
WT1 splice-site mutations are rarely associated with primary steroid-resistant focal and segmental...
5
Kidney International, Vol. 57 (2000), pp. 1868–1872 WT1 splice-site mutations are rarely associated with primary steroid-resistant focal and segmental glomerulosclerosis ERICK DENAMUR,NATHALIE BOCQUET,VERONIQUE BAUDOUIN,FRANC ¸ IS DA SILVA, REINER VEITIA,MICHEL PEUCHMAUR,JACQUES ELION,MARIE CLAIRE GUBLER, MARC FELLOUS,PATRICK NIAUDET, and CHANTAL LOIRAT INSERM U458, Ho ˆ pital Robert Debre ´, and Laboratoire d’Anatomopathologic and EA 3102, Paris, France associated. The identification of such WT1 mutations has prac- WT1 splice-site mutations are rarely associated with primary tical implications for the management of these patients. steroid-resistant focal and segmental glomerulosclerosis. Background. Donor splice-site de novo heterozygous muta- tions in intron 9 of the Wilms’ tumor gene (WT1) have been reported in Frasier syndrome, which is defined by the associa- Primary steroid-resistant nephrotic syndrome with fo- tion of focal and segmental glomerulosclerosis (FSGS), male cal and segmental glomerulosclerosis (FSGS) groups het- pseudohermaphroditism, and gonadoblastoma. These splice- erogenous conditions despite uniform renal histologic site mutations alter the WT1 alternative splicing leading to two WT1 isoforms, with (1) or without (2) three amino acids, findings. Besides the most common form considered as lysine-threonine-serine (KTS), between zinc fingers 3 and 4. secondary to the production of a proteinuric factor by T The aim of this work was to investigate the possibility that lymphocytes [1], familial forms with recessive autosomal some cases of primary steroid-resistant nephrotic syndrome transmission have been shown to be linked to chromo- associated with FSGS may be caused by WT1 splice-site muta- some 1q25-q31 [2], while at least three different genes tions. Methods. We analyzed WT1 exons 8 and 9 and the sur- have been found so far to cause autosomal dominant rounding exon/intron boundary DNA sequences in 37 children FSGS [3, 4]. with nonfamilial primary steroid-resistant nephrotic syndrome. It has been recently reported that Frasier syndrome, Semiquantitative reverse transcription-polymerase chain reac- a rare disease defined by FSGS associated with male tion (RT-PCR) was used to determine the relative ratio of pseudohermaphroditism and the frequent development 1KTS/2KTS transcripts from immortalized lymphocyte RNA. Results. One boy with FSGS and associated pathologies (di- of gonadoblastoma, is caused by de novo heterozygous aphragmatic hernia, proximal hypospadias, and unilateral tes- mutations in the donor splice-site in intron 9 (1228 12 ticular ectopia) was found to carry the heterozygous 1228 14 T→C, 1228 14C→T, 1228 15G→A or T, or 1228 16 C→T splice-site mutation. RT-PCR quantitation of the 1KTS/ T→A mutation) of the Wilms’ tumor gene (WT1) [5–7]. 2KTS transcripts from immortalized lymphocyte RNA of this WT1 is composed of 10 exons and encodes a zinc-finger patient showed a diminution of the 1KTS/2KTS isoform ratio (0.43), which is identical to that reported in patients with Fra- protein. During vertebrate development, WT1 is ex- sier syndrome. Using the same approach, healthy control sub- pressed in the kidneys, gonads, and spleen and at the jects have 1KTS/2KTS ratios ranging from 1.50 to 2.00. lining of the abdominal cavity (mesothelium) [8, 9]. Conclusions. This study expands the range of the phenotypic Within the kidney, WT1 is specifically expressed in the presentation of the intron 9 splice-site WT1 mutations and condensed mesenchyme, renal vesicle, and glomerular adds to the already reported heterogeneity of primary steroid- epithelial cells. Two alternative splicings generate four resistant nephrotic syndromes. We suggest that these mutations are not likely to be a common cause of isolated steroid-resistant WT1 isoforms. One of these pathways is conserved nephrotic syndrome, and recommend a WT1 exon 9/intron among vertebrates and uses two splice donor sites in the 9 splice-site study in children with primary steroid-resistant 59 region of intron 9, resulting in the presence (1) or nephrotic syndrome if genital or diaphragmatic anomalies are absence (2) of three amino acids, lysine-threonine-serine (KTS), between zinc fingers 3 and 4 [10]. The ratio of the WT1 isoforms is believed to be stable temporally Key words: focal segmental glomerulosclerosis, splice-site mutations, nephrotic syndrome, Frasier syndrome, Wilms’ tumor gene. and spatially, and whereas 2KTS proteins have a role in the regulation of transcription, 1KTS proteins seem Received for publication June 28, 1999 involved in RNA splicing [11]. In Frasier syndrome, the and in revised form November 23, 1999 Accepted for publication December 18, 1999 1KTS/2KTS isoform ratio is diminished because of the modified balance between isoforms [5–7]. 2000 by the International Society of Nephrology 1868
Transcript of WT1 splice-site mutations are rarely associated with primary steroid-resistant focal and segmental...

Kidney International, Vol. 57 (2000), pp. 1868–1872
WT1 splice-site mutations are rarely associated with primarysteroid-resistant focal and segmental glomerulosclerosis
ERICK DENAMUR, NATHALIE BOCQUET, VERONIQUE BAUDOUIN, FRANCIS DA SILVA,REINER VEITIA, MICHEL PEUCHMAUR, JACQUES ELION, MARIE CLAIRE GUBLER,MARC FELLOUS, PATRICK NIAUDET, and CHANTAL LOIRAT
INSERM U458, Hopital Robert Debre, and Laboratoire d’Anatomopathologic and EA 3102, Paris, France
associated. The identification of such WT1 mutations has prac-WT1 splice-site mutations are rarely associated with primarytical implications for the management of these patients.steroid-resistant focal and segmental glomerulosclerosis.
Background. Donor splice-site de novo heterozygous muta-tions in intron 9 of the Wilms’ tumor gene (WT1) have beenreported in Frasier syndrome, which is defined by the associa-
Primary steroid-resistant nephrotic syndrome with fo-tion of focal and segmental glomerulosclerosis (FSGS), malecal and segmental glomerulosclerosis (FSGS) groups het-pseudohermaphroditism, and gonadoblastoma. These splice-erogenous conditions despite uniform renal histologicsite mutations alter the WT1 alternative splicing leading to two
WT1 isoforms, with (1) or without (2) three amino acids, findings. Besides the most common form considered aslysine-threonine-serine (KTS), between zinc fingers 3 and 4. secondary to the production of a proteinuric factor by TThe aim of this work was to investigate the possibility that
lymphocytes [1], familial forms with recessive autosomalsome cases of primary steroid-resistant nephrotic syndrometransmission have been shown to be linked to chromo-associated with FSGS may be caused by WT1 splice-site muta-some 1q25-q31 [2], while at least three different genestions.
Methods. We analyzed WT1 exons 8 and 9 and the sur- have been found so far to cause autosomal dominantrounding exon/intron boundary DNA sequences in 37 children FSGS [3, 4].with nonfamilial primary steroid-resistant nephrotic syndrome. It has been recently reported that Frasier syndrome,Semiquantitative reverse transcription-polymerase chain reac-
a rare disease defined by FSGS associated with maletion (RT-PCR) was used to determine the relative ratio ofpseudohermaphroditism and the frequent development1KTS/2KTS transcripts from immortalized lymphocyte RNA.
Results. One boy with FSGS and associated pathologies (di- of gonadoblastoma, is caused by de novo heterozygousaphragmatic hernia, proximal hypospadias, and unilateral tes- mutations in the donor splice-site in intron 9 (1228 12ticular ectopia) was found to carry the heterozygous 1228 14 T→C, 1228 14 C→T, 1228 15 G→A or T, or 1228 16C→T splice-site mutation. RT-PCR quantitation of the 1KTS/
T→A mutation) of the Wilms’ tumor gene (WT1) [5–7].2KTS transcripts from immortalized lymphocyte RNA of thisWT1 is composed of 10 exons and encodes a zinc-fingerpatient showed a diminution of the 1KTS/2KTS isoform ratio
(0.43), which is identical to that reported in patients with Fra- protein. During vertebrate development, WT1 is ex-sier syndrome. Using the same approach, healthy control sub- pressed in the kidneys, gonads, and spleen and at thejects have 1KTS/2KTS ratios ranging from 1.50 to 2.00. lining of the abdominal cavity (mesothelium) [8, 9].
Conclusions. This study expands the range of the phenotypicWithin the kidney, WT1 is specifically expressed in thepresentation of the intron 9 splice-site WT1 mutations andcondensed mesenchyme, renal vesicle, and glomerularadds to the already reported heterogeneity of primary steroid-epithelial cells. Two alternative splicings generate fourresistant nephrotic syndromes. We suggest that these mutations
are not likely to be a common cause of isolated steroid-resistant WT1 isoforms. One of these pathways is conservednephrotic syndrome, and recommend a WT1 exon 9/intron among vertebrates and uses two splice donor sites in the9 splice-site study in children with primary steroid-resistant 59 region of intron 9, resulting in the presence (1) ornephrotic syndrome if genital or diaphragmatic anomalies are
absence (2) of three amino acids, lysine-threonine-serine(KTS), between zinc fingers 3 and 4 [10]. The ratio ofthe WT1 isoforms is believed to be stable temporallyKey words: focal segmental glomerulosclerosis, splice-site mutations,
nephrotic syndrome, Frasier syndrome, Wilms’ tumor gene. and spatially, and whereas 2KTS proteins have a rolein the regulation of transcription, 1KTS proteins seemReceived for publication June 28, 1999involved in RNA splicing [11]. In Frasier syndrome, theand in revised form November 23, 1999
Accepted for publication December 18, 1999 1KTS/2KTS isoform ratio is diminished because of themodified balance between isoforms [5–7]. 2000 by the International Society of Nephrology
1868

Denamur et al: WT1 splice-site mutation and FSGS 1869
The aim of this work was to investigate the possibility reached pubertal age had normal pubertal developmentthat some cases of nonfamilial primary steroid-resistant and menarche.nephrotic syndrome associated with FSGS may be caused
DNA sequencingby WT1 splice-site mutations affecting splicing of theKTS motif. Genomic DNA was extracted from white blood cells
according to standard techniques. Polymerase chain re-action (PCR) amplification and sequence analysis ofMETHODSWT1 exons 8 and 9, and surrounding exon/intron bound-
Patients aries were performed as described in Jeanpierre et alThirty-seven children (17 boys and 20 girls) with pri- [12] or Barbaux et al [5].
mary steroid-resistant nephrotic syndrome were studied.Semiquantitative reverse transcription-polymeraseThe mean age at first symptoms was four years and ninechain reactionmonths (from 1 year to 14 years 6 months). No familial
history of renal disease was found in any of the patients. Total RNA was extracted from a lymphoblastoid cellSteroid resistance was defined as the persistence of line of the patient with the WT1 splice-site mutation and
heavy proteinuria ($50 mg/kg/day) and plasma albumin of five control subjects without WT1 mutation. Reverselevel below 25 g/L after one month on 60 mg/m2/day transcription-PCR (RT-PCR) was performed on cDNAprednisone and three intravenous injections of 1000 mg/ as described by Barbaux et al [5], except that the WTRT11.73 m2 methylprednisolone. Eighteen patients subse- primer was 59 fluorescein labeled and that no radioactiv-quently received cyclosporine associated with predni- ity was used. The 113 and 104 bp fragments amplifiedsone during at least four months, and 14 received alkylat- from the 1KTS and 2KTS WT1 alternative transcriptsing agents (chlorambucil in 10, cyclophosphamide in 4, were resolved by capillary electrophoresis on an ABIwith 8 children receiving cyclosporine and alkylating Prism 310 (Perkin Elmer, Norwalk, CT, USA). RT-PCRsagents sequentially). None of these treatments induced were repeated at least three times, and the presenteda significantly beneficial effect. values correspond to the means.
At the last follow-up, nine children had persistent ne-phrotic syndrome, with chronic renal failure in five.Twenty-eight children reached end-stage renal failure RESULTSafter a delay of four years and two months (ranging from Characterization of WT1 exons 8 and 9 and the1 month to 18 years 7 months). Twenty-seven had a surrounding intronic regionskidney transplantation. One had primary nonfunction
Of the 37 patients with primary steroid-resistant ne-of the graft. In two cases, recurrence of the nephroticphrotic syndrome and FSGS, only one patient was foundsyndrome occurred after kidney graft. In the 24 otherto carry an heterozygous WT1 mutation in the exon 9children, no recurrence of the nephrotic syndrome hasand surrounding regions. This mutation located in thebeen observed after a mean follow-up of seven yearsalternative splice site (1228 14 C→T; Fig. 1) has beenand five months (ranging from 30 months to 15 years 5reported in Frasier syndrome [5–7]. The mutation wasmonths).not found in the child’s mother and sister. The fatherRenal biopsy performed within two months after onsetcould not be studied. This boy was born with left dia-(29 cases) or during the one to two following years (8phragmatic hernia and was operated on at the age ofcases) showed minimal change glomerular lesions in 14seven months. He also had proximal hypospadias withcases, FSGS in 20 cases, and isolated diffuse mesangialchordee, operated on at the ages of three and four years,proliferation in 3 cases. Two of the 20 patients with FSGSand right testicular ectopia, operated on at the age ofalso had diffuse mesangial proliferation. Repeated renalnine years. He developed steroid-resistant nephrotic syn-biopsies or histology of nephrectomy specimens showeddrome when 22 months old. Renal biopsy at age 23FSGS in all cases. Five patients had associated patholo-months showed minimal change glomerular lesions.gies. One boy had diaphragmatic hernia, proximal hypo-Cyclosporine treatment had no beneficial effect. Re-spadias with chordee, and unilateral testicular ectopy.peated renal biopsies showed minimal change glomeru-Three other boys had situs inversus, Noonan syndrome,lar lesions at the age of three years five months, andand sensorineural deafness, respectively. One girl hadFSGS at the age of four years nine months (Fig. 2). Themental retardation since infancy. Mitochondrial cytopa-child reached end-stage renal failure at the age of eightthy was not looked for in the two children with sensori-years and was transplanted one year later. A nephrec-neural deafness or mental retardation. Nevertheless, thistomy specimen at the age of eight years nine monthsdiagnosis is unlikely, as no symptoms suggestive of thisshowed diffuse FSGS. No recurrence of the nephroticdiagnosis have appeared in these patients, who are now
23 and 20 years old, respectively. The 15 girls who syndrome had occurred after two years of post-trans-
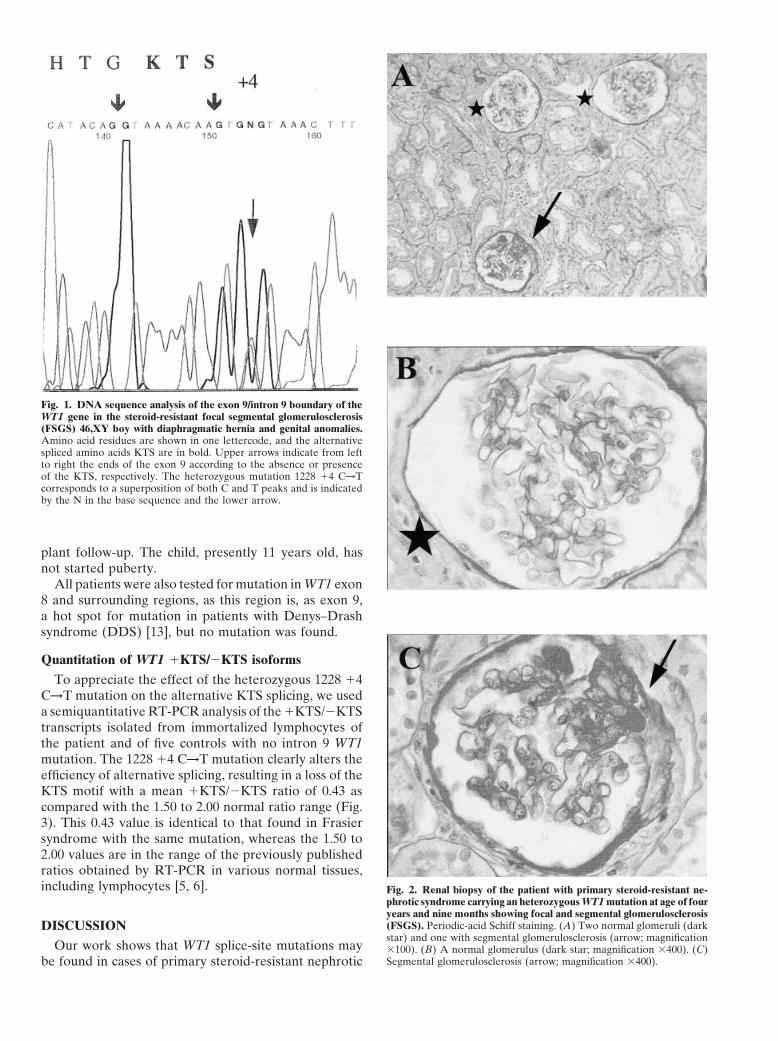
Fig. 1. DNA sequence analysis of the exon 9/intron 9 boundary of theWT1 gene in the steroid-resistant focal segmental glomerulosclerosis(FSGS) 46,XY boy with diaphragmatic hernia and genital anomalies.Amino acid residues are shown in one lettercode, and the alternativespliced amino acids KTS are in bold. Upper arrows indicate from leftto right the ends of the exon 9 according to the absence or presenceof the KTS, respectively. The heterozygous mutation 1228 14 C→Tcorresponds to a superposition of both C and T peaks and is indicatedby the N in the base sequence and the lower arrow.
plant follow-up. The child, presently 11 years old, hasnot started puberty.
All patients were also tested for mutation in WT1 exon8 and surrounding regions, as this region is, as exon 9,a hot spot for mutation in patients with Denys–Drashsyndrome (DDS) [13], but no mutation was found.
Quantitation of WT1 1KTS/2KTS isoforms
To appreciate the effect of the heterozygous 1228 14C→T mutation on the alternative KTS splicing, we useda semiquantitative RT-PCR analysis of the 1KTS/2KTStranscripts isolated from immortalized lymphocytes ofthe patient and of five controls with no intron 9 WT1mutation. The 1228 14 C→T mutation clearly alters theefficiency of alternative splicing, resulting in a loss of theKTS motif with a mean 1KTS/2KTS ratio of 0.43 ascompared with the 1.50 to 2.00 normal ratio range (Fig.3). This 0.43 value is identical to that found in Frasiersyndrome with the same mutation, whereas the 1.50 to2.00 values are in the range of the previously publishedratios obtained by RT-PCR in various normal tissues,including lymphocytes [5, 6]. Fig. 2. Renal biopsy of the patient with primary steroid-resistant ne-
phrotic syndrome carrying an heterozygous WT1 mutation at age of fouryears and nine months showing focal and segmental glomerulosclerosis(FSGS). Periodic-acid Schiff staining. (A) Two normal glomeruli (darkDISCUSSIONstar) and one with segmental glomerulosclerosis (arrow; magnification
Our work shows that WT1 splice-site mutations may 3100). (B) A normal glomerulus (dark star; magnification 3400). (C)Segmental glomerulosclerosis (arrow; magnification 3400).be found in cases of primary steroid-resistant nephrotic

Denamur et al: WT1 splice-site mutation and FSGS 1871
tions have been described in a wide range of diseases,including DDS (patient P18 in [12] and the index casein [17] with the 1228 15 mutation), Frasier syndrome[5–7], and isolated diffuse mesangial sclerosis (patientP4 in [12] with the 1228 14 mutation). Besides thesewell-defined diseases, splice-site heterozygous mutationshave also been reported in six patients with an atypicalpresentation: the case reported here, which cannot beclassified as classical Frasier syndrome nor DDS, three46,XX girls with FSGS but without genital abnormalities(1228 14 and 15 mutations) [6, 18, 19], a Frasier syn-drome patient with Wilms’ tumor (1228 14 mutation)[20], and a 46,XX woman with slowly evolving FSGSwho achieved normal pregnancy (index case’s mother in[17] with 1228 15 mutation). There appears to be no
Fig. 3. Quantitation of WT1 1KTS/2KTS isoforms in the patient with correlation between the clinical presentation and theheterozygous 1228 14 C→T mutation and a control without WT1 muta-
localization of the mutation within the intronic splicetion. The isoform ratio was calculated as the ratio of the area underthe curve of the two peaks (104 and 113 bp corresponding to the 2 site (either at position 12, 14, 15, or 16). Furthermore,and 1KTS transcripts, respectively) separated by 9 bp. The presented when studied, the 1KTS/2KTS transcript ratio was notvalues are means of at least three experiments.
significantly different between the patients, indicatingthat the observed phenotypic polymorphism cannot berelated to the WT1 mutation alone and that additionalloci are involved [5, 6, 17]. A molecular classificationsyndrome associated with urogenital and diaphragmatic
abnormalities not fulfilling the criteria of Frasier syn- based on the WT1 defects, that is, the intron 9 splice-site mutation, would thus consider all of these phenotypicdrome. We identified the 1228 14 C→T mutation at the
heterozygous state in one patient who, in association presentations as the polymorphic expression of the samegenetic entity.with the FSGS, had proximal hypospadias, unilateral
testicular ectopia, and diaphragmatic hernia. The male Our observation adds to the already demonstratedheterogeneity of steroid-resistant nephrotic syndromegenital phenotype of this patient excludes the diagnosis
of Frasier syndrome, which is classically characterized with FSGS and shows that some sporadic cases of FSGSmay have another genetic origin than the recognizedby female external genitalia. On the contrary, no such
a mutation was found in the 32 patients with strictly autosomal recessive or dominant familial forms, that is,intron 9 splice-site mutation in the WT1 gene on chromo-isolated FSGS or the 4 children with various associated
congenital abnormalities. In our patient, the dominant some 11. In a recent study, no WT1 mutation was foundin seven patients with isolated FSGS [21], but four ofcharacter of the WT1 intron 9 splice-site mutation is
probably a consequence of the 1KTS isoform haplo- these patients had a family history, and this is not, apriori, in favor of WT1 mutation. Schumacher et al re-insufficiency, as previously discussed in Frasier syndrome
[5]. Although testicular ectopia and hypospadias are fre- ported two patients with FSGS and WT1 exon 9 muta-tions (R394 W and R394Q) classified as DDS becausequent conditions, it can reasonably be assumed that in
our patient they are related to the WT1 mutation. Cryp- of associated genital anomalies and in one case becauseof Wilms’ tumor [14]. Our observation points out thattorchidism and hypospadias are common in DDS pa-
tients and are frequently reported in patients with Wilms’ genital and diaphragmatic anomalies must be carefullylooked for in children who present with steroid-resistanttumor [13]. DDS is usually due to heterozygous de novo
mutations within WT1 coding regions and is characterized nephrotic syndrome and minimal changes or FSGS le-sions. The presence of such anomalies justifies the searchby diffuse mesangial sclerosis in association with male
pseudohermaphroditism and/or Wilms’ tumor [12–14]. for WT1 splice-site mutations.Although primary steroid-resistant nephrotic syn-Diaphragmatic hernia in our patient is also probably
related to the WT1 mutation. WT1 is expressed in pleural drome cases with WT1 mutation clearly are uncommon,the demonstration of a WT1 mutation has practical impli-and abdominal mesothelium, and mice lacking both WT1
alleles exhibit diaphragmatic hernia [15]. Finally, the ob- cations in such cases. For instance, cyclosporine and al-kylating agents treatments are questionable and, in ourservation of a newborn child with DDS caused by a WT1
R366H heterozygous mutation associated to a diaphrag- opinion, unjustified. On the other hand, the risk of recur-rence of the nephrotic syndrome after kidney graft ismatic hernia has been reported [16].
The present observation points out the high pheno- probably zero in these patients. This is important, asmany units are reluctant to perform kidney graft fromtypic variability of WT1 splice-site mutations. Such muta-

Denamur et al: WT1 splice-site mutation and FSGS1872
splicing of WT1 leading to an altered ratio of WT1 1/2KTS spliceliving donors for recipients whose primary disease isisoforms. Hum Mol Genet 7:709–714, 1998
FSGS. Nevertheless, as some parents of children with 7. Kikuchi H, Takata A, Akasaka Y, Kukuzawa R, YoneyamaH, Kurosawa Y, Honda M, Kamiyama Y, Hata JI: Do intronicDDS have been shown to have the same WT1 mutationmutations affecting splicing of WT1 exon 9 cause Frasier syn-as their child [13, 17], we recommend looking for thedrome? J Med Genet 35:45–48, 1998
mutation in parents willing to give a kidney to their child 8. Armstrong JF, Pritchard-Jones K, Bickmore WA, Hastie ND,Bard JB: The expression of the Wilms’ tumour gene, WT1, in thewith FSGS and a WT1 mutation. It is not known whetherdeveloping mammalian embryo. Mech Dev 40:85–97, 1992unilateral nephrectomy, in an asymptomatic adult with
9. Pritchard-Jones K, Fleming S, Davidson D, Bickmore W, Por-a WT1 mutation, may favor the development of FSGS teous D, Gosden C, Bard J, Buckler A, Pelletier J, Housman
D, Van Heyningen V, Hastie N: The candidate Wilms’ tumourin the remaining kidney, but this possibility must begene is involved in genitourinary development. Nature 346:194–taken into consideration before such subjects decide197, 1990
upon kidney donation. Finally, regular gonadal and renal 10. Haber DA, Sohn RL, Buckler A, Pelletier J, Call KM, Hous-man DE: Alternative splicing and genomic structure of the Wilms’follow-up is needed in cases with the WT1 intron 9 muta-tumor gene WT1. Proc Natl Acad Sci USA 88:9618–9622, 1991tion because of the risk of gonadoblastoma, as observed
11. Larsson SH, Charlieu JP, Miyagawa K, Engelkamp D, Rassoul-in Frasier syndrome, and because of the recently re- zadegan M, Ross A, Cuzin P, Van Heyningen V, Hastie N:
Subnuclear localization of WT1 in splicing or transcription factorported occurrence of Wilms tumor in one patient withdomains is regulated by alternative splicing. Cell 81:391–401, 1995Frasier syndrome [20].
12. Jeanpierre C, Denamur E, Henry I, Cabanis MO, Luce S, CecilleA, Elion J, Peuchmaur M, Loirat C, Niaudet P, Gubler MC,Junien C: Identification of constitutional WT1 mutations, in pa-ACKNOWLEDGMENTStients with isolated diffuse mesangial sclerosis, and analysis of
N.B. was supported by a grant from the Fondation pour la Recherche genotype/phenotype correlations by use of a computerized muta-Medicale. We are grateful to Dr. J. Bedrossian, Hopital Saint Louis, tion database. Am J Hum Genet 62:824–833, 1998Paris, to have allowed us to test her patients. 13. Little M, Wells C: A clinical overview of WT1 gene mutations.
Hum Mutat 9:209–225, 1997Reprint requests to Erick Denamur, M.D., Ph.D., INSERM U 458, 14. Schumacher V, Scharer K, Wuhl E, Altrogge H, Bonzel KE,
Hopital Robert Debre, 48, boulevard Serurier, 75935 Paris cedex 19, Guschmann M, Neuhaus TJ, Pollastro RM, Kuwertz-BrokingFrance. E, Bulla M, Tonder AM, Mundel P, Helmchen U, WaldherrE-mail: [email protected] R, Weirich A, Royer-Pokora B: Spectrum of early onset nephrotic
syndrome associated with WT1 missense mutations. Kidney Int53:1594–1600, 1998REFERENCES
15. Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J,Housman D, Jaenisch R: WT1 is required for early kidney devel-1. Savin VJ, Sharma R, Sharma M, McCarthy ET, Swan SK, Ellis
E, Lovell H, Warady B, Gunwar S, Chonko AM, Artero M, opment. Cell 74:679–691, 199316. Devriendt K, Deloof E, Moerman P, Legius E, Vanhole C, DeVincenti F: Circulating factor associated with increased glomerular
permeability to albumin in recurrent focal segmental glomerulo- Zegher F, Proesmans W, Devlieger H: Diaphragmatic hernia inDenys-Drash syndrome. Am J Med Genet 57:97–101, 1995sclerosis. N Engl J Med 334:878–883, 1996
2. Fuchshuber A, Jean G, Gribouval O, Gubler MC, Broyer M, 17. Denamur E, Bocquet N, Mougenot B, Da Silva F, MartinatL, Loirat C, Elion J, Bensman A, Ronco PM: Mother-to-childBeckmann JS, Niaudet P, Antignac C: Mapping a gene (SRN1) to
chromosome 1q25-q31 in idiopathic nephrotic syndrome confirms a transmitted WT1 splice-site mutation is responsible for distinctglomerular diseases. J Am Soc Nephrol 10:2219–2223, 1999distinct entity of autosomal recessive nephrosis. Hum Mol Genet
4:2155–2158, 1995 18. Tsuda M, Owada M, Tsuchiya M, Murakami M, Sakiyama T:WT1 nephropathy in a girl with normal karyotype (46,XX). Clin3. Mathis BJ, Kim SH, Calabrese K, Haas M, Seidman JG, Seidman
CE, Pollak MR: A locus for inherited focal segmental glomerulo- Nephrol 51:62–63, 199919. Demmer L, Primack W, Loik V, Brown R, Therville N, McEl-sclerosis maps to chromosome 19q13. Kidney Int 53:282–286, 1998
4. Winn MP, Conlon PJ, Lynn KL, Howell DN, Slotterbeck BD, reavey K: Frasier syndrome: A cause of focal segmental glomerulo-sclerosis in a 46,XX female. J Am Soc Nephrol 10:2215–2218, 1999Smith AH, Graham FL, Bembe ML, Quarles LD, Pericak-Vance
MA, Vance JM: Linkage of a gene causing familial focal segmental 20. Barbosa AS, Hadjiathanasiou CG, Theodoridis C, Papathanas-iou A, Tar A, Merksz M, Gyorvari B, Sultan C, Dumas R,glomerulosclerosis to chromosome 11 and further evidence of ge-
netic heterogeneity. Genomics 58:113–120, 1999 Jaubert F, Niaudet P, Moreira-Filho CA, Cotinot C, FellousM: The same mutation affecting the splicing of WT1 gene is present5. Barbaux S, Niaudet P, Gubler MC, Grunfeld JP, Jaubert F,
Kuttenn F, Nihoul Fekete C, Souleyreau-Therville N, Thibaud on Frasier syndrome patients with or without Wilms’ tumour. HumMutat 13:146–153, 1999E, Fellous M, McElreavey K: Donor splice-site mutations in
WT1 are responsible for Frasier syndrome. Nat Genet 17:467–470, 21. Koziell AB, Grundy R, Barratt TM, Scambler P: Evidence forthe genetic heterogeneity of nephrotic phenotypes associated with1997
6. Klamt B, Koziell A, Poulat F, Wieacker P, Scambler P, Berta Denys-Drash and Frasier syndromes. Am J Hum Genet 64:1778–1781, 1999P, Gessler M: Frasier syndrome is caused by defective alternative


















