
SUPPLEMENTARY INFORMATION - Nature...excitation energies and oscillator strengths and later, all the...
17
NATURE CHEMISTRY | www.nature.com/naturechemistry 1 SUPPLEMENTARY INFORMATION DOI: 10.1038/NCHEM.1778 Metal-Free Binding and Coupling of Carbon Monoxide at a Boron-Boron Triple Bond Holger Braunschweig 1 *, Theresa Dellermann 1 , Rian D. Dewhurst 1 , William C. Ewing 1 , Kai Hammond 1 , J. Oscar C. Jimenez-Halla 1,2 , Thomas Kramer 1 , Ivo Krummenacher 1 , Jan Mies 1 , Ashwini K. Phukan 1,3 , Alfredo Vargas 1 Affiliations 1 Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany 2 Facultad de Química, Universidad de Guanajuato, Mexico 3 Department of Chemical Sciences, Tezpur University, Napaam 784028, Assam, India *Correspondence to: [email protected] Website: http://www-anorganik.chemie.uni-wuerzburg.de/Braunschweig/ © 2013 Macmillan Publishers Limited. All rights reserved.
Transcript of SUPPLEMENTARY INFORMATION - Nature...excitation energies and oscillator strengths and later, all the...

NATURE CHEMISTRY | www.nature.com/naturechemistry 1
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
1
Supplementary Information for:
Metal-Free Binding and Coupling of Carbon Monoxide at a Boron-Boron Triple Bond
Holger Braunschweig1*, Theresa Dellermann1, Rian D. Dewhurst1, William C. Ewing1, Kai
Hammond1, J. Oscar C. Jimenez-Halla1,2, Thomas Kramer1, Ivo Krummenacher1, Jan Mies1,
Ashwini K. Phukan1,3, Alfredo Vargas1
Affiliations
1 Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland,
97074 Würzburg, Germany
2 Facultad de Química, Universidad de Guanajuato, Mexico
3 Department of Chemical Sciences, Tezpur University, Napaam 784028, Assam, India
*Correspondence to: [email protected]
Website: http://www-anorganik.chemie.uni-wuerzburg.de/Braunschweig/
© 2013 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 2
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
2
Table of Contents
Synthetic Methods p3
Crystallographic Methods p5
Electrochemical Methods p7
Computational Methods p8
References p16
© 2013 Macmillan Publishers Limited. All rights reserved.
NATURE CHEMISTRY | www.nature.com/naturechemistry 3
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
3
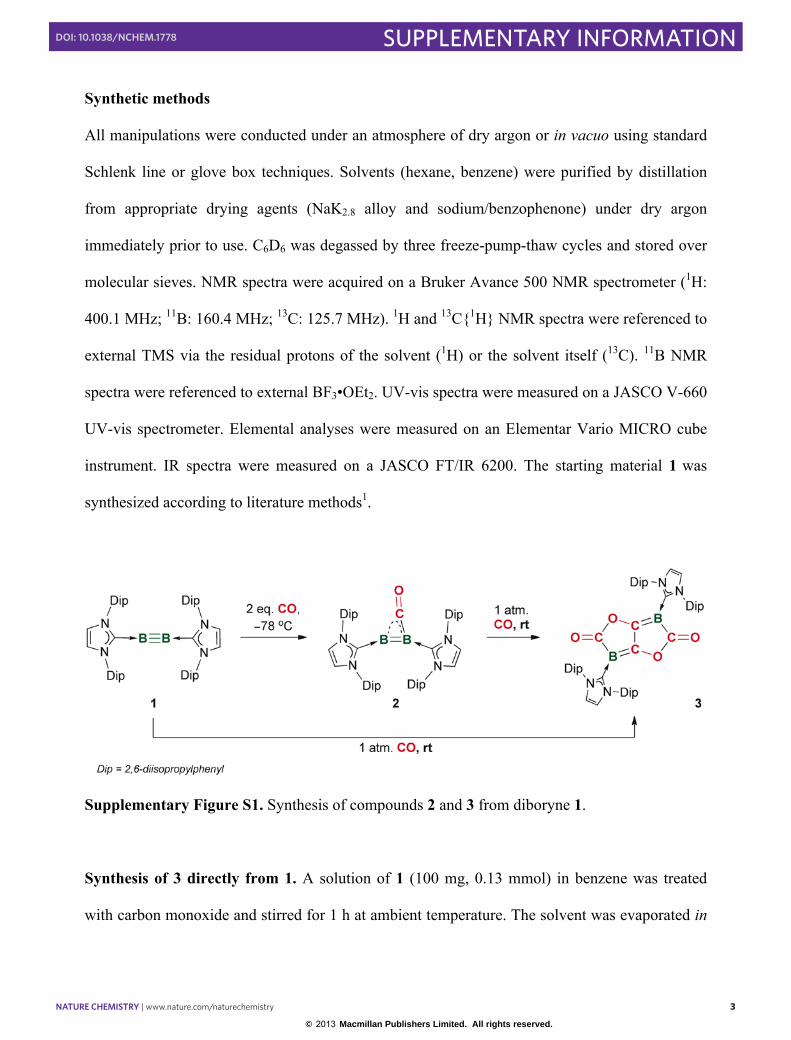
Synthetic methods
All manipulations were conducted under an atmosphere of dry argon or in vacuo using standard
Schlenk line or glove box techniques. Solvents (hexane, benzene) were purified by distillation
from appropriate drying agents (NaK2.8 alloy and sodium/benzophenone) under dry argon
immediately prior to use. C6D6 was degassed by three freeze-pump-thaw cycles and stored over
molecular sieves. NMR spectra were acquired on a Bruker Avance 500 NMR spectrometer (1H:
400.1 MHz; 11B: 160.4 MHz; 13C: 125.7 MHz). 1H and 13C{1H} NMR spectra were referenced to
external TMS via the residual protons of the solvent (1H) or the solvent itself (13C). 11B NMR
spectra were referenced to external BF3•OEt2. UV-vis spectra were measured on a JASCO V-660
UV-vis spectrometer. Elemental analyses were measured on an Elementar Vario MICRO cube
instrument. IR spectra were measured on a JASCO FT/IR 6200. The starting material 1 was
synthesized according to literature methods1.
Supplementary Figure S1. Synthesis of compounds 2 and 3 from diboryne 1.
Synthesis of 3 directly from 1. A solution of 1 (100 mg, 0.13 mmol) in benzene was treated
with carbon monoxide and stirred for 1 h at ambient temperature. The solvent was evaporated in
© 2013 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 4
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
4
vacuo and the crude product was washed with pentane (3 × 7 mL). Yield 83.0 mg (0.09 mmol,
70%, white solid). M.p.: 288 °C (decomp.). 1H NMR (500.1 MHz, C6D6, 293 K): δ = 7.24-7.21
(m, 4 H, CHaryl), 7.09-7.07 (m, 8 H, CHaryl), 6.19 (s, 4 H, CHNHC), 2.72-2.67 (m, 8 H, CHiPr),
1.31 (d, 24 H, CH3), 1.05 (d, 24 H, CH3). 13C{1H} NMR (125.8 MHz, C6D6, 293 K): δ = 189.0,
175.1, 161.3, 146.0, 134.5 (Cq), 130.6 (CHaryl), 124.3 (CHaryl), 123.0 (CHNHC), 29.2 (s, CHiPr),
25.0 (CH3), 24.0 (CH3). 11B NMR (160.4 MHz, C6D6, 293 K): δ = −3.6. IR: (solid) ν = 1670 (s),
1467 (s) cm-1. IR: (in benzene) ν = 1691 (s), 1428 (s), 1357 (s) cm-1. Elemental analysis calcd.
[%] for C58H72B2O4N4: C 76.31, H 7.92, N 6.15; found C 76.31 H 8.07, N 5.65.
Synthesis of 2. A solution of 1 (50 mg, 0.062 mmol) in hexane (3 mL) was treated under static
vacuum with carbon monoxide at –78 °C (2.8 mL, 1 bar, 0.12 mmol). The mixture was warmed
to ambient temperature within 3 h. The resulting brown suspension was filtered and washed with
hexane (2 × 3 mL) and dried in vacuo. Yield 35 mg (0.042 mmol, 68%). 1H NMR (400.1 MHz,
C6D6, 293 K): δ = 7.22-7.18 (m, 4 H, CHaryl), 7.06-7.04 (m, 8 H, CHaryl), 6.17 (s, 4 H, CHNHC),
3.07-3.00 (m, 8 H, CHiPr), 1.13 (vt, 48 H, CH3). 13C{1H} NMR (100.6 MHz, C6D6, 293 K): δ =
147.0, 135.5 (Cq), 129.6 (CHaryl), 123.8 (CHaryl), 121.2 (CHNHC), 28.7 (s, CHiPr), 24.9 (CH3),
24.2 (CH3) ppm. 11B NMR (128.4 MHz, C6D6, 293 K): δ = 18.4 ppm. IR: (in benzene) ν = 1926
(w), 1593 (m), 1568 (m), 1414 (m), 1353 cm-1 (s).
© 2013 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 5
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
5
Crystallographic Methods
The crystal data of 2 and 3 were collected on a Bruker X8-Apex II diffractometer with a CCD
area detector and multi-layer mirror monochromated MoKα radiation. The structure was solved
using direct methods, refined with the Shelx software package and expanded using Fourier
techniques. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were
included in structure factor calculations. All hydrogen atoms were assigned to idealised
geometric positions.
Selected bond lengths (Å) and angles (°) for 2: B1-B2 1.549(3), C1-B1 1.565(3), C1-B2
1.485(3), O1-C1 1.249(2); O1-C1-B2 155.38(17), O1-C1-B1 143.62(16), B2-C1-B1 61.00(13).
Crystal data for 2: C55H72B2N4O, Mr = 826.79, brown block, 0.34×0.25×0.07 mm3, monoclinic
space group P21, a = 12.8198(10) Å, b = 14.7557(11) Å, c = 13.2979(10) Å, β = 105.229(2)°,
V = 2427.2(3) Å3, Z = 2, ρcalcd = 1.131 g·cm–3, µ = 0.066 mm–1, F(000) = 896, T = 100(2) K,
R1 = 0.0526, wR2 = 0.0956, 10380 independent reflections [2θ≤53.62°] and 575 parameters.
Selected bond lengths (Å) and angles (°) for 3: B1-C1 1.4507(18), B1-C2 1.5742(18), B1-C11
1.5471(18), C1-C1’ 1.426(2), C1-O1 1.3853(14), C2-O1 1.4270(15), C2-O2 1.2152(15); C1-B1-
C11 131.25(11), C1-B1-C2 101.60(10), C11-B1-C2 126.99(11). Sum of angles around B1:
359.84°. Angle between NHC plane and C3BO plane: 45.65. Deviations from C6B2O4 plane:
0.002 (C11) - 0.050 (O2) Å. Crystal data for 3: C64H78B2N4O4, Mr = 988.92, yellow block,
0.31×0.25×0.22 mm3, monoclinic space group P21/n, a = 13.6446(6) Å, b = 14.3161(7) Å,
c = 15.0434(7) Å, β = 102.678(2)°, V = 2866.9(2) Å3, Z = 2, σcalcd = 1.146 g·cm–3,
© 2013 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 6
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
6
µ = 0.070 mm–1, F(000) = 1064, T = 173(2) K, R1 = 0.0527, wR2 = 0.1055, 6085 independent
reflections [2θ≤53.52°] and 354 parameters.
Crystallographic data have been deposited with the Cambridge Crystallographic Data Center as
supplementary publication nos. CCDC 935712 (2) and 913268 (3). These data can be obtained
free of charge from The Cambridge Crystallographic Data Centre via
www.ccdc.cam.ac.uk/data_request/cif
© 2013 Macmillan Publishers Limited. All rights reserved.
NATURE CHEMISTRY | www.nature.com/naturechemistry 7
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
7
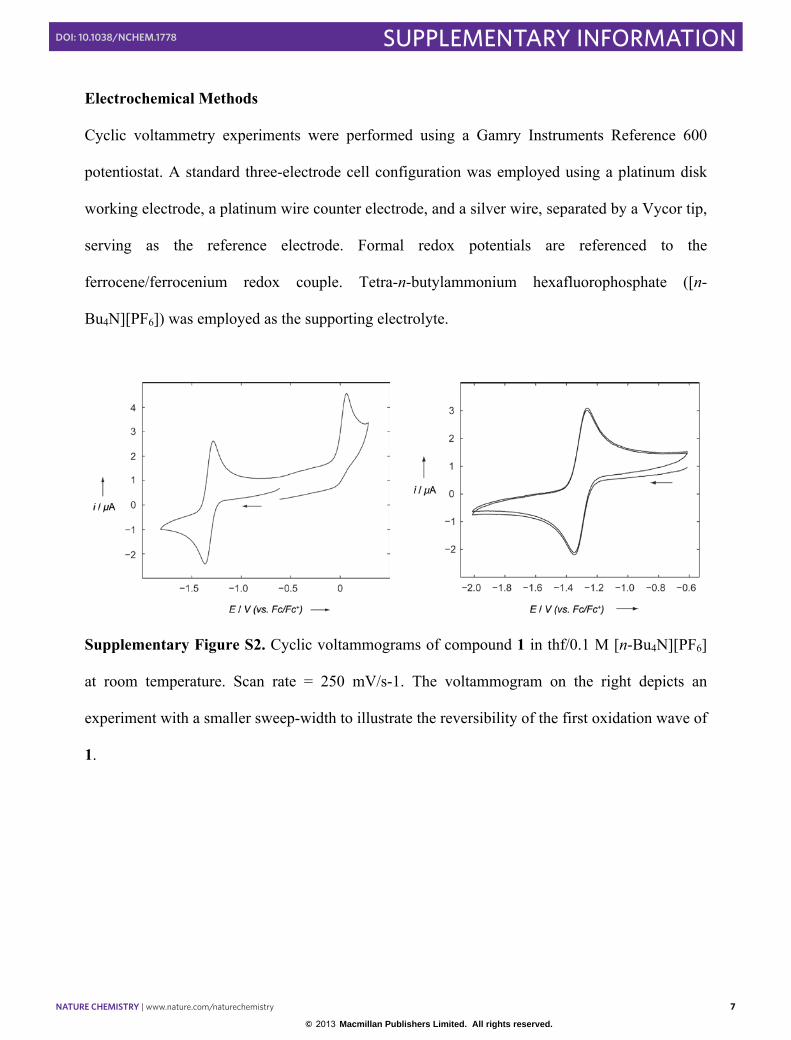
Electrochemical Methods
Cyclic voltammetry experiments were performed using a Gamry Instruments Reference 600
potentiostat. A standard three-electrode cell configuration was employed using a platinum disk
working electrode, a platinum wire counter electrode, and a silver wire, separated by a Vycor tip,
serving as the reference electrode. Formal redox potentials are referenced to the
ferrocene/ferrocenium redox couple. Tetra-n-butylammonium hexafluorophosphate ([n-
Bu4N][PF6]) was employed as the supporting electrolyte.
Supplementary Figure S2. Cyclic voltammograms of compound 1 in thf/0.1 M [n-Bu4N][PF6]
at room temperature. Scan rate = 250 mV/s-1. The voltammogram on the right depicts an
experiment with a smaller sweep-width to illustrate the reversibility of the first oxidation wave of
1.
© 2013 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 8
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
8
Computational Details
Modelling of the UV-vis and fluorescence spectra of 3
For the absorption and emission studies, calculations were conducted using the Gaussian09
program package2 at the ONIOM(M06-2X/6-311+G(d):PM6) level. In order to test the reliability
of our approach, we also carried out a geometry optimization (and harmonic frequency
calculations) on the product of the reaction at the M06-2X/6-31+G(d) level to compare the
performance of both methods with the experimental data (Table S1). The optimized geometrical
parameters of 3 are in good agreement with the crystallographic structure. Also, we have
computed the S-value test which is low (−0.038853) and therefore, our ONIOM2 partition
scheme is very suitable for this study. We performed TD-DFT calculations at the same combined
semi-empirical-DFT level of theory in order to provide some insight into the observed
fluorescence phenomenon in the bis(bora)lactone product. We computed the lowest single
excitation energies and oscillator strengths and later, all the fluorescence electronic transitions
that were calculated as vertical de-excitations based on the optimized geometry of S1 state. We
also checked for the energy difference between the T0 and S0 states, ΔH = 23.82 kcal/mol (ΔG =
22.65) so it is more likely that the photoluminescence proceeds from singlet states as discussed
below. We performed additional calculations in the basal and first singlet excited state geometry
of 3 (P1). We conducted geometry optimizations within the ONIOM methodology at the
CAS(4,3)/6-31+G(d) level over the bis(boralactone) core, i.e. using a multideterminantal
wavefunction method to incorporate all the possible configurations of HOMO-1, HOMO and
LUMO orbitals as they are the most relevant in the bonding picture along the B=C-C=B
fragment. Later, we refined the obtained orbital energies by applying the second-order
perturbational theory as CASPT2(4,3)/6-31+G(d) in a single-point calculation.
© 2013 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 9
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
9
Supplementary Figure S3. The selected two-layered ONIOM is shown for compound 3 (or P1),
where the inner layer is depicted using the ball & stick type and the outer layer is depicted by the
wireframe style. Boron atoms are colored in green, nitrogens are light blue, oxygens are red,
carbons are grey and hydrogens are white.
© 2013 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 10
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
10
Table S1. Comparison of the optimized geometries of 3 with our experimental data. Bond
distances are given in Angstrøms and angles in degrees.
Geometrical
Parameter
Experimental
value
ONIOM(M06-
2X:PM6)
M06-2X/6-
31+G(d)
B1-C1 1.4314 1.4524 1.4598
B1-C2 1.5720 1.5875 1.5986
B1-C3(NHC) 1.5417 1.5439 1.5491
C1-C4 central bond 1.4326 1.4438 1.4428
C2=O1 1.2127 1.2112 1.2095
C2-O2-C4 107.9 108.9 109.6
C2-B1-C3-N3 145.9 152.9 163.6
© 2013 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 11
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
11
Supplementary Figure S4. Relevant frontier molecular orbitals of compound 3 (P1), isosurface
value = 0.03 a.u.
© 2013 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 12
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
12
Supplementary Figure S5. Comparison of both optimized geometries of ground state (left) and
singlet first excited state (right) involved in the observed fluorescence process. Selected
geometrical parameters are shown: bond distances in Å, angles in degrees. Our CASPT2(4,3)
calculations in both optimized geometries also support the bonding situation of the ground state
and the biradical character of the first singlet excited state.
Supplementary Table S2. Vertical excitation energies, maximum wavelength and oscillator
strengths of both electronic transitions calculated at the specified level of theory.
Transition Excitation energy (eV) λmax (nm) Oscillator strength
S0→S1 2.80 542.2 0.90
S1→S0 2.49 567.6 0.89
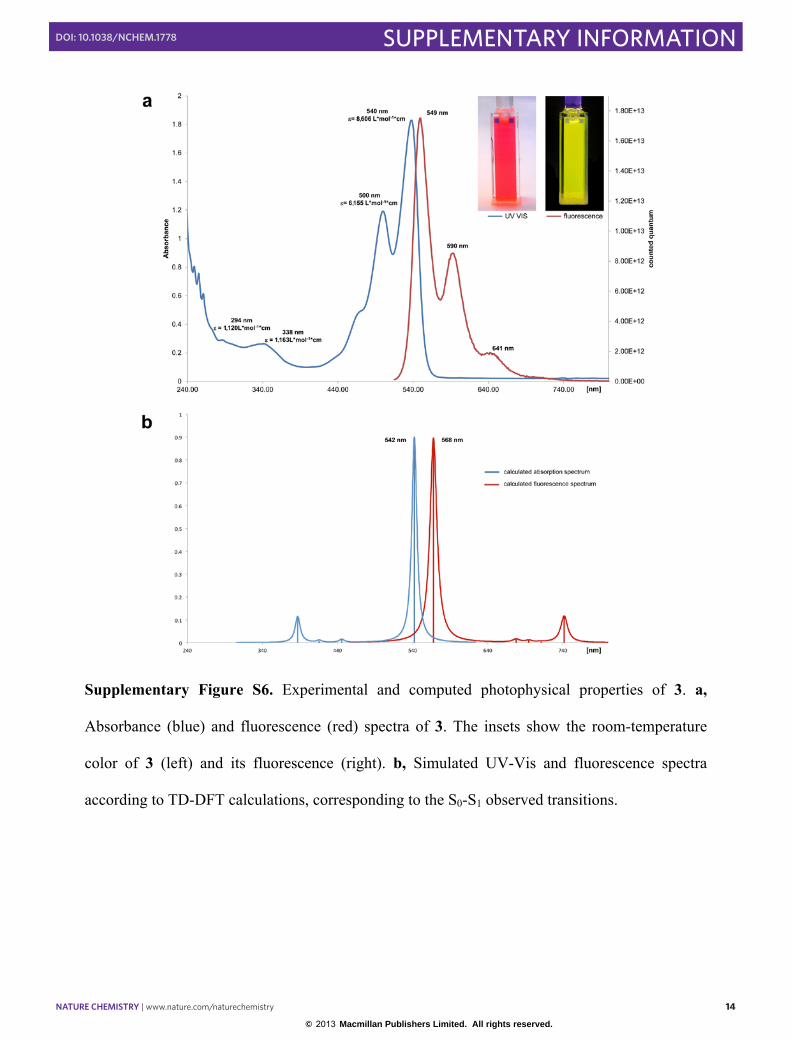
Compound 3 is red-orange in solution at room temperature, but appears yellow at −78 °C. Strong
absorption bands in the green-blue-violet region of the UV-vis spectrum of 3 explain this red-
orange color (Figure S6a). Pulvinic acid derivatives are partly responsible for the fluorescence of
lichens, including pulvinic acid lactone - which fluoresces yellow under UV light3,4. The
structural similarities of bis(boralactone) 3 to pulvinic acid lactone are striking - they differ only
© 2013 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 13
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
13
in the replacement of a [Ph-C] fragment with an isoelectronic [NHC→B] fragment. Likewise,
compound 3 fluoresces yellow under UV light; the fluorescence spectrum of 3 showed two major
bands at 549 and 590 nm (Figure S6b). Time-dependent DFT calculations reproduced the major
absorption (exp: 540 nm; calc: 542 nm) and emission bands (exp: 549 nm; calc: 568 nm) of 3
very well (see Figure S6a). Further calculation at the CASPT2(4,3) level indicates that upon UV
irradiation, compound 3 effectively forms a bis(base-stabilized boraalkene) with a B=C-C=B
core, is excited to a singlet diradical state with a B(�)-C=C-B(�) core. A small contraction in the
B-C(carbene) distances (1.5305 vs. 1.5440 Å), and an increase in the coplanarity of the NHC and
bis(boralactone) rings (torsion angle: 18.9 vs 27.4°), both indicate that a non-negligible amount
of B=C double bonding is present in the excited state molecule.
© 2013 Macmillan Publishers Limited. All rights reserved.
NATURE CHEMISTRY | www.nature.com/naturechemistry 14
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
14
Supplementary Figure S6. Experimental and computed photophysical properties of 3. a,
Absorbance (blue) and fluorescence (red) spectra of 3. The insets show the room-temperature
color of 3 (left) and its fluorescence (right). b, Simulated UV-Vis and fluorescence spectra
according to TD-DFT calculations, corresponding to the S0-S1 observed transitions.
© 2013 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 15
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
15
Geometry optimisations of 1, 2 and 3 for energy evaluations
Geometry optimizations were carried out in the gas phase using the Amsterdam Density
Functional (ADF)5,6 program at the OLYP/DZP7–11 level. To obtain the singlet state, spin-
restricted calculations were performed constraining the projection of the total electronic spin
along a reference axis to zero. The Jmol11 program was used for vizualisation purposes.
Supplementary Figure S7. Calculated and experimental (in italics) geometrical parameters for
1, 2 and 3. Bond distances in Å, calculations were performed at the OLYP/DZP level of theory.
© 2013 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 16
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
16
References
1. Braunschweig, H. et al. Ambient-Temperature Isolation of a Compound with a Boron-Boron
Triple Bond. Science 336, 1420-1422 (2012).
2. M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G.
Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P.
Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K.
Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T.
Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers,
K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.
C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox,
J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev,
A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G.
Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö.
Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09 Revision B.01,
Gaussian, Inc: Wallingford CT, 2010.
3. Gaylord, M. C., Benedict, R. G., Hatfield, G. M. & Brady, L. R. Isolation of Diphenyl-
Substituted Tetronic Acids from Cultures of Paxillus atrotomentosus. J. Pharm. Sci. 59,
1420-1423 (1970).
4. Lunak, S., Vynuchal, J. & Hrdina, R. Geometry and absorption of diketo-pyrrolo-pyrrole
isomers and their pi-isoelectronic furo-furanone analogues. J. Mol. Struct. 919, 239-245
(2009).
5. te Velde, G., Bickelhaupt, F. M., Baerends, E. J., Fonseca Guerra, C., van Gisbergen, S. J.
A., Snijders, J. G. & Ziegler, T. Chemistry with ADF. J. Comp. Chem., 22, 931-967 (2001).
© 2013 Macmillan Publishers Limited. All rights reserved.

NATURE CHEMISTRY | www.nature.com/naturechemistry 17
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NCHEM.1778
17
6. Amsterdam Density Functional, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The
Netherlands, http://www.scm.com.
7. Handy, N. C. & Cohen, A. Left-right correlation energy. J. Mol. Phys. 99, 403-412 (2001).
8. Raffenetti, R. C. Even-tempered atomic orbitals. II. Atomic SCF wavefunctions in terms of
even-tempered exponential bases. J. Chem. Phys. 59, 5936-5949 (1973).
9. van Lenthe, E. & Baerends, E. J. Optimized Slater-type basis sets for the elements 1–118. J.
Comp. Chem. 24, 1142-1156 (2003).
10. Chong, D. P., van Lenthe, E., van Gisbergen, S. J. A. & Baerends, E. J. Even-Tempered
Slater-Type Orbitals Revisited: From Hydrogen to Krypton. J. Comp. Chem. 25, 1030-1036
(2004).
11. Chong, D. P. Augmenting basis set for time-dependent density functional theory calculation
of excitation energies: Slater-type orbitals for hydrogen to krypton. Mol. Phys. 103, 749-761
(2005).
11. Jmol: an open-source Java viewer for chemical structures in 3D. http://www.jmol.org.
© 2013 Macmillan Publishers Limited. All rights reserved.


















