Unit 1_ Western Blotting
22
1/21/13 UNIT 1: WESTERN BLOTTING www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 1/22 UNIT 1: WESTERN BLOTTING INTRODUCTION: Western blotting is a method for identifying a specific protein in a complex mixture and simultaneously determining its molecular weight. The name came about this way: DNAblotting was first described by E. M. Southern and, hence, became Southern blotting. RNAblotting then was dubbed Northern blotting. Thus, it was inevitable that protein blotting would become Western blotting. The procedure can be broken down into a series of steps. 1. Size separation of the proteins in the mixture by Polyacrylamide Gel Elecrophoresis (PAGE). 2. Transfer of the separated proteins to a membrane while retaining their relative position. 3. Detecting the protein under investigation by its specific reaction with an antibody and determination of its size relative to standard proteins of known size. In practice there are usually a fewintermediate steps. Some workers stain the original gel to ensure that the protein separation was satisfactory. If this is the case, the method must be chosen carefully because most of commonly used protein staining methods precipitate the proteins, and hence preclude efficient transfer of the proteins to a membrane. Most experienced workers, however, usually omit this step because it is usually quite reliable. One way to confirmthat the proteins have separated properly on the gel is to use one of the commercially available colored molecular weight markers, which allowyou to followthe separation in real time without any staining at all. In any case, most workers do monitor the efficiency of the protein transfer by staining the membrane for total protein. This is important to ensure that the results seen are not unduly influenced by the failure of proteins in particular area of the gel fromtransferring. Background Information In polyacyrlamide gel electrophoresis, separation of charged molecules results fromboth differences in charge and in size. More highly charged species move more rapidly in an electric field; larger molecules, and those less spherical in shape, are retarded by the gel to a greater extent. These two effects may work together or in opposition. The proper choice of a gel systemwill maximize the differences in electrophoretic mobility and/or gel retardation to give the best possible resolution of particular macromolecules. The electrophoretic mobility of a protein molecule is a strong function of pHin the range of pH3-10, especially in the range close to its isoelectric point. At a pHbetween their respective isoelectric points, two proteins would move in opposite directions. Under non-denaturing conditions, the number of bands resolved froma complex mixture of proteins may vary considerably with pH. Separation due to size depends on the pore size of the gel matrix. In the case of agarose, the pore size simply varies inversely with the agarose concentration. The pore size of polyacrylamide gels, which are polymerized as the gel is formed, likewise decreases as the concentration of gel monomer (expressed as %T) increases; however, it is also affected by the extent of cross-linkage. Increasing the concentration of cross-linker relative to the total monomer (expressed as %C) up to 5%by weight decreases the pore size. Above 5%C, the pore size increases again because the cross-linker dimerizes with itself to formmore expanded gels. Most proteins are fractionated on polyacrylamide ranging from5%T to 20%T, containing 2-5%C. Very large proteins, such as immunoglobulins, or various protein aggregates, are resolved on agarose gels. Small polynucleotide fragments (up to 1 kb) are separated on polyacrylamide gels; larger nucleic acids, such as restriction fragments, on agarose. For optimal resolution, all species of interest should be retarded by the gel, but not be completely excluded. When the size range of the sample components is too wide to be sieved by a gel of a single pore size, a gradient gel may be used. Denaturing Vs. Nondenaturing Gels Electrophoresis of proteins may be carried out under either denaturing or non-denaturing conditions. Non-denaturing buffers are, of course, required whenever biological activity must be retained, although denaturing buffers can be used if there is a way to renature biological activity after electrophoresis. Adenaturing gel which allows detection of enzymatic activity following removal of the denaturant is called an activity gel. Non-denaturing buffers are also used when charge differences are known to exist which may give greater resolution than separation on the basis of size. An example would be the resolution of isozymes. Under non-denaturing conditions, the pHmust be carefully selected to maximize charge differences. It is useful to knowthe isoelectric points of the proteins of interest in order to choose the best pH. In contrast, if only the molecular size is relevant, the proteins are denatured with Sodiumdodecylsulfate SDS (Shapiro, 1967; Weber &Osborn, 1969). SDS is an anionic detergent which denatures proteins by wrapping around the polypeptide backbone. In so doing, SDS confers a net negative charge to the polypeptide in proportion to its length. When treated with SDS and a reducing agent, the polypeptides become rods of negative charges with equal "charge densities" or charge per unit length. Since the charge per unit length of the SDS- polypeptide complex is constant, the electrical force exerted on the complex per unit length is also constant. Thus the migration velocity is totally determined by the sieving effect of the acrylamide matrix so that proteins are separated according to their molecular weight. Although most proteins behave normally in SDS gel electrophoresis, it is important to recognize that the technique is an empirical one, and that if the assumptions underlying the method are not valid, the results will not be reliable. Thus for example, a protein with a large excess of acidic residues may well bind less SDS than a typical protein, and hence move abnormally in SDS gel electrophoresis. The major usefulness of this systemis to determine the molecular weights of impure mixtures of polypeptides. This is done by running a gel with standard proteins of known molecular weights along with the polypeptide to be characterized. Alinear relationship exists between the log 10 of the molecular weight of a polypeptide and its R f . By measuring Rf, that is, the distance fromthe sample well to the polypeptide, divided by the distance fromthe sample well to the dye front, a standard curve can be generated in which the log of the molecular weight standards is plotted against their Rf. The molecular weight of an unknown protein can then be determined by measuring its Rf in comparison to the standards. Further details on both denaturing and non-denaturing gel systems are provided in the Appendix to Protocol 1.3. The following table gives a general guideline for the acrylamide concentration needed to sieve SDS proteins of different sizes. MW Range for %T SDS Proteins 15%12,000-45,000
-
Upload
shiv-kumar -
Category
Documents
-
view
52 -
download
0
Transcript of Unit 1_ Western Blotting

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 1/22
UNIT 1: WESTERN BLOTTING
INTRODUCTION:
Western blotting is a method for identifying a specific protein in a complex mixture and simultaneously determining its molecular weight. The name came about this way: DNA blotting
was first described by E. M. Southern and, hence, became Southern blotting. RNA blotting then was dubbed Northern blotting. Thus, it was inevitable that protein blotting would
become Western blotting. The procedure can be broken down into a series of steps.
1. Size separation of the proteins in the mixture by Polyacrylamide Gel Elecrophoresis (PAGE).
2. Transfer of the separated proteins to a membrane while retaining their relative position.
3. Detecting the protein under investigation by its specific reaction with an antibody and determination of its size relative to standard proteins of known size.
In practice there are usually a few intermediate steps. Some workers stain the original gel to ensure that the protein separation was satisfactory. If this is the case, the method must be
chosen carefully because most of commonly used protein staining methods precipitate the proteins, and hence preclude efficient transfer of the proteins to a membrane. Most
experienced workers, however, usually omit this step because it is usually quite reliable. One way to confirm that the proteins have separated properly on the gel is to use one of the
commercially available colored molecular weight markers, which allow you to follow the separation in real time without any staining at all. In any case, most workers do monitor the
efficiency of the protein transfer by staining the membrane for total protein. This is important to ensure that the results seen are not unduly influenced by the failure of proteins in
particular area of the gel from transferring.
Background Information
In polyacyrlamide gel electrophoresis, separation of charged molecules results from both differences in charge and in size. More highly charged species move more rapidly in an
electric field; larger molecules, and those less spherical in shape, are retarded by the gel to a greater extent. These two effects may work together or in opposition. The proper choice
of a gel system will maximize the differences in electrophoretic mobility and/or gel retardation to give the best possible resolution of particular macromolecules.
The electrophoretic mobility of a protein molecule is a strong function of pH in the range of pH 3-10, especially in the range close to its isoelectric point. At a pH between their
respective isoelectric points, two proteins would move in opposite directions. Under non-denaturing conditions, the number of bands resolved from a complex mixture of proteins mayvary considerably with pH.
Separation due to size depends on the pore size of the gel matrix. In the case of agarose, the pore size simply varies inversely with the agarose concentration. The pore size of
polyacrylamide gels, which are polymerized as the gel is formed, likewise decreases as the concentration of gel monomer (expressed as %T) increases; however, it is also affected by
the extent of cross-linkage. Increasing the concentration of cross-linker relative to the total monomer (expressed as %C) up to 5% by weight decreases the pore size. Above 5%C,the pore size increases again because the cross-linker dimerizes with itself to form more expanded gels.
Most proteins are fractionated on polyacrylamide ranging from 5%T to 20%T, containing 2-5%C. Very large proteins, such as immunoglobulins, or various protein aggregates, are
resolved on agarose gels. Small polynucleotide fragments (up to 1 kb) are separated on polyacrylamide gels; larger nucleic acids, such as restriction fragments, on agarose.
For optimal resolution, all species of interest should be retarded by the gel, but not be completely excluded. When the size range of the sample components is too wide to be sieved
by a gel of a single pore size, a gradient gel may be used.
Denaturing Vs. Nondenaturing Gels
Electrophoresis of proteins may be carried out under either denaturing or non-denaturing conditions. Non-denaturing buffers are, of course, required whenever biological activity mustbe retained, although denaturing buffers can be used if there is a way to renature biological activity after electrophoresis. A denaturing gel which allows detection of enzymatic activity
following removal of the denaturant is called an activity gel. Non-denaturing buffers are also used when charge differences are known to exist which may give greater resolution than
separation on the basis of size. An example would be the resolution of isozymes.
Under non-denaturing conditions, the pH must be carefully selected to maximize charge differences. It is useful to know the isoelectric points of the proteins of interest in order tochoose the best pH.
In contrast, if only the molecular size is relevant, the proteins are denatured with Sodium dodecylsulfate SDS (Shapiro, 1967; Weber & Osborn, 1969). SDS is an anionic detergentwhich denatures proteins by wrapping around the polypeptide backbone. In so doing, SDS confers a net negative charge to the polypeptide in proportion to its length. When treated
with SDS and a reducing agent, the polypeptides become rods of negative charges with equal "charge densities" or charge per unit length. Since the charge per unit length of the SDS-
polypeptide complex is constant, the electrical force exerted on the complex per unit length is also constant. Thus the migration velocity is totally determined by the sieving effect of the
acrylamide matrix so that proteins are separated according to their molecular weight.
Although most proteins behave normally in SDS gel electrophoresis, it is important to recognize that the technique is an empirical one, and that if the assumptions underlying the
method are not valid, the results will not be reliable. Thus for example, a protein with a large excess of acidic residues may well bind less SDS than a typical protein, and hence move
abnormally in SDS gel electrophoresis.
The major usefulness of this system is to determine the molecular weights of impure mixtures of polypeptides. This is done by running a gel with standard proteins of known molecular
weights along with the polypeptide to be characterized. A linear relationship exists between the log10 of the molecular weight of a polypeptide and its Rf. By measuring Rf, that is, thedistance from the sample well to the polypeptide, divided by the distance from the sample well to the dye front, a standard curve can be generated in which the log of the molecular
weight standards is plotted against their Rf. The molecular weight of an unknown protein can then be determined by measuring its Rf in comparison to the standards. Further details on
both denaturing and non-denaturing gel systems are provided in the Appendix to Protocol 1.3.
The following table gives a general guideline for the acrylamide concentration needed to sieve SDS proteins of different sizes.
MW Range for
%T SDS Proteins
15% 12,000-45,000

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 2/22
10% 16,000-70,000
5% 60,000-200,000
Molecular Weight Markers
There are a number of different commercially available protein molecular weight markers which are used to estimate the molecular weight of proteins on Western blots. They generally
fall into two categories, prestained and unstained. Prestained or dyed markers either have all the proteins in the molecular weight ladder stained with a single dye or alternatively each
of the proteins in the ladder is stained with a different colored dye. The differently stained markers, while the most expensive, have the advantage of providing unambiguousidentification of the specific proteins in the ladder. This prevents confusion as to which molecular weight marker band you are looking at which can sometimes occur when some of the
singly dyed marker bands run off the gel. Dyed markers, therefore, have two main advantages. They allow the separation of the proteins on the gel to be readily followed during
electrophoresis and they indicate how efficiently proteins were transferred from the gel to the hybridization membrane. Dyed markers, however, also have several disadvantages. The
addition of dyes to the molecular weight markers does alter the overall mobility of these proteins, so that the accuracy of molecular weights determined from these markers may not be
as precise as desired. Additionally, the different protein molecules in any given band do not bind the same amount of dye which leads to a broadening of the protein peaks. The
stability of the dye-protein complex can also be marginal. Hence, colored markers, if boiled at all, should not be boiled more than one minute and should be loaded on the gel within
an hour after dilution from the stock to prevent loss of color in the bands.
Unstained markers are run with the protein samples of interest and then visualized after transfer to the hybridization membrane by staining with a protein stain. These markers have the
advantage of providing sharp bands whose mobility has not been altered by the addition of dyes. These standards therefore give the best estimates of molecular weight. They have theprimary disadvantage in that they do not allow one to monitor the progress of the gel or to assesses the transfer efficiency. To get around the relative disadvantages of the two major
types of molecular weight markers, many people run both prestained and unstained molecular weight markers on the same gel.
Separation Of DNA Or RNA
Polyacrylamide gels can also be used to separate small DNA fragments of <=1kb. The separation of double-stranded linear nucleic acid fragments is entirely based on size, since the
charge density is the same for pieces of different length. The same is true for single-stranded nucleic acid fragments when denatured by using high concentrations of urea in the gel or
by other means. Conformational differences can also be used to resolve polynucleotides under special circumstances. Closed circular DNA, for example, is retarded to different
extents depending on the degree of supercoiling, and the complementary strands of a duplex DNA fragment can sometimes be separated on a non-denaturing gel after sample
denaturation due presumably to differences in secondary structure.
Two-Dimensional Electrophoresis
In two-dimensional electrophoresis, proteins are fractionated first on the basis of one physical property, and, in a second step, on the basis of another. Most commonly, isoelectricfocusing is used for the first dimension, conveniently carried out in a tube gel, and SDS electrophoresis in a slab gel is used for the second dimension. The most widely used procedure
is that of O'Farrell (7). As many as 1100 distinct proteins have been resolved from a single sample of serum by this method.
Continuous and Discontinuous Buffer Systems
A continuous buffer system is one in which the same buffer is used throughout the gel and at both electrodes. Continuous buffer systems require only a single layer of gel. They are
simple to set up, and are often adequate for such purposes as monitoring an enzyme purification or for preparative electrophoresis of a partially purified protein. They are also used fornucleic acid electrophoresis. The main disadvantage is that the sample must be in a concentrated form, since each resolved band will be as wide as the depth of the original sample in
the well.
In contrast, a discontinuous buffer system concentrates each sample component into a narrow band known as the "stack". The original sample may therefore be much more dilute, and
components which band close together can be resolved more easily.
A discontinuous system uses buffers of different composition and pH to create a discontinuous voltage and pH gradient. The different buffer zones are stabilized by at least twodifferent gel layers.
The upper layer, through which the sample passes first, is known as the "stacking gel". It is a large-pore gel, which is non-restrictive to the protein sample. The buffer in which thestacking gel is made contains an ion (usually an anion) whose electrophoretic mobility is greater than that of the protein, while the tank buffer, or electrode buffer, must contain an ion
whose mobility is less than that of the protein. As electrophoresis begins, the "leading ion" in the stacking gel moves faster than the protein and leaves behind it a zone of lowerconductivity. The higher voltage gradient of this zone causes the protein to move faster and to "stack" at the boundary between the leading and trailing ions.
Below the stacking gel is a deeper layer of gel with a smaller pore size, known as the resolving or separating gel. This gel is prepared in a buffer of higher concentration and pH (in ananionic system). In this environment, the mobility of the trailing ion increases so that its boundary moves ahead of the protein. The protein is resolved into individual bands according to
size; and in the case of a non-denaturing gel, according to shape.
The Laemmli System
The most widely used system for electrophoresis of proteins is probably that described by Laemmli (5). It is a discontinuous system for resolving proteins denatured with SDS. The
leading ion in the Laemmli buffer system is chloride, and the trailing ion is glycine. Accordingly, the resolving gel and the stacking gel are made up in Tris-HCl buffers (of differentconcentration and pH), while the tank buffer is Tris-glycine. All buffers contain 0.1% SDS.
Membranes
Nitrocelulose and reinforced PVDF membranes are generally used in Western blotting. For larger molecular weight proteins both types of filters works well. However, if your protein
is less than 10 kb nitrocelluose should be used because it has a much higher binding capacity. The PVDF membranes will not work well under these circumstances.
General References

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 3/22
1. Anderson, NL and NG Anderson. 1977. Two-dimensional electrophoresis of human plasma proteins. Proc. Natl. Acad. Sci. USA 74:5421-5425.
2. Cohen, S et al. 1982. Example of non-denaturing gels with continuous buffer system for purification of a protein to be assayed by affinity binding. J. Biol. Chem. 257:1525-
1531.
3. Davis, BJ 1964. Description of discontinuous buffer system for non-denaturing gels in electrophoresis. Ann. NY. Acad. Sci. 209:373-381.
4. Jovin, TM 1973. Computer-derived selection of buffer recipes for multiphasic zone electrophoresis. Ann. NY Acad. Sci. 209:477-496.
5. Laemmli, UK 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680.
6. Matsudiara, PT and DR Burgess. 1987. SDS microslab linear gradient polyacrylamide gel electrophoresis. Anal. Biochem. 87:386-396.
7. O'Farrell, PH 1975. High resolution two-dimensional electrophoresis of proteins J. Biol. Chem. 250:4007-4021.
8. Ornstein, L 1964. Disc electrophoresis - I: Background and theory. Ann. NY Acad. Sci. 121:321-349.
9. Reisfeld, RA et al. 1962. Acidic buffer system for resolution of cationic proteins. Nature 195:281.
10. Strickland, TW and D Puett. 1982. Laemmli gels; used for separation of glycoprotein hormones. J. Biol. Chem. 257:2954-2960.
11. Towbin, H et al. 1979. Electrophoretic transfer of proteins from SDS and acid/urea gels to nitrocellulose. Proc. Natl. Acad. Sci. USA. 76:4350-4354.
12. Weber, K and J Osborn. 1969. Basic study of molecular weight determination by SDS electrophoresis in a continuous buffer system. J. Biol. Chem. 224:4406-4412.
PROTOCOL 1.1: ISOLATION OF PROTEIN FOR WESTERN BLOTTING AND PROTEIN QUANTITATION
INTRODUCTION:
Proteins can be easily prepared for Western blotting by multiple cycles of freeze thawing, sonication, or lysis with enzymes and detergents. Mammalian cells are usually quite easy tolyse and generally do not require freeze thawing or the enzymatic treatments that are necessary to lyse yeast and certain bacterial cells. While one can lyse mammalian cells directly in
the sample buffer used for SDS/PAGE electrophoresis, this buffer contains bromophenol blue, SDS and mercaptoethanol which at sufficient concentration can interfere with thecommonly used Bradford protein assay. Hence it is best to lyse the cells in a modified sample buffer which contains low enough levels of these interfering species to allow accurate
determination of the protein concentration. In our case we found that merely omitting bromophenol blue prevented any interference with the protein dye binding assay. Once the cellsare lysed, they should be boiled immediately in a screw cap tube to denature any proteases that may remain active in sample buffer.
In order to estimate how much protein to expect in our samples one should consider the following. For mammalian cells one obtains about 1 g wet weight of cells from a liter of
media. Assuming the cells have grown to about 106 cells/ml this gives 109 cells wet weight per gram. Since a cell is roughly 90% water and nearly everything else is protein, we canvery roughly estimate that 109 cells contain 0.1 g of protein. So if 100 ml of cells are resuspended in 1 ml of sample buffer the concentration will be roughly 10 mg/ml. A volume of
about 10 ml can then be taken for determination of the exact protein concentration.
PROCEDURE:
1. Start boiling water bath.
2. Spin down the cells in a conical centrifuge tube. Immediately remove the bulk of the supernatant and vortex to resuspend the pellet.. Proceed as quickly as possible
through the next steps in order to inactivate proteases that can rapidly degrade the protein(s) of interest.
3. Add a volume of room temperature 2X Sample Buffer that is equal to the volume of the resuspended cell pellet.
4. Vortex to resuspend the pellet and lyse cells, then transfer cells immediately to a clean screw cap microfuge tube using Pipettman. Cap and proceed to step 5immediately.
5. Place the screw cap microfuge or lock top microfuge tube in boiling water for 2-5 min., then chill it in ice until you are ready to use it. This treated sample can be
stored frozen for future runs.
6. The boiled extract tends to be very viscous due to the presence of high molecular weight chromosomal DNA. If the extract is too viscous to pipette easily, sonicate
the sample with two short pulses of ~ 5 seconds each using a microprobe set at about 50% full power. If the sample makes a steady "pssst" sound during each burst, theDNA will be sheared.
Alternatively, if a sonicator is not available, pass the extract back and forth several times through a 19 gauge hypodermic needle using a 1 ml syringe before loading an
aliquot of the sample on the SDS PAGE gel. If the sample is still viscous after the 19 gauge shearing, change needles and pull the sample back and forth through a 25gauge needle. The sonication method is faster and easier than using a syringe and generally leads to cleaner separations on the gels.
7. Spin the sample for 30 sec in a microfuge (14K), remove the supernatant, and place it in a clean microfuge tube.
Note: When samples are sonicated this step may not be necessary because the cells are so thoroughly disrupted that no pellet is formed after centrifuging the sample.Therefore, if you consistently observe no pellet after the spin, omit this step.
8. Determine the protein concentration of the supternatant (Protocol 1.2).
Note: The amount of protein needed for each lane depends on the sensitivity of the staining method to be used, and also on the distribution of protein among separate
bands. With Coomassie Blue, you can detect as little as 1 mg in a single band; with the 100-fold more sensitive silver stains, about 10 ng. The chemiluminescent
techniques used to detect antigenic protein are far more sensitive than these total protein stains.

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 4/22
MATERIALS:
1. 0.5 M Tris-Cl pH 6.8
Compound Amount/50 ml Amount/100 ml
Tris-Cl 3.0 g 6.0 g
Deionized H2O 45 ml 95 ml
Adjust to pH 6.8 with 1 M HCl.Add deionized H2O to 50 ml to 100 ml
Allow the solution to cool to room temperature before making the final adjustments to the pH. This is because the pH of Tris solutions are temperature-dependent and decrease approximately 0.03 pH
for each 1oC increase in temperature. For example, a 0.05 M solution has pH values of 9.5, 8.9, and 8.5 at 5oC, 25oC, and 37oC respectively.
Sterilize by autoclaving.
2. 10% Sodium Dodecyl Sulfate Sodium Salt (SDS, FW=288.4, also called Sodium Lauryl Sulfate)
Compound Amount/500 ml Amount/1000 ml
Deionized H2O 450 ml 900 ml
SDS 50 g 100 g
Heat to 68oC to assist dissolution.
Adjust pH to 7.2 by adding a few drops of concentrated HCl.Add deionized H2O to 1000 ml.
Dispense in aliquots. There is no need to sterilize 10% SDS.
Caution: Wear a mask when weighing SDS and wipe down the weighing area and balance after use because the fine crystals of SDS disperse easily.
3. 2X Sample buffer (Minus Bromophenol Blue)
Final Concentration Stock Solution Amount/10 ml Amount/50 ml
0.125 M Tris-Cl pH 6.8 0.5 M Tris-Cl pH 6.8 2.5 ml 12.5 ml
4 % SDS 10% SDS 4.0 ml 20 ml
10% Glycerol 2.0 ml 10 ml
10% 2-Mercaptoethanol 1.0 ml 5 ml
H2O 0.5 ml 3.5 ml
Divide in aliquots and freeze.
4. Boiling water bath.
5. Sonicator or 1 ml syringe with 19 and 25 gauge hypodermic needles.
6. Appropriate centrifuge tubes and screw cap microfuge tubes.
PROTOCOL 1.2: BRADFORD PROTEIN ASSAY (BIO-RAD REAGENTS)
REFERENCES: Ausubel et al. 10.1.1; BIO-RAD Protein Assay Kit Instructions; and Bradford, M.M. 1976. Anal. Biochem. 72:248-254
INTRODUCTION:
This very simple protocol for determining protein concentration has largely replaced the earlier widely used Lowry assay. The assay depends on the fact that when certain dyes (here,Coomassie Brilliant Blue R-250) bind to proteins they undergo a shift in their absorbance. By measuring the total absorbance at the shifted wavelength one can thus readily estimate
the concentration of the protein solution with respect to some standard protein. Note that not all proteins give an identical absorbance shift, so that the technique, like many techniques
used to measure protein concentration, is really only useful for comparing relative concentrations of the same protein. In reality, the amount of protein determined from the standard
curve may vary from the absolute protein concentration by a factor of two fold or more.
One of the most commonly used ways to determine protein concentration is with the dye Coomassie Brilliant Blue R-250. Generally, the standard curves used to determine protein
concentrations using this method cover a range from 0 to 100 mg of either lysozyme or BSA per standard 5 ml reaction. Again, it is important for you to understand that theCoomassie Brilliant Blue R-250 stain binds certain proteins more readily than others. Thus, the estimate of protein concentration in cell lysates varies with the particular protein being
used for the standard curve. BSA and lysozyme are generally chosen because they are inexpensive and readily obtainable in high purity. However, BSA, in particular, tends to
denature easily and does not give a particularly accurate estimate of the protein concentration in crude cell lysates. It is important to note that relative to water or TE, the lysis buffer
has a significant absorbance by itself. Hence the standard curve, should contain the same final Lysis Buffer concentration as the samples to prevent erroneous estimates of proteinconcentration.
Note: The technical sheet that comes with the Bradford assay lists a number of common buffer components, such as SDS, that interfere with the assay above a certain concentration.
Another commonly used assay using the dye bicinchonic acid (obtainable from the Pierce Chemical Company) is more resistant to SDS interference. Perhaps the best way to avoidpossible detergent interference in these assays is to omit detergents from the lysis buffers (or use detergents such as Triton which often interfere less). One can then use a sonicator or
homogenizer to prepare the extracts. However in this case it is important to provide adequate protease inhibitors in the lysis buffer to prevent degradation of the sample until it can be
assayed and boiled in sample lysis buffer.

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 5/22
PROCEDURE:
1. Dilute the dye with four volumes of D.I. water and filter out any remaining particulates. (This step will be done by the TAs).
2. Remove enough protein extract to give a reasonable color in the Bio-Rad Protein assay (typically 1-5 ml) and diluted to 100 ml with water in a glass test tube.
3. Add 5 ml of diluted dye mix to the diluted protein extract, mix. If the mixture immediately turns dark blue start over and use less extract. Conversely if you are unable
to detect by eye any difference between assay and blank add more extract.
4. Measure at OD595 nm against blank with buffer and dye.
Note: The Bradford reagent binds tightly to glass. These means that as you read consecutive samples the colored dye builds up on the cuvette walls and prevents and
accurate reading of the sample OD. Thus if you are using glass cuvettes, they must be rinsed out with ethanol between samples to remove the bound dye. An alternativeapproach is to use disposable plastic cuvettes which are relatively inexpensive.
5. By comparison with a standard curve (prepared by TAs), estimate the Y mg amount of protein in the X ml added to the standard 5 ml reaction. Then convert the
concentration estimate (Y mg/X ml) to mg/ml or mg/ml.
MATERIALS:
1. Bio-Rad Protein Assay Reagent (Proprietary Solution Of Coomassie Brilliant Blue R-250)
2. Spectrophotometer and disposable cuvettes.
3. Protein Standards from 0 to 100 mg per 5 ml reaction
You should note from the discussion in the introduction to this protocol that the sample buffer contains a number of compounds that can interfere with the assay-in particular SDS. It is important notto exceed the allowed concentrations of these interferring substances in the assay. For SDS the maximum concentration compatable with the assay is 0.1% in a 100 ul sample. (For a complete list ofinterferring substances see the BioRad manual). In addition, as in any assay it is important that the standard curve contain exactly the same components in the same concentrations as the samples-except of course for the substance one is measuring.
With these considerations in mind, here is a table for constructing a standard curve for the Biorad protein assay. Prepare BSA or Lysozme stock solutions at concentrations of 2 mg/ ml in water.
Note, the standards all contain a final concentration of 0.1 % SDS/100 ml sample which is the maximum amount of sample buffer (5 ul) compatable with the assay.
2 mg/ml stock (ml) 1X Sample buffer (<= 5 ml) H2O (ml) Final m�gs of protein being added to 5 ml assay
0 5 95 0
5 5 90 10
15 5 80 30
25 5 70 50
50 5 45 100
Then add each 100 ml stock solutions to a separate tube containing 5 ml of filtered protein reagent and read A595. after 5 min.
PROTOCOL 1.3: POURING AND RUNNING SDS POLYACRYLAMIDE GELS (SDS-PAGE GELS)
SEPARATION OF PROTEINS ON THE BASIS OF MOLECULAR WEIGHT: SDS GEL ELECTROPHORESIS
References: B. D. Hames and D. Rickwood Gel Electrophoresis of Proteins, A Practical Approach, David Garfin in Meth. Enzymol. (1990) 182:425-441., Sambrook et al.
Molecular Cloning 18:47-18.59.
SOURCE: HOEFER SCIENTIFIC INSTRUMENTS
INTRODUCTION:
There are two types of buffer systems in electrophoresis, continuous and discontinuous. A continuous system has only a single separating gel and uses the same buffer in the tanks and
the gel. In a discontinuous system, a method first developed by Ornstein (1964) and Davis (1964), and later popularized by Laemmli (1970), a nonrestrictive large pore gel, called a
stacking gel, is layered on top of a separating gel (also referred to as the resolving gel). Each gel layer is made with a different buffer, and the tank buffers are different from the gelbuffers. The resolution obtainable in a discontinuous system is much greater than that of a continuous one for reasons to be described. However, the simpler continuous system is a
little easier to set up.
In a discontinuous system, a protein's mobility, a quantitative measure of the migration rate of a charged species in an electrical field, is intermediate between the mobility of the bufferion in the stacking gel (leading ion) and the mobility of the buffer ion in the upper tank (trailing ion). When electrophoresis is started, the ions and the proteins start to migrate into the
stacking gel. The proteins concentrate in a very thin zone called the stack, between the leading ion and the trailing ion. The proteins continue to migrate in the stack until they reach the
separating gel. This concentration of proteins does not occur in a continuous system where the proteins enter the separating gel in a zone nearly as broad as the original sample. As a
result, a continuous system produces bands that are thick and poorly resolved.
An additional advantage of a discontinuous system is that the researcher can design a system to work with a particular protein. For example, he can use buffers which differ only
slightly in their mobilities so that a protein of interest will stack while other proteins or contaminants will not.
The procedure we give has been adapted to a minigel format. While larger gel formats give clearer resolution and have a larger capacity, they take longer to run and for many

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 6/22
purposes the increased resolution they provide is unnecessary.
PROCEDURE:
The SE 250 Mighty Small II is a miniature vertical slab gel unit intended for rapid electrophoresis of protein or nucleic acid samples of small volume. Two 7 x 8 cm gel sandwiches of
either polyacrylamide or agarose gels may be run simultaneously on the SE 250. Polyacrylamide gels are run using the glass and aluminum plates provided with the unit. The core of
the unit serves as a heat exchanger through which coolant may be circulated.
Preparing A Stacking Gel
The manual that comes with your model electrophoresis unit will describe how to pour gels using that specific equipment. The basic procedure, however, will be the same in most
instances. First depending on the particular apparatus being used, you must assemble either two glass plates, or a single glass plate and notched alumina plate, separated by two
plastic spacers which determine the thickness of the gel. Once the plates and spacers are assembled, the bottom has to be sealed with tape or agarose or clamped into a casting rigwhich prevents the gel assembly from leaking. Then you pour the resolving gel up to a predetermined depth that is dependent on the depth of sample in the well (see diagram below).
The general rule is that the distance between the bottom of the wells and the top of the resolving gel is 2 to -4 times the depth of samples in the wells. The increase in distance between
the bottom of the well and the top of the resolving gel with well depth of the sample simply reflects the fact that it takes more distance for the proteins to be focused into a band whenthe sample is spread out over a larger distance in the well.
The resolving gel should be poured quickly after the TEMED and ammonium persulfate is added to the acrylamide gel mixture. The best procedure is to withdraw the acrylamide mix
in an appropriate sized syringe or disposable pipette and to allow the gel mix to flow down one side of the assembled plates to avoid trapping bubbles. Once the resolving gel ispoured it should be covered with the Running Gel Overlay buffer or isobutanol to exclude oxygen which inhibits polymerization of the acrylamide at the top of the gel. Withdraw a
small amount of the acrylamide solution into a Pasteur pipette or place some in a microfuge tub and cap tightly so the progress of the polymerization can be readily followed. If theacrylamide in the pipette or tube fails to harden, then there is a problem with the acrylamide mix. A limited number of recipes for making the stacking and resolving gel are given in the
materials section.
Note: Many companies sell a casting tray that allows you to pour from 1 to 5 acrylamide gels at a time.
After the resolving gel has polymerized (i.e. when the polyacrylamide in the Pasteur pipette has hardened), pour off the isobutanol or buffer solution and assemble the stacking gel
acrylamide gel mix. Once complete, pour in the stacking gel mix until it reaches the top of the plates. The comb is then held at an angle and one corner inserted near the upper left or
right hand side of gel. Once one corner of the comb is inserted, gently lower the opposite corner of the comb being careful not to trap bubbles as the comb is inserted into the top of
the gel. Add additional stacking gel mix if some has been lost during the comb insertion. Cover any exposed gel mixture with Running Gel Overlay buffer or isobutanol and allow thestacking gel to polymerize. Place the remaining gel mixture in a Pasteur Pipette or microfuge tube to insure that polymerization is proceeding properly.
After the stacking gel polymerizes and the comb has been gently removed, the gel is ready to use. The gels can be wrapped in plastic wrap and placed at 4oC and used the next day if
necessary. There is generally only minimal loss of resolving ability the next day. However, it is not recommended that the gels be stored longer than one day as they begin to
deteriorate as they dry out and diffusion occurs between the stacking and resolving gel.
Sample Loading and Final Assembly of the SE 250
1. Clamp the sandwich against the core with the alumina plate against the gasket, and oriented with the notch up. Use the red clamps to clamp the gel in place.
2. Mark the location of the wells using a marker pen and then gently remove the comb.
3. Rinse the sample wells with tank buffer and drain by inverting the unit over a sink.
4. Fill the sample wells and the upper and lower buffer chambers with buffer. Each upper buffer chamber holds about 75 ml. The capacity of the lower buffer chamber is
approximately 150 ml.
5. Add 1/10 volume of 1X bromophenol blue solution to the protein samples and mix. This helps both in visualizing the sample during loading and in determining how
evenly and rapidly the proteins are being separated on the gel. Any other tracking dye, such as phenol red will work as well.
6. If cooling is desired, connect the tubing from the inner core to a cold water faucet or refrigeration unit, and begin to circulate coolant. Recommended coolants for use
with this cooling core are water or 50% water and 50% ethylene glycol. Do not use antifreeze or any alcohol based mixture as these will irreparably damage the unit.

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 7/22
7. Load the gel using flat tipped sequencing tips. The tips should curve toward you as you load. Be careful to depress the Pipettman slowly as you load the well so as not
to blow your sample out of the well. Well volumes formed by different combs are given in Table I below:
TABLE I
VOLUME OF SAMPLE PER 1 MM DEPTH
Thickness of Comb
Number of Wells 0.5 mm 0.75 mm 1.0 mm 1.5 mm
5-well comb 6.4 ml 9.5 ml 12.7 ml 19.1 ml
10-well comb 2.4 ml 3.6 ml 4.8 ml 7.2 ml
15-well comb 1.5 ml 2.2 ml 2.9 ml 4.4 ml
8. Place the safety lid on the unit and attach the leads to the power supply.
Running the SE 250
1. Gels may be run at either constant current or constant voltage. Constant current is traditionally used with a discontinuous buffer system so that the rate of
electrophoretic migration will remain constant throughout the run. Using a Laemmli gel system at 40 mA (20 mA per gel) constant current, with two 0.75 mm thick gels,
the run should be complete in just over an hour. The corresponding voltage and wattage settings should be approximately 160V (for either one or two gels) and 4
watts/gel or 8 watts/2 gels. For higher resolution, a lower current setting may be advisable. Novex recommends running their precat Tris/Glycine gel at a constant voltage
of 125 V for 1 or 2 gels.
2. When the tracking dye reaches the bottom of the gel (below the surface of the lower buffer), turn off the power supply, disconnect the leads, and remove the lid of the
unit.
3. Stop the flow of coolant through the inner core and disconnect the tubing. Remove the inner core from the lower buffer chamber and pour out the upper buffer by
inverting the core over a sink.
4. Remove the side clamps and lift each gel sandwich off the core.
5. Use plastic gel wedges to pry open the gel sandwich FROM THE BOTTOM to avoid breaking the ears of the notched alumina plate. The gel will usually stick to the
alumina plate. Remove the spacers and peel the gel off the plate into a tray of stain or fixative.
Care and Maintenance
1. Rinse the lower buffer chamber and inner core with distilled water after each use.
2. Glass and alumina plates should be cleaned in a strong detergent such a RBS -35, then rinsed thoroughly with tap and distilled water. Both plates can also be treated
with chromic acid cleaning solution.
3. Spacers should be washed with diluted RBS-35, then rinsed with tap and distilled water.
4. Do not autoclave or boil this unit or any of its plastic parts.
STOCK SOLUTIONS FOR LYSIS BUFFER AND RUNNING AND CASTING SDS PAGE GEL
Note: Use the free base forms of Tris and glycine when making these solutions. The term "Tris-Cl" used in these formulations is an indication that the pH of the solution is adjusted
with HCl.
1. 2X Sample buffer (Minus Bromophenol Blue)
Final Concentration Stock Solution Amount/10 ml Amount/50 ml
0.125 M Tris-Cl pH 6.8 0.5 M Tris-Cl pH 6.8 2.5 12.5
4 % SDS 10% SDS 4.0 20
10% Glycerol 2.0 ml 10
10% 2-Mercaptoethanol 1.0 ml 5
H2O to 10.0 ml to 50 ml
Divide in aliquots and freeze.
2. 1 X Bromophenol Blue
Compound Amount/1 ml Amount/5 ml
Bromophenol Blue 1 mg 5 mg
H2O to 1 ml to 5 ml
3. 10% SDS

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 8/22
Compound Amount/100 ml Amount/500 ml
SDS 10 g 50 g
H2O to 100 ml to 500 ml
4. 0.5 M Tris-Cl pH 6.8 (FW=121.1)
Compound Amount/50 ml Amount/100 ml
Tris base 3.0 g 6.0 g
Deionized H2O 45 ml 95 ml
Adjust to pH 6.8 with 1 M HCl.Add deionized H2O to 50 ml to 100 ml
Allow the temperature to cool to room temperature before making the final adjustments to the pH. This is because the pH of Tris solutions are temperature-dependent and decrease approximately 0.03
pH for each 1oC increase in temperature. For example, a 0.05 M solution has pH values of 9.5, 8.9, and 8.5 at 5oC, 25oC, and 37oC respectively.
Sterilize by autoclaving.
5. 0.5 M Tris, 8.8 (FW=121.1)
Compound Amount/50 ml Amount/100 ml
Tris base 3.0 g 6.0 g
Deionized H2O 45 ml 95 ml
Adjust to desired pH by adding concentrated HCl.
Allow the temperature to cool to room temperature before making the final adjustments to the pH. This is because the pH of Tris solutions are temperature-dependent and decrease approximately 0.03
pH for each 1oC increase in temperature. For example, a 0.05 M solution has pH values of 9.5, 8.9, and 8.5 at 5oC, 25oC, and 37oC respectively.
6. Initiator - 10% ammonium persulfate (FW=228.19)
Compound Amount/ 10 ml
(NH4)2S2O3 1 g
Deionized H2O to volume
The solution may be stored for several weeks at 4oC.
7. Running Gel Overlay (0.375 M Tris-Cl pH 8.8, 0.1% SDS, used to prevent oxygen from reaching gel while polymerizing)
Final Concentration Stock Solution Amount/100 ml Amount/500 ml
0.375 M Tris-Cl pH 8.8 0.5 M Tris-Cl pH 8.8 75 ml 375 ml
0.1% SDS 10 % SDS 1.0 5 ml
H2O to 100 ml to 500 ml
8. Tank Buffer (0.025M Tris pH 8.3, 0.192M glycine, 1% SDS)
Final Concentration Stock Solution Amount/1 L Amount/4 L
0.025 M Tris base pH 8.3 3.0 g 12 g

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 9/22
0.192 M Glycine 14.4 g 57.6 g
1 % SDS 10% SDS 10 ml 40 ml
H2O H2O to 1 L H2O to 4 L
Because of the pH of this solution need not be checked, it can be made up directly in large reagent bottles marked at 4.0 liters. 12-16 liters can be made up at a time. Reuse the buffer in the lowerbuffer chamber 4 or 5 times, but discard the upper buffer after each run.
9. TEMED (use required amount straight out of the bottle).
10. Casting the SDS PAGE Gel
The following recipes for SDS PAGE stacking and separating gels are for a 40% premade stock of 29:1 acrylaminde:bisacrylamide.
A. Preparation Of Separating Gel
In a 100 ml sidearm flask, mix 40% acrylamide solution (29:1) with water, Tris HCl (pH 8.8) and SDS, according to the chart below.
Optional. Degas under vacuum to 10 to 15 min. to remove oxygen which inhibits the polymerization.
Add 10% ammonium persulfate and TEMED according to the chart below. Swirl gently to mix. Use immediately. Draw up some of the excess mix into a disposable Pasteur pipette or placeabout 1 ml in a microfuge tube and cap tightly. If the gel fails to harden in the pipette or microfuge tube, there is usually a problem with the persulfate, TEMED or both.
ML Of Each Solution Needed To Make ~25 MLs Of SDS PAGE Resolving Gel
With The Following Final % Acrylamide Concentrations
Stock Solutions 5 6 7 8 10 12 13 15
H2O 15.17 14.55 13.93 13.3 12.05 10.8 10.17 8.92
40% Stock
29% acrylamide 3.13 3.75 4.37 5.0 6.25 7.5 8.13 9.38
1% Bisacrylamide
0.5 M Tris HCl 6.3 6.3 6.3 6.3 6.3 6.3 6.3 6.3
pH 8.8
10% SDS 250 ml 250 ml 250 ml 250 ml 250 ml 250 ml 250 ml 250 ml
Ammonium 125 ml 125 ml 125 ml 125 ml 125 ml 125 ml 125 ml 125 ml
persulfate
TEMED 25 ml 25 ml 25 ml 25 ml 25 ml 25 ml 25 ml 25 ml
There are a number of venders, such as Bio-Rad, that sell premixed acrylamide solutions at defined acrylamide:bis acrylamide ratios (e.g. 29:1). It is cost effective to buy these solutions and more healthy,too, because you, or your employee, does not have to breath acrylamide dust, a neurotoxin, during weighing.
B. Preparation Of Stacking Gel
In a 100 ml sidearm flask, mix 40% acrylamide solution (29 acrylamide:1.0 bisacrylamide) with water, Tris HCl (pH 6.8) and SDS, according to the chart below. Optional. Degas undervacuum to 10 to 15 min. to remove oxygen which inhibits the polymerization.
Add 10% ammonium persulfate and TEMED according to the chart below. Swirl gently to mix. Use immediately. Draw up some of the excess mix into a disposable Pasteur pipette or placeabout 1 ml in a microfuge tube and cap tightly. If the gel fails to harden in the pipette or microfuge tube, there is usually a problem with the persulfate, TEMED or both.
ML Needed To Make ~25 MLs Of SDS PAGE Resolving Gel
With The Following Final % Acrylamide Concentrations
Stock Solutions 4%
H2O 15.8
40% Stock 29% acrylamide: 1% Bisacrylamide 2.5
0.5 M Tris HCl, pH 6.8 6.3
10% SDS 250 ml
Ammonium persulfate 125 ml
TEMED 25 ml
PROTOCOL 1.4: PROTEIN TRANSFER FROM ACRYLAMIDE GEL TO MEMBRANE
INTRODUCTION:
Protein blotting is succinctly described by Therese Timmons and Bonnie Dunbar in Meth. Enzymol. (1990) 182:679-692. A description of the process can also be
found in Sambrook et al. on pages 18.60-18.75, and in Ausubel et al. 10.8.1. There are two major methods used to transfer proteins electrophoretically to a
membrane, semidry blotting and tank transfer blotting. Tank transfer blotting wastes large volumes of buffers and is not very versatile since only a single buffer can
be added to the tank. While many workers continue to use it, it seems to possess no advantage over semi-dry blotting. Transfer by diffusion blotting, which is

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 10/22
analogous to the original procedure developed by Southern for DNA, is possible. However, because of the highly cross-linked nature of the acrylamide gels used forprotein separations it is extremely slow (days).
Semidry Blotting
Reference: M. Lauriere, Anal. Biochem. 212: 206-211 (1993)
Semidry blotting is becoming increasingly popular as it is fast, uses little buffer, and the progress of the transfer can be readily checked. A wide variety of buffershave been used. The procedure we will adopt is more complicated than most since it uses 4 different buffers. However, it has the advantage that both high and low
molecular weight proteins can be quantitatively transferred. This is because the buffers are cleverly designed to efficiently elute the proteins out of the gel and
deposit them in high yield on the membrane (see the diagram below). Buffer I, closest to the cathode, contains a high concentration of SDS at an alkaline pH. This
buffer serves as a continuous source of SDS during electrophoresis and helps to solubilize proteins. The SDS-protein complexes will be negatively charged at the
alkaline pH and readily move to the anode. Buffer II, which is just underneath buffer I, reproduces the electrophoretic conditions of the SDS-PAGE separation with
respect to pH, ionic strength, and presence of SDS. Under these conditions the resulting SDS protein complexes in the gel continue to move rapidly to the anode.
However, as the proteins approach the membrane these conditions change rapidly. Buffer III contains no SDS and has a low pH and a high methanol concentration.This buffer is designed to stop most of the proteins at the surface of the membrane. The acidification of the medium promotes the dissociation of the SDS from the
proteins. Once the SDS is removed, most proteins become less soluble and lose their negative charge that stops them from migrating farther toward the anode. The
high concentration of methanol in this buffer also tends to favor protein precipitation at the membrane. Buffer IV raises the pH of the transferred proteins to 10.4 at
which pH basic arginine and lysine residues are positively charged. Thus, any protein that fails to stop at the pH 3.8 boundary, will acquire a net positive charge and
be directed back towards the cathode giving it a second chance to bind at the membrane. While this four buffer system may represent biochemical "overkill", it may
be useful for blotting very valuable samples or high molecular weight proteins which can be difficult to transfer quantitatively by standard methods.
THROUGHOUT THE PROCEDURE WEAR GLOVES TO AVOID GETTING OILY PRINTS ON THE MEMBRANES WHICH CAN INHIBIT TRANSFER.
ALSO BE SURE TO AVOID BUBBLES AND EXCESS LIQUID WITHIN THE GEL SANDWICH. TREAT THE BLOTTING APPARATUS GENTLY AS THE
GRAPHITE ELECTRODES ARE SOFT AND EASILY BROKEN.
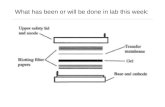
1. The gels and blotting membranes sandwiches are assembled with Whatman No. 3 paper and blotting pads in blotting sandwiches as shown in the figure
given below. The composition of the buffers is as given in Table I.
2. Dry blotting pads large enough to completely surround the gel to
be blotted are wetted with the desired buffer. The Whatman paper
is wetted with the desired buffer and the membrane filter with
deaerated water. (If using PVDF membranes, they must first be
wetted in methanol, then water.)
3. Equilibrate the gels for 15 minutes in Buffer II. (The gels will
swell slightly).
4. Assemble the gel sandwich as shown using a frame of polyester
transparency film (from overhead projectors) around each gel to
prevent buffer mixing and electric current leakage.
5. Transfer for 1 h/mm of gel thickness at a constant current of 1mA/cm2. Note: If you are transferring many gels, you can place a
wet sheet of dialysis membrane on top of the bottom stack, then
place a second stack on top of the first. The dialysis membrane will
serve to ensure that no proteins from the upper sandwich migrate
through to the lower sandwich.
6. After removing the blotted Immobilon-P filter you must first dry it to fix the proteins to the membrane before continuing on to any transillumination or
rapid immunodetection procedures to ensure optimum results. There are four drying options.
a) Soak the membrane in 100% methanol for 10 sec to drive out the water. Then place the blot on a piece of filter paper. Wait for the methanol to
evaporate (~15 Min)
b) Place the blot in a vacuum chamber (secure the blot between two sheets of filter paper (~30 min.)
c) Incubate the blot at 37oC (~1 hr)
d) Place the blot on a lab bench to let it dry at room temperature (~2 hr)
MATERIALS
1. 120 mM lactic acid (FW=90.08)
Compound Amount/500 ml Amount/1000 ml
dl-Lactic acid syrup (85% W/W, Sigma L-1250) 5.26 ml 10.52 ml
1.209 g/ml = 11.4 M
H2O to volume
2. 200 mM Tris base (FW=121.1)
Compound Amount/100 ml Amount/1000 ml

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 11/22
Deionized H2O 70 ml 700 ml.
Tris base 2.422 g 24.22 g
Allow the solution to cool to room temperature before making the final pH adjustments.
H2O to volume and sterilize by autoclaving.
NOTE: If the solution has a yellow color, discard it and obtain better-quality Tris
Also, make sure the pH electrode being used measures Tris accurately. Many electrodes will not measure Tris accurately.
3. Transfer Buffers I through IV can be made as follows:
Buffer preparation from stock solutions (for 100 ml final volume)
Buffer Stock A Stock B SDS Methanol Final composition of
Solution (ml) (ml) (g) (ml) buffers
I 50 50 0.4 -- 0.4% (w/v) SDS, 60
mM lactic acid, 100
mM Tris, pH 8.4
II 12.5 12.5 0.1 -- 0.1% (w/v) SDS ,
15 mM lactic acid, 25
mM Tris, pH 8.4
III 50 10 -- 20 20% v/v methanol,
60 mM lactic acid,
20 mM Tris, pH 3.8
IV -- 50 -- 20 20% (v/v) methanol,
100 mM Tris, pH 10.4
Note: Stock A, 120 mM lactic acid; Stock B, 200 mM Tris base.
Tank Blotting
This is a commonly used alternative to semidry blotting. Tank transfer uses much more buffer than the semidry procedure. The following protocol is written for a
large tank transfer apparatus (Hoefer) used for an overnight transfer. NOVEX makes a small combination apparatus that allows you to do both the Western and thesubsequent membrane transfer with the same device. If you do not already have a small acrylamide gel box and a semi-dry blotter or a tank blotter, then this is a
good buy because you get both pieces of equipment at a substantial net savings. NOVEX also sells very precise precast gels for about $8.00 each.
The Hoefer procedure is as follows.
1. Cut two sheets of Whatman #1 paper and one sheet of nitrocellulose to 8 cm x 5 cm. Wear gloves. Do not touch the paper or nitrocellulose with your
hands. Fingerprints can show up on blots (that way you know who has been touching the nitrocellulose).
2. Carefully wet the nitrocellulose in transfer buffer. Start from one corner and slowly lay the filter in to the transfer buffer.
3. Soak the Whatman paper and nitrocellulose in transfer buffer for at least 5 min.
4. Fill a large glass tray with transfer buffer and place one of the transfer blotting pads in it.
5. Build a blot sandwich on the sponge: Whatman paper, gel, nitrocellulose, Whatman paper. Carefully layer each piece onto the sandwich. Do not get
any bubbles in your sandwich.

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 12/22
6. Place sandwich into transfer tank. SDS in denaturing gel gives proteins a large net negative charge. Place sandwich such that proteins will move out ofthe gel and onto the nitrocellulose. Nitrocellulose side of sandwich should face positive pole.
7. Transfer proteins overnight at 0.2 Amp with cooling.
8. Turn off power and remove blot and membrane.
9. After removing the blotted Immobilon-P filter you must first dry it to fix the proteins to the membrane before continuing on to any transillumination orrapid immunodetection procedures to ensure optimum results. There are three drying options.
a) Soak the membrane in 100% methanol for 10 sec to drive out the water. Then place the blot on a piece of filter paper. Wait for the methanol toevaporate (~15. Min)
b) Place the blot in a vacuum chamber (secure the blot between two sheets of filter paper (~30 min.)
c) Incubate the blot at 37oC (~1 hr)
d) Place the blot on a lab bench to let it dry at room temperature (~2 hr)
SOLUTIONS:
1. 10x Running buffer
Compound Amount/100 ml Amount/1000 ml
Tris Base 3.0 g 30 g
Glycine 14.4 g 144 g
H2O to 1 liter, do not pH
2. Transfer Buffer
Final Concentration Stock Solution Amount/100 ml Amount/1000 ml
0.1% SDS 0.1 g 1.0 g
20 % methanol methanol 20 ml 200 ml
1X Running buffer 80 ml 800 ml
3. Methanol
PROTOCOL 1.5: STAINING OF WESTERN BLOTS FOR TOTAL PROTEIN
REFERENCE: D. BICKAR AND P. READ. ANAL. BIOCHEM. 203: 109-115 (1992)
INTRODUCTION:
Many different procedures have been published for staining total protein on Western blots after transfer. Some employ dyes such as Coomassie Brilliant Blue R250,Amido Black, Ponceau S, Fast green FCF or Eosin Y. Others used colloidal dyes such as India ink or colloidal gold or iron. Finally there are procedures using silver
staining or biotinylation. We have found the procedure below to be by far the fastest and easiest. It is also very inexpensive, sensitive, reversible, and applicable to a

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 13/22
wide variety of proteins. Finally, it works well on both nitrocellulose and PVDF (Immobilon) but not nylon type filters. (Remember that Immobilon filters always need
to be prewet in methanol and then water prior to carrying out any procedure using aqueous buffers!). The stain can also be used for staining gels prior to transfer, butin this application is not notably superior to Coomassie Brilliant Blue.
Staining
1. Submerge the blot for 30" in the staining solution. (If staining is not obvious, continue up to five minutes)
2. Rinse two or three times with 12 mM HCl to remove unbound dye. Protein will stain a vivid, turquoise-blue with little or no background staining of thefilter. On PVDF filters a rinse in 20% ethanol or methanol is necessary to remove all the unbound dye. (Immobilon P filters are more difficult to destainthan the original Immobilon filters).
3. Photograph the membrane through an orange filter in white light. Lay a ruler beside the filter so that the location of the molecular weight standardscan be readily determined if they fade during the hybridization process.
Destaining
1. Soak the blot in 0.5 M NaHCO3 for a few minutes and rinse repeatedly with deionized H2O to remove the bulk of the stain from the protein bands.
Not all the dye is removed, but that which remains does not appear to interfere with subsequent antibody detection methods.
NOTE: The sensitivity of the stain is about 10 ng of protein.
MATERIALS:
1. 12 mM HCl
Compound Amount/100 ml Amount/1000 ml
HCl (11.6 M, 36% ) 103 ml 1.03 ml
2. 0.05% CPTS Staining Solution
Final Concentration Stock Solution Amount/100 ml Amount/500 ml
CPTS (copper 50 mg 250 mg
phthalocyanin 3, 4', 4", 4"'-
tetrasulfonic acid tetrasodium
Dissolve salt
(Aldrich 24,535-6)
12 mM HCl to volume 12 mM HCL
3. 20% ethanol or methanol (for PVDF filters)
Compound Amount/100 ml Amount/1000 ml
Ethanol or Methanol 20 ml 200 mg
Deionized H2O to volume
4. 0.5 M NaHCO3
Compound Amount/500 ml Amount/1000 ml
NaHCO3 21 g 42 g
Deionized H2O to volume
PROTOCOL 1.6: RAPID IMMUNODETECTION PROTOCOL WITH ENHANCED CHEMILUMINESCENCE DETECTION (ECL)
SOURCE: ECL WESTERN BLOTTING PROTOCOLS, AMERSHAM CORP. 1994
This system is based on the detection of light emission from a secondary antibody linked to horseradish peroxidase which catalyzes the oxidation of luminol. Light
emission is detected on x-ray film. Although this system is more expensive and less convenient than the colorimetric alkaline phosphatase detection system, thesensitivity can be incredible.
The rapid protocol presented here is essentially that presented in Appendix I of the Amersham technical book on ECL. If time is in short supply the following
protocol allows the immunodetection using HRP-labeled antibodies to be completed in just over 2 hours, compared to 4 hours 15 minutes for the standard protocol. Ifdesired, the protocol can be further shortened by also optimizing the primary antibody for a shortened incubation.
Immediately following this protocol is a method for optimizing the ECL system before you even run an SDS-PAGE gel. If you are starting from scratch it is highly
recommended that you follow that protocol first.
PROCEDURE:
1. Electrophoresis and blotting should be done according to your favorite protocol.

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 14/22
2. Blocking the membrane. Non-specific antibody binding sites on membranes must be blocked before probing to prevent high background and/or
spurious binding to specific protein bands. Typical blocking reagents are 5% non-fat dry milk (blotto) or 10% BSA dissolved in phosphate bufferedsaline (PBS) or Tris buffered saline (TBS) solutions which contain 0.05% to 0.1% Trween 20.. Blocking may be accomplished in as little as 10 minutes or
may require overnight at 4oC depending on the antigen/antibody background and the specific membrane being used. Both the blocking times and the
optimal tween-20 concentrations have to be determined empirically.
3. During the blocking stage dilute the primary antibody in PBS-T or TBS-T.
Note: The optimum dilution of the primary antibody will vary and should be determined for each antibody used. See Protocol 1.8.
4. Rinse the membrane 3 X 15 seconds each in PBS-T or TBS-T. Slightly longer rinses are presumably acceptable.
5. Incubate the membrane in the diluted primary antibody for 1 hour (to overnight) at room temperature with agitation.
Note: A further shortening of the immunodetection is possible by increasing the primary antibody concentration. This allows a reduction in the incubation
time without compromising sensitivity. However, the optimal increase, which saves time, but does not cause severe background problems, must beoptimized for each new antibody.
6. Use PBS-T or TBS-T for the washing buffer. Briefly rinse the membrane using three changes of wash buffer, then wash twice for 10 minutes with fresh
changes of washing buffer, at room temperature. As large a volume of wash buffer as possible should be used at each time. As a guide, there should be at
least 4 ml per cm2 of membrane present.
7. During the wash steps dilute the HRP-labeled secondary antibody in PBS-T or TBS-T.
8. Incubate the membrane in the diluted secondary antibody for 15 minutes at room temperature with agitation.
9. Wash the membrane as described in step 6.
10. Signal Generation/Detection
Read through this whole section before proceeding. It is necessary to work quickly once the blots have been exposed to the detection solutions. All stepscan be carried out in a dark room; it is only necessary to switch off the light after step J below. Equipment that is needed includes an X-ray film cassette,
a roll of Saran Wrap (other 'cling-films' may not be suitable), a timer and blue-light sensitive autoradiography film, for example Hyperfilm-ECL (RPN2103). If possible, wear powder-free gloves because powder spots cause blank areas on films.
a. Mix an equal volume of detection solution 1 with detection solution 2 to sufficiently cover the blot, in this case 5 ml of solution 1 and 2.
b. Place a large piece of Saran Wrap on flat bench. Use clean Kimwipe to smooth out the Saran Wrap so that it is perfectly flat.
c. Use forceps to remove the blot from the hybridization tube. Be careful to only clamp the edges of the blot to prevent crushing the lane areas.Crushing sometimes causes high background.
d. Remove excess wash buffer by holding the blot vertically and touching one of the lower corners to a small pile of Kimwipes. Wait until all the
excess moisture is wicked from the blot.
e. Place the blot in the middle of the plastic sheet protector or Saran Wrap with the protein side up.
f. Using pipette draw up mixed developing solution and apply solution to the corners and middle of the blot. Add enough so that the entire blot is
covered and you can see a positive meniscus. Surface tension will hold the fluid on top. Stop adding the solution if it begins to flow out on thesurrounding page protector or Saran Wrap.
g. Incubate for 1 minute at room temperature.
h. While the sample is incubating put a clean piece of Saran Wrap (big enough to wrap the entire blot in) or page protector on the bench and spread
it out with a Kimwipe.
i. Using fresh Kimwipes, drain off excess detection buffer as described in Step 3.
j. If using Saran Wrap, place the blot protein side down onto the new piece of Saran Wrap. Fold the Saran Wrap over the back of the membrane so
the membrane is totally covered, and smooth out air pockets. Ideally the Saran Wrap on the protein side of the blot should remain even andsmooth using this procedure. As soon as the membrane is wrapped, flip it over so the protein side faces up, and attach fluorescent markers
asymmetrically to the side of the blot so that you can orient the autorad later. Alternatively, you can use the plastic sheet protectors. In this casethe membrane can be placed protein side up, then covered, and the fluorescent marker attached to the top of the sheet protectors. These aredifficult to clean adequately and should be thrown away after the Western has been developed.
k. Place the wrapped blot(s), protein side up, in the film cassette. Work as quickly as possible; minimize the delay between incubating the blots insubstrate and exposing them to the film.
l. Switch off the lights in the dark room and place a sheet of autoradiography film (for example Hyperfilm-ECL) on top of the blots, close the
cassette and expose for 15 seconds to 10 minutes or longer if necessary.
m. Interpret results.
11. For a Western blot detected with antibodies of known activity:
a. If a previous Western blot resulted in high background, dilute the HRP conjugate at least two-fold (when using biotin-streptavidin system,
dilute both biotinylated antibody and streptavidin-HRP by the same amount) for future detections.

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 15/22
b. If a previous Western blot resulted in non-specific banding, but clear background, dilute the primary antibody two-fold for futuredetections.
Background Correction
If a result has background or excess "banding" (cross-reactivity) these methods can be used to produce a result that is acceptable. While these three suggestionscan improve signal to noise, the best results are obtained after correct antibody concentrations are determined as described above.
1. Remove the membrane from the plastic wrap and repeat the final washing step. Background and unwanted bands (cross-reaction) are lower affinity
reactions and the antibodies will wash off faster than specifically-bound antibodies. Remember to re-incubate the blot in ECL detection reagents beforeexposing to film.
2. Allow the membrane to stand (not exposing to film) for several minutes; with time the HRP generated signal will decrease. In general, diffuse areas of
background will disappear faster than specific signal; resulting in improved signal to noise.
3. Treat membrane with 5-10% H2O2 in PBS for up to 30 minutes. Hydrogen peroxide treatment can help reduce backgrounds and will not affect themembrane. This treatment may not be suitable for rare antigen detection or weakly binding antibodies.
Materials:
1. Primary Antibody
2. Secondary Antibody
3. 1X Phosphate Buffered Saline, pH 7.2 (PBS)
Compound Amount/ 1000 ml
Deionized H2O 800 ml
NaCl 8 g
KCl 0.2 g
Na2HPO4 1.44 g
KH2PO4 0.24 g
Adjust pH to 7.2 with HCl. Add deionized H2O to 1 L.
Dispense the solution into aliquots and sterilize them by autoclaving for 20 minutes at 15 lb/sq. in. on liquid cycle.Store at room temperature.
4. PBST (0.1 % SDS) Wash Buffer
Compound Amount/1000 ml Amount/4000 ml
Polyoxethylene Sorbitan Monolaurate 1 g 4 g
(Tween 20) Sigma P-1379
1 X PBS, pH 7.2 to 1000 ml to 4000 ml
5. PBST (0.3 % ) Wash Buffer
Compound Amount/1000 ml Amount/4000 ml
Polyoxethylene Sorbitan Monolaurate 3 g 12 g
(Tween 20) Sigma P-1379
1 x PBS, pH 7.2 to 1000 ml to 4000 ml
6. Tris-Buffered Saline, pH 7.4 (PBS)
Compound Amount/ 1000 ml
Deionized H2O 800 ml
NaCl 8 g
KCl 0.2 g
Tris base 3 g
Adjust pH to 7.4 with HClAdd deionized H2O to 1 L
Dispense the solution into aliquots and sterilize them by autoclaving for 20 minutes at 15 lb/sq. in. on liquid cycle.Store at room temperature.
7. TBS-T is TBS buffer supplemented with the desired amount of Tween-20
8. 10% non-fat dried milk (Carnation) in PBS

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 16/22
Compound Amount/100 ml Amount/500 ml
Non-fat dried milk 10g 100 g
H2O to 100 ml to 100 ml
Stir vigorously on stir plate until dissolved which may take an hour or more.
9. ECL Western Kit with detection reagents
10. Saran Wrap or Page Protectors
11. Immobilon P (Millipore)
12. Plastic Page Protectors
13. 5-10% H2O2 in PBS
Compound Amount/100 ml Amount/500 ml
H2O2 5 to 10 ml 25 to 50 ml
Deionized H2O to volume
Homemade ECL Reagents
Note: Although the majority of workers continue to use the original ECL reagent developed by Amersham, Pierce now sells several alternative formulations which seem to offer the advantages ofa longer signal output and lower cost. Pierce also sells an ultrasensitive reagent that is very expensive, but works well. For routine use, you can use "homemade" ECL reagents. Two formulationsfrom the Internet are:
14. Stock Solutions For ECL - Formula 1
250 mM Luminol (Sigma A-8511, 5g)
Compound Amount/ 10 ml
DMSO 10 ml
Luminol 0.44 g\
Store in a dark bottle
90 mM p-Coumaric acid (Sigma C-9008, 10 g - there are smaller sizes)
Compound Amount/ 10 ml Amount/ 50 ml
DMSO 10 ml 50 ml
p-Coumaric acid 0.15 g .75 g
1 M Tris, 8.5 (FW=121.1)
Compound Amount/ ml Amount/ ml
Deionized H2O 800 ml
Tris base 121.1 g
Adjust to pH 8.5 by adding concentrated HCl.
Allow the temperature to cool to room temperature before making the final adjustments to the pH. This is because the pH of Tris solutions are temperature-dependent and decrease approximately
0.03 pH for each 1oC increase in temperature. For example, a 0.05 M solution has pH values of 9.5, 8.9, and 8.5 at 5oC, 25oC, and 37oC respectively.
Sterilize by autoclaving.
Solution A
Compound Amount/ 100 ml Amount/ 500 ml
Luminol Stock 1.0 ml 5.0 ml
p-Coumaric acid stock 0.44 ml 2.2 ml
1 M Tris-HCl, pH 8.5 10 ml 50 ml
Deionized H2O to volume.
Store at 4oC under light tight conditions.
Solution B
Compound Amount/ 10 ml Amount/ 100 ml
1 M Tris, pH 8.5 1.0 ml 10 ml

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 17/22
30% H2O2 6.1 ml 61 ml
Deionized H2O to volume
Store at 4oC, but goes bad quickly so make weekly.
Mix Solutions A and B 1:1 just prior to use. Do not allow any cross-contamination to occur.
15. Stock Solutions for Luminol - Formula 2
Luminol Stock (Sigma A-8511, 5g)
Compound Amount/ 10 ml Amount/ 50 ml
DMSO 10 ml 50 ml
4-Methylumbellierone 17.7 mg 88.5 mg
Store in a dark bottle
100 mM 4-Methylumbelliferone
Compound Amount/ 10 ml Amount/ 50 ml
DMSO 10 ml 50 ml
4-Methylumbellierone 176 mg 880 mg
Combined Solution
Compound Amount/ 10 ml Amount/ 50 ml
4-Methylumbelliferone Stock 40 ml 200 ml
Luminol Stock 75 ml 375 ml
30% H2O2 2.8 ml 14.0 ml
Deionized H2O to volume.
Make just prior to use. Works reliably and is reproducible, but is slightly less sensitive than the commercial formulations.
Alternative Combined Solution
Compound Amount/ 10 ml Amount/ 50 ml
4-Methylumbelliferone Stock 40 ml 200 ml
Luminol Stock 75 ml 375 ml
Sodium perborate 6.15 mg 30.75 mg
Deionized H2O to volume.
Make just prior to use. Works reliably and is reproducible, but is slightly less sensitive than the commercial formulations.
In our limited experience with the formulation given in 14, we found sensitivity similar to the Amersham ECL reagent. We have not tried formulation 15.
PROTOCOL 1.7: OPTIMIZING ECL WESTERN BLOTTING
SOURCE: AMERSHAM TECHTIP 129
INTRODUCTION:
This protocol is designed to provide a guideline for optimizing Western blot detection using the ECL Western blotting detection system (RPN.2106, RPN.2108,RPN.2109). Because ECL is extremely sensitive, determination of optimum antibody concentrations is important to ensure the best results. Generally, less primary
and secondary antibody is required for sensitive detection when using ECL compared to colorimetric detection. Reduction in antibody use is one of the many benefitsof ECL for Western blot detection. Additional benefits include, (1) greater sensitivity than traditional colorimetric methods; (2) rapid results in seconds to minutes;
(3) results on film, hard copy results which do not fade; (4) ability to make multiple copies of results.
Determination of Optimum Antibody Concentration
The key to sensitive detection is determining the optimal antibody concentrations. While proper blocking and washing are important, they are only temporarymeasures until the proper antibody concentration is determined. Outlined below are protocols for determining optimal antibody concentrations for the detection of
ECL Western blots.
1. For a primary antibody/antiserum with unknown activity, a dot blot is a quick and effective method for determining optimum dilution. Differentblocking and washing steps than those offered below may be required for some antibodies.
a. Spot approximately 1-5 mg of protein sample on nitrocellulose (e.g., Hybond ECL, RPN.2020D); allow to dry. Prepare one dot blot foreach primary antibody dilution to be tested.
b. Block for 11/2 hour (e.g. 10% non-fat dried milk in PBS plus 0.1% Tween 20) at room temperature. Wash 3 X 5 min. in 0.1% PBS Tween-
20.
c. Prepare several dilutions of primary antibody: 1/100, 1/500, 1/1000, 1/5000 for instance; incubate 1 blot with each antibody dilution for 1

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 18/22
hour at room temperature.
d. Wash (e.g., PBS plus 0.1% Tween 20) 3x for 5 minutes.
e. Dilute secondary antibody; incubate each test blot for 30 min. at room temperature.
f. Wash 3x for 5 minutes in 0.3% Tween 20 washes for 5 minutes each. Some weak binding antibodies can be adversely affected by PBS-
-0.3% Tween 20. In this case 3 X 5 min. washes in PBS--0.1% Tween are recommended.
g. Incubate in ECL detection reagents and expose to film as directed in protocol booklet.
2. For a secondary antibody with unknown activity, a dot blot is also effective.
a. Spot approximately 1-5 mg of protein sample on nitrocellulose (e.g., Hybond ECL, RPN.2020D); dry at room temperature. Prepare 1 dot
blot for each secondary antibody dilution to be tested.
b. Block for 1 hour (e.g., 10% non-fat dried milk in PBS plus 0.1% Tween 20) at room temperature.
c. Dilute primary antibody; incubate each test blot for 1 hour.
d. Wash (e.g., PBS plus 0.1% Tween 20) 3x for 5 minutes.
e. Prepare several dilutions of secondary antibody: for example 1/3,000, 1/5,000, 1/7,500, 1/10,000, 1/25,000, 1/50,000; incubate one blot with
each dilution for 1 hour at room temperature.
f. Wash 3x for 5 minutes each in PBS--0.1% Tween 20 followed by three PBS--0.3% Tween 20 washes for 5 minutes each. Some weakbinding antibodies can be adversely affected by PBS--0.3% Tween 20. In this case 6 washes in PBS--0.1% Tween are recommended.
g. Do not let the membrane dry out. Keep it moist by capping the tube again until time for signal detection.
3) Perform the detection with ECL reagents as outlined starting on step 3 in Protocol 1.7.
The Materials List Follows Protocol 1.7.
PROTOCOL 1.8: EXPRESSION OF CLONED DNA IN PET PLASMIDS AND DETECTION OF RESULTING PROTEIN
INTRODUCTION:
There are a large number of systems available for expressing cloned genes in a variety of hosts. Many of these hosts e.g. yeast or insect cells are capable of
producing functionally active mammalian proteins. Bacterial cells are also capable of producing functionally active mammalian proteins, but often bacterially-expressed proteins are non-functional either because they lack important post-translational modifications such as phosphorylation or glycosylation and/or because
the proteins fold improperly and aggregate. Despite these limitations, bacterial expression systems can still be useful for a variety of studies. For example, they canproduce large quantities of protein for antibody production or for use as substrates for post-translational modification enzymes.
Expression systems based on the expression of proteins from bacteriophage T7 promoters are especially popular because they can produce extremely high levels of
the desired protein amounting to as much as 25% of the total cell protein. The efficiency of these systems derives from the fact that the T7 RNA polymerase ishighly selective for transcription from its own promoters. As a result, in the presence of T7 RNA polymerase, nearly all the cell’s production of proteins comes fromthe cloned gene. Indeed, the system is so efficient that often the problem is ensuring that there is not premature expression of toxic proteins during the growth of the
cells.
Because of the problem of premature expression, the developers of the T7 system have developed ways of keeping tight control on the expression of the clonedgene. The first level of control is to control the level of the T7 RNA polymerase itself. This is usually accomplished by placing the polymerase under the control of
the highly inducible lacUV5 promoter which can be induced by IPTG. However, even though the basal expression from this promoter is quite low, modest amounts ofT7 RNA polymerase can still be expressed in such uninduced cells. To circumvent this low level of expression several procedures have been developed. One uses
the presence in the cell of a second plasmid that produces T7 lysozyme-an inhibitor of the polymerase. In another scenario, the cloned gene itself is placed under thecontrol of the lac operator which reduces expression in the absence of IPTG.
The Novagen Co. (Madison, WI) has developed a large number of such vectors under the name pET vectors. Many of these vectors have special features that
facilitate the purification of the expressed proteins such as affinity tags and protease sensitive sites for removing the affinity tags after purification.
In the present protocol, inserts that have been subcloned into the vector pET-28 in the host cell BL21(DE3) are induced with IPTG to provide expression of thepolymerase. The gene providing the polymerase is actually contained within a lambda phage DE3 that lysogenizes the host. Proteins from the induced cells are run
on SDS/PAGE and the fusion protein identified by its high level of expression and its molecular weight. A more definitive identification of the protein would involveWestern blotting to ensure that the proper gene had been cloned and expressed.
PROCEDURE:
1. For screening of protein expression, it is recommended to start with multiple colonies. In some circumstances some colonies may not be expressing ashigh as the others. Make a 1:100 dilution of an overnight culture of BL21(DE3)pLysS containing your gene cloned into the pET vector into 5 ml of LB
with appropriate antibiotics for plasmid selection.
2. Grow at 370C for 1.5-3 h until the OD600 is about 0.3.

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 19/22
3. The cultures then need to be split into two halves. Induce one half with IPTG to a final concentration of 0.2 mM. The other half should be leftuninduced as a control.
4. Continue the incubation for another 1-4 hours. The time depends on the particular protein.
5. Transfer the cultures into screw-cap or lock-top microfuge tubes and spin them in a microcentrifuge for 1 min.
6. Discard the supernatant and drain the pellets using a Pipetman tip.
7. You can freeze the pellet at -20oC to help lyse cells and to slow degradation. You can leave the pellet frozen indefinitely if your protein of interest is
fairly stable. However it is best to immediately proceed to step 8.
8. Immediately after the supernatant has been removed from the cell pellet (or the stored cell pellet has been thawed rapidly), add 50 - 100 ml of sterile water andresuspend cell pellet. You may have to pipette the cells up and down to resuspend the cells. It is important to resuspend the cells before adding the lysis buffer
to ensure complete lysis.9. Immediately add a volume of ice cold 2X sample buffer approximately equal to the estimated volume of the cell pellet plus the water added to resuspend the
pellet (~75 to 100 ml). Be sure to keep your final volume between 100-200 ml to keep your protein concentration sufficiently high.
9. Vortex to resuspend the pellet and immediately place the screw cap microfuge or lock top microfuge tube in boiling water bath for 2-5 min to preventdegradation. After boiling, you may again have to pipette up and down to resuspend the denatured proteins. If your prep is very viscous, sonicate the
sample with two short 5 sec. bursts at medium power and chill the sample on ice until you are ready to load it on the SDS/PAGE Gel. This treated samplecan be stored frozen for future runs.
10. The samples next need to be then analyzed on a SDS/PAGE gel, Protocol 1.3 starting at the section on Sample Loading and Final Assembly of the SE
250.. Usually about 5 ml of sample gives sufficient protein to allow visualization of the bands upon staining.
MATERIALS:
1. LB Medium (Luria-Bertani Medium)
In a clean dry 2L Erlenmeyer flask add the constituents listed below. If you turn the Bacto-tryptone and Yeast extract bottles on their side and gently tap them to loosen up the contents before youopen the bottle, the contents will shift so they can be more easily scooped out of the bottle without generating a large amount of dust.
Compound Amount/1000 ml
Bacto-tryptone 10 g
Yeast extract 5 g
NaCl 10 g
Add 900 ml H2O. Stir until the solutes have dissolved.
Adjust to pH 7.5 with 5 N NaOH.
Adjust the volume of the solution to 1 liter with deionized H2O.
Sterilize by autoclaving for 20 minutes at 15 lb/sq. in. on liquid cycle.
2. Appropriate antibiotics For Vector Being Used
Stock solution a Working concentration
Concentration storage stringent
plasmids
relaxed
plasmids
Ampicillin 50 mg/ml in H20 -200C 20 m g/ml 50 m g/ml
Carbenicillin 50 mg/ml in H20 -200C 20 m g/ml 50 m g/ml
Chloramphenicol 34 mg/ml in ethanol -200C 25 m g/ml 170 m g/ml
Kanamycin 10 mg/ml in H20 -200C 10 m g/ml 50 to 100 m g/ml
Streptomycin 10 mg/ml in H20 -200C 10 m g/ml 50 m g/ml
Tetracyclineb 5 mg/ml in ethanol -200C 10 m g/ml 50 m g/ml
a Stock solutions of antibiotics dissolved in H20 should be sterilized by filtration through a 0.22-micron filter.
Antibiotics dissolved in ethanol need not be sterilized. Store solutions in light-tight containers.
b Magnesium ions are antagonists of tetracycline. Use media without magnesium salts (e.g., LB medium) for selection of bacteria resistant to tetracycline.
3. 100 mM IPTG (FW = 238.3)
Compound Amount/1 ml Amount/10 ml
Isopropylthio-b-D-galactosidase 2.38 mg 23.83 mg
(IPTG)
Deionized H2O to volume.

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 20/22
Sterilize by filtration through a 0.22 mm disposable filter.
Dispense the solution into 1-ml aliquots and store them at -20oC.
4. SDS PAGE Gel of appropriate concentration
5. 0.5 M Tris-Cl pH 6.8
Compound Amount/50 ml Amount/100 ml
Tris-Cl 3.0 g 6.0 g
Deionized H2O 45 ml 95 ml
Adjust to pH 6.8 with 1 M HCl.Add deionized H2O to 50 ml to 100 ml
Allow the temperature to cool to room temperature before making the final adjustments to the pH. This is because the pH of Tris solutions are temperature-dependent and decrease approximately
0.03 pH for each 1oC increase in temperature. For example, a 0.05 M solution has pH values of 9.5, 8.9, and 8.5 at 5oC, 25oC, and 37oC respectively.
Sterilize by autoclaving.
6. 10% Sodium Dodecyl Sulfate Sodium Salt (SDS, FW=288.4, also called Sodium Lauryl Sulfate)
Compound Amount/500 ml Amount/1000 ml
Deionized H2O 450 ml 900 ml
SDS 50 g 100 g
Heat to 68oC to assist dissolution.
Adjust pH to 7.2 by adding a few drops of concentrated HCl.Adjust to volume with deionized H2O.
Dispense in aliquots. There is no need to sterilize 10% SDS.
Caution: Wear a mask when weighing SDS and wipe down the weighing area and balance after use because the fine crystals of SDS disperse easily.
7. 2X Sample buffer (Minus Bromophenol Blue)
Final Concentration Stock Solution Amount/10 ml Amount/50 ml
0.125 M Tris-Cl pH 6.8 0.5 M Tris-Cl pH 6.8 2.5 ml 12.5 ml
4 % SDS 10% SDS 4.0 ml 20 ml
10% Glycerol 2.0 ml 10 ml
10% 2-Mercaptoethanol 1.0 ml 5 ml
H2O 0.5 ml 3.5 ml
Divide in aliquots and freeze.
8. Boiling water bath.
9. Sonicator or 1 ml syringe with 19 and 25 gauge hypodermic needles.
10. Appropriate centrifuge tubes and screw cap microfuge tubes.
11. 10 X Bromophenol Blue
Compound Amount/1 ml Amount/5 ml
Bromophenol Blue 10 mg 50 mg
H2O to 1 ml to 5 ml
PROTOCOL 1.9 RAPID COOMASSIE BLUE STAINING OF PROTEIN GELS
REFERENCE: AUSUBEL ET AL. 10.6.1
INTRODUCTION:
There are innumerable variations on the use of Coomassie Brilliant Blue R-250 to stain proteins in gels. Some are faster but less sensitive. In addition there arevery sensitive silver staining procedures which are about 100 x as sensitive as the Coomassie procedures. In general, the silver stain procedures take somewhat
more hands on time, and require considerable attention to cleanliness. There are also some rapid staining procedures using Cu salts that are intermediate insensitivity between the Coomassie Blue and silver staining procedures. Ausubel et al. gives additional protocols for doing Coomassie and Silver staining of gels(10.6.1-10.6.2). The following is a rapid Coomassie stain protocol. Because the Coomassie blue concentration is lower than that used in the basic Coomassie staining
protocol, the gel background never stains very darkly and the bands can be seen even while the gel remains in the staining solution. Another difference from thebasic staining protocol is that isopropanol is substituted for methanol in the fixing and destaining solutions. This protocol is slightly less sensitive than the basic
staining protocol.
PROCEDURE:
1. Place the polyacrylamide gel in a clean plastic or glass container. Cover the gel with isopropanol fixing solution and shake gently at room temperaturefor 15 min. (0.7 mm thick gel) to 60 min. (1.5 mm thick gel).
2. Pour out fixing solution. Cover the gel with rapid Coomassie Blue staining solution and shake gently until desired intensity is reached. Bands becomevisible in 30 minutes and will become more pronounced with longer incubations of 2 h to overnight. A disadvantage of the longer staining times, however,
is that the gels require longer to destain.

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 21/22
3. Pour out staining solution. Cover gel with 10% acetic acid to destain and agitate gently ³ 2 h at room temperature until a clear background isobtained.
4. If a clear background is not obtained, pour out staining solution and repeat step 3. This step is generally unnecessary.
Note: You can speed up the destaining somewhat by including a Kimwipe or piece of sponge in the destaining solution to sop up the dye which diffuses
from the gel.
5. Store gel in 7% acetic acid or water, or in plastic wrap at 4oC.
6. If desired photograph or dry gel between membranes.
MATERIALS:
1. Isopropanol Fixing Solution
Final Concentration Stock Solution Amount/1000 ml Amount/2000 ml
25% isopropanol isopropanol 250 ml 500 ml
10% acetic acid acetic acid 100 ml 200 ml
Deionized H2O to volume.
2. Coomassie Brilliant Blue R-250 Stain: (10% acetic acid)
Final Concentration Stock Solution Amount/1000 ml Amount/2000 ml
0.012% wt/vol Coomassie 0.12 g 0.24 g
Brilliant Blue R-250
(BIO-RAD 161-0406)
10 % Acetic Acid Acetic Acid 100 ml 200 ml
Deionized H2O to volume.
3. Destaining solution
Final Concentration Stock Solution Amount/1000 ml Amount/2000 ml
10% acetic acid Acetic acid 100 ml 200 ml
Deionized H2O to volume.
PROTOCOL 1.10 STAINING OF POLYPEPTIDES WITH COOMASSIE BRILLIANT BLUE IN THE PRESENCE OF PICRATE
REFERENCE J.L. STEPHANO ET AL. ANAL. BIOCHEM. 152: 308-313 (1985)
INTRODUCTION:
Although this method of Coomassie staining is not appreciably more sensitive than standard methods, it is much faster for staining gels lacking detergents e.g. acetic
acid urea gels and considerably faster even for gels containing detergents like SDS or Triton. Furthermore one can sometimes stain low molecular weight peptidesthat are unstained by conventional Coomassie stain., and the stained gels are suitable for densitometry. About the only drawback to this technique is that bottles of
picric acid can explode if allowed to completely dry or if certain metal picrate salts are formed. Hence it is prudent to add a little water to the bottles after openingthem and wrap them tightly with Parafilm, and to avoid contact with metals. Waste picrate solutions should be washed down the drain with plenty of water. We give
here only the procedure of SDS gels which require a washing to remove detergent prior to staining. For other types of gels consult the original reference.
Procedure: (for standard Laemmli gels containing SDS)
1. Wash the detergent out of the gels by 3 washes in fixing solution for 20 minutes each.
2. Add the picrate Coomassie stain. The bands should begin to appear in 10 to 15 minutes although very minor bands may take up to several hours to
stain. The gels do not overstain even if left in the staining solution overnight. Although the paper reports that destaining is not needed, the staining willbe a bit more distinct if following the brief staining period the gels are washed for a half hour or so in water. The gels can then be dried andphotographed. The stain can be reused for multiple gels until staining is no longer satisfactory.
MATERIALS:
1. Fixing solution
Final Concentration Stock Solution Amount/1000 ml Amount/2000 ml
45% methanol Methanol 450 ml 900 ml
10% acetic acid Acetic Acid 100 ml 200 ml
Deionized H2O to volume 450 ml 900 ml
2. 0.2% Coomassie Brilliant Blue R-250 dissolved in fixing solution

1/21/13 UNIT 1: WESTERN BLOTTING
www.med.unc.edu/pmbb/PMBB web site/mbt/WESTERN.htm 22/22
Compound Amount/250 ml Amount/500 ml
Coomassie Brilliant Blue R-250 0.5 g 1.0 g
Fixing solution to volume
3. 0.1 M Picric Acid, pH 7.0 (FW=229.1)
Compound Amount/ 100 ml
Deionized water 75 ml
Picric acid 2.291 g
Adjust to pH 7.0 with 1M NaOHAdd deionized H2O to 100ml.
4. Picrate Coomassie Staining Solution
Compound Amount/120 ml Amount/1250 ml
0.1 M picric acid adjusted to pH 7.0 100ml 1000 ml
0.2% Coomassie Blue R dissolved in fixing solution 25 ml 250 ml

![Western Blotting BCH 462[practical] Lab#6. Objective: -Western blotting of proteins from SDS-PAGE.](https://static.fdocuments.in/doc/165x107/56649dc85503460f94abe06c/western-blotting-bch-462practical-lab6-objective-western-blotting-of.jpg)