
QUANTUM-MECHANICAL AND THERMODYNAMIC...
378
QUANTUM-MECHANICAL AND THERMODYNAMIC STUDY OF AMINES AND IONIC LIQUIDS FOR CO 2 CAPTURE A Thesis Submitted to the Faculty of Graduate Studies and Research In Partial Fulfillment of the Requirements For the Degree of Doctor of Philosophy in Industrial Systems Engineering University of Regina By Kazi Zamshad Sumon Regina, Saskatchewan December, 2013 Copyright © 2013: K. Z. Sumon
Transcript of QUANTUM-MECHANICAL AND THERMODYNAMIC...

QUANTUM-MECHANICAL AND THERMODYNAMIC STUDY
OF AMINES AND IONIC LIQUIDS FOR CO2 CAPTURE
A Thesis
Submitted to the Faculty of Graduate Studies and Research
In Partial Fulfillment of the Requirements
For the Degree of
Doctor of Philosophy
in
Industrial Systems Engineering
University of Regina
By
Kazi Zamshad Sumon
Regina, Saskatchewan
December, 2013
Copyright © 2013: K. Z. Sumon

UNIVERSITY OF REGINA
FACULTY OF GRADUATE STUDIES AND RESEARCH
SUPERVISORY AND EXAMINING COMMITTEE
Kazi Zamshad Sumon, candidate for the degree of Doctor of Philosophy in Industrial Systems Engineering, has presented a thesis titled, Quantum-Mechanical and Thermodynamic Study of Amines and Ionic Liquids for CO2 Capture, in an oral examination held on November 27, 2013. The following committee members have found the thesis acceptable in form and content, and that the candidate demonstrated satisfactory knowledge of the subject material. External Examiner: *Dr. Mert Atilhan, Qatar University
Co-Supervisor: Dr. Amr Henni, Industrial Systems Engineering
Co-Supervisor Dr. Allan East, Department of Chemistry
Committee Member: Dr. Renata Raina, Department of Chemistry
Committee Member: **Dr. David deMontigny, Industrial Systems Engineering
Committee Member: **Dr. Paitoon Tontiwachwuthikul, Industrial Systems Engineering
Committee Member: Dr. Stephanie Young, Environmental Systems Engineering
Chair of Defense: Dr. Warren Wessel, Faculty of Education *Participated via SKYPE **Not present at defense

i
Abstract
There is worldwide interest to develop improved CO2-capture solvents to help
reduce cost of capture and environmental footprint. This thesis aims to contribute to this
goal by studying computationally and experimentally a few aspects related to solvent
development, considering both aqueous amines and ionic liquids.
For pKa prediction of aliphatic amines, the performance of quantum-chemistry
(QM) continuum-plus-correction methods was evaluated by comparison with the 1981
pencil-and-paper group-additivity method of Perrin, Dempsey, and Serjeant (PDS). The
best continuum-plus-correction method has been achieved, and while it offers
improvement over the original 1981 PDS method, it is inferior to a trivial update of the
PDS method, and the latter is recommended for pKa prediction.
Static QM calculations using continuum-plus-explicit-water models were used to
examine the reaction mechanisms for CO2 capture by aqueous amines. For the first time,
carbamate anions are correctly predicted by QM to be lower in energy than zwitterion or
carbamic-acid forms. Zwitterions are most relevant at low amine concentrations (from
single-amine versus two-amine modeling). Activation energies vary with pKa in a
sufficient manner that the existence of the zwitterion may depend on pKa. QM-based ab
initio molecular dynamic simulations of aqueous zwitterions were also performed,
supporting the relevance of zwitterions at low concentrations, but also revealing 10-14-
atom H+ transfer relays from zwitterion to transient carbamic acid forms.

ii
A database of Henry’s law constants for CO2 in 2701 ionic liquids at 25°C was
prepared by computing predictions using the QM-based statistical-thermodynamic
method COSMO-RS. The predictions agree well with experimental values, although the
additionally computed predictions for selectivity and solvation enthalpy were not as
good. A new polarity descriptor of ions and ionic liquids is introduced. Henry’s law
constants are dissected into components to probe gas liquid interactions and compare the
solubility of a gas in different ionic liquids. Based on the analysis, a number of ionic
liquids are proposed for further experimental investigation, demonstrating the utility of
COSMO-RS in screening of ionic liquids for CO2 capture.
Based on the COSMO-RS study, three ionic liquids 1-alkyl-3-methyl
imidazolium tris(pentafluoroethyl)-trifluorophosphate (alkyl = ethyl, butyl, and hexyl)
were chosen for further experimental measurement of solubility of CO2 up to 2 MPa at
temperatures of 10, 25 and 50°C using a gravimetric microbalance. The Henry’s law
constants derived from experimental data compared favoribly with those predicted by
COSMO-RS, and with previous experimental values for alkyl = ethyl and hexyl.
Finally, the density, viscosity and excess molar enthalpy of the binary system
{[bmim][Ac]+water} were experimentally determined at atmospheric pressure and at
temperatures from 25 to 70°C. All the excess properties show strong negative deviation
from ideality. Viscosity of pure [bmim][Ac] decreases significantly with addition of
water and with increase in temperature. Excess enthalpies of equimolar binary mixtures
with pure amines {(MEA, DEA, MDEA, TEA, AMP) + [bmim][Ac]} were less negative
(less exothermic mixing) than the {[bmim][Ac] + water} system at 25°C.

iii
Acknowledgements
I am indebted to my co-supervisor Dr. Amr Henni for his constant
encouragements, moral and financial support, and invaluable guidance in every aspect of
my research throughout the study. I thank my co-supervisor Dr. Allan L. L. East for
introducing computational chemistry to me, sharing his enthusiasm and insights, his
support, and diligent efforts to improve the scientific quality of our work.
Drs. Renata J. Raina-Fulton, Paitoon Tontiwachwuthikul, David deMontigny,
and Stephanie Young are thanked for serving as committee members. Dr. Frank Eckert
is thanked for his invaluable assistance with COSMO-RS. Dr. Mark B. Shifflett is
thanked for a discussion on IGA at the 2009 AIChE conference. Dr. Esam Z Hamad is
thanked for his inspiration. I acknowledge the support of ITC, PTRC, WestGrid, and
Laboratory of Computational Discovery. FGSR is thanked for various scholarships.
I am thankful to Dr. Aravind V. Rayer for his support and friendship. Misbah,
Maruf, Firuz, Drs. Khan and Walid are thanked for their assistance. Thank you to Ani,
Shihab, Zahid, Faysal, Tina, Jeeshan, and Sujoy for your support. URBSA activities
were refreshing. Members of Henni and East group are thanked for their cooperation.
My gratitude to the many individuals and their families for helping us having a
great family life in Regina. Drs. M. H. Murad Chowdhury, Magfur Rahman and Messrs.
Md. Rokonuzzaman, Suhayeb Mir, Kalam Azad, and Malick Sohrab are also thanked.
I am grateful for the love and prayers of my parents Kazi Nazrul Islam and Kazi
Ayesha Siddiqa, brother Shabooj, sisters Munni and Roni, and other family members.
Finally, I thank my wife, Aklima S. Chowdhury for her love, patience and support.

iv
Dedication
This thesis is dedicated to my parents, wife, and children, Haya and Athir. Thank
you.

v
Table of Contents Abstract ............................................................................................................................. i
Acknowledgements ......................................................................................................... iii
Dedication ....................................................................................................................... iv
Table of Contents ............................................................................................................ v
List of Figures ............................................................................................................... viii
List of Tables ................................................................................................................ xvi
List of Abbreviations .................................................................................................... xx
List of Symbols ........................................................................................................... xxiii
Chapter 1: Introduction ................................................................................................ 1
1.1 CARBON CAPTURE: INDUSTRIAL CONTEXT ............................................ 1 1.1.1 Emission of carbon dioxide ....................................................................... 1 1.1.2 Emission control: opportunities and challenges ........................................ 3 1.1.3 CO2 separation methods ............................................................................. 6 1.1.4 Absorption technologies for CO2 capture .................................................. 7
1.2 AMINE TECHNOLOGY .................................................................................. 11 1.2.1 Amines ..................................................................................................... 11 1.2.2 Amine process .......................................................................................... 13 1.2.3 Effect of chemical reactions on solvent characteristics and cost ............. 16 1.2.4 Reaction mechanisms .............................................................................. 23 1.2.5 Basicity of amines .................................................................................... 24
1.3 IONIC LIQUIDS: PHYSICAL SOLVENT TECHNOLOGY .......................... 25
1.4 SOLVENT DEVELOPMENT ........................................................................... 27
1.5 OBJECTIVES AND SCOPE ............................................................................. 29
1.6 REFERENCES .................................................................................................. 31
Chapter 2: Computational Methods ........................................................................... 38
2.1 INTRODUCTION ............................................................................................. 38
2.2 MOLECULAR MODELING ............................................................................ 39 2.2.1 Schrodinger equation (SE) ....................................................................... 40 2.2.2 Potential energy surface (PES) ................................................................ 42 2.2.3 Solving the electronic schrodinger equation ............................................ 47 2.2.4 Molecular models for bulk liquid ............................................................ 58 2.2.5 Molecular dynamics (MD) simulation ..................................................... 67
2.3 APPLICATIONS IN THIS DISSERTATION .................................................. 70

vi
2.4 REFERENCES .................................................................................................. 71
Chapter 3: Predicting pKa of Amines ......................................................................... 75
3.1 INTRODUCTION ............................................................................................. 75
3.2 METHODS ........................................................................................................ 77 3.2.1 SHE method ............................................................................................. 77 3.2.2 PDS method ............................................................................................. 82 3.2.3 Experimentals .......................................................................................... 84
3.3 CONTINUUM-SOLVATION ISSUES ............................................................. 85 3.3.1 Choice of radii ......................................................................................... 85 3.3.2 Choice of conformer ................................................................................ 87
3.4 CONCLUSIONS .............................................................................................. 111
3.5 REFERENCES ................................................................................................ 112
Chapter 4: Reaction Mechansims in CO2/Aqueous Amine Systems ..................... 119
4.1 INTRODUCTION ........................................................................................... 119 4.1.1 Overview of competing mechanisms proposed ..................................... 119 4.1.2 Previous modeling studies ..................................................................... 123
4.2 METHOD ........................................................................................................ 128
4.3 RESULTS AND DISCUSSION ...................................................................... 129 4.3.1 Effect of spectator water molecules on ion solvation ............................ 129 4.3.2 Carbamate formation at neutral pH ....................................................... 133 4.3.3 Carbamate formation at basic pH .......................................................... 139 4.3.4 Discussion: Formation of carbamate ..................................................... 150 4.3.5 Formation of bicarbonate ....................................................................... 154 4.3.6 Other amines .......................................................................................... 156
4.4 CONCLUSIONS .............................................................................................. 167
4.5 REFERENCES ................................................................................................ 169
Chapter 5: Molecular Dynamics Simulation of CO2/Amine/Water Mixtures ..... 173
5.1 INTRODUCTION ........................................................................................... 173
5.2 METHOD ........................................................................................................ 175
5.3 RESULTS ........................................................................................................ 178 5.3.1 Group –I. 8-ps simulations of water-CO2 system .................................. 178 5.3.2 Group –II. 8-ps simulations of water-CO2-dimethyl amine system ...... 181 5.3.3 Group-III. longer zwitterion simulations ............................................... 185 5.3.4 Group-IV. simulations of zwitterion+amine .......................................... 197
5.4 REFERENCES ................................................................................................ 205

vii
Chapter 6: Screening of Ionic Liquids: A COSMO-RS Study ............................... 207
6.1 INTRODUCTION ........................................................................................... 207
6.2 THEORY ......................................................................................................... 210
6.3 IL DATABASE AND COMPUTATIONAL DETAILS ................................. 213
6.4 RESULTS AND DISCUSSION ...................................................................... 218 6.4.1 Henry’s law constants at 25°C ............................................................... 218 6.4.2 Quantitative evaluation of predicted HLC ............................................. 222 6.4.3 Trends in Henry’s law constant due to structural variations................. 227 6.4.4 Qualitative interpretations of molecular interactions ............................. 232 6.4.5 Activity coefficients at infinite dilutions ............................................... 242 6.4.6 Effect of molar volume and polarity on Henry’s law constant .............. 247 6.4.7 Effect of temperature on gas solubilities ............................................... 252 6.4.8 Selectivities ............................................................................................ 259
6.5 SCREENING AND DESIGNING OF ILS ...................................................... 265
6.6 CONCLUSIONS .............................................................................................. 269
6.7 REFERENCES ................................................................................................ 271
Chapter 7: Measurement of Solubility of CO2 in [eFAP]-Based Ionic Liquids ... 282
7.1 INRODUCTION .............................................................................................. 282
7.2 EXPERIMENTAL ........................................................................................... 282 7.2.1 Materials. ............................................................................................... 282 7.2.2 Apparatus and measurements. ............................................................... 283
7.3 MODELING .................................................................................................... 286
7.4 RESULTS AND DISCUSSION ...................................................................... 288
7.5 CONCLUSIONS .............................................................................................. 311
7.6 REFERENCES ................................................................................................ 312
Chapter 8: Density, Viscosity and Excess Enthalpy of { 1-Butyl-3-Methyl Imidazolium Acetate+Water} System ....................................................................... 314
8.1 INTRODUCION .............................................................................................. 314
8.2 EXPERIMENTAL ........................................................................................... 315
8.3 RESULTS AND DISCUSSION ...................................................................... 316
8.4 CONCLUSIONS .............................................................................................. 337
8.5 REFERENCES ................................................................................................ 338
Chapter 9: Conclusions, Recommendations, and Future Work ............................ 341
Appendix A. Experimental Determination of pKa ................................................... 347

viii
List of Figures Figure 1.1 Increasing trend in atmospheric CO2 concentration.. .................................... 2
Figure 1.2 Global temperature rise expressed by temperature anomaly relative to 20th
century average.. ............................................................................................................... 2
Figure 1.3 Structure of some amines. ............................................................................ 12
Figure 1.4 Typical amine-process for CO2 capture. ...................................................... 14
Figure 1.5 The dissolution of CO2 in aqueous ammonia is facilitated by the chemical
reaction.. .......................................................................................................................... 17
Figure 1.6 Comparison of loading (broken line) and absorption capacity (solid line) of
MEA and MDEA at low pressure.. ................................................................................. 19
Figure 1.7 Common cations and anions in conventional ionic liquids. ........................ 26
Figure 2.1 A one-dimensional projection of a potential energy surface showing the IRC
for a reaction that connects two minima. ........................................................................ 45
Figure 2.2 Continuum solvation model ......................................................................... 60
Figure 2.3 Semi-continuum solvation model. ............................................................... 63
Figure 2.4 Periodic boundary conditions (a two-dimensional periodic system). .......... 68
Figure 3.1 SHE results without empirical corrections (on conformers of ref. 7),
showing dramatic effects of cavity radii. . ..................................................................... 86
Figure 3.2 Cavity volumes of B·HOH complexes. See Figure 3.1 for legend. ........... 86
Figure 4.1 Conformers of X·(n H2O) complexes used in section 4.3.1 (B3LYP/6-
31G(d)/UFF-PCM). ....................................................................................................... 131

ix
Figure 4.2 Effect of explicit solvating water molecules on predicted ΔE values for
carbamate anion formation. ........................................................................................... 132
Figure 4.3 Reaction mechanisms observed in the modeling of eq (1) with B=H2O. .. 133
Figure 4.4 B3LYP/6-31G(d)/UFF-PCM results for 1-amine-1-H2O modeling. . ...... 134
Figure 4.5 B3LYP/6-31G(d)/UFF-PCM results for 1-amine-5-H2O modeling.. ........ 136
Figure 4.6 Results for 1-amine-20-H2O modeling. Energy profiles are at B3LYP/6-
31G(d) (square) and at MP2/6-31G(d,p) (triangle) level.. ............................................ 138
Figure 4.7 Reaction mechanisms observed in the modeling of eq (1) with B=amine. 139
Figure 4.8 6-atom cycle results from 2-amine-0-H2O modeling.. .............................. 141
Figure 4.9 6-atom cycle results for 2-amine-0-H2O modeling, but with gauche MEA
for maximal H-bonding at the outset. ........................................................................... 142
Figure 4.10 5-atom cycle results from 2-amine-0-H2O modeling.. ............................ 143
Figure 4.11 8-atom cycle with 2-amine-1-H2O modeling.. ........................................ 145
Figure 4.12 Effect of varying n in 2-amine-n-H2O modeling (zwitterion-to-carbamate
step).. ............................................................................................................................. 146
Figure 4.13 Results from 2-amine-18-H2O modeling.. ............................................... 148
Figure 4.14 Optimized transition structures for Figure 4.13 (results from 2-amine-18-
H2O modeling). ............................................................................................................. 149
Figure 4.15 Effect of level of modeling on zwitterion deprotonation step ................. 151
Figure 4.16 Mechanism for bicarbonic acid formation through 6-atom cycle ........... 154
Figure 4.17 Bicarbonate formation through 1-amine-1-H2O 6-atom cycle. ............... 155

x
Figure 4.18 1-amine-1-H2O models for comparison of amine pKa effects. ................ 156
Figure 4.19 Correlations of Ea at MP2/6-31g(d,p)/UFF-IEFPCM level of theory versus
pKa (upper plot) and transition state approach distance R(N-C) (lower plot), for
zwitterion formation. ..................................................................................................... 158
Figure 4.20 Potential energy surfaces for the formation of zwitterons in 1-amine-1-
H2O modeling. .............................................................................................................. 159
Figure 4.21 Correlation of Ea at MP2/6-31g(d,p)/UFF-IEFPCM level of theory versus
pKa, for bicarbonate formation. ..................................................................................... 160
Figure 5.1 Possible intermediates in the bicarbonate pathway in CO2/H2O system ... 174
Figure 5.2 Possible intermediates in the carbamate pathway in CO2/Me2NH/H2O
system ............................................................................................................................ 174
Figure 5.3 Starting geometry in simulation (b), showing only two of the neighboring
water molecules. ............................................................................................................ 178
Figure 5.4 Starting geometry in simulation (c), hydronium at bottom right. .............. 179
Figure 5.5 Evolution of two OH bond lengths in simulation (d), demonstrating
ionization of H2CO3 at 4.7 ps. ....................................................................................... 180
Figure 5.6 Starting geometry in simulation (e), showing only three of the neighboring
water molecules. .......................................................................................................... 181
Figure 5.7 Initial local geometry of carbamate and hydronium ion (left) and local
geometry of carbamic acid formed at t=5191fs (right) in simulation (f). ..................... 182
Figure 5.8 Evolution of the OH distance that demonstrates the interconversion of
carbamate with carbamic acid in simulation (f). . ........................................................ 182

xi
Figure 5.9 Initial geometry of carbamic acid (left); and geometry of carbamate anion at
t=8001fs (right) in simulation (g). ............................................................................... 183
Figure 5.10 Evolution of the two OH distances that demonstrate conversion of
carbamic acid into carbamate (see text) in simulation (g) ............................................ 184
Figure 5.11 The breaking of NH (blue) and simultaneous shrinking of NC(red) bond
lengths in simulation (h). .............................................................................................. 186
Figure 5.12 Evolution of two OH bond lengths in simulation (h), the oxygens being the
two in COO moiety. ...................................................................................................... 187
Figure 5.13 The 10-atom cycle at t=8ps in simulation (h) (all surrounding water
molecules are removed).. .............................................................................................. 188
Figure 5.14 Starting geometry in simulation (i), showing only five of the neighboring
water molecules. .......................................................................................................... 189
Figure 5.15 Plot of (a) NH bond (red line) demonstrating formation of carbamate from
zwitterion by NH bond cleavage and (b) OH bond (blue line) demonstrating formation
of carbamic acid from a carbamate intermediate in simulation (i). .............................. 190
Figure 5.16 t=1437fs of simulation (i), showing the 14-atom relay trajectory
(connected by broken lines), the surrounding water molecules are removed ............... 191
Figure 5.17 Starting geometry of MEA-zwitterion (left) and geometries of carbamate
and hydronium products at t=17203fs in simulation j. . .............................................. 192
Figure 5.18 Evolution of three OH bond lengths in the generated hydronium ion in
simulation (j). ................................................................................................................ 193
Figure 5.19 Starting geometry of AMP-zwitterion in simulation (k), only few
neighboring water molecules are shown. ...................................................................... 194

xii
Figure 5.20 Starting geometry of zwitterion in simulation (l): H22O2=3.1 Ǻ,
H21O5=1.79 Ǻ, H22O4=1.86 Ǻ. .................................................................................. 194
Figure 5.21 Starting geometry (left) of PPZ-zwitterion in simulation (m), few
surrounding water molecules are shown. 10-atom cycle at 15-ps (right). .................... 195
Figure 5.22 Difference in amplitude of vibration of two NH bond lengths of PPZ-
zwitterion in simulation (m), (red: 3-coordinated N, blue: 4-coordinated N). .............. 196
Figure 5.23 Initial geometry (left) and final geometry (right) in simulation (n), only
few water molecules are shown. ................................................................................... 197
Figure 5.24 Evolution of some important bond lengths in simulation (n). Red: breaking
of zwitterion NH bond. ................................................................................................. 199
Figure 5.25 Snapshot of simulation (n) at t=28213 fs showing the carbamate-
hydronium intermediate complex. ................................................................................ 200
Figure 5.26 Geometries in simulation (o) without the spectator water molecules. ..... 201
Figure 6.1 Comparison of Henry’s law constants of CO2: Predicted (grey); and
experimental (black). .................................................................................................... 223
Figure 6.2 Matching of sigma profiles of gases with those of ionic liquids.. ............. 234
Figure 6.3 Sigma-potentials of ionic liquids with different alkyl chain length.. ........ 235
Figure 6.4 Sigma-potentials of ionic liquids with different ring precursors:. ............. 237
Figure 6.5 Sigma-potentials of ionic liquids with non ring precursors: ...................... 238
Figure 6.6 Sigma-potentials of ionic liquids with different anions: ............................ 240
Figure 6.7 Comparison of CO2 sigma profiles with those of some ionic liquids. ....... 241

xiii
Figure 6.8 Effect of electrostatic polarity, sig2, of cations on residual activity
coefficients of CO2 in the ILs [cation][Tf2N]. .............................................................. 244
Figure 6.9 Effect of electrostatic polarity, sig2, of anions on residual activity
coefficients of CO2 in the ILs [bmim][anion]. .............................................................. 244
Figure 6.10 Effect of molar volume on the combinatorial activity coefficients of CO2
in the ILs [bmim][anion] and [cation][Tf2N]. .............................................................. 246
Figure 6.11 Trends in experimental Henry’s law constant of CO2.. ........................... 249
Figure 6.12 Comparison of the relative polarity parameter of ionic liquids.. ............. 249
Figure 6.13 Effect of molar volume of ILs with Henry’s law constant of CO2 in ionic
liquids [cation][Tf2N]. .................................................................................................. 251
Figure 6.14 Effect of polarity on the Henry’s law constant of CO2 in the ionic liquids
[cation][Tf2N]. .............................................................................................................. 251
Figure 6.15 Comparison of enthalpy of solvation at infinite dilution of CO2 in some
ILs.. ............................................................................................................................... 255
Figure 6.16 Relative effect of enthalpic and entropic contributions on the Henry’s law
constant for CO2 in ionic liquids [bmim][anion]. ......................................................... 255
Figure 6.17 Comparison of contribution in excess enthalpy (filled circle) due to CO2
with enthalpy of solvation (filled square) for CO2 dissolution in [bmim][anion]. ...... 257
Figure 6.18 Contribution in excess enthalpy due to CO2 in CO2-[bmim][anion]
mixture at infinite dilution of CO2 ................................................................................ 258
Figure 6.19 Effect of molar volume of ILs [cation][Tf2N] on CO2/CH4 selectivity. . 260
Figure 6.20 Effect of polarity of ILs [cation][Tf2N] on CO2/CH4 selectivity. ........... 260
Figure 6.21 Comparison of CO2/CH4 selectivity.Grey, experimental; black, prediction.262

xiv
Figure 6.22 Sigma profiles of some ionic liquids with [bmim] cation but with differnet
anions within the screening charge region between 0 and 0.4 e/nm2. .......................... 262
Figure 6.23 Comparison of CO2/N2 selectivity: grey, experimental; black, prediction.264
Figure 7.1 Structure of the ionic liquids [emim][eFAP], [bmim][eFAP] and
[hmim][eFAP] (R=C2H5, C4H9, C6H13) ........................................................................ 283
Figure 7.2 Computer-controlled integrated gravimetric microbalance ( IGA003 ). ... 285
Figure 7.3 Comparison of solubility of CO2 in [bmim][PF6] with literature data. ..... 291
Figure 7.4 Solubility of carbon dioxide in the ionic liquid [emim][eFAP]. ............... 298
Figure 7.5 Solubility of carbon dioxide in the ionic liquid [bmim][eFAP]. ............... 299
Figure 7.6 Solubility of carbon dioxide in the ionic liquid [hmim][eFAP]. ............... 300
Figure 7.7 Comparison of solubility of CO2 in [hmim][eFAP] with literature data. .. 301
Figure 7.8 Comparison of solubility of CO2 in [emim][eFAP] with literature data. .. 302
Figure 7.9 High-pressure phase behavior of CO2-[emim][eFAP] system at 323.15 K by
PR EoS. ......................................................................................................................... 306
Figure 7.10 High-pressure phase behavior of CO2-[emim][eFAP] system at 298.15 K
by PR EoS ..................................................................................................................... 307
Figure 7.11 Henry’s law constant of CO2 in [eFAP]-based ionic liquids as function of
temperatures. ................................................................................................................. 310
Figure 8.1 Comparison of density of [bmim][Ac] with literature data.4,11-14 .............. 319
Figure 8.2 Densities of binary mixture of water (1) with [bmim][Ac] (2) as a function
of ionic liquid mole fraction (upper plot) and water mole fraction (lower plot) at various
temperatures. ................................................................................................................. 320

xv
Figure 8.3 Excess molar volumes of binary mixture of water (1) with [bmim][Ac] (2)
as a function of ionic liquid mole fraction at various temperature ............................... 321
Figure 8.4 Effect of temperature on density of binary mixture of water + [bmim][Ac]
at various approximated percent mole fraction of water ............................................... 324
Figure 8.5 Comparison of viscosity data of pure [bmim][Ac] with literature data.. ... 327
Figure 8.6 Viscosity of binary mixture of water (1) with [bmim][Ac] (2) as a function
of water mole fraction at various temperature. ............................................................. 328
Figure 8.7 Viscosity deviations of binary mixture of water (1) with [bmim][Ac] (2) as
a function of ionic liquid mole fraction at various temperature. ................................... 329
Figure 8.8 Effect of temperature on viscosity of binary mixture of water + [bmim][Ac]
as a function of temperature at various approximated percent mole fraction of water. 332
Figure 8.9 Excess molar enthalpy of binary mixture of water with [bmim][Ac] as a
function of ionic liquid mole fraction at various temperatures ..................................... 335
Figure 8.10 Comparison of excess enthalpy of the binary mixture of [bmim][Ac] with
some common alkanolamines and water. ..................................................................... 336

xvi
List of Tables
Table 1.1 Typical Conditions for Post-Combustion Capture, Pre-Combustion
Capture and the Natural Gas Sweetening Process. ............................................................. 5
Table 1.2 Some Currently Operational Commercial CCS Plants. Data Taken from
Refs. 11 and 15. ................................................................................................................... 9
Table 1.3 Some Planned Demonstration Plants Using Physical Absorption
Process. . .......................................................................................................................... 10
Table 3.1 pKa Errors from Uncorrected SHE Procedure: Basis Set Dependencea ......... 79
Table 3.2 Terms in the Perrin-Dempsey-Serjeant Scheme for pKa Predictiona .............. 83
Table 3.3 Amines Used in Measurement of pKa ............................................................. 84
Table 3.4 pKa Results from Uncorrected SHE Procedure: Conformer Dependence ...... 89
Table 3.5 Optimized Structures of Geometries of Amines in Table 3.4 ......................... 90
Table 3.6 pKa Results: Comparison of Continuum-Solvation Procedures ...................... 95
Table 3.7 SHE vs. PDS Predictions for pKa of 32 Amines ............................................. 97
Table 3.8 Optimized Structures of Geometries of Amines in Table 3.7 ......................... 98
Table 3.9 Group Contributions in Old PDS Predictions for pKa of Amines in
Table 3.7 .......................................................................................................................... 105
Table 3.10 pKa Errors in SHE vs. PDS Predictions Outside the Training Set .............. 107
Table 3.11 Optimized Structures of Geometries of Amines in Table 3.10 ................... 108
Table 4.1 Reaction Pathways Observed in Single-Amine Modeling ............................ 124

xvii
Table 4.3 Optimized Structures for Figure 4.19 ............................................................ 161
Table 4.4 Optimized Structures for Amines in Figure 4.21 .......................................... 164
Table 5.1 Bond-Formation through 10-Atom Cycle Schematically Shown in
Figure 5.13 ...................................................................................................................... 188
Table 5.2 Evolution of Some Bond Lengths as a Function of Time Through the
14-atom Relay Shown in Figure 5.16 ............................................................................. 191
Table 6.1 List of Cations. ............................................................................................. 214
Table 6.2 List of Anions. ............................................................................................... 216
Table 6.3 Henry's Law Constants of CO2 (bar) at 298.15 K in Imidazolium-Based
Ionic Liquids ................................................................................................................... 219
Table 6.4 Henry's Law Constants of CO2 (bar) at 298.15 K in Pyridinium and
Pyrrolidinium-Based Ionic Liquids ................................................................................. 220
Table 6.5 Henry's Law Constants of CO2 (bar) at 298.15K in Ionic Liquids Based
on Ammonium, Phosphonium, Guanidinium, Uronium, Thiouronium,
Piperidinium, and Quinolinium Precursor ...................................................................... 221
Table 6.6 Comparison of Henry’s law constants of CO2 Predicted in this Work
with those Predicted by Zhang et al. (2008). .................................................................. 225
Table 6.7 Comparison of Henry’s law constants of CO2(MPa) Predicted in this
Work with those Predicted by G.-Miquel et al (2011) ................................................... 226
Table 6.8 Properties in the Ionic Liquids [cation][Tf2N] ............................................. 228
Table 6.9 Properties in the Ionic Liquids [bmim][anion] .............................................. 229
Table 6.10 Ranking of Anions for Some Fixed Cations and Vice Versa ...................... 266

xviii
Table 7.1 Measured Density of Ionic Liquids ............................................................... 289
Table 7.2 Number of Groups in the Ionic Liquids for Computation of Critical
Properties ........................................................................................................................ 290
Table 7.3 EoS constants for Ionic Liquids and CO2. ..................................................... 290
Table 7.4 Solubility of CO2 in [emim][eFAP] at Different Pressures and
Temperatures in the Mole-Fraction scale ........................................................................ 292
Table 7.5 Solubility of CO2 in [emim][eFAP] at Different Pressures and
Temperatures in the Molality Scale ................................................................................ 293
Table 7.6 Solubility of CO2 in [bmim][eFAP] at Different Pressures and
Temperatures in the Mole-Fraction Scale ....................................................................... 294
Table 7.7 Solubility of CO2 in [bmim][eFAP] at Different Pressures and
Temperatures in the Molality Scale ................................................................................ 295
Table 7.8 Solubility of CO2 in [hmim][eFAP] at Different Pressures and
Temperatures in the Mole Fraction Scale ....................................................................... 296
Table 7.9 Solubility of CO2 in [hmim][eFAP] at Different Pressures and
Temperatures in the Molality Scale ................................................................................ 297
Table 7.10 Estimated Binary Interaction Parameters and Modeling Results ................ 305
Table 7.11 The Mole-Fraction Based Henry’s law constant of CO2 in the [eFAP]
Ionic Liquids at Various Temperatures ........................................................................... 309
Table 7.12 The Enthalpy and Entropy of Solvation of CO2 in the [eFAP] Ionic
Liquids at Various Temperatures .................................................................................... 309
Table 8.1 Density of {[bmim][Ac]+water}System at (283.15 to 353.15) K ............... 319

xix
Table 8.2 Coefficients (cm3·mol-1) of the Redlich-Kister Equation for the
Correlation of the Excess Molar Volume (VE / cm3·Mol-1) of the System
[bmim][Ac] + Water, Along With the Standard Deviations (σ / cm3·mol-1) at
Various Temperatures ..................................................................................................... 322
Table 8.3 Parameters for the Empirical Polynomial Correlation of Density of
Aqueous [bmim][Ac] as a Function of Temperature (293.15 to 353.15K) at
Various Mole Fraction of Water. .................................................................................... 323
Table 8.4 Viscosity of {[bmim][Ac]+water}System at (298.15 to 343.15) K .............. 327
Table 8.5 Coefficients of the Redlich-Kister Equation for the Correlation of the
Viscosity Deviation (∆η / mPa·s) of the System [bmim][Ac]+Water, and the
Standard Deviations (σ / mPa·s). .................................................................................... 330
Table 8.6 Fit Parameters for the Correlation of Viscosity as a Function of
Temperature of aqueous [bmim][Ac] Using the Vogel-Fulcher-Tammann (VFT)
and Arrhenius-type Equation and Standard Deviation ................................................... 331
Table 8.7 Viscosity of {[bmim][Ac]+water}System at (298.15 to 343.15) K .............. 334
Table 8.8 Coefficients of the Redlich-Kister Equation for the Correlation of the
Excess Enthalpy (J/mol) of the Systems Aqueous [bmim][Ac], and the Standard
Deviations (σ )................................................................................................................. 335

xx
List of Abbreviations 3-MAPA 3-(Methylamino)Propylamine
AEEA Aminoethylethanolamine
AMP 2-Amino2-Methyl-Propanol
AO Atomic Orbital
ASC Apparent Surface Charges
B3LYP The Combination of Three-Parameter Exchange Functional of Becke
(B3) with The Correlation Functional of Larr, Yang, Perdew (LYP)
[bmim] 1-Butyl-3-Methyl-Imidazolium
[bmim][Ac] 1-Butyl-3-Methyl Imidazolium Acetate
CAPEX Captial Cost
CCS Carbon Capture And Storage
GTO Gaussian-type Orbital
COSMO Conductor-Like Screening Model
COSMO-RS Conductor-Like Screening Model for Realistic Solvents
CSM Continuum Solvation Models
DEA Diethanolamine
DEMEA N,N-Diethylethanolamine
DFT Density Functional Theory
DGA Diglycolamine
DIPA Diisopropanolamine

xxi
DMMEA Dimethylethanolamine
ECBM Enhanced Coal Bed Methane Production
EOR Enhanced Oil Recovery
EoS Equation of State
FILs Functionalized Ionic Liquids
HF Hartree-Fock
HLC Henry’s law constant
IEF-PCM Integral Equation Formalism for Polarisable Continuum Model
ILs Ionic Liquids
IRC Intrinsic Reaction Coordinate
KS Kohn-Sham
MD Molecular Dynamics
MDEA N-Methyl Diethanolamine
MEA Ethanolamine
MIPA Monoisopropanolamine
MMEA Dimethylamino Ethanol
MO Molecular Orbital
MOR Morpholine
MP Moller-Plesset
MPA Monopropanolamine
MW Molecular Weight
OPEX Operational Cost

xxii
PES Potential Energy Surface
PGTO Primitive Gaussian Type Orbitals
PPZ Piperazine
RTIL Room Temperature Ionic Liquid
SAS Solvent Accessible Surface
SCRF Self-Consistent Reaction Field
SES Solvent-Excluded Surface
TEA Triethanolamine
[Tf2N] Bis(trifluoromethylsulfonyl)imide
TS Transition State
TSILs Task-Specific Ionic Liquids
TZVP Triple-Zeta Valence Polarized Basis Set
VASP Vienna Ab Initio Molecular Dynamics Simulation Package
VFT Vogel-Fulcher-Tammann
VLE Vapor-Liquid Equilibrium
ZPVE Zero-Point Vibrational Energy

xxiii
List of Symbols
Adjustable parameters in Redlich-Kister equation
Activity of a base
Concentration of a base
Concentration of amine in the aqueous solvent
Liquid heat capacity
Ground-state energy
Activation energy
Enhancement factor
Exchange-correlation energy
Excess molar enthalpy
Henry’s law constant
Dissiciation constant for pure water
Molecular weight and density of the component
Critical pressure
Vapour pressure of pure component
Stripping energy
Heat of desorpton
Reboiler heat duty
Sensible heat

xxiv
Co2 caputre rate
CO2/CH4 selectivity
CO2/N2 selectivity
Fitting parameters in VFT equation (8.10)
Critical temperature
Excess molar volume
Molar volume of component
Surface area of a segment
Diameter of an ion
Mixture attractive parameter in Peng-Robinson equation
Mixture Co-volume parameter in Peng-Robinson equation
Rate constant for the formation of zwitterion
Gas phase mass transfer coefficient
Liquid phase mass transfer coefficient
Overall reaction rate constant for CO2 loss
Molal solubility of CO2
Number of segments of type on molecule
Rate of reaction of CO2
Mole fraction of CO2 in the binary framework
Mole fraction of CO2 in the ternary framework
Mole fraction of water and ionic liquids
Mole fraction of solute

xxv
Mole fraction of water and ionic liquids
H Hamiltonian operator
N Relative overall polarity
pKa Negative logarithm of the acid dissociation constant
q Internal coordinate
Sig2 Electrostatic polarity
Kinetic energy operator
Potential energy operator
VIL Molar volume of Ionic Liquid
Δ Different between stripper overhead and reboiler temperatures
Δ Gibbs free energy of solvation
Energy
Ionic strength
Equilibrium constant
Universal gas constant
Temperature
Molar volume of the liquid mixture
Pure component attractive parameter in Peng-Robinson EoS
Pure component co-volume parameters Peng-Robinson EoS
Concentration
Fugacity
Fitting parameters in equation (8.10)

xxvi
Partial pressure
Mole fraction
f Dihedral angle
Residual activity coefficient of molecule in the mixture
Ψ Electronic wave function
∞, Residual chemical potential at infinite dilution
∞, Combinatorial chemical potential at infinite dilution
∞ Activity coefficient of compound at infinite dilution
Activity coefficient of base
∞ Viscosity at infinite temperature
Viscosity of ionic liquid
Viscosity of water
, Gas phase chemical potential of solute
Chemical potential of in its pure liquid state
Pseudo-chemical potential of at infinite dilution
Ground-state electron density
Liquid density
Density of the component
Charge density on segment
Energetic interaction parameters between two segments and
∆ Viscosity deviation
Δ Difference between loadings at the top and bottom of absorption

xxvii
Ψ Wave function
Loading
Activity coefficient
Viscosity of mixture
Density of the liquid mixture
Standard deviation
Stoichiometirc coefficient
Molecular orbital
Basis function
Accentric factor

1
Chapter 1: Introduction _______________________________________________________________________
1.1 CARBON CAPTURE: INDUSTRIAL CONTEXT
1.1.1 Emission of carbon dioxide
The total concentration of atmospheric carbon dioxide (CO2), at present, is
rapidly increasing, at an unprecedented rate of 2 ppm/year (Figure 1.1).1 Thermal power
plants and many chemical industries around the world, while harnessing the raw
chemical energy of carbonaceous fossil-fuel through combustion, generate and then emit
CO2-containing flue gas streams to the atmosphere, thus contributing significantly to the
continued increase in the atmospheric concentration of CO2. Oil and gas industries
require purification of many fuel gas streams and generate streams of CO2 which are,
generally, vented to the atmosphere. Emitted CO2, being a heat-trapping gas, contributes
to global warming (Figure 1.2) through greenhouse gas effect, engendering significant
economic, environmental and humanitarian concerns (draught, flooding, extreme
weather, species extinction, food shortage, urban smog, acid rain, and health
problems).2,3

2
Figure 1.1 Increasing trend in atmospheric CO2 concentration. Data taken from ref. 4.
Figure 1.2 Global temperature rise expressed by temperature anomaly relative to 20th
century average. Data taken from ref. 5.
300
320
340
360
380
400
1950 1960 1970 1980 1990 2000 2010 2020
An
nu
al m
ean
CO
2co
nce
ntr
atio
ns
(pp
m)
obse
rved
at
the
Mau
na
Loa
Ob
serv
ator
y
Year
-0.6
-0.4
-0.2
0
0.2
0.4
0.6
0.8
1880
1886
1892
1898
1904
1910
1916
1922
1928
1934
1940
1946
1952
1958
1964
1970
1976
1982
1988
1994
2000
2006
2012
Ann
ual l
and
and
sea
mea
n te
mpe
ratu
re
anom
aly
(°C
) re
lativ
e to
20t
hce
ntur
y av
erag
e
Year

3
1.1.2 Emission control: opportunities and challenges
Carbon capture and storage (CCS), a key technology for emission control, allows
for the continued use of existing carbon-based energy infrastructure, but simultaneously
reduces CO2-emission; thus considered feasible for short-term (until renewable energy
sources mature).6-8 CCS consists of three steps: (i) capture of CO2 at the origin and (ii)
compression to liquid and transportation to a geological storage site, followed by (iii)
storage. For power plants, three major options for implementation of CCS are available:
post-combustion (capturing CO2 from flue gas streams after combustion of fuel with air);
pre-combustion (capturing CO2 from a high-pressure stream of synthetic fuel, mixture of
hydrogen and CO2, generated by coal-gasification), and oxy-fuel (burning fuel with pure
oxygen generating a N2-free stream).9 For currently operating power plant, the post-
combustion capture is feasible as no change in power plant design and operation is
required and a capture-unit can be retrofitted with the power-plant.9
More than 8000 large point sources including fossil-fuel-fired power plants, steel
mills, cement kilns, chemical plants and refineries (causing 60% of all human emission)
are amenable to CCS.7 At present, however, none but very few, employ CCS because
implementation of CCS is not, generally, economically beneficial (incur cost for new
installations for capture, transportation etc). Implementation of CCS is less expensive
(or even profitable) in few exceptional emission sources that are endowed with one or
more of the following criteria (i) relatively pure concentrated CO2 stream is available
(for example, CO2 streams generated in natural gas purification, the production of

4
hydrogen, ethylene oxide, and synthetic fuel (coal gasification)) and (ii) short
transportation distances for storage (iii) storage site with opportunities for concomitant
production of oil or gas through Enhanced oil Recovery (EOR) or Enhanced Coal Bed
Methane (ECBM) production.10,11 Unfortunately, major emission sources, such as coal
and gas-fired power plants lack definitely in the first criteria, with dilute flue gas streams
(15% CO2, Table 1.1), and thus necessitates the separation of CO2 from flue gas stream
to generate a concentrated CO2 stream (>85%) ready for compression and
transportation.11 Cost-effective separation is a challenge, as explained below.

5
Table 1.1 Typical Conditions for Post-Combustion Capture, Pre-Combustion Capture
and the Natural Gas Sweetening Process.
Flow conditionsa
Post-
combustion Pre-combustion
Natural gas
sweetening
Temperature (°C) 40-75 40 30-40
Pressure (bar) 1 30 5-120
Composition (by mole)
CO2 10-15% 38% 0.1-8%
H2O 5-10% 0.14%
H2 55.5%
O2 3-4%
CO 20 PPM 1.7%
N2 70-75% 3.9% 0-0.2%
NOX <800 PPM
SOX <200 PPM
H2S 0.4% 0-15%
CH4 70-95%
aData taken from Ref. 12.

6
1.1.3 CO2 separation methods
Separation of CO2 from various gas streams has been performed in oil and gas
industries for decades, and many separation methods have been developed based on the
principles of absorption (dissolution of a gas into a liquid solvent), adsorption (binding
of a gas on the surface of (porous) solid called adsorbent) and membrane permeation
(flow of a gas through a dense material, relying on the solubility and diffusivity).13
These methods, in their simple form, are pressure-driven, and when the feed gas stream
is available at sufficient pressure (and at low temperature), all of them could be
competitive and deserves consideration (Table 1.1). Conversely, if the feed gas is
available at low pressure; the driving force for separation must be provided by the
method itself, usually by employing a material reactive to CO2; but such chemical-
binding of CO2 creates difficulty in the regeneration of capture-material for cyclic use,
resulting in an energy-intensive, and consequently, cost-intensive capture process.
Therefore, considering the large quantity, low pressure and relatively high temperature,
the separation of CO2 from flue gas stream is a challenging task and constitutes the
major bottleneck in deployment of CCS at power-plants (this is especially so when the
separated CO2 has no future economic use, such as application in Enhanced oil Recovery
(EOR) or Enhanced Coal Bed Methane (ECBM).

7
1.1.4 Absorption technologies for CO2 capture
Absorption is widely used in CO2 separation from many industrial gas streams.
Typical feed gas streams prior to CO2 separation are given in Table 1.1 (application of
reactive absorption in CO2 separation is reviewed in ref. 14). Solvents considered for
CO2 separation may be categorized as
Traditional organic physical solvents (methanol, glycol ether, sulfolane
etc. )
Traditional organic chemical solvents (aqueous solution of ammonia,
K2CO3, amine )
Novel physical solvents (imidazolium, ionic liquids )
Novel chemical solvents (amine-functionalized ionic liquids, aqueous
amino acid salt, amino acid ionic liquid etc. )
Flue Gas CO2 Capture (FGCC): Although many separation methods based on
the principle of absorption, adsorption and membrane separation are vigorously pursued
at the R&D stage; the most advanced technology for FGCC is considered to be reactive
absorption and currently dominating in applications in the pilot-plant scale, full-scale
demonstration plants and commercial CCS projects (Table 1.2).3,15 The state-of-the art
for flue gas separation is considered to be reactive absorption by aqueous amine.16 But,
despite having many desirable characteristics and long industrial record, this energy-
intensive chemical absorption processes is estimated to increase the cost of electricity
between 70% and100% .17 This aspect has slowed down the integration of CO2-capture

8
units with existing power plants and calls for viable alternatives. Researchers worldwide
are critically examining the existing amine-based technologies as well as advanced
physical solvents to develop a process with reduced cost and environment footprint.
CO2 Capture from Syngas and Natural gas purification. For future power
plants, pre-combustion capture might be a feasible option, as it captures CO2 from a
high-pressure stream, as shown in Table 1.1 that facilitates adopting a less-energy
intensive method. Many planned full-scale demonstration units will be using physical
solvents (Table 1.3) such as Rectisol (proprietary solvent based on methanol) and
Selexol (proprietary solvent based on glycol ethers). Development of advanced physical
solvents better than the currently available ones will help them reduce cost and
environmental footprint, as in the case of FGCC.
Despite availability of a high pressure gas stream, natural gas industries mostly
employ chemical absorption due to the additional requirement of separating sulphur
compounds (such as the acid gas H2S) up to trace amount, for which some amines are
particularly suited. Generally, physical, chemical or hybrid solvents as well as membrane
separations are considered and sometimes applied in natural gas industries. 13

9
Table 1.2 Some Currently Operational Commercial CCS Plants. Data Taken from Refs.
11 and 15.
Project Name and Location
Plant and fuel Type
Year of Startup
Capture System (Vendor)
CO2 captured in Million tonnes/year (Storage site)
Benefit other than storage
Sleipner (Norway)
Natural gas separation
1996 Amine (Aker)
1(saline aquifers)
Avoid carbon tax
In Salah (Algeria)
Natural gas separation
2004 Amine (multiple)
1(saline aquifers)
Keep reservoir pressure high
Snohvit (Norway)
LNG plant 2008 Amine (Aker)
0.7(saline aquifers)
Avoid carbon tax
Weyburn-Midale (USA(capture)-Canada(storage))
Coal gasification plant
2000 Rectisol 3 (captured); 2(injected into oilfields), 1 vented.
EOR

10
Table 1.3 Some Planned Demonstration Plants Using Physical Absorption Process.
Data Taken from Ref. 15.
Project Name and Location
Plant and fuel Type
Year of Startup
Plant size CO2 capture System
CO2 captured in Million tonnes/year
Baard Energy Clean fuels (Ohio, USA)
Coal+biomass to liquid
2013 53000 barrels/day
Rectisol N/A
DKRW Energy (Medicine Bow, WY)
Coal to liquids
2014 20,000 barrels/day
Selexol
N/A
Summit Power (Penwell, Texas)
Coal IGCC and polygen (Urea)
2014 400 MW Rectisol 3
Dom Valley IGCC (UK)
Coal IGCC 2014 900MW Selexol 4.5

11
1.2 AMINE TECHNOLOGY
1.2.1 Amines
Amines are ammonia (NH3) derivatives. The workhorse amine is
monoethanolamine (NH2CH2CH2OH); the basic amino group (-NH2) is reactive to CO2,
the methylene groups(-CH2-) increases its boiling point relative to ammonia, and the
alcohol group (OH) promotes water solubility. Generally, the first, second and third
derivatives of ammonia generate the primary (NH2), secondary (-NH-) and tertiary
amino group which differ in basicity and reactivity. Generally, these three types of
functional groups build the library of amines often subjected to screening. Amines with
one, two or three amino groups are called mono, di, and triamine. Alkanolamines have
one or more alcohol groups. Sterically hindered amines usually have one or more methyl
group attached with the alpha-carbon (carbon attached with the amino group). Structures
of some amines are given in Figure 1.3. The final solvent is the aqueous solutions of an
amine (or blends of amines). For problems with degradation and corrosion, the solvent
strength is limited to 10-30 wt%. The reactive nature of amino site is also exploited
through a class of emerging solvents like amino acid salts and functionalized ionic
liquids. Amine solvents and their degradation product may have detrimental effect on
health and/or environment.3

12
Primary amine
Ethanolamine (MEA)
Secondary amine
Diethanolamine (DEA)
Tertiary amine
Methyldiethanolamine (MDEA)
Sterically hindered amine
2-amino-2-methyl-propanol (AMP)
Cyclic Diamine
Piperazine (AMP)
Figure 1.3 Structure of some amines.
NH2
CH2
CH2
OH
NH
CH2
CH2
OHCH2
CH2
OH
CH3
N
CH2
CH2
OHCH2
CH2
OH
NH2
C
CH2
OH
CH3
CH3
NH
NH
CH2
CH2CH2
CH2

13
1.2.2 Amine process
The basic process based on absorption was patented by Bottoms in 193012 and
still is in the heart of current amine based technologies. The capture unit has two primary
units: an absorber and stripper (Figure 1.4).
In the absorber, aqueous MEA solution (CO2-lean solvent) is brought into contact
with feed gas stream where CO2 is captured by one or more chemical reactions forming
carbamate and/or bicarbonate product (CO2-rich solvent). The principal reactions
occurring in aqueous amine solutions are usually represented by the following reversible
reactions.18
Ionization of water:
(1.1)
Hydration of carbon dioxide
2 (1.2)
Dissociation of bicarbonate ion
(1.3)
Dissociation of conjugate acid of amine (protonated amine)
(1.4)
Carbamate reversion to bicarbonate (for primary and secondary amines)
(1.5)

14
Figure 1.4 Typical amine-process for CO2 capture.

15
In the stripper, the rich solvent is heated with steam (generated in the re-boiler) to
break down the carbamate/bicarbonate to release CO2. The CO2-lean solution from the
stripper is re-circulated to the absorber and the released high-purity CO2 is compressed
to 100 to 150 bar for geologic sequestration. The reboiler heat duty ( ) required
to release CO2 is an important aspect in determining the cost of amine process. It has
three components, heat of desorpton ); the sensible heat ( required
to rise the temperature of the rich solvent to release CO2 and stripping energy ( .19
(1.6)

16
1.2.3 Effect of chemical reactions on solvent characteristics and cost
Chemical reactions are at the heart of amine-based capture processes and
influence the thermodynamic and kinetic characteristics of the solvent and thus the
process cost. We review the basic theoretical principles that link solvent characteristics
with process economics.
Equilibria and speciation. The maximum quantity (solubility) of a gas
transferred from the vapor phase to a liquid phase in contact, at a certain temperature and
pressure, is determined by the condition of thermodynamic equilibrium (equality of
fugacity or chemical potential of all components in both phases).20 With increase in
pressure, and generally with decrease in temperature, solubility increases. At a certain
temperature and pressure, physical solubility of a certain gas in different solvents varies
due to the differences in the solute-solvent interactions. The thermodynamic equilibrium
is called physical or chemical depending on whether the solute stays in molecular form
only (physical) or participates into chemically reactions (chemical) (Figure 1.5).
The physical absorption of gases in liquids at low pressure are commonly
quantified in terms of the Henry’s law constant (HLC), usually obtained from
experimental vapor-liquid equilibrium (VLE) measurement or estimation using a
thermodynamic correlation. The HLC of a gas in a solvent at a certain temperature may
be experimentally obtained from the low pressure solubility data as the linear slope of p-
x graph where p is the partial pressure of the gas in the vapor phase and x is its mole
fraction in the liquid phase.

17
Vapor phase
NH3 CO2 H2O
NH3 CO2 H2O
Liquid phase
Figure 1.5 The dissolution of CO2 in aqueous ammonia is facilitated by the chemical
reaction. The vertical and horizontal equilibria represent physical and
chemical equilibria respectively (modified from ref. 21).

18
If the molecular solute reacts with solvent and converts into other chemical
species, HLC decreases and the definition of Henry’s law constant in (1.1) needs to the
modified to take care of the additional driving force for CO2 dissolution (by depletion)
through the corresponding equilibrium constants of all reactions involved (coupled) in
CO2 capture. The mole-fraction based equilibrium constant of the reactions
∏ (1.1) to (1.5) are defined as
(1.7)
where , , and , represent the molefraction-based activity coefficient, mole
fraction, and stoichiometirc coefficient of species present in a reaction. Activity
coefficeint of a species in a liquid mixture is a measure of deviation of its behavior in
that mixtrue from its beahavior in an ideal solution.19
The concentration of captured CO2 is commonly expressed as loading, defined as
mole of CO2 captured in all chemical forms such as carbamte, bicarbonate, carbonate etc.
per mole of amine. The role of chemial reactions on the determination of loading and
absorption capacity is demonstrated in Figure 1.6 with the two most common amines. At
CO2 partial pressure of 0.1 atm (i.e., typical flue gas conditions), the loading in the
primary amime MEA, is more than double than in the tertiary amine MDEA. But, in
terms of absorption capacity, defined as the differnce in rich and lean loading (loading at
typical absorber and stripper conditions respectively, indicated by solid arrow in Figure
1.6), crucial in determining process cost, MDEA has much greater absorption capacity

19
than MEA. The performance of AMP, a sterically hindered primary amine, is in between
that produces mostly bicarbonate and some amount of carbamate (not shown in Figure).
Figure 1.6 Comparison of loading (broken line) and absorption capacity (solid line) of
MEA and MDEA at low pressure. The nubers 1, 2, 3, and 4 represent inlet or
outlet as shown in Figure 1.4. Loading is higher in MEA, but MDEA has
higher absorption capacity (data taken form ref. 19).
0
0.1
0.2
0.3
0.4
0.5
0.6
0.001 0.01 0.1 1 10
Loa
din
g (m
ol/m
ol M
EA
)
Carbon dioxide partial pressure (atm)
40°C, 5M MEA
100°C, 5M MEA
1
2 3
4
0
0.1
0.2
0.3
0.4
0.5
0.6
0.001 0.01 0.1 1 10
Loa
din
g (m
ol/m
ol M
DE
A)
Carbon dioxide partial pressure (atm)
40°C, 5M MDEA
100°C, 5M MDEA
1
2 3
4

20
Absorption capacity or cyclic capacity influences many parameters of the capture
plant and thus influences both the captial cost (CAPEX) and operational cost (OPEX).
An increase in capacity, corresponds to reduction of solvent mass flow rate, that in turn
favorably reduces the following
(i) solvent cost,
(ii) the diameter of the stripper and absorber column
(iii) the heat exchanger size and pumps size and duty (CAPEX) and blower
size
(iv) the sensitive heat ( ) required to rise the temperature rich solvent
inside the stripper defined as19,
ΔΔ
Δ
(1.8)
=liquid heat capacity
=liquid density
= the solvent circulation rate
Δ =different between stripper overhead and reboiler temperature
= concentration of amine in the solvent
=CO2 caputre rate
Δ =difference between loading at the top and bottom of absorption
=loading
Heat of absorption. The total heat of absorption is calculated as the sum of the
individual contributions including the heat of dissolution (physical) of CO2.
Δ (1.9)

21
Reaction rate. Reaction rate, coupled with other factors determines the height of
absorption column. The rate of absorption in a packed absorption column may be
expressed as22,
,,
1 1 (1.10)
The chemical equilibrium influences the concentration of solute in liquid, and
consequently, the driving force for diffusion (the concentration differences, numerator in
equation 1.10). The enhancement factor which is determined by the rate(s) of
chemical reaction(s), signifies the enhancement in absorption rate in addition to physical
solubility ( Henry’s constant, ). The gas and liquid phase mass transfer coefficients,
, and are defined as the ratio of mass flux divided by driving force (difference in
pressure and concentration between bulk and gas-liquid interphase) through the
following equaiton,
(1.11)
where = flux of the solute (quantity of the component transferred per unit time, per
unit area from vapor to liquid phase); and are the partial pressure of solute in main
body of gas adn at interphase; ); and are the concentration of solute in main body of
liquid and at interphase.
The absorption performance (i.e., the height of column necessary to reduce the
concentration of CO2 in the gas phase from 10% to zero) of a number of aqueous amine
solutions of same concentration was tested and, the amines are ranked in the following

22
order: MEA > DEA > AMP > DIPA > MDEA which corresponds with the rate of
reaction of these amines.23
Cost: Ranyanal et al. (2011)24 has identified different capital cost (CAPEX) and
operational cost (OPEX) related to the thermodynamic, kinetic and degradation
characteristics of the solvent for CO2-captrue (90%) from flue gas stream at 1 bar and at
45°C (15% CO2, flow rate 78480 kmol/h) from a 630MW coal-fired power plant using
30wt% MEA that shows that 20%, 39%, 22%, 8%, and 21% of the cost was associated
with heat of reaction, capacity, stripping, kinetics and degradation. 24

23
1.2.4 Reaction mechanisms
A reaction mechanism provides important details of the absorption process at the
molecular level by elucidating the sequence of elementary steps of a complex reaction
and thus helps to know the source of activation barrier, intermediates involved, heat of
reactions and the deduction of a rate law. Unfortunately, the reaction mechanisms of
aqueous alkanolamine solutions have been debated for years. Xie et al.25 summarized
three competing mechanisms: the zwitterion mechanism,26,27 termolecular mechanism28
and carbamic acid mechanism29 in reaction of CO2/MEA/H2O system. Considering a
primary or secondary amine ( ), these mechansims may be represented as,
Reaction Mechanism I (zwitterion mechanism)
Reaction Mechanism II (Single step termolecular mechanism)
Reaction Mechanism III (Carbamic acid reaction pathway)
where B represents a base that could be another amine, water or hydroxyl group.
The most widely-used mechanisms are the zwitterion-mechanism and the termolecular
mechanisms, the key difference being the occurrence of the reaction in two steps or one

24
step and the presence/absence of the intermediate called the ‘zwitterion’. Recently,
McCann and coworkers29 have proposed a two-step mechanism where the intermediate
is ‘carbamic acid’ instead of zwitterion.
1.2.5 Basicity of amines
The basicity of amine is quantified by the negative logarithm of the acid
dissociation constant (the pKa) of its conjugate acid (equilibrium constant of equation
1.4). Because it is a fundamental property, it is believed to have impact on all aspects of
CO2/aqueous amine chemistry. A linear correlation between the logarithm of the rate
constant for the formation of zwitterion ( in Equation 1.12) and pKa was found for
primary and secondary alkanolamines. 22
17.6 7188/ (1.12)
The total heat of absorption of CO2 is dominated by equation 1.5. Porcheron et
al. (2011) proposed a screening procedure of amines based on pKa and the pKc (negative
logarithm of the equilibrium constant of carbamate formation reaction).30
For the newer amino-functionalized ionic liquids (FILs), their stability, enthalpy
of absorption, and absorption capacity were controlled by the pKa of ILs,31 and pKa was
also proposed as a criteria to differentiate functionalized ILs from conventional ionic
liquids (ILs).32

25
1.3 IONIC LIQUIDS: PHYSICAL SOLVENT TECHNOLOGY
Room temperature ionic liquid (RTIL) is the generic name of a broad category of
solvents with melting point less than 100°C.33 Ionic liquids are composed of a bulky
organic cation and an organic or inorganic anion. Examples of common cations and
anions are shown in Figure 1.7. The substituent on the cations (the “R” groups) is
typically alkyl chains, but can contain any of a variety of other functional groups as well
(e.g., fluoroalkyl, alkenyl, methoxy, ether, etc.). The asymmetry in shape and bulkiness
in size of the ions results in loosely coordinating packing in ionic liquids responsible for
their low melting point (in contrast to solid NaCl crystal). But the ion-ions interaction in
room-temperature ionic liquids is stronger than other common intermolecular forces
present in organic solvents (e.g., London forces, ion-dipole interaction) that makes it
difficult for them to evaporate, creating no concern for fugitive loss (air pollution) for
process industries. Another appealing property of ionic liquids is their tunability - their
properties can be adjusted by adjusting the constituent ions to suit them for a particular
application. Other advantages include (i) broad liquid window which provides a greater
temperature range to work and (ii) non-flammability, and (iii) supportability on a
membrane. Such versatility has made their prolific growth in a number of potential
engineering applications. 33 However, ionic liquids could be non-biodegradable and/or
toxic that must be assessed before commercial applicaitons.12

26
In a 2009 paper in Nature, researchers at the university of Notre Dame showed
that the solubility of CO2 is very high in certain ionic liquids. This spurred interest to
investigate the possibility of using them in CO2 capture possibly as a physical solvent. 34
Recently, ionic liquids functionalized with reactive amino group or ionic liquids mixed
with amines have been considered for flue gas CO2 capture. 35,36
Some common cation
Some common Anions
Figure 1.7 Common cations and anions in conventional ionic liquids.

27
1.4 SOLVENT DEVELOPMENT
While the chemical landscape of potential solvents is vast, it is often the case that
no single solvent perform better in all desirable characteristics and a trade-off has to be
made. One or more of the following challenges are encountered:
(i) limited cyclic loading capacity of the solvent
(ii) high energy expenditure for solvent regeneration (reduce CO2 from rich
solvent to make lean solvent )
(iii) solvent is corrosive and limits solvent strength (wt% in aqueous solution)
(iv) slow absorption rate due to slow kinetics
(v) solvent is volatile at process conditions, leading to solvent loss and
requiring make-up solvent
(vi) solvent does not have other acceptable physicochemical properties such
as viscosity and operational characteristics
(vii) Solvent degrades with time (oxidative or thermal degradation)
(viii) Solvents (or degradation products) are toxic and not bio-degradable
Therefore, solvent development is a formidable task. The traditional approach
for solvent development is screening experiments followed by thorough investigation on
selected few for comparison with benchmark solvent which is usually the
monoethanolamine (MEA) of similar strength. Many solvents showing promise in early
experiments based on one or more characteristics turn out to be unsuitable later in
thorough investigation. One recent example is the fate of 3-(methylamino)propylamine

28
(3-MAPA) that showed promise in early studies (e.g., Kim et al. (2008))37 which later
was found not suitable when other properties were also considered (Voice et al.
(2013))38. Another example is that of 2-((2-aminoethyl) amino) ethanol (AEEA), which
was initially found to have both higher CO2 absorption rate and higher cyclic capacity
than those of MEA by a screening experiment39, but was later found to have problem
with degradation (thermally degrade at high temperature)40. Solvent development
through experimental screening of large number of possible solvents thus requires
significant experimental efforts.
Quantum-mechanical computational tools can be valuable in understanding
solvent chemistry and to develop new solvents.41 One computes chemical results either
as an alternative or complementary to experimental method for the following reasons.42
Accessibility
Economy
Understanding
Computation can reduce the cost and time for experiment by providing reliable a
priori guidelines. Computation also allows studying solvents not yet synthesized. For
example, Zhang et al. (2008) studied more than 700 ionic liquids virtually prepared
computationally, and then finally did measurement on selected few.43 Chemical
reactions in aqueous amine solutions are difficult to study experimentally due to
coupling of multiple reactions; and their fast nature makes it difficult to get mechanistic
details, therefore justifying using theory and computation for such study.44
Computational studies are suitable for interpreting phenomena at the molecular level that

29
help build novel solvent. For example, the computational mechanistic study of reaction
of amine with CO2 of Mindrup and Schneider (2009)45 led to the development of a
functionalized ionic liquid35 with high capture capacity (1 mole CO2 per mole of IL).
Similarly, computational study46 of the interaction of CO2 with the amino group led to
the development of two new TSILs47 with high absorption capacity.
1.5 OBJECTIVES AND SCOPE
The objectives of this dissertation is to use molecular modeling tools to predict
pKa of aqueous amines, address the CO2-capture mechanism debate and screen ionic
liquids as alternatives to aqueous amines as well as to measure (experimentally) the
solubility of CO2 in promising ionic liquids and thermophysical properties of an
important mixed solvent.
Due to the possible influence of basicity of amines on all aspects of CO2/amine
chemistry, an efficient computational protocol was developed to predict pKa (Chapter 3)
of amines of interest in CO2-capture. The pKa of some amines at 25°C were
experimentally measured to test the method. This method was contrasted with a pencil-
and-paper method based on linear free energy relationship. Reaction mechanisms for the
formation of carbamate and bicarbonate were studied to provide further fundamental
insight into the capture process, solve the debate over carbamate formation mechanism
and to study the effect of basicity on the mechanism of various amines. This work was
divided into two parts: static calculations (0 K) and dynamic calculations (313 K),

30
presented in Chapters 4 and 5 respectively, using computational tools of different levels
of sophistication (Chapter 2).
Room temperature ionic liquids (RTILs) for applications in CO2 separation from
flue gas or natural gas streams are computationally screened from a large database using
Henry’s law constant as screening criteria. The ‘structure-activity’ relationships are
explored to see how the properties of ILs affect the solubility and selectivity (Chapter 6).
Solubility of CO2 in three ionic liquids selected based on the screening study was
measured (Chapter 7) at (283.15,298.15 and 323.15) K and at (0.5 to 2) MPa. The data
were correlated with Peng-Robinson equation of state. The experimentally derived
Henry’s law constant was compared with COSMO-RS prediction.
In Chapter 8, the thermo-physical properties (density, viscosity, and excess
enthalpy) of aqueous solution of promising ionic liquid, 1-butyl-3-methyl imidazolium
acetate, [bmim][Ac], were studied. Excess enthalpy of ([bmim][Ac] + water) system at
25°C was compared with some (amine + [bmim][Ac]) systems.

31
1.6 REFERENCES
1. Maginn, E. J. What to Do With CO2? J. Phys. Chem. Lett. 2010, 1, 3478.
2. Stern, M. C.; Simeon F.; Herzog, H.; Hatton, T. A. Post-Combustion Carbon
Dioxide Capture Using Electrochemically Mediated Amine Regeneration.
Energy Environ. Sci. 2013, 6, 2505.
3. Li, B.; Duan Y.; Luebke D.; Morreale B. Advances in CO2 Capture Technology:
A patent review. Appl. Energy 2013, 102, 1439.
4. Scripps Institution of Oceanography (Scripps) and the National Oceanic and
Atmospheric Administration (NOAA)) (Available from:
http://CO2now.org/Current-CO2/CO2-Now/noaa-mauna-loa-CO2-data.html
Accessed June 23, 2013).
5. National Oceanic and Atmospheric Administration (NOAA) (Available from:
http://www.ncdc.noaa.gov/cag/time-series/global, (Accessed June 23, 2013).
6. Metz, B.; Davidson, O; de Coninck, H. C.; Loos; M.; Meyer, L. A. editors. IPCC
Special Report on Carbon Dioxide Capture and Storage. Prepared by Working
Group III of the Intergovernmental Panel on Climate Change. New York:
Cambridge University Press; 2005.
7. Dooley, J. J.; Davidson, C. L.; Dahowski, R. T. An Assessment of the
Commercial Availability of Carbon Dioxide Capture and Storage Technologies
as of June 2009. Pacific Northwest National Laboratory, U.S.DOE. 2009.

32
8. Hedin, N.; Andersson, L.; Bergström, L.; Yan, J. Adsorbents for the Post-
Combustion Capture of CO2 Using Rapid Temperature Swing or Vacuum Swing
Adsorption. Appl. Energy 2013, 104, 418.
9. Habib, M. A.; Nemitallah, M.; Ben-Mansour, R.. Recent Development in Oxy-
Combustion Technology and its Applications to Gas Turbine Combustors and
ITM Reactors. Energy Fuels 2013, 27, 2.
10. van Bergen, F.; Gale, J.; Damen, K. J.; Wildenborg, A. F. B. Worldwide
Selection of Early Opportunities for CO2-EOR and CO2-ECBM. Energy 2004,
29 , 1611.
11. Zakkour, P.; Cook, G. CCS Roadmap for Industry: High-purity CO2 Sources.
Carbon Counts Company (UK) Ltd., September, 2010.
12. Ramdin, M.; de Loos, T. W.; Vlugt, T. J. H. State-of-the-Art of CO2 Capture
With Ionic Liquids. Ind. Eng. Chem. Res. 2012, 51, 8149.
13. Kohl, A. L.; Nielsen, R. B. Gas Purification. (5th ed.). Gulf Professional
Publishing: Houston, 1997.
14. Yildirim, Ö.; Kiss, A. A.; Hüser, N.; Leßmann, K.; Kenig, E. Y. Reactive
Absorption in Chemical Process Industry: A Review on Current Activities.
Chem. Eng. J. 2012, 213, 371.
15. Rubin, E. S.; Mantripragada, H.; Marks, A.; Versteeg, P.; Kitchin, J. The Outlook
for Improved Carbon Capture Technology. Prog. Energy Combust. Sci. 2012,
38, 630.
16. Rochelle, G. T. Amine Scrubbing for CO2 Capture. Science 2009, 325, 1652.

33
17. Toth, F.L. Geological Disposal of Carbon Dioxide and Radioac-tive Waste: A
Comparative Assessment. Springer Verlag, Netherlands, 2011.
18. Kim, I.; Hoff, K. A.; Hessen, E. T.; Haug-Warberg T.; Svendsen, H. F. Enthalpy
of Absorption of CO2 With Alkanolamine Solutions Predicted From Reaction
Equilibrium constants. Chem. Eng. Sci. 2009, 64 , 2027.
19. Meldon, J. H. Amine Screening for Flue Gas CO2 Capture at Coal-Fired Power
Plants: Should the Heat of Desorption be High, Low or in Between? Curr. Opin.
Chem. Eng., 2011, 1, 55.
20. Prausnitz, J. M.; Lichtenthaler, R. M.; de Azevedo, E. G. Molecular
Thermodynamics of Fluid Phase Equilibria (3rd ed.). Prentice-Hall, Englewood
Cliffs, NJ. 1998.
21. Göppert, M.; Maurer G. Vapor-Liquid Equilibria in Aqueous Solutions of
Ammonia and Carbon Dioxide at Temperatures Between 333 and 393 K and
Pressures up to 7 MPa. Fluid Phase Equilib., 1988, 41, 153.
22. Versteeg, G. F.; van Dijck, L. A. J.; van Swaaij, W. P. M. On the Kinetics
between CO2 and Alkanolamines Both in Aqueous and Non-aqueous Solutions.
An Overview. Chem. Eng. Commun. 1996, 144, 113.
23. Aroonwilas, A.; Veawab, A.; Tontiwachwuthikul, P. Characterization and
Comparison of the CO2 Absorption Performance Into Single and Blended
Alkanolamines in a Packed Column. Ind. Eng. Chem. Res. 2004, 43, 2228.

34
24. Raynal, L.; Bouillon, P. A.; Gomez, A.; Broutin, P. From MEA to Demixing
Solvents and Future Steps, a Roadmap for Lowering the Cost of Post-
Combustion Carbon Capture. Chem. Eng. J. 2011, 171, 742.
25. Xie, H.; Zhou, Y.; Zhang, Y.; Johnson, J. K. Reaction Mechanism of Mono-
ethanolamine With CO2 in Aqueous Solution from Molecular Modeling. J. Phys.
Chem. A 2010, 114, 11844
26. Caplow, M. Kinetics of Carbamate Formation and Breakdown. J. Am. Chem.
Soc. 1968, 90, 6795.
27. Danckwerts, P. V.The Reaction of CO2 With Ethanolamines. Chem. Eng. Sci.
1979, 34, 443.
28. Crooks, J. E.; Donnellan, J. P. Kinetics and Mechanism of the Reaction between
Carbon Dioxide and Amines in Aqueous Solution. J. Chem. Soc., Perkin Trans.
2, 1989, 331.
29. McCann, N.; Phan, D.; Wang, X.; Conway, W.; Burns, R.; Attalla, M.; Puxty, G.;
Maeder, M. Kinetics and Mechanism of Carbamate Formation from CO2(aq),
Carbonate Species, and Monoethanolamine in Aqueous Solution. J. Phys. Chem.
A 2009, 113, 5022.
30. Porcheron, F.; Gibert, A.; Mougin, F.; Wender, A. High Throughput Screening of
CO2 Solubility in Aqueous Monoamines Solutions. Environ. Sci. Technol. 2011,
45, 2486.

35
31. Wang, C.; Luo, X.; Luo, H.; Jiang, D.-E.; Li, H. and Dai, S. Tuning the Basicity
of Ionic Liquids for Equimolar CO2 Capture. Angew. Chem. Int. Ed., 2011, 50,
4918.
32. Ren, S. H.; Hou, Y. C.; Tian, S. D.; Chen, X. M.; Wu, W. Z.What are Functional
Ionic Liquids for the Absorption of Acidic Gases? J. Phys. Chem. B 2013, 117,
2482.
33. Brennecke, J. F. and Maginn, E. J. Ionic liquids: Innovative Fluids for Chemical
Processing. AIChE J., 2001, 47: 2384.
34. Blanchard, L. A., Hancu, D., Beckman, E..J., Brennecke, J. F., Green Processing
Using Ionic Liquids and CO2. Nature 1999, 399, 28.
35. Gurkan, B. E.; de la Fuente, J. C.; Mindrup, E. M.; Ficke, L. E.; Goodrich, B. F.;
Price, E. A.; Schneider, W. F.; Brennecke, J. F. Equimolar CO2 Absorption by
Amine-Functionalized Ionic Liquids. J. Am. Chem. Soc. 2010, 132, 2116
36. Camper, D.; Bara, J. E.; Gin, D. L.; Noble, R. D. Room-Temperature Ionic
Liquid Amine Solutions Tunable Solvents for Efficient and Reversible Capture of
CO2. Ind. Eng. Chem. Res. 2008, 47, 8496.
37. Kim, I., Svendsen, H. F., Børresen, E., Ebulliometric Determination of Vapor–
Liquid Equilibria for Pure Water, Monoethanolamine, N-Methyldiethanolamine,
3-(Methylamino)-Propylamine, and their Binary and Ternary Solutions. J. Chem.
Eng. Data 2008, 53 , 2521.

36
38. Voice A. K.; Vevelstad S. J.; Chen X.; Nguyen T., Rochelle G. T.; Aqueous 3-
(methylamino)propylamine for CO2 Capture. Int. J. Greenhouse Gas Control
2013 15, 70.
39. Ma'mun, S.; Jakobsen, J. P.; Svendsen, H. F.; Juliussen, O. Experimental and
Modeling Study of the Solubility of Carbon Dioxide in Aqueous 30 Mass % 2-
((2-Aminoethyl)amino)ethanol Solution. Ind. Eng. Chem. Res. 2006, 45, 2505.
40. Davis, J.; Thermal Degradation of Aqueous Amines Used for Carbon Dioxide
Capture. Ph.D. Dissertation. The University of Texas, Austin, USA, 2009.
41. Maiti, A.; Bourcier W. L.; Aines, R. D. Atomistic Modeling of CO2 Capture in
Primary and Tertiary Amines – Heat of Absorption and Density Changes.
Chem.Phy. Lett. 2011, 509, 25.
42. Almlöf, J. Electronic Structure Theory. In Mathematical Frontiers in
Computational Chemical Physics, Truhlar D.G. Ed; Springer-Verlag, 1988; Vol.
15; p 18.
43. Zhang, X.; Liu, Z.; Wang, W. Screening of Ionic Liquids to Capture CO2 by
COSMO-RS and Experiments. AIChE J. 2008, 54, 2717.
44. Gallet, G. A.; Pietrucci, F.; Andreoni, W. Bridging Static and Dynamical
Descriptions of Chemical Reactions: an Ab Initio Study of CO2 Interacting With
Water Molecules. J. Chem. Theory Comput. 2012, 8, 4029.
45. Mindrup, E. M.; Schneider, W. F. Computational Comparison of Tethering
Strategies for Amine Functionalized Ionic Liquids. In ACS Symposium Series;
Seddon, K.; Rogers, R.; Plechkova, N., Eds.; American Chemical
Society: Washington, D.C., 2009, p 419.

37
46. Yu, G.; Zhang, S.; Yao, X.; Zhang, J.; Dong, K.; Dai, W.; Mori, R. Design of
Task-Specific Ionic Liquids for Capturing CO2: A Molecular Orbital Study. Ind.
Eng. Chem. Res. 2006, 45, 2875.
47. Zhang, J.; Zhang, S.; Dong, K.; Zhang, Y.; Shen, Y. ; Lv, X. Supported
Absorption of CO2 by Tetrabutylphosphonium Amino Acid Ionic Liquids.
Chem. -Eur. J. 2006, 12, 4021.

38
Chapter 2: Computational Methods _______________________________________________________________________
2.1 INTRODUCTION
Modeling bulk properties of a liquid phase has always been a challenge as they
arise from statistical behavior of a vast number of molecules, whose behavior, in turn,
stem from their electronic structure. The quest for the foundation of macroscopic
behavior of fluids in the microscopic realm of molecular properties (e.g., van der Waals
equation of state (1873)) 1 started at a time when even the existence of molecules was not
widely recognized. Today, more than a century later, such quest has turned into the
industrial practice of designing products from electronic level of matter.
Thermodynamic models have much evolved since van der Waals.
Thermodynamic models of liquid for industrial design and simulation (e.g., UNIFAC2 in
1975, SAFT3 in 1989) were developed by exploiting the link between macroscopic and
microscopic properties through classical statistical mechanics. More recently,
thermodynamic models with direct root at the electronic structure calculations appeared (
e.g., COSMO-RS (1995))4 and their applications to various chemical engineering
problems was facilitated by the engineers’ access to quantum-mechanical computational
tools (the application of quantum mechanics as a ‘new tool for engineering
thermodynamics’ was reviewed by Sandler (2003)5). The introduction of quantum

39
mechanics is necessary to treat the electronic structure of matter (because no classical
analog is present for it); and also for the ab initio (from the first principle) treatment of
atoms and molecules. John Pople and Walter Kohn received Nobel prize for their
contribution in making quantum-mechanical calculations possible for practical chemical
problems in 1998 (about 90 years later van der Waals received Nobel prize in 1910). At
present, with advancement in computer technologies, many powerful computational
tools have been developed that generally belong to the area of molecular modeling (also
known as computational chemistry).
2.2 MOLECULAR MODELING
“Molecular modeling is nowadays a well-established analytical tool exactly as
spectroscopies or other experimental methodologies, and we expect that its impact on
many research fields in chemistry, biology, material science, and even medicine will
enormously increase in the near future”.6 Molecular modeling provides a set of
techniques based on the principles of classical and quantum physics to compute chemical
and thermodynamic properties such as geometry, reactivity, reaction mechanisms,
spectra etc of a molecule or a system of molecules.7 The major tools of computational
chemistry (also termed as ‘molecular modeling’) include
Molecular mechanics methods,
Ab initio methods,
Semiempirical methods,
Density functional theory (DFT) methods and

40
Molecular dynamics methods.7
Selection of one or more methods depends on the nature of problem and the
chemical information sought. These methods are being increasingly applied by
researchers in the area of CO2 capture. We have used Ab initio and DFT based methods
for static calculations as well as molecular dynamics methods for dynamic calculations
in this dissertation to solve different aspects relevant to solvent development. We briefly
provide the overview of some key elements of quantum chemistry (The Schrodinger
equation, potential energy surface), basic computational methods and models frequently
used in molecular modeling below.
2.2.1 Schrodinger equation (SE)
Quantum-mechanics describes the state of a molecular system by a function that
depends on the coordinates and spin of the particle and time. This function is called the
wave function and contains all the information about the system.
Ψ Ψ coordinates and spin of all particles, time
The wave function is obtained by solving the Schrodinger equation (SE). In molecular
modeling, the time-independent, non-relativistic treatment of Schrodinger equation
(Equation 2.1)8 is normally sufficient.
Ψ , Ψ , (2.1)
Where H is the Hamiltonian operator, is energy and Ψ is the wave function. R and r
represent the set of spatial coordinates of the nuclei and electrons respectively. The spin-
dependence of wave function is introduced in an ad hoc manner in non-relativistic

41
treatment of Schrodinger Equation. Equation (2.1) is an eigen value equation as it has
the form where an operator acts on the wave function to produce a constant times the
wave function. Any physically observable quantity can be calculated from the wave
function with a corresponding eigen value equation using the corresponding quantum-
mechanical operator.
The Hamiltonian operator is formulated from the classical expression for kinetic
and potential energy. For a molecule in vacuum, the Hamiltonian ( ) consists of five
other operators associated with the kinetic ( ) and potential energy ( ) of the particles.
(2.2)
The subscripts ‘n’ and ‘e’ in equation (2.2) denotes nucleus and electron respectively;
and the five operators on the RHS of equation (2.2) are associated with kinetic energy of
nuclei ( ), kinetic energy of electrons ( ), electron-nuclei attraction ( ), neucleus-
neucleus repulsion ( ) and electron-electron repulsion ( ). The Hamiltonian is
augmented by additional terms if the system is in any external potential field (for
example to mimic the presence of solvents around a solute) or if other quantum-
mechanical or relativistic effects are necessary.
Since electrons are much lighter than the nuclei, the nuclear velocities are much
smaller than those of electrons. Thus, electrons adjust to nuclear motion instantly and
essentially see the nuclei as stationary. This enables to decouple the motions of electrons
from that of nuclei and thus Schrodinger equations can be applied for electrons and
nuclei separately. The electronic Schrodinger equation is written for fixed or clamed
nuclei as8

42
; Ψ ; Ψ ; (2.3)
where ; (later denoted as ) is the electronic energy
(with reference state at infinitely separated electrons and nuclei and at rest) of the system
and depends functionally on the spatial coordinates of the electrons, but only
parametrically on the nuclear coordinates. This approximation is known as Born-
Oppenheimer approximation.
2.2.2 Potential energy surface (PES)
Definition. Equation 2.3 is the cornerstone of computational chemistry and it
represent the energy-geometry relationship of a molecule or a group of molecules (The
BO approximation implies that a molecule has a shape or geometry defined by the
positions of the coordinates of the nuclei; electronic coordinates are not necessary to
define geometry as since the electron will adjust their motion instantly with a given
nuclear position). By plotting the energy (solutions of equation 2.3 for many given
nuclear configurations) as a function of nuclear coordinates (R), one obtains the
potential energy surface of the system. It may be mentioned that for each nuclear
configuration, many wave functions, each associated with an associated energy, will
satisfy equation 2.3, and among them, the one associated with lowest energy, called
ground-state energy are used in plotting the ground-state PES.

43
Dimensionality. For N atoms, 3N dimensions are required to define the
geometry; and leaving out the 6 degree of motion for overall (center-of-mass)
translational and rotational motion, there are 3N-6 degrees left for relative internal
movement of the nuclei. Therefore, the PES of a N-atom molecule with stationary or
frozen nuclei requires 3N-6 (or 3N-5 for linear molecules) nuclear coordinates. Different
possible geometries of a molecule can also be expressed in terms of internal coordinates
such as distance between two nuclei, angle among three nuclei, and dihedral angel
among four nuclei instead of the Cartesian coordinates of each nucleus.9 Mathematically,
the PES is thus the following multidimensional function,
, , … , (2.4)
Where, the , , . . are internal coordinates.
Stationary points. Due to large dimensionality of the complete PES for practical
systems, it is difficult to obtain information of full PES, rather one focuses on the
chemically relevant part of it (such as minima, maxima, saddle point).10 For example,
information of a bond breaking or forming during chemical reaction is obtained by
observing energetic variation along the coordinates directly involved. A minimum on
PES corresponds to a chemically stable configuration and the lowest energy path (also
called intrinsic reaction coordinate, IRC) connecting two minimum through a maxima
corresponds to an elementary reaction step (Figure 2.1); the minima could be
reactants/products/intermediate and the maximum is the transition state (TS) structure.
An ‘intermediate’ is a short-lived compound that may be isolated; whereas a TS has only
momentary existence. The value of the reaction coordinate (e.g., a bond length) in TS is
between the values of the coordinate in the two connecting minima; and such proximity

44
defines an early vs. late TS). The energy difference between a reactant and the transition
state of an elementary step is the activation energy of the step and the reactant will pass
over the maxima if it acquires kinetics energy higher than the activation energy.
Stationary points on a PES are identified with the following criteria.7
At a stationary point, 0 for all geometric coordinates (i.e. 0)
At a minimum, 0 for all geometric coordinates (along all directions) and
0 for all (along all directions)
For a transition state, 0 for all geometric coordinates (along all directions); and
0 for =the reaction coordinate, but 0 for all other (along all other
directions).

45
Figure 2.1 A one-dimensional projection of a potential energy surface showing the IRC
for a reaction that connects two minima.

46
Zero point energy. Calculations to locate minima and transition states on a PES
that requires the computations of first and second derivatives of energy with respect to
all the internal coordinates as discussed above and such calculations are called geometry
optimization. An initial geometry is given and the program moves the geometry towards
a stationary point using different numerical algorithms (e.g., Berny algorithm). Once a
minima or transition state is found, an additional frequency calculation is carried out to
get additional insight into the nature of that geometry (for example, to identify the
particular reaction coordinate involved in IRC). Such calculations find the normal-mode
frequency of all the modes of vibration (atoms vibrate even at 0 K and the associate
energy is called zero-point energy; the frozen-nuclei coordinates of the atoms on a PES
corresponds to the equilibrium position of such vibration). In a minima, all the bonds
vibrate periodically, a restoring force acts on the nuclei whenever it is out of equilibrium
position to bring it back to equilibrium; but in a transition state, one of the vibration will
occur without the restoring force, and therefore, it many fall to either direction towards
the reactants or products along the IRC.

47
2.2.3 Solving the electronic schrodinger equation
As mentioned earlier, geometry optimization (finding minima, TS on a PES)
requires the computation of electronic energy of the system for clamped nuclei by
solving equation (2.2). Methods for solving this equation are based on two types of
theory: the wave function theory (WFT) and the density functional theory (DFT). WFT-
based methods are called ab initio (from beginning) or first principle methods because
they use only the fundamental constants and do not use any experimental data directly.
In DFT-methods, ground-state energy is computed from ground-state electron density,
utilizing some notions of WFT to facilitate computation of the ground-state density.
Selection of a method depends on the trade-off between the two desired levels of cost
and accuracy.
Ab initio methods. All ab initio methods require the representation of the
complex N-electronic wave function (Ψ of equation 2.3) that depends on 4N variables
(three spatial coordinates and spin of each of the N electrons) by a number of simpler 1-
electron wave functions, known as spin-orbitals, that depends on 4 variables. This step,
that determines the mathematical relationship between the electronic wave function and
spin-orbitals (sprout) engenders many ab initio methods (discussed later).
Each spin-orbital is product of two functions, the molecular orbital (depends on the 3
coordinates of the electron) and the spin function (depends on the spin of the same
electron). Each of molecular orbital is further by linear combinations of some predefined
basis functions, representing the atomic orbitals of the constituent atoms.

48
; (2.5)
where, is the -th the basis function in the -th molecular orbital ( ); is the
corresponding coefficient. Thus, in effect, the problem of determining the wave function
turns into finding these coefficients (an easier numerical problem). The set of the basis
function is called a basis set (discussed later). The nature and number of basis functions
must be selected judiciously commensurate with a method and nature of chemical
interaction present. The method (also called ‘level of theory’) and basis-set chosen for a
problem together are called ‘model chemistry’.
Once an approximate mathematical form of the wave function is determined, the energy
is obtained from the newly constructed wave function by the postulates of quantum
mechanics (equation 2.6).
Ψ | |ΨΨ |Ψ
(2.6)
Equation (2.8) is written in the bra-ket notation used to represent multi-dimensional
integrals. Explicitly,
Ψ| |Ψ Ψ Ψ d (2.7)
Ψ |Ψ Ψ Ψ d (2.8)
where the integral sign represents multiple integration over all spatial variables and
summation over all spin variables. Ψ represents the complex conjugate of the function
Ψ (e.g., Ψ x iy if Ψ x iy , etc).
Finally, the variational principle, that any approximate wave function will always give
ground state energy higher than the true ground-state energy, is invoked and wave

49
function that corresponds to lowest possible energy is sought. An objective function by
means of Lagrange multipliers is constructed that additionally ensures that molecular
wave function be orthonormal (a property required of wave functions).
,
(2.9)
The objective function can also be formulated into basis functions using equation
(2.7) and then minimized. Minimization of equation (2.11) ( 0 ) leads to the a set of
equations that are solved iteratively and the solutions provide the molecular orbitals ,
orbital energies (the constant in equation 2.11 is generally interpreted as the energy
of orbital , designated as ) and the total electronic energy.
The basic scheme described above is common to all ab initio methods. They
differ in their construction of the approximate form of Ψ as a composite function of
spin-orbitals, (discussed before). Such construction has implication on the level of
rigor of the treatment of the quantum-mechanical nature of electronic motion in presence
of other electrons; rigorous treatment requires deployment of more molecular orbitals (to
represent many possible electronic configuration), but this increases the computational
cost.
The basic ab initio method is the Hartree-Fock (HF) method that satisfies some
crucial aspects of quantum-mechanical nature of electron motion (but not all) and the
starting point for more accurate (post-HF method) or less accurate (semiempirical)
methods. For a closed shell system (no unpaired electrons), the HF method approximates
the wave function with a single determinant called Slater determinant.

50
Ψ ~ΦSD (2.10)
ΦSD √N!
1 12 2
… 1… 2
…
; ;1,0, (2.11)
The Slater determinant is constructed from N spin-orbitals for a N-electron
closed-shell system, which is the minimum number of orbitals for its ground state
electronic configuration. Mathematical properties of Slater determinant automatically
accounts for the quantum-mechanical Pauli Exclusion Principle. Two-electrons may
occupy the same space only if they have opposite spin. Thus, the N spin-orbitals in the
Slater determinant are constructed from N/2 spatial orbital, each of them occupy two
electrons with different spin. If m basis functions are used, a total of m spatial molecular
orbitals are constructed by linear combination with different coefficients (a total of
coefficients arising from a mxm matrix). Out of the m molecular orbitals, only N/2
orbitals are occupied (filling in orbitals in ascending order of their energies), and the
remaining orbitals are called unoccupied or virtual orbitals. The energy difference
between the highest occupied molecular orbital (HOMO) and lowest unoccupied
molecular orbitals (LUMO), are important in the study of chemical reaction (HOMO and
LUMO, altogether are called frontier orbitals).
In Hartree Fock method, the electron-electron repulsion is treated in an average
manner: the ‘electron-electron’ repulsion is replaced with ‘electron-average field’
repulsion where the average electric field is generated by all other electrons and the bare
nuclei (instead of one to one electron-electron interaction). But the motion of an electron
at any instant is dynamically correlated with the motion of every other electrons. This

51
‘correlation energy’ (sometimes defined as the difference between the lowest possible
energy obtainable and the HF energy obtained by a given basis set) is treated in a better
way by Post-Hartree Fock methods. One such method is ‘MP2’ based on Moller-Plesset
(MP) perturbation theory where the HF energy is corrected to first order by adding the
‘correlation energy’ as a first order correction. Ψ is expanded into many wave functions
around the wavefunction of a reference system that can be treated very accurately.
Ψ Ψ Ψ .. (2.12)
The energy of the reference system is designated as MP0 and may be computed
very accurately, then corrections of increasing order are added to it (the corrected energy
is designated as MP1, MP2, MP3, MP4 etc). MP1 energy is the Hartree-Fock energy and
MP2 energy is the first correction beyond HF that is computed by promoting electrons
from occupied to unoccupied orbitals, in some sense, giving them more room to avoid
one another, thus recovering some part of the dynamic correlation.7

52
DFT methods. In Density functional theory (DFT) calculations, a functional
transforms ground-state electron density into the ground-state energy . A
functional converts a function into a number, like the definite integrals do (for example,
if the energy functional is defined as, ; then, 0.5 where
is the density function, the argument of the energy functional, is shown within square
brackets). The electron density function, , , , provides number of electrons per unit
volume. The number of electrons in a small volume element centered at a point
, , is , , , , , and when integrated over all space, gives
the total number of electrons of a system.
The ground-state energy is decomposed into three contributions, electronic
kinetic energy (T), nucleus-electron potential energy (Vne) and electron-electron
repulsion energy potential energy; all expressed as functional of density.
(2.13)
A reference system is imagined that has the same ground-state electron density
and ground-state energy of the actual system but consists of non-interacting electrons.
Due to the absence of interelectronic repulsion ( ), the reference
system can be exactly decomposed into molecular spin-orbitals by a Slater determinant
and the density and kinetic energy ( ) of the reference system can be expressed in
terms of the molecular orbitals known as Kohn-Sham orbitals. The second term in
equation (2.13) can be expressed as integral, by considering interaction between an
infinitesimal portion of charge cloud located in a small volume and a nucleus, and then
integrating over the volume and summing over all the nuclei. The third term is also

53
approximated as classical coulomb interaction between two charge cloud within two
separate infinitesimal volume elements and then integrating over the volume elements.
This electron-electron energy and the kinetic energy of the reference system is not same
as the energy for true system due to electron correlation, and the deviation is corrected
by a term called exchange-correlation energy ( ) . These consideration lead to the
total energy of actual system,
(2.14)
The exchange-correlation energy, , is unknown and different DFT-methods
differ in the approximate expression used for this functional. Using a suitable expression
for , constraint minimization of the ground state energy (equation 2.14) lead to the
solution of spatial molecular orbitals known as Kohn-Sham (KS) orbitals. The electron
density for a N electron closed shell system is computed as,
1 (2.15)
The approximate functional used in practical calculations are developed by
appealing to theoretical models and contain parameters that are fitted to experimental
data. These functionals are written as a summation of two parts, the exchange part
(dominating) and a correlation part ( ). In hybrid DFT method, another
possible improvement is to use a weighted sum of HF exchange energy and the DFT
exchange-correlation energy. The exchange-correlation functional also prevents
analytical solution of the integrals. The combination of three-parameter exchange
functional of Becke (1988) (abbreviated as B3) with the correlation functional of Perdew

54
(abbreviated as P88) or with that proposed by Larr, Yang, Perdew (abbreviated as LYP)
gives rise two popular DFT methods, BP88 and B3LYP, respectively.
Basis set. Selection of a basis-set (the set of basis functions) implies deciding on
(i) The type (minimal, split-valence, etc., discussed below) and number of basis
functions.
; , (2.7)
(ii) How to construct a basis function from linear combination of other known
functions. These known functions are, most instances, the primitive Gaussian Type
Orbitals (PGTOs). The constructed basis function is called a contracted GTO (cGTO).
; (2.16)
The PGTO are known function and has the following form.
, , , , , (2.17)
Where, indicates the type of orbitals ( 1 is a p-orbital etc) and zeta
represent the extent of the orbital (for example, a small zeta indicates electrons is held far
away from nucleus). is a normalized constant and is distance of the electron from a
nuclei.
In a minimal basis-set, one basis function for each atomic orbitals contained in
the core and valence shell of an atom are used. For example, one basis function is used
H atom (its electronic configuration require only the first shell, n=1) and five basis

55
functions are used for carbon, nitrogen or oxygen atom (their electronic configuration
require two shells, n=1 and n=2; and one basis function is allotted for each orbital in
these shell).
In a Split-valence basis set, one basis function for each of the core orbitals
(orbitals in the inner shells shell in an atom), more than one (two, three, four etc) basis
functions for each valence orbitals (orbitals in the outermost shell from nucleus) are
used. Thus, double-zeta, triple-zeta, quadruple-zeta respectively refer to the use of two,
three and four basis functions for representing the valence orbitals. For example, a split-
valence double zeta basis set uses two basis functions for hydrogen (for the 1s valence
orbital), and nine functions for carbon, nitrogen, and oxygen atoms (two functions for
each of the four orbitals in the second shell plus one for the core 1s orbital).
Split-valence basis set developed by Pople and coworkers are usually expressed
by acronyms such as ‘k-nlG’ (e.g., 6-31G). The numeral ‘k’ indicates the number of
PGTOs used to represent the core orbitals. The two numerals after the dash indicate a
double split valence orbitals respectively where ‘n’ and ‘l’ PGTOs will be used to
represent the first and second set of splited valence orbitals. PGTOs in these two sets
have different values of zeta and thus their linear combination allows for the possibility
for orbitals to be of different size. Similarly, ‘k-nlmG’ (e.g., 6-311G) represents split-
valence triple zeta basis set and can provide further flexibility in orbital sizes.
An orbital can be represented as a mixed orbital by adding other type of orbitals
to it. Adding polarization function to split-valence double zeta basis set generates the
‘polarized double-zeta’ basis set. Polarization functions add additional flexibility to an
orbital by permitting its electrons to be displaced in a certain direction necessary for an

56
interaction (for example when two atoms approach). They are denoted by a single
asterisk (*) if polarization function is added only to heavy atoms (atoms except H and
He) and double asterisk (**) is used to indicate polarization of all atoms. Instead of
asterisk the added orbitals is also used in the acronym. For example, 6-31G(d) or 6-31G*
indicate the same basis-set (add 6 d-orbitals to each of the valence orbitals of heavy
atom). To generate significant electron density far away from nucleus for example
necessary for an anion, diffuse functions can be added. They are expressed as (+ for
‘heavy atom’ or ++ for all atom) before the letter G in a split-valence basis set.
The basis-sets mentioned above are optimized at Hartree-Fock level.
‘Correlation-consistent split-valence’ basis sets were developed by Dunning and
coworkers by optimizing those using correlated wave functions for applications in post-
Hartree Fock methods. They are abbreviated as cc-pVXZ stands for correlated-
consistent polarized valence X zeta basis set with X=D, T,Q etc for double, triple,
quadruple split valence basis set.
The basis set mentioned above are atom-centered, and they originate with
reference to the orbitals for the simplest one-electron system, the hydrogen atom, for
which equation (2.2) can be exactly solved and the corresponding one-electron wave
functions are analytically known.8 Instead of building MOs from AOs which in turn
from PGTOs, one can build MOs directly from plane waves, without reference to AOs.
These waves stem from the solution of SE for a free electron.
. (2.18)

57
is a wave vector that play the same role as zeta is a GTO and is related to energy
(momentum of the wave, and energy, 2⁄ where, is the mass of
an electron;). They greatly simplifies calculation (involves integral of sine and cosine),
but a lot many of them are needed for proper presentation of an orbital. They are useful
for periodic systems extended to infinity such as in molecular dynamics simulation.

58
2.2.4 Molecular models for bulk liquid
In the very beginning of a quantum-mechanical (QM) computation one has to
decide on the following two major steps which affect the quality of subsequent results.
Physical modeling of the bulk system
Selection of model chemistry (a computational method plus a basis set,
e.g., B3LYP/6-31G(d), as discussed earlier)
The first step, physical modeling of the system, is necessary to reduce number of
particles to make it amenable to computation, but at the same time, must retain its
essential chemistry as well to obtain meaningful results. Using rigor in model chemistry
is not worthy when the physical model is poor.
It is often necessary, and sometimes vital, to include the effect of molecular
environments in solution-phase computations; particularly for the study of chemical
reactions and thermochemistry (both of them are of interest to us). The solvent may
behave quite differently towards the reactants, products, intermediates, transition states
and thus follow a different reaction path in different solvents or in vacuum. Solvent-
effect can be modeled in different ways: implicitly, explicitly or in a hybrid manner.
Thus, the number of solvent molecules to be explicitly treated, their conformers, vacuum
vs. solvent phase), and the level of computation should be properly chosen.
Results obtained at QM level are converted to bulk thermodynamic properties
through the framework of statistical mechanics. We will discuss the continuum
solvation models and an excess Gibbs energy model (called ‘COSMO-RS’), that are
used extensively, in chemistry and chemical engineering respectively.

59
Continuum Solvation Models (CSM)6,11 The focus of continuum solvation
models is a dilute liquid phase. Continuum solvation models retain the full geometric
structure of the solute but treat solvents as a structureless homogeneous medium
characterized by the dielectric constant of the solvent (Figure 2.2).12 Due to absence of
explicit solvents (thus reduction of many coordinates), the computations are much faster
and efficient (relative to explicit treatment of solvents). Over many years continuum
models have been refined and today, they are used widely in chemistry and
biochemistry.
The solvation process is defined as a transfer of a molecule from the gas phase to
the liquid phase and the Gibbs free energy of solvation (Δ ) is the difference between
the Gibbs free energy of solute in the liquid (solvent) and gas phase. The computation is
Δ is facilitated by viewing the solvation process consisting of the three following steps.
a) Creation of a cavity of inside the solvent to place the solute molecule. The
cavity acts as a mathematical boundary between the solute and solvent (now the
dielectric medium) and its shape and size strongly affect the solvation energies. Cavities
are defined empirically and many cavity models have been developed. 13
One method is to place sphere of specific radius around each atom or atomic group of the
solute to generate a solute-shape cavity; the outward surface of the interlocking sphere
thus represent the molecular surface, often called the van der Waals molecular surface
(WS). The radii of the sphere to define WS are usually the scaled up van der Waal
atomic radii (commonly by 20%); and generally, the cavity volume is 70% higher than
the vdW volume (van der Waals used an empirical constant to present volume of a
molecule in his famous equation of state).

60
Figure 2.2 Continuum solvation model

61
The crevices on vdW surface, where two or more surfaces cut each other, may be
smoothened in various ways, e.g., by inserting another sphere or by rolling a rigid sphere
rolling over the WS; such smoothed molecular surface is called a solvent excluded
surface (SES). The solvent radius may be added to unscaled vdW atomic radii and the
resulting radii can be used to define molecule surface; such a surface is called a solvent
accessible surface (SAS). In Gaussian, many cavity models are available: cavity models
such as UFF, Pauling, Bondi uses sphere for each atom (radii specific to each model),
but united-atom (UA) models (such as UA0, UAHF, UAKS) put sphere on all atoms
except hydrogen (which is grouped with the next the atom).
(b) Insertion of the solute with its gas phase charge distribution (unpolarized
solute) inside the cavity.
(c) Allowing mutual (two-way) solute-solvent polarization. The solute charges
will induce accumulation of opposite charges at the cavity surface, known as apparent
surface charges (ASC) or screening charges , which are opposite in nature to the original
charges on the surface segment, e.g., the negative charges on oxygen atom will be
screened by the positive charges of the conductor. These ASC will polarize the solute
charges (that might also affect geometry of solute; and solute is thought to be in the
‘reaction field’ of the solvent). At the end of mutual polarization, the solute is described
as ‘fully polarized solute’ and represented by the solvated wave function (the geometry
is also slightly changed).14
The electrostatic contribution of Δ (the dominating part for solvation in polar
solvents like water) is computed using previously discussed ab initio or DFT methods
using an effective Hamiltonian (gas phase Hamiltonian and half of the potential energy

62
due to solute charge-screening charge electrostatic interaction). An iterative procedure is
used to represent the mutual polarization until self-consistency is achieved and such
computation is called self-consistent reaction field (SCRF) method. The non-
electrostatic parts (cavitation, repulsion, dispersion) can either be included during the
iteration process or added separately.
Two important ASC dielectric continuum models relevant to this dissertation are
IEF-PCM (Integral Equation Formalism for Polarisable Continuum Model)15-16 and
COSMO (Conductor-like Screening Model)17. In IEF-PCM the solvent has a finite
dielectric constant; but in COSMO, the dielectric constant of the medium is assumed to
be infinity to provide a simple boundary condition (the total potential, sum of potential
due to solute charges plus apparent charges is zero at the boundary) for polarization
calculation based on Poisson or Poisson-Boltzmann equation, which calculates electric
potential from a charge distribution.
Some local solute-solvent energetic interactions, such as hydrogen bonding,
cannot be taken care of by continuum models; and thus pose a challenge in their
application in systems characterized by extensive hydrogen bonding such as aqueous
alkanolamines. There is one contribution of Gibbs free energy that cannot be taken care
of by any continuum models, called solvent reorganization, since the structure of
solvents themselves are absent. In semi-continuum solvation, some individual solvent
molecules are placed suitably around the solute (typically one to ten), and this
supermolecule is placed in a continuum (Figure 2.3).18 The calculation steps are same as
before, and the algorithm views the cluster as a supermolecule. The semi-continuum

63
approach thus may partially remedy for some specific solute-solvent interaction, but is
computationally more expensive.
Other useful quantities used in fluid phase thermodynamics such as equilibrium
constant of a reaction can be derived from Gibbs free energy of solvation by constructing
thermodynamic cycles. A master equation to compute pKa from Gibbs free energy of
solvation is derived by Khalili and East.19
Figure 2.3 Semi-continuum solvation model.

64
COSMO-RS. COSMO-RS20-22 (Conductor-like Screening Model for Realistic
Solvents) is an excess Gibbs energy (difference in Gibbs energy between the actual and
an idealized mixture) model, for mixture using a statistical-thermodynamic framework
that imports molecular level information dissolved in a conductor (screening charges,
energy, cavity surface and volume) obtained from COSMO model. The energy
computed by COSMO model serves as a new reference state for energy in
thermodynamic calculations where the conductor is replaced by the real solvents and
assumed that the solvents screen the solute perfectly, as the conductor does. In reality the
solvent molecules will not screen the solute perfectly and the deviation is accounted for
by modeling the attractive (electrostatic, H-bond, dispersive) interactions between
molecules as pair wise interaction of surface segments and here, the surface charges play
the key role in defining the electrostatic and H-bond interaction energies. Thus,
COSMO-RS is a surface-charge interaction model for excess Gibbs energy.
The molecular surface is divided into smaller pieces having a standard area,
where each surface piece is identified by its screening charge density. The total number
of surface segments on a molecule of compound is ∑ ; where = is
the total surface area of the molecule, and is the number of segments of type on
molecule and is the size of a thermodynamically independent surface contact
(adjustable parameter, of the order of 7Å ). A profile of area of portion of molecular
surface vs its charge density is called sigma-profile and provides much insight into the
electrostatic behavior of the molecule.

65
In terms of surface segment interactions, the solute-specific COSMO reference
state, the hypothetical state where the solute is imagined to be perfectly screened by the
surrounding solvents, refers to the idealized situation is where each solute-surface-
segment will find a solvent-surface-segment having equal and opposite charges to screen
it completely. The degree of deviation from the ideally-screened situation will differ by
the mismatch of the charges on the two interacting segments (and the contact statistics of
different segments). This electrostatic interaction energy is modeled as misfit energy,
2 (2.19)
where, is the surface area of the segment, is the charge density on segment
labelled as , is the charge density on segment labelled as , is a coefficient.
If, ; the misfit energy of interaction vanishes. Interaction energy due to
hydrogen-bonding is also defined in terms of the charge densities of the interacting
segments (some cut-off values of the charge is defined). The liquid phase (pure liquid or
mixture) is thus considered as an ensemble of interacting surfaces of different type and
the residual (due to attractive energetic interaction) surface activity coefficient due to
energetic interactions are obtained by solving the following coupled nonlinear equation
in an iterative manner (termed as COSMOSPACE23 equation; for derivation, see Klamt
(1995)). In terms of surface activity coefficients,
1Θ (2.20)
and are the activity coefficients of segment types and , respectively. Θ is the
relative amount (mole fraction) of segment type , defined as, Θ ; is the

66
number of segment of type and is the total number of segment in the mixture. is
energetic interaction parameters between two arbitrary segments labeled as and
defined by the total interaction energy between the segment pairs, and assuming
.
12
(2.21)
The residual part of the activity coefficient of molecule in the mixture is from
summing the contribution from their surface segments.
(2.22)
is the activity of segment type in the pure liquid of molecule (which is obtained by
considering the pure liquid as a mixture of its own surface segments and applying
equation (2.20)). The total number of surface segments on a molecule of compound is
∑ ; where = is the total surface area of the molecule, and is the
number of segments of type on molecule and is the size of a thermodynamically
independent surface contact (adjustable paprameter, of the order of 7 Å . The
combinatorial contribution due to shape and size effect of molecule is computed from the
surface area and volume obtained from cosmo calculations using Guggenheim-
Stavermann equation.
The computationally intensive COSMO calculation for a molecule is done only
once (one for each conformer if higher energy conformers are included in calculations)
and the results are stored in a ‘cosmo’ file which is subsequently used in all phase
equilibrium calculations. COSMO-RS can also be used for pure fluid.

67
2.2.5 Molecular dynamics (MD) simulation
A vivid account of a reaction-in-progress may be obtained by molecular
dynamics simulations, which explores the time evolution of a collection of atoms,
potentially demonstrating bond breaking and formation while including thermal, entropic
and solvation effects directly. In molecular dynamics simulation, a trajectory (position
of atoms as a function of time) of a complex molecular system is generated by solving
Newton’s equations of motion for all the atoms. In Born-Oppenheimer AIMD
simulations, the equations of motion solved involves only the nuclear coordinates, in
contrast to the Car-Parrinello AIMD method where the electronic degrees of freedom are
also involved of the equations of motion. We have applied the BO AIMD as
implemented in the software package Vienna Ab Initio Molecular Dynamics Simulation
Package (VASP) developed by mainly developed by G. Kresse and coworkers and
maintained at the University of Vienna in Austria.24-28
The system is modeled with some finite number of molecules (limited by
computational resources and time) enclosed in a cubic box and replicated infinitely in all
directions (known as periodic boundary conditions) (Figure 2.4). What happens in one
cell also happens simultaneously in all the replicants. If an atom enters the cell, an
image atom must leave the cell.
DFT-based methods are used to compute energy and the forces are calculated
from derivatives of Kohn-Sham energy with respect to nuclear coordinates where the
electrons are at their ground state (Hellman-Feynman theorem). Newton’s equation of
motion is solved for a small time step (1 to 3 fs) during which the force is assumed
constant.

68
Figure 2.4 Periodic boundary conditions (a two-dimensional periodic system).

69
Pseduopotentials are introduced to replace the actual strong nuclear potential and
the core electrons that act on psuedowave function of the valence electrons. The
pseudopotentials are weak and designed to generate pseudowave function for the valence
electrons that have smooth mathematical behavior (non-nodal, non-oscillatory). After a
certain nuclear distance (cut-off radius), the pseudofunction will be identical with the
standard all-electron (AE) wave function.29 The Kohn-Sham orbitals are expanded in
plane-wave basis sets suitable for representing systems with periodic replication and
with many electrons (explicit representation of solvents introduces many electrons for
the molecules in the cubic box). The size of the basis set is limited by the size of the
unit-cell. Simulations were performed with the NVT (canonical) ensemble, using a Nose
thermosatat.30 Temperature is related to particle velocities via the principle of
equipartition of energy.
23
(2.23)
Four input files are used for each simulation: INCAR (simulation procedure),
POSCAR (initial geometry), POTCAR (pseudo-potentials), KPOINTS (to generate a
mesh to solve the wave functions at some points using the plane waves). Many output
files are produced, and we were interested mostly in the XML file, that lists the new
coordinates of the atoms after each time-step.

70
2.3 APPLICATIONS IN THIS DISSERTATION
We have applied semi-continuum models in chapters 3 and 4 to model the
solvation effect of neutral and ionic species.
Computation of pKa involves the computation of Gibbs free energy of a base (B)
and its conjugate acid (BH+) in dilute aqueous solution. The water molecules in the
environment of the base and acid are modeled by with only one water molecule and the
rest with a dielectric (a homogeneous continuous media to represent the discrete solvent
molecule). In chapter 3, only one explicit water molecule was added with the solute
(neutral amine or protonated amine).
Proper consideration of solvent effect was crucial in studying reaction
mechanisms. In chapter 4, one to 20 water molecules were needed to properly model
different species in the CO2/amine/water reaction. AIMD molecular dynamics simulation
was employed to see stability and conversion of different species in the reaction in
Chapter 5. COSMO-RS method was applied in predicting low pressure solubility of
gases in ionic liquids in Chapter 6.

71
2.4 REFERENCES
1. Rowlinson, J. S. (Ed.) Studies in Statistical Mechanics, vol. 14, Amsterdam,
1988.
2. Fredenslund, A.; Jones, R. L.; Prausnitz, J. M. Group Contribution Estimation of
Activity Coefficients in Nonideal Liquid Mixtures. AIChE J. 1975, 21, 1086.
3. Chapman, W. G.; Gubbins, K. E.; Jackson, G.; Radosz, M. SAFT Equation-of-
state Solution Model for Associating Fluids. Fluid Phase Equilib. 1989, 52, 31.
4. Klamt, A. Conductor-like Screening Model for Real Solvents: A New Approach
to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem. 1995,
99, 2224.
5. Sandler, S. I. Quantum Mechanics: A New Tool for Engineering
Thermodynamics. Fluid Phase Equilib. 2003, 210, 147.
6. Mennucci, B. Continuum Solvation Models: What Else Can We Learn From
Them? J. Phys. Chem. Lett. 2010, 1, 1666.
7. Lewars, E. Computational Chemistry: Introduction to the Theory and
Applications of Molecular and Quantum Mechanics; Springer Science+Business
Media B. V., 2nd ed, 2011
8. Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley &
Sons: Chichester, England, 2007.

72
9. Allen, M. P. Introduction to Molecular Dynamics Simulation. In Computational
Soft Matter: From Synthetic Polymers to Proteins; Attig, N., Binder, R.,
Grubmuller, H., Kremer, K., Eds.; John von Neumann Institute for Computing:
Julich, Germany, 2004; Vol. 23, pp 1−28.
10. Leach, A. G.; Goldstein, E. Energy Contour Plots: Slices Through the Potential
Energy Surface That Simplify Quantum Mechanical Studies of Reacting
Systems. J. Chem. Educ. 2006, 83, 451
11. Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation
Models. Chem. Rev. 2005, 105, 2999
12. Woo, T. K.; Blochl, P. E.; Ziegler, T. Towards Solvation Simulations With a
Combined Ab Initio Molecular Dynamics and Molecular Mechanics Approach.
J. Mol. Struct. THEOCHEM 2000, 506, 313.
13. Pascual-Ahuir, J. L.; Silla, E.; Tuñón, I. The Solvent-Excluding Surface as a
Descriptor of Ionic Channels: Gramicidin-A. J. Mol. Struct. THEOCHEM 1998 ,
426 , 331.
14. Luque, F.; Curutchet, C.; Munoz-Muriedas, J.; Bidon-Chanal, A.; Sorietas, I.;
Morreale, A.; Gelpi, J. L.; Orozco, M. Continuum Solvation Models: Dissecting
the Free Energy of Solvation. Phys. Chem. Chem. Phys., 2003, 5, 3827.
15. Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a
Continuum. A Direct Utilization of ab Initio Molecular Potentials for the
Prevision of Solvent Effects. Chem. Phys. 1981, 55, 117.

73
16. Cancès, M. T.; Mennucci, B.; Tomasi, J.A. New Integral Equation Formalism for
the Polarizable Continuum Model: Theoretical Background and Applications to
Isotropic and Anisotropic Dielectrics. J. Chem. Phys. 1997, 107, 3032.
17. Klamt, A.; Schüürmann, G. COSMO: A New Approach to Dielectric Screening
in Solvents With Explicit Expressions for the Screening Energy and its Gradient.
J. Chem. Soc., Perkins Trans. 1993, 2, 799.
18. Sunoj, R. B.; Anand, M. Microsolvated Transition State Models for Improved
Insight Into Chemical Properties and Reaction Mechanisms. Phys. Chem. Chem.
Phys., 2012, 14, 12715.
19. Khalili, F.; Henni, A.; East, A. L. L. Entropy Contributions in pKa Computation:
Application to Alkanolamines and Piperazines. J. Mol. Struct. THEOCHEM
2009, 916, 1.
20. Klamt, A. Conductor-Like Screening Model for Real Solvents—A New Approach
to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem. 1995,
99, 2224.
21. Klamt, A. COSMO-RS: From Quantum Chemistry to Fluid Phase
Thermodynamics and Drug Design; Elsevier: Amsterdam, 2005.
22. Lucas, K. Molecular models for fluids, Cambridge University Press; 2007.
23. Klamt, A.; Krooshorf, G. J. P.; Taylor, R.. COSMOSPACE: Alternative to
Conventional Activity-Coefficient Models. AIChE J. 2002, 48, 2332.
24. Kresse G.; Hafner J. Ab Initio Molecular Dynamics for Liquid Metals. Phys.
Rev. B 1993, 47, 558.

74
25. Kresse G.; Furthmüller J. Efficient Iterative Schemes for Ab Initio Total-Energy
Calculations Using a Plane-Wave Basis Set. Phys. Rev B 1996, 54, 11169.
26. Kresse, G.; Joubert D. From Ultrasoft Pseudopotentials To the Projector
Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758.
27. Kresse G.; Hafner, J. Norm-Conserving and Ultrasoft Pseudopotentials for First-
row and Transition Elements. J. Phys. Condens. Matter 1994, 6, 8245.
28. Hafner, J. Ab-Initio Simulations of Materials Using VASP: Density-Functional
Theory and Beyond. J. Comput. Chem. 2008, 29: 2044.
29. Payne ; M. C.; Teter, M. P. ; Ailan; D. C.; Arias, T. A.; Joannopouios, J. D.
Iterative Minimization Techniques for Ab Initio Total-Energy Calculations:
Molecular Dynamics and Conjugate Gradients. Rev. Mod. Phys. 1992, 64 (4),
1045.
30. Nos’e, S. A Unified Formulation of the constant Temperature Molecular
Dynamics Methods. J. Chem. Phys. 1984, 81, 511.

75
Chapter 3: Predicting pKa of Amines† _______________________________________________________________________
3.1 INTRODUCTION
There is need for a simple procedure which can predict pKa to within ±0.3 for a
variety of amines important in CO2 capture.1,2 The pKa of a compound has been
estimated via quantum chemistry calculations using fairly routine continuum-solvation
approximations, but the accuracy of these models is often unsatisfactory:3,4 since 2005,
most such procedures for studies of ten or more amines have seen pKa errors of ±1 or
greater,5,6,7 and the addition of correction terms, either constant7,8 or proportional to the
pKa magnitude,5,6,9-13 has reduced errors but not to ±0.3 accuracy. At the moment the
best of these methods have pushed accuracy to ±0.4 for selected amines8,12 (±0.2 for the
subclass of benzimidazoles5). Some researchers have bypassed continuum-solvation
methods in favour of linear correlation formulae to other computable properties of the
amine,14,15 but to date have not improved upon ±0.4 accuracy.
_______________________________________________________________________ †This chapter contains material reproduced with permission from Sumon, K. Z.; Henni, A. East, A. L. L. Predicting pKa of Amines for CO2 Capture: Computer versus Pencil and-Paper. Ind. Eng. Chem. Res. 2012, 51, 11924-11930. Copyright © 2012 American Chemical Society.

76
Khalili, Henni and East published in 2009 a general continuum-plus-correction
procedure for pKa computation which produced rms errors of 0.68 for a test set of 17
amines.7 It employed Gaussian0316 self-consistent reaction-field (SCRF) computations in
a semicontinuum manner (“Model II”), in which an explicit water molecule is inside the
cavity with the solute molecule: the H2O is arranged to H-bond to the lone pair on N in
the neutral amine, but to receive an H-bond from the protonated amine. The procedure,
now labeled the KHE method, incorporated many terms in the calculation, including a
statistical entropy effect for multiple conformations, in an attempt to get as much
accuracy as one could out of the dielectric-continuum approximation. We have now
simplified the procedure drastically, for both Gaussian03 (G03)16 and Gaussian09
(G09)17 users, and pushed its accuracy down to a root-mean-squared error of 0.28 for a
training set of 32 CO2-relevant amines (acyclic amines including alkanolamines, and
substituted piperazines and morpholines). There remain problems, particularly with
other classes of ringed amines. The new procedure is labeled SHE to distinguish it from
our earlier one.
However, there is a computer-free group-additivity method for pKa prediction,
published in a book by Perrin, Dempsey, and Serjeant (PDS) in 1981,18 that was known
to provide accuracy to a few tenths for amines. Their ΔpKa method was based on the
knowledge that use of Taft parameters for organic substituents in an additive way can
account for trends in equilibrium constants and free energies of reaction within classes of
compounds.19 The current paper compares pencil-and-paper PDS results against those of
the computer-based SHE method.

77
3.2 METHODS
3.2.1 SHE method
The pKa of a base B is a scaled version of ∆rG(aq), the free energy change of the
acid-dissociation reaction BH+(aq) → B(aq) + H+
(aq). With energies in kcal mol-1, the
relation at T = 298.15 K is
pKa = ∆rG(aq) / RT ln 10 (3.1)
Expanding ∆rG(aq),
pKa = [ G(aq)(H+) + G(aq)(B) – G(aq)(BH+) ] / 1.3643 (3.2)
These Gaq quantities can be approximated with dielectric-continuum models, by
breaking each Gaq down into a sum of three components (Eel + ΔGnon-el + Enuc): Eel =
)()(21 fVHf is the internal energy of the solute in solution (including its
electrostatic interaction with the solvent continuum), ΔGnon-el is the correction due to
non-electrostatic contributions to solvation (cavitation, dispersion, repulsion, solvent-
structuring effects), and Enuc is the nuclear-motion energy of the solute in solution. Such
computations are problematic for G(aq)(H+) due to the strong covalent nature of H+ in
solution (H3O+, H5O2
+, etc.), and hence G(aq)(H+), a constant with respect to identity of
base, should be estimated differently. For the other two free energies, we consider the
half reaction free energy ∆hG(aq)(BH+ → B) and its components (∆hEel + ∆hΔGnon-el +
∆hEnuc), leading to

78
pKa = [ G(aq)(H+) + ∆hEel + ∆hΔGnon-el + ∆hEnuc ] / 1.3643 (3.3)
All four terms in eq 3 have been simplified in going from KHE to SHE, as follows.
G(aq)(H+) = −270.3 kcal mol-1. The KHE method used this G(aq)(H
+) term as a
fitting parameter, since it is difficult to predict ab initio. We now adopt a proper value,
the sum of G(g)(H+, 1 atm) = −6.3 kcal mol-1, computed via Sackur-Tetrode equation,
with ΔsolvG(H+, 1 atm gas 1 M aqueous) = −264.0 kcal mol-1, derived by Tissandier et
al.20 from experimental data. Using the Ben-Naim convention21 for ΔsolvG one obtains
the same value, summing G(g)(H+, 1 M) = −4.4 kcal mol-1 with ΔsolvG(H+, 1 M gas 1
M aqueous) = −265.9 kcal mol-1.
∆hEel now employs MP2/6-31G(d). The KHE method used MP2/6-311++G(d,p),
maximally trans conformers for acyclic amines, and UA0 radii for solute cavities.
However, further basis set testing by us revealed that the removal of diffuse functions
actually reduces errors for acyclic bases in both Model I and Model II calculations
(without or with an explicit H2O inside the cavity, respectively); Model II results are
shown in Table 3.1. We also decided to reduce the basis set from triple to double zeta,
possibly sacrificing some accuracy but gaining in speed for extension to larger amines.
The UA0 radii are default in G03; in G09 they are requested by adding G03Defaults to
the SCRF keyword.

79
Table 3.1 pKa Errors from Uncorrected SHE Procedure: Basis Set Dependencea
Basis Set
Amine 6-311++G(d,p) 6-311G(d,p) 6-31G(d)
NH3 -4.24 -0.06 -0.49
MeNH2 -2.81 0.78 0.52
Me2NH -2.14 1.14 0.67
Me3N -1.88 1.36 0.48
piperazine -0.77 2.48 2.37
a SHE method as described in this work, but omitting empirical corrections and allowing for basis set substitution in the SCRF MP2 geometry optimizations. Piperazine calculations employed conformer choice of Ref. 7. Experimental pKa values are 9.25, 10.66, 10.73, 9.80, and 9.73 for the five bases (CRC Handbook23).

80
ΔhEnuc = −9.4 kcal mol-1. We dramatically simplify this nuclear-motion energy
term. Enuc for a solute has an intrinsic term, Enuc,int , and a statistical entropy term −TSstat
due to multiple conformations and rotational symmetry.7 In the KHE method, Sstat was
derived for each solute (B and BH+) from symmetry considerations and conformer
counting, while Enuc,int was computed from vibrational frequencies of Model II gas-phase
complexes (i.e. with solute·H2O complexes). We now neglect TSstat effects altogether,
as the effects are less than 1 kcal mol-1 (Table 3 of ref. 7), and are a hassle to incorporate
for the non-specialist. We also now replace ΔhEnuc,int with the constant −9.4 kcal mol-1 to
eliminate the need for gas-phase opt+freq calculations. The value −9.4 was chosen to
mimic previous Model I (no explicit water) gas-phase frequency values, which were
consistently -9.4 ± 0.5 kcal mol-1 (Table 6 of ref. 7). Closer inspection revealed that this
amount is dominated by the zero-point vibrational energy (ZPVE) term: deprotonation
generally resulted in the loss of a 3400 cm-1 NH stretch and two 1600 cm-1 HNC bend
modes, and no net frequency shifts of other modes. Friesner and coworkers had already
commented on how constant this term likely is for a particular class of compounds.22
ΔhΔGnon-el = 0. We remind Gaussian users that three of the four non-electrostatic
effects (cavitation, dispersion, repulsion) are computed by default in G03 but must be
requested in G09, and in G03 the values appear only in the middle of the logfile. Their
effects are generally small. The sum of these three effects for the half-reaction, ΔhΔGnon-
el , were computed (G03) to be small and nearly constant: 0.2 and 0.6 kcal mol-1 in
Model I and Model II calculations, respectively (Tables 6 and 7 of ref. 7). These small
shifts tend to be swamped by remaining errors, and hence not of sufficient benefit for
inclusion in a simple procedure.

81
Empirical corrections. To make the errors uniformly small for multiple classes
of compounds, class-dependent empirical corrections C are needed (see Sec. 3.3.1).
Also, in general, optimal values for C would depend on the choices made for computing
∆hEel: level of theory, cavity radii, and molecular conformer. Attempts were made to
find theoretical justification for a choice of cavity radii and molecular conformer, but
these failed (Sec. 3.3). Given the choices we have made for ∆hEel, values for C deemed
optimal for a training set of 32 amines (Table 3.8, Sec. 3.4) are -0.7 for acyclic amines
and -1.7 for cyclic amines.
In summary, the SHE procedure is to compute
pKa = (1/1.3643) [ –270.3 + Eel(B·H2O) – Eel(BH+·OH2) – 9.4] + C (3.4)
where the Eel(B.H2O) and Eel(BH+·OH2) are MP2 energies, converted to kcal mol-1 (x
627.50955), from the bottom of Gaussian logfiles (“MP2=”) of
SCRF=(PCM,G03Defaults) MP2/6-31G(d) geometry optimizations of maximally trans
conformers. The C value is -1.7 for cyclic amines and -0.7 for acyclic amines.

82
3.2.2 PDS method
The empirical pencil-and-paper Perrin-Dempsey-Serjeant method18 uses only a
table (Table 3.2 here) of pKa “base” values and ΔpKa additive functional-group
corrections, thus offering much faster predictions. The original publication also offers
some temperature correction formulae for pKa. Since the parameter values in the PDS
scheme were chosen before much alkanolamine data had been studied, we provide an
updated set of these values (“new PDS”), obtained from a least-squares fit to
experimental pKa values of the same 32-amine training set used for SHE. Please note
that we have only updated the parameter values relevant for CO2-capture amines; the
original method offers terms for other amines, and carboxylic acids as well.18 Table 3.2
lists the old and new parameter values. Of the changes, note that (i) the base value for
primary and tertiary amines are now equal, and (ii) the correction for cyclic amines is
now zero. Our SHE results actually reveal a ring effect of -1.0 in the experimental pKa
values (Sec. 3.3.1); in the PDS method the effect is incorporated by counting a β group
twice if it occurs in a ring.
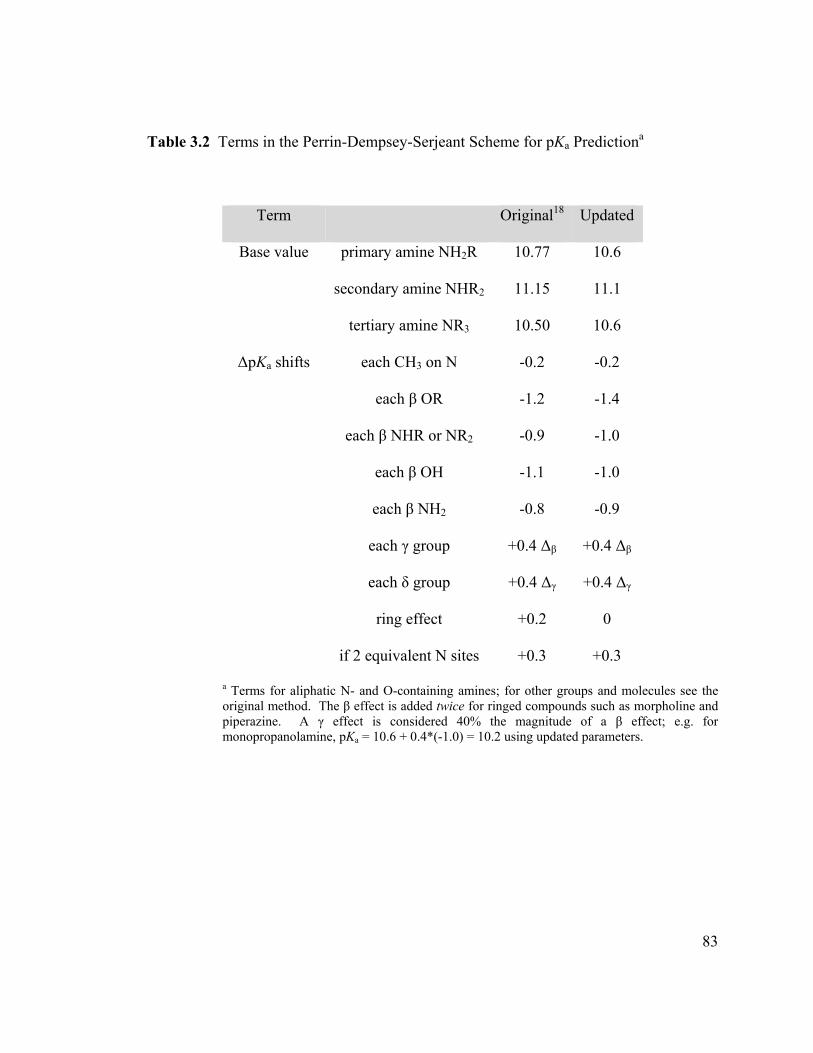
83
Table 3.2 Terms in the Perrin-Dempsey-Serjeant Scheme for pKa Predictiona
Term
Original18 Updated
Base value primary amine NH2R 10.77 10.6
secondary amine NHR2 11.15 11.1
tertiary amine NR3 10.50 10.6
ΔpKa shifts each CH3 on N -0.2 -0.2
each β OR -1.2 -1.4
each β NHR or NR2 -0.9 -1.0
each β OH -1.1 -1.0
each β NH2 -0.8 -0.9
each γ group +0.4 Δβ +0.4 Δβ
each δ group +0.4 Δγ +0.4 Δγ
ring effect +0.2 0
if 2 equivalent N sites +0.3 +0.3
a Terms for aliphatic N- and O-containing amines; for other groups and molecules see the original method. The β effect is added twice for ringed compounds such as morpholine and piperazine. A γ effect is considered 40% the magnitude of a β effect; e.g. for monopropanolamine, pKa = 10.6 + 0.4*(-1.0) = 10.2 using updated parameters.

84
3.2.3 Experimentals
The pKa values of some amines (Table 3.3), outside the training set used for the
development of SHE method and the determination of the updated PDS parameters, were
measured at 25°C to further assess these predictive methods. The potentiometric
titration method developed by Albert and Serjeant was followed (Appendix A). The pKa
values of MEA and MDEA were measured to validate the method.
Table 3.3 Amines Used in Measurement of pKa
Amine CAS # Purity
3-(methylamino)-1,2-propanediol 40137-22-2 ≥98.0%
3-(dimethylamino)-1,2-propanediol 623-57-4 98%
3-(diethylamino)-1,2-propanediol 621-56-7 98%
1,3 Bis(dimethylamino)-2-propanol 5966-51-8 97%
2-{[2-
(dimethylamino)ethyl]methylamino}ethanol] 2212-32-0 98%
Ethanolamine 141-43-5 ≥99.5%
N-methyl-diethanolamine 105-59-9 ≥99%

85
3.3 CONTINUUM-SOLVATION ISSUES
3.3.1 Choice of radii
The accuracy of continuum-solvation computational methods is, unfortunately,
very sensitive to the atomic radii used to define the solute cavity,24,25 and we noted that
the default radii choice changed (from unscaled “UA0” to “UFF” scaled by 1.1) when
Gaussian upgraded its software from G03 to G09. In Figure 3.1, the change shifts pKa
predictions down by an alarming amount: 3-4 units. (There was also a technical change
in cavity and solute-solvent-surface construction in going from G03 to G09 which may
have had a small effect upon predicted pKa.26,27) In Figure 3.1, good estimates for trans
conformers of all acyclic amines (NH3 to DEA) were achieved with the unscaled UA0
radii, and hence this choice was incorporated into the SHE procedure.
Although the UA0 choice provides the highest pKa estimates of the choices in
Figure 3.1, it provides only moderate total molecular volumes (Figure 3.2); the key
seems to be in the volume given to the reactive site, particularly primary and secondary
N atoms, where the UA0 choice results in the smallest volumes (23 and 19 Å3,
respectively; the other procedures use volumes ≥ 26 and 21 Å3, respectively).28

86
Figure 3.1 SHE results without empirical corrections (on conformers of ref. 7), showing
dramatic effects of cavity radii. Dashed line: experiment. Solid squares:
G03 (UA0). Open squares: G09 radii=UA0(x1.1). Solid triangles: G03
radii=UFF. Open triangles: G09 (UFFx1.1). Parentheses denote default
effects.
Figure 3.2 Cavity volumes of B·HOH complexes. See Figure 3.1 for legend.
5
6
7
8
9
10
11
12
13
NH
3
MeN
H2
Me2
NH
Me3
N
ME
A
MIP
A
MP
A
AM
P
AE
EA
DE
A
MO
R
PP
Z
2-M
ePP
Z
1-E
tPP
Z
1-M
ePP
Z
1-(E
tOH
)PP
Z
1,4-
Me2
PP
Z
pKa

87
Regardless of choice of algorithm and radii, there are class-dependent errors
evident in Figure 3.1, particularly the unique errors for cyclic compounds (morpholine
and the piperazines in our case) that have been seen before.7,29 Friesner and co-workers
hypothesized29 that the error shift for cyclic compounds is due to neglecting the 4th type
of non-electrostatic effect: a solvent-structuring effect. Such solvent-structuring may be
clathrate-like;30 solid clathrate structures are known to be stable around cyclic ethers for
example.31 This necessitated the use of separate values for the empirical shift C for
cyclic vs. acyclic amines (discussed in Sec. 3.2.1).
3.3.2 Choice of conformer
An additional unresolved issue is the choice of molecular conformer. This
commonly ignored problem is important because pKa results for alkanolamines are
heavily dependent upon conformer choice (Table 3.4).32 In the examples in the table,
conformer effects are ~0.5 for ethyl group rotation, but they are 2-4 for alkanolamine
internal rotation. The large effect for alkanolamines is due to gauche XCCY
conformations which place the polar X and Y groups in close proximity; this
preferentially stabilizes the BH+ cation in a continnum-solvation calculation, and shifts
up the pKa prediction roughly 2 units per polar gauche interaction.
Note that these large pKa variations with internal rotation of an XCCY unit are as
large as the variations in switching default cavity radii in Figure 3.1. In fact, if G09
default radii (UFF x 1.1) are used, Table 3.4 reveals that one gets the best agreement
with experiment if gauche conformers are used instead of trans ones!

88
We chose the trans + UA0 combination over the gauche + UFFx1.1 combination
mainly because UFF radii show less uniform errors than UA0 radii in Figure 3.1, which
translates into worse rms errors as one tests more amines. It would have been better, of
course, to choose the conformer based on which conformer dominates in aqueous
solutions. Unfortunately, theory has been unable to predict this to date: (i) the rule of
thumb that one should use the lowest-energy conformer in a conformer search cannot be
employed, because although gas-phase and dielectric-continuum optimizations both
predict the gauche forms of both B and BH+ to be lower in energy than the trans forms,
this prediction neglects the effects of explicit hydrogen bonding with solvating water
molecules;34 (ii) two nanosecond-scale classical dynamics simulations of aqueous
monoethanolamine (MEA) which purported to demonstrate preference for gauche
forms35,36 likely did not achieve full equilibration, since bizarrely asymmetric
distributions of observed NCCO dihedral angles were reported in a third such study.37 To
our knowledge the best relevant experimental study of this issue is a 1975 Raman
spectrum study38 which demonstrated that both conformers of neutral MEA are present
in solution; this calls into question the habit of choosing only one for quantum chemistry
computation.

89
Table 3.4 pKa Results from Uncorrected SHE Procedure: Conformer Dependence
Aminea Conformerb pKa G03c pKa G09c
AMP G 11.3 7.9 9.68 T 9.8 6.1
DEA GtgG 12.4 9.3 8.88 GttG 11.9 8.9
TtgG 11.2 7.6 TgtG 10.8 7.4 TttG 10.8 7.3 TggT 9.6 5.8 TgtT 9.5 5.7 TttT 9.5 5.7
DIPA GttG 12.5 9.1 8.84 TttG 10.5 7.3
TttT 8.6 5.3
MEA G 11.7 8.2 9.5 T 10.0 6.3
Et2NH GG 12.2 9.4 10.84 TG 12.1 9.1
TT 11.9 8.9
PPZ GG 12.5 9.2 9.73 TT ax/eq 12.1 8.5
TT ax/ax 12.1 8.5 TT eq/eq 11.7 8.0
TT eq/ax 11.6 7.9
rms error 0.9 1.1 a Experimental pKa values are listed: from Hamborg and Versteeg33 for AMP, DEA, DIPA, and from CRC Handbook23 for MEA, Et2NH, and piperazine. b Gauche and trans descriptors for NCCX dihedrals (G, T) and CNCC dihedrals (g,t) along the main atom chain. The HOCC and PNCC (P = lone pair) dihedrals are not specified in this notation; for T cases we took them to be trans, but for G cases they were chosen to arbitrarily maximize NH…O hydrogen-bond interaction. For PPZ, the axial and equatorial descriptors are for the protonating and spectator N atoms, respectively. c Bold values represent the best single-conformer predictions; the difference between G03 and G09 calculations is the default cavity radii used (UA0 and UFFx1.1, respectively).

90
Table 3.5 Optimized Structures of Geometries of Amines in Table 3.4
Amine/Conformer Neutral Cation
AMP G
AMP T
DEA GtgG
DEA GttG
DEA TtgG

91
Table 3.5 Optimized Structures of Geometries of Amines in Table 3.4 (Continued)
Amine/Conformer Neutral Cation
DEA TgtG
DEA TttG
DEA TggT
DEA TgtT
DEA TttT

92
Table 3.5 Optimized Structures of Geometries of Amines in Table 3.4 (Continued)
Amine/Conformer Neutral Cation
DIPA GttG
DIPA TttG
DIPA TttT
MEA G
MEA T

93
Table 3.5 Optimized Structures of Geometries of Amines in Table 3.4 (Continued)
Amine/Conformer Neutral Cation
Et2NH gg
Et2NH tg
Et2NH tt

94
Table 3.5 Optimized Structures of Geometries of Amines in Table 3.4 (Continued)
Amine/Conformer Neutral Cation
PPZ GG (twist-boat)
PPZ TT (chair) ax/eq
PPZ TT (chair) ax/ax
PPZ TT (chair) eq/eq
PPZ TT (chair) eq/ax

95
3.4 RESULTS
If one accepts the limitations inherent in a simple one-conformer procedure with
a traditional continuum-plus-correction methodology, the SHE method constitutes an
improvement within its class, as supported by a direct comparison of SHE results to
those of other continuum-solvation procedures (Table 3.6).
Table 3.6 pKa Results: Comparison of Continuum-Solvation Procedures
a Ref. 23. b Ref. 12. c Ref. 22. d,e Ref. 11 Table 3 “bare” and “H2O”, resp. f,g Ref. 10 Table 2 “pKa calc” and “pKa corrected”, resp.
COSMO JaguarCOSMO-
RSCOSMO-
RSCOSMO-
RSCOSMO-
RS
Amine Expt. a SHE 2010 b 2002 c 2010-1 d 2010-2 e 2006-1 f 2006-2 g
Methylamine 10.66 10.52 10.52 10.5 11.97 11.09 11.71 11.71Dimethylamine 10.73 10.74 10.71 10.9 10.94 9.64 10.67 11.67Trimethylamine 9.8 9.66 10.12 10.1 8.93 7.73 8.63 10.63
MEA 9.5 9.32 9.8 10.29 10.29Morpholine 8.5 8.25 9.5 8.36 9.36rms error 0.2 0.2 0.5 0.9 1.4 0.8 0.9

96
However, SHE is outperformed by the “New PDS” method. Table 3.7 presents
PDS and SHE results for the 32-amine training set. Both SHE and the “New PDS”
methods were trained on this set, and give rms errors of 0.28 and 0.18, respectively. The
optimized structures are given in Table 3.8 and the details of the computation of pKa by
PDS method from group contributions is illustrated in Table 3.9.

97
Table 3.7 SHE vs. PDS Predictions for pKa of 32 Amines
Amine Amine label SHE0a SHE Old PDS
New PDS
Expt. Ref. b
NH(C2H5)2 diethylamine 11.88 11.18 11.15 11.10 10.84 23
NH2(CH2)4NH2 1,4-
butanediamine 11.42 10.72 10.94 10.76 10.80 23
NH(CH3)2 dimethylamine 11.39 10.69 10.75 10.70 10.73 23 N(C2H5)3 triethylamine 11.78 11.08 10.50 10.60 10.75 23
NH2C(CH3)3 tert-butylamine 10.90 10.20 10.77 10.60 10.68 23 NH2CH2CH3 ethylamine 11.28 10.58 10.77 10.60 10.65 23
NH2CH(CH3)2 iso-propylamine 11.13 10.43 10.77 10.60 10.63 23 NH2CH2CH2CH2CH3 butylamine 11.48 10.78 10.77 10.60 10.56 23
NH2CH2CH2CH3 propylamine 11.39 10.69 10.77 10.60 10.54 23
NH2(CH2)3NH2 1,3-
propanediamine 11.34 10.64 10.75 10.54 10.55 23
NH2CH3 methylamine 11.17 10.47 10.57 10.40 10.66 23 NH2CH2CH2CH2OH MPA 10.90 10.20 10.33 10.20 9.96 42
N(CH3)3 trimethylamine 10.31 9.61 9.90 10.00 9.80 23
NH2(CH2)2NH2 1,2-
ethanediamine 10.99 10.29 10.27 10.00 9.92 23
NH(CH3)CH2CH2OH MMEA 10.75 10.05 9.85 9.90 9.85 33 N(C2H5)2CH2CH2OH DEMEA 10.66 9.96 9.40 9.60 9.75 33 NH2C(CH3)2CH2OH AMP 9.76 9.06 9.67 9.60 9.68 33
NH2CH2CH2OH MEA 9.97 9.27 9.67 9.60 9.50 23 NH2CH2CH(CH3)OH MIPA 9.76 9.06 9.67 9.60 9.45 33 N(CH3)2CH2CH2OH DMMEA 9.76 9.06 9.00 9.20 9.22 33 NH(CH2CH2OH)2 DEA 9.47 8.77 8.95 9.10 8.88 41
N(CH3)(CH2CH2OH)2 MDEA 8.64 7.94 8.10 8.40 8.56 40 N(CH2CH2OH)3 TEA 8.53 7.83 7.20 7.60 7.78 23
HN(CH2CH2)2NH PPZ (piperazine) 11.69 9.99 9.85 9.40 9.73 23
C4H9N2(CH3) 2-MePPZ (H+
on 4) 11.60 9.90 9.55 9.10 9.57 39
C4H9N2(C2H5) 1-EtPPZ 11.12 9.42 9.55 9.10 9.20 39 C4H9N2(CH3) 1-MePPZ 11.17 9.47 9.55 9.10 9.14 39
C4H9N2(C2H4OH) 1-(2-EtOH)PPZ 10.96 9.26 9.55 9.10 9.09 39 C4H8N2(CH3)2 1,4-Me2PPZ 9.94 8.24 9.00 8.70 8.38 39
HN(CH2CH2)2O MOR
(morpholine) 9.95 8.25 8.95 8.30 8.50 23
C2H5N(CH2CH2)2O 4-EtMOR 9.16 7.46 8.30 7.80 7.67 23 CH3N(CH2CH2)2O 4-MeMOR 8.76 7.06 8.10 7.60 7.38 23
rms error 1.11 0.28 0.33 0.18 a SHE procedure without empirical corrections C in eq. 3. b Reference for experimental value. Optimized structures are given in Table 3.8.

98
Table 3.8 Optimized Structures of Geometries of Amines in Table 3.7
Amine Neutral Cation
1.
diethylamine
2.
1,4-
butanediamine
3.
dimethylamine
4.
triethylamine
5.
tert-butylamine

99
Table 3.8 Optimized Structures of Geometries of Amines in Table 3.7 (Continued)
Amine Neutral Cation
6.
ethylamine
7.
iso-propylamine
8.
butylamine
9.
propylamine
10.
1,3-
propanediamine

100
Table 3.8 Optimized Structures of Geometries of Amines in Table 3.7 (Continued)
Amine Neutral Cation
11.
methylamine
12.
MPA
13
trimethylamine
14.
1,2-ethanediamine
15.
MMEA

101
Table 3.8 Optimized Structures of Geometries of Amines in Table 3.7 (Continued)
Amine Neutral Cation
16.
DEMEA
17.
AMP
18.
MEA
19.
MIPA
20.
DMMEA

102
Table 3.8 Optimized Structures of Geometries of Amines in Table 3.7 (Continued)
Amine Neutral Cation
21.
DEA
22.
MDEA
23.
TEA
24.
PPZ

103
Table 3.8 Optimized Structures of Geometries of Amines in Table 3.7 (Continued)
Amine Neutral Cation
25.
2-MePPZ
26.
1-EtPPZ
27.
1-MePPZ
28.
1-(2-EtOH)PPZ
29.
1,4-Me2PPZ

104
Table 3.8 Optimized Structures of Geometries of Amines in Table 3.7 (Continued)
Amine Neutral Cation
30.
MOR
(morpholine)
31.
4-EtMOR
32. 4-
MeMOR

105
Table 3.9 Group Contributions in Old PDS Predictions for pKa of Amines in Table 3.7
Amine label Old PDS Base Ring N-Me Stat β γ δ
diethylamine 11.15 11.15 0 0 0 0 0 0
1,4-butanediamine 10.94 10.77 0 0 0.3 0 0 -0.13
dimethylamine 10.75 11.15 0 -0.4 0 0 0 0
triethylamine 10.5 10.5 0 0 0 0 0 0
tert-butylamine 10.77 10.77 0 0 0 0 0 0
ethylamine 10.77 10.77 0 0 0 0 0 0
iso-propylamine 10.77 10.77 0 0 0 0 0 0
butylamine 10.77 10.77 0 0 0 0 0 0
propylamine 10.77 10.77 0 0 0 0 0 0
1,3-propanediamine 10.75 10.77 0 0 0.3 0 -0.32 0
methylamine 10.57 10.77 0 -0.2 0 0 0 0
MPA 10.33 10.77 0 0.0 0 0.0 -0.44 0
trimethylamine 9.9 10.5 0 -0.6 0 0 0 0
1,2-ethanediamine 10.27 10.77 0 0 0.3 -0.8 0 0
MMEA 9.85 11.15 0 -0.2 0 -1.1 0 0
DEMEA 9.4 10.50 0 0 0 -1.1 0 0
AMP 9.67 10.77 0 0 0 -1.1 0 0
MEA 9.67 10.77 0 0 0 -1.1 0 0
MIPA 9.67 10.77 0 0 0 -1.1 0 0
DMMEA 9 10.50 0 -0.4 0 -1.1 0 0
DEA 8.95 11.15 0 0 0 -2.2 0 0
MDEA 8.1 10.50 0 -0.2 0 -2.2 0 0
TEA 7.2 10.50 0 0 0 -3.3 0 0
PPZ (piperazine) 9.85 11.15 0.2 0 0 -1.8 0 0
2-MePPZ (H+ on 4) 9.55 11.15 0.2 0 0 -1.8 0 0
1-EtPPZ 9.55 11.15 0.2 0 0 -1.8 0 0
1-MePPZ 9.55 11.15 0.2 0 0 -1.8 0 0
1-(2-EtOH)PPZ 9.55 11.15 0.2 0 0 -1.8 0 0
1,4-Me2PPZ 9 10.50 0.2 -0.4 0 -1.8 0 0
MOR (morpholine) 8.95 11.15 0.2 0 0 -2.4 0 0
4-EtMOR 8.3 10.50 0.2 0 0 -2.4 0 0
4-MeMOR 8.1 10.5 0.2 -0.2 0 -2.4 0 0

106
Table 3.10 presents further comparisons of SHE and New PDS results, for 16
amines outside of the training set, and while the New PDS method is still performing
very well, the SHE method shows further weaknesses, particularly with piperidine and
N-methylpyrrolidine. Looking more closely, the SHE0 calculation (without empirical
shifts) reveals an error that is dependent on class of ringed compound: +1.9 for
piperazines, +1.4 for morpholines, +1.2 for piperidines, and +0.8 for pyrrolidines. It
seems that the entropy change of the surrounding water upon protonation of the amine
(the only term not in the calculation) is important in an ab initio calculation of pKa for
ringed compounds. Rather than introducing more empirical shifts onto a continuum-
solvation electronic structure procedure, one should turn to a simpler procedure that has
only empirical shifts and abandons the electronic structure calculation altogether, i.e. the
empirical PDS methods.

107
Table 3.10 pKa Errors in SHE vs. PDS Predictions Outside the Training Set
Amine pKa(Expt) Ref.a SHE
error
Old
PDS
error
New
PDS
error
piperidine 11.12 23 -0.49 0.23 -0.02 N-methylpyrrolidine 10.46 23 -0.88 0.04 -0.06
cis-2,5-dimethylpiperazine 9.66 23 0.09 0.19 -0.26 2-methoxyethylamine 9.4 23 -0.57 0.17 -0.2 2-(ethylamino)ethanol 9.99 13 -0.01 0.06 0.11 2-(proylamino)ethanol 9.9 13 0.18 0.15 0.2 2-(butylamino)ethanol 9.92 13 0.26 0.13 0.18
pentylamine 10.63 23 0.18 0.14 -0.03 hexylamine 10.56 23 0.27 0.21 0.04
diethylmethylamine 10.35 23 -0.05 -0.05 0.05 3-methyl-1-butylamine 10.6 23 0.27 -0.1 0
3-(methylamino)-1,2-propanediol 9.65 This work
0.12 -0.24 -0.15
3-(dimethylamino)-1,2-propanediol 9.04 This work
0.25 -0.48 -0.24
3-(diethylamino)-1,2-propanediol 9.76 This work
0.07 -0.80 -0.56
1,3 Bis(dimethylamino)-2-propanol 9.41 This work
0.06 -0.55 -0.31
2-{[2-dimethylamino)ethyl]methylamino}ethanol
9.04 This work
-0.54 0.16 0.46
0.35 0.31 0.24 a Reference for experimental value. Optimized structures are given in Table 3.11.

108
Table 3.11 Optimized Structures of Geometries of Amines in Table 3.10
Amine/Conformer Neutral Cation
piperidine
1-methylpyrrolidine
cis-2,5-
dimethylpiperazine
2-methoxyethylamine
2-(ethylalmino)ethanol
2-(propylamino)ethanol

109
Table 3.11 Optimized Structures of Geometries of Amines in Table 3.10(continued)
Amine/Conformer Neutral Cation
2-(butylamino)ethanol
pentylamine
hexylamine
diethylmethylamine
3-methyl-1-
butylamine

110
Table 3.11 Optimized Structures of Geometries of Amines in Table 3.10 (continued)
Amine/Conformer Neutral Cation
3-(methylamino)-
1,2-propanediol
3-(dimethylamino)-1,2-propanediol
3-(diethylamino)-1,2-propanediol
1,3 Bis(dimethylamino)-2-propanol
2-{[2-
(dimethylamino)ethyl]methylamino}
ethanol

111
3.4 CONCLUSIONS
For predicting aqueous pKa values of CO2-relevant amines, we have pushed the
continuum-plus-correction method about as far as we can push it, producing a method
(SHE) whose pKa predictions have a root-mean-square error of 0.28 for 32 such amines.
This appears to be an improvement over all other known computer-based methods.
However, the method employs 2 empirical corrections, which would need to be
expanded to accommodate other classes of ringed compounds. The 30-year-old
computer-free group-additivity-based scheme by Perrin, Dempsey, and Serjeant (PDS)
produced an rms error of 0.33 for the same amine set, and with updated parameter values
it produced an rms error of only 0.18. The updated PDS method outperforms the best
continuum-solvation methods in both speed and accuracy, can extend to other ringed
amines, and has no conformer or cavity-radii issues, and hence should be the method of
choice for aliphatic amines. Future work should aim at updating the values of other PDS
parameters for application to other bases and to acids.

112
3.5 REFERENCES
1. Versteeg, G. F.; van Dijck, L. A. J.; van Swaaij, W. P. M. On the Kinetics
Between CO2 and Alkanolamines Both in Aqueous and Non-Aqueous Solutions.
An Overview. Chem. Eng. Commun. 1996, 144, 113.
2. da Silva, E. F.; Svendsen, H. F. Prediction of the pKa Values of Amines Using ab
Initio Methods and Free-Energy Perturbations. Ind. Eng. Chem. Res. 2003, 42,
4414.
3. Alongi, K. S.; Shields, G. C. Theoretical Calculations of Acid Dissociation
constants: A Review Article. Ann. Rep. Comput. Chem. 2010, 6, 113.
4. Ho, J.; Coote, M. L. A Universal Approach for Continuum Solvent pKa
Calculations: Are We There Yet? Theor. Chem. Acc. 2010, 125, 3.
5. Brown T. N.; Mora-Diez N. Computational Determination of Aqueous pKa
Values of Protonated Benzimidazoles (Part 1). J. Phys. Chem. B 2006, 110,
9270.
6. Lu H.; Chen X; Zhan C.-G. First-Principles Calculation of pKa for Cocaine,
Nicotine, Neurotransmitters, and Anilines in Aqueous Solution. J. Phys. Chem.
B 2007, 111, 10599.
7. Khalili, F.; Henni, A.; East, A. L. L. Entropy Contributions in pKa Computation:
Application to Alkanolamines and Piperazines. J. Mol. Struct. THEOCHEM
2009, 916, 1.
8. Bryantsev, V. S.; Diallo, M. S.; Goddard, W. A. pKa Calculations of Aliphatic
Amines, Diamines, and Aminoamides via Density Functional Theory with a

113
Poisson-Boltzmann Continuum Solvent Model. J. Phys. Chem. A 2007, 111,
4422.
9. Ulander, J.; Broo, A. Use of Empirical Correction Terms in Calculating
Ionization constants. Int. J. Quant. Chem. 2005, 105, 866.
10. Eckert F.; Klamt A. Accurate Prediction of Basicity in Aqueous Solution with
COSMO-RS. J. Comput. Chem. 2006, 27, 11.
11. Eckert F.; Diedenhofen M.; Klamt A. Towards First Principles Prediction of pKa
and the Cluster-Continuum Approach. Mol. Phys. 2010, 108, 229.
12. Zhang, S.; Baker, J.; Pulay, P. A Reliable and Efficient First Principles-Based
Method for Predicting pKa Value. 1. Methodology. J. Phys. Chem. A 2010, 114,
425.
13. Yamada, H.; Shimizu, S.; Okabe, H.; Matsuzaki, Y.; Chowdhury, F. A.; Fujioka,
Y. Prediction of the Basicity of Aqueous Amine Solutions and the Species
Distribution in the Amine–H2O–CO2 System Using the COSMO-RS Method.
Ind. Eng. Chem. Res. 2010, 49, 2449.
14. Seybold, P. G. Analysis of the pKas of Aliphatic Amines Using Quantum
Chemical Descriptors. Int. J. Quant. Chem. 2008, 108, 2849.
15. Burger, S. K.; Liu, S.; Ayers, P. W. Practical Calculation of Molecular Acidity
with the Aid of a Reference Molecule. J. Phys. Chem. A 2011, 115, 1293.
16. Gaussian03, Revision E.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.;
Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven,
T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone,
V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji,

114
H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.;
Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.;
Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann,
R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala,
P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski,
V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.;
Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul,
A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.;
Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.;
Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.;
Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian Inc.: Wallingford,
CT, 2004.
17. Gaussian09, Revision B.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.;
Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.;
Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.
P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara,
M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.;
Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, Jr., J. A.; Peralta, J. E.; Ogliaro,
F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.;
Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.;
Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N. J.; Klene, M.; Knox,
J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.;
Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J.

115
W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.;
Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.;
Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian, Inc., Wallingford CT, 2010.
18. Perrin, D. D.; Dempsey, B.; Serjeant, E. P. pKa Prediction for Organic Acids and
Bases, Chapman and Hall: New York, 1981.
19. A relevant example is Hall, H. K. Correlation of the Base Strengths of Amines. J.
Am. Chem. Soc. 1957, 79, 5441.
20. Tissandier, M. D.; Cowen, K. A.; Feng, W. Y.; Gundlach, E.; Cohen, M. H.;
Earhart, A. D.; Coe, J. V.; Tuttle Jr., T. R. The Proton's Absolute Aqueous
Enthalpy and Gibbs Free Energy of Solvation from Cluster-Ion Solvation Data.
J. Phys. Chem. A 1998, 102, 7787.
21. Ben-Naim, A.; Mazo, R. M. Size Dependence of the Solvation Free Energies of
Large Solutes. J. Phys. Chem. 1993, 97, 10829.
22. Klicic, J. J.; Friesner, R. A.; Liu, S.-Y.; Guida, W. C. Accurate Prediction of
Acidity constants in Aqueous Solution via Density Functional Theory and Self-
Consistent Reaction Field Methods. J. Phys. Chem. A 2002, 106, 1327.
23. W. M. Haynes, ed., CRC Handbook of Chemistry and Physics, 92nd Edition,
CRC Press/Taylor and Francis, Boca Raton, FL, 2011.
24. Sadlej-Sosnowska, N. Calculation of Acidic Dissociation constants in Water:
Solvation Free Energy Terms. Their Accuracy and Impact. Theor. Chem. Acc.
2007, 118, 281.

116
25. Behjatmanesh-Ardakani, R.; Karimi, M. A.; Ebady, A. Cavity Shape Effect on
pKa Prediction of Small Amines. J. Mol. Struc. Theochem 2009, 910, 99.
26. Clemente, F. R., private communication.
27. Scalmani, G.; Frisch, M. Continuous Surface Charge Polarizable Continuum
Models of Solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110.
28. Atom volumes were determined by a group contribution scheme, fitted to the
total volumes of the 17 bases as calculated by Gaussian’s overlapping-spheres
procedure.
29. Marten, B.; Kim, K.; Cortis, C.; Friesner, R. A.; Murphy, R. B.; Ringnalda, M.
N.; Sitkoff, D.; Honig, B. New Model for Calculation of Solvation Free Energies:
Correction of Self-Consistent Reaction Field Continuum Dielectric Theory for
Short-Range Hydrogen-Bonding Effects. J. Phys. Chem. 1996, 100, 11775.
30. Head-Gordon, T. Is Water Structure around Hydrophobic Groups Clathrate-like?
Proc. Natl. Acad. Sci. USA 1995, 92, 8308.
31. Hawkins, R. E.; Davidson, D. W. Dielectric Relaxation in the Clathrate Hydrates
of Some Cyclic Ethers. J. Phys. Chem. 1966, 70, 1889.
32. Effects of up to 4 pKa units were reported in a continuum-solvation-based
computational study of serotonin: see Pratuangdejkul, J.; Nosoongnoen, W.;
Guérin, G.-A.; Loric, S.; Conti, M.; Launay, J.-M.; Manivet, P. Conformational
Dependence of Serotonin Theoretical pKa Prediction. Chem. Phys. Lett. 2006,
420, 538.

117
33. Hamborg, E. S.; Versteeg, G. F. Dissociation constants and Thermodynamic
Properties of Amines and Alkanolamines from (293 to 353) K. J. Chem. Eng.
Data 2009, 54, 1318.
34. Han, B.; Zhou, C.; Wu, J.; Tempel, D. J.; Cheng, H. Understanding CO2 Capture
Mechanisms in Aqueous Monoethanolamine Via First Principles Simulations. J.
Phys. Chem. Lett. 2011, 2, 522.
35. Lopez-Rendon, R.; Mora, M. A.; Alejandre, J.; Tuckerman, M. E. Molecular
Dynamics Simulations of Aqueous Solutions of Ethanolamines. J. Phys. Chem.
B 2006, 110, 14652.
36. da Silva, E. F.; Kuznetsova, T.; Kvamme, B.; Merz Jr., K. M. Molecular
Dynamics Study of Ethanolamine as a Pure Liquid and in Aqueous Solution. J.
Phys. Chem. B 2007, 111, 3695.
37. Gubskaya, A. V.; Kusalik, P. G. Molecular Dynamics Simulation Study of
Ethylene Glycol, Ethylenediamine, and 2-Aminoethanol. 2. Structure in Aqueous
Solutions. J. Phys. Chem. A 2004, 108, 7165.
38. Omura, Y.; Shimanouchi, T. Raman Spectra and Rotational Isomerism of
Ethylenediammonium and Monoethanolammonium Ions in Aqueous Solutions.
J. Mol. Spectrosc. 1975, 55, 430.
39. Khalili, F.; Henni, A.; East, A. L. L. pKa Values of Some Piperazines at (298,
303, 313, and 323) K. J. Chem. Eng. Data 2009, 54, 2914.

118
40. Hamborg, E. S.; Niederer, J. P. M.; Versteeg, G. F. Dissociation constants and
Thermodynamic Properties of Amino Acids Used in CO2 Absorption from (293
to 353) K. J. Chem. Eng. Data 2007, 52, 2491.
41. Bower, V. E.; Robinson, R. A.; Bates, R. G. Acidic Dissociation constant and
related Thermodynamic Quantities for Diethanolammonium Ion in Water from 0°
to 50 °C. J. Res. Natl. Bur. Stand. 1962, 66A, 71.
42. Schwabe, K.; Graichen, W.; Spiethoff, D. Physicochemical Investigations of
Alkanolamines. Z. Phys. Chem. (Munich) 1959, 20, 68.

119
Chapter 4: Reaction Mechanisms in CO2/Aqueous Amine Systems
_______________________________________________________________________
4.1 INTRODUCTION
Meaningful rate constant data for the important CO2-capture reaction
(4.1)
(B = {H2O, OH-, HXYN},) requires agreement about the rate law used to fit to the
experimental data. Two different but widely used mechanisms (the zwitterion1 and
termolecular2 mechanisms) have resulted in competing formalisms and impede
understanding and a third mechanism (the carbamic acid mechanism), a hypothesis
known to engineers in the 1970s,2 has recently been re-emphasized.4,5 A new and
improved modeling study of reaction (4.1) is needed because of various issues arising
from previous modeling efforts. This chapter reports the results of improved quantum
chemistry studies of this reaction, with hopes of clarifying the reaction mechanism.
4.1.1 Overview of competing mechanisms proposed
Zwitterion mechanism. This two-step mechanism, favored by Versteeg,6,7 was a
simplification by Danckwerts2 of a three-step mechanism originally proposed by
Caplow.1 The mechanism assumes a formation of an intermediate zwitterion which then
undergoes a deprotonation by basic molecules resulting in carbamate ion formation:

120
(4.2)
(4.3)
The mechanism only applies to primary and secondary amines due to the need for an NH
bond in the amine HXYN. Caplow’s original mechanism considered the zwitterion’s NH
group to be hydrogen-bonded to a water molecule, and featured an extra proton-transfer
middle step which allows the intermediate to be either H2O·H+XYNCOO− or
H3O+·XYNCOO−.
Applying the steady-state principle to the intermediate zwitterion in eqs 4.2 and
4.3, one obtains the following expression for the rate of reaction of CO2 ( :
∑
, (4.4)
where (4.5)
Here is the overall reaction rate constant for CO2 loss, and , , and are the
elementary rate constants in steps (2) and (3). B denotes any proton-accepting species
present; if one considers {H2O, OH-, HXYN}, then one has elementary rate constants kB
= {kw, kOH-, and kHXYN} and the corresponding composites KB from eq. (4.5).
Equation (4.4) is very general and can cover aqueous and non-aqueous amine
solutions. Fitting this equation to experimental data for aqueous amines would give
values for the three KB’s as well as , but this 4-parameter function often has
indeterminacy problems. For simplification, Danckwerts2 reiterated Caplow’s two

121
limiting cases (and generalized them to allow for other possibilities of B): simplifications
arise if k-1 << ∑ (eq. 4.6) or k-1 >> ∑ (eq. 4.7).
(4.6)
(4.7)
Equation (4.7), like (4.5), allows for fractional orders of amine between 1 and 2.
Termolecular mechanism. In 1990 Crooks and Donnellan3 challenged the belief
in an unobserved carbamate zwitterion intermediate, instead suggesting that overall
reaction (4.1) consists of one elementary termolecular step.4,5 The resulting rate law
matches eq. (4.7) but without the eq. (4.5) interpretation of the KB constants.
Meader and co-workers4,5 have frowned upon this mechanism, using the usual
theoretical argument that a termolecular elementary step requires simultaneous collision
of 3 entities. However, in solutions in which strong hydrogen bonding can occur,
complexes of 2 of the 3 entities pre-exist and this counter-argument is invalid. Crooks
and Donnellan did in fact suggest that the single step starts from an “encounter complex”
that is not zwitterionic. If formation of an encounter complex does not result in loss of
CO2 signal (like zwitterion formation would), the complexation stage would not affect
measured rate of CO2 loss, and hence CO2 loss would be solely due to the ensuing
concerted elementary step involving two bonds forming and two bonds breaking
amongst the three entities. This is what should be inferred from the label “termolecular
mechanism.”

122
Carbamic acid mechanism. Meader and co-workers4,5 favors the two-step
mechanism in which the intermediate is not the zwitterion but hydrogen carbamate
(“carbamic acid”) molecules:
(4.8)
(4.9)
Step (4.8) cannot be elementary on its own due to egregious bond strain that would exist
in a concerted transition state. It might be possible as a termolecular step from a Crooks-
Donnellan encounter complex with H2O or alcohol solvent molecules, in which case, 3
bonds are breaking and forming in a concerted manner.
Bicarbonate mechanisms. Equation (4.1) (carbamate formation) is not the only
CO2-capture reaction possible in amine solutions. It is known that ordinary basic
solutions can convert CO2 to bicarbonate in small amounts:
(4.10)
or (4.11)
Eq. (4.11), a base-catalyzed bicarbonate mechanism proposed by Donaldson and
Nguyen,8 is believed to be operative in the case of tertiary amines, which cannot form
carbamates. This base-catalyzed bicarbonate mechanism is, like the Crooks-Donaldson
base-catalyzed carbamate mechanism, a “termoleculecular” single-step mechanism, in
which two bonds are broken and two bonds are formed in a concerted step amongst the
three molecular entities within an encounter complex. The rate expression for (4.11) is:
(4.12)
where should be independent of amine concentration.

123
4.1.2 Previous modeling studies
Obtaining realistic mechanistic results using theoretical molecular models depend
on a number of factors – one being the use of a proper molecular model capable of
representing the essential chemistry of the real liquid solution. For liquid phase reactions
involving ions, one important modeling aspect is the stabilization of ions by polar
solvents: polar solvents will lower the energy of ionic intermediates and transition states
dramatically, thus affecting the rate and even course of reactions. A second aspect,
important in the current case of aqueous amines, is the local hydrogen-bonding solvation
effects.
A continuum solvation model (CSM), such as PCM,9 SMx,10 and COSMO,11 can
be used to incorporate most of the first effect. In CSM, the solute molecule or ion is
hypothetically placed in a semiempirically built cavity inside a continuum dielectric
having an adjustable polar strength parameter ε. Unfortunately, the use of a CSM does
not provide the effects of hydrogen bonding. Use of explicit solvent molecules in the
modeling (with or without CSM) is a common way of incorporating this second effect.
Previous computational chemistry studies of equation (1) have tried both of these
modeling strategies, but the results reveal problems which my work aims to address.
These problems are now discussed.
Non-observance of carbamate in single-amine modeling: It has been known
since 1925 that the dominant initial product upon reacting CO2 with aqueous amines is
carbamate ion12. To date, however, single-amine models (regardless of their use of CSM

124
or explicit water molecules) have been unable to produce carbamate as intermediates at
all (summarized in Table 4.1).
Table 4.1 Reaction Pathways Observed in Single-Amine Modeling
Reference Level of modeling
Results
13 (Arstad et al., 2007)
Gas phase with explicit water
CO2+MEA+H2O carbamic acid CO2+DEA+H2O carbamic acid
14 (Shim et al., 2009) IEFPCM CO2+MEA+H2O zwitterion
15 (Xie et al., 2010) CPCM CO2+MEA+H2Ozwitterion carbamic acid
16 (Han et al., 2011) COSMO CO2+MEA+H2O carbamic acid
17 (Ismael et al., 2009)
COSMO CO2+AMP+H2O carbamic acid
18 (Yamada et al., 2011)
IEFPCM-SMD CO2+AMP+H2O zwitterion carbamic acid
Altogether, the most common product in Table 4.1 is carbamic acid. We
speculated that more explicit water might properly stabilize the anionic product and we
will show that we were correct.
Disagreement on intermediate energies in double-amine modeling. Previous
two-amine-molecule studies (summarized in Table 4.2) have succeeded in finding
carbamate ions as intermediates, likely because these studies mimic very basic pH
conditions. These studies also generally observe zwitterion intermediates, but disagree
on the relative stability of zwitterion vs. carbamate ion.

125
Table 4.2 Reaction Pathways Observed in Double-Amine Modeling
Reference Level of modeling
Results
13 (Arstad et al., 2007)
Gas phase with explicit water
CO2+2MEA carbamic acid and CO2+2MEA carbamate
14 (Shim et al., 2009) IEFPCM CO2+2MEA carbamate
15 (Xie et al., 2010) CPCM MEA+MEA-zwitterioncarbamate
16 (Han et al., 2011) COSMO MEA+MEA-zwitterioncarbamate
18 (Yamada et al., 2011)
IEF-PCM-SMD MEA+AMP-zwitterioncarbamate
Shim et al. (2009)14 found a one-step mechanism with crude coordinate scanning,
which made them conclude that the overall reaction (CO2+2MEA) was termolecular,
although they stressed that a more concrete conclusion regarding zwitterion stability
warrants further consideration of solvation effect through explicit water molecules.
However, three later papers15,16,17 used proper transition state search and found a step
from zwitterion to carbamate ion. Puzzlingly, they disagree on the exothermicity of this
step. Again, we speculated that more explicit H2O in the modeling would help, and
properly show the consecutive exothermic steps.

126
Role of catalyst in two-amine modeling. Due to the non-observance of carbamate
in single-amine modeling, Shim et al. (2009)14 undermined the role of water as base, and
then emphasized its catalytic role in proton relay in two-amine modeling. They
hypothesized a quadrumolecular mechanism where a water molecule assists in amine-to-
amine proton-transfer. However, their hypothesized mechanistic pathway has not
received attention in subsequent studies. On the other hand, the catalytic role of amine in
two-amine modeling was emphasized by Arstad et al. (2007).13 They studied formation
of both carbamate and carbamic acid in separate pathways from (CO2+2MEA), and
thought that the amine-catalyzed ‘carbamic acid’ formation pathway was the most
plausible mechanism as it gave reaction order 1 with respect to MEA and had the right
activation energy. However, this has not been properly studied using a CSM. We will
study these mechanistic pathways at the same level of modeling as in other mechanisms.
Carbamic acid equilibria. None of the previous studies discuss the possible pH-
dependent equilibrium between “carbamic acid”/carbamate/zwitterion, relevant to the
carbamic acid hypothesis (eqs. 4.8 and 4.9).
Origin of activation energy barrier. da Silva et al. (2004)19 hypothesized that the
reaction barrier for CO2 absorption in aqueous monoethanol amine solution is caused by
the displacement of water molecules in the solvation shell of the amine group as CO2
approaches. The potential of mean force (PMF) calculations of Xie et al. (2010)15 also
suggest there is a small barrier due to solvent displacement in the formation of
zwitterion. However, PCM calculations published so far14-18 did not consider the solvent
displacement and hence drew different conclusions. We will study the reaction of CO2
with an amine which is N-HO and/or H-OH hydrogen-bonded with water (or N-HN

127
and/or NH-N hydrogen-bonded with other amine) using PCM calculations to see if a
transition state involving H2O displacement can be found.
Effect of pKa on mechanism. None of the previous studies have investigated the
effects of the basicity of the amine upon mechanism.
Thus, we still lack a good modeling study that can address the three hypotheses
for the mechanism of the CO2-capture reaction. In this work, a semi-continuum CSM-
plus-explicit-water model is used, addressing Shim et al.’s call for more explicit water
molecules in the modeling by adding as many as twenty in some instances. It will be
shown that such consideration will have important consequences in many aspects of the
reaction: for example, observance and relative stability of intermediates, activation
energies depending on degree of solvation of transition states or reactants, reaction
energy barrier due to solvent displacement, complexity in modeling bicarbonate
formation, and basicity (pKa) effects.

128
4.2 METHOD
In the pKa study (Chapter 3), hydrogen bonding was treated in a semi-continuum
manner: by including an explicit water molecule forming a hydrogen bond with the
neutral amine and protonated amine. Here, in studying the mechanistic pathway of
reaction of CO2 in aqueous alkanolamine, the same technique is used, since the cation,
anion, zwitterion, and neutrals are all capable of forming hydrogen bonds.
All calculations were done using the Gaussian0920 computational chemistry
software program on in-house supercomputer Dextrose. The electronic structure
approximation used was a density functional theory (DFT): the B3LYP 21,22 functional
combination, with the 6-31G(d) basis set for molecular orbital construction. The
polarization effect of solvation was modeled using a polarizable continuum model
(PCM), with UFF atomic radii used for overlapping-sphere solute cavity construction
within the continuum (algorithm: scrf=IEFPCM; solvent=water).23 Energies are
generally reported “raw” without zero-point and thermal-correction energies.
Full molecule geometry optimizations were performed. For determining
activation energies, the algorithm opt=(ts, calcfc, noeigentest)24 was used for transition
state (TS) optimization. Transition states were confirmed by vibrational frequency
computation (freq=noraman; one imaginary frequency needed) and by two energy
minimization runs, each starting from the TS geometry but with some atoms displaced
according to the direction shown in the imaginary-frequency normal mode.

129
Most results employ methylamine (MeNH2) or monoethanolamine (MEA) as the
amine HXYZ, with varying numbers of explicit H2O molecules in a hydrogen-bonded
cluster.
4.3 RESULTS AND DISCUSSION
This section is organized as follows. Demonstration of the need for many explicit
water molecules to determine the dominant intermediate occurs in section 4.3.1. This is
followed by reaction pathways originating from single-MEA modeling
(CO2+MEA+nH2O) and double-MEA modeling (CO2+2MEA+nH2O) in sections 4.3.2
and 4.3.3, with a cross comparison in section 4.3.4. The alternative bicarbonate-
formation pathways, and the effects of varying pKa (i.e. choosing other amines) on
reaction mechanisms, are discussed in sections 4.3.5 and 4.3.6 respectively.
4.3.1 Effect of spectator water molecules on ion solvation
The main problem with previous computational modeling of the CO2 + amine
reaction has been the peculiar non-observance of the well-known carbamate anion
product. To investigate why, a series of computations were performed, using
methylamine as a test amine, to determine the exothermicity of the carbamate-
producing reactions R1-R4 as functions of the number of water molecules explicitly
used to solvate the solute species. R1 and R3 form carbamate ions from the zwitterion,
whereas R2 and R4 form carbamate ions from carbamic acid molecules.

130
Single amine (neutral pH) model:
MeNH2COO·(n H2O) + H2O·(n H2O) MeNHCOO−·(n H2O) + H3O+·(n H2O)
(R1)
MeNHCOOH·(n H2O) + H2O·(n H2O) MeNHCOO−·(n H2O)) + H3O+·(n H2O)
(R2)
Double-amine (very basic pH) model:
MeNH2COO·(n H2O) + MeNH2·(n H2O) MeNHCOO−·(n H2O) + MeNH3+·(n H2O)
(R3)
MeNHCOOH·(n H2O) + MeNH2·(n H2O) MeNHCOO−·(n H2O) + MeNH3+·(n H2O)
(R4)
The B3LYP/6-31G(d)/UFF-PCM level of theory was used, and lowest-energy
conformers (Figure 4.1) were located from multiple trials. The raw ΔE results for these
reactions are graphically shown in Figure 4.2. As the number of explicit water
molecules was increased, a large lowering of ΔE was seen in Figure 4.2 for all four
reactions, due to improved solvation of the two created ions. Without explicit waters,
one would mistakenly conclude that the thermodynamically favored species in the
carbamate equilibria is either zwitterion or carbamic acid molecules. Thus, the peculiar
non-observance of carbamate anions in previously published work is due to improper
omission of explicit H2O molecules in the models.
Upon proper addition of explicit waters, Figure 4.2 reveals that the carbamate
anion becomes competitive at neutral pH (R1, R2), and dominant at basic pH (R3, R4).
This brings the modeling results into agreement with experimental observations,25 that
carbamates are quite unstable at neutral pH, but stable for hours in basic solution.

131
Figure 4.1 Conformers of X·(n H2O) complexes used in section 4.3.1 (B3LYP/6-
31G(d)/UFF-PCM).
n=0 n=1 n=2 n=3
Zwitterion
MeNH2COO
Carbamate
MeNHCOO−
Carbamic
Acid
MeNHCOOH
MeNH2
MeNH3+
H2O
H3O+
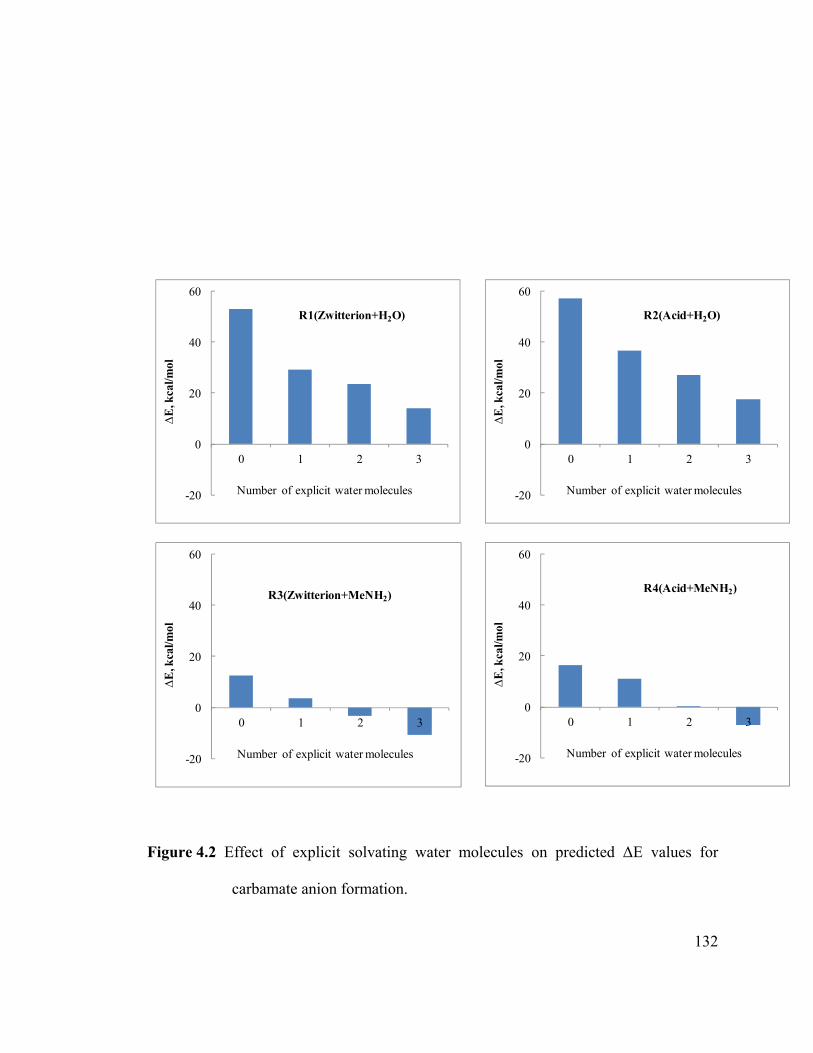
132
Figure 4.2 Effect of explicit solvating water molecules on predicted ΔE values for
carbamate anion formation.
-20
0
20
40
60
0 1 2 3
∆E
, kca
l/mol
Number of explicit water molecules
R1(Zwitterion+H2O)
-20
0
20
40
60
0 1 2 3
∆E
, kca
l/mol
Number of explicit water molecules
R2(Acid+H2O)
-20
0
20
40
60
0 1 2 3
∆E
, kca
l/mol
Number of explicit water molecules
R3(Zwitterion+MeNH2)
-20
0
20
40
60
0 1 2 3
∆E
, kca
l/mol
Number of explicit water molecules
R4(Acid+MeNH2)

133
4.3.2 Carbamate formation at neutral pH
The mechanistic pathway for the formation and breakdown of carbamate at
neutral pH (single-amine modeling) will be explored in this section, using MEA as the
amine. Results are presented for 1-H2O, 5-H2O and 20-H2O models. The mechanisms
observed appear in Figure 4.3. Clearly, mechanisms incorporating other numbers of
H2O in the proton relay are also possible.
6-atom cycle
(1-H2O modeling)
O C O
N
HR
RH
O
H
H
OH
H
OH
10-atom cycle
(5-H2O modeling)
12-atom cycle
(20-H2O modeling)
Figure 4.3 Reaction mechanisms observed in the modeling of eq (1) with B=H2O.
6-atom cycle. In 1-amine-1-H2O modeling, the 6-atom cycle in Figure 4.3
(involving 2 H transfers) occurred in a 2-step pathway, forming carbamic acid via
zwitterion intermediate. Structures and energies appear in Figure 4.4. This poor level of

134
modeling, employed by others 13-18 results in an overly high barrier between zwitterion
and acid forms, with no carbamate intermediate there.
TS1 reactant
TS1
TS1 product
TS2 reactant
TS2
TS3 product
Figure 4.4 B3LYP/6-31G(d)/UFF-PCM results for 1-amine-1-H2O modeling.
Elementary-step activation energies in parentheses.
-20
-15
-10
-5
0
5
10
15
20
Rel
ativ
e en
ergy
, kca
l/mol
TS1 reactant(Initial complex)
TS1 product/TS2 reactant(Zwitterion)
TS1 (0.94)
TS2(10)
TS2 product(Carbamicacid)

135
10-atom cycle. In 1-amine-5-H2O modeling, the pathway found was a 10-atom
cycle (involving 4 H transfers) occurring in a 3-step pathway. Structure and energies are
shown in Figure 4.5. Here, the first step is again simple zwitterion formation, but it
involves displacement by CO2 of an initial N…H hydrogen bond between amine N and
a water molecule (labeled as H23-O22-H24 in Figure 4.5) and consequently has a
higher activation energy barrier than in Figure 4.4. (Note that da Silva and Svendsen
believed that the activation energy barrier for CO2 capture by MEA arises from water
displacement.) In the second step, the zwitterion here converts not into a carbamic acid
directly, but first into a carbamate intermediate by the abstraction of the proton by
another water molecule (H17-O9-H15) that acts as base in this step. The presence of
additional explicit water molecules has considerably stabilized the creation of two
molecular ions here (the carbamate anion and a hydronium cation). The third step
produces the acid form via more H atom exchanges. So, now we see we have enough
H2O in the model to see a carbamate anion intermediate and a reduction of the overly
high barrier in 1-amine-1-H2O modeling. Unfortunately, this level of modeling is also
poor because of the predicted final step to an excessively stable carbamic acid, a form
which has not been experimentally observed.

136
TS3 reactant
TS3 product (zwitterion)
TS4 reactant (zwitterion)
TS4 product (carbamate)
TS5 reactant (carbamate)
TS5 product (acid)
Figure 4.5 B3LYP/6-31G(d)/UFF-PCM results for 1-amine-5-H2O modeling.
Elementary-step activation energies in parentheses.
-20
-15
-10
-5
0
5
10
15
20
Rel
ativ
e en
ergy
, kca
l/mol
TS3(7.58)
TS4(3.64)
TS4product(Carbamate)
TS3 product(Zwitterion)
TS5reactant(Carbamate)
TS5(0.49)
Carbamicacid
TS3reactnat(Initialcomplex)
TS4 reactant(Zwitterion)

137
12-atom cycle. In 1-amine-20-H2O modeling, the pathway found was a 12-atom
cycle (involving 5 H transfers). The initial step of zwitterion formation was omitted due
to the uncertainty in knowing how many H-bonds to displace in this bimolecular step.
Transition structures and energies for the 2nd and final steps appear in Figure 4.6. With
the H2O molecules now properly surrounding the amino and carboxyl groups, and the
addition of more explicit H2O, the carbamates are now seen to be thermodynamically
competitive with both the zwitterion and carbamic acid forms at the B3LYLP/6-
31G(d)/UFF-PCM level (Figure 4.6). The single-point energy calculations at the
alternative MP2/6-31G(d,p)/UFF-PCM level are presented to point out that the expected
accuracy of B3LYP and MP2 is perhaps 2-4 kcal/mol.
The 1-amine-20-H2O data should be the best to date for neutral-pH modeling. If
the encounter complex has similar raw energy to these intermediates, as suggested by
Figures 4.4 and 4.5, then it would have the lowest free energy of all these forms because
of the entropy benefit of dissociation; this agrees with the fact that carbamate in basic
solutions quickly decomposes to CO2 + amine when brought to pH = 7.25

138
TS6 (proton H57 is being transferred
from N23 to O15)
TS8 ((proton H38 is being
transferred from O8 to.O21)
Figure 4.6 Results for 1-amine-20-H2O modeling. Energy profiles are at B3LYP/6-
31G(d) (square) and at MP2/6-31G(d,p) (triangle) level. Elementary-step
activation energies at the B3LYP/6-31G(d) level in parentheses.
-20
-15
-10
-5
0
5
10
15
20
Rel
ativ
e p
oten
tial
en
ergy
, k
cal/m
ol
Zwitterion
TS6(3.56)
Carbamate Carbamate Acid
TS7(0.01)
TS8(4.46)

139
4.3.3 Carbamate formation at basic pH
In concentrated alkanolamine solutions of basic pH, carbamate ions can be stable
for several hours 25, so the energetics must be different than that of Figure 4.6. Basic pH
systems are better modeled by having two amine molecules in the model; thus, this
section deals with 2-amine-molecule modeling.
Initial modeling without spectator water molecules (B3LYP/6-31G(d)/UFF-PCM
level of theory) produced 3 different atom-cycle possibilities; the observed mechanisms
are summarized in Figure 4.7.
6-atom cycle
5-atom cycle
8-atom cycle
Figure 4.7 Reaction mechanisms observed in the modeling of eq (1) with B=amine.
5 and 6-atom cycle. The 6-atom cycle was first studied. A 2007 gas-phase study
13 predicted a single-step pathway forming carbamic acid products. However, results
here with the PCM continuum solvent shell predict this pathway to be multi-step. With
the usual all-trans conformer of the amine MEA, calculations here predict this pathway

140
to be two-step via zwitterion intermediate (Figure 4.8), just as in our 6-atom cycle in
neutral-pH modeling (Figure 4.4). Out of curiosity, the 6-atom cycle was also
investigated here with a maximally internally H-bonded initial complex, generated by
use of gauche (instead of trans) MEA. This gauche-MEA pathway produced a three-step
mechanism, with carbamate ion as the additional intermediate (Figure 4.9). Figure 4.9
reveals greater activation energy than Figure 4.8 (7 vs. 1 kcal/mol for the first step)
because there are more H-bonds to break to get to the first transition state. In addition, a
5-atom three-step cycle (Figure 4.10) was found by reorienting the all-trans MEA
molecules in Figure. 4.8 in a manner more consistent with the orientation used by
others15,16 for the zwitterion-to-carbamate step. This cycle is a 5-atom cycle because the
H atom donated to the catalytic amine in the second step is the same one that is returned
to the carbamate ion in the third step.
Most importantly, none of these 2-amine-0-H2O modeling results (Figures 4.8-
4.10) show a thermodynamic preference for the carbamate ion as the product, and hence
they do not agree with experiment (Wang et al, 1972).25 There are not enough explicit
H2O molecules in the model to show the expected stability of the carbamate ion.
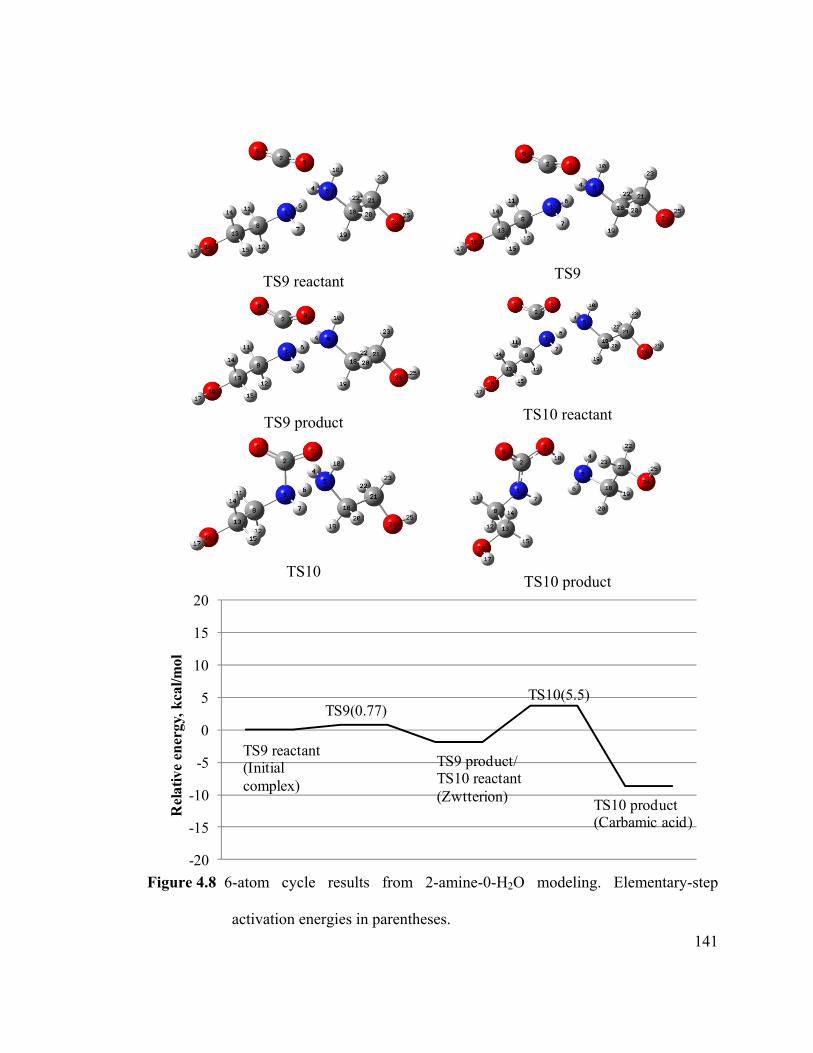
141
TS9 reactant
TS9
TS9 product
TS10 reactant
TS10
TS10 product
Figure 4.8 6-atom cycle results from 2-amine-0-H2O modeling. Elementary-step
activation energies in parentheses.
-20
-15
-10
-5
0
5
10
15
20
Rel
ativ
e en
ergy
, kca
l/mol
TS9 reactant(Initial complex)
TS9 product/TS10 reactant(Zwtterion)
TS9(0.77)TS10(5.5)
TS10 product(Carbamic acid)

142
TS11reactant
TS11
TS12
TS12 product
TS13
TS13 product
Figure 4.9 6-atom cycle results for 2-amine-0-H2O modeling, but with gauche MEA for
maximal H-bonding at the outset. Elementary-step activation energies in
parentheses.
-20
-15
-10
-5
0
5
10
15
20
Rel
ativ
e en
ergy
, kca
l/mol
TS11(InitialComplex)
TS11 (7)
TS13(0.64)
TS12(5.4)
TS11 product(Zwtterion) TS12 product
(Carbamate) TS13 product(Acid)

143
TS14 reactant TS14
TS15
TS15 product
TS16
TS16 product
Figure 4.10 5-atom cycle results from 2-amine-0-H2O modeling. Elementary-step
activation energies in parentheses.
-20
-15
-10
-5
0
5
10
15
20
Rel
ativ
e en
ergy
, kc
al/m
ol
TS14 reactant(InitialComplex)
TS16(3.7)
TS16 reactant(Carbamate)
TS16 product(Acid)
TS15 (5)TS14 (0.77)
TS15 product(Carbamate)TS14 product
(Zwtterion)

144
8-atom cycle. An 8-atom cycle (shown earlier in Figure 4.7, right) was found by
adding one water molecule between the two amines, to test a recent hypothesis (Shim et
al. 2009).14 The resulting structures and PEP of the three-step pathway are shown in
Figure 4.11. Comparison of the energetics here to the corresponding 6-atom cycle
(Figure 4.8) shows that adding an H2O molecule to the relay produced a slightly higher
energy pathway. More importantly, this 2-amine-1-H2O pathway, unlike the 2-amine-0-
H2O pathways of Figures 4.8-4.10, did produce the carbamate ion as the lowest-energy
form along the pathway, but not as an energetically dominant species. More H2O
molecules are probably required, as they were in the neutral-pH modeling of the previous
section.
As an initial check of the benefits of spectator H2O molecules, two were
sequentially added to the TS44 structure of Figure 4.10 and reoptimized, together with
the related reactants (zwitterion) and products (carbamate). The results (Figure 4.12)
show that the elementary reaction energy for this step is lowerd 4 kcal/mol for each of
the two waters added, confirming our suspicion that adding spectator waters would
improve the thermochemistry.

145
TS17 product
TS18
TS18 product
TS19 product
Figure 4.11 8-atom cycle with 2-amine-1-H2O modeling. Elementary-step activation
energies in parentheses.
-20
-15
-10
-5
0
5
10
15
20
Rel
ativ
e en
ergy
, kca
l/mol
TS19 product(Carbamicacid)
TS18(7.24)
TS17(0.72)
TS17 reactant(Initialcomplex) TS18 product
(Carbamate)
TS19(2.9)
TS17 product(Zwitterion)

146
TS15
TS15 product
TS20
TS20 product
TS21
TS21 product
Figure 4.12 Effect of varying n in 2-amine-n-H2O modeling (zwitterion-to-carbamate
step). Elementary-step activation energies in parentheses.
-4
0
4
8
Rel
ativ
e en
ergy
, kc
al/m
ol
No water molecule One water molecule Two water molecules
Carbamate
Zwitterion
Carbamate
Carbamate
TS15(5.06)
TS20(2.75)
TS21(0.99)

147
Finally, a 2-amine-18-H2O model (8-atom cycle) was attempted. Now the
carbamate ion intermediate is correctly predicted to be the thermodynamically dominant
product of reaction (Figures 4.13 and 4.14). Note the negligible activation barrier of
0.16 kcal/mol for its formation from zwitterion; this concurs with conclusions drawn
from lower-level modeling (Xie 2010).15 Extending the proton relay by adding a 19th
H2O between the two amines (as we did in going from Figure 4.8 to Figure 4.11) was
also performed but it increased the activation energy (to 2.1 kcal/mol) as it did before, so
the Figure 4.13 pathway represents the more plausible pathway at basic pH.

148
Figure 4.13 Results from 2-amine-18-H2O modeling. Elementary-step activation
energies in parentheses.
-20
-15
-10
-5
0
5
10
15
20
Rel
ativ
e en
ergy
, kca
l/mol
TS22 product (Carbamate)
TS23(B3LYP=18.7)
TS 22(B3LYP=0.16)
TS22 reactant(Zwitterion)
TS23 product(Carbamic Acid)
TS23 reactant(Carbamate)
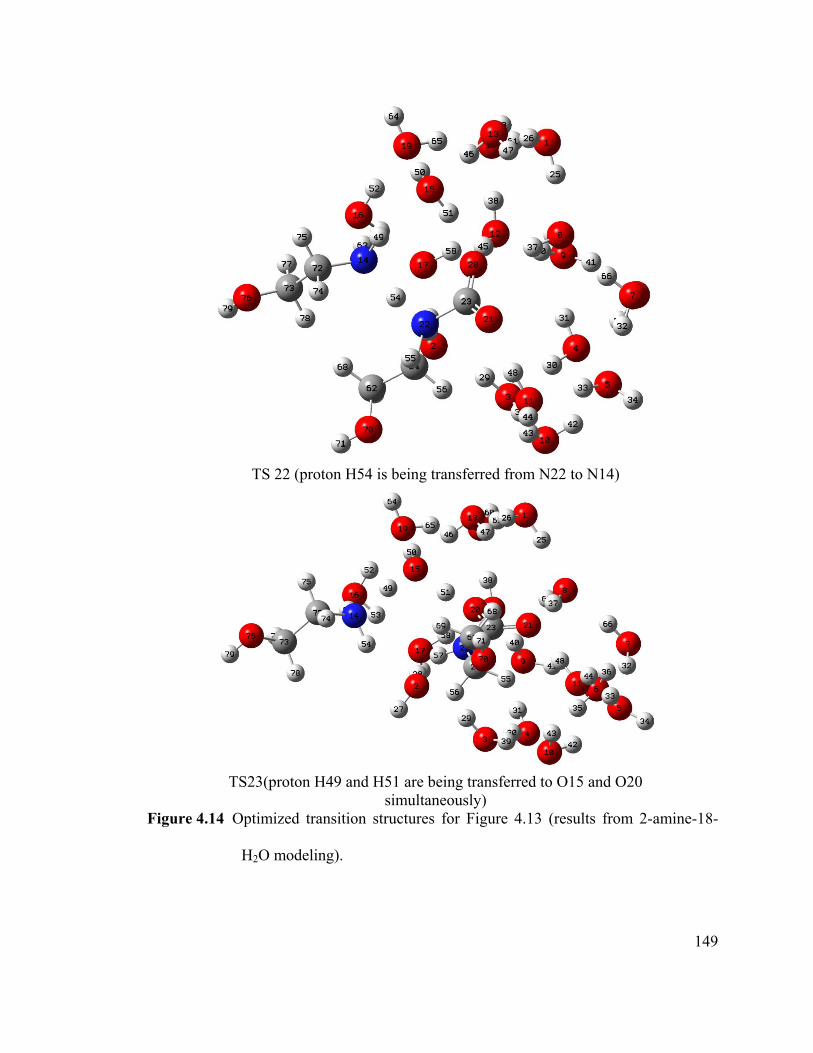
149
TS 22 (proton H54 is being transferred from N22 to N14)
TS23(proton H49 and H51 are being transferred to O15 and O20 simultaneously)
Figure 4.14 Optimized transition structures for Figure 4.13 (results from 2-amine-18-
H2O modeling).

150
4.3.4 Discussion: Formation of carbamate
It is apropos to compare the best results from basic-pH modeling (Figure 4.13) to
those of neutral-pH modeling (Figure 4.6). We focus on the zwitterion-to-carbamate
elementary step, whose exothermicity is significantly enhanced by additional explicit
water molecules in the modeling. The smaller activation barrier in two-amine modeling
vs. one-amine modeling is due to its transition state being “earlier” (Hammond’s
Postulate), which in turn is due to the step being more exothermic (Bell-Evans-Polanyi
Principle). To demonstrate the increasing “earliness” of the TS for this step in 2-amine
modeling, the activation energies from Figures 4.12 and 4.13 are plotted in Figure 4.15
against the N-H bond length of the zwitterion, which increases from 1.1 Å to 1.8 Å
during this step. As more H2O molecules are added (going left to right in the plot), the
increased exothermicity drives down the activation energy (triangle points), which
directly correlates to the transition-state N-H bond length occurring earlier (diamond
points). In fact, the increased exothermicity even impacts the N-H bond length in the
reactant zwitterion, mildly stretching it (square points). The activation energy barrier is
proportional to the vertical distance between the square and triangle points (i.e. to the
amount the N-H distance has to stretch to get to the TS), which shrinks with increasing
solvation of the system. For comparison, at the right side of Figure 4.15, the data from
the single-amine-20-water modeling (“A20”) previously presented in Figure 4.6 is
shown. Comparing A20 to B18 (double-amine-18-water modeling) data, the transition

151
state for A20 modeling is “later” (R (N-H) =1.35 vs. 1.20 Å), correlating with its larger
barrier (3.5 vs. 0.2 kcal/mol) for this exothermic step.
Figure 4.15 Effect of level of modeling on zwitterion deprotonation step
0
2
4
6
8
10
1
1.1
1.2
1.3
1.4
1.5
B0 B1 B2 B18 A20
Act
ivat
ion
en
ergy
(k
cal/m
ol)
N-H
bon
d le
ngt
h(Å
)
Level of modelling denoted by A (single-amine) and B (double-amine) followed by the number of water molecules in the model
N-H distance in TSN-H distance in zwitterionActivation energy

152
Comparison of Figure 4.6 (neutral pH) with Figure 4.13 (basic pH) suggests that
the zwitterion intermediate plays a role in dilute solutions where the amines are
completely solvated with water molecules (and perhaps for the ones Caplow studied,
using ordinary not-so-soluble amines) but would play essentially no role when an NH-N
hydrogen bond is present as might be the case in significantly basic solutions (modern-
day concentrated alkanolamine solutions). Therefore, both Zwitterion and Termolecular
mechanisms are valid mechanisms but the major route for the reactants will depend on
environment (concentration and nature of amines). This also explains that at low
concentration the reaction order in amine will be near unity, but with increasing
concentration, broken order of reaction greater than one will appear (Aboudhier et al.,
2003). Deprotonation by water molecules is statistically possible but would generally
be followed by relaying the proton to another amine so that overall heat of reaction will
be same as in the case of direct deprotonation by amine (Hess’s law). With slightly
more certainty one can say that our results do not support the Carbamic Acid
mechanism, since at the best levels of modeling this intermediate would appear after a
carbamate ion and would not be thermodynamically favored in any equilibrium.
Figure 4.13 suggests that the rate-limiting step for overall CO2 absorption (Eq.
4.1) in concentrated alkanolamine solutions would not be zwitterion deprotonation (in
agreement with Versteeg (Versteeg 1996))6 but rate of encounter of CO2 with an amine
molecule. This step would have an Arrhenius prefactor which would be affected by
things like rate of bubbling CO2 into the mixture, and rate of diffusing CO2 molecules
towards amine molecules within the solution (affected by stirring rate). This step might
also have activation energy, and to gain insight here, attempts were made to compute

153
zwitterion formation steps from encounter complexes within a large-water network. The
activation energies were often disturbingly large (as an example, 19.5 kcal/mol in a 1-
amine-19-H2O case), due to the reduction of total number of hydrogen bonds as CO2
heads towards amine. Since the number of broken H-bonds was dependent on starting
conformation, the quantitative values of the activation energy were dependent on
starting conformation and hence did not warrant presentation here.

154
4.3.5 Formation of bicarbonate
Figure 4.16 Mechanism for bicarbonic acid formation through 6-atom cycle
The bicarbonate-forming CO2 capture mechanism in Eq. 4.11 was also studied, in
hopes of understanding why this mechanism does not compete well with the carbamate-
forming reactions in the cases of primary and secondary amines. First, a 6-atom cycle
through to carbonic acid was studied (Figure 4.16), analogous to the 6-atom cycle
through to carbamic acid (Figure 3) used to study the carbamate-forming pathway. The
two-step energy profile and the optimized geometries are shown in Figure 4.17. The
transition state for the bicarbonate formation is the theoretical “carbonic acid
zwitterion” strongly complexed with the amine. No supporting water molecules were
necessary to observe the bicarbonate, but the activation energy and heat of reaction for
bicarbonate formation are 9.1 and -3.5 kcal/mol, whose magnitudes seem too high and
too low, respectively.
Use of one spectator water molecule to stabilize the CO2 in the transition state
reduced the barrier to 5.8 kcal/mol (TS 26 in Figure 4.17). Various pathways with
large-water network were attempted but the problem with this CO2-to-bicarbonate
reaction is that the important step starts right from the encounter complex, and this
complex is quite dependent on conformer which controls the H-bond differential in this
bimolecular step. Hence, the extension to more H2O caused insurmountable problems
with this reaction.
H
O H
CO
ON
H
R
R

155
TS24 reactant
TS24
TS24 product
TS25 reactant
TS25(activation energy=9.1kcal/mol)
TS25 product
TS26 reactant
TS26 (activation
energy=5.8 kcal/mol)
TS26 product
Figure 4.17 Bicarbonate formation through 1-amine-1-H2O 6-atom cycle.
-20
-15
-10
-5
0
5
10
15
20
Rel
ativ
e en
ergy
, kc
al/m
ol
TS24 reactant(Initialcomplex)
TS24(9.1)
TS24 product(Bicarbonate)
TS25 reactant(Bicarbonicacid)
TS25(0.2)

156
4.3.6 Other amines
Might the mechanism change depending on which amine is used? Since
conventional thinking is that the basicity (pKa) of the amine would probably be the
important factor, this thinking was tested with a simple 1-amine-1-H2O model, looking
at both zwitterion and bicarbonate formation (Figure 4.18). The effect of pKa on the
activation energies for the first step (formation of zwitterion and bicarbonate) was
studied at the MP2/6-31G(d,p)/UFF-IEFPCM level of theory. Optimized geometries
for zwitterion and bicarbonate formation steps are shown in Tables 4.3 and 4.4
respectively.
zwitterion
formation reaction
bicarbonate
formation reaction
Figure 4.18 1-amine-1-H2O models for comparison of amine pKa effects.
For zwitterion formation (left side of Figure 4.18), for both primary and
secondary amines, plots of activation energy (Ea) versus amine basicity (pKa) reveal
that, as pKa increases, Ea decreases (Figure 4.19 upper plot). This is as expected.
However, the two curves are not identical: for a given basicity, a secondary amine

157
requires a lower activation energy than a primary one. The transition state is "earlier"
for secondary amines versus primary amines (at R(N-C) values of 2.06-2.13 instead of
1.95-2.03 Å), correlating with lowered Ea (Figure 4.19 lower plot); both these effects
are due to the lowered reaction exothermicity (ΔE) for secondary amines (see PES in
Figure 20), which drags down the potential energy surface according to the Bell-Evans-
Polanyi principle. As for why the ΔE is lower for secondary amines than primary
amines of identical basicity, it could be that the negatively charged COO− group of the
product zwitterion is better stabilized in the secondary amine case because of the better
inductive effects of having more alkyl groups on the nitrogen atom. In summary,
secondary amines have inherently greater CO2 affinity than primary amines when
comparing amines of same H+ affinity (pKa).
For bicarbonate formation (right side of Figure 4.18), again we see the expected
trend that, as pKa increases, Ea decreases (Figure 4.21). However, in this case the
results for primary vs. secondary amines are identical. The reason for this is that the
amines in this mechanism are attacking H+, not CO2, and hence the pKa is the perfect
property for correlating to the process involved.
It is true that the proper mechanisms are likely not as simple as these 1-amine-1-
H2O models suggest. However, the smooth trends in Ea versus pKa will apply to the
initial amine attack regardless of how many additional molecules are involved. A
change in mechanism due to changing the amine is still possible, if for instance it leads
to loss of zwitterion as intermediate.

158
Figure 4.19 Correlations of Ea at MP2/6-31g(d,p)/UFF-IEFPCM level of theory versus
pKa (upper plot) and transition state approach distance R(N-C) (lower plot),
for zwitterion formation.
0
1
2
3
4
5
6
8 8.5 9 9.5 10 10.5 11
Act
ivat
ion
en
ergy
for
zw
itte
rion
fo
rmat
ion
m;
kca
l/mol
pKa of amines
Primary
Secondary
MOR
MEA
DEAPPZ
DMA
AMP
MA
PA
AP
0
1
2
3
4
5
6
1.80
1.85
1.90
1.95
2.00
2.05
2.10
2.15
Act
ivat
ion
en
ergy
for
zw
itte
rion
fo
rmat
ion
, kca
l/mol
N-C
bon
d le
ngt
h i
n t
he
tran
siti
on s
tate
fo
r zw
itte
rion
for
mat
ion
, Å
Amines (secondary or primary, pKa)
NC bond length in TSActivation energy

159
Figure 4.20 Potential energy surfaces for the formation of zwitterons in 1-amine-1-H2O
modeling.
-2
0
2
4
6
Pot
enti
al e
ner
gy,
E(k
cal/m
ol)
for
the
zwit
teri
on
form
atio
n
MAPAMEAAPAMPDMADEAMORPPZ
Zwitterions
Initial complex
Transition states

160
Figure 4.21 Correlation of Ea at MP2/6-31g(d,p)/UFF-IEFPCM level of theory versus
pKa, for bicarbonate formation.
13
13.5
14
14.5
15
15.5
16
8 8.5 9 9.5 10 10.5 11
Act
ivat
ion
ener
gy fo
r bi
carb
onat
e fo
rmat
ionm
; kca
l/mol
pKa of amines
MOR DEA
AMP
AP
MEAPPZ
PA
MA
DMA

161
Table 4.3 Optimized Structures for Figure 4.19
Amine Initial complex Transition state Zwitterion
Methyl
amine
(MA)
Propyl
amine
(PA)
Mono-
ethanol
amine
(MEA)

162
Table 4.3 Optimized Structures for Figure 4.19 (Continued)
Amine Initial complex Transition state Zwitterion
2-Amino
-1-
propanol
(AP)
2-
Amino-
2-
methyl-
1-
propanol
(AMP)
Dimethy
l
amine(D
MA)
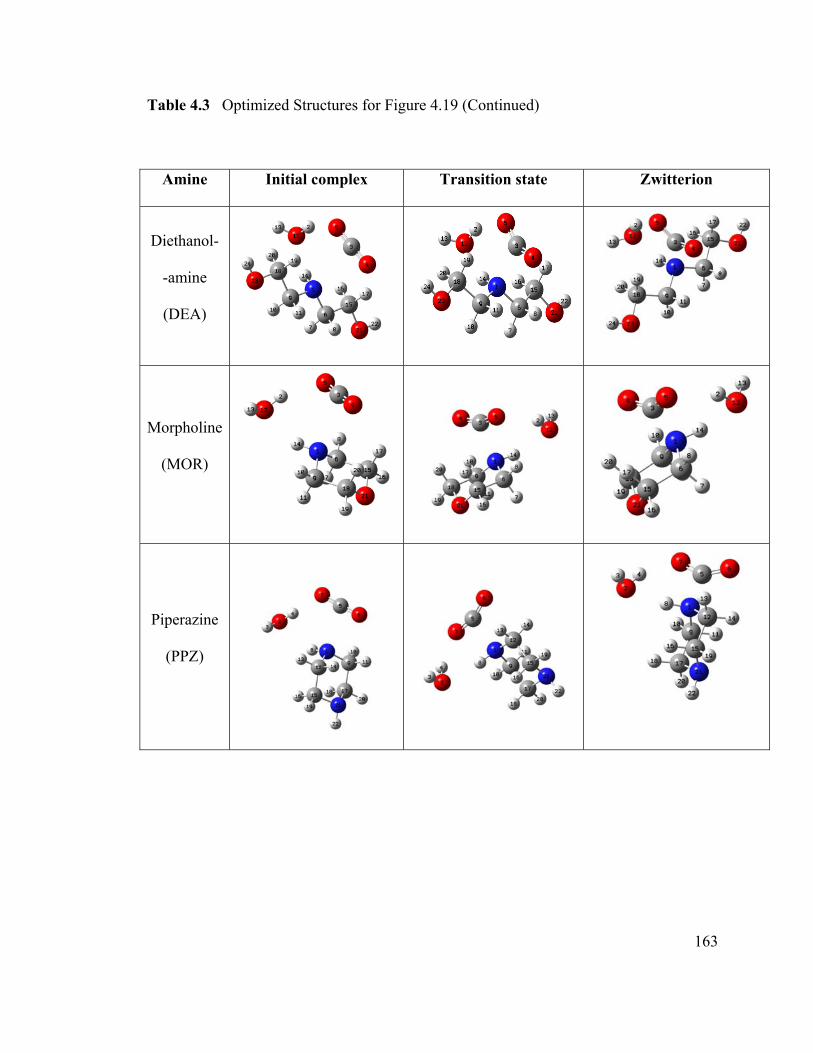
163
Table 4.3 Optimized Structures for Figure 4.19 (Continued)
Amine Initial complex Transition state Zwitterion
Diethanol-
-amine
(DEA)
Morpholine
(MOR)
Piperazine
(PPZ)

164
Table 4.4 Optimized Structures for Amines in Figure 4.21
Amine Initial complex Transition state Bicarbonate
Methyl
amine
(MA)
Propyl
amine
(PA)
Mono-
ethanol
amine
(MEA)

165
Table 4.4 Optimized Structures for Figure 4.21 (Continued)
Amine Initial complex Transition state Bicarbonate
2-Amino -1-
propanol (AP)
2-Amino-
2-methyl-
1-propanol (AMP)
Dimethyl Amine (DMA)

166
Table 4.4 Optimized Structures for Figure 4.21 (Continued)
Amine Initial complex Transition state Bicarbonate
Diethanol-
-amine
(DEA)
Morpholine
(MOR)
Piperazine
(PPZ)

167
4.4 CONCLUSIONS
Static calculations with PCM continuum model to determine dominant reaction
intermediates underscored the need for inclusion of explicit water molecules for realistic
modeling of the reaction pathways. Our DFT calculations with explicit water molecules
reveal, for the reaction involving one MEA molecule, that the CO2+MEA+nH2O reaction
proceeds as initial complex (IC)zwitterioncarbamatecarbamic acid. This 3-step
pathway was only seen when n, the number of explicit water molecules, is increased.
Instances of ICzwitterioncarbamic acid or ICcarbamic acid result with fewer
water molecules, and such results have been presented in the literature for years. Our
modeling is the first to correctly predict carbamate ions as the dominant product species.
The carbamate anion becomes thermodynamically competitive at neutral-pH and
dominant at basic-pH conditions, compared to both zwitterion and carbamic acid
intermediates, when properly solvated in the modeling.
Models involving two MEA molecules were deemed most relevant to modern-
day concentrated alkanolamine solutions. Such models, tried by others (Xie, Han,
Arstad), were improved incorporating further explicit water molecules. A tetramolecular
route (Shim 2008) featuring amine to amine proton transfer via water relay, was also
studied. Gradual incorporation of more water molecules shifted the zwitterion-
deprotonation transition state from “late” to “early,” and in a 2-amine-18-water model,
the predicted barrier is effectively non-existent (0.2 kcal/mol), suggesting that one could
consider the zwitterion as a species so short-lived that the Termolecular mechanism

168
would be dominant in concentrated alkanolamiane solutions. However, in dilute
solutions, when an amine is fully solvated by water molecules, single-MEA modeling
showed that zwitterion deprotonation will occur via a relay mechanism having a small
activation energy, making the Zwitterion mechanism more relevant. The relative
dominance of these mechanisms depends on amine concentration and thus explains
broken order kinetics. The results appear to dispel the Carbamic Acid mechanism,
revealing no thermodynamic drive for forming this intermediate.
To explore the idea of da Silva and Svendsen that the activation barrier of
carbamate formation comes from water displacement by CO2, the tricky zwitterion-
formation steps were searched for. As H2O molecules were explicitly added, activation
barriers rose, first due to breaking an initial amine…H2O hydrogen bond that impeded
the approach of CO2, and later due to differentials in total H-bonds in the model as CO2
formed the zwitterion. Thus their idea does seem plausible.
From the study with MEA we believe that the mechanism in aqueous
alkanolamine solution is heavily dependent on reaction environment of amine. For other
amines the effect of pKa on formation of zwitterion and bicarbonate was studied with
simple 6-atom cycles in neutral-pH modeling. It was discovered that secondary amines
have inherently greater CO2 affinity than primary amines when comparing amines of
same H+ affinity (pKa). Activation energies vary with pKa in a sufficient manner that this
effect could very well impinge on the importance (or non-importance) of a possible
zwitterion intermediate, and thus affect mechanism.

169
4.5 REFERENCES
1. Caplow, M. Kinetics of Carbamate Formation and Breakdown. J. Am. Chem.
Soc. 1968, 90,6795.
2. Dancwerts, P. V. The Reaction of CO2 with Ethanolamines. Chem. Eng. Sci.
1979, 34, 443.
3. Crooks, J. E.; Donnellan, J. P. Kinetics and Mechanism of the Reaction Between
Carbon Dioxide and Amines in Aqueous Solution. J. Chem. Soc, Perkin
Transactions 1989, 2, 331.
4. McCann, N.; Phan, D.; Wang, X.; Conway, W.; Burns, R.; Attalla, M.; Puxty, G.;
Maeder, M. Kinetics and Mechanism of Carbamate Formation from CO2 (Aq),
Carbonate Species, and Monoethanolamine in Aqueous Solution. J. Phys. Chem.
A 2009, 113, 5022.
5. Conway, W.; Wang, X.; Fernandes, D.; Burns, R.; lawrance, G.; Puxty, G.;
Maeder,M.Comprehensive Kinetic and Thermodynamic Study of the Reactions
of CO2(aq) and HCO3with Monoethanolamine (MEA) in Aqueous Solution. J.
Phys. Chem. A 2011, 115, 14340.
6. Versteeg, G. F.; van Swaaij, W. P. M. On the Kinetics Between CO2 and
Alkanolamines Both in Aqueous and Non-Aqueous Solutions-I. Primary And
Secondary Amines. Chem. Eng. Sci. 1987a, 43, 573..
7. Versteeg, G. F.; Van Dijck, L. A. J.; Van Swaaij, W. P. M. On the Kinetics
Between CO2 and Alkanolamines Both in Aqueous and Non-Aqueous Solutions.
An Overview. Chem. Eng. Commun. 1996, 144, 113.

170
8. Donaldson T. L.; Nguyen Y. N. Carbon Dioxide Reaction Kinetics and Transport
in Aqueous Amine Membranes. Ind. Eng. Chem. Fundam. 1980, 19, 260.
9. Tomasi J.; Mennucci B.; and Cammi R. Quantum Mechanical Continuum
Solvation Models. Chem. Rev., 2005, 105, 2999.
10. Cramer, C. J.; Truhlar D. G. A Universal Approach to Solvation Modeling. Acc.
Chem. Res. 2008, 41, 760.
11. Klamt A. and Schuurmann, G. COSMO: A New Approach to Dielectric
Screening in Solvents with Explicit Expressions for the Screening Energy and its
Gradient. J. Chem. Soc. Perkin Trans. 2 1993, 799.
12. Faurholt, C. Studies of Aqueous Solutions of Carbamates and Carbonates, J.
Chim. Phy. 1925, 22, 1-44.
13. Arstad, B.; Blom, R.; Swang O. CO2 Absorption in Aqueous Solutions of
Alkanolamines: Mechanistic Insight from Quantum Chemical Calculations. J.
Phys. Chem. A 2007, 111, 1222.
14. Shim, J. G.; Kim, J. H.; Jhon, Y. H.; Kim, J.; Cho, K. H. DFT Calculations on the
Role of Base in the Reaction Between CO2 and Monoethanolamine. Ind. Eng.
Chem. Res. 2009, 48, 2172.
15. Xie, H.-B.; Zhou, Y.; Zhang, Y.; Johnson, J. K. Reaction Mechanism of
Monoethanolamine with CO2 in Aqueous Solution from Molecular Modeling. J.
Phys. Chem. A 2010, 114, 11844.
16. Han, B.; Zhou, C.; Wu, J.; Tempel, J. T.; Cheng, H. Understanding CO2 Capture
Mechanisms in Aqueous Monoethanolamine Via First Principles Simulations. J.
Phys. Chem. Lett. 2011, 2,522.

171
17. Ismael, M.; Sahnoun, R.; Suzuki, A.; Koyama, M.; Tsuboi, H.; Hatakeyama,N.;
Endou, A.; Takaba, H.; Kubo, M.; Shimizu, S.;Carpio, C. A. D.; Miyamoto, A.
A DFT Sudy on the Carbamates Formation through the Absorption of CO2 by
AMP. Int. J. of Greenhouse Gas Control 2009, 3, 612.
18. Yamada, H.; Matsuzaki, Y.; Higashii, T.; Kazama, S. Density Functional Theory
Study on Carbon Dioxide Absorption into Aqueous Solutions of 2-Amino-2-
Methyl-1-Propanol Using A Continuum Solvation Model. J. Phys. Chem.
A 2011, 115, 3079.
19. da Silva, E. F., Svendsen, H. F. Ab Initio Study of the Reaction of Carbamate
Formation from CO2 and Alkanolamines. Ind. Eng. Chem. Res. 2004, 43, 3413.
20. Gaussian 09, Revision B.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E.
Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci,
G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F.
Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota,
R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai,
T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J.
Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K.
Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N.
Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J.
Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C.
Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A.
Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B.

172
Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc., Wallingford
CT, 2009.
21. Becke, A. D. Density-Functional Thermochemistry. III. The Role Of Exact
Exchange. J.Chem.Phys. 1993, 98, 5648.
22. Stephens, P. J. ; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. Ab Initio
Calculation of Vibrational Absorption and Circular Dichroism Spectra Using
Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623.
23. Cossi, M., Barone, V., Cammi, R., Tomasi, J. Ab Initio Study of Solvated
Molecules: A New Implementation of The Polarizable Continuum Model. Chem.
Phys. Lett. 1996, 255, 327.
24. Baker J. An Algorithm for the Location of Transition-States. J. Comp. Chem.
1986, 7, 385.
25. Wang, T-.T; Bishop S. H.; Himoe, A. Detection of Carbamate as A Product of
the Carbamate Kinase-Catalyzed Reaction by Stopped Flow Spectrophotometry.
J. Biol. Chem. 1972, 247, 14, 4437.
26. Aboudheir, A.; Tontiwachwuthikul, P.; Chakma, A.; Idem, R. Kinetics of the
Reactive Absorption of Carbon Dioxide in High CO2-Loaded, Concentrated
Aqueous Monoethanolamine Solutions. Chem. Eng. Sci. 2003, 50, 1071.

173
Chapter 5: Molecular Dynamics Simulation of CO2/Amine/Water Mixtures
_______________________________________________________________________
5.1 INTRODUCTION
The prediction of small, 4 kcal/mol activation energies (Ea) for interconversion of
zwitterion-to-carbamate-to-carbamic acid in the previous chapter (Figure 4.6) come with
an important caution: they are for only one pathway, and others might be possible with
lower Ea values. In this chapter, results from ab initio molecular dynamics (AIMD)
simulations are shown, providing a complementary study of intermediate stabilities.
AIMD includes the effect of temperature and searches for reaction paths in an unbiased
way. These AIMD simulations were performed in tandem with the calculations of the
previous chapter.
Recently two AIMD simulation papers have been published that observed the
fate of MEA-zwitterion. Han et al. (2011)1 studied the reaction of MEA-zwitterion in
presence of another MEA and 16 water molecules (approx. 30 wt% solution) and found
that the MEA-zwitterion reacted with MEA in 4 ps, the reaction barrier was 1.3 kcal/mol
and heat of absorption was -22 kcal/mol. Guido et al. (2013)2 studied the fate of a MEA-
zwitterion in 122 water molecules (approximately 3 wt% aqueous solution) and observed
that both forward reaction (deprotonation by water) and backward reaction (CO2 release)
are characterized by free-energy barriers of 6−8 kcal/mol and thus competitive. Both

174
researchers have underscored the importance of explicit water molecules in the model: to
reach quantitative agreement with the experimental heat of absorption value (Han, et
al,.2011)1 and to see the large entropic effect in the conversion of zwitterion (Guido et
al., 2013)2 both in forward and backward direction.
The first objective of the simulations was to see the fate of all possible
intermediates, in the bicarbonate (Group I) and carbamate (Group II) pathways (Figures
5.1 and 5.2), on an extremely limited 8 ps timescale, to see if all intermediates are indeed
separated by some sort of potential barrier. The second objective was to try some longer
simulations with a variety of amines to see if a complete transformation from carbamate
zwitterion to carbamic acid could be found (Group III). The third objective was to
explore the reactivity of the carbamate zwitterion in the presence of a nearby amine
molecule (Group IV).
Figure 5.1 Possible intermediates in the bicarbonate pathway in CO2/H2O system
Figure 5.2 Possible intermediates in the carbamate pathway in CO2/Me2NH/H2O system
+ H2O
HO
HC
O
O HOC
O
O HOC
O
OH
OC
O
+H
OH
+ H2O + H3O+ + H2O
carbonatezwitterion
bicarbonate carbonic acid
+ H2O
MeNH
Me
CO
O MeNC
O
O MeNC
O
OH
OC
O
+Me
HNMe
+ H2O + H3O+ + H2O
carbamatezwitterion
carbamate carbamic acid
Me Me

175
5.2 METHOD
The simulations were performed using the Vienna Ab Initio Simulation Package
(VASP)3,4 version 5.2.11 on the in-house supercomputer Dextrose. The following VASP
specifications were used in all simulations: the potpaw GGA plane-wave basis sets,5,6
standard precision (PREC=Normal); ENMAX=400eV; a Nosé thermostat for canonical
(NVT) conditions7 with 40 fs thermal oscillations (SMASS=0), a Verlet velocity
algorithm;8 a temperature of 313K (40°C), and a time step equaling 1 fs of real time.
The sample cell was cubic in shape and replicated using periodic boundary conditions to
mimic the bulk liquid. The forces used in the simulations were of the PW91 level of
density functional theory (DFT)9,10 but with a Grimme-style semi-empirical van der
Waals (vdW) attractive potential added on to the energy and force calculations.
First, 64 water molecules in a cubic box of width 12.45 Å were simulated for
8000 fs at 40°C, generating a pseudo-equilibrated water sample, Geometry A (step
8001). Another simulation was run for 24000 fs starting from geometry A, generating a
further equilibrated sample, Geometry B (step 32001). Then, starting with geometries A
and B respectively, starting ensembles for each group I and Group II “production run” at
40°C were made by substituting a solute molecule for a small number of water
molecules:

176
Group –I. 8-ps simulations of water-CO2 system (Figure 5.1)
a) Replace a H2O with a CO2
b) Replace two H2O with the carbonate zwitterion
c) Replace two H2O with bicarbonate, and one H2O with an H3O+
d) Replace two H2O with carbonic acid
Group –II. 8-ps simulations of water-CO2-dimethylamine system (Figure 5.2)
e) Replace four H2O with the carbamate zwitterion
f) Replace four H2O with carbamate, and one H2O with an H3O+
g) Replace four H2O with carbamic acid
A 8-ps continuation run (e2) of dimethyalamine-zwitterion was performed
starting with the final geometry obtained from simulation (e). The final geometry of run
e2 (geometry C) was the starting point of all the secondary amine zwitterion simulations
(h, l, m, and n) in Groups III and IV (prepared by suitable modification of geometry C).
Starting ensembles for Groups III and IV simulations (i, j, k, o) with primary amines
(methylamine, monoethanolamine and 2-methyl-2-amino-propanol (AMP)) were
prepared by solute replacement in geometry B.
Group –III. Longer zwitterion simulations
h) Dimethylamine (Me2NH) zwitterion, starting from geometry C
i) Replace four water molecules with methylamine (MeNH2) zwitterion
j) Replace five water molecules with monoethanolamine (MEA) zwitterion

177
k) Replace five water molecules with AMP-zwitterion and increase the cell
width to 12.59 Å (to match the density with that of experimental value)
l) Replace Me2NH-zwitterion with diethanolamine (DEA)-zwitterion
m) Replace Me2NH -zwitterion and one water molecule with PPZ-zwitterion
Group –IV. Zwitterion+amine simulation
n) Replace three water molecules with Me2NH
o) Replace seven water molecules with MeNH2-zwitterion and MeNH2

178
5.3 RESULTS
5.3.1 Group –I. 8-ps simulations of water-CO2 system
Results of 8-ps long simulations of pure CO2, carbonate zwitterion, bicarbonate
ion and bicarbonic acid (Figure 5.1) are presented.
a) Simulation of aqueous CO2
No reaction was observed in the 8 ps-long run.
b) Simulation of aqueous carbonate zwitterion
The carbonate zwitterion (Figure 5.3) immediately broke into water and CO2 by
O-C bond cleavage. The O-C bond length increases up to 4.25 Å in very short time
(within 20fs) and the CO2 and water does not return as close as 2.5 Å.
Figure 5.3 Starting geometry in simulation (b), showing only two of the neighboring
water molecules.

179
c) Simulation of aqueous bicarbonate anion:
No reaction of HCO3- was observed in 8 ps. The initial hydronium ion (Figure
5.4) was at a distance of 8.13Å from the bicarbonate (measured as the
C(bicarbonate)…..O(hydronium) distance), and after a few proton transfers, the final
hydronium ion was at 6.18Å from the same bicarbonate. Episodes of Zundel [(H2O5)+]
ions were observed.
Figure 5.4 Starting geometry in simulation (c), hydronium at bottom right.

180
d) Simulation of aqueous carbonic acid
H2CO3 dissociated into HCO3- + H+ after 4.7 ps (Figure 5.5). Initially placed
without hydrogen bonds, the H2CO3 formed simultaneous OH-O bonds with two
neighboring water molecules after 600 fs (Figure 5.5, inset).
Figure 5.5 Evolution of two OH bond lengths in simulation (d), demonstrating
ionization of H2CO3 at 4.7 ps.

181
5.3.2 Group –II. 8-ps simulations of water-CO2-dimethyl amine system
Results of simulations of intermediates that appear through pathway II (Figure
5.2) in CO2-aqueous dimethylamine (Me2NH) system are described.
e) Simulation of aqueous carbamate zwitterion
The zwitterion (initial geometry shown in Figure 5.6) persisted for 8ps.
Figure 5.6 Starting geometry in simulation (e), showing only three of the neighboring
water molecules. The N-C bond length is 1.6 Å and the NH..OH2 bond
length (broken line) is 2.31 Å

182
f) Simulation of aqueous carbamate anion
Carbamate interconverted with carbamic acid many times (Figures 5.7 and 5.8).
t=0fs, carbamate + H3O+
t=5191fs , carbamic acid
Figure 5.7 Initial local geometry of carbamate and hydronium ion (left) and local
geometry of carbamic acid formed at t=5191fs (right) in simulation (f).
Figure 5.8 Evolution of the OH distance that demonstrates the interconversion of
carbamate with carbamic acid in simulation (f). The proton, initially in the
hydronium ion, approaches from an initial distance of 3.01Å to the
carbamate anion.

183
g) Simulation of aqueous carbamic acid
The carbamic acid (CH3)2NCOOH dissociated into (CH3)2NCOO– and H+ at
t=6.5 ps. Figure 5.9 show the initial acid and final anion surrounded by water molecules
respectively. The conversion of carbamic acid into carbamate is captured in Figure 5.10:
The OH bond in the carbamic acid (blue line) becomes hydrogen bonded at 3ps which
causes the OH bond to vibrate more vigorously. Dissociation occurs at 6.5 ps to form
carbamate. The red line depicts the distance between the proton of carbamic acid and the
oxygen of a neighboring water molecule which finally abstracted the proton.
t=0,Carbamic acid
t=8001fs, Carbamate
Figure 5.9 Initial geometry of carbamic acid (left); and geometry of carbamate anion at
t=8001fs (right) in simulation (g). Only few water molecules are shown.
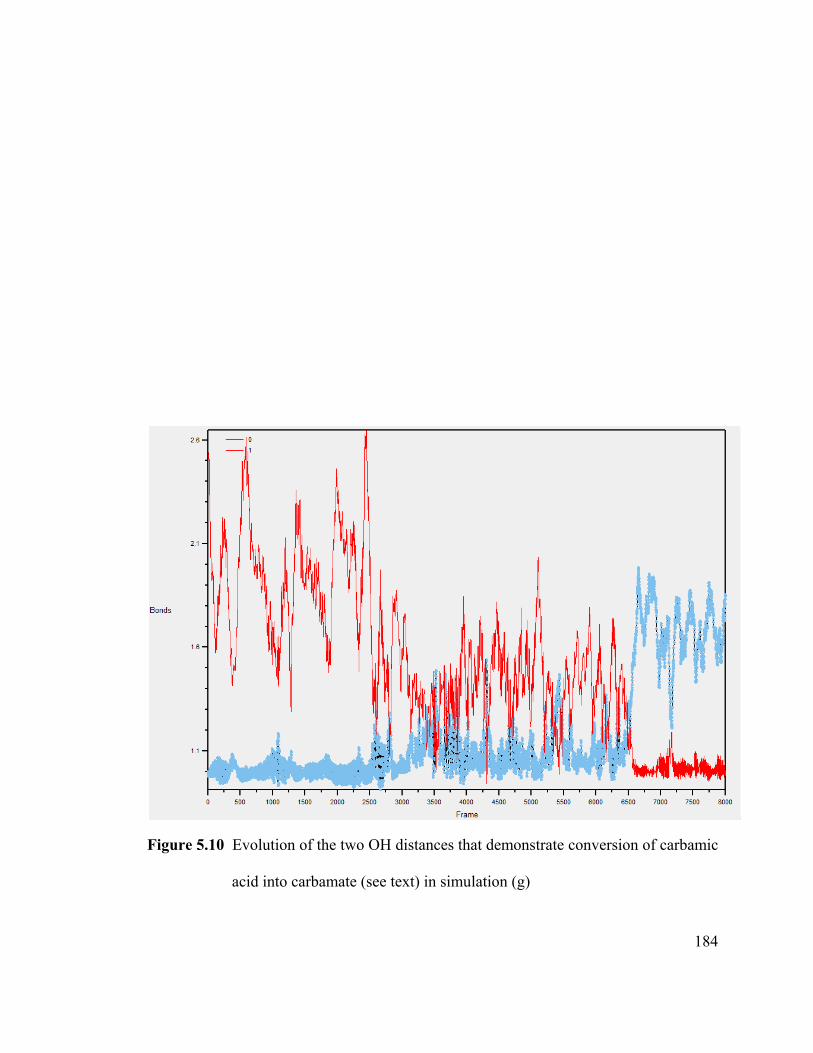
184
Figure 5.10 Evolution of the two OH distances that demonstrate conversion of carbamic
acid into carbamate (see text) in simulation (g)

185
5.3.3 Group-III. longer zwitterion simulations
Results for longer simulations, hoping to see a zwitterion ↔ carbamate transition,
are presented.
h) Dimethylamine zwitterion.
An 8 ps continuation run (Run e2) of simulation (e) also revealed no change, so a
further 100 ps continuation run (simulation (h)) was performed. In this 3rd run, the
zwitterion converted into carbamate at 8 ps (Figure 5.11), and then, the carbamate
converted into carbamic acid at 13 ps (Figure 5.12). The breaking of NH bond to form
carbamate also simultaneously caused the equilibrium NC bond-length to shrink (the
second plateau in the red line in Figure 5.11).
The reaction from zwitterion to carbamic acid via carbamate proceeded via
proton relay through a hydrogen-bonded 10-atom cycle formed by participation of three
solvent water molecules and the zwitterion (Figure 5.13). The lengths of four new OH
bonds that are formed during the relay are tabulated as a function of time in run (h) in
Table 2. Prior to zwitterion-deprotonation, at 8 ps, solvent molecules reoriented to
bridge the two polar ends of a OCNH segment of zwitterion; forming the “hydrogen-
bonded” cycle through which the two elementary reactions ‘zwitterion carbamate’
and ‘carbamate carbamic acid’ took place via proton transfer relay.
The carbamic acid formed at 13 ps lasted for only about 1 ps; then, it lost its
gained H+ at 14 ps (re-forming carbamate) and did not convert back into carbamic acid
until 95 ps where it stayed for the remaining 5 ps of run (h) (Figure 5.12).

186
Figure 5.11 The breaking of NH (blue) and simultaneous shrinking of NC(red) bond
lengths in simulation (h).

187
Figure 5.12 Evolution of two OH bond lengths in simulation (h), the oxygens being the
two in COO moiety.

188
Figure 5.13 The 10-atom cycle at t=8ps in simulation (h) (all surrounding water
molecules are removed). Some bond lengths are provided in Table 5.1.
Table 5.1 Bond-Formation through 10-Atom Cycle Schematically Shown in Figure
5.13
At t=6ps, all of these OH distances are too long to be considered covalent bond;
but at 13ps, all of them are considered to be covalent bonds. formation of bond O3H8 is
complete by 9 ps, bond O1 H 21 by 11 ps, bonds O2 H11 and O4H17 by 13 ps.
Time(ps) O3H18 O1H21 O2H11 O4H176 1.66 1.97 3.71 1.797 1.61 2.4 1.76 1.878 1.37 1.6 1.54 1.89 0.96 1.73 1.57 1.59
11 0.98 0.97 1.74 1.7212.5 0.98 1.05 1.13 1.5113 1 1.06 1 1.04
Distance (Å)

189
i) Methylamine (MeNH2) zwitterion
Only one 8-ps simulation ( run i) was needed to observe the MeNH2-zwitterion
(Figure 5.14) convert into carbamate at 0.9 ps and then into carbamic acid at 1.5 ps
(Figure 5.15). During the time period 1.5ps <t<3.8ps , carbamate interconverted with
carbamic acid and afterwards the carbamic acid persisted.
Figure 5.14 Starting geometry in simulation (i), showing only five of the neighboring
water molecules. The NH-OH2 bond length is 1.75 Å (H21O6) and the
O7H19 distance is 1.81 Å. The other NH bond was not hydrogen-bonded
to any water molecules in the starting geometry
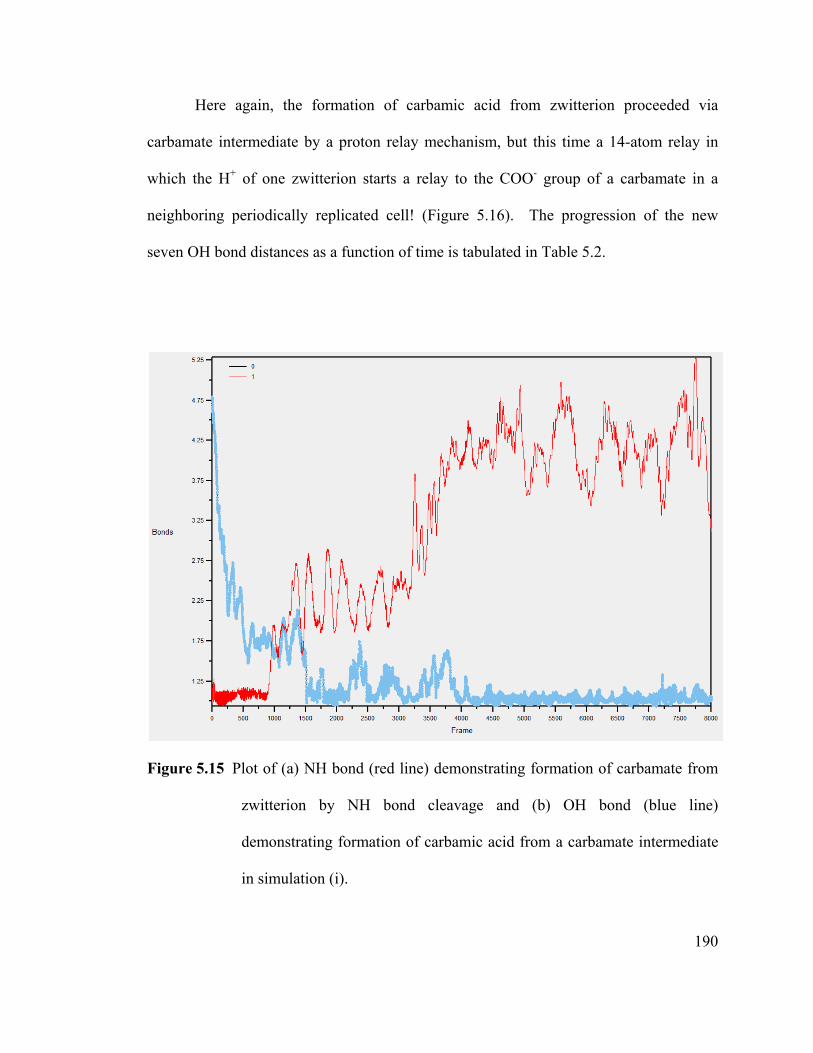
190
Here again, the formation of carbamic acid from zwitterion proceeded via
carbamate intermediate by a proton relay mechanism, but this time a 14-atom relay in
which the H+ of one zwitterion starts a relay to the COO- group of a carbamate in a
neighboring periodically replicated cell! (Figure 5.16). The progression of the new
seven OH bond distances as a function of time is tabulated in Table 5.2.
Figure 5.15 Plot of (a) NH bond (red line) demonstrating formation of carbamate from
zwitterion by NH bond cleavage and (b) OH bond (blue line)
demonstrating formation of carbamic acid from a carbamate intermediate
in simulation (i).

191
Table 5.2 Evolution of Some Bond Lengths as a Function of Time Through the 14-atom
Relay Shown in Figure 5.16
Figure 5.16 t=1437fs of simulation (i), showing the 14-atom relay trajectory (connected
by broken lines), the surrounding water molecules are removed
Time(ps) O28H43 O29H40 O5H41 O1H22 O2H12 O7H14 N8C90.3 1.69 1.72 1.9 2.12 1.58 2.47 1.830.5 1.45 1.87 1.68 1.75 1.81 1.86 1.560.9 1.35 1.48 1.87 2.23 1.7 1.8 1.51.1 0.99 1.2 1.52 1.96 1.67 1.69 1.37
1.437 1.03 0.96 1.02 1.05 1.2 1.62 1.441.6 0.96 0.98 0.97 1.01 1.03 1.03 1.347 0.94 1.02 0.99 0.99 1.02 1.04 1.38
Distance (Å)

192
j) Monoethanolamine (MEA) zwitterion
The MEA-zwitterion ionized to form carbamate at 0.8 ps but did no further
conversion in the 17-ps run. The created hydronium ion remained close to the carbamate
as an ion pair complex (Figures 5.17 and 5.18)
Initial geometry (t=0); broken line indicates
new bond later formed.
Final geometry (t=17203 fs) (carbamate-
proton complex)
Figure 5.17 Starting geometry of MEA-zwitterion (left) and geometries of carbamate
and hydronium products at t=17203fs in simulation j. Only few
neighboring water molecules are shown.

193
Figure 5.18 Evolution of three OH bond lengths in the generated hydronium ion in
simulation (j).

194
k) 2-amino-2-methyl propanolamine (AMP) zwitterion
An AMP-zwitterion (Figure 5.19) was simulated in 59 water molecules for 110
ps (initial run of 8ps, followed by two continuation runs of 8 ps and 94-ps) and no
change occurred. Two 12-atom H-bonded cycles formed at 80 ps in the final 94-ps run.
Figure 5.19 Starting geometry of AMP-zwitterion in simulation (k), only few
neighboring water molecules are shown.
l) Simulations with diethanolamine (DEA) zwitterion
A DEA-zwitterion (Figure 5.20) was simulated in 60 water molecules for 16203
fs, and no reaction was observed. A tight 6-atom cycle formed at 15 ps and lasted for the
remaining 1 ps.
Figure 5.20 Starting geometry of zwitterion in simulation (l): H22O2=3.1 Ǻ,
H21O5=1.79 Ǻ, H22O4=1.86 Ǻ.

195
m) Piperazine (PPZ) zwitterion
PPZ-zwitterion was simulated in 59 water molecules for 15 ps and no reaction
was observed. Cyclic arrangement of water molecules around the OCNH segment could
be identified after 11 ps. A 10-atom cycle around the OCNH segment (Figure 5.21, right)
and a 14-atom cycle connecting the same OC moiety to the other NH had formed by 15-
ps.
As a side note, the simulation data reveals the weakening of the NH bond upon
carboxylation, by a comparison of N- vibration amplitudes (Figure 5.22).
Initial geometry
t=15ps NH=1.1 Ǻ, NH-O=1.66 Ǻ
Figure 5.21 Starting geometry (left) of PPZ-zwitterion in simulation (m), few
surrounding water molecules are shown. 10-atom cycle at 15-ps (right).

196
Figure 5.22 Difference in amplitude of vibration of two NH bond lengths of PPZ-
zwitterion in simulation (m), (red: 3-coordinated N, blue: 4-coordinated
N).

197
5.3.4 Group-IV. simulations of zwitterion+amine
We did two simulations at higher amine concentration to see potential proton
transfer from zwitterion to amine.
n) Simulation of Me2NH+COO- + Me2NH
A dimethylamine-zwitterion was simulated in presence of another nearby neutral
dimethaylamine in 57 water molecules for 48 ps (24 ps and its continuation for another
24 ps). The initial geometry is shown in Figure 5.23, with the NH bond of zwitterion
pointing towards a water molecule (NH-O=2.31 Ǻ). The neutral amine was placed at a
N to N distance of 4.66 Ǻ from the zwitterion, and its lone pair was pointing towards
another water molecule (the N-HO distance is 1.66 Ǻ).
t=0, (dimethylamine-zwitterion+neutral
dimethylamine), N-N=4.66 Ǻ
t=48 ps (carbamate+protonated
dimethylamine), NH-O=1.74A, N-N=3.71Ǻ
Figure 5.23 Initial geometry (left) and final geometry (right) in simulation (n), only few
water molecules are shown.

198
Zwitterion deprotonated at 27.9 ps (Figure 5.24) . By 28.2 ps, a 14-atom H-
shuttling cycle stopped just short of forming a carbamic acid, staying as a carbamate-
hydronium complex (Figure 5.25). From 28.3 to 29.5 ps, an H-shuttling relay occurred
back-and-forth before finally resting with the extra H+ on the uncarboxylated amine
molecule. This final state persisted until the end of the 48 ps simulation. 11 water
molecules were involved in the relay of proton from the zwitterion to the neutral amine
and the time required was less than 2 ps.

199
Figure 5.24 Evolution of some important bond lengths in simulation (n). Red: breaking
of zwitterion NH bond; light blue: formation of NH bond in protonated
dimethylamine; green: shrinking of NC bond length immediately after
deprotonation (N becomes positively charged); dark blue: evolution of an
OH bond length in carbamate-hydronium complex.

200
Figure 5.25 Snapshot of simulation (n) at t=28213 fs showing the carbamate-hydronium
intermediate complex.

201
o) Simulation of MeNH2-zwitterion + MeNH2.
A MeNH2-zwitterion-like structure (Figure 5.26, top left) was simulated for 8-ps
in 57 water molecules plus a nearby MeNH2 molecule in cell of width 12.45 Ǻ.
Zwitterion was formed in 150 fs (NC bond length decreased to 1.54 Ǻ from initial 2 Ǻ)
which converted to carbamate by H-shuttling to neutral MeNH2 via a water molecule at
t~380 fs. The ions later approached each other and formed a cation-anion complex
(Figure 5.26, bottom right).
Starting geometry N5C7=2 Ǻ;H12O1=2.58 Ǻ H10N4=3.42 Ǻ
t=320fs,NC7=1.6Ǻ,H12O1=1.56Ǻ H10N4=1.67 Ǻ
t=380fs NC7=1.52 Ǻ;H12O1=1.06 Ǻ ; H10N4=1.10 Ǻ
t=8001fs,NC7=1.39 Ǻ;H12O1=1.02 Ǻ; H10N4=1.07 Ǻ;O2H17=1.58 Ǻ
Figure 5.26 Geometries in simulation (o) without the spectator water molecules.
Conversion of zwitterion to carbamate happens through proton relay to a
nearby amine.

202
5.4 CONCLUSIONS
Group I simulations of aqueous CO2, H2OCO2 zwitterion, HCO3- (with faraway
H3O+), and H2CO3 did the following: CO2 did not react, H2OCO2 fell back to CO2,
HCO3- did not react, and H2CO3 persisted for 5 ps before changing to HCO3
- for its final
3ps. What does this tell us? Firstly, the bicarbonate zwitterion seems to have no
stability at all, suggesting that a “wider” barrier exists between an encounter complex
and the bicarbonate/carbonic acid equilibrium, and secondly that the HCO3-/H2CO3
equilibration is too slow for us to conclude which one is dominant.
Group II simulations of Me2NHCO2 zwitterion, Me2NCO2- (with far away H3O
+),
and Me2NCOOH did the following: the zwitterion did not react, Me2NCO2- went back-
and-forth with the acid roughly once per ps, and the acid persisted 6.5 ps before
converting to the anion for 1.5 ps. What does this tell us? Firstly, that Me2NHCO2
zwitterion is more stable than the H2OCO2 zwitterion and thus deserves consideration as
an intermediate, and is separated from the anion/acid equilibrium pair by some sort of
barrier; and secondly that the Me2NCO2-/Me2NCOOH equilibration is too slow for us to
conclude which one is dominant. The anion and acid interconverted on a very short
timescale, suggesting that anion/acid equilibria are fairly barrierless, and hence
thermodynamics will determine the ratio of anion to acid.
Group III simulations of various carbamate-zwitterions revealed forward
conversion of zwitterions in 3 of the 6 cases: Me2NH-zwitterion, MeNH2-zwitterion and
MEA-zwitterion. Of these, only MEA-zwitterion failed to show carbamate/carbamic acid
equilibrium. Reaction of AMP-zwitterion, DEA-zwitterion and PPZ-zwitterion was not

203
observed in 110 ps, 16 ps, and 15 ps long runs, respectively. However, a common role
of solvent was identified in all six simulations: solvent molecules reoriented to bridge the
two polar ends of a OCNH segment of zwitterions, forming a “hydrogen-bonded” cycle
(if N water molecules participate, the cycle has 4+2N atoms, and 2+N covalent bonds
before any reaction takes place). These cycles likely occur in reality.
We highlight that the H-shuttling cycles seen in Group III simulations do not
exist in aqueous amine solution prior to absorption of CO2. The preexisting H-bond
networks must be reformed to connect to the negative OC segment of COO moiety. This
reorientation process can be considered as the minimum lifetime of zwitterions when a
nearby amine is absent. Both of the NH bonds of primary amines (and piperazine) were
seen active in formation of such cycles. The number of water molecules involved in
such cycles changes over course of time. Although the reaction was not observed in
simulations k-m, this could be due to the time limitation of our simulations.
The Group IV simulations of zwitterion in presence of nearby amine showed
H2O-mediated H+ transfer relays to form carbamate and protonated amine, products
which corroborate with experimental observation. The lengths of the relay were 11 H2O
(simulation ‘n’) and 1 H2O (simulation ‘o’). The previously described H-bonded cycle
for forming carbamic acid formed in simulation ‘n’. The speed of the reaction in
simulation ‘o’ (less than 0.5 ps) reveals that zwitterion lifetime and the formation of
carbamic acid is hindered by high amine concentrations, which put two amine molecules
in closer proximity. It also suggests that the zwitterion lifetime might be reduced even
further if the two amine molecules were directly bonded to each other.

204
An amine-to-amine proton transfer was observed in 0.5 ps in a simulation1 where
the neutral amine molecule was placed close to the zwitterion (the NH bond of zwitterion
pointed to the lone pair of neutral amine). But, classical MD simulations of aqueous
monoethanol amine showed significantly less frequent N-HN hydrogen bonding
interactions in aqueous solution than in pure liquid amine.11-15 Further work needs be
done in that area which will shed further light on zwitterion stability (lifetime) and the
fate of H-bonded cycles in case of direct proton transfer.
The observation of 10- to14-atom H+-shuttling cycles for formation of carbamic
acid (via carbamate from zwitterion) justifies our study of such multiple-water-mediated
pathways in our static calculations in Chapter 4. Indeed, such pentamolecular and
hexamolecular pathways have never been postulated, and it is hoped that these new paths
and the results of Chapter 4 will significantly advance the efforts to finally solve this
mechanism.

205
5.4 REFERENCES
1. Han, B.; Zhou, C.; Wu, J.; Tempel, J. T.; Cheng, H. Understanding CO2 Capture
Mechanisms in Aqueous Monoethanolamine Via First Principles Simulations. J.
Phys. Chem. Lett. 2011, 2, 522.
2. Guido C. A., Pietrucci F., Gallet G. A.; Andreoni W. The Fate of a Zwitterion in
Water from ab Initio Molecular Dynamics:Monoethanolamine (MEA)-CO2. J.
Chem. Theory Comput. 2013, 9, 28.
3. Kresse G.; Hafner J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev.
B 1993, 47, 558.
4. Kresse G.; Furthmüller J. Efficient Iterative Schemes for Ab Initio Total-Energy
Calculations Using A Plane-Wave Basis Set. Phys. Rev B 1996, 54, 11169.
5. Kresse G.; Hafner, J. Norm-Conserving and Ultrasoft Pseudopotentials for First-
Row and Transition Elements. J. Phys. Condens. Matter 1994, 6, 8245
6. Kresse, G.; Joubert D. From Ultrasoft Pseudopotentials to the Projector
Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758
7. Nos’e, S., A Unified Formulation Of The constant Temperature Molecular
Dynamics Methods. J. Chem. Phys. 1984, 81, 511.
8. Leach, A. R. Molecular Modeling: Principles & Applications, 2nd ed. Pearson,
Harlow, UK, 2001.
9. Perdew, J. P.; Chevary; J. A.; Vosko; S. H.; Jacson; K. A. ; Pederson; M. R. ;
Sing D. J.; Fiolhais; C. Atoms, Molecules, Solids, and Surfaces: Applications of

206
the Generalized Gradient Approximation for Exchange and Correlation. Phys.
Rev. B 1992, 46, 6671.
10. Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a
Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787.
11. Button, J. K.; Gubbins, K. E.; Tanaka, H.; Nakanishi, K. Molecular Dynamics
Simulation of the Hydrogen Bonding in Monoethanolamine. Fluid Phase
Equilib. 1996, 116, 320.
12. Alejandre, J.; Rivera, J. L.; Mora, M. A.; de la Garza, V. Force Field of
Monoethanolamine. J. Phys. Chem. B 2000, 104,1332.
13. Gubskaya, A. V.; Kusalik, P. G. Molecular Dynamics Simulation Study of
Ethylene Glycol, Ethylenediamine, and 2-Aminoethanol. 1. The Local Structure
in Pure Liquids. J. Phys. Chem. A 2004, 108, 7151.
14. da Silva, E. F.; Kuznetsova, T.; Kvamme, B.; Merz, K. M., Jr. Molecular
Dynamics Study of Ethanolamine as a Pure Liquid and in Aqueous Solution. J.
Phys. Chem. B 2007, 111, 695.
15. Gubskaya, A. V.; Kusalik, P. G. Molecular Dynamics Simulation Study of
Ethylene Glycol, Ethylenediamine, and 2-Aminoethanol. 2. Structure in Aqueous
Solutions. J. Phys. Chem. A 2004, 108, 7165.

207
Chapter 6: Screening of Ionic Liquids: A COSMO-RS Study†
_______________________________________________________________________
6.1 INTRODUCTION
The changing chemical composition of earth’s atmosphere, due to the colossal
rate of anthropogenic emission of CO2 produced from the burning of carbonaceous
fuel, warrants the necessity of the immediate abatement of CO2 emission.1 Such
concern, coupled with the prospect of the utilization of captured CO2 in enhanced oil
recovery,2 has propelled research efforts to develop affordable and environmentally
benign technologies for CO2 capture from large emission sources. The most
industrially advanced technology for CO2 capture, at present, is chemical absorption
of CO2 with aqueous alkanolamine solutions, which are, in general, volatile, prone to
degradation and equipment corrosion, and most importantly, energy-intensive to
regenerate.3 While energy and cost-efficient novel amine solvents are continually
investigated,4-6 the exploration of other advanced materials for CO2 are being
sought.7 Among the many innovative technologies, ionic liquids are identified as
potential solvents for developing green CO2 capture technology with significant cost
reduction benefit.8
_______________________________________________________________________ †This chapter contains material reproduced with permission from Sumon, K. Z.; Henni, A. Ionic liquids for CO2 capture using COSMO-RS: Effect of structure, properties and molecular interactions on solubility and selectivity. Fluid Phase Equilib. 2011, 310, 39-55. Copyright © 2011 Elsevier B.V.

208
Ionic liquids (ILs) are the generic names of a broad category of salts with
melting point less than 100 °C. They are usually composed of large, asymmetric and
loosely coordinating organic cations and inorganic or organic anions. Due to their
ionic nature and high thermal stability, they tend to have negligible vapour pressure
making them, in general, environmentally benign as well as suitable for gas
separation without solvent loss or contamination of the vapour phase. The potential
of ionic liquids for CO2 capture was recognized first by researchers at the University
of Notre Dame.9 Since then, research efforts in this area have been expanding.10-14
Opportunities exist to generate task-specific ionic liquids by chemical alteration of
the cation or anion such as tethering a specific functional group in one of the
comprising ions.15-20 Different combinations between a variety of cations and anions
result in a large array of ionic liquids with unique properties. Experimental
investigation of ionic liquids is a challenging task due to their increasing number, and
for now, high cost. Therefore, screening and designing of ionic liquids for CO2
capture with a reliable computational method would be of great value for subsequent
experimental work. The present work is a contribution to such an objective.
Many attempts were made to model and predict the solubility of CO2 in some
limited number of ionic liquids based on group contribution method,21 regular solution
theory,22-24 Quantitative Structure Property Relationship (QSPR) method,25 Equation of
State,26 Conductor-like Screening Model for Real Solvents (COSMO-RS) method,27
molecular dynamics (MD)28-34 and Monte Carlo35 simulations. Among them, COSMO-
RS is suitable for fast screening of a large number of novel solvents as it does not require
any compound or group specific interaction parameters. COSMO-RS was shown useful

209
for qualitative prediction of solubilities of gases by a number of studies.36-40 Henry’s law
constants for CO2 in many ionic liquids were compared with experimental values.27,36-39
Ionic liquids were screened for CO2 capture from a pool of 408 ionic liquids by Zhang et
al.,36 170 ILs by Palomar et al.38 and 224 ILs by Gozalez-Miquel et al.39 based on
COSMO-RS prediction of Henry’s law constants of CO236,38 and N2
39 at 25°C. A
combination of COMSO-RS approach with equation of state was successfully used for
prediction and screening of CO2solubility at from (20 to 60) °C and upto the critical
pressure of CO2.40 Anions with fluorine,36,38,41 bromine38 and cations based on
guanidinium and phosphonium41 were shown to have high CO2 absorption capabilities.
At the molecular level, high CO2 absorption was related to the strong vdW interaction
with IL38 and consequently with higher exothermicity. ILs with thiocyanate anions were
found39 to enhance CO2/N2 selectivity due to enhanced vdW interactions preferentially
with CO2 while showing almost no affinity for N2. Shimoyama et al.42 used the model
COMSO-SAC43 for prediction of solubilities, selectivities and permeabilities for CO2 in
ionic liquids with imidazolium cations.
In the present work, we further explore the capabilities and limitation of
COMSOtherm44 that implements COSMO-RS as an a priori auxiliary tool in
screening and designing of ILs for CO2 capture. Henry’s law constants (HLC) of
CO2, CH4 and N2 and selectivities45 for the CO2/CH4 and CO2/N2 separation as a
function of temperature in an extended database of 2701 ILs is predicted. Structural
modifications that promote or diminish the solubility and selectivity are categorized.
Molecular interactions is elucidated through COSMO-RS derived a priori solvent
properties of IL such as sigma profiles and sigma-potentials, and a posteriori

210
quantities such as activity coefficients and transfer properties like enthalpy and
entropies of solvation. A new polarity parameter is introduced and the effect of
molar volume and polarity of ILs on solubility and selectivity is discussed.
COSMOtherm predictions of Henry’s law constants, selectivities, enthalpies of
solvation are compared with experimental results. Henry’s law constants predicted
by different researchers are also compared.
6.2 THEORY
COSMO-RS is an excess Gibbs energy model for liquid mixtures based on
surface charge interaction. Details of COSMO and COSMO-RS model is given in
references.46-50 The surface of a molecule or ion is divided into segments of a
standard area. Each segment is identified by its average screening charge obtained
from COSMO calculation. A histogram that shows the number of segments versus
the corresponding screening charge density of a molecule or ion is called its sigma
profile. The sigma profile of a mixture is a mole fraction average of the sigma
profiles of the pure compounds. The liquid mixture is considered as an
incompressible mixture of surface segments that interact pairwise. The residual
pseudo-chemical potential of a compound is obtained through statistical mechanical
procedure. The combinatorial chemical potential of the compound is obtained from a
modified Guggenheim-Stavermann expression that takes into consideration both the
area and the volume of the molecules. Other thermodynamic properties can be
calculated from the chemical potentials, e.g. activity coefficient at infinite dilution is

211
calculated as,
ln (6.1)
where , , represent the activity coefficient of compound at infinite dilution;
pseudo-chemical potential of at infinite dilution; and chemical potential of in its
pure liquid state, respectively. Henry’s law constant ( ) of a solute in a solvent
can be estimated as37,38
(6.2)
where is the value of the vapour pressure of pure componenteither measured
experimentally or estimated (e.g., using correlation such as Antoine equation,
Wagner equation). can also be estimated by COSMOtherm from the ideal gas
phase chemical potential of solute, , , using the following relationship,49
ln /,
(6.3)
A reasonable estimation of , involves quantum-mechanical energies of
the solute in the gas and conductor with some additional empirical corrections.49,50
When the gas phase chemical potential is chosen for estimation of vapour pressure,
the definition of Henry’s law constant (HLC) reduces to the difference in pseudo-
chemical potential of solute in the solvent at infinite dilution and its ideal gas-phase
chemical potential at a standard pressure 0.1 MPa.
ln /,
(6.4)
HLC computed using equation (4), as is done in the present work, does not

212
contain any explicit reference to pure liquid state of solute and thus advantageous in
its equal applicability in the sub-critical and supercritical region.41 COSMOtherm is
based on the assumption of ideal gas phase and an incompressible liquid phase at all
temperatures. Above the supercritical temperature, COSMOtherm still computes the
chemical potential of a virtual liquid phase in predicting the activity coefficients and
vapour pressure and thus could be less accurate at that temperature. Similarly, the
estimation of gas phase chemical potential will not be sufficiently accurate in this
region. From classical thermodynamic viewpoint, and in equation (6.4)
correspond to the hypothetical chemical potential and fugacity of the pure solute
species as liquid extrapolated from its ideally dilute solution.51 Henry’s law constant
is inversely related to the mole fraction of solute ( ) in the dilute liquid phase
through its fugacity ( ) as,
lim (6.5)

213
6.3 IL DATABASE AND COMPUTATIONAL DETAILS
The IL database used in this work consists of the ions available in the BP-
COSMO-IL database (obtained from COSMOlogic). COSMO calculations were
performed with the software TURBOMOLE52 using density functional theory (DFT)
level of theory, utilizing the Becke and Perdew functional53,54 with triple-zeta valence
polarized (TZVP) basis set.55. 73 cations and 37 anions were used to increase the
variation in anionic and cationic structure to facilitate trend analysis as well as to
increase the possibility of obtaining any fortuitous combination that may result in
practically useful IL for CO2 capture. For most cations, three conformers are used.
The list of the cations and anions with abbreviations, molecular weights, second
sigma moment56 of lowest energy conformer (sig2), a polarity descriptor (later
introduced in section 4.1.5), and an identification number (ID) used in this work are
given in Tables 6.1 and 6.2.

214
Table 6.1 List of Cations.
ID # Cation Abbreviation MW/(g/mol) N sig 2
1 3-methyl-imidazolium [mim] 83.11 20.27 113.382 1,3-methyl-imidazolium [mmim] 97.14 29.27 87.643 1-ethyl-3-methyl-imidazolium [emim] 111.17 29.74 84.964 1-butyl-3-methyl-imidazolium [bmim] 139.22 32.79 84.405 1-pentyl-3-methyl-imidazolium [C5mim] 153.25 34.22 84.896 1-hexyl-3-methyl-imidazolium [hmim] 167.27 35.70 85.387 1-heptyl-3-methyl-imidazolium [C7mim] 181.30 37.11 86.188 1-octyl-3-methyl-imidazolium [omim] 195.33 38.51 86.889 1-decyl-3-methyl-imidazolium [dmim] 223.38 41.38 88.22
10 1-dodecyl-3-methyl-imidazolium [C12mim] 251.43 44.19 89.6611 1-tetradecyl-3-methyl-imidazolium [C14mim] 279.49 47.03 91.2912 1-hexadecyl-3-methyl-imidazolium [C16mim] 307.54 49.91 92.8413 1-octadecyl-3-methyl-imidazolium [C18mim] 335.59 52.71 94.3614 1-butyl-imidazolium [bim] 125.19 23.78 109.9715 1-(2-hydroxyethyl)-3-methylimidazolium [OC2mim] 127.17 24.46 119.9716 1-benzyl-3-methyl-imidazolium [bnmim] 173.24 33.59 104.0717 3-methyl-1-(3-phenyl-propyl)-imidazolium [PhPrmim] 201.29 39.33 109.2318 1-ethyl-2-3-methyl-imidazolium [emmim] 125.19 31.13 77.0619 1-propyl-2-3-methyl-imidazolium [Prmmim] 139.22 32.70 76.4220 1-butyl-2-3-methyl-imidazolium [bmmim] 153.25 34.28 76.5821 1-hexyl-2-3-methyl-imidazolium [hmmim] 181.30 37.16 78.0322 1-hexadecyl-2-3-methyl-imidazolium [C16mmim] 321.57 51.33 85.2923 pyridinium [py] 80.11 19.09 116.7624 1-ethyl-pyridinium [epy] 108.16 30.26 84.9325 1-butyl-pyridinium [bpy] 136.22 33.17 83.7426 1-hexyl-pyridinium [hpy] 164.27 36.01 84.8927 1-octyl-pyridinium [opy] 192.32 38.87 86.2628 N-(3-hxdroxypropyl)pyridinium [PrOpy] 138.19 23.11 123.0729 N-(3-sulfopropyl)pyridinium [sppy] 202.25 25.91 195.8630 1-hexyl-3-methyl-pyridinium [hm(3)py] 178.30 37.05 78.4431 1-butyl-3-ethyl-pyridinium [be(3)py] 164.27 34.20 76.6532 1-butyl-3-methyl-pyridinium [bm(3)py] 150.24 33.52 77.3433 1-octyl -3-methyl-pyridinium [om(3)py] 206.35 39.86 79.8634 4-methyl-n-butylpyridinium [bm(4)py] 150.24 34.42 77.4335 1-hexyl-4-methyl-pyridinium [hm(4)py] 178.30 37.16 78.6336 4-methyl-1-octyl-pyridinium [om(4)py] 206.35 39.98 80.2937 1-butyl-3,4-dimethyl-pyridinium [bmm(4)py] 164.27 34.44 72.4738 1-butyl-3,5-dimethyl-pyridinium [bmm(5)py] 164.27 35.44 71.0639 1,1-dimethyl-pyrrolidinium [mmpyrr] 100.18 30.20 77.0440 1-ethyl-1-methyl-pyrrolidinium [empyrr] 114.21 30.20 73.4541 1-butyl-1-methyl-pyrrolidinium [bmpyrr] 142.26 33.10 72.7942 1-hexyl-1-methyl-pyrrolidinium [hmpyrr] 170.32 35.97 74.0243 1-octyl-1-methyl-pyrrolidinium [ompyrr] 198.37 38.68 75.4544 1,1-dipropyl-pyrrolidinium [dppyrr] 156.29 34.70 68.3645 1-butyl-1-ethyl-pyrrolidinium [bepyrr] 156.29 34.79 69.4046 1-(2-ethoxyethyl)-1-methylpyrrolidinium [EtOEtpyrr] 158.26 32.71 99.13
imid
azol
ium
pyri
dini
um/p
yrro
lidi
nium

215
Table 6.1 List of Cations (Continued)
ID # Cation Abbreviation MW/(g/mol) N sig 247 ammonium [NH4] 18.04 -3.09 196.1948 tetra-methylammonium [Me4N] 74.15 28.67 70.1449 tetra-ethylammonium [Et4N] 130.26 32.50 85.3050 tetra-n-butylammonium [Bu4N] 242.47 43.40 67.3151 ethyl-dimethyl-propylammonium [EtMe2PrN] 116.23 32.03 76.0252 methyl-trioctyl-ammonium [MeOc3N] 368.71 56.78 79.3753 bis(2-methoxyethyl)ammonium [(MeOEt)2N] 134.20 20.69 152.7754 ethyl-dimethyl-2-methoxyethylammonium [EDMA] 132.22 31.27 88.4855 dimethylethanolammonium [Me2EtOHN] 90.14 18.46 123.9656 diethanolammonium [(EtOH)2N] 106.14 9.07 169.3957 butyl-diethanolammonium [Bu(EtOH)2N] 162.25 21.26 135.9258 dodecyl-dimethyl-3-sulfopropylammonium [SPA] 336.56 40.24 193.1259 triisobutyl-methyl-phosphonium [(iBu)3MeP] 217.35 39.12 68.8160 tetrabutyl-phosphonium [Bu4P] 259.43 43.81 68.6061 trihexyl-tetradecyl-phosphonium [thtdP] 483.86 66.57 80.6962 benzyl-triphenyl-phosphonium [BnPh3P] 353.42 47.19 102.5663 guanidinium [G] 60.08 -2.79 162.3164 hexamethylguanidinium [Me6G] 144.24 33.12 58.4065 N,N,N,N-tetramethyl-N-ethylguanidinium [Me4EtG] 144.24 30.66 67.4466 N,N,N,N,N-pentamethyl-N-propyl-guanidinium[Me5PrG] 172.29 35.50 57.5967 N,N,N,N,N-pentamethyl-N-isopropyl-guanidini[Me5(iPr)G] 172.29 34.77 57.6368 O-methyl-N,N,N,N-tetramethylisouronium [O-Me4MeU] 131.20 32.93 72.1969 O-ethyl-N,N,N,N-tetramethylisouronium [O-Me4EtU] 145.22 34.20 70.1570 S-ethyl-N,N,N,N-tetramethylisothiouronium [S-Me4EtT] 161.29 31.88 67.3471 N-butyl-isoquinolinium [BuQ] 186.27 36.63 82.3872 1-(3-methoxypropyl)-1-methylpiperidinium [MeOPrMePi] 172.29 33.08 99.7673 aniline cation [C6H8N] 94.14 6.38 179.07
amm
oniu
m/p
hosp
honi
umot
her
cati
ons

216
Table 6.2 List of Anions.
ID # Anions Abbreviation MW/(g/mol) N sig 21 acetate [Ac] 59.04 -69.72 204.242 decanoate [NnCOO] 171.26 -57.51 207.363 benzoic acid anion [PhCOO] 121.11 -53.33 193.984 tetrafluoroborate [BF4] 86.80 -22.18 111.975 tetracyanoborate [B(CN)4] 114.88 1.27 102.806 bis- oxalatoborate [BOXB] 186.85 1.31 134.377 bis- malonatoborate [BMB] 214.90 -14.51 179.658 bis- salicylatoborate [BSB] 283.02 -12.54 167.869 bis- biphenyldiolatoborate [BPhB] 379.20 -0.60 173.55
10 methylsulfate [MeSO4] 111.10 -36.76 160.7011 ethylsulfate [EtSO4] 125.12 -36.71 163.3512 butylsulfate [BuSO4] 153.18 -33.69 163.4813 octylsulfate [OcSO4] 209.28 -29.49 166.6714 methoxyethylsulfate [MeOEtSO4] 155.15 -36.75 185.1415 ethoxyethylsulfate [EtOEtSO4] 169.18 -36.22 185.5116 2-(2-methoxyethoxy)ethylsulfate [MDEGSO4] 199.20 -37.93 211.2917 trifluoromethane-sulfonate [TfO] 149.07 -19.66 119.0718 toluene-4-sulfonate [TOS] 171.19 -41.81 187.6819 dicyanamide [DCA] 66.04 -28.70 132.9520 bis (trifluoromethyl)imide [BTI] 152.02 2.99 85.9921 bis (trifluoromethylsulfonyle)imide [Tf2N] 280.15 6.22 99.1022 Tricyanomethanide or cyanoform [(CN)3C] 90.06 -11.55 113.1823 bis (trifluoromethylsulfonyl)methane [Tf2C] 279.16 2.35 107.1624 tris (trifluoromethylsulfonyl)methide [Tf3C] 411.22 17.19 96.3925 dimethylphosphate [Me2P] 125.04 -62.04 209.8226 hexaflurophsophate [PF6] 144.96 3.78 88.1127 tris (pentafluoroethyl)trifluorophosphate [eFAP] 445.01 24.81 51.9728 tris (nonafluorobutyl)trifluorophosphate [bFAP] 745.06 43.50 54.2729 bis (2,4,4-trimethylpentyl)phosphinate [(Me3p)2PO2] 289.42 -49.98 210.4230 bis -pentafluoroethyl-phosphinate [(C2F5)2PO2] 301.00 -7.61 103.7431 chloride [Cl] 35.45 -72.15 189.3632 bromide [Br] 79.90 -55.52 170.5633 iodide [I] 126.90 -52.52 147.8534 chlorate [ClO4] 99.45 -12.21 113.0535 2-chlorophenol anion [2-PhCl] 127.55 -35.42 154.8536 3-chlorophenol anion [3-PhCl] 127.55 -36.97 154.7437 4-chlorophenol anion [4-PhCl] 127.55 -39.28 160.82
halo
geni
deac
etat
ebo
rate
sulp
hate
/sul
phon
ate
imid
e/am
ide
phos
phot
e/ph
inat
e

217
Both conventional usage and IUPAC conventions were given preference in
writing the abbreviations. For structural elucidation, cations used in this work are
grouped according to precursors like imidazolium, pyridinium, pyrrolidinium,
guanidinium, uronium, thiouronium, piperidinium and quinolinium; whereas anions
are classified into acetate, borate, imide/amide and methide, sulfate and sulfonate,
phosphate and phosphinate. The following ions in our database can also be found in
similar COSMO-based studies: cations with ID # {2-6,30,32,41,60,64,66,69,70},36
{3,4,8,18,20,21,25,30,32,59},37 {3,4,6,8,9,15,16,25,41,70},38,39
{3,4,6,8,48,49,50,60,61,64,66},41{3,4,6},42 and anions with ID#
{4,10,21,26,27},36{4,10,11,17,18,19,21,24,26},37 {1,4,10,17,19,21,26,27},38
{1,4,5,10,17,19,21,22,27,31},39 {4,17,19,21,22,25,26,34},41 {4,17,19,21,26}42.
All thermodynamic calculations are performed with COSMOtherm with the
C21-0108 parameterization.44 The mole fraction in solution is defined with respect to
the distinct ions. A binary mixture of an ionic liquid with a dissolved gas is thus
treated as a ternary solution. Therefore, the activity coefficients or the Henry’s law
constants obtained directly from COSMOtherm ( ) are converted to binary
framework by scaling them with 0.5, in order to make them comparable with
experimental results where the ionic liquid is considered as a single entity.57 The
vapour phase is assumed to be free of ionic liquids.
Selectivities are defined as / and / for CO2/CH4
and CO2/N2 separation where 1, 2, 3, and 4 in the subscripts indicate ILs, CO2, CH4
and N2 respectively and is the Henry’s law constant of solute in an ionic liquid

218
in the binary framework. The molar volumes of ionic liquids at 25°C were calculated
from the densities of ionic liquids which predicted by COSMOtherm.
6.4 RESULTS AND DISCUSSION
Henry’s law constants of CO2 ( ) at 25°C and their relationship with
solvent properties and molecular interactions are discussed. Predicted at 25°C
and their quantitative evaluations are presented in sections 6.4.1 and 6.4.2. Trends in
due to structural variation are presented in section 6.4.3. The origin of these
trends in terms of solvent properties are discussed in section 6.4.4 and in terms of
activity coefficients in section 6.4.5. Effect of molar volume and polarity of ILs on
are discussed in section 6.4.6. Section 6.4.7 and 6.4.8 present solvation
properties of the three gases and selectivity.
6.4.1 Henry’s law constants at 25°C
Tables 6.3, 6.4 and 6.5 report Henry’s law constants of CO2 ( ) in 2701
ionic liquids. Table 6.3 reports in 814 imidazolium based ionic liquids (formed
from the 22 imidazolium cations and 37 anions). Di-substituted imidazolium cations
are arranged in order of increasing alkyl chain length which are followed by other
functional groups and tri-substituted cations. Table 6.4 reports in pyridinium and
pyrrolidinium based ionic liquids. Table 6.5 reports in ILs of precursors like
quinoline, piperidine, aniline and the non-ring cations such as ammonium,
phosphonium, guanidinium, thiouronium and isothiouronium..

219
Table 6.3 Henry's Law Constants of CO2 (bar) at 298.15 K in Imidazolium-Based Ionic Liquids
ID#
01_
[Ac]
ID#
02
_[N
nC
OO
]
ID#
03_[
Ph
CO
O]
ID#
04_
[BF
4]
ID#
05_
[B(C
N)4
]
ID#
06_[
BO
XB
]
ID#
07
_[B
MB
]
ID#
08_[
BS
B]
ID#
09_
[BP
hB
]
ID#
10_
[MeS
O4
]
ID#
11
_[E
tSO
4]
ID#
12_
[BuS
O4
]
ID#
13_
[OcS
O4
]
ID#
14
_[M
eO
EtS
O4]
ID#
15_
[EtO
EtS
O4
]
ID#
16
_[M
DE
GS
O4]
ID#
17_
[TfO
]
ID#
18_[
TO
S]
ID#
19
_[D
CA
]
ID#
20
_[B
TI]
ID#
21
_[T
f2N
]
ID#
22_
[(C
N)3
C]
ID#
23
_[T
f2C
]
ID#
24
_[T
f3C
]
ID#
25_
[Me2
P]
ID#
26_
[PF
6]
ID#
27_
[eF
AP
]
ID#
28_
[bF
AP
]
ID#
29
_[(M
e3p
)2P
O2
]
ID#
30_[
(C2
F5
)2P
O2
]
ID#
31
_[C
l]
ID#
32_
[Br]
ID#
33_
[I]
ID#
34
_[C
lO4]
ID#
35
_[2-
Ph
Cl]
ID#
36
_[3-
Ph
Cl]
ID#
37
_[4-
Ph
Cl]
ID# 01_[mim] 90 86 98 296 84 172 172 74 44 158 144 117 83 116 108 90 148 101 209 73 65 148 64 46 113 167 23 20 66 53 46 49 135 386 108 106 104
ID# 02_[mmim] 49 67 63 83 66 107 96 59 40 70 74 73 62 66 67 59 79 64 91 66 49 92 48 37 64 91 22 19 57 42 25 20 48 112 73 74 72
ID# 03_[emim] 57 64 60 71 55 84 81 52 37 66 67 64 56 60 60 53 64 58 79 49 42 73 42 34 63 62 21 19 55 38 39 29 60 93 64 65 64
ID# 04_[bmim] 65 60 58 61 46 66 70 47 34 65 62 58 51 57 55 50 53 54 68 37 36 58 36 30 63 44 20 18 53 34 61 42 73 78 57 57 57
ID# 05_[C5mim] 66 59 57 57 43 61 66 45 34 63 60 56 49 55 53 49 50 53 65 35 34 54 35 29 62 40 19 18 52 33 66 44 73 72 55 55 55
ID# 06_[hmim] 66 58 55 52 41 57 62 43 33 60 58 53 47 53 52 48 47 51 61 33 33 51 33 28 61 37 19 18 51 32 68 45 72 67 52 53 53
ID# 07_[C7mim] 66 56 54 49 39 54 59 42 32 58 56 52 46 52 50 47 45 50 58 31 32 49 32 28 60 35 19 18 50 31 70 45 70 62 51 51 52
ID# 08_[omim] 65 55 53 47 38 51 56 41 32 56 54 50 45 50 49 46 44 49 55 30 31 47 32 27 58 33 19 18 49 31 70 44 67 59 49 50 50
ID# 09_[dmim] 62 53 51 42 36 47 52 39 31 52 50 47 43 47 46 43 41 46 51 29 30 43 30 26 55 31 19 17 48 30 67 42 62 53 47 47 48
ID# 10_[C12mim] 59 51 49 39 35 44 48 37 30 49 47 45 41 45 44 42 39 44 48 28 29 41 30 26 53 29 19 17 47 29 64 40 58 49 45 45 46
ID# 11_[C14mim] 57 50 47 37 34 42 45 36 30 46 45 43 40 43 42 40 37 42 45 27 29 39 29 25 50 28 19 17 45 29 61 38 54 46 43 44 44
ID# 12_[C16mim] 55 48 45 35 33 40 43 35 29 44 43 41 39 41 40 39 36 41 44 27 28 38 28 25 48 27 19 18 44 28 58 36 51 43 42 42 43
ID# 13_[C18mim] 53 47 44 33 32 39 41 34 29 42 41 40 38 40 39 37 35 40 42 26 28 37 28 25 47 26 19 18 44 28 56 35 48 41 41 41 42
ID# 14_[bim] 100 75 76 99 55 89 98 56 38 101 91 78 63 80 75 67 74 73 100 42 44 77 45 35 91 58 20 19 61 40 115 84 134 129 70 70 70
ID# 15_[OC2mim] 71 75 76 120 74 115 111 64 42 95 94 87 70 82 80 70 92 77 118 67 53 105 53 40 82 99 24 20 61 46 47 40 86 152 83 83 82
ID# 16_[bnmim] 64 58 59 70 48 70 74 48 35 69 66 60 51 60 58 53 57 57 74 41 38 62 38 31 64 50 20 18 50 35 59 43 77 86 58 58 58
ID# 17_[PhPrmim] 56 51 50 52 41 57 60 42 32 55 53 50 44 50 48 45 46 48 58 34 33 50 33 28 54 39 19 17 45 31 55 38 62 65 49 50 49
ID# 18_[emmim] 44 53 47 47 46 66 63 45 33 49 50 50 46 47 47 43 48 46 56 40 36 55 36 30 48 45 20 18 48 32 31 20 43 64 51 52 51
ID# 19_[Prmmim] 48 52 47 46 42 59 59 42 32 49 49 48 44 46 46 42 45 45 55 35 34 51 34 29 49 39 19 18 47 31 39 24 49 62 49 50 50
ID# 20_[bmmim] 51 51 47 45 40 55 57 41 31 50 49 47 43 46 45 42 43 44 53 32 32 48 32 28 50 36 19 18 46 30 45 28 53 59 48 48 48
ID# 21_[hmmim] 54 50 47 42 37 49 53 38 30 49 47 45 41 44 44 41 40 43 50 29 30 44 30 27 50 32 19 18 45 28 53 32 55 54 45 46 46
ID# 22_[C16mmim] 48 43 40 31 31 37 39 33 27 39 38 37 35 37 36 35 33 36 39 25 27 34 27 24 43 25 19 18 40 26 49 29 44 39 38 38 39
halogenideim
ida
zoliu
m c
atio
ns
Anionsacetate borate sulfate & sulfonate imide/methide phosphate/phosphinate

220
Table 6.4 Henry's Law Constants of CO2 (bar) at 298.15 K in Pyridinium and Pyrrolidinium-Based Ionic Liquids
ID#
01
_[A
c]
ID#
02_
[NnC
OO
]
ID#
03_
[PhC
OO
]
ID#
04
_[B
F4]
ID#
05
_[B
(CN
)4]
ID#
06_[
BO
XB
]
ID#
07_
[BM
B]
ID#
08_
[BS
B]
ID#
09
_[B
PhB
]
ID#
10_
[Me
SO
4]
ID#
11
_[E
tSO
4]
ID#
12
_[B
uS
O4]
ID#
13
_[O
cSO
4]
ID#
14
_[M
eOE
tSO
4]
ID#
15
_[E
tOE
tSO
4]
ID#
16_
[MD
EG
SO
4]
ID#
17
_[T
fO]
ID#
18_
[TO
S]
ID#
19_
[DC
A]
ID#
20
_[B
TI]
ID#
21_
[Tf2
N]
ID#
22_
[(C
N)3
C]
ID#
23_
[Tf2
C]
ID#
24_
[Tf3
C]
ID#
25
_[M
e2P
]
ID#
26
_[P
F6]
ID#
27
_[e
FA
P]
ID#
28
_[b
FA
P]
ID#
29_
[(M
e3p
)2P
O2
]
ID#
30_
[(C
2F5)
2P
O2
]
ID#
31
_[C
l]
ID#
32
_[B
r]
ID#
33
_[I]
ID#
34
_[C
lO4
]
ID#
35
_[2
-PhC
l]
ID#
36
_[3
-PhC
l]
ID#
37
_[4
-PhC
l]
ID# 23_[py] 118 94 120 451 87 186 200 81 47 211 179 135 90 139 125 101 175 120 280 75 69 168 69 48 140 176 24 21 70 57 59 69 217 531 126 122 120
ID# 24_[epy] 61 68 66 84 59 91 89 56 39 75 75 71 59 66 65 58 72 64 90 56 45 81 45 35 69 72 22 19 57 40 41 32 68 107 71 71 70
ID# 25_[bpy] 71 64 62 67 47 69 73 49 36 70 67 62 53 60 58 53 57 58 74 40 38 62 38 31 67 48 20 19 55 35 66 48 80 84 61 61 61
ID# 26_[hpy] 72 61 59 57 42 59 65 45 34 64 61 57 49 56 54 50 50 55 65 34 34 54 35 29 65 39 20 18 53 33 75 52 78 70 55 56 56
ID# 27_[opy] 70 58 56 49 39 53 58 42 33 59 57 53 47 53 51 48 45 51 58 31 32 48 32 28 62 35 19 18 51 31 76 51 72 61 51 52 52
ID# 28_[PrOpy] 74 72 74 104 67 101 99 61 42 90 87 80 66 76 75 65 83 73 105 62 50 92 50 39 79 83 24 21 60 44 57 48 90 127 78 78 77
ID# 29_[sppy] 123 84 114 292 102 159 168 81 50 181 158 126 89 133 121 104 150 115 217 94 72 157 72 51 129 165 30 23 64 59 101 109 214 319 121 118 117
ID# 30_[hm(3)py] 59 53 50 44 38 51 55 40 32 52 50 47 43 47 45 43 42 46 53 31 31 46 31 27 53 33 19 18 48 30 60 37 60 57 48 49 49
ID# 31_[be(3)py] 59 55 52 49 41 57 60 43 33 54 53 50 45 49 48 44 46 48 58 34 33 51 34 29 55 38 20 18 49 32 55 35 61 64 51 52 52
ID# 32_[bm(3)py] 59 54 51 45 39 53 57 41 32 52 51 48 43 47 46 43 43 46 55 31 32 47 32 28 53 34 19 18 48 31 58 35 60 59 49 50 50
ID# 33_[om(3)py] 58 51 49 40 36 47 51 38 31 49 47 45 41 44 43 41 39 44 49 29 30 42 30 26 51 30 19 18 46 29 60 36 57 52 46 47 47
ID# 34_[bm(4)py] 59 55 52 49 41 57 60 43 33 54 53 50 45 49 48 44 46 48 58 34 33 51 34 29 55 37 20 18 49 32 56 35 61 64 52 52 52
ID# 35_[hm(4)py] 60 53 50 44 38 51 55 40 32 52 50 47 43 47 46 43 42 46 53 31 31 46 31 27 53 33 19 18 48 30 60 37 61 57 48 49 49
ID# 36_[om(4)py] 58 51 49 40 36 47 51 38 31 49 47 45 41 45 44 41 40 44 49 29 30 43 30 26 52 30 19 18 46 29 61 36 58 52 46 47 47
ID# 37_[bmm(4)py] 49 48 45 39 37 50 52 39 30 45 44 43 40 41 41 38 39 41 48 30 31 43 31 27 46 32 19 18 44 29 46 26 50 52 45 46 46
ID# 38_[bmm(5)py] 48 46 43 37 36 49 51 38 30 44 43 42 39 40 40 38 38 40 47 29 30 42 30 27 44 31 19 18 43 29 44 25 48 51 44 45 45
ID# 39_[mmpyrr] 38 61 50 50 57 85 74 50 35 50 56 59 54 52 54 49 54 51 63 48 39 70 39 32 51 57 19 17 53 33 19 13 34 74 59 61 59
ID# 40_[empyrr] 40 54 45 43 48 68 63 44 33 46 49 51 47 46 47 43 45 45 55 38 35 57 34 29 46 43 18 17 49 30 25 15 38 64 51 53 52
ID# 41_[bmpyrr] 50 52 46 43 41 57 58 40 31 49 48 47 43 45 45 42 41 43 53 31 31 49 31 27 49 35 18 17 47 28 42 24 52 62 48 49 48
ID# 42_[hmpyrr] 53 50 46 41 37 51 54 38 30 48 47 45 41 44 44 41 39 42 50 28 29 44 29 26 50 31 18 17 45 27 51 28 56 57 45 46 46
ID# 43_[ompyrr] 54 49 45 38 35 47 50 37 29 47 45 43 40 43 42 40 37 41 47 27 28 41 28 25 49 29 18 17 44 26 55 29 55 52 44 44 44
ID# 44_[dppyrr] 42 45 39 34 36 49 49 36 29 40 40 40 38 38 38 36 35 37 44 28 29 41 28 26 41 29 18 17 41 25 37 19 43 50 41 42 42
ID# 45_[bepyrr] 44 46 41 36 37 50 51 37 29 42 42 41 39 40 40 37 36 38 45 28 29 42 29 26 43 30 18 17 42 26 39 20 45 52 42 43 43
ID# 46_[EtOEtpyrr] 56 57 52 53 46 65 65 44 33 57 56 54 48 52 52 47 48 50 63 37 34 56 34 29 57 42 19 17 50 31 49 30 61 73 54 55 55
halogenide
Anionsacetate borate sulfate & sulfonate imide/methide phosphate/phosphinate
pyri
dini
um c
atio
nsp
yrro
lidin
ium
Ca
tions
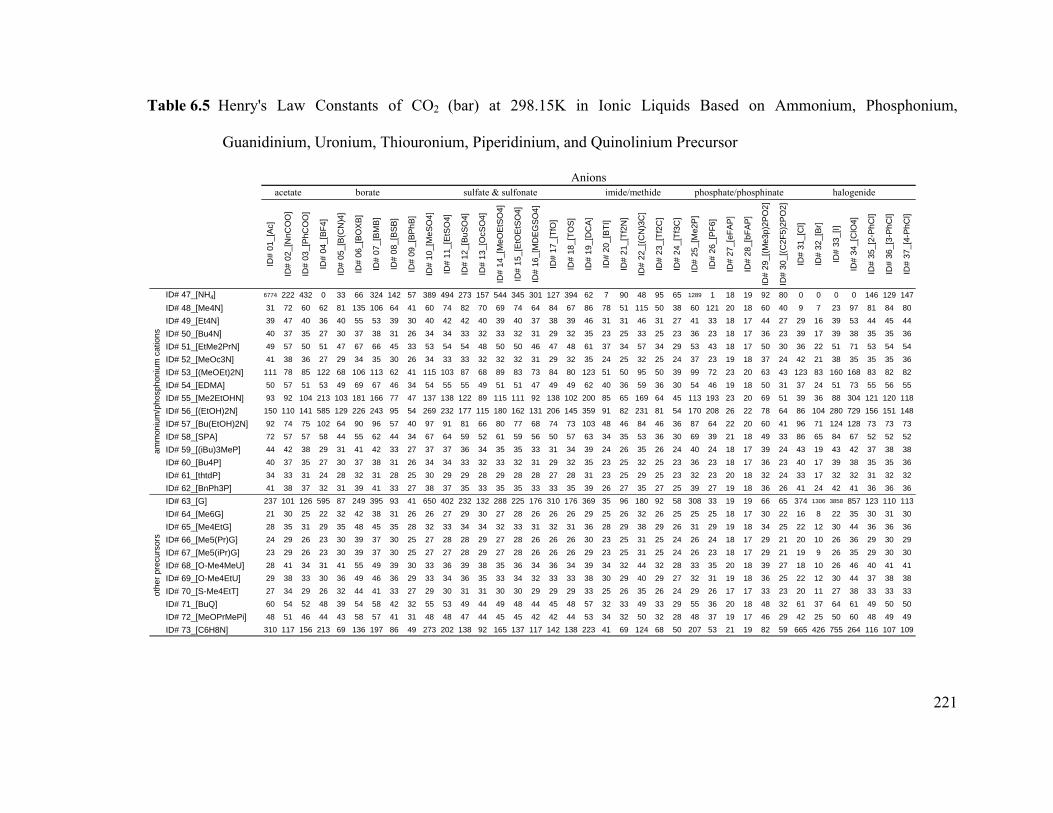
221
Table 6.5 Henry's Law Constants of CO2 (bar) at 298.15K in Ionic Liquids Based on Ammonium, Phosphonium,
Guanidinium, Uronium, Thiouronium, Piperidinium, and Quinolinium Precursor
ID#
01_[
Ac]
ID#
02_
[NnC
OO
]
ID#
03_[
PhC
OO
]
ID#
04_[
BF
4]
ID#
05_
[B(C
N)4
]
ID#
06_
[BO
XB
]
ID#
07_
[BM
B]
ID#
08_
[BS
B]
ID#
09_
[BP
hB
]
ID#
10_
[MeS
O4
]
ID#
11_
[EtS
O4]
ID#
12_
[BuS
O4]
ID#
13_[
OcS
O4]
ID#
14_[
MeO
EtS
O4
]
ID#
15_
[EtO
EtS
O4]
ID#
16_
[MD
EG
SO
4]
ID#
17_
[TfO
]
ID#
18_
[TO
S]
ID#
19_
[DC
A]
ID#
20_[
BT
I]
ID#
21_
[Tf2
N]
ID#
22_
[(C
N)3
C]
ID#
23_
[Tf2
C]
ID#
24_
[Tf3
C]
ID#
25_
[Me2
P]
ID#
26_[
PF
6]
ID#
27_
[eF
AP
]
ID#
28_
[bF
AP
]
ID#
29_
[(M
e3p)
2PO
2]
ID#
30_
[(C
2F
5)2
PO
2]
ID#
31_
[Cl]
ID#
32_
[Br]
ID#
33_
[I]
ID#
34_
[ClO
4]
ID#
35_
[2-P
hCl]
ID#
36_
[3-P
hCl]
ID#
37_
[4-P
hCl]
ID# 47_[NH4] 6774 222 432 0 33 66 324 142 57 389 494 273 157 544 345 301 127 394 62 7 90 48 95 65 1289 1 18 19 92 80 0 0 0 0 146 129 147
ID# 48_[Me4N] 31 72 60 62 81 135 106 64 41 60 74 82 70 69 74 64 84 67 86 78 51 115 50 38 60 121 20 18 60 40 9 7 23 97 81 84 80
ID# 49_[Et4N] 39 47 40 36 40 55 53 39 30 40 42 42 40 39 40 37 38 39 46 31 31 46 31 27 41 33 18 17 44 27 29 16 39 53 44 45 44
ID# 50_[Bu4N] 40 37 35 27 30 37 38 31 26 34 34 33 32 33 32 31 29 32 35 23 25 33 25 23 36 23 18 17 36 23 39 17 39 38 35 35 36
ID# 51_[EtMe2PrN] 49 57 50 51 47 67 66 45 33 53 54 54 48 50 50 46 47 48 61 37 34 57 34 29 53 43 18 17 50 30 36 22 51 71 53 54 54
ID# 52_[MeOc3N] 41 38 36 27 29 34 35 30 26 34 33 33 32 32 32 31 29 32 35 24 25 32 25 24 37 23 19 18 37 24 42 21 38 35 35 35 36
ID# 53_[(MeOEt)2N] 111 78 85 122 68 106 113 62 41 115 103 87 68 89 83 73 84 80 123 51 50 95 50 39 99 72 23 20 63 43 123 83 160 168 83 82 82
ID# 54_[EDMA] 50 57 51 53 49 69 67 46 34 54 55 55 49 51 51 47 49 49 62 40 36 59 36 30 54 46 19 18 50 31 37 24 51 73 55 56 55
ID# 55_[Me2EtOHN] 93 92 104 213 103 181 166 77 47 137 138 122 89 115 111 92 138 102 200 85 65 169 64 45 113 193 23 20 69 51 39 36 88 304 121 120 118
ID# 56_[(EtOH)2N] 150 110 141 585 129 226 243 95 54 269 232 177 115 180 162 131 206 145 359 91 82 231 81 54 170 208 26 22 78 64 86 104 280 729 156 151 148
ID# 57_[Bu(EtOH)2N] 92 74 75 102 64 90 96 57 40 97 91 81 66 80 77 68 74 73 103 48 46 84 46 36 87 64 22 20 60 41 96 71 124 128 73 73 73
ID# 58_[SPA] 72 57 57 58 44 55 62 44 34 67 64 59 52 61 59 56 50 57 63 34 35 53 36 30 69 39 21 18 49 33 86 65 84 67 52 52 52
ID# 59_[(iBu)3MeP] 44 42 38 29 31 41 42 33 27 37 37 36 34 35 35 33 31 34 39 24 26 35 26 24 40 24 18 17 39 24 43 19 43 42 37 38 38
ID# 60_[Bu4P] 40 37 35 27 30 37 38 31 26 34 34 33 32 33 32 31 29 32 35 23 25 32 25 23 36 23 18 17 36 23 40 17 39 38 35 35 36
ID# 61_[thtdP] 34 33 31 24 28 32 31 28 25 30 29 29 28 29 28 28 27 28 31 23 25 29 25 23 32 23 20 18 32 24 33 17 32 32 31 32 32
ID# 62_[BnPh3P] 41 38 37 32 31 39 41 33 27 38 37 35 33 35 35 33 33 35 39 26 27 35 27 25 39 27 19 18 36 26 41 24 42 41 36 36 36
ID# 63_[G] 237 101 126 595 87 249 395 93 41 650 402 232 132 288 225 176 310 176 369 35 96 180 92 58 308 33 19 19 66 65 374 1306 3858 857 123 110 113
ID# 64_[Me6G] 21 30 25 22 32 42 38 31 26 26 27 29 30 27 28 26 26 26 29 25 26 32 26 25 25 25 18 17 30 22 16 8 22 35 30 31 30
ID# 65_[Me4EtG] 28 35 31 29 35 48 45 35 28 32 33 34 34 32 33 31 32 31 36 28 29 38 29 26 31 29 19 18 34 25 22 12 30 44 36 36 36
ID# 66_[Me5(Pr)G] 24 29 26 23 30 39 37 30 25 27 28 28 29 27 28 26 26 26 30 23 25 31 25 24 26 24 18 17 29 21 20 10 26 36 29 30 29
ID# 67_[Me5(iPr)G] 23 29 26 23 30 39 37 30 25 27 27 28 29 27 28 26 26 26 29 23 25 31 25 24 26 23 18 17 29 21 19 9 26 35 29 30 30
ID# 68_[O-Me4MeU] 28 41 34 31 41 55 49 39 30 33 36 39 38 35 36 34 36 34 39 34 32 44 32 28 33 35 20 18 39 27 18 10 26 46 40 41 41
ID# 69_[O-Me4EtU] 29 38 33 30 36 49 46 36 29 33 34 36 35 33 34 32 33 33 38 30 29 40 29 27 32 31 19 18 36 25 22 12 30 44 37 38 38
ID# 70_[S-Me4EtT] 27 34 29 26 32 44 41 33 27 29 30 31 31 30 30 29 29 29 33 25 26 35 26 24 29 26 17 17 33 23 20 11 27 38 33 33 33
ID# 71_[BuQ] 60 54 52 48 39 54 58 42 32 55 53 49 44 49 48 44 45 48 57 32 33 49 33 29 55 36 20 18 48 32 61 37 64 61 49 50 50
ID# 72_[MeOPrMePi] 48 51 46 44 43 58 57 41 31 48 48 47 44 45 45 42 42 44 53 34 32 50 32 28 48 37 19 17 46 29 42 25 50 60 48 49 49
ID# 73_[C6H8N] 310 117 156 213 69 136 197 86 49 273 202 138 92 165 137 117 142 138 223 41 69 124 68 50 207 53 21 19 82 59 665 426 755 264 116 107 109
halogenideborate sulfate & sulfonate imide/methide phosphate/phosphinate
Anionsacetate
oth
er p
recu
rsor
sam
mon
ium
/ph
osph
oniu
m c
atio
ns

222
6.4.2 Quantitative evaluation of predicted HLC
Henry’s law constants around room temperature for 26 ILs are compared with
experimental data12,24,33,36,58-63 in Figure 6.1. The absolute average deviation and root
mean square deviation (defined below) are 15% and 9.1 respectively.
%1 100
(6.6)
1 (6.7)
Greater discrepancies appear in the prediction of [bmpyrr][eFAP], [S-
Me4EtT][eFAP], [thtdPh][Cl], [MeOc8N][Tf2N], [bmmim][BF4], [bmmim][PF6],
[bmim][MeSO4], [emim][EtSO4]. However, the deviations are within the overall
accuracy of the method and considering the fact that Henry’s law constant obtained
from experiment can vary to a large extent, the quantitative prediction can be
considered satisfactory. Rigorous evaluation of COSMOtherm prediction of gas
solubilities at different temperatures and pressures in ionic liquids was done by
Manan et al.37.

223
Figure 6.1 Comparison of Henry’s law constants of CO2: Predicted (grey); and
experimental (black).
0
1020
30
40
50
6070
80
90
100
[C5m
im][
bF
AP
](29
8.15
K)
[hm
im][
eFA
P](
298.
6K)
[MeO
c8N
][T
f2N
](30
3.15
K)
[bm
pyr
r][e
FA
P](
298.
6K)
[th
tdP
][D
CA
](30
3.15
K)
[S-M
e4E
tT][
eFA
P](
298.
6K)
[om
im][
Tf2
N](
298.
15K
)[h
mim
][T
f2N
](29
8.15
K)
[hm
py]
[Tf2
N](
298.
15K
)[b
mim
][T
f2N
](29
8K)
[th
tdP
][C
l](3
03.1
5K)
[em
im][
Tf2
N](
298.
15K
)[t
htd
P][
Tf2
N](
303.
15K
)[b
nm
im][
Tf2
N](
295K
)[b
mp
yrr]
[Tf2
N](
298K
)[e
mm
im][
Tf2
N](
298.
15K
)[b
mim
][P
F6]
(298
K)
[bm
im][
BF
4](2
98K
)[b
mm
im][
BF
4](2
98.1
5K)
[bm
mim
][P
F6]
(298
.15K
)[b
mim
][O
cSO
4](3
13.1
5K)
[em
im][
TfO
](30
3K)
[bm
im][
MeS
O4]
(293
.2K
)[e
mim
][D
CA
](30
3K)
[em
im][
BF
4](3
03K
)[e
mim
][E
tSO
4](3
03.3
8K)
H21
/ 0.1
MP
a

224
The Henry’s law constants predicted in this work are also compared with
those predicted by standard COSMOtherm procedure in earlier studies.27,36,37 For
example, at 10°C, the predicted solubility of CO2 in [bmim][PF6] in our work is 3.9
MPa and matches very well with experimental values of 3.8 and 3.9 MPa reported
respectively by Jacquemin et al.77 and Anthony et al.60 . Zhang et al.36 predicted a
lower value (approximately 3 MPa as read from graph in ref. [36]) whereas Marsh et
al.27 predicted a greater value (approximately 4.5 MPa, read from graph). The
average of the absolute deviation between the two sets (ref. [36] and this work) of
predicted at 25°C in 65 ILs is only 0.34 MPa (Table 6.6). A number of factors
such as minor differences in COSMO files, treatment of conformers, the
parameterizations used in the different versions of the COSMOtherm program may
result in such variation. The Henry’s law constants obtained from COSMOtherm
were further corrected39 by a linear correlation obtained from experimental values.
The AAD(%) (equation 6.6) of the predicted at 25°C in 19 ILs is 12% by the
optimized method38 compared to 16% predicted in this work (Table 6.7).

225
Table 6.6 Comparison of Henry’s law constants of CO2 Predicted in this Work with
those Predicted by Zhang et al. (2008) (Ref. 36).
Anions
[BF4]
(ID#4)
[PF6]
(ID#26)
[MeSO4]
(ID#10)
[Tf2N]
(ID#21)
[eFAP]
(ID#27)
Cations Ref.
36
This
work
Ref.
36
This
work
Ref.
36
This
work
Ref.
36
This
work
Ref.
36
This
work
[dmim] (ID#2) 64 83 86 91 72 70 49 49 23 22
[Emim](ID#3) 59 71 60 62 59 66 42 42 22 21
[Bmim](ID#4) 54 61 44 44 50 65 37 36 20 20
[pmim](ID#5) 51 57 40 40 47 63 35 34 20 19
[hmim](ID#6) 48 52 37 37 45 60 34 33 20 19
[bmpy](ID#32) 42 45 36 34 42 52 33 32 20 19
[hmpy](ID#30) 39 44 32 33 39 52 31 31 20 19
[bmpyrr](ID#41) 39 43 46 35 39 49 31 31 19 18
[HMG](ID#64) 21 22 25 25 27 26 26 26 19 18
[PPrG](ID#66) 22 23 24 24 26 27 25 25 19 18
[ETU](ID#69) 27 30 31 31 32 33 29 29 20 19
[ETT](ID#70) 20 26 23 26 24 29 23 26 17 17
[P(C4)4](ID#60) 25 27 23 23 27 34 25 25 20 18
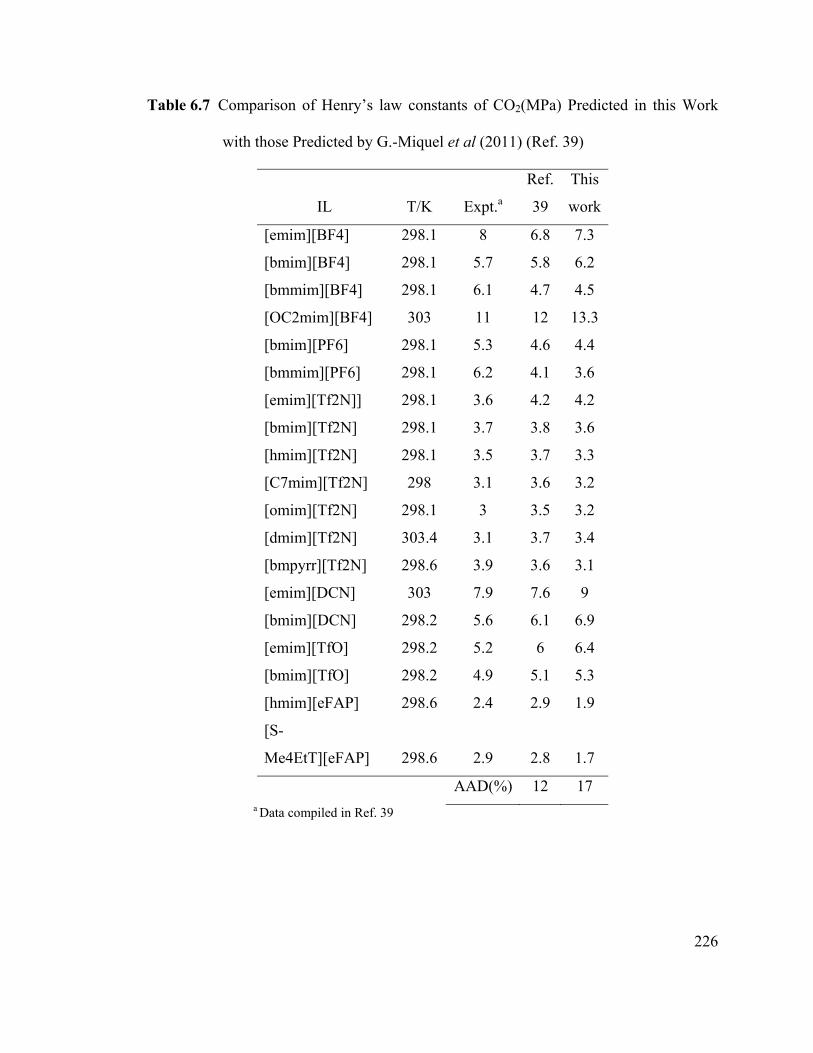
226
Table 6.7 Comparison of Henry’s law constants of CO2(MPa) Predicted in this Work
with those Predicted by G.-Miquel et al (2011) (Ref. 39)
IL T/K Expt.a
Ref.
39
This
work
[emim][BF4] 298.1 8 6.8 7.3
[bmim][BF4] 298.1 5.7 5.8 6.2
[bmmim][BF4] 298.1 6.1 4.7 4.5
[OC2mim][BF4] 303 11 12 13.3
[bmim][PF6] 298.1 5.3 4.6 4.4
[bmmim][PF6] 298.1 6.2 4.1 3.6
[emim][Tf2N]] 298.1 3.6 4.2 4.2
[bmim][Tf2N] 298.1 3.7 3.8 3.6
[hmim][Tf2N] 298.1 3.5 3.7 3.3
[C7mim][Tf2N] 298 3.1 3.6 3.2
[omim][Tf2N] 298.1 3 3.5 3.2
[dmim][Tf2N] 303.4 3.1 3.7 3.4
[bmpyrr][Tf2N] 298.6 3.9 3.6 3.1
[emim][DCN] 303 7.9 7.6 9
[bmim][DCN] 298.2 5.6 6.1 6.9
[emim][TfO] 298.2 5.2 6 6.4
[bmim][TfO] 298.2 4.9 5.1 5.3
[hmim][eFAP] 298.6 2.4 2.9 1.9
[S-
Me4EtT][eFAP] 298.6 2.9 2.8 1.7
AAD(%) 12 17 a Data compiled in Ref. 39

227
6.4.3 Trends in Henry’s law constant due to structural variations
To establish trends due to structural variations in the cations and anions,
in the model ionic liquids [cation][Tf2N] (represent 73 ILs) and [bmim][anion]
(represent 37 ILs) are used. Henry’s law constants of CO2, ; CO2/CH4
selectivity, ; CO2/N2 selectivity, ; residual activity coefficients at infinite
dilutions ( , ), enthalpy (∆ ) and entropy (∆S ) of solvation with their molar
volume(VIL),molecularweight (MWIL) and relative overall polarity parameter at
298.15K (NIL) are given in Table 6.8 for the 73 ionic liquids [cation][Tf2N] and in
Table 6.9 for the 37 ionic liquids [bmim][anion].

228
Table 6.8 Properties in the Ionic Liquids [cation][Tf2N]
Cations CO2 CH4 N2 CO2 CH4 N2 CO2 CH4 N2
ID#01_[mim] 363.3 0.220 26.49 65 16.5 37.7 -16.35 -3.55 -4.43 -89.5 -69.9 -79.7 -0.43 1.21 0.51ID#02_[mmim] 377.3 0.239 35.49 49 15.8 45.5 -16.78 -3.87 -3.84 -88.6 -68.2 -76.9 -0.69 0.91 0.44ID#03_[emim] 391.3 0.256 35.96 42 14.0 45.7 -17.13 -4.72 -4.12 -88.5 -68.9 -76.6 -0.82 0.66 0.32ID#04_[bmim] 419.4 0.290 39.00 36 11.6 44.9 -17.35 -5.70 -4.43 -88.0 -69.3 -76.3 -0.94 0.36 0.19ID#05_[C5mim] 433.4 0.307 40.44 34 10.8 44.6 -17.38 -6.00 -4.48 -87.7 -69.3 -76.0 -0.96 0.26 0.15ID#06_[hmim] 447.4 0.324 41.92 33 10.1 44.4 -17.40 -6.25 -4.51 -87.4 -69.3 -75.8 -0.99 0.17 0.13ID#07_[C7mim] 461.4 0.341 43.32 32 9.5 44.1 -17.38 -6.45 -4.53 -87.2 -69.2 -75.5 -1.00 0.10 0.11ID#08_[omim] 475.5 0.358 44.73 31 9.0 43.9 -17.37 -6.63 -4.53 -86.9 -69.2 -75.3 -1.01 0.04 0.09ID#09_[dmim] 503.5 0.392 47.60 30 8.2 43.5 -17.34 -6.91 -4.52 -86.5 -69.0 -74.9 -1.02 -0.07 0.08ID#10_[C12mim] 531.6 0.426 50.41 29 7.6 43.2 -17.30 -7.13 -4.50 -86.1 -68.8 -74.5 -1.02 -0.15 0.07ID#11_[C14mim] 559.6 0.460 53.25 29 7.1 42.9 -17.25 -7.30 -4.46 -85.7 -68.7 -74.1 -1.02 -0.21 0.06ID#12_[C16mim] 587.7 0.493 56.13 28 6.7 42.5 -17.19 -7.45 -4.42 -85.4 -68.5 -73.7 -1.01 -0.27 0.07ID#13_[C18mim] 615.7 0.527 58.93 28 6.3 42.2 -17.13 -7.57 -4.37 -85.1 -68.3 -73.4 -1.00 -0.31 0.07ID#14_[bim] 405.3 0.271 30.00 44 11.8 39.6 -17.05 -5.59 -4.83 -88.8 -70.8 -78.3 -0.74 0.56 0.25ID#15_[OC2mim] 407.3 0.261 30.68 53 14.3 39.9 -16.47 -4.01 -4.07 -88.3 -68.6 -77.4 -0.57 0.93 0.43ID#16_[bnmim] 453.4 0.303 39.81 38 12.6 52.6 -17.13 -4.96 -3.55 -87.7 -67.9 -75.0 -0.87 0.51 0.41ID#17_[PhPrmim] 481.4 0.337 45.55 33 11.7 54.6 -17.57 -5.60 -3.79 -88.0 -68.4 -75.0 -0.97 0.34 0.35ID#18_[emmim] 405.3 0.272 37.35 36 13.3 47.6 -17.59 -5.21 -4.22 -88.8 -68.8 -76.0 -0.96 0.47 0.22ID#19_[Prmmim] 419.4 0.290 38.92 34 12.2 46.9 -17.70 -5.67 -4.39 -88.6 -69.0 -75.9 -1.01 0.33 0.16ID#20_[bmmim] 433.4 0.307 40.50 32 11.2 46.4 -17.73 -6.00 -4.49 -88.3 -69.0 -75.8 -1.04 0.22 0.12ID#21_[hmmim] 461.4 0.341 43.38 30 9.9 45.5 -17.71 -6.45 -4.57 -87.7 -69.0 -75.4 -1.07 0.07 0.07ID#22_[C16mmim] 601.7 0.510 57.55 27 6.6 42.9 -17.43 -7.55 -4.49 -85.8 -68.3 -73.6 -1.05 -0.31 0.03ID#23_[py] 360.3 0.215 25.31 69 15.8 36.0 -16.17 -3.63 -4.54 -89.5 -70.3 -80.3 -0.36 1.23 0.53ID#24_[epy] 388.3 0.251 36.48 45 14.1 45.8 -16.87 -4.50 -3.94 -88.2 -68.7 -76.6 -0.76 0.73 0.38ID#25_[bpy] 416.4 0.285 39.39 38 11.6 45.2 -17.20 -5.59 -4.30 -87.8 -69.3 -76.3 -0.90 0.39 0.23ID#26_[hpy] 444.4 0.319 42.23 34 10.1 44.7 -17.27 -6.18 -4.42 -87.3 -69.3 -75.8 -0.96 0.20 0.16ID#27_[opy] 472.5 0.353 45.09 32 9.0 44.3 -17.28 -6.58 -4.45 -86.8 -69.2 -75.3 -0.99 0.05 0.12ID#28_[PrOpy] 418.3 0.273 29.32 50 13.2 41.5 -16.54 -4.43 -3.98 -88.0 -68.8 -76.8 -0.62 0.80 0.42ID#29_[sppy] 482.4 0.308 32.13 72 14.2 31.3 -15.57 -3.26 -4.39 -87.8 -68.6 -78.9 -0.22 1.28 0.54ID#30_[hm(3)py] 458.4 0.336 43.27 31 9.5 44.7 -17.54 -6.54 -4.56 -87.4 -69.3 -75.5 -1.03 0.06 0.09ID#31_[be(3)py] 444.4 0.312 40.42 33 10.8 45.5 -17.54 -6.09 -4.49 -88.0 -69.4 -76.0 -1.00 0.22 0.14ID#32_[bm(3)py] 430.4 0.309 39.74 32 10.1 44.9 -17.59 -6.37 -4.59 -87.8 -69.4 -75.8 -1.03 0.13 0.10ID#33_[om(3)py] 486.5 0.370 46.08 30 8.6 44.2 -17.50 -6.86 -4.58 -86.9 -69.1 -75.1 -1.05 -0.05 0.06ID#34_[bm(4)py] 430.4 0.303 40.63 33 10.7 45.4 -17.54 -6.10 -4.50 -88.0 -69.4 -76.0 -1.00 0.22 0.13ID#35_[hm(4)py] 458.4 0.336 43.38 31 9.5 44.7 -17.54 -6.54 -4.57 -87.4 -69.3 -75.5 -1.03 0.06 0.09ID#36_[om(4)py] 486.5 0.370 46.20 30 8.6 44.1 -17.49 -6.87 -4.58 -86.9 -69.2 -75.1 -1.04 -0.05 0.06ID#37_[bmm(4)py] 444.4 0.318 40.66 31 10.2 45.6 -17.79 -6.47 -4.64 -88.1 -69.4 -75.7 -1.07 0.09 0.07ID#38_[bmm(5)py] 444.4 0.320 41.66 30 10.0 45.3 -17.85 -6.56 -4.70 -88.2 -69.5 -75.8 -1.08 0.07 0.05ID#39_[mmpyrr] 380.3 0.253 36.42 39 15.6 45.7 -17.66 -4.62 -4.42 -89.7 -68.8 -77.0 -0.90 0.69 0.24ID#40_[empyrr] 394.4 0.269 36.42 35 14.1 45.8 -17.97 -5.30 -4.65 -89.7 -69.2 -76.8 -1.00 0.48 0.14ID#41_[bmpyrr] 422.4 0.303 39.32 31 11.8 44.8 -18.03 -6.05 -4.84 -89.0 -69.3 -76.4 -1.08 0.23 0.04ID#42_[hmpyrr] 450.5 0.337 42.19 29 10.3 44.1 -17.96 -6.47 -4.86 -88.3 -69.2 -75.9 -1.10 0.08 0.01ID#43_[ompyrr] 478.5 0.371 44.90 28 9.2 43.5 -17.88 -6.78 -4.84 -87.8 -69.0 -75.4 -1.10 -0.04 -0.01ID#44_[dppyrr] 436.4 0.317 40.92 29 11.0 44.9 -18.27 -6.47 -4.99 -89.1 -69.5 -76.2 -1.15 0.10 -0.02ID#45_[bepyrr] 436.4 0.319 41.01 29 11.0 44.7 -18.22 -6.42 -4.97 -89.0 -69.4 -76.2 -1.13 0.11 -0.01ID#46_[EtOEtpyrr] 438.4 0.309 38.93 34 12.2 44.6 -17.65 -5.61 -4.52 -88.6 -69.0 -76.2 -0.96 0.38 0.15ID#47_[NH4] 298.2 0.174 3.13 90 11.4 12.9 -17.44 -7.15 -10.17 -95.9 -81.7 -92.8 -0.17 1.09 -0.31ID#48_[Me4N] 354.3 0.237 34.89 51 18.3 44.7 -16.87 -3.24 -3.91 -89.2 -67.7 -77.4 -0.66 1.08 0.45ID#49_[Et4N] 410.4 0.299 38.72 31 12.4 46.0 -18.21 -6.00 -4.82 -89.5 -69.5 -76.5 -1.10 0.26 0.05ID#50_[Bu4N] 522.6 0.436 49.62 25 7.7 42.4 -18.11 -7.48 -5.06 -87.6 -69.0 -75.0 -1.17 -0.27 -0.09ID#51_[EtMe2PrN] 396.4 0.285 38.25 34 13.1 45.5 -17.81 -5.48 -4.60 -89.1 -69.2 -76.6 -1.00 0.42 0.14ID#52_[MeOc3N] 648.9 0.589 63.00 25 5.9 40.9 -17.52 -7.85 -4.66 -85.7 -68.1 -73.4 -1.05 -0.42 -0.01ID#53_[(MeOEt)2N] 414.3 0.285 26.91 50 12.1 35.0 -17.09 -5.23 -5.12 -89.9 -70.8 -79.3 -0.61 0.72 0.26ID#54_[EDMA] 412.4 0.293 37.48 36 13.2 44.8 -17.66 -5.30 -4.49 -89.0 -68.9 -76.4 -0.95 0.47 0.17ID#55_[Me2EtOHN] 370.3 0.241 24.68 65 16.3 34.3 -16.29 -3.26 -4.57 -89.3 -68.8 -79.4 -0.41 1.22 0.44ID#56_[(EtOH)2N] 386.3 0.249 15.29 82 14.7 26.9 -15.72 -3.34 -5.13 -89.4 -70.2 -81.2 -0.16 1.37 0.45ID#57_[Bu(EtOH)2N] 442.4 0.315 27.48 46 11.2 33.4 -16.83 -5.25 -5.04 -88.3 -69.5 -77.9 -0.67 0.59 0.16ID#58_[SPA] 616.7 0.495 46.46 35 8.3 36.1 -16.80 -6.39 -4.73 -86.0 -68.7 -75.3 -0.78 0.18 0.13ID#59_[(iBu)3MeP] 497.5 0.401 45.34 26 8.8 43.3 -18.14 -7.12 -5.03 -88.0 -69.1 -75.3 -1.17 -0.14 -0.08ID#60_[Bu4P] 539.6 0.447 50.03 25 7.7 42.4 -18.08 -7.48 -5.03 -87.5 -68.9 -74.9 -1.16 -0.28 -0.09ID#61_[thtdP] 764.0 0.717 72.79 25 5.2 39.9 -17.40 -8.14 -4.57 -85.1 -67.6 -72.7 -1.00 -0.51 0.02ID#62_[BnPh3P] 633.6 0.446 53.41 27 8.8 56.5 -17.63 -6.54 -3.55 -86.6 -67.5 -72.9 -1.06 -0.04 0.30ID#63_[G] 340.2 0.203 3.43 96 20.2 25.0 -15.77 -3.01 -5.81 -90.8 -73.0 -84.2 -0.07 1.78 0.46ID#64_[Me6G] 424.4 0.310 39.34 26 11.6 46.4 -18.93 -6.84 -5.31 -90.6 -70.4 -76.8 -1.25 0.04 -0.09ID#65_[Me4EtG] 424.4 0.307 36.88 29 11.3 44.6 -18.50 -6.62 -5.20 -90.0 -70.3 -77.0 -1.15 0.12 -0.03ID#66_[Me5PrG] 452.4 0.343 41.72 25 10.2 45.1 -18.82 -7.16 -5.35 -89.9 -70.1 -76.4 -1.26 -0.10 -0.13ID#67_[Me5(iPr)G] 452.4 0.341 40.99 25 10.5 45.3 -18.87 -7.11 -5.38 -90.1 -70.1 -76.5 -1.26 -0.07 -0.13ID#68_[O-Me4MeU] 411.3 0.286 39.15 32 13.0 44.9 -18.24 -5.72 -4.84 -89.9 -69.3 -76.6 -1.07 0.34 0.05ID#69_[O-Me4EtU] 425.4 0.304 40.42 29 11.9 45.4 -18.38 -6.21 -4.94 -89.8 -69.5 -76.4 -1.13 0.19 0.00ID#70_[S-Me4EtT] 441.4 0.311 38.10 26 12.7 49.8 -18.78 -6.36 -4.98 -90.1 -69.5 -76.3 -1.25 0.13 -0.02ID#71_[BuQ] 466.4 0.323 42.85 33 10.1 47.8 -17.37 -6.13 -4.15 -87.3 -68.9 -75.1 -0.99 0.17 0.20ID#72_[MeOPrMePi] 452.4 0.321 39.30 32 11.9 45.8 -17.89 -5.92 -4.61 -88.9 -69.4 -76.1 -1.01 0.31 0.13ID#73_[C6H8N] 374.3 0.235 12.60 69 11.0 29.5 -16.65 -5.69 -5.86 -91.0 -74.2 -83.0 -0.35 0.88 0.34
S23 S24H21/0.1
MPaVIL/(m3/
kmol)
MWIL/(g/
mol)NIL
imid
azol
ium
pyrid
iniu
m/p
yrro
lidin
ium
amm
oniu
m/p
hosp
honi
umot
her
catio
ns∆H∞(kJ/mol) ∆S∞(J/mol/K) ln γ ∞,r

229
Table 6.9 Properties in the Ionic Liquids [bmim][anion]
Cations CO2 CH4 N2 CO2 CH4 N2 CO2 CH4 N2
ID#01_[Ac] 198.3 0.193 -36.93 65 5.7 89.7 -16.85 -9.35 -2.25 -91.3 -80.5 -79.7 -0.47 0.09 1.33ID#02_[NnCOO] 310.5 0.327 -24.72 60 4.6 69.8 -15.33 -8.22 -1.45 -85.5 -74.4 -74.3 -0.39 -0.02 1.17ID#03_[PhCOO] 260.3 0.237 -20.55 58 6.9 108.4 -16.83 -7.98 -1.11 -90.2 -76.6 -76.4 -0.53 0.25 1.47ID#04_[BF4] 226.0 0.189 10.61 61 13.3 123.8 -16.71 -6.04 -0.39 -90.2 -76.0 -75.5 -0.54 0.88 1.59ID#05_[B(CN)4] 254.1 0.246 34.06 46 10.4 94.9 -16.42 -5.96 -1.05 -86.8 -71.2 -73.1 -0.76 0.42 1.11ID#06_[BOXB] 326.1 0.238 34.09 66 8.2 47.9 -15.84 -5.65 -2.35 -88.0 -71.2 -74.9 -0.38 0.56 0.81ID#07_[BMB] 354.1 0.269 18.27 70 8.3 67.7 -15.84 -6.23 -1.19 -88.4 -73.7 -74.3 -0.29 0.66 1.24ID#08_[BSB] 422.2 0.328 20.25 47 7.6 92.0 -16.37 -6.48 -0.53 -86.8 -70.6 -71.3 -0.62 0.25 1.22ID#09_[BPhB] 518.4 0.419 32.19 34 7.8 120.1 -17.16 -6.77 -0.27 -87.0 -69.2 -70.1 -0.84 0.06 1.27ID#10_[MeSO4] 250.3 0.209 -3.98 65 8.3 95.1 -16.60 -7.62 -1.18 -90.3 -77.7 -76.5 -0.45 0.49 1.41ID#11_[EtSO4] 264.3 0.226 -3.92 62 7.9 91.5 -16.37 -7.47 -1.09 -89.2 -76.5 -75.5 -0.47 0.43 1.36ID#12_[BuSO4] 292.4 0.259 -0.91 58 7.3 84.2 -16.08 -7.24 -1.05 -87.7 -74.6 -74.1 -0.50 0.33 1.25ID#13_[OcSO4] 348.5 0.327 3.29 51 6.5 75.1 -15.84 -7.21 -1.17 -85.8 -72.3 -72.5 -0.56 0.14 1.07ID#14_[MeOEtSO4] 294.4 0.253 -3.96 57 8.0 88.3 -16.69 -7.59 -1.41 -89.5 -76.3 -75.5 -0.53 0.39 1.26ID#15_[EtOEtSO4] 308.4 0.269 -3.43 55 7.7 85.2 -16.47 -7.42 -1.31 -88.6 -75.2 -74.7 -0.54 0.34 1.22ID#16_[MDEGSO4] 338.4 0.298 -5.14 50 7.8 84.3 -16.84 -7.68 -1.64 -89.1 -75.4 -75.0 -0.60 0.29 1.15ID#17_[TfO] 288.3 0.217 13.12 53 11.1 68.8 -16.78 -6.09 -2.54 -89.3 -73.4 -76.7 -0.64 0.61 0.91ID#18_[TOS] 310.4 0.267 -9.02 54 7.6 102.5 -16.53 -7.54 -0.88 -88.7 -75.3 -74.7 -0.55 0.32 1.40ID#19_[DCA] 205.3 0.197 4.08 68 9.0 115.5 -16.17 -6.81 -0.32 -89.4 -76.3 -75.7 -0.41 0.62 1.64ID#20_[BTI] 291.2 0.226 35.77 37 16.4 85.9 -17.90 -4.89 -2.54 -90.1 -69.7 -75.6 -0.98 0.65 0.78ID#21_[Tf2N] 419.4 0.290 39.00 36 11.6 44.9 -17.35 -5.70 -4.43 -88.0 -69.3 -76.3 -0.94 0.36 0.19ID#22_[(CN)3C] 229.3 0.220 21.23 58 9.7 110.0 -16.00 -6.05 -0.12 -87.4 -73.0 -73.3 -0.55 0.56 1.47ID#23_[Tf2C] 418.4 0.295 35.14 36 11.6 48.5 -17.34 -5.72 -4.15 -88.0 -69.4 -76.1 -0.92 0.37 0.28ID#24_[Tf3C] 550.4 0.356 49.98 30 11.3 36.7 -17.71 -5.83 -5.37 -87.7 -68.0 -76.3 -1.05 0.22 -0.12ID#25_[Me2P] 264.3 0.237 -29.25 63 6.3 85.9 -16.36 -8.50 -1.69 -89.3 -78.2 -77.1 -0.45 0.22 1.31ID#26_[PF6] 284.2 0.212 36.56 44 17.7 108.7 -17.45 -4.51 -1.03 -90.0 -70.5 -73.9 -0.84 0.87 1.16ID#27_[eFAP] 584.2 0.371 57.59 20 14.4 45.9 -19.31 -6.23 -6.27 -89.5 -67.8 -77.5 -1.48 0.03 -0.33ID#28_[bFAP] 884.3 0.529 76.29 18 12.9 34.2 -18.96 -6.30 -6.88 -87.7 -66.5 -76.5 -1.43 -0.02 -0.57ID#29_[(Me3p)2PO2] 428.6 0.452 -17.20 53 4.2 62.9 -15.18 -8.12 -1.46 -83.9 -72.1 -72.3 -0.42 -0.15 1.04ID#30_[(C2F5)2PO2] 440.2 0.304 25.18 34 11.4 45.7 -17.40 -6.21 -4.88 -87.6 -70.3 -77.4 -0.99 0.29 0.15ID#31_[Cl] 174.7 0.161 -39.36 61 5.4 89.6 -17.10 -10.48 -3.16 -91.6 -83.4 -82.2 -0.57 -0.06 1.22ID#32_[Br] 219.1 0.163 -22.74 42 9.9 143.9 -17.85 -9.77 -2.70 -90.9 -82.8 -81.4 -0.95 0.16 1.32ID#33_[I] 266.1 0.167 -19.73 73 7.3 112.3 -16.53 -8.60 -1.51 -91.1 -81.0 -80.0 -0.38 0.43 1.64ID#34_[ClO4] 238.7 0.187 20.58 78 9.5 76.2 -16.31 -6.18 -1.00 -90.9 -75.7 -75.6 -0.29 0.80 1.35ID#35_[2-PhCl] 266.8 0.228 -2.63 57 7.9 101.6 -16.91 -7.13 -1.18 -90.3 -74.7 -76.0 -0.54 0.36 1.39ID#36_[3-PhCl] 266.8 0.228 -4.19 57 7.6 95.4 -16.83 -7.10 -1.33 -90.1 -74.4 -76.0 -0.53 0.34 1.34ID#37_[4-PhCl] 266.8 0.228 -6.50 57 7.5 94.4 -16.90 -7.23 -1.44 -90.4 -74.7 -76.3 -0.54 0.32 1.33
MWIL/(g/
mol)VIL/(m3/
kmol)NIL
H21/0.1
MPaS23 S24
hal
ogen
ide
ace
tate
bora
tesu
lph
ate/
sulp
hona
teim
ide/
amid
eph
osph
ote/
phin
ate
∆H∞(kJ/mol) ∆S∞(J/mol/K) ln γ ∞,r

230
The cation [bmim] is regarded as a model cation for the analysis of structural
variations in the ring cations. Structural modifications relative to [bmim] that
enhance CO2 solubility (decrease ) include (i) increase in the alkyl chain lengthin
ring-precursor (ii) alkylation of ammonium and phosphonium cation
([Et4N][Tf2N]<[Bu4N][Tf2N] and [(iBu)3MeP][Tf2N<[Bu4P][Tf2N]), (iii) change
in cation family (e.g., for ring cation:[bmim] [Tf2N] <[bm(3)py][Tf2N]<[bmpyrr]
[Tf2N] and for non-ring cation: [O-Me4EtU]~[Me4EtG]<[S-Me4EtT], (iv)
substitution of methyl group in C2 position (e.g., [bmim][Tf2N]<[bmmim][Tf2N]).
Cation modifications that diminish CO2 solubility include the presence of the
following groups: hydroxyl (e.g., [bmim][Tf2N]>[OC2mim][Tf2N]), phenyl
([hmim][Tf2N]>[bnmim][Tf2N]), and ether ([bepyrr]][Tf2N]>[EtOEtpyrr]][Tf2N]).
Zhang et al. [36] showed that COSMO-RS predicts a reduction of CO2 solubility due
to the presence of electronegative fluorine in the cation. In general, the effect of
cation modification on is rather low. in the IL [cation][Tf2N] is around 3MPa
except some extreme cases (e.g., [NH4], [G], [mim], [Py] that may result in solid ILs
at room temperature when paired with relatively small anions).
Anions are less amenable to systematic structural variation but usually have
larger impact on gas solubility as can be seen from the large variation in the
magnitude of in Table 6.9 (1.8 to 7.8 MPa). Presence of fluorine-containing
anions results in smaller . Acetate, chloride, some large borate, dimethyl
phosphate, dicyanamide anions result in high (~6MPa) when paired with [bmim]
cation. Longer alkyl chain length generally enhances CO2 solubility
([bmim][MeSO4]<[bmim][OcSO4]; [bmim][Ac]<[bmim][NnCOO]). The presence

231
of ether group causes slight increase in the solubility of
CO2([bmim][BuSO4]<[bmim][MeOEtSO4]). Among the seven sulphate anions
considered, [MDEGSO4] has the lowest . Presence of hydroxyl group also results
in high Henry’s law constant as shown for the case of [bmim][Lactate] (6.2 MPa).36
The dominance of cation can be dramatically influenced by some anions. For
example, Tables 6.3, 6.4 and 6.5 reveal that all ILs with fluoroalkylphosphate (FAP)
anions have very similar indicating negligible effect of companion cations. On
the other hand, in ILs with chloride/bromide anion is highly influenced by the
counterpart cation. For example, in [O-Me4EtU][Br] and [dmim][Br] are (1.2
and 4.2) MPa respectively.
Comparison with experimental trends shows that COSMOtherm accurately
predicts the effect of alkyl chain length, phenyl and ether group in the cations relative
to the model [bmim] cation. However, its prediction is opposite to experimental
trend for the effect of methyl substitution on the C2 position of an imidazolium ring
and cation family.37 Cation fluorination was found59 to increase CO2 solubility
slightly but the COSMOtherm prediction shows the opposite trend36. For the [bmim]
cation, COSMOtherm predicts the correct order of the five anions as shown in Figure
6.1, i.e. [MeSO4]<[BF4]<[OcSO4]<[PF6]<[Tf2N]. In phosphonium ILs,
COSMOtherm incorrectly predicts the ordering between [thtdP][Cl] and
[thtdP][DCA] but correctly predicts that CO2 is less soluble in both of these ILs than
[thtdP][Tf2N]. COSMOtherm correctly predicts that increased fluorination on the
anion enhances CO2 solubility. Manan et al.37 found that COSMOtherm ordering of
five anions (nitrate<[BF4]<[TfO]<[PF6]<[Tf2N]) matched with experimental results

232
except the order between [PF6] and [TfO].
6.4.4 Qualitative interpretations of molecular interactions
The above mentioned trends in solubility are a consequence of the variations
in molecular interactions of ILs with CO2 which originate from the variations in the
chemical constituents, shapes and sizes of ILs. Effect of structural variations in the
ILs can be visualized through their sigma profiles. Corresponding sigma-potentials
of the solvents can be used to ascertain the affinity of solvents to a solute [24]. The
sigma profiles of four imidazolium cations ([bmim], [bnmim], [omim] and
[OC2mim]) paired with [Tf2N] anion along with CO2 and CH4 are shown in Figure.
6.2.
Effect of benzyl, hydroxyl and alkyl chain length. The sigma profiles of the
ILs shown in Figure 6.2 extend from (+2 to -2) e/nm2. The similarity of the IL sigma
profiles within the range (0.5 to 2) e/nm2 is due to the common imidazolium ring and
within the range (-1 to -2) e/nm2is due to the common [Tf2N] anion. Major
differences among them are found around the neutral regions. Of the four ILs,
[omim][Tf2N], which has the highest CO2 solubility, is seen to have a lot more
nonpolar surfaces than others where sigma is 0e/nm2. Due to the apolar and
symmetric benzyl ring, [bnmim][Tf2N] is seen to have slightly more surfaces around
-0.7e/nm2 and 0.7 e/nm2. [OC2mim][Tf2N] has slightly more polar surface pieces
due to the polar oxygen at sigma +1.4 e/nm2. All four ILs have enough negative
surface pieces that will interact favourably with the positive surface pieces of CO2.

233
The affinity of these ILs to CO2 can be compared by their sigma-potentials which is a
measure of the response of the solvent to a molecular surface of polarity sigma. ILs
with a more negative potential at a particular sigma will be more favorable to the
corresponding surface segment.
The sigma-potentials of [bmim][Tf2N], [bnmim][Tf2N], [OC2mim][Tf2N]
and [omim][Tf2N] at 25°C are shown in Figure 6.3 in the sigma region between (-0.1
and +0.8) e/nm2 that corresponds to the screening charges of CO2.Among the three
ILs ([bmim][Tf2N], [bnmim][Tf2N], [OC2mim][Tf2N]), almost all the surface pieces
of CO2 will interact most favorably with [bmim][Tf2N] which has the most negative
sigma potential of the three, followed by [bnmim][Tf2N] and [OC2mim][Tf2N].
Thus, the activity coefficients is expected to be lesser in [bmim][Tf2N] than either in
[bnmim][Tf2N] or in [OC2mim][Tf2N]. For most of the surfaces of CO2,
[omim][Tf2N] will be more favorable than the other three, even though in the most
positive (>0.5 e/nm2) region [bmim][Tf2N] seems to be more favorable. The final
solubility trend will be a result of all interactions between the positive and negative
surface pieces of ILs with those of CO2 at equilibrium combined with shape and size
effect.
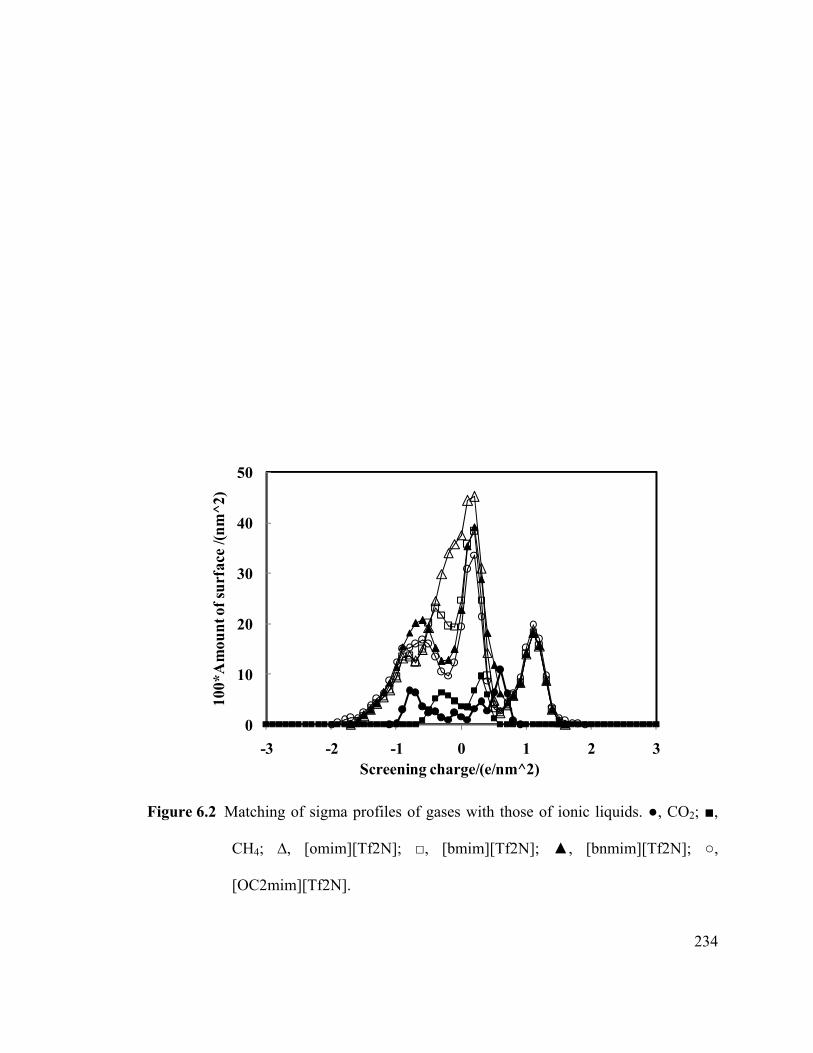
234
Figure 6.2 Matching of sigma profiles of gases with those of ionic liquids. ●, CO2; ■,
CH4; ∆, [omim][Tf2N]; □, [bmim][Tf2N]; ▲, [bnmim][Tf2N]; ○,
[OC2mim][Tf2N].
0
10
20
30
40
50
-3 -2 -1 0 1 2 3
100*
Am
oun
t of
surf
ace
/(n
m^
2)
Screening charge/(e/nm^2)
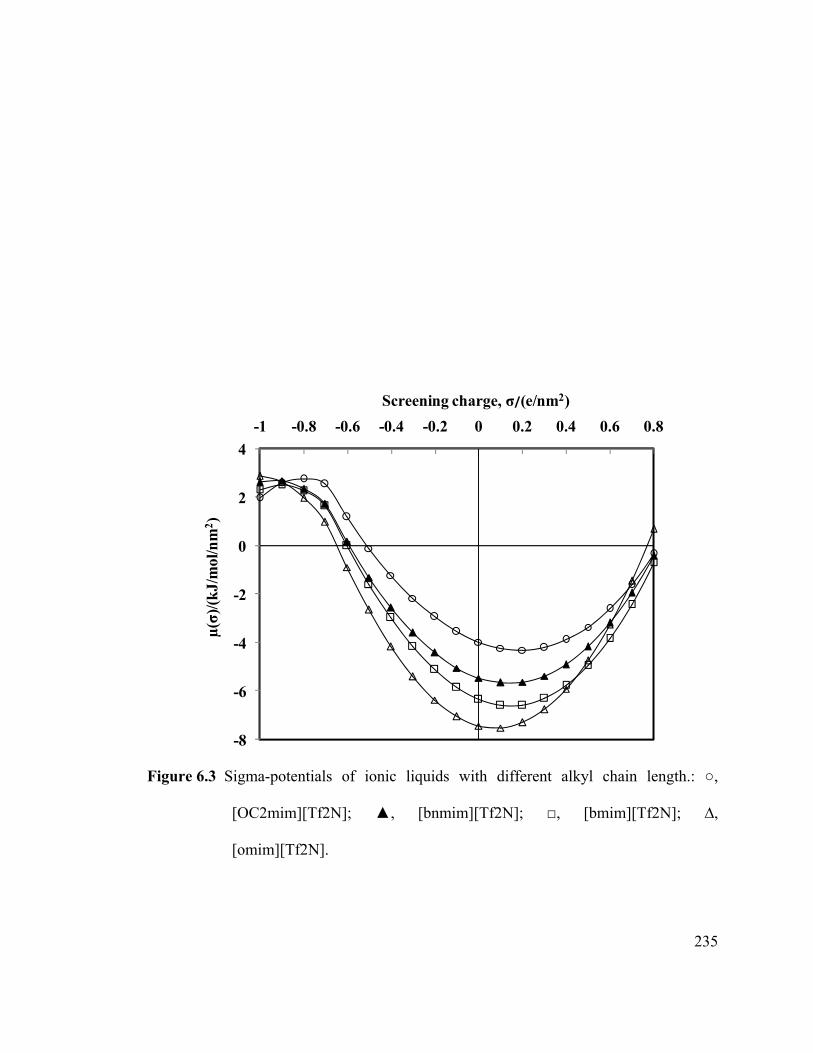
235
Figure 6.3 Sigma-potentials of ionic liquids with different alkyl chain length.: ○,
[OC2mim][Tf2N]; ▲, [bnmim][Tf2N]; □, [bmim][Tf2N]; ∆,
[omim][Tf2N].
-8
-6
-4
-2
0
2
4
-1 -0.8 -0.6 -0.4 -0.2 0 0.2 0.4 0.6 0.8
µ(σ
)/(k
J/m
ol/n
m2 )
Screening charge, σ/(e/nm2)

236
Effect of C2 substitution and change in cation family. Sigma-potentials of
[bmim][Tf2N], [bmmim][Tf2N], [bm(3)py][Tf2N] and [bmpyrr][Tf2N] are shown in
supplementary information (Figure 6.4). The sigma-potentials of [bmim][Tf2N] and
[bmmim][Tf2N] reveal that substitution of acidic hydrogen in the C2 position of the
imidazolium ring with a more basic methyl group increase the affinity of the ionic
liquids towards CO2. Similarly, among the three ring cations with same alkyl chain
length, the affinity increases in the following order
[bmpyrr]>[bm(3)py]>[bmim].Sigma-potentials of phosphonium and ammonium
cation with same alkyl chain length, [Bu4P][Tf2N] and [Bu4N][Tf2N], in the sigma
region (-0.1 and +0.8) e/nm2 are identical (Figure 6.5) indicating similar degree of
interaction; and therefore, the trend between these two cations will be determined by
shape and size effect. The difference between the guanidinium, uranium and
thiouronium cation based ILs with [Tf2N] are also small, the thiouronium cation will
have slightly more affinity toward CO2 (Figure 6.5).

237
Figure 6.4 Sigma-potentials of ionic liquids with different ring precursors: □,
[bmim][Tf2N]; ○, [bm(3)py][Tf2N]; ∆, [bmmim][Tf2N]; ▲,
[bmpyrr][Tf2N].
-8
-6
-4
-2
0
2
4
-1 -0.8 -0.6 -0.4 -0.2 0 0.2 0.4 0.6 0.8
µ(σ
)/(k
J/m
ol/n
m2 )
Screening charge, σ (e/nm2)

238
Figure 6.5 Sigma-potentials of ionic liquids with non ring precursors: □, [bmim][Tf2N];
○, [Bu4P][Tf2N]; ∆, [Bu4N][Tf2N]; ▲, [Me4EtG][Tf2N]; ■, [O-
Me4EtU][Tf2N]; ●, [S-Me4EtT][Tf2N].
-10
-8
-6
-4
-2
0
2
4
-1 -0.8 -0.6 -0.4 -0.2 0 0.2 0.4 0.6 0.8
µ(σ
)/(k
J/m
ol/n
m2 )
Screening charge, σ (e/nm2)

239
Effect of anions. The differences between the sigma-potentials of ionic
liquids are much more pronounced when different anions are paired with a same
cation as can be seen in Figure 6.6. Among the ILs shown, [bmim][EtSO4] will
favour the surfaces having charge (0 to 0.8) e/nm2, but disfavours the surfaces with
charge (-0.2 to -1) e/nm2. The final solubility will be the result between the two
competing effects at equilibrium. Due to these competitive effects, it is more
difficult to categorize the anion effect through their sigma-potentials.CO2 has the
highest solubility in ILs containing FAP anions which has negative sigma potential
within the whole range of CO2 sigma charge. The reason for the high miscibility of
CO2 with FAP anions can also be explained by the complementarity of the sigma
profiles of [bmim][eFAP] and CO2 shown in Figure 6.7. Almost all the surface
pieces of CO2will find opposite surface pieces when dissolved in [bmim][eFAP] and
thus will have low misfit energy. The sigma profile of [bmim][Cl] is also shown in
Figure 6.7. Since halogens lie in the extreme polar regions, slight difference in
polarity results in significant difference in misfit energy and hydrogen-bonding
among the surface pieces of ionic liquids and thus making the mixture environment
very different in different ILs.

240
Figure 6.6 Sigma-potentials of ionic liquids with different anions: ●, [bmim][BF4]; ∆,
[bmim][DCA]; □, [bmim][Tf2N]; ▲, [bmim][EtSO4]; ■, [bmim][eFAP];
○, [bmim][TfO].
-10
-8
-6
-4
-2
0
2
4
6
8
10
-1 -0.8 -0.6 -0.4 -0.2 0 0.2 0.4 0.6 0.8
µ(σ
)/(k
J/m
ol/n
m2 )
Screening charge, σ (e/nm2)

241
Figure 6.7 Comparison of CO2 sigma profiles with those of some ionic liquids: ●, CO2;
□, [bmim][Tf2N]; ▲, [bmim][eFAP]; ○, [bmim][Cl].
0
10
20
30
40
50
60
70
-3 -2 -1 0 1 2 3
100*
Su
rfac
e A
rea
/(n
m^
2)
Screening charges, σ/(e/nm^2)

242
6.4.5 Activity coefficients at infinite dilutions
Activity coefficients are useful quantitative means for studying the solubility
of single gas in different solvents. According to equation 6.2, trends in Henry’s law
constants are identical to the trends in as the vapour pressure of solute remains
same. The activity coefficient of a solute in a mixture is a measure of the degree of
non-ideality in the liquid mixture with reference to its pure liquid state. The residual
chemical potential ( , ) reflects non-ideality due to energetic interactions, and was
obtained by switching off the combinatorial contributions for all compounds. ln , ,
that reflects non-ideality due to solute-solvent size and shape differences, is defined
to be the difference between ln and ln , . These quantities for CO2, CH4 and N2
in the model ionic liquids are given in Tables 6.10 and 6.11 to show the effect of
structural variation in cations and anions. Henry’s law constants can be expressed in
terms of these quantities as,
, , (6.10)
According to the above equation, more negative values of ln , and ln ,
tend to decrease Henry’s law constants. These values are all negative for CO2. The
ln , of CO2 in [OC2mim][Tf2N], [bnmim][Tf2N], [bmim][Tf2N], [omim][Tf2N]
are (-0.84, -1.18, -1.23, -1.37) and consistent with our discussions in section 6.4.3
(Qualitative interpretations of molecular interactions). Among the anions, ln ∞, is
more negative in case of fluorine-containing anions. Since the screening charge of

243
CO2 lies in the electrostatic misfit region, attempt is made to correlate , with the
electrostatic polarity, quantified as second sigma moment (sig2)56, of the varying ions
for a fixed counterpart ion. The trend is not linear and, in general, activity
coefficients seem to increase with increase in sig2 (Figures 6.8 and 6.9). For
example, among the anions, the FAP anions has the lowest sig2 values that results in
smaller values of residual activity coefficients, and consequently in low .
However, there are a lot of exceptions to this relationship that undermines the
significance of sig2 as a tool for solvent characterization.

244
Figure 6.8 Effect of electrostatic polarity, sig2, of cations on residual activity
coefficients of CO2 in the ILs [cation][Tf2N].
Figure 6.9 Effect of electrostatic polarity, sig2, of anions on residual activity
coefficients of CO2 in the ILs [bmim][anion].
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
0 100 200 300Res
idua
l act
ivit
y co
effic
ient
of C
O2
in
[cat
ion]
[Tf2
N]
Electrostatic polarity (sig2) of cationsin the ILs [cation][Tf2N]
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
0 100 200 300Res
idua
l act
ivit
y co
effic
ient
of C
O2
in
[bm
im][
anio
n]
Electrostatic polarity (sig2) of anions in the ILs [bmim][anion]

245
The values of ln ∞, are negative for the three gases which favors their
solubility as per equation 6.3. Since the combinatorial contribution ln ∞, reflects
the difference of area and volume of an ionic liquid with those of the solute, it may
be correlated with a size parameter of the solvent. For example, ∞, of CO2 was
correlated with molar volume of the IL with the relationship ∞, 0.22
0.467 with a correlation coefficient 0.99 where represents the molar volume
(m3/kmol) (Figure 6.10). Prediction of activity coefficients of some liquid solutes in
ionic liquids using COSMO-RS was found satisfactory.57

246
Figure 6.10 Effect of molar volume on the combinatorial activity coefficients of CO2 in
the ILs [bmim][anion] and [cation][Tf2N].
0.5
0.6
0.7
0.8
0.9
1.0
0.0 0.2 0.4 0.6 0.8Com
bin
ator
ial A
ctiv
ity
Coe
ffic
ein
t at
infi
nit
e d
iluti
on o
f C
O2
in I
Ls
[bm
im][
anio
n]
and
[cat
ion
][T
f2N
]
Molar Volume of ILs (m3/kmol)

247
6.4.6 Effect of molar volume and polarity on Henry’s law constant
Molar volume and polarity of ionic liquids are used in the literature to
correlate or explain the solubility of CO2 in ILs. For example, in predictive models
based on regular solution theories, Henry’s law constant of gases are correlated with
molar volume of solvent through the solubility parameters.12,64 According to scaled
particle theory,65 Henry’s law constant of a gas should decrease with increase in
molar volume of ILs when the cavity work and gas-liquid interaction remains same.
This implies that in ILs could be correlated with molar volume when a cation is
varied with a fixed anion as the effect of anion is more pronounced than that of a
cation on the gas-liquid interactions. On the other hand, the polarity, a key property
of a solvent, reflects the overall solvation capability due to varieties of solute-solvent
interactions and used often to characterize solvents.66 Bara et al.67 discussed the
effect of polarity of oligo(ethylene glycol)-functionalized imidazolium ionic liquids
on solubility and selectivity. Solvation of nonpolar, polar, and associating solutes in
imidazolium-based ionic liquids was studied by Lopes et al.68. Polarity of ILs was
studied by other researchers.69-72
In the context of COSMO-RS, an operational temperature-dependent relative
polarity descriptor that can be easily calculated by COSMOtherm would be the
residual pseudo-chemical potential of an ion at its infinite dilution, , in a protic
polar solvent like water. We define a polarity parameter, , / 1 . ,
where is an ion. The polarity of ILs is considered to be the linear sum of the
polarity of its constituent ions. It quantifies the degree of chemical affinity of water

248
with the ion due to all kinds of interactions (vdW, misfit as well as H-bond) and can,
at least qualitatively, be used as its polarity descriptor. The more negative the value
of N, the more polar the ion or the ionic liquid is. This polarity descriptor is
compared with experimental in Figure 6.11 to find its relationship with solubility
and it is seen that solubility decreases with increase in polarity as represented by N,
with few exceptions. One of the reasons for such deviations could be a poor
prediction of their polarity by COSMOtherm. On the other hand, the interactions
between cation and anion within an IL which will alter the sigma profile of the IL are
ignored in this work. Palomar et al.73 have discussed such impact on sigma profiles
that will also affect their polarity. N is compared with experimental measures of
polarity (Figure 6.12).

249
Figure 6.11 Trends in experimental Henry’s law constant of CO2. ○, Henry’s law
constant, ●,. relative overall polarity descriptor.
Figure 6.12 Comparison of the relative polarity parameter of ionic liquids. ●,
calculated; ○, experimental ENT values (taken from Ref. 74).
-20
-10
0
10
20
30
40
50
60
70
80
90
0
10
20
30
40
50
60
70
80
90
100
[C5m
im][
bF
AP
](29
8.15
K)
[hm
im][
eFA
P](
298.
6K)
[MeO
c8N
][T
f2N
](30
3.15
K)
[bm
pyr
r][e
FA
P](
298.
6K)
[th
tdP
][D
CA
](30
3.15
K)
[S-M
e4E
tT][
eFA
P](
298.
6K)
[om
im][
Tf2
N](
298.
15K
)[h
mim
][T
f2N
](29
8.15
K)
[hm
py]
[Tf2
N](
298.
15K
)[b
mim
][T
f2N
](29
8K)
[th
tdP
][C
l](3
03.1
5K)
[em
im][
Tf2
N](
298.
15K
)[t
htd
P][
Tf2
N](
303.
15K
)[b
nm
im][
Tf2
N](
295K
)[b
mp
yrr]
[Tf2
N](
298K
)[e
mm
im][
Tf2
N](
298.
15K
)[b
mim
][P
F6]
(298
K)
[bm
im][
BF
4](2
98K
)[b
mm
im][
BF
4](2
98.1
5K)
[bm
mim
][P
F6]
(298
.15K
)[b
mim
][O
cSO
4](3
13.1
5K)
[em
im][
TfO
](30
3K)
[bm
im][
MeS
O4]
(293
.2K
)[e
mim
][D
CA
](30
3K)
[em
im][
BF
4](3
03K
)[e
mim
][E
tSO
4](3
03.3
8K)
Rel
ativ
e p
olar
ity
par
amet
er,
N
H21
/ 0.1
MP
a
0.5
0.55
0.6
0.65
0.7
0
10
20
30
40
50
[bm
im][
ClO
4]
[bm
im][
BF
4]
[bm
im][
TfO
]
[bm
im][
PF
6]
[bm
im][
Tf2
N]
[om
im][
PF
6]
[om
im][
Tf2
N]
[bm
mim
][T
f2N
]
Exp
erim
enta
l EN
Tva
lues
Rel
ativ
e po
lari
ty p
aram
eter
, N
pred
iced
by
CO
SMO
ther
m

250
It is observed that a single solvent property such as molar volume or polarity can
qualitatively describe the trend in in the model ILs [cation][Tf2N], i.e., when the
anion is fixed. The calculated in [cation][Tf2N], in general, decrease with increase
in molar volume and N (higher N indicates less polar IL) (Figures 6.13 and 6.14). For
example, as seen from the data in Table 6.8, with increase in alkyl chain length in the
imidazolium cation, decreases and both molar volume and N increases (polarity
decreases). The trend of in ILs [bmim][anion] with a single property of ILs such as
molar volume and polarity could not be clearly related. Nevertheless, CO2 is seen to be
more soluble in the less polar fluorine-containing anions (e.g., N is very high for [eFAP]
and [bFAP]) and less soluble in more polar sulphate anions with more negative values of
N. As a whole, one single parameter was insufficient to establish a general trend in
many ILs knowing that the simultaneous effects of both properties determine the trend.

251
Figure 6.13 Effect of molar volume of ILs with Henry’s law constant of CO2 in ionic
liquids [cation][Tf2N].
Figure 6.14 Effect of polarity on the Henry’s law constant of CO2 in the ionic liquids
[cation][Tf2N].
0
20
40
60
80
100
120
0.0 0.2 0.4 0.6 0.8
H21
/ 0.1
MP
a
Molar volume (m3/kmol) of ILs [cation][Tf2N]
0
20
40
60
80
100
120
0 20 40 60 80
H21
/ 0.1
MP
a
Relative overall polarity parameter of ILs [cation][Tf2N] at 298.15K

252
6.4.7 Effect of temperature on gas solubilities
Enthalpic and entropic contributions to the solvation process of a gas at
infinite dilution in a solvent can be obtained from the temperature dependence of
Henry’s law constants. The partial molar thermodynamic quantities like Gibbs
energy of solvation, ∆ ; enthalpy of solvation, ∆ ; and entropy of solvation,
∆ ; were calculated at 25°C using the following relationships37 by assuming linear
temperature dependence of the Henry’s law constants on temperature within the
range (10 to 50)°C.
∆ ln ⁄ (6.11)
∆ / ln ⁄ (6.12)
∆ ∆ ∆ ⁄ (6.13)
These properties correspond to the transfer of gas from ideal gas state at
0.1MPa to the standard state of hypothetical pure liquid state of solute
extrapolated from its ideally dilute behaviour.

253
Comparison with experimental temperature dependence. COSMOtherm
predicts the correct trend that the solubility of CO2 decreases with increase in
temperature. Quantitative prediction27 is better around room temperature. Enthalpy
of solvation as calculated by COSMOtherm is compared with experimental data33,60,63
for 10 ILs (Figure 6.15). AAD in the absolute values of enthalpies of solvation is
35% and the RMSD is 4.6. COSMO-RS is not expected to perform satisfactorily in
the supercritical region as mentioned in section 6.2. Moreover, COSMOtherm uses
empirical temperature dependence of the van der Waals (vdW) and hydrogen bond
energy. Instead of using COSMOtherm estimation for gas phase chemical potential,
Maiti et al.41 obtained a temperature-dependent empirical correlation for gas phase
chemical potential by fitting with experimental data.
Effect of IL structure on solvation of various gases. Enthalpy and entropy of
solvation of the gases in the [cation][Tf2N] and [bmim][anion] are provided in
Tables 6.10 and 6.11. These quantities are negative for all gases and from (0 to -20)
kJ/mol referring to the physical exothermic solvation process.11 For a single gas, the
variation in ∆ , due to structural variation is not pronounced. For example, the
∆ values for CO2 in the 73 model ILs, as given in Table 6.8, fall within the range
{-18.93, -15.57} kJ/mol (if [NH4] is not considered) and, in the 37 ILs
[bmim][anion], as given in Table 6.9, fall within {-19.31,-15.18} kJ/mol. Similarly,
the range (minimum value, maximum value) of ∆ for CH4 is {-8.14,-3.01} kJ/mol
in the ILs [cation][Tf2N] and {-10.48,-4.89} kJ/mol in the ILs [bmim][anion].
Finally, the range of ∆ for N2 are {-5.81,-3.84} kJ/mol in [cation][Tf2N] and {-
6.88,-0.12} kJ/mol in [bmim][anion] that indicates anions have more appreciable

254
effect (slightly wider range) on the degree of exothermicity for N2. The solvation
process of CO2 is most exothermic in [bmim][eFAP] among the [bmim][anion] ILs.
For N2, the solvation process is least exothermic in [bmim][(CN)3C].
The entropic (– ∆ ) and enthalpic (∆ ) components of Gibbs free energy
change ( ln ⁄ ) for the model ILs [bmim][anion] are compared to determine
their relative contribution in Henry’s law constant (Figure 6.16). The – ∆
component varies within 2.3 kJ/mol (from 25 to 27.3 kJ/mol) which is even narrower
than the range of ∆ , from (-19.31 to -15.18) kJ/mol. Slightly greater variation in
∆ with change in anions for the CO2 dissolution process indicates the dominance
of enthalpic contribution in the ranking of ILs in terms of solubility. However, in
any particular IL, – ∆ ∆ and therefore plays a key role in determing the
magnitude of ∆ and thus the solubility of CO2 in an individual IL. Strength of
interaction of these gases CO2, CH4 and N2 in a fixed solvent can be compared
through enthalpy of solvation at the infinite dilution. The order of exothermicity is
CO2>CH4>N2 in any ILs.

255
Figure 6.15 Comparison of enthalpy of solvation at infinite dilution of CO2 in some ILs.
Grey, experimental; black, prediction.
Figure 6.16 Relative effect of enthalpic and entropic contributions on the Henry’s law
constant for CO2 in ionic liquids [bmim][anion]. ■, enthalpic; □, and
entropic
-20-18-16-14-12-10-8-6-4-20
[em
im][
EtS
O4]
[em
mim
][T
f2N
]
[bm
mim
][B
F4]
[bm
im][
PF
6]
[em
im][
Tf2
N]
[bm
im][
BF
4]
[bm
mim
][P
F6]
[bm
im][
Tf2
N]
[bm
pyrr
][T
f2N
]
[bm
im][
OcS
O4]
∆H∞
( kJ/
mol
)
Ionic Liquids
-20
-19
-18
-17
-16
-15
-14
-13
-12
-11
-10
20
21
22
23
24
25
26
27
28
29
30
ID#0
1_[A
c]ID
#02_
[NnC
OO
]ID
#03_
[PhC
OO
]ID
#04_
[BF4
]ID
#05_
[B(C
N)4
]ID
#06_
[BO
XB
]ID
#07_
[BM
B]
ID#0
8_[B
SB]
ID#0
9_[B
PhB
]ID
#10_
[MeS
O4]
ID#1
1_[E
tSO
4]ID
#12_
[BuS
O4]
ID#1
3_[O
cSO
4]ID
#14_
[MeO
EtS
O4]
ID#1
5_[E
tOE
tSO
4]ID
#16_
[MD
EG
SO4]
ID#1
7_[T
fO]
ID#1
8_[T
OS]
ID#1
9_[D
CA
]ID
#20_
[BT
I]ID
#21_
[Tf2
N]
ID#2
2_[(
CN
)3C
]ID
#23_
[Tf2
C]
ID#2
4_[T
f3C
]ID
#25_
[Me2
P]ID
#26_
[PF6
]ID
#27_
[eFA
P]ID
#28_
[bFA
P]ID
#29_
[(M
e3p)
2PO
2]ID
#30_
[(C
2F5)
2PO
2]ID
#31_
[Cl]
ID#3
2_[B
r]ID
#33_
[I]
ID#3
4_[C
lO4]
ID#3
5_[2
-PhC
l]ID
#36_
[3-P
hCl]
ID#3
7_[4
-PhC
l] ΔH
∞(k
J/m
ol) a
t 298
.15K
, (fi
lled
rect
angl
e)
-TΔ
S∞
(kJ/
mol
) at
298
.15K
, (op
en r
ecta
ngl
e)
Ionic liquids [bmim][anion]

256
Interaction enthalpies due to specific interaction. The domminanat
molecular interactions affecting the solubility of CO2 in IL solvents was identified
with COSMOtherm derived a posteriori quantities like interaction enthalpies due to
specific molecular interactions.38,39,75 First, we compared (Figure 6.17) the effect of
anion variation in the ionic liquids on two similar terms (i) , ,
representing the contribution of CO2 in the excess enthalpy of the liquid mixture (in
ternary framework) at equilibrium with gaseous CO2 at 0.1MPa and 25°C; where,
, and are the partial molar enthalpy of solute in the liquid phase at infinite
dilution in ternary framework and the molar enthalpy of solute in its own
hypothetical pure liquid state which is related to its vapour phase enthalpy through
heat of vaporization76 and (ii) solvation enthalpy (∆ ), which is the enthalpic
component of the quantity ln ⁄ that is a scaled version of solubility. The
trends (Figure 6.17), are similar with some exceptions e.g., halogenide and alkyl
sulphate anions (possibly due to inaccuracies in the predictions of either of the
quantities for such anions). The components of , due to mistfit, vdW
and H-bond interactions (Figure 6.18) reveal the general dominance of vdW
interactions. This also indicates the dominance of vdW interaciton (among the three
attractive interactions considered) in determining solubility. The high misfit energy
between CO2 and [bmim][Cl] was anticipated from their sigma profiles in section
6.4.2.

257
Figure 6.17 Comparison of contribution in excess enthalpy (filled circle) due to CO2
with enthalpy of solvation (filled square) for CO2 dissolution in
[bmim][anion].
-0.04
-0.03
-0.02
-0.01
0
0.01
0.02
-20
-19
-18
-17
-16
-15
-14
-13
-12
-11
-10
ID#0
1_[A
c]ID
#02_
[NnC
OO
]ID
#03_
[PhC
OO
]ID
#04_
[BF4
]ID
#05_
[B(C
N)4
]ID
#06_
[BO
XB
]ID
#07_
[BM
B]
ID#0
8_[B
SB]
ID#0
9_[B
PhB
]ID
#10_
[MeS
O4]
ID#1
1_[E
tSO
4]ID
#12_
[BuS
O4]
ID#1
3_[O
cSO
4]ID
#14_
[MeO
EtS
O4]
ID#1
5_[E
tOE
tSO
4]ID
#16_
[MD
EG
SO4]
ID#1
7_[T
fO]
ID#1
8_[T
OS]
ID#1
9_[D
CA
]ID
#20_
[BT
I]ID
#21_
[Tf2
N]
ID#2
2_[(
CN
)3C
]ID
#23_
[Tf2
C]
ID#2
4_[T
f3C
]ID
#25_
[Me2
P]ID
#26_
[PF6
]ID
#27_
[eFA
P]ID
#28_
[bFA
P]ID
#29_
[(M
e3p)
2PO
2]ID
#30_
[(C
2F5)
2PO
2]ID
#31_
[Cl]
ID#3
2_[B
r]ID
#33_
[I]
ID#3
4_[C
lO4]
ID#3
5_[2
-PhC
l]ID
#36_
[3-P
hCl]
ID#3
7_[4
-PhC
l] Con
trib
uti
on o
f C
O2
in e
xces
s en
thal
py,
kJ/
mol
En
thal
py
of s
olva
tion
, k
J/m
ol

258
Figure 6.18 Contribution in excess enthalpy due to CO2 in CO2-[bmim][anion] mixture
at infinite dilution of CO2 as linear sum of contributions from specific
interactions: vdW interaction (∆), misfit-interaction (▲) and hydrogen-
bond interaction (■), total (●)
-0.05
-0.04
-0.03
-0.02
-0.01
0
0.01
0.02
0.03
0.04
0.05
ID#0
1_[A
c]ID
#02_
[NnC
OO
]ID
#03_
[PhC
OO
]ID
#04_
[BF4
]ID
#05_
[B(C
N)4
]ID
#06_
[BO
XB
]ID
#07_
[BM
B]
ID#0
8_[B
SB]
ID#0
9_[B
PhB
]ID
#10_
[MeS
O4]
ID#1
1_[E
tSO
4]ID
#12_
[BuS
O4]
ID#1
3_[O
cSO
4]ID
#14_
[MeO
EtS
O4]
ID#1
5_[E
tOE
tSO
4]ID
#16_
[MD
EG
SO4]
ID#1
7_[T
fO]
ID#1
8_[T
OS]
ID#1
9_[D
CA
]ID
#20_
[BT
I]ID
#21_
[Tf2
N]
ID#2
2_[(
CN
)3C
]ID
#23_
[Tf2
C]
ID#2
4_[T
f3C
]ID
#25_
[Me2
P]ID
#26_
[PF6
]ID
#27_
[eFA
P]ID
#28_
[bFA
P]ID
#29_
[(M
e3p)
2PO
2]ID
#30_
[(C
2F5)
2PO
2]ID
#31_
[Cl]
ID#3
2_[B
r]ID
#33_
[I]
ID#3
4_[C
lO4]
ID#3
5_[2
-PhC
l]ID
#36_
[3-P
hCl]
ID#3
7_[4
-PhC
l]
Con
trib
uti
on o
f C
O2
in e
xces
s en
thal
py,
kJ/
mol

259
6.4.8 Selectivities
Selectivities in all ionic liquids at 25°C are predicted and presented in
supplementaryinformation.CO2/CH4 selectivities at 25°C in the ionic liquids
[cation][Tf2N] is around 10 in the common ring cations as well as in the guanidinium,
uronium and thiouroniumcations. is relatively low in phophonium and high in
ammonium cations. Increased alkylation slightly reduces the separation factor .
Presence of ether and benzyl functionality enhances . For similar degree of
alkylation, the ranking of cation families having a ring is: [bmpyrr]>[bmim]>[bm(3)py]
and without a ring is: [S-EtMe4T] ]>[O-EtMe4U]>[Me4EtG]. In general, for a fixed
anion, the effect of cation is similar on and meaning that ILs that absorbs more
CO2 tend to absorb more methane. Fluorine-containing anions have a high solubility as
well as high CO2/CH4 selectivity. Increase in alkyl chain length in the anion decreases
the CO2/CH4 selectivity. The presence of ether group in the sulphonate anions slightly
enhances selectivity. In general, with an increase in molar volume and N, decreases
in the ILs [cation][Tf2N] (Figures 6.19 and 6.20). However, anionic effect was difficult
to correlate with a single property of ILs. slightly decreases with increase in
temperature. For example, at (10, 25, 50)°C, in the IL [bmim][Tf2N] are (15, 12 and
8) respectively.

260
Figure 6.19 Effect of molar volume of ILs [cation][Tf2N] on CO2/CH4 selectivity.
Figure 6.20 Effect of polarity of ILs [cation][Tf2N] on CO2/CH4 selectivity.
0
5
10
15
20
25
0.0 0.2 0.4 0.6 0.8
CO
2/C
H4
sele
ctiv
ity,
S23
Molar volume (m3/kmol) of ILs [cation][Tf2N]
0
5
10
15
20
25
0 20 40 60 80
CO
2/C
H4
sele
ctiv
ity,
S23
Relative overall polarity parameter of ILs [cation][Tf2N] at 298.15K

261
COSMOtherm predictions of arecompared with experimentally reported
values12,77,78 in 10 ionic liquids and the AAD and RMSD are 23% and 8, respectively
(Figure 6.21). Quantitative prediction is satisfactory in most cases and greater deviation
is seen in the case of [emim][BF4], [emim][DCA], [emim][TfO]. The trend in CO2/CH4
selectivity does not match with experimental ranking for the four ILs with [emim] cation
paired with [DCA], [BF4], [TfO], [Tf2N]. The contrasting feature of the sigma profiles
of CH4 and CO2 (Figure 6.2) is the presence of the peak of hydrogen around -0.4 e/nm2
in CH4where CO2 has almost no surface segments. Therefore, anions that can provide
more surfaces containing opposite charges (0 to 0.4 e/nm2) will favor the dissolution of
methane. Observation of the sigma profiles of the four anions in this charge region
(Figure 6.22), reveals that [BF4] has almost no surface pieces in that region whereas
[Tf2N] has lots of surface pieces. Hence, from the sigma profiles, we may predict
selectivity in this order: [BF4]>[DCA]>[TfO]>[Tf2N] which better represents the
experimental trend: [DCA]~[BF4]>[TfO]>[Tf2N]. Shimoyama et al.53 successfully
reproduced this experimental trend using the model COSMO-SAC.

262
Figure 6.21 Comparison of CO2/CH4 selectivity.Grey, experimental; black, prediction.
Figure 6.22 Sigma profiles of some ionic liquids with [bmim] cation but with differnet
anions within the screening charge region between 0 and 0.4 e/nm2. ○,
[BF4]; ∆, [DCA]; ●, [Tf2N]; ▲, [TfO]
0
5
10
15
20
25
30
35
40
[em
im][
BF
4](2
98K
)
[em
im][
BF
4](3
13K
)
[em
im][
DC
A](
313K
)
[em
im][
TfO
](31
3K)
[em
im][
Tf2
N](
298K
)
[em
im][
Tf2
N](
313K
)
[hm
im][
Tf2
N](
298K
)
[hm
im][
Tf2
N](
313K
)
[bnm
im][
Tf2
N](
295K
)
[dm
im][
Tf2
N](
313K
)
[bm
im][
BF
4](3
04K
)
[bm
im][
PF
6](3
03K
)
CO
2/C
H4
sele
ctiv
ity,
S23
0
5
10
15
20
25
30
35
0 0.1 0.2 0.3 0.4
100*
Are
a/ n
m2
screening charge, e/nm^2

263
COSMOtherm predictionof was not satisfactory as it predicts much higher
than experimentally reported values12,77,78 in 9 ionic liquids and the AAD and
RMSD error are 111% and 44 (Figure 6.23). From the data in Tables 6.10 and 6.11,
is seen to be much greater than (about 4 times in most cases) due to very low
solubility of nitrogen in ILs. is rather insensitive to cation and comparatively
high in cations with benzyl ([bnmim], [PhPrmim]) and isothiouronium([S-Me4EtT])
groups. Anions have appreciable effect on and they are comparatively low in
fluorine-containing anions. decreases appreciably with an increase in
temperature. For example, at (10, 25, 50)°C, in the IL [bmim][Tf2N] are (60, 45
and 30) respectively. No further analysis was carried out with .

264
Figure 6.23 Comparison of CO2/N2 selectivity: grey, experimental; black, prediction.
0
20
40
60
80
100
120
140
160
[em
im][
BF
4](2
98K
)
[em
im][
BF
4](3
13K
)
[em
im][
DC
A](
313K
)
[em
im][
TfO
](31
3K)
[em
im][
Tf2
N](
298K
)
[em
im][
Tf2
N](
313K
)
[hm
im][
Tf2
N](
298K
)
[hm
im][
Tf2
N](
313K
)
[bnm
im][
Tf2
N](
295K
)
[dm
im][
Tf2
N](
313K
)
[bm
im][
BF
4](3
04K
)
[bm
im][
PF
6](3
03K
)
CO
2/N
2se
lect
ivit
y,S
CO
2/N
2

265
6.5 SCREENING AND DESIGNING OF ILS
For screening and designing of ILs, structural variations and properties of ILs
that enhance CO2 solubility and selectivity could be utilized. For example, an
increase in alkylation increases both and . Since it is desirable to have low
and high , a balance in alkyl chain in ionic liquid would be required. Similar
consideration should be given for other functionalities present in the cation. It is
easy to tune physico-chemical properties of ILs by modifying the cations rather than
the anions. However, much variation was not observed in due to structural
variation in the cation. A less polar IL with high molar volume is ideal for high CO2
solubility. Henry’s law constants presented here may be used for initial screening of
ionic liquids for CO2 capture since most ionic liquids are used as physical solvents.
For a fixed cation, its counterpart anions are ranked in ascending order of Henry’s
law constant of CO2 and vice versa, and are presented in Table 6.10. The non-ring
guanidinium, uronium, thiouronium, [thtdP] cations, long-chain ring cations and
fluorine-containing anions dominate the ranking.

266
Table 6.10 Ranking of Anions for Some Fixed Cations and Vice Versa
Rank ID# 04_[bmim] ID# 34_[bm(4)py] ID# 41_[bmpyrr] ID# 50_[Bu4N] ID# 61_[thtdP] ID# 64_[Me6G] ID# 68_[O-Me4MeU]
1 ID# 28_[bFAP],18a
ID# 28_[bFAP],18 ID# 28_[bFAP],17 ID# 28_[bFAP],17 ID# 32_[Br],17 ID# 32_[Br],8 ID# 32_[Br],10
2 ID# 27_[eFAP],20 ID# 27_[eFAP],20 ID# 27_[eFAP],18 ID# 32_[Br],17 ID# 28_[bFAP],18 ID# 31_[Cl],16 ID# 31_[Cl],18
3 ID# 24_[Tf3C],30 ID# 24_[Tf3C],29 ID# 32_[Br],24 ID# 27_[eFAP],18 ID# 27_[eFAP],20 ID# 28_[bFAP],17 ID# 28_[bFAP],18
4 ID# 30_[(C2F5)2PO2],34 ID# 30_[(C2F5)2PO2],32 ID# 24_[Tf3C],27 ID# 26_[PF6],23 ID# 26_[PF6],23 ID# 27_[eFAP],18 ID# 27_[eFAP],20
5 ID# 09_[BPhB],34 ID# 09_[BPhB],33 ID# 30_[(C2F5)2PO2],28 ID# 20_[BTI],23 ID# 20_[BTI],23 ID# 01_[Ac],21 ID# 33_[I],26
Anions
Rank ID# 04_[BF4] ID# 09_[BPhB] ID# 13_[OcSO4] ID# 21_[Tf2N] ID# 24_[Tf3C] ID# 32_[Br] ID# 28_[bFAP]
1 ID# 47_[NH4],~0 ID# 61_[thtdP],25 ID# 61_[thtdP],28 ID# 61_[thtdP],25 ID# 60_[Bu4P],23 ID# 47_[NH4],~0 ID# 70_[S-Me4EtT],17
2 ID# 64_[Me6G],22 ID# 66_[Me5PrG],25 ID# 66_[Me5PrG],29 ID# 67_[Me5(iPr)G],25 ID# 50_[Bu4N],23 ID# 48_[Me4N],7 ID# 43_[ompyrr],17
3 ID# 67_[Me5(iPr)G],23 ID# 67_[Me5(iPr)G],25 ID# 67_[Me5(iPr)G],29 ID# 66_[Me5PrG],25 ID# 61_[thtdP],23 ID# 64_[Me6G],8 ID# 42_[hmpyrr],17
4 ID# 66_[Me5PrG],23 ID# 60_[Bu4P],26 ID# 64_[Me6G],30 ID# 60_[Bu4P],25 ID# 67_[Me5(iPr)G],24 ID# 67_[Me5(iPr)G],9 ID# 59_[(iBu)3MeP],17
5 ID# 61_[thtdP],24 ID# 50_[Bu4N],26 ID# 70_[S-Me4EtT],31 ID# 50_[Bu4N],25 ID# 52_[MeOc3N],24 ID# 66_[Me5PrG],10 ID# 41_[bmpyrr],17
Cations
Ani
ons
Cat
ions

267
Volumetric solubility12 or molal solubility41 may also be used as selection
criteria. For example, based on predicted molal solubility at 30 bar and 40°C,
[Me5PrG][BF4] was found to be approximately 80% more efficient than
[Me5PrG][eFAP].41 The HLC presented in Tables 6.3, 6.4 and 6.5 can be readily
converted to molal solubility ( ) using the expression,
1000.1
(6.14)
As an illustration, we compare the molal solubility of the above two ILs at 1
bar and 25°C. Reading molecular weights from Tables 6.1 and 6.2 for cation ID#66
and anions ID#4 and 27, HLC from Table 6.5 (2.3 and 1.8 MPa respectively), and
using equations (6.8), (6.7) and (6.14), the molality of CO2 in [Me5PrG][BF4] and
[Me5PrG][eFAP] are 0.172 mol/Kg and 0.093 mol/Kg, respectively; the former is ca.
85% more efficient in molality scale according to our prediction, which also
corroborates well with the results published by Maiti et al.41
Relatively low temperature would enhance the solubility of carbon dioxide
and its selectivity. It may be noted that even though we have not focused on
solubility of gases in mixed IL solvents58,79, which can also be studied using
COSMOtherm. Since COSMO-RS prediction is qualitatively correct for a broader
range of ionic liquids and not quantitatively satisfactory for all kinds of anions,
intuition and judgement of the experimentalist is indispensible along with the
computational prediction.

268
As observed, COSMOtherm cannot predict many of the experimental trends
accurately and therefore the quality of prediction need be improved. In this work, no
fugacity correction was incorporated. Incorporation of fugacity correction, as was
done by Shimoyama et al.,53 may improve the prediction. Mixtures of amines and
ILs are recently proposed as an economical candidate for CO2 capture80. Such
system may not be well modelled with COSMO-RS.36 Being a surface interaction
model, COSMO-RS neither uses nor provides explicit three-dimensional geometric
information of the microscopic solute-solvent interactions. Such information could
be obtained, for example, through molecular dynamics simulations for detailed
examination of liquid structure and the solvation process.
Rigorous screening of ionic liquids must include consideration of their
thermo-physical properties,81-83 mass transfer coefficients, desorption characteristics,
toxicity and cost. Not all combinations of the cations and anions considered here
would exist as room temperature ionic liquids in reality. Ionic liquids with
symmetric and smaller ions will, in general, have higher melting point.

269
6.6 CONCLUSIONS
We have explored the capabilities of COSMOtherm as an auxiliary tool for
screening and design of ILs for CO2 capture. Henry’s law constants ( ) of CO2,
CH4, and N2 in 2701 ionic liquids are predicted using COSMO-RS at (10, 25 and
50)°C. Trends in solubility and selectivity as a function of the chemical structure of
cations and anions were analysed. Sigma profiles and sigma-potentials of solvents
are valuable tools for a priori solvent characterization. Gas liquid interactions were
described qualitatively through sigma-potentials of ILs and quantitatively through
activity coefficients. Enthalpy and entropy of solvation were obtained from
temperature dependence of Henry’s law coefficients. Overall polarity of Ilsis
expressed through a temperature-dependent polarity descriptor, N, and Henry’s law
constants of CO2 were found to increase with increase in polarity. The components
of Henry’s law constants of CO2 were dissected and the trend in residual contribution
was roughly related to the electrostatic polarities of the ions as quantified by second
sigma moment (sig2) when different ionic liquids with a fixed opposite ion are
considered. Residual activity coefficients increased with increase in sig2 of the
varying ions in ionic liquids [cation][Tf2N] and [bmim][anion]. The combinatorial
activity coefficients were correlated with the molar volume of ILs. Solubility of CO2
and selectivities for CO2/CH4 and CO2/N2 separations decrease with an increase in
temperature.CO2 is much more soluble than methane and nitrogen and therefore the
solubility of CO2 in ionic liquid and temperature will play the key role in solvent
selection for CO2 capture. ILs with a fixed cation or anion are ranked based on their

270
. For a fixed anion, the solubility and CO2-IL interaction is in general, stronger in
guanidium, isouronium and pyrrolidinium based cations and fluorine-containing
anions than the commonly used imidazolium-based ones. COSMOtherm is a
promising preliminary tool for fast screening and design of ILs for such purpose as it
readily provides a number of pertinent information at both molecular and bulk level.

271
6.7 REFERENCES
1. Maginn, E. J. What to Do with CO2? J. Phys. Chem. Lett. 2010, 1, 3478.
2. Thomas, S. Enhanced Oil Recovery - An Overview. Oil & Gas Sci.
Technol. 2008, 63 ( 1) 9.
3. Rochelle, G. T. Amine Scrubbing for CO2 Capture. Science 2009, 325, 1652.
4. Puxty, G.; Rowland, R.; Allport, A.; Yang, Q.; Bown, M.; Burns, R.;
Maeder, M.; Attalla,M. Carbon Dioxide Postcombustion Capture: A Novel
Screening Study of the Carbon Dioxide Absorption Performance of 76 Amines.
Environ. Sci. Technol. 2009, 43, 6427.
5. Chowdhury, F. A.; Okabe, H.; Shimizu, S.; Onoda, M.; Fujioka, Y. Development
of Novel Tertiary Amine Absorbents for CO2 Capture. Energy
Procedia 2009, 1, 1241.
6. Rayer, A. V.; Sumon, K. Z.; Henni, A.; Tontiwachwuthikul, P. Kinetics of The
Reaction of Carbon Dioxide (CO2) with Cyclic Amines Using the Stopped-Flow
Technique. Energy Procedia 2011, 4, 140.
7. D’Alessandro, D. M.; Smit, B.; Long, J. R. Carbon Dioxide Capture: Prospects
for New Materials. Angew. Chem., Int. Ed. 2010, 49, 6058.
8. Figueroa, J. D.; Fout, T.; Plasynski, S.; McIlvried, H.; Srivastava, R. D.Advances
in CO2 capture technology-The U.S. Department of Energy’s Carbon
Sequestration Program. Int. J. Greenhouse Gas Control 2008, 2, 9.

272
9. Blanchard, L. A.; Hancu, D.; Beckman, E. J.; Brennecke, J. F. Green Processing
Using Ionic Liquids and CO2. Nature 1999, 398 ( 6731) 28.
10. Keskin, S.; Kayrak-Talay, D.; Akman, U.; Hortaçsu, O. A Review of Ionic
Liquids Towards Supercritical Fluid Applications. J. Supercrit. Fluids 2007, 43 (
1) 150.
11. Anderson, J. L.; Dixon, J. K.; Brennecke, J. F.Solubility of CO2, CH4, C2H6,
C2H4, O2, and N2 in 1- yl-3-methylpyridinium
Bis(trifluoromethylsulfonyl)imide: Comparison to Other Ionic Liquids. Acc.
Chem. Res. 2007, 40, 1208.
12. Bara, J. E.; Carlisle, T. K.; Gabriel, C. J.; Camper, D.; Finotello, A.; Gin, D. L.;
Noble, R. D.Guide to CO2 Separations in Imidazolium-Based Room-Temperature
Ionic Liquids. Ind. Eng. Chem. Res. 2009, 48, 2739.
13. Karadas, F.; Atilhan, M.; Aparicio, S. Review on the Use of Ionic Liquids (Ils) as
Alternative Fluids for CO2 Capture and Natural Gas Sweetening. Energy Fuel.
2010, 24, 5817.
14. Hasib-ur-Rahman M., Siaj M., Larachi F. Ionic Liquids for CO2 Capture-
Development and Progress. Chem. Eng. and Process. 2010, 49, 313.
15. Bates, E. D.; Mayton, R. D.; Ntai, I.; Davis, J. H. CO2 Capture by A Task-
Specific Ionic Liquid. J. Am. Chem. Soc. 2002, 124, 926.
16. Sánchez, L. M.; Meindersma, G. W.; de Haan, A. B. Solvent Properties of
Functionalized Ionic Liquids for CO2 Absorption. Chem. Eng. Res. Des. 2007,
85 ( A1) 31.

273
17. Gutowski, K. E.; Maginn, E. J.Amine-Functionalized Task-Specific Ionic
Liquids: A Mechanistic Explanation for the Dramatic Increase in Viscosity upon
Complexation with CO2 from Molecular Simulation. J. Am. Chem. Soc. 2008,
130, 14690.
18. Goodrich, Brett F.; de la Fuente, Juan C.; Gurkan, Burcu E.; Zadigian, David J.;
Price, Eric A.; Huang, Yong; Brennecke, Joan F.Experimental Measurements of
Amine-Functionalized Anion-Tethered Ionic Liquids with Carbon Dioxide. Ind.
Eng. Chem. Res. 2011, 50, 111.
19. Gurkan, B.; Goodrich, B. F.; Mindrup, E. M.; Ficke, L. E.; Massel, M.; Seo, S.;
Senftle, T. P.; Wu, H.; Glaser, M. F.; Shah, J. K.; Maginn, E. J.; Brennecke, J. F.;
Schneider, W. F. Molecular Design of High Capacity, Low Viscosity,
Chemically Tunable Ionic Liquids for CO2 Capture. J. Phys. Chem. Lett. 2010,
1, 3494.
20. Wang, C.; Luo, H.; Jiang, D.-E.; Li, H.; Dai, S., Carbon dioxide capture by
superbase-derived protic ionic liquids. Angew. Chem. Int. Ed., 2010, 49, 5978.
21. Kim, Y. S.; Choi, W. Y.; Jang, J. H.; Yoo, K. P.; Lee, C. S. Solubility
Measurement and Prediction of Carbon Dioxide in Ionic Liquids. Fluid Phase
Equilib. 2005, 228, 439.
22. Scovazzo, P.; Camper, D.; Kieft, J.; Poshusta, J.; Koval, C.; Noble, R. Regular
Solution Theory and CO2 Gas Solubility in Room-Temperature Ionic Liquids.
Ind. Eng. Chem. Res. 2004, 43, 6855.

274
23. Finotello, A.; Bara, J. E.; Camper, D.; Noble, R. D. Room-Temperature Ionic
Liquids: Temperature Dependence of Gas Solubility Selectivity. Ind. Eng. Chem.
Res. 2007, 47, 3453.
24. Kilaru, P. K.; Condemarin, R. A.; Scovazzo, P. Correlations of Low-Pressure
Carbon Dioxide and Hydrocarbon Solubilities in Imidazolium-, Phosphonium-,
And Ammonium-Based Room-Temperature Ionic Liquids. Part 1. Using Surface
Tension. Ind. Eng. Chem. Res. 2008, 47, 900.
25. Eike, D. M.; Brennecke, J. F.; Maginn, E. J. Predicting Infinite-Dilution Activity
Coefficients of Organic Solutes in Ionic liquids. Ind. Eng. Chem. Res. 2004, 43,
1039.
26. Kroon, M. C.; Karakatsani, E. K.; Economou, I. G.; Witkamp, G.-J.; Peters, C.
J.Modeling of the Carbon Dioxide Solubility in Imidazolium-Based Ionic Liquids
with the tPC-PSAFT Equation of State. J. Phys. Chem. B 2006, 110, 9262.
27. Marsh, K. N.; Boxall, J. A.; Lichtenthaler, R. Room Temperature Ionic Liquids
and Their Mixtures A Review. Fluid Phase Equilib. 2004, 219, 93.
28. Kerlé, D.; Ludwig, R.; Geiger, G.; Paschek, D.Temperature Dependence of the
Solubility of Carbon Dioxide in Imidazolium-Based Ionic Liquids. J. Phys.
Chem. B 2009, 113, 12727.
29. Gomes, M. F. C.; Padua, A. A. H. Gas–Liquid Interactions in Solution. Pure
Appl. Chem. 2005, 77, 653.
30. Shi, W.; Maginn, E. J. Molecular Simulation and Regular Solution Theory
Modeling of Pure and Mixed Gas Absorption in the Ionic Liquid 1-N-hexyl-3-

275
Methylimidazolium Bis(Trifluoromethylsulfonyl)Amide ([Hmim][Tf2N]). J.
Phys. Chem. B 2008, 112, 16710.
31. Huang, X. H.; Margulis, C. J.; Li, Y. H.; Berne, B. J. Why is the Partial Molar
Volume of CO2 So Small When Dissolved in a Room Temperature Ionic Liquid?
Structure and Dynamics of CO2 Dissolved in [Bmim+] [PF6-]. J. Am. Chem. Soc.
2005, 127, 17842.
32. Urukova, I.; Vorholz, J.; Maurer, G.Solubility of CO2, CO, and H2 in the Ionic
Liquid [bmim][PF6] from Monte Carlo simulations. J. Phys. Chem. B 2005, 109,
12154.
33. Cadena, C.; Anthony, J. L.; Shah, J. K.; Morrow, T. I.; Brennecke, J. F.; Maginn,
E. J. Why is CO2 So Soluble in Imidazolium-Based Ionic Liquids? J. Am. Chem.
Soc. 2004, 126, 5300.
34. Bhargava, B.; Balasubramanian, S.Probing Anion-Carbon Dioxide Interactions in
Room Temperature Ionic Liquids: Gas Phase Cluster Calculations. Chem. Phys.
Lett. 2007, 444, 242.
35. Shah, J. K.; Maginn, E. J.Monte Carlo Simulations of Gas Solubility in the Ionic
Liquid 1-n-Butyl-3-methylimidazolium afluorophosphate. J. Phys. Chem. B
2005, 109, 10395.
36. Zhang, X. C.; Liu, Z. P.; Wang, W. C. Screening of Ionic Liquids to Capture CO2
by COSMO-RS and Experiments. AIChE J. 2008, 54, 2717.

276
37. Manan, N. A.; Hardacre, C.; Jacquemin, J.; Rooney, D. W.; Youngs, T. G.
Evaluation of Gas Solubility Prediction in Ionic Liquids Using COSMOthermX.
J. Chem. Eng. Data 2009, 54, 2005.
38. Palomar, J.; Gonzalez-Miquel, M.; Polo, A.; Rodriguez, F. Understanding the
Physical Absorption of CO2 in Ionic Liquids Using the COSMO-RS Method.
Ind. Eng. Chem. Res. 2011, 50, 3452.
39. Gonzalez-Miquel, M.; Palomar, J.; Omar, S.; Rodriguez, F. CO2/N2 Selectivity
Prediction in Supported Ionic Liquid Membranes (SILMs) by COSMO-RS. Ind.
Eng. Chem. Res. 2011, 50, 5739.
40. Miller, M. B.; Chen, D. L.; Xie, H. B.; Luebke, D. R.; Johnson, J. K.; Enick, R.
M. Solubility of CO2 in CO2-Philic Oligomers; COSMOtherm Predictions and
Experimental Results. Fluid Phase Equilib. 2009, 287, 26.
41. Maiti, A. Theoretical Screening Of Ionic Liquid Solvents for Carbon Capture.
ChemSusChem. 2009, 2: 628.
42. Shimoyama, Y.; Ito, A. Predictions of Cation and Anion Effects on Solubilities,
Selectivities and Permeabilities for CO2 in Ionic Liquid Using COSMO Based
Activity Coefficient Model. Fluid Phase Equilib. 2010, 297, 178.
43. Lin, S.; Sandler, S. A Priori Phase Equilibrium Prediction From A Segment
Contribution Solvation Model. Ind. Eng. Chem. Res. 2002, 41, 899.
44. Eckert, F.; Klamt, A. COSMOtherm, version C2.1, release 01.08; Cosmologic
GmbH & Co. KG, Leverkusen, Germany, 2006.

277
45. Henni, A.; Tontiwachwuthikul, P.; Chakma, A. Solubility of Methane and Ethane
in Promising Physical Solvents for Natural Gas Sweetening Operations. J. Chem.
Eng. Data 2006, 51, 64.
46. Klamt, A.Conductor-Like Screening Model for Real Solvents—A New Approach
to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem. 1995,
99, 2224.
47. Klamt, A.; Schüürmann, G. COSMO: A New Approach to Dielectric Screening
in Solvents with Explicit Expressions for the Screening Energy and its Gradient.
J. Chem. Soc., Perkins Trans. 2 1993, 799.
48. Klamt, A.; Eckert, F. COSMO-RS: A Novel and Efficient Method for the A
Priori Prediction of Thermophysical Data of Liquids. Fluid Phase Equilib. 2000,
172, 43.
49. Eckert, F.; Klamt, A. Fast Solvent Screening Via Quantum Chemistry: COSMO-
RS Approach. AIChE J., 2002, 48: 369.
50. Klamt, A. COSMO-RS: From Quantum Chemistry to Fluid Phase
Thermodynamics and Drug Design; Elsevier: Amsterdam, 2005.
51. Lucas, K. ; Molecular Models for Fluids, Cambridge University Press; 2007.
52. Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C.; Electronic Structure
Calculations on Workstation Computers: The Program System Turbomole.
Chem. Phys. Lett. 1989, 162, 165.
53. Becke, A. D. Density-Functional Exchange-Energy Approximation with Correct
Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098−3100.

278
54. Perdew, J. P. Density-Functional Approximation for the Correlation Energy of
the Inhomogeneous Electron Gas. Phys. Rev. B 1986, 33, 8822.
55. Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis
sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100,
5829.
56. Zissimos, A. M.; Abraham, M. H.; Klamt, A.; Eckert, F.; Wood, J. J. A
Comparison between the Two General Sets of Linear Free Energy Descriptors of
Abraham and Klamt Chem. Inf. Comput. Sci. 2002, 42, 1320.
57. Diedenhofen, M.; Eckert, F.; Klamt, A.Prediction of Infinite Dilution Activity
Coefficients of Organic Compounds in Ionic Liquids Using COSMO-RS. J.
Chem. Eng. Data 2003, 48, 475.
58. Baltus, R. E.; Counce, R. M.; Culbertson, B. H.; Luo, H. M.; DePaoli, D. W.;
Dai, S.; Duckworth, D. C. Examination of the Potential of Ionic Liquids for Gas
Separations. Sep. Sci. Technol. 2005, 40, 525−541.
59. Muldoon, M. J.; Aki, S. N. V. K.; Anderson, J. L.; Dixon, J. K.; Brennecke, J.
F.Improving carbon dioxide solubility in ionic liquids. J. Phys. Chem. B 2007,
111, 9001.
60. Anthony, J. L.; Anderson, J. L.; Maginn, E. J.; Brennecke, J. F.Anion Effects on
Gas Solubility in Ionic Liquids. J. Phys. Chem. B 2005, 109, 6366.
61. Kumelan, J.; Perez-Salado, K. A.; Tuma, D.; Maurer, G.Solubility of CO2 in the
ionic liquids [bmim][CH3SO4] and [bmim][PF6]. J. Chem. Eng. Data 2006, 51,
1802.

279
62. Camper, D.; Scovazzo, P.; Koval, C.; Noble, R.Gas Solubilities in Room-
Temperature Ionic Liquids. Ind. Eng. Chem. Res. 2004, 43, 3049.
63. Jacquemin, J.; Husson, P.; Majer, V.; Padua, A. A. H.; Costa Gomes, M. F.
Thermophysical Properties, Low Pressure Solubilities and Thermodynamics of
Solvation of Carbon Dioxide and Hydrogen in Two Ionic Liquids Based on the
Alkylsulfate Anion. Green Chem. 2008, 10, 944.
64. Camper, D.; Bara, J.; Koval, C.; Noble, R. Bulk-Fluid Solubility and Membrane
Feasibility of Rmim-Based Room-Temperature Ionic Liquids. Ind. Eng. Chem.
Res. 2006, 45, 6279.
65. Pierotti, R. A. Scaled Particle Theory of Aqueous and Nonaqueous Solutions.
Chem. Rev. 1976, 76, 717.
66. Reichardt, C. Solvatochromic Dyes as Solvent Polarity Indicators. Chem. Rev.
1994, 94, 2319.
67. Bara, J. E.; Gabriel, C. J.; Lessmann, S.; Carlisle, T. K.; Finotello, A.; Gin, D. L.;
Noble, R. D. Enhanced CO2 Separation Selectivity in Oligo(Ethylene Glycol)
Functionalized Room-Temperature Ionic Liquids. Ind. Eng. Chem. Res. 2007,
46, 5380.
68. Lopes, J. C.; Gomes, M. F. C.; Padua, A. A. H. Nonpolar, Polar, and Associating
Solutes in Ionic Liquids. J. Phys. Chem. B 2006, 110, 16816.
69. Singh, T.; Kumar, A. Static Dielectric constant of Room Temperature Ionic
Liquids: Internal Pressure and Cohesive Energy Density Approach. J. Phys.
Chem. B 2008, 112, 12968.

280
70. Izgordina, E. I.; ; Forsyth, M.; ; MacFarlane, D. R. On the Components of the
Dielectric constants of Ionic Liquids: Ionic Polarization? Phys. Chem. Chem.
Phys. 2009, 11, 2452.
71. Wakai, C.; Oleinikova, A.; Ott, M.; Weingaertner, H. How Polar Are Ionic
Liquids? Determination of the Static Dielectric constant of an Imidazolium-
Based Ionic Liquid by Microwave Dielectric Spectroscopy. J. Phys. Chem. B
2005, 109, 17028.
72. Znamenskiy, V.; Kobrak, M. N. Molecular Dynamics Study of Polarity in Room-
Temperature Ionic Liquids. J. Phys. Chem. B 2004, 108, 1072.
73. Palomar, J.; Torrecilla, J. S.; Ferro, V. R.; Rodríguez, F.Development of an a
Priori Ionic Liquid Design Tool. 1. Integration of a Novel COSMO-RS
Molecular Descriptor on Neural Networks. Ind. Eng. Chem. Res. 2008, 47, 4523.
74. Wasserscheid, P.; Welton, T. (Eds) Ionic Liquids in Synthesis, 1st ed.; Wiley-
VCH: Weinheim, Germany, 2008.
75. Guo, Z.; Lue, B. M.; Thomasen, K.; Meyer, A. S.; Xu, X. Predictions of
Flavonoid Solubility in Ionic Liquids by COSMO-RS: Experimental Verification,
Structural Elucidation, and Salvation Characterization. Green Chem. 2007, 9,
1362.
76. Prausnitz, J. M. and Shair, F. H. A Thermodynamic Correlation of Gas
Solubilities. AIChE J., 1961, 7: 682.

281
77. Jacquemin, J., Husson, P., Majer, V., Gomes, M.F.C. Low-Pressure Solubilities
And Thermodynamics of Solvation Of Eight Gases in 1-Butyl-3-
Methylimidazolium hexafluorophosphate. Fluid Phase Equilib. 2006, 240, 87.
78. Jacquemin, J.; Gomes, M. F. C.; Husson, P.; Majer, V. Solubility of Carbon
Dioxide, Ethane, Methane, Oxygen, Nitrogen, Hydrogen, Argon, and Carbon
Monoxide in 1-Butyl-3-Methylimidazolium Tetrafluoroborate Between
Temperatures 283 K and 343 K and at Pressures Close to Atmospheric. J. Chem.
Thermodyn. 2006, 38, 490.
79. Shiflett, M. B.; Yokozeki, A. Phase Behavior of Carbon Dioxide in Ionic
Liquids: [emim][Acetate], [emim][Trifluoroacetate], and [emim][Acetate] +
[emim][Trifloroacetate] Mixtures. J. Chem. Eng. Data 2009, 54, 108.
80. Camper, D.; Bara, J. E.; Gin, D. L.; Noble, R. D.Room-Temperature Ionic
Liquid–Amine Solutions: Tunable Solvents for Efficient and Reversible Capture
of CO2. Ind. Eng. Chem. Res. 2008, 47, 8496.
81. Huang, J.; Rüther, T.Why are Ionic Liquids Attractive for CO2 Absorption? An
Overview. Aust. J. Chem. 2009, 62, 298.
82. Aparicio, S.; Atilhan, M.; Karadas, F. Thermophysical Properties of Pure Ionic
Liquids: Review of Present Situation. Ind. Eng. Chem. Res. 2010, 49, 9580.
83. Wasserscheid, P.; Welton, T. (Eds) Ionic Liquids in Synthesis, 2nd ed.; Wiley-
VCH: Weinheim, Germany, 2008

282
Chapter 7: Measurement of Solubility of CO2 in [eFAP]-Based Ionic Liquids
_______________________________________________________________________
7.1 INTRODUCTION
The screening study based on COSMO-RS prediction in Chapter 6 showed that
irrespective of the nature of cation, ionic liquids with [eFAP] anion had low Henry’s law
constant. We therefore chose three imidazolium based ionic liquids with [eFAP] anion:
[emim][eFAP], [bmim][eFAP], [hmim][eFAP] for measurement of solubility of CO2 at
temperatures (10, 25, 50)°C and at pressures up to 2 MPa. Solubility data were
correlated with the Peng-Robinson cubic equation of state. These ionic liquids have
excellent hydrolytic and thermal stability.1
7.2 EXPERIMENTAL
7.2.1 Materials.
Ionic liquids [emim][eFAP] (CAS No. 377739-43-0, Lot No. S5204301 907),
[bmim][eFAP] (CAS No. 917762-91-5, Lot No. S5204932 943), [hmim][eFAP] (CAS
No. 713512-19-7, Lot No. S5202278 845) were obtained from EMD Chemicals Inc. and
[bmim][PF6] (CAS No.174501-64-5, assay ) were obtained from Sigma-Aldrich and
were used without further purification.

283
Figure 7.1 Structure of the ionic liquids [emim][eFAP], [bmim][eFAP] and
[hmim][eFAP] (R=C2H5, C4H9, C6H13)
7.2.2 Apparatus and measurements.
All the measurements were performed using a gravimetric microbalance (Hiden
Isochema Ltd., Integrated Gravimetric Analyzer (IGA003)). The equipment enables
fully-automatic computer-controlled measurement; and run by a software called
IGASwin. Real-time measurements of pressure, temperature and weight change (0.1
microgram) are recorded.
The microbalance consists of an electrobalance with two arms, one arm holding
the sample container, and the other arm holding the counterweight components. The
balance is inside the cabinet and both arms are covered from outside by stainless steel
tubular pressure-vessels (SS316N). The stainless steel vessel covering the sample arm is
called a reactor and could be heated or cooled with an external water-jacket connected to
a water bath (Polyscience). The gas is introduced to the microbalance chamber through
a multi flow-meter (MFC) from an outside cylinder. The microbalance chamber could
also be evacuated to about 20 mbar by first using a coarse diaphragm pump (Vacuum
brand) and then to deeper vacuum (< 5 mbar) by a turbo pump (Pfeiffer). The sample

284
temperature was measured with a platinum resistance thermocouple (± 0.1 K), and
pressure was measured with a pressure transducer (Druck PDCR 4010, ± 8 mbar).
Sample between 50-80 mg were loaded into the sample container and properly
sealed. The samples were heated to 348.2 K under deep vacuum in the chart mode to
outgas water and other impurities until the weight was stabilized. The temperature was
then reset to the experimental temperature. Once the desired temperature was reached,
CO2 was introduced to the reactor until the first isotherm pressure was reached.
Continuous absorption data was recorded until 99% of the predicted equilibrium weight
was reached. After that, measurement at the next isotherm pressure is continued and
continues until measurement is done at all the pressure points under the isotherm.
An important step in obtaining accurate data from gravimetric method using IGA
is buoyancy correction to the weight as the sample remains immersed in the gas in the
pressure vessel. The gas solubilities in ILs were determined from real-time equilibrium
mass uptake at a given pressure and temperature with appropriate buoyancy correction.
A blank experiment (without any sample) was run for each isotherm and at all pressures
with the same bucket. The real-time weights obtained from these runs were used to
nullify the buoyancy corrections due to all the components of the balance and the bucket
and balance sensitivity.
Density of ionic liquid, required for buoyancy correction due to solvent was
measured using Anton Paar digital density meter DMA 4500 (accuracy ± .00005 g/cm3)
in the temperature range between 283.15 K and 353.15 K with 5 K interval at
atmospheric pressure. It measures liquid density based on the oscillating U-tube
principle. The machine was calibrated with air and water following the instruction

285
manual and was deemed acceptable if the density of water provided in the manual could
be reproduced within ± .00005 g/cm3. The U-tube of the density meters was filled with
about 1 ml ionic liquid slowly without forming air bubbles and then electronically
excited during the measurement and density is determined from the period of oscillation.
Figure 7.2 Computer-controlled integrated gravimetric microbalance ( IGA003 ).

286
7.3 MODELING
CO2-IL system is a complex non-ideal system characterized by the large
differences in shape and size and the presence of ionic interactions. Cubic equations of
state, originally developed for modeling phase equilibrium for non-electrolyte systems,
were recently used successfully for modeling many ionic liquids+CO2 systems. While
not deeply rooted in statistical mechanics, their relatively simple algebraic form capable
of representing two phases and the need for few adjustable parameters have made them
popular choice in design and analysis of many industrial complex VLE process. The
solubility data were correlated with the Peng-Robinson equation of state (equation 7.1)
with two-fluid mixing rule.2
2
(7.1)
Where and are the mixture attractive and co-volume parameters, respectively and
related to the corresponding pure component parameters with van der Waals quadratic
two parameter mixing rules.
(7.2)
(7.3)
where
1
with and 1
(7.4)

287
1
2
with and 1
(7.5)
And the pure component attractive and co-volume parameters and are related to the
critical temperature ( ) , critical pressure ( ) and accentric factor ( ) as
(7.6)
0.45724
(7.7)
0.37464 1.5422 0.26992 (7.8)
0.0778
(7.9)
Critical properties of ionic liquids were estimated using Group-contribution
methods.3 The binary interaction parameters were regressed using the software Phase
Equilibrium 2000 (PE2000)4 developed by Brunner and coworkers that uses a Simplex-
Nelder-Mead algorithm for regression. The average absolute relative deviation (equation
7.10) in the liquid phase mole fraction was minimized at experimental temperature and
pressure.
100% (7.10)

288
7.4 RESULTS AND DISCUSSION
The densities of the ionic liquids were measured between 283.15 K and 353.15 K
in 5 K interval and the data reported in Table 7.1. The number of various functional
groups present in the ionic liquids required for computation of critical properties is
tabulated in Table 7.2 and the estimated critical properties of the ionic liquids required
for modeling are presented in Table 7.3. To verify our experimental method, solubility
data of CO2 in a common ionic liquid [bmim][PF6] at 25°C were measured and
compared with those reported by Anthony et al. (2002)5 and Shiflett et al. (2005)6 in
Figure 7.3. The average absolute deviation between the data of Shiflett et al.6 and this
work is 0.001 in mole fraction of CO2.
The solubility data of CO2 in the ionic liquids [emim][eFAP], [bmim][eFAP] and
[hmim][eFAP] were then measured and reported in Tables 7.4, 7.6 and 7.8 in mole-
fraction scale and in Tables 7.5, 7.7 and 7.9 in molality scale (mol CO2/kg IL)
respectively. The solubility data presented in Tables 7.4, 7.6 and 7.8 are also plotted in
Figures 7.4, 7.5, and 7.6 respectively with modeling results.
The measured solubility data of CO2 in [hmim][eFAP] was compared with
literature data in Figure 7.7. At low pressure, good agreement is found with the data of
both Muldoon et al.(2007)7 and Zhang et al. (2008)8; but, at higher pressure, slight
discrepancies appear from the data of Zhang et al. (2008). The measured solubility data
of CO2 in [emim][eFAP] was in good agreement with the high-pressure bubble-point
data of Althuluth et al. (2012)9 (Figure 7.8).

289
Table 7.1 Measured Density of Ionic Liquids
Ionic Liquids
T(K) [bmim][PF6] [emim][eFAP] [bmim][eFAP] [hmim][eFAP]
283.15 1.37892 1.72709 1.64218 1.56636
288.15 1.37464 1.72114 1.63654 1.56059
293.15 1.37037 1.71519 1.63089 1.55553
298.15 1.36611 1.70923 1.62524 1.5501
303.15 1.36185 1.7033 1.61959 1.54467
308.15 1.35775 1.69738 1.61395 1.53927
313.15 1.35365 1.69148 1.60833 1.53387
318.15 1.34954 1.68559 1.60272 1.52848
323.15 1.34544 1.67972 1.59713 1.52311
328.15 1.34135 1.67387 1.59155 1.51775
333.15 1.33727 1.66803 1.58598 1.51241
338.15 1.3332 1.66221 1.58044 1.50708
343.15 1.32914 1.65641 1.57491 1.50176
348.15 1.32509 1.65063 1.56939 1.49647
353.15 1.32107 1.64487 1.56391 1.49119
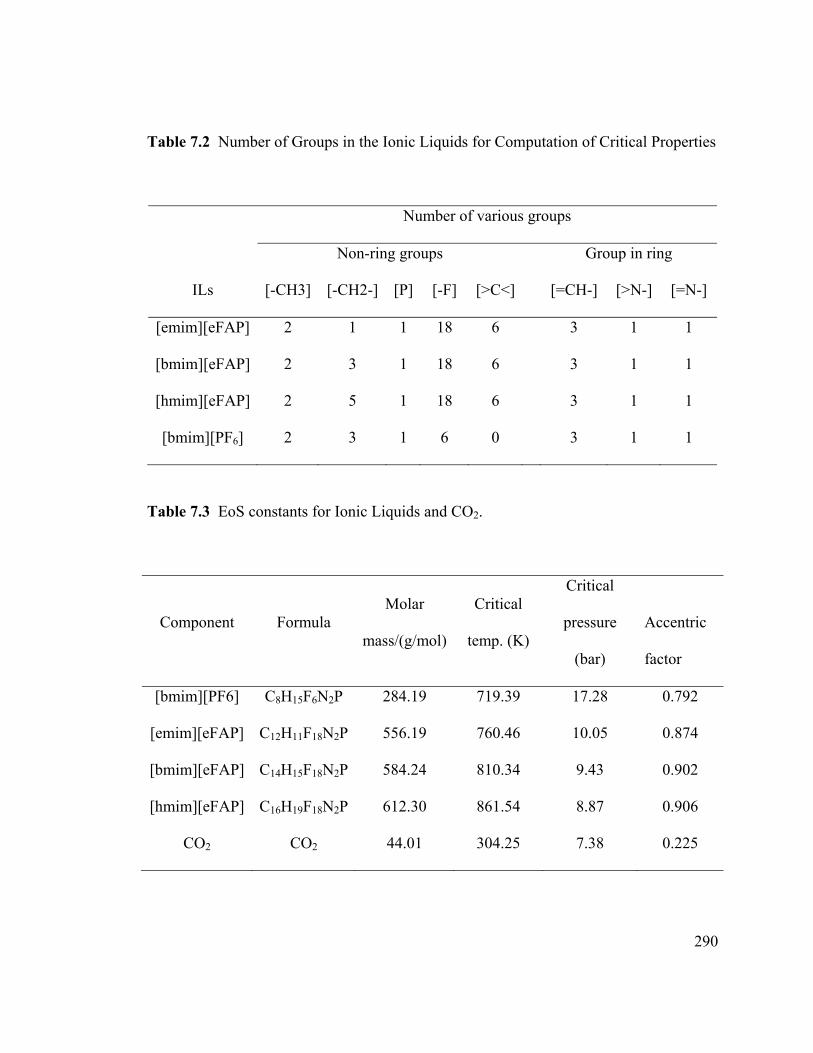
290
Table 7.2 Number of Groups in the Ionic Liquids for Computation of Critical Properties
Number of various groups
Non-ring groups Group in ring
ILs [-CH3] [-CH2-] [P] [-F] [>C<] [=CH-] [>N-] [=N-]
[emim][eFAP] 2 1 1 18 6 3 1 1
[bmim][eFAP] 2 3 1 18 6 3 1 1
[hmim][eFAP] 2 5 1 18 6 3 1 1
[bmim][PF6] 2 3 1 6 0 3 1 1
Table 7.3 EoS constants for Ionic Liquids and CO2.
Component Formula Molar
mass/(g/mol)
Critical
temp. (K)
Critical
pressure
(bar)
Accentric
factor
[bmim][PF6] C8H15F6N2P 284.19 719.39 17.28 0.792
[emim][eFAP] C12H11F18N2P 556.19 760.46 10.05 0.874
[bmim][eFAP] C14H15F18N2P 584.24 810.34 9.43 0.902
[hmim][eFAP] C16H19F18N2P 612.30 861.54 8.87 0.906
CO2 CO2 44.01 304.25 7.38 0.225

291
Figure 7.3 Comparison of solubility of CO2 in [bmim][PF6] with literature data.
0
0.5
1
1.5
2
2.5
0.0 0.1 0.2 0.3 0.4
Tot
al p
ress
ure
, PM
Pa
Mole fraction of CO2 in [bmim][PF6]
Anthony et al. (2002) (Ref. 5)Shiflett et al. (2005) (Ref. 6)This work

292
Table 7.4 Solubility of CO2 in [emim][eFAP] at Different Pressures and Temperatures
in the Mole-Fraction scale
P (MPa) Mole
Fraction P (MPa)
Mole
Fraction P (MPa)
Mole
Fraction
T=283.15K T=298.15K T=323.15K
0.05 0.023 0.05 0.019 0.05 0.012
0.10 0.048 0.10 0.037 0.10 0.024
0.30 0.137 0.30 0.103 0.30 0.065
0.50 0.213 0.50 0.162 0.50 0.112
0.70 0.280 0.70 0.217 0.70 0.150
1.00 0.364 1.00 0.287 1.00 0.203
1.20 0.413 1.20 0.330 1.20 0.236
1.40 0.456 1.40 0.365 1.40 0.267
1.60 0.495 1.60 0.402 1.60 0.294
1.80 0.530 1.80 0.440 1.80 0.321
2.00 0.562 1.99 0.462 2.00 0.345
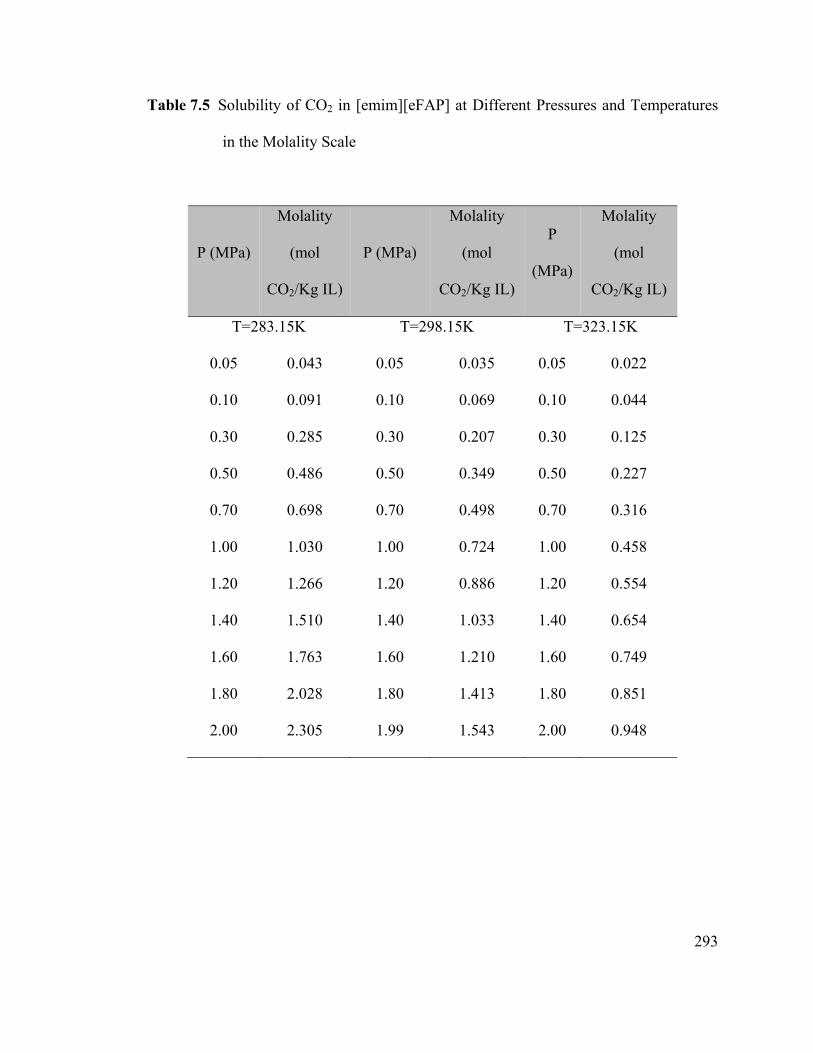
293
Table 7.5 Solubility of CO2 in [emim][eFAP] at Different Pressures and Temperatures
in the Molality Scale
P (MPa)
Molality
(mol
CO2/Kg IL)
P (MPa)
Molality
(mol
CO2/Kg IL)
P
(MPa)
Molality
(mol
CO2/Kg IL)
T=283.15K T=298.15K T=323.15K
0.05 0.043 0.05 0.035 0.05 0.022
0.10 0.091 0.10 0.069 0.10 0.044
0.30 0.285 0.30 0.207 0.30 0.125
0.50 0.486 0.50 0.349 0.50 0.227
0.70 0.698 0.70 0.498 0.70 0.316
1.00 1.030 1.00 0.724 1.00 0.458
1.20 1.266 1.20 0.886 1.20 0.554
1.40 1.510 1.40 1.033 1.40 0.654
1.60 1.763 1.60 1.210 1.60 0.749
1.80 2.028 1.80 1.413 1.80 0.851
2.00 2.305 1.99 1.543 2.00 0.948

294
Table 7.6 Solubility of CO2 in [bmim][eFAP] at Different Pressures and Temperatures
in the Mole-Fraction Scale
P (MPa) Mole
Fraction P (MPa)
Mole
Fraction P (MPa)
Mole
Fraction
T=283.15K T=298.15K T=323.15K
0.05 0.027 0.05 0.021 0.05 0.013
0.10 0.052 0.10 0.040 0.10 0.025
0.30 0.144 0.30 0.109 0.30 0.071
0.50 0.223 0.50 0.176 0.50 0.118
0.70 0.290 0.70 0.229 0.70 0.157
0.90 0.342 0.90 0.274 0.90 0.189
1.00 0.377 1.00 0.301 1.00 0.214
1.20 0.426 1.20 0.345 1.20 0.249
1.40 0.470 1.40 0.384 1.40 0.281
1.60 0.509 1.60 0.419 1.60 0.310
1.80 0.544 1.80 0.452 1.80 0.338
2.00 0.576 2.00 0.481 2.00 0.363

295
Table 7.7 Solubility of CO2 in [bmim][eFAP] at Different Pressures and Temperatures
in the Molality Scale
P (MPa)
Molality
(mol
CO2/Kg
IL)
P (MPa)
Molality
(mol
CO2/Kg
IL)
P (MPa)
Molality
(mol
CO2/Kg
IL)
T=283.15K T=298.15K T=323.15K
0.05 0.047 0.05 0.036 0.05 0.023
0.10 0.094 0.10 0.071 0.10 0.044
0.30 0.288 0.30 0.210 0.30 0.130
0.50 0.490 0.50 0.366 0.50 0.229
0.70 0.701 0.70 0.509 0.70 0.320
0.90 0.891 0.90 0.645 0.90 0.400
1.00 1.036 1.00 0.737 1.00 0.467
1.20 1.271 1.20 0.900 1.20 0.566
1.40 1.518 1.40 1.066 1.40 0.670
1.60 1.774 1.60 1.236 1.60 0.771
1.80 2.041 1.80 1.409 1.80 0.876
2.00 2.325 2.00 1.586 2.00 0.975

296
Table 7.8 Solubility of CO2 in [hmim][eFAP] at Different Pressures and Temperatures
in the Mole Fraction Scale
P (MPa)
Mole
Fraction P (MPa)
Mole
Fraction P (MPa)
Mole
Fraction
T=283.15K T=298.15K T=323.15K
0.05 0.029 0.05 0.023 0.05 0.016
0.10 0.055 0.10 0.044 0.10 0.029
0.30 0.150 0.30 0.115 0.30 0.078
0.50 0.230 0.50 0.185 0.50 0.127
0.70 0.300 0.70 0.239 0.70 0.167
1.00 0.388 1.00 0.314 1.00 0.224
1.20 0.438 1.20 0.358 1.20 0.258
1.40 0.482 1.40 0.397 1.40 0.291
1.60 0.521 1.60 0.433 1.60 0.320
1.80 0.556 1.80 0.465 1.80 0.348
2.00 0.588 2.00 0.494 2.00 0.374

297
Table 7.9 Solubility of CO2 in [hmim][eFAP] at Different Pressures and Temperatures
in the Molality Scale
P (MPa)
Molality
(mol
CO2/Kg
IL)
P (MPa)
Molality
(mol
CO2/Kg
IL)
P (MPa)
Molality
(mol
CO2/Kg
IL)
T=283.15K T=298.15K T=323.15K
0.05 0.048 0.05 0.039 0.05 0.027
0.10 0.095 0.10 0.075 0.10 0.049
0.30 0.288 0.30 0.213 0.30 0.137
0.50 0.488 0.50 0.371 0.50 0.237
0.70 0.699 0.70 0.514 0.70 0.328
1.00 1.035 1.00 0.746 1.00 0.472
1.20 1.272 1.20 0.910 1.20 0.569
1.40 1.519 1.40 1.075 1.40 0.672
1.60 1.775 1.60 1.245 1.60 0.769
1.80 2.042 1.80 1.417 1.80 0.872
2.00 2.329 2.00 1.592 2.00 0.976
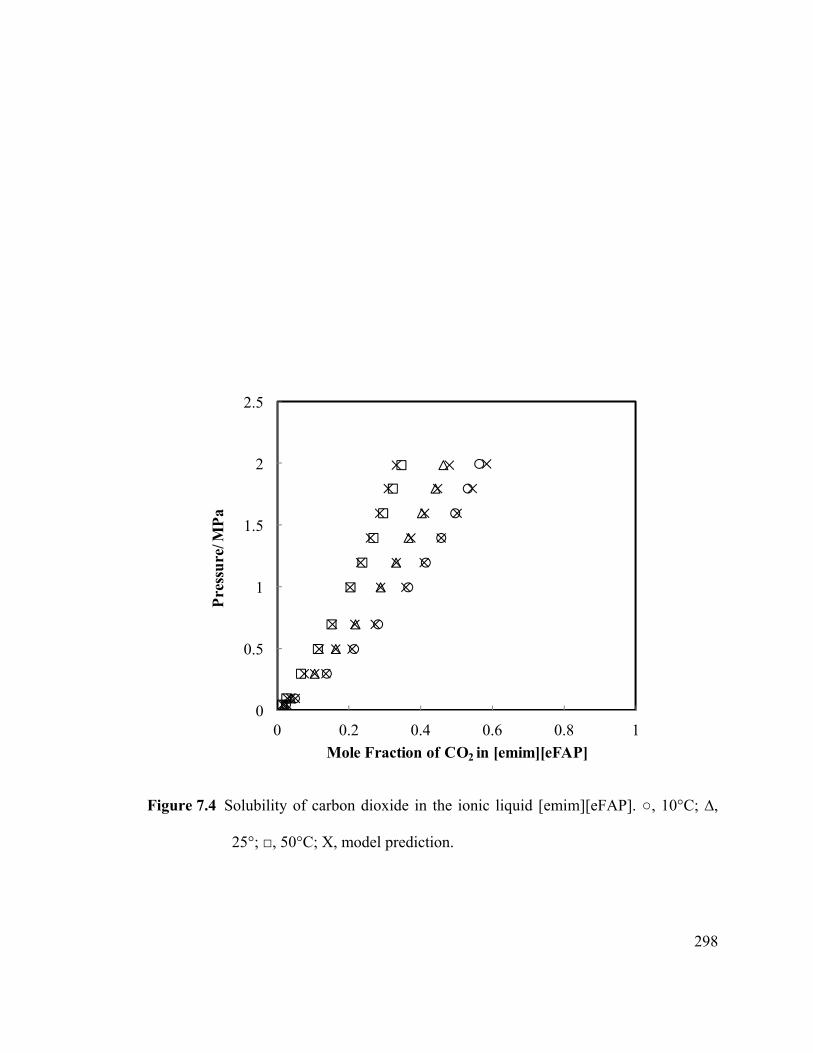
298
Figure 7.4 Solubility of carbon dioxide in the ionic liquid [emim][eFAP]. ○, 10°C; ∆,
25°; □, 50°C; X, model prediction.
0
0.5
1
1.5
2
2.5
0 0.2 0.4 0.6 0.8 1
Pre
ssu
re/ M
Pa
Mole Fraction of CO2 in [emim][eFAP]

299
Figure 7.5 Solubility of carbon dioxide in the ionic liquid [bmim][eFAP]. ○, 10°C; ∆,
25°C; □, 50°C, X, model prediction.
0
0.5
1
1.5
2
2.5
0 0.2 0.4 0.6 0.8 1
Pre
ssu
re/ M
Pa
Mole fraction of CO2 in [bmim][eFAP]

300
Figure 7.6 Solubility of carbon dioxide in the ionic liquid [hmim][eFAP]: ○, 10°C; ∆,
25°C; □, 50°C, X, model prediction.
0
0.5
1
1.5
2
2.5
0 0.2 0.4 0.6 0.8 1
Pre
ssu
re/ M
Pa
Mole fraction of CO2 in [hmim][eFAP]
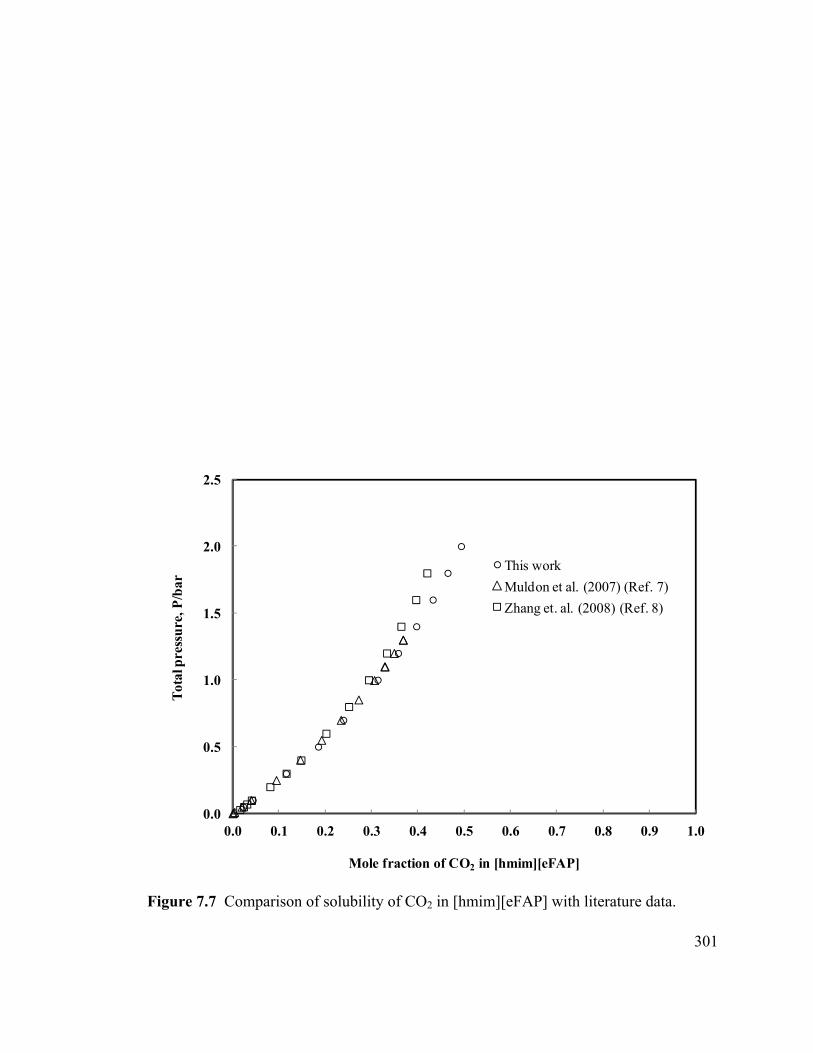
301
Figure 7.7 Comparison of solubility of CO2 in [hmim][eFAP] with literature data.
0.0
0.5
1.0
1.5
2.0
2.5
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
Tot
al p
ress
ure
, P/b
ar
Mole fraction of CO2 in [hmim][eFAP]
This work
Muldon et al. (2007) (Ref. 7)
Zhang et. al. (2008) (Ref. 8)

302
Figure 7.8 Comparison of solubility of CO2 in [emim][eFAP] with literature data.
0
0.5
1
1.5
2
2.5
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
Tot
al p
ress
ure
, P/b
ar
Mole Fraction of CO2 in [hmim][eFAP]
Althuluth et al. (2012) (Ref. 9), 284 K
This work, 283K
Althuluth et al. (2012) (Ref. 9), 324 K
This work, 323K

303
The temperature-dependent binary interaction parameters for Peng-Robinson EoS
and the corresponding absolute average deviation calculated using the objective function
(equation 7.10) is given in Table 7.10. The solubility data was correlated well with one
binary interaction parameter within the pressure and temperature range of the
experimental data used in correlation. Solubility increases quickly with pressure. The
effect of temperature is less at low pressure but gets more pronounced as the pressure
increases.
For many ionic liquids, experimental measurements at high pressure showed that
after a certain concentration of CO2 is dissolved in the liquid phase, very high pressure is
required to further dissolve CO2 in the ionic liquid (Aki et al., 2004).10 This behavior is
explained by free-volume theory that solubility of CO2 increases with pressure fast at
low pressure when CO2 starts occupying the free-spaces in the ionic liquid structure, but
once the free-space is filled, it requires very high-pressure to increase the solubility even
nominally resulting in an infinite P-x slope (diverging bubble point and dew point
pressure curve) (Blanchard. et al., 2001).11 The present EoS with one optimized binary
interaction parameter did not reproduce this behavior, it predicts a mixture critical point
where bubble point and dew point curves meet at high CO2 mole fraction (Figure 7.9).
Similar modeling results was also found by Ren et al. (2010)12 for {carbon dioxide (CO2)
+ n-hexyl-imidazolium bis[(trifluoromethyl)sulfonyl]amide} system who correlated
high-pressure data by the PR EoS with two binary interaction parameters. Re-optimizing
the binary parameters using one high-pressure data point from the measurement of
Althuluth et al. (2012)9 for the ionic liquids [emim][eFAP] reveals the typical high-
pressure phase behavior that the pressure-composition curve diverges after a certain

304
amount of CO2 dissolution (Figure 7.9). In this case, two binary interaction parameters
were required (kij=0.0832, lij=0.0392) to correlate the solubility data. Moreover,
prediction of high-pressure phase behavior using these parameters (without re-
optimizing) at 25°C, also reveals a VLLE phase at high pressure (Figure 7.10), and such
phase behavior is also predicted by cubic EoS modeling for other ionic liquids such as
[bmim][PF6] (Shiflett et al.,2005), [bmim][acetate] (Shiflett et al., 2008)13 and
[hmim][Tf2N] (Ren et al., 2010)12 below the critical temperature of CO2. However,
such extrapolation is cautioned in absence of experimental evidence due to the inherent
weaknesses of cubic equation of state for the present system due to the poor theoretical
basis of cubic EoS for polar and associating fluids in general, and relevant to the present
systems, there is no explicit ionic interaction term in the EoS (Reissi et al., 2010).14

305
Table 7.10 Estimated Binary Interaction Parameters and Modeling Results
Ils+CO2 T/K
Number
of data
points
Binary interaction
parameters
kij lij AARD%
[emim][eFAP] 283.15 11 - 0.021 1.92
298.15 13 - 0.020 1.24
323.15 11 - 0.014 1.52
[bmim][Tf2N] 283.15 12 - 0.023 1.16
298.15 12 - 0.022 0.75
323.15 12 - 0.018 0.71
[hmim][Tf2N] 283.15 11 - 0.023 1.33
298.15 11 - 0.024 0.61
323.15 11 - 0.019 1.82
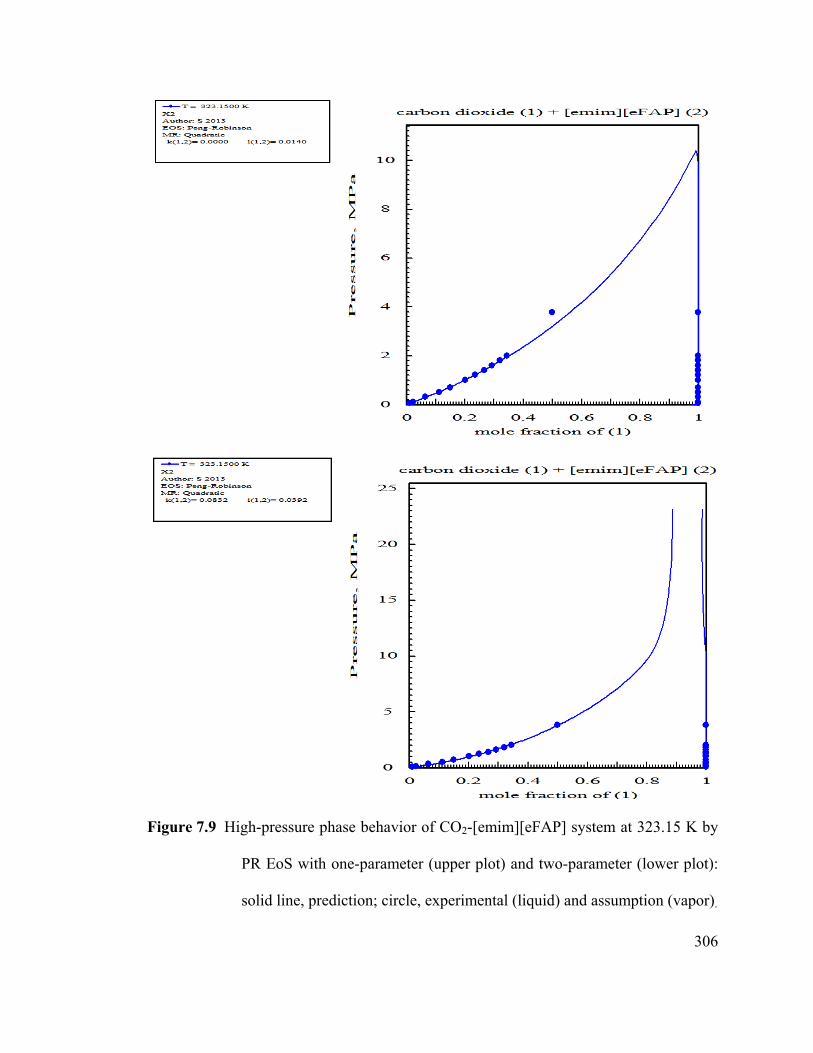
306
Figure 7.9 High-pressure phase behavior of CO2-[emim][eFAP] system at 323.15 K by
PR EoS with one-parameter (upper plot) and two-parameter (lower plot):
solid line, prediction; circle, experimental (liquid) and assumption (vapor).

307
Figure 7.10 High-pressure phase behavior of CO2-[emim][eFAP] system at 298.15 K by
PR EoS with one-parameter (upper plot) and two-parameter (lower plot):
solid line, prediction; circle, experimental (liquid) and assumption (vapor).

308
The mole-fraction based Henry’s law constant is defined in equation 7.11. At
equilibrium, the fugacities of the solute gas in both phases are equal (equation 7.12).
Moreover, assuming the gas phase is essentially free of ionic liquid, the fugacity of vapor
phase is assumed to be equal to that of pure carbon dioxide at the same temperature and
pressure which is computed using the Peng-Robinson equation of state. Thus Henry’s
law constant is computed from the linear slope of fugacity versus mole-fraction curve at
low pressure (equation 7.14).
lim
(7.11)
, , , , (7.12)
, , , (7.13)
lim lim
,
(7.14)
The calculated Henry’s law constants are presented in Table 7.11 and in Figure
7.11. Henry’s law constants decrease with both increase in alkyl chain length and
increase in temperature. The Henry’s law constant of [emim][eFAP] at 283.15 is 2.14
MPa, comparable with 2.24 MPa reported by Althuluth et al. (2012)9 and Henry’s law
constant of [hmim][eFAP] at 25°C are comparable with those of 2.52 MPa by Muldoon
et al. (2007)7 and 2.37 MPa by Zhang et al. (2008)8. Henry’s law constants at 25°C in
[emim][eFAP], [bmim][eFAP], and in [hmim][eFAP] are slightly higher than the
corresponding COSMO-RS prediction of 2.1, 2.0 and 1.9 MPa presented in the previous
chapter. The effect of temperature on gas solubility is expressed in terms of enthalpy and
entropy of solvation in Table 7.12. Both values become less negative with increase in

309
alkyl chain length and their magnitudes are similar to common room temperature ionic
liquids such as [bmim][PF6].
Table 7.11 The Mole-Fraction Based Henry’s law constant of CO2 in the [eFAP] Ionic
Liquids at Various Temperatures
/MPa
T/K [emim][eFAP] [bmim][eFAP] [hmim][eFAP]
283.15 2.14 2.02 1.93
298.15 2.83 2.67 2.5
323.15 4.47 4.13 3.72
Table 7.12 The Enthalpy and Entropy of Solvation of CO2 in the [eFAP] Ionic Liquids
at Various Temperatures
[emim][eFAP] [bmim][eFAP] [hmim][eFAP]
∆solH
(J/mol) -14.1 -13.6 -12.5
∆solS
(J/mol/K) -46.4 -45.0 -41.2
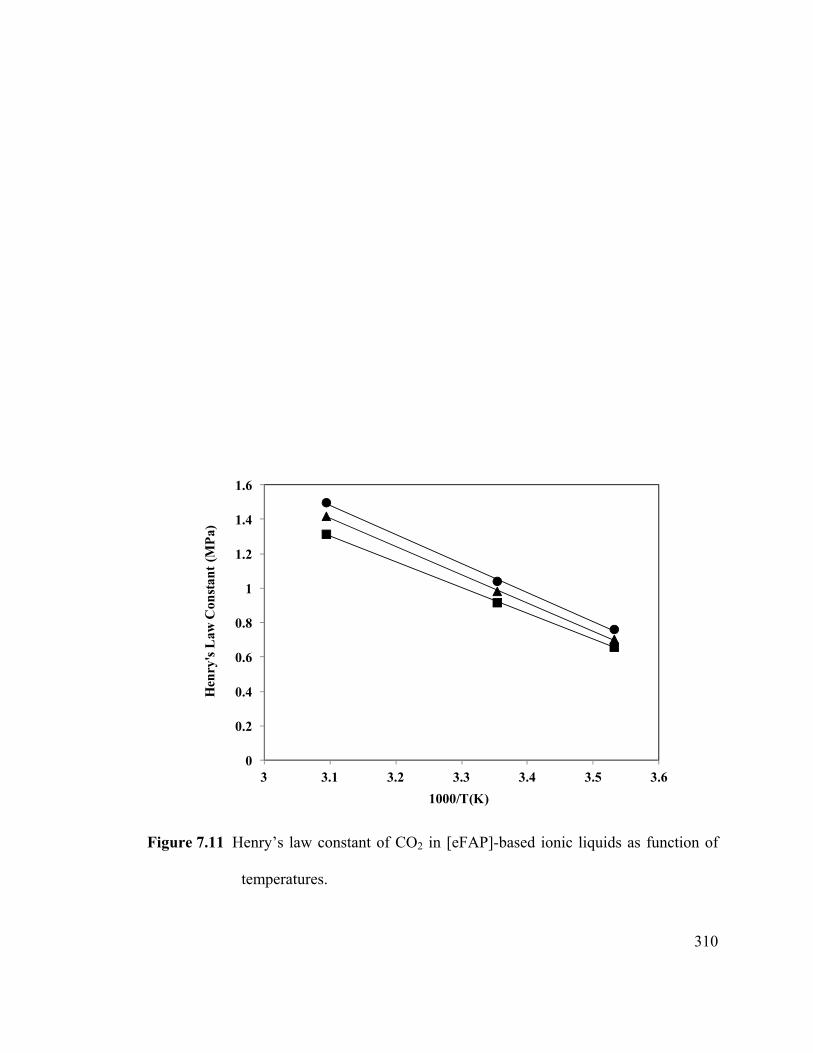
310
Figure 7.11 Henry’s law constant of CO2 in [eFAP]-based ionic liquids as function of
temperatures.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
3 3.1 3.2 3.3 3.4 3.5 3.6
Hen
ry's
Law
Con
stan
t (M
Pa)
1000/T(K)

311
7.5 CONCLUSIONS
The solubility of CO2 in the ionic liquids based on [eFAP] is presented at
temperatures (283.15, 298.15 and 323.15) K and at pressures (0.5 to 2) MPa. The
solubility data were in good agreement with literature data. The solubility data were
correlated with Peng-Robinson equation of state. Henry's law constants of CO2 in these
ionic liquid are derived and was in good agreement with literature data and COSMO-RS
prediction. The measured Henry’s law constants at 25°C in [emim][eFAP],
[bmim][eFAP], and in [hmim][eFAP] are 2.83, 2.67 and 2.5 MPa.

312
7.6 REFERENCES
1. Ignat’ev, N. V.; Welz-Biermann, U.; Kucheryna, A.; Bissky, G.; Willner, H. New
Ionic Liquids with Tris(perfluoroalkyl)trifluorophosphate (FAP) Anions. J.
Fluorine Chem. 2005, 126, 1150.
2. Peng, D. Y.; Robinson, D. B.A New Two-constant Equation of State. Ind. Eng.
Chem. Res. 1976, 15, 59.
3. Valderrama, J. O.; Rojas, R. E. Critical Properties of Ionic Liquids. Revisited.
Ind. Eng. Chem. Res. 2009, 48, 6890.
4. Pfohl, O.; Petkov, S.; Brunner, G. PE 2000 – A Powerful Tool to Correlate
Phase Equilibria. Herbert Utz Verlag, München, 2000.
5. Anthony, J. L.; Maginn, E. J.; Brennecke, J. F. Solubilities and Thermodynamic
Properties of Gases in the Ionic Liquid 1-n-Butyl-3-methylimidazolium
hexafluorophosphate. J. Phys. Chem. B 2002, 106, 7315.
6. Shiflett, M. B.; Yokoseki, A. Solubilities and Diffusivities of Carbon Dioxide in
Ionic Liquids: [bmim][PF6] and [bmim][BF4]. Ind. Eng. Chem.
Res. 2005, 44, 4453.
7. Muldoon, M. J.; Aki, S. N. V. K.; Anderson, J. L.; Dixon, J. K.; Brennecke, J. F.
Improving Carbon Dioxide Solubility in Ionic Liquids. J. Phys. Chem. B 2007,
111, 9001.
8. Zhang, X. C.; Liu, Z. P.; Wang, W. C. Screening of Ionic Liquids to Capture CO2
by COSMO-RS and Experiments. AIChE J. 2008, 54, 2717.

313
9. Althuluth, M.; Mota-Martinez, M.; Kroon, M. C.; Peters, C. J. Solubility of
Carbon Dioxide in the Ionic Liquid 1-Ethyl-3-methylimidazolium
tris(pentafluoroethyl)trifluorophosphate. J. Chem. Eng. Data 2012, 57 (12),
3422.
10. Aki, S. N. V. K.; Mellein, B. R.; Saurer, E. M.; Brennecke, J. F. High-Pressure
Phase Behavior of Carbon Dioxide with Imidazolium-Based Ionic Liquids. J.
Phys. Chem. B 2004, 108, 20355.
11. Blanchard L. A.; Gu Z. Y.; Brennecke J. F. High-pressure Phase Behavior of
Ionic Liquid/CO2 Systems. J Phys Chem. B. 2001, 105, 2437.
12. Ren, W.; Sensenich1, B.; Scurto, A. M. High-pressure Phase Equilibria of
{Carbon Dioxide (CO2) + n-Alkyl-Imidazolium
Bis(trifluoromethylsulfonyl)amide} Ionic Liquids. J. Chem. Thermodyn. 2010,
42, 305.
13. Shiflett, M. B.; Kasprzak, D. J.; Junk, C. P.; Yokozeki, A. Phase Behavior of
{Carbon Dioxide + [bmim][Ac]} Mixtures. J. Chem. Thermodyn. 2008, 40, 25.
14. Raeissi, S.; Florusse, L.; Peters, C. J. Scott–Van Konynenburg Phase Diagram of
Carbon Dioxide + Alkylimidazolium-Based Ionic Liquids. J. of Supercritical
Fluids 2010, 55, 825.

314
Chapter 8: Density, Viscosity and Excess Enthalpy of { 1-Butyl-3-Methyl Imidazolium
Acetate+Water} System _______________________________________________________________________
8.1 INTRODUCION
Aqueous mixture of the ionic liquid 1-butyl-3-methyl imidazolium acetate
([bmim][Ac]) is a patented solvent for CO2 separation.1 The {CO2+([bmim][Ac]}
system has spurred much interest in recent years due to high solubility of CO2 in
[bmim][Ac] and has been subject to many computational and experimental
investigations. The solubility of CO2 in 1-butyl-3-methylimidazolium acetate as a
function of temperature and pressure was measured by various researchers.2-5 The
distinct phase behavior of CO2 with [bmim][Ac] was attributed to chemical absorption
rather than physical absorption by spectroscopic and computational study.6-8 An
economic evaluation of CO2 capture using [bmim][Ac] was carried out using process
simulation.9 Effect of water on solubility of CO2 and physical properties of [bmim][Ac]
was recently studied.4 Addition of water decreases solubility of CO2, but dramatically
reduces the viscosity and thus expected to reduce overall cost of capture.4
Knowledge of various thermo physical properties is required for engineering
design and subsequent operations. We have measured the density, viscosity, and excess
enthalpies of {[bmim][Ac]+water} at various temperatures over the whole composition

315
range. The temperature and composition dependence of these properties is analyzed and
correlated. Excess enthalpy of {water+[bmim][Ac]} system was compared with those of
{amine+[bmim][Ac]} system.
8.2 EXPERIMENTAL
1-butyl-3-methyl-imidazolium acetate (CAS No. 284049-75-8) was obtained
from Sigma-Aldrich (assay ≥95% mas percent) and was used without further
purification. The water content of the ionic liquid changed during experiments and was
measured on average to be 6000 ppm (Karl-Fisher coulometric titration). The amines
monoethanolamine (≥98% mass percent) (CAS 141-43-5), diethanolamine, (≥98% mass
percent) (CAS 111-42-2), triethanolamine (≥99% mass percent) (CAS 102-71-6) , 2-
amino-2-methyl-1-propanol (≥99% mass percent) (CAS 124-68-5), N-methyl
diethanolamine (≥99% mass percent) (CAS 105-59-9) were obtained from Sigma Adrich
and was used without further purification. The solutions were prepared by mass on an
analytical balance (model Ap 205D, Ohaus, Florham Park, NJ) with ± 0.01 mg accuracy.
Densities of the binary mixtures were measured with an Anton Paar DMA-4500
density meter as described in chapter 7. Density was adjusted with air and bidistilled
degassed waster for the full temperature range as is recommend by the manufacturer
(uncertainties are about ± 5E-5 g.cm3). Kinematic viscosities (ν) were determined with
a number of Cannon-Ubbelohde viscometers (Cole-Parmer) to cover the whole
composition and temperature range (25°C to 70°C). The temperature was controlled by
means of a digital controller (± 0.004°C) in a well-stirred bath within ±0.01°C as

316
measured by a Cole-Parmer resistance thermometer (model H-01158-65, Anjou, Québec,
Canada). The efflux time was averaged over repeated measurements with a handhold
digital stopwatch capable of measuring time within ±0.01s. The value of the dynamic
viscosity (η) was obtained by multiplying the measured kinematic viscosities by the
density (ρ). Excess enthalpy of mixing was measured using a C80 Calvet type
calorimeter (Setaram Instrumentation) following procedure described in details in earlier
work.10
8.3 RESULTS AND DISCUSSION
The densities of {[bmim][Ac]+water} system are reported in Table 1 and the
densities of pure [bmim][Ac] at various temperatures are compared with literature values
in Figure 8.1. The experimentally measured densities of the aqueous [bmim][Ac]
solutions at (283.15 to 353.15) °C throughout the whole concentration range are
presented in Figure 8.2. Density of the binary mixture slightly increases upon addition
of water and a maximum is seen in the mixture density at nearly 75 mol% water at all
temperature. After the maximum, the density to decrease sharply upon further addition of
water. This behavior is observed at all temperatures but the maximum get less
pronounced with increase in temperature.
The excess molar volume ) was calculated from the raw data by the following
equation and shown in Figures 8.3. All the excess properties display negative deviation,
(8.1)

317
where ( ) is the molar volume of the liquid mixture, and ( ) and ( ) are the mole
fraction and molar volume of the component ( ) respectively. In terms of density, the
excess molar volume is expressed as,
1 1 (8.2)
where is the density of the liquid mixture, and and are the molecular weight and
density of the component . The values of as a function of addition of water are
shown in Figure 8.3. All excess volumes are negative, that is the actual mixture volume
is less than the linear mole-fraction average of the molar volumes of the pure
components. This indicates stronger attractive interaction between water and [bmim][Ac]
to form a more closely packed liquid structure in the binary system than the liquid
structures of each pure component. The degree of close packing kept increasing (
became more negative) with increase in water content and reached a pronounced
minimum at around 70 mol% water at all temperature. After further addition of water,
the excess volumes started to be less native than the minimum. The temperature-
dependence of ( ) is very weak up to 40 mol% water in the mixture and greatly
influence the degree of volume contraction roughly between 70 mol% water and 90
mol% water.
The excess molar volumes were fitted to the Redlich-Kister equation,
(8.3)

318
where and are the mole fraction of water and ionic liquids respectively and
are the temperature-dependent adjustable parameters that were obtained by minimizing
the standard deviation ( )
, , / (8.4)
where is the number of experimental points and is the number of parameters used in
the regression. The regressed values of the coefficients and along with standard
deviation are given in Table 8.2.
To show the effect of temperature on density, the experimental data were also
correlated as a function of temperature for various compositions using the following
polynomial equation. The regressed parameters are presented in Table 8.3 with standard
deviations. Density decreases with increase in temperature at all compositions (Figure
8.3).
(8.5)
The volume expansivity of pure [bmim][Ac] can be calculated by the following equation
(-0.00057K-1 at 298.15 K and -0.00058K-1 at 313.15 K).
1 (8.6)

319
Table 8.1 Density of {[bmim][Ac]+water}System at (283.15 to 353.15) K
Figure 8.1 Comparison of density of [bmim][Ac] with literature data.4,11-14
Mole Fraction of [bmim][Ac]
283.15 K 293.15 K 298.15 K 303.15 K 313.15 K 323.15 K 333.15 K 343.15 K 353.15 K
0.0000 0.99970 0.99820 0.99704 0.99565 0.99221 0.98803 0.98319 0.97776 0.971790.0533 1.04435 1.03874 1.03583 1.03285 1.02672 1.02031 1.01367 1.00678 0.999970.1003 1.06247 1.05580 1.05241 1.04900 1.04211 1.03508 1.02791 1.02064 1.013200.2008 1.07346 1.06686 1.06352 1.06018 1.05341 1.04655 1.03962 1.03260 1.025500.2997 1.07406 1.06777 1.06458 1.06138 1.05493 1.04841 1.04182 1.03517 1.028460.4004 1.07233 1.06625 1.06316 1.06007 1.05382 1.04753 1.04119 1.03480 1.028370.4983 1.06979 1.06372 1.06072 1.05769 1.05161 1.04548 1.03933 1.03315 1.026940.5968 1.06720 1.06109 1.05813 1.05516 1.04916 1.04314 1.03710 1.03106 1.025000.6942 1.06508 1.05896 1.05590 1.05296 1.04702 1.04105 1.03507 1.02910 1.023130.7997 1.06315 1.05704 1.05399 1.05094 1.04504 1.03910 1.03316 1.02724 1.021340.9070 1.06154 1.05534 1.05228 1.04921 1.04325 1.03731 1.03138 1.02547 1.019581.0000 1.06110 1.05472 1.05165 1.04858 1.04259 1.03664 1.03070 1.02480 1.01889
T/K
1.01
1.02
1.03
1.04
1.05
1.06
1.07
1.08
280 300 320 340 360 380
Den
sity
of p
ure
[bm
im][
Ac]
(g/c
m3 )
T/K
Pinkert et al. (2011)
Tariq et al. (2009)
Bogolitsyn et al.(2009)
Almeida et al. (2012)
Stevanovic et al. (2012)
This work

320
Figure 8.2 Densities of binary mixture of water (1) with [bmim][Ac] (2) as a function of
ionic liquid mole fraction (upper plot) and water mole fraction (lower plot)
at various temperatures : ▲, 293.15 K; ●, 298.15 K; □, 303.15 K; ∆, 313.15
K; ○, 323.15 K; ◊, 333.15 K; +,343.15 K; x, 353.15 K; ..., Redlick-Kister
prediction.
0.96
0.98
1
1.02
1.04
1.06
1.08
0 0.2 0.4 0.6 0.8 1
Den
sity
of
aqu
eou
s [b
mim
][A
c](g
/cm
3 )
Mole fraction of water

321
Figure 8.3 Excess molar volumes of binary mixture of water (1) with [bmim][Ac] (2) as
a function of ionic liquid mole fraction at various temperature : ▲, 293.15
K; ●, 298.15 K; □, 303.15 K; ∆, 313.15 K; ○, 323.15 K; ◊, 333.15 K;
+,343.15 K; x, 353.15 K. ..., Redlick-Kister prediction.
-1.6
-1.35
-1.1
-0.85
-0.6
-0.35
-0.1
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
Exc
ess
mol
ar v
olu
me
(cm
3 /m
ol)
Mole fraction of water
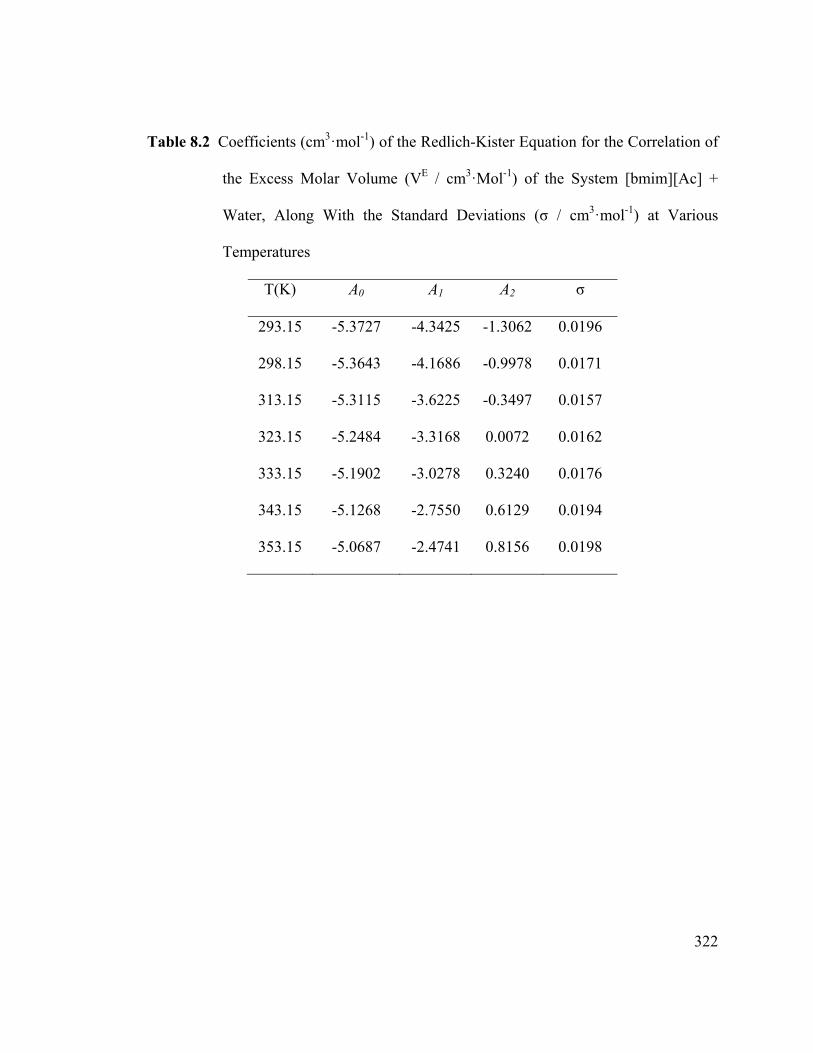
322
Table 8.2 Coefficients (cm3·mol-1) of the Redlich-Kister Equation for the Correlation of
the Excess Molar Volume (VE / cm3·Mol-1) of the System [bmim][Ac] +
Water, Along With the Standard Deviations (σ / cm3·mol-1) at Various
Temperatures
T(K) A0 A1 A2 σ
293.15 -5.3727 -4.3425 -1.3062 0.0196
298.15 -5.3643 -4.1686 -0.9978 0.0171
313.15 -5.3115 -3.6225 -0.3497 0.0157
323.15 -5.2484 -3.3168 0.0072 0.0162
333.15 -5.1902 -3.0278 0.3240 0.0176
343.15 -5.1268 -2.7550 0.6129 0.0194
353.15 -5.0687 -2.4741 0.8156 0.0198
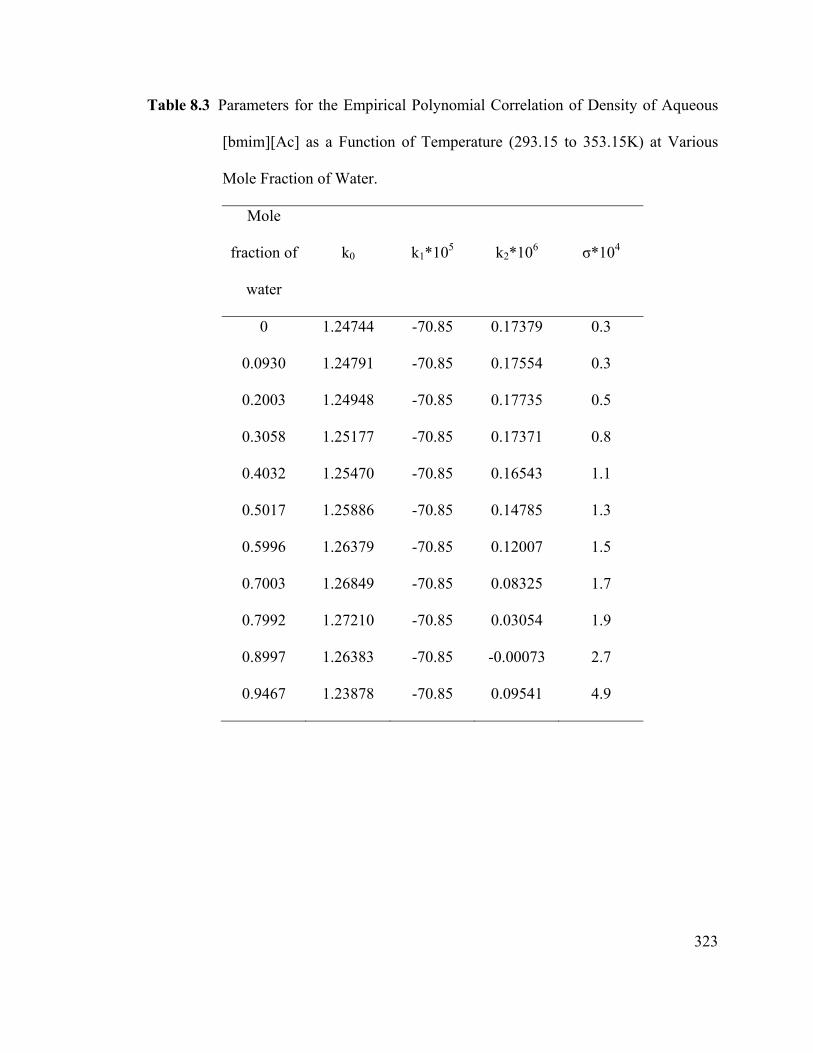
323
Table 8.3 Parameters for the Empirical Polynomial Correlation of Density of Aqueous
[bmim][Ac] as a Function of Temperature (293.15 to 353.15K) at Various
Mole Fraction of Water.
Mole
fraction of
water
k0 k1*105 k2*106 σ*104
0 1.24744 -70.85 0.17379 0.3
0.0930 1.24791 -70.85 0.17554 0.3
0.2003 1.24948 -70.85 0.17735 0.5
0.3058 1.25177 -70.85 0.17371 0.8
0.4032 1.25470 -70.85 0.16543 1.1
0.5017 1.25886 -70.85 0.14785 1.3
0.5996 1.26379 -70.85 0.12007 1.5
0.7003 1.26849 -70.85 0.08325 1.7
0.7992 1.27210 -70.85 0.03054 1.9
0.8997 1.26383 -70.85 -0.00073 2.7
0.9467 1.23878 -70.85 0.09541 4.9

324
Figure 8.4 Effect of temperature on density of binary mixture of water + [bmim][Ac] at
various approximated percent mole fraction of water: ▲, 0%; □, 9%;
+,31%; ○, 50%; ◊, 70%; ∆, 95%; ●, 100%;..., polynomial prediction.
0.96
0.98
1
1.02
1.04
1.06
1.08
290 300 310 320 330 340 350 360
Den
sity
(g/c
m3 )
Temperatrue (K)

325
The experimental data of the viscosities for the {[bmim][Ac]+water} system are
reported in Table 8.4 and the viscosity of pure [bmim][Ac] as a function of temepratrue
is compared with literature data in Figure 8.5. The experimental data of the viscosity for
the binary mixture at different temperatures for the full composition range is presented in
Figure 8.5 as a function of temperature and mole fraction of water. The viscosity values
of pure ionic liquid is high, but decreases quickly with addition of water. This
considerable decrease in viscosity might compensate for the diminished ability of the IL
to dissolve more CO2 in presence of water and at higher temperature. The viscosity
deviation,
∆ (8.7)
were fitted to the Redlich-Kister equation,
∆ (8.8)
where and are the mole fraction of water and ionic liquids respectively and
are the temperature-dependent adjustable parameters that were obtained by minimizing
the standard deviation ( ) . Viscosity deviations from an ideal mixture viscosity are
negative at all composition. At 298.15K, the viscosity deviation becomes more negative
with addition of water, reaches a minimum at around 50 mole% water, and then becomes
less negative. With increase in temperature, the observed viscosity approaches more
towards the ideal mixture viscosity and at 70°C, the mixture viscosity is only slightly
deviated from an ideal mixture viscosity.
The commonly used two-parameter Arrhenius-type equation was tested to
correlate the temperature-dependence of the observed viscosity data

326
ln ln
(8.9)
where, ∞, is the viscosity at infinite temperature, is the universal gas constant (8.314
J.mol-1.K-1), is the activation energy to flow and is temperature. The parameters
were obtained from a linear plot and presented in Table 8.5. The activation energy to
flow for pure [bmim][Ac] was about 51.4 kJ/mol.
The following Vogel-Fulcher-Tammann (VFT) equation
(8.10)
where, , and 0 are fitting parameters and is temperature, widely used to correlate
liquid viscosity data, was more suitable to correlate the experimental viscosity data. The
parameters are presented in Table 8.6. The value of is 188.9 K, comparable to the
glass-transition temperature of the [bmim][Ac] (203.5K). Viscosity decreases
dramatically with rise in temperature at all concentration (Figure 8.8).17

327
Table 8.4 Viscosity of {[bmim][Ac]+water}System at (298.15 to 343.15) K
Figure 8.5 Comparison of viscosity data of pure [bmim][Ac] with literature data.
Experimental data taken from Ref. 11, 14-16.
Mole Fraction of
[bmim][Ac]298.15 303.15 313.15 323.15 333.15 343.15
0.0533 3.7 3.2 2.4 1.8 1.5 1.20.1003 7.9 6.4 4.6 3.4 2.6 2.10.2008 22.9 18.0 11.9 8.3 6.1 4.60.2997 44.7 34.7 22.0 14.8 10.5 7.70.4004 76.3 58.0 35.4 23.2 16.0 11.50.4983 115.5 86.4 51.4 32.7 22.0 15.60.5968 163.8 120.3 69.4 43.1 28.4 19.80.6942 216.1 155.9 87.5 53.2 34.6 23.70.7997 281.9 200.0 109.1 64.8 41.3 27.80.9070 357.2 249.4 132.0 76.7 48.0 31.91.0000 433.7 297.6 154.0 87.5 54.0 35.3
T/K
0
50
100
150
200
250
300
350
300 320 340 360 380
Vis
cosi
ty(m
Pa.
s)
Temperature/K
Crosthwaite et al. (2005)
Pinket et al. (2012)
Xu et al. (2012)
Almeida et al. (2012)
This work

328
Figure 8.6 Viscosity of binary mixture of water (1) with [bmim][Ac] (2) as a function
of water mole fraction at various temperature : ●, 298.15 K; □, 303.15 K; ∆,
313.15 K; ○, 323.15 K; ◊, 333.15 K; +,343.15 K. ..., Redlick-Kister
prediction.
0
50
100
150
200
250
300
350
400
450
0 0.2 0.4 0.6 0.8 1
Vis
cosi
ty (m
Pa.
s)
Mole fraction of water

329
Figure 8.7 Viscosity deviations of binary mixture of water (1) with [bmim][Ac] (2) as a
function of ionic liquid mole fraction at various temperature : ●, 298.15 K;
□, 303.15 K; ∆, 313.15 K; ○, 323.15 K; ◊, 333.15 K; +,343.15 K. ...,
Redlick-Kister correlation.
-110
-100
-90
-80
-70
-60
-50
-40
-30
-20
-10
00 0.2 0.4 0.6 0.8 1
Vis
cosi
ty d
evia
tion
(m
Pa.
s)
Mole fraction of water

330
Table 8.5 Coefficients of the Redlich-Kister Equation for the Correlation of the
Viscosity Deviation (∆η / mPa·s) of the System [bmim][Ac]+Water, and the
Standard Deviations (σ / mPa·s).
T(K) B0 B1 B2 B3 σ
298.15 -402.3368 -20.2604 -16.5342 55.6913 0.42
313.15 -248.4730 -35.7401 -10.3502 34.6251 0.30
323.15 -102.6328 -36.7245 -9.4603 23.0090 0.17
333.15 -44.6907 -23.7759 -6.5823 6.6493 0.21
343.15 -20.4567 -19.8377 -5.6889 8.7746 0.06
353.15 -8.8674 -14.1451 -3.4183 4.3190 0.06
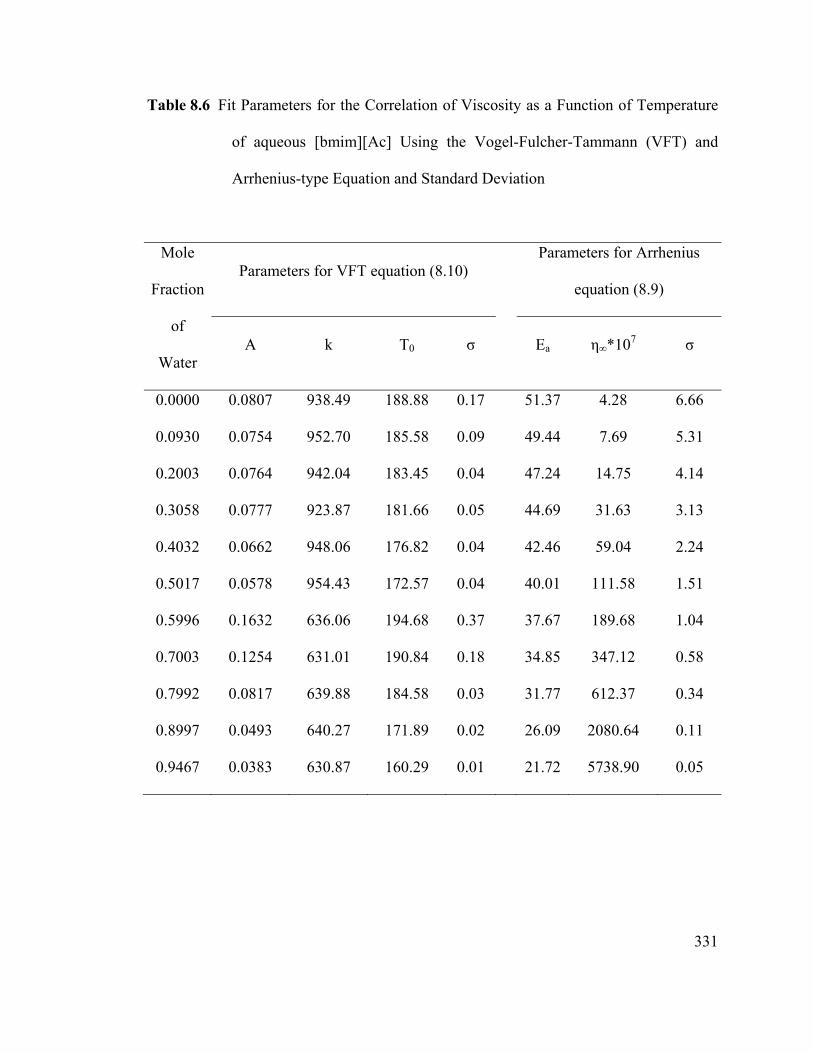
331
Table 8.6 Fit Parameters for the Correlation of Viscosity as a Function of Temperature
of aqueous [bmim][Ac] Using the Vogel-Fulcher-Tammann (VFT) and
Arrhenius-type Equation and Standard Deviation
Mole
Fraction
of
Water
Parameters for VFT equation (8.10)
Parameters for Arrhenius
equation (8.9)
A k T0 σ Ea η∞*107 σ
0.0000 0.0807 938.49 188.88 0.17
51.37 4.28 6.66
0.0930 0.0754 952.70 185.58 0.09
49.44 7.69 5.31
0.2003 0.0764 942.04 183.45 0.04
47.24 14.75 4.14
0.3058 0.0777 923.87 181.66 0.05
44.69 31.63 3.13
0.4032 0.0662 948.06 176.82 0.04
42.46 59.04 2.24
0.5017 0.0578 954.43 172.57 0.04
40.01 111.58 1.51
0.5996 0.1632 636.06 194.68 0.37
37.67 189.68 1.04
0.7003 0.1254 631.01 190.84 0.18
34.85 347.12 0.58
0.7992 0.0817 639.88 184.58 0.03
31.77 612.37 0.34
0.8997 0.0493 640.27 171.89 0.02
26.09 2080.64 0.11
0.9467 0.0383 630.87 160.29 0.01 21.72 5738.90 0.05
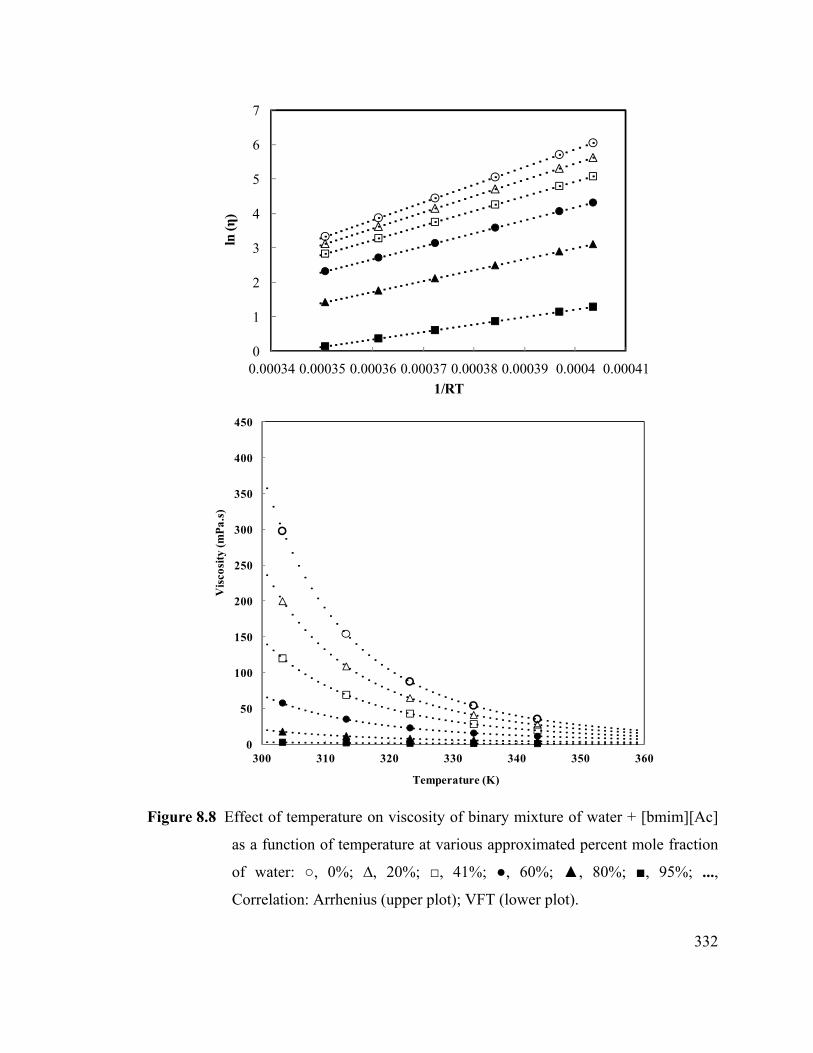
332
Figure 8.8 Effect of temperature on viscosity of binary mixture of water + [bmim][Ac]
as a function of temperature at various approximated percent mole fraction
of water: ○, 0%; ∆, 20%; □, 41%; ●, 60%; ▲, 80%; ■, 95%; ...,
Correlation: Arrhenius (upper plot); VFT (lower plot).
0
1
2
3
4
5
6
7
0.00034 0.00035 0.00036 0.00037 0.00038 0.00039 0.0004 0.00041
ln (η
)
1/RT
0
50
100
150
200
250
300
350
400
450
300 310 320 330 340 350 360
Vis
cosi
ty (m
Pa.
s)
Temperature (K)

333
The molar excess enthalpy of the mixtures {[bmim][Ac]+amine} at 25°C and
60°C are shown in Figure 8.9 and reported in Table 8.7. The system
{water+[bmim][Ac]} is highly exothermic indicating hydrogen bonding between the
ionic liquid and water. The exothermicity decreases only slightly with increase in
temperature.
The excess enthalpy defined as,
(8.11)
were fitted to the Redlich-Kister equation:
∆ (8.12)
where and are the mole fraction of water and ionic liquids respectively and
are the temperature-dependent adjustable parameters that were obtained by minimizing
the standard deviation ( ) and presented in Table 8.8.
The excess enthalpies of the systems {amine+[bmim][Ac]} were found less
exothermic than the system {water+[bmim][Ac]} (Figure 8.10). Based on exothermicity
of the systems at 0.4 mole fraction of [bmim][Ac], the solvents can be ranked as water
(most exothermic)>TEA>MDEA>DEA>AMP>MEA (least exothermic). The system
{MEA+[bmim][Ac]} was found slightly endothermic at MEA-rich region.

334
Table 8.7 Viscosity of {[bmim][Ac]+water}System at (298.15 to 343.15) K
Mole
Fraction of
[bmim][Ac]
Excess
Enthalpy
(J/mol) at
25°C
Mole
Fraction of
[bmim][Ac]
Excess
Enthalpy
(J/mol) at 60°C
0.0294 -1259 0.0301 -1093
0.0496 -2008 0.0505 -1445
0.1008 -3536 0.1034 -3284
0.2022 -4943 0.2023 -4968
0.2984 -5876 0.3033 -5611
0.3977 -6011 0.3982 -5751
0.4947 -5833 0.5042 -5712
0.6057 -4886 0.5962 -4835
0.7092 -3774 0.6997 -4040
0.7906 -2841 0.8000 -2497
0.9067 -1545 0.8935 -1497

335
Table 8.8 Coefficients of the Redlich-Kister Equation for the Correlation of the Excess
Enthalpy (J/mol) of the Systems Aqueous [bmim][Ac], and the Standard
Deviations (σ )
T/K C0 C1 C2 (σ)
298.15 -22443.0 -12124.6 -7163.6 165.40
333.15 -21968.7 -11160.7 -4216.8 175.10
Figure 8.9 Excess molar enthalpy of binary mixture of water with [bmim][Ac] as a
function of ionic liquid mole fraction at various temperatures : ●, 298.15 K;
◊, 333.15 K; ..., Redlick-Kister prediction.
-7000
-6000
-5000
-4000
-3000
-2000
-1000
0
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
Exc
ess
enth
alpy
of a
queo
us [b
mim
][A
c] (J
/mol
)
Mole fraction of water

336
Figure 8.10 Comparison of excess enthalpy of the binary mixture of [bmim][Ac] with
some common alkanolamines and water (solid lines are polynomial fit to
guide the eyes).
-7000
-6000
-5000
-4000
-3000
-2000
-1000
0
10000.0 0.2 0.4 0.6 0.8 1.0
HE
of
{[b
mim
][[A
c]+
amin
e} m
ixtu
res
at 2
98.1
5K
(J/
mol
)
Mole fraction of ionic liquid [bmim][Ac]
MEAAMPDEAMDEATEAwater

337
8.4 CONCLUSIONS
We have measured the density, viscosity and excess molar enthalpy at
atmospheric pressure and at temperatures from (298.15 to 343.15) K. Excess molar
volume and viscosity deviation were calculated from experimental data and correlated
with Redlich-Kister equation. The density data were correlated with a polynomial
function of temperature to discuss the effect of temperature. Excess molar volumes were
derived from experimental density data to discuss the effect of composition and were
correlated with Redlich-Kister equation. The viscosity of the mixture was fitted with
both Arrhenius and Vogel-Fulcher-Tammann equation to discuss the effect of
temperature. Viscosity deviation was derived and correlated with Redlich-Kister
equation. Viscosity of pure [bmim][Ac] decreases significantly with addition of water
and with increase in temperature. All excess properties show strong negative deviation
from ideality. The molar excess enthalpies of binary mixture of [bmim][Ac] with the
following amines {Monoethanolamine (MEA), Diethanolamine (DEA), N-N-
dimethylethanolamine (MDEA), 2-amino-2-methyl-1-propanol (AMP)} were measured
and compared with {water+[bmim][Ac]} system. The solvents added to the IL can be
ordered as MEA(least exothermic) <AMP<DEA<MDEA<H2O(most exothermic) in
terms of ascending order of exothermic molar excess enthalpy at 25°C of equimolar
mixture of these solvents with [bmim][Ac].

338
8.5 REFERENCES
1. Chinn, D.; Vu, D. Q.; Driver, M. S.; Boudreau., L. C. CO2 Removal from Gas
Using Ionic Liquid Absorbents. U.S. Patent 0251558 A1, 2006.
2. Maginn, E. J. Design and Evaluation of Ionic Liquids as Novel CO2 Absorbents,
Quarterly Technical Reports to DOE, 2004−2006.
3. Shiflett, M. B.; Kasprzak, D. J.; Junk, C. P.; Yokozeki, A. Phase Behavior of
{Carbon dioxide + [bmim][Ac]} Mixtures. J. Chem. Thermodyn. 2008, 40, 25.
4. Stevanovic, S.; Podgoršek, A.; Pádua, A. A. H; Gomes, M.F.C. Effect of Water
on the Carbon Dioxide Absorption by 1‑Alkyl-3-methylimidazolium Acetate
Ionic Liquids. J. Phys. Chem. B 2012, 116, 14416.
5. Carvalho, P. J.; Alvarez, V. C. H.; Schroder, B.; Gil, A. M.; Marrucho, I. M.;
Aznar, M. N.; Santos, L. M. N. B. F.; Coutinho, J. A. P. Specific Solvation
Interactions of CO2 on Acetate and Trifluoroacetate Imidazolium Based Ionic
Liquids at High Pressures. J. Phys. Chem. B 2009, 113, 6803.
6. Cabaço, M. I.; Besnard, M.; Danten, Y.; Coutinho, J.A.P.;Carbon Dioxide in 1-
Butyl-3-methylimidazolium Acetate. I. Unusual Solubility Investigated by
Raman Spectroscopy and DFT Calculations. J. Phys. Chem. A 2012, 116, 1605.
7. Gurau, G.; Rodriguez, H.; Kelley, S. P.; Janiczek, P.; Kalb, R. S.; Rogers, R. D.
Demonstration of Chemisorption of Carbon Dioxide in 1,3-Dialkylimidazolium
Acetate Ionic Liquids. Angew. Chem., Int. Ed. 2011, 123, 12230.

339
8. Besnard, M.; Cabaço, M. I.; Chavez, F. V.; Pinaud, N.; Sebastiao, P. J.;
Coutinho, J. A. P.; Danten, Y. On the Spontaneous Carboxylation of 1-butyl-3-
methylimidazolium Acetate by Carbon Dioxide. Chem. Commun. 2012, 48,
1245.
9. Shiflett, M. B.; Drew, D. W.; Cantini, R. A.; Yokozeki, A. Carbon Dioxide
Capture Using Ionic Liquid 1-Butyl-3-methylimidazolium Acetate. Energy Fuels
2010, 24, 5781.
10. Rayer, A. V. Screening of solvents for CO2: Kinetics, Solubility and Calorimetric
Studies, PhD thesis, University of Regina, 2012.
11. Pinkert, A.; Ang, K. L.; Marsh, K.N.; Pang, S. Density, Viscosity and Electrical
Conductivity of Protic Alkanolammonium Ionic Liquids. Phys. Chem. Chem.
Phys., 2011, 13, 5136.
12. Tariq, M.; Forte, P.A.S.; Gomes, M.F.C.; Lopes, J.N.C.; Rebelo, L.P.N. Densities
and Refractive Indices of Imidazolium- and Phosphonium-based Ionic Liquids:
Effect of temperature, alkyl chain length, and anion. J. Chem. Thermodynamics
2009, 41, 790.
13. Bogolitsyn, K. G.; Skrebets, T. E.; Makhova, T. A. Physicochemical Properties
of 1-butyl-3-methylimidazolium Acetate. Russ. J. Gen. Chem. 2009, 79, 125.
14. Almeida, H. F. D. ; Passos, H.; Lopes-da-Silva, J. A. ; Fernandes, A. M. ; Freire,
M. G.; Coutinho, J. A. P. Thermophysical Properties of Five Acetate-Based
Ionic Liquids. J. Chem. Eng. Data 2012, 57, 3005.
15. Crosthwaite, J.M.; Muldoon, M.J.; Dixon, J.K.; Anderson, J.L., Brennecke, J.F.;
Phase Transition and Decomposition Temperatures, Heat Capacities and

340
Viscosities Of Pyridinium Ionic Liquids. J. Chem. Thermodynamics 2005, 37,
559.
16. Xu, A.; Zhang, Y.; Li, Z.; Wang, J. Viscosities and Conductivities of 1-Butyl-3-
methylimidazolium Carboxylates Ionic Liquids at Different Temperatures. J.
Chem. Eng. Data, 2012, 57 , 3102.
17. Strechan, A.A. ; Paulechka, Y.U. ; Blokhin, A.V. ; Kabo, G.J. Low-Temperature
Heat Capacity of Hydrophilic Ionic Liquids [BMIM][CF3COO] and
[BMIM][CH3COO] and A Correlation Scheme for Estimation of Heat Capacity
of Ionic Liquids. J. Chem. Thermodynamics 2008, 40, 632.
18. Maham, Y.; Mather, A.E.; Hepler, L. G. Excess Molar Enthalpies of
(Water+Alkanolamine) Systems and Some Thermodynamic Calculations. J.
Chem. Eng. Data, 1997, 42 , 988.

341
Chapter 9: Conclusions, Recommendations, and Future Work
_______________________________________________________________________
Several important aspects related to the understanding of CO2-solvent chemistry
and development of novel solvents were addressed. We optimized a continuum-plus-
correction strategy (SHE method) for predicting pKa of amines relevant to industrial CO2
capture, as well as updated the parameter values in the pencil-and-paper group-additivity
method of Perrin, Dempsey, and Serjeant (PDS). The PDS method outperformed the
continuum-based method: root-mean-square errors for a sample of 32 amines are 0.28
for the continuum-based method, 0.33 for the original PDS method, and 0.18 for the
updated PDS method. Considering also that there is ambiguity in choice of cavity radii
and molecular conformer for continuum-based methods, we recommend the pencil-and-
paper PDS method over such methods.
Static calculations with PCM continuum model to determine dominant reaction
intermediates underscored the need for inclusion of explicit water molecules for realistic
modeling of the reaction pathways. Our DFT calculations with explicit water molecules
revealed, for the reaction involving one MEA molecule, that the CO2+MEA+nH2O
reaction proceeds as initial complex (IC)zwitterioncarbamate(carbamic acid).
This was only seen when n, the number of explicit water molecules, was increased;
instances of ICzwitterioncarbamic acid or ICcarbamic acid resulted with fewer
water molecules, and such results have been presented in the literature for years. Our
modeling is the first to correctly predict carbamate ions as the dominant product species.

342
The carbamate anion becomes thermodynamically competitive at neutral-pH and
dominant at basic-pH conditions, compared to both zwitterion and carbamic acid
intermediates, when properly solvated in the modeling.
Models involving two MEA molecules were deemed most relevant to modern-
day concentrated alkanolamine solutions. Such termolecular models, tried by others,
were improved incorporating further explicit water molecules. A tetramolecular route
(Shim 2008) featuring amine to amine proton transfer via water relay was also studied.
Gradual incorporation of more water molecules shifted the zwitterion-deprotonation
transition state from “late” to “early,” and in a 2-amine-18-water model, the predicted
barrier is effectively non-existent (0.2 kcal/mol), suggesting that one could consider the
zwitterion as a species so short-lived that the Termolecular mechanism would be
dominant in concentrated alkanolamine solutions. However, in dilute solutions, when an
amine is fully solvated by water molecules, single-MEA modeling showed that
zwitterion deprotonation will occur via a relay mechanism having small activation
energy, making the Zwitterion mechanism more relevant.
From the study with MEA we believe that the mechanism in aqueous
alkanolamine solution is heavily dependent on reaction environment of amine. For other
amines the effect of pKa on formation of zwitterion and bicarbonate was studied with
simple 6-atom cycles in neutral-pH modeling. It was discovered that secondary amines
have inherently greater CO2 affinity than primary amines when comparing amines of
same H+ affinity (pKa). Activation energies vary with pKa in a sufficient manner that this
effect could very well impinge on the importance (or non-importance) of a possible
zwitterion intermediate, and thus affect mechanism.

343
Ab initio molecular dynamic simulations of aqueous CO2, H2OCO2 zwitterion,
HCO3- (with faraway H3O
+), and H2CO3 revealed that the bicarbonate zwitterion seems
to have no stability at all, suggesting that a “wider” barrier exists between an encounter
complex and the bicarbonate/carbonic acid equilibrium, and secondly that the HCO3-
/H2CO3 equilibration is too slow for us to conclude which one is dominant.
AIMD simulations of Me2NHCO2 zwitterion, Me2NCO2- (with far away H3O
+),
and Me2NCOOH revealed that Me2NHCO2 zwitterion is more stable than the H2OCO2
zwitterion and thus deserves consideration as an intermediate, and is separated from the
anion/acid equilibrium pair by some sort of barrier. The Me2NCO2-/Me2NCOOH
equilibration is too slow for us to conclude which one is dominant. The anion and acid
interconverted on a very short timescale, suggesting that anion/acid equilibria are fairly
barrierless, and hence thermodynamics will determine the ratio of anion to acid.
Simulations of various carbamate-zwitterions revealed forward conversion of
zwitterions in 3 of the 6 cases: Me2NH-zwitterion, MeNH2-zwitterion and MEA-
zwitterion. Of these, only MEA-zwitterion failed to show carbamate/carbamic acid
equilibrium. Reaction of AMP-zwitterion, DEA-zwitterion and PPZ-zwitterion was not
observed in 110 ps, 16 ps, 15 ps long runs, respectively. However, a common role of
solvent was identified in all six simulations: solvent molecules reoriented to bridge the
two polar ends of a OCNH segment of zwitterions, forming a “hydrogen-bonded” cycle
(if N water molecules participate, the cycle has 4+2N atoms, and 2+N covalent bonds
before any reaction takes place). These cycles likely occur in reality. We highlight that
the H-shuttling cycles seen in these simulations do not exist in aqueous amine solution

344
prior to absorption of CO2. The number of water molecules involved in such cycles
changes over course of time.
Simulations of zwitterion in presence of nearby amine showed H2O-mediated H+
transfer relays to form carbamate and protonated amine, products which corroborate with
experimental observation. An amine-to-amine proton transfer was observed in 0.5 ps in
a simulation where the neutral amine molecule was placed close to the zwitterion (the
NH bond of zwitterion pointed to the lone pair of neutral amine).
The observation of 10- to14-atom H+-shuttling cycles for formation of carbamic
acid (via carbamate from zwitterion) justifies our study of such multiple-water-mediated
pathways in our static calculations (Chapter 4). Indeed, such pentamolecular and
hexamolecular pathways have never been postulated, and it is hoped that these new paths
and the results of Chapter 4 will significantly advance the efforts to finally solve this
mechanism.
The statistical thermodynamic method COSMO-RS was used to predict Henry’s
law constant of CO2 in a database of 2701 ionic liquids virtually formed from the
combination of 73 cations with 37 anions of different chemical structure at 25 °C.
Trends in solubility of the gases and selectivity in the separation of CO2 from CO2/CH4
and CO2/N2 mixture due to systematic variation in the structure and property of ionic
liquids were analyzed. The residual chemical potential of the ions at infinite dilution in
water was introduced as a qualitative polarity descriptor of ions and ionic liquids.
Solubility of CO2 is found to decrease with decrease in the polarity of ionic liquids.
Henry’s law constants were dissected into components to probe gas liquid interactions
and compare the solubility of a gas in different ionic liquids. Based on the

345
computational study, the ionic liquids 1-alkyl-3-methyl imidazolium
tris(pentafluoroethyl)-trifluorophosphate ([Cnmim][eFAP]) where n = 2,4,6), were
chosen for further experimental measurement of solubility of CO2 using a gravimetric
microbalance at temperatures (10, 25 and 50)°C in the pressure range upto 2 MPa. The
Henry’s law constant derived from experimental data compared well with those
predicted by COSMO-RS.
We measured the density, viscosity and excess molar enthalpy of the binary
system {[bmim][Ac]+water} at atmospheric pressure and at temperatures from (298.15
to 343.15) K. The density data were correlated with a polynomial function of
temperature to discuss the effect of temperature. Excess molar volumes were derived
from experimental density data to discuss the effect of composition and were correlated
with Redlich-Kister equation. The viscosity of the mixture was fitted with both
Arrhenius and Vogel-Fulcher-Tammann equations to discuss the effect of temperature.
Viscosity deviation was derived and correlated with Redlich-Kister equation. Viscosity
of pure [bmim][Ac] decreases significantly with addition of water and with increase in
temperature. All the excess properties show strong negative deviation from ideality. The
molar excess enthalpies of binary mixture of [bmim][Ac] with the following amines
{Monoethanolamine (MEA), Diethanolamine (DEA), N-N-dimethylethanolamine
(MDEA), 2-amino-2-methyl-1-propanol (AMP)} were measured and compared with
{water+[bmim][Ac]} system. The solvents added to the IL can be ordered as MEA(least
exothermic) <AMP<DEA<MDEA<H2O(most exothermic) in terms of ascending order
of exothermic molar excess enthalpy at 25°C of equimolar mixture of these solvents with
[bmim][Ac].

346
Future work arising from the amine studies here would be in developing a master
rate law for CO2 absorption. Further study of aqueous amine solutions are needed to find
the percent of amine molecules in amine-amine H-bonded complexes, for this likely to
be a decisive factor in merging the termolecular and zwitterion mechanisms into a master
rate law.
Future work arising from the ionic liquid studies here could be in using COSMO-
RS to screen other ionic liquids with environmentally friendly functional groups, and
developing molecular dynamics simulations to model ionic liquids and predict their
viscosity and/or solubility.

347
Appendix A. Experimental Determination of pKa
Theory. The potentiometric titration method for pKa measurement, developed by
Albert and Serjeant was followed. The pertinent reactions in an aqueous amine solution
are amine-protonation (eq A.1) and dissociation of water (eq A.2)
(A.1)
(A.2)
The acid dissociation constant is expressed in terms of activities of the species that are
related with their concentration through activity coefficient.
(A.3)
where, , , represent the activity, concentration and activity coefficient of base
at equilibrium. Using definition of pKa and pH as negative common logarithm of and
, respectively, the following relationship is obtained that will be used to determine
pKa from measured values of pH.
(A.4)
For dilute aqueous solution of a base, Equation (A.4) is used for pKa determination.
Apparatus. A pH meter, model 270 Denver Instrument, was employed to
determine the pH values of the aqueous solutions. Three buffer solutions with an
accuracy of (+0.01) for pH 4.00 and 7.00 and (+ 0.02) for pH 10.00 and Hydrochloric
acid solution (HCl) 0.1M (+0.002) were supplied by VWR International. Nitrogen gas
having a high purity (> 99.99%) was purchased from PRAXAIR. The chemicals in

348
Table 3.3 (Chapter 3) were purchased from Sigma-Aldrich and were used without further
purification.
Experimental. Dilute aqueous solutions of amines at 0.01M (+ 0.005M) were
prepared using deionized double distilled water in a 100 ml conical flask with a magnetic
stirrer. A jacket beaker connected to a external water bath was used for acid-base
titration. The pH meter electrode was calibrated at the desired temperature using the
buffer solutions. 50 mL amine solution was then titrated in 10-steps with 5 ml of the
titrant, 0.1 M aqueous solution of hydrochloric acid. An amount of 0.5 ml of titrant was
added in each step from a burette. The pH value was recorded as soon the equilibrium
reached after the addition of the titrant. A slow stream of nitrogen was used to blanket
the solution from atmospheric carbon-dioxide.
Determination of values. An amine solution of known concentration and
volume is titrated with a dilute hydrochloric acid solution of known concentration and
volume and equation (A. 4) is used to determine pKa. The pH of the solution is
measured using a pH meter as described above, the concentration of and are
obtained from standard treatment of chemical equilibria; that is, simultaneous solution of
equations representing charge-neutrality of solution (eq A. 5); mole balances (equations
A.6 and A.7) and extended Debye-Huckel equation (eq A.8).
Charge Balance:
(A.5)
Mole Balance
(A.6)

349
(A.7)
Activity coefficients
The activity coefficient of molecular species is assumed to be unity. The reference state
for ions are hypothetical 1M aqueous solution at standard where the ions behave as they
would do in a real solution but extrapolated to infinitely dilute solution. For ionic
species,
√
1 √ (A.8)
where, the ionic strength,
0.5 (A.9)
and the constants A and B are temperature dependent Debye-Huckel parameters, and at
25°C, assumed to be A=0.5092 mol-1/2L1/2, B=3.29E-09 mol-1/2L1/2 cm-1/2; is the
diameter of an ion and the average ion diameter is taken to be 4.50E-08 cm. An iterative
scheme is used to calculate the activity coefficient since the concentration of hydroxide
ion is unknown. First it is assumed that and activity of hydroxide ion
can be calculated from known values of pH and . Initial guess for ionic strength is
then calculated using equations (A.9) in conjunction with (A.6) and (A.7). from known
values of pH and (dissiciation constant for pure water, equation A.2). This initial
guess is used to calculate the activity coefficient of hydroxyl ion, and then to calculate
new / which is then used to calculate the ionic strength again, this
process is repeated until the difference in ionic strength is less than 0.001. The pKa
values MEA and MDEA were determined to be to be 9.49 and 8.55 with a maximum
uncertainty of ±0.1. The pKa of other chemicals in Table 3.3 were then determined.


















