
Sequence Alignment - Baylor ECSweb.ecs.baylor.edu/faculty/cho/3350/2_SequenceAlignment.pdf · PAM...
30
9/1/2015 1 Young-Rae Cho Associate Professor Department of Computer Science Baylor University BINF 3350, Genomics and Bioinformatics Sequence Alignment BINF 3350, Chapter 4, Sequence Alignment 1. Sequence Alignment 2. Dynamic Programming 3. Scoring Alignments 4. Gap Penalty 5. Global vs. Local Alignment 6. Pairwise vs. Multiple Sequence Alignment 7. Sequence Homolog Search 8. Motif Search
-
Upload
phamnguyet -
Category
Documents
-
view
223 -
download
0
Transcript of Sequence Alignment - Baylor ECSweb.ecs.baylor.edu/faculty/cho/3350/2_SequenceAlignment.pdf · PAM...

9/1/2015
1
Young-Rae Cho
Associate Professor
Department of Computer Science
Baylor University
BINF 3350, Genomics and Bioinformatics
Sequence Alignment
BINF 3350, Chapter 4, Sequence Alignment
1. Sequence Alignment
2. Dynamic Programming
3. Scoring Alignments
4. Gap Penalty
5. Global vs. Local Alignment
6. Pairwise vs. Multiple Sequence Alignment
7. Sequence Homolog Search
8. Motif Search

9/1/2015
2
Sequence Homology
Homologs
Similar sequence and Common ancestor
Similar sequence and Same function (in divergent evolution)
Orthologs
Homologous sequences in different species by species divergence
Paralogs
Homologous sequences in the same species by gene duplication
Analogs
Similar sequence and No common ancestor (in convergent evolution)
Sequence Similarity
Importance of finding similar (DNA or protein) sequences
Evolutionary closeness
Relationship between sequences and evolution
Functional similarity
Relationship between sequences and functions
How to measure sequence similarity
(Method 1) Counting identical letters on each position
(Method 2) Inserting gaps to maximize the number of identical letters
Sequence alignment
A T G T T A T
T C G T A C T| | |
A T ‐ G T T A ‐ T
‐ T C G T ‐ A C T| | | | |

9/1/2015
3
Sequence Alignment
Sequence Alignment
Aligning two or more sequences to maximize their similarity including gaps
How to find sequence alignment?
(1) Measuring edit distance
Edit Distance (1)
Definition
Edit distance between two sequences x and y : the minimum number of
editing operations (insertion, deletion, substitution) to transform x into y
Example
x=“TGCATAT” (m=7), y=“ATCCGAT” (n=7)
TGCATAT
ATGCATATinsertion of “A”
ATCCATATsubstitution of “G” with “C”
ATCCGATATinsertion of “G”
ATCCGATTdeletion of “A”
ATCCGATdeletion of “T”
edit distance = 5 ?

9/1/2015
4
Edit Distance (2)
Example
x=“TGCATAT” (m=7), y=“ATCCGAT” (n=7)
Can it be done in 3 steps?
How to measure edit distance efficiently?
TGCATAT
ATGCATATinsertion of “A”
ATGCAATdeletion of “T”
ATCCAATsubstitute of “G” with “C”
ATCCGATsubstitute of “A” with “G”
edit distance = 4 ?
A T G T T A T G C A A T G T A C T T A
T C G T A C T C A G T T C A A G T C A
Edit Distance (3)
Example in 2-Row Representation
x=“ATCTGATG” (m=8), y=“TGCATAC” (n=7)
A T C T G A T G
T G C A T A C
x
y
4 matches
1 substitutions
3 insertions2 deletions
A T C T G A T G
T G C A T A C
x
y
4 matches4 insertions3 deletions
Edit distance = #insertions + #deletions + #substitutions

9/1/2015
5
Hamming Distance vs. Edit Distance
Hamming Distance
Compares the letters on the same position between two sequences
Not good to measure evolutionary distance between DNA sequences
Edit Distance
Compares the letters between two sequences after inserting gaps
Allows comparison of two sequences of different lengths
Good to measure evolutionary distance between DNA sequences
Example
x=“ATATATAT” , y=“TATATATA”
Hamming distance between x and y ?
Edit distance between x and y ?
Sequence Alignment
Sequence Alignment
Aligning two or more sequences to maximize their similarity including gaps
How to find sequence alignment?
(1) Measuring edit distance
(2) Finding longest common subsequence

9/1/2015
6
Longest Common Subsequence (1)
Subsequence of x
An ordered sequence of letters from x
Not necessarily consecutive
e.g., x=“ATTGCTA”, “AGCA” ?, “TCG” ?, “ATCT” ?, “TGAT” ?
Common Subsequence of x and y
e.g., x=“ATCTGAT” and y=“TGCATA”, “TCTA” ?, “TGAT” ?, “TATA” ?
Longest Common Subsequence (LCS) of x and y ?
Longest Common Subsequence (2)
Example
x=“ATCTGATG” (m=8), y=“TGCATAC” (n=7)
LCS of X and Y ?
2-row representation
How to find LCS efficiently?
A T G T T A T G C A A T G T A C T T A G A C T C A A G T G C C A T T T G A C
T C G T A C T C A G T T C A A G T C A G T T A C G A G T A C A T G C A A A C
A T G T T A T G C A A T G T A C T T A
T C G T A C T C A G T T C A A G T C A
A T G T T A T G C A
T C G T A C T C A G
A T G T T
T C G T A

9/1/2015
7
Sequence Alignment
Sequence Alignment
Aligning two or more sequences to maximize their similarity including gaps
How to find sequence alignment?
(1) Measuring edit distance
(2) Finding longest common subsequenceDynamic programming
BINF 3350, Chapter 4, Sequence Alignment
1. Sequence Alignment
2. Dynamic Programming
3. Scoring Alignments
4. Gap Penalty
5. Global vs. Local Alignment
6. Pairwise vs. Multiple Sequence Alignment
7. Sequence Homolog Search
8. Motif Search

9/1/2015
8
Definition
An algorithm to solve complex problems by breaking them down into simpler sub‐problems
The result of a sub‐problem is used to solve the next sub‐problem
Features
Optimization
• Finding an optimal solution
• Saving memory space
Examples
Binary search tree
Sequence alignment
Dynamic Programming
Edit Graph
2‐D grid structure having a diagonal
on the position of the same letter
Weight the diagonal lines as 1
Weight the other lines as 0
Goal
Finding the strongest path from
source to sink
Algorithm
(1) Compute the max score for each node
(The max score means the max counts of identical letters from source to each node)
(2) When reaching the sink, trace backward to find LCS
Dynamic Programming for Sequence Alignment
sink
source A T C G T A C
A
T
G
T
T
T
A
+1
+1
+1
+1
+1
+1
+1
+1
+1
+1
+1
+1
+1
9/1/2015
9
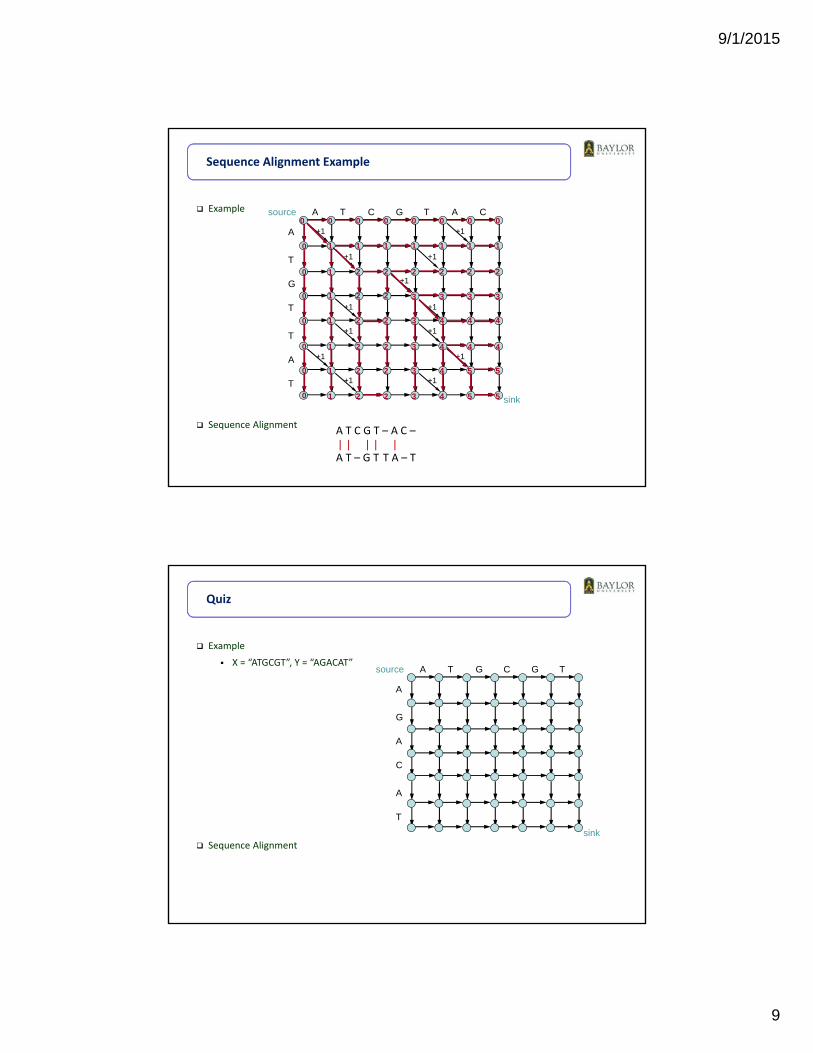
Example
Sequence Alignment
Sequence Alignment Example
source
sink
A T C G T A C
A
T
G
T
T
T
A
+1
+1
+1
+1
+1
+1
+1
+1
+1
+1
+1
+1
+1
0 0 0 0 0 0 0 0
0
0
0
0
0
0
0
1 1 1 1 1 1 1
1 2 2 2 2 2 2
1 2 2
4
3 3 3
1 2 3
3
4 4
1 2 2 3 4 4 4
1 2 2 3 4 5 5
1 2 2 3 4 5 5
2
A T C G T – A C –
A T – G T T A – T| | | | |
Example
X = “ATGCGT”, Y = “AGACAT”
Sequence Alignment
Quiz
sink
source A T G C G T
A
G
A
C
A
T

9/1/2015
10
BINF 3350, Chapter 4, Sequence Alignment
1. Sequence Alignment
2. Dynamic Programming
3. Scoring Alignments
4. Gap Penalty
5. Global vs. Local Alignment
6. Pairwise vs. Multiple Sequence Alignment
7. Sequence Homolog Search
8. Motif Search
Scoring Alignments: Percent Identity (1)
Identity
Degree of identical matches between sequences
Percent Identity
Percentage of identical matches
Dot-plot representations
Visualization method of identity

9/1/2015
11
Scoring Alignments: Percent Identity (2)
Dot-plot representations of self alignment
The background noise can be removed by setting a threshold of the min
identity score in a fixed window
Scoring Alignments: Percent Similarity
Percent Similarity
Percentage of similar amino acid pairs in biochemical structure (Protein)
Percentage of similar nucleotide pairs in biochemical structure (DNA)
Advanced Scoring Schemes
Varying scores in similarity of biochemical structures
Penalties (negative scores) for strong mismatches
Relative likelihood of evolutionary relationship
Probability of mutations
Minimum Acceptance Score
90% of sequence pairs with more than 30% sequence identity: homolog
20~30% sequence identity: twilight zone

9/1/2015
12
Substitution Matrices (1)
Substitution Matrix
Score matrix among nucleotides or amino acids
4 × 4 array representation for DNA sequences
or (4+1) × (4+1) array
20 × 20 array representation for protein sequences
or (20+1) × (20+1) array
Entry of δ(i,j) has the score between i and j,
i.e., the rate at which i is substituted with j over time
Substitution Matrices (2)
PAM (Point Accepted Mutations)
For protein sequence alignment
Amino acid substitution frequency in mutations
Logarithmic matrix of mutation probabilities
PAM120: Results from 120 mutations per 100 residues
PAM120 vs. PAM240
BLOSUM (Block Substitution Matrix)
For protein sequence alignment
Applied for local sequence alignments
Substitution frequencies between clustered groups
BLOSUM-62: Results with a threshold (cut-off) of 62% identity
BLOSUM-62 vs. BLOSUM-50

9/1/2015
13
Substitution Matrices (3)
Substitution Matrix Examples
BLOSUM-62 PAM120
Theory of Scoring Alignments
Random model
Non-random model
Odds ratio
Odds ratio for each position
Odds ratio for entire alignment
log-odds ratio (a score in a substitution matrix)
Expected score

9/1/2015
14
BINF 3350, Chapter 4, Sequence Alignment
1. Sequence Alignment
2. Dynamic Programming
3. Scoring Alignments
4. Gap Penalty
5. Global vs. Local Alignment
6. Pairwise vs. Multiple Sequence Alignment
7. Sequence Homolog Search
8. Motif Search
Gap Penalty (1)
Gaps
Contiguous sequence of spaces in one of the aligned sequences
Gaps inserted as the results of insertions and deletions (indels)
Gap Penalties
High penalties vs. Low penalties
Fixed penalties vs. Flexible penalties depending on residues
No penalty on start gaps and end gaps
Finding optimal number of gaps for the best score in sequence alignment
Dynamic Programming

9/1/2015
15
Gap Penalty (2)
Examples of high penalties and low penalties
Affine Gap Penalty (1)
Motivation
-σ for 1 gap (insertion or deletion)
-2σ for 2 consecutive gaps (insertions or deletions)
-3σ for 3 consecutive gaps (insertions or deletions), etc.
→ too severe penalty for a series of 100 consecutive gaps
Example
x=“ATAGC”, y=“ATATTGC”
x=“ATAGGC”, y=“ATGTGC”
single event

9/1/2015
16
Affine Gap Penalty (2)
Linear Gap Penalty
Score for a gap of length x : -σ x
Constant Gap Penalty
Score for a gap of length x : -ρ
Affine Gap Penalty
Score for a gap of length x : - (ρ + σ x)
ρ : gap opening penalty / σ : gap extension penalty ( ρ σ )
BINF 3350, Chapter 4, Sequence Alignment
1. Sequence Alignment
2. Dynamic Programming
3. Scoring Alignments
4. Gap Penalty
5. Global vs. Local Alignment
6. Pairwise vs. Multiple Sequence Alignment
7. Sequence Homolog Search
8. Motif Search

9/1/2015
17
Global vs. Local Alignment
Global Alignment
Finding sequence alignment across the whole length of sequences
Local Alignment
Finding significant similarity in a part of sequences
Example
x = “TCAGTGTCGAAGTTA”
y = “TAGGCTAGCAGTGTA”
T C A G – – T – G T C G A A G T – T A
T – A G G C T A G – C – A – G T G T A| | | | | | | | | | |
T C A G T G T C G A A G T T A
T A G G C T A G C A G T G T A| | | | | |
Dynamic Programming (Needleman-Wunch algorithm)
Dynamic Programming (Smith-Waterman algorithm)
Local Alignment Example
Local Alignment
Applied for multi-domain
protein sequences
Protein domain
• Basic functional block
• Evolutionary conserved

9/1/2015
18
BINF 3350, Chapter 4, Sequence Alignment
1. Sequence Alignment
2. Dynamic Programming
3. Scoring Alignments
4. Gap Penalty
5. Global vs. Local Alignment
6. Pairwise vs. Multiple Sequence Alignment
7. Sequence Homolog Search
8. Motif Search
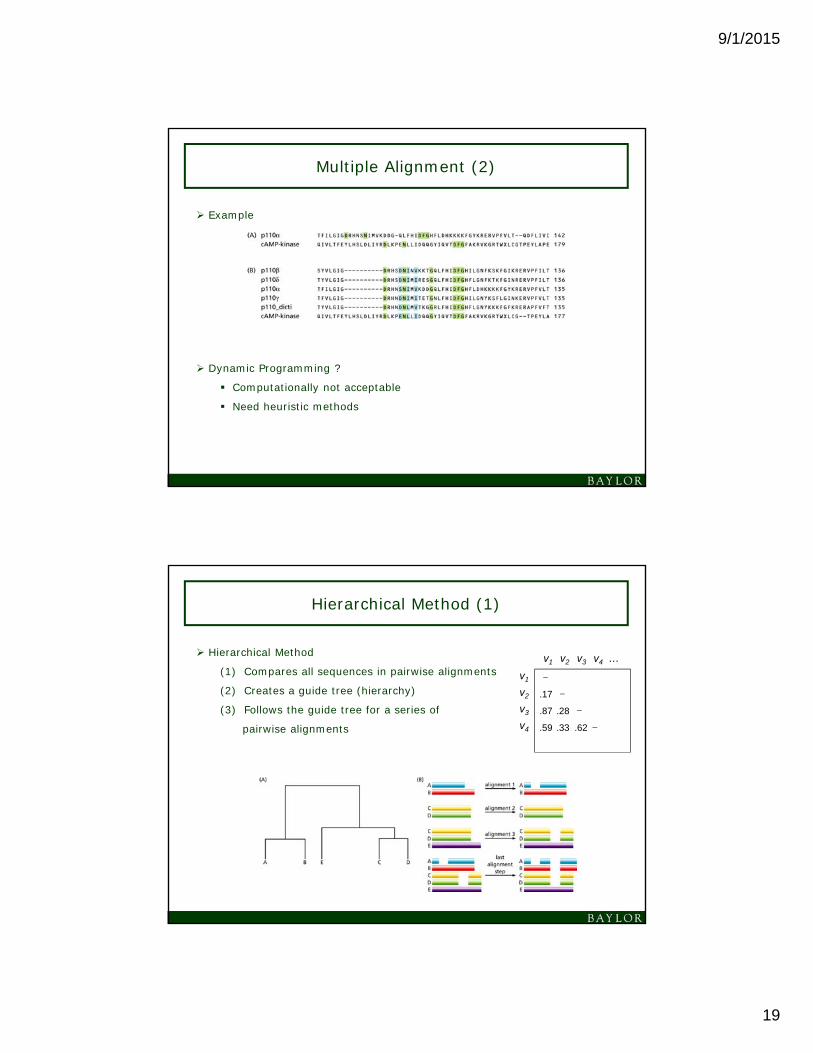
Multiple Alignment (1)
Pairwise Alignment
Alignment of two sequences
Sometimes two sequences are functionally
similar or have common ancestor although
they have weak sequence similarity
Multiple Alignment
Alignment of three or more sequences simultaneously
Finds similarity which is invisible in pairwise alignment
9/1/2015
19
Multiple Alignment (2)
Example
Dynamic Programming ?
Computationally not acceptable
Need heuristic methods
Hierarchical Method (1)
Hierarchical Method
(1) Compares all sequences in pairwise alignments
(2) Creates a guide tree (hierarchy)
(3) Follows the guide tree for a series of
pairwise alignments
-
.17 –
.87 .28 –
.59 .33 .62 –
v1 v2 v3 v4 …
v1
v2
v3
v4

9/1/2015
20
Hierarchical Method (2)
Features
Also called progressive alignment
More intelligent strategy on each step
Use of consensus sequence to compare groups of sequences
Gaps are permanent (“once a gap, always a gap”)
Works well for close sequences
Application Tools
ClustalW
• Comparing residues one pair at a time and imposing gap penalties
DIALIGN
• Finding pairs of equal-length gap-free segments
Divide-and-Conquer Method
Process
Features
Fast aligning of long sequences

9/1/2015
21
Multiple Alignment Results
Examples
Summary of PSA & MSA Algorithms
Rigorous Algorithms
Heuristic Algorithms

9/1/2015
22
BINF 3350, Chapter 4, Sequence Alignment
1. Sequence Alignment
2. Dynamic Programming
3. Scoring Alignments
4. Gap Penalty
5. Global vs. Local Alignment
6. Pairwise vs. Multiple Sequence Alignment
7. Sequence Homolog Search
8. Motif Search
Searching Databases
Sequence Homolog Search
Search similar sequences to a query sequence in a database
Computational issues
• Dynamic programming (N-W / S-W algorithms) are rigorous
• But inefficient in searching a huge database
• Need heuristic approaches
Sequence Homolog Searching Tools
FASTA
BLAST

9/1/2015
23
FASTA (1)
FASTA
DNA / protein sequence alignment tool (local alignment)
Applies dynamic programming in scoring selected sequences
Heuristic method in candidate sequence search
Algorithm
(1) Finding all pairwise k-tuples (at least k contiguous matching residues)
(2) Scoring the k-tuples by a substitution matrix
(3) Selecting sequences with high scores for alignment
FASTA (2)
Indexing (or Hashing)
Indexing Process in FASTA
(1) Find all k-tuples from a query sequence and calculate ci
(2) Build an index table

9/1/2015
24
FASTA Package
FASTA package
ssearch : applies dynamic programming (S-W algorithm)
query sequence database
fasta protein protein
fasta DNA DNA
fastx / fasty DNA (all reading frames) protein
tfastx / tfasty protein DNA (all reading frames)
BLAST (1)
BLAST (Basic Local Alignment Search Tool)
DNA / protein sequence alignment tool
Finds local alignments
Heuristic method in sequence search
Runs faster than FASTA
Algorithm
(1) Makes a list of words (word pairs) from the query sequence
(2) Chooses high-scoring words
(3) Searches database for matches (hits) with the high-scoring words
(4) Extends the matches in both directions to find high-scoring segment pair
(HSP)
(5) Selects the sequence which has two or more HSPs for S-W alignment

9/1/2015
25
BLAST (2)
Deterministic Finite Automata (DFA)
DFA Analysis Process in BLAST
(1) Build DFA using high-scoring words
(2) Read sequences in database and trace DFA
(3) Output the positions for hits
BLAST Package
BLAST programs
query sequence database
blastp protein protein
blastn DNA DNA
blastx DNA (all reading frames) protein
tblastn protein DNA (all reading frames)
tblastx DNA (all reading frames) DNA (all reading frames)

9/1/2015
26
Search Results
BLAST Search Results
FASTA Search Results
E-value
E-value
Average number of alignments with a score of at least S that would be
expected by chance alone in searching a database of n sequences
Ranges of E-value:
High alignment score S
Low alignment score S
Factors
• Alignment score
• The number of sequences in the database
• Sequence length
Default E-value threshold: 0.01 ~ 0.001
Low E-value
High E-value
0 ~ n

9/1/2015
27
Filtering
Low-Complexity Region
Highly biased amino acid composition
Lowers significant hits in sequence alignment
BLAST filters the query sequence for low-complexity regions and mark “X”
Summary of Homolog Search Algorithms
Rigorous Algorithms
Heuristic Algorithms

9/1/2015
28
BINF 3350, Chapter 4, Sequence Alignment
1. Sequence Alignment
2. Dynamic Programming
3. Scoring Alignments
4. Gap Penalty
5. Global vs. Local Alignment
6. Pairwise vs. Multiple Sequence Alignment
7. Sequence Homolog Search
8. Motif Search
Motifs
Motifs
Short sequence patterns
Functionally related sequences share similarly distributed patterns (motifs)
of critical functional residues
Types of Motif Search
Search a query sequence in a motif database
Search a pattern in a sequence database
Find a pattern from a set of sequences
Motif Finding
Consensus method by global multiple alignment

9/1/2015
29
Motif Search Tools (1)
BLOCKS
Logos
• Size of letters: conservation levels
• Color of letters: biochemical properties
Motif Search Tools (2)
MEME
Summary motif information
• Location of motifs in sequences

9/1/2015
30
Motif Databases
PROSITE
Code for patterns
• Each letter represents an amino acid residue
• All positions are separated by “-”
Code description Example
X any amino acid G-X-L-M-S-A-D-F-F-F
[] two or more possible amino acid G-[LI]-L-M-S-A-D-F-F-F
{} disallowed amino acid G-[LI]-L-M-S-A-{RK}-F-F-F
(n) repetition by n of the amino acid G-[LI]-L-M-S-A-{RK}-F(3)
(n,m) a range: only allowed with X G-[LI]-L-M-S-A-{RK}-X(1,3)


















