
Protein Sequence Motifs
75
www.bioinformatics.nl Protein Sequence Motifs Aalt-Jan van Dijk Plant Research International, Wageningen UR Biometris, Wageningen UR [email protected]
-
Upload
damon-parrish -
Category
Documents
-
view
59 -
download
0
description
Protein Sequence Motifs. Aalt-Jan van Dijk Plant Research International, Wageningen UR Biometris , Wageningen UR [email protected]. Plant Bioinformatics. Integrated analysis of omics datasets Transcriptomics Alternative splicing EST analysis Proteomics - PowerPoint PPT Presentation
Transcript of Protein Sequence Motifs

www.bioinformatics.nl
Protein Sequence Motifs
Aalt-Jan van DijkPlant Research International, Wageningen UR
Biometris, Wageningen [email protected]

www.bioinformatics.nlwww.bioinformatics.nl
Genomics Next Generation Sequencing Genome assembly & annotation (Comparative) genome analysis SNP analysis, marker development
Technology Computational infrastructure Database development Webbased analysis tools Software- development Workflow management systems machine learning
Integrated analysis of omics datasets Transcriptomics
• Alternative splicing• EST analysis
Proteomics• Data (pre-)processing pipelining• Alternative splicing• Protein interactions networks
Metabolomics• Database- development• Data (pre-)processing pipelining• Metabolite and pathway-identification
Systems biology network modelling (bottom-up)
• Protein interactions networks
Plant Bioinformatics

www.bioinformatics.nlwww.bioinformatics.nl
Protein complex structures Protein-protein docking Correlated mutations
Interaction site prediction/analysis Protein-protein
interactions Protein-DNA interactions
Motif search Enzyme active sites
My research

www.bioinformatics.nlwww.bioinformatics.nl
Overview
Protein Motif Searching Hydrophobicity & Transmembrane Domains Protein Interactions Sequence-motifs to predict interaction sites
Secondary Structure Prediction

www.bioinformatics.nl
Protein Motif Searching

www.bioinformatics.nlwww.bioinformatics.nl
What is a motif?
A motif is a description of a particular element of a protein that contains a specific sequence pattern
Motifs are identified by 3D structural alignment Multiple sequence alignment Pattern searching programs

www.bioinformatics.nlwww.bioinformatics.nl
What is a motif?
A motif is a description of a particular element of a protein that contains a specific sequence pattern
Motifs are identified by 3D structural alignment Multiple sequence alignment Pattern searching programs

www.bioinformatics.nlwww.bioinformatics.nl
C C P C
Protein Motif Searching
Strict consensus pattern use only strictly conserved residues
C--QASCDGIPLKMNDCC---VTCEGLPMRMDQCCERTLGCQPMPVH---C
CxxxxxCxxxPxxxxxC

www.bioinformatics.nlwww.bioinformatics.nl
C C P C
Protein Motif Searching
Strict consensus pattern use only strictly conserved residues
C--QASCDGIPLKMNDCC---VTCEGLPMRMDQCCERTLGCQPMPVH---C
CxxxxxCxxxPxxxxxC

www.bioinformatics.nlwww.bioinformatics.nl
C C P C
Protein Motif Searching
Strict consensus pattern use only strictly conserved residues
But what about: variable residues? gaps?
C--QASCDGIPLKMNDCC---VTCEGLPMRMDQCCERTLGCQPMPVH---C
CxxxxxCxxxPxxxxxC

www.bioinformatics.nlwww.bioinformatics.nl
C C P CCxxxxxCxxxPxxxxxC
Protein Motif Searching
Strict consensus patterns contain no alternative residues no flexible regions no mismatches no gaps C--QASCDGIPLKMNDC
C---VTCEGLPMRMDQCCERTLGCQPMPVH---C

www.bioinformatics.nlwww.bioinformatics.nl
Protein Motif Searching
Most motifs defined as regular expressions Motifs can contain
alternative residues flexible regions
C-x(2,5)-C-x-[GP]-x-P-x(2,5)-C
CXXXCXGXPXXXXXC | | | | | FGCAKLCAGFPLRRLPCFYG

www.bioinformatics.nlwww.bioinformatics.nl
The PROSITE Syntax
A-[BC]-X-D(2,5)-{EFG}-H A B or C anything 2-5 D’s not E, F, or G H

www.bioinformatics.nlwww.bioinformatics.nl
PROSITE entries
Mandatory motifs characterise a protein (super-) family
ID SUBTILASE_ASP; PATTERN.
DE Serine proteases, subtilase family, aspartic acid active site.
PA [STAIV]-x-[LIVMF]-[LIVM]-D-[DSTA]-G-[LIVMFC]-x(2,3)-[DNH].
ID SUBTILASE_HIS; PATTERN.DE Serine proteases, subtilase family, histidine active site.PA H-G-[STM]-x-[VIC]-[STAGC]-[GS]-x-[LIVMA]-[STAGCLV]-[SAGM].
ID SUBTILASE_SER; PATTERN.DE Serine proteases, subtilase family, serine active site.PA G-T-S-x-[SA]-x-P-x(2)-[STAVC]-[AG].

www.bioinformatics.nlwww.bioinformatics.nl
Exercise
Find the three subtilase motifs in prosite (prosite.expasy.org)
Compare the lists of proteins in which the motifs occur – what does this tell you?
Similarly, compare protein structures in which the motifs occur
Have a look at the “sequence logo”

www.bioinformatics.nlwww.bioinformatics.nl
Protein Motif Searching
Some motifs occur frequently in proteins; they may not actually be present, such as Post-translational modification sites
ID ASN_GLYCOSYLATION; PATTERN.DE N-glycosylation site.PA N-{P}-[ST]-{P}.
www.bioinformatics.nlwww.bioinformatics.nl
Exercise
Use a glycosylation site predictor such as http://www.cbs.dtu.dk/services/NetNGlyc/
Input: your favorite set of sequences
Do you observe that some N-{P}-[ST] sites are likely to be glycosylated and others not?

www.bioinformatics.nlwww.bioinformatics.nl
Profiles
Many motifs cannot be easily defined using simple patterns
Such motifs can be defined using profiles A profile is constructed from a multiple
sequence alignment. For each position, each amino acid is given a score depending on how likely it is to occur

www.bioinformatics.nlwww.bioinformatics.nl
Calculating a Profile
For each alignment position: take the (weighted) average of the appropriate rows from the scoring matrix
An (extremelysimple) example:
seq_01 A A A A A A A A A A Wseq_02 A A A A A A A A A W Wseq_03 A A A A A A A A W W Wseq_04 A A A A A A A W W W Wseq_05 A A A A A A W W W W Wseq_06 A A A A A W W W W W Wseq_07 A A A A W W W W W W Wseq_08 A A A W W W W W W W Wseq_09 A A W W W W W W W W Wseq_10 A W W W W W W W W W W

www.bioinformatics.nlwww.bioinformatics.nl
A C D E F G H I K L M 10A: 4.0 0.0 -2.0 -1.0 -2.0 0.0 -2.0 -1.0 -1.0 -1.0 -1.0 N P Q R S T V W Y -2.0 -1.0 -1.0 -1.0 1.0 0.0 0.0 -3.0 -2.0
A C D E F G H I K L M 5A+5W: 1.0 -2.0 -6.0 -4.0 -1.0 -2.0 -4.0 -4.0 -4.0 -3.0 -2.0 N P Q R S T V W Y -6.0 -5.0 -3.0 -4.0 -2.0 -2.0 -3.0 8.0 0.0
A C D E F G H I K L M 10W: -3.0 -2.0 -4.0 -3.0 1.0 -2.0 -2.0 -3.0 -3.0 -2.0 -1.0 N P Q R S T V W Y -4.0 -4.0 -2.0 -3.0 -3.0 -2.0 -3.0 11.0 2.0
A R N D C Q E G H I L K M F P S T W Y VA 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0W -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11 2 -3
prophecy (EMBOSS), using Henikoff profile type, and BLOSUM62 matrix;
Excerpt from the EBLOSUM62 matrix:

www.bioinformatics.nlwww.bioinformatics.nl
Pattern Searching
Short linear motifs: e.g. http://dilimot.russelllab.org/
Profiles: memehttp://meme.sdsc.edu/meme/cgi-bin/meme.cgi

www.bioinformatics.nlwww.bioinformatics.nl
Exercise
Use a number of sequences wich contain the prosite subtilase motif and find motifs in those sequences with MEME

www.bioinformatics.nl
Hydropathy Plot
Prediction hydrophobic and hydrophilic regions in a protein

www.bioinformatics.nlwww.bioinformatics.nl
Partition Coefficients
Water
Oil
Hydrophilic Hydrophobic

www.bioinformatics.nlwww.bioinformatics.nl
Hydrophobicity/Hydrophilicity ValuesFauchere & Pliska Kyte & Doolittle Hopp & Woods Eisenberg
R -1.37 -4.50 3.00 -2.53K -1.35 -3.90 3.00 -1.50D -1.05 -3.50 3.00 -0.90Q -0.78 -3.50 0.20 -0.85N -0.85 -3.50 0.20 -0.78E -0.87 -3.50 3.00 -0.74H -0.40 -3.20 -0.50 -0.40S -0.18 -0.80 0.30 -0.18T -0.05 -0.70 -0.40 -0.05P 0.12 -1.60 0.00 0.12Y 0.26 -1.30 -2.30 0.26C 0.29 2.50 -1.00 0.29G 0.48 -0.40 0.00 0.48A 0.62 1.80 -0.50 0.62M 0.64 1.90 -1.30 0.64W 0.81 -0.90 -3.40 0.81L 1.06 3.80 -1.80 1.06V 1.08 4.20 -1.50 1.08F 1.19 2.80 -2.50 1.19I 1.38 4.50 -1.80 1.38
hydrophilic hydrophobic

www.bioinformatics.nlwww.bioinformatics.nl
ki
kin
ni Hk
H12
1
Hydrophobicity Plot
Sum amino acid hydrophobicity values in a given window
Plot the value in the middle of the window Shift the window one position

www.bioinformatics.nlwww.bioinformatics.nl
Sliding Window Approach
Calculate property for first sub-sequence
Use the result (plot/print/store)
Move to next residue position, and repeat

www.bioinformatics.nlwww.bioinformatics.nl
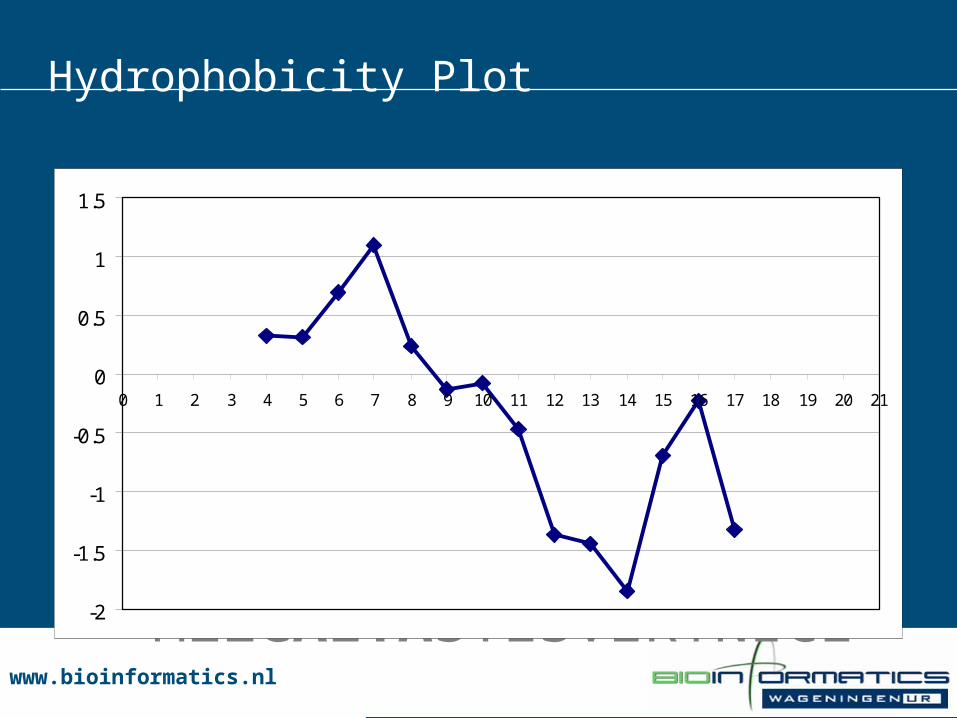
Hydrophobicity Plot
MEZCALTASTESVERYNICE

www.bioinformatics.nlwww.bioinformatics.nl
Hydrophobicity Plot
-2
-1.5
-1
-0.5
0
0.5
1
1.5
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
MEZCALTASTESVERYNICE
www.bioinformatics.nlwww.bioinformatics.nl
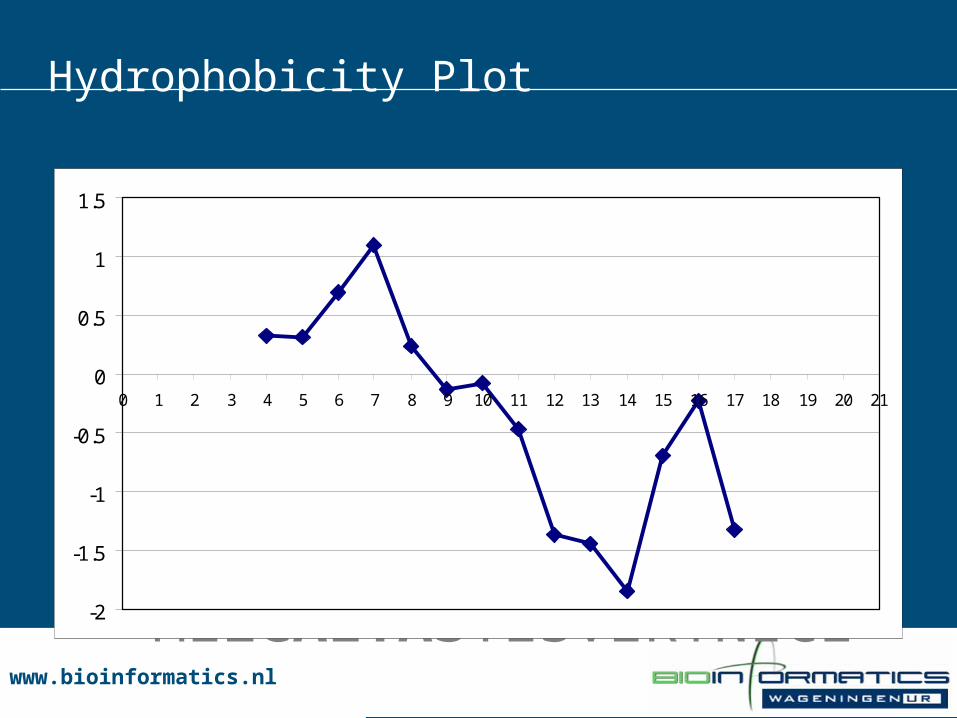
Hydrophobicity Plot
-2
-1.5
-1
-0.5
0
0.5
1
1.5
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
MEZCALTASTESVERYNICE
www.bioinformatics.nlwww.bioinformatics.nl
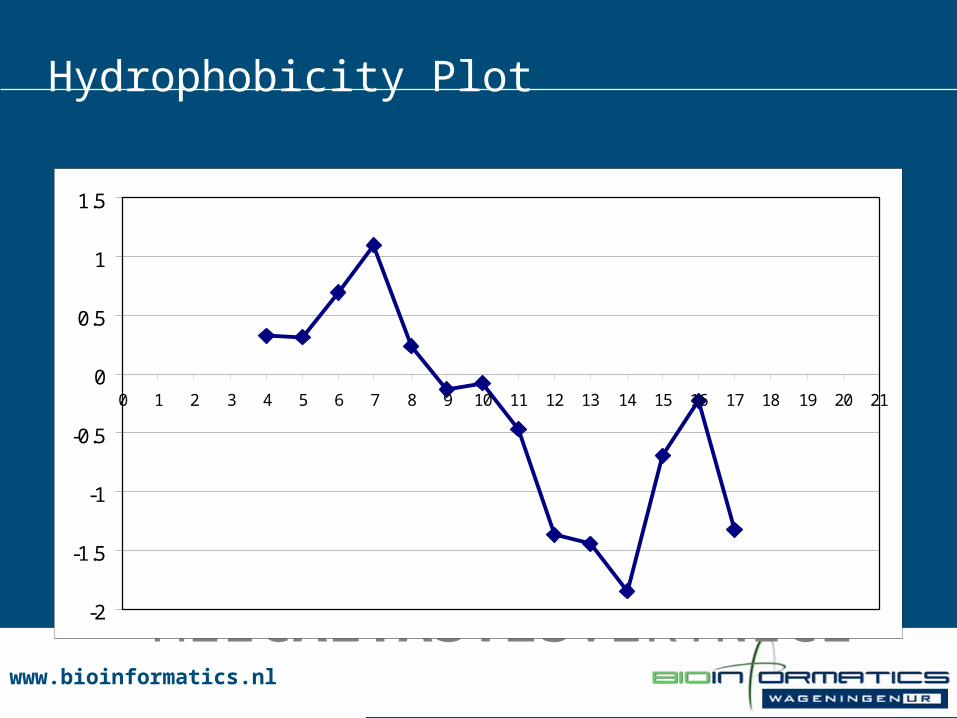
Hydrophobicity Plot
-2
-1.5
-1
-0.5
0
0.5
1
1.5
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
MEZCALTASTESVERYNICE

www.bioinformatics.nlwww.bioinformatics.nl
Hydrophobicity Plot
-2
-1.5
-1
-0.5
0
0.5
1
1.5
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
MEZCALTASTESVERYNICE
www.bioinformatics.nlwww.bioinformatics.nl
Hydrophobicity Plot
-2
-1.5
-1
-0.5
0
0.5
1
1.5
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
MEZCALTASTESVERYNICE

www.bioinformatics.nlwww.bioinformatics.nl
Hydrophobicity Plot
-2
-1.5
-1
-0.5
0
0.5
1
1.5
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
MEZCALTASTESVERYNICE

www.bioinformatics.nlwww.bioinformatics.nl
Transmembrane Regions
Rotation is 100 degrees per amino acid
Climb is 1.5 Angstrom per amino acid residue

www.bioinformatics.nlwww.bioinformatics.nl
Transmembrane Regions
30 angstrom
So we need approx. 30 / 1.5 = 20 amino acids to span the membrane

www.bioinformatics.nlwww.bioinformatics.nl

www.bioinformatics.nlwww.bioinformatics.nl
Adapting the window size to the size of the membrane
spanning segment makes the picture easier to interpret
Adapting the window size to the size of the membrane
spanning segment makes the picture easier to interpret
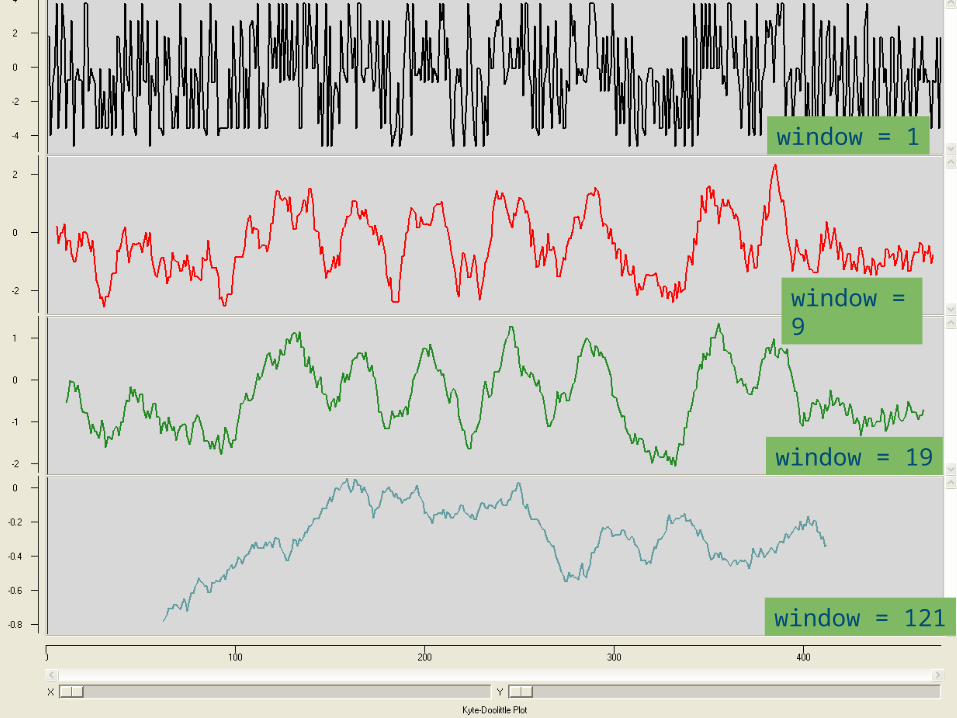
www.bioinformatics.nlwww.bioinformatics.nl
window = 1
window = 9
window = 19
window = 121

www.bioinformatics.nl
Protein Interactions
www.bioinformatics.nlwww.bioinformatics.nl
Protein Interactions
Obligatory
hemoglobin
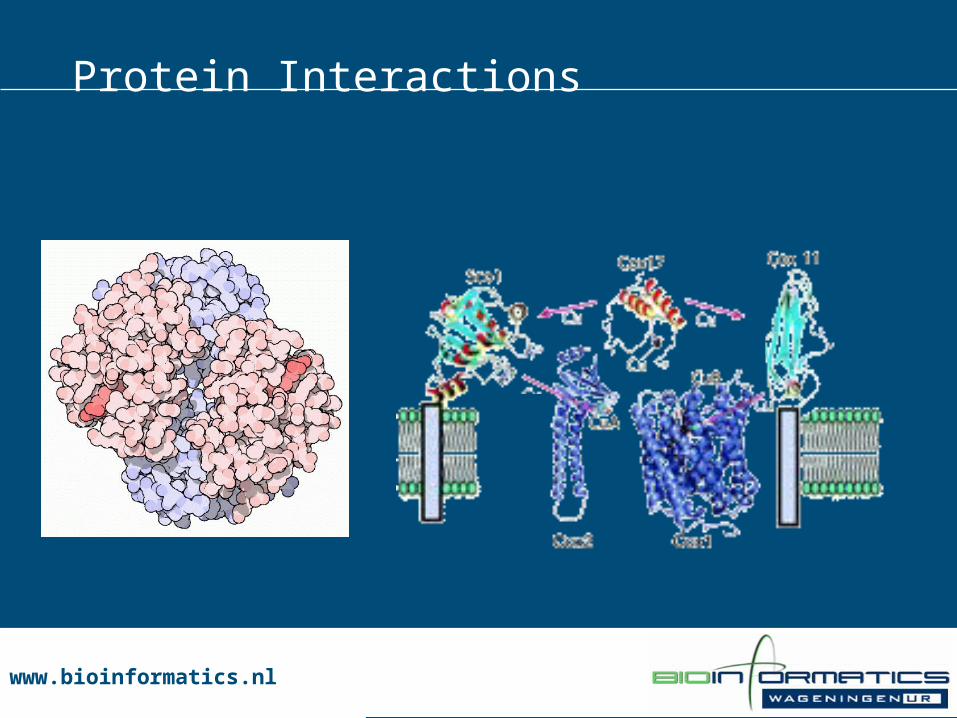
www.bioinformatics.nlwww.bioinformatics.nl
Obligatory Transient
hemoglobin Mitochondrial Cu transporters
Protein Interactions

www.bioinformatics.nlwww.bioinformatics.nl
Yeast two-hybrid (Y2H)
Experimental approaches (1)

www.bioinformatics.nlwww.bioinformatics.nl
Affinity Purification + mass spectrometry (AP-MS)
Experimental approaches (2)

www.bioinformatics.nlwww.bioinformatics.nl
STRING http://string.embl.de/
Interaction Databases

www.bioinformatics.nlwww.bioinformatics.nl
Interaction Databases

www.bioinformatics.nlwww.bioinformatics.nl
STRING http://string.embl.de/
HPRD http://www.hprd.org/
Interaction Databases

www.bioinformatics.nlwww.bioinformatics.nl
Interaction Databases

www.bioinformatics.nlwww.bioinformatics.nl
STRING http://string.embl.de/
HPRD http://www.hprd.org/ InteroPorc
http://biodev.extra.cea.fr/interoporc/Default.aspx
Many others….E.g. see
http://nar.oxfordjournals.org./content/39/suppl_1.toc
Interaction Databases

www.bioinformatics.nlwww.bioinformatics.nl
Yeast protein interaction network

www.bioinformatics.nl
Sequence-based Protein Binding Site Prediction

www.bioinformatics.nlwww.bioinformatics.nl
Binding site
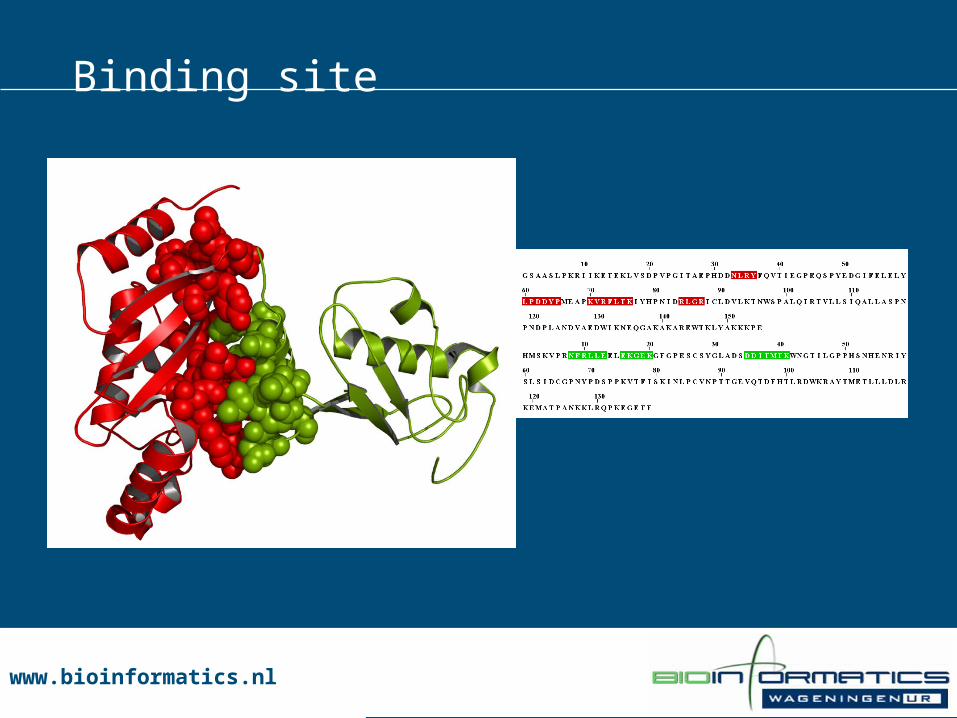
www.bioinformatics.nlwww.bioinformatics.nl
Binding site

www.bioinformatics.nlwww.bioinformatics.nl
Predefined motifs

www.bioinformatics.nlwww.bioinformatics.nl
Predefined motifs

www.bioinformatics.nlwww.bioinformatics.nl
Predefined motifs
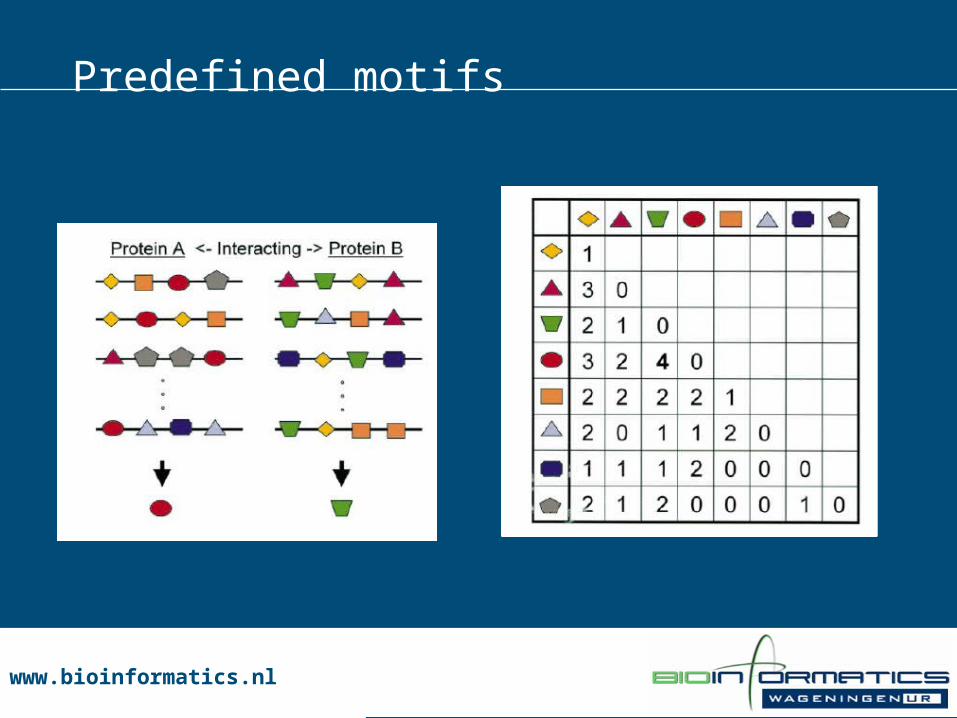
www.bioinformatics.nlwww.bioinformatics.nl
Predefined motifs
www.bioinformatics.nlwww.bioinformatics.nl
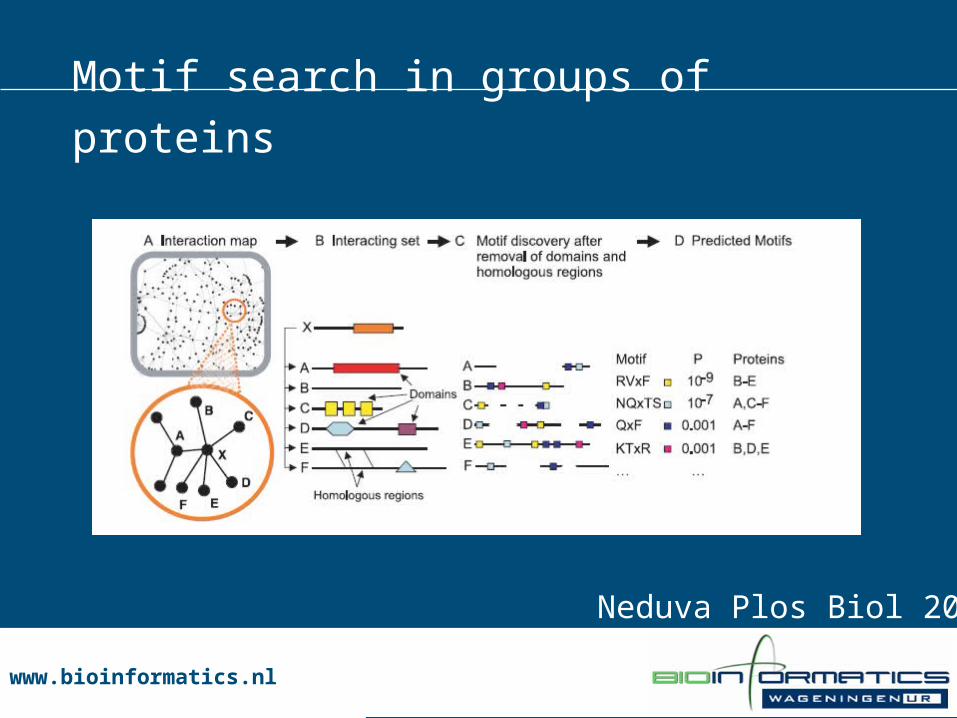
• Group proteins which have same interaction partner• Use motif search, e.g. find PWMs
Neduva Plos Biol 2005
Motif search in groups of proteins
www.bioinformatics.nlwww.bioinformatics.nl
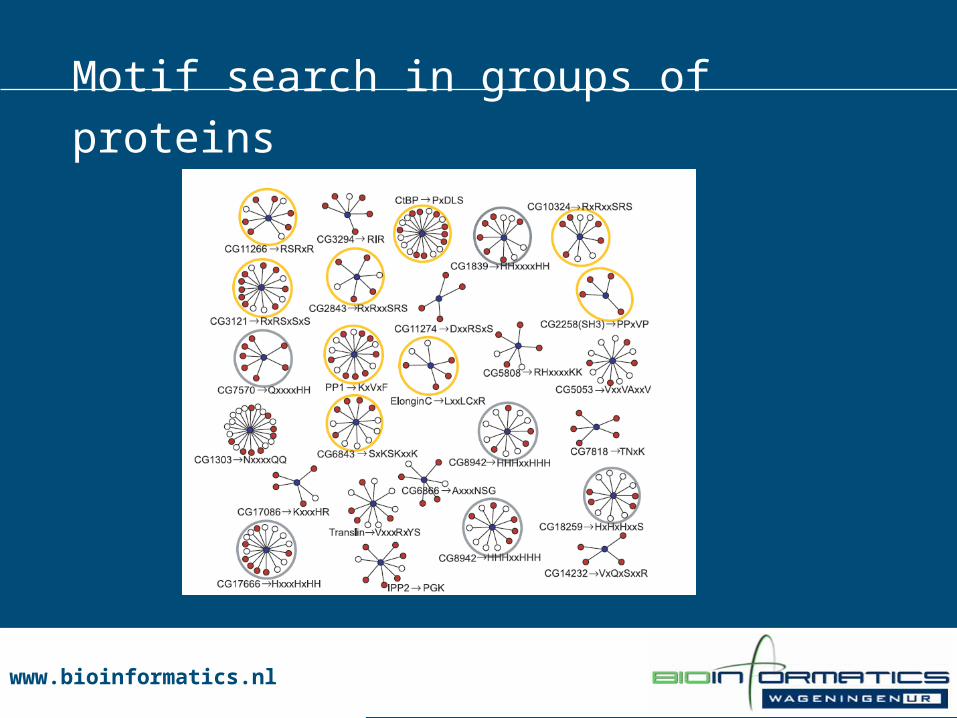
• Group proteins which have same interaction partner• Use motif search
Motif search in groups of proteins

www.bioinformatics.nlwww.bioinformatics.nl
Correlated Motif Search

www.bioinformatics.nlwww.bioinformatics.nl
Interactors Non-interactorsAARLL PLTEQ AARLL MARLTMARLT DLTEP VVRLM MARLTVVRLM MMTER PLTEQ DLTEP
Correlated Motif Pair: (RL,TE)
Correlated Motif Search

www.bioinformatics.nlwww.bioinformatics.nl
Van Dijk et al, Plos Comp Biol 2010
Experimental validation

www.bioinformatics.nlwww.bioinformatics.nlBoyen et al, IEEE/ACM Trans Comput Biol Bioinform. 2011
• Faster approach genome wide searching for interaction motifs• Improve mining algorithm with a priori biological knowledge
(conservation score, surface accessibility)
New approach: slider

www.bioinformatics.nlwww.bioinformatics.nl
THE END….. Questions?

www.bioinformatics.nlwww.bioinformatics.nl

www.bioinformatics.nl
Secondary Structure Prediction

www.bioinformatics.nlwww.bioinformatics.nl
Secondary Structure Prediction
Traditional methods (statistical and/or rule-based) E.g. Garnier, Osguthorpe & Robson
• Statistical method
Accuracy ~ 60%

www.bioinformatics.nlwww.bioinformatics.nl
i-8 i-6 i-4 i-2 i i+2 i+4 i+6 i+8Gly -5 -10 -15 -20 -30 -40 -50 -60 -86 -60 -50 -40 -30 -20 -15 -10 -5ala 5 10 15 20 30 40 50 60 65 60 50 40 30 20 15 10 5val 0 0 0 0 0 0 5 10 14 10 5 0 0 0 0 0 0leu 0 5 10 15 20 25 28 30 32 30 28 25 20 15 10 5 0ile 5 10 15 20 25 20 15 10 6 0 -10 -15 -20 -25 -20 -10 -5ser 0 -5 -10 -15 -20 -25 -30 -35 -39 -35 -30 -25 -20 -15 -10 -5 0thr 0 0 0 -5 -10 -15 -20 -25 -26 -25 -20 -15 -10 -5 0 0 0asp 0 -5 -10 -15 -20 -15 -10 0 5 10 15 20 20 20 15 10 5glu 0 0 0 0 10 20 60 70 78 78 78 78 78 70 60 40 20asn 0 0 0 0 -10 -20 -30 -40 -51 -40 -30 -20 -10 0 0 0 0gln 0 0 0 0 5 10 20 20 10 -10 -20 -20 -10 -5 0 0 0lys 20 40 50 55 60 60 50 30 23 10 5 0 0 0 0 0 0his 10 20 30 40 50 50 50 30 12 -20 -10 0 0 0 0 0 0arg 0 0 0 0 0 0 0 0 -9 -15 -20 -30 -40 -50 -50 -30 -10phe 0 0 0 0 0 5 10 15 16 15 10 5 0 0 0 0 0tyr -5 -10 -15 -20 -25 -30 -35 -40 -45 -40 -35 -30 -25 -20 -15 -10 -5trp-10 -20 -40 -50 -50 -10 0 10 12 10 0 -10 -50 -50 -40 -20 -10cys 0 0 0 0 0 0 -5 -10 -13 -10 -5 0 0 0 0 0 0met 10 20 25 30 35 40 45 50 53 50 45 40 35 30 25 20 10pro-10 -20 -40 -60 -80-100-120-140 -77 -60 -30 -20 -10 0 0 0 0
GOR Helix Parameters
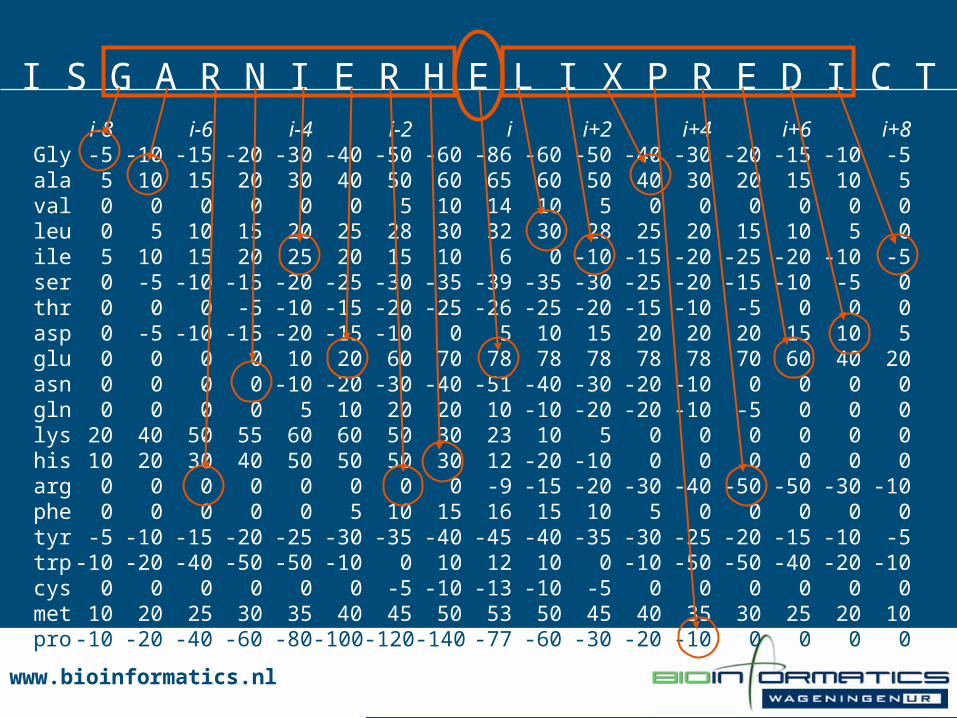
www.bioinformatics.nlwww.bioinformatics.nl
i-8 i-6 i-4 i-2 i i+2 i+4 i+6 i+8Gly -5 -10 -15 -20 -30 -40 -50 -60 -86 -60 -50 -40 -30 -20 -15 -10 -5ala 5 10 15 20 30 40 50 60 65 60 50 40 30 20 15 10 5val 0 0 0 0 0 0 5 10 14 10 5 0 0 0 0 0 0leu 0 5 10 15 20 25 28 30 32 30 28 25 20 15 10 5 0ile 5 10 15 20 25 20 15 10 6 0 -10 -15 -20 -25 -20 -10 -5ser 0 -5 -10 -15 -20 -25 -30 -35 -39 -35 -30 -25 -20 -15 -10 -5 0thr 0 0 0 -5 -10 -15 -20 -25 -26 -25 -20 -15 -10 -5 0 0 0asp 0 -5 -10 -15 -20 -15 -10 0 5 10 15 20 20 20 15 10 5glu 0 0 0 0 10 20 60 70 78 78 78 78 78 70 60 40 20asn 0 0 0 0 -10 -20 -30 -40 -51 -40 -30 -20 -10 0 0 0 0gln 0 0 0 0 5 10 20 20 10 -10 -20 -20 -10 -5 0 0 0lys 20 40 50 55 60 60 50 30 23 10 5 0 0 0 0 0 0his 10 20 30 40 50 50 50 30 12 -20 -10 0 0 0 0 0 0arg 0 0 0 0 0 0 0 0 -9 -15 -20 -30 -40 -50 -50 -30 -10phe 0 0 0 0 0 5 10 15 16 15 10 5 0 0 0 0 0tyr -5 -10 -15 -20 -25 -30 -35 -40 -45 -40 -35 -30 -25 -20 -15 -10 -5trp-10 -20 -40 -50 -50 -10 0 10 12 10 0 -10 -50 -50 -40 -20 -10cys 0 0 0 0 0 0 -5 -10 -13 -10 -5 0 0 0 0 0 0met 10 20 25 30 35 40 45 50 53 50 45 40 35 30 25 20 10pro-10 -20 -40 -60 -80-100-120-140 -77 -60 -30 -20 -10 0 0 0 0
I S G A R N I E R H E L I X P R E D I C T

www.bioinformatics.nlwww.bioinformatics.nl
GOR Prediction
beta sheet
helix

www.bioinformatics.nlwww.bioinformatics.nl
Secondary Structure Prediction
Recent methods Neural networks = flexible statistics Multiple alignments = variability Heuristics = common sense
Or a combination of the above
Accuracy ~ 70%

www.bioinformatics.nlwww.bioinformatics.nl
Heuristics
Conserved parts are structurally and/or functionally important
Segments with many gaps must be in loop regions

www.bioinformatics.nlwww.bioinformatics.nl
Secondary Structure Prediction
Strategy
Use as many methods as possible
Use homologous sequences
Combine predictions into consensus prediction

www.bioinformatics.nlwww.bioinformatics.nl
Why can’t it be 100% correct?
All current 2D prediction schemes are based upon observation of occurrence of 2D elements in 3D structures
Deduction of 2D elements from structures is ambiguous! DSSP, Stride, and the PDB (human) annotation
do not always agree upon the assigned elements

www.bioinformatics.nlwww.bioinformatics.nl
Do these residues still belong to the helix?


















