
THE OP BIOLOGICAL CHEMISTRY Vol. No. 24. 12060 … · Co-induction of Fatty Acid Reductase and...
6
THE JOURNAL OP BIOLOGICAL CHEMISTRY Vol. 255, No. 24. Issue of December 25. pp. 12060-12065, 1980 Printed ~n 11. S A Co-induction of Fatty Acid Reductase and Luciferase during Development of Bacterial Bioluminescence* (Received for publication, April 30, 1980) Denis Riendeau and Edward Meighen From the Department of Biochemistry, McGdl University, Montreal, Quebec, Canada The luminescent bacterium Photobacteriumphospho- mum has been shown to possess a fatty acid reductase based on the stimulation of the aldehyde-dependent luminescent reaction on incubation of the enzyme with ATP, NADPH, and tetradecanoic acid (Riendeau, D., and Meighen, E. (1979) J. Biol. Chem 254, 7488-7490). A direct, luciferase-independent assay for the fatty acid reductase has now been developed using [’Hltetra- decanoic acid as substrate and thin layer chromatog- raphy to separate and identify the products of the reaction. Tritiated aldehyde was the only product of the reaction at early times of assay, and the amount produced was linearly dependent on time and extract concentration. The labeled aldehyde was further re- duced to alcohol after prolonged incubation, indicating that long chain aldehyde reductase(s) are also present in bioluminescent bacteria. Measurement of the fatty acid reductase activity in extracts during growth and development of the bioluminescent bacteria showed that the fatty acid reductase activity is co-induced with luciferase, suggesting that these enzymes are coordi- nately regulated and directly implicating the fatty acid reductase in aldehyde biosynthesis in the bacterial bio- luminescent system. The enzymatic synthesis of long chain aldehydes has been shown to occur in a number of different systems (1-13). Long chain aldehydes are produced during degradation of alkyl- glycerols (3, 4) and dihydrosphingosine (5), as intermediates in the synthesis of long chain alcohols involved in the forma- tion of wax esters (6, 7) and alkylglycerolipids (8), and as intermediates during the oxidation of hydrocarbons by certain microorganisms (9). In luminescent bacteria, a long chain aldehyde is required for the light emission catalyzed by luciferase (14, 15) and is oxidized to the corresponding acid during the light-emitting reaction (16, 17). The expression of bioluminescence occurs only at a late stage of the bacterial growth, and is accompanied by an increase in the relative amount of luciferasein the bacteria (18, 19). Information about the mechanism of synthesis of long chain aldehydes in these bacteria has only been obtained recently. The characterization of certain “dark” mutants of Benechea harueyi, the luminescence of which was stimulated by fatty acids as well as long chain aldehydes, has indicated that these bacteria are able to synthesize aldehydes from fatty acids in vivo (20). The first in vitro evidence for the mechanism of aldehyde synthesis was obtained when it was found that * This work was supported by Grant MT-4314 from the Medical Hesearch Council of Canada. The costs of publication of this article were defrayed in part by the payment of page charges. This article mustthereforebeherebymarked“aduertisement”inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact. NADPH and ATP could stimulate light emission by luciferase in the absence of added aldehyde in extracts of Photobacter- ium phosphoreum (21). Partial purification of the enzyme responsible for thisstimulationhasdemonstratedthatits efficiency in stimulating luciferase activity was dependent on tetradecanoic acid as well as NADPH and ATP, thus indicat- ing that it functioned as a fatty acid reductase (22). Although these initial studies showed that extracts of P. phosphoreum catalyze the synthesis of aldehydes from fatty acids, it could not be established whether aldehyde was the only or even the major product obtained from tetradecanoic acid or whether the aldehydewas further metabolized. Furthermore, compar- ison of fatty acid reductase levels in extracts containing vari- able amounts of luciferase was not possible using this assay system. In the presentwork, we have developed a direct and independent assay for fatty acid reductase activity by meas- uring the conversion of [”Hltetradecanoic acid to tritiated aldehyde. The assay was used to study the variations of the fatty acid reductase activity at different stages of bacterial growth in order to determine whether it correlates with the expression of the bioluminescent system. The present experi- ments have also shown that the fatty acid is initially converted only to aldehyde and then further metabolized to alcohol by extracts of the bioluminescent bacteria. EXPERIMENTAL PROCEDURES Materials-The following products were obtained from Sigma Chemical Co.: ATP (disodium, crystalline), NADPH (tetrasodium, type HI), FMN, tetradecanoic acid (99 to 100%). cis-9-tetradecenoic acid, tetradecyl alcohol, and horse liver alcohol dehydrogenase. Do- decyl and tetradecyl aldehyde were purchased from Aldrich Chemical Co. [“HITetradecanoic acid (150 mCi/mg) was prepared by New England Nuclear by reduction of cis-9-tetradecenoic acid with tritium gas in the presence of a 5% palladium on carbon catalyst. Cell Growth and Lysis-P. phosphoreum (NCMB 844) was grown at 20°C with limited aeration in complex medium containing 5 g of 30 g of NaCl, 3.7 g of Na,HPO,, 1 g of KHJ’OI, 0.5 g of (NHI)JHI’OI, Difco Bacto-Tryptone, 0.5 g of Difco yeast extract, 2 ml of glycerol, and 0.1 g of MgSO, per liter. Extracts were prepared from cells grown to peak luminescence (3,000 to 5,000 LU’/ml of culture). The cells were harvested by centrifugation at 12,OOO X g for 10 min, resuspended in 1 mM 8-mercaptoethanol (2% by volume of the original cell culture), and lysed by sonication at 4°C to give a uniform suspension. After removalof cellular debris by centrifugation at 27,000 X g for 15 min, the supernatant was made 0.1 M in 8-mercaptoethanol and 0.05 M in phosphate, pH 7.0, by the addition of 2 M /3-mercaptoethanol, 1 M phosphate, pH 7.0. Phosphate buffers were prepared by mixing appropriate amounts of K,HPO? and NaHrPOd. For the study of the expression of the luminescence and enzyme activityasafunction of growth,thecellsweregrownina New Brunswick Magnaferm incubator containing 9 liters of complex me- dium. The growth was initiated with an inoculum of luminescent cells to give an initial absorbance at 660 nm (AtKd,) of 0.05. Bacterial growth and luminescence invivo were followed by the increase in AMX, and in ‘ The abbreviations used are: LU, lightunits;TLC,thinlayer - .~ . chromatography. 12060
Transcript of THE OP BIOLOGICAL CHEMISTRY Vol. No. 24. 12060 … · Co-induction of Fatty Acid Reductase and...

T H E JOURNAL OP BIOLOGICAL CHEMISTRY Vol. 255, No. 24. Issue of December 25. pp. 12060-12065, 1980 Printed ~n 11. S A
Co-induction of Fatty Acid Reductase and Luciferase during Development of Bacterial Bioluminescence*
(Received for publication, April 30, 1980)
Denis Riendeau and Edward Meighen From the Department of Biochemistry, McGdl University, Montreal, Quebec, Canada
The luminescent bacterium Photobacteriumphospho- mum has been shown to possess a fatty acid reductase based on the stimulation of the aldehyde-dependent luminescent reaction on incubation of the enzyme with ATP, NADPH, and tetradecanoic acid (Riendeau, D., and Meighen, E. (1979) J. Biol. Chem 254, 7488-7490). A direct, luciferase-independent assay for the fatty acid reductase has now been developed using [’Hltetra- decanoic acid as substrate and thin layer chromatog- raphy to separate and identify the products of the reaction. Tritiated aldehyde was the only product of the reaction at early times of assay, and the amount produced was linearly dependent on time and extract concentration. The labeled aldehyde was further re- duced to alcohol after prolonged incubation, indicating that long chain aldehyde reductase(s) are also present in bioluminescent bacteria. Measurement of the fatty acid reductase activity in extracts during growth and development of the bioluminescent bacteria showed that the fatty acid reductase activity is co-induced with luciferase, suggesting that these enzymes are coordi- nately regulated and directly implicating the fatty acid reductase in aldehyde biosynthesis in the bacterial bio- luminescent system.
The enzymatic synthesis of long chain aldehydes has been shown to occur in a number of different systems (1-13). Long chain aldehydes are produced during degradation of alkyl- glycerols (3, 4) and dihydrosphingosine (5), as intermediates in the synthesis of long chain alcohols involved in the forma- tion of wax esters (6, 7) and alkylglycerolipids (8), and as intermediates during the oxidation of hydrocarbons by certain microorganisms (9).
In luminescent bacteria, a long chain aldehyde is required for the light emission catalyzed by luciferase (14, 15) and is oxidized to the corresponding acid during the light-emitting reaction (16, 17). The expression of bioluminescence occurs only at a late stage of the bacterial growth, and is accompanied by an increase in the relative amount of luciferase in the bacteria (18, 19).
Information about the mechanism of synthesis of long chain aldehydes in these bacteria has only been obtained recently. The characterization of certain “dark” mutants of Benechea harueyi, the luminescence of which was stimulated by fatty acids as well as long chain aldehydes, has indicated that these bacteria are able to synthesize aldehydes from fatty acids in vivo (20). The first in vitro evidence for the mechanism of aldehyde synthesis was obtained when it was found that
* This work was supported by Grant MT-4314 from the Medical Hesearch Council of Canada. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
NADPH and ATP could stimulate light emission by luciferase in the absence of added aldehyde in extracts of Photobacter- ium phosphoreum (21). Partial purification of the enzyme responsible for this stimulation has demonstrated that its efficiency in stimulating luciferase activity was dependent on tetradecanoic acid as well as NADPH and ATP, thus indicat- ing that it functioned as a fatty acid reductase (22). Although these initial studies showed that extracts of P. phosphoreum catalyze the synthesis of aldehydes from fatty acids, it could not be established whether aldehyde was the only or even the major product obtained from tetradecanoic acid or whether the aldehyde was further metabolized. Furthermore, compar- ison of fatty acid reductase levels in extracts containing vari- able amounts of luciferase was not possible using this assay system. In the present work, we have developed a direct and independent assay for fatty acid reductase activity by meas- uring the conversion of [”Hltetradecanoic acid to tritiated aldehyde. The assay was used to study the variations of the fatty acid reductase activity at different stages of bacterial growth in order to determine whether it correlates with the expression of the bioluminescent system. The present experi- ments have also shown that the fatty acid is initially converted only to aldehyde and then further metabolized to alcohol by extracts of the bioluminescent bacteria.
EXPERIMENTAL PROCEDURES
Materials-The following products were obtained from Sigma Chemical Co.: ATP (disodium, crystalline), NADPH (tetrasodium, type HI), FMN, tetradecanoic acid (99 to 100%). cis-9-tetradecenoic acid, tetradecyl alcohol, and horse liver alcohol dehydrogenase. Do- decyl and tetradecyl aldehyde were purchased from Aldrich Chemical Co. [“HITetradecanoic acid (150 mCi/mg) was prepared by New England Nuclear by reduction of cis-9-tetradecenoic acid with tritium gas in the presence of a 5% palladium on carbon catalyst.
Cell Growth a n d Lysis-P. phosphoreum (NCMB 844) was grown a t 20°C with limited aeration in complex medium containing 5 g of
30 g of NaCl, 3.7 g of Na,HPO,, 1 g of KHJ’OI, 0.5 g of (NHI)JHI’OI, Difco Bacto-Tryptone, 0.5 g of Difco yeast extract, 2 ml of glycerol,
and 0.1 g of MgSO, per liter. Extracts were prepared from cells grown to peak luminescence (3,000 to 5,000 LU’/ml of culture). The cells were harvested by centrifugation a t 12,OOO X g for 10 min, resuspended in 1 mM 8-mercaptoethanol (2% by volume of the original cell culture), and lysed by sonication a t 4°C to give a uniform suspension. After removal of cellular debris by centrifugation at 27,000 X g for 15 min, the supernatant was made 0.1 M in 8-mercaptoethanol and 0.05 M in phosphate, pH 7.0, by the addition of 2 M /3-mercaptoethanol, 1 M phosphate, pH 7.0. Phosphate buffers were prepared by mixing appropriate amounts of K,HPO? and NaHrPOd.
For the study of the expression of the luminescence and enzyme activity as a function of growth, the cells were grown in a New Brunswick Magnaferm incubator containing 9 liters of complex me- dium. The growth was initiated with an inoculum of luminescent cells to give an initial absorbance at 660 nm (AtKd,) of 0.05. Bacterial growth and luminescence in vivo were followed by the increase in AMX, and in
‘ The abbreviations used are: LU, light units; TLC, thin layer - .~ .
chromatography.
12060

Co-induction of Fatty Acid light units (LU) per ml of culture medium, respectively, where 1 LU is equivalent to 6 X 10'' quanta/s, based on the standard of Hastings and Weber (23). One unit of absorbance at 660 nm is equivalent to approximately 5 X 10" cells/ml.
A constant amount of cells was collected a t different intervals (milliliters of culture X Aw, = 20) as growth proceeded, and was stored froaen at -20°C. Each sample of cells was lysed a t 4'C in 2 ml of 1 mM P-mercaptoethanol by sonication and the cellular debris was removed by centrifugation as described above. For each extract, part of the supernatant was mixed with 2 M /I-mercaptoethanol in 1 M phosphate, pH 7.0 (0.05 ml/ml of extract) and was used for assays of the fatty acid reductase and luciferase while the activities of malate dehydrogenase and glucose-6-phosphate dehydrogenase and the pro- tein content were measured in the untreated fraction. P.phosphoreum (NCMB 844) was obtained from the National Collection of Marine Bacteria, Aberdeen, Scotland. Strain A-13 of P. phosphoreum was generously supplied by Dr. J . Lee, University of Georgia.
Luciferase Purification a n d Assay-P. phosphoreum luciferase was purified by anion exchange Chromatography and gel fi1tration.l Luciferase activity was determined from the maximal light intensity measured upon injection of 1 ml of 5 X 10 '' M FMNHr (catalytically reduced with HI over platinized asbestos) into 1 ml of 0.05 M phos- phate buffer, pH 7.0, containing 0.2%. bovine serum albumin, 20 pI of 0.5% dodecvl aldehyde (in isopropyl alcohol), and an aliquot of the enzyme fraction (24).
Luminescence Assay for Fatty Acid Reductase-The samples to be analyzed were incubated a t 22 ? 2°C in a final volume of 1 ml of 0.05 M phosphate buffer, pH 7.0, containing 1 mM /3-mercaptoethanol, 1 mM ATP, 1 mM NADPH, and 0.58 to 10 p~ tetradecanoic acid from a 0.01 M stock solution prepared in ethanol. A constant amount of purified luciferase (at least a 6- to 10-fold excess over that endoge- nously present in any sample to be analyzed) was added at the end of the incubation period and the luminescent activity was measured on injection of 5 X 10 " M FMNH?. Activities are given in light units (LU) as defined above after correction for background luminescence in the absence of added substrates (ATP, NADPH, tetradecanoic acid).
Radioactice Assay for Fatty Acid Reductase-The samples were incubated under conditions identical with those described for the luminescence assay above, except that 0.58 p~ [.'H]tetradecanoic acid (150 mCi/mg) was used as substrate. The reaction was stopped by the addition of 3 pl of ethanol containing 120 pg of tetradecanoic acid, 80 pg of tetradecyl aldehyde, and 80 pg of tetradecyl alcohol, and the mixture was then blended on a Vortex mixer for 30 s with 1 ml of cold diethyl ether. Centrifugation of the sample for 10 min in a clinical centrifuge resulted in clear separation of the two phases with the proteins precipitated at the interface of the organic and aqueous phases. Less than 5% of the radioactivity was found to remain in the aqueous phase after extraction.
The ether phase was transferred to a glass tube and a 5-pl aliquot was spotted for thin layer chromatography on silica gel plates (Mach- ery-Nagel Co.. N-HH-UVI:,,) activated at 120°C for 1 h and prespotted with 120 pg of tetradecanoic acid, 80 pg of tetradecyl alcohol, and.60 pg of tetradecyl aldehyde dissolved in ethanol or benzene. The chro- matogram was developed in benzene/ether/acetic acid (90:10:2) until the solvent front reached approximately 13 cm from the origin. The plates were allowed to dry at room temperature (10 min) and sprayed with a 0.5% Malachite green solution (25). Upon drying (10 to 20 min), the spots gradually appeared; the spots corresponding to alco- hol, aldehyde, and acid were then excised, incubated for 16 to 20 h in scintillation mixture (Econofluor. New England Nuclear) at 4OC in the dark, and then counted with an efficiency of 36%. The recovery of the radioactivity from the plates was greater than 90'3. In many experiments, the other regions of the chromatogram were analyzed in an identical fashion.
Malate Dehydrogenase Assay-Malate dehydrogenase activity was determined from the initial rate of oxidation of NADH in the presence of oxaloacetate as followed by the decrease in (26). The incubation mixture contained 0.2 mM NADPH, 1 mM oxaloacetate, and 50 p1 of extract in 1 ml of 0.05 M phosphate, pH 7.0.
Glucose-6-phosphate Dehydrogenase Assay-Glucose-6-phos- phate dehydrogenase activity was determined from the initial rate of increase in A:I.,,, upon addition of 50 pI of extract to 1 ml of 0.05 M Tris/chloride, pH 7.8, containing 3 mM glucose 6-phosphate, 3 mM MgCI2, and 0.2 mM NADP' (27).
' E. Meighen. M. Ziegler, and T. Baldwin, manuscript in prepara- tion.
Reductase and Luciferase 12061
Protein Assay-Protein content of the extracts was determined according to the method of Lowry et 01. (28). using bovine serum albumin as a standard.
RESULTS
Deoelopment of a Direct Assay for Fatty Acid Reductase- The use of a radioactive fatty acid (["Hltetradecanoic acid) in the assay for fat ty acid reductase not only permits one to determine the fate of the fat ty acid, but also provides the basis for development of a direct method for measurement of fatty acid reductase activity. A necessary requirement for such a direct assay is a rapid method for separation of any labeled products (eg . aldehyde, alcohol) from the radioactive precur- sor. Earlier studies showed that decanal, decanol, and deca- noic acid could be resolved by thin layer chromatography using a chloroform/ether solvent system (16). However, this system gave insufficient resolution of tetradecyl aldehyde, tetradecyl alcohol, a n d tetradecanoic acid, and consequently a better solvent system had to be developed.
Fig. 1 shows that thin layer chromatography on silica gel in benzene/ether/acetic acid (90 10:2) gave good resolution of tetradecyl alcohol (RF. = 0.4), tetradecanoic acid (RF. = 0.6). and tetradecyl aldehyde (RF. = 0.8). Determination of t h e puri ty of ['lH]tetradecanoic acid after TLC showed that greater than 95% of the radioactivity migrated with the unla- beled acid. Between 2 a n d 4 6 of the radioactivity was detected at the position of tetradecyl alcohol in the chromatogram, apparent ly as a result of some trailing and consequent overlap of the fat ty acid with the position of the long chain alcohol. In contrast, less than 0.3% of the radioactivity was found at the position of tetradecyl aldehyde, which has a higher mobility than the fat ty acid. No significant radioactivity ( ~ 2 4 ) was found at a n y other position of equivalent area on the chro- matogram.
When the extract of the bioluminescent bacteria containing fat ty acid reductase act ivi ty was incubated with the tri t iated tetradecanoic acid in the presence of NADPH a n d ATP, 6.47 of the radioactivity now appeared at the position of tetradecyl a ldehyde after TLC of an ether extract of the reaction mixture
- Solvent front
Tetradecyl aldehyde
)Tetradecanoic acid
Tetradecyl alcohol
-Origin
A B C D
FIG. 1. Separation of aliphatic long chain alcohol, aldehyde, and acid by thin layer chromatography in benzene/ether/ acetic acid (90:10:2). The silica gel plate was spotted with 80 pg of tetradecyl alcohol (A), 120 pg of tetradecanoic acid (B), 60 p g of tetradecyl aldehyde ( C ) . or a mixture of the three (Dl. The com- pounds were visualized by spraying with a Malachite green solution as described under "Experimental Procedures."

12062 Co-induction of Fatty Acid Reductase and Luciferase
(Table I). The conversion was not observed if the reaction was immediately stopped after mixing of the assay components or if the reaction mixture did not contain ATP and NADPH. Attempts to detect this activity in the pelleted cellular debris were unsuccessful, indicating that the fatty acid reductase is completely solubilized by the extraction procedure. In addi- tion, the insoluble material had little effect on the activity of the fatty acid reductase in the extract.
Evidence that a long chain aldehyde was produced and not a non-aldehyde component co-migrating with tetradecyl al- dehyde is also given in Table I. The addition of horse liver alcohol dehydrogenase and NADH to the incubation mixture resulted in a decrease in radioactivity at the aldehyde position to background levels (0.2%) and an increase in radioactivity at the alcohol position (13%), whereas the addition of only NADH to the reaction mixture had little effect. Since horse liver alcohol dehydrogenase catalyzes the reduction of long chain aldehydes ( 5 ) , the radioactivity at the position of tetra- decyl aldehyde must have arisen by conversion of [”Hltetra- decanoic acid to tritiated aldehyde by the fatty acid reductase.
Euidence for Long Chain Aldehyde Reduction in Extracts of Bioluminescent Bacteria-An appreciable conversion of aldehyde to alcohol can also be achieved in the absence of horse liver alcohol dehydrogenase if the extract is incubated for longer periods of time in the reaction mixture. Fig. 2 gives the percentage of aldehyde and alcohol produced from [“’HI- tetradecanoic acid as a function of time of incubation. Al- though aldehyde is the only detectable product in the first 20 min of the reaction, an appreciable amount of conversion to long chain alcohol can be observed after longer periods of incubation. Maximum aldehyde levels are reached after 1 h of reaction, whereas at this time, the alcohol level is still rising and now exceeds the aldehyde level. The overall rate of reduction of tetradecanoic acid, however, stays relatively con- stant over this period with a linear increase with time in the total amount of aldehyde and alcohol. For any assay, between 94 and 98% of the radioactivity was recovered at the aldehyde, alcohol, and acid positions, with very low amounts ( t l to 2%) elsewhere on the chromatogram, indicating that only aldehyde and alcohol were produced in significant amounts in the reaction mixture. These results not only c o n f i i t h e existence of a fatty acid reductase in extracts, but provide the fist evidence for the existence of an aldehyde reductase(s) in
TABLE I Conversion of [’H]tetradecanoic acid to aldehyde and alcohol
Radioactivity” Components added“ Incubation
time
rnm
Aldehyde Alcohol
ATP, NADPH 0 0.6 4.1 None 20 0.3 3.7 ATP, NADPH 20 6.4 ATP, NADPH, NADH
4.6 20 4.6 4.8
ATP, NADPH, NADH, al- 20 0.2 13.3 coho1 dehydrogenase
‘‘ The extract (100 pl) was incubated with [’Hltetradecanoic acid (0.58 p ~ ) and the indicated components at the following concentra- tions: ATP ( 1 mM), NADPH ( 1 mM), NADH (0.05 mM), and horse liver alcohol dehydrogenase (50 pg/ml), as described for the radioac- tive assay for fatty acid reductase under “Experimental Procedures.” The extract was prepared from cells grown to A,,,,, = 2.6 (3,000 LU/ ml).
’After incubation, the samples were extracted with diethyl ether and analyzed by TLC. The amount of radioactivity co-migrating at the position of tetradecanoic acid, tetradecyl aldehyde, and tetradecyl alcohol was measured and the percentages of total radioactivity found at the aldehyde and alcohol positions were calculated. Additional details are given under “Experimental Procedures.”
301
FIG. 2. Time dependence of the synthesis of long chain al- dehyde and alcohol from [3H]tetradecanoic acid in bacterial extracts. One hundred microliters of the extract (Table I ) was incubated for various periods of time with [ ’Hltetradecanoic acid, ATP, and NADPH in a final volume of 1 ml as described under “Radioactive Assay for Fatty Acid Reductase” (see under “Experi- mental Procedures”). The percentage of conversion (% Concvrsion) into long chain aldehyde and alcohol was determined from the fraction of radioactivity co-migrating on TLC with tetradecyl aldehyde and tetradecyl alcohol, respectively, after correction for the amount of radioactivity found at zero time at these positions (see Table I). Alc, alcohol; Ald, aldehyde.
extracts of luminescent bacteria capable of reducing long chain aldehydes to alcohols.
Dependence of Aldehyde Synthesis on the Amount of Er- tract-Previous studies on fatty acid reductase using an assay system coupled to bacterial luciferase have demonstrated that the activity in the coupled assay was dependent on the square of the amount of extract analyzed (22). Since the activity of luciferase is dependent on the amount of extract, the fatty acid reductase activity should also be linearly dependent on the extract concentration. The direct measurement of the conversion of [3H]tetradecanoic acid to aldehyde by fatty acid reductase as a function of the amount of extract clearly establishes this point (Fig. 3). Furthermore, it is also demon- strated in Fig. 3 for the coupled assay that the addition of a large excess of luciferase to the reaction mixture eliminates the square dependence on the amount of extract, so that the response now follows closely the amount of long chain alde- hyde produced. It may be noted that the assay was only conducted for a short period of time (20 min) and that no appreciable amount of alcohol was produced for the different amounts of extracts analyzed. Both assay systems, therefore, allow one to measure the relative amounts of fatty acid reductase activity in different extracts and provide the oppor- tunity to determine whether or not the fatty acid reductase activity is co-induced with luciferase during the development of bacterial bioluminescence.
Induction of Luciferase and Fatty Acid Reductase-The change in growth and luminescence in vivo of P. phosphoreum as a function of time is illustrated in Fig. 4. The luminescence in vivo initially decreases, and then at a later stage of cell growth, it increases over 10,000-fold. In contrast, cellular growth increases only 6-fold during the development of bio- luminescence. The final level of luminescence for this P. phosphoreum strain is almost ten times higher than that observed for B. harveyi, the bioluminescent strain that has
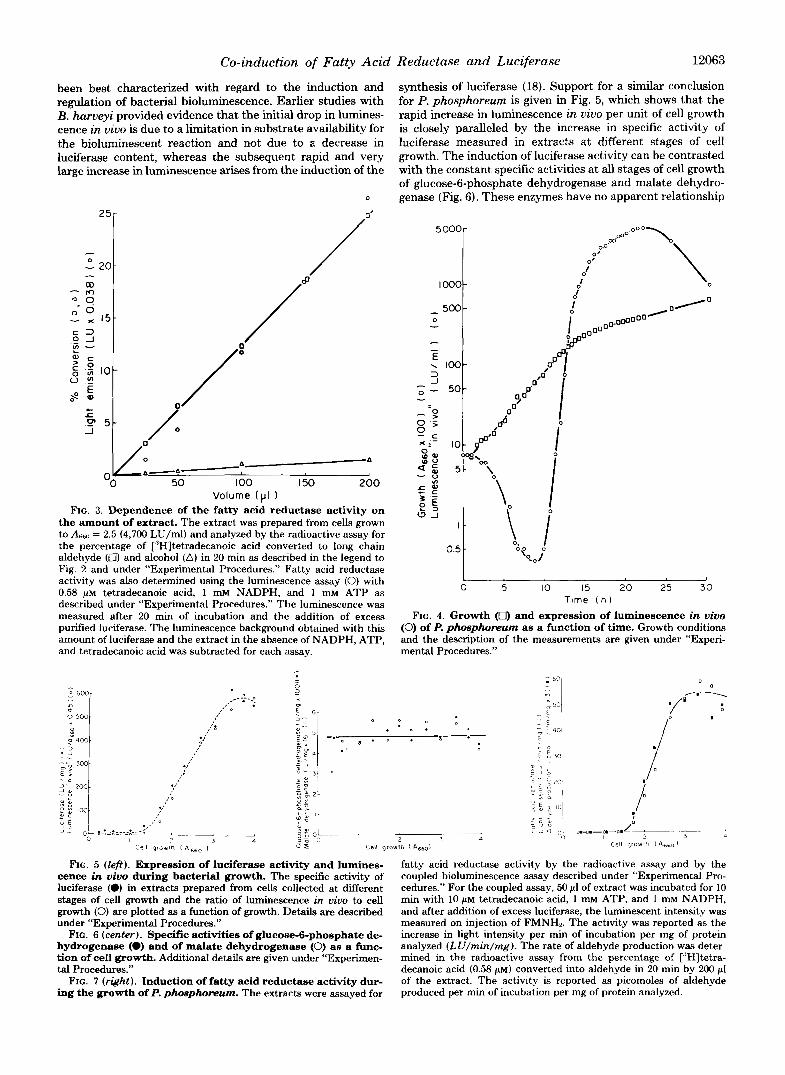
Co-induction of Fatty Acid Reductase and Luciferase 12063
been best characterized with regard to the induction and regulation of bacterial bioluminescence. Earlier studies with B. harveyi provided evidence that the initial drop in lumines- cence in vivo is due to a limitation in substrate availability for the bioluminescent reaction and not due to a decrease in luciferase content, whereas the subsequent rapid and very large increase in luminescence arises from the induction of the
0
Volume (P I ) FIG. 3. Dependence of the fatty acid reductase activity on
the amount of extract. The extract was prepared from cells grown to AM, = 2.5 (4,700 LU/ml) and analyzed by the radioactive assay for the percentage of I:’H]tetradecanoic acid converted to long chain aldehyde (0) and alcohol (A) in 20 min as described in the legend to Fig. 2 and under “Experimental Procedures.” Fatty acid reductase activity was also determined using the luminescence assay (0) with 0.58 p~ tetradecanoic acid, 1 m~ NADPH, and 1 m~ ATP as described under “Experimental Procedures.” The luminescence was measured after 20 min of incubation and the addition of excess purified luciferase. The luminescence background obtained with this amount of luciferase and the extract in the absence of NADPH, ATP, and tetradecanoic acid was subtracted for each assay.
synthesis of luciferase (18). Support for a similar conclusion for P. phosphoreum is given in Fig. 5, which shows that the rapid increase in luminescence in vivo per unit of cell growth is closely paralleled by the increase in specific activity of luciferase measured in extracts at different stages of cell growth. The induction of luciferase activity can be contrasted with the constant specific activities at all stages of cell growth of glucose-6-phosphate dehydrogenase and malate dehydro- genase (Fig. 6). These enzymes have no apparent relationship
0 5 IO 15 2 0 25 3 0 Tlme ( h )
FIG. 4. Growth (0) and expression of luminescence in vivo (0) of P. phosphomum as a function of time. Growth conditions and the description of the measurements are given under “Experi- mental Procedures.”
FIG. 5 (left). Expression of luciferase activity and lumines- cence in vivo during bacterial growth. The specific activity of luciferase (0) in extracts prepared from cells collected at different stages of cell growth and the ratio of luminescence in vivo to cell growth (0) are plotted as a function of growth. Details are described under “Experimental Procedures.”
FIG. 6 (center). Specific activities of glucose-6-phosphate de- hydrogenase (0) and of malate dehydrogenase (0) as a func- tion of cell growth. Additional details are given under “Experimen- tal Procedures.”
FIG. 7 (right). Induction of fatty acid reductase activity dur- ing the growth of P. phosphorewn. The extracts were assayed for
fatty acid reductase activity by the radioactive assay and by the coupled bioluminescence assay described under “Experimental Pro- cedures.” For the coupled assay, 50 pl of extract was incubated for 10 min with 10 ~ L M tetradecanoic acid, 1 m~ ATP, and 1 mM NADPH, and after addition of excess luciferase, the luminescent intensity was measured on injection of FMNH2. The activity was reported as the increase in light intensity per min of incubation per mg of protein analyzed (LU/min/mg). The rate of aldehyde production was deter- mined in the radioactive assay from the percentage of [’Hltetra- decanoic acid (0.58 p ~ ) converted into aldehyde in 20 min by 200 pl of the extract. The activity is reported as picomoles of aldehyde produced per min of incubation per mg of protein analyzed.

12064 Co-induction of Fatty Acid Reductase and Luciferase
TABLE I1 Effect of extracts of noninduced cells on fatty acid reductase
actiuity Components" Activityh
'ib Extract 1 Extract 2 Extract 3 Extracts 3 and 1 Extracts 3 and 2 Extract 3 and boiled Extract 1 Extract 3 and boiled Extract 3
<l 2
100 76 83 76 81
" Extracts were prepared as described under "Experimental Pro- cedures" using a constant amount of cells collected at different stages of bacterial growth: Extract 1, A,, = 0.7, LU/ml = 1.5 Extract 2, Awl = 1.4 LU/ml = 18; Extract 3, A,**, = 3.3, LU/ml = 2,300.
"The fatty acid reductase activity was determined with 50 p1 of each extract using the coupled luminescence assay as described in the legend to Fig. 7. The activity is given relative to the luminescence response obtained with the extract prepared from luminescent cells (Extract 3) . Additional details are included under "Experimental Procedures."
to the bioluminescent system and thus serve as internal con- trols to attest to the uniformity of the procedure for cell collection and extraction.
The fatty acid reductase activity was measured as a function of cell growth in the same extracts as luciferase and the control enzymes (Fig. 7). Both the production of aldehyde from [3H]tetradecanoic acid in the radioactive assay and the lumi- nescence response in the coupled assay with excess luciferase were used, since the fatty acid reductase activity is directly proportional to the responses in these two assays (see Fig. 3). No fatty acid reductase activity could be detected with either assay at the early stages of cell growth. However, a rapid increase in the specific activity of fatty acid reductase in both assays occurred at higher cell densities (Am > 1.5) (Fig. 7). The specific activity of fatty acid reductase in extracts reached a maximum level a t about the same absorbance (Aw = 3.0) as that of luciferase (see Fig. 5), after which the specific activity stayed relatively constant. Very little, if any, long chain alcohol production (0 to 2.2%) was observed in any sample under the conditions used for the radioactive assay.
The possibility that the relative increase in fatty acid re- ductase activity with the cell growth reflected a growth-de- pendent loss of an inhibitor was investigated by mixing ex- tracts from cells before and after induction of luminescence (Table 11). Extracts from noninduced cells prepared a t two different stages of cell growth and containing very little fatty acid reductase activity caused only a small degree of inhibition of fatty acid reductase activity in extracts from induced (lu- minescent) cells. A similar degree of inhibition was observed using a boiled extract of either induced or noninduced cells. These results thus show that the increase in the fatty acid reductase activity is not due to the removal of an inhibitor, but rather arises from induction of the activity of the enzyme during the growth of the bioluminescent bacteria. A compar- ison of the specific activities of luciferase (Fig. 5) and fatty acid reductase (Fig. 7) as a function of growth clearly suggests that the activity of both enzymes are induced simultaneously and are under similar regulation.
DISCUSSION
The expression of bioluminescence in uivo has been shown to occur at a late stage of growth for bioluminescent bacteria (18). Using two isotopes to differentially label the proteins, Michaliszyn and Meighen (19) have shown that at least seven polypeptides were synthesized specifically during the induc- tion of bioluminescence in B. harueyi. In addition, the synthe-
sis of certain membrane proteins has also been reported to be induced during the growth of these bacteria (29).
At the present time, the function of only three of these polypeptides in B. harveyi have been directly identified in vitro. Two polypeptides correspond to the a and j3 subunits of luciferase (19) and one corresponds to an NAD'-dependent aldehyde dehydrogenase (30,31).
In viuo studies with temperature-sensitive dark mutants of B. harveyi, the luminescence of which was stimulable by long chain aldehydes, have also indicated that the expression of at least two genes, involved in aldehyde biosynthesis, are re- quired for light emission in bioluminescent bacteria (32). The characterization of dark mutants, the luminescence of which was also stimulable by fatty acids, indicated that a t least one of the gene products was an enzyme involved in the conversion of fatty acid to aldehyde (33).
The present work has demonstrated that fatty acid reduc- tase activity can be measured directly in extracts of P. phos- phoreum by analysis of the tritiated aldehyde produced from ["H]tetradecanoic acid. Furthermore, the levels of fatty acid reductase activity increased only during the development of bacterial bioluminescence, clearly implicating this enzyme in the bioluminescent system and suggesting that its synthesis is under the same regulation as luciferase. We have not yet been able to detect this activity in 8. harveyi, and extracts of this bacterium inhibit the fatty acid reductase activity of P. phos- phoreurn.:' This inhibition may arise from the presence of a long chain aldehyde dehydrogenase in extracts of B. harueyi (30). However, we have recently detected the fatty acid re- ductase activity in another bioluminescent bacterium (P . phosphoreum, strain A-13).
A coupled bioluminescent assay to measure aldehyde bio- synthesis was also shown in the present experiments to give a response proportional to the amount of fatty acid reductase in the extracts. It was necessary to add a constant high amount of luciferase to these assays, since extracts contain variable amounts of luciferase as well as fatty acid reductase. It should be noted that an absolute relationship between the two differ- ent assays for fatty acid reductase has not yet been estab- lished, although activities in both assays are directly propor- tional to the amount of enzyme. In this regard, it is important to point out that the reduction of [,"H]tetradecanoic acid to aldehyde is not an absolute measure of the total reduction of fatty acids in the radioactive assay, since aldehyde biosyn- thesis in the absence of exogenous fatty acids can be demon- strated in the extracts by the use of the coupled luminescent assay. The fatty acid reductase activity in extracts using the coupled luminescent assay with NADPH and ATP alone is about 60 to 70% of the activity obtained if tetradecanoic acid is also added (21). This result suggests that extracts contain high levels of endogenous fatty acids, and thus the conversion of ["Hltetradecanoic acid to tritiated aldehyde measures only part of the fatty acid reductase activity. The concentration of ["Hltetradecanoic acid used in these assays was selected so that on one hand, a significant and measurable conversion (per cent) to aldehyde occurred during the course of the assay and, on the other hand, the decrease in concentration of [3H]tetradecanoic acid during the assay did not significantly alter the rate of reaction. Consequently, the amount of [3H]tetradecanoic acid converted to aldehyde was propor- tional to the amount of extract analyzed; this is an important and necessary feature for investigation of the level of fatty acid reductase activity in different extracts as a function of bacterial growth.
These results also demonstrated that the aldehyde pro- duced from tetradecanoic acid could be further reduced to
D. Riendeau and E. Meighen, unpublished data.

Co-induction of Fatty Acid Reductase and Luciferase 12065
alcohol, providing the first evidence that these bacteria con- tain aldehyde reductases capable of reducing long chain al- dehydes. The existence of aldehyde reductases in the extracts shows that caution must also be exercised in using the coupled luminescence assay to measure fatty acid reductase activity. Since only the relative level of aldehyde is measured in the luminescence assay, the fatty acid reductase activity would be underestimated unless it is measured under assay conditions where there is no conversion of aldehyde to alcohol. If a purification procedure can be designed to remove both the aldehyde reductase activity and the endogenous fatty acids from the fatty acid reductase, then a direct quantitative rela- tionship between the radioactive and luminescence assays should be achieved. Preliminary experiments have shown that the aldehyde reductase activity can be partially resolved from fatty acid reductase by the same gel filtration procedure used for resolution from luciferase ( 2 2 ) .
The present results have thus demonstrated the enzymatic reduction of tetradecanoic acid to long chain aldehyde and alcohol in extracts of bioluminescent bacteria. The develop- ment of the assay for measuring the conversion of tetradeca- noic acid to aldehyde will provide a useful tool for studying the properties of the fatty acid reductase, having the advan- tage over the luminescence-coupled assay that measurements of activity are independent of any variation in luciferase content and activity. Thus, the properties and activity of fatty acid reductase can be determined over a broader range of conditions. This work also presents evidence that the fatty acid reductase is co-induced with luciferase, indicating that the in vivo function of the reductase is the synthesis of the long chain aldehyde required for the bacterial luminescent reaction. Further studies are important to determine the role of the fatty acid reductase in the regulation and mechanism of the induction of bacterial luminescence.
Acknowledgment-We would like to thank Irene Bartlet for her valuable assistance in these experiments.
REFERENCES 1. Ferrel, W. d . , and Radloff. J . F. (1969) Bioscience 19, 243 2. Day, J. I. E., Goldfine. H., and Hagen, P.-0. (1970) Biochim.
3. Tietz, A., Lindberg, M., and Kennedy, E. P. (1964) J. Biol. Chem.
4. Pfleger, R. C., Piantadosi, C., and Snyder, F. (1967) Biochim.
Biophys. Acta 218, 179-182
239,4081-4090
Biophys. Acta 144, 633-648
5. Stoffel, W., LeKim, D., and Heyn, G. (1970) Hoppe-Seyler’,s Z.
6. Kolattukudy, P. E. (1970) Biochemistry 9, 1095-1102 7. Wang, L., Tokayama, K., Goldman, D. S., and Schnoes, H. K.
8. Rock, C. O., Fitzgerald, V., and Snyder, F. (1978) Arch. Biochem.
9. Lebeault, J . M., Roche, B., Durnjak, Z., and Azoulay, E. (1970)
10. Kolattukudy, P. E. (1971) Arch. Biochem. Biophys. 142, 701-709 11. Johnson, K. C., and Gilbertson, J. R. (1972) J. Biol. Chem. 247,
12. Natarajan, V., and Sastry, 1’. S. (1976) J. Neurochem. 26, 107-
13. Bishop, J. E., and Hajra, A. K. (1978) J . Neurochem. 30.643-647 14. Cormier, M. J., and Strehler, B. L. (1953) J . Am. Chem. Soc. 75,
15. Strehler, B. L., and Cormier, M. J . (1954) J . B i d Chem. 211,
16. Dunn, D. K., Michaliszyn, G. A., Bogacki, I. G., and Meighen. E.
17. Shimomura, O., Johnson, F. H., and Ko.yama, Y. (1972) Proc.
18. Nealson, K. H., Platt, T., and Hastings, J. W. (1970) J. Bncferzol.
19. Michaliszyn, G. A., and Meighen, E. A. (1976) J . Bioof Chem. 251,
20. Ulitzur, S., and Hastings, J . W. (1978) Proc. Natl. Acad. Sci. U.
21. Meighen, E. A. (1979) Biochent. Biophys. Res. Commun. 87,
22. Hiendeau, D., and Meighen, E. (1979) J . B ~ o l . Chem. 254, 7488-
23. Hastings, .J. W., and Weber, G. (1963) J. Opt. Soc. Am. 53, 1410-
24. Gunsalus-Miguel, A., Meighen, E. A., Nicoli, M. Z., Nealson, K.
25. Teichman, R. J., Takei, G. H., and Cummins, J. M. (1974) J .
26. Kitto, G. B. (1969) Methods Enzymol. 13, 106-116 27. DeMoss, K. D. (1955) Methods Enzymol. 1, 328-334 28. Lowry, 0. H., Kosebrough, N. J . , Farr, A. I,., and Randall, H. .J.
29. Ne’eman, Z., Ulitzur, S., Branton, D., and Hastings, J. W. (1977)
30. Meighen, E. A., Bogacki, I. G., Bognar, A., and Michaliszyn, G. A.
31. Bognar, A., Michaliszyn, G., and Meighen, E. A. (1978) Can. J .
32. Cline, T . W., and Hastings, J. W. (1974) J. Bacteriol. 118, 1059-
33. Ulitzur, S., and Hastings, J . W. (1979) J. Bacteriol. 137, 854-859
Physiol. Chem. 351,875-883
(1972) Biochim. Biophys. Acta 260, 41-48
Biophys. 186, 77-83
Biochim. Biophys. Acta 220, 373-385
6991-6998
113
4864-4865
213-225
A. (1973) Biochemistry 12, 4911-4918
Natl. Acad. Sci. U. S. A . 69, 2086-2089
104,313-322
2541-2549
S. A . 75, 266-269
1080- 1086
7490
1415
H., and Hastings, J . W. (1972) J. Biol. Chem. 247, 398-404
Chromatogr. 88,425-427
(1951) J . Biol. Chem. 193, 265-275
J. Biol. Chem. 252,5150-5154
(1976) Biochem. Biophys. Res. Commun. 69,423-430
Biochem. 56, 605-610
1066


















