
“Sleeping Beauty” Phenomenon: SuFEx-Enabled Discovery of ...
107
doi.org/10.26434/chemrxiv.7842020.v1 “Sleeping Beauty” Phenomenon: SuFEx-Enabled Discovery of Selective Covalent Inhibitors of Human Neutrophil Elastase Qinheng Zheng, Jordan L. Woehl, Seiya Kitamura, Diogo Santos-Martins, Christopher J. Smedley, Gencheng Li, Stefano Forli, John E. Moses, Dennis W. Wolan, K. Barry Sharpless Submitted date: 14/03/2019 • Posted date: 15/03/2019 Licence: CC BY-NC-ND 4.0 Citation information: Zheng, Qinheng; Woehl, Jordan L.; Kitamura, Seiya; Santos-Martins, Diogo; Smedley, Christopher J.; Li, Gencheng; et al. (2019): “Sleeping Beauty” Phenomenon: SuFEx-Enabled Discovery of Selective Covalent Inhibitors of Human Neutrophil Elastase. ChemRxiv. Preprint. Sulfur-Fluoride Exchange (SuFEx) has emerged as the new generation of click chemistry. We report here a SuFEx-enabled approach exploiting the “sleeping beauty” phenomenon of sulfur fluoride compounds in the context of the serendipitous discovery of selective covalent human neutrophil elastase (hNE) inhibitors. Evaluation of an ever-growing collection of SuFExable compounds toward various biological assays unexpectedly yielded a selective and covalent hNE inhibitor, benzene-1,2-disulfonyl fluoride. Derivatization of the initial hit led to a better agent, 2- triflyl benzenesulfonyl fluoride, itself made through a SuFEx trifluoromethylation process, with IC50 = 1.1 μM and ~200-fold selectivity over the homologous neutrophil serine protease, cathepsin G. The optimized probe only modified active hNE and not its denatured form, setting another example of the “sleeping beauty” phenomenon of sulfur fluoride capturing agents for the discovery of covalent medicines. File list (2) download file view on ChemRxiv Zheng et al Sleeping Beauty 2019 ChemRxiv.pdf (3.27 MiB) download file view on ChemRxiv Zheng et al Sleeping Beauty 2019 SI.pdf (4.60 MiB)
Transcript of “Sleeping Beauty” Phenomenon: SuFEx-Enabled Discovery of ...

doi.org/10.26434/chemrxiv.7842020.v1
“Sleeping Beauty” Phenomenon: SuFEx-Enabled Discovery of SelectiveCovalent Inhibitors of Human Neutrophil ElastaseQinheng Zheng, Jordan L. Woehl, Seiya Kitamura, Diogo Santos-Martins, Christopher J. Smedley, GenchengLi, Stefano Forli, John E. Moses, Dennis W. Wolan, K. Barry Sharpless
Submitted date: 14/03/2019 • Posted date: 15/03/2019Licence: CC BY-NC-ND 4.0Citation information: Zheng, Qinheng; Woehl, Jordan L.; Kitamura, Seiya; Santos-Martins, Diogo; Smedley,Christopher J.; Li, Gencheng; et al. (2019): “Sleeping Beauty” Phenomenon: SuFEx-Enabled Discovery ofSelective Covalent Inhibitors of Human Neutrophil Elastase. ChemRxiv. Preprint.
Sulfur-Fluoride Exchange (SuFEx) has emerged as the new generation of click chemistry. We report here aSuFEx-enabled approach exploiting the “sleeping beauty” phenomenon of sulfur fluoride compounds in thecontext of the serendipitous discovery of selective covalent human neutrophil elastase (hNE) inhibitors.Evaluation of an ever-growing collection of SuFExable compounds toward various biological assaysunexpectedly yielded a selective and covalent hNE inhibitor, benzene-1,2-disulfonyl fluoride. Derivatization ofthe initial hit led to a better agent, 2- triflyl benzenesulfonyl fluoride, itself made through a SuFExtrifluoromethylation process, with IC50 = 1.1 μM and ~200-fold selectivity over the homologous neutrophilserine protease, cathepsin G. The optimized probe only modified active hNE and not its denatured form,setting another example of the “sleeping beauty” phenomenon of sulfur fluoride capturing agents for thediscovery of covalent medicines.
File list (2)
download fileview on ChemRxivZheng et al Sleeping Beauty 2019 ChemRxiv.pdf (3.27 MiB)
download fileview on ChemRxivZheng et al Sleeping Beauty 2019 SI.pdf (4.60 MiB)

“Sleeping beauty” phenomenon: SuFEx-enabled discovery of selective covalent
inhibitors of human neutrophil elastase
Qinheng Zhenga,1 Jordan L. Woehlb,1 Seiya Kitamurab, Diogo Santos-Martinsc, Christopher J.
Smedleyd, Gencheng Lia, Stefano Forlic, John E. Mosesd, Dennis W. Wolanb,2, and K. Barry
Sharplessa,2
aDepartment of Chemistry; bDepartment of Molecular Medicine; cDepartment of Integrative
Structural and Computational Biology, The Scripps Research Institute, La Jolla, California 92037,
United States; dLa Trobe Institute for Molecular Science, La Trobe University, Bundoora,
Melbourne, VIC 3086, Australia
1Q.Z. and J.L.W. contributed equally to this work.
2To whom correspondence may be addressed. Email: [email protected] or
Keywords:
click chemistry; SuFEx; sulfonyl fluoride; elastase; covalent inhibitor

Abstract:
Sulfur-Fluoride Exchange (SuFEx) has emerged as the new generation of click chemistry. We
report here a SuFEx-enabled approach exploiting the “sleeping beauty” phenomenon of sulfur
fluoride compounds in the context of the serendipitous discovery of selective covalent human
neutrophil elastase (hNE) inhibitors. Evaluation of an ever-growing collection of SuFExable
compounds toward various biological assays unexpectedly yielded a selective and covalent hNE
inhibitor, benzene-1,2-disulfonyl fluoride. Derivatization of the initial hit led to a better agent, 2-
triflyl benzenesulfonyl fluoride, itself made through a SuFEx trifluoromethylation process, with IC50
= 1.1 μM and ~200-fold selectivity over the homologous neutrophil serine protease, cathepsin G.
The optimized probe only modified active hNE and not its denatured form, setting another
example of the “sleeping beauty” phenomenon of sulfur fluoride capturing agents for the discovery
of covalent medicines.

Sulfur fluoride exchange (SuFEx)—the new generation click chemistry, since first introduced in
2014 (1), has quickly found diverse applications across an array of fields including chemical
synthesis (2-12), material science (13-19), chemical biology (20-31), and drug discovery (32, 33).
SuFEx creates robust intermolecular links between modules. The extreme fidelity stems from the
ability of otherwise, very stable higher oxidation state sulfur fluorides (34-38) to exchange S–F
with incoming nucleophiles under SuFEx catalysis conditions, forming stable and irreversible
linkages united through a sulfur center. These SuFEx reactions are made possible by the unique
requirements set to stabilize a departing fluoride ion in its transit away from a strong covalent
bond, with “H+” or “R3Si+” mediators, and especially favored by DBU-type amine catalysts (13, 39,
40), and also thought to involve bifluoride counter ion species (16, 17, 41).
In the context of selective in vitro or in vivo covalent capture of proteins by SuFExable*
compounds, examples are accumulating rapidly (20-31), but we are still reluctant to make strong
a priori inferences about the factors which determine a given capture’s occurrence. This said,
there is one remarkable fact shared by all SuFEx-based protein captures that the probes’ special
S–F links are among the most demure electrophiles known† (1, 34-38). Only a correctly folded
and functionally active protein can serve as a catalyst‡ for a SuFEx capture event upon one of its
own nucleophilic amino acid sidechains (Fig. 1A). These bio-compatible SuFEx reactions are
presumably mediated by environmental factors unique to the “live” system, thereby facilitating
displacement of the otherwise stable sulfur-fluoride by the partner protein. In contrast, the
denatured proteins are completely inert to the same S–F probes (24, 32). We name this special
relationship between functional living proteins and SuFExable probes the “sleeping beauty”
phenomenon§, which mirrors very closely our earlier protein enabled Huisgen azide-alkyne
cycloaddition (i.e. in situ click chemistry) (42, 43), in terms of the “live” enzyme requirement.
Taking inspiration from the original click chemistry manifesto (44); that molecular diversity can
be achieved with ease through the connection of small modules, using just a few good reactions,
in sequences of usually no more than three steps, we demonstrate here two distinct but logically

connected, sequential SuFEx-enabled entities: (1) efficient construction of a pool of SuFExable
compounds via click chemistry principles, and (2) bioprospecting this library for covalent capture
agents for important protein targets via “sleeping beauty” reactivity. The expectation, based on
previous experience, is that evaluation of a SuFEx library will often result in multiple lead
compounds. The latter can usually be followed up by easily deployed SAR-based (structure-
activity-relationship-based) lead studies. This “compound-bank” mode of the SuFEx-enabled
platform, itself contributes to the ever-growing library of SuFExable modules and candidates, and
like “compounding-interest”, rewards the principle library with expanding opportunities for
discovering new kinds of phenotypic modulators; including useful new functions for medicines in
the long term (Fig. 1B).
Over the past five years, we (1, 5, 8, 45) and others (12, 46-49) have developed a valuable
collection of efficient methods to synthesize SuFExable compounds from either simple or complex
organic molecules using highly connective SuFEx core electrophiles (e.g. SO2F2, O=SF4,
CH2=CHSO2F). Collective work within the Sharpless laboratory continues to build a library of
SuFExable small modules. There are >1000 compounds in this collection¶. Thanks to the great
stability of the SuFExable S–F links toward water and oxygen, the oldest library deposits (as
DMSO solutions) are still pure. The individual compounds are dissolved as 10 mM DMSO stock
solutions and stored in –20 ºC freezer, with most being stable at –20 ºC as evidenced by periodic
inspection by LC-MS of randomly sampled compound over 2 years. To date, the library and/or its
sub-libraries have been screened with collaborators at Scripps Research and other institutes
against multiple targets. Here we report a case study using the SuFEx-enabled approach to the
discovery of selective, covalent inhibitors of human neutrophil elastase (hNE).
Human neutrophil elastase; a member of the serine protease superfamily, is aberrantly active
in cystic fibrosis (CF), chronic obstructive pulmonary disease (COPD), and inflammatory bowel
diseases (IBD) (50-66). This protease is therefore a key target for the development of anti-
inflammatory agents to combat these diseases. Alkyl and aryl sulfonyl fluorides have a long

history as rather promiscuous covalent inhibitors of serine proteases (67-76). In the late 1970’s,
the Powers group demonstrated the intrinsic reactivity differences across several serine
proteases, including elastase, with 2-amido(peptido) benzenesulfonyl fluoride inhibitors (77, 78).
Their results encouraged us to investigate our library of SuFExable compounds as potential
covalent inhibitors of hNE.
Results and Discussion
A set of 105 compounds (SI Appendix, Table S1), many of which appear to be new
compounds, were selected without bias, to form a primary library for the screen against hNE. The
selected compounds can be categorized into four subsets by the nature of S–F functional groups
(Fig. 1C). Each group tends to have its own intrinsic zone of reactivity in acid-base environments.
To give a general impression, the relative rate measured on a representative molecule of each
subset under SuFEx catalysis are 28.5 (I), 1.0 (II, reference), 14.1 (III), and 4.1 (IV), respectively
(SI Appendix, Figure S6). Aryl sulfonyl fluorides (I), despite their own extreme resistance toward
nucleophiles*, top the SuFEx reactivity hierarchy among the four subsets, while aryl fluorosulfates
(II) lie at the bottom.
Elastase (5 nM) was incubated with each entry of the primary SuFEx library (final compound
concentration 200 μM) for 10 min at room temperature prior to the addition of peptide substrate
MeOSuc-AAPV-AMC (50 μM). Increase in fluorescence was measured for 30 min at 30 sec
intervals. The assay was well behaved, as evidenced by a Z’ of 0.86, signal-to-background of
~3000:1, and a hit cutoff of >95% inhibition. Reasoning that a covalent inhibitor, which presumably
targets a catalytic residue, should completely inactivate the enzyme at high compound
concentration (200 μM), a high threshold (95%) was chosen for hit identification. Under this
criterion, the screen yielded 7 hits as probable covalent inhibitors (Fig. 2A, 6.7% overall hit rate
and 23% hit rate within subset I). All 7 compounds belong to subset I, which suggests a rough cut
off based on the S-link’s inherent reactivity. After validation by NMR, LC-MS, and dose-dependent

response, benzene-1,2-disulfonyl fluoride (1) proved to be the leading candidate, inhibiting hNE
with IC50 = 3.3 ± 1.0 μM (Fig. 2B,C).
The covalent inhibition of hNE by 1 was examined by high-resolution MALDI-TOF mass
spectrometry (Fig. 3A). Incubation of 1 (exact mass 242) with hNE yielded a peak shift from the
protein mass by ~223 Da. Increased mass corresponds to: (1) a single molecule of 1 covalently
captured by hNE; (2) the loss of one hydrogen from the protease (possibly from the catalytic
serine); (3) the loss of one fluorine from 1.
We also subjected hNE to co-crystallization with 1 in order to corroborate the covalent binding.
The structure was determined using molecular replacement with PDB ID 5adw and the co-
complex was refined to 2.33 Å resolution (SI Appendix, Table S2). Importantly the naïve Fo-Fc
electron density maps contoured to 4σ clearly position 1, as a result of the strong diffraction of
sulfurs (Fig. 3B). The aryl group of 1 is nestled into a hydrophobic pocket consisting of residues
Phe192 and Val216 and the compound is covalently bound to the catalytic Ser195, as highlighted
by continuous electron density and a bond distance of ~1.6 Å (Fig. 3C,D). The covalent inhibition
of hNE via sulfonylation by 1 appeared to be permanent—dialyzing away small molecules after
incubation did not recover enzyme function (SI Appendix, Figure S2).
Considering the mono-covalent attachment mode of 1, with the second –SO2F intact, it was
envisaged that one of the two sulfonyl fluoride groups could be substituted so as to perhaps
improve the capture rate, and/or selective binding. A set of benzenesulfonyl fluoride cores
carrying ortho-substituents (8–23) was therefore examined. Compounds 8–18 were synthesized
by the efficient aqueous potassium bifluoride exchange procedure from commercially available
sulfonyl chlorides but showed poorer reactivity/binding with hNE#. Of the two improved
compounds found in Table 1, 19, the mono-vinylogous derivative of 1, was a more active inhibitor
with IC50 = 2.2 ± 0.7 μM. The ortho-sulfamoyl benzenesulfonyl fluorides (20, 21) led to
considerably lower activity. A recently developed potassium bifluoride catalyzed SuFEx
trifluoromethylation of aryl sulfonyl fluorides (79) enabled us to quickly convert 1 to the

perfluoroalkyl sulfones (22, 23). Compound 22 with the ortho-triflyl group emerged as the best
lead molecule to date with IC50 = 1.1 ± 0.1 μM.
Compound R IC50 (µM)||
1 SO2F 3.3 ± 1.0
8 F ~120
9 Cl 82 ± 16
10 Br 20 ± 10
11 I 9.7 ± 1.2
12 Me >200
13 OMe 73 ± 4
14 CN 13.3 ± 0.5
15 CF3 60 ± 8
16 NO2 20 ± 1
17 CO2Me 37 ± 2
18 Ph 27 ± 1
19
2.2 ± 0.7
20
84.4 ± 0.6
21
>200
22 SO2CF3 1.1 ± 0.1
23 SO2(CF2)2CF3 48 ± 2
SO2F
R
SO2F
S NO
OO
S NO
ON S F
O
O

Table 1. Efforts of optimization of lead compound 1 with alternative ortho-substitutions.
High resolution MALDI-TOF mass spectrometry study supports the covalent inhibition
mechanism of 22 to be sulfonylation of hNE (+ ~273 Da) possibly at its active site serine (Fig.
4A,B). In contrast, the incubation of compound 22 with inactive denatured hNE led to no
detectable covalent modification of the enzyme (Fig. 4C). The requirement for the naturally folded
enzyme represents another example of the unusual “sleeping beauty” phenomenon of SuFEx,
wherein the sulfur fluoride probes are inert in biological fluids/buffer if the capture protein or
proteins presented are denatured (24, 32).
Intriguingly, testing the lead compounds (1, 19, 22) against a panel of serine proteases, we
found that two (1, 22) among the three effective hNE inhibitors did not inactivate the homologous
serine protease, human cathepsin G (hCG), which has 37% sequence identical to hNE and highly
similar crystal structure (root-mean-square deviation (rmsd) = 0.82 Å, max rmsd = 5.89 Å for 180
out of 218 Cα residues of hNE). Unlike PMSF (PhCH2SO2F) long known for ablating the hydrolytic
activity of almost all serine proteases, the compounds 1 and 22 identified in this study showed 58
and 182-fold specificity for hNE over hCG, respectively (Table 2). The selective inhibition of hNE
could be partly attributed to a proximity factor as suggested by molecular modeling using a
reactive docking protocol (SI Appendix, Figure S5).
Compound Structure hNE IC50 (µM)|| hCG IC50 (µM)|| S**
1
3.3 ± 1.0 190 ± 40 58
19
2.2 ± 0.7 6.0 ± 0.7 2.7
22
1.1 ± 0.1 >200 >182
SO2F
SO2F
SO2F
SO2F
SO2F
SO2CF3

PMSF
24 ± 1 69 ± 6 2.5
Table 2. Selectivity of the most potent compounds 1, 19, 22 against hNE and hCG.
To conclude, we have demonstrated a SuFEx library-enabled approach to discover covalent
deactivators of an enzyme’s function, the protein at hand being human neutrophil elastase. Its
structure is known, including complexed with reversible inhibitors in the active site, but the library
of sulfonyl fluorides used in the screen was chosen without regard to any enzyme•potential ligand
relationships. In other words, agnostic of structural considerations, the library yielded two more
examples of the “sleeping beauty” phenomenon, and in this instance its successful outcome is
distinguished by lack of known design elements around the pendant S–F electrophiles on the
candidate “ligand” scaffolds. This successful sulfur fluoride library is being used and augmented
regularly at Scripps Research, and it will hopefully contribute to future SuFEx-driven covalent drug
discovery endeavors.
SO2F

Fig. 1. Overview of SuFEx-enabled covalent drug discovery. (A) The “sleeping beauty”
phenomenon: sulfur fluoride probes only capture a naturally folded protein where a nucleophilic
sidechain (Nu) and activating sidechains (Act) are correctly positioned. (B) Cartoon schematic of
the SuFEx-enabled covalent drug discovery process. (C) Efficient SuFEx derivatization of
abundant building blocks yields a SuFExable library with four subset groups, categorized
according to their sulfur-fluoride based functionalities.

Fig. 2. Screen of the primary SuFEx library toward elastase inhibitory activity. (A) Initial screen
with 105 SuFExable compounds yielded 7 hits with >95% inhibition at 200 µM. (B) Dose-response
curves of hit compounds against hNE (AAPV-AMC fluorescence assay). Each compound was
assessed over a two-fold logarithmic dilution series. (C) Structures and IC50 values of compounds
1–7 and PMSF (phenylmethane sulfonyl fluoride). IC50 values were measured based on 10 min
incubation and are shown in mean ± SD (n ≥ 3).

Fig. 3. Compound 1 is a covalent inhibitor of hNE. (A) MALDI-TOF mass spectrometry evidence
of covalent complex 1:hNE formation. (B) Naïve Fo-Fc map contoured at 1.5s (green) and 4s
(magenta) clearly delineate the binding orientation of 1 to Ser195 and the specific location of the
sulfur groups, respectively. Residue of 1 is shown as a stick model with yellow carbon, red oxygen,
light blue fluorine, mustard sulfur. (C) Active site residues that provide potential hydrogen bonds
(black dashes), repulsive interactions (brown dash), and hydrophobic residues that bind 1 (grey
elastase carbon, nitrogen blue). (D) Schematic of bond distances between 1 and elastase with
potential hydrogen bonds (black dashes) and negative repulsive interactions (brown dash) with
bond distances in Å. The covalent bond (red) between Ser195 and the sulfur of 1 were set at 1.57
Å during structure refinement.

Fig. 4. The “sleeping beauty” phenomenon of sulfonyl fluoride 22 demonstrated by the MALDI-
TOF mass spectra of (A) naturally folded hNE; (B) naturally folded hNE incubated with 22; (C)
denatured hNE (boiled) incubated with 22.

Footnotes:
*SuFExable are defined to describe those compounds containing a sulfur fluoride link that can
undergo SuFEx reaction.
†For example, no reaction occurred in the neat mixture of refluxing benzenesulfonyl fluoride (4
mL, ~33 mmol) and aniline (45 mL, ~500 mmol) at 184 ºC for 3 h. Under the same conditions,
electrophiles commonly studied as covalent “warheads” in medicinal chemistry including epoxide,
acrylamide, vinyl sulfone, chloroacetamide, chloromethyl ketone, β-lactam, maleimide, and
fluorophosphate are not stable. (SI Appendix, Table S3).
‡As long as the accelerating AA sidechains (including but not limited to His57) and reactive moiety
(Ser195) are separately considered, and the former remain overall unchanged, the intramolecular
activation mode can be seen as “catalytic”, even if the TON equals 1. For a similar case, see:
Kassem S, et al. (2017) Stereodivergent synthesis with a programmable molecular machine.
Nature 549(7672):374-378.
§“Sleeping beauty” here defines the phenomenon of the remarkable acceleration of a SuFEx
reaction for otherwise very stable higher valent sulfur fluoride compounds, by the right, naturally
folded protein catalyst for its own covalent capture. The term has been used by Izsvák and co-
workers in molecular biology to describe the use of a specifically designed transposon system
(i.e. Tc1/mariner-type transposase and transposon) that can selectively insert genes into the
genomes of vertebrates. The genome of humans as well as of other mammals encodes for non-
functional transposases and the introduction of an active transposase from lower vertebrate fish
and amphibian species “awakens the system from an evolutionary sleep”. For the original paper
on sleeping beauty transposon, see: Ivics Z, Hackett PB, Plasterk RH, & Izsvak Z (1997)
Molecular reconstruction of Sleeping beauty, a Tc1-like transposon from fish, and its transposition
in human cells. Cell 91(4):501-510.
¶We are very grateful to the following co-workers for their contribution to the SuFExable compound
library: Hua Wang (manager), Peng Wu and his group, Scripps Research, Jiajia Dong and his

group, SIOC, Larisa Krasnova, Suhua Li, Qinheng Zheng, Bing Gao, Grant A. L. Bare, John E.
Moses and his Group, La Trobe University, Hua-Li Qin and his group, Wuhan University of
Technology, En-Xuan Zhang and his group, AsymChem Inc., Gerui Ren, Gencheng Li, Feng Zhou,
Feng Liu, Hamid R. Safaei.
#At the time of manuscript preparation, Murthy and co-workers reported a study on 2-
nitrobenzenesulfonyl fluoride (19) and derivatives as covalent capture agents for new antibiotics
development. Compound 17 showed only moderate inhibitory potency in our elastase activity
assay. For Murthy’s paper, see: Sadlowski C, et al. (2018) Nitro sulfonyl fluorides are a new
pharmacophore for the development of antibiotics. Mol Syst Des Eng 3(4):599-603.
||IC50 values were measured based on 10 min incubation and are shown in mean ± SD (n ≥ 3).
**S value denotes the selectivity, defined by the ratio of IC50 (hCG) over IC50 (hNE).
Acknowledgements:
The authors gratefully acknowledge financial support from NIH R01 GM117145 (K.B.S.), The
Scripps Research Institute (D.W.W.), NIH T32 AI7354-27 (J.L.W.), NIH R01 GM069832 (D.S.M.,
S.F.). We thank H. Rosen for access to instrumentation and the staff of the Stanford Synchrotron
Radiation Lightsource. We thank Y. Su of the Molecular Mass Spectrometry Facility of UCSD for
assistance on APCI-MS.

Reference:
1. Dong JJ, Krasnova L, Finn MG, & Sharpless KB (2014) Sulfur(VI) Fluoride Exchange
(SuFEx): Another Good Reaction for Click Chemistry. Angew Chem Int Edit 53(36):9430-
9448.
2. Hanley PS, Ober MS, Krasovskiy AL, Whiteker GT, & Kruper WJ (2015) Nickel- and
Palladium-Catalyzed Coupling of Aryl Fluorosulfonates with Aryl Boronic Acids Enabled
by Sulfuryl Fluoride. Acs Catal 5(9):5041-5046.
3. Qin HL, Zheng QH, Bare GAL, Wu P, & Sharpless KB (2016) A Heck-Matsuda Process
for the Synthesis of -Arylethenesulfonyl Fluorides: Selectively Addressable Bis-
electrophiles for SuFEx Click Chemistry. Angew Chem Int Edit 55(45):14155-14158.
4. Barrow AS & Moses JE (2016) Synthesis of Sulfonyl Azides via Lewis Base Activation of
Sulfonyl Fluorides and Trimethylsilyl Azide. Synlett 27(12):1840-1843.
5. Li SH, Wu P, Moses JE, & Sharpless KB (2017) Multidimensional SuFEx Click Chemistry:
Sequential Sulfur(VI) Fluoride Exchange Connections of Diverse Modules Launched From
An SOF4 Hub. Angew Chem Int Edit 56(11):2903-2908.
6. Schimler SD, et al. (2017) Nucleophilic Deoxyfluorination of Phenols via Aryl
Fluorosulfonate Intermediates. J Am Chem Soc 139(4):1452-1455.
7. Zelli R, Tommasone S, Dumy P, Marra A, & Dondoni A (2016) A Click Ligation Based on
SuFEx for the Metal-Free Synthesis of Sugar and Iminosugar Clusters. Eur J Org Chem
(30):5102-5116.
8. Zha GF, et al. (2017) Palladium-Catalyzed Fluorosulfonylvinylation of Organic Iodides.
Angew Chem Int Edit 56(17):4849-4852.
9. Ren GR, Zheng QH, & Wang H (2017) Aryl Fluorosulfate Trapped Staudinger Reduction.
Org Lett 19(7):1582-1585.

10. Smedley CJ, et al. (2017) Sulfur-Fluoride Exchange (SuFEx)-Mediated Synthesis of
Sterically Hindered and Electron-Deficient Secondary and Tertiary Amides via Acyl
Fluoride Intermediates. Chem-Eur J 23(42):9990-9995.
11. Gao B, Li SH, Wu P, Moses JE, & Sharpless KB (2018) SuFEx Chemistry of Thionyl
Tetrafluoride (SOF4) with Organolithium Nucleophiles: Synthesis of Sulfonimidoyl
Fluorides, Sulfoximines, Sulfonimidamides, and Sulfonimidates. Angew Chem Int Edit
57(7):1939-1943.
12. Guo TJ, et al. (2018) A New Portal to SuFEx Click Chemistry: A Stable Fluorosulfuryl
Imidazolium Salt Emerging as an "F-SO2+" Donor of Unprecedented Reactivity,
Selectivity, and Scope. Angew Chem Int Edit 57(10):2605-2610.
13. Dong JJ, Sharpless KB, Kwisnek L, Oakdale JS, & Fokin VV (2014) SuFEx-Based
Synthesis of Polysulfates. Angew Chem Int Edit 53(36):9466-9470.
14. Yatvin J, Brooks K, & Locklin J (2015) SuFEx on the Surface: A Flexible Platform for
Postpolymerization Modification of Polymer Brushes. Angew Chem Int Edit 54(45):13370-
13373.
15. Oakdale JS, Kwisnek L, & Fokin VV (2016) Selective and Orthogonal Post-Polymerization
Modification using Sulfur(VI) Fluoride Exchange (SuFEx) and Copper-Catalyzed Azide-
Alkyne Cycloaddition (CuAAC) Reactions. Macromolecules 49(12):4473-4479.
16. Gao B, et al. (2017) Bifluoride-catalysed sulfur(VI) fluoride exchange reaction for the
synthesis of polysulfates and polysulfonates. Nat Chem 9(11):1083-1088.
17. Wang H, et al. (2017) SuFEx-Based Polysulfonate Formation from Ethenesulfonyl
Fluoride-Amine Adducts. Angew Chem Int Edit 56(37):11203-11208.
18. Gahtory D, et al. (2018) Quantitative and Orthogonal Formation and Reactivity of SuFEx
Platforms. Chem-Eur J 24(41):10550-10556.
19. Brooks K, et al. (2018) SuFEx Postpolymerization Modification Kinetics and Reactivity in
Polymer Brushes. Macromolecules 51(2):297-305.

20. Grimster NP, et al. (2013) Aromatic Sulfonyl Fluorides Covalently Kinetically Stabilize
Transthyretin to Prevent Amyloidogenesis while Affording a Fluorescent Conjugate. J Am
Chem Soc 135(15):5656-5668.
21. Baranczak A, et al. (2015) A Fluorogenic Aryl Fluorosulfate for Intraorganellar
Transthyretin Imaging in Living Cells and in Caenorhabditis elegans. J Am Chem Soc
137(23):7404-7414.
22. Narayanan A & Jones LH (2015) Sulfonyl fluorides as privileged warheads in chemical
biology. Chem Sci 6(5):2650-2659.
23. Hett EC, et al. (2015) Rational Targeting of Active-Site Tyrosine Residues Using Sulfonyl
Fluoride Probes. Acs Chem Biol 10(4):1094-1098.
24. Chen WT, et al. (2016) Arylfluorosulfates Inactivate Intracellular Lipid Binding Protein(s)
through Chemoselective SuFEx Reaction with a Binding Site Tyr Residue. J Am Chem
Soc 138(23):7353-7364.
25. Chen WT, et al. (2016) Synthesis of Sulfotyrosine-Containing Peptides by Incorporating
Fluorosulfated Tyrosine Using an Fmoc-Based Solid-Phase Strategy. Angew Chem Int
Edit 55(5):1835-1838.
26. Hoppmann C & Wang L (2016) Proximity-enabled bioreactivity to generate covalent
peptide inhibitors of p53-Mdm4. Chem Commun 52(29):5140-5143.
27. Fadeyi O, et al. (2016) Chemoselective Preparation of Clickable Aryl Sulfonyl Fluoride
Monomers: A Toolbox of Highly Functionalized Intermediates for Chemical Biology Probe
Synthesis. Chembiochem 17(20):1925-1930.
28. Li SH, et al. (2016) Direct introduction of R-SO2F moieties into proteins and protein-
polymer conjugation using SuFEx chemistry. Polymer 99:7-12.
29. Fadeyi OO, et al. (2017) Covalent Enzyme Inhibition through Fluorosulfate Modification of
a Noncatalytic Serine Residue. Acs Chem Biol 12(8):2015-2020.

30. Wang NX, et al. (2018) Genetically Encoding Fluorosulfate-L-tyrosine To React with
Lysine, Histidine, and Tyrosine via SuFEx in Proteins in Vivo. J Am Chem Soc
140(15):4995-4999.
31. Yang B, et al. (2018) Proximity-enhanced SuFEx chemical cross-linker for specific and
multitargeting cross-linking mass spectrometry. P Natl Acad Sci USA 115(44):11162-
11167.
32. Mortenson DE, et al. (2018) "Inverse Drug Discovery" Strategy To Identify Proteins That
Are Targeted by Latent Electrophiles As Exemplified by Aryl Fluorosulfates. J Am Chem
Soc 140(1):200-210.
33. Liu ZL, et al. (2018) SuFEx Click Chemistry Enabled Late-Stage Drug Functionalization.
J Am Chem Soc 140(8):2919-2925.
34. Suter C (1944) Derivatives of Aromatic Sulfonic Acids. 1 Sulfonyl Halides, Esters, and
Anhydrides. The Organic Chemistry of Sulfur: Tetracovalent Sulfur Compounds, (Wiley,
New York), pp 452–458.
35. Steinkopf W (1927) Aromatic sulphuric flouride. J Praktische Chemie 117(1/3):1-82.
36. Steinkopf W (1930) On aromatic sulpho-fluoride. J Praktische Chemie 128(1/3):63-88.
37. Davies W & Dick JH (1932) Aliphatic sulphonyl flurorides. J Chem Soc:483-486.
38. Davies W & Dick JH (1932) Benzenesulphonyl flouride derivatives. J Chem Soc:2042-
2046.
39. Gembus V, Marsais F, & Levacher V (2008) An efficient organocatalyzed interconversion
of silyl ethers to tosylates using DBU and p-toluenesulfonyl fluoride. Synlett (10):1463-
1466.
40. Choi EJ, Jung D, Kim JS, Lee Y, & Kim BM (2018) Chemoselective Tyrosine
Bioconjugation through Sulfate Click Reaction. Chem-Eur J 24(43):10948-10952.
41. Hmissa T, et al. (2018) Autocatalytic Synthesis of Bifluoride Ionic Liquids by SuFEx Click
Chemistry. Angew Chem Int Edit 57(49):16005-16009.

42. Lewis WG, et al. (2002) Click chemistry in situ: Acetylcholinesterase as a reaction vessel
for the selective assembly of a femtomolar inhibitor from an array of building blocks.
Angew Chem Int Edit 41(6):1053-+.
43. Agnew HD, et al. (2009) Iterative In Situ Click Chemistry Creates Antibody-like Protein-
Capture Agents. Angew Chem Int Edit 48(27):4944-4948.
44. Kolb HC, Finn MG, & Sharpless KB (2001) Click chemistry: Diverse chemical function from
a few good reactions. Angew Chem Int Edit 40(11):2004-+.
45. Zheng QH, Dong JJ, & Sharpless KB (2016) Ethenesulfonyl Fluoride (ESF): An On-Water
Procedure for the Kilogram-Scale Preparation. J Org Chem 81(22):11360-11362.
46. Veryser C, Demaerel J, Bieliunas V, Gilles P, & De Borggraeve WM (2017) Ex Situ
Generation of Sulfuryl Fluoride for the Synthesis of Aryl Fluorosulfates. Org Lett
19(19):5244-5247.
47. Zhou H, et al. (2018) Introduction of a Crystalline, Shelf-Stable Reagent for the Synthesis
of Sulfur(VI) Fluorides. Org Lett 20(3):812-815.
48. Smedley CJ, et al. (2018) 1-Bromoethene-1-sulfonyl fluoride (BESF) is another good
connective hub for SuFEx click chemistry. Chem Commun 54(47):6020-6023.
49. Leng J & Qin HL (2018) 1-Bromoethene-1-sulfonyl fluoride (1-Br-ESF), a new SuFEx
clickable reagent, and its application for regioselective construction of 5-sulfonylfluoro
isoxazoles. Chem Commun 54(35):4477-4480.
50. Birrer P, et al. (1994) Protease-antiprotease imbalance in the lungs of children with cystic
fibrosis. Am J Respir Crit Care Med 150(1):207-213.
51. Cantin AM, Hartl D, Konstan MW, & Chmiel JF (2015) Inflammation in cystic fibrosis lung
disease: Pathogenesis and therapy. J Cyst Fibros 14(4):419-430.
52. Gehrig S, et al. (2014) Lack of neutrophil elastase reduces inflammation, mucus
hypersecretion, and emphysema, but not mucus obstruction, in mice with cystic fibrosis-
like lung disease. Am J Respir Crit Care Med 189(9):1082-1092.

53. Gibson RL, Burns JL, & Ramsey BW (2003) Pathophysiology and management of
pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 168(8):918-951.
54. Mayer-Hamblett N, et al. (2007) Association between pulmonary function and sputum
biomarkers in cystic fibrosis. Am J Respir Crit Care Med 175(8):822-828.
55. Nakamura H, Yoshimura K, McElvaney NG, & Crystal RG (1992) Neutrophil elastase in
respiratory epithelial lining fluid of individuals with cystic fibrosis induces interleukin-8 gene
expression in a human bronchial epithelial cell line. J Clin Invest 89(5):1478-1484.
56. Nichols DP & Chmiel JF (2015) Inflammation and its genesis in cystic fibrosis. Pediatr
Pulmonol 50 Suppl 40:S39-56.
57. Sagel SD, Chmiel JF, & Konstan MW (2007) Sputum biomarkers of inflammation in cystic
fibrosis lung disease. Proc Am Thorac Soc 4(4):406-417.
58. Sagel SD, Wagner BD, Anthony MM, Emmett P, & Zemanick ET (2012) Sputum
biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am J
Respir Crit Care Med 186(9):857-865.
59. Twigg MS, et al. (2015) The Role of Serine Proteases and Antiproteases in the Cystic
Fibrosis Lung. Mediators Inflamm 2015:293053.
60. Wagner CJ, Schultz C, & Mall MA (2016) Neutrophil elastase and matrix metalloproteinase
12 in cystic fibrosis lung disease. Mol Cell Pediatr 3(1):25.
61. Barnes PJ (1994) Cytokines as mediators of chronic asthma. Am J Respir Crit Care Med
150(5 Pt 2):S42-49.
62. Barnes PJ (2004) Mediators of chronic obstructive pulmonary disease. Pharmacol Rev
56(4):515-548.
63. Pandey KC, De S, & Mishra PK (2017) Role of Proteases in Chronic Obstructive
Pulmonary Disease. Front Pharmacol 8:512.

64. Qiu Y, et al. (2003) Biopsy neutrophilia, neutrophil chemokine and receptor gene
expression in severe exacerbations of chronic obstructive pulmonary disease. Am J
Respir Crit Care Med 168(8):968-975.
65. Belaaouaj A, et al. (1998) Mice lacking neutrophil elastase reveal impaired host defense
against gram negative bacterial sepsis. Nat Med 4(5):615-618.
66. Motta JP, et al. (2012) Food-grade bacteria expressing elafin protect against inflammation
and restore colon homeostasis. Sci Transl Med 4(158):158ra144.
67. Myers DK & Kemp A (1954) Inhibition of Esterases by the Fluorides of Organic Acids.
Nature 173(4392):33-34.
68. Fahrney DE & Gold AM (1963) Sulfonyl Fluorides as Inhibitors of Esterases .1. Rates of
Reaction with Acetylcholinesterase, Alpha-Chymotrypsin, and Trypsin. J Am Chem Soc
85(7):997-&.
69. Gold AM & Fahrney D (1964) Sulfonyl Fluorides as Inhibitors of Esterases .2. Formation
+ Reactions of Phenylmethanesulfonyl Alpha-Chymotrypsin. Biochemistry-Us 3(6):783-&.
70. Gold AM (1965) Sulfonyl Fluorides as Inhibitors of Esterases .3. Identification of Serine as
Site of Sulfonylation in Phenylmethanesulfonyl Alpha-Chymotrypsin. Biochemistry-Us
4(5):897-&.
71. Baker BR & Hurlbut JA (1968) Irreversible Enzyme Inhibitors .114. Proteolytic Enzymes .4.
Additional Active-Site-Directed Irreversible Inhibitors of Alpha-Chymotrypsin Derived from
Phenoxyacetamides Bearing a Terminal Sulfonyl Fluoride. J Med Chem 11(2):241-&.
72. Baker BR & Hurlbut JA (1968) Irreversible Enzyme Inhibitors .113. Proteolytic Enzymes .3.
Active-Site-Directed Irreversible Inhibitors of Alpha-Chymotrypsin Derived from
Phenoxyacetamides with an N-Fluorosulfonylphenyl Substituent. J Med Chem 11(2):233-
&.
73. Baker BR & Erickson EH (1968) Irreversible Enzyme Inhibitors .115. Proteolytic
Enzymes .5. Active-Site-Directed Irreversible Inhibitors of Trypsin Derived from P-

(Phenoxyalkoxy)Benzamidines with a Terminal Sulfonyl Fluoride. J Med Chem 11(2):245-
&.
74. Baker BR (1970) Specificic Irreversible Enzyme Inhibitors. Annu Rev Pharmacolog 10:35-
+.
75. Laura R, Robison DJ, & Bing DH (1980) (Para-Amidinopheny)Methanesulfonyl Fluoride,
an Irreversible Inhibitor of Serine Proteases. Biochemistry-Us 19(21):4859-4864.
76. Shannon DA, et al. (2012) Sulfonyl Fluoride Analogues as Activity-Based Probes for
Serine Proteases. Chembiochem 13(16):2327-2330.
77. Lively MO & Powers JC (1978) Specificity and Reactivity of Human Granulocyte Elastase
and Cathepsin-G, Porcine Pancreatic Elastase, Bovine Chymotrypsin and Trypsin toward
Inhibition with Sulfonyl Fluorides. Biochim Biophys Acta 525(1):171-179.
78. Yoshimura T, Barker LN, & Powers JC (1982) Specificity and Reactivity of Human-
Leukocyte Elastase, Porcine Pancreatic Elastase, Human Granulocyte Cathepsin-G, and
Bovine Pancreatic Chymotrypsin with Arylsulfonyl Fluorides - Discovery of a New Series
of Potent and Specific Irreversible Elastase Inhibitors. J Biol Chem 257(9):5077-5084.
79. Smedley CJ, et al. (2019) Bifluoride Ion Mediated SuFEx Trifluoromethylation of Sulfonyl
Fluorides and Iminosulfur Oxydifluorides. Angew Chem Int Ed Engl.

download fileview on ChemRxivZheng et al Sleeping Beauty 2019 ChemRxiv.pdf (3.27 MiB)

S1
Supporting Information
“Sleeping beauty” phenomenon: SuFEx-enabled discovery of
selective covalent inhibitors of human neutrophil elastase
Qinheng Zhenga,1 Jordan L. Woehlb,1 Seiya Kitamurab, Diogo Santos-Martinsc, Christopher J.
Smedleyd, Gencheng Lia, Stefano Forlic, John E. Mosesd, Dennis W. Wolanb,2, and K. Barry
Sharplessa,2
aDepartment of Chemistry; bDepartment of Molecular Medicine; cDepartment of Integrative
Structural and Computational Biology, The Scripps Research Institute, La Jolla, California 92037,
United States; dLa Trobe Institute for Molecular Science, La Trobe University, Bundoora,
Melbourne, VIC 3086, Australia
1Q.Z. and J.L.W. contributed equally to this work.
2To whom correspondence may be addressed. Email: [email protected] or

S2
Table of Contents
Contents Page
1. General 4
2. Compound synthesis and characterizations 5
2.1. General procedures 5
2.2. Naphthalene-1,3,6-trisulfonyl trifluoride (2) 8
2.3. Naphthalene-2,3-disulfonyl difluoride (3) 9
2.4. Methyl 3-(fluorosulfonyl)thiophene-2-carboxylate (4) 11
2.5. 2,6-dichlorobenzenesulfonyl fluoride (5) 12
2.6. 3,4-dichlorobenzenesulfonyl fluoride (7) 13
2.7. 2-Iodobenzenesulfonyl fluoride (11) 14
2.8. [1,1'-biphenyl]-2-sulfonyl fluoride (18) 15
2.9. (E)-2-(2-(fluorosulfonyl)vinyl)benzenesulfonyl fluoride (19) 16
2.10. 2-(Morpholinosulfonyl)benzenesulfonyl fluoride (20) 17
2.11 4-((2-(Fluorosulfonyl)phenyl)sulfonyl)piperazine-1-sulfonyl fluoride (21) 19
2.12. 2-((Trifluoromethyl)sulfonyl)benzenesulfonyl fluoride (22) 21
2.13. 2-((Perfluoropropyl)sulfonyl)benzenesulfonyl fluoride (23) 22
3. Protease activity assays and library screen 23
3.1. Methods 23
3.2. Screen results 24
3.3. Validation of permanent binding 35
3.4. Inhibitory activity of hit molecules against hCG. 36
4. Mass Spectrometry 37
4.1. Methods 37
4.2. Results 37
5. X-ray crystal structure 38
5.1. Methods 38
5.2. Results 39
6. Reactive docking 40
6.1. Methods 40
6.2. Results and discussion 40
7. Reactivities of SuFExable functional groups 42
8. NMR spectra 43
8.1. Naphthalene-1,3,6-trisulfonyl trifluoride (2) 43

S3
8.2. Naphthalene-2,3-disulfonyl difluoride (3) 46
8.3. Methyl 3-(fluorosulfonyl)thiophene-2-carboxylate (4) 49
8.4. 2,6-dichlorobenzenesulfonyl fluoride (5) 52
8.5. 3,4-dichlorobenzenesulfonyl fluoride (7) 55
8.6. 2-Iodobenzenesulfonyl fluoride (11) 58
8.7. [1,1'-biphenyl]-2-sulfonyl fluoride (18) 61
8.8. (E)-2-(2-(fluorosulfonyl)vinyl)benzenesulfonyl fluoride (19) 64
8.9. 2-(Morpholinosulfonyl)benzenesulfonyl fluoride (20) 67
8.10. 4-((2-(Fluorosulfonyl)phenyl)sulfonyl)piperazine-1-sulfonyl fluoride (21) 70
8.11. 2-((Trifluoromethyl)sulfonyl)benzenesulfonyl fluoride (22) 73
8.12. 2-((Perfluoropropyl)sulfonyl)benzenesulfonyl fluoride (23) 76
9. References 80

S4
1. General Synthetic reagents, catalysts, and solvents were used as purchased without further
purification, unless otherwise indicated. The extent of reaction was monitored by thin-layer
chromatography (TLC), performed on 250 μm silica gel G plates with F254 indicator. The TLC
plates were visualized by ultraviolet light (254 nm) and treatment with potassium permanganate
stain followed by gentle heating. Flash chromatography was performed using 40−63 μm (230−400
mesh) silica gel.
Unless otherwise noted, 1H, 13C, and 19F NMR spectra were recorded on Bruker DRX-500,
Bruker DRX-600, Bruker AMX-400 instruments. Data for 1H NMR spectra is reported as follows:
chemical shift (ppm, referenced to residual solvent peak), coupling constant (Hz), and integration.
Data for 13C NMR is reported in terms of chemical shift, δ (ppm) relative to residual solvent peak
(CDCl3 singlet at 77.0 ppm, DMSO multiplet at 39.5 ppm). Data for 19F NMR is reported in terms
of chemical shift (ppm) relative to added internal standard (CFCl3 at 0.65 ppm) (1). Accurate mass
spectrometry (a.k.a. HRMS) spectra were recorded using electrospray ionization (ESI) or
atmosphere-pressure chemical ionization (APCI) with a time-of-flight (TOF) analyzer. For some
entries (compounds 5, 7, 11, and 18), soft ionization like ESI and APCI failed to give a molecular
ion signal. Hence, GC-MS with electron impact ionization (EI) and quadrupole analyzer was used
and gave strong signals at [M]+ for these compounds. Melting points were measured on a
Barnstead Electrothermal 9300 digital capillary melting point apparatus and are uncorrected.

S5
2. Compound synthesis and characterizations 2.1. General procedures
Unless otherwise noted, compounds in the SuFEx library were synthesized by the following
general procedures (I, II, III, IV). Synthesis and characterizations of compounds 1, 6, 8–10, and
12–17 have been reported in the literature (2-6).
(I) Synthesis of aryl sulfonyl fluorides
Procedure I (2): Aryl sulfonyl chloride (from commercial sources or synthesized by established
methods) dissolved in acetonitrile (0.5–1 M) was treated with saturated potassium bifluoride
aqueous solution (~5 M, 1.5–2.5 equiv). The emulsion was stirred vigorously for 1–4 h before
partitioned between ethyl acetate and water. The organic solution was collected, dried over
anhydrous sodium sulfate, concentrated and purified by column chromatography, if necessary, to
yield desired aryl sulfonyl fluoride (33 examples, 90–100% isolated yield).
(II) Synthesis of aryl fluorosulfates
Procedure IIA (2): (CAUTION: the reaction must be performed in a well vented fume hood.
Sulfuryl fluoride gas is toxic.) Phenols (from commercial sources), and triethylamine (1.5 equiv)
were dissolved in DCM. The flask sealed with a rubber septum was evacuated to gentle vacuum
evidenced by bubbling of solvent, and a balloon filled with sulfuryl fluoride gas was introduced to
the flask via a needle. The reaction was stirred vigorously for 2 h. Upon completion, solvent was
removed in vacuo. Residue was partitioned between ethyl acetate and water. The organic phase
Ar SO
OCl CH3CN/H2O
rt, 1–6 h
KFHF Ar SO
OF+
33 samples90–100% yield
OH
F S F
O O
RO S
FO
O
RNN S
O
OF
OTf 32 examples82–99% yield
NEt3DCMrt, 2 h
NEt3acetonitrile
rt, 1 h
IIA
IIB

S6
was washed with brine, dried over anhydrous sodium sulfate, then concentrated and purified by
flash column chromatography to give desired aryl fluorosulfate (32 examples, 82–99% isolated
yield).
Procedure IIB (7): Phenols (from commercial sources), and N-fluorosulfonyl N’-methyl-2-
methylimidazolium triflate (1.2 equiv) were dissolved in acetonitrile (0.1–1 M). Triethylamine (1.5
equiv) was added dropwise via a syringe. The reaction was stirred at room temperature for 1 h.
Upon completion, solvent was removed in vacuo. Residue was partitioned between ethyl acetate
and water. The organic phase was washed with brine, dried over anhydrous sodium sulfate, then
concentrated and purified by flash column chromatography to give desired aryl fluorosulfate (32
examples, 82–99% isolated yield).
(III) Synthesis of alkyl sulfonyl fluorides by Michael addition
Procedure IIIA (2, 8): To a solution of primary or secondary alkyl amine in DCM (0.5–1 M),
ESF (2.2 equiv) was added dropwise (exotherm). The mixture was stirred at autogenous
temperature for 6–12 h. Upon completion, volatiles were removed in vacuo. The residue was
purified by flash column chromatography to give desired sulfonyl fluoride adducts of primary or
secondary amines. (30 examples, 85–98% isolated yield).
Procedure IIIB (2, 8): Anilines and ESF (2.2 equiv) were dissolved (or suspended at starting
point) in glacial acetic acid (10 mmol per 1 mmol). The mixture was heated to 50 ºC (or 80 ºC if
necessary) for 12 h. Upon completion, volatiles were removed in vacuo. The residue was
purified by either recrystallization in ethanol or flash column chromatography to give desired
sulfonyl fluoride adducts of anilines (30 examples, 85–98% isolated yield).
(IV) Synthesis of vinyl sulfonyl fluorides
+ S F
O
OR N S
FO
ORR
NHR IIIA: DCM, rt, 2–6 h
IIIB: AcOH, rt, 12 h
30 examples85–98% yield
R I + S F
O
O R SFO
O
Pd(OAc)2AgTFAacetone
60 ºC, 6–12 h10 examples59–99% yield

S7
Procedure IV (9): An oven-dried vessal was charged with (hetero)aryl iodide, AgTFA (1.2
equiv), Pd(OAc)2 (2 mol%), acetone, and ethenesulfonyl fluoride (ESF, 2 equiv) were added.
The resulting mixture was refluxed at 60 ºC. Upon full conversion of (hetero)aryl iodide (6–12 h),
solvent was removed in vacuo. Crude was purified by flash column chromatography to give
desired product (10 examples, 59–99% yield).

S8
2.2. Naphthalene-1,3,6-trisulfonyl trifluoride (2)
A 100-mL round bottom flask equipped with a condenser was charged with naphthalene-
1,3,(6,7)-trisulfonic acid trisodium salt hydrate (4.34 g, < 10 mmol), and phosphorus pentachloride
(21 g, 100 mmol). Upon shaken of the flask, gas (hydrochloride with moist) evolution and
exotherm were observed. The mixture liquefied after heated at 110 ºC for about 0.5 h. The
reaction was stirred at 110 ºC overnight and cooled to room temperature. The yellow suspension
was poured with care onto crushed ice (200 g). The mixture was extracted with chloroform (150
mL x 3). The combined organic phase was washed by brine and dried over anhydrous sodium
sulfate before evaporated to a yellow solid crude (2-int-mix).
The crude sulfonyl chloride was mixed with potassium fluoride (11.6 g, 200 mmol) in a 500-mL
round bottom flask. Acetone (200 mL) was added and the resulting suspension was stirred
overnight. Volatiles were removed in vacuo, and the solid was partitioned with chloroform/water.
Organic phase was collected, concentrated and purified by column chromatography (SiO2, eluted
with hexanes to 20% ethyl acetate in hexanes). The titled compound was isolated as a white
crystalline (1.53 g, 41% yield over 2 steps, not corrected for the contamination of water in starting
material). 1H NMR (500 MHz, CDCl3) δ 9.17 (d, J = 1.3 Hz, 1H), 9.01 (d, J = 1.9 Hz, 1H), 8.99 (d, J = 1.8
Hz, 1H), 8.96 (dd, J = 9.1, 1.8 Hz, 1H), 8.50 (dd, J = 9.1, 2.0 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 139.6, 134.8 (d, JCF = 27.7 Hz), 133.5, 133.1, 132.9, 132.2, 132.0,
131.1 (d, JCF = 2.5 Hz), 130.1, 127.6. 19F NMR (377 MHz, CDCl3) δ 67.7, 66.8, 64.9.
TLC Rf = 0.65 (17% ethyl acetate in hexanes).
Melting point 124 – 126 ºC.
Mass Spectrometry APCI-TOF accurate mass calculated for C10H5F3O6S3 [M]– 373.9206, found
373.9213.
HPLC purity (280 nm) 96.8% (tR = 5.320 min).
SO3Na
SO3NaNaO3S •xH2O(6,7) PCl5
110 ºC
SO2Cl
SO2ClClO2S(6,7) KF
acetonert
SO2F
SO2FFO2S
22-int-mix

S9
2.3. Naphthalene-2,3-disulfonyl difluoride (3)
A 250-mL round bottom flask equipped with a stir bar was charged with 2,3-
dibromonaphthalene (1.1 g, 3.82 mmol), and sodium 2-propylthiolate (90% tech. grade, 1.66 g,
15.3 mmol). Hexamethylphosphoramide (HMPA, 20 mL, anhydrous, stored over 4 Å molecular
sieves) was added under N2. Upon heated to 100 ºC, the suspension was rapidly stirred into an
orange solution, and about 15 min later to another suspension. The suspension was stirred for 8
h and cooled to room temperature. Partition the mixture between ether (100 mL) and water (100
mL). Aqueous phase was further extracted by ether (50 mL x 2). Combine all organic phase, wash
with water (150 mL x 2) and brine (150 mL). Solvent was removed in vacuo giving crude thioether
(3-i). 1H NMR (500 MHz, Chloroform-d) δ 7.76 (s, 2H), 7.72 (dd, J = 6.2, 3.3 Hz, 2H), 7.42 (dd, J
= 6.2, 3.2 Hz, 2H), 3.59 (hept, J = 6.7 Hz, 2H), 1.39 (d, J = 6.7 Hz, 12H). 13C NMR (126 MHz,
CDCl3) δ 135.8, 132.3, 129.2, 127.0, 126.2, 37.4, 23.0.
The crude product (>95 % purity by 1H NMR) was dissolved in HMPA (20 mL). Under N2 stream,
freshly cut sodium strip (about 0.3 g, 10 mmol) was added with care over 15 min. A dark solution
was obtained, which was further stirred at 100 ºC overnight. The reaction gradually turned light-
yellow. Cool the mixture to room temperature, concentrated HCl was added dropwise (Be very
careful!) to adjust the pH lower than 3. The resulting suspension was partitioned between tert-
butyl methyl ether (100 mL) and water (100 mL). The organic phase was washed by water (100
mL x 2) and brine (100 mL) and concentrated to a yellow solid crude (3-ii, 0.83 g, 4.3 mmol, >100%
over 2 steps, HMPA contaminated). 1H NMR (400 MHz, Chloroform-d) δ 7.90 (s, 2H), 7.66 (dd, J
= 6.3, 3.2 Hz, 2H), 7.41 (dd, J = 6.3, 3.2 Hz, 2H), 3.88 (s, 2H).
The yellow solid was suspended on methanol (10 mL). Gentle heating at 50 ºC helped to
dissolve the thiolphenol. Cooled back to room temperature, into the solution was added hydrogen
peroxide (30%, 10 mL, ~ 40 equiv). This process was found exotherm, and precipitates formed
instantly. The yellow suspension was stirred at room temperature, turned into a thick porridge
after 3 h, and then a clear yellow solution overnight. After 24 h, the clean conversion to 3-iii was
Br
Br
3
NaSiPrHMPA100 ºC
S
S
Na (metal)HMPA100 ºC
SH
SH
H2O2H2O/MeOH
SO
O OH
SO
O
OH
NaOHH2O
SO
O ONa
SO
O
ONa
PCl5POCl3110 ºC
SO
O Cl
SO
O
Cl
KFHFCH3CN/H2O
rt
SO
O F
SO
O
F
3-i 3-ii 3-iii
3-iv 3-v

S10
determined by 1H NMR. 1H NMR (400 MHz, Methanol-d4) δ 8.68 (s, 1H), 8.01 (dd, J = 6.1, 3.2
Hz, 1H), 7.67 (dd, J = 6.2, 3.2 Hz, 1H).
Adjust the pH of the acid solution with 2 mol L-1 NaOH (3 mL) to over 8 (exotherm). Catalytic
amount of manganese dioxide (25 mg) was added to digest excess amount of hydrogen peroxide.
When gas evolution ceased, the solvent was removed in vacuo, and crude product being
azeotropically dried by toluene (25 mL x 3).
Naphthalene-2,3-disulfonic acid disodium salt, with MnO2 contaminated, was mixed with
phosphorus pentachloride (1.5 g, 7.2 mmol) in a 100-mL round bottom flask. Reaction occurred
instantly releasing heat and fume. The mixture was heated at 110 ºC for 6 h, before cooled to
room temperature and poured onto crushed ice (50 g). Sulfonyl chloride was extracted by
chloroform (100 mL), and washed sequentially by cold water (100 mL), brine (100 mL). The
sulfonyl chloride was not stable on silica gel, neither TLC nor column was applicable. 1H NMR
showed > 90% purity. 1H NMR (400 MHz, Chloroform-d) δ 8.98 (s, 1H), 8.21 (dd, J = 6.2, 3.3 Hz,
1H), 7.98 (dd, 1H).
Dissolved in acetone (20 mL, insoluble in acetonitrle), naphthalene-2,3-disulfonyl chloride (3-v) was treated by finely powdered potassium fluoride (464 mg, 8.00 mmol). The suspension was
stirred overnight, and then concentrated. Partition the crude between chloroform and water.
Organic phase was collected, concentrated, and purified by column chromatography (SiO2, 10%
to 33% ethyl acetate in hexanes) to give the titled compound as a yellow solid (208 mg, 0.711
mmol, 19% over 6 steps). 1H NMR (600 MHz, CDCl3) δ 9.00 – 8.96 (m, 2H), 8.20 (dt, J = 6.4, 3.2 Hz, 2H), 8.02 – 7.97 (m,
2H). 13C NMR (151 MHz, CDCl3) δ 137.2, 133.7, 132.9, 130.2, 126.8 (d, JCF = 30.2 Hz). 19F NMR (377 MHz, CDCl3) δ 72.8.
TLC Rf = 0.52 (33% ethyl acetate in hexanes, UV).
Melting point 156 – 158 ºC (ether).
Mass spectrometry APCI-TOF accurate mass calculated for C10H6F2O4S2NH4 [M + NH4]+
310.0014, found 310.0013.
HPLC purity (280 nm) 92.3% (tR = 5.210 min).

S11
2.4. Methyl 3-(fluorosulfonyl)thiophene-2-carboxylate (4)
The title compound was synthesized by General Procedure I. Saturated potassium bifluoride
solution effected the conversion of methyl 3-(chlorosulfonyl)thiophene-2-carboxylate (Combi-
Blocks, 1.20 g, 5.00 mmol) to methyl 3-(fluorosulfonyl)thiophene-2-carboxylate (4, 1.06 g, 4.73
mmol, 95%) as a brown solid. 1H NMR (600 MHz, CDCl3) δ 7.65 (dd, J = 5.3, 0.6 Hz, 1H), 7.61 (d, J = 5.3 Hz, 1H), 3.99 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 159.1, 137.3, 134.6 (d, JCF = 30.2 Hz), 130.9 (d, JCF = 1.5 Hz), 130.6
(d, JCF = 1.5 Hz), 53.6. 19F NMR (377 MHz, CDCl3) δ 63.0.
TLC Rf = 0.26 (17% ethyl acetate in hexanes, UV).
Melting point 78 – 80 ºC (decompoused).
Mass spectrometry ESI-TOF accurate mass calculated for C6H5FO4S2Na [M + Na]+ 246.9505,
found 246.9505.
HPLC purity (254 nm) 100% (tR = 4.307 min).
SO
O Me
SO
O Cl
SO
O Me
SO
O F
KFHFacetonitrile/H2O
rt2 h
4

S12
2.5. 2,6-dichlorobenzenesulfonyl fluoride (5)
The title compound was synthesized by General Procedure I. Saturated potassium bifluoride
solution effected the conversion of 2,6-dichlorobenzenesulfonyl chloride (Combi-Blocks, 1.23 g,
5.00 mmol) to 2,6-dichlorobenzenesulfonyl fluoride (5, 1.11 g, 4.85 mmol, 97%) as a yellow
crystalline. 1H NMR (600 MHz, CDCl3) δ 7.63 – 7.44 (m, 3H). 13C NMR (151 MHz, CDCl3) δ 136.2, 135.0, 131.6, 131.0 (d, JCF = 24.2 Hz). 19F NMR (377 MHz, CDCl3) δ 68.2.
TLC Rf = 0.47 (17% ethyl acetate in hexanes, UV).
Melting point 70 – 72 ºC (hexane/ethyl acetate).
Mass spectrometry EI-Q mass calculated for C6H3Cl2FO2S [M]+ 227.92, found 227.9 (100), 132.9
(80), 229.9 (78), 109.0 (61), 74.0 (53), 160.9 (38), 144.9 (43).
HPLC purity (280 nm) 97.8% (tR = 4.775 min).
Cl
Cl
SO
OFCl
Cl
SO
OClKFHF
acetonitrile/H2Ort
2 h5

S13
2.6. 3,4-dichlorobenzenesulfonyl fluoride (7)
The title compound was synthesized by General Procedure I. Saturated potassium bifluoride
solution effected the conversion of 3,4-dichlorobenzenesulfonyl chloride (Combi-Blocks, 1.23 g,
5.00 mmol) to 3,4-dichlorobenzenesulfonyl fluoride (7, 1.03 g, 4.50 mmol, 90%) as a colorless
liquid. 1H NMR (600 MHz, CDCl3) δ 8.10 (d, J = 2.2 Hz, 1H), 7.85 (dd, J = 8.5, 2.2 Hz, 1H), 7.73 (dd, J
= 8.5, 1.0 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 140.7, 134.2, 132.0 (d, JCF = 27.2 Hz), 131.3, 129.8, 126.9. 19F NMR (377 MHz, CDCl3) δ 66.89, 0.65.
TLC Rf = 0.50 (9% ethyl acetate in hexanes, UV).
Mass spectrometry EI-MS calculated for C6H3Cl2FO2S [M]+ 227.92, found 227.9.
HPLC purity (254 nm) 98.0% (tR = 4.977 min).
Cl
SO
O F
7
ClCl
SO
O Cl
Cl
KFHFCH3CN/H2O
rt2 h

S14
2.7. 2-Iodobenzenesulfonyl fluoride (11)
Neutralizing 2-iodobenzene sulfonic acid hydrate with sodium bicarbonate gave 2-
iodobenzene sulfonic acid sodium salt (11-i) as a brown crystalline. A 100-mL round bottom flask
equipped with a condenser and a tail gas absorbing water tank was charged with 2-iodobenzene
sulfonic acid sodium salt (5.00 g, 16.3 mmol) and phosphorus pentachloride (5.10 g, 24.5 mmol).
The solid mixture liquefied partially after shaken with exotherm. Phosphoryl chloride (2 mL) was
added to help the stirring of the syrup-like viscous mixture. The reaction was heated at 110 ºC for
4 h before cooled to room temperature. The dark red mixture was poured onto ice (150 g) and
extracted with ether (100 mL x 3). The organic phase was washed with cold water and dried over
anhydrous sodium sulfate. TLC indicated the purity of the crude product (11-ii) about 90%. The
ether solution was concentrated and remaining thick oil was re-dissolved into acetonitrile (10 mL).
Saturated potassium bifluoride solution (~ 5 mol L-1, 6.5 mL) was added, and the resulting biphasic
mixture being stirred into an emulsion for 3 h. Partition the mixture with water (100 mL) and ethyl
acetate (100 mL). The organic phase was concentrated and purified by column chromatography
(SiO2, hexanes to 17% ethyl acetate in hexanes) to give an off-white crystalline (2.73 g, 9.54
mmol, 59% over 3 steps). 1H NMR (500 MHz, CDCl3) δ 8.18 (dd, J = 7.9, 1.1 Hz, 1H), 8.15 (dd, J = 8.0, 1.5 Hz, 1H), 7.59
(tt, J = 7.9, 1.3 Hz, 1H), 7.38 (td, J = 7.7, 1.6 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 143.3, 137.8 (d, JCF = 28.7 Hz), 135.8, 132.1 (d, JCF = 1.3 Hz),
128.8, 92.3. 19F NMR (377 MHz, CDCl3) δ 56.9.
TLC Rf = 0.58 (17% ethyl acetate in hexanes, UV).
Melting point 61 – 62 ºC (MeOH).
Mass spectrometry EI-MS calculated for C6H4FIO2S [M]+ 285.9, found 285.6.
HPLC purity (280 nm) 96.2% (tR = 5.088 min).
IS
O
O FIS
O
O OH NaOHH2O
IS
O
O ONa PCl5POCl3110 ºC
IS
O
O Cl KFHFCH3CN/H2O
rt1111-i 11-ii

S15
2.8. [1,1'-biphenyl]-2-sulfonyl fluoride (18)
The title compound was synthesized by General Procedure I. Saturated potassium bifluoride
solution effected the conversion of [1,1'-biphenyl]-2-sulfonyl chloride (Alfa-Aesar, 1.25 g, 5.00
mmol) to [1,1'-biphenyl]-2-sulfonyl fluoride (18, 1.10 g, 4.66 mmol, 93%) as a white crystalline. 1H NMR (600 MHz, CDCl3) δ 8.19 (d, J = 8.0 Hz, 1H), 7.76 (t, J = 7.6 Hz, 1H), 7.60 (t, J = 7.8 Hz,
1H), 7.51 – 7.43 (m, 5H), 7.41 – 7.35 (m, 3H). 13C NMR (151 MHz, CDCl3) δ 143.2, 138.0, 134.9, 133.2 (d, JCF = 1.5 Hz), 132.5 (d, JCF = 22.7
Hz), 130.1 (d, JCF = 1.5 Hz), 129.1 (d, JCF = 1.5 Hz), 128.7, 128.2 (d, JCF = 10.6 Hz). 19F NMR (377 MHz, CDCl3) δ 67.6.
TLC Rf = 0.60 (17% ethyl acetate in hexanes).
Melting point 76 – 77 ºC (CH3CN/H2O)
Mass spectrometry EI-MS calculated for C12H9FO2S [M]+ 236.0, found 235.8.
HPLC purity (280 nm) 98.8% (tR = 5.320 min).
SO
O Cl SO
O FKFHF
CH3CN/H2Ort
18

S16
2.9. (E)-2-(2-(fluorosulfonyl)vinyl)benzenesulfonyl fluoride (19)
The title compound was synthesized by General Procedure IV. Palladium acetate/silver
trifluoroacetate effected the conversion of 11 (286 g, 1.00 mmol) and ESF (220 mg, 2.00 mmol)
to (E)-2-(2-(fluorosulfonyl)vinyl) benzenesulfonyl fluoride (19, 173.8 mg, 0.648 mmol, 65%) as a
white crystalline. 1H NMR (600 MHz, CDCl3) δ 8.50 (d, J = 15.3 Hz, 1H), 8.22 (d, J = 7.9 Hz, 1H), 7.88 (t, J = 7.6
Hz, 1H), 7.78 (dd, J = 12.8, 7.7 Hz, 3H), 6.98 – 6.91 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 143.2, 136.1, 132.8 (d, JCF = 25.7 Hz), 132.3, 131.6, 131.3, 129.7,
124.8 (d, JCF = 30.2 Hz), 117.4 (d, JCF = 336.73 Hz). 19F NMR (377 MHz, CDCl3) δ 67.1, 61.6.
TLC Rf = 0.50 (33% ethyl acetate in hexanes, UV).
Melting point 91 – 92 ºC (hexanes/ethyl acetate).
Mass spectrometry APCI-TOF accurate mass calculated for C8H6F2O4S2 [M]– 267.9681, found
267.9678.
HPLC purity (280 nm) 98.3% (tR = 5.013 min).
SO
O F
SO
O FIS
O
O F+ S F
O
O
Pd(OAc)2AgOC(O)CF3
acetone60 ºC
19

S17
2.10. 2-(Morpholinosulfonyl)benzenesulfonyl fluoride (20)
A 4-mL vial equipped with an egg-shape stir bar was charged with 2-nitrobenzenesulfonyl
chloride (222 mg, 1.00 mmol), morpholine (95.8 mg, 1.10 mmol), and dichloromethane (1.0 mL).
Triethylamine (152 mg, 1.50 mmol) was added dropwise to give a yellow suspension. The mixture
was stirred at room temperature for 4 h, before partitioned between hydrochloric acid (1 mol L-1,
50 mL) and ethyl acetate (50 mL). The organic phase was collected and concentrated to give
virtually pure sulfonamide (20-i, 270 mg, 0.99 mmol).
Into a reaction tube charged with the crude sulfonamide (270 mg), palladium on carbon (27
mg, 10% wt.), tetrahydrofuran (2 mL) and ethanol (2 mL) were added. Hydrogen gas was
introduced by a balloon, and bubbled for 10 min. The suspension was stirred under hydrogen for
another 6 h, before diluted by methanol (50 mL). The methanolic solution was passed through a
pad of celite and concentrated to give a yellow oil (20-ii). A 20-mL scintillation vial was charged with the crude aniline. Concentrated hydrochloric acid
(0.4 mL) was added, and the suspension was cooled to 0 ºC. At the same temperature, sodium
nitrite solution (40%, 0.4 mL) was added dropwise via a syringe. The diazotization process took
30 min to complete at 0 ºC. In another 20-mL scintillation vial, copper(I) chloride (30 mg, 0.31
mmol) was dissolved in hydrochloric acid (1 mL), and to which, sodium bisulfite solution (40%, 1
mL) was added dropwise to make a yellow suspension. Add the diazonium solution to the SO2
solution at 0 ºC via a glass pipette dropwise. The resulting mixture was allowed to be warmed to
5 – 10 ºC and stirred for a further 30 min, before extracted by dichloromethane (20 mL).
Concentration of the dichloromethane solution gave crude sulfonyl chloride (20-iii, ~ 80% purity).
N
O
SO
O
SO
O
F
20
N
O
SO
O
SO
O
Cl
KFHFCH3CN/H2O
rt
NaNO2, HClthen CuCl, NaHSO3
H2O0 ºC
NO2
S ClO
O
NH
O
NEt3, DCMrt NO2
SNO
O
OH2, Pd/C
THF/EtOHrt
NH2
SNO
O
O
20-i 20-ii
20-iii

S18
The crude sulfonyl chloride was dissolved into acetonitrile (2 mL) and treated with saturated
potassium bifluoride solution (1 mL). The biphasic mixture was stirred at room temperature for 2
h and partitioned between ethyl acetate (50 mL) and water (50 mL). The organic phase was
concentrated and purified by chromatography to give the titled compound as a yellow solid (142
mg, 0.459 mmol, 46% over 4 steps). 1H NMR (600 MHz, CDCl3) δ 8.35 (dd, J = 7.9, 1.4 Hz, 1H), 8.24 (dd, J = 7.9, 1.4 Hz, 1H), 7.91
(td, J = 7.7, 1.4 Hz, 1H), 7.84 (tt, J = 7.7, 1.3 Hz, 1H), 3.75 – 3.71 (m, 4H), 3.34 – 3.29 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 138.5, 135.4, 133.5, 133.2, 132.9 (d, J = 1.5 Hz), 132.1 (d, J = 27.2
Hz), 66.46, 46.14. 19F NMR (377 MHz, CDCl3) δ 64.0.
TLC Rf = 0.32 (50% ethyl acetate in hexanes, UV).
Melting point 135 – 138 ºC.
Mass spectrometry Accurate mass (ESI-TOF) calculated for C10H13FNO5S2 [M + H]+ 310.0214,
found 310.0212.
HPLC purity (280 nm) 96.8% (tR = 4.257 min).

S19
2.11. 4-((2-(Fluorosulfonyl)phenyl)sulfonyl)piperazine-1-sulfonyl fluoride (21)
A 4-mL vial equipped with an egg-shape stir bar was charged with 2-nitrobenzenesulfonyl
chloride (222 mg, 1.00 mmol), N-Boc-piperazine (205 mg, 1.10 mmol), and dichloromethane (1.0
mL). Triethylamine (152 mg, 1.50 mmol) was added dropwise to give a yellow suspension. The
mixture was stirred at room temperature for 4 h, before partitioned between hydrochloric acid (0.1
mol L-1, 50 mL) and ethyl acetate (50 mL). The organic phase was collected and concentrated to
give virtually pure sulfonamide (21-i, 364 mg, 0.98 mmol).
Treating the crude sulfonamide with hydrochloric acid in 1,4-dioxane (4 mol L-1, 10 mL)
overnight cleanly cleaved the Boc group. The resulting hydrochloride salt was obtained in >98%
purity by evaporation, and then mixed with 2,3-dimethyl-1-fluorosulfonylimidozolium triflate (492
mg, 1.50 mmol) in a 20-mL scintillation vial. Acetonitrile (5 mL) and triethylamine (303 mg, 3.00
mmol) were added, and the solution being stirred for 3 h at room temperature before quenched
by water. The sulfamoyl fluoride (21-ii) was isolated by column chromatography as a white
crystalline (328 mg, 0.921 mmol, 92% over 2 steps). The structure of this compound was
determined by NMR and LRMS.
Into a reaction tube charged with the crude sulfamoyl fluoride (328 mg, 0.921 mmol), palladium
on carbon (27 mg, 10% wt.), tetrahydrofuran (2 mL) and ethanol (2 mL) were added. Hydrogen
gas was introduced by a balloon, and bubbled for 10 min. The suspension was stirred under
hydrogen for another 6 h, before diluted by methanol (50 mL). The methanolic solution was
passed through a pad of Celite and concentrated to give a yellow solid (21-iii). A 20-mL scintillation vial was charged with the crude aniline (21-iii). Concentrated hydrochloric
acid (0.4 mL) was added, and the suspension was cooled to 0 ºC. At the same temperature,
sodium nitrite solution (40%, 0.4 mL) was added dropwise via a syringe. The diazotization process
took 30 min to complete at 0 ºC. In another 20-mL scintillation vial, copper(I) chloride (30 mg, 0.31
NN
SO
OS
O
O F
SO
O
F21
KFHFCH3CN/H2O
rt
NaNO2, HClthen CuCl, NaHSO3
H2O0 ºC
NO2
S ClO
O
NH
BocN
NEt3, DCMrt NO2
SNO
O
NBocH2, Pd/C
THF/EtOHrt
NH2
SNO
O
N
HCl (dioxane)then NEt3
N N SO2F
OTf
NO2
SNO
O
N SFO
O
NN
SO
OS
O
O F
SO
O
Cl
SFO
O
21-i 21-ii
21-iii 21-iv

S20
mmol) was dissolved in hydrochloric acid (1 mL), and to which, sodium bisulfite solution (40%, 1
mL) was added dropwise to make a yellow suspension. Add the diazonium solution to the SO2
solution at 0 ºC via a glass pipette dropwise. The resulting mixture was allowed to be warmed to
5 – 10 ºC and stirred for a further 30 min, before extracted by dichloromethane (20 mL).
Concentration of the dichloromethane solution gave crude sulfonyl chloride (21-iv, ~ 80% purity).
The crude sulfonyl chloride (21-iv) was dissolved into acetonitrile (2 mL) and treated with
saturated potassium bifluoride solution (1 mL). The biphasic mixture was stirred at room
temperature for 2 h and partitioned between ethyl acetate (50 mL) and water (50 mL). The organic
phase was concentrated and purified by chromatography to give the titled compound as an off-
white solid (210 mg, 0.538 mmol, 58% over 3 steps). 1H NMR (600 MHz, Chloroform-d) δ 8.36 (dd, J = 7.9, 1.4 Hz, 1H), 8.31 (dd, J = 7.8, 1.4 Hz, 1H),
7.94 (td, J = 7.7, 1.4 Hz, 1H), 7.90 – 7.86 (m, 1H), 3.54 (dd, J = 7.2, 3.1 Hz, 4H), 3.51 (dd, J = 6.6,
3.6 Hz, 4H). 13C NMR (151 MHz, CDCl3) δ 138.4, 135.7, 134.0, 133.4, 133.2 (d, J = 1.5 Hz), 131.8 (d, J = 25.7
Hz), 46.8, 45.0. 19F NMR (377 MHz, CDCl3) δ 64.1, 41.4.
TLC Rf = 0.34 (33% ethyl acetate in hexanes, UV).
Melting point 175 – 177 ºC.
Mass spectrometry APCI-TOF accurate mass calculated for C10H12F2N2O6S3NH4 [M + NH4]+
408.0164, found 408.0160.
HPLC purity (254 nm) 96.4% (tR = 4.594 min).

S21
2.12. 2-((Trifluoromethyl)sulfonyl)benzenesulfonyl fluoride (22)
The reaction condition was adapted from Smedley et al (10). A 10-mL vial sealed with a
septum was charged with benzene-1,2-disulfonyl fluoride (242 mg, 1.00 mmol), finely powdered
potassium bifluoride (1.6 mg, 0.02 mg). Under N2 atmosphere, anhydrous DMSO (4.0 mL) and
trifluoromethyl trimethylsilane (200 mg, 1.4 mmol) were added via syringe. The resulting mixture
was stirred at room temperature for 2 h (distribution of starting material, mono-trifluoromethylated
product, di-trifluoromethylated product was estimated by analytical HPLC as 41/52/7), before
directly loaded onto a prep-HPLC. The titled compound was isolated as a white crystalline (104
mg, 0.304 mmol, 30%). 1H NMR (600 MHz, Chloroform-d) δ 8.53 – 8.47 (m, 1H), 8.47 – 8.42 (m, 1H), 8.16 – 8.02 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 137.0, 136.6, 136.1, 134.9 (d, JCF = 28.7 Hz), 133.8 (d, JCF = 1.5
Hz), 132.2 (d, JCF = 1.5 Hz), 118.6 (qd, JCF3 = 336.7 Hz, JCF = 359.4 Hz). 19F NMR (376 MHz, CDCl3) δ 67.1 (s, 1F), -73.5 (s, 3F).
TLC Rf = 0.45 (33% ethyl acetate in hexanes, UV)
Melting point 91 – 93 ºC (MeOH).
Mass spectrometry APCI-TOF accurate mass calculated for C7H4F4O4S2NH4 [M + NH4]+
309.9825, found 309.9821.
HPLC purity (280 nm) 97.1% (tR = 4.949 min).
SO
O
SO
O F
FF
F
22
SO
O
SO
O F
F
+ SiF
FF
Me
MeMe KFHF
DMSOrt

S22
2.13. 2-((Perfluoropropyl)sulfonyl)benzenesulfonyl fluoride (23)
The reaction condition was adapted from Smedley et al (10). A 10-mL vial sealed with a septum
was charged with benzene-1,2-disulfonyl fluoride (242 mg, 1.00 mmol), finely powdered
potassium bifluoride (1.6 mg, 0.02 mg). Under N2 atmosphere, anhydrous DMSO (2.0 mL) and
perfluoropropyl trimethylsilane (242 mg, 1.0 mmol) were added via syringe. The resulting mixture
was stirred at room temperature for 2 h (distribution of starting material, mono-perfluoropropylated
product, di-perfluoropropylated product was estimated by analytical HPLC as 50/45/5). The titled
compound was isolated by prep-TLC (SiO2, 33% ethyl acetate in hexanes) as a white crystalline
(43 mg, 0.110 mmol, 11%). 1H NMR (600 MHz, CDCl3) δ 8.53 – 8.48 (m, 1H), 8.46 – 8.42 (m, 1H), 8.12 – 8.05 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 136.5, 136.2, 135.5, 134.46 (d, JCF = 28.7 Hz), 133.2 (JCF = 1.5 Hz),
131.9, 120 – 106 (m, perfluoropropyl). 19F NMR (377 MHz, CDCl3) δ 67.1 (s, 1F), -80.8 (t, J = 9.3 Hz, 3F), -107.38 (qt, J = 8.8, 4.2 Hz,
2F), -124.78 – -124.81 (m, 2F).
TLC Rf = 0.45 (33% ethyl acetate in hexanes, UV).
Melting point 78 – 79 ºC (hexanes/ethyl acetate).
Mass spectrometry APCI-TOF accurate mass calculated for C9H3F8O4S2 [M]– 390.9350, found
390.9339.
HPLC purity (280 nm) 95.2% (tR = 5.368 min).
SO
O
SO
O F
FF
F FFFF
SO
O
SO
O F
FFF
F FFFF
Me3SiKFHFDMSOrt
+
23

S23
3. Protease activity assays and library screen 3.1. Methods
Activity of hNE was measured in a total volume of 100 µL in a reaction buffer of PBS (pH
7.4) and 0.05% (v/v) NonidetTM P 40 Substitute (Sigma). Final composition of each reaction was
5 nM hNE (Elastin Products Corp.), 50 µM AAPV-aminomethylcoumarin (AMC) substrate
(Millipore), ~2.5% dimethyl sulfoxide (Fisher), and various concentrations of compounds as
inhibitors. hNE was incubated with inhibitors for 10 min at room temperature before addition of
AAPV-AMC (11, 12). Residual proteolytic activity was measured kinetically at 25 °C using an
Envision microplate reader for a total of 30 min at 30 sec intervals. Only data points reflecting
linear substrate conversion were used to determine relative protease activity. IC50 values were
obtained by fitting the data to a dose-response inhibition, log (inhibitor) vs. response – variable
slope (four parameters) using GraphPad Prism. Human Cathepsin G (hCG) activity was
measured in a comparable manner to hNE, except that the final concentration of protease in
each reaction was 15 nM and 50 µM of the substrate, AAPF-AMC (Millipore) (11, 12).

S24
3.2. Screen results
Table S1. Randomly picked SuFExable molecules and corresponding hNE inhibitory activity.
BBS Code* (cmpd # in text) Subset Structure % inhibition†
(IC50)‡
BBS-63 III
20%
BBS-64 III
12%
BBS-65 III
22%
BBS-66 III
17%
BBS-67 III
39%
BBS-68 III
19%
BBS-70 III
12%
BBS-244 III
11%
BBS-245 III
7%
BBS-246 III
37%
BBS-247 III
28%
FO2SN
SO2F
S
FO2SN
SO2F
FO2SN
SO2F
FO2SN
SO2F
FO2SN
SO2F
FO2SN
SO2F
O
FO2SN
SO2F
NH
FO2SN N
SO2F
SO2F SO2F
NSO2F
NSO2FFO2S
N
NSO2FFO2S
SO2FFO2S

S25
BBS-248 III
25%
BBS-249 III
31%
BBS-250 III
10%
BBS-254 III
0%
BBS-255 III
36%
BBS-256 III
36%
BBS-257 III
9%
BBS-258 III
16%
BBS-268 III
12%
BBS-297 III
5%
BBS-298 III
10%
BBS-299 III
41%
BBS-300 III
3%
BBS-310 III
40%
N
SO2F
SO2FN
SO2F
FO2S
NSO2F
N CO2H
SO2F
NH
F
SO2F
NH
F3C
SO2F
NH
O2N
SO2F
NH
NC
SO2F
NH
F5S
SO2F
NH
Br
SO2F
NN
SO2FFO2S
SO2F
SO2F
N
NSO2F
FO2S
N
SO2F
SO2F
FO2S
SO2F
O2N
SO2F
N
O

S26
BBS-311 III
24%
BBS-418 III
26%
BBS-421 III
1%
BBS-422 III
1%
BBS-424 III
6%
BBS-430 III
44%
BBS-432 III
14%
BBS-420 III
25%
BBS-216 III
0%
BBS-219 IV
24%
BBS-222 IV
0%
BBS-224 IV
10%
BBS-225 IV
21%
BBS-226 IV
15%
O2N
SO2F
N
NHBoc
N
N
FO2S
SO2F
Boc
N
O
SO2F
N
NSO2F
Boc
N
O
N
N
Cl
SO2F
SO2F
FO2S CN
MeO2C
SO2F
FO2S CN
NC
NSO2FFO2S
OMe
MeO
Cl
SO2F
SO2F
Br
SO2F
SO2F
F3CO
SO2F
HO2C
SO2F
F3C

S27
BBS-235 IV
9%
BBS-228 IV
40%
BBS-232 IV
18%
BBS-417 IV
12%
BBS-241 I
0%
BBS-295 I
(>200)
BBS-305 (1) I
(3.3 ± 1.0)
BBS-306 I
(20 ± 10)
BBS-416 I
(>150)
BBS-433 (10) I
(20 ± 10)
BBS-434 I
(>150)
BBS-114 II
0%
BBS-116 II
0%
BBS-240 II
0%
BBS-263 II
34%
BBS-266 II
12%
BBS-272 II
0%
BBS-274 II
0%
SO2F
OSO2F
O2N
SO2FF3C
SO2F
F5S
SO2F
SO2F
SO2F
SO2F
SO2F
Br
SO2F
O2N
SO2F
Br
SO2F
I
OSO2F
N
OSO2F
OSO2F
CO2H
OSO2FFO2SO
H2N
OSO2F
OSO2F
OCH3
OSO2F
OCH3
SO O
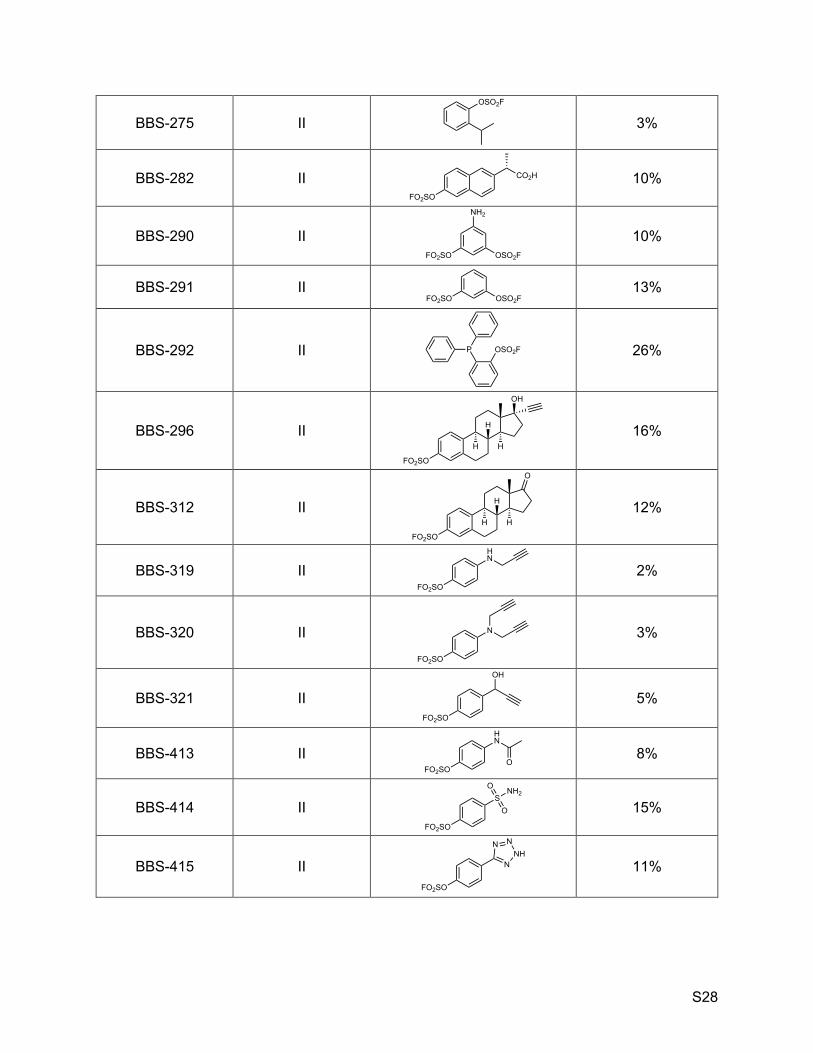
S28
BBS-275 II
3%
BBS-282 II
10%
BBS-290 II
10%
BBS-291 II
13%
BBS-292 II
26%
BBS-296 II
16%
BBS-312 II
12%
BBS-319 II
2%
BBS-320 II
3%
BBS-321 II
5%
BBS-413 II
8%
BBS-414 II
15%
BBS-415 II
11%
OSO2F
CO2H
FO2SO
NH2
OSO2FFO2SO
OSO2FFO2SO
P OSO2F
FO2SO
H
OH
HH
FO2SO
H
HH
O
FO2SO
HN
FO2SO
N
FO2SO
OH
FO2SO
HN
O
FO2SO
SNH2
O
O
FO2SO
NNH
NN

S29
BBS-419 II
11%
BBS-423 II
11%
BBS-425 II
10%
BBS-426 II
15%
BBS-427 II
3%
BBS-428 II
7%
BBS-429 II
17%
BBS-301 II
(9.5 ± 3.5)
BBS-1001 I
(~200)
BBS-740 I
(10 ± 4)
BBS-743 I
(>200)
BBS-1002 II
(>400)
BBS-1003 (8) I
(~120)
BBS-1004 (9) I
(82 ± 16)
FO2SO
NH
SO
O O
PPhO
Ph
FO2SO
OH
O
FO2SOOH
O
FO2SO
NH
NH
O N
O
F3C CF3
OSO2FFO2SO
FO2SO
N3
FO2SO
MeO
N
N
FO2S
O
O
S OO
F
S OO
F
SO
O
F
S OO
F
NSO
O F
F
SO
O F
Cl
SO
O F

S30
BBS-1005 (12) I
(>200)
BBS-1006 (13) I
(73 ± 4)
BBS-1007 (14) I
(13.3 ± 0.5)
BBS-1008 (15) I
(60 ± 8)
BBS-1009 (16) I
(20 ± 1)
BBS-1010 (17) I
(37 ± 2)
BBS-1011 (2) I
(17.5 ± 1.1)
BBS-1012 (18) I
(27 ± 2)
BBS-1013 (22) I
(1.1 ± 0.1)
BBS-1014 (11) I
(9.7 ± 1.2)
BBS-1015 (19) I
(2.2 ± 0.7)
BBS-1016 I
(7.7 ± 0.7)
CH3SO
O F
O
SO
O F
H3C
SO
O F
N
SO
O F
FF
F
SO
O FN+
O-O
SO
O F
OO
CH3
S OO
F
SO
O
FS
O
OF
SO
O F
SO
O
SO
O F
FF
F
I
SO
O F
SO
O F
SO
O F
S
O
O
F S
O
O
F

S31
BBS-1017 I
(51 ± 7)
BBS-1018 I
(>200)
BBS-1019 II
(>200)
BBS-1020 II
(>200)
BBS-1021 I
(~200)
BBS-1022 I
(~200)
BBS-1023 I
(>200)
BBS-1024 N/A
(~200)
BBS-1025 N/A
(~200)
BBS-1026 N/A
(~200)
BBS-1027 (23) I
(48 ± 2)
BBS-1028 I
(36 ± 3)
S
O
O
F S
O
O
N
O
O
S OO
F
SO O
F
S
FF
FF
F
OS
O
O
F
S
FF
FF
FO
SO
O F
OS
O
O
F
SO
OF
SO
O
FSO
O
F
S OO
F
SO
OF
O
OSO
O
O
OSO
O
O
OSO
O
SO
O
SO
O F
FF
F FF
FF
S OO
F
SO
O
FS
O
OF

S32
BBS-1029 II
(~200)
BBS-1030 (3) I
(6.8 ± 1.1)
BBS-1031 I
(>200)
BBS-1032 I
(>200)
BBS-1033 N/A
(>200)
BBS-1034 N/A
(59 ± 15)
BBS-1035 II
(>200)
BBS-1036 N/A
(>200)
BBS-1037 I
(>200)
BBS-1038 I
(67 ± 19)
BBS-1039 (4) I
(17 ± 2)
BBS-1040 (5) I
(5.4 ± 0.5)
OS
O
O
FF
F
O
SO O
F
SO
O F
SO
O
F
SF
FF
F
F
SO
O
F
SF
FF
F
F
S OO
F
O
OS O
O
O
OSO
O
OO
CH3
O O
OSO
O
F
OS
O O
SOO
F
Br
SO
O F
F
F SO
O F
SO
O Me
SO
O F
Cl
Cl
SO
OF

S33
BBS-1041 I
(105 ± 17)
BBS-1042 I
(>200)
BBS-1043 (6) I
(5.9 ± 1.1)
BBS-406 I
(89 ± 4)
BBS-1044 (20) I
(84 ± 1)
BBS-1045 II
(>200)
BBS-1048 I
(60 ± 10)
BBS-1049 (7) I
(49 ± 3)
BBS-1050 (21) I
(>200)
PMSF (ref.) N/A
(24 ± 1)
*BBS-numbering is assigned for every SuFExable compound for internal (within TSRI) reference. †Per cent inhibition of hNE activity was measured based on a compound concentration of 200 μM. ‡IC50 values were measured based on 10 min incubation and are shown in mean ± SD (n ≥ 3).
F
F
F
SO
O F
S Cl
Cl
S
O
O
F
N+O
O-
N+
O
O-S O
O
F
OH
SO
OF
SO
O
F
SOO
F
N
O
SO
O
SO
O
F
SF
FF
F
FO
SO
O
F
Cl
Cl
SO
O F
Cl
SO
O F
Cl
N
N
SO
O
SO
O F
SO
O
F
SO
O
F

S34
Fig S1. Scatterplot of in-house sulfonyl fluoride and fluorosulfate compounds screened against human neutrophil elastase. Residual hNE activity toward a fluorescent-peptide
substrate (AAPV-AMC) after incubation of 5 nM hNE with 200 µM compound from a 10mM DMSO
stock. Percent residual activity normalized against a positive control of hNE plus substrate and
negative control of substrate alone.
1 11 21 31 41 51 61 71 81 91 101
111
121
131
0
25
50
75
100
125
Arbitrary Cmpd #
Nor
mal
ized
hN
E A
ctiv
ity (%
)
candidates of covalent inhibitors
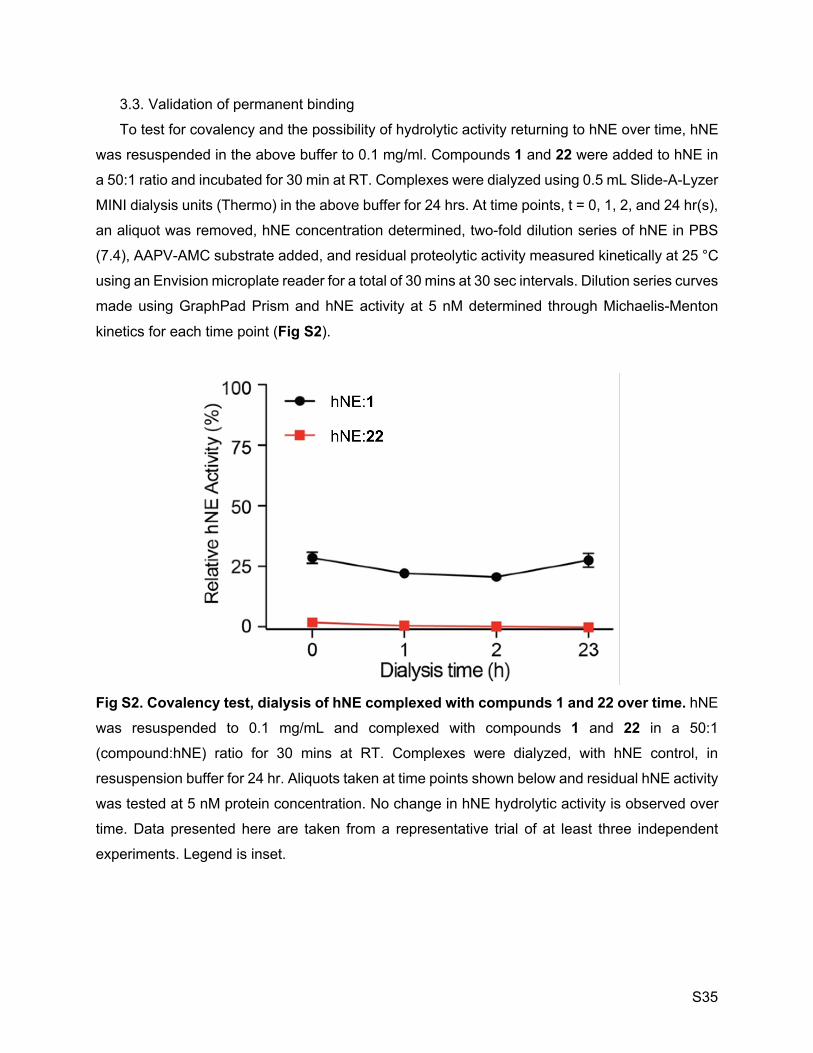
S35
3.3. Validation of permanent binding
To test for covalency and the possibility of hydrolytic activity returning to hNE over time, hNE
was resuspended in the above buffer to 0.1 mg/ml. Compounds 1 and 22 were added to hNE in
a 50:1 ratio and incubated for 30 min at RT. Complexes were dialyzed using 0.5 mL Slide-A-Lyzer
MINI dialysis units (Thermo) in the above buffer for 24 hrs. At time points, t = 0, 1, 2, and 24 hr(s),
an aliquot was removed, hNE concentration determined, two-fold dilution series of hNE in PBS
(7.4), AAPV-AMC substrate added, and residual proteolytic activity measured kinetically at 25 °C
using an Envision microplate reader for a total of 30 mins at 30 sec intervals. Dilution series curves
made using GraphPad Prism and hNE activity at 5 nM determined through Michaelis-Menton
kinetics for each time point (Fig S2).
Fig S2. Covalency test, dialysis of hNE complexed with compunds 1 and 22 over time. hNE
was resuspended to 0.1 mg/mL and complexed with compounds 1 and 22 in a 50:1
(compound:hNE) ratio for 30 mins at RT. Complexes were dialyzed, with hNE control, in
resuspension buffer for 24 hr. Aliquots taken at time points shown below and residual hNE activity
was tested at 5 nM protein concentration. No change in hNE hydrolytic activity is observed over
time. Data presented here are taken from a representative trial of at least three independent
experiments. Legend is inset.

S36
3.4. Inhibitory activity of hit molecules against hCG.
Fig S3. Dose-response curves of top hNE inhibitors against human cathepsin G. Each
compound was assessed over a two-fold logarithmic dilution series. Data presented here are
taken from a representative trial of at least three independent experiments. Legend is inset.

S37
4. Mass Spectrometry 4.1. Methods
Enzyme hNE was resuspended in 50mM sodium acetate (pH 4.5), 100 mM NaCl to 0.2 mg/ml
final concentration. DMSO solution of each compound (10 mM) was diluted 1:10 in the above
buffer, then added in a compound:hNE ratio = 3:1 and incubated at RT for 1 h prior to analysis by
MALDI-TOF mass spectrometry.
4.2. Results
Fig S4. MALDI-TOF mass spectrometry analysis of hNE with or without compounds, 1 and 22. hNE was incubated alone (top left) and in a 1:3 ratio with compounds 1 (top right) and 22
(bottom) for 1 hr at RT and analyzed by high accuracy MALDI-TOF mass spectrometry. Peaks
correspond to the mass of hNE apo and ± compound in Da.

S38
5. X-ray crystal structure 5.1. Methods
Crystallization and x-ray data collection. Crystallization of hNE in complex with our
covalent capture agent was set up similarly to Hansen et al. and their structure of a
dihydropyrimidone inhibitor (13). In short, inhibitor 1 was added in a 1.2 molar excess to human
neutrophil elastase (Elastin Products Co.) [10 mg/ml in 10 mM HEPES (pH 6.5)], incubated for 1
hr at 25°C and immediately used for crystallization. Crystals were grown by sitting drop-vapor
diffusion by mixing equal volumes (1.5 µL) of hNE:1 complex and reservoir solution consisting of
0.3 M ammonium citrate (pH 5.0), 14% (w/v) PEG 3350 at 25°C. Data was collected on single,
flash-cooled crystals at 100 K in cryoprotectant consisting of 0.2 M ammonium citrate (pH 5.0),
20% (w/v) PEG3350, and 20% (v/v) glycerol, and were processed with HKL2000 in orthorhombic
space group P212121. The calculated Matthews’ coefficient (VM = 2.77 Å3Da-1) suggested four
monomers per asymmetric unit with a solvent content of 56%. X-ray data was collected to 2.33 Å
resolution on beamline 12.2 at the Stanford Synchrotron Radiation Lightsource (SSRL) (Menlo
Park, CA). Data collection and processing statistics are summarized in Table S2. Structure solution and refinement. The hNE:1 structure was determined by molecular
replacement (MR) with Phaser (14) using the previously published apo structure (PDB ID: 5abw)
as the initial search model. The structure was manually built with Coot (15) and iteratively refined
using Phenix (16) with cycles of conventional positional refinement with isotropic B-factor
refinement. Non-crystallographic symmetry (NCS) constraints were applied for the initial rounds
of refinement. The electron density maps clearly identified that 1 was covalently attached to
Ser195 within the active site (Fig. 3B-D). Water molecules were automatically positioned by
Phenix using a 2.5σ cutoff in Fo-Fc maps and manually inspected. The final Rcryst and Rfree are
19.3% and 24.1%, respectively (Table S2). The model was analyzed and validated with
PROCHECK (17), WHATCHECK (18), and Molprobity (19) on the JCSG webserver. Analysis of
backbone dihedral angles with the program PROCHECK indicated that all residues are located in
the most favorable and additionally allowed regions in the Ramachandran plot. Coordinates and
structure factors have been deposited in the PDB with accession entry 6e69. Structure refinement
and statistics are shown in Table S2. X-ray structure data deposition. The atomic coordinates and structure factors have been
deposited in the Protein Data Bank, www.wwpdb.org (PDB ID: 6e69).

S39
5.2. Results
Table S2. Data collection and refinement statistics.
Structure 6E69 Space group P 212121
Cell dimensions a, b, c; Å 69.5, 124.6, 126.7 α, β, γ; ⁰ 90, 90, 90
Data Processing Resolution, Å (outer shell) 50.0-2.33 (2.37-2.33)
Completeness, % 96.8 (72.5) Unique reflections 46,266 (1,697)
Redundancy 2.7 (2.6) Rmeas (%)* 6.7 (44.9) Rmerge (%)† 4.7 (31.7) Rp.i.m. (%)‡ 7.1 (31.2)
Average I / Average s (I) 13.0 (3.2) CC1/2 87.4 (100)
Refinement Resolution, Å (outer shell) 50.0-2.33 (2.38-2.33) No. reflections (test set)§ 46,245 (2,386)
Rcryst (%)¶ 19.3 (21.3) Rfree (%) 24.1 (28.5)
Protein atoms / Waters 6,711 / 267 CV coordinate error (Å)# 0.28
Rmsd bonds (Å) / angles ⁰ 0.006 / 0.9 B-values protein/waters/ligands (Å2) 39 / 34 / 53
Ramachandran Statistics (%) Preferred 97.1 Allowed 2.9 Outliers 0
*Rmeas = /ΣhklΣi Ii(hkl), where Ii(hkl) are the observed intensities, <I(hkl)> are the average intensities and
N is the multiplicity of reflection hkl. †Rmerge = ΣhklΣi|Ii(hkl) -<I(hkl)>|/ ΣhklΣiIi(hkl) where Ii(hkl) is the ith
measurement of reflection h and < I(hkl)> is the average measurement value. ‡Rp.i.m. (precision-
indicating Rmerge) = Σhkl[1/( N hkl – 1) ]1/2Σi|Ii(hkl) - <I(hkl)>|/ΣhklΣiIi(hkl). §Reflections with I > 0 were used
for refinement (15). ¶Rcryst = Σh||Fobs|-|Fcalc||/Σ|Fobs|, where Fobs and Fcalc are the calculated and
observed structure factor amplitudes, respectively. Rfree is Rcryst with 5.0% test set structure factors. #Cross-validated (CV) Luzzati coordinate errors.

S40
6. Reactive docking 6.1. Methods
The structures of hNE (PDB ID: 6e69) and hCG (PDB ID: 1t32) (20) were superposed and
prepared according to the standard AutoDock protocol (21). Hydrogens, protonation states, and
side chain conformations of Gln, Asn, and His residues were calculated with Reduce (22). Ligand
3D coordinates were generated from SMILES strings using custom Python scripts based on
OpenBabel (23). The reactive docking protocol was applied, as described previously (24, 25). The
reactive potential was defined for the sulfonyl-fluoride sulfur and the serine Og, with e value of
0.241. For each ligand, 10 independent ga_runs were performed with the default GA settings.
Results were clustered, and the lowest energy result was selected.
6.2. Results and discussion
Fig S5. Reactive docking results. Residues defining binding sites are shown as sticks, and
volumes represented as semi-transparent surfaces. Ligands are shown as sticks. (a) Compound
1 (yellow) docked in hNE (white; PDB ID 6e69); pseudo-covalent bond between 1 and Ser195 is
shown as dashed lines (green). (b). Compound 1 (yellow) docked in hCG (green; PDB ID 1t32);

S41
(c) Compound 19 (yellow) docked in hCG (green; PDB ID 1t32), and the experimental coordinates
of beta-ketophosphonate 1 (JNJ-10311795; purple), a potent non-covalent hCG inhibitor.
Superposition of hCG (PDB ID: 1t32) and our hNE:1 co-complex (PDB ID: 6e69) crystal
structures provides insight as to how 1 is selective for hNE over hCG. The active sites are
conserved in shape, topography, and sequence. However, key differences in residues flanking
the catalytic site are likely responsible for selectivity of 1 for hNE, including substitutions of Phe192
to Lys, Val216 to Gly, and that hCG lacks the disulfide bond formed by hNE Cys191 and Cys220
(Fig S5a). We applied a reactive docking protocol to identify the determinants of selectivity by 1
and the promiscuity of 19. Modeling the near-attack conformations (NAC), which precede
formation of irreversible adducts, suggests that the Val216 side chain of hNE is important for
binding the free compound and stabilizing the proper reactive geometry. The side chain of Val216
engages the aromatic ring of 1 and 19 and orients the sulfonyl fluoride at an optimal distance for
nucleophilic attack by Ser195. The substitutions of hNE’s Val216 for a Gly in hCG and Ala213 for
a Val, expand the active site cavity and promote repulsion with the compound’s aromatic ring,
respectively (Fig S5b). Our docking results suggest the combined substitutions in hCG result in
a shift of the aromatic ring binding interaction away from Ser195 and a significant increase in
distance for nucleophilic attack by the active site serine (from 2.2 ± 0.2 Å in hNE to 3.4 ± 0.1 Å in
hCG). Importantly, compounds with the reactive warhead attached to the phenyl group via one or
more carbons (i.e., 19, and PMSF) have optimal distance between the sulfonyl fluoride and
aromatic ring to react with the serine and interact with the larger hydrophobic pocket of hCG (1.8
± 0.1 Å for 19), respectively, as demonstrated by the orientation of the naphthyl group of JNJ-
10311795 in the x-ray co-complex structure with hCG (Fig S5c) (20).
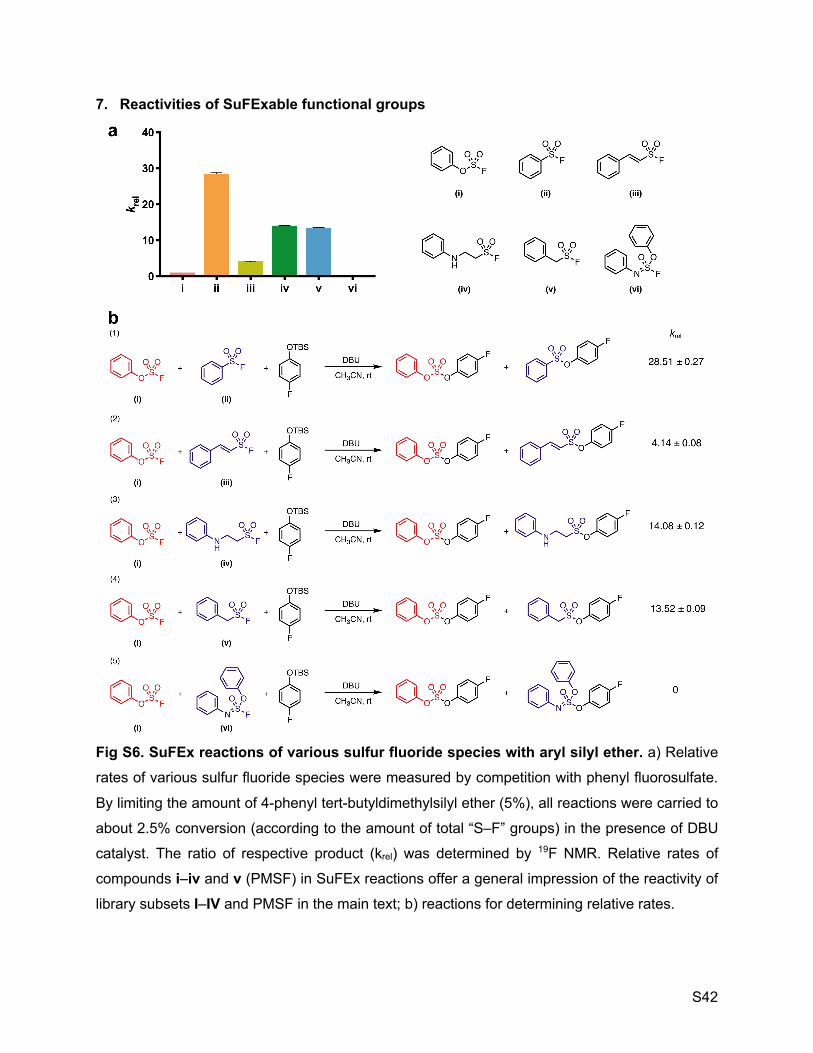
S42
7. Reactivities of SuFExable functional groups
Fig S6. SuFEx reactions of various sulfur fluoride species with aryl silyl ether. a) Relative
rates of various sulfur fluoride species were measured by competition with phenyl fluorosulfate.
By limiting the amount of 4-phenyl tert-butyldimethylsilyl ether (5%), all reactions were carried to
about 2.5% conversion (according to the amount of total “S–F” groups) in the presence of DBU
catalyst. The ratio of respective product (krel) was determined by 19F NMR. Relative rates of
compounds i–iv and v (PMSF) in SuFEx reactions offer a general impression of the reactivity of
library subsets I–IV and PMSF in the main text; b) reactions for determining relative rates.

S43
8. NMR spectra 8.1. Naphthalene-1,3,6-trisulfonyl trifluoride (2)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5
f1(ppm)
-200000
0
200000
400000
600000
800000
1000000
1200000
1400000
1600000
1800000
2000000
2200000
2400000
2600000
2800000
zqh-1684-500.1.fid—50mMcyclosporine
7.26
8.49
8.49
8.51
8.51
8.95
8.96
8.97
8.97
8.99
8.99
9.01
9.01
9.17
9.18
8.48.58.68.78.88.99.09.19.29.3f1(ppm)
8.49
8.49
8.51
8.51
8.95
8.96
8.97
8.97
8.99
8.99
9.01
9.01
9.17
9.18
SO2F
SO2FFO2S
2

S44
-100102030405060708090100110120130140150160170180190200210220230
f1(ppm)
-200000
-100000
0
100000
200000
300000
400000
500000
600000
700000
800000
900000
1000000
1100000
1200000
1300000
1400000
1500000
1600000
1700000
1800000
zqh-1684-500.2.fid—
76.91
77.16
77.41
127.62
130.08
131.12
131.14
132.01
132.25
132.91
133.13
133.52
134.73
134.95
139.55
126128130132134136138140142f1(ppm)
127.62
130.08
131.12
131.14
132.01
132.25
132.91
133.13
133.52
134.73
134.95
139.55
SO2F
SO2FFO2S
2

S45
-15-10-505101520253035404550556065707580f1(ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
800
850
zqh-1684-40-F.33.fid—
0.65
64.85
66.79
67.65
64.565.065.566.066.567.067.568.0f1(ppm)
0
100
200
300
400
500
600
64.85
66.79
67.65 SO2F
SO2FFO2S
2

S46
8.2. Naphthalene-2,3-disulfonyl difluoride (3)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5
f1(ppm)
0
50
100
150
200
250
300
350
400
450
zqh-1782-600.1.fid—H-1Routine,CP-DCH,AVIIIHD-600
2.0
72.
05
2.0
0
7.26
7.9
87.
99
8.0
08
.00
8.0
18
.01
8.1
98
.19
8.2
08
.20
8.2
18
.21
8.9
68
.97
8.9
88
.98
8.9
9
3
SO
O F
SO
O
F

S47
0102030405060708090100110120130140150160170180190200210220
f1(ppm)
-100
0
100
200
300
400
500
600
700
800
900
zqh-1782-600.2.fid—
76.95
77.16
77.37
126.66
126.86
130.16
132.87
133.72
137.17
3
SO
O F
SO
O
F
S48
-10-50510152025303540455055606570758085f1(ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
zqh-1782-400-F.1.fid—F-19,CDCl3,DPX-400QNPProbe.CF3ClasRefat0ppm.
7.26
72.84
3
SO
O F
SO
O
F
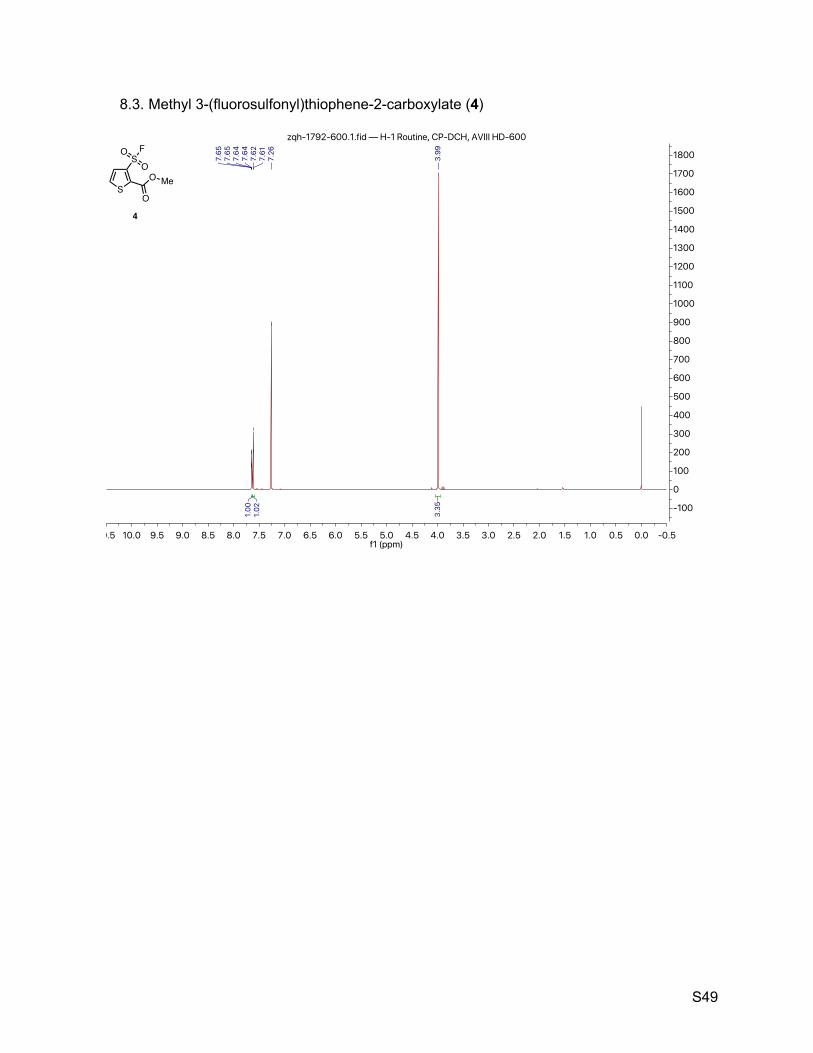
S49
8.3. Methyl 3-(fluorosulfonyl)thiophene-2-carboxylate (4)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5
f1(ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500
1600
1700
1800
zqh-1792-600.1.fid—H-1Routine,CP-DCH,AVIIIHD-600
3.3
5
1.0
21.
00
3.9
9
7.26
7.6
17.
62
7.6
47.
64
7.6
57.
65
SO
O Me
SO
O F
4
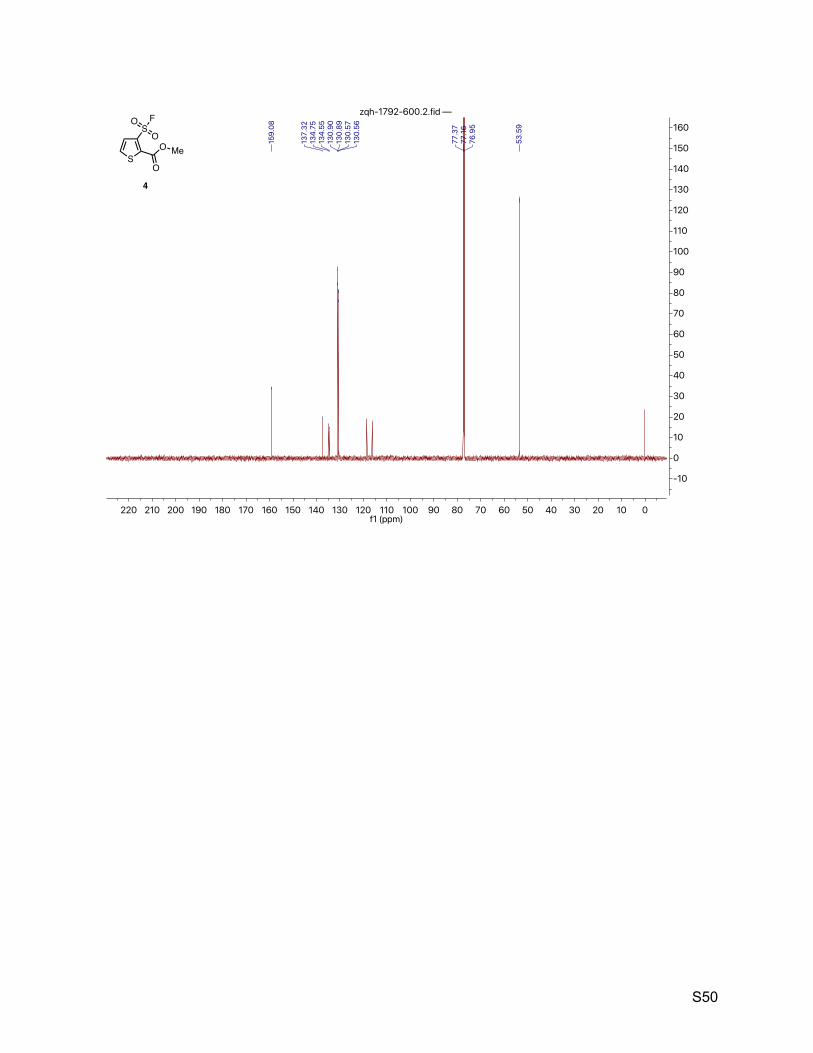
S50
0102030405060708090100110120130140150160170180190200210220
f1(ppm)
-10
0
10
20
30
40
50
60
70
80
90
100
110
120
130
140
150
160
zqh-1792-600.2.fid—
53.59
76.95
77.16
77.37
130.56
130.57
130.89
130.90
134.55
134.75
137.32
159.08
SO
O Me
SO
O F
4
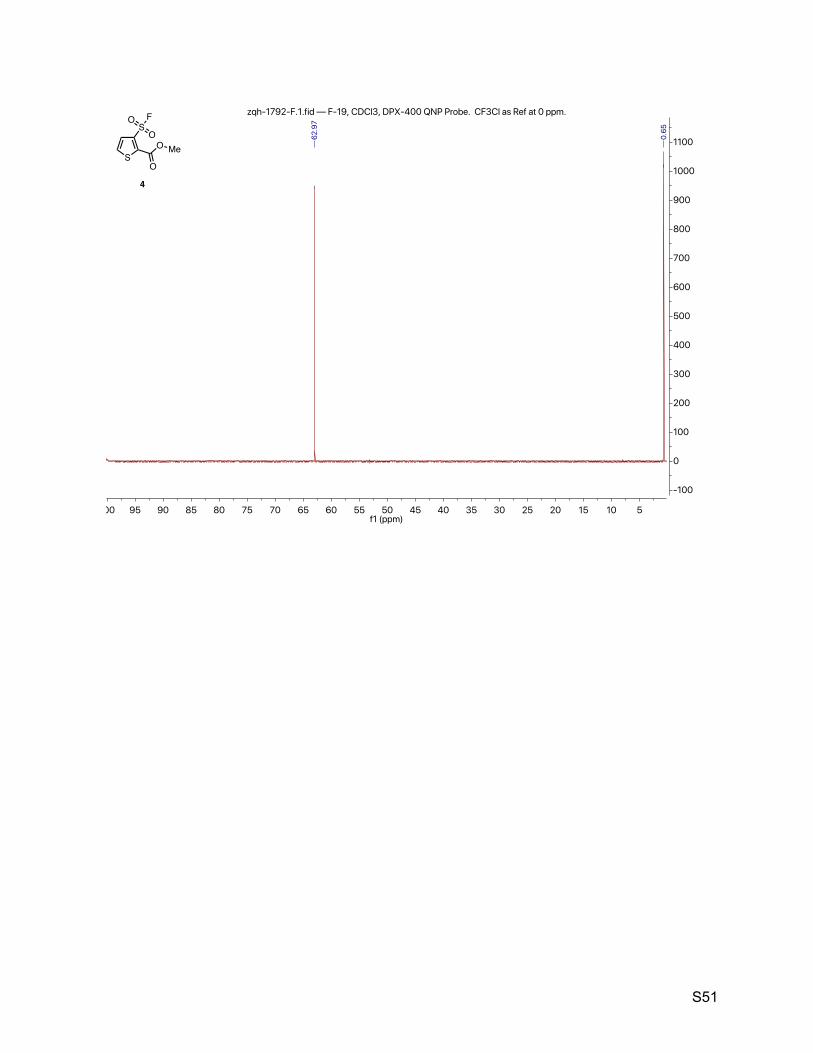
S51
5101520253035404550556065707580859095100f1(ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
zqh-1792-F.1.fid—F-19,CDCl3,DPX-400QNPProbe.CF3ClasRefat0ppm.
0.65
62.97
SO
O Me
SO
O F
4

S52
8.4. 2,6-dichlorobenzenesulfonyl fluoride (5)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5
f1(ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
340
zqh-1793-600.1.fid—H-1Routine,CP-DCH,AVIIIHD-600
1.0
0
7.26
7.50
7.52
7.53
7.56
7.57Cl
Cl
SO
OF
5

S53
0102030405060708090100110120130140150160170180190200210220
f1(ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
zqh-1793-600.2.fid—C-13Routine1D,CPDCHCryoProbe,AVIII-600
76.95
77.16
77.37
130.95
131.11
131.60
134.97
136.15
Cl
Cl
SO
OF
5
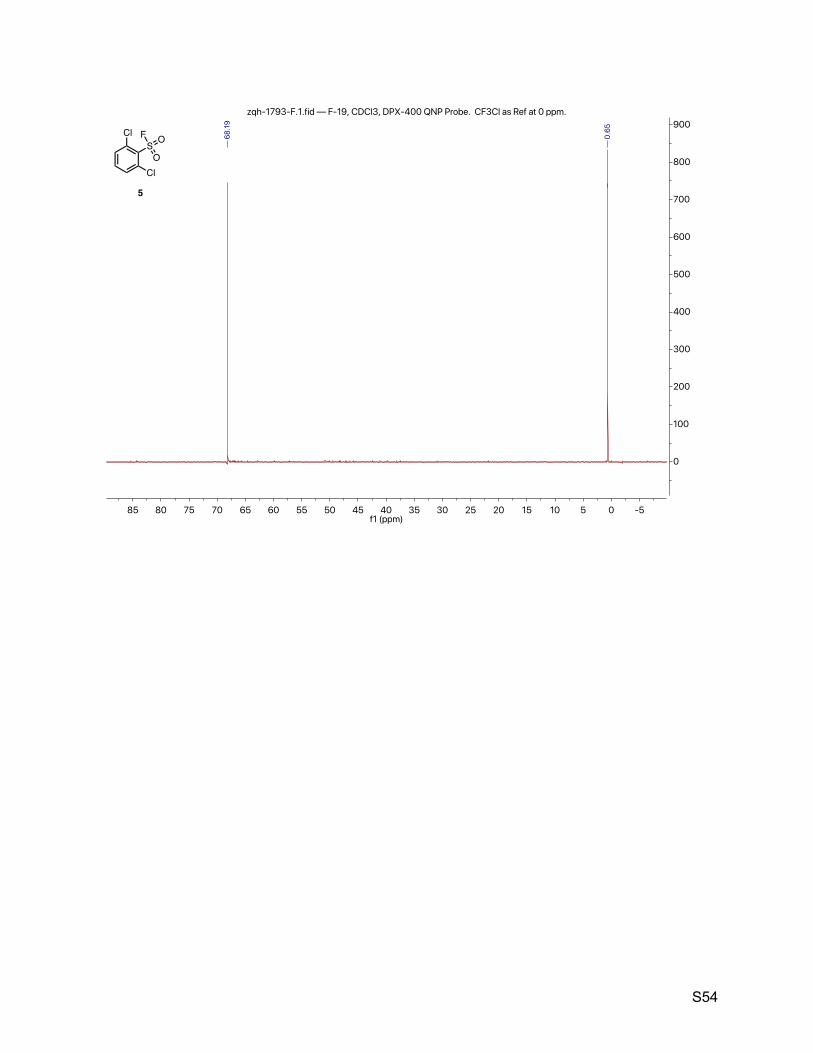
S54
-50510152025303540455055606570758085f1(ppm)
0
100
200
300
400
500
600
700
800
900zqh-1793-F.1.fid—F-19,CDCl3,DPX-400QNPProbe.CF3ClasRefat0ppm.
0.65
68.19
Cl
Cl
SO
OF
5

S55
8.5. 3,4-dichlorobenzenesulfonyl fluoride (7)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5
f1(ppm)
-200
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
2600
2800
3000
3200
3400
3600
zqh-1824-600.1.fid—H-1Routine,CP-DCH,AVIIIHD-600
1.0
31.
02
1.0
0
7.26
7.72
7.72
7.74
7.74
7.8
47.
84
7.8
57.
86
8.1
08
.10
Cl
SO
O F
7
Cl
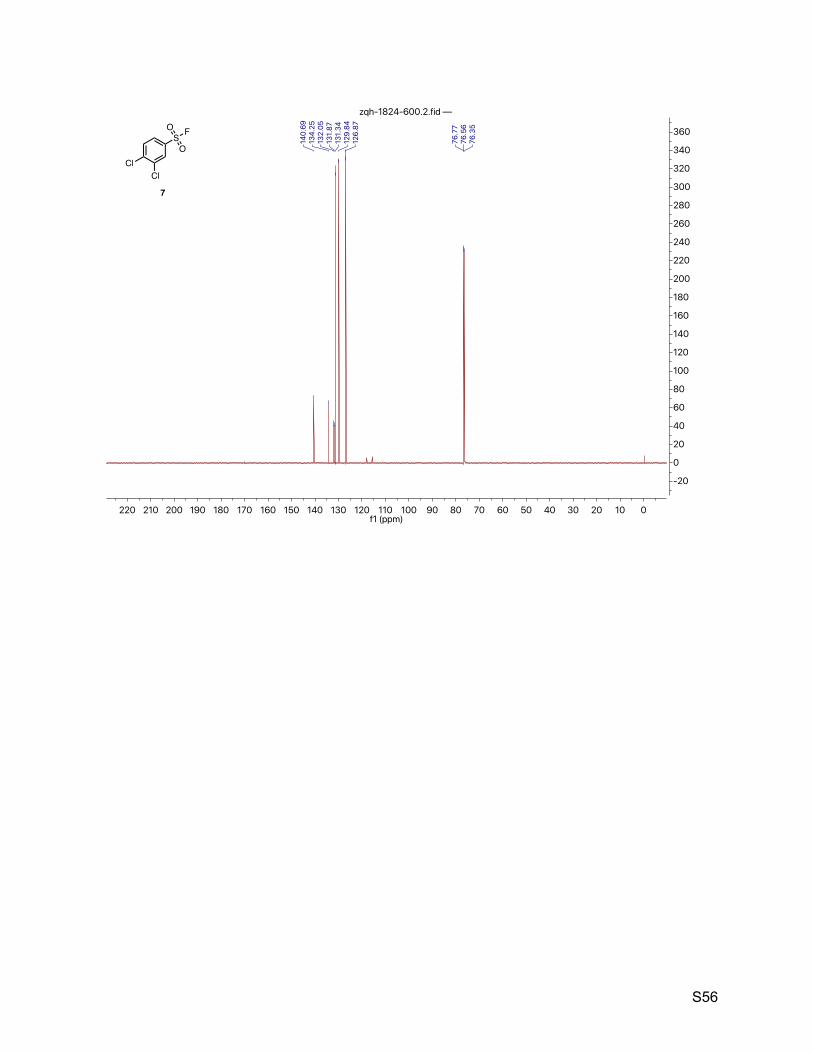
S56
0102030405060708090100110120130140150160170180190200210220
f1(ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
320
340
360
zqh-1824-600.2.fid—
76.35
76.56
76.77
126.87
129.84
131.34
131.87
132.05
134.25
140.69
Cl
SO
O F
7
Cl
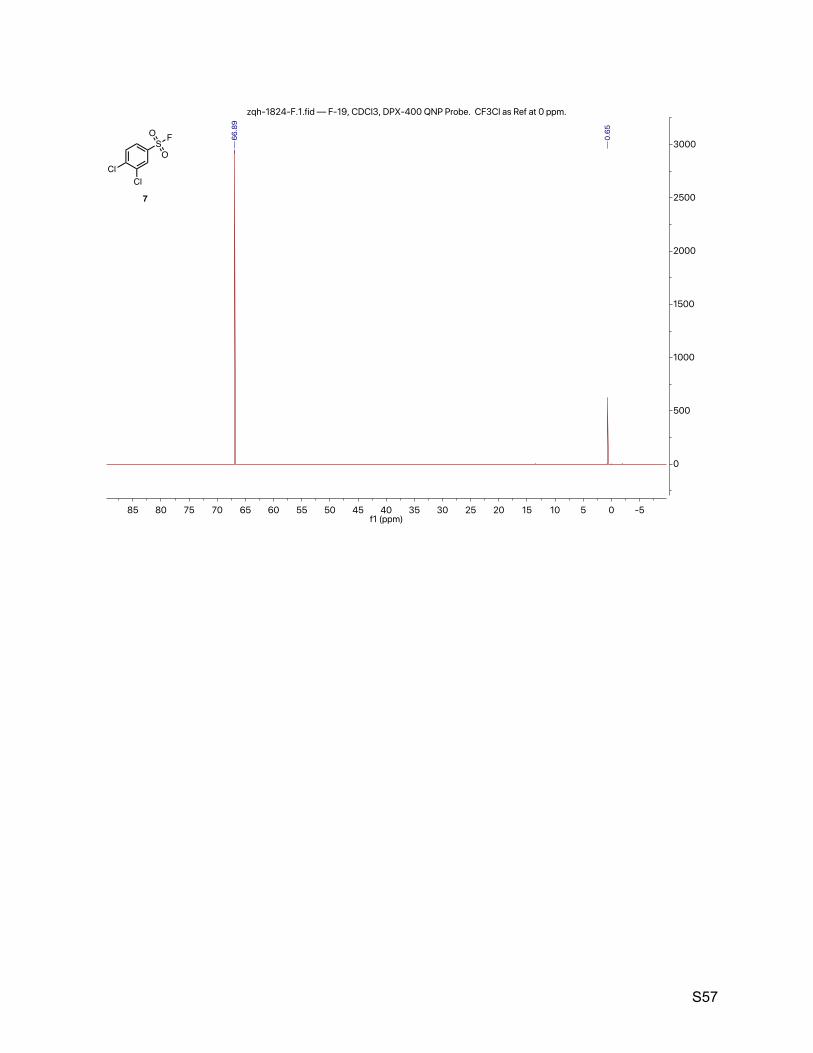
S57
-50510152025303540455055606570758085f1(ppm)
0
500
1000
1500
2000
2500
3000
zqh-1824-F.1.fid—F-19,CDCl3,DPX-400QNPProbe.CF3ClasRefat0ppm.
0.65
66.89
Cl
SO
O F
7
Cl
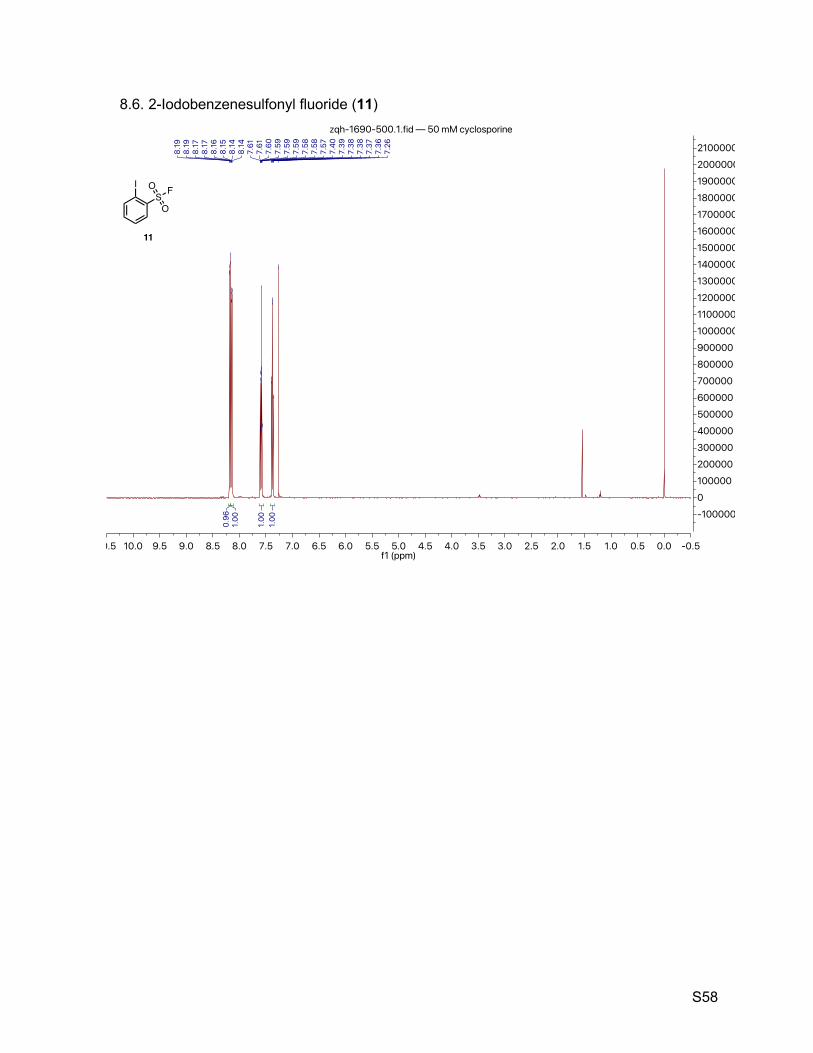
S58
8.6. 2-Iodobenzenesulfonyl fluoride (11)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5
f1(ppm)
-100000
0
100000
200000
300000
400000
500000
600000
700000
800000
900000
1000000
1100000
1200000
1300000
1400000
1500000
1600000
1700000
1800000
1900000
2000000
2100000
zqh-1690-500.1.fid—50mMcyclosporine
1.00
1.00
1.00
0.96
7.26
7.36
7.37
7.38
7.38
7.39
7.40
7.57
7.58
7.58
7.59
7.59
7.59
7.60
7.61
7.61
8.14
8.14
8.15
8.16
8.17
8.17
8.19
8.19
ISO
O F
11

S59
-100102030405060708090100110120130140150160170180190200210220230
f1(ppm)
-200000
-100000
0
100000
200000
300000
400000
500000
600000
700000
800000
900000
1000000
1100000
1200000
1300000
1400000
1500000
1600000
1700000
1800000zqh-1690-500.2.fid—AVNEO500,C-13Routine,5-25-2017
76.91
77.16
77.41
92.28
128.77
132.07
132.08
135.82
137.66
137.85
143.30
ISO
O F
11
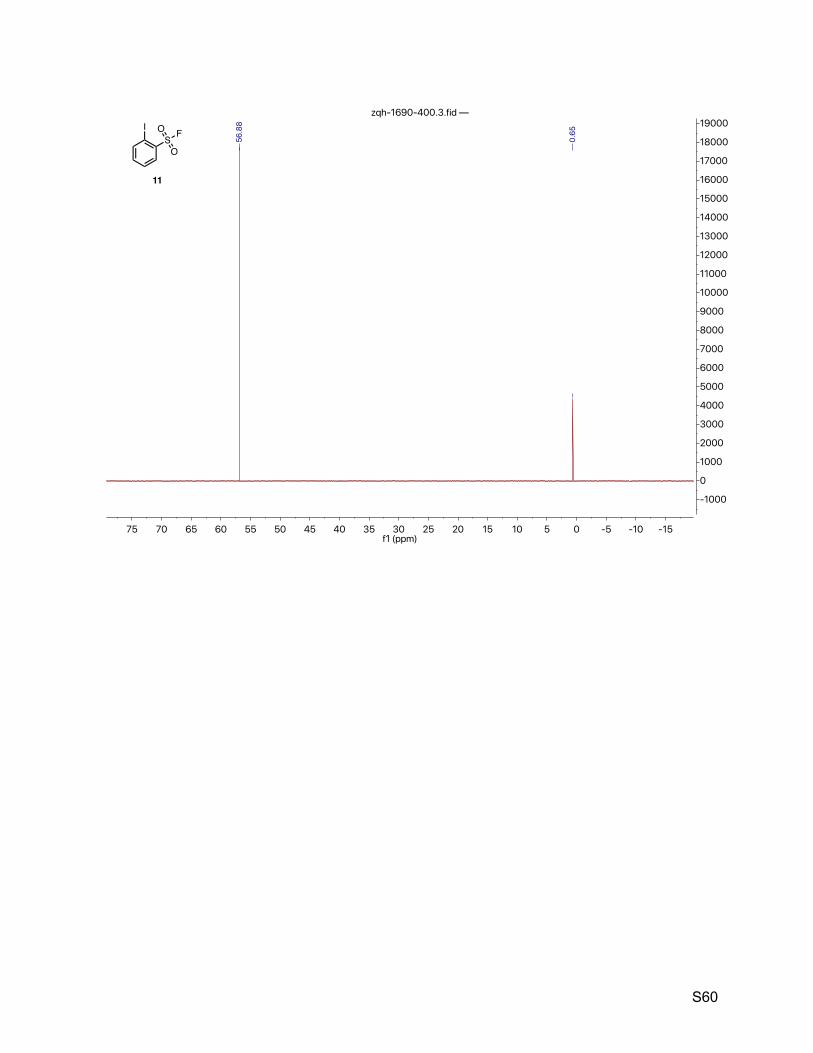
S60
-15-10-5051015202530354045505560657075
f1(ppm)
-1000
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
15000
16000
17000
18000
19000zqh-1690-400.3.fid—
0.65
56.88I
SO
O F
11
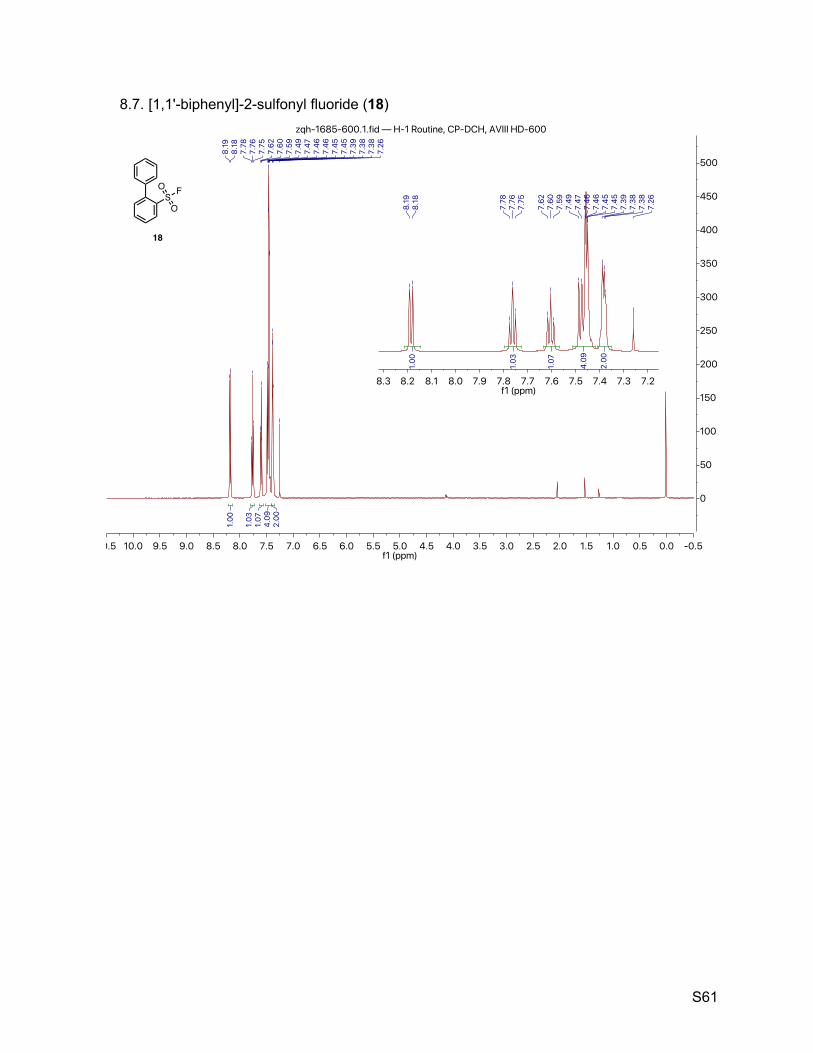
S61
8.7. [1,1'-biphenyl]-2-sulfonyl fluoride (18)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5
f1(ppm)
0
50
100
150
200
250
300
350
400
450
500
zqh-1685-600.1.fid—H-1Routine,CP-DCH,AVIIIHD-600
2.0
04
.09
1.0
71.
03
1.0
0
7.26
7.3
87.
38
7.3
97.
45
7.4
57.
46
7.4
67.
47
7.4
97.
597.
60
7.6
27.
757.
767.
788
.18
8.1
9
7.27.37.47.57.67.77.87.98.08.18.28.3f1(ppm)
2.0
0
4.0
9
1.0
7
1.0
3
1.0
0
7.26
7.3
87.
38
7.3
97.
45
7.4
57.
46
7.4
67.
47
7.4
97.
597.
60
7.6
2
7.75
7.76
7.78
8.1
88
.19S
O
O F
18
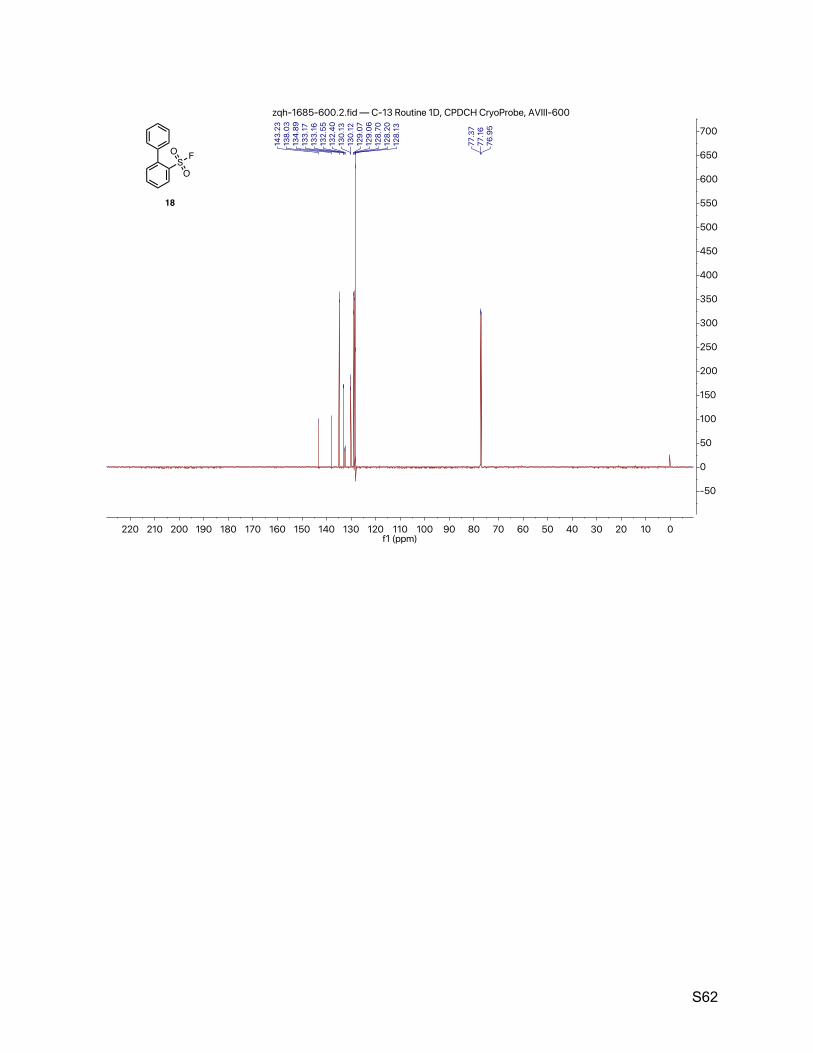
S62
0102030405060708090100110120130140150160170180190200210220
f1(ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
zqh-1685-600.2.fid—C-13Routine1D,CPDCHCryoProbe,AVIII-600
76.95
77.16
77.37
128.13
128.20
128.70
129.06
129.07
130.12
130.13
132.40
132.55
133.16
133.17
134.89
138.03
143.23
SO
O F
18
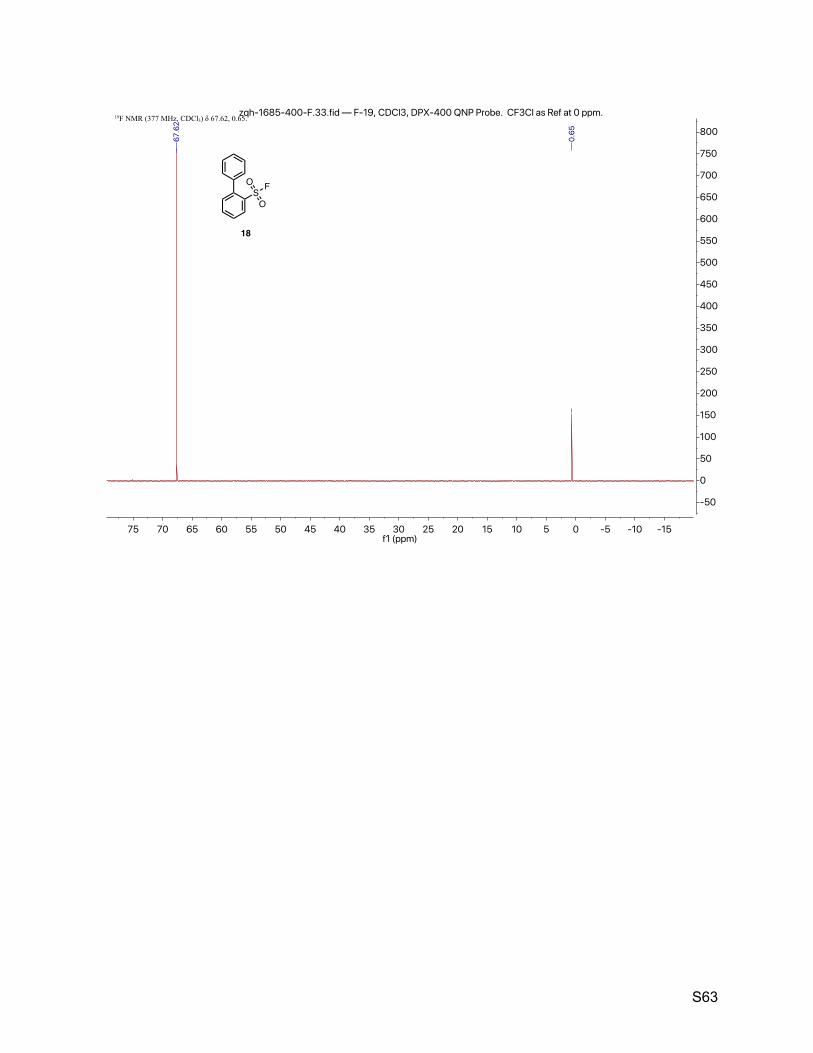
S63
-15-10-5051015202530354045505560657075
f1(ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
800
zqh-1685-400-F.33.fid—F-19,CDCl3,DPX-400QNPProbe.CF3ClasRefat0ppm.
0.65
67.62
19FNMR(377MHz,CDCl3)δ67.62,0.65.
SO
O F
18

S64
8.8. (E)-2-(2-(fluorosulfonyl)vinyl)benzenesulfonyl fluoride (19)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5
f1(ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
300
zqh-1691-600.1.fid—H-1Routine,CP-DCH,AVIIIHD-600
1.0
4
1.9
61.
01
0.9
4
1.0
0
6.9
36
.93
6.9
56
.96
7.26
7.76
7.78
7.79
7.8
07.
87
7.8
87.
90
8.2
28
.23
8.4
98
.51
SO
O F
SO
O F
19

S65
0102030405060708090100110120130140150160170180190200210220
f1(ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
zqh-1691-600.2.fid—
76.95
77.16
77.37
116.33
124.70
124.90
129.73
131.30
131.62
132.25
132.76
132.93
136.14
143.23
115120125130135140145f1(ppm)
116.33
124.70
124.90
129.73
131.30
131.62
132.25
132.76
132.93
136.14
143.23
SO
O F
SO
O F
19

S66
-15-10-5051015202530354045505560657075
f1(ppm)
-1000
-500
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
5500
6000
6500
7000
7500
8000
zqh-1691-400-F.3.fid—AV400BBOFProbe,F-19inCDCl3,CFCl3=0ppm.
0.65
61.56
67.07
SO
O F
SO
O F
19

S67
8.9. 2-(Morpholinosulfonyl)benzenesulfonyl fluoride (20)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5
f1(ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
zqh-1806-600.1.fid—H-1Routine,CP-DCH,AVIIIHD-600
3.8
0
3.8
0
1.0
11.
02
1.0
01.
00
3.3
13
.32
3.3
23
.32
3.3
23
.33
3.7
33
.73
3.7
4
7.26
7.8
47.
84
7.8
47.
85
7.9
07.
90
7.9
17.
91
7.9
27.
93
8.2
38
.23
8.2
48
.24
8.3
48
.35
8.3
68
.36
N
O
SO
O
SO
O
F
20

S68
0102030405060708090100110120130140150160170180190200210220
f1(ppm)
0
50
100
150
200
250
300
350
400
450
zqh-1806-600.2.fid—C-13Routine1D,CPDCHCryoProbe,AVIII-600
46.14
66.46
76.95
77.16
77.37
132.02
132.20
132.89
132.90
133.17
133.46
135.44
138.47
N
O
SO
O
SO
O
F
20

S69
-15-10-505101520253035404550556065707580
f1(ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400
1500zqh-1806-400-f.1.fid—F-19,CDCl3,DPX-400QNPProbe.CF3ClasRefat0ppm.
0.65
64.00
N
O
SO
O
SO
O
F
20

S70
8.10. 4-((2-(Fluorosulfonyl)phenyl)sulfonyl)piperazine-1-sulfonyl fluoride (21)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5
f1(ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300
1400zqh-1828-600.1.fid—H-1Routine,CP-DCH,AVIIIHD-600
3.9
03
.85
1.10
1.12
1.0
21.
00
3.5
03
.50
3.5
13
.51
3.5
43
.54
3.5
53
.55
7.26
7.8
77.
87
7.8
87.
88
7.8
87.
89
7.8
97.
89
7.9
37.
93
7.9
47.
94
7.9
57.
96
8.3
08
.30
8.3
18
.32
8.3
68
.36
8.3
78
.37
NN
SO
OSO
O F
SO
O
F21

S71
0102030405060708090100110120130140150160170180190200210220
f1(ppm)
0
50
100
150
200
250
zqh-1828-600.2.fid—C-13Routine1D,CPDCHCryoProbe,AVIII-600
44.95
46.83
76.95
77.16
77.37
131.70
131.87
133.17
133.18
133.36
133.97
135.70
138.40
NN
SO
OSO
O F
SO
O
F21

S72
-15-10-5051015202530354045505560657075
f1(ppm)
-5
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85zqh-1828-400-f.1.fid—F-19,CDCl3,DPX-400QNPProbe.CF3ClasRefat0ppm.
0.65
41.41
64.11
NN
SO
OSO
O F
SO
O
F21

S73
8.11. 2-((Trifluoromethyl)sulfonyl)benzenesulfonyl fluoride (22)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5
f1(ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
zqh-1688-p2-600.1.fid—H-1Routine,CP-DCH,AVIIIHD-600
2.0
0
1.0
01.
01
7.26
8.0
78
.08
8.0
88
.09
8.1
08
.11
8.4
58
.45
8.4
68
.46
8.4
78
.48
8.4
98
.49
8.5
08
.50
8.5
08
.51
7.98.08.18.28.38.48.58.6f1(ppm)
2.0
0
1.0
0
1.0
1
8.0
78
.07
8.0
88
.08
8.0
98
.10
8.1
18
.11
8.4
48
.45
8.4
58
.46
8.4
68
.47
8.4
88
.49
8.4
98
.50
8.5
08
.50
8.5
1
SO
O
SO
O F
FF
F
22

S74
0102030405060708090100110120130140150160170180190200210220
f1(ppm)
-50
0
50
100
150
200
250
300
350
400
450
500
550
600
650
700
750
800
zqh-1688-p2-600.2.fid—
76.95
77.16
77.37
116.33
116.59
118.56
118.77
120.71
120.94
123.12
132.16
132.17
133.83
133.84
134.78
134.97
136.10
136.63
136.96
115120125130135f1(ppm)
115.70
115.96
117.92
118.13
120.08
120.31
122.48
131.52
131.54
133.20
133.21
134.15
134.34
135.99
136.33
114115116117118119120121122123124f1(ppm)
116.33
116.59
118.56
118.77
120.71
120.94
123.12
CF3
SO
O
SO
O F
FF
F
22

S75
-110-100-90-80-70-60-50-40-30-20-10010203040506070
f1(ppm)
-200
0
200
400
600
800
1000
1200
1400
1600
1800
2000
2200
2400
zqh-1688-p2-400.1.fid—F-19,CDCl3,DPX-400QNPProbe.CF3ClasRefat0ppm.
2.81
1.00
-73.48
0.65
67.11
SO
O
SO
O F
FF
F
22
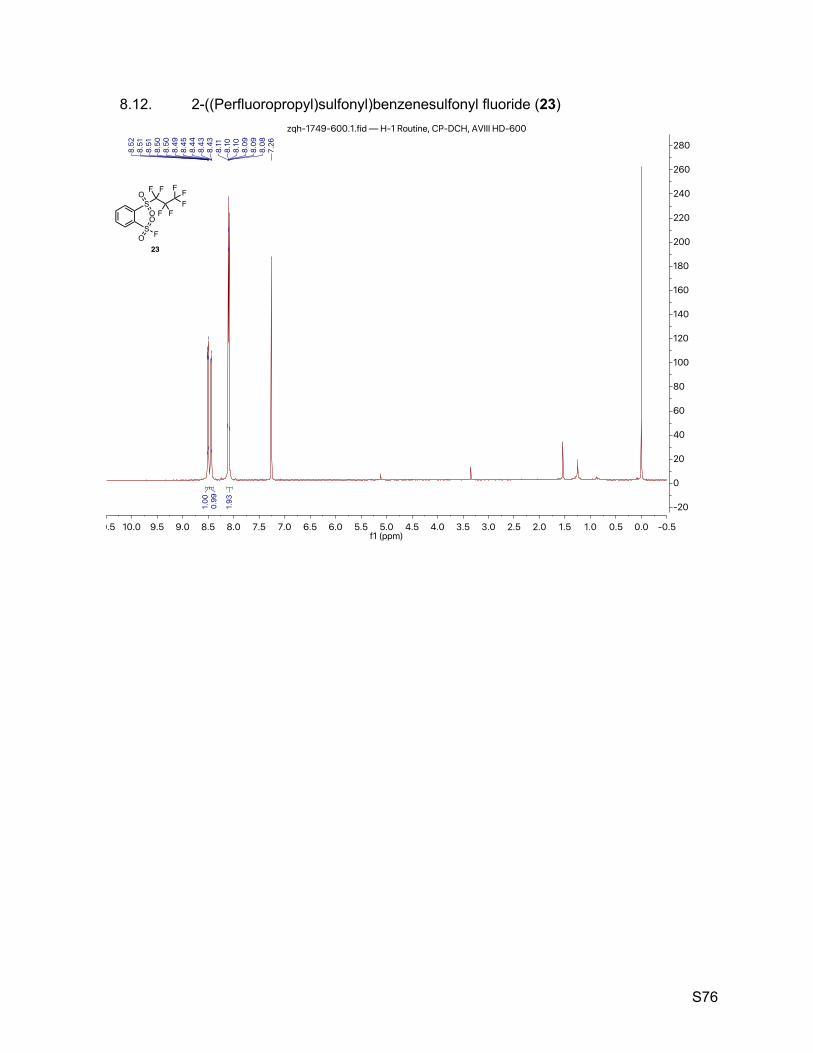
S76
8.12. 2-((Perfluoropropyl)sulfonyl)benzenesulfonyl fluoride (23)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.09.510.010.5
f1(ppm)
-20
0
20
40
60
80
100
120
140
160
180
200
220
240
260
280
zqh-1749-600.1.fid—H-1Routine,CP-DCH,AVIIIHD-600
1.9
3
0.9
91.
00
7.26
8.0
88
.09
8.0
98
.10
8.1
08
.11
8.4
38
.43
8.4
48
.45
8.4
98
.50
8.5
08
.51
8.5
18
.52
SO
O
SO
O F
FF
F FFFF
23
S77
0102030405060708090100110120130140150160170180190200210220
f1(ppm)
-100
0
100
200
300
400
500
600
700
800
900
1000
1100
1200
1300zqh-1749-600.2.fid—C-13Routine1D,CPDCHCryoProbe,AVIII-600
76.34
76.55
76.76
106.33
106.59
106.80
107.86
107.91
108.12
108.17
108.33
108.38
108.59
108.64
109.70
109.91
110.17
111.81
112.04
112.28
113.78
113.84
114.07
114.30
115.47
115.69
115.87
115.91
116.10
116.33
117.39
117.61
117.83
117.95
119.53
119.76
131.88
133.20
133.21
134.37
134.56
135.48
136.22
136.45
104106108110112114116118120f1(ppm)
106.59
108.12
108.17
108.38
108.59
110.17
111.81
112.04
113.84
114.07
114.30
115.47
115.69
115.87
115.91
116.10
116.33
117.39
117.61
117.83
117.95
119.53
SO
O
SO
O F
FF
F FFFF
23

S78
-15-10-5051015202530354045505560657075
f1(ppm)
0
50
100
150
200
250
300
350
400
zqh-1749-400.1.fid—F-19,CDCl3,DPX-400QNPProbe.CF3ClasRefat0ppm.
0.65
67.08
SO
O
SO
O F
FF
F FFFF
23

S79
-140-135-130-125-120-115-110-105-100-95-90-85-80-75-70-65-60-55-50-45f1(ppm)
0
50
100
150
200
250
300
350
400
450
zqh-1749-400.4.fid—F-19,CDCl3,DPX-400QNPProbe.CF3ClasRefat0ppm.
-124.80
-124.79
-107.43
-107.42
-107.41
-107.40
-107.39
-107.37
-107.36
-107.35
-107.34
-80.84
-80.81
-80.79
-107.55-107.45-107.35-107.25f1(ppm)
-107.43
-107.42
-107.41
-107.40
-107.39
-107.37
-107.36
-107.35
-107.34
-80.95-80.85-80.75-80.65f1(ppm)
-80.84
-80.81
-80.79
SO
O
SO
O F
FF
F FFFF
23

S80
9. References
1. Rosenau CP, Jelier BJ, Gossert AD, & Togni A (2018) Exposing the Origins of
Irreproducibility in Fluorine NMR Spectroscopy. Angew Chem Int Edit 57(30):9528-9533.
2. Dong JJ, Krasnova L, Finn MG, & Sharpless KB (2014) Sulfur(VI) Fluoride Exchange
(SuFEx): Another Good Reaction for Click Chemistry. Angew Chem Int Edit 53(36):9430-
9448.
3. Gakh AA, Romaniko SV, Ugrak BI, & Fainzilberg AA (1991) N-Fluorination with Cesium
Fluoroxysulfate. Tetrahedron 47(35):7447-7458.
4. Nielsen MK, Ugaz CR, Li WP, & Doyle AG (2015) PyFluor: A Low-Cost, Stable, and
Selective Deoxyfluorination Reagent. J Am Chem Soc 137(30):9571-9574.
5. Tribby AL, Rodriguez I, Shariffudin S, & Ball ND (2017) Pd-Catalyzed Conversion of Aryl
Iodides to Sulfonyl Fluorides Using SO2 Surrogate DABSO and Selectfluor. J Org Chem
82(4):2294-2299.
6. Vanderpuy M (1988) Potassium Fluoride Catalyzed Fluorodesulfonylations of Aryl
Sulfonyl Fluorides. J Org Chem 53(18):4398-4401.
7. Guo TJ, et al. (2018) A New Portal to SuFEx Click Chemistry: A Stable Fluorosulfuryl
Imidazolium Salt Emerging as an "F-SO2+" Donor of Unprecedented Reactivity,
Selectivity, and Scope. Angew Chem Int Edit 57(10):2605-2610.
8. Krutak JJ, Burpitt RD, Moore WH, & Hyatt JA (1979) Chemistry of Ethenesulfonyl Fluoride
- Fluorosulfonylethylation of Organic-Compounds. J Org Chem 44(22):3847-3858.
9. Zha GF, et al. (2017) Palladium-Catalyzed Fluorosulfonylvinylation of Organic Iodides.
Angew Chem Int Edit 56(17):4849-4852.
10. Smedley CJ, et al. (2019) Bifluoride Ion Mediated SuFEx Trifluoromethylation of Sulfonyl
Fluorides and Iminosulfur Oxydifluorides. Angew Chem Int Ed Engl.
11. Stapels DAC, et al. (2014) Staphylococcus aureus secretes a unique class of neutrophil
serine protease inhibitors. P Natl Acad Sci USA 111(36):13187-13192.
12. Stapels DAC, et al. (2018) Evidence for multiple modes of neutrophil serine protease
recognition by the EAP family of Staphylococcal innate immune evasion proteins. Protein
Sci 27(2):509-522.
13. Hansen G, et al. (2011) Unexpected Active-Site Flexibility in the Structure of Human
Neutrophil Elastase in Complex with a New Dihydropyrimidone Inhibitor. J Mol Biol
409(5):681-691.
14. Mccoy AJ, et al. (2007) Phaser crystallographic software. J Appl Crystallogr 40:658-674.

S81
15. Emsley P, Lohkamp B, Scott WG, & Cowtan K (2010) Features and development of Coot.
Acta Crystallogr D 66:486-501.
16. Adams PD, et al. (2010) PHENIX: a comprehensive Python-based system for
macromolecular structure solution. Acta Crystallogr D 66:213-221.
17. Laskowski RA, Moss DS, & Thornton JM (1993) Main-Chain Bond Lengths and Bond
Angles in Protein Structures. J Mol Biol 231(4):1049-1067.
18. Hooft RWW, Vriend G, Sander C, & Abola EE (1996) Errors in protein structures. Nature
381(6580):272-272.
19. Davis IW, et al. (2007) MolProbity: all-atom contacts and structure validation for proteins
and nucleic acids. Nucleic Acids Res 35:W375-W383.
20. de Garavilla L, et al. (2005) A novel, potent dual inhibitor of the leukocyte proteases
cathepsin G and chymase - Molecular mechanisms and anti-inflammatory activity in vivo.
J Biol Chem 280(18):18001-18007.
21. Forli S, et al. (2016) Computational protein-ligand docking and virtual drug screening with
the AutoDock suite. Nat Protoc 11(5):905-919.
22. Word JM, Lovell SC, Richardson JS, & Richardson DC (1999) Asparagine and glutamine:
Using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol
285(4):1735-1747.
23. O'Boyle NM, et al. (2011) Open Babel: An open chemical toolbox. J Cheminformatics 3.
24. Backus KM, et al. (2016) Proteome-wide covalent ligand discovery in native biological
systems. Nature 534(7608):570-+.
25. Mortenson DE, et al. (2018) "Inverse Drug Discovery" Strategy To Identify Proteins That
Are Targeted by Latent Electrophiles As Exemplified by Aryl Fluorosulfates. J Am Chem
Soc 140(1):200-210.

download fileview on ChemRxivZheng et al Sleeping Beauty 2019 SI.pdf (4.60 MiB)


















