
REPÚBLICA BOLIVARIANA DE VENEZUELA ... -...
71
REPÚBLICA BOLIVARIANA DE VENEZUELA UNIVERSIDAD DEL ZULIA FACULTAD DE ODONTOLOGÍA DIVISIÓN DE ESTUDIOS PARA GRADUADOS POSTGRADO DE PERIODONCIA HELICOBACTER PYLORI, Y SU RELACION CON LA ENFERMEDAD PERIODONTAL. REVISION DE LA LITERATURA. Trabajo Especial de Grado presentado ante la División de Estudios para Graduados de la Facultad de Odontología, Universidad del Zulia, Maracaibo, Venezuela, para optar al Grado de Especialista en Periodoncia. AUTOR: Od Milagros Morán Boscán C.I. 13.529.404 TUTOR ACADÉMICO: Esp. Luis Ernesto López Maracaibo, Marzo 2007
Transcript of REPÚBLICA BOLIVARIANA DE VENEZUELA ... -...

REPÚBLICA BOLIVARIANA DE VENEZUELA
UNIVERSIDAD DEL ZULIA FACULTAD DE ODONTOLOGÍA
DIVISIÓN DE ESTUDIOS PARA GRADUADOS POSTGRADO DE PERIODONCIA
HELICOBACTER PYLORI, Y SU RELACION CON LA ENFERMEDAD PERIODONTAL. REVISION DE LA LITERATURA.
Trabajo Especial de Grado presentado ante la División de Estudios para Graduados de la Facultad de Odontología, Universidad del Zulia, Maracaibo,
Venezuela, para optar al Grado de Especialista en Periodoncia.
AUTOR: Od Milagros Morán Boscán
C.I. 13.529.404
TUTOR ACADÉMICO: Esp. Luis Ernesto López
Maracaibo, Marzo 2007

MORÁN BOSCÁN MILAGROS DEL CARMEN. “HELICOBACTER PYLORI, Y SU RELACION CON LA ENFERMEDAD PERIODONTAL” TRABAJO ESPECIAL DE GRADO PARA OPTAR AL TITULO DE ESPECIALISTA EN PERIODONCIA. UNIVERSIDAD DEL ZULIA. FACULTAD DE ODONTOLOGIA. DIVISIÓN DE ESTUDIOS PARA GRADUADOS PROGRAMA DE POSTGRADO DE PERIODONCIA. NIVEL: ESPECIALIDAD. MARACAIBO, VENEZUELA, 2007. p 70.
“HELICOBACTER PYLORI, Y SU RELACION CON LA ENFERMEDAD PERIODONTAL”
AUTOR: MORÁN BOSCÁN, MILAGROS DEL CARMEN
__________________
C.I. 13.529.404.
TUTOR ACADEMICO: ESP. LUIS ERNESTO LÓPEZ
___________________
C.I. 11.451.564
Urbanización los Sauces Calle 88 # 71 A-55 Maracaibo, Edo. Zulia
Tlf. (0416)4689366
Email: [email protected]

VEREDICTO Quienes suscribimos, miembros de jurado designado por el Comité Académico del Programa de Periodoncia de la División de Estudios para Graduados de la Facultad de Odontología de la Universidad del Zulia, para evaluar el Trabajo
Especial de Grado titulado: “HELICOBACTER PYLORI, Y SU RELACION CON LA ENFERMEDAD PERIODONTAL” REVISIÓN BIBLIOGRAFICA; presentado por la
Od. Milagros del Carmen Morán Boscán, Cédula de identidad N° 13.529.404, para
optar al Título de Especialista en Periodoncia, después de haber leído y estudiado
el referido trabajo y evaluada la defensa por su autor, consideran que el mismo
reúne los requisitos señalados por las Normas y reglamentos de la División de Estudios para Graduados de la Facultad de Odontología y además constituye
desde el punto de vista Académico, Científico y Profesional un aporte a la
transformación del conocimiento requerido por la mencionada Especialidad. Por lo
tanto el veredicto del jurado es “APROBADO”. Para que conste, se firma en
Maracaibo a los 29 días del mes de Marzo del año dos mil siete.
JURADO: _________________________________FIRMA:________________
Nombres y Apellidos
CI.
JURADO: _________________________________ FIRMA: _______________
Nombres y Apellidos
CI.
JURADO: _________________________________ FIRMA: _______________
Nombres y Apellidos
CI.

AGRADECIMIENTO
A Dios todopoderoso por darme salud, sabiduría y
mucha fortaleza.
A mis dos grandes amores Javier y José Javier por
estar siempre a mí lado brindándome su amor y apoyo
en todo momento.
A mis padres por darme la educación y principios
necesarios para lograr las metas propuestas.
A mi madre y hermanas por ofrecerme su apoyo
absoluto y por ayudarme a cuidar a mi más preciado
tesoro.
A mis suegros y cuñada por su apoyo incondicional,
palabras de aliento, paciencia y comprensión.
A mis amigas Marisol, Soraya, Noely, Linda y Heliana
por haber estado ahí cuando más las necesite
brindándome su amistad, apoyo, y por haber sido
participes de este gran sueño.
A mis profesores, y especialmente a mi comité asesor
por haber sido la mejor guía en este proceso de
aprendizaje, a todos… mil gracias

Morán Boscán, Milagros del Carmen. HELICOBACTER PYLORI, Y SU RELACION CON LA ENFERMEDAD PERIODONTAL Trabajo Especial de Grado para Optar al Titulo de Especialista en Periodoncia. Universidad del Zulia. Facultad de Odontología. División de Estudios para Graduados Programa de Postgrado de Periodoncia. Nivel: Especialidad. Maracaibo, Estado Zulia, Venezuela, 2007. p 73.
RESUMEN
La infección por Helicobacter pylori (Hp) es una de la más frecuentes a nivel mundial, datos epidemiológicos han demostrado que a nivel mundial la prevalencia es aproximadamente de un 50 a 60%; y se ha indicado que entre el 44 y 57% de los adultos de países en vías de desarrollo padecen de Periodontitis moderada, y cerca del 10% de Periodontitis avanzada. Se ha referido que Hp puede estar presente en la cavidad bucal como consecuencia del reflujo gastroesofágico y quizás se encuentre como una parte de la microbiota transitoria, más que como un residente normal. Igualmente se ha reportado que en algunos pacientes la colonización bucal de la bacteria, podría constituir un factor de riesgo para la reinfección gastrointestinal posterior a la terapia antibiótica. Así mismo, si la mucosa gástrica es recolonizada por la bacteria, este puede hacerse inaccesible a la terapia antibiótica; razón por la cual se indica el control de placa en pacientes con enfermedades del aparato digestivo causadas por Hp previo al tratamiento con la terapia antibiótica.
Palabras claves: Helicobacter pylori, Enfermedad Periodontal, Patógenos periodontales, Placa bacteriana.

Morán Boscán, Milagros del Carmen. HELICOBACTER PYLORI, AND HIS RELATION WITH THE ILLNESS PERIODONTAL. I Work Special of Degree to opt al the Specialist's title in Periodoncy. University of Zulia. Odontology Faculty. División of Graduated Studies for. Postgraduate of Periodontology. Maracaibo. Zulia State. Year 2007. Level: Especialidad Maracaibo, Zulia State Venezuela, 2007. p 73.
Abstract
The infection for Helicobacter pylori (Hp) is one of the most frequent on a global scale, epidemiologic information has demonstrated that worldwide the prevalence is approximately 50 to 57%; and moderate has indicated herself that between 44 and 57% of the developing countries suffer of Periodontitis, and next to 10% of advanced Periodontitis. One has recounted that the Hp can be present in the mouth cavity as consequence of the ebb gastroesofágico and perhaps be as part of the transitory microbiota more than as a normal resident. Equally it has been brought that in some patients the mouth colonization of the bacterium might constitute a factor of risk for the gastrointestinal reinfection later to the antibiotic therapy. Likewise, if the mucous gastric one is re-colonized by the bacterium, this one can become inaccessible to the antibiotic therapy; reason by which the control of badge is indicated in patients with illnesses of the digestive tract caused for Helicobacter pylori before to the treatment with the antibiotic therapy.
Key Words: Helicobacter pylori, periodontal disease, pathogenic periodontales, dental plaque.

III.-INTRODUCCION
La infección por Helicobacter pylori (Hp) es la enfermedad bacteriana
gastrointestinal más frecuente del mundo, la cual a partir de su descubrimiento, esta
implicada en diferentes patologías gástricas (dispepsia, gastritis, ulceras gástricas y
duodenales y como factor de riesgo para el cáncer gástrico), y la Organización
Mundial de la Salud (OMS) la ha considerado como un agente Carcinógeno tipo 1. (1,
2, 3)
El ambiente del estómago humano se ha considerado no propicio para la
colonización microbiana del Hp debido al pH ácido del mismo, (4,5) hasta el año 1982
cuando Marshall y Waren detectaron en el estómago humano un microorganismo
con características de Campylobacter el cual llamaron Pyloridis, pero poco después
en 1989 Goodwin y col. Observaron que la bacteria no pertenecía a ese género y la
denominaron Hp. (6)
Se hace referencia a que hay condiciones que puedan facilitar la colonización
oral de Hp tales como: Reflujo gastroesofágico, malos hábitos de higiene y la
infección intrafamiliar entre otros. (7)
La función de la microbiota oral es impedir la implantación de patógenos
oportunistas, colaborando con los mecanismos de defensa del hospedero para
controlar el crecimiento y reproducción de los microecosistemas que habitan en la
cavidad bucal. La comunidad bacteriana de la superficie dentaria forma parte de la
microflora, residente o transitoria, del cuerpo humano. Se organiza formando una
película (Biofilm) a la que se suman especies bacterianas que instituyen relaciones
entre ellas: mediante receptores, estructuras y compuestos adherentes, e
interacciones iónicas, hasta formar una capa densa que trasciende de la
colonización primaria de bacterias que conforman la Placa bacteriana (PB). (8, 9) Las
bacterias Gram negativas pueden colonizar la placa dental y competir por lo
nutrientes necesarios para la microflora local; también pueden aprovechar ciertas
condiciones de menor presión de oxígeno en algunas regiones de la cavidad bucal. (10, 11)
Cuando nos referimos a la PB, estamos hablando del principal factor
etiológico de la Enfermedad Periodontal (EP), la cual afecta a gran parte de la

población, en dicha enfermedad ocurren una cadena de procesos patológicos que
afectan al periodonto (tejidos que rodean y soportan al diente). Durante mucho
tiempo se ha intentado clasificar las diferentes formas de la enfermedad, lo que ha
resultado bastante complejo debido a la falta de conocimientos sobre las
Interrelaciones en el equilibrio hospedero-microorganismo que se establece en los
procesos patológicos asociados a la EP. (12)
En la actualidad se reconoce que las distintas formas de presentación de
dicha enfermedad obedecen a las posibilidades de quiebre del equilibrio antes
mencionado, que se revelan, sobre todo, en las pautas de destrucción periodontal,
con mecanismos patogénicos, manifestaciones clínicas y anatomopatológicas
similares. Dentro de este contexto, los conocimientos sobre los procesos reactivos
inherentes al receptor, al microorganismo atacante y formación de PB, permiten la
realización de diagnósticos acertados y planes de tratamiento dirigidos directamente
sobre el agente etiológico. (12)
Aunado a ello, es posible que el Hp se pueda encontrar en la cavidad bucal
mas como parte de la microbiota transitoria que como un residente normal, (13, 14) lo
que podría representar un factor de riesgo para la reinfección gastrointestinal
posterior a la terapia antibiótica. (14, 15, 16)
Por consiguiente, se abre la posibilidad de considerar que la cavidad bucal y
específicamente la PB puedan servir como reservorio para el Hp, (17, 18, 19, 20, 21, 22)
siendo interesante analizar a los efectos del presente estudio, las evidencias
documentales existentes que determinen una posible asociación entre el
Helicobacter pylori y la placa bacteriana. En consideración a los planteamientos
antes formulados, se procederá a centrar el problema de estudio en determinar la
relación del Helicobacter pylori y la enfermedad periodontal.

IV.-MARCO TEÓRICO
4.1.- Helicobacter Pylori:
Es un bacilo gram negativo, curvado y microaerofílico que se encuentra en la
mucosa gástrica del estómago humano y que se ha asociado con diferentes
enfermedades digestivas. Dicho microorganismo posee una morfología espiral en
forma de sacacorchos cuando se encuentra en la mucosa gástrica y menos espiral
cuando crece en medios artificiales. Presenta unas dimensiones de 0,5 a 1,0 um de
ancho y de 3 um de largo. Tiene de 4 a 8 flagelos polares primordiales para su
movilidad, los cuales están recubiertos por una vaina de estructura lipídica, al igual
que su membrana externa, la cual parece tener la misión de proteger a los flagelos de
su degradación por el medio ácido. Su característica bioquímica más importante es la
producción de UREASA, la cual junto con otra enzima, la catalasa son útiles para su
identificación cuando crecen en medios de cultivo. (23, 24,)
La ureasa le protege frente al ácido al catalizar la hidrólisis de la urea para
producir amonio, esto significa que la ureasa desempeña una función en la
colonización y también puede ser importante en el mantenimiento de la infección. (6)
Existen numerosas especies, la mayor parte de las cuales coloniza
huéspedes animales específicos. El Hp infecta de forma natural al ser humano y al
mono. (6)
Hp también se ha presentado en una forma cocoide viable pero no cultivable;
de esta manera puede sobrevivir en microambientes acuáticos como forma viable,
aunque en estado silente; y poseen una resistencia mayor que la forma bacteriana
normal, lo que las ayuda a sobrevivir en ambientes hostiles durante largos periodos
de tiempo gracias al desarrollo de un estricto metabolismo endógeno. Se especula
que la forma cocoide sea una forma de resistencia; capaces de transformarse de
nuevo en partículas infectantes helicoidales, cuando el ambiente se vuelve más
propicio. (25)
Uno de los principales interrogantes por responder sobre la infección por Hp
es conocer por qué sólo el 10-20 % de las personas infectadas por esta bacteria
desarrollan enfermedades gástricas. Parece ser que depende de la compleja

interacción existente entre diversos factores ambientales y los factores de virulencia
de la bacteria. (26)
4.1.1.- Epidemiología:
Los datos recopilados hasta la fecha, indican que Hp tiene una amplia
distribución mundial y puesto que un porcentaje muy elevado de sujetos son
portadores, se considera que la infección es adquirida durante la infancia y persistirá
a lo largo de la vida, contribuyendo al desarrollo de enfermedad ulcero péptica y
cáncer, en ciertos individuos susceptibles. La tasa de infección aumenta con la edad,
de manera que mientras alrededor de 10% de los individuos menores de 30 años
están infectados, tal cifra asciende a 60%, entre los mayores de 60 años.
Estudios epidemiológicos nos revelan que la prevalencia a nivel mundial de la
infección por Hp es aproximadamente de un 50 a 60%. (2, 27, 28, 29, 30, 31) En países
desarrollados es de un 25 a 30%, no obstante hay autores que reflejan un 70% en
dichas zonas, (31) y en los países en vías de desarrollo varía de un 70 a un 90%, (31,
32, 33, 34) lo cual suele asociarse con la insatisfacción de las necesidades básicas de
salud, bajo nivel socioeconómico, malos hábitos higiénico-dietético y hacinamiento. (24, 28,29) En Venezuela no es desigual con respecto al resto de los países en
desarrollo, habiéndose divisado su presencia en un 90 a 96%. (35)
En los Estados Unidos la incidencia anual de infección se presenta entre el
0.5% y el 1% para menores de 10 años y la infección aumenta hasta en un 50% en
adultos, con un promedio de edad de 60 años. Por otro lado, en el grupo de afro-
americanos, hispanos e indios nativos de ese país, la infección por el
microorganismo se observa desde edades tempranas y la transmisión intrafamiliar
es alta. Por el contrario, se estima que en los países en desarrollo la mayoría de las
personas (el 80% aproximadamente) se infectan con Hp a una edad promedio de 10
años; fenómeno que al parecer está relacionado con los factores anteriormente
señalados. Se han investigado muchos factores, pero todos tienen como
denominador común el bajo nivel económico. El hacinamiento, la vivienda insalubre,
el agua contaminada, la promiscuidad y la consanguinidad están involucrados. El
residir en comunidades cerradas, tales como hogares para pacientes con retardo
mental, hospitales de estancia prolongada para enfermos crónicos y orfanatos es

otro factor de incidencia; en estas circunstancias, el contacto entre individuos es más
cercano que el normal y las normas de higiene pueden ser menores. (2)
En países latinoamericanos como Costa Rica y Brasil se reporta una
incidencia anual de 45 enfermos de cáncer gástrico asociado a Hp por cada 100 000
habitantes. En algunos países como México se han observado regiones de mayor
riesgo, como las zonas altas del estado de Chiapas donde existen grupos indígenas
que presentan una alta incidencia de cáncer gástrico asociado al microorganismo. (2)
La prevalencia publicada en España va desde el 36% en una determinada
comarca valenciana, hasta una prevalencia del 53% en la zona de Madrid. (36)
Según los estimativos recientes, 90% a 95% de los pacientes con úlcera
duodenal y 60% a 70% de aquellos con úlcera gástrica están colonizados por el
germen. Los estudios epidemiológicos han demostrado una fuerte correlación entre
incidencia de cáncer gástrico y prevalencia de infección por la bacteria, e indican
que las personas infectadas tienen entre tres y seis veces mayor probabilidad de
desarrollar cáncer gástrico, que quienes no lo están. También se ha demostrado la
influencia de Hp en el desarrollo de algunas neoplasias de tipo linfoproliferativo, tales
como el linfoma asociado a la mucosa gástrica (MALT del inglés Mucosa-Associated
Lymphoid Tissue). (26)
4.1.2.- Patogenia y Factores de Virulencia: Los mecanismos patógenos principalmente implicados son:
1) Producción de toxinas (citotoxinas, ureasas, mucinasa, lipasa, lipolisacaridasa,
hemolisinas, Fosfolipasa A.
2) Mediadores de inflamación (activación de neutrófilos, activación de monocitos y
macrófagos, leucotrienos, fenómenos autoinmunes, infiltración y degranulación de
eosinófilos).
3) Capacidad de contribuir con el incremento de la actividad ácido-péptica gástrica.
(25)
Dentro de los factores de virulencia se encuentran:

La forma y los movimientos espirales: Hp es muy móvil y activo, lo cual es
resultado de su forma espiral y de los flagelos unipolares que le confiere una gran
movilidad y le permite introducirse a través de la capa de moco gástrico actuando de
forma similar a un sacacorchos y favoreciendo por lo tanto el acercamiento a las
células epiteliales gástricas; y además la ayuda a no ser eliminado por los
mecanismos defensivos del huésped. Actualmente se ha sugerido que el flagelo es
determinante como sensor de cambios de pH entre la luz gástrica y la capa de
moco. (25)
Enzimas y proteínas de adaptación (ureasas, catalasas, proteína inhibidora
de la secreción de ácido gástrico): La ureasa es producida en cantidades muy altas
por Hp, es esencial para sobrevivir, puesto que bacterias mutantes ureasa-
deficientes no logran colonizar el estómago. La actividad de la ureasa permite el
desdoblamiento rápido y permanente de la urea en el jugo gástrico, moco y mucosa,
a substratos principales en la producción de amonio; aparentemente estos procesos
proveen a la bacteria de la habilidad de exportar iones de hidrógeno desde dentro
del citoplasma, regulando así el pH, y también crea un micro ambiente alcalino que
lo protege de la acidez del estómago. (25)
Habilidad para adherirse a las células de la mucosa gástrica y al moco
(adhesinas bacterianas y receptores para células epiteliales): Además de la ureasa,
también produce superóxido-dismutasa y otras moléculas diversas que se
encuentran involucradas en la adhesión específica a las células epiteliales del
estómago. El Hp posee gran variedad de adhesinas que reconocen de forma
específica a los receptores de la mucosa gástrica y se unen a ellos comenzando la
colonización bacteriana. (25)
Las adhesinas del Hp están constituidas por una hemaglutinina radiante
desde la superficie de la bacteria con estructura de tipo afrimbial y un diámetro de 2
mm y perteneciente al grupo de los sialoconjugados. Es detectable utilizando
eritrocitos de una variedad de especies con las propiedades de una sustancia
antigénica termolábil y sensible a la acción de la pronasa, la papaína y la
neuraminidasa; resistente a la pepsina y parcialmente a la tripsina. En general, la
adhesión es ventajosa para la supervivencia del patógeno y para favorecer la

liberación de las toxinas de los gérmenes directamente sobre las células epiteliales. (25)
Junto a los factores antes mencionados, los cuales son producidos por todas
las cepas de Hp, existen algunos otros que son producidos por un subgrupo de
bacterias aisladas. Clínicamente estas son proteínas asociadas a Citotoxina (CagA)
y la Citotoxina Vacuolizante (VacA). La proteína VacA forma vacuolas en células
epiteliales aisladas y ulceraciones superficiales en estómagos de animales de
estudios. (25)
Según análisis de expresión de estas proteínas y sus genes, Hp puede ser
clasificado en dos tipos: el tipo I donde la bacteria tiene el gen que codifica para
CagA y expresa la proteína CagA y la citotoxina vacuolizante. El tipo II donde la
bacteria no tiene el gen que codifica para el CagA y no expresa por consiguiente la
proteína CagA o la citotoxina vacuolizante. El tipo I y II representa aproximadamente
el 56 y 16% respectivamente, y el restante posee un fenotipo intermedio que
expresa CagA independientemente de VacA o viceversa lo que hace pensar que
CagA no es necesario para la expresión de la citotoxina vacuolizante. (25)
La citotoxina vacuolizante: Se ha descrito la presencia de una toxina que
produce la formación de grandes vacuolas en las células eucarióticas. Este efecto lo
producen más de la mitad de los aislamientos clínicos de Hp y aquellos que poseen
la toxina se han asociado con cuadros más graves de enfermedad. La toxina está
codificada por un gen denominado vacA que está presente en todos los
aislamientos, produzcan o no la toxina, aunque la secuencia del gen parece ser
diferente. (37)
La proteína CagA: Se ha descrito la presencia de una proteína codificada por
el gen cagA que podría estar implicada en el proceso de activación de la toxina
vacuolizante (cagA= gen asociado a citotoxina). La presencia de esta proteína
podría influir en la respuesta inflamatoria y aumentar la secreción de interleukina.
Algunos autores han observado una clara diferencia en cuanto al proceso de
enfermedad que se produce cuando se trata de una cepa cagA+ o cagA-, sin
embargo, para otros autores la diferencia no es tan importante. (37)

El gen cagA se encuentra localizado en una región del cromosoma que se
conoce como Isla de patogenicidad (PAI). (37)
La presencia del gen CagA y la expresión de su proteína, son reportados
significativamente en pacientes con úlcera duodenal y gastritis severa, incluyendo
también gastritis atrófica y gastritis superficial. Recientemente dos genes coligados
cercanamente han sido identificados y a su inicio se llamaron CagB y CagC,
posteriormente fueron renombrados como PicB y PicC, siendo la terminología Pic
referida a la habilidad de promover la inducción de citoquinas en el huésped; este
clauster de nuevos genes podrían definir una importante región del genoma
relacionada con la patogenicidad de la bacteria y podría llegar a representar la
diferencia entre cepas patógenas y cepas comensales de Hp. El conocimiento del
genoma de Hp ha permitido comprobar que dentro de la misma persona y al mismo
tiempo hay muchos diferentes tipos de Helicobacter que compiten unos con otros
para ocupar el mismo nicho. En esa competencia se desarrollan cambios tanto
intragenómicos (dentro del mismo germen) como intergenómicos (entre gérmenes
distintos). (25)
La infección por cepas de Hp con el gen de antígeno A asociado a citotoxina
(CagA) está asociada con el carcinoma gástrico. En Taiwán, Hp con vacA s1a, cagA
y iceA1 están presentes en la mayoría de los pacientes con cáncer gástrico y
gastritis crónica. Se ha demostrado que VacA interrumpe la maduración de los
fagosomas en los macrófagos como también modula e interfiere con las funciones
inmunológicas de los linfocitos T; adicionalmente, las células infectadas por Hp
expresan receptores A y B para interleukina 8 lo cual puede contribuir al aumento de
la respuesta inflamatoria. Numerosas evidencias relacionan la variación genética de
la respuesta inflamatoria producida por Hp con el mayor riesgo de carcinoma
gástrico. (25)
La infección por Hp induce una respuesta inmunológica humoral sistémica y a
nivel de la mucosa gástrica. La producción de anticuerpos no conduce a la
erradicación de la infección pero puede contribuir al daño tisular. Algunos pacientes
infectados tienen autoanticuerpos directamente contra la ATPasa de las células
parietales lo cual se correlaciona con incremento de la atrofia del cuerpo gástrico. (25)

En adición al daño asociado con traslocación de proteínas mediada a cag-
PAI, la injuria epitelial de Hp puede ser secundaria a otros mecanismos como
liberación de especies de oxígeno y nitrógeno reactivos producidos por la activación
de neutrófilos. La inflamación crónica también incrementa el recambio de células
epiteliales y la apoptosis. (25)
Después de la ingestión, Hp coloniza predominantemente el antro gástrico,
facilitado por los factores de virulencia y de patogenicidad antes mencionados, una
vez allí, la infección persiste por años y tal vez de por vida, desencadenando y
manteniendo una marcada respuesta inflamatoria que produce daños sobre la
mucosa gástrica (gastritis superficial a gastritis crónica atrófica), además de producir
alteración en los mecanismos de regulación de la secreción ácida gástrica por
inhibición de la relación entre la somatostatina y gastrina, lo cual trae como
consecuencia, cambios de la motilidad antro-piloro-duodenal, pudiendo ocasionar
síntomas, aunque un gran número de individuos se mantiene asintomático. (25)
La gastrinemia basal aumenta aproximadamente en un 50% y la postprandial
en un 100% en los pacientes infectados. De algún modo Hp altera los mecanismos
reguladores antrales, propiciando la producción inapropiada de gastrina. Además de
la hipótesis de que esta alteración sea resultado de la actividad de la ureasa podría
deberse también a una alteración de la liberación de gastrina inducida por los
mecanismos que causan la gastritis antral. La alteración estructural que Hp en la
mucosa gástrica puede potenciar los mecanismos ulcerogénicos como la inhibición
de la somatostatina. Una tercera posibilidad sería que la hipergastrinemia sea
secundaria a una alteración de los mecanismos locales que regulan las células
neuroendocrinas G y D. El rápido descenso de los niveles de gastrina como
respuesta al tratamiento de erradicación se correlaciona mejor con una alteración de
la regulación entre las células G y D que con un cambio en el número de estas. (25)
4.1.3.- Manifestaciones Clínicas:
Los pacientes portadores de dicha infección pueden o no tener síntomas:
Dolor abdominal generalmente epigástrico y con menos frecuencia
periumbilical,

Dispepsia o indigestión
Distensión y llenura
Náusea leve que se puede aliviar al vomitar
Eructos y regurgitación
Sentir mucha hambre de 1 a 3 horas después de comer
En menor proporción de anorexia con pérdida de peso,
Pirosis y sensación de plenitud posprandial,
Lo que constituye el motivo de consulta habitual. (24)
4.1.4.- Vías de Transmisión
Existen diversas vías de transmisión que han sido consideradas en la cadena
epidemiológica dentro de las cuales se encuentran:
Transmisión Oral-Oral: Se desconoce cual es el modo exacto de transmisión,
pero parece que resulta imprescindible que se produzca un contacto estrecho entre
personas para que tenga lugar. Aunque se ha aislado Hp de la saliva y de la placa
dental, lo que podría sugerir la posibilidad de que la cavidad bucal sea un reservorio
natural de la bacteria, pero se piensa que podría ocurrir una colonización transitoria
en casos de reflujo o en pacientes sometidos a endoscopia; utilizando la técnica de
PCR para la detección de la bacteria, los resultados de prevalencia en este tipo de
muestras fluctúan desde valores elevados, hasta resultados próximos a cero, (6) y en
algunos casos, las cifras son difícilmente correlacionables con la prevalencia, que
por el mismo método, se obtenía en esos estudios cuando se analizaban muestras
de mucosa gástrica. (38)
La transmisión oral-oral posible ha sido investigada en la comida
premasticada entre algunos grupos étnicos, el uso de la misma cuchara tanto por
madre como por el niño, contacto oral-oral íntimo, y aspiración del vómito. (39)
La transmisión intrafamiliar tampoco está suficientemente esclarecida y
aunque numerosos estudios señalan una mayor prevalencia de infección en
familiares de niños o cónyuges, infectados y en algunos casos, se ha determinado
por técnicas de PCR que se trata del mismo tipo de cepa infectiva, siempre queda la

explicación alternativa de la existencia de una fuente común de infección. Estudios
de transmisión horizontal en tres generaciones de una misma familia identifican tanto
cepas comunes, como cepas no relacionadas entre los distintos miembros de la
misma. (40) Otros estudios señalan que la transmisión vertical es poco probable y
justifican esta aseveración señalando que los anticuerpos que se detectan en los
primeros meses de vida desaparecen paulatinamente y que además, no confieren
protección frente a la bacteria durante esa primera etapa de la vida. (41)
Transmisión Fecal-Oral: El cultivo de formas viables de la bacteria en
muestras de heces, apoya la hipótesis de esta vía de transmisión. (38) Ha sido
aislado en los excrementos de niños infectados, pero el aislamiento de los
excrementos de los adultos ha sido raro. (39) Sin embargo, los resultados de los
diferentes trabajos son poco coherentes, El fracaso de recuperar la bacteria de
excrementos podría ser debido al efecto tóxico de excrementos; a la presencia en la
muestra de sustancias capaces de disminuir o inhibir el crecimiento bacteriano
(sales biliares, polisacáridos), o bien, por la existencia de características ambientales
(presencia de nitrógeno y carbono) que hacen que la bacteria adopte su forma
resistente (cocoide) de difícil replicación en cultivo. Utilizando PCR como método de
detección, los problemas aparecen como consecuencia de la presencia en la
muestra de interferentes para esta técnica, por lo que los resultados dependen en
gran medida del método de obtención del DNA desde las heces. Otro problema que
parece dificultar la detección por cualquiera de los métodos, es lo que se conoce
como fenómeno de excreción intermitente que determina que la bacteria se elimina
en heces fundamentalmente durante la fase aguda de la infección, ya que la
excreción se ve facilitada por la hipoclorhidria transitoria de este periodo, lo que
convierte el inicio de la infección y el tratamiento con fármacos antisecretores, en
factores de riesgo para la transmisión de la bacteria. (38)
El agua contaminada por excrementos puede ser una fuente de infección; una
asociación entre Hp y la ausencia del agua potable fue encontrada en algunos
estudios. (39)
Además, un riesgo aumentado de la infección fue observado en niños que
nadaron en ríos, corrientes, o piscinas en los Andes colombianos del sur. Sin
embargo, el organismo no ha sido aislado del agua, excepto en dos casos en los

cuales fue descubierto usando el efecto de dominó polymerase en muestras de
Aldana, Colombia, y Lima, Perú. En Suecia, la exposición a aguas residuales entre
trabajadores de aguas residuales no causó un riesgo aumentado de la infección. (39)
En Chile, el consumo de verduras no cocinadas que habían sido irrigadas con
el agua contaminada o residual no tratadas tuvo que ver con la presencia de Hp,
más del 60 % de 1815 chilenos menores de 35 años y de grupos socioeconómicos
inferiores fue encontrado seropositivo a Hp. Los niños que obtuvieron su agua
potable de corrientes locales en los Andes Colombianos también fueron encontrados
con un alto predominio de Hp. (39)
En el centro médico Santa Isabel de Boston (Estados Unidos) se demostró
que la participación de la mosca doméstica como transportador de la bacteria al
tomarla de comidas contaminadas o heces. A partir de las heces de los portadores la
bacteria se puede ahilar viva y con potencial para infectar, lo cual es evidencia a
favor de la teoría que considera a la vía fecal-oral como la principal ruta de
transmisión .La difusión a partir de reservorio de agua de consumo, es otra
posibilidad y está respaldada por la alta prevalencia de la infección en comunidades
que carecen del servicio de agua potable, y por la presencia de brotes epidémicos,
atribuidos a la contaminación de los acueductos. (6, 42)
Situaciones de hacinamiento o de estrecha convivencia sí se asocian en casi
todos los estudios, a una mayor prevalencia de la infección, lo que también apoyaría
los mecanismos de transmisión persona-persona, de tipo oral-oral y/o fecal-oral o
bien apuntaría de nuevo hacia la existencia de una fuente común de infección. (38)
Iatrogenia: Es primer, y el más frecuente, modo de transmisión, en el cual
tubos o endoscopios que han estado en el contacto con la mucosa gástrica de un
individuo son usados para otro paciente, sin previa o debida técnica de desinfección.
Las infecciones ocupacionalmente adquiridas, por lo general en que la infección es
transmitida de un paciente al empleado, también han sido relatadas, sobre todo
entre endoscopistas y gastroenterólogos. Sin embargo, en términos cuantitativos se
piensa que la ruta iatrogénica es secundaria. (39)
La transmisión instrumental, esta perfectamente documentada; estudios de
prevalencia en gastroenterólogos endoscopistas arrojan resultados más elevados
que la encontrada en población general e incluso, que en otros profesionales

sanitarios como neumólogos u odontólogos que, por otra parte, están expuestos de
forma continua a aerosoles orales, lo que lleva a pensar que el riesgo de infección
no está tanto en las secreciones salivares como en las gástricas, aseveración
apoyada por un trabajo reciente que detecta DNA en el canal del endoscopio, por
trabajos que consiguen cultivar la bacteria a partir de muestras de jugo gástrico y por
estudios que concluyen que el moco gástrico expulsado durante el vómito constituye
una vía muy importante de transmisión de la infección en población infantil. (38)
4.2.- Clasificación de las Enfermedades del Aparato Digestivo Causadas por
Helicobacter Pylori.
4.2.1.- Dispepsia no Ulcerosa o Funcional:
Tiene como sinónimo mala digestión, o indigestión. Su nombre viene de una
raíz Griega dis que significa malo y pepsia que se utiliza como referente a tracto
gastrointestinal alto. Ya que los síntomas de la dispepsia se asemejan mucho a los
síntomas causados por una úlcera, si se confirma que éstos pacientes carecen de
una úlcera, se les diagnostica con una dispepsia no ulcerosa. (43)
El término dispepsia es utilizado para describir a un conjunto de síntomas que
tienen su causa en el tracto gastrointestinal superior (esófago, estómago o la
primera parte del intestino delgado (duodeno) y el cual cursa con dolor o
incomodidad, que afecta la parte superior del abdomen, dicha incomodidad es una
sensación muy subjetiva que el paciente no interpreta como dolor, y puede abarcar
una gran variedad de síntomas, como náuseas, saciedad prematura, plenitud
abdominal, distensión y eructos. Esta definición comprende a los pacientes con
síntomas continuos o intermitentes y que no explican de ninguna forma la duración
de los mismos los cuales pueden ser de corta o larga duración. (44) Este tipo de
patología es totalmente benigna (no se asocia con ningún tumor). (43)
Epidemiología: su prevalencia a nivel mundial es de aproximadamente 25%,
de los cuales solo el 25% busca atención médica, que es cerca del 5% de todas las
primeras consultas. En Estados Unidos tiene una prevalencia anual de 25% (+
pirosis: 40%). Menos del 50% consultan al médico, 2-5% de consultan a los

médicos de atención primaria. Representan del 40-70% de las molestias
Gastrointestinales. (44)
En cuanto a su fisiopatología se han propuesto varios mecanismos, los cuales
no son absolutos; algunos de los postulados son; Vaciamiento gástrico
retardado, alteración de la acomodación del estómago proximal al alimento ,
hipersensibilidad visceral, motilidad duodenoyeyunal anormal, infección por Hp,
hipersensibilidad a ácidos y nutrimentos, causas iatrogénicas como esofagitis por
medicamentos o intolerancia a analgésicos antiinflamatorios no esteroideos (AINES)
y disminución del sistema nervioso central. Existe una hipótesis de que la infección
por Hp induce cambios en la motilidad gastrointestinal y en el umbral sensorial, que
el paciente puede percibir como dolor epigástrico. (44)
De acuerdo con los llamados «criterios de Roma II» (Roma, 1999), para
establecer el diagnóstico de dispepsia funcional se debe cumplir lo siguiente:
1. Síntomas con una duración mínima de 12 semanas (que no tienen que ser
consecutivas) durante los últimos 12 meses.
2. Presencia de dispepsia (dolor o molestia abdominal localizada en la parte
central del abdomen superior) de forma persistente o recurrente.
3. Ausencia de enfermedades orgánicas (debe incluirse la realización de
endoscopia digestiva alta) que puedan explicar los síntomas.
4. Sin evidencia de que la dispepsia se alivia exclusivamente con la defecación
o se asocia con cambios en la frecuencia o consistencia de las deposiciones
(para diferenciarla del síndrome del intestino irritable). (44)
En los pacientes con historia previa de enfermedad ulcerosa péptica crónica
no se debe establecer el diagnóstico de dispepsia funcional, aunque es posible que,
en algunos casos, la persistencia de los síntomas tras la erradicación de Hp se deba
a esta causa.
El abordaje de un paciente con dispepsia no ulcerosa debe incluir como
mínimo una historia clínica, exploración física, una endoscopia de tracto digestivo
alto durante el período sintomático sin tratamiento antisecretor. (44)

4.2.2.- Gastritis:
Condición de la mucosa gástrica que se caracteriza por inflamación cuyo
agente etiológico más común es el Hp, aunque también puede ser causada por otros
agentes infecciosos, reacciones autoinmunitarias, reacciones de hipersensibilidad,
alergias. (44)
4.2.2.1.- Gastritis Aguda: Es una inflamación súbita del revestimiento del estómago con dolor
abdominal inespecífico, entre sus causas se pueden mencionar: el consumo de
medicamentos, alcohol y sustancias corrosivas el estrés fisiológico severo y las
infecciones. Con frecuencia, la GA se asocia con una enfermedad severa aguda o
con un trauma. Los factores de riesgo son entre otros, el uso de medicamentos
antiinflamatorios no esteroideos (AINES), el consumo de alcohol reciente y en
grandes cantidades y el estrés fisiológico como una cirugía mayor, trauma de
cabeza, insuficiencia renal, insuficiencia hepática e insuficiencia respiratoria, suele
presentarse con frecuencia en pacientes hospitalizados con enfermedades graves, y
sus síntomas pueden verse opacados por otros más severos. (45)
Dentro de sus síntomas se encuentran: Indigestión abdominal, pérdida del
apetito, náuseas, vómitos, vómito con sangre o con un material similar a granos de
café, heces negras e hipo. Para su diagnóstico se recomienda la realización de
exámenes como: series del tracto Gastrointestinal superior y del intestino delgado,
examen de guayacol en heces, gastroscopia que revela gastritis, conteo sanguíneo
completo que revela anemia. (45)
El tratamiento depende de la causa de la gastritis. Por lo general, los
antiácidos y otros medicamentos que disminuyen o neutralizan el ácido gástrico en
el estómago, eliminan los síntomas y contribuyen a la curación. Se deben suspender
los medicamentos que causan la gastritis. Puede existir una úlcera gástrica que
requiera tratamiento. (45)
La gastritis ocasionada por el estrés se trata mejor mediante la prevención. A
los pacientes hospitalizados y sometidos a estrés se les deben suministrar

medicamentos que disminuyan la producción de ácido gástrico tales como los
inhibidores de la bomba de protones, en cuanto a las expectativas; la mayoría de las
gastritis mejoran rápidamente con el tratamiento y una complicación es la pérdida
severa de sangre. (45)
4.2.2.1.1.- Gastritis Erosiva y Hemorrágica:
Las erosiones son lesiones superficiales de la mucosa, no penetran en la
capa muscular de la misma y miden menos de 5mm de diámetro, su causa puede
ser; hipoxia, lesión directa de los fármacos u otros agentes lesivos (antiinflamatorios
no esteroideos, etanol), reducción del flujo sanguíneo (estrés, traumatismos,
quemaduras, agentes físicos o sepsis) o infección por Hp. (44)
La fase aguda se define como una infección con inicio repentino de síntomas
e hipoclorhidria que es difícil de encontrar en biopsias gástricas. Cuando se
sospecha la enfermedad las pruebas serológicas resultan útiles. Las biopsias dejan
ver cambios degenerativos en la superficie del epitelio, como disminución del moco,
exfoliación celular y cambios regenerativos, infiltración de polimorfonucleares
foveolares y exudado polimorfo adherente en la superficie; estos cambios suceden
tanto en el cuerpo como en el antro gástrico. (44)
4.2.2.2.- Gastritis Crónica:
Inflamación del revestimiento del estómago que se presenta gradualmente y
que persiste durante un tiempo prolongado, (45) que se caracteriza por un infiltrado
difuso con linfocitos y plasmocitos que se extiende a la lámina propia y el epitelio sin
células atípicas. Puede ser atrófica y no atrófica, entendiéndose por atrofia la
desaparición de las glándulas normales en algún área del estómago. (44)
4.2.2.2.1.- Gastritis por Helicobacter Pylori:
Se caracteriza por inflamación superficial crónica, infiltración de leucocitos,
células plasmáticas, macrófagos y eosinófilos. En ocasiones esta inflamación
crónica se acompaña de inflamación aguda con infiltrado de neutrófilos. De igual

forma con frecuencia se observan folículos linfoides que son agregados de linfocitos
con un centro germinal de células mononucleares pálidas patognomónicas de
gastritis por Hp. (44)
Con respecto al cuadro clínico y el diagnóstico, puede haber síntomas típicos
o de certeza solo si esta relacionada con enfermedad ulcerosa. Pero los síntomas
pueden ser de dispepsia no ulcerosa o bien es posible que curse asintomática en un
80 a 90% de los casos. Los pacientes con alta secreción de ácido tienen gastritis
predominantemente de antro, con predisposición a las úlceras duodenales. Los que
tienen baja secreción suelen tener gastritis del cuerpo gástrico, con predisposición a
úlcera gástrica y en raras ocasiones puede iniciarse una secuencia de fenómenos
que resulta en cáncer gástrico. La infección por Hp induce la formación de tejido
linfoide vinculado con la mucosa (MALT o mucosa-associated lynphoid tisue), que es
maligno. (44)
4.2.2.2.1.- Gastritis Atrófica Metaplásica Ambiental (GAME):
Es más frecuente en Japón, China y América del Sur. Resulta de proliferación
bacteriana en el estómago y esta relacionad con Hp, tabaquismo, exposición a polvo
de carbón y consumo de sal y de nitratos o nitritos. La secreción de ácido gástrico es
baja o normal. Este tipo de gastritis no esta relacionada con anemia perniciosa y no
es posible detectar anticuerpos contra las células parietales y contra el factor
intrínseco. (44)
La GAME se caracteriza por afectar principalmente el antro gástrico, donde
se observan áreas de inflamación, atrofia y metaplasma. El cuerpo gástrico resulta
afectado en menor grado. Para establecer el diagnóstico la mucosa normal del antro
debe estar sustituida en un 20% por atrofia o metaplasma. Este proceso patológico
puede cursar con pólipos hiperplásico, úlceras gástricas, cáncer gástrico y tumores
carcinoides. (44)

4.2.3.- Ulceras Pépticas: Cuando hablamos de úlceras nos referimos a los defectos de la mucosa
gastrointestinal que se extienden por la lámina muscular de la mucosa y que
persisten como consecuencia de la actividad acidopéptica del jugo gástrico, las
cuales ocurren por una infección con Hp, por el consumo de AINES, sin olvidar la
aspirina. (44)
4.2.3.1.- Ulcera Gástrica:
Es una ruptura en el tejido normal que recubre el estómago. La mayor parte
de las úlceras gástricas se presentan en el epitelio gástrico que no produce ácido
(antro gástrico), o bien, cerca de su unión con la mucosa oxíntica productora de
ácido (cuerpo y fondo), fenómeno que apunta a que la mucosa no productora de
ácido es inherentemente más susceptible a la ulceración péptica. La gastritis crónica
secundaria a infección por Hp causa atrofia de la mucosa oxíntica, desarrollo
posterior de metaplasma intestinal y extensión del epitelio de tipo no productor de
ácido hacia el estómago proximal. Por consiguiente, es posible establecer que las
úlceras en estómago proximal (cuerpo y fondo) son úlceras secundarias a gastritis
crónica y atrofia gástrica relacionadas con infección por Hp, y se denominan úlceras
gástricas tipo I. los pacientes cursan con niveles elevados de pepsinógeno II
(proteasa encontrada en mucosa antral y oxíntica). En cambio, los pacientes que
presentan úlceras en el cuerpo gástrico y el duodeno (denominadas úlceras tipo II),
así como los pacientes con úlceras gástricas confinadas al antro prepilórico (úlceras
gástricas tipo III) manifiestan anormalidades en la homeostasis de la secreción ácida
similares a las de aquellos pacientes con úlcera duodenal, a menudo con
concentraciones elevadas de pepsinógeno tipo I. (44)
Una úlcera gástrica benigna es una enfermedad causada por un desequilibrio
entre la secreción de ácido y una enzima llamada pepsina y las defensas del
revestimiento de la mucosa estomacal. Esto lleva a que se presente inflamación que
puede empeorar por el uso de aspirina y de medicamentos antiinflamatorios no
esteroideos (AINES), como el ibuprofeno. Teniendo como factores de riesgo el
consumo de aspirina y AINES, infección por Hp, Gastritis Crónica, consumo de
cigarrillo, edad avanzada, ventilación mecánica (ser puesto en un respirador). (46)

En cuanto a sus síntomas tenemos; Dolor abdominal que puede despertar a
la persona durante la noche, se puede aliviar con antiácidos o leche, se presenta
de 2 a 3 horas después de una comida y puede empeorar por no comer; náuseas,
vómito (especialmente sanguinolento), heces sanguinolentas o negras
(alquitranadas), indigestión abdominal, pérdida involuntaria de peso y fatiga no
obstante es posible que no haya síntomas. (46)
Para su diagnóstico se recomienda la realización de exámenes como: Una
serie del tracto gastrointestinal superior muestra la úlcera gástrica, una
esofagogastroduodenoscopia (EGD) y una biopsia muestran una úlcera gástrica
benigna. (46)
Tratamiento: Entre los pacientes con la infección por Hp, el objetivo principal
es la erradicación del organismo causante del problema. Existen múltiples
regímenes efectivos y usualmente incluyen ya sea un antagonista de receptor H2,
como famotidina (Pepcid) o nizatidina (Axid) o un inhibidor de la bomba de protones,
como omeprazol (Prilosec) o esomeprazol (Nexium) para suprimir el ácido,
combinado con dos antibióticos. (46)
En las personas sin infección por Hp, los medicamentos para curar la úlcera,
como los antiácidos, antagonistas de los receptores H2 o los inhibidores de la
bomba de protones, generalmente son efectivos y es posible que sea necesaria la
terapia a largo plazo. (46)
En caso de que se presente sangrado de la úlcera, se puede controlar en la
mayoría de los pacientes con una terapia endoscópica.
Se puede recomendar la intervención quirúrgica para las personas que no
responden a la terapia médica o a la terapia endoscópica para el sangrado. Así
mismo, es posible que se requiera una vagotomía (escisión del nervio vago que
controla la producción de ácido gástrico en el estómago) o una gastrectomía parcial
(extirpación de una parte del estómago). (46)

Las medidas de autoayuda son, entre otras: Evitar el consumo de tabaco, el
té, el café y las bebidas gaseosas que contengan cafeína, el consumir alcohol, el
consumo de AINES y aspirina, Consumir varias porciones de comidas pequeñas al
día a intervalos regulares. (46)
4.2.3.2- Ulcera Duodenal:
Es una lesión deprimida en el revestimiento del duodeno, la primera parte del
intestino delgado que se conecta con el estómago. (46)
Una úlcera duodenal es un tipo de enfermedad péptica causado por un
desequilibrio entre la secreción de ácido y pepsina (una enzima) y las defensas del
revestimiento de la mucosa (ya sea producido por el microorganismo o por el
sistema inmunológico del hospedero), que neutralizan los sistemas de defensa de la
mucosa gástrica, permiten la penetración tisular y estimulan la inflamación local,
todo lo cual hace posible que la colonización evolucione hacia infección, y de allí a
gastritis y ulceración. Aunque se requiere la confluencia de otros factores, Hp es,
también, el principal agente etiológico de la úlcera duodenal. En esta entidad, como
resultado de fenómenos de metaplasia, células epiteliales gástricas aparecen en el
duodeno y con ellas, viene la bacteria, causando una intensa inflamación local que
deteriora la resistencia de la mucosa, permitiendo el ataque del ácido clorhídrico y
conduciendo finalmente a la ulceración. La inflamación puede precipitarse por el
consumo de aspirina y de medicamentos antiinflamatorios no esteroides (AINES). (44)
Las úlceras duodenales están asociadas comúnmente con la presencia de la
bacteria Hp en el estómago. Los factores de riesgo son, el consumo de aspirina y
AINES, el consumo de cigarrillo y la edad avanzada. La úlcera duodenal se ha
presentado históricamente con más frecuencia en los hombres, pero datos más
recientes sugieren tasas similares en hombres y mujeres. La prevalencia durante la
vida de una úlcera péptica es 5 a 10% y se aproxima de 10 a 20% en pacientes que
son positivos para Hp. (44)
La infección por Hp que infecta predominantemente el antro, con preservación
relativa de la porción secretora gástrica (cuerpo y fondo), predispone al desarrollo de

úlcera duodenal. En cambio, la infección por Hp que causa inflamación intensa de la
mucosa parietal ocasiona atrofia gástrica con menor producción de ácido y
predisposición al desarrollo de úlcera gástrica y cáncer. (44)
Los pacientes con úlcera duodenal tienden a ser hipersecretores de ácido
gástrico, con una mayor cantidad de células parietales determinada en autopsias,
así como una producción de ácido basal mayor que en los pacientes control. Esto se
debe tal vez a que estos pacientes presentan en ayunas concentraciones de
gastrina más elevadas (en lo cual el Hp tiene cierta participación, puesto que
inducen un incremento de las citocinas que estimulan a las células G a liberar
gastrina y disminuyen la expresión mucosa de somatostatina, la cual inhibe la
producción de gastrina) La marcada hiperproducción de gastrina, se produce de
igual modo, ya que el amonio generado por la bacteria interfiere con la señal ácida
que, en condiciones normales, sirve como mensaje de retroalimentación negativa
sobre las células G del antro, creando un falso ambiente de alcalinidad. Así,
aumenta la liberación de gastrina y es mayor el estímulo sobre las células parietales,
de manera que éstas liberan altas cantidades de ácido clorhídrico. Asimismo, las
concentraciones de pepsinógeno I (proteasa producida por las células principales en
la mucosa gástrica oxíntica) está elevada en un 50% de los pacientes con úlcera
duodenal. Existen del mismo modo anormalidades en el control vagal de la
secreción ácida en pacientes con úlcera duodenal, ya que estos manifiestan una
disminución extraordinaria de la producción de ácido después de una vagotomía o
luego de la administración de atropina. Estos pacientes muestran un mayor
vaciamiento gástrico a alimentos líquidos, lo cual induce al duodeno a un
vaciamiento rápido de ácido gástrico. Dicho vaciamiento lo predispone a la
formación de úlcera. Además, estos pacientes presentan una menor producción de
bicarbonato duodenal. (44)
4.2.4. – Cáncer Gástrico (CG)
Es una neoplasia maligna que se desarrolla en el estómago. Este carcinoma
es el segundo tumor maligno más frecuente en el mundo. En países orientales,
este de Europa y Sur de América la incidencia es epidémica, incluso es superior en
Japón (100/100 000 hab.) y constituye la primera causa de muerte por cáncer. En
Navarra (España) se sitúa en el quinto lugar en orden de frecuencia de incidencia en

varones y en mujeres. En este registro se observa que, al igual que en el resto de la
población, la incidencia de cáncer gástrico ha ido disminuyendo en las últimas
décadas, ya que en los años 70 era el tumor más frecuente en varones y el segundo
en mujeres. (44)
En los países con una incidencia elevada se han podido identificar algunos
factores epidemiológicos relacionados con la alimentación tales como el consumo de
alimentos ahumados, de carnes procesadas (jamón salchichas) que contienen
nitritos, los cuales en el estómago pueden formar nitrosaminas, que son compuestos
carcinógenos; y la capsaicina, principio activo que le dar picor a los chiles (es
mutágena y carcinógena) y la baja ingesta de vitamina C y vitamina A. Otros
factores que se han relacionado con la etiopatogénesis del carcinoma gástrico son la
gastritis atrófica y la infección prolongada por Hp. (44)
La infección por Hp induce hipoclorhidria y atrofia gástrica, los cuales son
acontecimientos precursores del CG, no obstante, existe aún una paradoja: por una
parte, la infección por Hp incrementa el riesgo de úlcera duodenal, que es un
padecimiento que se caracteriza por gastritis predominantemente antral, con
hiperclorhidria y sin atrofia del cuerpo, pero también con la infección hay mucho
incremento de riesgo de CG, situación que se caracteriza por gastritis
predominantemente del cuerpo del estómago, atrofia gástrica e hiperclorhidria. Las
razones de esta divergencia no son claras; podría ser que algunos factores de
virulencia de la bacteria sean los que determinen estos cursos clínicos
contrastantes. Las cepas más virulentas que producen un daño más agresivo en el
tejido son las que se relacionan con los hechos clínicos más extremos, tal es el
caso de las que codifican a las proteínas CagA, VacA y BabA. (44)
Hay diferentes tipos de cáncer que pueden ocurrir en el estómago y el más
común es el adenocarcinoma, que se refiere a la forma como se ve el cáncer bajo el
microscopio y existen diversos tipos. El adenocarcinoma del estómago es un cáncer
común del tracto digestivo que se presenta en todo el mundo, aunque es
relativamente raro en los Estados Unidos. Ocurre con mayor frecuencia en hombres
mayores de 40 años y su incidencia es extremadamente alta en Japón, Chile e
Islandia. En los Estados Unidos, ha disminuido la incidencia de la mayoría de los
tipos de adenocarcinomas gástricos con el pasar de los años y dicha disminución,

según los expertos, puede estar relacionada con la reducción en la ingesta de
alimentos curados, salados y ahumados, así como con el incremento en el
consumo de Vit C. (47)
Sin embargo, la incidencia del cáncer en las áreas superiores del estómago,
donde éste se conecta con el esófago, ha aumentado de manera considerable, junto
con un aumento en cánceres en la parte baja del esófago. La razón de esto se
desconoce. (47)
El diagnóstico de este tipo de cáncer a menudo se retrasa por la ausencia de
síntomas en las etapas tempranas de la enfermedad o por el autotratamiento de
síntomas que pueden ser comunes a otros trastornos gastrointestinales menos
serios, como distensión, gases y sensación de llenura.
Los factores de riesgo del CG son: antecedentes familiares de esta
enfermedad, infección por Hp, grupo sanguíneo tipo A, antecedentes de anemia
perniciosa, antecedentes de gastritis atrófica crónica, una enfermedad que
disminuye el ácido gástrico, y antecedentes de pólipos gástricos adenomatosos. (47)
Presentando síntomas como; pérdida del apetito, disfagia, si se localiza en la
unión gastro- esofágica; llenura abdominal vaga, náuseas y vómitos, vómitos con
sangre, dolor abdominal, eructos excesivos, halitosis, exceso de gases (flatulencia),
pérdida involuntaria de peso, deterioro de la salud en general, llenura abdominal
prematura después de las comidas. (47)
En cuanto a los exámenes se requiere una serie del tracto gastrointestinal
superior que muestre el cáncer gástrico, EGD (esofagogastroduodenoscopia) y
biopsia que muestren el cáncer gástrico, CSC que revele anemia (aunque hay
muchas otras razones para la anemia), Un examen de materia fecal positivo para
sangre. (47)
Su tratamiento puede ser la extirpación quirúrgica del estómago
(gastrectomía) es el único procedimiento curativo que existe, aunque la terapia por
radiación y la quimioterapia pueden traer beneficios. Un estudio reciente mostró que
para algunos pacientes, la radioterapia y la quimioterapia administradas después de
la cirugía mejoran las posibilidades de curación. (47)

Para los pacientes en los cuales la cirugía no es una opción, la quimioterapia
y la radioterapia pueden mejorar los síntomas. En algunos pacientes, un
procedimiento de derivación (bypass) quirúrgica puede brindar alivio a los síntomas. (47)
Hay una amplia variación en el pronóstico de tumores gástricos. Los tumores
en la parte inferior del estómago suelen curarse más a menudo que los tumores en
el área superior (cardias gástrico o la unión gastroesofágica). La profundidad a la
cual los tumores invaden la pared estomacal y si hay compromiso de los ganglios
linfáticos influencia la probabilidad de la curación. (47)
En circunstancias en las cuales el tumor se ha diseminado por fuera del
estómago, la curación no es posible y el tratamiento está dirigido a mejorar los
síntomas.
En Japón, donde el riesgo de CG es muy alto, los programas de exámenes
masivos han tenido éxito en la detección de la enfermedad en las etapas tempranas.
El valor de los exámenes en los Estados Unidos y en otros países con tasas más
bajas de cáncer gástrico no es claro. El hecho de evitar el consumo de cigarrillo
puede reducir el riesgo. (47)
El CG pude ser incipiente, el cual esta limitado a la mucosa o submucosa,
independientemente de la presencia de metástasis; y CG avanzado que
corresponde a un CG con invasión de la capa muscular o más allá, sin que importe
la presencia de metástasis. (44)
4.2.5. – Linfoma MALT:
El linfoma MALT “Tejido Linfoide Asociado a Mucosas” es un Linfoma no
Hodkin de células B, extranodal, encuadrado dentro del grupo de los linfomas de la
zona marginal (junto a los Linfomas B Esplénico y Linfoma B Ganglionar de la zona
marginal), siendo posible el compromiso de los nodos linfáticos y de otros órganos
durante su evolución. (48) Actualmente es reconocido como una entidad clínico-

patológica distinta, los linfomas MALT transformados en difusos de alto grado
(LMTAG) y los linfomas difusos de células grandes B (LDCGB). (49)
Se puede definir como Linfoma MALT o Maltoma a la proliferación neoplásica
monoclonal de linfocitos B que infiltran las glándulas gástricas, con típicas lesiones
linfoepiteliales. (48)
Se diagnostica más frecuentemente a partir de los 50 años, con un
predominio de los varones sobre las mujeres de 1,7:1. En el momento de su
diagnóstico es un tumor de bajo grado en un 70-85% de los casos. Asienta
preferentemente en el antro: 41%, pudiendo ser multifocal en un 33%. El número de
diagnósticos de linfoma gástrico primario es cada vez más frecuente; representa
aproximadamente 7% de todas las neoplasias malignas del estómago y alrededor
del 2% de todos los linfomas, siendo la localización gástrica la más frecuente entre
los linfomas MALT. (48)
En cuanto a su etiología; suele existir antecedente de enfermedad
autoinmune o inflamatoria. En concreto al linfoma MALT se le asocia con el
Síndrome de Sjögren, tiroiditis de Hashimoto y gastritis asociada a Hp. (50) El hecho
de que en la célula tumoral aparezcan mutaciones somáticas en la región variable
del gen de las inmunoglobulinas aboga por un contacto previo entre linfocitos y
antígeno. (51)
El estómago normal carece de tejido linfoide organizado, siendo la infección
crónica por la bacteria Hp la responsable de la aparición de tejido MALT en la
mucosa gástrica. Existe una estrecha relación entre la infección por el Hp y el
desarrollo de linfoma MALT gástrico, la cual está basada en datos epidemiológicos,
histológicos, inmunológicos-experimentales y en la respuesta del tumor tras la
erradicación del Hp. En estudios epidemiológicos se ha observado una significativa
prevalencia de infección por Hp en pacientes afectados con el linfoma MALT, con
una tasa de infección cercana al 100%. (51)
Las primeras relaciones que se establecieron entre Hp y linfoma MALT fueron
de tipo epidemiológico y corresponden al año 1991 con los trabajos de Parsonnet,
que describe un 90,9% de prevalencia de Hp en 11 casos de linfoma gástrico tipo

MALT. Ese mismo año Wotherspoon presenta una serie mucho más amplia con 110
linfomas gástricos MALT, entre los que registra un 92% de prevalencia de Hp
posteriormente han aparecido nuevas publicaciones que confirman estas altísimas
tasas de prevalencia, (52) llegándose incluso a cifras del 100%. (53)
El proceso comienza con una colonización e inflamación aguda de la mucosa
gástrica por el Hp con destrucción de foveolas gástricas. El Hp se aloja en ellas
creando una nube de amonio gracias a que posee una enzima, la ureasa, para
defenderse del medio ácido. Allí actúa extracelularmente sobre las vacuolas de
mucina, provocando en muchos casos una erosión de la mucosa. (50)
La inflamación aguda evoluciona a una inflamación crónica, con aumento de
linfocitos, células plasmáticas y eosinófilos. Inicialmente puede existir una gastritis
difusa y los linfocitos emigran al territorio gástrico, hasta los capilares de la lámina
propia. (50) En el curso de la gastritis crónica pueden aparecer folículos linfoides y
agregados linfáticos en la base de la mucosa gástrica, lo que constituye el llamado
tejido MALT. (51)
El diagnóstico de linfoma MALT se basa en la gastroscopia, con toma de
biopsias. La radiología baritada aporta datos inespecíficos. La gastroscopia muestra
una mucosa de aspecto “gastrítico” inespecífico, con la presencia de ulceraciones,
con pliegues engrosados y/o masas irregulares o polipoideas. El hallazgo de estas
lesiones junto a úlceras múltiples, estrelladas y en ocasiones confluentes, (que
incluso pueden rebasar el píloro y afectar al duodeno) es sugestivo de lesión
linfomatosa. (51)
Los pacientes suelen presentar síntomas como dolor abdominal, perdida de
peso, fatiga, sensación de plenitud, nauseas y vómitos y hasta en un 10-20% de
casos, según algunos autores es posible palpar masa abdominal. (51)
Tratamiento erradicador de la infección por Hp, quirúrgico, y oncológico no
quirúrgico con quimio y/o radioterapia.

4.3.-Enfermedad Periodontal. El concepto de “enfermedad periodontal” (EP) es muy extenso e inicialmente,
incorporaría a todas aquellas condiciones clínico-patológicas relacionadas con la
encía, ligamento periodontal, cemento dentario y hueso alveolar. Se ha
caracterizado a la EP como un proceso de causalidad compleja, de naturaleza
inflamatoria debido a infecciones y de múltiples manifestaciones, cuyo estado inicial,
en la gran mayoría de los casos, es de carácter reversible. (8, 54)
Se considera que, la EP es de origen multifactorial donde se involucran
factores del medio como la microbiota del surco; genéticos, inmunológicos y
sistémicos que dependen del individuo; y otros que se relacionan con los estilos de
vida como el hábito de fumar y la higiene bucal, sin dejar de mencionar la
organización de los servicios de salud, o sea, todo lo que depende de la atención
primaria y especializada en relación con esta enfermedad, que participa en la
promoción de salud, la prevención, el tratamiento, y la limitación de los daños. (55)
El término EP se utiliza para identificar a la enfermedad que va afectando
progresivamente a los tejidos de soporte del diente, avanzando desde los más
superficiales (encía) hasta los más profundos (hueso). Es una afección
particularmente grave, ya que en los grados más avanzados, conduce
irremediablemente a la movilidad y pérdida dentaria. (56)
Se debe reconocer que las enfermedades periodontales son procesos de
causa infecciosa, lo que es importante en la comprensión de su patogenia. Como en
toda infección, las bacterias y los tejidos afectados están en intima relación ya sea
mediante invasión bacteriana a los tejidos o por la acción de los productos tóxicos
bacterianos, que vencen los sistemas locales de defensa y producen destrucción
celular o activan mecanismos de reacción que condicionan la destrucción tisular. (56)
Anterior a los años setenta existían varias teorías que explicaban el inicio de
la EP. Se enseñó durante mucho tiempo que el 70 % de la población padecía de EP,
y el modelo que se definía sobre la naturaleza de esa afección era la acumulación
de bacterias de la placa sobre el diente y subgingivalmente, produciendo el cálculo.
La combinación de placa y cálculo formaban el saco, con perdida de hueso y
subsiguiente perdida del diente. Todos los factores en la superficie dentaria eran

peligrosos. Para esa época, los investigadores demostraron que un inadecuado
control de placa iniciaba la gingivitis y luego, se podría convertir en periodontitis.
Posteriormente la investigación concluyó que no todo el mundo era igualmente
susceptible al proceso de la enfermedad. Hoy es evidente que la mayoría de los
adultos padecen solamente de gingivitis y los casos de periodontitis severa
generalizada afectan solamente entre el 8 al 13 % de la población. (56)
4.3.1.-Epidemiología de la Enfermedad Periodontal.
En la década de los sesenta, se revisó la literatura disponible sobre la
epidemiología de la EP y se concluyó que la misma parece ser un problema grave
de salud pública global que afecta a la mayoría de la población adulta después de
los 35-40 años de edad. (8)
El 53% de los adultos tienen por lo menos un sitio de pérdida de inserción. Es
más probable que el aumento de la prevalencia con la edad sea un reflejo del efecto
acumulativo de la pérdida de inserción y no una mayor propensión a la periodontitis. (57)
Por lo general la periodontitis crónica adquiere importancia clínica después de
los 30 años de edad, y se caracteriza por la pérdida de inserción. Las observaciones
principales de varios estudios transversales en adultos de áreas geográficas
diferentes que analizaron muestras de un tamaño bastante grande. La mayoría de
los estudios se concentraron en las evaluaciones de la prevalencia de la EP
avanzada, parece ser que las formas graves de la misma afectan a una minoría de
sujetos de países desarrollados, sin exceder el 10-15% de la población. La
proporción de dichos sujetos aumenta considerablemente con la edad y parece
alcanzar su pico a los 50-60 años. (8)
El estudio del perfil epidemiológico bucal del distrito Maracaibo, realizado en
el año 1985 mostró, que en los grupos sociales mayoritarios (obreros, capas media y
sub empleados) entre 15-44 años estaban afectados en forma leve, los pacientes
mayores de 45 años estaban afectados en forma moderada y después de los 55
años en forma severa destructiva. (58)

En los Estados Unidos de América (EUA), alrededor de 25% a 30% de los
sujetos mayores de 30 años, sufren de algún grado de EP, desde ligera inflamación
con sangrado al sondeo hasta pérdida de inserción severa, incluyendo 3.1% de
periodontitis severa . Los datos de Brasil, Argentina y Chile muestran cifras de
periodontitis severa de 5.5%, 10% a 49% y 50%, respectivamente. En Colombia, se
presenta una pérdida de inserción periodontal de 50.2% que incluye 1.2% de pérdida
de inserción severa en sujetos con más de 30 años. En África, los mayores de 25
años muestran una pérdida de inserción periodontal severa entre 18% y 99%. (59)
En los caucásicos, la enfermedad parece afectar a las mujeres con mayor
frecuencia que a los varones y la prevalencia es baja. En otras razas y en particular
negros, la periodontitis crónica es más prevalente (1%) y la proporción por sexos
parece invertida, pues los varones son afectados con mayor frecuencia. (8)
En un estudio realizado en la Facultad de Odontología de la Universidad del
Zulia en la ciudad de Maracaibo, se analizó la prevalencia de la EP, indicando que la
misma se manifiesta mayormente en los pacientes adultos entre 36 – 60 años de
edad, donde en un 82% de la población estudiada exhibo algún tipo de periodontitis,
de las cuales un 84% concierne a Periodontitis Crónica y 14% a Periodontitis
Agresiva. El sexo femenino resulto ser el que más acude a recibir tratamiento
periodontal, (60) en el Zulia la Periodontitis es una de las enfermedades más
comúnmente diagnosticada ocupando una prevalencia del 60%. (61)
4.1.2.-Etiopatogenia de la Enfermedad Periodontal
Se reconoce que el factor etiológico primario y esencial en la patología
inflamatoria periodontal es la placa bacteriana (PB) y se sabe que ésta se organiza
en forma de biopelícula lo que aumenta su resistencia. Dentro de este contexto, el
conocimiento de los procesos reactivos inherentes al huésped y al atacante
(microorganismo) y a los procesos de formación de placa dental permite la
realización de diagnósticos acertados y la instauración de planes de tratamiento
adecuados, asociados con la prevención de la enfermedad atacando directamente al
agente etiológico y optimiza los resultados en el manejo de la patología en curso.
Durante años han existido diversas definiciones expresadas por diferentes

investigadores acerca del término de PB, pero actualmente, se define como una
comunidad microbiana compleja que se encuentra en la superficie de los dientes,
embebida en una matriz de origen bacteriano y salival. (12)
Se considera que la salud periodontal es un estado de equilibrio en el cual la
población de bacterias coexiste con el receptor y no hay daño a los tejidos del
mismo. La ruptura de ese equilibrio genera destrucción de los tejidos conectivos del
periodonto con la subsecuente aparición de la enfermedad. Si bien la EP es
multifactorial, esta no se produce en ausencia de placa. La eliminación de la PB
conduce a la desaparición de los signos y síntomas. La PB por si sola no produce
daño y su relación con la EP es compleja y va a estar determinada por las
características individuales del huésped y su capacidad de respuesta. (12)
Sabemos que el comienzo y la progresión de una determinada EP son
modificados por condiciones locales o sistémicas. Los procesos patológicos que
afectan a las estructuras del periodonto, incluyen a la gingivitis y a la periodontitis,
los cuales se producen como consecuencia de un desequilibrio entre los
microorganismos y los mecanismos de defensa del hospedero. Dicho desequilibrio
puede ser consecuencia de un cambio en el tipo de microorganismos, aunque a
veces se producen alteraciones en los mecanismos de defensa que permiten el
desarrollo de cambios patológicos con pequeñas modificaciones de la placa.
Asimismo, los factores del medio pueden establecer un ambiente propicio para que
se produzca este desequilibrio. (62)
Para que el ciclo infeccioso se desarrolle tienen que reunirse ciertas
condiciones tales como la presencia de bacterias patógenas, ausencia de bacterias
protectoras, presencia de una situación favorable para el desarrollo de bacterias
virulentas, y la deficiencia en la inmunidad innata o adquirida. En los paradigmas
actuales se establece que la presencia de especies bacterianas patogénicas es
necesaria pero no suficiente para desarrollar una periodontitis, destacando la
existencia de tipos de colonias virulentas y factores genéticos cromosomales y
extracromosomales para iniciar la enfermedad. (62)
Recientes estudios, han determinado que las bacterias son responsables de
la iniciación y progresión de la EP como del daño tisular que se produce como

consecuencia de la activación de los mecanismos de defensa dentro de los tejidos
periodontales. (62)
Aunque varias formas de EP están relacionadas, con bacterias específicas,
aún no se ha podido demostrar la relación causa-efecto. Se admite que la EP no
tiene una única causa, sino que es multifactorial y que las múltiples variables pueden
interactuar entre sí. A la fecha son más de 500 especies microbianas las que han
podido identificarse en la cavidad bucal, y un pequeño grupo de complejas bacterias
están relacionadas a la etiopatogenia de las diversas entidades de la EP, por esta
razón el término de PB puede resultar inespecífico para determinar su papel en el
desarrollo de la EP. (62)
Por lo tanto, se puede indicar que la EP es una consecuencia de la
interacción compleja de diversos factores de riesgo con los microorganismos y el
sistema inmunológico del paciente, existiendo una gran cantidad de especies que
participan, las cuales han hecho a la microbiología de la EP una de las mas
complejas áreas de estudio de todas las microbiologías clínicas. (63)
La etiología de la EP implica la participación de ciertas bacterias facultativas y
anaerobias que se localizan junto con otras en el espacio subgingival del diente para
conformar la denominada microbiota subgingival. En ella, reconocidos patógenos
periodontales como Actinobacillus actinomycetemcomitans, Porphyromonas
gingivalis, Tannerella forsythia (Bacteroides forsythus), Campylobacter rectus,
Eubacterium nodatum, Fusobacterium nucleatum, Peptostreptococcus micros,
Prevotella intermedia, Prevotella nigrescens, Dialister pneumosintes y Treponema
denticola, actúan en conjunto o independientemente para iniciar el proceso
inflamatorio que produce la periodontitis. Sin embargo, no se conocen todas las
especies comprometidas en el desarrollo de la enfermedad periodontal. Así, desde
hace algunos años se muestra interés por especies bacterianas poco frecuente en la
microbiota subgingival y que podrían contribuir con la patogenia periodontal en
personas que no responden adecuadamente a la terapia convencional. Dentro de
esos microorganismos se citan especies de la familia Enterobacteriaceae, bacilos
Gram negativos no fermentadores como especies de Pseudomonas, y también
algunos otros como levaduras y estafilococos. La característica que comparten los
microorganismos inusuales es la de ser patógenos oportunistas, es decir, que se

consideran parte de la microbiota normal en determinados sitios anatómicos y
aprovechan las condiciones de inmunodepresión para generar o agravar una
enfermedad. (60)
En estado de salud, la microflora aislada en el surco gingival consiste en una
asociación compleja de bacterias, con predominio de una microbiota residente
formada por microorganismos de los géneros Actinomyces y Streptococcus. Estas
bacterias son fundamentalmente aerobias o anaerobias facultativas. Cuando las
medidas de higiene no son muy efectivas, se acumula la PB, se consume la mayor
parte del oxígeno y comienzan a aparecer gérmenes anaerobios y especies Gram
negativas, entre los que predominan Fusobacterium nucleatum, Veillonella parvula y
Treponema sp. (64, 65, 66)
Si la placa formada sobre la superficie del diente no es removida, las
bacterias allí presentes se acumulan e inician una reacción inflamatoria en la encía;
el establecimiento de un proceso infeccioso provoca la disminución de la tensión de
oxígeno molecular y en consecuencia del potencial redox, con lo cual se favorece la
supervivencia y crecimiento de la flora bacteriana anaeróbica, prevaleciendo ésta
sobre la flora aeróbica y anaerobia facultativa. (67)
El acumulo de bacterias a lo largo del margen gingival lleva a la gingivitis.
Cuando queda establecida la gingivitis, el cultivo de las bacterias de los puntos
infectados revela un incremento en la cantidad de bacterias anaerobias estrictas en
relación con las anaerobias facultativas. (66)
El cultivo de muestras de casos avanzados de EP, demuestra el predominio
de bacilos anaerobios Gram negativos. (63) También hay cantidades importantes de
espiroquetas. La ausencia total de PB en la cavidad bucal no es alcanzable; se trata
de una ilusión, que ni siquiera es fisiológica. Sin embargo, es posible mantener
sanas la encía y el periodonto cuando la cantidad de placa es pequeña, la flora
bacteriana es poco virulenta (Gram. positivos y anaerobios facultativos) y el sistema
de defensa del receptor es suficientemente efectivo. (66)
Respecto a los posibles patógenos periodontales, el Actinobacillus
actinomycetemcomitans, es un bacilo pequeño no móvil, Gram negativo,

sacarolítico, capnofílico, que ha sido reconocido como posible patógeno, por la alta
frecuencia con que se detecta la gran concentración observada en las lesiones de
periodontitis de este microorganismo (8) A. actinomycemcomitans, se ha relacionado
principalmente con la Periodontitis Juvenil, el cual se ha encontrado colonizando la
mucosa bucal, paladar, lengua, así como en placa supra y subgingival de individuos,
tanto sanos como enfermos periodontalmente. (67)
El segundo patógeno periodontal potencial más intensamente estudiado es P.
gingivalis. Los cultivos de esta especie demuestran que es un microorganismo Gram
negativo, anaeróbio, no móvil, asacarolítico que suele presentarse en forma de
cocos o bacilos cortos. Produce respuestas inmunitarias elevadas generales y
locales en sujetos con diversas formas de periodontitis. (8)
Van Winkelhoff (1988); Marhs (2000); Liebana (2002), señalan que el poder
patógeno de esta bacteria en la colonización, destrucción del tejido periodontal y
evasión de las defensas del hospedero, tiene relación con un gran número de
factores de virulencia, tales como: Fimbrias, que se comportan como adhesinas, que
intervienen en el proceso de adhesión a tejidos del hospedero y en la coagregación
bacteriana; Hemaglutininas, que participan en la aglutinación de hematíes en los
inicios de la colonización tisular; Residuos proteicos, glucídicos y de
lipopolisacáridos, que contribuyen a los procesos de adhesión a células epiteliales, y
a la coagregación con otras bacterias; Cápsula, cuya acción es fundamentalmente
antifagocítica; Vesículas superficiales, que participan en la captación de nutrientes. (68)
También esta bacteria, es capaz de producir una variedad de enzimas que
causan alteraciones directamente en los tejidos del periodonto. Entre éstas, se
encuentran: la colagenasas o gingipainas, que actúa sobre el colágeno del ligamento
periodontal y hueso alveolar; las enzimas trípsicas, que lo hacen sobre el colágeno
alterado; hialuronidasa, que actúa sobre el ácido hialurónico del tejido favoreciendo
la difusión del microorganismo; fosfatasa ácida y alcalina, que determinan la pérdida
del hueso alveolar, y otras enzimas como fosfolipasa A, heparinasa,
desoxirribonucleasa, condroitinsulfatasa, gelatinasa y queratinasa, que de alguna
manera contribuyen a la destrucción tisular. (68)

En relación al Bacteroides forsythus, es un bacilo pleomórfico, anaerobio
estrictos, inmóvil, no esporulado, no fermentativo y no crece en bilis. Su crecimiento
in Vitro es difícil, sin embargo, el desarrollo de sus colonias en placas de
agar sangre es favorecido por la presencia de N-acetilmuramato, hemina y
vitamina K. El B. forsythus, se ha asociado frecuentemente con periodontitis
refractaria. Al parecer, su virulencia está relacionada con la producción de
neuraminidasas y enzimas tripsicas con especificidad sobre proteínas con residuos
de arginina. (68)
Se ha encontrado en cantidades mayores en EP destructiva y fue detectado
con mayor frecuencia lesiones periodontales activas que en inactivas. Los estudios
de Gmur y colaboradores en 1989 sugirieron que era una especie más común de lo
usualmente observado en estudios de cultivos y que sus niveles estaban
fuertemente relacionados con un incremento en la profundidad de la bolsa
periodontal. Grossi y colaboradores en 1995 consideraron que este patógeno es el
factor de riesgo microbiano más significativo, que distingue a los sujetos con
periodontitis de los periodontalmente sanos. Por su parte la Prevotella intermedia es
otro bacteroide, que parece tener propiedades virulentas como las mostradas
P.gingivalis. Se ha demostrado que por inyección induce infecciones mixtas en
animales de laboratorio. (8)
Otras especies patógenas han sido asociadas a la PB en la EP, tales como
fusobacterium nucleatum, Campylobacter rectus, Eikenella corrodens,
Peptostreptococcus micros, Streptococcus intermedius, Espiroquetas y ciertas
especies de Selenomonas y Eubacterium. No obstante, los datos limitados
existentes indican un posible papel etiológico para estas especies. (8)
La reacción inflamatoria consiste en una serie de fenómenos fisiológicos y
morfológicos en los que toman parte principalmente los vasos sanguíneos, células
de la sangre y tejidos conectivos adyacentes, en respuesta a la agresión de las
bacterias y sus productos con el propósito de destruir al agente extraño, iniciándose
simultáneamente procesos de reparación en el área afectada. (69, 70)
Ahora bien, para que un patógeno periodontal cause enfermedad es esencial
que sea capaz de colonizar el área subgingival y de producir factores que,

directamente, dañen los tejidos del receptor o indirectamente, conduzcan a la lesión
del tejido del receptor. Para colonizar sitios gingivales, una especie debe ser capaz
de adherirse a una o más superficies disponibles, multiplicarse, y competir
satisfactoriamente con otras especies de ese hábitat y defenderse contra
mecanismos hostiles. (8)
4.3.3.-Reacción del Receptor en la Enfermedad Periodontal
Los mecanismos de respuesta del huésped son básicamente defensivos
pero pueden ser responsables indirectamente del daño a los tejidos periodontales.
Los mecanismos del proceso de defensa pueden ser específicos e inespecíficos.
Los mecanismos inespecíficos son las barreras físicas del huésped como las
superficies epiteliales cutáneas y mucosas. Los mecanismos específicos son
resultado de la interacción entre el huésped y el agente atacante (respuesta
inmunológica del organismo). (12)
La distribución irregular de las bacterias subgingivales en la bolsa
periodontal causa una respuesta histológica de los tejidos blandos. Esta respuesta
del receptor muestra variaciones similares al modelo de destrucción y a pesar de los
esfuerzos de la investigación, no ha sido posible identificar el modelo de respuesta
histológica que caracterizaría una lesión, que desarrolle la progresión a EP. Se
considera que la gingivitis es la precursora de la periodontitis pero no es conocido
que la gingivitis se convierta en periodontitis o si las dos enfermedades sólo
ocurren simultáneamente. La lesión del tejido blando en la gingivitis y la periodontitis
se caracteriza por tres tipos de reacciónes principales. (54)
a) Respuesta Vascular: Los vasos laterales del epitelio de unión coexisten en
un plexo venoso post capilar. Estos vasos se agrandan y proliferan en la
inflamación periodontal y gingival. Estas alteraciones en su mayoría se observan en
la vecindad del epitelio de la bolsa y cerca de la placa subgingival.
b) Respuesta Celular. Clínicamente en la encía saludable hay una migración
continua de leucocitos de los vasos capilares al epitelio de unión y a la región del
surco ó hendidura gingival. Esta migración leucocitaria se refuerza en la inflamación

gingival y periodontal; y las células aumentan en el epitelio del surco y de la bolsa.
Las células participan en la defensa del tejido contra la invasión franca de bacterias.
c) Respuesta Inmune: El epitelio de unión y el epitelio de la bolsa son muy
permeables a moléculas grandes, que activan una reacción inmune en el tejido
conjuntivo gingival. En las fases tempranas de esta reacción, predominan los
linfocitos para luego aparecer las células plasmáticas y en forma tardía predomina
la infiltración inmune. La reacción inmune genera los mediadores que refuerzan la
inflamación y el daño al tejido. En particular la producción de citoquinas es
prominente. Estas sustancias son moléculas muy potentes que activan varios
eventos perjudiciales en el tejido conjuntivo. (53)
La respuesta inflamatoria y la respuesta inmune en el hombre, juegan un rol
en el mantenimiento de la integridad del receptor. Ante la constante amenaza de
invasión y colonización de microorganismos, la respuesta inflamatoria e inmune
provee al receptor de una defensa en contra de los microorganismos. Pero un
fracaso de la respuesta, sea ésta una respuesta disminuida o exuberante, resulta en
un progreso de la enfermedad. (71)
Una vez que los elementos de protección en el periodonto han sido atacados
por los mecanismos de virulencia bacteriana, un número de procesos destructivos
mediados por el receptor en los tejidos revelan la inducción, estimulación o
activación de sus defensas o de factores humorales que después causan
destrucción del tejido periodontal del lugar. (71)
La destrucción de tejidos asociados con EP, ocurre como una consecuencia
del intento del receptor para eliminar la bacteria del surco gingival. Los mecanismos
inmunorreguladores así como el incremento en la producción de mediadores
inflamatorios han sido implicados en estos procesos patológicos. (71)
Por otra parte, si bien la EP es causada por infección crónica de anaerobios
Gram negativos, la severidad de la afección parece ser conducida principalmente
por la magnitud de la respuesta inflamatoria del receptor. Los patógenos de la EP
son necesarios, pero no son suficientes para la expresión de la enfermedad. El rol
de la respuesta inflamatoria del receptor parece ser el determinante crítico de

susceptibilidad y severidad. (71)
Las células involucradas en los procesos de defensa del huésped son: Leucocitos Polimorfonucleares (PMN): Los PMN de la hendidura gingival
constituyen la primera línea de defensa contra los patógenos periodontales, los
cuales pueden contribuir a la patología de la EP. La elastasa, una proteasa de
serina, que es un constituyente granular primario de los PMN causa degradación
tisular y está presente con mayor actividad en sitios de inflamación gingival (Murray y
cols. 1995). (12) Hay evidencia, de que la mayor parte de la destrucción de tejidos en
periodontitis es un resultado de la movilización y la activación de monocitos,
linfocitos y fibroblastos y otras células en el tejido del receptor. El compromiso de
estos elementos celulares por factores bacterianos, en particular los
Lipopolisacaridos, se cree que estimula la producción tanto de citoquinas y
mediadores inflamatorios, además de metabolitos del ácido araquidónico como
prostaglandinas E2, entre las citoquinas y los mediadores inflamatorios más
consistentemente asociados con periodontitis se encontraron los siguientes:
prostaglandinas E2 (PGE2), Interleukina 1 (IL – 1), Interleukina 6 (IL – 6), 8
Interleukina (IL – 8), y factor de necrosis tumoral alfa (FNT.-α ).(71)
Macrófagos: Tienen una función directa importante en la inmunidad celular.
Estas células grandes notablemente fagocíticas forman parte del sistema
reticuloendolelial depurador. Su actividad fagocítica mejora por receptores de
superficie para la porción Fe de la inmunoglobulina G (IgG), que provee mayor
contacto de los antígenos con el macrófago luego de la interacción antígeno-
anticuerpo, Participan con los linfocitos T al favorecer la respuesta de los linfocitos B
ante muchos inmunógenos. Se considera que los macrófagos procesan el antígeno
para el linfocito B, En las lesiones inflamatorias, los macrófagos se forman por la
diferenciación de monocitos transportados a la lesión por la sangre. Dichas células
actúan de modo no específico con los antígenos que les aportan la capacidad para
destruir unos grupos diversos de bacterias, sin nexo antigénico. (12)

Linfocitos: Incluyen tres tipos de células: Linfocitos T, o células T. derivados del timo y con una función en la inmunidad
celular: 2) linfocitos B. o células B. derivados del hígado, el bazo y la médula ósea.
Precursores de las células plasmáticas que intervienen en la inmunidad humoral, y
3) células asesinas naturales (NK, por sus siglas en inglés natural killer) y asesina.
Se reconoce que las células T están compuestas por varios subconjuntos que
modulan la reacción humoral, Estos incluyen células T cooperadoras-inductoras
(células TH) (CD4 positivas), que ayudan en la reacción celular de las células B para
que se diferencien en células plasmáticas y produzcan anticuerpos y células T
supresoras-citotóxicas (células Ts) (CD8 positivas), que estimulan la actividad
citotóxica y microbicida de las células inmunitarias. Las células TH liberan IL-2 e IFN-
g. en tanto que las células Ts liberan IL-4 c IL-5. Las células T se subdividen aún
más en tres subgrupos (T H1, TH2, TH O), que se distinguen por sus perfiles de
producción de citocinas. En la periodontitis del adulto las células TH aumentan y las
células TS decrecen con el incremento de la inflamación gingival. (8)
Las células B se reconocen por su inmunoglobulina de superficie celular, a
menudo inmunoglobulina M (IgM) o (IgD). Sin embargo, algunas células B prensan
igG. IgA o E. Estas inmunoglobulinas de superficie sirven como receptores para
antígenos. Las células NK se caracterizan por su falta de receptores para células T
(TCR. Por sus siglas en inglés T-cell receptors) e inmunoglobulina de superficie. La
interacción entre antígenos y macrófagos conocida como procesamiento de
antígenos conduce a la activación de las células NK. (11)
Citoquinas: Las citoquinas son proteínas solubles, segregadas por células,
que actúan como moléculas mensajeras que transmiten señales a las otras células.
Las interleucinas son miembros importantes del grupo de las citoquinas y están
involucradas primariamente en la comunicación entre los leucocitos y las otras
células implicadas en los procesos inmunitarios e inflamatorios. El control de la
liberación y acción de la citoquina es complejo y depende de inhibidores y
receptores. Muchas citoquinas son capaces de actuar sobre la célula que las
produjo, de manera que autoestimulan su propia producción y la producción de otras
citoquinas. (12)

Citoquinas proinflamatorias: Las citoquinas interleucina-1 (IL), IL-6 y factor de
necrosis tumoral (TNF) estimulan la reabsorción ósea in Vitro e in vivo. Tanto el A.
Actinomicetemcomitans como P. gigivalis inducen la liberación de IL-1 de los
macrófagos la cual juega un papel importante en la reabsorción del hueso. (12)
Citoquinas quimiotácticas: Han sido identificadas una serie de más de 20
moléculas entre las cuales la más famosa y mejor caracterizada es la interleucina 8
(IL-8). (12)
La reacción del huésped es determinante en el desarrollo o la progresión de
la enfermedad periodontal. Es importante tener presente la existencia de
condiciones que afecten la reacción normal del huésped y la respuesta a estímulos
negativos.
Puede indicarse que, aún cuando las EP son procesos infecciosos cuyo factor
etiológico esencial es la biopelícula de PB, no obstante, se encuentran
condicionadas por una cantidad de factores no clarificados en su totalidad. Entre las
causas de la EP podrían encontrarse procesos intrínsecos al ser humano, como la
respuesta del receptor. Se ha señalado que entre los factores asociados a tomarse
en cuenta están la edad, la situación económica y la biología de la PB. A estos
factores asociados se suman como factores de riesgo. (62)
Son diversos los factores de riesgo de la EP entre ellos se mencionan los
siguientes. (61)
a) Innatos: raza, sexo, factores genéticos, inmunodeficiencias congénitas,
disfunción fagocítica, síndrome de Down, síndrome de Papillon-Lefevre, síndrome de
Ehler-Danlos.
b) Adquiridos y ambientales: mala higiene oral, edad, medicamentos,
tabaquismo, estrés, defectos inmune adquiridos, enfermedades endocrinas
adquiridas, enfermedad inflamatorias adquiridas, deficiencias nutricionales,
osteoporosis, y medicación con esteroides por períodos prolongados entre otros. (58,
61)

En las últimas décadas se ha discutido la importancia relativa de las bacterias,
de los factores locales que facilitan su desarrollo, así como también las influencias
sistémicas en la etiología de la EP. Se conoce que el comienzo y la progresión de
una determinada EP es modificada por condiciones locales o sistémicas. Así mismo,
los factores del medio pueden establecer un ambiente propicio para que se produzca
un desequilibrio. Las bacterias son necesarias pero no suficientes para que se
desarrolle la enfermedad. (62)
Es importante recordar que cualquier condición que comprometa los
mecanismos de defensa del receptor, incluyendo la irradiación y enfermedades de
transmisión genética, predispone a los pacientes a episodios tempranos de
periodontitis severa. (72)
4.3.4.-Clasificación de Enfermedades y Lesiones Periodontales:
La clasificación presentada a continuación se basa en la opinión consensuada
internacional mas reciente respecto a las enfermedades que afectan los tejidos del
periodonto. Se presentó y analizó en el International Workshop for the Classification
of Periodontal Diseases de 1999, organizado por la American Academy of
Periodontology: (73)
I. Enfermedad Gingival:
Enfermedad Gingival Inducida por Placa Dental.
I. Gingivitis asociada con Placa Dental únicamente.
A. Sin otros factores locales asociados.
B. Con otros factores locales asociados (Ver VIII-A).
II. Enfermedad Gingival Modificada por Factores Sistémicos.
A. Asociada con el Sistema Endocrino.
1. Gingivitis Asociada con la Pubertad.
2. Gingivitis Asociada con el Ciclo Menstrual.
3. Gingivitis Asociada con el Embarazo.
a. Gingivitis.

b. Granuloma Piógeno
4. Gingivitis Asociada a Diabetes Mellitus.
B. Asociada con Discrasias Sanguíneas.
1. Gingivitis Asociada con Leucemia.
2. Otros.
III. Enfermedad Gingival Modificada por Medicamentos.
A. Enfermedad Gingival Influenciada por Fármacos.
1. Agrandamientos Gingivales Influenciados por Drogas.
2. Gingivitis Influenciada por Drogas.
a. Gingivitis Asociada a Anticonceptivos Orales.
b. Otras.
IV. Enfermedad Gingival Modificada por Malnutrición.
A. Gingivitis Asociada a Deficiencia de Ácido Ascórbico.
B. Otras.
Lesiones Gingivales No Inducidas por Placa.
I. Enfermedad Gingival de Origen Bacteriano Específico.
A. Lesiones Asociadas con Neisseria Gonorrhoeae.
B. Lesiones asociadas con Traponema Pallidum.
C. Lesiones Asociadas a Especies Streptocócicas.
D. Otros.
II. Enfermedad Gingival de Origen Viral.
A. Infecciones por el Herpes Virus.
1. Gingivoestomatitis Herpética Primaria.
2. Herpes Oral Recurrente.
3. Infecciones por Varicella Zoster.
B. Otras.
III. Enfermedad Gingival de Origen Fúngico.
A. Infecciones por Especies de Candida.
B. Eritema Gingival Lineal.
C. Histoplasmosis.

D. Otras.
IV. Lesiones Gingivales de Origen Genético.
A. Fibromatosis Gingival Hereditaria.
B. Otros.
V. Manifestaciones Gingivales de Condiciones Sistémicas.
A. Desórdenes Mucocutáneos.
1. Liquen Plano.
2. Penfigoide.
3. Pénfigo Vulgar.
4. Eritema Multiforme.
5. Lupus Eritematoso.
6. Inducidas por Drogas.
7. Otras.
B. Reacciones Alérgicas.
1. Reacciones a los materiales restaurativos dentales.
a. Mercurio.
b. Níquel.
c. Acrílico.
d. Otros.
2. Reacciones atribuidas a
a. Cremas Dentales.
b. Enjuagues Dentales.
c. Aditivos de Gomas de Mascar.
d. Aditivos de los Alimentos.
3. Otras.
VI. Lesiones Traumáticas.
A. Lesiones Químicas.
B. Lesiones Físicas.
C. Lesiones Térmicas.
VII. Reacciones a Cuerpo Extraño.
VIII. Otras no Específicas.

II. Lesiones Periodontales
II. Periodontitis Crónica.
A. Localizada.
B. Generalizada.
III. Periodontitis Agresiva
A. Localizada.
B. Generalizada.
IV. Periodontitis como manifestación de enfermedades sistémicas.
A. Asociada con Desórdenes Hematológicos.
1. Neutropenia Adquirida.
2. Leucemia.
3. Otros.
B. Asociada con Desórdenes Genéticos.
1. Neutropenia Cíclica Familiar.
2. Síndrome de Down.
3. Síndromes de Deficiencia de Adhesión Leucocitaria.
4. Síndrome Papillon-Lefèvre.
5. Síndrome de Chediak-Higashi.
6. Histiocitosis.
7. Enfermedad de Almacenamiento de Glicógeno.
8. Agranulocitosis Genética Infantil.
9. Síndrome de Cohen.
10. Síndrome de Ehlers-Danlos.
11. Hipofosfatasia.
12. Otros.
C. Otros no específicos.
V. Enfermedad Periodontal Necrotizante.
A. Gingivitis Ulceronecrotizante.
B. Periodontitis Ulceronecrotizante.
VI. Absceso Periodontal.
A. Absceso Gingival.

B. Absceso Periodontal.
C. Absceso Pericoronal.
VII. Periodontitis Asociada con Lesiones Endodónticas.
A. Lesiones Combinadas Endo-Periodontales.
VIII. Condiciones o deformidades del desarrollo o adquiridas
A. Factores Localizados Relacionados a los Dientes que Modifican o
Predisponen a la Enfermedad Gingival Inducida por Placa o
Periodontitis.
1. Factores Anatómicos Dentales.
2. Aparatos y Restauraciones Dentales.
3. Fracturas Radiculares.
4. Reabsorción radicular cervical y Lágrimas de Cemento.
B. Condiciones y Deformidades Mucogingivales Adyacentes a los
Dientes.
1. Resección de los Tejidos Gingivales Blandos.
a. Superficies Lingual o Vestibular.
b. Interproximal (Papilar).
2. Ausencia de Encía Queratinizada.
3. Profundidad Vestibular Disminuida.
4. Posición Aberrante de Músculos/Frenillo.
5. Exceso Gingival.
a. Seudobolsas.
b. Margen Gingival Inconsistente.
c. Gran exceso Gingival.
d. Agrandamiento Gingival.
6. Color Anormal.
C. Condiciones y Deformidades Mucogingivales en Rebordes
Edentulos.
1. Deficiencia de Reborde Horizontal y/o Vertical.
2. Ausencia de Tejido Queratinizado/Encía.
3. Agrandamiento de Tejido Blando/Gingival.

4. Posición Aberrante de músculos/Frenillo.
5. Profundidad Vestibular Disminuida.
6. Color Anormal.
D. Trauma Oclusal.
1. Trauma Oclusal Primario.
2. Trauma Oclusal Secundario.
4.4.- Relación entre Helicobacter Pylori y Enfermedad Periodontal:
El Helicobacter pylori es un patógeno gastrointestinal importante asociado con
gastritis, úlceras pépticas y aumenta el riesgo de Carcinoma gástrico. La cavidad
bucal se ha indicado como posible depósito de Hp, y puede por lo tanto estar
implicada en la reinfección del estómago posterior a la terapia de erradicación de la
bacteria (18). Estudios realizados indican que el Hp está presente en la PB, aunque
el número de microorganismos en muestras individuales es muy bajo, estos
números parecen variar de un sitio a otro dentro de la cavidad bucal. La presencia
de este microorganismo en la placa puede ser intermitente, quizás ocurriendo como
resultado del reflujo gastroesofágico. (74)
La eficacia de la terapia de erradicación para la infección gástrica de Hp es
afectada por la presencia de Hp en la cavidad bucal (16). La erradicación de Hp
después de dicha terapia es más efectiva para el estómago que para la boca. (75)
4.4.1.- Estudios de Intervención:
En un estudio realizado en el Postgrado de la clínica estomatológica de la
Facultad de Odontología UCV por Berroteran y cols. en 2001, se evaluó el potencial
de la cavidad bucal en los procesos patogénesis por Hp, la presencia de este
microorganismo fue determinada en 40 pacientes provenientes de Hospital Clínico
de la Universidad Central de Venezuela, quienes asistían para rutina endoscópica, y
en 20 pacientes asintomáticos (grupo control). Fueron tomadas biopsias gástricas y
muestras de placa dental y sembradas en medios selectivos y no selectivos. El Hp
fue detectado en 45% (18/40) de las biopsias de estómago y en 17,5% (7/40) de las
muestras de PB por medio del cultivo microbiológico. Concluyendo que la PB puede

ser un importante reservorio para Hp y la detección de esta bacteria en la cavidad
bucal de pacientes con gastritis podría sugerir la vía bucal como una importante
fuente de transmisión. (76)
Scarano Pereira y cols. en el año 2005 en su estudio intentaron determinar la
presencia de Helicobacter pylori en placa dental y en mucosa gástrica de pacientes
adultos, para lo cual se seleccionaron 48 pacientes, que habían de someterse a
endoscopia por indicación médica. En todos los pacientes se recogió placa dental
antes de la endoscopia. A partir de la placa dental se realizó extracción de ADN, con
finalidad de identificar Hp por el método de Reacción en Cadena de la Polimerasa
(RCP o PCR). Posteriormente, los pacientes fueron sometidos a endoscopia
digestiva, mediante la cual se tomaron muestras de mucosa gástrica. A continuación
se extrajo ADN para la posterior identificación de Hp por el método PCR. La
presencia de Hp fue observada en el 100% de las muestras de placa dental y en el
66,67% de las de mucosa gástrica. De los 48 pacientes se constató que 42 (87,5%)
presentaban alteraciones gastroduodenales y, entre ellos, 29 (69,05%) presentaron
reacción positiva para Hp en placa dental y en mucosa gástrica. Entre los 6
pacientes con endoscopia normal se encontraron 3 casos (50%) con presencia
simultánea de Hp en placa dental y mucosa gástrica. Los resultados de este estudio
sugieren la presencia simultánea de Hp en placa dental y mucosa gástrica en
elevadas proporciones. Estos resultados permiten sugerir además que la placa
dental puede constituir un reservorio importante de este microorganismo. (77)
Moromi Nakata y cols. en el 2001 realizaron un estudio con el objetivo de
determinar la prevalencia de helicobacter pylori en pacientes con gingivitis y
enfermedad periodontal, se colectó y procesó muestras de placa subgingival de 117
pacientes categorizados según los siguientes antecedentes registrados: 1) úlcera y
gingivitis, 2) gastritis y gingivitis, 3) gingivitis y 4) periodontitis, aleatoriamente
seleccionados en la Clínica a Odontológica de la Universidad de San Marcos. Se
recupero Hp en 9,4% de la población en estudio, más frecuentemente en mujeres
(6,8%) que en hombres (2,6%). Hp se recupero en el 7,7 % de pacientes con
gingivitis, y en el 1, 7 % de pacientes con gastritis y gingivitis. No se recuperó en
pacientes con úlcera y gingivitis, ni en pacientes con periodontitis. Hp se recuperó
mayormente en los estratos de 20 a 30 años, y más frecuente en mujeres que
hombres. Discusión: En el presente estudio, el porcentaje de Hp hallado (9.4%) en

pacientes con procesos inflamatorios moderados, mediante cultivo en medio
selectivo, puede considerarse dentro de los márgenes de porcentajes hallados por
otros autores a partir de la placa dental. Es importante mencionar que el mayor
porcentaje de las muestras de pacientes positivos para Hp se da en casos con
gingivitis y sin que el paciente mencione gastritis; es probable que los síntomas no
son tan evidentes o que en su defecto son portadores que podrían ser colonizados
favorablemente si las condiciones son propicias, como la existencia del stress. Otro
aspecto que llama la atención es el mayor porcentaje de positivos hallados en la
población de adultos jóvenes y no en los de mayor edad. Sin embargo la carencia
de resultados positivos en los cultivos se debe atribuir a varios causas, entre ellas a
la presencia de células de formas cocoides no viables, pero que pueden sobrevivir
en la placa dental. (78)
En un estudio más reciente, realizado por Gebara y cols. en el 2006 se
propuso evaluar si la cavidad oral de pacientes con Periodontitis crónica puede
albergar Hp después de la terapia sistémica de erradicación. Se evaluaron las
muestras de 30 pacientes (15 con gingivitis y 15 con Periodontitis crónica) positivo
para Hp en el estómago. Las muestras fueron tomadas de la saliva, microbiota del
dorso de la lengua, y placa de supra- y sub-gingival así como biopsias gástricas y
se reunieron 3 meses después que se triplica la terapia antibiótica sistémica; el ADN
de cada muestra fue extraído por el método hirviente y utilizado como una plantilla
en el reacción en cadena de polymerase con el primers JW22/23. Los resultados:
Dieciocho pacientes (60%) albergaron Hp en bocas. Cinco pacientes (16.6 %) eran
positivo en la saliva, dos (6.6 %) en el dorso de la lengua, nueve (30%) en la placa
de supra-gingival, 14 (46.6 %) en la placa de sub-gingival y tres (10%) en el
estómago. No hubo diferencia estadísticamente significativa entre grupos de estudio.
Conclusión: La erradicación de H. pylori después que la terapia era más efectiva
para el estómago que para la boca (p<0.001). Las bocas de pacientes con gingivitis
o con Periodontitis crónica, que eran positivos para Hp en estómagos, se pueden
considerar como depósitos de estas bacterias. (79)
Con el propósito de determinar si placa dental, la higiene oral pobre, y la
enfermedad periodontal eran los factores del riesgo para infección de Hp, Anand, P.
S y cols. en el 2006 realizaron un estudio con 134 pacientes, a los cuales se, les
clasificó como casos, 65 pacientes que dieron positivo a la prueba de serología de

Hp o prueba de ureasa, evidencia histológica rápida positiva para la presencia de
Hp en especimenes de biopsia antral. Quedando como grupo control 69 pacientes
que eran negativos para Hp en la prueba serología, rápida de ureasa, e histológica.
En los resultados se encontró que la asociación de la enfermedad periodontal, la
higiene oral pobre y la infección con Hp no era significativa. Había una frecuencia
más alta de H. pylori en la placa dental de pacientes con infección gástrica por Hp
que en controles, pero agrupándolos tuvieron una taza sorprendentemente alto con
la prueba ureasa positiva para Hp en la placa (89% y 71%, respectivamente).
Conclusiones: Hp en la placa dental rara vez es eliminado con la terapia antibiótica
de erradicación de Hp, y esto puede actuar como una fuente de futura reinfección.
De ahí, la importancia de acompañar la terapia de erradicación de Hp en el
estomago con lo terapia periodontal. (75)
Campuzano, S. y cols. en el 1996 realizaron un estudio en el cual se plantea
aislar el Hp a partir de muestras de placa dental y para este fin, se estudiaron 300
adultos de diferentes sitios de Santa fe de Bogotá; todos los pacientes estudiados
fueron asintomáticos. La colonización por Hp se puede determinar por varias
técnicas. La prueba de mayor rapidez es la detección de la actividad de ureasa la
cual se aplicó a todas las muestras. La ventaja del cultivo es la posibilidad de
determinar la sensibilidad de las cepas aisladas frente a los antibióticos; situación
que es importante para el manejo del paciente. Hp crece como colonias pequeñas,
claras y circulares. La identificación bacteriana fue realizada en base a morfología,
tinción Gram y reacciones positivas de ureasa, catalasa y oxidasa. En este estudio
se demuestra que Hp se puede aislar de la placa dental, hallazgo que tiene
implicaciones fundamentales desde el punto de vista epidemiológico con respecto a
las vías posibles de diseminación de este importante patógeno responsable de gran
parte del espectro de la enfermedad acido-péptica y probablemente implicada en la
patogenia del cáncer gástrico. (80)
Chumpitaz, J y cols. en el 2006 en Perú estudiaron 115 pacientes cuyas
edades fluctuaron entre 20 y 70 años, habiendo un predominio en mayores de 50
años y del sexo femenino. La población estudiada correspondió a los pacientes con
síntomas de gastritis que acudieron al consultorio de gastroenterología y que fueron
programados para una endoscopia. A todos estos pacientes se diagnosticó gastritis
leve o moderada según la observación del gastroenterólogo de turno. Los pacientes

que acuden a este consultorio son asegurados de la zona sur correspondiente a los
distritos de Surquillo, Miraflores, San Isidro. Se ha tomado como criterio de exclusión
los casos de pacientes con otra patología como úlcera gástrica o duodenal,
esofagitis, pacientes que han tenido tratamiento previo con antibióticos para
erradicar el Hp y los que presentan dentadura postiza completa. La muestra de sarro
dentario se tomo después de la endoscopia, mediante raspado de la superficie
dentaria con cureta de Gracey colocándose la muestra en medio de transporte.
Luego de sembrarse y cultivarse en medio selectivo con antibióticos igual al medio
de Skirrow para Campylobacter microaerófilo a 37ºC. Después de 5-10 días se
observó colonias sospechosas haciéndose coloración y la pruebas de ureasa,
oxidasa y catalasa para la identificación de Hp. Al mismo tiempo se hizo la biopsia
de las zonas de antro y fondo del estómago que se recolectaron en frascos con
formol y transportaron a patología para estudio histopatológico, coloración de
hematoxilina eosina para el estudio anatopatológico y para diagnóstico de casos de
gastritis con o sin atrofia o metaplasia, además de la presencia de Hp por la
observación microscópica de su morfología. (81)
De Sousa, L. y cols., estudiaron 97 pacientes dispépticos y 50 pacientes sin
dispepsia a quienes se les tomó muestras de biopsia gástrica, placa dental y saliva.
Las muestras de biopsia gástrica fueron evaluadas mediante métodos
microbiológicos e histológicos. A las muestras de cavidad oral se les realizó cultivo y
prueba de la ureasa y se incluyó un método de pretratamiento, usando urea y HCl.
La prevalencia de infección por Hp fue de 75,5% en la totalidad de los pacientes
evaluados. No se logró aislar Hp en saliva y placa dental de ninguno de los dos
grupos estudiados, con o sin pretratamiento de la muestra. La prueba de la ureasa
en placa dental fue positiva en 99,3% de los pacientes y 89,8% en saliva. No hubo
diferencia estadísticamente significativa entre la frecuencia de infección por Hp en
pacientes dispépticos y sin dispepsia. Los resultados obtenidos sugieren que la
metodología empleada para la detección de Hp no es suficientemente sensible para
la determinación del microorganismo en cavidad oral. (82)
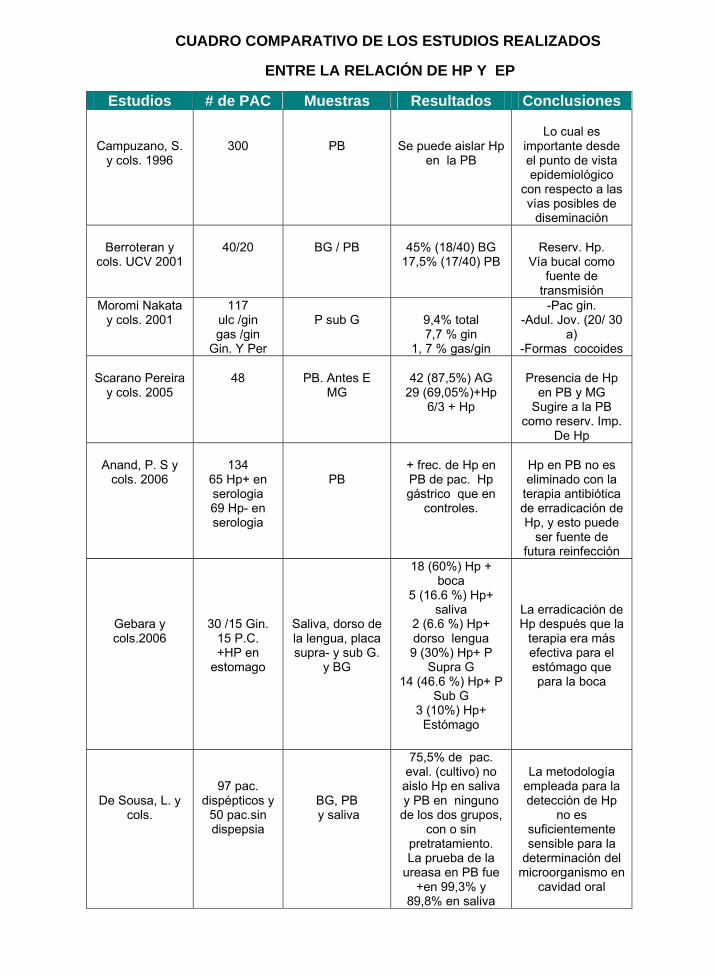
CUADRO COMPARATIVO DE LOS ESTUDIOS REALIZADOS
ENTRE LA RELACIÓN DE HP Y EP
Estudios # de PAC Muestras Resultados Conclusiones
Campuzano, S. y cols. 1996
300
PB
Se puede aislar Hp en la PB
Lo cual es
importante desde el punto de vista epidemiológico
con respecto a las vías posibles de
diseminación
Berroteran y cols. UCV 2001
40/20
BG / PB
45% (18/40) BG
17,5% (17/40) PB
Reserv. Hp.
Vía bucal como fuente de
transmisión Moromi Nakata
y cols. 2001 117
ulc /gin gas /gin
Gin. Y Per
P sub G
9,4% total 7,7 % gin
1, 7 % gas/gin
-Pac gin. -Adul. Jov. (20/ 30
a) -Formas cocoides
Scarano Pereira
y cols. 2005
48
PB. Antes E
MG
42 (87,5%) AG
29 (69,05%)+Hp 6/3 + Hp
Presencia de Hp
en PB y MG Sugire a la PB
como reserv. Imp. De Hp
Anand, P. S y
cols. 2006
134
65 Hp+ en serologia 69 Hp- en serologia
PB
+ frec. de Hp en PB de pac. Hp gástrico que en
controles.
Hp en PB no es eliminado con la
terapia antibiótica de erradicación de Hp, y esto puede
ser fuente de futura reinfección
Gebara y cols.2006
30 /15 Gin. 15 P.C. +HP en
estomago
Saliva, dorso de la lengua, placa supra- y sub G.
y BG
18 (60%) Hp + boca
5 (16.6 %) Hp+ saliva
2 (6.6 %) Hp+ dorso lengua 9 (30%) Hp+ P
Supra G 14 (46.6 %) Hp+ P
Sub G 3 (10%) Hp+
Estómago
La erradicación de Hp después que la
terapia era más efectiva para el estómago que para la boca
De Sousa, L. y cols.
97 pac. dispépticos y
50 pac.sin dispepsia
BG, PB y saliva
75,5% de pac. eval. (cultivo) no aislo Hp en saliva y PB en ninguno
de los dos grupos, con o sin
pretratamiento. La prueba de la
ureasa en PB fue +en 99,3% y
89,8% en saliva
La metodología
empleada para la detección de Hp
no es suficientemente sensible para la
determinación del microorganismo en
cavidad oral

Significado de las abreviaturas utilizadas en este cuadro:
Hp: Helicobacter pylori. BG: Biopsia Gástrica. PB: Placa Bacteriana. MG: Mucosa Gástrica. E: Endoscopia. Ulc: Ulcera. Gas: Gastritis. Gin: Gingivitis. Per: Periodontitis. Pac: Paciente. P Sub G: Placa sub gingival. P Supra G Placa supra gingival. 4.4.2 Perfil del paciente susceptible a padecer la asociación de estas enfermedades.
Pacientes con alteraciones gastroduodenales Helicobacter pylori positivos
en el estomago
Pacientes con enfermedad periodontal que pueden o no manifestar síntomas
gástricos
Pacientes que asistan a hogares de cuidado diario, que vivan en
hacinamiento, asilos o en sanatorios.

V.- OBJETIVOS DE LA INVESTIGACIÓN
Objetivo General:
• Se analizó a través de la literatura científica la relación del Helicobacter Pylori
y la enfermedad periodontal
Objetivos Específicos:
• Se consideró la estructura y mecanismos patógenos del Helicobacter pylori.
• Se describieron los aspectos clínicos epidemiológicos y etiopatogénicos de la
enfermedad periodontal y de las enfermedades del aparato digestivo
causadas por Helicobacter Pylori.
• Se analizó la relación entre la enfermedad periodontal y las enfermedades
del aparato digestivo causadas por Helicobacter Pylori.
• Se describió el perfil del paciente susceptible a padecer la asociación de
estas enfermedades.

VI.- MATERIALES Y MÉTODOS Tipo de investigación:
El estudio realizado fue de tipo documental.
Tipo de Diseño:
En este estudio se utilizó un diseño no experimental, pues se dirigió a analizar
a través de la literatura científica la relación del Helicobacter Pylori y la Enfermedad
Periodontal
Unidad de información:
La realización de la revisión bibliográfica se centró en dos líneas de compilación
de información:
Búsqueda de artículos a través de Internet en servidores especializados como
son: PUBMED, National Institute of Dental and Craneofacial Research
(NIDCR), Biblioteca Cochrane Plus, SCIELO, EMBASE, Odontología-Online,
Acta Odontológica Venezolana, DOCMEDICAL.
Búsqueda de textos y revistas especializadas en bibliotecas y hemerotecas
de la Facultad de Odontología y Medicina Universidad del Zulia.

VII.- CONCLUSIONES
La mayor parte de los estudios analizados reportan que la placa bacteriana
puede servir como reservorio importante de Helicobacter pylori.
Se hace referencia a que las condiciones que puedan facilitar la colonización
oral de Helicobacter pylori son:
Reflujo gastroesofágico
Malos hábitos de higiene
Infección intrafamiliar entre otros
Se indicó que el Helicobacter pylori en la placa dental rara vez es
eliminado con la terapia antibiótica de erradicación del mismo en el estómago,
y esto puede actuar como una fuente de futura reinfección.
Algunos de los resultados obtenidos sugieren que la metodología empleada
para la detección de Hp no es suficientemente sensible para la determinación
del microorganismo en cavidad oral.
La asociación de la Enfermedad Periodontal y las Enfermedades del aparato
digestivo causadas por Helicobacter pylori pueden suceder en pacientes
Helicobacter pylori positivos en el estomago, pacientes con enfermedad
periodontal que pueden o no manifestar síntomas gástricos, pacientes que
asistan a hogares de cuidado diario, que vivan en hacinamiento, asilos o en
sanatorios.
Es importancia acompañar la terapia de erradicación de Hp en el estomago
con lo terapia periodontal.

VIII.- RECOMENDACIONES
Concientizar al paciente sobre la importancia de la salud periodontal y de sus
repercusiones en el estado de salud general.
Sensibilizar a los profesionales de la salud sobre la posible relación que
puedan tener la Enfermedad Periodontal y las Enfermedades del aparato
digestivo causadas por Helicobacter pylori.
Fortalecer el trabajo en equipo multidisciplinario, para brindarle a los
pacientes con riesgo de una posible asociación de estas patologías, el
manejo y la atención adecuada.
Realizar ensayos controlados aleatorios para determinar con precisión las
bases biológicas y evaluar la posible asociación causal entre la Enfermedad
Periodontal y las Enfermedades del aparato digestivo causadas por
Helicobacter pylori.
Diseñar programas de educación y prevención para informar a la población
acerca de la etiología, factores de riesgo y consecuencias de la Enfermedad
Periodontal y las Enfermedades del aparato digestivo causadas por
Helicobacter pylori.
Realizar un mayor número de investigaciones que diluciden que aclaren el
papel de la placa bacteriana como reservorio de Helicobacter pylori.
Promover más investigaciones que evalúen el papel de la placa bacteriana y
cavidad bucal como posibles fuentes de reinfección gástrica, posterior al
tratamiento antibiótico para la erradicación de Helicobacter pylori.
Promover más investigaciones que evalúen el efecto de la higiene oral
adecuada sobre la colonización de Helicobacter pylori en la placa bacteriana.
Promover la higiene y cuidados orales particularmente en pacientes que vivan
en hacinamiento, en hogares de cuidado diario, en asilos, sanatorios.

INDICE GENERAL
Portada………………………………………………………………………………….I
Frontispicio……………………………………………………………………………..II
Veredicto…….…………………………………………………………………………III
Agradecimiento...……………………………………………………………………..IV
I.- Resumen……………………………………………………………………………V
II.- Abstract…………………………………………………………………………….VI
III.- Introducción…………….………………..………………………………...........VII
4.-Marco Teórico………………………………………………………………………10
4.1.-Helicobacter pylori……………………………………………………………..10
4.1.1.- Epidemiología de la infección por helicobacter pylori……………...11
4.1.2.- Patogenia y factores de virulencia…………………………………..12
4.1.3.- Manifestaciones clínicas……………………………………………….17
4.1.4.- Vías de transmisión…………………………………………………….17
4.2.- Clasificación de las enfermedades del aparato digestivo causadas por
Helicobacter Pylori….....................................................................................20
4.2.1.- Dispepsia no ulcerosa o funcional……………………………….……20
4.2.2.- Gastritis…………………………………………………………………..22
4.2.2.1Gastritis Aguda…………………………………………………..22
4.2.2.1.1.- Gastritis erosiva y hemorrágica……………………23
4.2.2.2.- Gastritis Crónica………………………………………………24
4.2.2.2.1.- Gastritis por Helicobacter pylori…………………..24
4.2.2.2.2.-Gastritis atrófica metaplásica ambiental (GAME)…....25
4.2.3.- Ulceras pépticas……………………………………………………....25
4.2.3.1.- Ulcera gástrica……………………………………………….25
4.2.3.2.- Ulcera duodenal……………………………………………...27
4.2.4. – Cáncer Gástrico……………………………………………………….29
4.2.5. – Linfoma MALT…………………………………………………….......32
4.3.-Enfermedad Periodontal…………………………………………………………..34
4.3.1.- Epidemiología de la Enfermedad Periodontal……………………….35

4.3.2.- Etiopatogenia de la Enfermedad Periodontal………………………...37
4.3.3.- Reacción del receptor en la Enfermedad Periodontal……………….43
4.3.4.- Clasificación de Enfermedades y Lesiones Periodontales………….48
4.4.- Relación entre Helicobacter pylori y Enfermedad Periodontal………………..53
4.4.1.- Estudios de Intervención………………………………………………..53
4.4.1.1.-Cuadro Comparativo de los estudios realizados entre
La relación de Helicobacter Pylori y la Enfermedad
Periodontal……………………………………………………..58
V.- Objetivos………………………………………………………………………………60
VI.- Materiales y Métodos………………………………………………………...……..61
VII.- Conclusiones……………..……………………………………………...………….62
VIII.- Recomendaciones………………………………………………………………….63
IX. - Índice General……………………………………………………………………….64
X. - Bibliografía…………..………………………………………………………………..66

BIBLIOGRAFIA.
1. – Xie, H.G.; Richard, B. 2001. Molecular basis of ethnic differences in drug
disposition and response. Annu Rev Pharmacol Toxicol, 11:815-50.
2. – Versalovic, J.; Lewandrowsk, K. 1998. Helicobacter pylori Update. Clin Microbiol
Newletter; 20(13): 107-113.
3.- Chelimsky, G.; Zcinn, S. 2002. Enfermedad ulceropéptica en la infancia.Pediatri
Rev; 22 (10):349-358.
4.- Mattana, C. M.; Vega, A. E.; Flores, A. G. de Domeniconi, Centorbi. 1998. ONP
de. Aislamiento de Helicobacter pylori en placa dental. Rev Arg de Microbiol; 30: 93-
95.
5.- Moroni, N. H. 1999. Helicobacter pylori en flora bacteriana oral. Odontol
Sanmarquina, Univ. San Marcos (Lima); (3) 37-38.
6.- Fernández Céspedes, N. A.; Fochesatto, N. A.; Guayán, V. A.; Haro Ríos, P. M.;
Aragués Alegre, V. 2004. HELICOBACTER PYLORI: ¿Un riesgo cardiovascular?
Alumnos del Internado Rotatorio de la Facultad de Medicina de la UNNE. Corrientes.
Argentina http://med.unne.edu.ar/revista/revista 139/ hpylori.pdf. Revisión Marzo -
2006.
7.- Gardner, J. D.; Perdomo, C.; Sloan, S.; Hahne, W. F.; Barth, J. A.; Rodríguez-
Stanley, S.; Robinson, M. 2002. Integrated acidity and rabeprazole pharmacology.
Aliment Pharmacol Ther; 16 (3): 455-64.
8.- Lindhe, J. 2001. Periodontología Clínica e implantodontología Odontológica. 3ra
Edición. Madrid- España. Editorial Médica Panamericana. Pág. 75, 81, 146, 150,
151, 152, 153, 154, 155, 166, 139.
9. – Marsh, P. D. 2003. Plaque as a biofilm: pharmacological principles of drug
delivery and action in the sub- and supragingival environment. Oral disease; 9: 16-
22.
10. – Nguyen, A. M. 1993. Engstrand L, Genta RM, Graham DY. Detection of
Helicobacter pylori in dental plaque by reverse transcription-polymerase chain
reaction. J Clin Microbiol; 31(4):783-787.
11. – Okuda, K. Kimizuka, R. Katakura, T. Ishihara, K. 2003. Ecological and
immunopathological implication of oral bacteria in Helicobacter pylori infected
disease. J Periodontol: 123-128.
12. - Martínez Tellez, J. L. 2003. Factores de Riesgo de la Enfermedad

Periodontal. República Bolivariana de Venezuela. Universidad Santa María
Facultad de Odontología.http://www.odontologia-online.com/casos/part/JMLT/JMLT
03/jmlt03. html. Revisión Febrero-2006.
13. – Allaker, R. P.; Young, K. A.; Hardie, J. M.; Domizio, P. 2002. Prevanlence of
Helicobacter pylori at oral and gastrointestinal sites in children: evidence for possible
oral to oral transmission. J Med Microbiol; 51 (4):312-317.
14. - Madinier, I. M.; Fosse, T. M.; Monteil, R. A. 1997. "Oral carriage of Helicobacter
pylori a review". Journal of Periodontology; 68:2-6.
15.- Pignataro, S. 1996. Helicobacter pylori: reservorios no humanos. Acta
Gastroenterol. Latinoam.; 26(5): 34-35.
16.- Hou, H. L.; Meng, H. X.; Hu, W. J.; Wang, J. W. 2003. The relationship between
Helicobacter pylori in oral cavity and the Hp infection in stomach. Zhonghua Kou
Qiang Yi Xue Za Zhi; 38(5):327-9 PMID: 14680575
17. - Identification by PCR of Helicobacter pylori in subgingival plaque of adult
periodontitis patients. Infection Research Group, University of Glasgow Dental
School.J Med Microbiol 1999 Mar; 48(3): 317- 22.
18. – Hu, W.; Cao, C.; Meng, H. 1999. Helicobacter pylori in dental plaque of
periodontitis and gastric disease patients Zhonghua Kou Qiang Yi Xue Za Zhi. Oral
Microbiol Immunol; 34(1):49-51.
19.- Gebara, E. C.; Pannuti, C.; Faria, C. M.; Chehter, L.; Mayer, M. P. Lima, L. A.
2004. Prevalence of Helicobacter pylori detected by polymerase chain reaction in the
oral cavity of periodontitis patients. Oral Microbiol Immunol; 19(4):277-80.
20. – Andersen, R. N.; Ganeshkumar, N. 1998. Helicobacter pylori adheres
selectively to Fussobacterium spp. Oral Microbiol Immunol; 13 (1): 51-4.
21. - Muñiz, C.; Jane, M.; Gonzalez-Cuevas, A. 1999. Evaluation of a simple rapad
polymerase Caín reactin (PCR) technique for the diagnosis of H. pylori infection in
childhood. Enferm. Infecc. Microbiol. Clin; 17 (3):119-25.
22. - Nabwera, H. M.; Logan, R. 1999. Epidemiology of Helicobacter pylori:
transmission, translocation and extragastric reservoir. J. Phisiol. Pharmacol ; 50
(9):711-22.
23. – Dore, M. P.; Sepúlveda, A. R.; El-Zimaity, H.; et al. 2001. Isolation of
Helicobacter pylori from sheep-implications for transmission to humans. Am J
Gastroenterol; 96: 1396-401.

24.- Medina, M. G.; Medina, M. L.; Merino, L. A. Revisión: Lo que se conoce sobre
infección por Helicobacter pylori en niños: http://www.odontologia-
online.com/casos/part/MLM/MLM02/mlm02.html. Revisión Marzo -2006.
25.- Rivera, M.; Contreras, F.; Terán, A.; Fouillioux, C. 2004. Helicobacter Pylori:
Enteropatógeno frecuente del ser humano. Archivos Venezolanos de
FarmacologíayTerapéutica;Caracas jul. v.23 n.2 http://www.scielo.org.ve/scielo.php?
pid=S0798-02642004000200003&script=sci_arttext&tlng=es Revisión Marzo 2006.
26. - Hocker, M.; Hohenberger, P. 2003. Helicobacter pylori virulence factors - one
part of a big picture. Lancet; 362: 1231-1233.
27. - Cave, D. R. 1997. How is Helicobacter pylori transmitted? Gastroenterol; 113:
S9-S14.
28. - Porter, S. R.; Barrer, G. R.; Scully, C.; MacFarlance, G.; Bain. L. 1997. FERUM
IgG antibodies to Helicobacter pylori in patients with recurrent aphtous stomatitis and
other oral disorders. Oral Surg Oral Med Oral Pathol Oral Radio Endod; 83 (3):325-8.
29.– Nakamura, R. M. 2001. Laboratory Tests for the Evaluation of Helicobacter
pylori Infections. J Clin Lab Ana; 15: 301-7.
30. – Fredricks, D. 2002. Novel pathogens and chronic diseases. Clin Microbiol
Newletter; 24(6): 41-44.
31. - Hernández-Nieto, R. 2002. Contribuciones al Análisis Estadístico: Análisis
comparativo de la sensibilidad (estabilidad y consistencia) de varios coeficientes de
variación relativa y el Coeficiente de Variación Proporcional (Cvp) en diferentes
prototipos de distribuciones aleatorias. El coeficiente de Validez de Contenido (Cvc)
y el Coeficiente KAPPA, en la determinación de la Validez de contenido, según la
Técnica de Juicio de Expertos. Mérida, Venezuela: Universidad de Los Andes -
IESINFO (Instituto de Estudios en Informática).
32. – Forman, D. 1995. The prevalence of Helicobacter pylori infection in gastric
cancer. Aliment Pharmacol Ther, 9 (suppl. 2): 71-76.
33. – Atherton, J. C. 1998. H. pylori virulence factors. British Medical Bulletin;
54:105-120.
34.- Díaz, A. R. 2002. ”Enfermedad por Helicobacter pylori”. Rev. Elec. de Med.
Intensiva sep. Artículo. n▫461. Vol. 2 Nº 9, URL http//
www.iladiba.com./upr/1997/N▫41997/htm/helic.asp. Revisión Febrero -2006

35. - Goodman, K. J.; Correa, P.; Tengana Aux, H. J.; et al. 1996. Helicobacter Pylori
infection in the Colombian Andes; a population-based study of transmisión pathways.
Am J Epidemiol; 144:290- 5.
36.- Pueyo, A.; Huarte, M. P,; Jiménez, C. Epidemiología de la infección por
Helicobacter pylori. ANALES Sistema Sanitario de Navarra.
http://www.cfnavarra.es/salud/anales/textos/vol21/biblio2/bsuple2.html. Revisión
Abril-2006.
37.- López-Brea, M.; Alarcón, T.; Domingo, D.; Pérez-Pérez, G.; Correa, P.; Skirrow,
M. B.; Marshall, B. J. Helicobacterspain retos para el siglo XXI.
http://www.helicobacterspain.com/micro/caraviruM.htm. Revisión Abril 2006.
38.- Parra, T.; Carballo, F. Reservorios y vías de transmisión de la infección por
Helicobacter pylori. ANALES, Vol.21, Suplemento 2.
39.- Van Duynhoven ,Y. T.; De Jorge, R. 2001. Transmission of Helicobacter pylori:
a role for food?. Bull World Health Organ v.79 n.5 Genebra.
http://www.scielosp.org/scielo.php?script=sci_arttext&pid=S0042-
96862001000500012 &lng= es&nrm= iso& tlng=en. Revisión Abril 2006.
40.- Bonamico, M.; Monti, S.; Luzzi, I.; Magliocca, F. M.; Cipolletta, E.; Calvani, L.;
et al. 1996. Helicobacter pylori infection in families of Helicobacter pylori-positive
children. Ital J Gastroenterol; 28(9): 512-517.
41. - Ashorn, M.; Ruuska, T.; Miettinen, A. 1996. Seroepidemiology of Helicobacter
pylori infection in infancy. Arch Dis Child; 74: F141-F142.
42. – Grubel, P. 1997. Vector potential of houseflies (Musca domestica) for
Helicobacter pylori. Journal of Clinical Microbiology, 35:1300–1303.
43.- Talley, N. J.; Silverstein, M. D.; Agreus, L.; Nyren, O.; Sonnenberg, A.;
Holtmann, G. 1998. Gastroenterología; 114:582-95.
44. – Méndez-Sánchez, N.; Uribe Esquivel, M. 2005. Gastroenterología. Mc Graw Hill
Interamericana; México Pag. 317,318, 319, 320, 327, 328, 329, 333, 334, 335, 336,
353, 354, 355, 356.
45. – Courtney, W.; Houchen, M. D. Division of Gastroenterology, Washington
University School of Medicine, St. Louis, MO. Review provided by VeriMed
Healthcare Network.http://www.nlm.nih.gov/medlineplus/spanish/ency/article
/000232. htm#top. Revision Mayo -2006.

46.- Christian Stone, M. D.; Division of Gastroenterology, Washington University in
St. Louis School of Medicine, St. Louis, MO. Review provided by VeriMed
HealthcareNetwork. http://www.nlm.nih.gov/medlineplus/spanish/ency/article
/000213.htm Revision Mayo- 2006.
47. – Marcia, S.; Brose, M. D.; Ph.D., Assistant Professor, Hematology/Oncology, the
University of Pennsylvania Cancer Center, Philadelphia, PA. Review provided by
VeriMed Healthcare Network. http://www.nlm.nih.gov/medlineplus/spanish/ency/
article/000223.htm. Revisión Mayo- 2006.
48. - Pajares García, J. 2000 Linfoma Malt. Actualización diagnóstica y terapéutica.;
www.caded.org/linfomas_malt.htm Revisión Mayo-2006.
49. - Harris, N. L.; Jaffe, E. S.; Diebold, J.; et al. 1997. The World Health
Organization classification of neoplasms of the hematopoietic and lymphoid tissues:
report of the Clinical Advisory Committee meeting—Airlie House, Virginia, Hematol J;
1: 53-66.
50. – Garcia- Sipido, A. P. 1999. Linfoma tipo MALT de localización gástrica.
Infección por Helicobacter Pylori.; www.helicobacterspain.com/clinica/linfoma_
MALT_revisión.htm Revisión Mayo- 2006.
51. – Ferreira, P. A.; Teramoto, S. A. 2002. Linfoma MALT Gástrico: Revisión.
Revista de Posgrado de la VIa Cátedra de Medicina - N° 119.
52. – Martin de Argila, C.; Boixeda, D.; Gisbert, J. P. 1996. Epidemiología de la
infección por Helicobacter pylori. En: Boixeda D., Gisbert J.P., Martin de Argila C.
Eds. Infección por Helicobacter pylori. ¿Dónde estaacute; el límite?. Pros Science,
Barcelona, 1407-1410.
53.- Savio, B. A.; Franzin, G.; Wotherspoon, A. G.; Zamboni, G.; Negroni, R.;
Buffoli, F.; et al. 1996. Diagnosis and post-treatment follow-up of Helicobacter pylori
positive gastric lymphoma of mucosa-associated lymphoid tissue. Histology,
polymerase chain reaction or both? Blood, 87: 1255-1260.
54. - Williams, R. C. 1990. Periodontal disease. New England Journal of Medicine.
322:373-382.
55.- Osorio Núñez, M.; Harteman Díaz, L.; López Ramón, R. 2002. Epidemiología
de la enfermedad periodontal en dos policlínicas de Ciudad Habana.
http://www.ucmh.sld.cu/rhab/articulo_rev7/ maritza_osorio.pdf. Revisión Junio 2006
56. - Calatrava, L. A. 2002. Crecimiento Científico Contemporáneo, Escenario
Epidemiológico Actual de las Enfermedades Bucales y Currículo Odontológico. Acta

Odontológica Venezolana VOL 40 Nº 2. http://www.actaodontologica.com/402
2002/crecimientocientificocontemporaneo .asp. Revisión Mayo -2006.
57. – Carranza, F.; Newman, M.; Takei, H. 2003. Carranza. Periodontología Clínica.
9 Edición. Mexico. Ediciones Mc Graw - Hill Interamericana. Pag. 91, 104, 256,257.
58. – Morón, A.; Vanegas, W.; Salazar, C.R. 1986. Estudio del Perfil Epidemiológico
Bucal del Distrito Maracaibo, Edo, Zulia, Maracaibo: Universidad del Zulia, Facultad
de odontología (I. I. O) Consejo de Desarrollo Científico y Humanístico (CONDES),
59. – Betancourt, M.; Arce, R.; Botero, J.; Jaramillo, A.; Cruz, C.; Contreras, A. 2006.
Microorganismos inusuales en surcos y bolsas periodontales. Volumen 37N°1
http://colombiamedica.univalle.edu.co/Vol37No1/Cm37n1%20html/Cm 37n1 a1.htm.
Revisión Mayo- 2006.
60.- Paz, M.; 2000. Categorización de la enfermedad periodontal en los pacientes
que acuden al postgrado de periodoncia de la División de Estudios para Graduados
de la Facultad de Odontología. Maracaibo. Tesis doctoral. Universidad del Zulia.
Pag. 32-34.
61.- Morón, A.; Hernández, H.; Rivera, L.; y col. 1995. Epidemiología Bucal del
Zuliano. Facultad de Odontología LUZ. Maracaibo- Venezuela.
62.- Medina, M. L.; Merino, L. A.; Gorodner, J. O. 2002. Evidencias clínicas y
aspectos microbiológicos en enfermedades gingivoperiodontales.
http://www.unne.edu.ar/cyt/2002/03-Medicas/M-031.pdf. Revisión Mayo-2006
63. - Slots, J. 1979. Subgingival microflora and periodontal disease. J. Clin.
Periodontol; 6:35-82.
64. - Leknes, K. 1997. A correlation Study of Inflammatory Cell Mobilzation in
Response Subgingival Microbial Colonization. J Periodontol; 68: 67-72.
65. - Timmerman, M.; Van der Weijden, G.; Arief, E.; Armand, S.; Abbas, F.; Winkel,
E.; Van Winkelhoff, A.; Van der Velden, U. 2001. Untreated periodontal disease in
Indonesian Adolescents. Subgingival microbiota in relation to experienced
progression of periodontitis. J. Clin Periodontol. 28: 617-627.
66. – Guilarte, C.; Perrone, M. 2004. Microorganismos de la placa dental
relacionados con la etiologia de la periodontitis. Acta Odontologica Venezolana.
Volumen 42. No.3. http://www.actaodontologica.com/42_3_2004/
microorganismos_placa_dental_ etiologia_periodontitis.asp. Revisión Marzo-2006.

67.- Guilarte, C. 2001. Patógenos Periodontales. Acta Odontológica Venezolana
VOL 39 Nº3. http://www.actaodontologica.com/39_3_2001/
patogenos_periodontales. asp. Revision Marzo-2006.
68.- Guilarte, C.; Perrone, M. 2003. Bacterias Periodontopatógenas: Bacilos
Anaerobios Gram Negativos Como Agentes Etiológicos de la Enfermedad
Periodontal. Acta Odontológica Venezolana VOL 43 Nº 2.
http://www.actaodontologica.com/43_2_2005/bacterias_periodontopatogenas_
enfermedad_periodontal.asp Revisión Marzo-2006.
69. - Scannapieco, F. A. 2004. Periodontal inflammation: from gingivitis to systemic
disease? Compend Contin Educ Dent; 25(7 Suppl 1)16-25.
70. - Page, R. C. 2002. The etiology and patogénesis of periodontitis. Compend
Contin Educ Dent. May: 23(5 Suppl):11-4.
71. – Maita, V. L.; Maita, C. L. 2004. Tratamiento periodontal No Quirúrgico.
Enfoque Biológico. Odontología Sanmarquina. UNMSM; 8 (1): 51-56.
http://sisbib.unmsm.edu.pe/BVRevistas/odontologia/vol8_N1/a12.htm. Revisión
Mayo- 2006.
72. – Scannapieco, F. A. 1998. Periodontal disease as a potential risk factor for
systemic diseases. J. Periodontol 69:841-850.
73. – Armitage Gary, C. 1999. Development of a Classification System for
Periodontal Diseases and Conditions. Ann Periodontol; 4:1-6.
74. – Kilmartin, C. M. 2002. Dental Implications of Helicobacter pylori. Journal Can
Dent Assoc; 68(8):489-93.
75. – – Anand, P. S.; Nandakumar, K.; Shenoy, K. T. 2006. Are Dental Plaque, Poor
Oral Hygiene, and Periodontal Disease Associated with Helicobacter pylori
Infection? J. Periodontol. Apr; 77(4):692-698.
76. – Berroteran, A.; Perrone, M.; Correnti, M.; Cavazza, M. E.; Tombazzi, C.;
Lecuna, V.; Goncalvez, R. 2001. Prevalencia de Helicobacter pylori en el estómago
y la placa dental de una muestra de la población en Venezuela. Caracas abr. Acta
Odontol. Venez v.39 n.2
77. Scarano Pereira, G. A.; Correia de Medeiros, A.; Marques Soares, M. S.;
Chimenos Küstner, E.; De Castro Barreto. R.; Perdomo Lovera, M. 2005. Detección
de Helicobacter pylori en placa dental y en mucosa gástrica de pacientes sometidos
a endoscopia digestiva. Acta Odontológica Venezolana.
venez v.43 n.2 Caracas mayo.

78.- Moromi Nakata, H.; Calle Espinoza, S.; Zambrano de la Pena, S. 2001.
Prevalencia de Helicobacter pylori en pacientes con Gingivitis y Enfermedad
periodontal. Odontología Sanmarquina. Enero-junio Vol. 1. N° 7.
79.- Gebara, E. C.; Faria, C. M.; Pannuti, C.; Chehter, L.; Mayer, M. P.; Lima, L. A.
2006. Persistence of Helicobacter pylori in the oral cavity after systemic eradication
therapy. J Clin Periodontol. May; 33(5):329-33.
80.- Campuzano, S.; Lombana, I.; Argúello, E.; Herrera, M. 1996. Aislamiento de
Helicobacter pylori en placa dental. Trib. Méd. (Bogotá); 93(4):169-79, abr.
81.- Chumpitaz, J.; Gutiérrez, J.; Córdova, R.; Sánchez, M.; Vásquez, N.; Rivadeira,
C.; Beteta, D.; Solano, L.; Marocho, L.; Pareja, E.; Huamán, A.; Valencia, B. 2006.
Aislamiento de Helicobacter pylori en Sarro Dental de pacientes con Gastritis del
Policlínico "Angamos". Revista Gastroenterol Perú 26:373-376.
http://sisbib.unmsm.edu.pe/BVRevistas/gastro/vol26n4/PDF/a05v 26n4.pd
Revisión Enero- 2007
82.- De Sousa, L.; Vásquez, L.; Velasco, J.; y Parlapiano, D. Aislamiento de
Helicobacter pylori en mucosa gástrica, placa dental y saliva en una población de los
Andes venezolanos. http://www.serbi.luz.edu.ve/pdf/ic/v47n2/art_02.pdf. Revisión
Febrero 2007.


















