
The Recombination Protein RAD52 Cooperates with the Excision ...
Upload
somnath-ghoshCategory
view
214download
1
Ra
SR
a
ARR2AA
KFABDR
1
nestcibrbufltfi
dsb
(
0d
Mutation Research 729 (2012) 61– 72
Contents lists available at SciVerse ScienceDirect
Mutation Research/Fundamental and MolecularMechanisms of Mutagenesis
jo ur n al hom ep a ge: www.elsev ier .com/ locate /molmutC om mun i ty a ddress : www.elsev ier .com/ locate /mutres
ole of Rad52 in fractionated irradiation induced signaling in A549 lungdenocarcinoma cells
omnath Ghosh, Malini Krishna ∗
adiation Biology and Health Sciences Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400085, India
r t i c l e i n f o
rticle history:eceived 16 June 2011eceived in revised form2 September 2011ccepted 27 September 2011vailable online 4 October 2011
a b s t r a c t
The effect of fractionated doses of �-irradiation (2 Gy per fraction over 5 days), as delivered in cancerradiotherapy, was compared with acute doses of 10 and 2 Gy, in A549 cells. A549 cells were found tobe relatively more radioresistant if the 10 Gy dose was delivered as a fractionated regimen. Microarrayanalysis showed upregulation of DNA repair and cell cycle arrest genes in the cells exposed to fractionatedirradiation. There was intense activation of DNA repair pathway-associated genes (DNA-PK, ATM, Rad52,MLH1 and BRCA1), efficient DNA repair and phospho-p53 was found to be translocated to the nucleus of
eywords:ractionated irradiationTMRCA1NA repairad52
A549 cells exposed to fractionated irradiation. MCF-7 cells responded differently in fractionated regimen.Silencing of the Rad52 gene in fractionated group of A549 cells made the cells radiosensitive. The aboveresult indicated increased radioresistance in A549 cells due to the activation of Rad52 gene.
© 2011 Elsevier B.V. All rights reserved.
. Introduction
Cellular response to ionizing radiation is a complex phe-omenon where the dose, dose rate and fractionation play anqually important role in deciding the fate of the cell. Chronic expo-ure of cells to ionizing radiation induces an adaptive responsehat results in the increased tolerance to subsequent cytotoxicityaused by the same [1–4]. Evidence from a number of studies hasndicated that a potential cause of radiation treatment failure maye that multifraction irradiation selects a population of radiationesistant cells from which regrowth of tumor progresses [5–7]. Aetter understanding of how resistance is promoted at the molec-lar level can form the basis for novel treatment strategies in theuture. The targets have been chosen based on their activation fol-owing a single dose of radiation. However, more information abouthe response of these signaling factors at clinically relevant dosesollowing a fractionated regimen is needed to understand their role,f any, in the development of a radioresistant phenotype.
Mammalian cells have evolved a number of repair systems to
eal with the DNA double-strand breaks (DSBs) induced by expo-ure to ionizing radiation [8]. The two major types of double-strandreak repair are homologous recombination and nonhomologous∗ Corresponding author. Tel.: +91 22 2559 0416; fax: +91 22 2550 5151.E-mail addresses: [email protected] (S. Ghosh), [email protected]
M. Krishna).
027-5107/$ – see front matter © 2011 Elsevier B.V. All rights reserved.oi:10.1016/j.mrfmmm.2011.09.007
end-joining. The relative contribution of each of these types ofrepair is controversial [9–11].
The phosphatidyl-inositol kinase-related protein ATM is themost proximal signal transducer initiating cell cycle changes afterthe DNA damage induced by ionizing radiation [12]. Likewise,the rapid induction of ATM serine/threonine protein kinase activ-ity after ionizing radiation has also suggested that ATM acts atan early stage of signal transduction in mammalian cells [13,14].Mammalian ATM is a member of a family of protein kinases thatinclude ATM-Rad3-related (ATR), DNA-dependent protein kinase,and FRAP, which are related because they have a similar carboxy-terminal kinase domain [15,16]. Recently, Bakkenist and Kastan[17] showed that, ATM activation may result from changes in thestructure of chromatin brought about by intermolecular autophos-phorylation and ATM dimer dissociation. Once dissociated, ATMcan then potentially phosphorylate numerous downstream targets,including p53, MDM2, CHK2, NBS1, RAD9, and BRCA1. ATM actsspecifically in the cellular responses to ionizing radiation and DNADSBs, rather than having a more general function in DNA repair.It resides in a complex with BRCA1 and phosphorylates BRCA1in a region that contains clusters of serine-glutamine residues[18]. Phosphorylation of this domain appears to be functionallyimportant because a mutated BRCA1 protein that lacks these key
phosphorylation sites is unable to rescue the radiation hypersen-sitivity of BRCA1-deficient cell lines [18]. Cells deficient in BRCA1show genetic instability, defective G2/M checkpoint control, andreduced homologous recombination [19,20]
6 tion R
ifDoRsMaYr[etbt[etwtdwci
aMDmhqtiriccml
wmm
2
2
c(
2
crotAgm2dCdpw
2 S. Ghosh, M. Krishna / Muta
Repair of DNA damage is critical for the maintenance of genomentegrity and cell survival. Living organisms have developed dif-erent pathways of DNA repair to deal with various types ofNA damage. In Saccharomyces cerevisiae, three epistasis groupsf DNA-damage-repair genes have been identified [21,22]. Thead52 epistasis group, which is mainly responsible for double-trand break (DSB) repair contains several genes: Rad50–Rad57,RE11, and XRS2. Among these genes, mutations in Rad51, Rad52,
nd Rad54 cause the most severe and pleiotropic defects [23–25].east strains lacking a functional Rad52 gene are extremely X-ay sensitive and deficient in mitotic and meiotic recombination26]. Mutations in different regions of Rad52 often result in differ-nt phenotypes [27]. Shinohara et al. proposed that the product ofhe Rad52 gene is not required for the initiation of recombination,ut is essential for an intermediate stage following the forma-ion of DSBs but before the appearance of stable recombinants28]. Recently, homologs of the Rad52 gene were found in severalukaryotic organisms. Sequence analysis has revealed that the N-erminal amino acid sequence of Rad52 protein is highly conservedhile the C-terminal region is less conserved [29–32]. Rad52 pro-
ein interacts through its C-terminal domain with the N-terminalomain of Rad51 protein in a species specific manner [33–35]. Itas reported that the over expression of human Rad52 HsRad52
onferred enhanced resistance to �-rays and induced homologousntrachromosomal recombination in cultured monkey cells [36]
MLH1, another protein involved in Mismatch repair (MMR), haslso been suggested to be involved in the DSB repair pathway [37].LH1 modulates error prone NHEJ by inhibiting the annealing ofNA ends containing noncomplementary base pairs [38]. Further-ore, the influence of MLH1 on IR-induced autosomal mutations
as been studied in a mouse Mlh1−/− kidney cell line. A high fre-uency of IR induced mutations was found in Mlh1−/− cells dueo increased crossover events of mitotic recombination, suggest-ng that MLH1, or MMR may serve as a modulator in mitotic HRepair [39]. Previous study showed that the expression of MLH1 wasnduced by IR, and the loss of MLH1 expression not only elevatedell cycle progression but also increased the yield of IR-inducedhromosomal translocations, suggesting that this gene, involvedostly in MMR, may play a role in DSB repair and cell cycle regu-
ation [40]The present study was conducted to look at how repair path-
ays respond to different irradiation regimens with aim to identifyarkers of radioresistance which may serve as future targets forodulation to enhance the efficacy of radiotherapy.
. Materials and methods
.1. Cell culture
Human Lung Adenocarcinoma A549 cells and human breast carcinoma MCF-7ells were grown in DMEM (Sigma, USA), supplemented with 10% fetal calf serumSigma). Cells were maintained at 37 ◦C in humidified atmosphere with 5% CO2.
.2. Irradiation of cells
Before designing the experiment, there were two aspects that had to be takenare of. One the stochastic effects of radiation and second the deterministic effects ofadiation. Radiation dose as well as confluency of cells could influence the stochasticr deterministic effects of radiation. Exposure of cells (≥2 Gy) with confluency morehan 80%, deterministic effects will be more and stochastic effects will be minimal.t this confluency of cells and radiation dose, it is expected that all the cells willet threshold number of hits. Deterministic rather than stochastic factors explainost of the variation in radio-responsiveness following fractionated irradiation of
Gy per fraction [41]. A549 and MCF-7 cells with 80% confluency were exposed to
ifferent doses of � irradiation using Gamma Cell 220 irradiator (Atomic Energy ofanada Ltd.), at a dose rate of 3.5 Gy/min; the first set was irradiated with acuteose of 2 Gy or 10 Gy. The second set was exposed to 5 fraction of 2 Gy each over aeriod of five days, after irradiation cells were trypsinized and seeded, so that cellsill reach 80% confluency by next day.esearch 729 (2012) 61– 72
2.3. Clonogenic cell survival assay
After irradiation, clonogenic assay was done as described earlier [42]. Absoluteplating efficiency of A549 cells at 0 Gy (sham irradiated control) was 32.3 ± 2.4%.Absolute plating efficiency of MCF-7 cells at 0 Gy (sham irradiated control) was45.2 ± 2.4%.
2.4. Immunofluorescence staining
Immunofluorescence staining was done as described earlier [42]. The primaryantibodies used were mouse anti-pATM (S1981), rabbit anti-�-H2AX (S139), rabbitanti-pBRCA1 (Ser1524) and mouse anti-phospho-p53 (S15) (Cell Signaling). Sec-ondary antibodies used were Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor488 goat anti-mouse IgG (Molecular Probe, USA).
2.5. Image analysis using ImageJ software
The captured images were analyzed for relative quantification of phosphoryla-tion using ImageJ software [43]. DAPI stained nucleus of each cell was selected andthe same frame was used for relative quantification of phosphorylation using ImageJsoftware as described earlier [42]. All the foci were counted manually; at least 100cells per experiment were analyzed from three independent experiments.
2.6. Western Blotting
Immunoblotting was done as described previously [4]. Primary antibody usedwas mouse anti-phospho-p53 (S15) (Cell Signaling). The bound HRP labeled sec-ondary antibody was then detected using BM Chemiluminiscence Western BlottingKit (Roche Molecular Bio chemicals, Germany). The band intensity was quantified byImage J software. The total intensity of bands obtained in ponceau staining was usedas a protein loading and transfer control. All other band intensities were divided bythe intensity of their respective ponceau blot.
2.7. Microarray
For microarray analysis, 4 h after irradiation was selected as the time pointto monitor the early response of cells to irradiation and to identify differentiallyexpressed early genes that mediate cellular events such as DNA repair and apopto-sis. Two independent series of experiments were performed with Human GenomeU-133 Plus 2.0 microarrays containing 54,675 probe sets (Affymetrix, Santa Clara,CA). Microarray analysis was carried out at Oscimum Biosolution, Hyderabad on paidbasis. Total RNA was isolated from ∼3 × 106 A549 cells with RNeasy Mini Kits (Qia-gen) including digestion with RNase-free DNase I, and its quantity and integrity werechecked spectrophotometrically and by electrophoresis in 1% agarose gels. Materialsand methods for microarrays were from Affymetrix (Santa Clara); double-strandedcDNA was prepared with the GeneChip Expression 3′-Amplification One-Cycle cDNASynthesis Kit, cleaned using the Sample Cleanup Module, and biotinylated cRNAwas synthesized with GeneChip Expression 3′-Amplification Reagents for IVT Label-ing, cleaned on RNA Sample Cleanup columns, and fragmented at 94 ◦C for 35 minin Fragmentation Buffer. The biotinylated cRNA was hybridized first to a controlTest3 microarray to evaluate its quality and then to a Human Genome U-133Plus 2.0 microarrays containing 54,675 probe sets, using GeneChip Expression 3′-Amplification Reagents. Microarrays were stained with streptavidin–phycoerythrinconjugate and scanned in a Gene Array G2500A scanner (Agilent) and the expres-sion data on human Affymetrix chip was generated. Since the numbers of samplesin each treatment type were sparse, we used a bivariate simulation approach toidentify differentially expressed (DE) genes for the specified threshold fold change(FC). The DE candidates across each comparison were further evaluated for theirbiological relevance through Gene Ontology and Pathways studies. The statisticalanalysis was carried out using R package and the biological analysis was carried outusing GenowizTM software.
For comparative evaluation, an un-irradiated control cell was used. The expres-sion data on Affymetrix Human 133AB chip was obtained for each sample. Theprimary objective of the study was to obtain differentially expressed (DE) probesets (genes) across various comparisons. Also, the interest was to study the cluster-ing of genes and samples and the functional relevance of the differentially expressedgenes.
Upon identifying the DE genes, the next interest was to study the biologicalrelevance of selected marker genes through GO and Pathway analysis.
The data analysis process involves Robust Multichip Average (RMA) normaliza-tion, quality check analysis, differential expression analysis and the gene enrichmentanalysis.
The correlation between the expression values for samples was studied throughPearson’s coefficient. A pairwise correlation analysis was performed to generate
a correlation matrix indicating how the samples are related with each other. Thecoefficient tells about the similarity of expression trend between the two arrays.A coefficient value close to 1.0 indicates the linear expression between the arrays.The minimum correlation between the samples was 0.987, which was above theacceptable criterion of 0.8.
S. Ghosh, M. Krishna / Mutation Research 729 (2012) 61– 72 63
Table 1The sequences of gene specific primers and their annealing temperature (Tm) usedin RT-PCR experiments.
Gene Primer sequence Tm (◦C)
�-Actin F 5′-TGGAATCCTGTGGCATCCATGAAA-3′ 55R 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′
ATM F 5′-GGACAGTGGAGGCACAAAAT-3′ 57R 5′-GTGTCGAAGACAGCTGGTGA-3′
DNA-PK F 5′-TGCCAATCCAGCAGTCATTA-3′ 64R 5′-GGTCCATCAGGCACTTCACT-3′
Rad52 F 5′-AGTTTTGGGAATGCACTTGG-3′ 50R 5′-TCGGCAGCTGTTGTATCTTG-3′
MLH1 F 5′-GAGGTGAATTGGGACGAAGA-3′ 52R 5′-TCCAGGAGTTTGGAATGGAG-3′
CDKN1A (p21) F 5′-CAGCAGAGGAAGACCATGTG-3′ 59R 5′-GGCGTTTGGAGTGGTAGAAA-3′
GADD45� F 5′-TGCGAGAACGACATCAACAT-3′ 58R 5′-TCCCGGCAAAAACAAATAAG-3′
TLR3 F 5′-GCCTCTTCGTAACTTGACCA-3′ 57R 5′-AAGGATGTGGAGGTGAGACA-3′
FDXR F 5′-AGAGAACGGACATCACGAAG-3′ 57
F
2
cs
2
baawasm(
2
OpP
3
3
aTtto
3
G
a3Sfwrs
0Gy 2Gy 5X2Gy 10Gy
0
20
40
60
80
100
Perc
enta
ge S
urvi
val
Radiation Dose
**
Fig. 1. Clonogenic cell survival of A549 cells irradiated with 2 Gy, 5 fractions of 2 Gy
R 5′-GTCCTGGAGACCCAAGAAAT-3′, forward and R, reverse.
.8. Semiquantitative reverse transcriptional polymerase chain reaction (RT-PCR)
Total RNA from 1 × 106 cells was extracted 4 h after irradiation and RT-PCR wasarried out as described earlier [44]. Primer sequence and annealing temperature ofpecific primers are given in Table 1.
.9. Transfection with ShRNA
MLH1, Rad52 and Control shRNA plasmids were procured from Santa cruziotechnology (Cat No. sc-35943-SH, sc-37399-SH and sc-108060 respectively). In
six well tissue culture plate, A549 cells were grown to 50–70% confluency inntibiotic-free normal growth medium supplemented with FBS and transfectionas carried out as per supplier’s instruction. 48 h post-transfection, medium was
spirated and replaced with fresh medium containing puromycin (2 �g/ml) forelection of stably transfected cells. Semi-quantitative RT-PCR was performed toonitor MLH1 and Rad52 gene expression knockdown using gene specific primers
Table 1).
.10. Statistical analysis
The data were imported to excel work sheets, and graphs were made usingrigin version 5.0. One-way ANOVA with Tukey–Kramer Multiple Comparisons asost-test for P < 0.05 used to study the significant level. Data were insignificant at
> 0.05. Each point represented as mean ± SE (standard error of the mean).
. Results
.1. Clonogenic cell survival
To determine the sensitivity of cells to different doses of radi-tion, their ability to form colonies after irradiation was observed.here was 60 ± 2.5% survival of A549 cells that had been exposedo 2 Gy acute dose where as there was 48 ± 2.8% of cells survivedhat had been exposed to 5 fractions of 2 Gy radiations and 4 ± 0.5%f cells survived that had been exposed to 10 Gy acute dose (Fig. 1).
.2. Human genome U-133 Plus 2.0 microarray data analysis
Global expression analysis of 54,675 probe set using Humanenome U-133 Plus 2.0 microarray was performed in A549 cells.
On comparison of control vs 5X2 Gy (henceforth called fraction-ted dose), 461 genes were identified as differentially expressed;12 genes up-regulated (defined as a 2.0-fold or greater increase,upplementary data) and 149 down-regulated (defined as a 2.0-
old or greater decrease, Supplementary data). Up-regulated genesere associated with p53 signaling pathway, cytokine–cytokineeceptor interaction, hematopoietic cell lineage, Toll-like receptorignaling pathway, Jak-STAT signaling pathway, MAPK signaling
or 10 Gy �-radiation. Data represent means ± SE of three independent experiments;*P < 0.05, **P < 0.01 compared with 10 Gy acute dose.
pathway, cell-cycle check point, B cell receptor signaling pathway.Down-regulated genes were associated with TGF-beta signalingpathway and tyrosine metabolism.
In the second group (2 Gy vs 5X2 Gy), 234 genes were iden-tified as differentially expressed; 172 genes as up-regulated(Supplementary data) and 62 as down-regulated (Supplementarydata). Genes involved in cytokine–cytokine receptor interaction,toll-like receptor signaling pathway, hematopoietic cell lineage,Jak-STAT signaling pathway, MAPK signaling pathway were up-regulated. Gene involved in folate biosynthesis, glycine, serine andthreonine metabolisms were down-regulated.
In the 10 Gy vs fractionated group, 289 genes were iden-tified as differentially expressed; 211 genes as up-regulated(Supplementary data) and 78 as down-regulated (Supplementarydata). Genes involved in cytokine–cytokine receptor interaction,toll-like receptor signaling pathway, cell cycle, DNA replication,mismatch repair, homologous recombination were up-regulated.Genes involved in regulation of autophagy, base excision repair,folate biosynthesis were down-regulated.
Cluster analysis of the expression of these genes showed thatthe fractionated group differed most in gene expression comparedto the other three groups (control, 2 Gy or 10 Gy) (Supplementarydata, TCS2 means 2 Gy treatment group, TCS3 means fractionatedgroup, TCS4 means 10 Gy treatment group).
3.3. Validation of microarray data
In order to verify the microarray data, semi-quantitative RT-PCRwas performed for four genes from control and fractionated groupof A549 cells. These results corresponded very well with those forthe microarray data for all four genes, strongly supporting the reli-ability and rationale of our strategy (Fig. 2A and B).
3.4. Activation of repair related genes
When compared to the controls there was a significant up-regulation of ATM, DNA-PK, Rad52 and MLH1 genes in A549 cellsthat had been exposed to fractionated irradiation (Fig. 3A–D, Lane3). However, in A549 cells that had been exposed to 10 Gy acutedose there was marginal up-regulation of ATM, DNA-PK and Rad52
genes although it was significantly lower than the fractionatedgroup (Fig. 3A–D, Lane 4). In A549 cells which had been exposedto 2 Gy acute dose there was marginal up-regulation of Rad52 and
64 S. Ghosh, M. Krishna / Mutation R
2 83.03.23.43.6
rray
) Con trol 5X2Gy
1 01.21.41.61.82.02.22.42.62.8
Cha
nge
(Mic
roar
(A)
CDK1A(p21 ) GADD45 A TLR3 FDXR0.00.20.40.60.81.0
Fold
C
Genes
2 83.03.23.43.6
CR
)
Con trol 5X2 Gy
1.21.41.61.82.02.22.42.62.8
Cha
nge
(RT-
P C
(B)
CDK1A(p 21) GADD45A TLR3 FDXR0.00.20.40.60.81.01.2
Fold
C
GenesGenes
Fig. 2. Validation of microarray data by semi-quantitative RT-PCR. (A) Fold changesof genes as determined by microarray analysis. (B) Fold changes of genes as deter-mined by RT-PCR. Four genes from control and 5X2 Gy treatment group of A549 cellswia
Mt
3
abffotfD(
3A
riDlAci
ere selected and performed three sets of semi-quantitative RT-PCR as describedn Section 2. Four genes were CDKN1A (p21), GADD45�, Toll-like receptor 3 (TLR3)nd Ferredoxin reductase (FDXR).
LH1 genes but these were also significantly lower than the frac-ionated group (Fig. 3A–D, Lane 2).
.5. Efficient repair of DNA
Repair kinetics was determined by counting the �-H2AX focit 15 min and 4 h after irradiation. As expected maximum num-ers of foci were seen at 15 min in A549 cells exposed to 10 Gyollowed by cells exposed to fractionated irradiation and very fewoci were seen in cells exposed to 2 Gy acute dose. By 4 h the numberf foci in cells exposed to fractionated irradiation had been reducedo almost control but in the cells exposed to 10 Gy the number ofoci was still very high, indicating that there was efficient repair ofNA in A549 cells that had been exposed to fractionated irradiation
Fig. 4).
.6. Radiation induced foci of DNA damage response proteinsTM and BRCA1
ATM, an important DNA damage response protein to ionizingadiation and a part of irradiation induced foci (IRIF), is mainlynvolved in cell cycle arrest and repair. It gets activated after DNASB by phosphorylation at ser 1981 and activates many key cellu-
ar proteins that include H2AX, BRCA1, Chk1/2 and p53. PhosphoTM foci and intensity were looked at 4 h after irradiation. All theells either exposed to 10 Gy (23 ± 3 foci per cell) or fractionatedrradiation (25 ± 2.8 foci per cell) showed ATM foci but only 10%
esearch 729 (2012) 61– 72
of cells exposed to 2 Gy showed ATM foci and the number of fociper cell was 2 ± 0.08 (Fig. 5A). The intensity of phosphorylation ofATM was quantified by ImageJ software, was significantly higher inthe fractionated group as compared to any of the treatment groups(control, 2 Gy or 10 Gy) (Supplementary data, Fig. 5C).
BRCA1, which is phosphorylated by ATM, is a part of IRIF andshares many downstream substrates of ATM. 4 h after irradiation,A549 cells that had been exposed to 2 Gy acute dose did not showany BRCA1 foci where as 55% of cells that had been exposed to frac-tionated irradiation showed BRCA1 foci and the number of foci percell was 24 ± 4. 22% of cells that had been exposed to 10 Gy acuteshowed BRCA1 foci and the number of foci per cell was 15 ± 2.1(Fig. 5B). The intensity of phosphorylation of BRCA1 as determinedby ImageJ software was significantly higher in the fractionatedgroup as compared to any of the treatment groups (control, 2 Gyor 10 Gy) (Supplementary data, Fig. 5D).
3.7. Translocation of phospho-p53
18 h after irradiation, phospho-p53 was found to be translocatedinto the nucleus of A549 cells exposed to fractionated irradiation(Fig. 6A). The intensity of phosphorylation was significantly higherin the cells exposed to fractionated irradiation (Supplementarydata, Fig. 6C). Phosphorylation of p53 was further confirmed byWestern Blot (Fig. 6B, Lane 3).
3.8. Response of A549 and MCF-7 cells exposed to fractionatedirradiation
There was 48 ± 2.8% survival of A549 cells exposed to fraction-ated irradiation where as only 10 ± 1.2% of MCF-7 cells survivedexposed to fractionated irradiation (Fig. 7A). 4 h after irradiation,there was a significant up-regulation of ATM, DNA-PK, Rad52 andMLH1 genes in A549 cells but not in MCF-7 cells (Fig. 7B, Lane3). ATM and BRCA1 phosphorylation was found to be significantlyhigher 4 h after irradiation in A549 cells but not in MCF-7 cells(Fig. 7C). 18 h after irradiation, phospho-p53 was found to betranslocated into the nucleus of A549 cells but not in MCF-7 cells(Fig. 7C). There was efficient repair of DNA by 4 h in A549 cells asdetermined by counting �-H2AX but not in MCF-7 cells (Fig. 7D).
3.9. Role of Rad52 and MLH1
Since there was an intense activation of Rad52 and MLH1 inA549 cells exposed to fractionated irradiation, it was of interest tolook for their role in survival of A549 cells. MLH1 and Rad52 geneexpression knockdown was carried out in A549 cells using MLH1and Rad52 shRNA plasmids. Stably transfected cells were selectedand the transfection efficiency was found to be 85% for MLH1 geneand 80% for Rad52 gene (Fig. 8A).
There was 48 ± 2.8% survival of A549 cells exposed to fraction-ated irradiation. The survival of A549 cells that had been transfectedwith MLH1 shRNA plasmid and exposed to fractionated irradiationwas found to be 40 ± 1.8% where as the survival of A549 cells thathad been transfected with Rad52 shRNA plasmid and exposed tofractionated irradiation was found to be 23 ± 1.5% (Fig. 8B and C).The survival of cells transfected with control shRNA plasmid wasthe same as unirradiated control cells (data not shown).
4. Discussion
Chronic exposure of cells to IR has been reported to induce
an adaptive response that results in an enhanced tolerance to thecytotoxicity of subsequent doses of IR [1–4]. There could be manypossible mechanisms for the induction of the adaptive responsewhich would eventually determine the cell fate. In the present
S. Ghosh, M. Krishna / Mutation Research 729 (2012) 61– 72 65
Fig. 3. Gene expression of ATM, DNA-PK, Rad52 and MLH1 in A549 cells irradiated with different doses of �-radiation. Total RNA from A549 cells was isolated and reversetranscribed 4 h after irradiation. RT-PCR analysis of ATM, DNA-PK, Rad52 and MLH1 genes was carried out as described in Section 2. PCR products were resolved on 1.5%agarose gels containing ethidium bromide. �-Actin gene expression in each group was used as an internal control. Ratio of intensities of (A) ATM, (B) DNA-PK, (C) Rad52 and(D) MLH1 band to that of respective �-actin band as quantified from gel pictures are shown above each gel picture. Key: Lane 1, control unirradiated A549 cell line; Lane 2,2 60 ion; L 60
( SE of*
sm2rtftsagcipcr
Gy acute dose (single dose that was delivered in each fraction) of Co �-irradiatwhich is the sum total of all the fraction) 60Co �-irradiation. Data represent means ±P < 0.05, **P < 0.01.
tudy human lung adenocarcinoma A549 cells were found to beore radioresistant if 10 Gy dose was delivered as 5 fractions of
Gy dose. Although fractionation is supposed to play an importantole in inducing an adaptive response and deciding the fate of cells,he mechanism by which it does so can be many and innumerableactors could be responsible. DNA microarray analysis is a powerfulool for obtaining comprehensive information concerning expres-ion of thousands of genes in cancer cells [45] DNA microarraynalysis revealed variable expression of many genes. Up-regulatedenes in A549 cells exposed to fractionated irradiation were asso-iated with p53 signaling pathway, cytokine-cytokine receptor
nteraction, hematopoietic cell lineage, toll-like receptor signalingathway, Jak-STAT signaling pathway, MAPK signaling pathway,ell-cycle check point, B cell receptor signaling pathway, DNAeplication, mismatch repair, homologous recombination. Toll-likeane 3, 2 Gy of Co �-irradiation daily over a period of 5 days; Lane 4, 10 Gy acute three independent experiments; significantly different from unirradiated controls:
receptor signaling pathway is responsible for survival and prolif-eration of the cells [46]. The Janus kinase-signal transducer andactivator of transcription (JAK-STAT) pathway mediate signalingby cytokines, which control survival, proliferation and differen-tiation of several cell types [47]. MAPK signaling pathways areknown to be involved in various processes of growth, differen-tiation and cell death [48]. Most of the pathways which wereup-regulated in A549 cells exposed to fractionated irradiation inall comparisons were associated with the survival or repair ofthe A549 cells. This indicates that fractionated irradiation couldinduce up-regulation of survival/repair related pathways in cancer
cells.To the authors’ knowledge, this is the first report indi-cating the differential gene expression profiles of A549 cellsexposed to fractionated irradiation. Previous reports concerning

66 S. Ghosh, M. Krishna / Mutation Research 729 (2012) 61– 72
Fig. 4. Repair kinetics as determined by �-H2AX foci in different treatment group of A549 cells at 15 min and 4 h after irradiation. Cells were fixed 15 min and 4 h afterirradiation in 4% paraformaldehyde, permeabilized in 0.25% Triton X-100 and labeled with specific antibodies as described in Section 2. Each phospho-site antibody wasindirectly labeled with Molecular Probe 488 secondary antibody (green) and cells were mounted with ProLong Gold antifede with DAPI (blue). The encircled area roughlyrepresents cell nuclei as seen by DAPI staining. All images were captured using Carl Zeiss confocal microscope with the same exposure time. Representative image of threei lyzedi
ram[pt
ndependent experiments is shown here; at least 100 cells per experiment were anan this figure legend, the reader is referred to the web version of the article.)
adioresistant non-small-cell lung cancer and HeLa cellslso indicated that cell cycle signaling pathways, mis-
atch repair, homologous recombination were up-regulated49,50]. In the current study, some of these genes werereviously known to be associated with responsivenesso radiation, such as p21 and GADD45˛, but others were
from three independent experiments. (For interpretation of the references to color
novel. These novel genes can be expected to be involved inradioresistance, but the precise function of each gene remains
unclear; so further study is necessary to clarify the nature ofassociations.Microarray data had shown that p53 signaling pathway wasup-regulated in A549 cells that had been exposed to fractionated
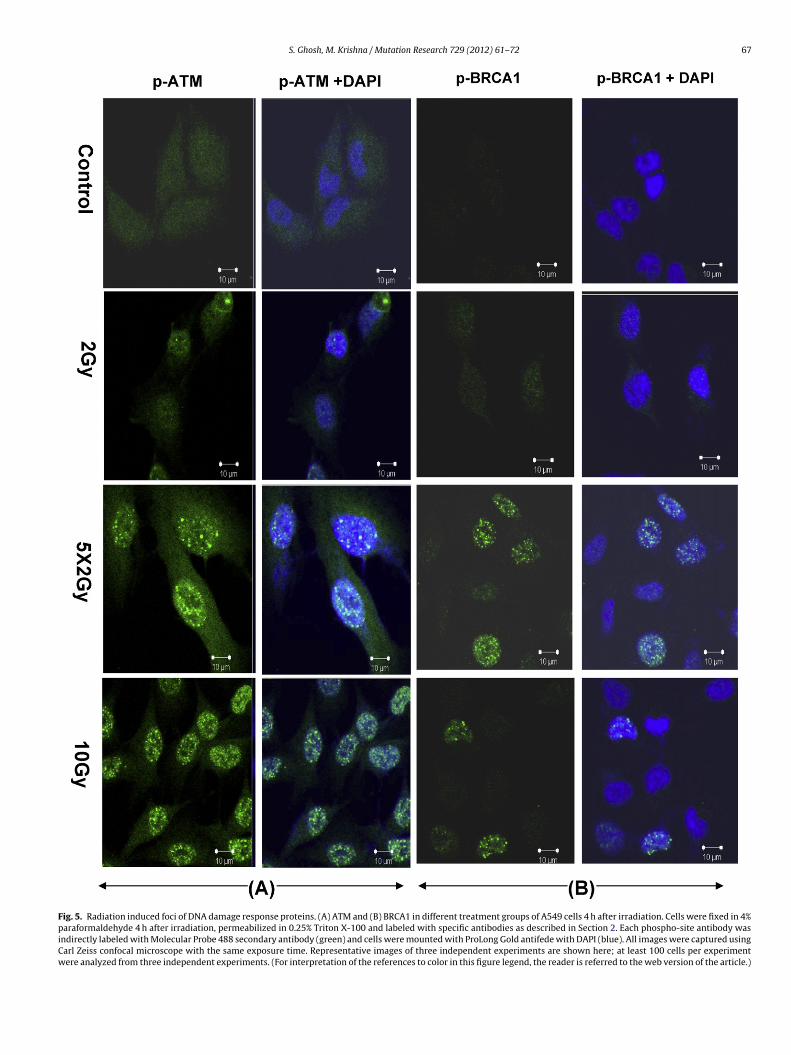
S. Ghosh, M. Krishna / Mutation Research 729 (2012) 61– 72 67
Fig. 5. Radiation induced foci of DNA damage response proteins. (A) ATM and (B) BRCA1 in different treatment groups of A549 cells 4 h after irradiation. Cells were fixed in 4%paraformaldehyde 4 h after irradiation, permeabilized in 0.25% Triton X-100 and labeled with specific antibodies as described in Section 2. Each phospho-site antibody wasindirectly labeled with Molecular Probe 488 secondary antibody (green) and cells were mounted with ProLong Gold antifede with DAPI (blue). All images were captured usingCarl Zeiss confocal microscope with the same exposure time. Representative images of three independent experiments are shown here; at least 100 cells per experimentwere analyzed from three independent experiments. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)

68 S. Ghosh, M. Krishna / Mutation Research 729 (2012) 61– 72
Fig. 6. Translocation of phospho-p53 into the nucleus of A549 cells 18 h after irradiation. (A) Representative image of three independent experiments showing phosphorylationand translocation of p53 into nucleus of A549 cells. Each phospho-site antibody was indirectly labeled with Molecular Probe 488 secondary antibody (green) and cells weremounted with ProLong Gold antifede with DAPI (blue). All images were captured using Carl Zeiss confocal microscope with the same exposure time. At least 100 cells perexperiment were analyzed from three independent experiments. (B) Representative immunoblot image of three independent experiments depicting phosphorylation of p53in different treatment group. Graph represents phospho-p53 band intensities (divided with the respective loading control intensities and normalized). Key: Lane 1, controlunirradiated A549 cell line; Lane 2, 2 Gy acute dose (single dose that was delivered in each fraction) of 60Co �-irradiation; Lane 3, 2 Gy of 60Co �-irradiation daily over a periodof 5 days; Lane 4, 10 Gy acute (which is the sum total of all the fraction) 60Co �-irradiation. Data represent means ± SE of three independent experiments. (For interpretationof the references to color in this figure legend, the reader is referred to the web version of the article.)

S. Ghosh, M. Krishna / Mutation Research 729 (2012) 61– 72 69
Fig. 7. Response of A549 and MCF-7 cells exposed to fractionated irradiation. (A) Clonogenic cell survival of A549 and MCF-7 cells exposed to fractionated irradiation.Data represent means ± SE of three independent experiments; **P < 0.01 compared with MCF-7. (B) Representative image of three independent experiments showing geneexpression of ATM, DNA-PK, Rad52 and MLH1 in A549 and MCF-7 cells exposed to fractionated irradiation. �-Actin gene expression in each group was used as an internalcontrol. (C) Phosphorylation of ATM, BRCA1 and p53 in A549 and MCF-7 cells exposed to fractionated irradiation. (D) Repair kinetics as determined by �-H2AX foci in A549and MCF-7 cells exposed to fractionated irradiation at 15 min and 4 h after irradiation. Each phospho-site antibody was indirectly labeled with Molecular Probe 488 secondaryantibody (green) and cells were mounted with ProLong Gold antifede with DAPI (blue). The encircled area roughly represents cell nuclei as seen by DAPI staining. All theimages were taken using a 100× objective lens except the images of p53 phosphorylation. The images of p53 phosphorylation were taken using 40× objective lens for A549cells and MCF-7 cells. All images were captured using Carl Zeiss confocal microscope with the same exposure time. Representative images of three independent experimentsare shown here; at least 100 cells per experiment were analyzed from three independent experiments. (For interpretation of the references to color in this figure legend, thereader is referred to the web version of the article.)

70 S. Ghosh, M. Krishna / Mutation R
Fig. 8. Effect of inhibition of Rad52 and MLH1 on the growth and radioresistance ofA549 cells which had been exposed to 5 fractions of 2 Gy radiation. A549 cells weretransfected with either MLH1 or Rad52 ShRNA as described in Section 2. (A) Trans-fection efficiency as determined by semi-quantitative RT-PCR. Gene expression ofRad52 and MLH1 in A549 cells transfected with Control ShRNA, Rad52 ShRNA orMLH1 ShRNA. Total RNA from A549 cells was isolated and reverse transcribed 7days after transfection. RT-PCR analysis of Rad52 and MLH1 genes were carried outas described in Section 2. PCR products were resolved on 1.5% agarose gels containingethidium bromide. �-Actin gene expression in each group was used as an internalcontrol. Ratio of intensities of Rad52 or MLH1 band to that of respective �-actin bandas quantified from gel pictures are shown above each gel picture. Key: Rad52, Geneexpression of Rad52 to that of �-actin in A549 cells transfected with control ShRNA;−ve Rad52, gene expression of Rad52 to that of �-actin in A549 cells transfectedwith Rad52 ShRNA; MLH1, gene expression of MHL1 to that of �-actin in A549 cellstransfected with control ShRNA; −ve MLH1, gene expression of MLH1 to that of �-actin in A549 cells transfected with MLH1 ShRNA. (B) Clonogenic survival assay ofA549 cells. Key: control, control unirradiated A549 cells; 5X2 Gy, A549 cells exposedto 2 Gy of 60Co �-irradiation daily over a period of 5 days; 5X2 Gy + M, A549 cellstransfected with MLH1 ShRNA and exposed to 2 Gy of 60Co �-irradiation daily over aperiod of 5 days; 5X2 Gy + R, A549 cells transfected with Rad52 ShRNA and exposedto 2 Gy of 60Co �-irradiation daily over a period of 5 days. (C) Representative imageof three independent experiments. Data represent means ± SE of three independentexperiments; *P < 0.05, **P < 0.01 compared with 5X2 Gy treatment group.
esearch 729 (2012) 61– 72
irradiation; it was our interest to look at the phosphorylation ofp53 and its translocation to the nucleus of A549 cells. Lung ade-nocarcinoma A549 cells typically express wild-type p53 protein[51] and would be expected to be sensitive to the DNA-damagingagents used for cancer therapy; however, these cells are radiore-sistant if the radiation dose is delivered as fractionated irradiation.p53 classically considered to be a tumor suppressor protein, but inthis study the results are contradictory. Even though the p53 levelsare up-regulated following fractionated doses (Fig. 6A and B), theapoptotic function of the p53 appears to be compromised. p53 isconsidered to be a classic “gatekeeper” of cellular fate [52,53]. p53is activated in response to genotoxic stress such as radiation. Andonce activated, it initiates cell cycle arrest, senescence or apopto-sis via pathways involving the transactivation of p53 target genes[52,53]. There was no change in the expression of pro apoptotictargets of p53 such as Bax, Perp, Puma and Noxa in the cells thathad been exposed to fractionated irradiation as compared to con-trol cells (Microarray data in Supplementary Files). However, therewas higher expression of cell cycle target genes of p53 such as p21and GADD45� in the cells that had been exposed to fractionatedirradiation as compared to control cells (Fig. 2).
Since DNA repair pathways were up-regulated in A549 cells thathad been exposed to fractionated irradiation, it was of interest tolook at the activation of genes and proteins involved in DNA repairpathways. Our results indicate that there was intense activation ofrepair genes and protein in A549 cells exposed to fractionated irra-diation (Figs. 3 and 5). Our results on DNA-PK are consistent withearlier works where authors have also shown the role of DNA-PK inthe radioresistance of lung carcinoma cells [54,55]. However, theseauthors have looked at the response only after single dose of irra-diation and it is logical to expect these pathways to get activated.In this study we reiterate the fact that DNA-PK is also involved inradioresistance if the cells are subjected to fractionated irradiationbut the reason for the development of radioresistance still remainsan enigma.
ATM and BRCA1 have been regarded as primary regulators ofhomologous recombination repair (HRR) [56]. In this study therewas intense activation of ATM gene and ATM foci were seen in allthe cells exposed to fractionated irradiation and most of the cellsshowed BRCA1 foci (Figs. 3 and 5) indicating the fact that HRR path-way was activated in A549 cells and therefore, these proteins couldbe involved in HRR which can take advantage of the other strandthat may be intact. ATM and BRCA1 reside in macromolecular com-plex and form foci after irradiation. Our results are in agreementwith earlier works where authors have also shown the role ATMin radioresistance of cancer cells [57,58]. However, these authorshave studied using single dose of irradiation.
By 4 h there was an efficient repair of DNA in A549 cells exposedto fractionated irradiation but not in cells that had been exposed to10 Gy acute dose as determined by the disappearance of �-H2AXfoci (Fig. 4). It has been established recently that H2AX at the DNADSB sites are immediately phosphorylated upon irradiation, andthe phosphorylated H2AX (�-H2AX) can be visualized in situ byimmunostaining with a �-H2AX specific antibody [59,60]. �-H2AXinduction can be measured quantitatively at physiological doses,and the numbers of residual �-H2AX foci can be used to estimatethe kinetics of DSB rejoining. This has become the gold standard forthe detection of DSB [60,61].
A clear cause and effect relationship was established betweenDNA damage signaling, gene expression and efficient DNA repairand was evident in the form of cell survival in A549 cells that hadbeen exposed to fractionated irradiation.
It has been reported that A549 cells are relatively moreradioresistant than MCF-7 cells if radiation dose was delivered asfractionated regimen [62]. But the mechanism of such response hasnot been studied. To the authors’ knowledge this is the first report

tion R
epia
mfaAsAawR2gsiic
5
mdDuiatr
C
A
KL
A
t
R
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
S. Ghosh, M. Krishna / Muta
xplaining the mechanism of such response as activation of repairathway, efficient DNA repair and translocation of phospho-p53
nto the nucleus of A549 cells exposed to fractionated irradiationnd not in MCF-7 cells exposed to fractionated irradiation (Fig. 7).
Numerous reports have implicated DNA repair genes as beingainly responsible for the development of radioresistance due to
ractionated irradiation [54,55,57,58]. To investigate what factorsre involved in fractionated irradiation induced radioresistance in549 cells, the cells were transfected with either Rad52 or MLH1hRNA plasmid and exposed to 5 fractions of 2 Gy. The survival of549 cells that had been transfected with MLH1 shRNA plasmidnd exposed to fractionated irradiation was found to be 40 ± 1.8%here as the survival of A549 cells which had been transfected withad52 shRNA plasmid and exposed to fractionated irradiation was3 ± 1.5% (Fig. 8). The above results demonstrate that the Rad52ene is one of the factors responsible for the increased radiore-istance in A549 cells if 10 Gy dose was delivered as fractionatedrradiation. These observations strongly argue for the functionalmportance of Rad52 in DSB repair in lung adenocarcinoma A549ells.
. Conclusions
In summary, our results indicated that A549 cells were relativelyore radioresistant if these cells were exposed to fractionated irra-
iation. The reason for radioresistance could be the activation ofNA repair pathways and Rad52 gene is an important factor mod-lating radioresistance in A549 cells. This study contributes basic
nformation about some of the genes involved in radioresistancend may eventually contribute to clinical investigations of predic-ion markers and prognostic factors, as well as the development ofadiosensitive molecules for therapeutic use.
onflict of interest statement
The authors declare that there are no conflicts of interest.
cknowledgments
Authors would like to thank Mr. Manjoor Ali and Mr. Pareshhadilkar, RB&HSD, BARC for their help in Confocal Microscopy andab work respectively.
ppendix A. Supplementary data
Supplementary data associated with this article can be found, inhe online version, at doi:10.1016/j.mrfmmm.2011.09.007.
eferences
[1] J. Russell, T.E. Wheldon, P. Stanton, A radioresistant variant derived from ahuman neuroblastoma cell line is less prone to radiation-induced apoptosis,Cancer Res. 55 (1995) 4915–4921.
[2] W.K. Dahlberg, E.I. Azzam, Y. Yu, J.B. Little, Response of human tumor cells ofvarying radiosensitivity and radiocurability to fractionated irradiation, CancerRes. 59 (1999) 5365–5369.
[3] A.G. Pearce, T.M. Segura, A.C. Rintala, N.D. Rintala-Maki, H. Lee, The generationand characterization of a radiation-resistant model system to study radioresis-tance in human breast cancer cells, Radiat. Res. 156 (2001) 739–750.
[4] A.K. Mitra, M. Krishna, Fractionated and acute irradiation induced signaling ina murine tumor, J. Cell. Biochem. 101 (2007) 745–752.
[5] R.R. Weichselbaum, M.A. Beckett, W. Dahlberg, A. Dritschilo, Heterogeneity ofradiation response of a parent human epidermoid carcinoma cell line and fourclones, Int. J. Radiat. Oncol. Biol. Phys. 14 (1988) 907–912.
[6] M.C. Joiner, Evidence for induced radioresistance from survival and other end
points: an introduction, Radiat. Res. 138 (1994) S5–S8.[7] S. Henness, M.W. Davey, R.M. Harvie, R.A. Davey, Fractionated irradiation of H69small-cell lung cancer cells causes stable radiation and drug resistance withincreased MRP1, MRP2, and topoisomerase IIalpha expression, Int. J. Radiat.Oncol. Biol. Phys. 54 (2002) 895–902.
[
[
esearch 729 (2012) 61– 72 71
[8] K. Valerie, L.F. Povirk, Regulation and mechanisms of mammalian double-strand break repair, Oncogene 22 (2003) 5792–5812.
[9] R. Kanaar, J.H. Hoeijmakers, D.C. van Gent, Molecular mechanisms of DNA dou-ble strand break repair, Trends Cell Biol. 8 (1998) 483–489.
10] J.E. Haber, Partners and pathwaysrepairing a double-strand break, TrendsGenet. 16 (2000) 259–264.
11] R.D. Johnson, M. Jasin, Double-strand-break-induced homologous recombina-tion in mammalian cells, Biochem. Soc. Trans. 29 (2001) 196–201.
12] T. Samuel, H.O. Weber, J.O. Funk, Linking DNA damage to cell cycle checkpoints,Cell Cycle 1 (2002) 162–168.
13] S. Banin, L. Moyal, S. Shieh, Y. Taya, C.W. Anderson, L. Chessa, N.I. Smorodinsky,C. Prives, Y. Reiss, Y. Shiloh, Y. Ziv, Enhanced phosphorylation of p53 by ATMin response to DNA damage, Science 281 (1998) 1674–1677.
14] C.E. Canman, D.S. Lim, K.A. Cimprich, Y. Taya, K. Tamai, K. Sakaguchi, E. Appella,M.B. Kastan, J.D. Siliciano, Activation of the ATM kinase by ionizing radiationand phosphorylation of p53, Science 281 (1998) 1677–1679.
15] C.H. Westphal, Cell-cycle signaling: Atm displays its many talents, Curr. Biol. 7(1997) R789–R792.
16] M.F. Hoekstra, Responses to DNA damage and regulation of cell cycle check-points by the ATM protein kinase family, Curr. Opin. Genet. Dev. 7 (1997)170–175.
17] C.J. Bakkenist, M.B. Kastan, DNA damage activates ATM through intermolecularautophosphorylation and dimer dissociation, Nature 421 (2003) 499–506.
18] D. Cortez, Y. Wang, J. Qin, S.J. Elledge, Requirement of ATM-dependent phos-phorylation of brca1 in the DNA damage response to double-strand breaks,Science 286 (1999) 1162–1166.
19] X. Xu, Z. Weaver, S.P. Linke, C. Li, J. Gotay, X.W. Wang, C.C. Harris, T. Ried, C.X.Deng, Centrosome amplification and a defective G2-M cell cycle checkpointinduce genetic instability in BRCA1 exon 11 isoform-deficient cells, Mol. Cell 3(1999) 389–395.
20] M.E. Moynahan, J.W. Chiu, B.H. Koller, M. Jasin, Brca1 controls homology-directed DNA repair, Mol. Cell 4 (1999) 511–518.
21] E. Friedberg, W. Siede, A.J. Cooper, The Molecular and Cellular Biology of theYeast Saccharomyces, Cold Spring Harbor Laboratory Press, Plainview, NY,1991.
22] J. Game, Radiation-sensitive Mutants and Repair in Yeast, Springer-Verlag, NewYork, 1983.
23] M. Ajimura, S.H. Leem, H. Ogawa, Identification of new genes requiredfor meiotic recombination in Saccharomyces cerevisiae, Genetics 133 (1993)51–66.
24] E.L. Ivanov, V.G. Korolev, F. Fabre, XRS2, a DNA repair gene of Saccharomycescerevisiae, is needed for meiotic recombination, Genetics 132 (1992) 651–664.
25] T. Petes, R.E. Malone, I.S. Symington, Recombination in Yeast, Cold Spring Har-bor Laboratory Press, New York, 1991.
26] M.A. Resnick, Genetic control of radiation sensitivity in Saccharomyces cere-visiae, Genetics 62 (1969) 519–531.
27] K.L. Boundy-Mills, D.M. Livingston, A Saccharomyces cerevisiae RAD52 alleleexpressing a C-terminal truncation protein: activities and intragenic comple-mentation of missense mutations, Genetics 133 (1993) 39–49.
28] A. Shinohara, H. Ogawa, T. Ogawa, Rad51 protein involved in repair and recom-bination in S. cerevisiae is a RecA-like protein, Cell 69 (1992) 457–470.
29] O.Y. Bezzubova, H. Schmidt, K. Ostermann, W.D. Heyer, J.M. Buerstedde,Identification of a chicken RAD52 homologue suggests conservation of theRAD52 recombination pathway throughout the evolution of higher eukaryotes,Nucleic Acids Res. 21 (1993) 5945–5949.
30] D.F. Muris, K. Vreeken, A.M. Carr, B.C. Broughton, A.R. Lehmann, P.H. Lohman, A.Pastink, Cloning the RAD51 homologue of Schizosaccharomyces pombe, NucleicAcids Res. 21 (1993) 4586–4591.
31] C. Bendixen, I. Sunjevaric, R. Bauchwitz, R. Rothstein, Identification of a mousehomologue of the Saccharomyces cerevisiae recombination and repair gene,RAD52, Genomics 23 (1994) 300–303.
32] Z. Shen, K. Denison, R. Lobb, J.M. Gatewood, D.J. Chen, The human and mousehomologs of the yeast RAD52 gene: cDNA cloning, sequence analysis, assign-ment to human chromosome 12p12.2-p13, and mRNA expression in mousetissues, Genomics 25 (1995) 199–206.
33] G.T Milne, D.T. Weaver, Dominant negative alleles of RAD52 reveal a DNArepair/recombination complex including Rad51 and Rad52, Genes Dev. 7 (1993)1755–1765.
34] J.W. Donovan, G.T. Milne, D.T. Weaver, Homotypic and heterotypic proteinassociations control Rad51 function in double-strand break repair, Genes Dev.8 (1994) 2552–2562.
35] Z. Shen, K.G. Cloud, D.J. Chen, M.S. Park, Specific interactions between thehuman RAD51 and RAD52 proteins, J. Biol. Chem. 271 (1996) 148–152.
36] M.S. Park, Expression of human RAD52 confers resistance to ionizing radiationin mammalian cells, J. Biol. Chem. 270 (1995) 15467–15470.
37] Y. Habraken, O. Jolois, J. Piette, Differential involvement of thehMRE11/hRAD50/NBS1 complex, BRCA1 and MLH1 in NF-kappaB activationby camptothecin and X-ray, Oncogene 22 (2003) 6090–6099.
38] L.A. Bannister, B.C. Waldman, A.S. Waldman, Modulation of error-prone double-strand break repair in mammalian chromosomes by DNA mismatch repairprotein Mlh1, DNA Repair (Amst) 3 (2004) 465–474.
39] Q. Wang, O.N. Ponomareva, M. Lasarev, M.S. Turker, High frequency inductionof mitotic recombination by ionizing radiation in Mlh1 null mouse cells, Mutat.Res. 594 (2006) 189–198.
40] Y. Zhang, L.H. Rohde, K. Emami, D. Hammond, R. Casey, S.K. Mehta, A.S. Jeevara-jan, D.L. Pierson, H. Wu, Suppressed expression of non-DSB repair genes inhibits

7 tion R
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[61] O. Fernandez-Capetillo, A. Lee, M. Nussenzweig, A. Nussenzweig, H2AX: thehistone guardian of the genome, DNA Repair (Amst) 3 (2004) 959–967.
[62] J.H. Matthews, B.E. Meeker, J.D. Chapman, Response of human tumor cell lines
2 S. Ghosh, M. Krishna / Muta
gamma-radiation-induced cytogenetic repair and cell cycle arrest, DNA Repair(Amst) 7 (2008) 1835–1845.
41] A. Safwat, S.M. Bentzen, I. Turesson, J.H. Hendry, Deterministic rather thanstochastic factors explain most of the variation in the expression of skin telang-iectasia after radiotherapy, Int. J. Radiat. Oncol. Biol. Phys. 52 (2002) 198–204.
42] S. Ghosh, N.N. Bhat, S. Santra, R.G. Thomas, S.K. Gupta, R.K. Choudhury, M.Krishna, Low energy proton beam induces efficient cell killing in A549 lungadenocarcinoma cells, Cancer Invest. 28 (2010) 615–622.
43] W.S. Rasband, J.U.S. Image, National Institutes of Health, Bethesda, USA.http://rsb.info.nih.gov/ij/, 1997–2006.
44] S. Ghosh, D.K. Maurya, M. Krishna, Role of iNOS in bystander signaling betweenmacrophages and lymphoma cells, Int. J. Radiat. Oncol. Biol. Phys. 72 (2008)1567–1574.
45] S.M. Bentzen, Preventing or reducing late side effects of radiation therapy:radiobiology meets molecular pathology, Nat. Rev. Cancer 6 (2006) 702–713.
46] F.J. Barrat, R.L. Coffman, Development of TLR inhibitors for the treatment ofautoimmune diseases, Immunol. Rev. 223 (2008) 271–283.
47] S.N. Constantinescu, M. Girardot, C. Pecquet, Mining for JAK-STAT mutations incancer, Trends Biochem. Sci. 33 (2008) 122–131.
48] M. Krishna, H. Narang, The complexity of mitogen-activated protein kinases(MAPKs) made simple, Cell. Mol. Life Sci. 65 (2008) 3525–3544.
49] Y.S. Lee, J.H. Oh, S. Yoon, M.S. Kwon, C.W. Song, K.H. Kim, M.J. Cho, M.L. Mol-lah, Y.J. Je, Y.D. Kim, C.D. Kim, J.H. Lee, Differential gene expression profilesof radioresistant non-small-cell lung cancer cell lines established by fraction-ated irradiation: tumor protein p53-inducible protein 3 confers sensitivity toionizing radiation, Int. J. Radiat. Oncol. Biol. Phys. 77 (2010) 858–866.
50] Y. Qing, X.Q. Yang, Z.Y. Zhong, X. Lei, J.Y. Xie, M.X. Li, D.B. Xiang, Z.P. Li, Z.Z. Yang,G. Wang, D. Wang, Microarray analysis of DNA damage repair gene expressionprofiles in cervical cancer cells radioresistant to 252Cf neutron and X-rays, BMC
Cancer 10 (2010) 71.51] T.A. Lehman, W.P. Bennett, R.A. Metcalf, J.A. Welsh, J. Ecker, R.V. Modali, S.Ullrich, J.W. Romano, E. Appella, J.R. Testa, et al., p53 mutations, ras mutations,and p53-heat shock 70 protein complexes in human lung carcinoma cell lines,Cancer Res. 51 (1991) 4090–4096.
esearch 729 (2012) 61– 72
52] P. May, E. May, Twenty years of p53 research: structural and functional aspectsof the p53 protein, Oncogene 18 (1999) 7621–7636.
53] B. Vogelstein, D. Lane, A.J. Levine, Surfing the p53 network, Nature 408 (2000)307–310.
54] S.P. Lees-Miller, R. Godbout, D.W. Chan, M. Weinfeld, R.S. Day, G.M. 3rd,J. Barron, Allalunis-Turner, Absence of p350 subunit of DNA-activated pro-tein kinase from a radiosensitive human cell line, Science 267 (1995)1183–1185.
55] F. Sirzen, A. Nilsson, B. Zhivotovsky, R. Lewensohn, DNA-dependent proteinkinase content and activity in lung carcinoma cell lines: correlation with intrin-sic radiosensitivity, Eur. J. Cancer 35 (1999) 111–116.
56] S.E. Golding, E. Rosenberg, A. Khalil, A. McEwen, M. Holmes, S. Neill, L.F. Povirk,K. Valerie, Double strand break repair by homologous recombination is regu-lated by cell cycle-independent signaling via ATM in human glioma cells, J. Biol.Chem. 279 (2004) 15402–15410.
57] S. Tribius, A. Pidel, D. Casper, ATM protein expression correlates with radiore-sistance in primary glioblastoma cells in culture, Int. J. Radiat. Oncol. Biol. Phys.50 (2001) 511–523.
58] S.J. Collis, M.J. Swartz, W.G. Nelson, T.L. DeWeese, Enhanced radiation andchemotherapy-mediated cell killing of human cancer cells by small inhibitoryRNA silencing of DNA repair factors, Cancer Res. 63 (2003) 1550–1554.
59] E.P. Rogakou, D.R. Pilch, A.H. Orr, V.S. Ivanova, W.M. Bonner, DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139, J. Biol.Chem. 273 (1998) 5858–5868.
60] P.L. Olive, J.P. Banath, Phosphorylation of histone H2AX as a measure ofradiosensitivity, Int. J. Radiat. Oncol. Biol. Phys. 58 (2004) 331–335.
in vitro to fractionated irradiation, Int. J. Radiat. Oncol. Biol. Phys. 16 (1989)133–138.


















