
Regulators of Hedgehog signaling in chondrocytes: Sufu ... · Shu-Hsuan Claire Hsu Doctor of...
155
Regulators of Hedgehog signaling in chondrocytes: Sufu, Kif7, and primary cilium By Shu-Hsuan Claire Hsu Doctor of Philosophy Institute of Medical Science University of Toronto © Copyright by Shu-Hsuan Claire Hsu 2012
Transcript of Regulators of Hedgehog signaling in chondrocytes: Sufu ... · Shu-Hsuan Claire Hsu Doctor of...

Regulators of Hedgehog signaling in chondrocytes: Sufu, Kif7, and primary cilium
By
Shu-Hsuan Claire Hsu
Doctor of Philosophy
Institute of Medical Science
University of Toronto
© Copyright by Shu-Hsuan Claire Hsu 2012

ii
Regulators of Hedgehog signaling in chondrocytes: Sufu, Kif7, and primary cilium
Shu-Hsuan Claire Hsu
Doctor of Philosophy
Institute of Medical Science
University of Toronto
2012
Abstract
The Hedgehog (Hh) signaling pathway has received attention regarding its important role
in embryonic development, however the mechanism by which pathway regulators, such as
Suppressor of fused (Sufu), Kinesin family member 7 (Kif7), and primary cilium, mediate Hh
signaling transduction is not entirely understood. The work presented here examines the roles of
Sufu and Kif7 in regulating Hh signaling in growth plate chondrocytes, as well as how they
mediate parathyroid hormone-like hormone (Pthlh) signaling during chondrocyte development. I
show here that Sufu and Kif7 are essential regulators of Indian hedgehog (Ihh) signaling. While
Sufu negatively regulates Gli transcription factors, Kif7 functions both positively and negatively
in chondrocytes. Kif7 plays a role in Sufu protein degradation and the exclusion of Sufu-Gli
complexes from the primary cilium. Importantly, halving the dosage of Sufu restores normal Hh
pathway activity and chondrocyte development in Kif7-null mice, demonstrating that the positive
role of Kif7 is to restrict the inhibitory function of Sufu. Furthermore, Kif7 exerts inhibitory
function on Gli transcriptional activity in chondrocytes when Sufu function is absent. Therefore,
Kif7 regulates the activity of Gli transcription factors through both Sufu-dependent and Sufu-
independent mechanisms. I show that Sufu is crucial for mediating the negative effect of Pthlh

iii
on Gli transcriptional activity and chondrocyte hypertrophic differentiation, whereas Kif7 and
primary cilium are dispensable in this process. Although primary cilium is required for Hh
ligand-mediated activation of Gli transcription, Pthlh negatively controls Gli transcriptional
activity in a cilia-independent manner. The results of this work provide insight into how Hh
signaling is regulated by Sufu and Kif7 in the context of primary cilium, but also suggest Sufu
serves as an important link between Ihh and Pthlh signaling during growth plate chondrocyte
development.

iv
Acknowledgments
I sincerely thank my supervisor Dr. Benjamin Alman for his guidance and
encouragement. Enormous thanks to Dr. Chi-Chung Hui for his invaluable suggestions and
guidance. I am also very grateful to my committee members, Dr. David Bazett-Jones and Dr.
Martin Post, for their comments and suggestions. Thanks to Xiaoyun Zhang from the Hui lab for
helping me with the western analysis. Thanks to the entire Alman lab, past and present for their
scientific and moral support. Thanks to my family and friends for their love, for cheering me on,
and their patience with me these past few years. Finally thanks to my boyfriend Robert for his
support and for putting a smile on my face when things did not work at the bench.

v
Table of contents
Abstract ..................................................................................................................................... ii
Acknowledgments ................................................................................................................... iv
Table of contents ....................................................................................................................... v
List of abbreviations ................................................................................................................. x
List of figures .......................................................................................................................... xii
Chapter 1 Introduction .............................................................................................................. 1
1.1 Summary ......................................................................................................................... 2
1.2 Overview of growth plate development .......................................................................... 3
1.3 Indian hedgehog (Ihh) and Parathyroid hormone-like hormone (Pthlh) signaling in
growth plate development ........................................................................................................... 4
1.3.1 Ihh/Pthlh feedback loop ............................................................................................ 4
1.3.2 Pthlh-dependent and –independent effects of Ihh .................................................... 4
1.3.3 Actions of Pthlh in the growth plate ......................................................................... 5
1.4 Ihh and Pthlh signaling pathways regulate Gli transcription factors .............................. 7
1.5 Regulators in Hedgehog (Hh) signaling pathway: Sufu and Kif7 .................................. 9

vi
1.6 The primary cilium in Hh signaling .............................................................................. 11
1.7 Hypothesis & Specific aims .......................................................................................... 14
1.8 Figures ........................................................................................................................... 15
1.9 References ..................................................................................................................... 19
Chapter 2 Kif7 promotes Hh signaling in growth plate chondrocytes by restricting the
inhibitory function of Sufu ........................................................................................................... 25
2.1 Summary ....................................................................................................................... 26
2.2 Introduction ................................................................................................................... 27
2.3 Results ........................................................................................................................... 31
2.3.1 Sufu is differentially expressed in the growth plate and is required for normal
skeletal development ............................................................................................................. 31
2.3.2 Sufu acts as a negative regulator of Hh signaling during chondrocyte
differentiation ........................................................................................................................ 32
2.3.3 Loss of Kif7 in growth plate chondrocytes results in reduced Hh pathway activity
............................................................................................................................................... 33
2.3.4 Sufu-Gli complexes are localized to the ciliary tip in the absence of Kif7 ............ 34
2.3.5 Removal of one copy of Sufu rescues the Kif7 mutant growth plate phenotype .... 37

vii
2.3.6 Kif7 and Sufu share overlapping functions in Hh signaling during chondrocyte
development........................................................................................................................... 37
2.4 Discussion ..................................................................................................................... 39
2.5 Materials & Methods ..................................................................................................... 44
2.6 Figures ........................................................................................................................... 49
2.7 References ..................................................................................................................... 66
Chapter 3 Sufu mediates the effect of Pthlh on chondrocyte differentiation in the growth plate
...................................................................................................................................................... 70
3.1 Summary ....................................................................................................................... 71
3.2 Introduction ................................................................................................................... 72
3.3 Results ........................................................................................................................... 75
3.3.1 Sufu regulates the effect of Pthlh on chondrocyte differentiation .......................... 75
3.3.2 Kif7 is not required for Pthlh to inhibit chondrocyte differentiation ..................... 76
3.3.3 Pthlh regulates Sufu protein level ........................................................................... 76
3.3.4 Sufu is required for the ability of Pthlh to process Gli transcription factors .......... 77
3.4 Discussion ..................................................................................................................... 79
3.5 Materials & Methods ..................................................................................................... 82

viii
3.6 Figures ........................................................................................................................... 87
3.7 References ..................................................................................................................... 94
Chapter 4 Conclusions & future directions ............................................................................. 97
4.1 Summary ....................................................................................................................... 98
4.2 Sufu expression is tightly regulated by multiple factors in the growth plate ................ 99
4.2.1 Lessons from genetically modified mice and Pthlh activation ............................... 99
4.2.2 Transcriptional regulation of Sufu ........................................................................ 100
4.2.3 How does Sufu regulate subsequent steps of endochondral ossification? ............ 101
4.2.4 Sufu and osteoarthritis .......................................................................................... 102
4.3 The role of Sufu in Wnt/ß-catenin signaling pathway during chondrocyte development
................................................................................................................................................. 105
4.3.1 Wnt/ß-catenin signaling in skeletal development ................................................. 105
4.3.2 The role of Sufu in mediating Wnt/ ß-catenin signaling ...................................... 106
4.4 Conclusions ................................................................................................................. 109
4.5 Figures ......................................................................................................................... 110
4.6 References ................................................................................................................... 112

ix
Appendix A: Pthlh negatively regulates Hh signaling activity in chondrocytes through a cilia-
independent mechanism .............................................................................................................. 116
A.1 Summary .................................................................................................................... 117
A.2 Introduction ................................................................................................................ 118
A.3 Results ........................................................................................................................ 121
A.3.1 Ift88ORPK/ ORPK
mice show skeletal defects and dysregulated Hh signaling in
chondrocytes ........................................................................................................................ 121
A.3.2 Pthlh downregulates Col10a1 expression and Hh signaling activity through a cilia-
independent mechanism in chondrocytes ............................................................................ 121
A.3.3 Ciliary tip localization of Sufu is induced by Pthlh ................................................ 122
A.4 Discussion .................................................................................................................. 125
A.5 Materials & Methods .................................................................................................. 128
A.6 Figures ........................................................................................................................ 132
A.7 References .................................................................................................................. 139
x
List of abbreviations
APC Adenomatous polyposis coli gene product
ChIP Chromatin immunoprecipitation
Ci Cubitus interruptus
CHX cycloheximide
DAPI 4 ,6-diamidino-2-phenylindole (DAPI)
Gli1 Glioma-associated oncogen family member 1
Gli2 Glioma-associated oncogen family member 2
Gli3 Glioma-associated oncogen family member 3
Gli3FL Full-length of glioma-associated oncogene family member 3
Gli3R C-terminally truncated transcriptional repressor form of glioma-associated oncogene
family member 3
GSK3ß Glycogen synthase kinase 3ß
Hh Hedgehog
H&E Hematoxylin and eosin

xi
IFT Intraflagellar transport
Ihh Indian hedgehog
IP Immunoprecipitate
Kif7 Kinesin family member 7
Lef Lymphoid enhancer factor
PBS Phosphate-buffered saline
PKA Protein kinase A
Ptch1 Patched 1
Pthlh Parathyroid hormone-like hormone
PTHrP Parathyroid hormone-related protein
PTHR1 Parathyroid hormone 1 receptor
Pur Purmorphamine
Shh Sonic hedgehog
Smo Smoothened
Sufu Supppressor of fused
TCF Transcription factors T-cell factor

xii
List of figures
FIGURE 1-1 SCHEMATIC REPRESENTATION OF ENDOCHONDRAL OSSIFICATION. ............................. 15
FIGURE 1-2 IHH/PTHLH SIGNALING REGULATION IN THE GROWTH PLATE. ..................................... 16
FIGURE 1-3 HH SIGNALING IN VERTEBRATES. ................................................................................ 18
FIGURE 2-1 SUFU IS DIFFERENTIALLY EXPRESSED IN THE GROWTH PLATE. .................................... 49
FIGURE 2-2 GENERATING CHONDROCYTE-SPECIFIC KNOCKOUT OF SUFU. ..................................... 50
FIGURE 2-3 SKELETAL PHENOTYPE ANALYSIS OF COL2A1-CRE; SUFUF/F
. ...................................... 52
FIGURE 2-4 SUFU NEGATIVELY REGULATES GROWTH PLATE CHONDROCYTE DIFFERENTIATION. ... 54
FIGURE 2-5 IN SITU HYBRIDIZATION ANALYSIS OF PTCH1 AND COL10A1 EXPRESSION IN VARIOUS
GENOTYPES. ............................................................................................................................ 56
FIGURE 2-6 KIF7 INACTIVATION RESULTED IN OPPOSITE EFFECTS ON CHONDROCYTE
PROLIFERATION AND DIFFERENTIATION TO THOSE OF SUFU INACTIVATION. ........................... 57
FIGURE 2-7 INCREASED LEVEL OF SUFU IN KIF7-DEIFICENT CHONDROCYTES. ............................... 59
FIGURE 2-8 INCREASED LEVEL OF SUFU-GLI COMPLEXES IN KIF7-DEIFICENT CHONDROCYTES. .... 60
FIGURE 2-9 A DUAL FUNCTION FOR KIF7 IN HH SIGNALING MODULATION. .................................... 62

xiii
FIGURE 2-10 PROPOSED MODEL FOR HOW SUFU AND KIF7 REGULATE HH SIGNALING IN GROWTH
PLATE CHONDROCYTES. .......................................................................................................... 64
FIGURE 3-1 SUFU IS REQUIRED FOR THE NEGATIVE EFFECT OF PTHLH ON CHONDROCYTE
DIFFERENTIATION. .................................................................................................................. 87
FIGURE 3-2 KIF7 IS NOT REQUIRED FOR THE NEGATIVE EFFECT OF PTHLH ON CHONDROCYTE
DIFFERENTIATION. .................................................................................................................. 89
FIGURE 3-3 PTHLH INCREASES SUFU PROTEIN LEVEL. .................................................................... 90
FIGURE 3-4 PTHLH SIGNALING ACTIVATION PROMOTES GLI2 AND GLI3 PROTEIN PROCESSING VIA A
SUFU-MEDIATED MANNER. ..................................................................................................... 91
FIGURE 3-5 PROPOSED MODEL FOR HOW PTHLH REGULATES GLI TRANSCRIPTIONAL FACTORS
THROUGH A SUFU-MEDIATED MECHANISM. ............................................................................ 93
FIGURE 4-1 PROPOSED MODEL BY WHICH SUFU PROTEIN STABILITY SERVES AS A COMMON
MECHANISM THAT REGULATES GLI-MEDIATED TRANSCRIPTION. .......................................... 110
FIGURE 4-2 PROPOSED MECHANISM BY WHICH PTHLH AND IHH SIGNALING COORDINATE SUFU
EXPRESSION IN THE GROWTH PLATE. ..................................................................................... 111
FIGURE A- 1 THE PRIMARY CILIA ARE REQUIRED FOR NORMAL EMBRYONIC SKELETAL
DEVELOPMENT. ..................................................................................................................... 132

xiv
FIGURE A- 2 ASYMMETRICAL HYPERTROPHY PHENOTYPE IN IFT88ORPK/ ORPK
MICE IS NOT SEEN AT
E18.5. ................................................................................................................................... 133
FIGURE A- 3 THE ROLE OF PRIMARY CILIA IN CHONDROCYTE HH LIGAND RESPONSIVENESS........ 134
FIGURE A- 4 THE ROLE OF PRIMARY CILIA IN MEDIATING PTHLH SIGNALING IN CHONDROCYTE
DIFFERENTIATION. ................................................................................................................ 135
FIGURE A- 5 INCREASED SUFU CILIARY LOCALIZATION IN PTHLH-TREATED WILD-TYPE
CHONDROCYTES.................................................................................................................... 136
FIGURE A- 6 A PROPOSED MODEL FOR CILIA-MEDIATED REGULATION OF GLI ACTIVITY. ............. 138

1
Chapter 1 Introduction

2
1.1 Summary
The work presented in the subsequent chapters is aimed at investigating Hh regulators,
Sufu, Kif7 and the primary cilium, in mediating Hh signaling as well as Pthlh signaling during
growth plate chondrocyte development. I examined the role of Sufu and Kif7 in regulating Hh
signaling in developing growth plate, and also proposed a model by which Sufu and Kif7 exert
distinct and overlapping functions in Hh pathway regulation. Furthermore, I examined how
these Hh regulators mediate the effects of Pthlh in chondrocytes. By dissecting the roles that
Sufu, Kif7 and the primary cilium play in regulating Hh and Pthlh signaling in the growth plate
chondrocytes. I provided insight into the mechanism by which Hh signaling is regulated and
highlighted Sufu as a common link between Ihh and Pthlh signaling in growth plate chondrocyte
development.

3
1.2 Overview of growth plate development
Endochondral bone growth, which is the process of the cartilaginous skeletal template
progressively being replaced by bone, is precisely regulated by a number of signaling pathways
during development. Chondrocyte proliferation and differentiation are tightly controlled, and
dysregulation of these processes causes chondrodysplasias (generalized skeletal malformation
diseases), cartilaginous tumors, and osteoarthritis, a common degenerative joint disease. At the
onset of endochondral bone formation, mesenchymal cells condense and form a template of the
future skeletal element. This cartilage template is surrounded by the perichondrium, which is a
thin layer of flattened fibroblastic cells. Centrally, cells differentiate to chondrocytes and
proliferate and then mature from resting/low-proliferating chondrocytes into high-proliferating
cells that typically arrange in columns. The proliferating chondrocytes then undergo
differentiation successively into prehypertrophic and eventually hypertrophic chondrocytes. In
parallel, cells in the perichondrium adjacent to the hypertrophic region differentiate into
osteoblasts, which form the bone collar. The hypertrophic chondrocytes undergo programmed
cell death, and osteoblasts invade along with blood vessels and osteoclasts, leading to the
replacement of the cartilage template with trabecular bone (Kronenberg, 2003; Olsen et al., 2000;
Provot and Schipani, 2005). This process is illustrated in Figure 1-1. These layers of
chondrocytes proceeding in a staggered manner through steps of hypertrophic differentiation
constitute the growth plate, which is responsible for the longitudinal growth of the bones during
embryonic development and in the postnatal long bones.

4
1.3 Indian hedgehog (Ihh) and Parathyroid hormone-like hormone
(Pthlh) signaling in growth plate development
1.3.1 Ihh/Pthlh feedback loop
A feedback loop formed by Ihh and Pthlh signaling functions to regulate the pace of
growth plate chondrocyte differentiation (Karp et al., 2000; Kobayashi et al., 2002; Lanske et
al., 1996; Minina et al., 2001). Ihh, which is expressed by prehypertrophic chondrocytes,
stimulates Pthlh expression in the articular region of the growth plate. Pthlh, which is a secreted
protein that acts on PTH/Pthlh receptor (PTHR1) bearing chondrocytes to maintain cells in a
proliferative state, thereby delaying the production of Ihh by prehypertrophic chondrocytes
(Kronenberg, 2003; Long et al., 2001) (Fig.1-2A). The levels of Ihh and Pthlh signaling
synchronize and regulate the pool of proliferating chondrocytes and the pace of chondrocyte
hypertrophic differentiation, thus determining the distance from the joint to which chondrocyte
hypertrophic differentiation take place (Karaplis et al., 1994; Lanske et al., 1996; St-Jacques et
al., 1999; Vortkamp et al., 1996).
1.3.2 Pthlh-dependent and –independent effects of Ihh
Loss of Ihh results in severe dwarfism due to accelerated chondrocyte hypertrophic
differentiation, reduced chondrocyte proliferation, and absence of osteoblasts in the bone collar
(Chung et al., 2001; Lanske et al., 1996; Long et al., 2004; Long et al., 2001; Maeda et al., 2007;
Razzaque et al., 2005; St-Jacques et al., 1999). Activation of Ihh signaling in Pthlh or PTHR1

5
mutant limbs does not result in a delay in chondrocyte hypertrophic differentiation, suggesting
that the inhibitory effect of Ihh on chondrocyte hypertrophic differentiation is Pthlh-dependent
(Fig.1-2A). However, Ihh mutant mice (Ihh-/-
) exhibit a more severe reduction in the size of the
bone compared with Pthlh (Pthlh-/-
) or PTHR1 (PTHR1-/-
) mutants, whereas expression of
PTHR1 in Ihh mutant chondrocytes delays the accelerated onset of hypertrophic differentiation,
but does not possess any effect on the proliferation defect observed in Ihh mutant mice (Karp et
al., 2000). The data reveal a Pthlh-independent function of Ihh in chondrocyte proliferating (St-
Jacques et al., 1999) (Fig.1-2A). Further studies reveal that Ihh stimulates chondrocyte
proliferation in part through a cyclin D1-mediated mechanism (Duman-Scheel et al., 2002; Long
et al., 2001). The absence of bone collar in the Ihh-deficient bones reveals a positive role of Ihh
in regulating osteoblasts differentiation in the perichondrium. Interestingly, Ihh-/-
mice do not
exhibit severe defects in intramembranous bone formation (St-Jacques et al., 1999), which is
another essential process of mammalian skeletal development that contains the direct conversion
of mesenchymal tissue into bone. During later stages of osteoblast differentiation, inactivation of
Ihh signaling does not result in defects in the osteoblast differentiation process. The data suggest
that Ihh plays a role in initiating the differentiation of osteoblasts during endochondral skeletal
development (Fig.1-2A).
1.3.3 Actions of Pthlh in the growth plate
The binding of Pthlh to its receptor, PTHR1, stimulates G protein activation, which leads
to the production of cyclic AMP and activation of protein kinase A (PKA). The mechanism by
which cyclic AMP and PKA maintain chondrocytes in a proliferative state and delay

6
chondrocyte hypertrophic differentiation is not completely understood. During mouse
embryogenesis, Sox9 plays an important role in regulating chondrocyte differentiation (de
Crombrugghe et al., 2001). Phosphorylation of Sox9 by Pthlh/PKA results in a more potent
activator function in upregulating target genes (Huang et al., 2001); thus, Sox9 phosphorylation
could contribute to the negative effect of Pthlh on chondrocyte hypertrophic differentiation. In
addition to its inhibitory role in regulating chondrocyte hypertrophic differentiation, Pthlh also
exhibits a stimulatory effect on chondrocyte proliferation. In contrast to the PKA-mediated
action of Pthlh on chondrocyte hypertrophic differentiation, its pro-proliferative effect is not
mimicked by PKA activation. Deletion of p57 results in a growth plate phenotype mimicking
what was observed in Pthlh-null mice, including increased chondrocyte proliferation and delayed
chondrocyte hypertrophic differentiation. Double knockout of Pthlh and p57 attenuates the
growth plate defects in Pthlh-null mice (MacLean et al., 2004). The data suggest that the
stimulatory effect of Pthlh on chondrocyte proliferation is mediated in part through decreasing
p57 expression in the growth plate (MacLean et al., 2004).

7
1.4 Ihh and Pthlh signaling pathways regulate Gli transcription factors
Ihh, a member of the Hedgehog (Hh) family of signaling molecules, regulates Gli
transcriptional activity through binding to its receptor Patched 1 (Ptch1) and derepressing the
signal transducer Smoothened (Smo). Three Gli zinc finger proteins Gli1, Gli2 and Gli3 (glioma-
associated oncogene family member 1, 2 and 3) are transcription factors mediating Hh pathway
in mammalian cells (Jiang & Hui, 2008). Analysis of genetically modified mice show that Gli2
and Gli3 act as essential mediators in Hh signaling, whereas Gli1 is dispensable for embryonic
development and acts as a secondary mediator of Hh signaling transduction (Bai et al., 2004;
Ding et al., 1998; Mo et al., 1997; Motoyama et al., 1998; Park et al., 2000). Although all three
Gli proteins can function as activators of the pathway, Gli2 and Gli3 are the major transcriptional
activator and repressor of the mammalian Hh signaling, respectively (Bai et al., 2004; Buttitta et
al., 2003; McDermott et al., 2005; Motoyama et al., 1998).
In the absence of Hh ligand, Gli3 is phosphorylated sequentially by PKA, glycogen
synthase kinase 3ß (GSK3ß) and casein kinase 1 (CK1) and targeted to ubiquitin/proteasome-
mediated proteolysis to generate a C-terminally truncated transcriptional repressor (Gli3R)
(Tempe et al., 2006; Wang et al., 2000; Wang and Li, 2006). Gli2 is also phosphorylated by
these kinases, although in the case of processing is inefficient and leads to mostly degradation by
the proteasome (Pan et al., 2006). The specific domain in the C-terminus has been suggested to
be responsible for the differential processing and degradation of Gli2 and Gli3 (Pan and Wang,
2007). Hh signaling activation blocks the proteolytic cleavage of Gli2 and promotes the activator
function of Gli2 and Gli3. However the mechanism is undefined.

8
Data from mutant mouse analyses suggest that Gli2 and Gli3 are involved in Ihh-
dependent growth plate chondrocyte development. Ihh-null mice show reduced chondrocyte
proliferation, increased zone of hypertrophic chondrocytes and lack of ossification in
endochondral bones (St-Jacques et al., 1999). Similarly, Gli2 knockout mice display expanded
hypertrophic zone and reduced bone formation, suggesting that Gli2 functions as an activator in
regulating Hh signaling in chondrocytes and reduced Gli2 function is in part responsible for the
Ihh-null phenotype (Miao et al., 2004). Genetic studies in mice show that Gli3 functions as a
major repressor regulating Hh signaling in growth plate chondrocytes. Loss of function of Gli3
rescues the growth plate developmental defects in Ihh-null mice, which supports the notion that
Gli3 acts as a negative regulator of Hh-mediated signaling in chondrocytes and Ihh possess
inhibitory effect on the repressor function of Gli3 in this process (Koziel et al., 2005).
Besides the Hh-mediated regulation, the Gli transcription factors may also be regulated
through an Hh/Smo-independent mechanism. Studies of explants lacking the Gli genes suggest
that Gli3, but not Gli2, is involved in the inhibitory effect of Pthlh on growth plate chondrocyte
hypertrophic differentiation. Pthlh signaling activation inhibits Gli-mediated transcription
through processing Gli3 via a PKA-dependent manner (Mau et al., 2007; Miao et al., 2004;
Tukachinsky et al., 2010; Tuson et al., 2011; Vortkamp et al., 1996; Zeng et al., 2010 (Fig1-2B).
These results suggest another level of feedback control between Ihh and Pthlh signaling and raise
the possibility that the Gli transcription factors act as integrators of multiple signaling pathways
during growth plate chondrocyte differentiation. However, our knowledge of the regulation of
Gli-mediated pathway activity is still limited.

9
1.5 Regulators in Hedgehog (Hh) signaling pathway: Sufu and Kif7
In mammalian Hh signaling, Suppressor of fused (Sufu) and Kinesin family member 7
(Kif7) are two evolutionarily conserved regulators of Gli transcription factors. Deletion of Sufu
results in embryonic lethality at E9.5 with severe ectopic Hh signaling activation, indicating Sufu
functions as a major repressor of mammalian Hh pathway (Cooper et al., 2005; Svard et al.,
2006). Western analysis revealed a severe reduction in the levels of Gli2 and full length Gli3 as
well as a lack of Gli3 repressor in Sufu-/-
embryos (Humke et al.; Wang et al., 2010), suggesting a
role of Sufu in controlling Gli transcriptional repressor processing. However, the mechanism of
action of Sufu remains controversial. Sufu interacts directly with all three Gli proteins and
sequesters them in the cytoplasm in the absence of Hh ligand, which prevents Hh pathway
activation (Barnfield et al., 2005; Ding et al., 1999; Kogerman et al., 1999; Murone et al., 2000).
Sufu acts to recruit a co-repressor complex to inhibit Gli activator function (Cheng and Bishop,
2002) and promotes Gli3R formation through interacting with GSK3ß (Kise et al., 2009). Recent
studies further reveal a role for Sufu in maximal Gli-dependent transcriptional activation,
suggesting that Sufu also acts positively in Hh signaling (Chen et al., 2009). Hh signaling
activation promotes Sufu protein turnover via the ubiquitin-proteasome system (Yue et al.,
2009). PKA and GSK3ß phosphorylate Sufu at Ser-346 and Ser-342, respectively, and
phosphorylation stabilizes Sufu against Hh signaling-induced degradation (Chen et al., 2011).
Taken together, the data suggest that Sufu plays an important role in regulating Gli activities, and
Sufu itself is tightly regulated to achieve a gradient level of Hh signaling activity during
development.

10
In contrast, less is known about the action of Kif7. Loss of Kif7 results in perinatal death
and phenotypes, such as polydactyly, mimicking Gli3 knockout mice (Chen et al., 2009; Cheung
et al., 2009; Hui and Joyner, 1993). Kif7 also interacts with all three Gli proteins and elevated
Gli2 and reduced Gli3 levels are found in Kif7-null mice, suggesting that it acts as a negative
regulator of the mammalian Hh pathway. Kif7-/-
embryos exhibit defects in floor plate
development, which depends on maximal level of Hh pathway activation, indicating a potential
positive role of Kif7 in Hh signaling (Cheung et al., 2009). The mechanism by which Sufu and
Kif7 execute dual functions and/or whether they possess cooperative regulatory functions is not
known.

11
1.6 The primary cilium in Hh signaling
In vertebrates, the primary cilium, which is a non-motile microtubule-based organelle,
serves as a focal point in Hh signaling regulation (Corbit et al., 2005; Goetz and Anderson, 2010;
Huangfu et al., 2003). The formation and maintenance of the primary cilium depend on
intraflagellar transport (IFT) of protein complexes driven by kinesin and dynein motors for
anterograde and retrograde IFT movements, respectively. Mutation in proteins involved in IFT,
such as IFT88, IFT172, and the motor protein Kif3a, prevents functional cilia formation and
attenuates Hh signaling (Huangfu et al., 2003). To date, many components of the Hh pathway are
found to be localized to the cilia either in the absence of Hh ligand or during pathway activation
(Corbit et al., 2005; Haycraft et al., 2005; Huangfu and Anderson, 2005; Liu et al., 2005). Ptch1
is localized to the primary cilia where it prevents the ciliary localization of Smo in the absence of
Hh ligand. Binding of Hh ligand to Ptch1 induces its export from the cilia, which allows the
ciliary translocation of Smo. Through an unknown mechanism, ciliary localization of Smo relays
Hh signals to the cytoplasm, which leads to Gli-mediated transcription activation (Rohatgi et al.,
2007) (Fig. 1-3).
Hh pathway activation in the absence of Sufu is independent of cilia (Chen et al., 2009;
Jia et al., 2009), suggesting that Smo may activate Gli proteins at the cilia by inhibiting Sufu.
PKA downregulates Smo-mediated Hh signaling activation via a Sufu-dependent mechanism
(Chen et al., 2009; Svard et al., 2006; Wu et al., 2004), suggesting that Smo and PKA both
modulate Hh signaling thorough Sufu. Therefore, a model by which Hh signaling activates Gli
protein at the cilia through relieving the inhibitory function of Sufu, resulting in Gli-mediated

12
transcriptional activation was proposed. This model is supported by recent studies carried out in
cultured fibroblasts, showing that Smo plays a positive role in dissociation of inhibitory Sufu-Gli
complexes at the primary cilium upon Hh pathway activation. Furthermore, Gli2 and Gli3 are
required to recruit Sufu to cilia, but Gli proteins can localize to cilia in the absence of Sufu
(Humke et al., 2010; Tukachinsky et al., 2010). However, how Hh signaling and/or other
signaling pathways regulate Sufu in the context of primary cilia is unknown. Mammalian Sufu
bears four conserved cAMP-dependent PKA recognition sites, and phosphorylation by PKA and
GSK3ß stabilizes Sufu and promotes its tendency to stay longer at the ciliary tip (Chen et al.,
2011) (Fig. 1-3). This stabilizing effect of PKA on Sufu is consistent with its inhibitory function
on Hh signaling (Dai et al., 1999; Tempe et al., 2006). A carboxy-terminal region of Sufu
interacts and recruits GSK3ß for efficient Gli3 processing (Kise et al., 2009). It is plausible that
Sufu, stabilized through phosphorylation by PKA and GSK3ß, forms a complex with Gli2/3 and
is transported into the primary cilium where further modification takes place, allowing formation
of the truncated transcriptional repressors via proteolytic processing in the cytoplasm (Fig.1-3).
We do not yet know how the integrity of the Sufu-Gli complex is maintained or how the
phosphorylation state of Sufu and/or Gli regulates this complex. We do not yet know how Hh
signaling promotes Sufu-Gli complexes dissociation or whether this process can take place
outside of the primary cilium. Additionally, it will be interesting to know if Kif7, which shows
dynamic ciliary localization (Liem et al., 2009), plays a regulatory role in the formation and/or
dissociation of Sufu-Gli complexes in the primary cilium. Furthermore, during growth plate
chondrocyte differentiation, Pthlh exerts regulatory functions on Gli proteins as well as Gli-
mediated transcriptional activity, and it functions mainly through PKA activation. Thus, Sufu
regulation may serve as a common and important regulatory mechanism between Ihh and Pthlh

13
signaling during growth plate chondrocyte development.

14
1.7 Hypothesis & Specific aims
Proper regulation of Hh and Pthlh signaling is crucial in chondrogenesis. Hh signaling
mediators, such as Sufu, Kif7, and primary cilium, may play important roles in controlling the
process of chondrocyte differentiation. I hypothesize that Sufu, Kif7, and primary cilium play
distinct and overlapping roles in regulating Hh signaling activity during chondrocyte
development.
There are three specific aims set out for this work:
Aim 1: What are the regulatory roles of Sufu and Kif7 in Hh signaling during growth plate
chondrocyte development?
Aim 2: What is the role of Kif7 in the formation and/or dissociation of Sufu-Gli complexes?
Aim 3: What are the potential roles of Sufu, Kif7, and primary cilium in Pthlh action during
chondrocyte development?
15
1.8 Figures
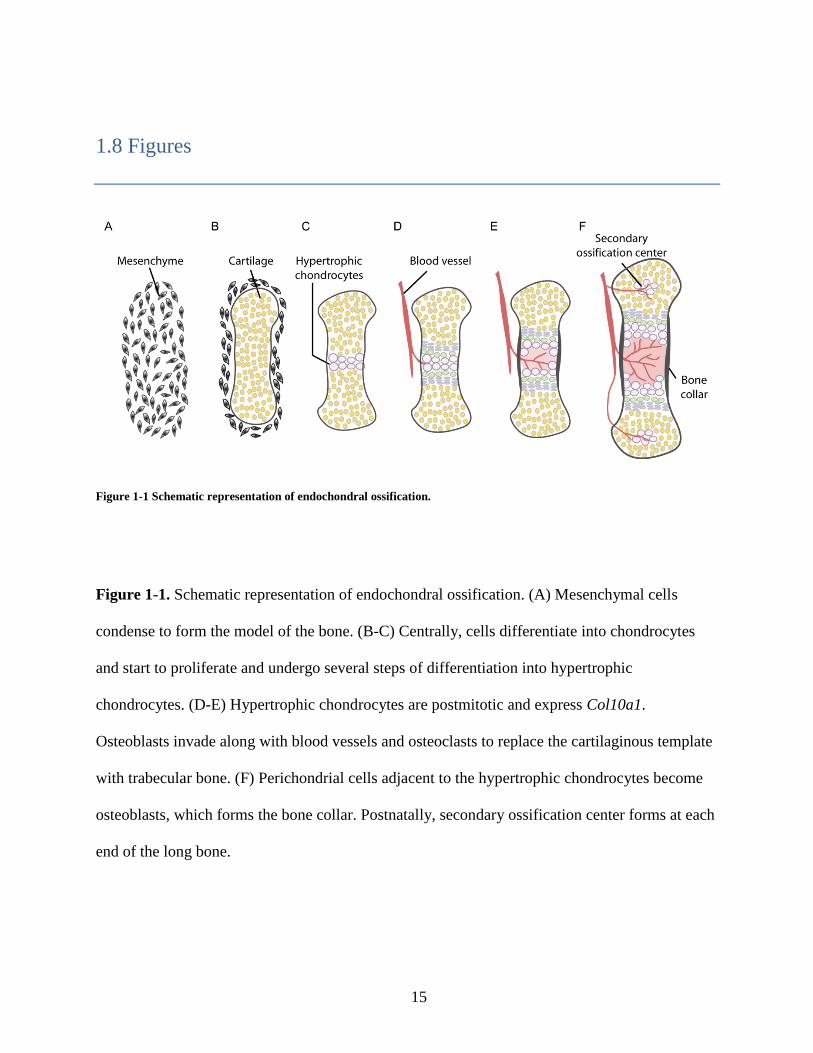
Figure 1-1 Schematic representation of endochondral ossification.
Figure 1-1. Schematic representation of endochondral ossification. (A) Mesenchymal cells
condense to form the model of the bone. (B-C) Centrally, cells differentiate into chondrocytes
and start to proliferate and undergo several steps of differentiation into hypertrophic
chondrocytes. (D-E) Hypertrophic chondrocytes are postmitotic and express Col10a1.
Osteoblasts invade along with blood vessels and osteoclasts to replace the cartilaginous template
with trabecular bone. (F) Perichondrial cells adjacent to the hypertrophic chondrocytes become
osteoblasts, which forms the bone collar. Postnatally, secondary ossification center forms at each
end of the long bone.

16
Figure 1-2 Ihh/Pthlh signaling regulation in the growth plate.

17
Figure 1-2. Ihh/Pthlh signaling regulation in the growth plate. (A) Growth plate chondrocytes
undergo a coordinated differentiation process, ending in chondrocyte hypertrophy and apoptosis.
1) Ihh, which is expressed by prehypertrophic chondrocytes, stimulates Pthlh expression at the
periarticular region that functions to inhibit chondrocyte hypertrophic differentiation and Ihh
expression (illustrated by 4.). 2) Ihh positively regulates chondrocyte proliferation (circled P) via
a Pthlh-independent manner. 3) Ihh initiates osteoblast differentiation at the perichondrium
adjacent to its expressing region. 4) Pthlh inhibits chondrocyte hypertrophic differentiation
mainly through PKA activation. 5) Pthlh possesses stimulatory effect on chondrocyte
proliferation. Solid lines show the regulation of Pthlh, and dashed lines show the regulation of
Ihh. (B) Both Gli2 and Gli3 are involved in the regulation of chondrocyte hypertrophic
differentiation. Pthlh negatively regulates this differentiation process through processing Gli3FL
into its truncated form (Gli3R) via a PKA-dependent mechanism.

18
Figure 1-3 Hh signaling in vertebrates.
Figure 1-3. Hh signaling in vertebrates. In the absence of the Hh ligand, Ptch1 localizes to the
primary cilium and prevents Smo ciliary accumulation. Phosphorylation by PKA and GSK3ß
stabilizes Sufu and promotes its tendency to stay longer at the ciliary tip. Sufu-Gli complexes
traffic through the cilium at low level and promote Gli repressor (GliR) formation. Hh ligand
binding to Ptch1 leads to the translocation of Smo into the cilium. Smo-mediated dissociation of
Smo-Gli complexes results in Gli transcriptional activation.

19
1.9 References
Bai, C. B., Stephen, D. and Joyner, A. L. (2004). All mouse ventral spinal cord patterning by
hedgehog is Gli dependent and involves an activator function of Gli3. Dev Cell 6, 103-15.
Barnfield, P. C., Zhang, X., Thanabalasingham, V., Yoshida, M. and Hui, C. C. (2005).
Negative regulation of Gli1 and Gli2 activator function by Suppressor of fused through multiple
mechanisms. Differentiation 73, 397-405.
Buttitta, L., Mo, R., Hui, C. C. and Fan, C. M. (2003). Interplays of Gli2 and Gli3 and their
requirement in mediating Shh-dependent sclerotome induction. Development 130, 6233-43.
Chen, M. H., Wilson, C. W., Li, Y. J., Law, K. K., Lu, C. S., Gacayan, R., Zhang, X., Hui,
C. C. and Chuang, P. T. (2009). Cilium-independent regulation of Gli protein function by Sufu
in Hedgehog signaling is evolutionarily conserved. Genes Dev 23, 1910-28.
Chen, Y., Yue, S., Xie, L., Pu, X. H., Jin, T. and Cheng, S. Y. (2011). Dual phosphorylation of
Suppressor of fused by PKA and GSK3beta regulates its stability and localization in the primary
cilium. J Biol Chem.
Cheng, S. Y. and Bishop, J. M. (2002). Suppressor of Fused represses Gli-mediated
transcription by recruiting the SAP18-mSin3 corepressor complex. Proc Natl Acad Sci U S A 99,
5442-7.
Cheung, H. O., Zhang, X., Ribeiro, A., Mo, R., Makino, S., Puviindran, V., Law, K. K.,
Briscoe, J. and Hui, C. C. (2009). The kinesin protein Kif7 is a critical regulator of Gli
transcription factors in mammalian hedgehog signaling. Sci Signal 2, ra29.
Chung, U. I., Schipani, E., McMahon, A. P. and Kronenberg, H. M. (2001). Indian hedgehog
couples chondrogenesis to osteogenesis in endochondral bone development. J Clin Invest 107,
295-304.
Cooper, A. F., Yu, K. P., Brueckner, M., Brailey, L. L., Johnson, L., McGrath, J. M. and
Bale, A. E. (2005). Cardiac and CNS defects in a mouse with targeted disruption of suppressor
of fused. Development 132, 4407-17.
Corbit, K. C., Aanstad, P., Singla, V., Norman, A. R., Stainier, D. Y. and Reiter, J. F. (2005). Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018-21.
Dai, P., Akimaru, H., Tanaka, Y., Maekawa, T., Nakafuku, M. and Ishii, S. (1999). Sonic
Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J Biol Chem 274, 8143-
52.

20
de Crombrugghe, B., Lefebvre, V. and Nakashima, K. (2001). Regulatory mechanisms in the
pathways of cartilage and bone formation. Curr Opin Cell Biol 13, 721-7.
Ding, Q., Fukami, S., Meng, X., Nishizaki, Y., Zhang, X., Sasaki, H., Dlugosz, A.,
Nakafuku, M. and Hui, C. (1999). Mouse suppressor of fused is a negative regulator of sonic
hedgehog signaling and alters the subcellular distribution of Gli1. Curr Biol 9, 1119-22.
Ding, Q., Motoyama, J., Gasca, S., Mo, R., Sasaki, H., Rossant, J. and Hui, C. C. (1998).
Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice.
Development 125, 2533-43.
Duman-Scheel, M., Weng, L., Xin, S. and Du, W. (2002). Hedgehog regulates cell growth and
proliferation by inducing Cyclin D and Cyclin E. Nature 417, 299-304.
Goetz, S. C. and Anderson, K. V. (2010). The primary cilium: a signalling centre during
vertebrate development. Nat Rev Genet 11, 331-44.
Haycraft, C. J., Banizs, B., Aydin-Son, Y., Zhang, Q., Michaud, E. J. and Yoder, B. K. (2005). Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for
processing and function. PLoS Genet 1, e53.
Huang, W., Chung, U. I., Kronenberg, H. M. and de Crombrugghe, B. (2001). The
chondrogenic transcription factor Sox9 is a target of signaling by the parathyroid hormone-
related peptide in the growth plate of endochondral bones. Proc Natl Acad Sci U S A 98, 160-5.
Huangfu, D. and Anderson, K. V. (2005). Cilia and Hedgehog responsiveness in the mouse.
Proc Natl Acad Sci U S A 102, 11325-30.
Huangfu, D., Liu, A., Rakeman, A. S., Murcia, N. S., Niswander, L. and Anderson, K. V. (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426,
83-7.
Hui, C. C. and Joyner, A. L. (1993). A mouse model of greig cephalopolysyndactyly
syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat Genet 3,
241-6.
Humke, E. W., Dorn, K. V., Milenkovic, L., Scott, M. P. and Rohatgi, R. The output of
Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and
the Gli proteins. Genes Dev 24, 670-82.
Humke, E. W., Dorn, K. V., Milenkovic, L., Scott, M. P. and Rohatgi, R. (2010). The output
of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused
and the Gli proteins. Genes Dev 24, 670-82.
Jia, J., Kolterud, A., Zeng, H., Hoover, A., Teglund, S., Toftgard, R. and Liu, A. (2009).
Suppressor of Fused inhibits mammalian Hedgehog signaling in the absence of cilia. Dev Biol
330, 452-60.

21
Karaplis, A. C., Luz, A., Glowacki, J., Bronson, R. T., Tybulewicz, V. L., Kronenberg, H.
M. and Mulligan, R. C. (1994). Lethal skeletal dysplasia from targeted disruption of the
parathyroid hormone-related peptide gene. Genes Dev 8, 277-89.
Karp, S. J., Schipani, E., St-Jacques, B., Hunzelman, J., Kronenberg, H. and McMahon, A.
P. (2000). Indian hedgehog coordinates endochondral bone growth and morphogenesis via
parathyroid hormone related-protein-dependent and -independent pathways. Development 127,
543-8.
Kise, Y., Morinaka, A., Teglund, S. and Miki, H. (2009). Sufu recruits GSK3beta for efficient
processing of Gli3. Biochem Biophys Res Commun 387, 569-74.
Kobayashi, T., Chung, U. I., Schipani, E., Starbuck, M., Karsenty, G., Katagiri, T., Goad,
D. L., Lanske, B. and Kronenberg, H. M. (2002). PTHrP and Indian hedgehog control
differentiation of growth plate chondrocytes at multiple steps. Development 129, 2977-86.
Kogerman, P., Grimm, T., Kogerman, L., Krause, D., Unden, A. B., Sandstedt, B.,
Toftgard, R. and Zaphiropoulos, P. G. (1999). Mammalian suppressor-of-fused modulates
nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol 1, 312-9.
Koziel, L., Wuelling, M., Schneider, S. and Vortkamp, A. (2005). Gli3 acts as a repressor
downstream of Ihh in regulating two distinct steps of chondrocyte differentiation. Development
132, 5249-60.
Kronenberg, H. M. (2003). Developmental regulation of the growth plate. Nature 423, 332-6.
Lanske, B., Karaplis, A. C., Lee, K., Luz, A., Vortkamp, A., Pirro, A., Karperien, M.,
Defize, L. H., Ho, C., Mulligan, R. C. et al. (1996). PTH/PTHrP receptor in early development
and Indian hedgehog-regulated bone growth. Science 273, 663-6.
Liem, K. F., Jr., He, M., Ocbina, P. J. and Anderson, K. V. (2009). Mouse Kif7/Costal2 is a
cilia-associated protein that regulates Sonic hedgehog signaling. Proc Natl Acad Sci U S A 106,
13377-82.
Liu, A., Wang, B. and Niswander, L. A. (2005). Mouse intraflagellar transport proteins
regulate both the activator and repressor functions of Gli transcription factors. Development 132,
3103-11.
Long, F., Chung, U. I., Ohba, S., McMahon, J., Kronenberg, H. M. and McMahon, A. P. (2004). Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton.
Development 131, 1309-18.
Long, F., Zhang, X. M., Karp, S., Yang, Y. and McMahon, A. P. (2001). Genetic
manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the
regulation of chondrocyte proliferation. Development 128, 5099-108.

22
MacLean, H. E., Guo, J., Knight, M. C., Zhang, P., Cobrinik, D. and Kronenberg, H. M. (2004). The cyclin-dependent kinase inhibitor p57(Kip2) mediates proliferative actions of PTHrP
in chondrocytes. J Clin Invest 113, 1334-43.
Maeda, Y., Nakamura, E., Nguyen, M. T., Suva, L. J., Swain, F. L., Razzaque, M. S.,
Mackem, S. and Lanske, B. (2007). Indian Hedgehog produced by postnatal chondrocytes is
essential for maintaining a growth plate and trabecular bone. Proc Natl Acad Sci U S A 104,
6382-7.
Mau, E., Whetstone, H., Yu, C., Hopyan, S., Wunder, J. S. and Alman, B. A. (2007). PTHrP
regulates growth plate chondrocyte differentiation and proliferation in a Gli3 dependent manner
utilizing hedgehog ligand dependent and independent mechanisms. Dev Biol 305, 28-39.
McDermott, A., Gustafsson, M., Elsam, T., Hui, C. C., Emerson, C. P., Jr. and Borycki, A.
G. (2005). Gli2 and Gli3 have redundant and context-dependent function in skeletal muscle
formation. Development 132, 345-57.
Miao, D., Liu, H., Plut, P., Niu, M., Huo, R., Goltzman, D. and Henderson, J. E. (2004).
Impaired endochondral bone development and osteopenia in Gli2-deficient mice. Exp Cell Res
294, 210-22.
Minina, E., Wenzel, H. M., Kreschel, C., Karp, S., Gaffield, W., McMahon, A. P. and
Vortkamp, A. (2001). BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte
proliferation and differentiation. Development 128, 4523-34.
Mo, R., Freer, A. M., Zinyk, D. L., Crackower, M. A., Michaud, J., Heng, H. H., Chik, K.
W., Shi, X. M., Tsui, L. C., Cheng, S. H. et al. (1997). Specific and redundant functions of Gli2
and Gli3 zinc finger genes in skeletal patterning and development. Development 124, 113-23.
Motoyama, J., Takabatake, T., Takeshima, K. and Hui, C. (1998). Ptch2, a second mouse
Patched gene is co-expressed with Sonic hedgehog. Nat Genet 18, 104-6.
Murone, M., Luoh, S. M., Stone, D., Li, W., Gurney, A., Armanini, M., Grey, C., Rosenthal,
A. and de Sauvage, F. J. (2000). Gli regulation by the opposing activities of fused and
suppressor of fused. Nat Cell Biol 2, 310-2.
Olsen, B. R., Reginato, A. M. and Wang, W. (2000). Bone development. Annu Rev Cell Dev
Biol 16, 191-220.
Pan, Y., Bai, C. B., Joyner, A. L. and Wang, B. (2006). Sonic hedgehog signaling regulates
Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol 26,
3365-77.
Pan, Y. and Wang, B. (2007). A novel protein-processing domain in Gli2 and Gli3
differentially blocks complete protein degradation by the proteasome. J Biol Chem 282, 10846-
52.

23
Park, H. L., Bai, C., Platt, K. A., Matise, M. P., Beeghly, A., Hui, C. C., Nakashima, M. and
Joyner, A. L. (2000). Mouse Gli1 mutants are viable but have defects in SHH signaling in
combination with a Gli2 mutation. Development 127, 1593-605.
Provot, S. and Schipani, E. (2005). Molecular mechanisms of endochondral bone development.
Biochem Biophys Res Commun 328, 658-65.
Razzaque, M. S., Soegiarto, D. W., Chang, D., Long, F. and Lanske, B. (2005). Conditional
deletion of Indian hedgehog from collagen type 2alpha1-expressing cells results in abnormal
endochondral bone formation. J Pathol 207, 453-61.
Rohatgi, R., Milenkovic, L. and Scott, M. P. (2007). Patched1 regulates hedgehog signaling at
the primary cilium. Science 317, 372-6.
St-Jacques, B., Hammerschmidt, M. and McMahon, A. P. (1999). Indian hedgehog signaling
regulates proliferation and differentiation of chondrocytes and is essential for bone formation.
Genes Dev 13, 2072-86.
Svard, J., Heby-Henricson, K., Persson-Lek, M., Rozell, B., Lauth, M., Bergstrom, A.,
Ericson, J., Toftgard, R. and Teglund, S. (2006). Genetic elimination of Suppressor of fused
reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell
10, 187-97.
Tempe, D., Casas, M., Karaz, S., Blanchet-Tournier, M. F. and Concordet, J. P. (2006).
Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3
ubiquitination by SCFbetaTrCP. Mol Cell Biol 26, 4316-26.
Tukachinsky, H., Lopez, L. V. and Salic, A. (2010). A mechanism for vertebrate Hedgehog
signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol 191,
415-28.
Tuson, M., He, M. and Anderson, K. V. (2011). Protein kinase A acts at the basal body of the
primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube.
Development.
Vortkamp, A., Lee, K., Lanske, B., Segre, G. V., Kronenberg, H. M. and Tabin, C. J. (1996). Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related
protein. Science 273, 613-22.
Wang, B., Fallon, J. F. and Beachy, P. A. (2000). Hedgehog-regulated processing of Gli3
produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100,
423-34.
Wang, B. and Li, Y. (2006). Evidence for the direct involvement of {beta}TrCP in Gli3 protein
processing. Proc Natl Acad Sci U S A 103, 33-8.

24
Wang, C., Pan, Y. and Wang, B. (2010). Suppressor of fused and Spop regulate the stability,
processing and function of Gli2 and Gli3 full-length activators but not their repressors.
Development 137, 2001-9.
Wu, X., Walker, J., Zhang, J., Ding, S. and Schultz, P. G. (2004). Purmorphamine induces
osteogenesis by activation of the hedgehog signaling pathway. Chem Biol 11, 1229-38.
Yue, S., Chen, Y. and Cheng, S. Y. (2009). Hedgehog signaling promotes the degradation of
tumor suppressor Sufu through the ubiquitin-proteasome pathway. Oncogene 28, 492-9.
Zeng, H., Jia, J. and Liu, A. (2010). Coordinated translocation of Mammalian gli proteins and
suppressor of fused to the primary cilium. PLoS One 5, e15900.

25
Chapter 2 Kif7 promotes Hh signaling in growth plate
chondrocytes by restricting the inhibitory function of Sufu
Shu-Hsuan C. Hsu, Xiaoyun Zhang, Chunying Yu, Zhu Juan Li, Jay S. Wunder, Chi-Chung Hui,
and Benjamin A. Alman
All experiments and analysis described in this chapter were conducted by Shu-Hsuan C. Hsu
with the exception of the western and in situ analysis by Xiaoyun Zhang and Chunying Yu. This
study has been published in Development (Hsu et al., 2011).

26
2.1 Summary
Proper regulation of Indian hedgehog (Ihh) signaling is vital for chondrocyte proliferation
and differentiation in the growth plate. Its dysregulation causes skeletal dysplasia, osteoarthritis,
or cartilaginous neoplasia. Here, I show that Suppressor of fused (Sufu) and Kif7 are essential
regulators of Ihh signaling. While Sufu acts as a negative regulator of Gli transcription factors,
Kif7 functions both positively and negatively in chondrocytes. Kif7 plays a role in the turnover
of Sufu and the exclusion of Sufu-Gli complexes from the primary cilium. Importantly, halving
the dosage of Sufu restores normal Hedgehog pathway activity and chondrocyte development in
Kif7 null mice demonstrating that the positive role of Kif7 is to restrict the inhibitory activity of
Sufu. Furthermore, Kif7 also inhibits Gli transcriptional activity in the chondrocytes when Sufu
function is absent. Therefore, Kif7 regulates the activity of Gli transcription factors through both
Sufu-dependent and -independent mechanisms.

27
2.2 Introduction
The precise regulation of chondrocyte proliferation and differentiation is critical for
normal bone growth. During endochondral bone development, growth plate chondrocyte
differentiation is governed by the spatial and temporal regulation of a number of signaling
pathways. Dysregulation of these processes during development is responsible for skeletal
dysplasias, which are characterized by short stature (Karsenty et al., 2009). Inhibition of
differentiation of growth plate chondrocytes can cause cartilaginous tumors (Bovee et al., 2010)
and aberrant activation of signaling pathways normally involved in the regulation of growth plate
chondrocytes is associated with osteoarthritis, a common degenerative joint disease (Lin et al.,
2009). Therefore, abnormal chondrocyte proliferation and differentiation have profound
negative effects on overall health.
Ihh, a member of the Hedgehog (Hh) family of signaling molecules, regulates the
transcriptional activity of Gli proteins through binding to its receptor Patched1 (Ptch1) and
derepression of the signaling receptor Smoothened (Smo). Three Gli zinc finger proteins (Gli1-3)
are transcription factors mediating Hh signaling in mammalian cells (Jiang & Hui, 2008). In
mice, Gli2 and Gli3 are essential genes, whereas Gli1 is dispensable for embryonic development
and encodes a secondary mediator of Hh signaling. Gli2 and Gli3 are the major transcriptional
activator and repressor of the mammalian Hh pathway, respectively, though all three Gli proteins
can activate the expression of Hh target genes, such as Ptch1 and Gli1 itself. Gli3 is processed
efficiently by the proteasome into a C-terminally truncated transcriptional repressor. Through ill-
defined mechanisms, Hh signaling blocks the proteolytic cleavage of Gli3 and promotes the

28
transcriptional activator function of Gli2 and Gli3. Mutant mouse analysis indicates that Gli2 and
Gli3 are involved in Ihh-dependent chondrocyte development. Mice lacking Ihh are
characterized by reduced chondrocyte proliferation, an expanded hypertrophic zone in the
growth plate, and lack of ossification in endochondral bones (St-Jacques et al., 1999). Similar to
Ihh knockout mice, Gli2 knockout mice show an expanded hypertrophic zone and reduced bone
formation, suggesting that the Ihh mutant phenotype is in part due to a reduction of Gli2
activator function (Miao et al., 2004). Loss of Gli3 rescues the chondrocyte proliferation and
differentiation defects in Ihh mutant mice indicating that a major action of Ihh is to limit the
repressor function of Gli3 in growth plate chondrocytes (Koziel et al., 2005). These observations
indicate that Ihh-dependent regulation of Gli2 and Gli3 plays a critical role in chondrocyte
differentiation.
In mammalian Hh signaling, Sufu and Kif7 are two evolutionarily conserved regulators
of Gli transcription factors (Wilson et al., 2009). In mice, Sufu is a major negative regulator of
Hh signaling and inactivation of Sufu leads to embryonic lethality at E9.5 with severe ectopic Hh
pathway activation similar to those observed in Ptch1-null embryos (Cooper et al., 2005; Svard
et al., 2006). Sufu forms complexes with all three Gli proteins and inhibits their transcriptional
activity (Barnfield et al., 2005; Ding et al., 1999). Recent studies in cultured fibroblasts have led
to the suggestion that Hh signaling promotes the nuclear translocation and transcriptional activity
of Gli2 and Gli3 through dissociation of cytoplasmic Sufu-Gli complexes (Humke et al., 2010;
Tukachinsky et al., 2010). In addition, Sufu-/-
cells exhibit a drastic reduction in the levels of full
length Gli2 and Gli3 as well as a lack of Gli3 repressor suggesting that it also plays a critical role
in the stabilization of Gli activators and the formation of Gli3 repressor (Humke et al., 2010;
Wang et al., 2010). In contrast, less is known about the action of Kif7, which is a kinesin motor

29
protein recently shown to play regulatory roles in mammalian Hh signaling. Kif7-null mice die at
birth and exhibit a phenotype (Chen et al., 2009; Cheung et al., 2009) similar to that of Gli3-null
mice (Hui and Joyner, 1993). Kif7-null embryos show mild ectopic Hh pathway activation with
ectopic formation of ventral neurons in the spinal cord as well as elevated Gli2 and reduced Gli3
levels, suggesting that it acts negatively in Hh signaling. Interestingly, Kif7 also functions
positively in controlling Hh pathway activity (Cheung et al., 2009; Liem et al., 2009). For
example, floor-plate development, which is induced by maximal level of Hh pathway activity, is
compromised in the absence of Kif7 function (Adolphe et al., 2006; Cheung et al., 2009; Endoh-
Yamagami et al., 2009). How Kif7 acts both negatively and positively in mammalian Hh
signaling is not understood and whether Kif7 possesses cooperative regulatory function with
Sufu is unclear.
The primary cilium acts as a focal point in the processing of Hh signaling (Corbit et al.,
2005; Goetz and Anderson, 2010; Huangfu et al., 2003). Recent studies have suggested that,
when the Hh pathway is activated, Smo promotes the dissociation of inhibitory Sufu-Gli
complexes at the primary cilium (Tukachinsky et al., 2010). Kif7 has been shown to translocate
to the tip of primary cilium in cultured fibroblasts upon pathway stimulation (Liem et al., 2009).
However, it is unknown whether Kif7 plays a regulatory role in the formation and/or dissociation
of Sufu-Gli complexes in the primary cilium. In this study, I explored the roles of Sufu and Kif7
in Ihh-dependent chondrocyte development using genetically modified mice. Our results indicate
that while Sufu is a major negative regulator of Hh pathway activity, Kif7 plays dual roles in the
control of chondrocyte development. Intriguingly, Kif7 is localized to the ciliary tip of
proliferating chondrocytes in vivo and appears to exclude Sufu-Gli complexes from the primary
cilium. I speculate that Kif7 functions positively in Hh signaling to promote Smo-induced

30
dissociation of Sufu-Gli complexes at the primary cilium.

31
2.3 Results
2.3.1 Sufu is differentially expressed in the growth plate and is required for normal
skeletal development
To determine if Sufu might play a role in growth plate chondrocyte function, Sufu
expression was examined in fetal limb cartilage. qRT-PCR analysis on micro-dissected sections
of the growth plate and in situ hybridization were used to localize its RNA expression. Sufu is
highly expressed in articular and resting chondrocytes (also known as reserve chondrocytes; the
early stages of chondrocyte differentiation), while its expression is downregulated when cells
undergo differentiation to hypertrophic chondrocytes (Fig. 2-1A, D). Immunohistochemical
staining of fetal limbs revealed high level of Sufu protein expression in resting and proliferating
chondrocytes, and that fewer than 10% of the prehypertrophic and hypertrophic chondrocytes are
Sufu-positive (Fig. 2-1C, E). The pattern of Sufu expression is similar to that of Pthlh (an Hh
regulated gene in the growth plate), but in contrast, Ihh expression is highest in the
prehypertrophic chondrocytes (Fig. 2-1B). Thus, Sufu is differentially expressed in the growth
plate, with its highest level in early stages of chondrocyte differentiation.
To bypass the embryonic lethality of Sufu-null mice at E9.5 (Cooper et al., 2005; Svard et
al., 2006), I generated chondrocyte-specific knockout mice (Col2a1-Cre; Sufuf/f
) to investigate
the role of Sufu in growth plate chondrocyte development. Immunohistochemical staining and
western analysis demonstrated efficient deletion of Sufu in the growth plate of Col2a1-Cre;
Sufuf/f
mice (Fig. 2-2A-C). Chondrocyte-specific knockout of Sufu in mice resulted in perinatal
death with a few exceptions that survived for 10 days. Body length and weight measurements

32
demonstrated a ~24% and ~50% reduction in the mutants, respectively, when compared with
wild-type littermates (Fig. 2-2D-F). Alcian blue/Alizarin red staining showed a significant
reduction of bone ossification in Col2a1-Cre; Sufuf/f
mice (Fig. 2-3A, D). Histological analysis
revealed delayed formation of secondary ossification centers, expansion of proliferating zone as
well as reduction of hypertrophic zone in the Sufu-deficient tibial and vertebral growth plates
(Fig. 2-3B). By Ki67 and Phospho-H3 immunostaining, a higher percentage of proliferating cells
was found in the mutant growth plate (Fig. 2-3E, F; Fig. 2-6J). In contrast, immunostaining of
active Caspase-3 and TUNEL analysis did not reveal a significant difference of apoptosis in the
mutants (Fig. 2-3G). These data indicate that Sufu is required for normal endochondral skeletal
development, where it regulates growth plate chondrocyte proliferation and differentiation.
2.3.2 Sufu acts as a negative regulator of Hh signaling during chondrocyte
differentiation
Ptch1 inhibits Smo in the absence of Hh signals and acts as a negative regulator of Hh
signaling. Chondrocyte-specific knockout of Ptch1 (Col2a1-Cre; Ptch1f/f
) leads to elevated Hh
pathway activity and results in a phenotype similar to those observed in Sufu mutants, including
an expansion of proliferative zone and a reduction of hypertrophic zone (Mak et al., 2008) (Fig.
2-3C; Fig. 2-4A-C). I found that the Sufu knockout phenotype is consistently milder than the
Ptch1 knockout phenotype (Fig. 2-4A-C). To determine whether knockout of Sufu also results in
Hh pathway activation in the chondrocytes, I performed qRT-PCR analysis. Knockout of Ptch1
leads to increased expression of Hh target genes, such as Gli1, Ptch1, and Hhip1 (Fig. 2-4D).
Interestingly, I detected up-regulation of Ptch1 and Hhip1, but not Gli1, in Sufu-deficient

33
chondrocytes (Fig. 2-4D). Analysis by in situ hybridization revealed Ptch1 transcripts in the
proliferating chondrocytes. In wild-type mice, cells adjacent to Ihh-producing prehypertrophic
chondrocytes show highest levels of Ptch1 transcripts and Ptch1 expression decreases toward the
end of the bone. In addition to proliferative zone, Sufu-deficient mice exhibited higher levels of
Ptch1 transcripts also in the resting chondrocytes (Fig. 2-5A, C). Furthermore, loss of Sufu
resulted in a reduction of the hypertrophic zone as verified by in situ hybridization analysis for
collagen X expression (Fig. 2-5E, G). Western analysis revealed an increase (2-fold) in the level
of Gli2 protein as well as an increase (1.8-fold) in the ratio of full-length versus repressor form
of Gli3 (Gli3FL:Gli3R) in Sufu-deficient chondrocytes (Fig. 2-4E). Together, these data suggest
that Sufu, like Ptch1, functions as a negative regulator of Hh signaling in chondrocytes but its
inactivation only leads to partial pathway activation.
2.3.3 Loss of Kif7 in growth plate chondrocytes results in reduced Hh pathway
activity
As Sufu inactivation did not lead to a phenotype as severe as that of Ptch1 inactivation, I
reasoned that other pathway components might cooperate with Sufu in regulating Hh signaling.
One potential candidate is Kif7, which was recently shown to be a negative regulator of Gli
transcription factors (Cheung et al., 2009). Kif7 is expressed at high levels in the articular/resting
chondrocytes and its expression is drastically down-regulated in proliferating, prehypertrophic,
and hypertrophic chondrocytes demonstrated by qRT-PCR on micro-dissected sections of the
growth plate, in situ hybridization, and immunostaining (Fig. 2-6A-C). To investigate whether it
functions as a regulator of Ihh signaling, I examined the chondrocyte phenotype of Kif7-null

34
mice (Cheung et al., 2009). Kif7-/-
mice die at birth and as such, the growth plates of E16.5 mice
were analyzed. Contrary to that observed in chondrocyte-specific Sufu knockout mice, I found
that Kif7-null mice exhibit a reduction in the size of the proliferative zone and an expansion of
hypertrophic zone (Fig. 2-6D, F, G). To rule out the possibility that the growth plate phenotype is
due to secondary effects caused by Kif7 inactivation in other cells types, I generated
chondrocyte-specific Kif7 knockout mice (Col2a1-Cre; Kif7f/f
). Col2a1-Cre; Kif7f/f
mice appear
normal and do not exhibit any gross defects (Fig. 2-6E). No obvious phenotypic difference was
found in tibial growth plates of P10 Kif7-deficient mice compared to their wild-type counterparts
(Fig. 2-6H). However, histological and in situ hybridization analyses of E16.5 Col2a1-Cre;
Kif7f/f
tibia revealed a reduction of proliferative zone and an expansion of hypertrophic zone (Fig.
2-6F, G; Fig. 2-5E, F) similar to those observed in Kif7-/-
mice. Furthermore, the growth plates
of Kif7-/-
and Col2a1-Cre; Kif7f/f
mice showed a reduction in cell proliferation demonstrated by
Ki67 and Phospho-H3 immunostaining (Fig. 2-6I, J). These results indicate that the effects of
Kif7 inactivation on chondrocyte proliferation and differentiation are opposite to those of Sufu
inactivation, and suggest that Kif7 acts as a positive regulator of Ihh signaling. Consistent with
this notion, Kif7 inactivation leads to a down-regulation of Hh target genes, Gli1 and Ptch1, as
revealed by qRT-PCR analysis (Fig. 2-9D), and reduced Ptch1 mRNA expression in the
proliferating region as illustrated by in situ hybridization analysis (Fig. 2-5A, B). Therefore, in
contrast to its role as a negative regulator of Hh signaling in early mouse embryos (Cheung et al.,
2009), Kif7 functions positively in Ihh signaling during growth plate chondrocyte development.
2.3.4 Sufu-Gli complexes are localized to the ciliary tip in the absence of Kif7

35
To investigate whether loss of Kif7 affects the functional activity of Sufu in chondrocytes
and vice versa, I first examined their expression in Kif7 and Sufu mutant mice. While no
difference in Kif7 protein or RNA levels was detected in Sufu-deficient chondrocytes (Fig. 2-7A,
B), I found a substantial increase of Sufu protein levels in Kif7-deficient chondrocytes (Fig. 2-
7C). To determine if this is caused by altered RNA expression or protein stability, I examined
Sufu transcript levels using qRT-PCR (Fig. 2-7D) and Sufu protein levels using pulse chase
experiments in the presence of cycloheximide (Fig. 2-7E, F). These experiments demonstrated
that elevated Sufu protein levels are due to increased stability in Kif7-deficient chondrocytes.
Furthermore, treatment with MG132, an inhibitor of proteasome degradation, resulted in a higher
level of Sufu protein in wild-type, but not in Kif7-deficient chondrocytes, indicating that Sufu
protein is rapidly degraded by the proteasome in wild-type chondrocytes (Fig. 2-7G, H).
Together, these data suggest that Kif7 plays a role in the turnover of Sufu in wild-type
chondrocytes.
The observations that Smo activation promotes the dissociation of Sufu-Gli complexes at
the primary cilium prompted us to investigate the ciliary localization of Sufu and Kif7 in the
growth plate chondrocytes and to examine whether Kif7 influences Sufu-Gli complexes in their
primary cilia. In Gli2-/-
;Gli3-/-
embryonic fibroblasts, where all Gli proteins are absent, Sufu is
not detected in the cilia even upon Shh stimulation demonstrating that Gli proteins are necessary
to recruit Sufu to the cilia (Tukachinsky et al., 2010; Zeng et al., 2010). Thus, Sufu
immunostaining could serve as an indicator of Sufu-Gli complexes at the primary cilium. In
proliferating chondrocytes (where the Hh pathway is active), Kif7 was mostly localized to the
ciliary tip, whereas Sufu was found at the basal bodies in ~50% of these cells but was rarely
detected at the ciliary tip (Fig. 2-8A, B). Similarly, Gli2 and Gli3 staining were rarely found in

36
the primary cilium of these chondrocytes (Fig. 2-8A, B). Both Sufu and Kif7 signals are specific
since they are absent in Sufu-deficient and Kif7-deficient chondrocytes, respectively. Together,
these results indicate that there is very little Sufu-Gli2 or Sufu-Gli3 complexes present in the
primary cilium of proliferating wild-type chondrocytes.
Loss of Sufu has no apparent effects on the ciliary localization of Kif7 (Fig. 2-8A, C).
Consistent with previous studies done in cultured fibroblasts (Humke et al., 2010; Tukachinsky
et al., 2010), Gli2 and Gli3 could be detected at the ciliary tip in ~10% of the Sufu-deficient
chondrocytes (Fig. 2-8A, C), suggesting that Gli proteins can translocate to the cilia independent
of Sufu. Strikingly, in the absence of Kif7, ~20% and ~60% of proliferating chondrocytes
showed Gli2 and Gli3 staining at the ciliary tip, respectively (Fig. 2-8A, D). Consistent with the
notion that Sufu and Glis are transported to the cilia as a complex, Sufu is localized to the ciliary
tip of ~90% of Kif7-deficient chondrocytes (Fig. 2-8A, D). These observations indicate that both
Sufu-Gli2 and Sufu-Gli3 complexes accumulate in the primary cilium of Kif7-deficient
chondrocytes, and suggest that when the Hh pathway is activated in chondrocytes, Kif7 plays a
key role in excluding them from the primary cilia. I propose that Smo promotes the dissociation
of Sufu-Gli complexes in the primary cilium through the action of Kif7.
To determine whether Ihh signaling promotes the dissociation of Sufu-Gli complexes in
chondrocytes, primary chondrocyte cultures were treated with cyclopamine (Smo antagonist),
purmorphamine (Smo agonist) or Shh, and Sufu-Gli2 complexes were quantified using western
analysis of Gli2 followed by immunoprecipitation with Sufu-specific antibodies. There was a
similar level of Gli2 in all samples. While Gli2 levels were similar in the Sufu-
immunoprecipitate from control and cyclopamine-treated chondrocytes, there was a significant
reduction of Gli2 levels in those of purmorphamine- and Shh-treated chondrocytes (Fig. 2-8E,

37
F). These results indicate that, similar to those observed in cultured fibroblasts, Smo activation
also promotes the dissociation of Sufu-Gli2 complexes in the chondrocytes.
2.3.5 Removal of one copy of Sufu rescues the Kif7 mutant growth plate phenotype
To investigate whether increased Sufu protein levels contribute to the reduced Hh
pathway activity in Kif7-deficient chondrocytes, I generated Col2a1-Cre; Sufuf/+
; Kif7f/f
mice.
Strikingly, these mice develop a normal growth plate (Fig. 2-9A). The expansion of hypertrophic
zone and the reduction of proliferative zone observed in Col2a1-Cre; Kif7f/f
mice are almost
completely rescued by the simultaneous removal of one dose of Sufu (Fig. 2-9A-C). Importantly,
Gli1 and Ptch1 expression was also restored to near wild-type levels in Col2a1-Cre; Sufuf/+
;
Kif7f/f
chondrocytes (Fig. 2-9D). These data clearly indicate that the reduced Hh pathway activity
in Kif7-deficient chondrocytes is due to increased Sufu protein level and suggest that the positive
role of Kif7 in Ihh signaling during chondrocyte differentiation is in part through the down-
regulation of Sufu protein expression as well as the dissociation of Sufu-Gli complexes (Fig. 2-
10A, B).
2.3.6 Kif7 and Sufu share overlapping functions in Hh signaling during
chondrocyte development
The co-expression of Sufu and Kif7 in articular/resting chondrocytes prompted us to
analyze whether they possess additional overlapping functions in Ihh signaling and chondrocyte

38
development. I generated Col2a1-Cre; Sufuf/f
mice with additional deletion of one allele of Kif7
(Col2a1-Cre; Sufuf/f
; Kif7f/+
) as well as Sufu; Kif7 double knockout mice (Col2a1-Cre; Sufuf/f
;
Kif7f/f
). While deletion of one allele of Kif7 in Sufu-deficient growth plate resulted in a similar
phenotype as Col2a1-Cre;Sufuf/f
mice, Col2a1-Cre;Sufuf/f
;Kif7f/f
mice showed a more severe
phenotype (Fig. 2-9A-C; Fig. 2-5E, H), including further reduction of hypertrophic zone and
expansion of proliferative zone demonstrated by immunohistochemical staining and in situ
hybridization analyses, similar to those observed in Col2a1-Cre;Ptch1f/f
mice (Fig. 2-4A).
Importantly, I found that inactivation of both Sufu and Kif7 leads to augmented expression of all
three Hh target genes Ptch1 and Hhip1 as well as Gli1 (Fig. 2-9D). By in situ hybridization, I
found that Ptch1 expression is elevated in both the resting and proliferating regions of Sufu;Kif7-
deficient mice when compared with that of Sufu-deficient mice (Fig. 2-5A, C, D). Thus, in the
absence of Sufu, Kif7 functions instead as a negative regulator of the Hh pathway suppressing
the expression of Hh target genes. I examined the localization of Gli2 and Gli3 in the primary
cilium of Sufu; Kif7-deficient chondrocytes. Interestingly, neither Gli2 nor Gli3 is localized to
the ciliary tip of Sufu; Kif7-deficient chondrocytes suggesting that Gli proteins cannot be
processed in the cilium in the absence of both Sufu and Kif7 (data not shown). Together, these
observations indicate that both Sufu and Kif7 could contribute to the negative regulation of Gli
transcription factors and their concomitant deletion results in maximal Hh pathway activation,
similar to that observed in cells lacking Ptch1. Therefore, Kif7 possesses both positive and
negative roles in the regulation of Ihh signaling during chondrocyte development (Fig. 2-10A,
B).

39
2.4 Discussion
In this study, I demonstrated distinct and overlapping functions of Sufu and Kif7 in Hh
signaling during chondrocyte development. Recent studies established that Sufu and Kif7 are
evolutionarily conserved regulators of Gli transcription factors and that they both play negative
regulatory roles in Hh signaling during early mouse embryogenesis (Cheung et al., 2009; Cooper
et al., 2005; Svard et al., 2006). There is evidence suggesting that Sufu and Kif7 are also required
for high levels of Hh pathway activity (Chen et al., 2009), but the underlying mechanism is not
defined. Here I provide genetic and molecular data indicating that Sufu is a major negative
regulator of the Hh pathway in the growth plate and that Kif7 plays dual roles in controlling Ihh
signaling and chondrocyte development (Fig. 2-10A).
Phenotypic analysis of conditional knockout mice indicates that Sufu and Kif7 normally
play opposing roles in Ihh signaling in the developing chondrocytes. Sufu-deficient chondrocytes
showed augmented Hh pathway activity, increased proliferation and delayed differentiation. In
contrast, Kif7-deficient chondrocytes exhibited lower Hh pathway activity, a decrease in
proliferation, and an expansion of the hypertrophic zone. This could be due to the negative effect
of proliferation or the stimulatory effect on chondrocyte hypertrophy. I showed that Sufu protein
levels are elevated in Kif7-deficient chondrocytes and that reduction of Sufu gene dosage
restores Hh pathway activity and growth plate development. These results suggest that a major
role for Kif7 in the growth plate is to maintain Hh signaling activity by lowering the level of
Sufu, the major negative regulator of the pathway. Importantly, Kif7 also plays a negative role in
suppressing Hh signaling activity in Sufu-deficient chondrocytes as revealed by further pathway
augmentation in Sufu;Kif7-deficient chondrocytes. This partly explains why the Hh pathway is

40
not fully activated in Sufu-deficient chondrocytes and why the Sufu knockout growth plate
phenotype is not as severe as those of Ptch1 knockout mice (Mak et al., 2008). Interestingly, I
also found that Hh pathway activation in Sufu; Kif7-deficient chondrocytes is not as robust as
those observed in Ptch1-deficient chondrocytes. It remains to be determined whether this is due
to the requirement of Sufu and/or Kif7 for full transcriptional activity of Gli proteins, or related
to the ability of Ptch1 to sequester Hh ligands in the extracellular environment. Nevertheless, our
data clearly revealed a cross-regulation of Sufu and Kif7 in Ihh signaling during chondrocyte
development and demonstrated that Kif7 can positively modulate Hh pathway activity through
down-regulation of Sufu. These results also represent to date the first genetic evidence that Sufu
and Kif7 play overlapping regulatory roles in the negative control of the Hh signaling pathway.
Hh pathway activation promotes degradation of Sufu in some cancer cell lines (Yue et al.,
2009). Our results here show that Kif7 plays a key role in controlling the stability of Sufu protein
in chondrocytes. Strikingly, removal of one copy of Sufu could restore normal chondrocyte
development in Kif7-deficient growth plates indicating that the control of Sufu protein/activity
by Kif7 is a critical regulatory step in chondrocyte proliferation and differentiation. Recent
studies suggest that Hh stimulation promotes the dissociation of Sufu-Gli protein complexes at
the ciliary tip and that this dissociation is important for Gli activation (Humke et al., 2010;
Tukachinsky et al., 2010). Interestingly, I found that Sufu is mostly excluded from the ciliary tip
in wild-type chondrocytes, whereas Sufu appear to be stabilized in the ciliary tip in the absence
of Kif7. This is consistent with the observation that Hh pathway activity is reduced in Kif7-
deficient chondrocytes (due to inefficient dissociation of the Sufu-Gli complexes) and supports
the notion that Kif7 plays a direct or indirect role in the Hh-stimulated dissociation of Sufu-Gli
protein complexes and Gli activation. However, whether the ciliary tip localization contributes to

41
the stabilization of Sufu in Kif7-deficient chondrocytes or is related to the increased Sufu protein
level awaits further investigation. Nonetheless, our results clearly demonstrated that Sufu is a
regulatory target of Kif7 in Ihh signaling during chondrocyte development.
Previous in vitro data show that Hh stimulation promotes the translocation of Kif7 to
the tip of the cilia (Liem et al., 2009). Here I found that Kif7 localizes to the ciliary tip in both
the resting and proliferating chondrocytes in vivo (data not shown). Since the Hh pathway is
inactive (as indicated by the lack of Hh target gene expression) in the resting zone, these results
suggest that Kif7 might have a functional role in the primary cilium, even in the absence of Hh
ligand activation. Such a role is likely important in the growth plate, allowing for the normal
regulation of Gli proteins and other Hh pathway components, such as Sufu. This also hints that
dynamic localization of Kif7 in the primary cilium itself is likely not the key control in
regulating Hh ligand-mediated transcription in the growth plate chondrocytes. In embryonic
fibroblasts, Gli2 and Gli3 do not localize to the cilium in the absence of Sufu (Chen et al., 2009).
However, I found that in the Sufu-deficient chondrocytes, both Gli2 and Gli3 can still be found in
the cilia. This supports the concept that Sufu is not required for the Gli proteins to localize to the
cilia (Tukachinsky et al., 2010) and also raises the possibility that other proteins or processes in
the cilium are important for the regulation of Gli proteins, and their ability to regulate
transcription.
During endochondral bone development, the coordinated differentiation of growth plate
chondrocytes regulates the pace of long bone growth. Hh signaling plays a critical role in this
process, but how the cells are able to escape their proliferative state and undergo terminal
differentiation is unclear. The proliferation and differentiation defects observed in Kif7- and
Sufu-deficient mice suggest that Sufu and Kif7 expression plays an important role in how cells in

42
various regions of the developing growth plate process Hh signals as they progress from the
resting to hypertrophic zones. In wild-type mice, Sufu and Kif7 are highly expressed in the
periarticular/resting region of the growth plate where minimal Ptch1 expression was observed
However, in the proliferating and hypertrophic regions, the level of expression of Sufu and Kif7
decreases and Ptch1 expression increases. Thus, the gradient of expression of Sufu and Kif7
negatively correlates with Ptch1 expression and Hh activity. Deletion of both Sufu and Kif7
resulted in a significant expansion of the Ptch1 expression, notably in the resting region of the
growth plate, suggesting that the level of Sufu and Kif7 plays an important role in how cells
process Hh signals in the growth plate. Intriguingly, the expression pattern of Sufu and Kif7 is
very similar to the expression pattern of Parathyroid hormone-like protein (Pthlh; previously
known as PTHrP), which raises the possibility that interaction between those molecules may
control how Pthlh regulates Hh signaling activity during development.
Taken together, I show here a novel mechanism by which Sufu and Kif7 interact to
mediate Hh signaling in the growth plate (Fig. 2-10, B). In wild-type chondrocytes, Kif7
positively regulates Gli-mediated transcription by downregulating Sufu protein levels, but also
inhibits Gli-mediated transcription through a Sufu-independent mechanism (Fig. 2-10Aa). Its
role in the primary cilium allows for normal Sufu-Gli complex localization and processing (Fig.
2-10B). In Sufu-deficient chondrocytes, the regulatory role of Kif7 on Sufu is lost; Gli protein
localization in the cilium does not differ much from the situation in wild-type chondrocytes,
resulting in a sub-maximal level of Hh pathway activation (Fig. 2-10Ab). In Kif7-deficient
chondrocytes, Sufu acts unopposedly as a negative regulator of Gli-mediated transcription, and
there is an increased level of Sufu-Gli complexes in the primary cilium, resulting in a greater
inhibition of Hh signaling than in the wild-type situation (Fig. 2-10Ac). In the absence of both

43
Sufu and Kif7, Gli proteins will not be bound to Sufu and prevented from becoming
transcriptionally active. The inhibitory effect of Kif7 is relieved, leading to further augmentation
of the Hh pathway activity (Fig. 2-10Ad), showing that the regulation of Hh signaling by Kif7
and Sufu play critical roles in the growth plate.

44
2.5 Materials & Methods
Ethics Statement
A mouse protocol describing the experimental procedures used in the study was approved
by the Animal Care Committee of The Hospital for Sick Children.
Mice
The generation of Kif7-deficient mice was previously reported (Cheung et al., 2009).
Conditional Sufu-deficient mice (Col2a1-Cre;Sufuf/f
) were generated by crossing Col2a1-Cre
mice expressing Cre-recombinase under type II collagen regulatory elements specific to
chondrocytes with Sufu-floxed mice containing loxP sites flanking exons 4 to exon 8 of Sufu
(Pospisilik et al., 2010). Conditional Ptch1 deficient mice (Col2a1-Cre; Ptch1f/f
) were generated
from Ptch1-floxed mice, which contain loxP sites flanking exon 3 of Ptch1 (Adolphe et al.,
2006; Ellis et al., 2003). Embryonic mice were obtained from timed pregnancies and the
genotypes of the various mice were determined as described (Cheung et al., 2009; Ding et al.,
1999). In all cases, littermate mice were used as controls. The recombination efficiency in all
conditional mutants was confirmed through PCR, western analysis, and by examining the Cre-
drivers crossed with a Rosa-26 reporter line. All mice are on the 129/Sv background.
Skeletal staining

45
Mice were fixed in 95% ethanol after removal of skin and viscera. Bone samples were
incubated in Alcian Blue solution (15% Alcian Blue in 80% ethanol and 20% glacial acetic acid)
for 2-3 days at room temperature. Samples were then rehydrated and cleared in 1% KOH
overnight or till clear. Samples were stained with Alizarin Red solution (7.5% Alizarin Red in
1% KOH) for 1-2 days and immersed in glycerol for storage (Mau et al., 2007).
Microdissection of the growth plate
Hindlimbs were obtained from E18.5 mouse embryos. Sections of the growth plate were
dissected out using the assistance of a microscope followed by RNA isolation using Trizol
reagent (Invitrogen). RNA concentration was determined by Nanodrop. cDNA was synthesized
from 500 ng of total RNA using qScript cDNA SuperMix (Quanta Biosciences) for Real time
PCR analysis.
Histological analysis and Immunohistochemistry
Samples were fixed in 4% paraformaldehyde overnight, embedded in paraffin, and
sectioned for histological evaluation. Sections were examined for Col10a1, a marker for
hypertrophic growth plate chondrocytes, by immunohistochemistry using previously reported
techniques and antibodies (Hu et al., 2006; Linsenmayer et al., 1988; Saika et al., 2004; Tiet et
al., 2006; Wang et al., 2000). The proximal tibial growth plate was used for all analysis to
minimize morphological variations due to anatomic location. Hematoxylin and eosin (H&E),
Safranin O and Alcian blue staining were performed using standard techniques. Proliferation was

46
evaluated by immunostaining using antibodies against Ki-67 (DakoCytomation M7249) at a 1:50
dilution and Phospho-H3 (Sigma) at a 1:200 dilution at 4 °C overnight. The proportion of
positively stained cells were calculated in the proliferative zone of the growth plate by counting
the number of positive and negative cells over 10X high powered field. Apoptotic cells were
detected by using an antibody against active Caspase-3 (Promega, Madison, WI, Cat # G7481)
and TUNEL assay as previously reported (Tiet et al., 2006). Positively stained cells were
analyzed in a similar manner as for Ki67 staining. Sufu protein was detected using a rabbit anti-
mouse antibody (Santa Cruz Biothechnology, sc-28847) incubated at a 1:100 dilution at 4 °C
overnight.
Western analysis and Co-immunoprecipitation
Western blot analysis was performed using standard protocols. Immunoblotting was
performed overnight at 4 °C with the following primary antibodies: Gli3 antibody from Santa
Cruz Biotechnology (1:800), actin antibody from Oncogene (1: 10000), Sufu antibody (Meng et
al., 2001; 1:3000), Kif7 antibody (Cheung et al., 2009; 1:1000), and Gli2 antibody (Hu et al.,
2006; 1:1000). Precipitation of Sufu protein was performed using Dynabeads Protein A
(Invitrogen following manufacture’s protocol.
Real-time quantitative PCR
RNA isolated from at least three independent experiments was analyzed by qRT-PCR in
triplicate for each treatment condition and primer set. The reactions were made up in TaqMan

47
Universal PCR master mix (Applied Biosystems) with TaqMan Gene Expression Assays for
mouse Gli1, Ptch1, Hhip1, Gli2, Gli3, Ihh, Kif7 and Sufu (Applied Biosystems). The gene
expression levels between samples were analyzed using the 2 ΔΔCt
method (Livak and
Schmittgen, 2001). Either GAPDH or β-actin (Applied Biosystems) was used as endogenous
control for target gene normalization.
Primary growth plate chondrocyte cultures
Chondrocyte isolation protocol was modified from previous published methods (Gosset
et al., 2008). Growth plates of the hindlimbs from E16.5 embryos were isolated and incubated in
collagenase type 4 (Worthington) solution (3mg/ml) for 45 min at 37°C incubator, under 5%
CO2 in a petri dish. Soft tissues were detached by pipetting. The growth plates were placed in a
clean petri dish with 0.5 mg/ml collagenase type 4 solution and were incubated overnight at
37°C. Collagenase type 4 solution containing chondrocytes was collected. Cells were washed
with PBS and seeded at a density of 8x103 cells per cm
2.
Measuring protein turnover
Cells were cultured till reach confluency before cycloheximide (20 µM) (Calbiochem)
treatment to block de novo protein synthesis for the time indicated. At the end of each time point,
cells were collected for western analysis.

48
Immunofluorescence and in situ hybridization
Samples were fixed in 4% paraformaldehyde overnight, embedded in paraffin and
sectioned. Sections (5 µm) of growth plate were cut parallel to the longitudinal axis of the bone.
The sections were stained with a monoclonal antibody against acetylated α-tubulin (clone 6-11b-
1, 1:1000; Sigma- -
tubulin (Dyomics647) (1:100; Abcam), and/or a monoclonal antibody against Sufu (1:100;
Abcam), and/or a polyclonal antibody against Kif7 (1:100) at 4°C overnight. Secondary
antibodies conjugated with Alexa Fluor 488 (1:100; Invitrogen) and TRITC (1:100 Jackson
ImmunoResearch Laboratories) were applied for 45 minutes to detect the primary antibody.
Finally the sections were mounted in 4 ,6-diamidino-2-phenylindole (DAPI) containing
Vectashield (Vector Laboratories). Confocal images were acquired using a spinning disc
confocal microscope. In situ hybridization of embryonic sections was performed as described
previously (Mo et al., 1997).
Statistical analysis
For all of the data, the mean, 95 % confidence interval, and standard deviation were
calculated for each condition, and Student’s t test was utilized to compare data sets. A p value of
< 0.05 was used as a threshold for statistical significance.

49
2.6 Figures
Figure 2-1 Sufu is differentially expressed in the growth plate.
Figure 2-1. Sufu is differentially expressed in the growth plate. (A, B) Expression of Sufu and
Ihh in wild-type mouse growth plates at E18.5 assessed by qRT-PCR. (AC/RC: articular
chondrocytes/resting chondrocytes; PC/PRC: proliferating chondrocytes/prehypertrophic
chondrocytes; HC: hypertrophic chondrocytes) Data are shown as means and 95% confidence
intervals (n=3). (C) Immunohistochemical analysis of E16.5 wild-type mouse growth plates
showed that Sufu is highly expressed by in AC/RC and PC/PRC regions and fewer than 10 % of
the HC are stained positive for Sufu. (D) In situ hybridization analysis of Sufu expression in
wild-type mouse growth plate at E16.5. (E) Analysis of immunohistochemical studies. ~25% of

50
the cells in the AC/RC and PC/PRC regions are Sufu-positive. Less than 10% of the HC are
Sufu-positive. Data are shown as means and 95% confidence intervals (n=3).
Figure 2-2 Generating chondrocyte-specific knockout of Sufu.

51
Figure 2-2. Generating chondrocyte-specific knockout of Sufu. (A) A lack of
immunohistochemical staining for Sufu was found in Col2a1-Cre; Sufuf/f
growth plate
demonstrating efficient deletion of Sufu in chondrocytes. (B) Effective deletion of Sufu in growth
plate chondrocytes was also verified using western analysis. (C) a and b: Immunohistochemical
staining of Sufu in the growth plates of wild-type and Col2a1-Cre; Sufuf/f
mice at E16.5; c and d:
magnified images of the indicated areas in a and b, respectively (same images used in A). Due to
the loss of glycogen and lipid during tissue preparation, the chondrocyte (Ch) display little more
than the nucleus residing in a lacuna (L). (D) P10 Col2a1-Cre; Sufuf/f
mutant mice showed
significant reduction in body length compared with wild-type littermates. (E and F) Analysis of
P10 Col2a1-Cre; Sufuf/f
mutant mice showed significant reduction in body length and body
weight compared with littermate wild-type controls. Data are shown as means with 95%
confidence intervals indicated. (n=3). *P<0.05.

52
Figure 2-3 Skeletal phenotype analysis of Col2a1-Cre; Sufuf/f.

53
Figure 2-3. Skeletal phenotype analysis of Col2a1-Cre; Sufuf/f
. (A) Alcian Blue/ Alizarin Red
staining showed a delay in ossification in the P10 tibial bones and P0 lumbar vertebrae of the
Col2a1-Cre; Sufuf/f
mice. (B) Safranin O staining revealed an expansion of the proliferative zone
(PZ), a decrease in the length of the hypertrophic zone (HZ), and a delayed secondary
ossification (SOC) formation in the tibia of P10 Col2a1-Cre; Sufuf/f
mice (upper panels). An
increased length of the PZ and a reduction of the HZ were also found in the P10 mutant vertebral
growth plates as revealed by H&E staining (lower panels). (C) H&E staining revealed an
expansion of the proliferative zone, which is represented by the length from the end of the tibia
to the beginning of the hypertrophic zone (black bar), and a reduction of the hypertrophic zone
(red bar) in the Sufu-deficient tibial growth plates compared with wild-type littermates at E16.5.
Lower panel: magnified images of the hypertrophic zone (red bar) seen in the upper panel. (D)
Analysis of skeletal staining of Col2a1-Cre; Sufuf/f
mice exhibited delayed ossification
demonstrated by the decreased percentage of the length of the mineralized zone to the length of
the tibia. Data are shown as means with 95% confidence intervals indicated. (n=3). *P<0.05. (E
and F) Deletion of Sufu in chondrocytes resulted in a significant increase in the percentage of
Ki67-positive cells (indicated by red arrows at lower panel) in the E16.5 growth plate. Data are
shown as means with 95% confidence intervals indicated. (n=3). *P<0.05. (G) No significant
difference was found in apoptosis rate in the E16.5 Sufu-deficient growth plates demonstrated by
TUNEL staining and active caspase-3 staining. Positive cells were indicated by red arrows. Data
are shown as means with 95% confidence intervals indicated. (n=3). *P<0.05.

54
Figure 2-4 Sufu negatively regulates growth plate chondrocyte differentiation.

55
Figure 2-4. Sufu negatively regulates growth plate chondrocyte differentiation. (A-C)
Histological and immunohistochemical analysis of the E16.5 mouse tibia showed the length of
the hypertrophic zone (red bar), which is characterized by Col10a1 staining, is reduced in
Col2a1-Cre; Sufuf/f
embryos compared to wild-type littermates. An increased size of the
proliferative zone, which represented by the length from the end of the tibia to the beginning of
the Col10a1 expression zone (black bar) was found in the Col2a1-Cre; Sufuf/f
embryos. This
phenotype resembles the phenotype displayed by deletion of Ptch1, another repressor of Hh
signaling, although less severe. Data are shown as means and 95% confidence intervals (n=3). *
p<0.05. (D) Expression of Hh target genes, Gli1, Ptch1, and Hhip1, were assessed by qRT-PCR.
Data are shown as means and 95% confidence intervals (n=3-6). * p<0.05. (E) Protein levels of
Gli2, full-length Gli3 (Gli3FL), and repressor form of Gli3 (Gli3R) in wild-type and Sufu-
deficient growth plates were assessed by western analysis. Actin was used as loading control.
Sufu-deficient chondrocytes exhibited elevated Gli2 level and an increase in the Gli3FL:Gli3R
ratio (*: non-specific band).

56
Figure 2-5 In situ hybridization analysis of Ptch1 and Col10a1 expression in various genotypes.
Figure 2-5. In situ hybridization analysis of Ptch1 and Col10a1 expression in various genotypes.
(A-D) In situ hybridization for Ptch1 expression in the growth plates of wild-type, Col2a1-Cre;
Sufuf/f
, Col2a1-Cre; Kif7f/f
, and Col2a1-Cre; Sufuf/f
; Kif7f/f
mice at E16.5; E-F: In situ
hybridization for Col10a1 expression in the growth plates of wild-type, Col2a1-Cre; Sufuf/f
,
Col2a1-Cre; Kif7f/f
, and Col2a1-Cre; Sufuf/f
; Kif7f/f
mice at E16.5. Studies using a sense probe as
control showed no hybridization.

57
Figure 2-6 Kif7 inactivation resulted in opposite effects on chondrocyte proliferation and differentiation to those of Sufu
inactivation.

58
Figure 2-6. Kif7 inactivation resulted in opposite effects on chondrocyte proliferation and
differentiation to those of Sufu inactivation. (A) Immunohistochemical analysis of the tibia of
E16.5 wild-type embryos showed that Kif7 is highly expressed in the AC/RC region, and its
expression progressively decreases as chondrocytes differentiate. (B) Expression of Kif7 in wild-
type mouse growth plates at E16.5 assessed by qRT-PCR. Data are shown as means with 95%
confidence intervals as indicated (n=3). (C) In situ hybridization analysis of Kif7 expression in
the growth plate. (D) Safranin O and Col10a1 staining revealed an expansion of the hypertrophic
zone (red bar), and reduced length of the proliferative zone (black bar) in the Kif7 knockout mice
(Kif7-/-
) compared to wild-type littermates. (E) Alcian Blue/ Alizarin Red staining of wild-type
and Col2a1-Cre; Kif7f/f
littermates at E18.5 and P10. Col2a1-Cre; Kif7f/f
mice exhibited no gross
abnormalities in skeletal development. (F, G) An increase in the length of the hypertrophic zone
and a reduction in the length of the proliferative zone were found in Kif7-/-
and Col2a1-Cre;
Kif7f/f
mice compared to wild-type littermates. Data are shown as means and 95% confidence
intervals (n=3). * p<0.05. (H) H&E staining on the tibial sections of P10 wild-type and Col2a1-
Cre; Kif7f/f
mice (SOC: secondary ossification center; PZ: proliferative zone; HZ; hypertrophic
zone). (I) A significant decrease in the percentage of proliferating cells was found in Kif7-/-
and
Col2a1-Cre; Kif7f/f
growth plates as measured by immunohistochemical staining for Ki67. Data
are shown as means and 95% confidence intervals (n=3). * p<0.05. (J) Sufu and kIf7 possess
positive and negative effects on growth plate chondrocyte proliferation, respectively,
demonstrated by immunohistochemical staining for Phospho H3.

59
Figure 2-7 Increased level of Sufu in Kif7-deificent chondrocytes.
Figure 2-7. Increased level of Sufu in Kif7-deficient chondrocytes. (A) Sufu protein levels in
wild-type and Kif7-deficient growth plates were assessed by western analysis. Actin was used as
loading control. (B) Sufu mRNA expression level was found comparable with the wild-type
controls. Data are shown as means and 95% confidence intervals (n=3). (C) A similar Kif7
protein level was found in Sufu-deficient chondrocytes. (D) The mRNA expression level of Kif7
remained comparable with the wild-type controls. (E, F) Western analysis of Sufu in wild-type
and Kif7-deficient chondrocytes following 20µM cycloheximide (CHX) treatment. (G, H)
Western analysis of Sufu in wild-type and Kif7-deficient chondrocytes following 25µM MG132
treatment. Actin was used as loading control.

60
Figure 2-8 Increased level of Sufu-Gli complexes in Kif7-deificent chondrocytes.

61
Figure 2-8. Increased level of Sufu-Gli complexes in Kif7-deficient chondrocytes. (A)
Fluorescence micrographs of cilia from wild-type, Col2a1-Cre; Sufuf/f
, and Col2a1-Cre; Kif7f/f
growth plate chondrocytes. Gli2 (ciliary tip localization of Gli2 is indicated by an asterisk), Gli3
(ciliary tip localization of Gli3 is indicated by double asterisks), Sufu (ciliary base localization of
Sufu is indicated by a yellow arrow; ciliary tip localization of Sufu is indicated by a green
arrow), and Kif7 (ciliary tip localization of Kif7 is indicated by a white arrow) are detected in the
green channel. Cilia are detected by staining against acetylated α-tubulin (red channel).
Centrioles are detected by staining against γ-tubulin (blue channel) to label the base of the cilia.
(B-D) 25-50 ciliated cells from the proliferative zone of wild-type, Sufu-deficient and Kif7-
deficient growth plates were examined for Gli2, Gli3, Sufu, and Kif7 ciliary localization. Data
are shown as percentages. (E, F) Western analysis of total Gli2 protein and Gli2 protein
immunoprecipitated (IP) with anti Sufu antibody from control (C), cyclopamine- (Cy),
purmorphamine- (Pur), and Shh-treated primary chondrocyte cultures (*: rabbit IgG band). The
level of total Gli2 is comparable in all samples. The level of Gli2 co-immunoprecipitated with
Sufu is similar in cyclopamine-treated and control cells. Significantly less Gli2 co-
immunoprecipitates with Sufu in purmorphamine- and Shh-treated chondrocyte cultures. Data
are shown as means and 95% confidence intervals. ** p<0.01.

62
Figure 2-9 A dual function for Kif7 in Hh signaling modulation.

63
Figure 2-9. A dual function for Kif7 in Hh signaling modulation. (A-C) Histological and
immunohistochemical analysis of the mouse tibial growth plates. Upper panel: Alcian blue
staining on E16.5 tibial sections. Lower panel: Col10a1 immunostaining on E18.5 tibial sections
(black bar: proliferative zone; red bar: hypertrophic zone). Data are shown as means and 95%
confidence intervals (n=3). * p<0.05. (D) Expression of Gli1, Ptch1, and Hip1 in mouse growth
plates of various mutants at E16.5 assessed by qRT-PCR. Data are shown as means and 95%
confidence intervals (n=3-6). * p<0.05. See also Figure S1-3.

64
Figure 2-10 Proposed model for how Sufu and Kif7 regulate Hh signaling in growth plate chondrocytes.

65
Figure 2-10. Proposed model for how Sufu and Kif7 regulate Hh signaling in growth plate
chondrocytes. (A) Schematic representation of Hh signaling regulation in chondrocytes. a) In
wild-type cell, Sufu acts as a repressor in controlling Hh pathway activity. Kif7 possesses dual
functions in regulating Gli transcription activity. b) In the absence of Sufu, Kif7 acts as a
repressor, resulting in a submaximal level of Hh pathway activation. c) Loss of Kif7 resulted in
increased Sufu activity, resulting in reduced Hh pathway activity. d) Further augmentation of the
Hh pathway activity is found in the absence of both Sufu and Kif7. (B) Schematic representation
of Kif7-mediated dissociation of Sufu-Gli protein complexes at the primary cilium. Kif7 plays a
functional role at the tip of the primary cilium in growth plate chondrocytes; it positively
regulates Hh signaling activity via promoting the dissociation of Sufu-Gli complexes, leading to
Gli-mediated transcriptional activation. Kif7 also function negatively in Hh signaling possibly
through binding with Gli proteins in the cytoplasm.

66
2.7 References
Adolphe, C., Hetherington, R., Ellis, T. and Wainwright, B. (2006). Patched1 functions as a
gatekeeper by promoting cell cycle progression. Cancer Res 66, 2081-8.
Barnfield, P. C., Zhang, X., Thanabalasingham, V., Yoshida, M. and Hui, C. C. (2005).
Negative regulation of Gli1 and Gli2 activator function by Suppressor of fused through multiple
mechanisms. Differentiation 73, 397-405.
Bovee, J. V., Hogendoorn, P. C., Wunder, J. S. and Alman, B. A. (2010). Cartilage tumours
and bone development: molecular pathology and possible therapeutic targets. Nat Rev Cancer
10, 481-8.
Chen, M. H., Wilson, C. W., Li, Y. J., Law, K. K., Lu, C. S., Gacayan, R., Zhang, X., Hui,
C. C. and Chuang, P. T. (2009). Cilium-independent regulation of Gli protein function by Sufu
in Hedgehog signaling is evolutionarily conserved. Genes Dev 23, 1910-28.
Cheung, H. O., Zhang, X., Ribeiro, A., Mo, R., Makino, S., Puviindran, V., Law, K. K.,
Briscoe, J. and Hui, C. C. (2009). The kinesin protein Kif7 is a critical regulator of Gli
transcription factors in mammalian hedgehog signaling. Sci Signal 2, ra29.
Cooper, A. F., Yu, K. P., Brueckner, M., Brailey, L. L., Johnson, L., McGrath, J. M. and
Bale, A. E. (2005). Cardiac and CNS defects in a mouse with targeted disruption of suppressor
of fused. Development 132, 4407-17.
Corbit, K. C., Aanstad, P., Singla, V., Norman, A. R., Stainier, D. Y. and Reiter, J. F. (2005). Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018-21.
Ding, Q., Fukami, S., Meng, X., Nishizaki, Y., Zhang, X., Sasaki, H., Dlugosz, A.,
Nakafuku, M. and Hui, C. (1999). Mouse suppressor of fused is a negative regulator of sonic
hedgehog signaling and alters the subcellular distribution of Gli1. Curr Biol 9, 1119-22.
Ellis, T., Smyth, I., Riley, E., Graham, S., Elliot, K., Narang, M., Kay, G. F., Wicking, C.
and Wainwright, B. (2003). Patched 1 conditional null allele in mice. Genesis 36, 158-61.
Endoh-Yamagami, S., Evangelista, M., Wilson, D., Wen, X., Theunissen, J. W., Phamluong,
K., Davis, M., Scales, S. J., Solloway, M. J., de Sauvage, F. J. et al. (2009). The mammalian
Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during
development. Curr Biol 19, 1320-6.
Goetz, S. C. and Anderson, K. V. (2010). The primary cilium: a signalling centre during
vertebrate development. Nat Rev Genet 11, 331-44.

67
Gosset, M., Berenbaum, F., Thirion, S. and Jacques, C. (2008). Primary culture and
phenotyping of murine chondrocytes. Nat Protoc 3, 1253-60.
Hu, M. C., Mo, R., Bhella, S., Wilson, C. W., Chuang, P. T., Hui, C. C. and Rosenblum, N.
D. (2006). GLI3-dependent transcriptional repression of Gli1, Gli2 and kidney patterning genes
disrupts renal morphogenesis. Development 133, 569-78.
Huangfu, D., Liu, A., Rakeman, A. S., Murcia, N. S., Niswander, L. and Anderson, K. V. (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426,
83-7.
Hui, C. C. and Joyner, A. L. (1993). A mouse model of greig cephalopolysyndactyly
syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat Genet 3,
241-6.
Humke, E. W., Dorn, K. V., Milenkovic, L., Scott, M. P. and Rohatgi, R. (2010). The output
of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused
and the Gli proteins. Genes Dev 24, 670-82.
Jiang, J. and Hui, C. C. (2008). Hedgehog signaling in development and cancer. Dev Cell 15,
801-12.
Karsenty, G., Kronenberg, H. M. and Settembre, C. (2009). Genetic control of bone
formation. Annu Rev Cell Dev Biol 25, 629-48.
Koziel, L., Wuelling, M., Schneider, S. and Vortkamp, A. (2005). Gli3 acts as a repressor
downstream of Ihh in regulating two distinct steps of chondrocyte differentiation. Development
132, 5249-60.
Liem, K. F., Jr., He, M., Ocbina, P. J. and Anderson, K. V. (2009). Mouse Kif7/Costal2 is a
cilia-associated protein that regulates Sonic hedgehog signaling. Proc Natl Acad Sci U S A 106,
13377-82.
Lin, A. C., Seeto, B. L., Bartoszko, J. M., Khoury, M. A., Whetstone, H., Ho, L., Hsu, C.,
Ali, S. A. and Alman, B. A. (2009). Modulating hedgehog signaling can attenuate the severity
of osteoarthritis. Nat Med 15, 1421-5.
Linsenmayer, T. F., Eavey, R. D. and Schmid, T. M. (1988). Type X collagen: a hypertrophic
cartilage-specific molecule. Pathol Immunopathol Res 7, 14-9.
Livak, K. J. and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-
time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402-8.
Mak, K. K., Kronenberg, H. M., Chuang, P. T., Mackem, S. and Yang, Y. (2008). Indian
hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development
135, 1947-56.

68
Mau, E., Whetstone, H., Yu, C., Hopyan, S., Wunder, J. S. and Alman, B. A. (2007). PTHrP
regulates growth plate chondrocyte differentiation and proliferation in a Gli3 dependent manner
utilizing hedgehog ligand dependent and independent mechanisms. Dev Biol 305, 28-39.
Meng, X., Poon, R., Zhang, X., Cheah, A., Ding, Q., Hui, C. C. and Alman, B. (2001).
Suppressor of fused negatively regulates beta-catenin signaling. J Biol Chem 276, 40113-9.
Miao, D., Liu, H., Plut, P., Niu, M., Huo, R., Goltzman, D. and Henderson, J. E. (2004).
Impaired endochondral bone development and osteopenia in Gli2-deficient mice. Exp Cell Res
294, 210-22.
Mo, R., Freer, A. M., Zinyk, D. L., Crackower, M. A., Michaud, J., Heng, H. H., Chik, K.
W., Shi, X. M., Tsui, L. C., Cheng, S. H. et al. (1997). Specific and redundant functions of Gli2
and Gli3 zinc finger genes in skeletal patterning and development. Development 124, 113-23.
Pospisilik, J. A., Schramek, D., Schnidar, H., Cronin, S. J., Nehme, N. T., Zhang, X.,
Knauf, C., Cani, P. D., Aumayr, K., Todoric, J. et al. (2010). Drosophila genome-wide
obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell
140, 148-60.
Saika, S., Muragaki, Y., Okada, Y., Miyamoto, T., Ohnishi, Y., Ooshima, A. and Kao, W.
W. (2004). Sonic hedgehog expression and role in healing corneal epithelium. Invest Ophthalmol
Vis Sci 45, 2577-85.
St-Jacques, B., Hammerschmidt, M. and McMahon, A. P. (1999). Indian hedgehog signaling
regulates proliferation and differentiation of chondrocytes and is essential for bone formation.
Genes Dev 13, 2072-86.
Svard, J., Heby-Henricson, K., Persson-Lek, M., Rozell, B., Lauth, M., Bergstrom, A.,
Ericson, J., Toftgard, R. and Teglund, S. (2006). Genetic elimination of Suppressor of fused
reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell
10, 187-97.
Tiet, T. D., Hopyan, S., Nadesan, P., Gokgoz, N., Poon, R., Lin, A. C., Yan, T., Andrulis, I.
L., Alman, B. A. and Wunder, J. S. (2006). Constitutive hedgehog signaling in
chondrosarcoma up-regulates tumor cell proliferation. Am J Pathol 168, 321-30.
Tukachinsky, H., Lopez, L. V. and Salic, A. (2010). A mechanism for vertebrate Hedgehog
signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol 191,
415-28.
Wang, B., Fallon, J. F. and Beachy, P. A. (2000). Hedgehog-regulated processing of Gli3
produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100,
423-34.
Wang, C., Pan, Y. and Wang, B. (2010). Suppressor of fused and Spop regulate the stability,
processing and function of Gli2 and Gli3 full-length activators but not their repressors.
Development 137, 2001-9.

69
Wilson, C. W., Nguyen, C. T., Chen, M. H., Yang, J. H., Gacayan, R., Huang, J., Chen, J.
N. and Chuang, P. T. (2009). Fused has evolved divergent roles in vertebrate Hedgehog
signalling and motile ciliogenesis. Nature 459, 98-102.
Yue, S., Chen, Y. and Cheng, S. Y. (2009). Hedgehog signaling promotes the degradation of
tumor suppressor Sufu through the ubiquitin-proteasome pathway. Oncogene 28, 492-9.
Zeng, H., Jia, J. and Liu, A. (2010). Coordinated translocation of Mammalian gli proteins and
suppressor of fused to the primary cilium. PLoS One 5, e15900.

70
Chapter 3 Sufu mediates the effect of Pthlh on chondrocyte
differentiation in the growth plate
Shu-Hsuan C. Hsu, Xiaoyun Zhang, Chi-Chung Hui, and Benjamin A. Alman
All experiments and analysis described in this chapter were conducted by Shu-Hsuan C. Hsu
with the exception of the western analysis by Xiaoyun Zhang. This study is submitted to EMBO
Reports.

71
3.1 Summary
Growth plate chondrocytes undergo a coordinated process of proliferation and
differentiation regulating normal long bone growth. Parathyroid hormone-like hormone (Pthlh)
acts to regulate chondrocyte hypertrophic differentiation and the activation of hedgehog (Hh)
signaling regulated Gli transcription factors. The mechanism by which Pthlh regulates Hh
signaling is incompletely elucidated. Pthlh downregulates Gli transcriptional activity, and
inhibits hypertrophic differentiation in the growth plate chondrocytes. In the absence of
Suppressor of fused (Sufu), Pthlh treatment resulted in upregulated Hh activity and increased
size of the hypertrophic zone. Pthlh treatment resulted in phosphorylation of Sufu and increased
Sufu stability in wild-type chondrocytes through a protein kinase A (PKA)-mediated mechanism.
In contrast, the regulation of Pthlh on Gli transcriptional activity and chondrocyte differentiation
was independent of Kinesin family member 7 (Kif7), another mediator in the Hh signaling
cascade. Thus, Sufu is a critical regulator in the ability of Pthlh to regulate Gli transcription
activity and chondrocyte hypertrophic differentiation. Pthlh regulates Hh signaling activity and
chondrocyte hypertrophy by promoting Sufu stability through PKA-mediated phosphorylation,
allowing Gli2 degradation and Gli3 processing to a repressor form (Gli3R) in the growth plate
chondrocytes.

72
3.2 Introduction
Long bones grow through a process of endochondral ossification, in which growth plate
cartilage undergoes a coordinated process of proliferation and differentiation, ultimately
resulting in replacement by osteoblasts and new bone. Two signaling pathways that play a
critical role coordinating growth plate chondrocyte differentiation are Pthlh (also called PTHrP)
and Indian Hedgehog (Ihh). They act in a feedback loop, regulating each other’s activity. Pthlh
inhibits chondrocyte hypertrophic differentiation, while Ihh regulates the onset of hypertrophic
differentiation, in part by signaling the periarticular chondrocytes to upregulate the expression of
Pthlh. Ihh also regulates chondrocyte proliferation and induces ossification of the perichondrium
in a Pthlh-independent manner. These two pathways interact with each other at several levels in
their signaling cascades (Karp et al., 2000; Kobayashi et al., 2002; Lanske et al., 1996; Minina et
al., 2001).
Hh signaling is activated in response to an appropriate Hh ligand binding to the Patched 1
(Ptch1) receptor. This binding leads to Smoothened (Smo) protein activation, ultimately resulting
in the transcription of target genes. In Drosophila, Hh signaling activates the transcription factor
Cubitus interruptus (Ci). In the absence of Hh binding, Ci is cleaved to a transcriptional
repressor, while upon Hh binding, Smo activation allows uncleaved Ci to translocate into the
nucleus where it functions as a transcriptional activator. In mammals, there are three
transcription factors that mediate Hh signaling, Gli1, Gli2 and Gli3 (Jiang & Hui, 2008). Data
from studies in mice in the context of the development of major organ systems including the
limb suggest that Gli1 is functionally redundant. Gli2 is not processed and as such functions as

73
an activator, while Gli3 is processed similar to Ci, and can act as an activator or repressor of Hh-
mediated transcriptional activation (Bai et al., 2004; Ding et al., 1998; Mo et al., 1997;
Motoyama et al., 1998; Park et al., 2000). I elucidated the role of Sufu and Kif7, essential
regulators of Hh signaling, in the growth plate (Hsu et al., 2011; see also chapter 2). Sufu acts as
a repressor of the Hh signaling pathway (Cooper et al., 2005; Svard et al., 2006), while Kif7
functions both positively and negatively in the Hh signaling (Chen et al., 2009; Cheung et al.,
2009; Hui and Joyner, 1993). Kif7 positively regulates Hh signaling activity in part by regulating
Sufu protein level and the exclusion of Sufu-Gli complexes from the primary cilium (Hsu et al.,
2011). Thus, Sufu and Kif7 play important roles regulating Hh signaling activity and
chondrocyte differentiation.
Previous studies show that Pthlh and the transcription factors Gli2 and Gli3 interact
during growth plate chondrocyte differentiation and proliferation (Mau et al., 2007). Limb
explant cultures showed that Pthlh treatment inhibited Col10a1 expression, a marker of growth
plate chondrocyte differentiation, and increased chondrocyte proliferation. This effect was
substantially enhanced in Gli2-/-
limbs, was absent in Gli3-/-
limbs, and was only partially
inhibited by Hh ligand blockade. Pthlh negatively regulates Gli-mediated transcription in cell
cultures, and controls the level of the repressor form of Gli3 (Gli3R) in a PKA-dependent
manner (Mau et al., 2007; Miao et al., 2004; Tukachinsky et al., 2010; Tuson et al., 2011;
Vortkamp et al., 1996; Zeng et al., 2010). Thus, the interaction between Pthlh and Gli
transcription factors plays an important role regulating growth plate chondrocyte differentiation.
Sufu and Kif7 are critical mediators of Hh signaling and the regulation of chondrocyte
hypertrophic differentiation; they are highly expressed by articular and resting chondrocytes in
the growth plate, the same region as where Pthlh is expressed (Hsu et al., 2011). This co-

74
expression pattern raises the possibility that Pthlh and these Hh signaling intermediates interact
in these growth plate cells. Furthermore, since Sufu and Kif7 mediate Hh signaling and regulate
chondrocyte hypertrophic differentiation, I examined how the interaction of Pthlh, Sufu, and
Kif7 in the growth plate regulates chondrocyte hypertrophic differentiation.

75
3.3 Results
3.3.1 Sufu regulates the effect of Pthlh on chondrocyte differentiation
To determine if Sufu mediates the effect of Pthlh on growth plate chondrocyte
differentiation, I examined the effects of Pthlh treatment on chondrocyte hypertrophic
differentiation in E16.5 tibial explants from wild-type and Sufu-deficient mice (Col2a1-Cre;
Sufuf/f
). I found that Pthlh treatment reduced the length of the hypertrophic zone (represented by
Col10a1 staining) in explants from wild-type limbs (Fig. 3-1A, B). This finding is consistent
with the known inhibitory role of Pthlh on chondrocyte hypertrophic differentiation.
Unexpectedly, Pthlh treatment of Sufu-deficient explants resulted in an expansion of the
hypertrophic zone (Fig. 3-1A, B). Thus, Sufu is required for the normal inhibitory effect of Pthlh
on growth plate chondrocyte differentiation. Pthlh also possess a stimulatory effect on
chondrocyte proliferation (Beier et al., 2001). I found that Pthlh treatment resulted in an
expansion of the proliferative zone (represented by the length from the end of the tibia to the
beginning of the hypertrophic zone) in wild-type limbs (Fig. 3-1A, C). Similar increase in the
proliferative zone was observed in the Pthlh-treated Sufu-deficient limbs (Fig. 3-1A, C); showing
that in contrast to its role in regulating chondrocyte hypertrophic differentiation, Sufu is not
required for Pthlh to activate growth plate chondrocyte proliferation.
The effects of Pthlh on normal growth plate chondrocytes is primarily mediated by PKA
(Guo et al., 2002; Mau et al., 2007; Wang et al., 2000). A PKA-specific inhibitor H-89 abolished
the effects of Pthlh on both wild-type and Col2a1-Cre; Sufuf/f
explants (Fig. 3-1A-C). Taken

76
together, the data suggest that Pthlh signaling inhibits chondrocyte hypertrophic differentiation
through the activation of PKA, and it functions upstream of Sufu.
3.3.2 Kif7 is not required for Pthlh to inhibit chondrocyte differentiation
Kif7 plays a role in controlling Hh activity through restricting Sufu function in the
growth plate chondrocytes (Hsu et al., 2011). To determine if Kif7 is required for the effect of
Pthlh on chondrocyte differentiation, I performed tibial explants from E16.5 Col2a1-Cre; Kif7f/f
embryos. Similar to wild-type explants, Pthlh exerts an inhibitory effect on chondrocyte
hypertrophic differentiation in Kif7-deficient limbs (Fig. 3-2A, B), showing that Pthlh negatively
regulates chondrocyte hypertrophic differentiation through a Kif7-independent mechanism. In
growth plate chondrocytes, Kif7 demonstrates a negative function in Hh signaling in the absence
of Sufu (Hsu et al., 2011). Since Kif7 plays a different role regulating Hh pathway activity in the
absence of Sufu, I examined mice lacking both Sufu and Kif7. Pthlh treatment resulted in an
expansion of the hypertrophic zone in these mice lacking both Sufu and Kif7 (Fig. 3-2A, B),
similar to those observed in Col2a1-Cre; Sufuf/f
explants (Fig. 3-1A, B), indicating that Sufu is
indeed required for the inhibitory effect of Pthlh on chondrocyte hypertrophic differentiation.
3.3.3 Pthlh regulates Sufu protein level
Given the important role of Sufu in Pthlh-mediated regulation of chondrocyte
hypertrophy, I set out to investigate how Pthlh and Sufu might interact. To investigate whether

77
Sufu is controlled in response to Pthlh signaling activation, I analyzed Sufu levels in Pthlh-
treated wild-type explant cultures. In wild-type chondrocytes, Pthlh treatment resulted in a
significant increased level of Sufu protein. The increase of Sufu in Pthlh-treated cells is likely
due to post-translational regulation because the level of Sufu transcript in these cells remained
comparable with that of the controls as determined by qRT-PCR analysis (Fig. 3-3A, B). As
expected, Sufu protein was not found in cell lysates derived from Col2a1-Cre; Sufuf/f
explants,
demonstrating efficient deletion of Sufu in mutant chondrocytes (Fig. 3-3A).
Phosphorylation of Sufu protein by PKA can positively regulate its stability in cultured
fibroblasts (Chen et al., 2011). Since Pthlh functions mainly through PKA in the growth plate
chondrocytes, it is possible that Pthlh promotes phosphorylation of Sufu, leading to its
stabilization. To determine whether Pthlh stimulates Sufu phosphorylation in chondrocytes,
primary chondrocyte cultures were treated with Pthlh, and the level of serine phosphorylation of
Sufu was quantified using western analysis of phosphor-serine followed by immunoprecipitation
with Sufu-specific antibodies. There was a higher level of serine phosphorylation of Sufu in the
Pthlh-treated chondrocytes compared to the untreated controls (data not shown). Taken together,
these data suggest that, similar to those observed in cultured fibroblasts, Pthlh activation
promotes phosphorylation of Sufu and stabilizes Sufu protein in the chondrocytes.
3.3.4 Sufu is required for the ability of Pthlh to process Gli transcription factors
One mechanism by which Pthlh negatively regulates Gli-mediated transcription is
through processing of Gli3 (Mau et al., 2007). To investigate the interaction between Pthlh and
Gli transcription factors, I examined the levels of Gli2, full-length Gli3 (Gli3FL), and truncated

78
repressor form of Gli3 (Gli3R) in wild-type and Col2a1-Cre; Sufuf/f
growth plate chondrocytes
with or without the treatment of Pthlh. qRT-PCR analysis revealed comparable levels of Gli2
and Gli3 mRNA in wild-type and Sufu-deficient chondrocytes (Fig. 3-4A). In wild-type limbs,
Pthlh treatment resulted in a decreased level of Gli2. However, this effect was not seen in the
Sufu-deficient chondrocytes (Fig. 3-4B). Consistent with previous data, Western analysis
revealed a decrease in the ratio of full-length versus repressor form of Gli3 (Gli3FL:Gli3R) upon
Pthlh signaling activation in wild-type chondrocytes (Fig. 3-4C, D). However, Pthlh treatment
resulted in an increase in the ratio of Gli3FL:Gli3R in chondrocytes lacking Sufu (Fig. 3-4C, D).
To determine whether Sufu influences Pthlh-mediated regulation of Hh signaling activity,
primary chondrocytes from wild-type and Sufu-deficient mice were treated with Pthlh and the
expression of Gli1 and Ptch1 was analyzed using qRT-PCR. Consistent with the repressor role
of Sufu in Hh signaling pathway, significant increases in Gli1 and Ptch1 expression levels were
found in Sufu-deficient chondrocytes. In wild-type cells, Pthlh treatment resulted in reduced
expression level in Gli1 and Ptch1. In the absence of Sufu, Pthlh treatment poses a positive
effect on Gli1 and Ptch1 expression (Fig. 3-4E, F), which suggests that Sufu is required for the
inhibitory function of Pthlh on Hh signaling activity in the chondrocytes.

79
3.4 Discussion
Here, I found that Sufu is specifically required for the inhibitory effect of Pthlh on
chondrocyte hypertrophic differentiation. Because its absence results in a reversal of the sequels
of Pthlh stimulation of chondrocyte hypertrophic differentiation, it serves as a molecular switch
in Pthlh function. In contrast to the role I found for Sufu, Kif7 is not required for Pthlh to
influence chondrocyte hypertrophy in the growth plate. Thus, Sufu appears to be a critical link
between Hh and Pthlh signaling in the control of chondrocyte hypertrophic differentiation, and a
number of molecules play important roles regulating Sufu stability, which in turn is likely a
central node in Hh signaling activation.
Phosphorylation of Sufu through PKA activation promotes Sufu protein stability (Chen et
al., 2011). Here, I found that Pthlh increases Sufu protein level as well as its phosphorylation
level. Thus, our data support the notion that phosphorylation stabilizes Sufu and Pthlh plays a
role regulating this phosphorylation in the growth plate. Intriguingly, Kif7 functions positively
in regulating Hh pathway activity through restricting Sufu level (Hsu et al., 2011). Therefore,
the regulation of Sufu level may be a common mechanism regulating Hh-mediated transcription.
In the absence of Hh ligand, Sufu sequesters Gli proteins in the cytoplasm, preventing Hh
pathway activation (Barnfield et al., 2005; Ding et al., 1999; Kogerman et al., 1999; Murone et
al., 2000). Sufu also functions to promote full length Gli3 to be processed into it repressor form
(Gli3R) in concert with GSK3ß (Kise et al., 2009). Here, I found that the stimulatory effect of
Pthlh on the formation of the Gli3R is absent in Sufu-deficient chondrocytes, which supports the
notion that Sufu plays an important role in Gli3 protein processing. Although, Pthlh inhibits Gli

80
transcriptional activity and chondrocyte hypertrophy independent of Gli2 (Mau et al., 2007), I
found that Pthlh promotes Gli2 degradation in wild-type chondrocytes. However, this effect of
Pthlh on Gli2 is not seen in chondrocytes lacking Sufu, suggesting that Sufu is required for this
process. Thus, these data suggest that Pthlh stimulates Gli2 degradation and the formation of
Gli3R through a Sufu-dependent mechanism. Taken together, I propose a model by which Sufu
mediates the effect of Pthlh signaling in the growth plate (Fig. 3-5). Pthlh positively regulates
Sufu protein stability in part through phosphorylation and promotes Sufu-mediated Gli2
degradation and Gli3 protein processing, which inhibits chondrocyte hypertrophic
differentiation.
Ihh and Pthlh maintain chondrocytes in a proliferative state and premature differentiation
occurs in the absence of a functional Ihh/Pthlh feedback loop (Karp et al., 2000; Kobayashi et al.,
2002; Lanske et al., 1996; Minina et al., 2001; Kronenberg, 2003; Long et al., 2001). However,
what controls the transition from a proliferative state to terminal differentiation is not elucidated.
Here, I found that Pthlh treatment delays hypertrophic differentiation in Kif7-deficient limb
explants, but not in mutant limb explants lacking Sufu (Col2a-Cre;Sufuf/f
as well as Col2a-
Cre;Sufuf/f
;Kif7f/f
). The requirement of Sufu for the effect of Pthlh on chondrocyte hypertrophic
differentiation raises the possibility that Sufu serves as the molecular switch regulating the
transition from proliferative to hypertrophic chondrocytes.
In contrast to the PKA-mediated action of Pthlh on chondrocyte hypertrophic differentiation,
its stimulatory effect on chondrocyte proliferation is not mimicking by PKA activation. Notably,
in the absence of Sufu, Pthlh treatment promotes chondrocyte proliferation and subsequently
increases the number of growth plate chondrocytes. Thus, the stimulatory effect of Pthlh on
chondrocyte proliferation is not dependent on Sufu, suggesting that Sufu is involved in the PKA-

81
mediated action of Pthlh. Taken together, Sufu appears to link Ihh and Pthlh signaling in the
control of chondrocyte proliferation versus differentiation. Indeed, I showed here that Sufu
protein expression is regulated by Pthlh. It is important to note that Sufu is highly expressed in
the articular and resting chondrocytes (Hsu et al., 2011). It is plausible that phosphorylation of
Sufu by Pthlh near the articular region acts to maintain the level of Sufu protein, thus, preventing
Hh pathway activation mediated by Ihh that is expressed by the prehypertrophic chondrocytes.
Therefore, the low level of Sufu in prehypertrophic and hypertrophic chondrocytes may be
involved in determining the transition from proliferation to differentiation.
Furthermore, a germline mutation of Sufu has been identified in a family of Gorlin
syndrome, which is characterized by a range of developmental abnormalities and increased risk
of developing of basal cell carcinoma (Pastorino et al., 2009). The pathogenesis of basal cell
carcinoma, which is the most common skin tumor, involves aberrant activation of the Hh
signaling pathway (Epstein, 2008). Elevated Hh signaling in basal cell carcinomas can be
attributed to loss of Ptch1, independent overexpression of Gli1 and Gli2 (Dahmane et al., 1997;
Gailani et al., 1996; Grachtchouk et al., 2000; Reifenberger et al., 2005). In mice, deletion of
one allele of Sufu resulted in a basal cell carcinoma-like neoplastic growth. Interestingly, Pthlh
expression does not seem to be altered in basal cell carcinomas (Philbrick et al., 1996). Thus, it is
tempting to speculate that Sufu functions as a tumor suppressor through mediating Gli
processing downstream of Pthlh signaling activation in the skin. Therefore, proper regulation of
Sufu-mediated Gli processing plays important role in both embryogenesis and tumorgenesis.

82
3.5 Materials & Methods
Ethics Statement
A mouse protocol describing the experimental procedures used in the study was approved
by the Animal Care Committee of The Hospital for Sick Children.
Mice
The generation of Kif7-deficient mice was previously reported (Cheung et al., 2009).
Conditional Sufu-deficient mice (Col2a1-Cre; Sufuf/f
) were generated by crossing Col2a1-Cre
mice expressing Cre-recombinase under type II collagen regulatory elements specific to
chondrocytes with Sufu-floxed mice containing loxP sites flanking exons 4 to exon 8 of Sufu.
Conditional Ptch1-deficient mice (Col2a1-Cre; Ptch1f/f
) were generated from Ptch1-floxed mice,
which contain loxP sites flanking exon 3 of Ptch1 (Adolphe et al., 2006; Ellis et al., 2003).
Embryonic mice were obtained from timed pregnancies and the genotypes of the various mice
were determined as described (Cheung et al., 2009; Ding et al., 1999). In all cases, littermate
mice were used as controls. The recombination efficiency in all conditional mutants was
confirmed through PCR, western analysis, and by examining the Cre-drivers crossed with a
Rosa-26 reporter line. All mice are on the 129/Sv background. A mouse protocol describing the
above experimental procedures was approved by the Animal Care Committee of The Hospital
for Sick Children.

83
Skeletal staining
Mice were fixed in 95% ethanol after removal of skin and viscera. Bone samples were
incubated in Alcian Blue solution (15% Alcian Blue in 80% ethanol and 20% glacial acetic acid)
for 2-3 days at room temperature. Samples were then rehydrated and cleared in 1% KOH
overnight or till clear. Samples were stained with Alizarin Red solution (7.5% Alizarin Red in
1% KOH) for 1-2 days and immersed in glycerol for storage (Mau et al., 2007).
Explant cultures
Hindlimb organ cultures from E16.5 embryos were established as previously described
(Ho et al., 2009; Lanske et al., 1996; Mau et al., 2007; Minina et al., 2001). i bs ere placed
on a ucleopore filter inside a ell of - ell tissue culture plate, ith the li b at the li uid air
interface. The li b e plants ere then cultured for days at in a 2 incubator in
DMEM containing 0.1% BSA, Vitamin C (50 mg/ ml), and Antibiotics-Antimycotic Solution
(Wisent). One limb from each embryo was treated with various agents whereas the other acted as
a control. 10-7
M Pthlh (Bachem, King of Prussia, Pennysylvania, USA) was added to activate
Pthlh signaling. The PKA inhibitor, H-89 dihydrochloride (Alexia Biochemicals), was used at
20µM. These reagents were used alone or in combination for two consecutive days, with media
changed each day replenishing the specific agent utilized. Each explant experiment was
undertaken three times or more using limbs from three different fetal mice and littermate
controls.

84
Histological analysis and Immunohistochemistry
Samples were fixed in 4% paraformaldehyde overnight, embedded in paraffin, and
sectioned for histological evaluation. Sections were examined for Col10a1, a marker for
hypertrophic growth plate chondrocytes, by immunohistochemistry using previously reported
techniques and antibodies (Hu et al., 2006; Linsenmayer et al., 1988; Saika et al., 2004; Tiet et
al., 2006; Wang et al., 2000). The proximal tibial growth plate was used for all analysis to
minimize morphological variations due to anatomic location. Safranin O staining was performed
using standard techniques. Proliferation was evaluated by immunostaining using antibodies
against Ki-67 and Phospho-H3 (DakoCytochemical M7249 (from Clone Tec-3) at a 1:50 dilution
or (from Sigma) at a 1:200 dilution) at 4 °C overnight. The proportion of positively stained cells
were calculated in the proliferative zone of the growth plate by counting the number of positive
and negative cells over 10X high powered field. Apoptotic cells were detected by using an
antibody against active Caspase-3 (Promega, Madison, WI, Cat # G7481) and TUNEL assay as
previously reported (Tiet et al., 2006). Positively stained cells were analyzed in a similar manner
as for Ki67 staining. Sufu protein was detected using a rabbit anti-mouse antibody (Santa Cruz
Biothechnology, sc-28847) incubated at a 1:100 dilution at 4 °C overnight.
Western analysis
Western blot analysis was performed using standard protocols. Immunoblotting was
performed overnight at 4 °C with the following primary antibodies: Gli3 antibody from Santa
Cruz Biotechnology (1:800), actin antibody from Oncogene (1: 10000), Sufu antibody (Meng et

85
al., 2001), Kif7 antibody (Cheung et al., 2009), and Gli2 antibody (Hu et al., 2006), and
phosphor-serine antibody from Cell Signaling (1:1000).
Real time quantitative PCR
RNA isolated from at least three independent experiments was analyzed by qRT-PCR in
triplicate for each treatment condition and primer set. The reactions were made up in TaqMan
Universal PCR master mix (Applied Biosystems) with TaqMan Gene Expression Assays for
mouse Gli1, Ptch1, Gli2, Gli3, and Sufu (Applied Biosystems). The gene expression levels
between samples were analyzed using the 2 ΔΔ t
method (Livak and Schmittgen, 2001). Either
GAPDH or β-actin (Applied Biosystems) was used as endogenous control for target gene
normalization.
Primary growth plate chondrocyte cultures
Chondrocyte isolation protocol was modified from previously published methods (Gosset
et al., 2008). Growth plates of the hindlimbs from E16.5 embryos were isolated and incubated in
collagenase type 4 (Worthington) solution (3mg/ml) for 45 min at 37°C incubator, under 5%
CO2 in a petri dish. Soft tissues were detached by pipetting. The growth plates were placed in a
clean petri dish with 0.5 mg/ml collagenase D solution and were incubated overnight at 37°C.
Collagenase D solution containing chondrocytes was collected. Cells were washed with PBS and
seeded at a density of 8*103 cells per cm
2.

86
Statistical analysis
For all of the data, the mean, 95 % confidence interval, and standard deviation were
calculated for each condition, and the Student’s t test was utilized to compare data sets. The
threshold for statistical significance was p < 0.05.
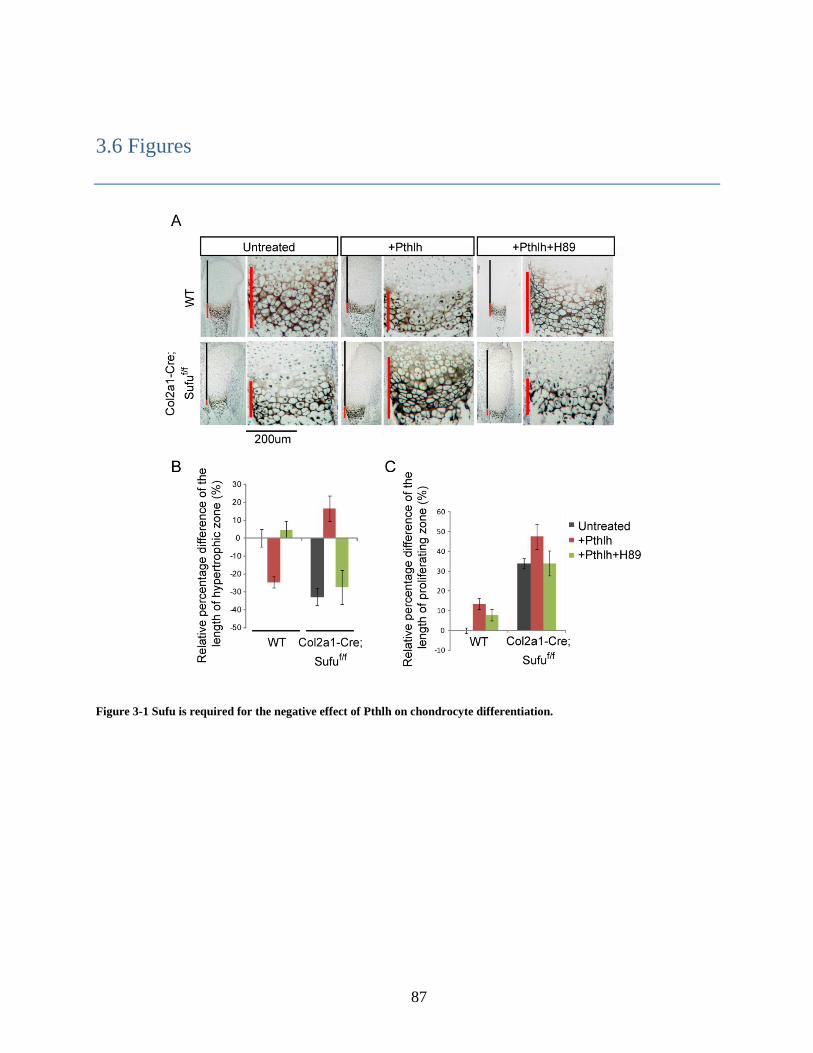
87
3.6 Figures
Figure 3-1 Sufu is required for the negative effect of Pthlh on chondrocyte differentiation.

88
Figure 3-1. Sufu is required for the negative effect of Pthlh on chondrocyte differentiation. (A)
Histological analysis of tibial explants from wild-type and Col2a1-Cre; Sufuf/f
mice in the
absence or presence of Pthlh or Pthlh+H89. (B) Relative percentage difference of the length of
the hypertrophic zone of Pthlh or Pthlh+H89 treated tibial explants compared to the controls. (C)
Relative percentage difference of the proliferative zone of Pthlh or Pthlh+H89 treated tibial
explants compared to the controls. Data are shown as means and 95% confidence intervals (n=3).

89
Figure 3-2 Kif7 is not required for the negative effect of Pthlh on chondrocyte differentiation.
Figure 3-2. Kif7 is not required for the negative effect of Pthlh on chondrocyte differentiation.
(A) Immunohistochemical staining for Col10a1 and Safranin O staining on tibial explants of
Col2a1-Cre; Kif7f/f
and Col2a1-Cre; Sufuf/f
; Kif7f/f
mice with or without Pthlh treatments. (B)
Relative percentage difference of the length of the hypertrophic zone of Pthlh-treated tibial
explants compared to the controls. Data are shown as means and 95% confidence intervals (n=3).
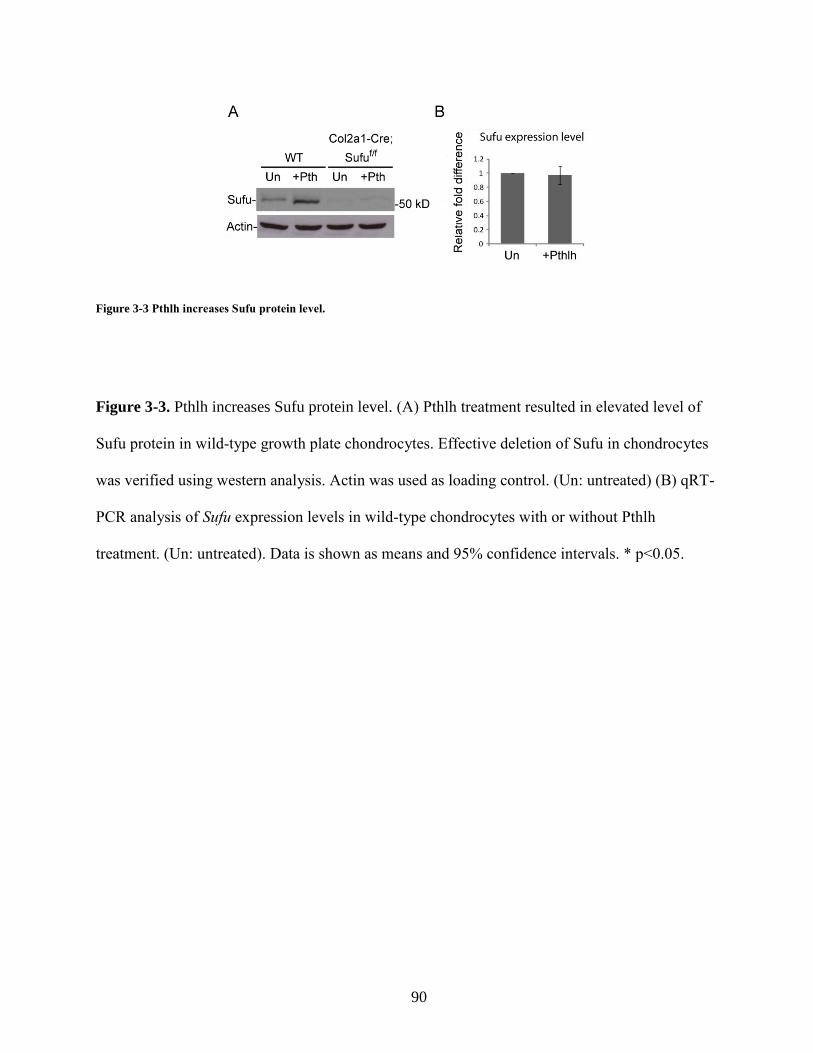
90
Figure 3-3 Pthlh increases Sufu protein level.
Figure 3-3. Pthlh increases Sufu protein level. (A) Pthlh treatment resulted in elevated level of
Sufu protein in wild-type growth plate chondrocytes. Effective deletion of Sufu in chondrocytes
was verified using western analysis. Actin was used as loading control. (Un: untreated) (B) qRT-
PCR analysis of Sufu expression levels in wild-type chondrocytes with or without Pthlh
treatment. (Un: untreated). Data is shown as means and 95% confidence intervals. * p<0.05.

91
Figure 3-4 Pthlh signaling activation promotes Gli2 and Gli3 protein processing via a Sufu-mediated manner.

92
Figure 3-4. Pthlh signaling activation promotes Gli2 and Gli3 protein processing via a Sufu-
mediated manner. (A) qRT-PCR analysis of the expression level of Gli2 and Gli3 in Wild-type
and Sufu-deficient chondrocytes. (B) Western analysis for Gli2 of wild-type and Sufu-deficient
chondrocytes with or without Pthlh. Actin was used as loading control. (C) Western analysis for
full-length Gli3 (Gli3FL) and truncated Gli3 (Gli3R) of wild-type and Sufu-deficient
chondrocytes with or without Pthlh treatments. (D) Graphical evaluation of the Gli3FL:Gli3R
ratio in wild-type and Sufu-deficient chondrocytes with or without Pthlh treatment. (E) qRT-PCR
analysis of Gli1 expression in wild-type and Sufu-deficient growth plate chondrocytes with or
without Pthlh treatment. (F) qRT-PCR analysis of Ptch1 expression in wild-type and Sufu-
deficient growth plate chondrocytes with or without Pthlh treatment. Data is shown as means and
95% confidence intervals. * p<0.05.

93
Figure 3-5 Proposed model for how Pthlh regulates Gli transcriptional factors through a Sufu-mediated mechanism.
Figure 3-5. Proposed model for how Pthlh regulates Gli transcriptional factors through a Sufu-
mediated mechanism. Pthlh promotes Sufu protein level through phosphorylation, and facilitates
Gli2 degradation and Gli3R formation, inhibiting chondrocyte hypertrophic differentiation in the
growth plate.

94
3.7 References
Adolphe, C., Hetherington, R., Ellis, T. and Wainwright, B. (2006). Patched1 functions as a
gatekeeper by promoting cell cycle progression. Cancer Res 66, 2081-8.
Barnfield, P. C., Zhang, X., Thanabalasingham, V., Yoshida, M. and Hui, C. C. (2005).
Negative regulation of Gli1 and Gli2 activator function by Suppressor of fused through multiple
mechanisms. Differentiation 73, 397-405.
Beier, F., Ali, Z., Mok, D., Taylor, A. C., Leask, T., Albanese, C., Pestell, R. G. and
LuValle, P. (2001). TGFbeta and PTHrP control chondrocyte proliferation by activating cyclin
D1 expression. Mol Biol Cell 12, 3852-63.
Chen, Y., Yue, S., Xie, L., Pu, X. H., Jin, T. and Cheng, S. Y. (2011). Dual phosphorylation of
Suppressor of fused by PKA and GSK3beta regulates its stability and localization in the primary
cilium. J Biol Chem.
Cheung, H. O., Zhang, X., Ribeiro, A., Mo, R., Makino, S., Puviindran, V., Law, K. K.,
Briscoe, J. and Hui, C. C. (2009). The kinesin protein Kif7 is a critical regulator of Gli
transcription factors in mammalian hedgehog signaling. Sci Signal 2, ra29.
Dahmane, N., Lee, J., Robins, P., Heller, P. and Ruiz i Altaba, A. (1997). Activation of the
transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature
389, 876-81.
Ding, Q., Fukami, S., Meng, X., Nishizaki, Y., Zhang, X., Sasaki, H., Dlugosz, A.,
Nakafuku, M. and Hui, C. (1999). Mouse suppressor of fused is a negative regulator of sonic
hedgehog signaling and alters the subcellular distribution of Gli1. Curr Biol 9, 1119-22.
Ellis, T., Smyth, I., Riley, E., Graham, S., Elliot, K., Narang, M., Kay, G. F., Wicking, C.
and Wainwright, B. (2003). Patched 1 conditional null allele in mice. Genesis 36, 158-61.
Epstein, E. H. (2008). Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer 8, 743-54.
Gailani, M. R., Stahle-Backdahl, M., Leffell, D. J., Glynn, M., Zaphiropoulos, P. G.,
Pressman, C., Unden, A. B., Dean, M., Brash, D. E., Bale, A. E. et al. (1996). The role of the
human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet 14, 78-81.
Gosset, M., Berenbaum, F., Thirion, S. and Jacques, C. (2008). Primary culture and
phenotyping of murine chondrocytes. Nat Protoc 3, 1253-60.
Grachtchouk, M., Mo, R., Yu, S., Zhang, X., Sasaki, H., Hui, C. C. and Dlugosz, A. A. (2000). Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat Genet 24, 216-7.

95
Guo, J., Chung, U. I., Kondo, H., Bringhurst, F. R. and Kronenberg, H. M. (2002). The
PTH/PTHrP receptor can delay chondrocyte hypertrophy in vivo without activating
phospholipase C. Dev Cell 3, 183-94.
Ho, L., Stojanovski, A., Whetstone, H., Wei, Q. X., Mau, E., Wunder, J. S. and Alman, B. (2009). Gli2 and p53 cooperate to regulate IGFBP-3- mediated chondrocyte apoptosis in the
progression from benign to malignant cartilage tumors. Cancer Cell 16, 126-36.
Hsu, S. H., Zhang, X., Yu, C., Li, Z. J., Wunder, J. S., Hui, C. C. and Alman, B. A. (2011).
Kif7 promotes hedgehog signaling in growth plate chondrocytes by restricting the inhibitory
function of Sufu. Development 138, 3791-801.
Hu, M. C., Mo, R., Bhella, S., Wilson, C. W., Chuang, P. T., Hui, C. C. and Rosenblum, N.
D. (2006). GLI3-dependent transcriptional repression of Gli1, Gli2 and kidney patterning genes
disrupts renal morphogenesis. Development 133, 569-78.
Karp, S. J., Schipani, E., St-Jacques, B., Hunzelman, J., Kronenberg, H. and McMahon, A.
P. (2000). Indian hedgehog coordinates endochondral bone growth and morphogenesis via
parathyroid hormone related-protein-dependent and -independent pathways. Development 127,
543-8.
Kise, Y., Morinaka, A., Teglund, S. and Miki, H. (2009). Sufu recruits GSK3beta for efficient
processing of Gli3. Biochem Biophys Res Commun 387, 569-74.
Kobayashi, T., Chung, U. I., Schipani, E., Starbuck, M., Karsenty, G., Katagiri, T., Goad,
D. L., Lanske, B. and Kronenberg, H. M. (2002). PTHrP and Indian hedgehog control
differentiation of growth plate chondrocytes at multiple steps. Development 129, 2977-86.
Kogerman, P., Grimm, T., Kogerman, L., Krause, D., Unden, A. B., Sandstedt, B.,
Toftgard, R. and Zaphiropoulos, P. G. (1999). Mammalian suppressor-of-fused modulates
nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol 1, 312-9.
Kronenberg, H. M. (2003). Developmental regulation of the growth plate. Nature 423, 332-6.
Kronenberg, H. M. (2006). PTHrP and skeletal development. Ann N Y Acad Sci 1068, 1-13.
Lanske, B., Karaplis, A. C., Lee, K., Luz, A., Vortkamp, A., Pirro, A., Karperien, M.,
Defize, L. H., Ho, C., Mulligan, R. C. et al. (1996). PTH/PTHrP receptor in early development
and Indian hedgehog-regulated bone growth. Science 273, 663-6.
Linsenmayer, T. F., Eavey, R. D. and Schmid, T. M. (1988). Type X collagen: a hypertrophic
cartilage-specific molecule. Pathol Immunopathol Res 7, 14-9.
Livak, K. J. and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-
time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402-8.

96
Long, F., Zhang, X. M., Karp, S., Yang, Y. and McMahon, A. P. (2001). Genetic
manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the
regulation of chondrocyte proliferation. Development 128, 5099-108.
Mau, E., Whetstone, H., Yu, C., Hopyan, S., Wunder, J. S. and Alman, B. A. (2007). PTHrP
regulates growth plate chondrocyte differentiation and proliferation in a Gli3 dependent manner
utilizing hedgehog ligand dependent and independent mechanisms. Dev Biol 305, 28-39.
Meng, X., Poon, R., Zhang, X., Cheah, A., Ding, Q., Hui, C. C. and Alman, B. (2001).
Suppressor of fused negatively regulates beta-catenin signaling. J Biol Chem 276, 40113-9.
Minina, E., Wenzel, H. M., Kreschel, C., Karp, S., Gaffield, W., McMahon, A. P. and
Vortkamp, A. (2001). BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte
proliferation and differentiation. Development 128, 4523-34.
Murone, M., Luoh, S. M., Stone, D., Li, W., Gurney, A., Armanini, M., Grey, C., Rosenthal,
A. and de Sauvage, F. J. (2000). Gli regulation by the opposing activities of fused and
suppressor of fused. Nat Cell Biol 2, 310-2.
Pastorino, L., Ghiorzo, P., Nasti, S., Battistuzzi, L., Cusano, R., Marzocchi, C., Garre, M.
L., Clementi, M. and Scarra, G. B. (2009). Identification of a SUFU germline mutation in a
family with Gorlin syndrome. Am J Med Genet A 149A, 1539-43.
Philbrick, W. M., Wysolmerski, J. J., Galbraith, S., Holt, E., Orloff, J. J., Yang, K. H.,
Vasavada, R. C., Weir, E. C., Broadus, A. E. and Stewart, A. F. (1996). Defining the roles of
parathyroid hormone-related protein in normal physiology. Physiol Rev 76, 127-73.
Reifenberger, J., Wolter, M., Knobbe, C. B., Kohler, B., Schonicke, A., Scharwachter, C.,
Kumar, K., Blaschke, B., Ruzicka, T. and Reifenberger, G. (2005). Somatic mutations in the
PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br J Dermatol 152,
43-51.
Saika, S., Muragaki, Y., Okada, Y., Miyamoto, T., Ohnishi, Y., Ooshima, A. and Kao, W.
W. (2004). Sonic hedgehog expression and role in healing corneal epithelium. Invest Ophthalmol
Vis Sci 45, 2577-85.
Tiet, T. D., Hopyan, S., Nadesan, P., Gokgoz, N., Poon, R., Lin, A. C., Yan, T., Andrulis, I.
L., Alman, B. A. and Wunder, J. S. (2006). Constitutive hedgehog signaling in
chondrosarcoma up-regulates tumor cell proliferation. Am J Pathol 168, 321-30.
Wang, B., Fallon, J. F. and Beachy, P. A. (2000). Hedgehog-regulated processing of Gli3
produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100,
423-34.

97
Chapter 4 Conclusions & future directions

98
4.1 Summary
Although the signaling cascades in the regulation of the growth plate chondrocyte
development have been identified, we know little about the precise mechanism by which they
act. This study provides insight into a number of open questions in the Hh signaling field as well
as into the development of growth plate; how is the dual function of Kif7 mediated, how does
Kif7 control Sufu activity, how is Hh pathway activity regulated by Sufu and Kif7 in the context
of primary cilium, and how is Sufu activity coupled to Pthlh signaling? In order to develop
effective therapeutic approaches to target diseases caused by Hh pathway dysregulation,
revealing the molecular basis underlying the Hh signaling cascade in various tissues will be
necessary.

99
4.2 Sufu expression is tightly regulated by multiple factors in the growth
plate
4.2.1 Lessons from genetically modified mice and Pthlh activation
In chapter 2, I confirmed that Sufu serves as a negative regulator of the Hh pathway, and
it is differentially expressed in the growth plate. Kif7 promotes Hh signaling in chondrocytes by
restricting the inhibitory function of Sufu in part through a proteasome-mediated pathway. Kif7
also negatively regulates Hh activity through a Sufu-independent mechanism. Previous studies in
cultured fibroblasts have demonstrated that Hh signaling activation accelerates Sufu protein
turnover through promoting its degradation in the proteasome (Yue et al., 2009). These data
suggest that controlling Sufu protein stability serves as a common mechanism that connects the
action of Smo to downstream Gli-mediated transcription (Fig. 4-1).
In chapter 3, I demonstrated that Pthlh increases Sufu protein levels possibly through
phosphorylation (Fig. 3-3). Studies have shown that Sufu is phosphorylated at Ser-342 and Ser-
346 by GSK3ß and PKA, respectively, which leads to Sufu stabilization against Hh signaling-
induced degradation (Chen et al., 2011). Although an increased level of phospho-serine upon
Pthlh treatment was observed in this study, it will be necessary to confirm the phosphorylation
site(s) of Sufu in those Pthlh-treated chondrocytes by examining their Ser-342 and Ser-346
phosphorylation levels. I found that Sufu is required for Pthlh to promote Gli2 degradation and
the processing of Gli3, it is tempting to speculate that phosphorylation of Sufu by PKA stabilizes
Sufu-Gli2/3 complexes, allowing proteolytic processing of Gli2 and Gli3. Further investigation
of this issue requires examination of mice with mutated PKA phosphorylation site(s) of Sufu.

100
Thus, controlling Sufu activity by regulating its protein stability may again serve as a common
mechanism between Ihh and Pthlh signaling in the developing growth plate (Fig. 4-2). In turn
this gradual inactivation of Sufu with chondrocyte differentiation could establish a differential
responsiveness of differentiating chondrocytes to various signaling cues in the growth plate, such
as Ihh itself.
4.2.2 Transcriptional regulation of Sufu
The necessity for proper control of Sufu activity during chondrocyte differentiation raises
the question of whether Sufu activity is also regulated by transcriptional mechanisms in the
growth plate in addition to post-translational regulation. One potential candidate is Sox9, which
is a key regulator for chondrogenesis, chondrocyte proliferation, and expression of cartilage
matrix, such as Col2a1, Col9a1, Col11a1, Aggrecan. Sox9 also inhibits the transition of
chondrocyte into hypertrophic chondrocyte and subsequent endochondral ossification. I
hypothesize that Sox9 inhibits the expression of Sufu at the mRNA level. This hypothesis is
supported by several lines of evidence. In the wild-type growth plate, Sox9 is expressed in
resting and proliferating chondrocytes, with a maximum of expression in prehypertrophic
chondrocyte, but its expression is abolished in hypertrophic chondrocytes (Zhao et al., 1997).
Chondrocyte-specific knockout of Sox9 (Col2a1-Cre; Sox9f/f
) results in severe chondrodysplasia,
which is a characteristic of defective Hh signaling, as well as reduced expression of Ihh and its
downstream target genes, Pthlh and Ptch1 (Akiyama et al., 2002). Furthermore, studies done in
cultured multipotent neural precursor cells show that Sufu is a direct target gene of Sox10, which
together with Sox8 and Sox9 belong to the SoxE family of transcription factors (Pozniak et al.,

101
2010). Examination of Sufu transcripts via in situ hybridization in the Col2a1-Cre; Sox9f/f
mutant
growth plate will provide preliminary information to this hypothesis. Identification of potential
Sox9-binding sites at the promoter region of Sufu through in silico analysis combined with
chromatin immunoprecipitation (ChIP) will be necessary to further determine whether Sox9
modulates Sufu expression level in chondrocytes.
4.2.3 How does Sufu regulate subsequent steps of endochondral ossification?
The lack of Sufu expression in the hypertrophic region of the growth plate is of particular
interest since it suggests that the downregulation of Sufu might be permissive for subsequent
transitional changes from the cartilage template to bone, such as cartilage absorption,
angiogenesis, and trabecular bone formation. At the lower end of the hypertrophic zone in the
growth plate, hypertrophic chondrocytes further mature to become terminally differentiated
chondrocytes, a transition which is marked by upregulation of osteopontin (Farzan et al., 2008),
Vegfa (Gerber et al., 1999), and Mmp13 (Johansson et al., 1997). Vegfa is required for capillary
invasion into the hypertrophic zone in the diaphysis from the perichondrium (Carlevaro et al.,
2000; Gerber et al., 1999; Maes et al., 2004; Zelzer et al., 2002). Mmp13, which is a matrix
metalloproteinase, loosens up the matrix secreted by hypertrophic chondrocytes in order to
facilitate blood vessel invasion (Inada et al., 2004; Johansson et al., 1997; Selvamurugan et al.,
2004). In the Col2a1-Cre; Sufuf/f
mice, a delay in ossification was observed in both the tibial and
vertebral growth plates (Fig. 2-2), suggesting that Sufu may play a role in cartilage
vascularization. It will be interesting to generate transgenic mouse lines misexpressing Sufu in
hypertrophic chondrocytes under the Col10a1 promoter. In situ hybridization and qRT-PCR

102
studies will be needed to demonstrate the level of osteopontin, vegfa and Mmp13 expression in
the growth plate as a result of Sufu overexpression.
4.2.4 Sufu and osteoarthritis
Activated Hh signaling in chondrocytes is associated with predisposition to the
development of osteoarthritis and the level of Hh signaling activity in chondrocytes is associated
with the severity of osteoarthritis (Lin et al., 2009). In this condition, articular chondrocytes
undergo phenotypic and gene expression changes that are reminiscent of those that occur in the
growth plate. Such a process at the articular surface could contribute to the onset of
osteoarthritis.
However, the molecular mechanism underlying the Hh pathway activation in osteoarthritis
development is unclear. I observed the expression of Sufu at the articular surface of wild-type
tibia (Fig. 2-1C). It is tempting to speculate that, similar to what was observed in chondrocyte
hypertrophic differentiation, downregulation of Sufu is required at the articular surface for the
development of osteoarthritis. If this hypothesis is correct, the next question would be does
overexpression of Sufu attenuate the progression of osteoarthritis in mice in vivo? The role of
Sufu and Kif7 in the development of osteoarthritis has not been tested, but can be, as described
below.
Consistent with the notion that Hh signaling activation is associated with a predisposition
to osteoarthritis, reduced Alcian blue staining was observed in the superficial layers of the
articular cartilage of Col2a1-Cre; Sufuf/f
mice and further reduction was observed in the Col2a1-

103
Cre; Sufuf/f
;Kif7f/f
mice at E16.5. Sufu is negatively regulated by Kif7 in chondrocytes. Thus, in
the absence of Kif7, the increased level of Sufu may be involved in maintaining chondrocyte
characteristics at the articular surface. Indeed, Alcian blue-stained cartilage was observed
distributed evenly at the articular surface of Kif7-deficient growth plates at E16.5. The next step
to test the significance of Sufu and Kif7 in developing osteoarthritis would be to look at Sufu
expression level in osteoarthritic cartilage samples from mouse and/or human. The perinatal
lethality in Col2a1-Cre; Sufuf/f
precludes comprehensive studies on the role of Sufu in
osteoarthritis. To investigate whether Sufu inactivation predisposes mice to osteoarthritis,
examination of the joint phenotype of genetically modified mice in which Sufu is deleted
postnatally will be needed. Sufu level is negatively regulated by Kif7 in chondrocytes. Thus, the
effect of overexpression of Sufu on developing osteoarthritis could be tested in Kif7-deficient
mice (Col2a1-Cre;Kif7f/f
). To generate conditional knockout of Sufu, Sufuf/f
mice could be
crossed with Col2a1-rtTA-Cre mice, which express Cre recombinase under Col2a1 regulatory
elements inducible by doxycycline so that Sufu can be deleted in chondrocytes when we
administer doxycycline (Grover and Roughley, 2006; Jeong et al., 2004). Doxycyclin inducible
chondrocyte specific knockout of Sufu and Kif7 (Col2a1-rtTA-Cre;Sufuf/f
;Kif7f/f
) could be
generated in the same manner. Radiographic and histological analysis could be used to assess
the severity of osteoarthritis on wild-type, Col2a1-rtTA-Cre;Sufuf/f
, Col2a1-rtTA-
Cre;Sufuf/f
;Kif7f/f
, and Col2a1-Cre; Kif7f/f
mice at 18-week-old. qRT-PCR analysis would be
needed for genes known to be upregulated in osteoarthritis, such as Adamts5, Col10a1 and
Mmp13 in all samples. Overall, it would be interesting to see if Sufu inactivation in articular
chondrocytes signals the cells to undergo hypertrophic differentiation. If so, can Sufu

104
overexpression help to maintain their chondrocyte characteristics and therefore serve as a
protective mechanism against cartilage degeneration over time?

105
4.3 The role of Sufu in Wnt/ß-catenin signaling pathway during
chondrocyte development
4.3.1 Wnt/ß-catenin signaling in skeletal development
Similar to Ihh and Pthlh signaling, the Wnt/ß-catenin pathway plays an important role in
several aspects of endochondral ossification, such as chondrocyte and osteoblast differentiation.
ß-catenin is a major mediator of Wnt signaling (Kikuchi, 2000; Miller et al., 1999; Waltzer and
Bienz, 1999) which has an important role in chondrocyte differentiation, as demonstrated by the
growth plate phenotype in mutants lacking ß-catenin in their chondrocytes. Studies have shown
that overexpression of ß-catenin inhibits the differentiation of chondrocyte progenitors, enhances
hypertrophic chondrocyte differentiation and promotes the expression of markers for terminally
differentiated chondrocytes, including MMP13 and vegfa (Hartmann and Tabin, 2000; Ryu et al.,
2002). However, the precise function and regulation of ß-catenin in chondrocyte differentiation
still needs to be clarified.
ß-catenin functions as a downstream intracellular signaling molecule in the canonical
Wnt pathway, which regulates cell proliferation and cell differentiation, and cell migration in
many aspects of embryogenesis (Huelsken and Birchmeier, 2001; Moon et al., 2002). A large
multiprotein complex that includes axin and adenomatous polyposis coli gene product (APC)
normally facilitates ß-catenin phosphorylation by glycogen synthase kinase 3ß (GSK3ß) (Ikeda
et al., 1998; Kishida et al., 1999; Liu et al., 2002). Phosphorylated ß-catenin is ubiquitinated and
targeted for degradation through the proteasome-mediated pathway (Kitagawa et al., 1999). In
the absence of Wnt ligand, ß-catenin phosphorylation and subsequent degradation are inhibited.

106
ß-catenin then accumulates in the cell and translocates into the nucleus, where it binds to the
transcription factors T-cell factor (TCF) and lymphoid enhancer factor (Lef), leading to the
activation of pathway target genes. The molecular mechanism underlying the cross-talk between
Ihh, Pthlh, and Wnt/ß-catenin signaling pathways during chondrocyte differentiation remains
elusive. Investigating the genetic interaction between Ihh, Pthlh, and Wnt/ß-catenin signaling
pathways will provide further insight into the means by which different signaling cascades
orchestrate cartilage development.
4.3.2 The role of Sufu in mediating Wnt/ ß-catenin signaling
Sufu and ß-catenin are present in the same complex and Sufu can bind ß-catenin and
export it from the nucleus thereby negatively regulating TCF-dependent transcription in the
human colon cancer SW480 cell line (Meng et al., 2001). Immunofluorescence studies have
shown that whereas much of the endogenous ß-catenin is localized in the cytoplasm of
proliferating and prehypertrophic chondrocytes, nuclear localization of ß-catenin was observed
in hypertrophic chondrocytes (Enomoto-Iwamoto et al., 2002). In chapter 2 I described the
expression pattern of Sufu in the growth plate. Sufu was absent in hypertrophic chondrocytes,
where ß-catenin resides in the nucleus. This result corroborates the notion that Sufu inhibits the
nuclear localization of ß-catenin. Thus, the downregulation of Sufu may be required for ß-
catenin nuclear localization in the hypertrophic chondrocytes. Consistent with an inhibitory role
of Sufu in regulating Wnt/ß-catenin signaling activity, I found a ~40% increase in ß-catenin
levels in Sufu-deficient chondrocytes demonstrated by western analysis. To clarify the role of
Sufu in regulating ß-catenin activity and cellular localization, it will be necessary to examine the

107
expression level of Wnt/ß-catenin pathway target genes by qRT-PCR and to visualize the
subcellular localization of ß-catenin through immunohistochemical or immunofluorescence
analysis in the Sufu-deficient growth plate chondrocytes.
In chapter 3 I showed that treatment of growth plate chondrocytes with Pthlh increases
Sufu protein level through phosphorylation. Interestingly, phosphorylation of ß-catenin at Ser-
675 by PKA promotes ß-catenin stability by inhibiting its ubiquitination (Hino et al., 2005).
Thus, activation of Pthlh/PKA signaling increases the level of ß-catenin, which interacts with
Sufu. I speculate that Sufu plays a role in mediating the stabilization effect of Pthlh on ß-catenin.
To test that, western analysis was performed on lysates derived from wild-type and Sufu-
deficient growth plates with or without Pthlh treatment. As expected, Pthlh treatment resulted in
increased levels of ß-catenin (~2-fold) in chondrocytes. However, this effect of Pthlh on ß-
catenin protein level is absent in the Sufu-deficient cells, suggesting that Sufu is required for the
positive effect of Pthlh on the level of ß-catenin. It is necessary to confirm the role of PKA as a
major effector of Pthlh signaling activation in this process by analyzing the level of ß-catenin in
cells treated with Pthlh and H89, a PKA inhibitor. The mechanism by which Sufu mediates the
stabilization of -catenin is unknown. One possibility is that the Ser-675 residue of ß-catenin
may be masked by changes of its three-dimensional structure in the absence of Sufu.
Alternatively, Sufu may be required for the cytoplasmic localization of ß-catenin where it can be
phosphorylated and stabilized.
Wnt/ß-catenin signaling promotes chondrocyte hypertrophic differentiation, whereas
Pthlh signaling inhibits it. Thus, activation of Pthlh in chondrocytes opposes Wnt/ß-catenin
signaling. Although Wnt/ ß-catenin signaling has been shown to regulate osteoblast
differentiation from the perichondrium at late stage chondrocyte differentiation, the function of

108
high levels of ß-catenin upon Pthlh/PKA stimulation in articular/resting and proliferating
chondrocytes in vivo is unknown. This complicated pattern may allow regulatory cross-talk with
other signaling pathways that are useful. One possible scenario is that Pthlh upregulates ß-
catenin and Sufu levels to ensure sufficient amounts of ß-catenin throughout the course of
chondrocyte hypertrophic differentiation; upregulation of Sufu levels may serve as an important
mechanism by which cells maintain low Wnt signaling such that chondrocytes can differentiate.
Later, in hypertrophic chondrocytes, Sufu inhibition is relieved, ß-catenin translocates into the
nucleus and activates expression of target genes, such as MMP13 and vegfa, which are essential
for subsequent bone formation.
The involvement of Sufu in the regulation of Wnt/ß-catenin signaling provides evidence
that multiple levels of cross-talk exist between Ihh, Pthlh, and Wnt/ß-catenin signaling. It also
highlights the importance of Sufu as a general mediator for multiple signaling pathways during
chondrocyte development. To this end, it will be interesting to further investigate the function of
ß-catenin in Sufu-expressing chondrocytes by generating Col2a1-Cre;Sufuf/f
; Catnbtm1Tak
mice,
which express stabilized ß-catenin in the absence of Sufu in growth plate chondrocytes.

109
4.4 Conclusions
Proper coordinate control of cell proliferation and differentiation through cell-cell
signaling is a major strategy during endochondral ossification. The well-orchestrated cross-talk
between genes spatially and temporally is essential to guide the patterning of each of the skeletal
elements. I demonstrated that the molecular mechanism by which Sufu and Kif7 coordinate Ihh
signaling and further pinpointed the role of Sufu in mediating Pthlh signaling in chondrocytes.
These discoveries suggest that Sufu levels could be critical for cells to respond to multiple
signaling pathways in chondrogenesis. Thus, understanding the mechanism underlying Sufu
regulation may provide insight into cartilage regeneration in vitro. As other signaling pathways,
such as Wnt/ ß-catenin signaling, also play important roles in controlling skeletal development,
further investigation of how Sufu is integrated with various signaling pathways will help us to
gain a full picture of the molecular regulatory network in this process.

110
4.5 Figures
Figure 4-1 Proposed model by which Sufu protein stability serves as a common mechanism that regulates Gli-mediated
transcription.
Figure 4-1. Proposed model by which Sufu protein stability serves as a common mechanism that
regulates Gli-mediated transcription. Sufu protein stability is negatively regulated by Smo-
mediated Hh activation and Kif7 through a proteasome-mediated pathway. Phosphorylation of
Sufu upon Pthlh signaling activation promotes Sufu stabilization resulted in reduced expression
of Hh target genes.

111
Figure 4-2 Proposed mechanism by which Pthlh and Ihh signaling coordinate Sufu expression in the growth plate.
Figure 4-2. Proposed mechanism by which Pthlh and Ihh signaling coordinate Sufu level in the
growth plate. Pthlh positively regulates Sufu expression through phosphorylation, and Ihh
negatively regulates Sufu level thorough promoting its degradation. Sox9 may negatively
regulate Sufu expression at the mRNA level.

112
4.6 References
Akiyama, H., Chaboissier, M. C., Martin, J. F., Schedl, A. and de Crombrugghe, B. (2002).
The transcription factor Sox9 has essential roles in successive steps of the chondrocyte
differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev 16, 2813-
28.
Carlevaro, M. F., Cermelli, S., Cancedda, R. and Descalzi Cancedda, F. (2000). Vascular
endothelial growth factor (VEGF) in cartilage neovascularization and chondrocyte
differentiation: auto-paracrine role during endochondral bone formation. J Cell Sci 113 ( Pt 1),
59-69.
Chen, Y., Yue, S., Xie, L., Pu, X. H., Jin, T. and Cheng, S. Y. (2011). Dual phosphorylation of
Suppressor of fused by PKA and GSK3beta regulates its stability and localization in the primary
cilium. J Biol Chem.
Enomoto-Iwamoto, M., Kitagaki, J., Koyama, E., Tamamura, Y., Wu, C., Kanatani, N.,
Koike, T., Okada, H., Komori, T., Yoneda, T. et al. (2002). The Wnt antagonist Frzb-1
regulates chondrocyte maturation and long bone development during limb skeletogenesis. Dev
Biol 251, 142-56.
Farzan, S. F., Ascano, M., Jr., Ogden, S. K., Sanial, M., Brigui, A., Plessis, A. and Robbins,
D. J. (2008). Costal2 functions as a kinesin-like protein in the hedgehog signal transduction
pathway. Curr Biol 18, 1215-20.
Gerber, H. P., Vu, T. H., Ryan, A. M., Kowalski, J., Werb, Z. and Ferrara, N. (1999).
VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during
endochondral bone formation. Nat Med 5, 623-8.
Grover, J. and Roughley, P. J. (2006). Generation of a transgenic mouse in which Cre
recombinase is expressed under control of the type II collagen promoter and doxycycline
administration. Matrix Biol 25, 158-65.
Hartmann, C. and Tabin, C. J. (2000). Dual roles of Wnt signaling during chondrogenesis in
the chicken limb. Development 127, 3141-59.
Haycraft, C. J., Zhang, Q., Song, B., Jackson, W. S., Detloff, P. J., Serra, R. and Yoder, B.
K. (2007). Intraflagellar transport is essential for endochondral bone formation. Development
134, 307-16.
Hino, S., Tanji, C., Nakayama, K. I. and Kikuchi, A. (2005). Phosphorylation of beta-catenin
by cyclic AMP-dependent protein kinase stabilizes beta-catenin through inhibition of its
ubiquitination. Mol Cell Biol 25, 9063-72.

113
Huelsken, J. and Birchmeier, W. (2001). New aspects of Wnt signaling pathways in higher
vertebrates. Curr Opin Genet Dev 11, 547-53.
Ikeda, S., Kishida, S., Yamamoto, H., Murai, H., Koyama, S. and Kikuchi, A. (1998). Axin,
a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-
catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J 17, 1371-
84.
Inada, M., Wang, Y., Byrne, M. H., Rahman, M. U., Miyaura, C., Lopez-Otin, C. and
Krane, S. M. (2004). Critical roles for collagenase-3 (Mmp13) in development of growth plate
cartilage and in endochondral ossification. Proc Natl Acad Sci U S A 101, 17192-7.
Jeong, J., Mao, J., Tenzen, T., Kottmann, A. H. and McMahon, A. P. (2004). Hedgehog
signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes
Dev 18, 937-51.
Johansson, N., Saarialho-Kere, U., Airola, K., Herva, R., Nissinen, L., Westermarck, J.,
Vuorio, E., Heino, J. and Kahari, V. M. (1997). Collagenase-3 (MMP-13) is expressed by
hypertrophic chondrocytes, periosteal cells, and osteoblasts during human fetal bone
development. Dev Dyn 208, 387-97.
Kikuchi, A. (2000). Regulation of beta-catenin signaling in the Wnt pathway. Biochem Biophys
Res Commun 268, 243-8.
Kishida, M., Koyama, S., Kishida, S., Matsubara, K., Nakashima, S., Higano, K., Takada,
R., Takada, S. and Kikuchi, A. (1999). Axin prevents Wnt-3a-induced accumulation of beta-
catenin. Oncogene 18, 979-85.
Kitagawa, M., Hatakeyama, S., Shirane, M., Matsumoto, M., Ishida, N., Hattori, K.,
Nakamichi, I., Kikuchi, A. and Nakayama, K. (1999). An F-box protein, FWD1, mediates
ubiquitin-dependent proteolysis of beta-catenin. EMBO J 18, 2401-10.
Lin, A. C., Seeto, B. L., Bartoszko, J. M., Khoury, M. A., Whetstone, H., Ho, L., Hsu, C.,
Ali, S. A. and Alman, B. A. (2009). Modulating hedgehog signaling can attenuate the severity
of osteoarthritis. Nat Med 15, 1421-5.
Liu, C., Li, Y., Semenov, M., Han, C., Baeg, G. H., Tan, Y., Zhang, Z., Lin, X. and He, X. (2002). Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell
108, 837-47.
Maes, C., Stockmans, I., Moermans, K., Van Looveren, R., Smets, N., Carmeliet, P.,
Bouillon, R. and Carmeliet, G. (2004). Soluble VEGF isoforms are essential for establishing
epiphyseal vascularization and regulating chondrocyte development and survival. J Clin Invest
113, 188-99.
McGlashan, S. R., Haycraft, C. J., Jensen, C. G., Yoder, B. K. and Poole, C. A. (2007).
Articular cartilage and growth plate defects are associated with chondrocyte cytoskeletal

114
abnormalities in Tg737orpk mice lacking the primary cilia protein polaris. Matrix Biol 26, 234-
46.
Meng, X., Poon, R., Zhang, X., Cheah, A., Ding, Q., Hui, C. C. and Alman, B. (2001).
Suppressor of fused negatively regulates beta-catenin signaling. J Biol Chem 276, 40113-9.
Miller, J. R., Hocking, A. M., Brown, J. D. and Moon, R. T. (1999). Mechanism and function
of signal transduction by the Wnt/beta-catenin and Wnt/Ca2+ pathways. Oncogene 18, 7860-72.
Moon, R. T., Bowerman, B., Boutros, M. and Perrimon, N. (2002). The promise and perils of
Wnt signaling through beta-catenin. Science 296, 1644-6.
Poole, C. A., Zhang, Z. J. and Ross, J. M. (2001). The differential distribution of acetylated
and detyrosinated alpha-tubulin in the microtubular cytoskeleton and primary cilia of hyaline
cartilage chondrocytes. J Anat 199, 393-405.
Pozniak, C. D., Langseth, A. J., Dijkgraaf, G. J., Choe, Y., Werb, Z. and Pleasure, S. J. (2010). Sox10 directs neural stem cells toward the oligodendrocyte lineage by decreasing
Suppressor of Fused expression. Proc Natl Acad Sci U S A 107, 21795-800.
Ryu, J. H., Kim, S. J., Kim, S. H., Oh, C. D., Hwang, S. G., Chun, C. H., Oh, S. H., Seong,
J. K., Huh, T. L. and Chun, J. S. (2002). Regulation of the chondrocyte phenotype by beta-
catenin. Development 129, 5541-50.
Scherft, J. P. and Daems, W. T. (1967). Single cilia in chondrocytes. J Ultrastruct Res 19, 546-
55.
Selvamurugan, N., Kwok, S. and Partridge, N. C. (2004). Smad3 interacts with JunB and
Cbfa1/Runx2 for transforming growth factor-beta1-stimulated collagenase-3 expression in
human breast cancer cells. J Biol Chem 279, 27764-73.
Song, B., Haycraft, C. J., Seo, H. S., Yoder, B. K. and Serra, R. (2007). Development of the
post-natal growth plate requires intraflagellar transport proteins. Dev Biol 305, 202-16.
Waltzer, L. and Bienz, M. (1999). The control of beta-catenin and TCF during embryonic
development and cancer. Cancer Metastasis Rev 18, 231-46.
Yoder, B. K., Tousson, A., Millican, L., Wu, J. H., Bugg, C. E., Jr., Schafer, J. A. and
Balkovetz, D. F. (2002). Polaris, a protein disrupted in orpk mutant mice, is required for
assembly of renal cilium. Am J Physiol Renal Physiol 282, F541-52.
Yue, S., Chen, Y. and Cheng, S. Y. (2009). Hedgehog signaling promotes the degradation of
tumor suppressor Sufu through the ubiquitin-proteasome pathway. Oncogene 28, 492-9.
Zelzer, E., McLean, W., Ng, Y. S., Fukai, N., Reginato, A. M., Lovejoy, S., D'Amore, P. A.
and Olsen, B. R. (2002). Skeletal defects in VEGF(120/120) mice reveal multiple roles for
VEGF in skeletogenesis. Development 129, 1893-904.

115
Zhang, Q., Murcia, N. S., Chittenden, L. R., Richards, W. G., Michaud, E. J., Woychik, R.
P. and Yoder, B. K. (2003). Loss of the Tg737 protein results in skeletal patterning defects. Dev
Dyn 227, 78-90.
Zhao, Q., Eberspaecher, H., Lefebvre, V. and De Crombrugghe, B. (1997). Parallel
expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev Dyn 209, 377-86.

116
Appendix A: Pthlh negatively regulates Hh signaling activity
in chondrocytes through a cilia-independent mechanism
Shu-Hsuan C. Hsu, Xiaoyun Zhang, and Benjamin A. Alman
All experiments and analysis described here were conducted by Shu-Hsuan C. Hsu with the
exception of the western analysis by Xiaoyun Zhang.

117
A.1 Summary
Chondrocyte proliferation and differentiation are tightly controlled by Indian hedgehog
(Ihh) and parathyroid hormone-like hormone (Pthlh) signaling pathways in the growth plate.
Pthlh is known to function as a negative regulator of chondrocyte hypertrophic differentiation
and of the Hh signaling pathway. Smo-mediated Hh pathway activation depends on the primary
cilium, which plays a positive role in regulating Hh activity in chondrocytes. Here I show that
Pthlh functions in chondrocytes through a cilia-independent mechanism. The data suggest a
model in which Pthlh promotes Sufu-Gli complexes accumulation at the cilium, resulting in
reduced Hh pathway activity. However, in the absence of cilia, Pthlh treatment resulted in
reduced Hh pathway activity, suggesting that the ciliary tip localization of Sufu-Gli complexes is
not required for their ability to regulate transcription.

118
A.2 Introduction
Cilia are microtubule-based organelles that project from the surface of most eukaryotic
cells (Davenport and Yoder, 2005; Pan et al., 2005; Satir and Christensen, 2007). The cilium
extends from a basal body, which is a centriole-derived microtubule organizing center. Cilia are
assembled and maintained through intraflagellar transport (IFT), a process involving anterograde
and retrograde transportation of IFT particles, mediating by kinesin and dynein motors,
respectively (Bisgrove and Yost, 2006; Davenport and Yoder, 2005; Pan et al., 2005; Satir and
Christensen, 2007). Disruption of the expression and function of IFT particles, such as IFT88,
results in the loss of cilia (Haycraft et al., 2007; Yoder et al., 2002). Ift88ORPK/ ORPK
mouse, which
was developed from a large-scale transgene-induced insertional mutagenesis project at the Oak
Ridge National Laboratory harbors in 1994, exhibits scruffy fur, growth retardation, and
polydactyly (Moyer et al., 1994).
During growth plate development, Indian Hedgehog (Ihh) is secreted by the
prehypertrophic chondrocytes and it regulates chondrocyte hypertrophic differentiation through
upregulating parathyroid hormone-like hormone (Pthlh) expression at the periarticular region
(Chung et al., 1998; St-Jacques et al., 1999; Vortkamp et al., 1996). Pthlh functions to maintain
chondrocytes at the proliferative state and delay hypertrophic differentiation thus reducing the
level of Ihh expression in prehypertrophic chondrocytes (Karaplis et al., 1994; Lanske et al.,
1996). This negative feedback loop formed by Ihh and Pthlh plays an important role in
coordinating chondrocyte proliferation and differentiation in the growth plate (Chung et al.,
1998; Vortkamp et al., 1996).

119
In vertebrates, the effects of Hh signaling are mediated by three Gli transcription factors
(Gli1-3). Upon Hh ligand binding to its receptor, patched 1(Ptch1), Ptch1 inhibition on
smoothened (Smo) is released, leading to activation of Gli-mediated transcription in the nucleus.
Suppressor of fused (Sufu) and kinesin member 7 (Kif7) are two evolutionary conserved
regulators of Gli transcription factors in mammalian Hh signaling (Wilson et al., 2009). During
growth plate development, Sufu acts as a major negative regulator of Gli transcription factors
whereas Kif7 functions both positively and negatively in chondrocytes (Hsu et al., 2011). The
primary cilium serves as the focal point of hedgehog (Hh) signaling regulation in vertebrates.
Important components of the Hh pathway, suppressor of fused (Sufu) and the Gli proteins,
localize to cilia and Smo and Kif7 translocate to cilia upon pathway activation (Corbit et al.,
2005; Haycraft et al., 2005). Recent studies carried out in cultured fibroblasts showed that Smo
plays a positively role in dissociating of inhibitory Sufu-Gli complexes at the primary cilium
upon Hh pathway activation. Therefore a model by which Hh signaling activates Gli protein at
the cilia through relieving the inhibitory function of Sufu, leading to Gli-mediated transcription
activation was proposed.
Studies of explants lacking Gli genes suggest the involvement of Gli3 in the inhibitory
effect of Pthlh on growth plate chondrocyte differentiation. Pthlh represses Gli-mediated
transcription via a protein kinase A (PKA)-dependent mechanism (Mau et al., 2007; Miao et al.,
2004; Vortkamp et al., 1996). These data suggest that besides the Hh-mediated regulation, the
Gli transcription factors may also be regulated by Pthlh signaling. However, the molecular
mechanism as well as the role of cilia in the Pthlh-mediated regulation of Gli activity is
unknown.

120
In this study, I explored the role of primary cilium in regulating the effect of Pthlh on Hh
signaling activity during chondrocytes development using Ift88ORPK/ ORPK
mice. My results
indicate that the primary cilia are not required for the inhibitory effect of Pthlh on chondrocyte
hypertrophic differentiation or Gli-mediated transcription activity. Sufu is localized to the ciliary
tip of resting chondrocytes in vivo upon Pthlh stimulation, which is consistent with previous
studies showing that phosphorylation of Sufu by PKA regulates its ciliary localization (Chen et
al., 2011). However, the ciliary tip localization of Sufu is not required for the negative regulation
of Pthlh on downstream Gli transcription activity.

121
A.3 Results
A.3.1 Ift88ORPK/ ORPK
mice show skeletal defects and dysregulated Hh signaling in
chondrocytes
To investigate the role of primary cilia in growth plate chondrocyte development, I
examined the skeletal phenotype of Ift88ORPK/ ORPK
mice. Consistent with previous studies,
Ift88ORPK/ ORPK
mice survive into young adulthood and exhibit polydactyly and shortening of the
bone in the limbs due to defects in endochondral bone formation (Fig. A-1). Histological analysis
revealed asymmetric chondrocyte hypertrophy as well as a delay in vascularization of the
primary ossification center in Ift88ORPK/ ORPK
mice compared with wild-type littermates (Fig. A-
1). Notably, this asymmetric hypertrophy phenotype does recover with time, and two opposite
growth plates were observed in the tibia of Ift88ORPK/ ORPK
mice at E18.5 (Fig. A-2). In support of
the role of cilia in mediating Hh signaling during development, I found reduced Ptch1 and Gli1
mRNA expression in the cilia-deficient chondrocytes compared with wild-type controls.
Activation of Ptch1 and Gli1 upon Hh ligand stimulation is lost in Ift88ORPK/ ORPK
chondrocytes,
suggesting that cilia are required for Hh ligand responsiveness in chondrocytes (Fig. A-3).
A.3.2 Pthlh downregulates Col10a1 expression and Hh signaling activity through a
cilia-independent mechanism in chondrocytes
In chapter 2 I demonstrated that Pthlh represses Hh signaling activity in part through
increasing Sufu levels and facilitating Gli2 degradation and Gli3R formation in chondrocytes.

122
However, questions remain regarding the role of primary cilia in mediating the effect of Pthlh on
Hh activity regulation. In particular, the involvement of cilia in Pthlh signaling in chondrocytes
needs to be tested. To begin to explore the role of primary cilia in mediating Pthlh signaling
during chondrocyte differentiation, I performed Pthlh treatment on wild-type and Ift88ORPK/ ORPK
micromass cultures at various time points followed by qRT-PCR analysis for Col10a1 and Ptch1,
as readouts for hypertrophy and Hh activity, respectively. The level of Col10a1 expression
increased with time in wild-type cultures, demonstrating chondrocyte hypertrophy in vitro.
Deletion of Ift88 resulted in reduced levels of Col10a1 expression in all three time points
examined (Fig.A-4). Pthlh treatment resulted in a reduction in the Col10a1 expression levels in
differentiated chondrocytes cultures (at Day 6 and 9) from both wild-type and Ift88ORPK/ ORPK
mice (Fig.A-4), suggesting that Pthlh inhibits Col10a1 expression in chondrocytes in a cilia-
independent manner. In wild-type cultures, Ptch1 expression progressively increases with time,
which mimics what was seen during chondrocyte differentiation in the growth plate. In the
absence of Ift88, Ptch1 expression is compromised, although increased Ptch1 expression can still
be observed over time (Fig.A-4). Pthlh treatment resulted in decreased levels of Ptch1 expression
in differentiated chondrocytes in both wild-type and Ift88ORPK/ ORPK
cultures (Fig.A-4). These data
suggest that cilia are not required for the regulation of Gli transcription activity and chondrocyte
hypertrophic differentiation by Pthlh.
A.3.3 Ciliary tip localization of Sufu is induced by Pthlh
In chapter 2 I demonstrated that Kif7 plays a role in the exclusion of Sufu-Gli complexes
from the primary cilium. To investigate whether primary cilium is required for Sufu and Kif7

123
protein and/or RNA expression, I examined Sufu and Kif7 expression in Ift88ORPK/ ORPK
mice. No
significant difference in protein or RNA levels of Sufu and Kif7 was detected in Ift88ORPK/ ORPK
cells (Fig A-5A, B), suggesting that primary cilia are not required for the expression of Sufu and
Kif7. The observations that Pthlh represses Hh signaling activity independent of cilia prompted
us to investigate the ciliary localization of Sufu and Kif7 upon Pthlh signaling activation in the
presence or absence of primary cilium. Studies have shown that Sufu and Gli proteins are
transported into the primary cilium as a complex (Hsu et al., 2011; Humke et al., 2010;
Tukachinsky et al., 2010). In Gli2-/-
; Gli3-/-
embryonic fibroblasts, where all Gli proteins are
absent, Sufu is not detected in the cilia, even upon Shh stimulation, suggesting that Gli proteins
are necessary to recruit Sufu to the cilia (Tukachinsky et al., 2010; Zeng et al., 2010). Thus, the
immunostaining of Sufu could serve as an indicator of Sufu-Gli complexes at the primary cilium.
In resting chondrocytes (where the Hh pathway is inactive), Kif7 was mostly localized to the
ciliary tip, whereas Sufu was found at the basal bodies in ~75% of these cells but was rarely
detected at the ciliary tip (Fig A-5C-E). Both Sufu and Kif7 signals are specific as they are
absent in Sufu-deficient and Kif7-deficient chondrocytes, respectively (Fig. 2-8A). Together,
these results indicate that there is very little Sufu-Gli complexes in the primary cilium of resting
wild-type chondrocytes. Pthlh treatment resulted in a ~20% decrease in the ciliary tip
localization of Kif7 (Fig. A-5C-E). Consistent with recent studies carried out in cultured
fibroblasts showing that PKA phosphorylation of Sufu promotes its ciliary localization (Chen et
al., 2011), Sufu was detected at the ciliary tip in ~50% of the Pthlh-treated wild-type
chondrocytes (Fig. A-5C-E), suggesting that Pthlh stimulates Sufu ciliary tip localization
possibility through promote phosphorylation of Sufu by PKA. Interestingly, in the absence of the
primary cilium, ~80% of resting chondrocytes showed Sufu and Kif7 staining at the basal body

124
(Fig. A-5C, F, G). Although no change in the Sufu basal body localization was detected, a ~40%
decrease of Kif7 basal body localization was found upon Pthlh stimulation (Fig. A-5C, F, G).
These observations suggest that Pthlh promotes Sufu-Gli complexes ciliary tip localization in
wild-type chondrocytes, and the ciliary tip localization of Sufu-Gli complexes is not required for
their downstream inhibitory function on Gli-mediated transcription activity. In chapter 3 I
demonstrated that Kif7 is not required for the negative effect of Pthlh on growth plate
chondrocytes. Thus, the changes in Kif7 ciliary localization may play a minor role in regulating
the effect of Pthlh on chondrocyte Hh signaling activity.

125
A.4 Discussion
In chapter 2 I demonstrated distinct and overlapping functions of Sufu and Kif7 in Hh
signaling during chondrocyte development. In chapter 3 I established that Sufu plays an
important role in regulating the negative effect of Pthlh on chondrocyte hypertrophic
differentiation, Gli proteins processing, and Hh signaling activity. There is evidence suggesting
that the primary cilium serves as a focal point in Hh signaling regulation in vertebrates. Here, I
provided data to indicate that Pthlh functions to inhibit chondrocyte hypertrophic differentiation
and Hh signaling activity in chondrocytes through a cilia-independent mechanism.
Phenotypic analysis of Ift88ORPK/ ORPK
mice indicates that the primary cilia are required
for normal skeletal development. Ift88ORPK/ ORPK
chondrocytes showed reduced Hh pathway
activity and lack of response to Hh ligand stimulation compared to wild-type controls. Pthlh
possess inhibitory effect on the expression of Col10a1 and Ptch1 in wild-type micromass
cultures. This is consistent with its negative role in regulating chondrocyte hypertrophic
differentiation and Hh signaling activity in the growth plate in vivo. Similarly, reduced Col10a1
and Ptch1 expression were observed upon Pthlh signaling activation in cilia-deficient (Ift88ORPK/
ORPK) micromass cultures. The data suggest that primary cilia are not required for Pthlh ligand
responsiveness or the negative effect of Pthlh on chondrocyte differentiation and Hh activity
regulation.
Hh pathway activation stimulates degradation of Sufu in some cancer cell lines (Yue et
al., 2009). Recent studies suggest that Hh stimulation promotes the dissociation of Sufu-Gli
protein complexes at the ciliary tip and that this dissociation is important for Gli activation
(Humke et al., 2010; Tukachinsky et al., 2010). In chapter 3 I showed that Pthlh plays a positive

126
role in controlling the stability of Sufu protein as well as regulating Gli2 protein degradation and
Gli3 repressor (Gli3R) formation in the growth plate chondrocytes. Interestingly, here I found
that Sufu is mostly excluded from the ciliary tip in wild-type chondrocytes, whereas Sufu
appears to be stabilized in the ciliary tip in the presence of Pthlh. This is consistent with the
observation that Hh pathway activity is reduced in Pthlh-treated cells, owing to inefficient
dissociation of the Sufu-Gli complexes. Sufu is found to be localized at the basal body in the
absence of the primary cilium and there is no change in its basal body localization observed upon
Pthlh activation. The data suggest that the ciliary tip localization is not required for the inhibitory
effect of Sufu-Gli complexes in chondrocytes. However, whether the ciliary tip localization
contributes to the stabilization of Sufu in Pthlh-treated chondrocytes or its related to the
increased level of Sufu protein awaits further investigation. Nevertheless, results here
demonstrated that Pthlh functions independent of primary cilia in chondrocytes.
Previous in vitro studies show that Hh ligand stimulation promotes Kif7 ciliary tip
translocation (Liem et al., 2009). In chapter 2 I found that Kif7 localizes to the ciliary tip in the
proliferating chondrocytes in vivo. Here I reported that Kif7 localizes also to the ciliary tip in the
resting chondrocytes where the Hh pathway is inactive (as indicated by the lack of Hh target
gene expression). These results suggest that Kif7 might have a functional role in the primary
cilium, even in the absence of Hh ligand activation which supports the proposed model in
chapter 2 (Fig. 2-10B). In the cilia-deficient (Ift88ORPK/ ORPK
) chondrocytes, Sufu localizes to the
basal body in the absence or presence of Pthlh, suggesting that the ciliary tip localization is not
the key control for the inhibitory function of Sufu-Gli complexes, and their ability to regulate
transcription. This notion is consistent with the previous reports showing that Sufu functions
independent of the primary cilium in controlling Gli protein levels (Chen et al., 2009). Taken

127
together, the data suggest that despite the importance of the primary cilium in controlling Hh
pathway activation, the primary cilium is not required for Sufu-mediated negative regulation of
Hh pathway activity in chondrocytes.
Recent studies have shown that PKA, the main mediator of Pthlh signaling in
chondrocytes, localizes to the basal body in cultured fibroblasts (Barzi et al., 2010; Tuson et al.,
2011). This colocalization of PKA and Sufu prompts me to propose a model for cilia-mediated
regulation of Gli activity (Fig. A-6). In the presence of Hh ligand, Sufu-Gli complexes enter the
cilium and are modified within the cilium, either by covalent modification or through interaction
with other proteins such as Kif7, to allow dissociation of the complexes and activation of the Hh
pathway. In the presence of Pthlh ligand, Sufu is phosphorylated by PKA; p-Sufu-Gli complexes
enter the cilium. After exiting the cilium Gli2 complexed with Sufu is recognized and
phosphorylated by PKA, which leads to Gli2 degradation. Phosphorylation by PKA targets Gli3
complexed with Sufu to proteasome-mediated processing into Gli3R.
Taken together, I show here a cilia-independent mechanism by which Pthlh mediates
chondrocyte hypertrophic differentiation and Hh signaling activity regulation in chondrocytes.
Therefore, I propose that in wild-type chondrocytes, Pthlh inhibits hypertrophic differentiation
and Gli-mediated transcription. There is an increased level of Sufu-Gli complexes in the primary
cilium upon Pthlh activation, resulting in an inhibition of Hh signaling. In the absence of the
primary cilium, the inhibitory function of Sufu-Gli complexes remains. Pthlh treatment results in
an increased level of Sufu-Gli complexes localized at the basal body, resulting in reduced Hh
pathway activity.

128
A.5 Materials & Methods
Ethics Statement
A mouse protocol describing the experimental procedures used in the study was approved
by the Animal Care Committee of The Hospital for Sick Children.
Mice
Ift88ORPK
(Tg737ORPK
) mutants were received from Dr. Yoder (The University of
Alabama). The mice were genotyped as described previously (Yoder et al., 1996). In all cases,
littermate mice were used as controls. A mouse protocol describing the above experimental
procedures was approved by the Animal Care Committee of The Hospital for Sick Children.
Skeletal staining
Mice were fixed in 95% ethanol after removal of skin and viscera. Bone samples were
incubated in Alcian Blue solution (15% Alcian Blue in 80% ethanol and 20% glacial acetic acid)
for 2-3 days at room temperature. Samples were then rehydrated and cleared in 1% KOH
overnight or till clear. Samples were stained with Alizarin Red solution (7.5% Alizarin Red in
1% KOH) for 1-2 days and immersed in glycerol for storage (Mau et al., 2007).
Histological analysis and Immunohistochemistry

129
Samples were fixed in 4% paraformaldehyde overnight, embedded in paraffin, and
sectioned for histological evaluation. Sections were examined for Col10a1, a marker for
hypertrophic growth plate chondrocytes, by immunohistochemistry using previously reported
techniques and antibodies (Hu et al., 2006; Linsenmayer et al., 1988; Saika et al., 2004; Tiet et
al., 2006; Wang et al., 2000). The proximal tibial growth plate was used for all analysis to
minimize morphological variations due to anatomic location. Hematoxylin and eosin (H&E)
staining were performed using standard techniques.
Real-time quantitative PCR
RNA isolated from at least three independent experiments was analyzed by qRT-PCR in
triplicate for each treatment condition and primer set. The reactions were made up in TaqMan
Universal PCR master mix (Applied Biosystems) with TaqMan Gene Expression Assays for
mouse Gli1, Ptch1, and Col10a1 (Applied Biosystems). The gene expression levels between
samples were analyzed using the 2 ΔΔCt
method (Livak and Schmittgen, 2001). Either GAPDH
or β-actin (Applied Biosystems) was used as endogenous control for target gene normalization.
Primary growth plate chondrocyte cultures
Chondrocyte isolation protocol was modified from previous published methods (Gosset
et al., 2008). Growth plates of the hindlimbs from E16.5 embryos were isolated and incubated in
collagenase type 4 (Worthington) solution (3mg/ml) for 45 min at 37°C incubator, under 5%
CO2 in a petri dish. Soft tissues were detached by pipetting. The growth plates were placed in a

130
clean petri dish with 0.5 mg/ml collagenase type 4 solution and were incubated overnight at
37°C. Collagenase type 4 solution containing chondrocytes was collected. Cells were washed
with PBS and seeded at a density of 8x103 cells per cm
2.
Micromass cultures
Fore- and hind-limbs were isolated from E11.5 mouse embryos and collected in growth
medium (DMEM + 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin),
washed twice in PBS, and digested in 0.1% trypsin and 0.1% collagenase D (Worthington
Biochemical) in PBS at 37 °C for 20 min. 20 μl of a single cell suspension (2 × 107 cells/ml) was
plated in the center of 9.6 cm2 dishes. The cultures were incubated at 37 °C in a humidified
incubator with 5% CO2 for 1 h, after which they were gently flooded with 2 ml of growth
medium.
Immunofluorescence
Samples were fixed in 4% paraformaldehyde overnight, embedded in paraffin and
sectioned. Sections (5 µm) of growth plate were cut parallel to the longitudinal axis of the bone.
The sections were stained with a monoclonal antibody against acetylated α-tubulin (clone 6-11b-
1, 1:1000; Sigma-Aldrich, Steinheim, Germany), and/or a monoclonal antibody against γ-tubulin
(Dyomics647) (1:100; Abcam), and/or a monoclonal antibody against Sufu (1:100; Abcam),
and/or a polyclonal antibody against Kif7 (1:100) at 4°C overnight. Secondary antibodies
conjugated with Alexa Fluor 488 (1:100; Invitrogen) and TRITC (1:100 Jackson

131
ImmunoResearch Laboratories) were applied for 45 minutes to detect the primary antibody.
Finally the sections were mounted in 4 ,6-diamidino-2-phenylindole (DAPI) containing
Vectashield (Vector Laboratories). Confocal images were acquired using a spinning disc
confocal microscope.
Statistical analysis
For all of the data, the mean, 95 % confidence interval, and standard deviation were
calculated for each condition, and Student’s t test was utilized to compare data sets. A p value of
< 0.05 was used as a threshold for statistical significance.

132
A.6 Figures
Figure A- 1 The primary cilia are required for normal embryonic skeletal development.
Figure A-1. The primary cilia are required for normal embryonic skeletal development. Upper
panel: Alcian blue/Alizarin red stained hind limbs from E16.5 wild-type and Ift88ORPK/ ORPK K
mice. Note polydactyly (arrowhead) and shortening of the long bones in the Ift88ORPK/ ORPK
limb.
Lower panel: H&E staining showed asymmetrical chondrocyte hypertrophy and delayed in
primary ossification center formation in the Ift88ORPK/ ORPK
mice compared to wild-type
littermates at E16.5.

133
Figure A- 2 Asymmetrical hypertrophy phenotype in Ift88ORPK/ ORPK mice is not seen at E18.5.
Figure A-2. Asymmetrical hypertrophy phenotype in Ift88ORPK/ ORPK K
mice is not seen at E18.5.
Histological and immunohistochemical analysis for Col10a1 expression of the Ift88ORPK/ ORPK K
mouse tibia showed the asymmetrical hypertrophy phenotype observed at E16.5 is not seen at
E18.5.

134
Figure A- 3 The role of primary cilia in chondrocyte Hh ligand responsiveness.
Figure A-3. The role of primary cilia in chondrocyte ligand responsiveness. Reduced level of
Ptch1 and Gli1 expression was found in cilia-deficient chondrocytes assessed by qRT-PCR. Shh
treatment resulted in increased Ptch1 and Gli1 mRNA expression in wild-type chondrocytes. In
cilia-deficient chondrocytes, Shh treatment did not result in Hh signaling activation. Data are
shown as means with 95% confidence intervals indicated (n=3). *P<0.05.

135
Figure A- 4 The role of primary cilia in mediating Pthlh signaling in chondrocyte differentiation.
Figure A-4. The role of primary cilium in mediating Pthlh signaling in chondrocyte
differentiation. Expression of Col10a1 and Ptch1 in micromass cultures of wild-type and
Ift88ORPK/ ORPK K
mice at various time points (Day3, Day6, and Day9) with or without Pthlh
treatment was assessed by qRT-PCR. Data are shown as means with 95% confidence intervals
indicated (n=2). *P<0.05.

136
Figure A- 5 Increased Sufu ciliary localization in Pthlh-treated wild-type chondrocytes.

137
Figure A-5. Increased Sufu ciliary localization in Pthlh-treated wild-type chondrocytes. (A) Sufu
and Kif7 protein levels in wild-type and Ift88ORPK/ ORPK
mice were assessed by western analysis.
Actin was used as loading control. (B) Sufu and Kif7 mRNA expression levels in Ift88ORPK/ ORPK
mice were found to be comparable with wild-type controls. Data are shown as means with 95%
confidence intervals indicated (n=3). (C) Fluorescence micrographs of cilia or basal bodies from
wild-type and Ift88ORPK/ ORPK
growth plate chondrocytes in the present or absence of Pthlh
treatment. Sufu (ciliary tip localization of Sufu is indicated by white arrow; basal body
localization of Sufu is indicated by white asterisk) and Kif7 (ciliary tip localization of Kif7 is
indicated by green arrow; basal body localization of Kif7 is indicated by green asterisk) are
detected in the green channel. ilia are detected by staining against acetylated α-tubulin (red
channel). Centrioles are detected by staining against γ-tubulin (blue channel) to label the base of
the cilia. (D-G) 25 to 50 ciliated cells from the resting zone of wild-type and Ift88ORPK/ ORPK
growth plates with (+Pthlh) or without (Un: untreated) Pthlh treatment were examined. Data are
shown as percentages.

138
Figure A- 6 A proposed model for cilia-mediated regulation of Gli activity.
Figure A-6. A proposed model for cilia-mediated regulation of Gli activity. In this model, PKA-
mediated Gli processing is determined by the appropriate Gli substrate, which becomes available
only when Gli is complexed with phosphorylated Sufu (p-Sufu) in the cytoplasm. In the presence
of Hh ligand, active Smo localizes to the cilia and dissociation of Sufu-Gli complexes in the
cilium is promoted by Smo and Kif7; free Gli proteins enter the nucleus and activate the
transcription. In the presence of Pthlh ligand, Sufu is phosphorylated by PKA and p-Sufu-Gli
complexes enter the cilium. Gli2 complexed with p-Sufu is recognized and phosphorylated by
PKA leading to Gli2 degradation. Gli3 complexed with p-Sufu is targeted for phosphorylation
and proteasome-mediated processing into Gli3R.

139
A.7 References
Barzi, M., Berenguer, J., Menendez, A., Alvarez-Rodriguez, R. and Pons, S. (2010). Sonic-
hedgehog-mediated proliferation requires the localization of PKA to the cilium base. J Cell Sci
123, 62-9.
Bisgrove, B. W. and Yost, H. J. (2006). The roles of cilia in developmental disorders and
disease. Development 133, 4131-43.
Chen, M. H., Wilson, C. W., Li, Y. J., Law, K. K., Lu, C. S., Gacayan, R., Zhang, X., Hui,
C. C. and Chuang, P. T. (2009). Cilium-independent regulation of Gli protein function by Sufu
in Hedgehog signaling is evolutionarily conserved. Genes Dev 23, 1910-28.
Chen, Y., Yue, S., Xie, L., Pu, X. H., Jin, T. and Cheng, S. Y. (2011). Dual phosphorylation of
Suppressor of fused by PKA and GSK3beta regulates its stability and localization in the primary
cilium. J Biol Chem.
Chung, U. I., Lanske, B., Lee, K., Li, E. and Kronenberg, H. (1998). The parathyroid
hormone/parathyroid hormone-related peptide receptor coordinates endochondral bone
development by directly controlling chondrocyte differentiation. Proc Natl Acad Sci U S A 95,
13030-5.
Corbit, K. C., Aanstad, P., Singla, V., Norman, A. R., Stainier, D. Y. and Reiter, J. F. (2005). Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018-21.
Davenport, J. R. and Yoder, B. K. (2005). An incredible decade for the primary cilium: a look
at a once-forgotten organelle. Am J Physiol Renal Physiol 289, F1159-69.
Gosset, M., Berenbaum, F., Thirion, S. and Jacques, C. (2008). Primary culture and
phenotyping of murine chondrocytes. Nat Protoc 3, 1253-60.
Haycraft, C. J., Banizs, B., Aydin-Son, Y., Zhang, Q., Michaud, E. J. and Yoder, B. K. (2005). Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for
processing and function. PLoS Genet 1, e53.
Haycraft, C. J., Zhang, Q., Song, B., Jackson, W. S., Detloff, P. J., Serra, R. and Yoder, B.
K. (2007). Intraflagellar transport is essential for endochondral bone formation. Development
134, 307-16.
Hsu, S. H., Zhang, X., Yu, C., Li, Z. J., Wunder, J. S., Hui, C. C. and Alman, B. A. (2011).
Kif7 promotes hedgehog signaling in growth plate chondrocytes by restricting the inhibitory
function of Sufu. Development 138, 3791-801.
Humke, E. W., Dorn, K. V., Milenkovic, L., Scott, M. P. and Rohatgi, R. (2010). The output
of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused
and the Gli proteins. Genes Dev 24, 670-82.

140
Karaplis, A. C., Luz, A., Glowacki, J., Bronson, R. T., Tybulewicz, V. L., Kronenberg, H.
M. and Mulligan, R. C. (1994). Lethal skeletal dysplasia from targeted disruption of the
parathyroid hormone-related peptide gene. Genes Dev 8, 277-89.
Lanske, B., Karaplis, A. C., Lee, K., Luz, A., Vortkamp, A., Pirro, A., Karperien, M.,
Defize, L. H., Ho, C., Mulligan, R. C. et al. (1996). PTH/PTHrP receptor in early development
and Indian hedgehog-regulated bone growth. Science 273, 663-6.
Liem, K. F., Jr., He, M., Ocbina, P. J. and Anderson, K. V. (2009). Mouse Kif7/Costal2 is a
cilia-associated protein that regulates Sonic hedgehog signaling. Proc Natl Acad Sci U S A 106,
13377-82.
Livak, K. J. and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-
time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402-8.
Mau, E., Whetstone, H., Yu, C., Hopyan, S., Wunder, J. S. and Alman, B. A. (2007). PTHrP
regulates growth plate chondrocyte differentiation and proliferation in a Gli3 dependent manner
utilizing hedgehog ligand dependent and independent mechanisms. Dev Biol 305, 28-39.
Miao, D., Liu, H., Plut, P., Niu, M., Huo, R., Goltzman, D. and Henderson, J. E. (2004).
Impaired endochondral bone development and osteopenia in Gli2-deficient mice. Exp Cell Res
294, 210-22.
Moyer, J. H., Lee-Tischler, M. J., Kwon, H. Y., Schrick, J. J., Avner, E. D., Sweeney, W. E.,
Godfrey, V. L., Cacheiro, N. L., Wilkinson, J. E. and Woychik, R. P. (1994). Candidate gene
associated with a mutation causing recessive polycystic kidney disease in mice. Science 264,
1329-33.
Pan, J., Wang, Q. and Snell, W. J. (2005). Cilium-generated signaling and cilia-related
disorders. Lab Invest 85, 452-63.
Satir, P. and Christensen, S. T. (2007). Overview of structure and function of mammalian cilia.
Annu Rev Physiol 69, 377-400.
St-Jacques, B., Hammerschmidt, M. and McMahon, A. P. (1999). Indian hedgehog signaling
regulates proliferation and differentiation of chondrocytes and is essential for bone formation.
Genes Dev 13, 2072-86.
Tukachinsky, H., Lopez, L. V. and Salic, A. (2010). A mechanism for vertebrate Hedgehog
signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol 191,
415-28.
Tuson, M., He, M. and Anderson, K. V. (2011). Protein kinase A acts at the basal body of the
primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube.
Development.

141
Vortkamp, A., Lee, K., Lanske, B., Segre, G. V., Kronenberg, H. M. and Tabin, C. J. (1996). Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related
protein. Science 273, 613-22.
Wilson, C. W., Nguyen, C. T., Chen, M. H., Yang, J. H., Gacayan, R., Huang, J., Chen, J.
N. and Chuang, P. T. (2009). Fused has evolved divergent roles in vertebrate Hedgehog
signalling and motile ciliogenesis. Nature 459, 98-102.
Yoder, B. K., Richards, W. G., Sommardahl, C., Sweeney, W. E., Michaud, E. J.,
Wilkinson, J. E., Avner, E. D. and Woychik, R. P. (1996). Functional correction of renal
defects in a mouse model for ARPKD through expression of the cloned wild-type Tg737 cDNA.
Kidney Int 50, 1240-8.
Yoder, B. K., Tousson, A., Millican, L., Wu, J. H., Bugg, C. E., Jr., Schafer, J. A. and
Balkovetz, D. F. (2002). Polaris, a protein disrupted in orpk mutant mice, is required for
assembly of renal cilium. Am J Physiol Renal Physiol 282, F541-52.
Yue, S., Chen, Y. and Cheng, S. Y. (2009). Hedgehog signaling promotes the degradation of
tumor suppressor Sufu through the ubiquitin-proteasome pathway. Oncogene 28, 492-9.
Zeng, H., Jia, J. and Liu, A. (2010). Coordinated translocation of Mammalian gli proteins and
suppressor of fused to the primary cilium. PLoS One 5, e15900.


















