
Psicologia sviluppo psicomotorio, ritardo mentale, epilessia
61
UNIVERSITÀ DEGLI STUDI - AZIENDA USL - CHIETI - www.unich.it/cliped/ Department of Women and Children’s Health (Head: Prof. Francesco Chiarelli) Sviluppo Psicomotorio, Ritardo Mentale, Convulsioni Febbrili ed Epilessia Dr Cosimo Giannini Clinica Pediatrica, Università di Chieti
-
Upload
iva-zigghyova-martini -
Category
Internet
-
view
390 -
download
6
Transcript of Psicologia sviluppo psicomotorio, ritardo mentale, epilessia

UNIVERSITÀ DEGLI STUDI - AZIENDA USL- CHIETI -
www.unich.it/cliped/
Department of Women and Children’s Health(Head: Prof. Francesco Chiarelli)
Sviluppo Psicomotorio, Ritardo Mentale, Convulsioni
Febbrilied EpilessiaDr Cosimo Giannini
Clinica Pediatrica, Università di Chieti

Sommario

Esame neurologico
L’EO neurologico pediatrico si distingue per
una serie di difficoltà legate a due peculiarità:
•Le variazioni dello sviluppo psicomotorio,
specie nel I anno di vita.
•La scarsa collaborazione del bambino.

Sviluppo psicomotorioLo sviluppo psicomotorio fa riferimento alle variazioni delle capacità
cognitive, comportamentali, motorie e sociali di un bambino a partire dal periodo fetale fino a quello adolescenziale.
Viene suddiviso in:
Motilità grossolana (riflessi arcaici -> postura -> deambulazione -> corsa e salto )
Motilità fine (prensione e manipolazione)
Funzioni cognitive superiori (linguaggio, ragionamento, memoria e apprendimento);
Funzioni di interazione sociale ed affettività (sorriso, capacità d’attaccamento, gioco di gruppo, senso del sé).
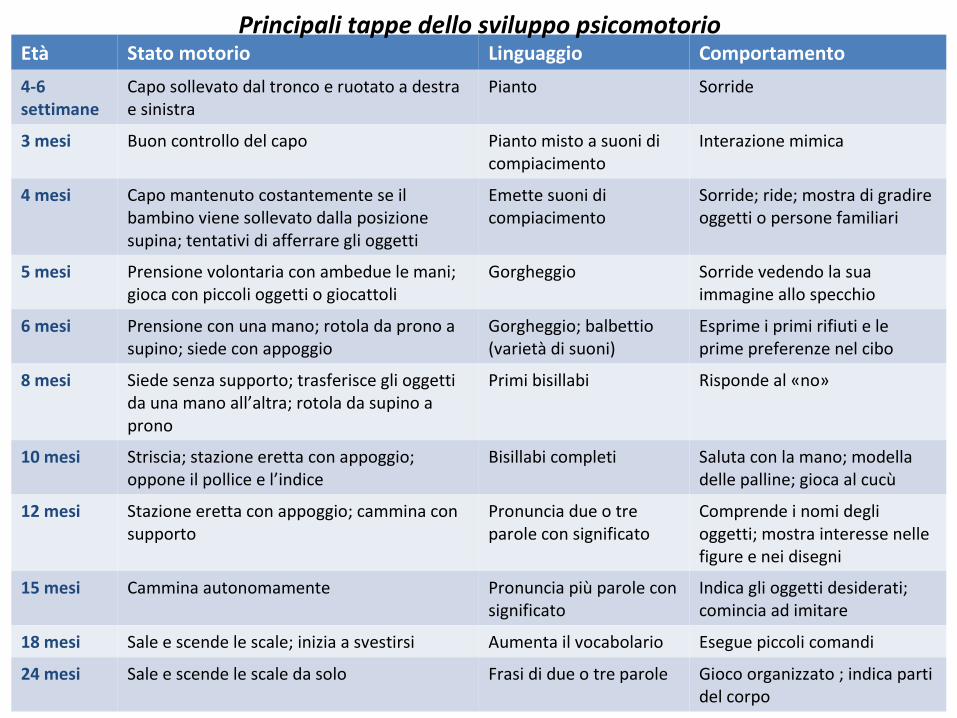
Età Stato motorio Linguaggio Comportamento
4-6 settimane
Capo sollevato dal tronco e ruotato a destra e sinistra
Pianto Sorride
3 mesi Buon controllo del capo Pianto misto a suoni di compiacimento
Interazione mimica
4 mesi Capo mantenuto costantemente se il bambino viene sollevato dalla posizione supina; tentativi di afferrare gli oggetti
Emette suoni di compiacimento
Sorride; ride; mostra di gradire oggetti o persone familiari
5 mesi Prensione volontaria con ambedue le mani; gioca con piccoli oggetti o giocattoli
Gorgheggio Sorride vedendo la sua immagine allo specchio
6 mesi Prensione con una mano; rotola da prono a supino; siede con appoggio
Gorgheggio; balbettio (varietà di suoni)
Esprime i primi rifiuti e le prime preferenze nel cibo
8 mesi Siede senza supporto; trasferisce gli oggetti da una mano all’altra; rotola da supino a prono
Primi bisillabi Risponde al «no»
10 mesi Striscia; stazione eretta con appoggio; oppone il pollice e l’indice
Bisillabi completi Saluta con la mano; modella delle palline; gioca al cucù
12 mesi Stazione eretta con appoggio; cammina con supporto
Pronuncia due o tre parole con significato
Comprende i nomi degli oggetti; mostra interesse nelle figure e nei disegni
15 mesi Cammina autonomamente Pronuncia più parole con significato
Indica gli oggetti desiderati; comincia ad imitare
18 mesi Sale e scende le scale; inizia a svestirsi Aumenta il vocabolario Esegue piccoli comandi
24 mesi Sale e scende le scale da solo Frasi di due o tre parole Gioco organizzato ; indica parti del corpo
Principali tappe dello sviluppo psicomotorio
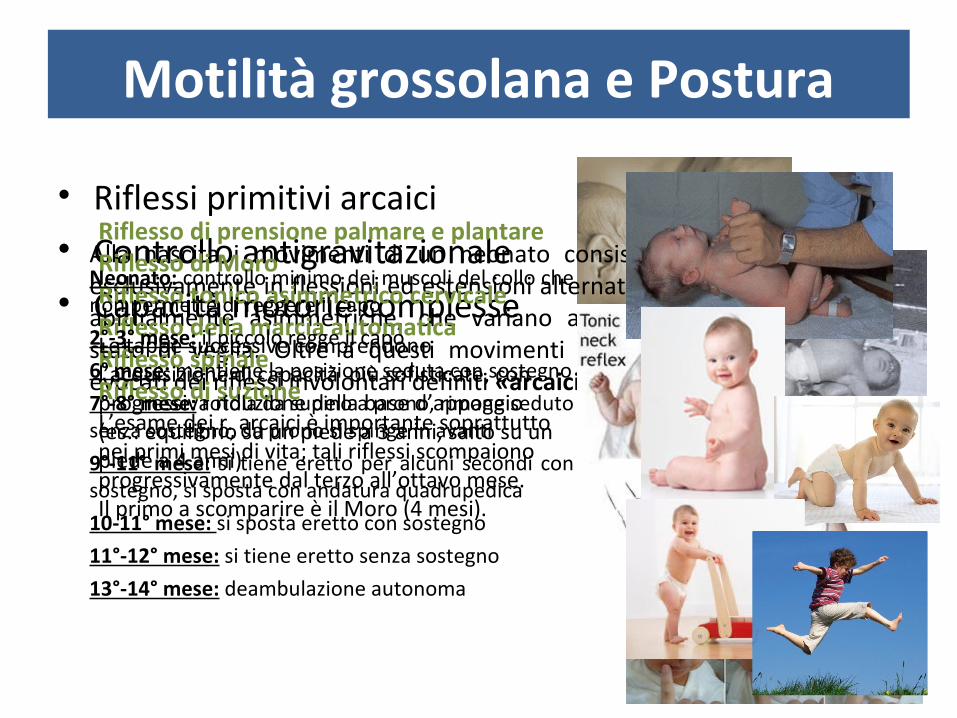
Motilità grossolana e Postura
• Riflessi primitivi arcaici• Controllo antigravitazionale• Capacità motorie complesse
Alla nascita i movimenti di un neonato consistono quasi esclusivamente in flessioni ed estensioni alternate dei 4 arti, abitualmente asimmetriche, che variano a seconda dello stato di veglia. Oltre a questi movimenti possono essere evocati dei riflessi involontari definiti «arcaici».
Riflesso di prensione palmare e plantareRiflesso di MoroRiflesso tonico asimmetrico cervicaleRiflesso della marcia automaticaRiflesso spinaleRiflesso di suzioneL’esame dei r. arcaici è importante soprattutto nei primi mesi di vita; tali riflessi scompaiono progressivamente dal terzo all’ottavo mese. Il primo a scomparire è il Moro (4 mesi).
Neonato: controllo minimo dei muscoli del collo che non permette di reggere il capo
2°-3° mese: il piccolo regge il capo
6° mese: mantiene la posizione seduta con sostegno
7°-8° mese: rotola da supino a prono, rimane seduto senza sostegno, da prono si spinge in avanti
9°-11° mese: si tiene eretto per alcuni secondi con sostegno, si sposta con andatura quadrupedica
10-11° mese: si sposta eretto con sostegno
11°-12° mese: si tiene eretto senza sostegno
13°-14° mese: deambulazione autonoma
Le tappe successive comprendono l’acquisizione di capacità più sofisticate, con progressiva riduzione della base d’appoggio (es.: equilibrio su un piede a 3 anni, salto su un piede a 4 anni)

Reazioni di equilibrio
Nell’evoluzione posturo–motoria verso la capacità di mantenere la posizione eretta, oltre le modificazioni del tono, gioca un ruolo fondamentale lo
sviluppo dell’equilibrio.
Può accadere che il mancato raggiungimento di una tappa posturale sia dovuto non ad anomalie del tono, ma a turbe della funzione dell’equilibrio.
È importante valutare sempre le reazioni di paracadute
Riflesso del paracadute anteriore compare fin dal 4 mese Riflesso del paracadute laterale compare intorno al 6 mese Riflesso del paracadute posteriore compare intorno al 9 mese

Motilità fine
• Prensione La motilità fine si sviluppa in senso prossimo-distale, parallelamente allo sviluppo
delle capacità sensitive.
3° mese: il piccolo tiene un oggetto con una mano senza saperlo prendere o
lasciare volontariamente
4°-5° mese: prensione a “rastrello”
5°-7° mese: presa palmare
7°-8° mese: presa con tutta la mano
9° mese: presa a pinzetta
10°-12° mese: prensione fine acquisita
• Capacità motorie fini complesse

Funzioni cognitive
Le capacità sensitive innate (vista, udito, ecc.) permettono al neonato di costruire le prime basi dello sviluppo cognitivo.VISTA -> acuità visiva del neonato è di circa 20-40/200; fissa il volto di un esaminatore a 30 cm.
UDITO -> già alla nascita il bambino risponde ai suoni voltando il capo verso la fonte del suono.
•Primi 2 anni -> sviluppo dell’intelligenza di tipo sensitivo- motorio:-sviluppa il concetto di permanenza di un oggetto;-apprende che un oggetto esiste anche quando non è direttamente visualizzato ; -comprende la relazione tra causa ed effetto.
•2-3 anni -> sviluppo delle capacità simboliche•Età prescolare e scolare -> sviluppo del pensiero logico

Linguaggio
“Capacità di generare suoni riproducibili o gesti riconosciuti da altri come rappresentativi di concetti”
Neonato: riconosce determinate voci2-3 mesi: localizza la fonte di un suono, è capace di emettere suoni musicali o lunghi6 mesi: vocalizza emettendo sillabe composte da consonanti e vocali (ma-ma, pa-pa), risponde se chiamato col proprio nome9-12 mesi: segue comandi verbali di routine, emette dapprima bisillabi completi, poi pronuncai le prime paroline18 mesi: aumenta il vocabolario (20-50 parole), esegue comandi sempre più comlplessi24 mesi: prime frasi di 2-3 parole, inizia a distinguere tra “tu” e “me”, inizia a comporre giudizi negativi36 mesi: usa pronomi, preposizioni e plurali, capisce il passato e il futuro

Il concetto di ritardo mentale non è applicabile ai primi 3 anni di vita per i quali si parla di ritardo psicomotorio.
In questa fascia di età si valuta, con appositi test, il quoziente di sviluppo (QS) che privilegia le funzioni motorie e non è predittivo del successivo QI.
Test utilizzati:•Test d’esame dello sviluppo di Denver• Scale di Bayley• Scale di Brunet-Lezine
Ritardo psicomotorio e Ritardo mentale

Indagini
Test di Denver
Test di Denver: un utile strumento di screening per identificare i pazienti con sospette anomalie dello sviluppo da sottoporre a indagini più approfondite.
Altri sistemi di
valutazione prevedono
l’utilizzo di scale come
quelle di Brunet-Lézine,
Bayley e Griffiths ed
altri tests che valutano
i singoli campi cognitivi
(es. Early Language
Milestone Scale 2).

Deficit significativo in due o più delle seguenti aree di sviluppo:
• motricità fine/grossolana• motricità grossolana• linguaggio• capacità sociali/interpersonali• attività della vita quotidiana
American Academy of Neurology
Ritardo Mentale

Caratteristiche diagnostiche (DSM-IV):• funzionamento intellettivo significativamente inferiore alla norma.
• concomitanti deficit o compromissioni nel funzionamento adattivo attuale, (cioè la capacità del soggetto di adeguarsi agli standard propri della sua età e del suo ambiente culturale), in almeno due delle seguenti aree:
“comunicazione, cura della propria persona, vita in famiglia, capacità sociali/interpersonali, uso delle risorse della comunità,
autodeterminazione, capacità di funzionamento scolastico, lavoro, tempo libero, salute e sicurezza”
• età di esordio prima dei 18 anni.
Ritardo Mentale

Il funzionamento intellettivo generale è definito dal quoziente di intelligenza (QI) ottenuto tramite la
valutazione con uno o più test psicometrici standardizzati somministrati individualmente.
I test più utilizzati sono:
• Lo Standford-Binet;• Le scale di Wechsler che comprendono:
- WISPP (Wechsler Intelligence Scale for the Preschool Period) applicabile dai 4 ai 6 anni.
- WISC (Wechsler Intelligence Scale for Children) per bambini e ragazzi dai 5 ai 16 anni e mezzo.
Metodi di Valutazione

Il QI esprime come le capacità intellettive di un soggetto si collochino rispetto ai propri coetanei in un continuum dove la prestazione media cade sul punteggio 100.
Rappresenta una misura della deviazione della prestazione individuale rispetto alla media del gruppo d’età di appartenenza del soggetto.
Un funzionamento intellettivo significativamente al di sotto della media è definito da un QI di circa 70 o inferiore (circa 2 DS al di sotto della media).
Quoziente Intellettivo

Possono essere specificati 4 gradi di gravità del ritardo mentale, che riflettono il livello della compromissione intellettiva:
• Ritardo Mentale Lieve (livello del QI da 50-55 a circa 70).• Ritardo Mentale Moderato (livello del QI da 35-40 a 50-55)• Ritardo Mentale Grave (livello del QI da 20-25 a 35-40).• Ritardo Mentale Gravissimo (livello del QI sotto 20 o 25)
Livelli di Ritardo Mentale

• Comprende circa un 1-2% dei soggetti con ritardo mentale.
• Durante la prima infanzia, questi pazienti mostrano considerevole compromissione del funzionamento sensomotorio.
• L’autonomia delle condotte della vita quotidiana (alimentazione, pulizia, controllo sfinterico) è parziale. Il linguaggio è limitato a pochi fonemi o inesistente.
• I pazienti restano dipendenti dagli adulti.
Ritardo Mentale Gravissimo

• Comprende il 3-4% dei soggetti con ritardo mentale.
• Durante la prima fanciullezza questi individui acquisiscono un livello minimo di linguaggio comunicativo, o non lo acquisiscono affatto.
• Durante il periodo scolastico possono imparare a parlare e possono essere addestrati alle attività elementari di cura della propria persona.
• Il bambino necessita comunque di un ambiente protettivo e nel contempo stimolante.
Ritardo Mentale Grave

• Comprende circa il 10% dei soggetti con ritardo mentale. • La maggior parte di questi soggetti acquisisce capacità
comunicative durante la prima fanciullezza. • Possono provvedere alla cura della propria persona con una
moderata supervisione e possono imparare a spostarsi da soli in luoghi familiari.
• Possono beneficiare dell’addestramento alle attività sociali e lavorative, ma difficilmente progrediscono oltre il livello della seconda elementare nelle materie scolastiche.
• Durante l’adolescenza, le loro difficoltà nel riconoscere le convenzioni sociali possono interferire nelle relazioni con i coetanei.
Ritardo Mentale Moderato

• Comprende la parte più ampia (circa l’85%) dei soggetti con ritardo mentale.
• Questi bambini sembrano avere avuto uno sviluppo psicomotorio sostanzialmente normale prima della scolarizzazione e il loro comportamento in famiglia e con i coetanei è soddisfacente. Il linguaggio non presenta anomalie grossolane.
• Le difficoltà riscontrate nelle abituali prestazioni scolastiche rivelano la mancata acquisizione del pensiero formale.
• Prima dei 20 anni, possono acquisire capacità scolastiche corrispondenti all’incirca alla quinta elementare.
Ritardo Mentale Lieve

• Il ritardo mentale lieve è tanto più frequente quanto più basse sono le condizioni della vita socio-economica e la stimolazione dell’ambiente familiare.
• La deprivazione ambientale da sola non è però sufficiente a spiegare il ritardo mentale.
• Ogni bambino reagisce all’ambiente in modo diversificato in base alle proprie capacità biologiche.
Ritardo Mentale Lieve

PRENATALI: infezioni (Rosolia, CMV, Toxo) malattie materne non infettive. droghe e alcool irradiazione materno-fetale malnutrizione.
PERINATALI: anossia traumi Infezioni turbe metaboliche prematurità
POSTNATALI: Infezioni Traumi Turbe metaboliche Intossicazioni (piombo) Carenze ormonali (ipotiroidismo) Encefalopatie epilettiche Epilessie farmaco-resistenti
Eziologia Ritardo Mentale:Fattori acquisiti

ABERRAZIONI
CROMOSOMICHE: Anomalie degli autosomi (s. di Down,
s. di Williams, …….) Anomalie dei cromosomi sessuali (s.
di Klinefelter, s. di Turner, s. dell’X fragile)
TRASMISSIONE
DOMINANTE: Sclerosi tuberosa Neurofibromatosi……
TRASMISSIONE
RECESSIVA: Fenilchetonuria Galattosemia Malattie lisosomiali Malattia di Tay-Sachs ………
TRASMISSIONE
POLIGENICA: Malformazioni cerebrali varie
Eziologia Ritardo Mentale:Fattori acquisiti

• Disturbi generalizzati dello sviluppo;
• Disturbo da deficit di attenzione ed iperattività;
• Disturbi della condotta;
• Disturbi dell’umore.
Ritardo mentale: COMORBIDITÀ

OBIETTIVI PER L’ASSISTENZA ALBAMBINO CON RITARDO MENTALE
1. Intraprendere terapia mirata e tempestiva
2. Sviluppare l’approccio multidisciplinare al bambino con handicap
3. Favorire l’inserimento sociale del bambino con handicap

Sommario
Convulsioni Febbrili - Definizione
Convulsioni Febbrili: episodi critici convulsivi, o non, che si manifestano in età compresa fra 6 mesi e 6 anni in bambini che presentano rialzo febbrile oltre i 38°C (o brusco decremento di TC) in assenza di segni di precedenti neurologici e
in assenza di precedenti convulsioni afebbrili.

Convulsioni Febbrili - Patogenesi
Ruolo del rapido rialzo (o calo) febbrile
Immaturità del SNC
Genetica (non sono stati mai identificati loci specifici né pattern di ereditarietà, probabilmente di tipo multifattoriale; gene IMPA2 in uno studio su 59 nuclei familiari giapponesi)

Convulsioni Febbrili Manifestazioni cliniche
• Semplici (basso rischio): crisi singole generalizzate che si presentano durante un episodio febbrile, di durata non superiore a 15* minuti, non ripetuta nelle 24 ore, non dovuta ad un’affezione acuta del SNC, in un bambino senza precedenti neurologici.
• Complesse (alto rischio): crisi subentranti nelle 24 ore e/o di durata superiore a 15 minuti*, associate a segni focali o seguite da anomalie neurologiche transitorie.
• Stato di male febbrile: convulsione febbrile >30 min o crisi seriate subentranti, più brevi, senza ripristino della coscienza.

Convulsioni FebbriliCriteri per il ricovero
- Primo episodio di CF semplice: pz< 18 mesi o clinicamente instabile o pz che richiede approfondimenti diagnostici
- CF complessa o stato di male febbrile- Condizioni socio-ambientali o culturali inadeguate (es.
contesto familiare poco affidabile)

Convulsioni Febbrili Indagini di Laboratorio
Non sono raccomandateNon sono raccomandate
La decisione circa la necessità di eseguire le suddette indagini deve essere volta esclusivamente all’identificazione della causa
della febbre.

Convulsioni FebbriliIndagini strumentali
Convulsione febbrile semplice:
Non sono raccomandati esami strumentali quali EEG o RMN, perchè di limitato valore diagnostico.
(La diagnosi è essenzialmente clinica).
La puntura lombare va eseguita SOLO in presenza di segni meningei e va considerata in caso di pz<18 mesi (per la clinica sfumata) e in caso di soggetti già in trattamento con antibiotici.
Convulsione febbrile complessa:È raccomandata l’indagine EEG e l’esecuzione di RMN o (in assenza di quest’ultima) di TC encefalo; Va ricercata la patologia cerebrale sottostante per distinguere le forme sintomatiche (es. encefalite virale) da quelle a predisposizione genetica.La punura lombare va effettuata quando si sospetti una patologia infettiva del SNC.

Convulsioni FebbriliDiagnosi differenziale
• Brividi scuotenti• Meningite asettica• Meningoencefalite virale o batterica, o altra infezione del SNC• Meningite neonatale• Convulsioni neonatali• Sindromi epilettiche, crisi generalizzate o parziali• Ipoglicemia• Pseudocrisi (PNES)• Disturbi parossistici non epilettici (spasmus nutans, mioclono
benigno della prima infanzia, …)• Evento vascolare acuto: trombosi arteria o vena cerebrale,
emorragia cerebrale

Convulsioni Febbrili - Trattamento
Trattamento dell’episodio acuto
Valutazione criteri di “rischio” Indagini Profilassi Informazione dei genitori

Convulsioni Febbrili - TrattamentoLa maggior parte delle Convulsioni Febbrili Semplici termina
spontaneamente entro 2-3 minuti; di conseguenza esse non richiedono alcun trattamento.
Sporadicamente la Convulsione Febbrile Semplice può durare oltre i tre minuti; in tali casi non essendo prevedibile la durata spontanea sarà opportuno intervenire
farmacologicamente.
DIAZEPAM per via rettale alla dose di 0,5 mg/kg
in caso di convulsione prolungata oltre 2-3 min

Convulsioni FebbriliPrognosi e Terapia preventiva
Le CFS hanno prognosi benigna, non lasciano danni permanenti e tendono a scomparire spontaneamente con l’età.
Per tale motivo e a causa dei numerosi effetti collaterali potenzialmente legati alla terapia anticonvulsivante, non è raccomandato l’uso di alcuna terapia, continuativa o intermittente, per la profilassi delle recidive di CFS.
La profilassi intermittente o continua, non impedisce l’insorgenza di un’eventuale successiva epilessia idiopatica.

Convulsioni FebbriliEducazione sanitaria per i familiari
- Descrivere nel modo più dettagliato possibile le caratteristiche delle convulsioni febbrili;
- Verificare la comprensione delle indicazioni per il controllo della febbre;- Fornire le misure per la gestione dell’eventuale recidiva:
mantenere la calma allentare l'abbigliamento, in particolare intorno al collo se il bambino è incosciente metterlo in decubito laterale per evitare
l'inalazione di saliva ed eventuale vomito non forzare l'apertura della bocca osservare il tipo e la durata della crisi non dare farmaci o liquidi per via orale somministrare DIAZEPAM alla dose di 0,5 mg/kg per via rettale in caso di convulsione prolungata oltre 2-3 min. (il Diazepam somministrato per via
rettale impiega circa 3 minuti a raggiungere una concentrazione cerebrale efficace).
in ogni caso contattare il pediatra curante o altro sanitario un intervento medico è necessario nei seguenti casi: crisi di durata >10
minuti o che non cessa con la terapia, crisi ripetute, crisi focali, presenza di prolungato disturbo della coscienza e/o paralisi post-critica

Convulsioni FebbriliRischio di recidiva
Rischio di recidiva per una CFS: 20 - 30%.Il rischio individuale è molto variabile in relazione a fattori genetici e ambientali.Fattori prognostici: •Prematurità •l’età precoce d’insorgenza della prima CF (<12 mesi);• la familiarità per epilessia idiopatica o per CF, nei parenti di primo grado;• numerosi episodi di CF semplice o un CF di tipo complesso;•massimo rischio di ricaduta nel primo anno dopo l’esordio•Esordio in corso di episodio febbrile a temperatura relativamente bassa
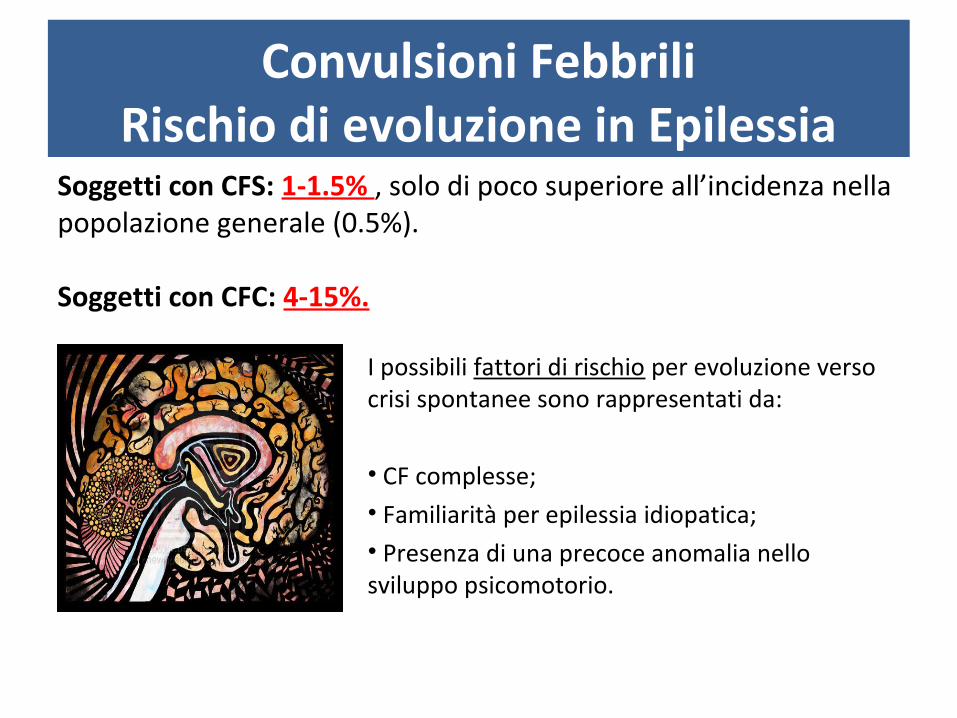
Convulsioni FebbriliRischio di evoluzione in Epilessia
Soggetti con CFS: 1-1.5% , solo di poco superiore all’incidenza nella popolazione generale (0.5%).
Soggetti con CFC: 4-15%.
I possibili fattori di rischio per evoluzione verso crisi spontanee sono rappresentati da:
• CF complesse;• Familiarità per epilessia idiopatica;• Presenza di una precoce anomalia nello sviluppo psicomotorio.

Epilessia - Definizione
EPILESSIAEPILESSIA: : sindrome neurologica cronica caratterizzata dalla predisposizioni a generare crisi epilettiche.
Per la diagnosi occorre che il paziente abbia almeno 2 crisi non provocate a più di 24 ore di distanza.
Per “crisi” epilettica intendiamo un evento conseguente ad un'attività abnorme, sincrona ed eccessiva di un gruppo di neuroni cerebrali che si estrinseca in un fenomeno improvviso e transitorio, correlato alle aree cerebrali coinvolte dall'origine della scarica epilettica e dal suo schema di propagazione.
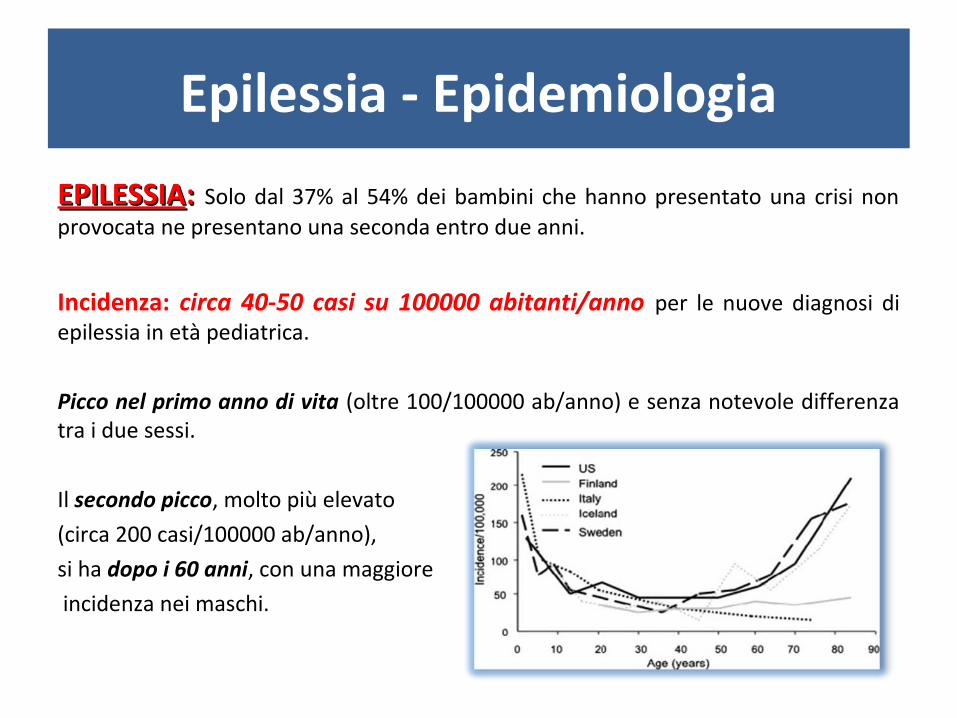
Epilessia - Epidemiologia
EPILESSIAEPILESSIA: : Solo dal 37% al 54% dei bambini che hanno presentato una crisi non provocata ne presentano una seconda entro due anni.
Incidenza: circa 40-50 casi su 100000 abitanti/anno per le nuove diagnosi di epilessia in età pediatrica.
Picco nel primo anno di vita (oltre 100/100000 ab/anno) e senza notevole differenza tra i due sessi.
Il secondo picco, molto più elevato
(circa 200 casi/100000 ab/anno),
si ha dopo i 60 anni, con una maggiore
incidenza nei maschi.
Epilessia - Eziologia

Epilessia - Classificazione
A causa dell’eterogeneità delle manifestazioni epilettiche,l’ILAE (lega internazionale contro l’epilessia) ha sviluppato nel corso degli anni un sistema classificativo tutt’ora in via di aggiornamento e perfezionamento.
Tale classificazione raggruppa le forme di epilessia tenendo conto:Delle manifestazioni cliniche e delle caratteristiche EEG delle crisiDell’età di comparsaDell’eziologia delle crisi
A causa dell’eterogeneità delle manifestazioni epilettiche,l’ILAE (lega internazionale contro l’epilessia) ha sviluppato nel corso degli anni un sistema classificativo tutt’ora in via di aggiornamento e perfezionamento.
Tale classificazione raggruppa le forme di epilessia tenendo conto:Delle manifestazioni cliniche e delle caratteristiche EEG delle crisiDell’età di comparsaDell’eziologia delle crisi

EpilessiaManifestazioni cliniche
• Crisi focali: originano da una singola area di un solo emisfero;
possono pertanto avere manifestazioni motorie localizzate,
somatosensoriali, autonomiche o “psichiche” (aura).
– CRISI FOCALI SEMPLICI (senza alterazione della coscienza)
– CRISI FOCALI COMPLESSE (coscienza compromessa o persa, spesso associata ad automatismi motori)
• Crisi focali secondariamente generalizzate

EpilessiaManifestazioni cliniche
• Crisi generalizzate: esordisce con compromissione della coscienza per il coinvolgimento esteso della corteccia cerebrale. Manifestazioni bilaterali e simmetriche classificabili in:
- Crisi tipo assenza- Crisi a tipo assenza atipica- Crisi miocloniche- Crisi cloniche- Crisi toniche- Crisi tonico cloniche - Crisi atoniche- Crisi non classificabili

EpilessiaIndagini strumentali
ELETTROENCEFALOGRAMMAStrumento di notevole supporto al neurologo pediatra per la diagnosi
differenziale e il follow-up. Tuttavia non è fondamentale, data la bassa sensibilità.Le alterazioni eeg (punte, polipunte, punte-onda ecc…) possono essere
evidenziate in veglia e/o sonno a seconda della sindrome.
VIDEO-EEGNei casi in cui la clinica e l’EEG standardnon siano sufficienti per definire unadiagnosi differenziale tra crisi epilettiche enon epilettiche, è necessario documentarel’evento critico mediante video-eeg.

EpilessiaIndagini strumentali
RMN ENCEFALOHa un ruolo fondamentale al momento della
diagnosi per evidenziare alterazioni strutturali.
Permette inoltre di individuare la tipologia dello sviluppo corticale, lo stato della mielinizzazione e le sue alterazioni sia di maturazione sia conseguenti a danni infiammatori, vascolari, tossici, genetici o metabolici.
Sarà quindi indispensabile anche in caso di intervento neurochirurgico.
S. di Aicardi, agenesia corpo calloso
S. di Aicardi, agenesia corpo calloso

Principali sindromi epiletttiche
SINDROME DI WESTIncidenza 2,9-4,5/10.000 /annoEncefalopatia più conosciuta e frequente della prima infanzia. Età d’esordio: dal 3°mese al 1°anno di vita.Eziologia: nel 90% dei casi sintomatica (encefalopatie pre, peri, postnatali, malformazioni encefaliche, sindromi neurocutanee etc.).Caratteristiche principali: Spasmi in flessione o esetensione/Arresto Psicomotorio/Ipsaritmia all’EEG.Prognosi: è legata all’eziologia; il 60% dei casi sviluppa una successiva epilessia (Lennox-Gastaut); il 25% va in remissione spontanea.Trattamento: ACTH, corticosteroidi, vigabatrim, benzodiazepine, valproato.

Principali sindromi epiletttiche
SINDROME DI DRAVET
(epilessia mioclonica severa)
Incidenza: 1/30.000/anno
Età d’esordio: nel 1 anno di vita.
Eziologia: sconosciuta, mutazioni del canale voltaggio-dip del Na.
Caratteristiche principali: Crisi febbrili e afebbrili cloniche o tonico-cloniche, assenze atipiche e crisi focali. Frequenti stati di male. Ritardo psicomotorio.
Prognosi: crisi farmacoresistenti con compromissione cognitiva grave.
EEG: anomalie polimorfe a seconda del tipo di crisi.

Principali sindromi epiletttiche
Epilessia benigna dell’infanzia con punte centro-temporali (BECTS o Rolandica)
Incidenza: 10.7-21/100.000/anno tra i 0 e i 15 anniEtà d’esordio: tra 2 e 14 anni, picco tra gli 8 e I 9 anni.Eziologia: sconosciuta, può essere presente familiarità per epilessia.Caratteristiche principali: crisi con sintomi sensitivo-motori facciali unilaterali, manifestazioni orofaringee, disartria e arresto del linguaggio, a volte secondaria generalizzazione, spesso durante il sonno.Prognosi: remissione spontanea in 2-4 anni.EEG: tracciato intercritico con P centro-temporali o PO lentte di alto voltaggio.Trattamento: data la remissione spontanea entro I 14-16 anni si preferisce non trattare questa sindrome; eventualemente si può usare carbamazepina o valproato.

Principali sindromi epiletttiche
Epilessia occipitale benigna a insorgenza precoce (Panayiotopoulos)
Prevalenza: 13% dei pazienti epilettici tra i 3 e i 6 anniEtà d’esordio: tra 1 e 14 anni, picco tra I 4 e i 5 anni.Eziologia: sconosciuta, può essere presente familiarità per epilessia [mutaz SNC1A], antecedenti per convulsioni febbrili.Caratteristiche principali: crisi notturne rare, nel 30% dei casi un solo episodio, di lunga durata, caratterizzate da sintomi vegetativi, deviazione laterale dello sguardo, perdita di coscienza.Prognosi: eccellente.EEG: in fase interciritica, complessi PO lenta, posteriori, di ampio voltaggio; in fase critica attività theta o delta ritmica con piccole P.Trattamento: data la remissione spontanea, si preferisce non trattare questa sindrome, se crisi ricorrenti si raccomanda la carbamazepina.

Principali sindromi epiletttiche
Epilessia occipitale idiopatica a insorgenza tardiva (Gastaut)
2–7% delle epilessie focali benigne dell’infanziaEtà d’esordio: tra 3 e 15 anni.Eziologia: sconosciuta, può essere presente familiarità per epilessia o emicrania.Caratteristiche principali: crisi frequenti, brevi, durante il giorno, con allucinazioni visive o amaurosi, associate a cefalea postcritica.Prognosi: remissione in 2-4 anni, nel 50% buona risposta alla terapia.EEG: in fase intercritica, ad occhi chiusi, P o PO occipitali bilaterali, sincrone o asincrone.Trattamento: carbamazepina.

Principali sindromi epiletttiche
Sindrome di Lennox-GastautIncidenza: 2/100.000/annoEtà d’esordio: tra 1 e 7 anni, segue spesso una s. di West.Eziologia: lesionale nel 66% dei casi, fattori eziolgici pre/peri/postnatali, familiarità, segnalate forme idiopatiche.Caratteristiche principali: ritardo mentale, crisi polimorfe, toniche assiali, atoniche, assenze atipiche, miocloniche, generalizzate tonico-cloniche.Prognosi: sfavorevole, farmacoresistente con compromissione cognitiva, variazione della semeiologia delle crisi nel tempo, mortalità 4,2-7%.Trattamento: politerapie (mediche e chirurgiche), valproato e benzodiazepine in prima linea, etosuccimide , lamotrigina e carbamazepina, ACTH e corticosteroidi efficaci solo transitoriamente.

Principali sindromi epiletttiche
Epilessia assenza dell’infanzia (Piccolo Male)
Incidenza: 6,3 casi/100.000/anno 10% di tutte le epilessie, f>mEtà d’esordio: tra 4 e 10 anni.Eziologia: genetica multifattoriale, familiare nel 13% dei casi, nel 8,6% dei bambini anamnesi positiva per convulsioni febbrili.Caratteristiche principali: assenze semplici, caratterizzate da breve (5-10 sec.) sospensione della coscienza e sguardo fisso nel vuoto, talvolta si associano fini mioclonie palpebrali o più raramente automatismi motori.L’inizio e la fine sono bruschi (il bambino riprende l’attività in corso e di solito non ha memoria delle crisi).Le crisi sono pluriquotidiane, fino a 200 al giorno nelle forme sintomatiche.

Principali sindromi epiletttiche
Epilessia assenza dell’infanzia (Piccolo Male)
Prognosi: risoluzione prima dei 12 anni, nel 10% dei casi si sviluppano crisi tonico-clonico generalizzate o assenze da adulto. Tra I fattori prognostici negativi: presenza di segni neuropsicologici, associazioni con crisi GM frequenti, età tardiva di insorgenza, resistenza al trattamento.EEG: scariche di complessi PO ritmici a 3 c/s sincrone sui due emisferi, con massima ampiezza delle punte, in genere, sulle regioni frontali. Inizio e fine bruschi o seguite da lievi modificazioni del tracciato. La crisi clinica e le alterazioni dell’EEG possono essere scatenate dall’iperpnea. In sonno, le scariche possono frammentarsi e includere, anche, PP generalizzate.In fase intercritica: attività di fondo normale, attività lenta posteriore a 3-4 c/s facilitata dalla chiusura degli occhi. Rare O aguzze o brevi scariche di complessi PO più o meno tipici, diffusi, dominanti anteriormente, facilitate dall’iperpnea.Trattamento: valproato ed etosuccimide in prima linea, lamotrigina, topiramato.

Principali sindromi epiletttiche
Epilessia mioclonica giovanile (S. di Janz)Incidenza: 1/2000/annoEtà d’esordio: tra 12 e 18 anni.Eziologia: sconosciuta, alcuni geni implicati sono EFHC1, CACNB4, GABRA1, CLCN2, GABRD.Caratteristiche principali: improvvise scosse di entrambi gli arti superiori, a frequenza e intensità variabili, raramente lateralizzate, prevalentemente al mattino, subito dopo il risveglio, senza perdita di coscienza.Mioclonie singole, a intervalli regolari, o organizzate in cluster fino a dar luogo a uno stato di piccolo male mioclonico. Più spesso sono di intensità minima, a volte sono violente. Agli arti inferiori le scosse sono più rare. Frequenti anche le scosse tonico-cloniche. Fattori scatenanti: deprivazione di sonno, abuso di alcolici, stress, risvegli precociPrognosi: crisi ben controllate dalla terapia, ma frequenti recidive alla sospensione.Trattamento: valproato in prima linea, lamotrigina, topiramato, levetiracetam.

Principali sindromi epiletttiche
Sindrome di RasmussenPrevalenza: <1/1.000.000Età d’esordio: tra 1 e 10 anni.Eziologia: sconosciuta, probabile encefalite cronica di natura autoimmune. Emiatrofia progressiva all’RM encefalo.Caratteristiche principali: esordio con crisi focali motorie, seguono epilessia parziale continua, crisi focali polimorfe, tonico-cloniche di un emilato o generalizzate ed emiplegia.Prognosi: decorso progressivo con aumento nella frequenza delle crisi, deficit mentali e neurologici permanenti e progressivi. Scarsa risposta alla terapiaEEG: graduale comparsa di O delta ampie polimorfe, prima da un lato, poi bilaterali, costanti P e PO intercritiche.Terapia: emisferectomia o emisferotomia.

Epilessia - Trattamento
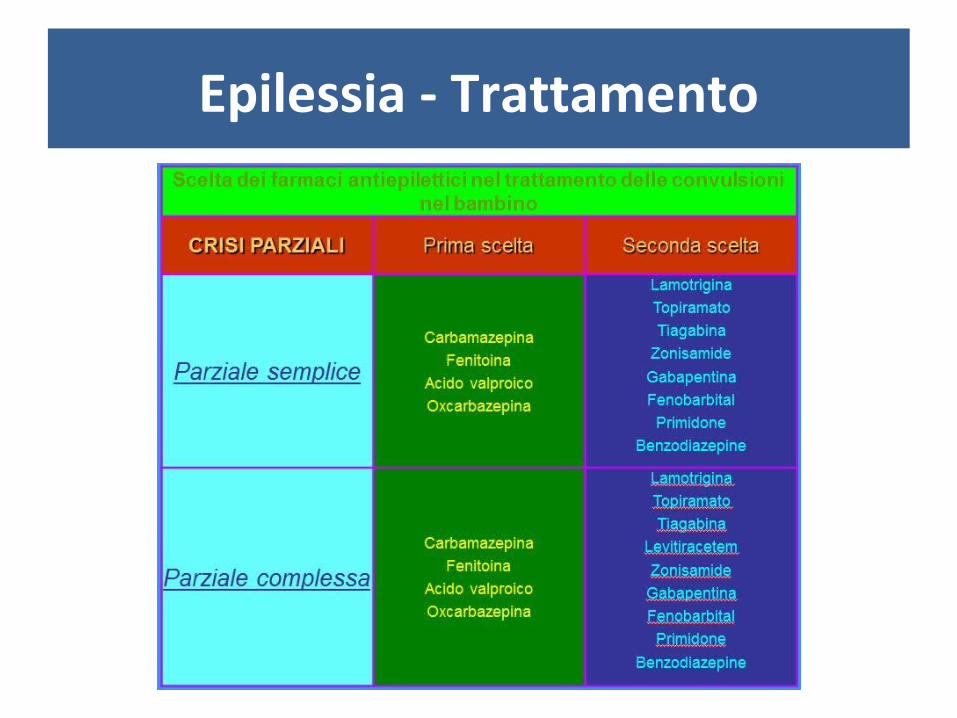
Epilessia - Trattamento
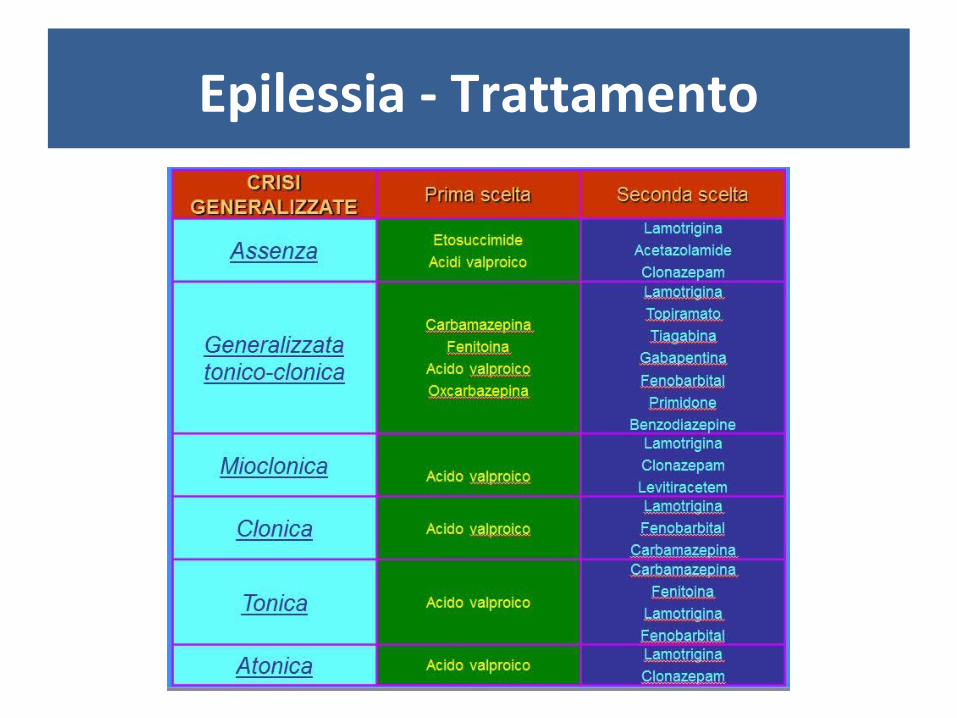
Epilessia - Trattamento
















![THALIA - bft-automation.com · PRESET DEFAULT ar sr ac sc ind PARAMETRI Tempo ritardo apertura motore 2 [s] 3 3 3 3 3 3 Tempo di ritardo chiusura motore 1 [s] 3 3 3 3 3 3 Tempo chiusura](https://static.fdocuments.in/doc/165x107/5c6930f109d3f263648cc312/thalia-bft-preset-default-ar-sr-ac-sc-ind-parametri-tempo-ritardo-apertura.jpg)

