
Preformulation Studies VL
141
1 Pre-Formulation Studies VARIOUS FACTORS INFLUENCING FORMULATION INCLUDING DRUG- EXCIPIENT INTERACTION & INCOMPATIBILITIES
-
Upload
ketantchaudhari -
Category
Documents
-
view
136 -
download
4
Transcript of Preformulation Studies VL

1
Pre-Formulation Studies
VARIOUS FACTORS INFLUENCING FORMULATION INCLUDING DRUG-
EXCIPIENT INTERACTION & INCOMPATIBILITIES

2
Preformulation
•What does it convey?
•How important is it?
•When is it required?
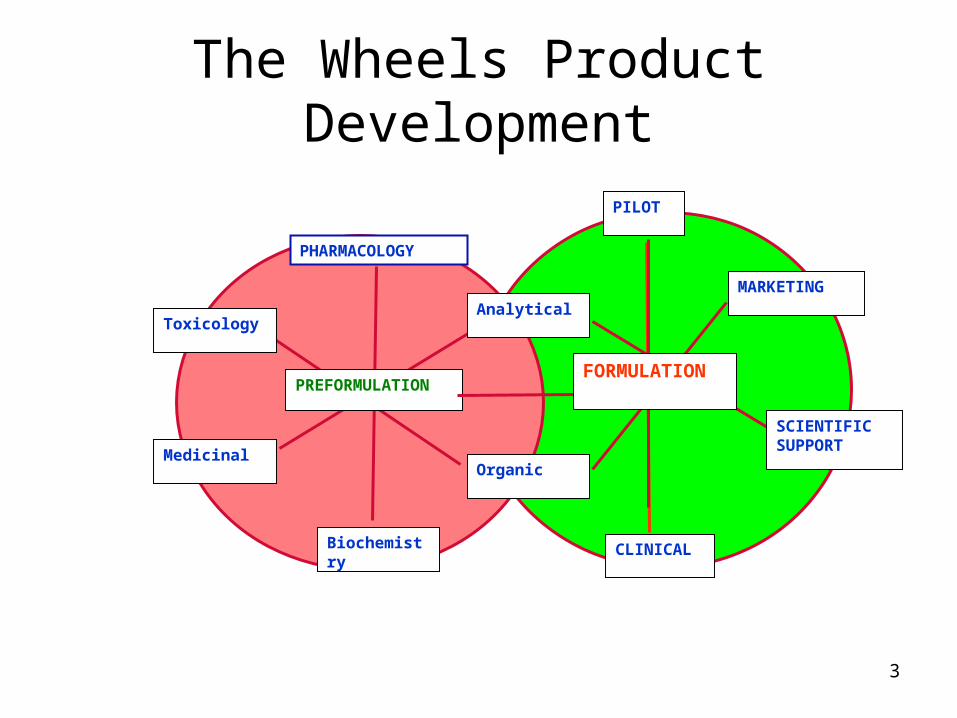
3
The Wheels Product Development
PHARMACOLOGY
Toxicology
Medicinal
Biochemistry
Organic
Analytical
PREFORMULATION
PILOT
MARKETING
SCIENTIFIC SUPPORT
CLINICAL
FORMULATION

4
• Preformulation begins or shall be updated immediately after the synthesis and initial toxicity screening of a new drug
• when a newly synthesised drug shows pharmacological evidence that requires further evaluation in man
• when formulation and dosage form changes are requiredCharacterization of all Preformulation bulk lots is necessary to avoid misleading predictions of stability or solubility, which depend on particular crystalline form.

5
• These studies should focus on:Physicochemical properties that affect drugperformance and development of efficaciousdosage form.
• These properties may provide:Support the need for Molecular modificationsA rationale for Formulation Design

6
Preformulation
Preformulation investigations are designed to deliver all necessary data (especially physicochemical, Physio-mechanical and biopharmaceutical properties of drug substances, excipients and packaging materials) which may Influence
formulation designmethod of manufacture of drug substance and drug productpharmacokinetic/biopharmaceutic properties of the resulting
productpackaging of the product

7
Direct Benefit
Gives directions for development of formulation in choice of dosage form, excipients, composition, physical structure...
Helps in adjustment of pharmacokinetic and biopharmaceutical properties
Support for process development of drug substance (yield, filtration...)
Support for PAT (Process Analytical Technology) (critical process parameters...)
Produce necessary and useful data for development of analytical methods.

8
Where to begin?
Desk work……….
• Active substance• Additives• Process conditions• Analytical means
………………Protocols………………..

9
I- To Generate the physical and chemical properties information that will assist the formulator in developing:
*Stable
*Safe
*Effective Dosage Forms that can be mass produced with:
– Maximum Bioavailability– (Support Dosage Form Design)

10
• II- To provide an initial working model of behaviour of dosage form in vitro and in vivo
• III- To establish the compatibility of the drug with common excipients.
• IV- To establish the kinetic rate profile of the drug.
• V- To Provide a scientific data that support the dosage form design and evaluation of the product efficacy, stability and bioavailability

11
Evaluation Phase
Calculation/Prediction ‘in silico’ of many of the important physicochemical characteristics of NCEsolubilitypolymorphismmorphology of the particlesacute toxicityintestinal permeation

12
Evaluation Phase
Literature search of similar or related compounds to provide an understandingof the degradation processany adverse conditions relevant to the drugbioavailabilitypharmacokinetics and formulation of similar or related compounds

13
Dosage form of choice
Dosage Form Considerations– IV vs. oral formulations– High dose vs. low dose– Excipient compatibility– Interaction with other actives in potential combination
formulations Salts and Other Solubilization Techniques– Effect of Salts on Complexation Binding Constants– Effect of Salts on Solublization by Surfactants– Solubility of Salts in Non-aqueous SolventsToxicological Considerations

14
Choosing the right route
Solid dosage forms– Effect of micronization and processing such as granulation on solid stateproperties and chemical stability– Effect of excipients on crystallization/nucleation– Powder flow properties: bulk density, compression properties and particlesize and shapes
Parenteral Dosage Forms– Injection site precipitation– Pain upon injection– Toxicity of new excipients– Effect of excipients on crystallization/nucleation
Suspensions– Effect of processing and formulation on the physical and chemical stability– Effect of excipients on crystallization/nucleation

15
API
Study what?
16

17
Index of studies Stress Testing of API Impact of Impurities on API Specifications Pre-Formulation Investigations Solid State Degradation & Stability Assessment Role of Excipients in API Instability
Hydrolysis Oxidation Photolysis
API Solubility/Solution-state Stability Assessment Selection of API & Drug Product Processing Methods Degradation Issues for Combination Products Role of API Processing in Product Instability

18
Analytical Preformulation
Know what you start with…..Identity
NMR, UV, TLC, DSC
Purity
Moisture Content, Impurity Profile, DSC, Heavy element, Inorganic elements.
Assay
UV, HPLC
Quality
Appearance, Odour, pH of Slurry, Melting Point

19
Potentially critical attributes of API
Cross reference to stress testing (forced degradation):
1. Sensitivity to temperature (wet granulation, sterilization)
2. Sensitivity to moisture (wet granulation, hygroscopicity)
3. Sensitivity to light (packing materials)
4. Sensitivity to oxidation (inert gas atmosphere in ampoules)
5. Sensitivity to pH (FDC with HCL salts of weak bases)
6. Sensitivity to metal ions (internal peroxide bond)
Expected degradants, manufacturing conditions, etc.
This information is partially available from the Open Part of the DMF

20
Potentially critical attributes of API
Key physicochemical characteristics:
1. Polymorphic or solid state form (amorphous, hydrate, solvate)2. Solubility at 37 oC over the physiological pH range (e.g., BCS,
dissolution testing, cleaning validation)3. Permeability (octanol-water partition) (BCS)4. Crystal habit, particle shape and size (pharmaceutical and
bioequivalence, processability)5. Bulk density, untapped and tapped (processability)6. Flowability (processability)7. Color, odor, taste, consistency (choice of dosage form)should be
discussed and supported by experimental data.

21
Stress Testing of API
Deliberate forced degradation of API - serves several purposes:
To facilitate development of a ‘stability indicating’ analytical method’, e.g. HPLC
To aid in development of the first API specification To understand the degradation pathways of the API to facilitate
rational product development To screen for possible formation of potential genotoxins
Initially performed over a short period of time (28-days) using accelerated or stress conditions (so that reactions proceed more rapidly); target ~10% degradation.

22
Stress Testing of API
Deliberate forced degradation of API
Typical conditions for API in solid-state might be: 80°/75%RH, 60°C/ambient RH, 40°/75%RH, Light irradiation
Typical conditions for API in solution state might be: pH 1-9 in buffered media with peroxide (and/or free radical initiator) Light irradiation

23
Impurities: Impact on API Specification
The allowable level of any given impurity or impurities that are permitted in API/drug product, without explicit non-clinical safety testing, are defined by ICH Q3A/B.
The amounts of impurities that are allowable are based on the total daily intake of the drug product.
There are separate limits (or thresholds) for reporting, identification and qualification of API impurities.
The reporting threshold is defined as the level that must be reported to regulatory agencies to alert them to the presence of a specified impurity.

24
Impurities: Impact on API Specification
The identification threshold is defined as the level that requires analytical identification of a specified impurity.
Finally, the qualification threshold is defined as the level where the specified impurity must be subjected to non-clinical toxicological testing to demonstrate safety.
Threshold limits are defined as a percentage of the total daily intake (TDI) of the drug product, or in absolute terms as the total allowable amount, whichever is lower.

25
Threshold Maximum Daily Dose of API in Drug Product
Threshold Limit Based on TDI
Reporting ≤1g>1g
0.1%TDI0.05%TDI
Identication <1mg1mg-10mg10mg-2g
>2g
1.0%TDI or 5µg
0.5%TDI or 20 µg
0.2%TDI or 2mg
0.1%TDI
Qualification <10mg10mg-100mg
100mg-2g>2g
1.0%TDI or 50µg0.5%TDI or 200µg0.2%TDI or 3mg
0.1%TDI
Impurities: Impact on API SpecificationImpurities: Impact on API Specification

26
API solid-state stability study • An early indication of stability challenges for product
development:
– Accelerated stability challenge but not unrealistically severe and so allows confident extrapolation to provide an indication of API shelf-life
– Conditions less extreme than API stress testing:• 40ºC/75%RH open vial• 50ºC closed vial• At least l month storage data; typically 1w, 2w, 4w, 3m (potentially
supporting 12m shelf-life at RT)• Light stability (ICH conditions); typically 1week• HPLC analysis• Monitor solid-state form (crystallinity) - DSC, TGA, pXRD

27
API solid-state stability study • An early indication of stability challenges for
product development:
Allows comparison with other versions & forms of same API
Provides a control baseline for excipient compatibility studies
Important to bear in mind that API development is ongoing so API batch used in this early stability study may become unrepresentative of final selected API version & form.

28
API degradation pathways
• Hydrolysis and Oxidation are the most common pathways for API degradation in the solid-state and in solution
• Photolysis and trace metal catalysis are secondary processes of degradation

29
API degradation pathways
• Temperature affects each of the above chemical degradation pathways; the rate of degradation increases with temperature. Extrapolation of accelerated temperature data to different temperatures, e.g. proposed storage conditions, is common practice (e.g. using Arrhenius plots) but we must be mindful of the pit-falls – the influence of the various degradation pathways and mechanisms can change with temperature.

30
API degradation pathways
• It is well understood that pH, particularly extremes, can encourage hydrolysis of API when ionised in aqueous solution. This necessitates buffer control if such a dosage form is required. pH within the micro-environment of a solid oral dosage form can also impact on the stability of the formulation where the API degradation is pH sensitive; through understanding the aqueous pH imparted by typical excipients, a prudent choice can overcome this issue.

31
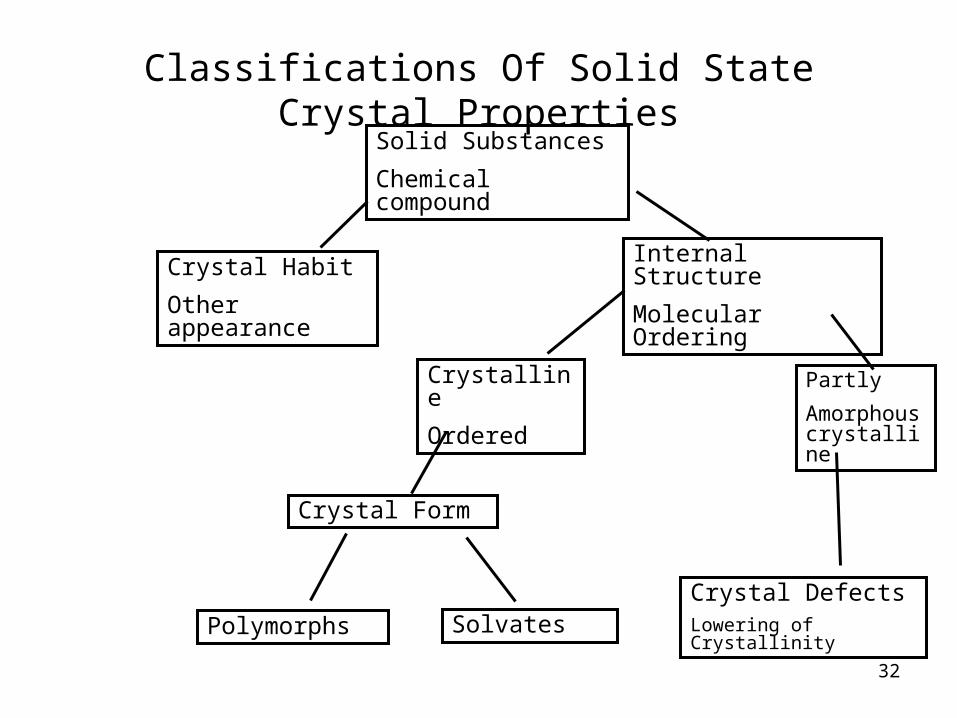
Polymorphism
What is it?
32
Classifications Of Solid State Crystal PropertiesSolid Substances
Chemical compound
Crystal Habit
Other appearance
Crystalline
Ordered
Crystal Form
Polymorphs
Internal Structure
Molecular Ordering
Partly
Amorphous crystalline
Crystal DefectsLowering of CrystallinitySolvates

33
Polymorphism
• Polymorphism is a solid state property of organic molecule which can be obtained in more than one distinct crystal form.
• Polymorphism may also include solvation or hydration products and amorphous forms
• enantiotropic – one polymorph can be reversibly changed into another one by varying the temperature or pressure
• monotropic – the change between the two forms is irreversible

34
Importance of Pharmaceutical Polymorphism
Polymorphic forms of a drug substance can have different chemical and physical properties, including melting point, chemical reactivity, apparent solubility, dissolution rate, optical and mechanical properties, vapor pressure, and density.
These properties can have a direct effect on the ability to process and/or manufacture the drug substance and the drug product, as well as on drug product stability, dissolution, and bioavailability.
Thus, polymorphism can affect the quality, safety, and efficacy of the drug product.

35
Characterization of polymorphs
• X- Ray Diffraction• Microscopy, • Thermal analysis
Differential scanning calorimetry, thermal gravimetric analysis, and hot-stage microscopy
• Spectroscopy
Infrared [IR], Raman, solid-state nuclear magnetic resonance [ssNMR]

36
Why this is Important
• Most important question that likely arises during the registration process is “What assurance can be provided that no other crystalline forms of this compound exist?”.
–The manufacturer of a new drug substance is obliged to show that “due diligence” has been employed to isolate and characterize the various solid-state forms of a new chemical entity

37
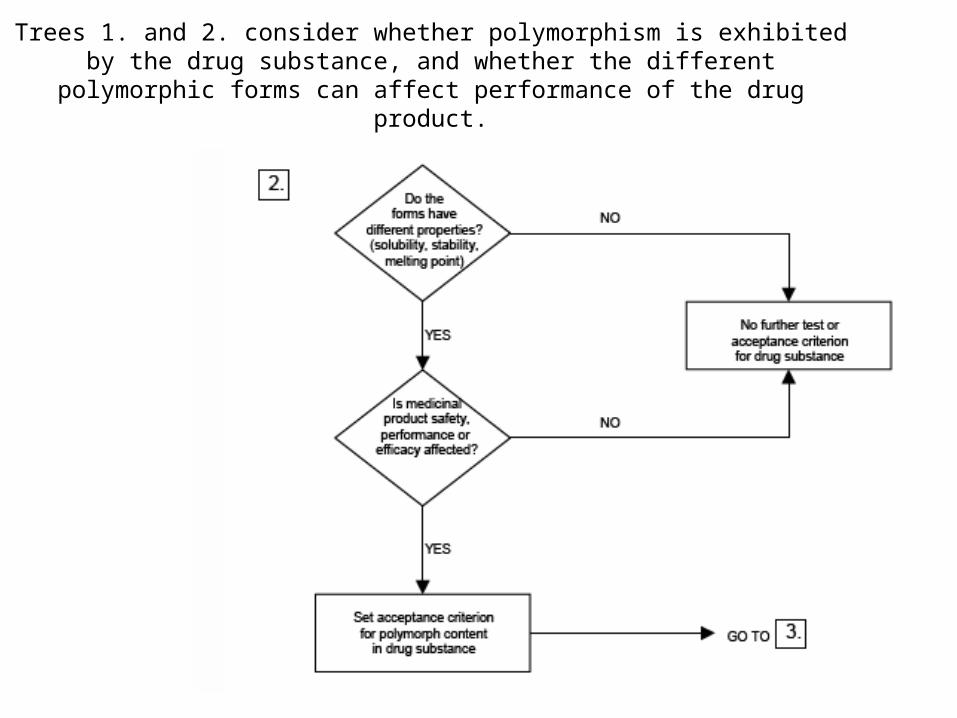
Decision Tree for Acceptance of Polymorphism
38
Trees 1. and 2. consider whether polymorphism is exhibited by the drug substance, and whether the different polymorphic forms can affect performance
of the drug product.
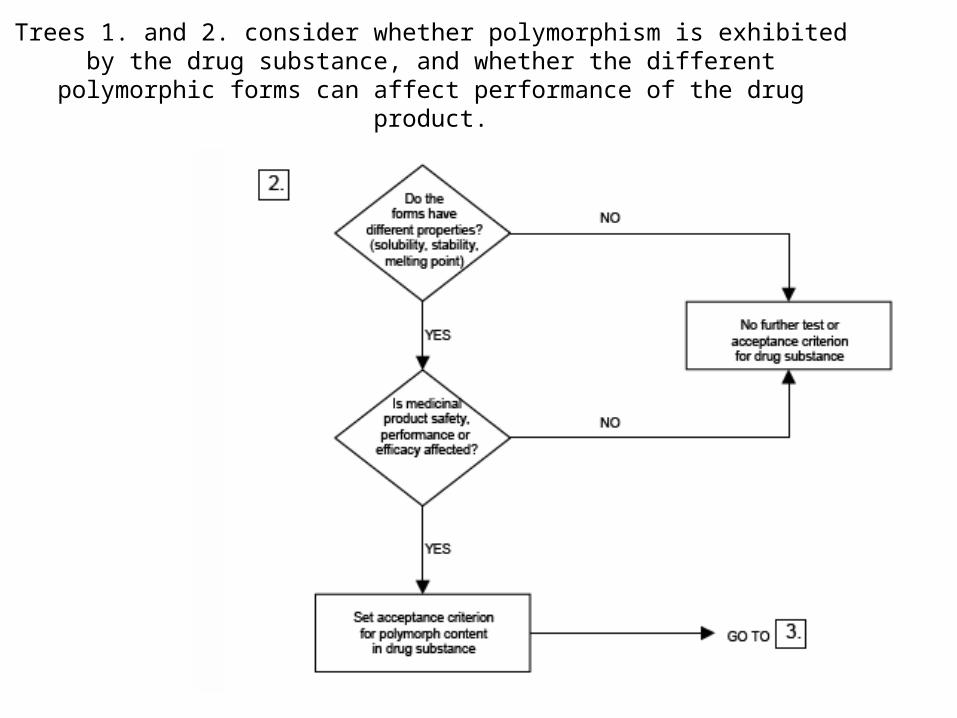
39
Trees 1. and 2. consider whether polymorphism is exhibited by the drug substance, and whether the different polymorphic forms can affect performance
of the drug product.

40
Why This IS Important
Preformulation studies gives answers for critical phenomena so that:
• the desired forms can be consistently manufactured;
• the effects of pharmaceutical manipulations are understood,
e.g., granulation, milling and compression; and
• the effect of storage conditions on the dosage form can be evaluated and predicted

41
Influence of Polymorphism on Drug Substances and Drug products
• Solubility and Dissolution - polymorphic forms differ in their internal solid-state structure, a drug substance that exists in various polymorphic forms can have different aqueous solubilities and dissolution rates.
• BA/BE

42
Influence of Polymorphism on Drug Substances and Drug products
• Influence on manufacturing of Drug products
• Drug substance polymorphic forms can also
exhibit different physical and mechanical properties, including hygroscopicity, particle shape, density, flowability and compactibility, which in turn may affect processing of the drug substance and/or manufacturing of the drug product.
43
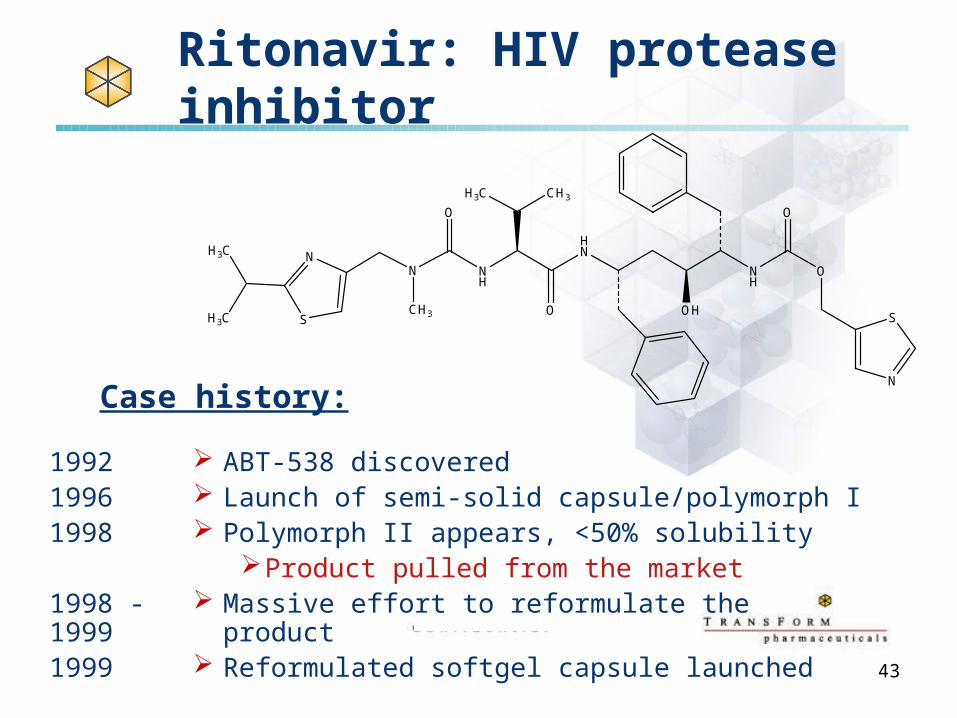
Ritonavir: HIV protease inhibitor
ONH
HN
NH
N
CH3
O
OHO
CH3H3C
O
N
SS
NH3C
H3C
ABT-538 discovered Launch of semi-solid capsule/polymorph I Polymorph II appears, <50% solubility
Product pulled from the market Massive effort to reformulate the product Reformulated softgel capsule launched
Case history:
199219961998
1998 - 19991999

44
Summary of Ritonavir Crystal Forms
IV
mp 122 °C mp 125 °C mp 80 °C mp 97 °C mp 116 °C
Launch in 1996
Summer of 1998
TransForm 2002 – 6 week effort
Launch in 1996
Summer of 1998 Morissette et al. PNAS 100, (2003).
2002 5 forms found

45
API Processing

46
Role of API Processing in Product Instability
• High energy processes (milling, lyophilisation, granulating, roller-compaction, drying) can introduce a degree of amorphicity into otherwise highly crystalline material. This can lead to increased local levels of moisture and increased chemical reactivity in these areas.
• With some materials, ball milling causes irregularity, surface faults and imperfections in crystals. The degree of crystal damage can be directly correlated with the energy of the milling process.

47
Selection of Product Processing
• Understanding of degradation pathways of API will help to decide on most appropriate process:
– For APIs showing severe moisture mediated degradation pathways, choose direct compression or dry granulation

48
Selection of Product Processing• Understanding of physical properties of API will help to
decide on most appropriate process:– For APIs showing flow issues, choose a granulation approach (wet
or dry granulation)– For APIs showing reduced crystallinity after processing e.g. milling,
micronisation, etc., choose wet granulation (presence of water will anneal (crystallise) amorphous API)
– For APIs with low melting point, choose an encapsulation approach (high speed rotary presses will generate significant frictional forces that could melt API)

49
Degradation Issues For Combination Products
• Objective is to minimise incompatibilities. Degradation pathways of the two APIs could well be different, so a stabilisation strategy for API #1 could destabilise API #2.
• In this situation, first intent strategy could be to prepare separate compression blends of each individual API and compress as a bi-layer tablet
–Disadvantages: adds complexity and bi-layer rotary presses are expensive

50
Degradation Issues For Combination Products
• Objective is to minimise incompatibilities. • Alternatively, could compress one of the APIs and over-
encapsulate this into a capsule product, along with the powder blend from the second API
–Disadvantage are that capsule size could be large, it requires specialised encapsulation equipment to fill tablets and blend… process is more complex and expensive
• If however, simplicity and cost are significant issues, look to produce a common blend (particle size of APIs should be similar), and by understanding of degradation pathways stabilise the blend and compress or encapsulate.

51
API related…..
Preformulation studies are an important foundation tool early in the development of both API and drug products. They influence….
Selection of the drug candidate itself Selection of formulation components API & drug product manufacturing processes Determination of the most appropriate container closure system Development of analytical methods Assignment of API retest periods The synthetic route of the API Toxicological strategy

52
Studies designed for Insight
Methodology

Steps in Preformulation Process Pharmaceutical ResearchSteps in Preformulation Process Pharmaceutical Research1. Stability i. Solubility
a. Solid State (1) Water and Other Solvents (1) Temperature (2) pH-Solubility Profile (2) Light (3) Salt Forms (3) Humidity (4) Cosolventsb. Solution (5) Complexation (1) Solvent (6) Prodrug (2) pH j. Effect of pH on UV Spectra (3) Light k. Ionization Constant
2, Solid State Compatibility l. Volatilitya. TLC Analysis m. Optical Activity b. DRS Analysis n. Polymorphism Potential
3. Physico-chemical Properties o. Solvate Formationa. Molecular Structure and Weight 4. Physico-mechanical Propertiesb. Color a. Bulk and Tapped Densityc. Odor b. Compressibilityd. Particle size, Shape, and Crystallinity c. Photomicrographe. Melting Point 5. In Vitro Availability Propertiesf. Thermal Analysis Profile a. Dissolution of Drug Crystal Per se (1) DTA b. Dissolution of Pure Drug Pellet (2) DSC c. Dissolution Analysis of Pure Drug (3) TGA d. Rat Everted Gut Techniqueg. Hygroscopicity Potential 6. Other Studiesh. Absorbance Spectra a. Plasma Protein Binding (1) UV b. Effect of Compatible Excipients (2) IR on Dissolution
c. Kinetic Studies of Solution Degradation 53

54
Preformulation Steps
• Solubility (aqueous, pKa, log P, log D)• Molecular optimization (salt, hydrate, solvate, new
analogs,…)• Crystal Engineering (polymorph, habit, size, surface
characteristics…)• Crystal structure determination• Biopharmaceutical classification (BCS) (solubility,
dissolution, absorption)

55
Preformulation Steps
• Complete Physical and Chemical Characterization• Drug stability evaluation (physical, chemical, solution
phase, solid phase, …)• Compatibility analyses (drug substance, excipient,
packaging materials) • Technical characterization (Flowability,
Compactability, ...

56
Solubility• Solubility may be defined as the
maximum concentration of a substance that may be completely dissolved in a given solvent at a given temperature and pressure.
Drug candidates are becoming more lipophilic and poorly soluble
A Market Survey of 257 Marketed Drug and their liphophilicity

57
definition
• The USP/NF generally expresses the solubility in terms of the volume of solvent required to dissolve 1 gram of the drug at a specified temperature (eg. 1 g ASA in 300 ml H2O, 5 ml ethanol at 25°C).

58
SATURATION SOLUBILITY
• SATURATION SOLUBILITY is understood as a maximum amount of solute that dissolves in a solvent at equilibrium.
• Equilibrium is a state where reactants and products reach a balance - no more solute can be dissolved in the solvent in the set conditions (temerature, pressure). Such a solution is called a saturated solution.

59
Solubility parametersParts of solvent needed for 1 part
of solute
• < 1
• 1 to 10
• 10 to 30
• 30 to 100
• 100 to 1000
• 1000 to 10,000
> 10,000
• Descriptive term
• Very Soluble
• Freely Soluble
• Soluble
• Sparingly soluble
• Slightly soluble
• Very Slightly Soluble
• Practically insoluble or insoluble

60
Formulation challenge in Poorly Soluble Drugs
• Poor dissolution rate• Low and variable bioavailability• More potential for food effect• Inability to deliver high doses for tox studies• Difficulty in developing parenteral formulations
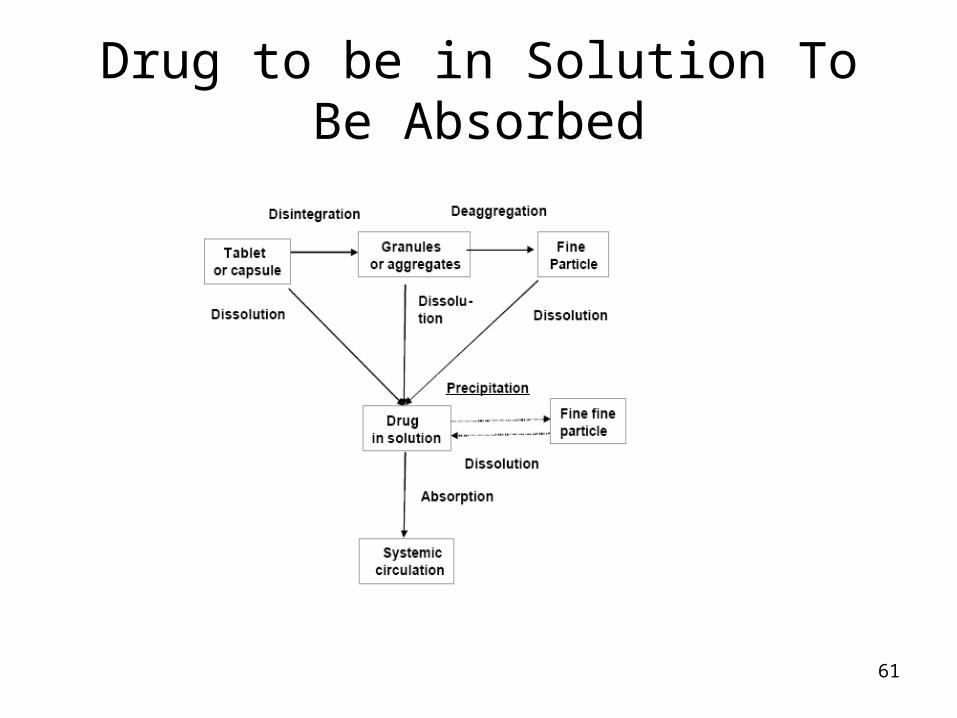
61
Drug to be in Solution To Be Absorbed

62
Biopharmaceutical Classification System
Classification Class I - High Permeability, High Solubility Class II - High Permeability, Low Solubility Class III - Low Permeability, High Solubility Class IV - Low Permeability, Low Solubility Class Boundaries• Highly soluble: the highest dose is soluble in <250 ml
water over a pH range of 1 to 7.5• Highly permeable: >90% dose absorbed in humans• Rapidly dissolving: >85% of labeled amount of drug
substance dissolves within 30 minutes

63
Solvent for Solubility StudiesFor “developability” assessment:– Simulated gastric fluid (SGF)– Simulated intestinal fluid (SIF)– pH 7.4 buffer– Intrinsic solubility to estimate pH-solubility profile For Formulation Development:– pH solubility profile

64
Factor Causing poor solubility
• High Crystallinity/high MP
– Zwitterion formation
– Insoluble salts
– H-bonding networks
• Hydrophobicity/High LogP
– Lack of ionizable groups
– High molecular weight

65
Dissolution
• Dissolution rate for poorly soluble compounds may often be the rate limiting step to absorption
• Examples of drugs with dissolution rate limited absorption:- Digoxin - Penicillin V - Phenytoin
- Quinidine - Tetracyclines

66
Approaches to improving solubility
Buffers Co-solvents (GRAS approved) Additives/complexes -surfactants, polymers,
cyclodextrins Lipid-based systems (solutions, emulsions) Solid-state modification (particle size reduction, salt
formation, solid-state stabilisation of the amorphous state)

67
Particle Size
Important?

68
Micromeritics
Micromeritics is the science of particle sizes, particle shapes, size distributions, porosity, density and surface areas.Methods
• Microscopy
• Sieving
• Laser diffraction
• Others: Sedimentation, Coulter counter etc.
69
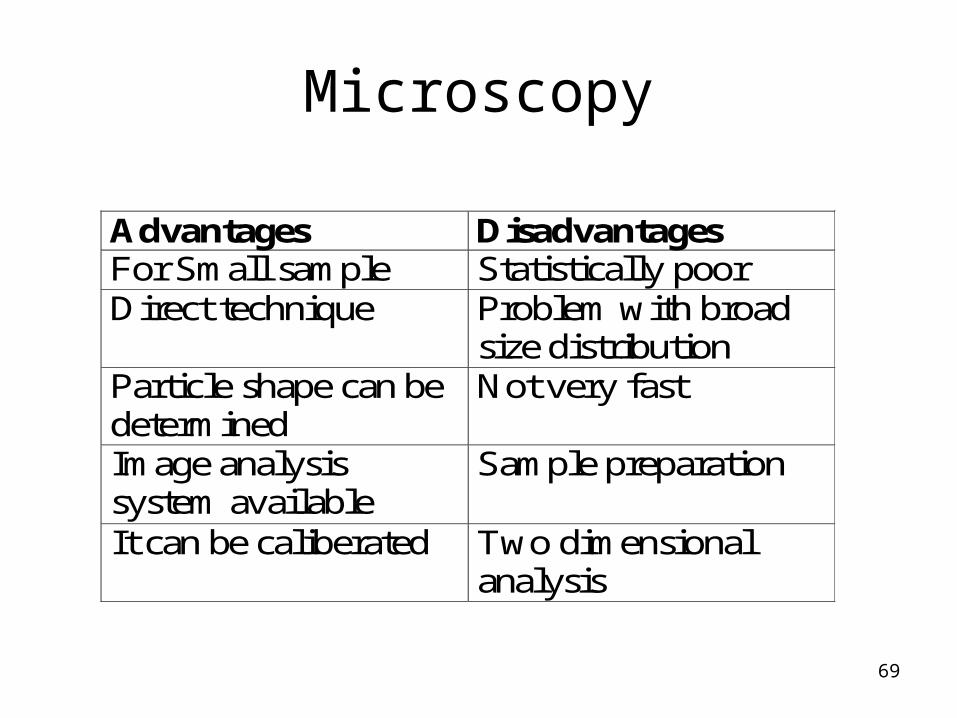
Microscopy
Advantages Disadvantages For Small sample Statistically poor Direct technique Problem with broad
size distribution Particle shape can be determined
Not very fast
Image analysis system available
Sample preparation
It can be caliberated Two dimensional analysis

70
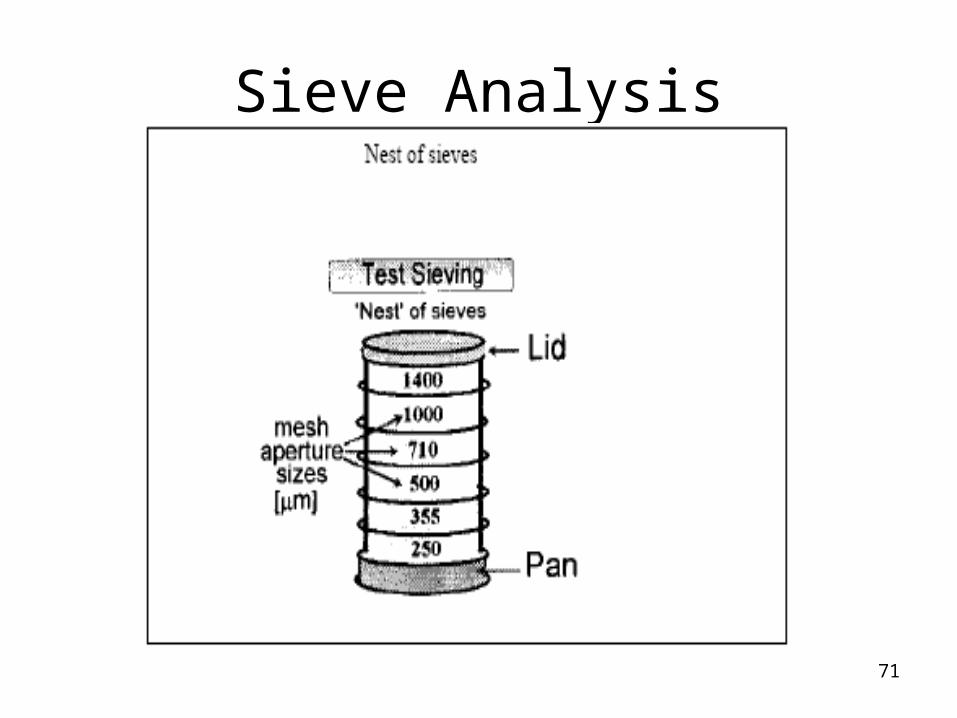
Sieve Analysis
Advantages
Determining samples having broad size distribution.
Easy to operate
Calibrated sieves
Disadvantages
Inaccuracy increases in the size of < 75 um.
Large sample size.
71
Sieve Analysis

72
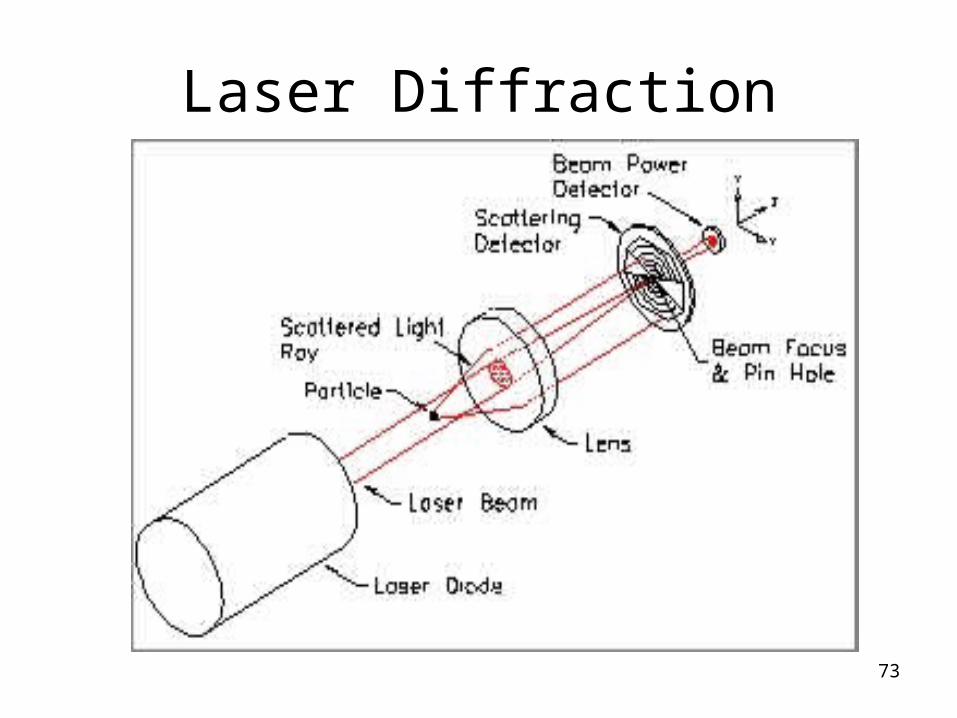
Laser Diffraction
Advantages: Precise Fast Analysis.Small sample amount Well automated and statistically good method Accepted by various regulatory authorities.Disadvantages:Needs expertise.Expensive.Sample is destroyed.
73
Laser Diffraction

74
Volume Particle size distribution
75
ACCEPTANCE CRITERIA FOR DRUG SUBSTANCE PARTICLE SIZE DISTRIBUTION
76
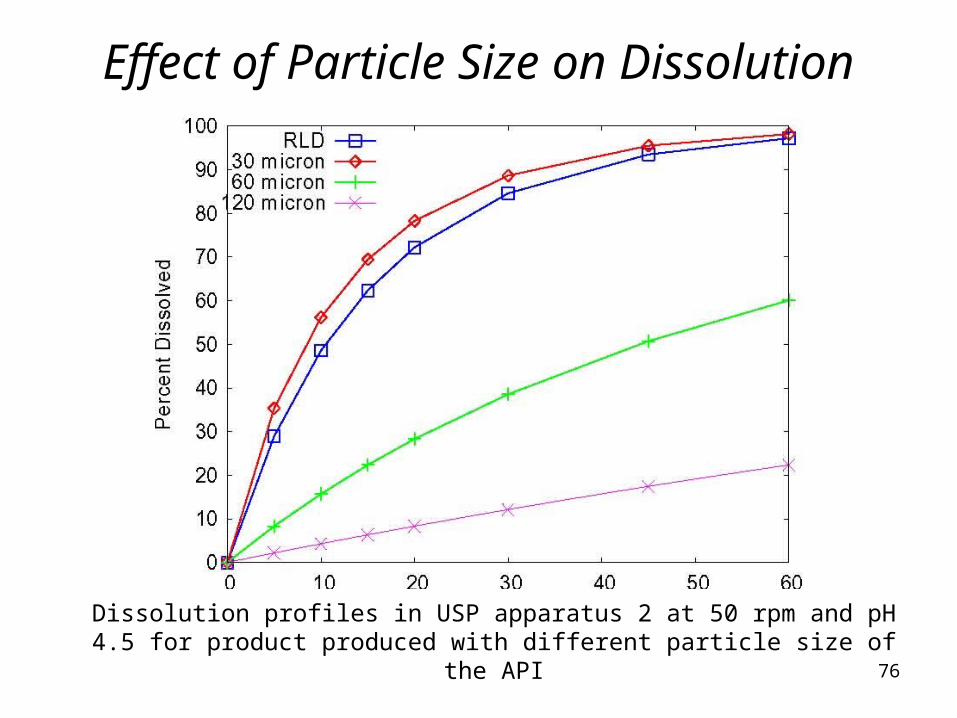
Effect of Particle Size on Dissolution
Dissolution profiles in USP apparatus 2 at 50 rpm and pH 4.5 for product produced with different particle size of the API
77
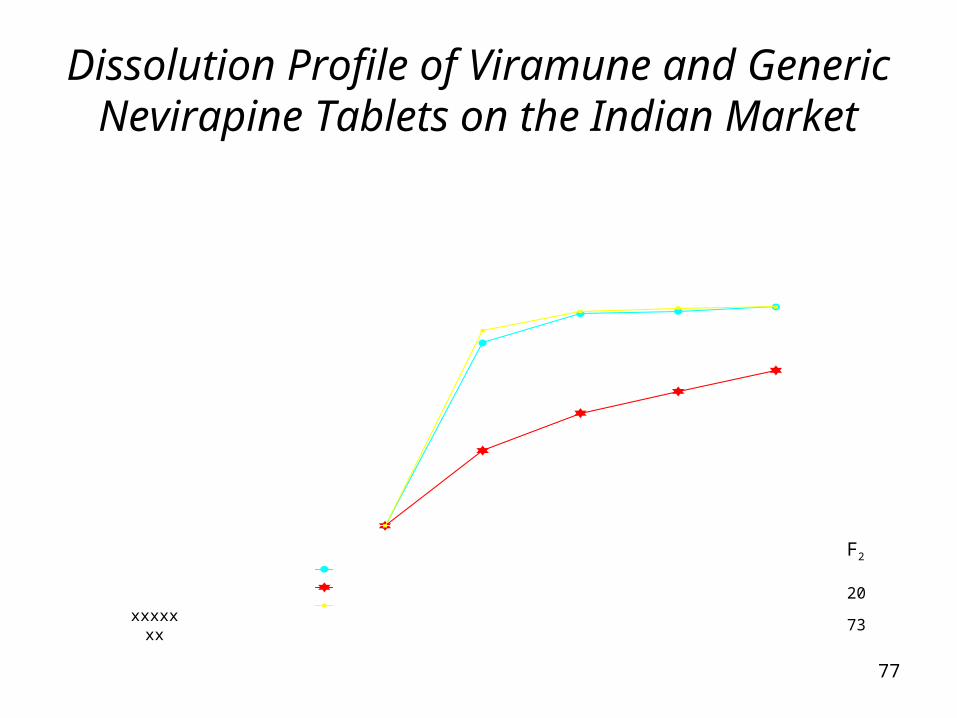
Dissolution Profile of Viramune and Generic Nevirapine Tablets on the Indian Market
USP Type II / 0.01N HCl 50 RPM / 900 ml
0
20
40
60
80
100
120
Time (Min)Viramune B.No.992633BBrand C B.No.C00139Ranbaxy B.No.(1024)17
0 10 20 30 450.0 83.3 96.6 97.7 99.70.0 34.3 51.3 61.2 70.80.0 88.9 97.6 99.2 99.7
% D
rug
Dis
so
lve
d
F2
20
73xxxxxxx

78
Flow
How important?

79
Flowability
Primary forces at work:• Friction
– Particle size and shape• Cohesion
– Moisture, fats, oils– Molecular forces, electrostatic forces
• Environmental– Temperature, pressure, time
• Physical– Bin angle, aperture diameter– Wall conditions

80
Flowability methods
Flowing time

81
Flowability Tester Model BEP-Auto
• Copley-Scientific

82
Flowability Tester Model BEP-Auto
• The BEP Series can be used in 4 modes: - • Determination of the flow time of a
predetermined sample weight • Determination of the flow time of a
predetermined sample volume • Determination of the weight of sample in a
predetermined time • Plot of time against sample weight
(Weight/Time)

83
Flowability Tester Model BEP-Auto
• According to EP, the flowability is expressed in seconds and tenths of seconds, related to 100 grams of sample.
• The results can be expressed as: -(1) the mean of the three results, if none of the individual values
deviate from the mean value by more than 10%
(2) as a range, if the individual values deviate from the mean value by more than 10%
(3) as a plot of the mass against the flow time or
(4) as an infinite time, if the entire sample fails to flow through.

84
Angle of Repose
• Ө = 2H tan 1
D
• H = Height of cone [4.1 cm]
• D = Base of cone [9.8 cm]
• Ө = Angle of repose
Ө » 2 x 4.1 tan 1 = 39.93 o
9.8

85
Angle of reposeFlow property Angle of repose {degrees}
Excellent 25 - 30
Good 31 – 35
Fair-aid not needed 36 - 40
Passable- may hang up/ block 41 - 45
Poor- must agitate vibrate 46 - 55
Very Poor 56 - 65
Very, very poor > 66

86
Carr IndexCarr Index = Tapped Density- Bulk Density
Tapped Density
X 100
Carr Index (%) Type of Flow
5-15 Excellent
12-16 Good
18-21 Fair to Passable*
23-35 Poor*
33-38 Very poor
> 40 Extremely Poor

87
Hygroscopicity
Environment control

88
• Slightly hygroscopic: Increase in mass is less than 2 percent m/m and equal to or
• greater than 0.2 percent m/m.• • Hygroscopic: Increase in mass is less than 15
percent m/m and equal to or greater than• 0.2 percent m/m.• • Very hygroscopic: Increase in mass is equal to
or greater than 15 percent m/m.• • Deliquescent: Sufficient water is absorbed to
form a liquid.
89
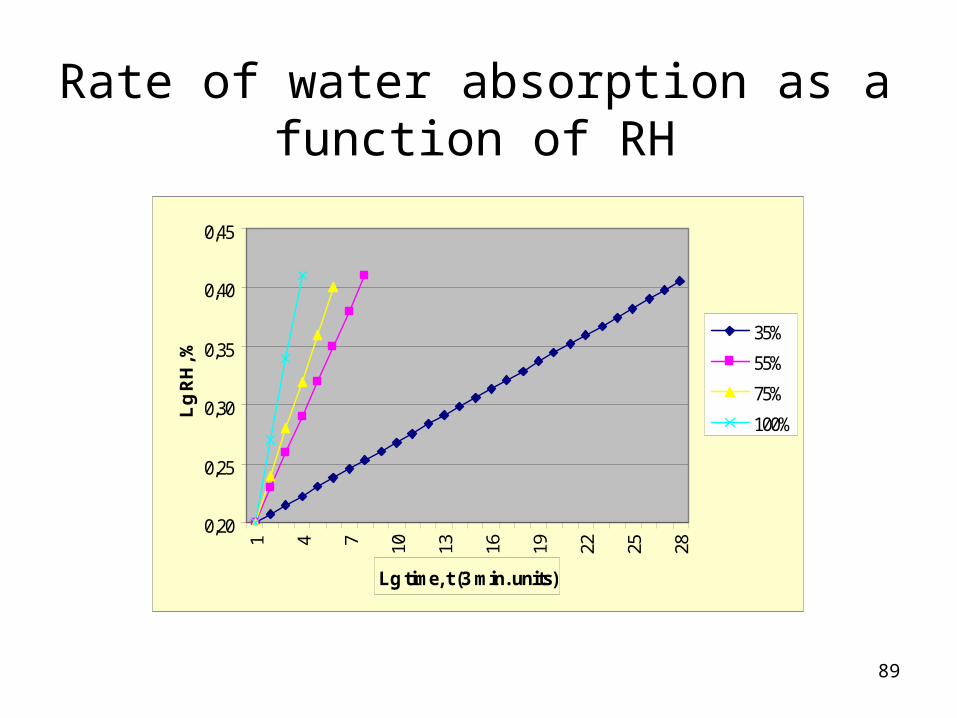
Rate of water absorption as a function of RH
0,20
0,25
0,30
0,35
0,40
0,451 4 7 10 13 16 19 22 25 28
Lg time, t (3 min. units)
Lg
RH
, %
35%
55%
75%
100%

90
Dynamic Vapour Sorption (DVS)• The relative humidity is generated by bubbling nitrogen through a
water reservoir where it is saturated with moisture. • Using a mixing chamber, the moist nitrogen is mixed with dry
nitrogen in a fixed ratio, thus producing the required relative humidity.
• The moist nitrogen is then passed over the sample, and the instrument is programmed such that the increase in weight due to moisture is monitored with time using an ultrasensitive microbalance.
• The compound takes up moisture and reaches equilibrium, at which point the next relative humidity stage is programmed to start.
• The adsorption and desorption of moisture can be studied using this instrument, and the effect of temperature can be investigated as well.
• Using this technique, a quantity as small as 1 mg can be assessed.

91
Corrosive potential
Affects which stage?

92
Corrosivity
• Personnel safety
• Equipment
• Pack
• Scale of operations
• Cleaning process
• Handling procedures

93
What is covered ?
– Bulk propertiescrystallinity and polymorphismHygroscopicity (water adsorption)Fine Particle Characterization
(particle size, shape, and surface area)powder flow Properties
(Bulk Density, angle of repose, compressibility)

Ionization Constant
• The unionized species are more lipid-soluble and hence more readily absorbed.
• The gi absorption of weakly acidic or basic drugs is related to the fraction of unionized drug in solution.
• Factors affecting absorption:
- pH at the site of absorption
- Ionization constant
- Lipid solubility of unionized species
“pH-partition theory”94

95
What is covered?
solubility analysisIntrinsic Solubilityionization ConstantpH solubility profilepartition coefficientsSaltsSolventsDissolution

96
Dissolution testing
• Dissolution testing is used for the selection of the formulation and comparison of the dissolution profiles with that of the innovator product and clinical batches. This should be a basic strategy in pharmaceutical development to maximize the chances of bioequivalence.
• Limits should be set for each API in fixed-dose FP’s. • The dissolution method should be incorporated into the
stability and quality control programs. • Multipoint dissolution profiles of both the test and the
reference FP’s should be compared.

97
Dissolution profile testing
• Three media - 900 ml or less - all at 37°C – Buffer pH 1.2, SGF without enzymes or 0.1M HCl– Buffer pH 4.5– Buffer pH 6.8 or SIF without enzymes
Water may be used additionally (not instead of)2. Paddle at 50 or basket at 100 rpm3. Twelve units of each product in all 3 media4. Dissolution samples collected at short intervals, e.g.
– 10, 15, 20, 30, 45 and 60 minutes– Analyse samples for all APIs, when applicable

98
Stability Analysis……..
Serves two purposes:• To evaluate the specificity of the ‘stability indicating’
method’, e.g. LC• To understand the degradation pathways of the API to
facilitate rational product development i.e.Hydrolysis, Oxidation, Photolysis and the role of pH
• Wherever possible, commercial pharmaceutical products should have shelf-life of Three years.
• The potency should not fall below 90 or 95% under the recommended storage conditions and the product should still look and perform as it did when first manufactured.

99
Drug degradation processes
Drug degradation occurs by four main processes:• Hydrolysis due to H2O, H3O+, OH-, pH• Oxidation O2• Photolysis UV and visible• Trace metal ion catalysis Fe2+, Fe3+, Cu2+,
Co2+, etc
Hydrolysis and oxidation are the most common pathways!

100
Typical Stress Condition for FP/s in Solid State
Storage conditions Testing period*
40°C, 75 % RH; open
storage** 3 months
50-60 °C, ambient RH; open
storage 3 months
Photostability; according to ICH
according to ICH
* 3 months or 5-15% degradation, whatever comes first** For API1-API2, or API-excipient, or FPP (finished pharmaceutical product) without packing material, typically a thin layer of material is spread in a Petri dish. Open storage is recommended, if possible.

101
Stress Testing of API in Solution
Storage conditions Testing period*
pH ± 2, room temperature 2 weeks
pH ± 7, room temperature 2 weeks
pH ± 10-12, room temperature 2 weeks
H2O2, 0.1-2% at neutral pH,
room temperature
24 hours
* Storage times given or 5-15% degradation, whatever comes first

102
Arrhenius Equation
Thermal effects are superimposed on all four chemical processes mentioned above.
Typically a 10ºC increase produces a two- to five fold increase in the rate of reaction.
Example. Storing b-lactam penicillins in a refrigerator reduces the hydrolysis rate by 90% of that at room temperature.
K = A e –Ea/RT or
log K = log A – (Ea/2.303R)/T.
Plotting the rate of reaction (K) against 1/T K allows the calculation of rate at any temperature (Ea, activation energy; R, gas constant), and therefore a prediction of shelf-life (t90, time to 90% potency).
This forms the basis of many accelerated stability tests!

103
PHOTOSTABILITY• Many pharmaceutical drug molecules are known to be
sensitive to light and degrade chemically under light exposure.
• During the development we have to evaluate this risk and how we can stabilise a potential photo-labile drug with packaging material, films and/or colorants.
• This evaluation of the drug molecule can be done by Isothermal Microcalorimetry (IMC) in real time.
• Photosensitivity of molecules is often studied in solutions, and the changes in concentrations have been examined by chromatography and spectrophotometry. Discoloration of powders has been examined by colorimetry.

Photostability studies
A systematic approach to photostability testing is recommended covering, as appropriate, studies such as:
(i) Tests on the drug substance; (ii) Tests on the exposed drug product outside
of the immediate pack; and if necessary,(iii) Tests on the drug product in the immediate
pack; and if necessary,(iv) Tests on the drug product in the marketing
pack.104

Photostability As Per ICH
Two types of studies, exposure is cumulative from the light sources
• Forced degradation study to generate potential degradation products
– 2 X exposure to UV and fluorescent sources
• Confirmatory study to confirm product and package performance
– NLT 1.2 million Lux hours + 200 watts/hrs per sq. meter
105

Photostability studies
Light Sources:Option 1• Dual output light sources, such as D65/ID65• Simulates artificial day light fluorescent lamp• Use Xenon or Metal HalideOption 2• Tandem exposure to single light source types• Cool white fluorescent lamp + near UV fluorescent
lamp (320-400 nm)• Accumulate exposure under one, then the other
106

Solid state studies
107
• Essential

108
Solid State Physical Stability & compatibility of
pharmaceutical Substances
Incompatibility is an undesirable interaction between two or more substances
drug – drug,
drug – excipient,
drug – packing material,
excipient – packing material…
• Interaction can be chemical, physical or both
• Physical/chemical stability data of pure components obtained at the same time

109
Advantages
• Prerequisite of the stable and effective formulation
• Saves time
- facilitate formulation process (exclude high-risk excipients)
• Lowers risk
- support process and packaging development decisions
• Saves money
- minimize the number of needless, formal (expensive)
stability studies

• Dissolution• Crystallization• Growth of crystals• Crystallinity changes• Phase transitions• Degradation• Degradation by nucleation via the gaseous phase {a contracting surface due to nucleation with coverage by
the breakdown products}• Degradation mediated by surface moisture• Oxidation• Photolysis
110
Potential changes due to Incompatibility

111
Methods
• Applied procedures
– Desktop working
– Traditional way
– Microcalorimetric way
• Application of Phys. analytical methods
– X-Ray Powder Diffraction, XRPD
– Differential Scanning Calorimetry, DSC
– Isothermal Microcalorimetry, IMC
– Inverse Gas Chromatography, IGC
– Chemical methods (HPLC, TLC,…)

112
Desktop Working
PROCEDURE molecular formula of active substance and potential
excipient are compared to each others potential interaction points are listed molecule modeler typically hunts ‘compatible
molecules’ with target molecule as a purpose getting strong interaction with the binding site of the target molecule
pharmaceutical formulator decides on the basis of the list of excipients which have a minimum number of potential interaction points

113
Traditional ProcedureOne option Binary Mix Compatibility Testing: samples or of drug and excipients are intimately mixed
(1:1), but other mixtures used as well samples can be either powder blends or compressed
discs one set of samples are moistened and sealed into ampoules to prevent moisture loss
study design stored under room and accelerated conditions
– at least 1 week in accelerated conditions 40ºCRH75% or 60 ºC RH75%
– 1-3 month in room conditions (25ºCRH60%) analysed at various time points using HPLC, DSC and
TGA (NIR, Raman, XRD,…) and visually

114
Traditional Procedure• However, the binary mix approach takes time and resources and….it
is well known that the chemical compatibility of an API in a binary
mixture may differ completely from a multi-component prototype
formulation.
• An alternative is to test “prototype” formulations. The amount of API in
the blend can be modified according to the anticipated drug-excipient
ratio in the final compression blend.
– Platform prototypes can be used for specific dosage forms, e.g.
DC vs. wet granulation in tablets
– There is better representation of likely formulation chemical and
physical stability
– However, this is a more complex system to interpret
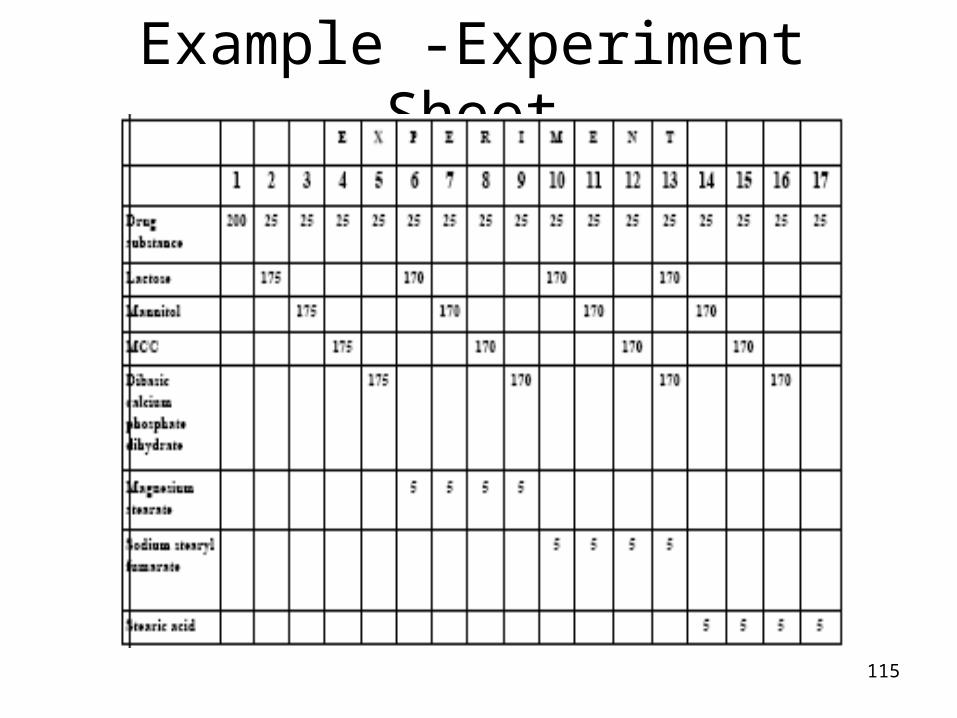
115
Example -Experiment Sheet

116
Drug excipient….
• Can apply statistical models (e.g. 2n factorial design) to determine the chemical interactions in more complex systems such as prototype formulations, with a view towards establishing which excipients cause incompatibility within a given mixture.

117
Microcalorimetric Procedure
• Most chemical or physical processes are due to changes in free energy and are usually accompanied by heat change.
• ISOTHERMAL MICROCALORIMETRY (IMC) can monitor these changes
• ISOTHERMAL MICROCALORIMETRY (IMC) is exceptionally sensitive technique to see any reactions in the sample:
- It has been estimated that a change that would take more than 200 years to run to completion could be easily detected with IMC- temperature difference sample versus reference < 10-6 ºC detectable- degradation rate 0.01%/annum detectable
• Non-specific method- for fully interpretation of the data it is necessary to investigate the origin of the thermal events by additional methods

118
Method
• about 1 gram of sample into the specific sample ampoule
• heat flow curves of pure substances are compared to those of the binary mixtures
• incompatibility is revealed if the features of the heat flow curve of the mixture doesn’t resemble the sum curve of the components of the binary mixture studied.
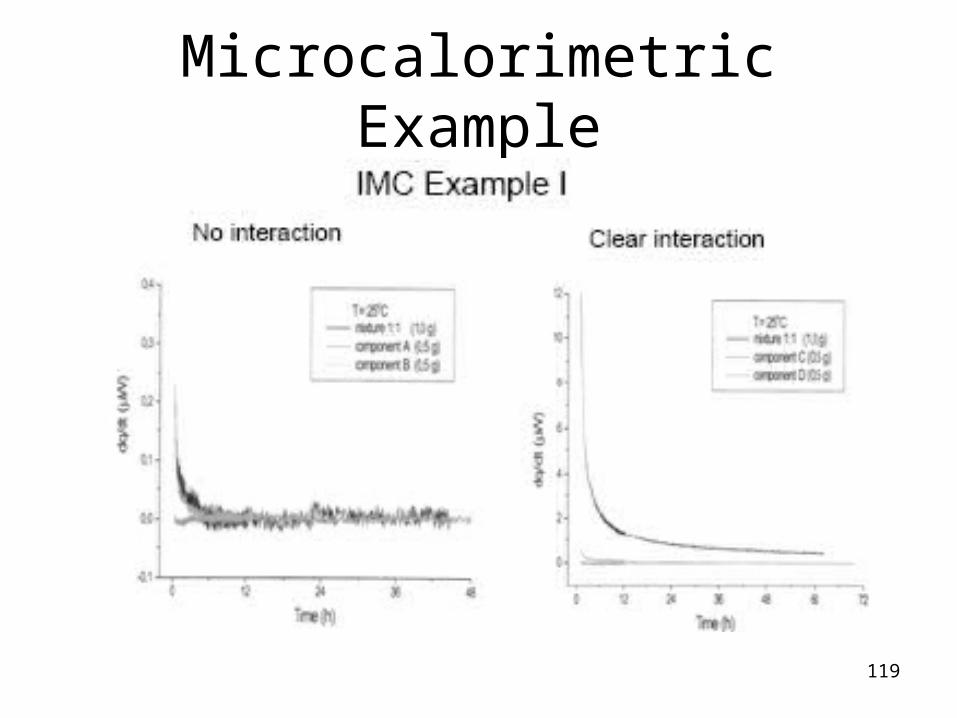
119
Microcalorimetric Example

120
XRD Application/Methods
X-Ray Powder Diffraction• powerful equipment to evaluate solid state properties
- polymorphism
- solvation state (solvates, hydrates)
- Crystallinity• useful method to reveal any other phase transition or
solid state impurities of the samples.• non-ambient XRD can give valuable supporting data for
stability prediction
- humidity and temperature can be controlled
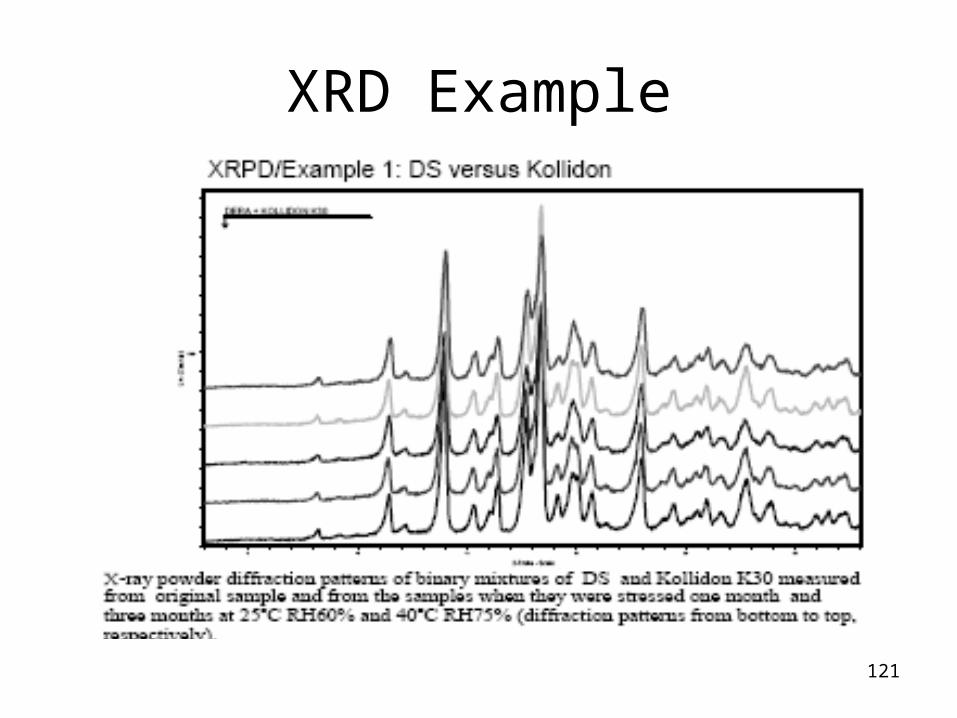
121
XRD Example

122
DSC Application Method
Differential Scanning Calorimetry- DSC
• requires 5 mg of drug, in a 50% mixture with excipient• mixtures should be examined under nitrogen to eliminate
oxidative and pyrrolytic effects• standard heating rate (2,5 or 10ºC min-1) over a
temperature range on the DSC apparatus• the melting range and any transitions of the drug will be
known from earlier investigations into purity, polymorphism and solvates.
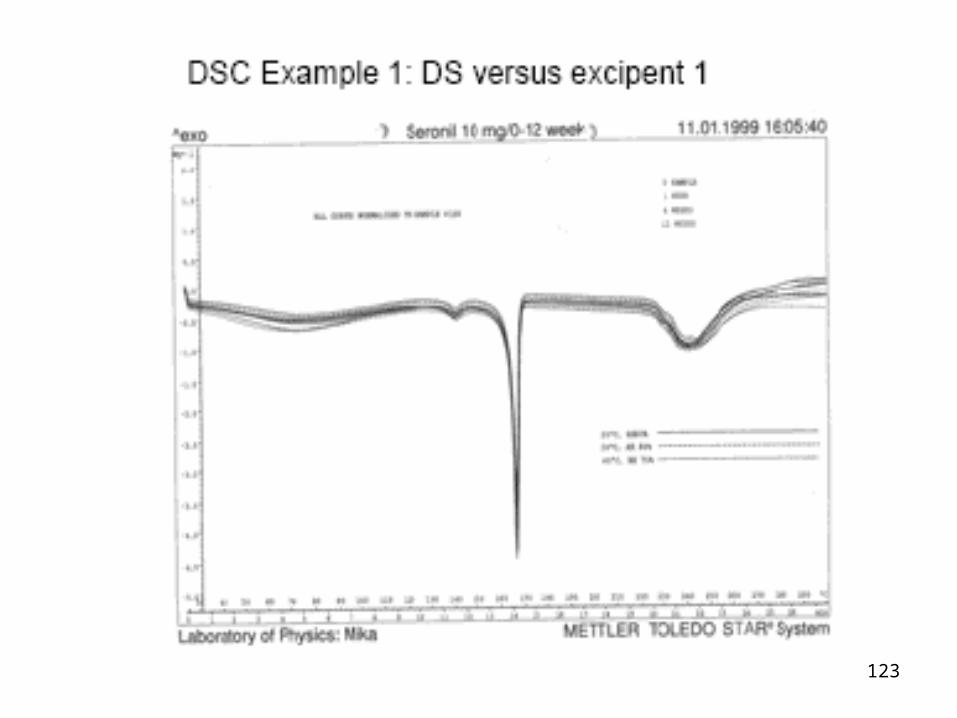
123

124
Excipient Compatibility
API: EXC
EXC:EXC
API: PACK

Excipients:API Interaction Whereas excipients are usually biologically inactive, the
same cannot be said from a chemical perspective.
Excipients, and any impurities present, can stabilise and/or destabilise drug products.
Considerations for the formulation scientist:– the chemical structure of the API – the type of delivery system required– the proposed manufacturing process
125

Excipients:API Interaction Initial selection of excipients should be based on:
– expert systems; predictive tools– desired delivery characteristics of dosage form– knowledge of potential mechanisms of degradation, e.g.
Maillard reaction– Prior development experience
The objective of drug/excipient compatibility considerations and practical studies is to delineate, as quickly as possible, real and possible interactions between potential formulation excipients and the API. This is an important risk reduction exercise early in formulation development.
126

Excipient Compatibility Studies
One option….Binary Mix Compatibility Testing:
In the typical drug/excipient compatibility testing program, binary (1:1 or customised) powder mixes are prepared by triturating API with the individual excipients.
These powder samples, usually with or without added water and occasionally compacted or prepared as slurries, are stored under accelerated conditions and analysed by stability-indicating methodology, e.g. HPLC.
(The water slurry approach allows the pH of the drug-excipient blend and the role of moisture to be investigated.)
127

Excipient Compatibility Studies
128
One option….Binary Mix Compatibility Testing:
Alternatively, binary samples can be screened using thermal methods, such as DSC/ITC. No need for stability set-downs; hence cycle times and sample consumption are reduced. However, the data obtained are difficult to interpret and may be misleading; false positives and negatives are routinely encountered.
Also sensitive to sample preparation.

129April 8, 2023 129
However, the binary mix approach takes time and resources and it is well known that the chemical compatibility of an API in a binary mixture may differ completely from a multi-component prototype formulation.
An alternative is to test “prototype” formulations. The amount of API in the blend can be modified according to the anticipated drug-excipient ratio in the final compression blend.
• Platform prototypes can be used for specific dosage forms, e.g. DC vs. wet gran tablets• There is better representation of likely formulation chemical and physical stability• However, this is a more complex system to interpret
Excipient Compatibility StudiesExcipient Compatibility Studies

Drug excipient studies
130
Drug-excipient interactions can be studied using both approaches in a complementary fashion. The first tier approach is to conduct short-term (1-3m) stability studies using generic prototype formulations under stressed conditions, with binary systems as diagnostic back-up: Chemical stability measured by chromatographic methods
Physical stability measured by microscopic, particle analysis, in vitro dissolution methods, etc.
The idea is to diagnose any observed incompatibility from the prototype formulation work then hopefully identify the “culprit” excipients from the binary mix data.
Hopefully, a prototype formulation can then be taken forward as a foundation for product development.

• Can apply statistical models (e.g. 2n factorial design) to determine the chemical interactions in more complex systems such as prototype formulations, with a view towards establishing which excipients cause incompatibility within a given mixture.
131

132
EXCIPIENTS - STARCH
All starches are hygroscopic and rapidly absorb atmospheric moisture. Approximate equilibrium moisture content values at 50% relative humidity are:
• 11% FOR MAIZE (CORN) STARCH,• 18% FOR POTATO STARCH,• 14% FOR RICE STARCH, AND
• 13% FOR WHEAT STARCH.
Between 30-80% relative humidity, corn starch is the least hygroscopic starch and potato starch is the most hygroscopic starch.

133
Oxidation and the Role of Excipients
• Oxidation is broadly defined as a loss of electrons in a system, but it can be restated as an increase in oxygen or a decrease in hydrogen content.
• Oxidation always occurs in tandem with reduction; the so-called REDOX reaction couple.
• Oxidation reactions can be catalysed heavy metals, light, leading to free radical formation (initiation). Free radicals then react with oxygen to form peroxy radicals, which react with the oxidative substrate to yield further complex radicals (propagation), finally the reaction ceases (termination).

134
Oxidation and the Role of Excipients
• Excipients play a key role in oxidation; either as a primary source of oxidants, trace amounts of metals, or other contaminants.
• E.g. Peroxides are a very common impurity in many excipients, particularly polymeric excipients. They are used as initiators in polymerisation reactions, but are difficult to remove.

135
Hydrolysis and the Role of Excipients• One of the most common pathways of drug product
degradation is hydrolysis. • The source of the excipients can greatly influence
hydrolytic reactions.
• For example, talc obtained from different sources impacts markedly on the overall stability of the aspirin tablet formulation.
• This is possibly attributable to the affect of different types and amounts of surface impurities, which are dissolved in the adsorbed moisture layer, where they subsequently react with the API. It could also influence the pH of the micro-environment.

136
Drug Excipient Incompatibilities
• Ahlneck and Lundgren studied the compatibility of aspirin in the presence of 3 common diluents; – lactose– microcrystalline cellulose – dicalcium phosphate
• Dicalcium phosphate despite having much lower moisture pick up levels than microcrystalline cellulose, had a greater de-stabilising effect.
• Attributed to the alkalinity of the dicalcium phosphate in the solid state (pH 7.4). The increase in the pH adversely affects the stability of the formulation, despite minimal solubility in water.

137
Photolysis and the Role of Excipients• Sunlight (both in the UV and visible regions) may degrade
drug products and excipients; and consequently photolabile APIs can raise many formulation (& phototoxicity) issues.
• The addition of light absorbing agents is a well known approach to stabilising photolabile products. – Order of effectiveness: pigments > colorants > UV
absorbers• However, beware variable performance between
grades/sources. e.g. Surface-treated titanium dioxide is inferior to the untreated excipient as an opacifier.

138
More examples….
• Lactose can react with primary amines.
• Silicone dioxide acts as Lewis acid

139
Conclusion
Preformulation Study is Major tool for pharmaceutical product Development - it saves time as well as cost for development of Dosage form.

140
• Pharmaceutical Preformulation and Formulation by Mark Gibson
• The theory and practice of Industrial pharmacy by Lachman
• Pharmaceutics by Aulton
• Handbook of Preformulation by S. Niazi

141
Thank you!


















