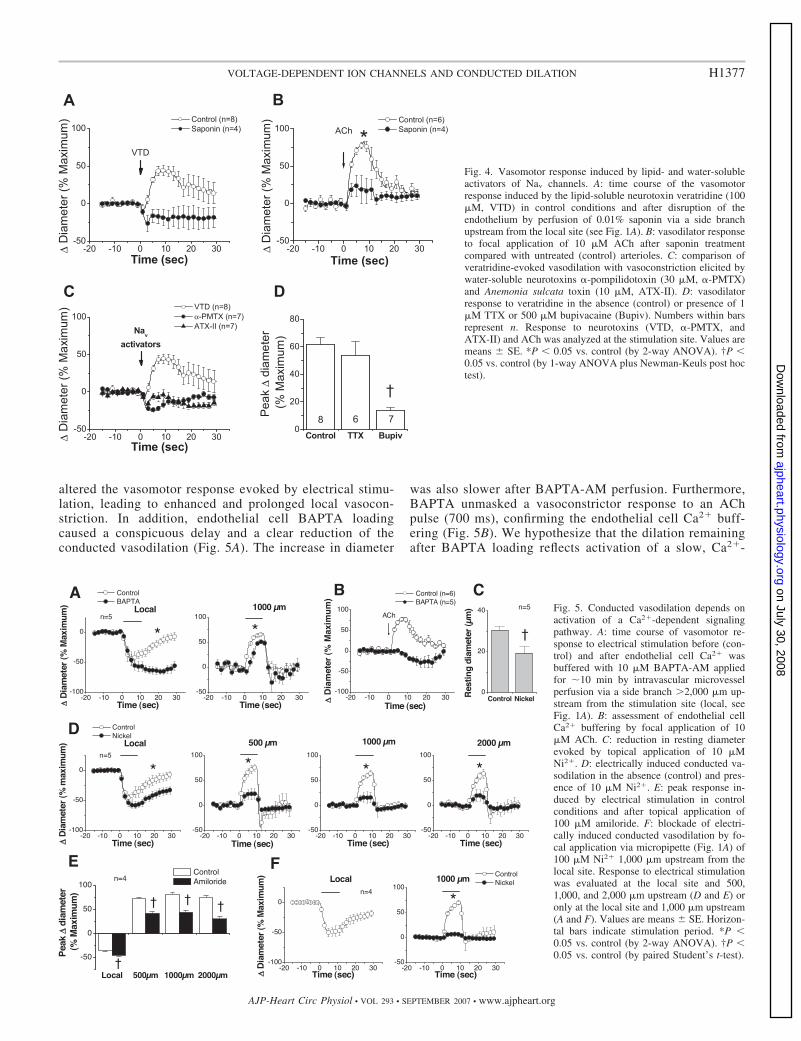
altered the vasomotor response evoked by electrical stimu-lation, leading to enhanced and prolonged local vasocon-striction. In addition, endothelial cell BAPTA loadingcaused a conspicuous delay and a clear reduction of theconducted vasodilation (Fig. 5A). The increase in diameter
was also slower after BAPTA-AM perfusion. Furthermore,BAPTA unmasked a vasoconstrictor response to an AChpulse (700 ms), confirming the endothelial cell Ca2� buff-ering (Fig. 5B). We hypothesize that the dilation remainingafter BAPTA loading reflects activation of a slow, Ca2�-
Fig. 4. Vasomotor response induced by lipid- and water-solubleactivators of Nav channels. A: time course of the vasomotorresponse induced by the lipid-soluble neurotoxin veratridine (100�M, VTD) in control conditions and after disruption of theendothelium by perfusion of 0.01% saponin via a side branchupstream from the local site (see Fig. 1A). B: vasodilator responseto focal application of 10 �M ACh after saponin treatmentcompared with untreated (control) arterioles. C: comparison ofveratridine-evoked vasodilation with vasoconstriction elicited bywater-soluble neurotoxins �-pompilidotoxin (30 �M, �-PMTX)and Anemonia sulcata toxin (10 �M, ATX-II). D: vasodilatorresponse to veratridine in the absence (control) or presence of 1�M TTX or 500 �M bupivacaine (Bupiv). Numbers within barsrepresent n. Response to neurotoxins (VTD, �-PMTX, andATX-II) and ACh was analyzed at the stimulation site. Values aremeans SE. *P � 0.05 vs. control (by 2-way ANOVA). †P �0.05 vs. control (by 1-way ANOVA plus Newman-Keuls post hoctest).
Fig. 5. Conducted vasodilation depends onactivation of a Ca2�-dependent signalingpathway. A: time course of vasomotor re-sponse to electrical stimulation before (con-trol) and after endothelial cell Ca2� wasbuffered with 10 �M BAPTA-AM appliedfor �10 min by intravascular microvesselperfusion via a side branch 2,000 �m up-stream from the stimulation site (local, seeFig. 1A). B: assessment of endothelial cellCa2� buffering by focal application of 10�M ACh. C: reduction in resting diameterevoked by topical application of 10 �MNi2�. D: electrically induced conducted va-sodilation in the absence (control) and pres-ence of 10 �M Ni2�. E: peak response in-duced by electrical stimulation in controlconditions and after topical application of100 �M amiloride. F: blockade of electri-cally induced conducted vasodilation by fo-cal application via micropipette (Fig. 1A) of100 �M Ni2� 1,000 �m upstream from thelocal site. Response to electrical stimulationwas evaluated at the local site and 500,1,000, and 2,000 �m upstream (D and E) oronly at the local site and 1,000 �m upstream(A and F). Values are means SE. Horizon-tal bars indicate stimulation period. *P �0.05 vs. control (by 2-way ANOVA). †P �0.05 vs. control (by paired Student’s t-test).
H1377VOLTAGE-DEPENDENT ION CHANNELS AND CONDUCTED DILATION
AJP-Heart Circ Physiol • VOL 293 • SEPTEMBER 2007 • www.ajpheart.org
on July 30, 2008 ajpheart.physiology.org
Dow
nloaded from
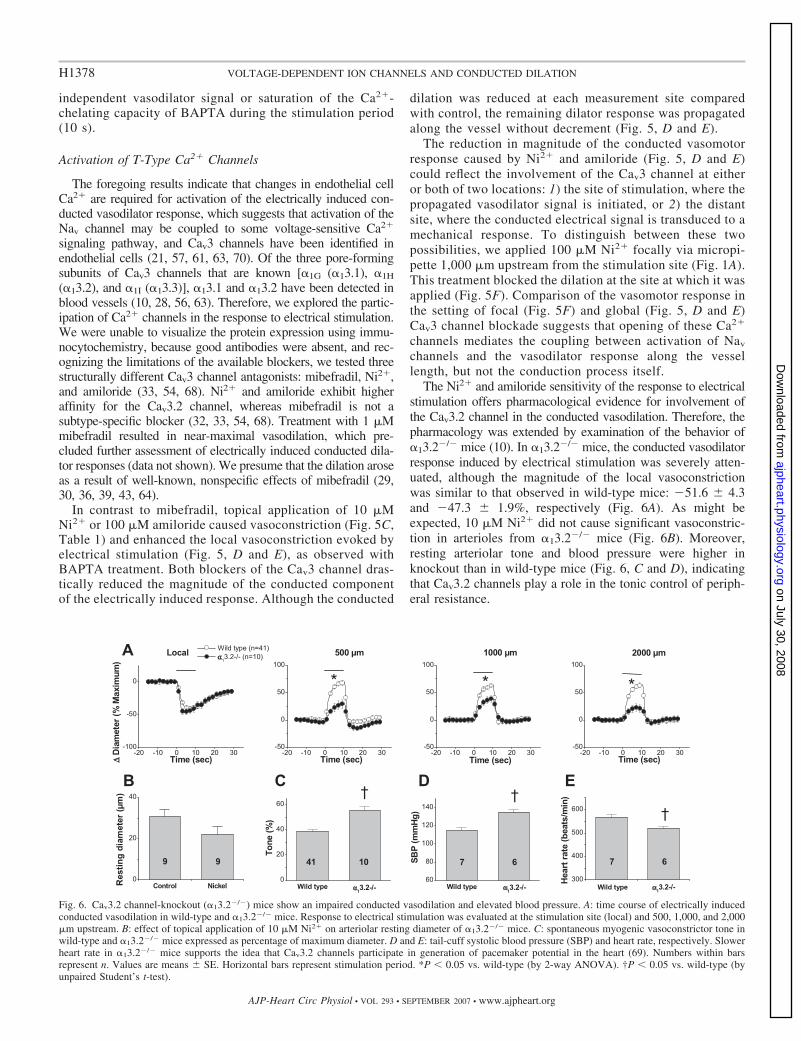
independent vasodilator signal or saturation of the Ca2�-chelating capacity of BAPTA during the stimulation period(10 s).
Activation of T-Type Ca2� Channels
The foregoing results indicate that changes in endothelial cellCa2� are required for activation of the electrically induced con-ducted vasodilator response, which suggests that activation of theNav channel may be coupled to some voltage-sensitive Ca2�
signaling pathway, and Cav3 channels have been identified inendothelial cells (21, 57, 61, 63, 70). Of the three pore-formingsubunits of Cav3 channels that are known [�1G (�13.1), �1H
(�13.2), and �1I (�13.3)], �13.1 and �13.2 have been detected inblood vessels (10, 28, 56, 63). Therefore, we explored the partic-ipation of Ca2� channels in the response to electrical stimulation.We were unable to visualize the protein expression using immu-nocytochemistry, because good antibodies were absent, and rec-ognizing the limitations of the available blockers, we tested threestructurally different Cav3 channel antagonists: mibefradil, Ni2�,and amiloride (33, 54, 68). Ni2� and amiloride exhibit higheraffinity for the Cav3.2 channel, whereas mibefradil is not asubtype-specific blocker (32, 33, 54, 68). Treatment with 1 �Mmibefradil resulted in near-maximal vasodilation, which pre-cluded further assessment of electrically induced conducted dila-tor responses (data not shown). We presume that the dilation aroseas a result of well-known, nonspecific effects of mibefradil (29,30, 36, 39, 43, 64).
In contrast to mibefradil, topical application of 10 �MNi2� or 100 �M amiloride caused vasoconstriction (Fig. 5C,Table 1) and enhanced the local vasoconstriction evoked byelectrical stimulation (Fig. 5, D and E), as observed withBAPTA treatment. Both blockers of the Cav3 channel dras-tically reduced the magnitude of the conducted componentof the electrically induced response. Although the conducted
dilation was reduced at each measurement site comparedwith control, the remaining dilator response was propagatedalong the vessel without decrement (Fig. 5, D and E).
The reduction in magnitude of the conducted vasomotorresponse caused by Ni2� and amiloride (Fig. 5, D and E)could reflect the involvement of the Cav3 channel at eitheror both of two locations: 1) the site of stimulation, where thepropagated vasodilator signal is initiated, or 2) the distantsite, where the conducted electrical signal is transduced to amechanical response. To distinguish between these twopossibilities, we applied 100 �M Ni2� focally via micropi-pette 1,000 �m upstream from the stimulation site (Fig. 1A).This treatment blocked the dilation at the site at which it wasapplied (Fig. 5F). Comparison of the vasomotor response inthe setting of focal (Fig. 5F) and global (Fig. 5, D and E)Cav3 channel blockade suggests that opening of these Ca2�
channels mediates the coupling between activation of Nav
channels and the vasodilator response along the vessellength, but not the conduction process itself.
The Ni2� and amiloride sensitivity of the response to electricalstimulation offers pharmacological evidence for involvement ofthe Cav3.2 channel in the conducted vasodilation. Therefore, thepharmacology was extended by examination of the behavior of�13.2�/� mice (10). In �13.2�/� mice, the conducted vasodilatorresponse induced by electrical stimulation was severely atten-uated, although the magnitude of the local vasoconstrictionwas similar to that observed in wild-type mice: �51.6 4.3and �47.3 1.9%, respectively (Fig. 6A). As might beexpected, 10 �M Ni2� did not cause significant vasoconstric-tion in arterioles from �13.2�/� mice (Fig. 6B). Moreover,resting arteriolar tone and blood pressure were higher inknockout than in wild-type mice (Fig. 6, C and D), indicatingthat Cav3.2 channels play a role in the tonic control of periph-eral resistance.
Fig. 6. Cav3.2 channel-knockout (�13.2�/�) mice show an impaired conducted vasodilation and elevated blood pressure. A: time course of electrically inducedconducted vasodilation in wild-type and �13.2�/� mice. Response to electrical stimulation was evaluated at the stimulation site (local) and 500, 1,000, and 2,000�m upstream. B: effect of topical application of 10 �M Ni2� on arteriolar resting diameter of �13.2�/� mice. C: spontaneous myogenic vasoconstrictor tone inwild-type and �13.2�/� mice expressed as percentage of maximum diameter. D and E: tail-cuff systolic blood pressure (SBP) and heart rate, respectively. Slowerheart rate in �13.2�/� mice supports the idea that Cav3.2 channels participate in generation of pacemaker potential in the heart (69). Numbers within barsrepresent n. Values are means SE. Horizontal bars represent stimulation period. *P � 0.05 vs. wild-type (by 2-way ANOVA). †P � 0.05 vs. wild-type (byunpaired Student’s t-test).
H1378 VOLTAGE-DEPENDENT ION CHANNELS AND CONDUCTED DILATION
AJP-Heart Circ Physiol • VOL 293 • SEPTEMBER 2007 • www.ajpheart.org
on July 30, 2008 ajpheart.physiology.org
Dow
nloaded from

Activation of NO Production and K� Channels
We hypothesize that an Nav channel-mediated depolariza-tion triggers influx of Ca2� into the endothelial cells byopening Cav3.2 channels, thereby activating Ca2�-dependent,endothelium-mediated dilator mechanisms. NO is a criticalendothelial cell Ca2�-dependent vasodilator, and we used theNO synthase (NOS) antagonist L-NNA and eNOS�/� mice toassess the participation of NO in the response to electricalstimulation. As anticipated, topical application of 100 �ML-NNA caused a reduction of resting arteriolar diameter (Table1). In addition, this treatment enhanced the local vasoconstric-tion and reduced the conducted vasodilation induced by elec-trical stimulation (Fig. 7A), indicating that the endothelial cellCa2� signal initiated by electrical stimulation leads to NOproduction. A similar reduction in eNOS�/� mice (Fig. 7B)strongly supports an endothelial origin for the NO released.
Because NO itself does not evoke conducted responses (14)and because Ca2� channels participate, it seems likely that theelectrically induced, NO-dependent vasodilation representsCa2�-mediated activation of eNOS along the vessel length.
Blockade of NO production reduced but did not abolish theelectrically induced conducted vasodilation. We thereforetested the participation of K� channels in this response. TEA(1–10 mM), Ba2� (50 �M), and glibenclamide (1 �M) reducedbaseline diameter (Table 1). TEA enhanced the local vasocon-striction and inhibited the conducted vasodilation in a concen-tration-dependent manner (Fig. 8A), suggesting that the KCa
channel is involved in the response to electrical stimulation.Neither Ba2� nor glibenclamide affected the electrically in-duced vasomotor responses (Fig. 8B). Furthermore, focal ap-plication via micropipette of the KCa channel blocker ChTX (1�M) 1,000 �m upstream from the stimulation site inhibited the
Fig. 7. Activation of endothelial nitric oxide synthase (eNOS) participates in electrically induced conducted vasodilation. A: time course of electrically inducedconducted vasodilation in control conditions and after treatment with 100 �M NG-nitro-L-arginine (L-NNA). B: conducted vasodilator response elicited byelectrical stimulation in wild-type and eNOS�/� mice. Response to electrical stimulation was evaluated at the stimulation site (local) and 500, 1,000, and 2,000�m upstream. Values are means SE. Horizontal bars indicate stimulation period. *P � 0.05 vs. control or wild-type (by 2-way ANOVA).
Fig. 8. Opening of Ca2�-activated K� channels contributes toelectrically induced conducted vasodilation. A: peak of vaso-motor response to electrical stimulation in control conditionsand during superfusion with 1–10 mM tetraethylammonium(TEA). B: peak of vasomotor response induced by electricalstimulation in control conditions and during superfusion with1 �M glibenclamide (Glib) or 50 �M Ba2�. Because neitherglibenclamide nor Ba2� altered local vasoconstriction or con-ducted vasodilation, involvement of ATP-sensitive K� chan-nels or inward rectifier K� channels in the response wasexcluded. C: focal application of 1 �M charybdotoxin (ChTX)at the conduction pathway (1,000 �m upstream from the localsite; see Fig. 1A) of the electrically induced vasodilation.Local, 500 �m, 1,000 �m, and 2,000 �m denote stimulation(local) and conducted (500, 1,000, and 2,000 �m) sites, whereresponse to electrical stimulation was evaluated. Values aremeans SE. Horizontal bars indicate stimulation period.*P � 0.05 vs. control (by 2-way ANOVA). †P � 0.05 vs.control (by 1-way ANOVA plus Newman-Keuls post hoctest).
H1379VOLTAGE-DEPENDENT ION CHANNELS AND CONDUCTED DILATION
AJP-Heart Circ Physiol • VOL 293 • SEPTEMBER 2007 • www.ajpheart.org
on July 30, 2008 ajpheart.physiology.org
Dow
nloaded from

vasodilator response at the site of ChTX application but not thepropagation of the electrically induced vasodilation to moredistant vessel segments (Fig. 8C).
The activation of eNOS (Fig. 7) strongly supports the ideathat the Cav3.2 channel-dependent conducted vasodilation wasactivated in the endothelial cells. However, we note that theCav3.2 channels could be functionally coupled to a large-conductance KCa (BKCa) channel in the smooth muscle cells(10). It is known that the �1-subunit of the KCa channel isessential for effective coupling of Ca2� to the BKCa channel insmooth muscle cells (4, 42); we therefore used �1
�/� mice (4)to evaluate the possible direct activation of smooth muscleBKCa channels by Cav3.2 channels. Deletion of the �1-subunitdid not affect the electrically induced conducted vasodilation(Fig. 9), which further supports an endothelial origin for theCav3.2 channel-dependent vasodilator signal.
DISCUSSION
Blood vessels are complex, multicellular structures in whichcell and vessel functions must be coordinated. Functionalcoupling within the vascular network is manifested by conduc-tion of vasomotor signals along the vessel length (27, 48, 49),and electrotonic spread of changes in membrane potential givesrise to many conducted vasomotor responses (27, 50, 62). Ithas also been observed that conduction may extend over longerdistances than can be explained by passive electrotonic currentspread (11, 12, 16, 20, 37); thus some active process must beinvoked. Our results show that depolarizing electrical stimuliapplied to a short arteriolar segment activate the vessel’scontractile machinery at the site of stimulation, as would beexpected (17, 20). In addition, activation of a nondecremental,nerve-independent conducted vasodilation propagates for re-markably long distances through the endothelium (Figs. 1 and2). The behavior of this conducted response is not consistentwith simple electrotonic conduction along the vessel axis;rather, the data argue that electrical activation of an endothelialNav channel initiates a depolarizing signal that spreads in aregenerative manner along the vessel length (Figs. 1–8). Fur-thermore, the propagating depolarization appears to be coupledto activation of Cav3.2 channel-dependent signaling pathwaysat distant sites (Figs. 5 and 6) that are responsible for thedilation. Among the Ca2�-dependent pathways linking open-ing of the Ca2� channel to dilation is activation of eNOS andthe KCa channel (Figs. 7, 8, and 10). This linkage is indicatedby the finding that damage of the endothelium or inhibition ofthe Nav channel disrupted the regenerative component of the
electrically induced conducted vasodilation (Fig. 2), whereasblockade of Cav3.2 channels, eNOS, or the KCa channel re-duced the magnitude of the conducted dilation without affect-ing the propagation of the response (Figs. 5–8).
Although the pharmacology implicating a role for Nav chan-nels is provocative, the uncertainty in specificity and access ofthe activators and inhibitors limits the interpretation of thedata. The propositions advanced here must ultimately be testedwith appropriate knockout animals and patch-clamp methodsto define the electrical signature of the channels involved. Thisrepresents a serious technical challenge, however, since avail-able data indicate that the microvessels are quite different fromthe conduit vessels, and our findings indicate that understand-ing this system will ultimately require investigation of intactmicrovessels in vivo.
The reduced magnitude of the conducted vasodilation ob-served after buffering the endothelial cell Ca2� (Fig. 5), as wellas the behavior of arterioles from eNOS-deficient mice (Fig. 7),supports the idea that there is coupling between Cav3.2 chan-nels and Ca2�-dependent endothelial cell function in thisresponse. However, in our experiments, the KCa channel mayhave been activated indirectly in the smooth muscle cells,rather than in the endothelial cells by an NO-independentmediator such as the endothelium-derived hyperpolarizing fac-tor (EDHF) (22, 51). The indirect activation of the KCa channelby an EDHF would be further supported by the reported lackof functional myoendothelial gap junctions in cremaster mi-crovessels (52), and this possibility demands further explora-tion, including more detailed channel analysis in microvesselswith selective study of smooth muscle and endothelial cellssingly and in situ (31).
Although our data strongly support an endothelial origin ofthe Cav3.2 channel-dependent conducted vasodilation, it is inprinciple possible that the Ca2� channel-dependent responsewas triggered in the smooth muscle cells. Recently, Chen et al.(10) proposed that the vasodilator response to ACh in thecoronary artery relies on Cav3.2 channels functionally coupledto BKCa channels in the smooth muscle cells. However, thispossibility is unlikely in our system, inasmuch as we found thatthe electrically induced conducted vasodilation is not altered inanimals in which the �1-subunit of the KCa channel has beendeleted (4) (Fig. 9). Although this subunit plays a critical rolein the coupling of Ca2� to the BKCa channel in smooth musclecells (4, 42), it does not seem to be required for the directactivation of this channel by NO (1) or another endothelium-derived relaxing factor such as EDHF (8).
Fig. 9. Cav3.2 channels do not activate large-conductance Ca2�-activated K� (BKCa) channels directly in smooth muscle cells. Time course of electricallyinduced conducted vasodilation in wild-type and �1-subunit-knockout (�1
�/�) mice is shown. Response to electrical stimulation was evaluated at the stimulationsite (local) and 500, 1,000, and 2,000 �m upstream. The �1-subunit of the BKCa channel is essential for BKCa channel activation by Ca2� and is only expressedin smooth muscle cells (4, 41, 42). Deletion of the �1-subunit did not change the vasomotor response induced by electrical stimulation, arguing against a directfunctional coupling between Cav3.2 and BKCa channels in smooth muscle cells. Values are means SE. Horizontal bars indicate stimulation period.
H1380 VOLTAGE-DEPENDENT ION CHANNELS AND CONDUCTED DILATION
AJP-Heart Circ Physiol • VOL 293 • SEPTEMBER 2007 • www.ajpheart.org
on July 30, 2008 ajpheart.physiology.org
Dow
nloaded from
Deletion of Cav3.2 channels caused an increase in arte-riolar vasomotor tone and blood pressure (Fig. 6), observa-tions that highlight the critical importance of conductedvasomotor responses and the likely involvement of Cav3.2channels in the tonic control of peripheral resistance. Theconducted vasodilation was not eliminated in �13.2�/�
mice, although it was eliminated with a high concentrationof Ni2� (Fig. 5F), which nonselectively blocks Cav3 chan-nels. This might suggest that other channels, such as Cav3.1,which have been detected in the endothelium (61, 63, 70),are also involved in the vasodilator response. Interestingly,it has been observed that in myoblasts from �13.2�/� micethere is compensation of function by upregulation of Cav3.1channels (10), which may have also occurred in the endo-thelial cells of the cremaster arterioles, masking the effect ofthe deletion of Cav3.2 channels.
To summarize, we report here several lines of evidencethat support the conclusion that there is a rapid, conductedvasodilator signal that propagates along the endotheliumover thousands of micrometers and that the propagation isnot blocked by any of several K� or Ca2� channel blockers.We present pharmacological evidence for the involvementof one or more Nav channel isoforms in electrically induceddilations of arterioles. Furthermore, we advance the inter-pretation of the data that activation of the Nav channel leadsto a regenerative, depolarizing current that propagates alongthe vessel axis through the endothelium. The data supportthe proposition that the spreading depolarization is function-
ally coupled to eNOS and the KCa channel via activation ofCav3.2 channels. We note that an agonist such as ACh canlikely trigger the same mechanism, since ACh-induced Ca2�
entry in freshly isolated endothelial cells is sensitive tomembrane potential in a manner that cannot be simplyexplained by changes in the electrical driving force for Ca2�
(60). In addition, after buffering the endothelial cell Ca2�
with BAPTA, ACh evokes membrane depolarization (40)and an endothelium-dependent vasoconstriction (Figs. 2A,4B, and 5B). Moreover, the response to electrical stimula-tion (Fig. 7) and the ACh-induced conducted vasodilationare coupled to Ca2�-dependent NO production (5).
Our data are thus consistent with the discovery of a novel,vasodilator mechanism that relies on interplay between twovoltage-sensitive ion channels in the endothelium (Fig. 10).Expression of functional voltage-dependent ion channels inthe endothelial cells has enormous implications for cardio-vascular homeostasis and offers new opportunities for un-derstanding the key role of the endothelium in vascularfunction. These findings make it imperative that a greaterunderstanding of the subtype distribution and function ofvascular Nav and Cav3 channels be developed, inasmuch asthis pathway is likely to play an essential role in the tonic,endothelial control of arterial blood pressure, which mayprovide clues to the design of new therapeutic strategies forthe treatment of cardiovascular diseases such as hyperten-sion.
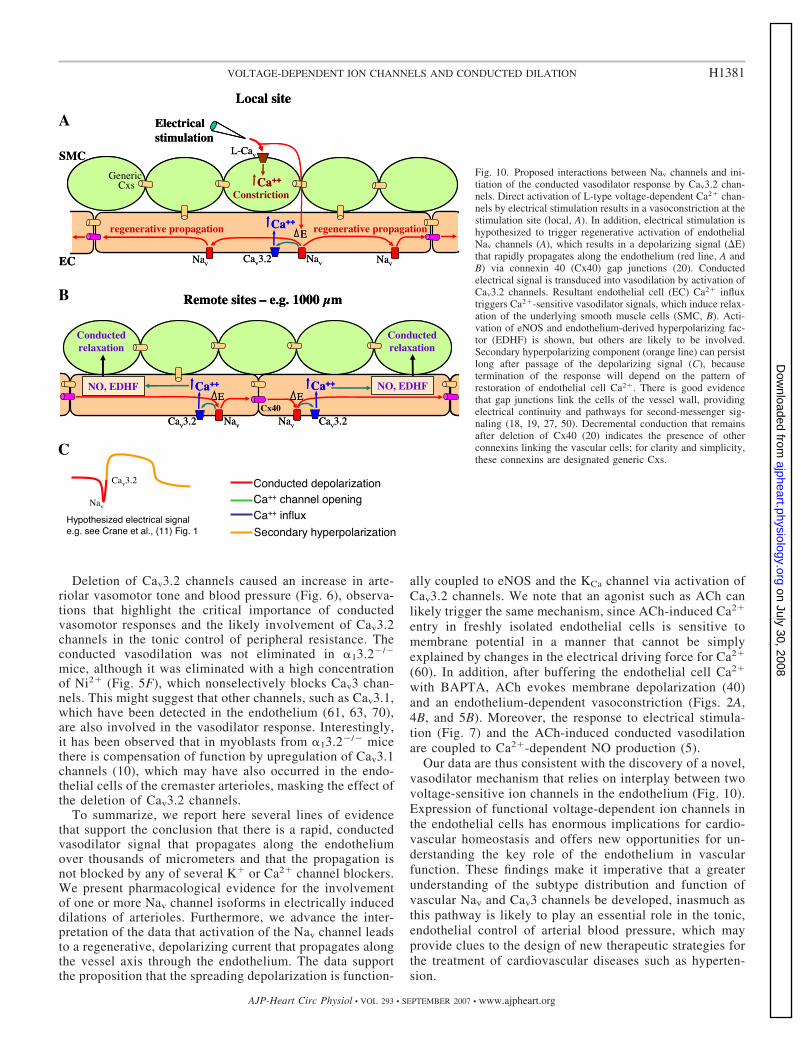
Fig. 10. Proposed interactions between Nav channels and ini-tiation of the conducted vasodilator response by Cav3.2 chan-nels. Direct activation of L-type voltage-dependent Ca2� chan-nels by electrical stimulation results in a vasoconstriction at thestimulation site (local, A). In addition, electrical stimulation ishypothesized to trigger regenerative activation of endothelialNav channels (A), which results in a depolarizing signal (�E)that rapidly propagates along the endothelium (red line, A andB) via connexin 40 (Cx40) gap junctions (20). Conductedelectrical signal is transduced into vasodilation by activation ofCav3.2 channels. Resultant endothelial cell (EC) Ca2� influxtriggers Ca2�-sensitive vasodilator signals, which induce relax-ation of the underlying smooth muscle cells (SMC, B). Acti-vation of eNOS and endothelium-derived hyperpolarizing fac-tor (EDHF) is shown, but others are likely to be involved.Secondary hyperpolarizing component (orange line) can persistlong after passage of the depolarizing signal (C), becausetermination of the response will depend on the pattern ofrestoration of endothelial cell Ca2�. There is good evidencethat gap junctions link the cells of the vessel wall, providingelectrical continuity and pathways for second-messenger sig-naling (18, 19, 27, 50). Decremental conduction that remainsafter deletion of Cx40 (20) indicates the presence of otherconnexins linking the vascular cells; for clarity and simplicity,these connexins are designated generic Cxs.
H1381VOLTAGE-DEPENDENT ION CHANNELS AND CONDUCTED DILATION
AJP-Heart Circ Physiol • VOL 293 • SEPTEMBER 2007 • www.ajpheart.org
on July 30, 2008 ajpheart.physiology.org
Dow
nloaded from

ACKNOWLEDGMENTS
We thank Dr. Victor E. Laubach for generously supplying the eNOS�/�
mice and Dr. Richard W. Aldrich for sharing the �1-subunit-knockout mice.All the tissue embedment was performed by the Research Histology Core atthe University of Virginia.
GRANTS
This work was supported by National Heart, Lung, and Blood InstituteGrants HL-53318 and HL-72864 (to B. R. Duling), American Heart Associ-ation Postdoctoral Fellowship Grant 0325730U (to X. F. Figueroa), a grantfrom the Vicerrectorıa Adjunta de Investigacion y Doctorado de la PontificiaUniversidad Catolica de Chile (to X. F. Figueroa), and Fondo Nacional deDesarrollo Cientıfico y Tecnologico Grant 11060289 (to X. F. Figueroa). K. P.Campbell is an investigator of the Howard Hughes Medical Institute and issupported by the Muscular Dystrophy Association.
REFERENCES
1. Abderrahmane A, Salvail D, Dumoulin M, Garon J, Cadieux A,Rousseau E. Direct activation of KCa channel in airway smooth muscle bynitric oxide: involvement of a nitrothiosylation mechanism? Am J RespirCell Mol Biol 19: 485–497, 1998.
2. Bossu JL, Elhamdani A, Feltz A. Voltage-dependent calcium entry inconfluent bovine capillary endothelial cells. FEBS Lett 299: 239–242,1992.
3. Bossu JL, Feltz A, Rodeau JL, Tanzi F. Voltage-dependent transientcalcium currents in freshly dissociated capillary endothelial cells. FEBSLett 255: 377–380, 1989.
4. Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW,Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the �1-subunit of the calcium-activated potassium channel. Nature 407: 870–876, 2000.
5. Budel S, Bartlett IS, Segal SS. Homocellular conduction along endothe-lium and smooth muscle of arterioles in hamster cheek pouch: unmaskingan NO wave. Circ Res 93: 61–68, 2003.
6. Budel S, Schuster A, Stergiopoulos N, Meister JJ, Beny JL. Role ofsmooth muscle cells on endothelial cell cytosolic free calcium in porcinecoronary arteries. Am J Physiol Heart Circ Physiol 281: H1156–H1162,2001.
7. Buga GM, Ignarro LJ. Electrical-field stimulation causes endothelium-dependent and nitric oxide-mediated relaxation of pulmonary artery. Am JPhysiol Heart Circ Physiol 262: H973–H979, 1992.
8. Campbell WB, Gauthier KM. What is new in endothelium-derivedhyperpolarizing factors? Curr Opin Nephrol Hypertens 11: 177–183,2002.
9. Cestele S, Catterall WA. Molecular mechanisms of neurotoxin action onvoltage-gated sodium channels. Biochimie 82: 883–892, 2000.
10. Chen CC, Lamping KG, Nuno DW, Barresi R, Prouty SJ, Lavoie JL,Cribbs LL, England SK, Sigmund CD, Weiss RM, Williamson RA,Hill JA, Campbell KP. Abnormal coronary function in mice deficient in�1H T-type Ca2� channels. Science 302: 1416–1418, 2003.
11. Crane GJ, Neild TO, Segal SS. Contribution of active membrane pro-cesses to conducted hyperpolarization in arterioles of hamster cheekpouch. Microcirculation 11: 425–433, 2004.
12. De Wit C, Roos F, Bolz SS, Kirchhoff S, Kruger O, Willecke K, PohlU. Impaired conduction of vasodilation along arterioles in connexin40-deficient mice. Circ Res 86: 649–655, 2000.
13. Dora KA, Doyle MP, Duling BR. Elevation of intracellular calcium insmooth muscle causes endothelial cell generation of NO in arterioles. ProcNatl Acad Sci USA 94: 6529–6534, 1997.
14. Doyle MP, Duling BR. Acetylcholine induces conducted vasodilation bynitric oxide-dependent and -independent mechanisms. Am J Physiol HeartCirc Physiol 272: H1364–H1371, 1997.
15. Ellerkmann RK, Remy S, Chen J, Sochivko D, Elger CE, Urban BW,Becker A, Beck H. Molecular and functional changes in voltage-depen-dent Na� channels following pilocarpine-induced status epilepticus in ratdentate granule cells. Neuroscience 119: 323–333, 2003.
16. Emerson GG, Neild TO, Segal SS. Conduction of hyperpolarizationalong hamster feed arteries: augmentation by acetylcholine. Am J PhysiolHeart Circ Physiol 283: H102–H109, 2002.
17. Emerson GG, Segal SS. Electrical activation of endothelium evokesvasodilation and hyperpolarization along hamster feed arteries. Am JPhysiol Heart Circ Physiol 280: H160–H167, 2001.
18. Figueroa XF, Isakson BE, Duling BR. Connexins: gaps in our knowl-edge of vascular function. Physiology Bethesda 19: 277–284, 2004.
19. Figueroa XF, Isakson BE, Duling BR. Vascular gap junctions in hyper-tension. Hypertension 48: 804–811, 2006.
20. Figueroa XF, Paul DL, Simon AM, Goodenough DA, Day KH, DamonDN, Duling BR. Central role of connexin40 in the propagation ofelectrically activated vasodilation in mouse cremasteric arterioles in vivo.Circ Res 92: 793–800, 2003.
21. Fisher AB, Al Mehdi AB, Manevich Y. Shear stress and endothelial cellactivation. Crit Care Med 30: S192–S197, 2002.
22. Fleming I. Cytochrome P450 epoxygenases as EDHF synthase(s). Phar-macol Res 49: 525–533, 2004.
23. Frank GW, Bevan JA. Electrical stimulation causes endothelium-depen-dent relaxation in lung vessels. Am J Physiol Heart Circ Physiol 244:H793–H798, 1983.
24. Geary GG, Maeda G, Gonzalez RR Jr. Endothelium-dependent vascularsmooth muscle relaxation activated by electrical field stimulation. ActaPhysiol Scand 160: 219–228, 1997.
25. Gordienko DV, Tsukahara H. Tetrodotoxin-blockable depolarization-activated Na� currents in a cultured endothelial cell line derived from ratinterlobar artery and human umbilical vein. Pflugers Arch 428: 91–93,1994.
26. Gosling M, Harley SL, Turner RJ, Carey N, Powell JT. Humansaphenous vein endothelial cells express a tetrodotoxin-resistant, voltage-gated sodium current. J Biol Chem 273: 21084–21090, 1998.
27. Gustafsson F, Holstein-Rathlou N. Conducted vasomotor responses inarterioles: characteristics, mechanisms and physiological significance.Acta Physiol Scand 167: 11–21, 1999.
28. Hansen PB, Jensen BL, Andreasen D, Skott O. Differential expressionof T- and L-type voltage-dependent calcium channels in renal resistancevessels. Circ Res 89: 630–638, 2001.
29. Hayashi K, Ozawa Y, Wakino S, Kanda T, Homma K, Takamatsu I,Tatematsu S, Saruta T. Cellular mechanism for mibefradil-inducedvasodilation of renal microcirculation: studies in the isolated perfusedhydronephrotic kidney. J Cardiovasc Pharmacol 42: 697–702, 2003.
30. Hermsmeyer K, Miyagawa K. Protein kinase C mechanism enhancesvascular muscle relaxation by the Ca2� antagonist, Ro 40-5967. J VascRes 33: 71–77, 1996.
31. Jackson WF. Potassium channels in the peripheral microcirculation.Microcirculation 12: 113–127, 2005.
32. Lacinova L, Klugbauer N, Hofmann F. Low voltage activated calciumchannels: from genes to function. Gen Physiol Biophys 19: 121–136, 2000.
33. Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E. Nickel block of threecloned T-type calcium channels: low concentrations selectively block �1H.Biophys J 77: 3034–3042, 1999.
34. Lew MJ, Rivers RJ, Duling BR. Arteriolar smooth muscle responses aremodulated by an intramural diffusion barrier. Am J Physiol Heart CircPhysiol 257: H10–H16, 1989.
35. Manabe I, Owens GK. Recruitment of serum response factor and hyper-acetylation of histones at smooth muscle-specific regulatory regions dur-ing differentiation of a novel P19-derived in vitro smooth muscle differ-entiation system. Circ Res 88: 1127–1134, 2001.
36. Moosmang S, Haider N, Bruderl B, Welling A, Hofmann F. Antihy-pertensive effects of the putative T-type calcium channel antagonistmibefradil are mediated by the L-type calcium channel Cav1.2. Circ Res98: 105–110, 2006.
37. Neild TO, Crane GJ. Cellular coupling and conducted vasomotor re-sponses. Clin Exp Pharmacol Physiol 29: 626–629, 2002.
38. Nilius B, Droogmans G. Ion channels and their functional role in vascularendothelium. Physiol Rev 81: 1415–1459, 2001.
39. Nilius B, Prenen J, Kamouchi M, Viana F, Voets T, Droogmans G.Inhibition by mibefradil, a novel calcium channel antagonist, of Ca2�- andvolume-activated Cl� channels in macrovascular endothelial cells. Br JPharmacol 121: 547–555, 1997.
40. Ohashi M, Satoh K, Itoh T. Acetylcholine-induced membrane potentialchanges in endothelial cells of rabbit aortic valve. Br J Pharmacol 126:19–26, 1999.
41. Papassotiriou J, Kohler R, Prenen J, Krause H, Akbar M, EggermontJ, Paul M, Distler A, Nilius B, Hoyer J. Endothelial K� channel lacksthe Ca2� sensitivity-regulating �-subunit. FASEB J 14: 885–894, 2000.
42. Patterson AJ, Henrie-Olson J, Brenner R. Vasoregulation at the mo-lecular level: a role for the �1-subunit of the calcium-activated potassium(BK) channel. Trends Cardiovasc Med 12: 78–82, 2002.
H1382 VOLTAGE-DEPENDENT ION CHANNELS AND CONDUCTED DILATION
AJP-Heart Circ Physiol • VOL 293 • SEPTEMBER 2007 • www.ajpheart.org
on July 30, 2008 ajpheart.physiology.org
Dow
nloaded from

43. Potocnik SJ, Murphy TV, Kotecha N, Hill MA. Effects of mibefradiland nifedipine on arteriolar myogenic responsiveness and intracellularCa2�. Br J Pharmacol 131: 1065–1072, 2000.
44. Prorock AJ, Hafezi-Moghadam A, Laubach VE, Liao JK, Ley K.Vascular protection by estrogen in ischemia-reperfusion injury requiresendothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol 284:H133–H140, 2003.
45. Rivers RJ, Duling BR. Arteriolar endothelial cell barrier separates twopopulations of muscarinic receptors. Am J Physiol Heart Circ Physiol 262:H1311–H1315, 1992.
46. Scholz A, Vogel W. Tetrodotoxin-resistant action potentials in dorsal rootganglion neurons are blocked by local anesthetics. Pain 89: 47–52, 2000.
47. Segal SS. Integration of blood flow control to skeletal muscle: key role offeed arteries. Acta Physiol Scand 168: 511–518, 2000.
48. Segal SS, Damon DN, Duling BR. Propagation of vasomotor responsescoordinates arteriolar resistances. Am J Physiol Heart Circ Physiol 256:H832–H837, 1989.
49. Segal SS, Duling BR. Communication between feed arteries and mi-crovessels in hamster striated muscle: segmental vascular responses arefunctionally coordinated. Circ Res 59: 283–290, 1986.
50. Segal SS, Duling BR. Conduction of vasomotor responses in arterioles: arole for cell-to-cell coupling. Am J Physiol Heart Circ Physiol 256:H838–H845, 1989.
51. Shimokawa H, Morikawa K. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J Mol Cell Cardiol39: 725–732, 2005.
52. Siegl D, Koeppen M, Wolfle SE, Pohl U, de Wit C. Myoendothelialcoupling is not prominent in arterioles within the mouse cremaster micro-circulation in vivo. Circ Res 97: 781–788, 2005.
53. Sullivan JC, Davison CA. Effect of age on electrical field stimulation(EFS)-induced endothelium-dependent vasodilation in male and femalerats. Cardiovasc Res 50: 137–144, 2001.
54. Tang CM, Presser F, Morad M. Amiloride selectively blocks the lowthreshold (T) calcium channel. Science 240: 213–215, 1988.
55. Traub O, Ishida T, Ishida M, Tupper JC, Berk BC. Shear stress-mediated extracellular signal-regulated kinase activation is regulated bysodium in endothelial cells. Potential role for a voltage-dependent sodiumchannel. J Biol Chem 274: 20144–20150, 1999.
56. Vanbavel E, Sorop O, Andreasen D, Pfaffendorf M, Jensen BL.Role of T-type calcium channels in myogenic tone of skeletal muscle
resistance arteries. Am J Physiol Heart Circ Physiol 283: H2239 –H2243, 2002.
57. Vinet R, Vargas FF. L- and T-type voltage-gated Ca2� currents inadrenal medulla endothelial cells. Am J Physiol Heart Circ Physiol 276:H1313–H1322, 1999.
58. Walsh KB, Wolf MB, Fan J. Voltage-gated sodium channels in cardiacmicrovascular endothelial cells. Am J Physiol Heart Circ Physiol 274:H506–H512, 1998.
59. Wang SY, Wang GK. Voltage-gated sodium channels as primary targetsof diverse lipid-soluble neurotoxins. Cell Signal 15: 151–159, 2003.
60. Wang X, van Breemen C. Depolarization-mediated inhibition of Ca2�
entry in endothelial cells. Am J Physiol Heart Circ Physiol 277: H1498–H1504, 1999.
61. Wei Z, Manevich Y, Al Mehdi AB, Chatterjee S, Fisher AB. Ca2� fluxthrough voltage-gated channels with flow cessation in pulmonary micro-vascular endothelial cells. Microcirculation 11: 517–526, 2004.
62. Welsh DG, Segal SS. Endothelial and smooth muscle cell conduction inarterioles controlling blood flow. Am J Physiol Heart Circ Physiol 274:H178–H186, 1998.
63. Wu S, Haynes J Jr, Taylor JT, Obiako BO, Stubbs JR, Li M, StevensT. Cav3.1 (�1G) T-type Ca2� channels mediate vaso-occlusion of sicklederythrocytes in lung microcirculation. Circ Res 93: 346–353, 2003.
64. Wu S, Zhang M, Vest PA, Bhattacharjee A, Liu L, Li M. A mibefradilmetabolite is a potent intracellular blocker of L-type Ca2� currents inpancreatic �-cells. J Pharmacol Exp Ther 292: 939–943, 2000.
65. Xia J, Duling BR. Electromechanical coupling and the conducted vaso-motor response. Am J Physiol Heart Circ Physiol 269: H2022–H2030,1995.
66. Yakubu MA, Leffler CW. L-type voltage-dependent Ca2� channels incerebral microvascular endothelial cells and ET-1 biosynthesis. Am JPhysiol Cell Physiol 283: C1687–C1695, 2002.
67. Yashiro Y, Duling BR. Integrated Ca2� signaling between smoothmuscle and endothelium of resistance vessels. Circ Res 87: 1048–1054,2000.
68. Yunker AM. Modulation and pharmacology of low voltage-activated(“T-type”) calcium channels. J Bioenerg Biomembr 35: 577–598, 2003.
69. Yunker AM, McEnery MW. Low-voltage-activated (“T-type”) calciumchannels in review. J Bioenerg Biomembr 35: 533–575, 2003.
70. Zhou C, Wu S. T-type calcium channels in pulmonary vascularendothelium. Microcirculation 13: 645– 656, 2006.
H1383VOLTAGE-DEPENDENT ION CHANNELS AND CONDUCTED DILATION
AJP-Heart Circ Physiol • VOL 293 • SEPTEMBER 2007 • www.ajpheart.org
on July 30, 2008 ajpheart.physiology.org
Dow
nloaded from


















