
Prediction of Functional Quantitative Traits in Plants...
91
Prediction of Functional Quantitative Traits in Plants using a Whole-Genome Population- Based Approach Tatiana Tatarinova
-
Upload
hoangduong -
Category
Documents
-
view
218 -
download
1
Transcript of Prediction of Functional Quantitative Traits in Plants...

Prediction of Functional Quantitative Traits in Plants using a Whole-Genome Population-
Based ApproachTatiana Tatarinova

What is the SNP
• SINGLE • NUCLEOTIDE • POLIMORPHISM
Human genome has about 3 billion nucleotides Two randomly selected individuals differ by
<0.5% of a genome (<15 mil)

Why do we care about SNPs?
• History • Personalized medicine • Clinical trials • Forensics • Agriculture and breeding • Monitoring rare species

DNA
…TCAGGTCACAGTCT…
…TCAGGTCACAGTCT……TCAGGCCACAGTCT……TCAGGCCACAGTCT…
Individual 1
Individual 2
Individual 3
DNA
Reference sequence
Katarzyna Bryc, 2013

Single Nucleotide Polymorphism (SNP)
…TCAGGTCACAGTCT……TCAGGCCACAGTCT……TCAGGCCACAGTCT…
SNP A.k.a. allele, locus, marker, variant
Katarzyna Bryc, 2013
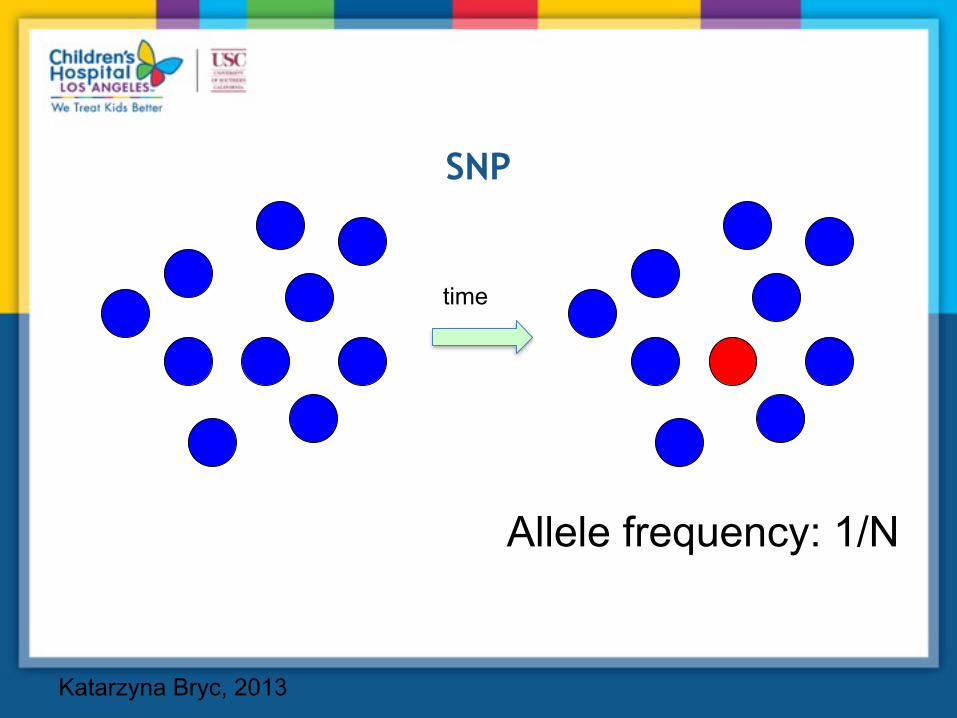
SNP
Allele frequency: 1/N
time
Katarzyna Bryc, 2013
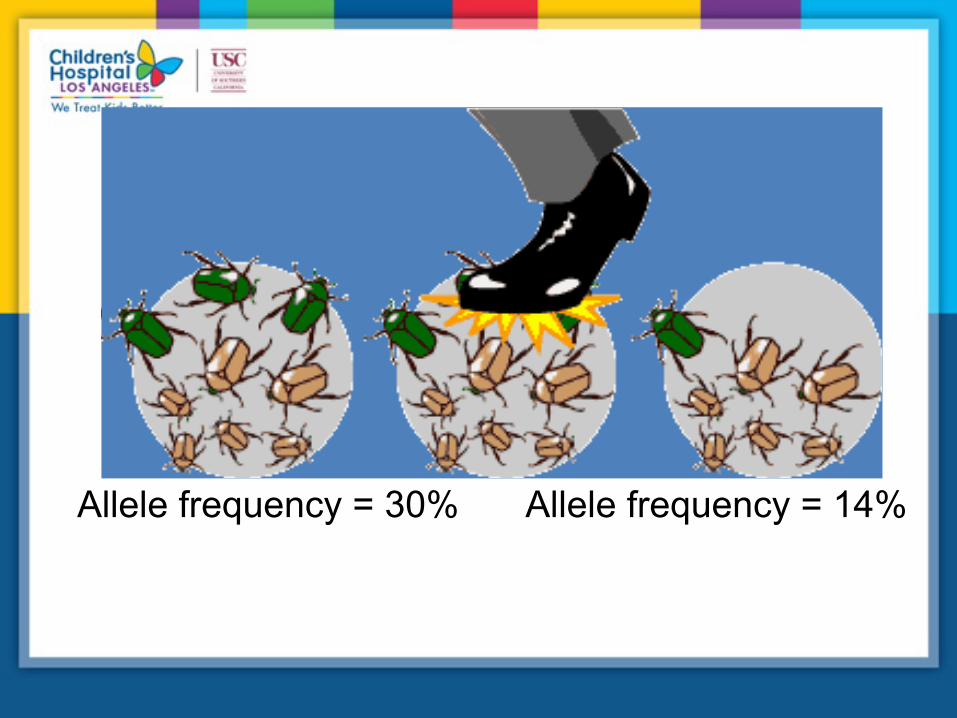
Genetic drift
Allele frequency = 30% Allele frequency = 14%
time
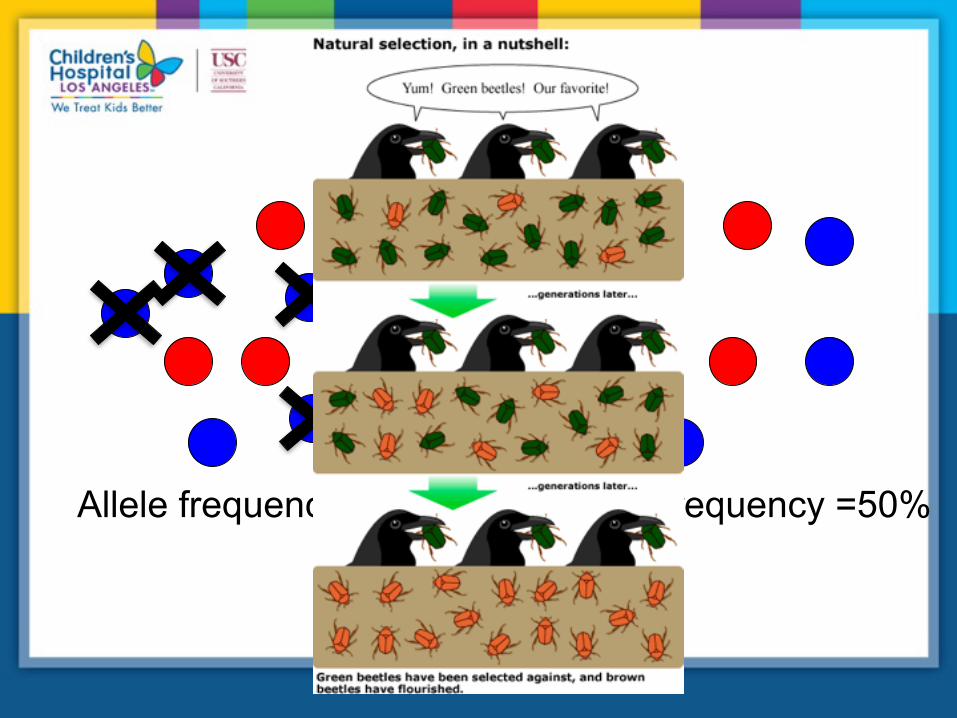
Natural selection
Allele frequency = 30% Allele frequency =50%

Population structure
67%
30%
17%
Population 1
Population 2
Barrier

Pigmentation example - SLC45A2
ALFRED: The ALlele FREquency Database
http://alfred.med.yale.edu/alfred/Katarzyna Bryc, 2013

Genes mirror geography -PCA
Novembre et al. 2008, Nature

Genotyping Sequencing
Potentially, every position on a genome is studied However, quality is variable, lower than for the SNP chip (0.1% error is achieved for 75% of bases) Some areas require read depths of 100 or more.
Large, but limited number of high-quality calls. 1 million SNPs can be genotyped for $100 Error rate (wrong calls) <1% (reported by 23 and me)
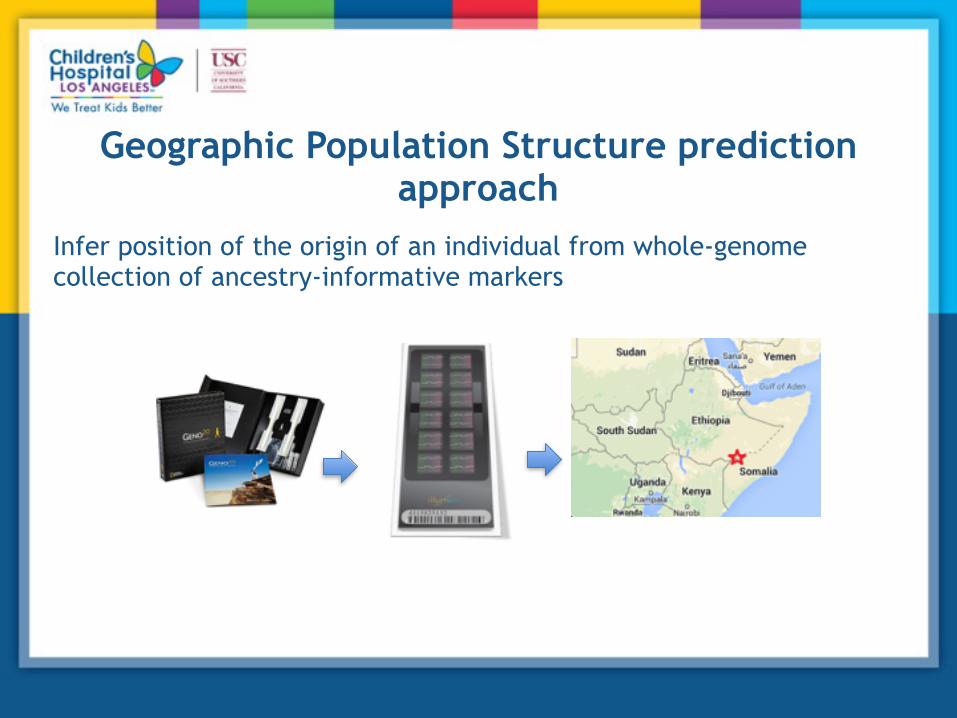
Geographic Population Structure prediction approach
Infer position of the origin of an individual from whole-genome collection of ancestry-informative markers

National Genographic Project
• Goal: Assemble world-wide collection of reference genotypes • How is it done?

Machu Picchu

Solstice festival in Cusco

Uros people

Taquile island, Titicaca lake

Amazon river
Urarina


IMG_1947.CR2


~700 km
Turuhansk
Bor
Kellog


Nganasan
300 km
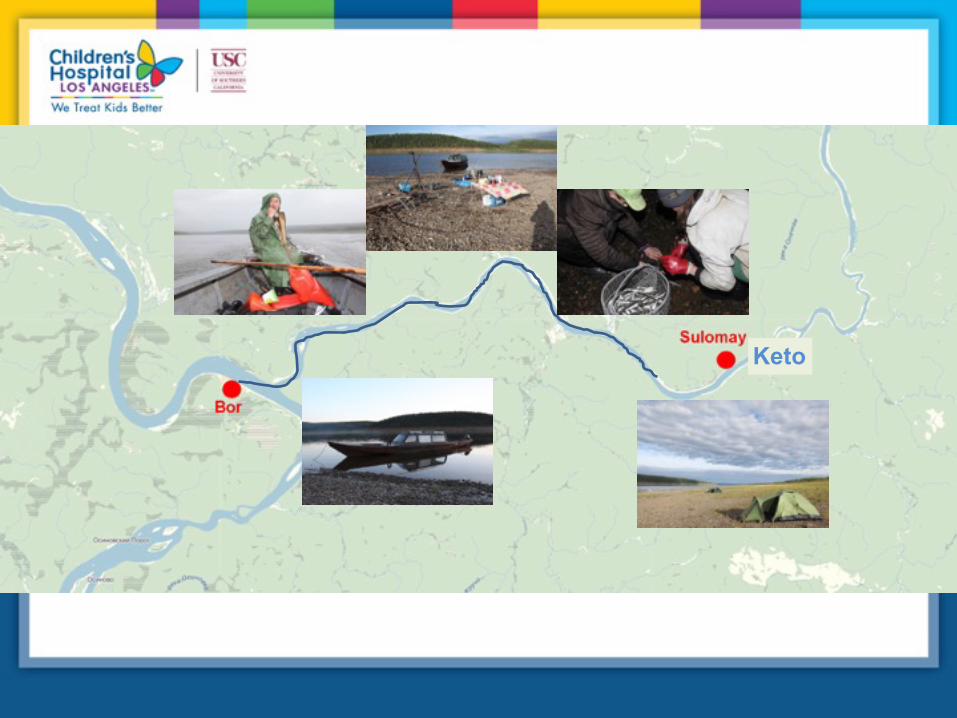
67 km
Keto



K e t o – Yeniseian language

Discoveries
• Peruvians from Lima were not typical Peruvians • Many self-identified Keto were not Keto
–Median Keto component in self-identified Keto is 71%
–Only 13 of 67 have over 90% of Keto ancestry (pure Keto)
• Why is it so difficult?

How to collect data
• Approach village nurse, elders, shamans • Request to select unrelated people of “pure” origin, who remember
their grandparents • Distribute a survey requesting information about place of birth and
ethnicity of the individual, his/her parents and grandparents
All >75% <=75%

• In pairs
– Obtain informed consent • Find out:
– Parents’ names – Parents’ dates of birth – Parents’ place of birth – Parents’ ethnicities
• Then:
– Grandparents’ names – Grandparents’ dates of birth – Grandparents’ place of birth – Grandparents’ ethnicities
Exercise

How to find Ancestry Informative Markers (AIMs)?
• How to find Ancestry Informative Markers? • For each position on the genome, we have information from K
populations. Each locus can have up to N alleles. • In - informativeness for assignment. For a given set of populations,
the minimal of In=0 occurs when all alleles have equal frequencies in all populations. The maximal value, log(K), occurs when N > K and no allele is found in more than one population
Example: K=2 and N=2

How to find Ancestry Informative Markers (AIMs)?
Example: K=2 and N=2. Let’s assume that we have A or C in the selected position. 1. Informative marker p1A=1, p1C=0, p2A=0, p2C=1
PA=PC=0.5
Informativeness=(-0.5log2(0.5)+0-0.5log2(0.5)+0)=1=log2(K)
1. Non-Informative marker p1A=0.5, p1C=0.5, p2A=0.5, p2C=0.5
PA=PC=0.5
Informativeness=(-0.5log2(0.5)-0.5log2(0.5) +4*(0.5*log2(0.5)/2)) =1-1=0

HOW TO PROCESS SAMPLES

To infer population structure from genotype data, it is necessary to first reduce the dimensionality of the dataset due to the thousands of SNPs it encompasses.
From SNPs to Admixture
Thousands of ancestry informative SNPs
North East Asian Mediterranian South African
South West Asian
Native American Oceanian
South East Asian
Northern European
Sub-Saharan African
HGDP00985 0.5253 0.0202 0 0.2222 0.0404 0.0101 0.0101 0.1717 0
HGDP01094 0.04 0.04 0 0.03 0.83 0 0.01 0.05 0
HGDP00982 0.0102 0.1531 0.0306 0.0714 0.0408 0 0.0102 0.2041 0.4796
ADMIXTURE
Admixture proportions in geographically adjacent populations, such as Italian and Greeks, and populations sharing similar history, like British and Germans, are similar.
36

Selection of K
Identify the best K value as judged by prediction of systematically withheld data points

Question
• How to link genetic and geographic divergence?
39
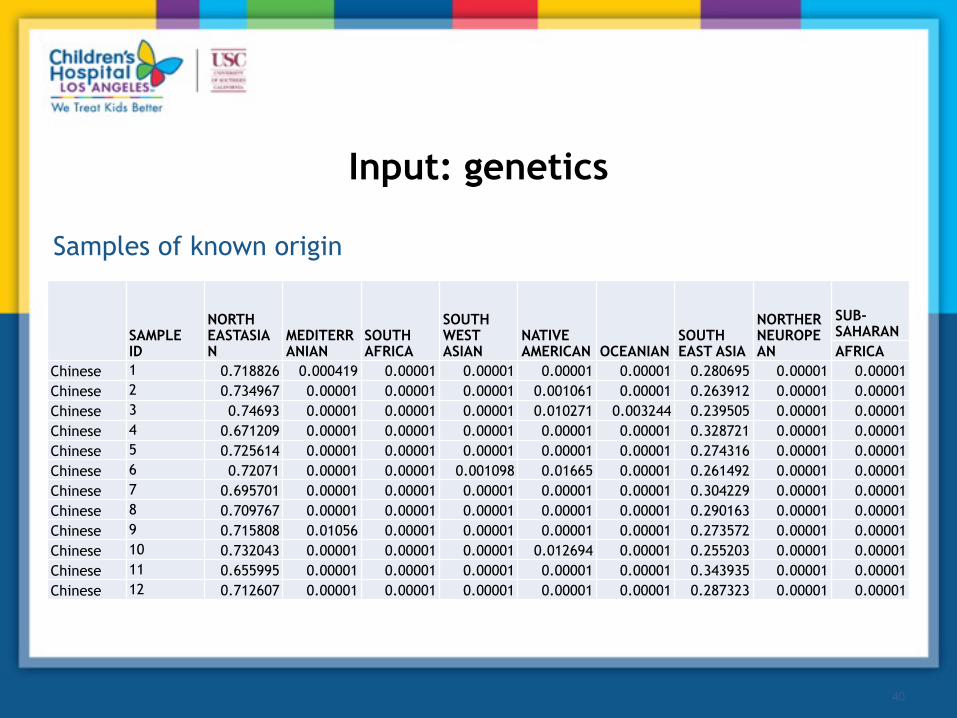
Input: genetics
Samples of known origin
40
SAMPLE ID
NORTH EASTASIAN
MEDITERRANIAN
SOUTH AFRICA
SOUTH WEST ASIAN
NATIVE AMERICAN OCEANIAN
SOUTH EAST ASIA
NORTHERNEUROPEAN
SUB-SAHARANAFRICA
Chinese 1 0.718826 0.000419 0.00001 0.00001 0.00001 0.00001 0.280695 0.00001 0.00001Chinese 2 0.734967 0.00001 0.00001 0.00001 0.001061 0.00001 0.263912 0.00001 0.00001Chinese 3 0.74693 0.00001 0.00001 0.00001 0.010271 0.003244 0.239505 0.00001 0.00001Chinese 4 0.671209 0.00001 0.00001 0.00001 0.00001 0.00001 0.328721 0.00001 0.00001Chinese 5 0.725614 0.00001 0.00001 0.00001 0.00001 0.00001 0.274316 0.00001 0.00001Chinese 6 0.72071 0.00001 0.00001 0.001098 0.01665 0.00001 0.261492 0.00001 0.00001Chinese 7 0.695701 0.00001 0.00001 0.00001 0.00001 0.00001 0.304229 0.00001 0.00001Chinese 8 0.709767 0.00001 0.00001 0.00001 0.00001 0.00001 0.290163 0.00001 0.00001Chinese 9 0.715808 0.01056 0.00001 0.00001 0.00001 0.00001 0.273572 0.00001 0.00001Chinese 10 0.732043 0.00001 0.00001 0.00001 0.012694 0.00001 0.255203 0.00001 0.00001Chinese 11 0.655995 0.00001 0.00001 0.00001 0.00001 0.00001 0.343935 0.00001 0.00001Chinese 12 0.712607 0.00001 0.00001 0.00001 0.00001 0.00001 0.287323 0.00001 0.00001

For every reference population, calculate mean admixture vectors
NORTH EAST ASIA
MEDI-TERRANIA
SOUTH AFRICA
SOUTH WEST ASIA
NATIVE AMERICA OCEANIA
SOUTH EAST ASIA
NORTHERN EUROPE
SUB-SAHARANAFRICA
Chinese 0.711681 0.000923 1.00E-05 0.000101 0.003396 0.00028 0.283589 1.00E-05 1.00E-05
Russian 0.068867 0.265222 0.001241 0.224659 0.035011 0.008622 0.031844 0.363107 0.001419
Tatar 0.15794 0.209897 1.00E-05 0.210957 0.011902 0.002605 0.005703 0.400975 1.00E-05
41

Input: geography
• For every reference population find the corresponding coordinates.
Latitude Longitude
Chinese 39.55 116.2
Russian 55.75 37.62
Tatar 55.55 50.93
Moscow
42

Prediction of provenance
• Knowing relationship between geographic and genetic distances, is it possible to find a geographic origin of a person of known genotype?
• We decided to try a simple approach
43
A B
X

Dealing with individuals of unknown origin
NORTH EAST ASIA
MEDI-TERRANIA
SOUTH AFRICA
SOUTH WEST ASIA
NATIVE AMERICA OCEANIA
SOUTH EAST ASIA
NORTHERN EUROPE
SUB-SAHARANAFRICA
Unknown 0.711681 0.000923 1.00E-05 0.000101 0.003396 0.00028 0.283589 1.00E-05 1.00E-05
• Find distances between the Unknown vector and all reference vectors
• Sort reference populations by distance from smallest to largest
44
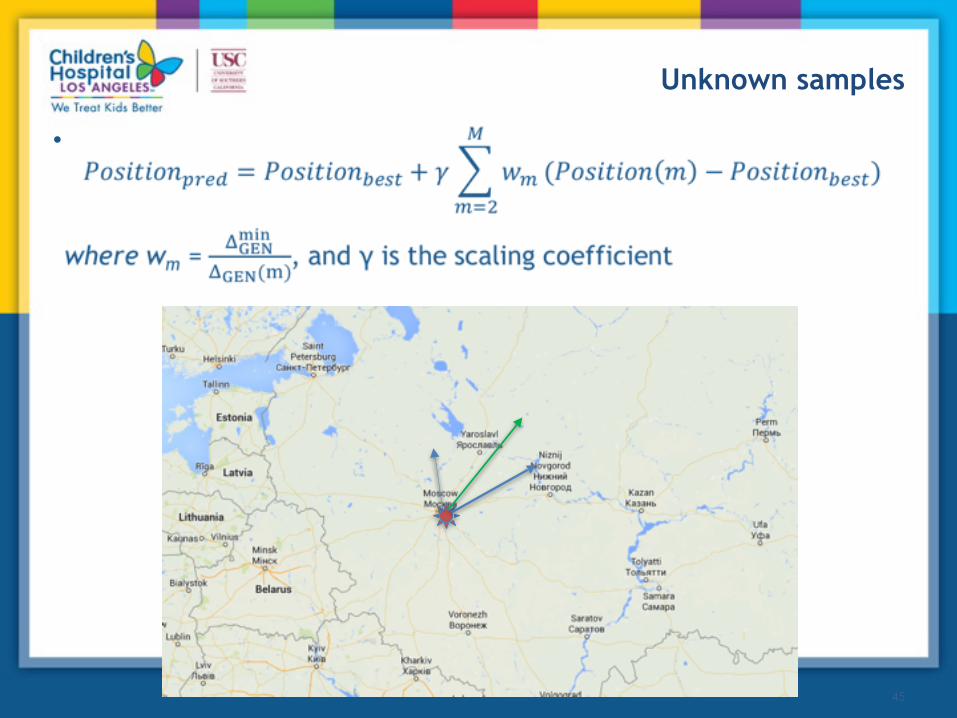
Unknown samples
•
45
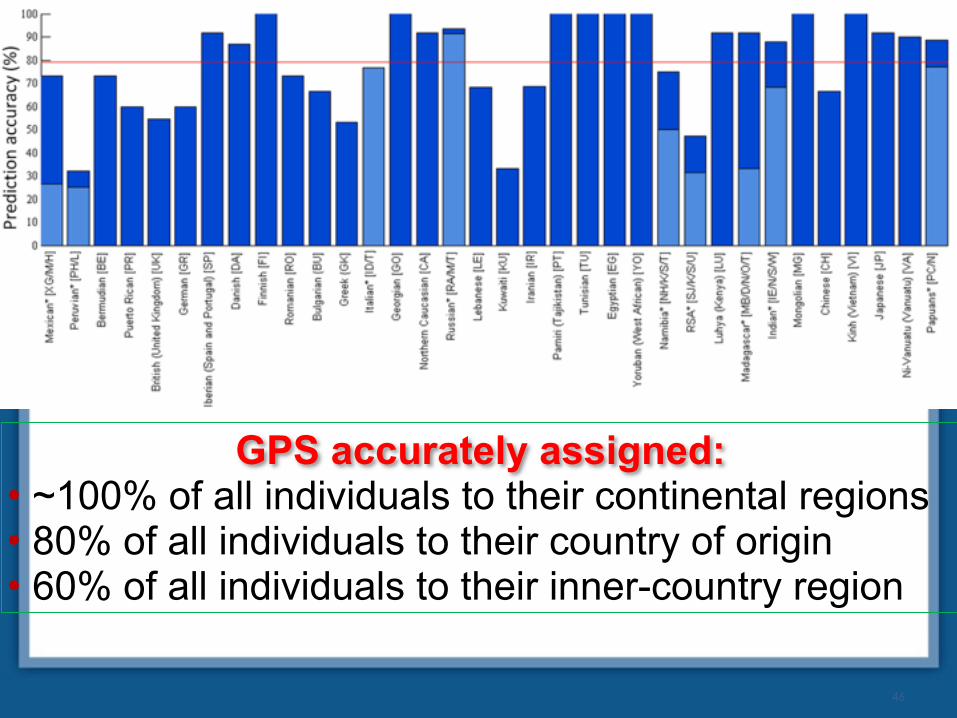
GPS accurately assigned: • ~100% of all individuals to their continental regions • 80% of all individuals to their country of origin • 60% of all individuals to their inner-country region
46

True Predicted
GPS
SPA
47

.
GPS achieved high assignment accuracy for Southeast Asian and Oceanian populations (87.5%) and subpopulations (77%).
We traced the origins of Remote Oceanian populations to Near Oceania.
Reich et al. (2011). Denisova admixture and the first modern human dispersals into Southeast Asia and Oceania, AJHG
243 Southeast Asian and Oceanian samples
48

OTHER ORGANISMS WHOGEM
Application of GPS to

Prediction of bio-origin
• Knowing relationship between geographic and genetic distances, is it possible to find a geographic origin of a person of known genotype?
• We decided to try a simple approach
50
A B
X

Application to Arabidopsis
51
•Percent of populations that are perfectly mapped: 60% •Average distance to correct population: 200 km
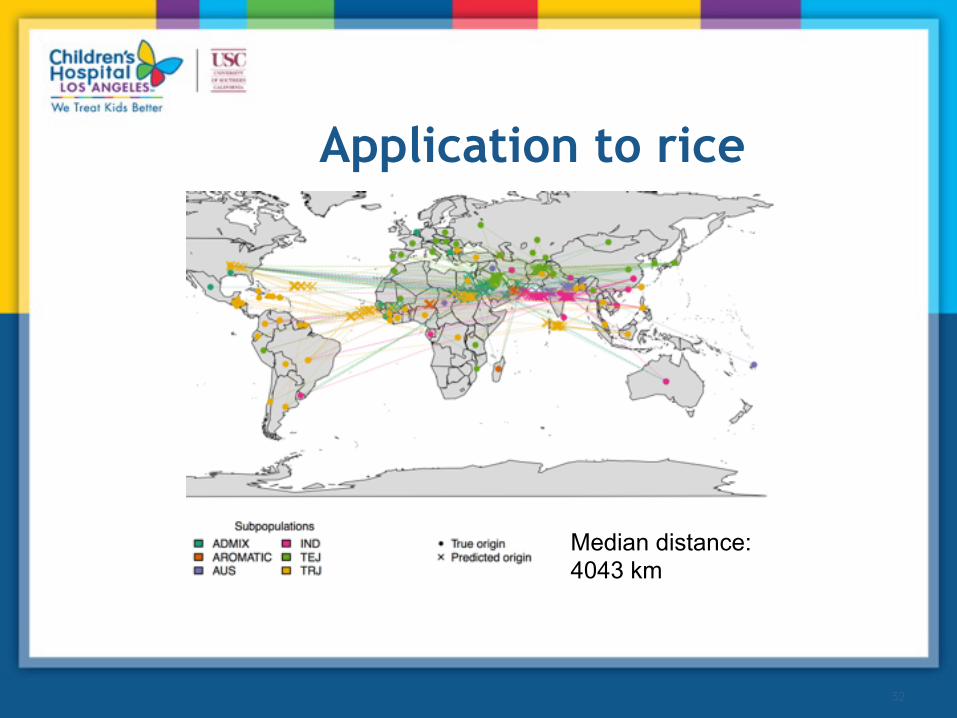
Application to rice
52
Median distance: 4043 km

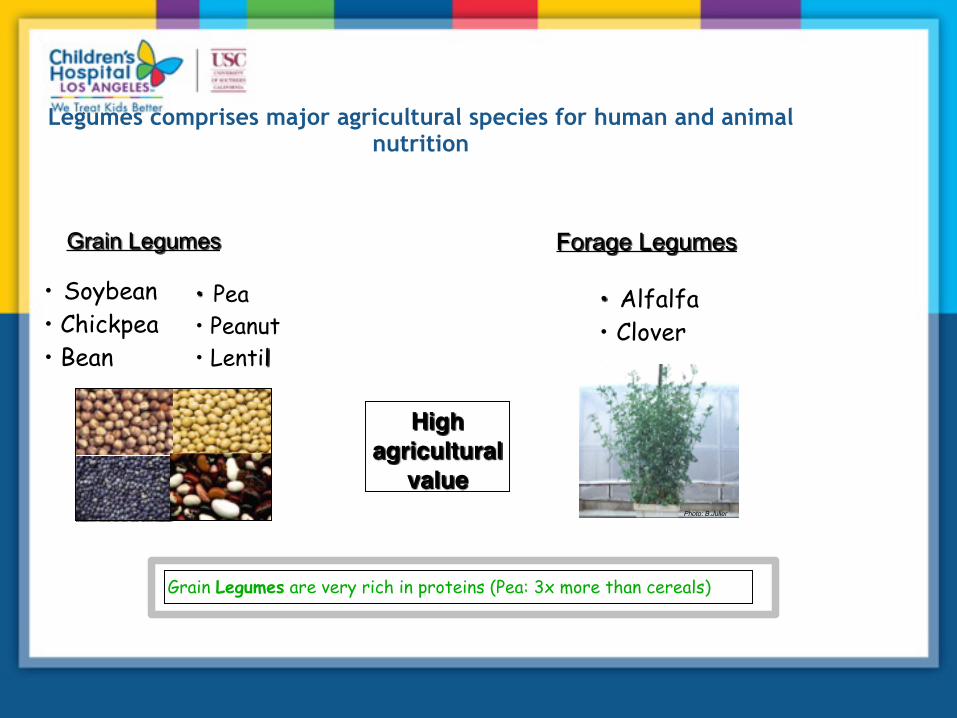
■Legumes (fabaceae) plant family Ability to establish nodule symbiosis High synteny to legume crops
■Small diploid genome (500Mb) sequenced Tang et al., 2014 (BMC Genomics)
■Numerous genomic resources • RIL populations • Mutant collections • Large EST and RNASEQ databases • HAPMAP resources Branca et al., 2011
(PNAS) • 288 genomes already sequenced
■Biotechnology tools for validation ■Large biodiversity Gentzbittel et al., 2015 (Front. Pl. Sci)
Medicago truncatula
M. truncatula - a model legume plant
01/08/2017
Grain Legumes Forage Legumes
• Alfalfa • Clover
• Soybean • Chickpea • Bean
High agricultural
value
• Pea • Peanut • Lentil
Grain Legumes are very rich in proteins (Pea: 3x more than cereals)
Photo: B.Julier
Legumes comprises major agricultural species for human and animal nutrition

HapMap data for M. truncatulaCurrent state: 288 sequenced accessions
# SNPs Chr1 : 1.508.346 Chr2 : 1.964.419 Chr3 : 2.472.365 Chr4 : 2.225.537 Chr5 : 3.251.290 Chr6 : 1.172.980 Chr7 : 2.262.094 Chr8 : 1.659.690
Total : 16.516.721 ~ 40% of SNPs in coding regions
18 sister taxa
Here are the data available for the 288 Medicago accessions that have been already sequenced leading to a total number of more than 16.5 million SNP. Among those, 18 accessions which highly diverged from others have been re-classified to some sister taxa despite perfect botanic similarities

56
M. truncatula is spontaneously occurring all around the Mediterranean basin
Several collections are available: DZ, TN, FR , AU, US, …. for thousands of accessions
Photo: A.Abdelguerfi, INA, Alger, AlgeriaPhoto: J-M Prospéri, INRA Montpellier, France
Photo: J-M Prospéri, INRA Montpellier, France Photo: E.Aouani, CBBC, Tunisia Photo: L. Gentzbittel, ENSAT, Toulouse, France
Photo: L. Gentzbittel, ENSAT, Toulouse, France

57
NSF HapMap project
Current state: 288 sequenced accessions Total: 16.516.721 SNPs 30 lines at > 20XThe rest at least 5X
Branca et al., 2011 (PNAS)
58
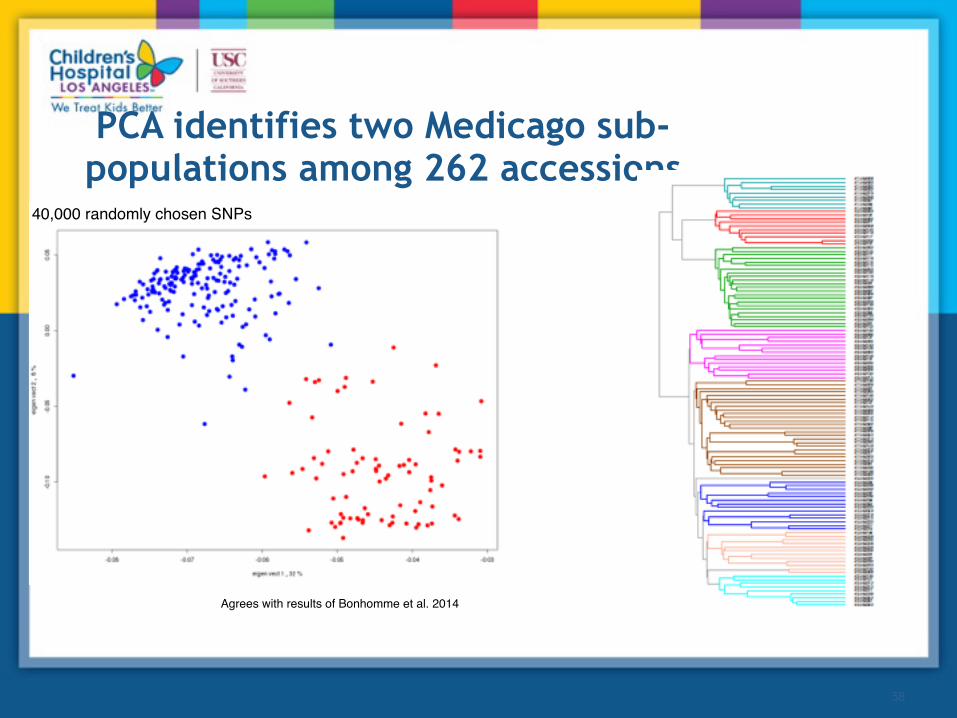
PCA identifies two Medicago sub-populations among 262 accessions
Agrees with results of Bonhomme et al. 2014
40,000 randomly chosen SNPs

A refined population structure for Medicago species using GPS algorithm
Medicago is likely to be structured in 8 ancestral genomesElhaik, E., Tatarinova, T., et al. 2014. Geographic population structure analysis of worldwide human populations infers their biogeographical origins. Nat Commun 5, article 313
Fig. 2: Predicted distance from true origin for each M. truncatula accession using t h e l e a v e - o n e - o u t procedure, for increasing values of K.
'leave-one-out' test for assignation of accessions to actual populations, using inferred ancestral genomes proportions.
840K randomly selected SNPs (~100,000 per chromosome)
Admixture analysis
Choice of # putative ancestral populations
Fig. 1: BIC as a function of increasing values of K, using Discriminant Analysis of Principal Components (DAPC) applied on the 8 4 0 K S N P d a t a s e t . (Jombart et al 2010)
K=8
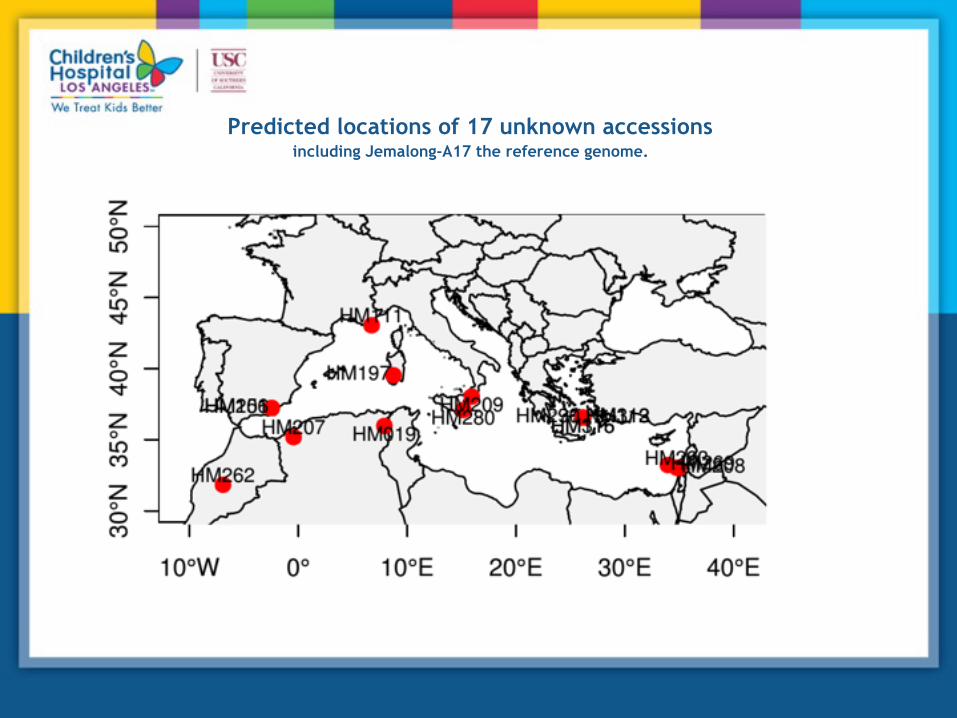
Predicted locations of 17 unknown accessions including Jemalong-A17 the reference genome.

Examples of GPS analysis with various levels of accuracy
'Leave-one Out' procedure accuracy is <300 kilometers
Linear relationship between geographical and genetic distances when d < 950 km (R=0.8, p-value=10-4, Mantel's test)
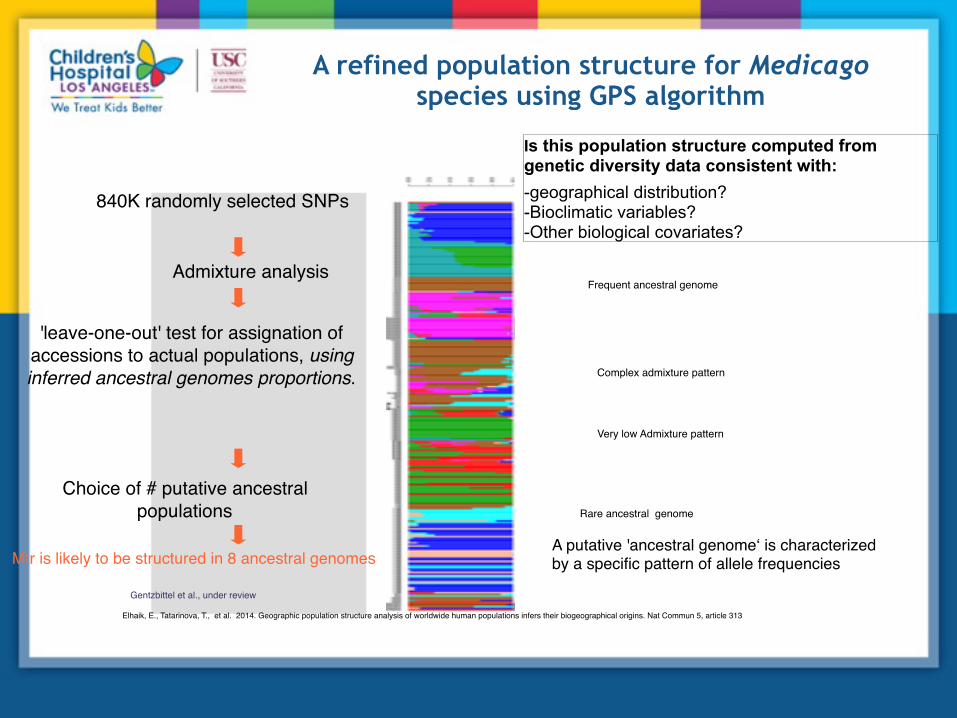
A refined population structure for Medicago species using GPS algorithm
Mtr is likely to be structured in 8 ancestral genomes
Elhaik, E., Tatarinova, T., et al. 2014. Geographic population structure analysis of worldwide human populations infers their biogeographical origins. Nat Commun 5, article 313
'leave-one-out' test for assignation of accessions to actual populations, using inferred ancestral genomes proportions.
840K randomly selected SNPs
Admixture analysis
Choice of # putative ancestral populations Rare ancestral genome
Complex admixture pattern
Very low Admixture pattern
Frequent ancestral genome
A putative 'ancestral genome‘ is characterized by a specific pattern of allele frequencies
Gentzbittel et al., under review
Is this population structure computed from genetic diversity data consistent with: -geographical distribution? -Bioclimatic variables? -Other biological covariates?

Geographic distribution of the putative eight ancestral genomes of M. truncatula
Spanish coastalAlgiers
North Tunisian coastal Atlas
French
Greek Spanish-Moroccan Inland
South Tunisian coastal
Gentzbittel et al., under review
5 out of 8 populations are present in Mahgreb
Algiers (K1)
Spanish coastal (K2)
North Tunisian coastal (K3)Atlas (K4)
South Tunisian coastal (K5)
French (K6)
Greek (K7)
Spanish-Moroccan Inland (K8)
Maghreb as a putative center of diversity for M. truncatula species

Some ancestral genomes may be adapted to local bio-climatic variables
We looked for putative association with the 19 bio-climatic variables, as defined by WorldClim (http://www.worldclim.org).
The “South Tunisian Coastal" genome is positively correlated to annual mean T°C (BIO1) and Mean T°C of colder quarter (BIO11) and negat ive ly cor re la ted to annua l precipitation (BIO12).
The “Spanish coastal" genome is
negatively correlated to T°C seasonality (BIO4), and T°C annual range (BIO7).
The “Greek" genome is positively correlated to precipitation of the wettest month (BIO13), precipitation seasonality (BIO15), precipitation of the wettest quarter (BIO16) and precipitation of coldest quarter (BIO19).
Spanish coastal
Greek
BIO1: r=0.26*** BIO11: r=-0.24** BIO12: r=--0.28***
BIO4: r=-0.42*** BIO7: r=-0.31***
BIO13: r=0.39*** BIO15: r=0.45*** BIO16: r=0.33*** BIO19: r=0.29***
The “North Tunisian Coastal" genome is not significantly correlated to any bioclimatic covariables.

Climate and geography explain less <50% of the variation in genome admixture component proportions
Other putative explanatory variables?
-soil structure and composition?
-Biotic interactions?
Gentzbittel et al., under review
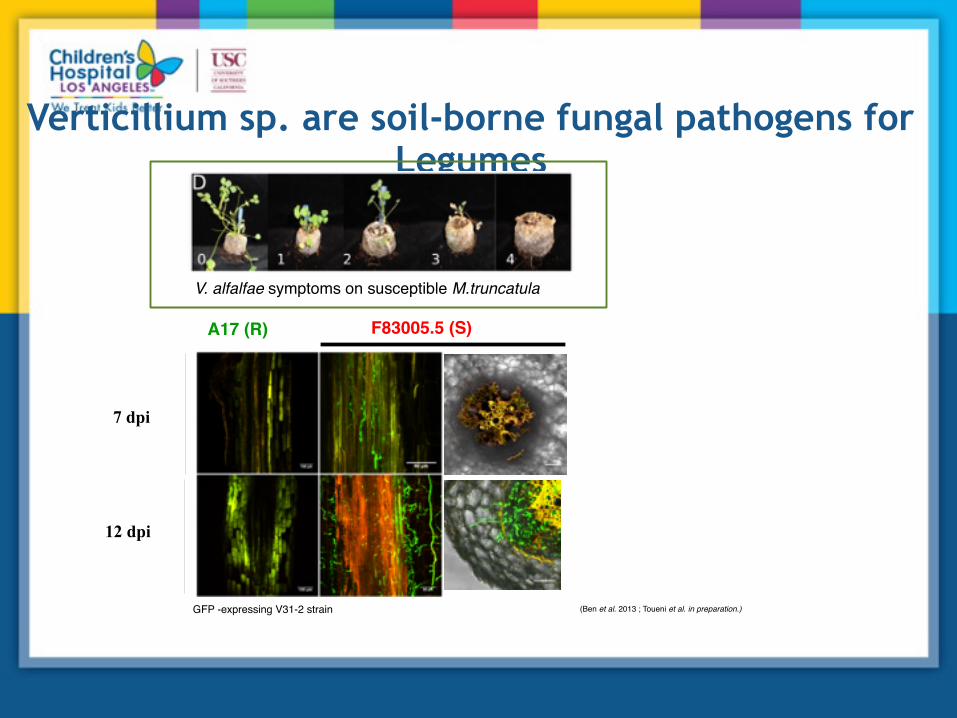
Verticillium sp. are soil-borne fungal pathogens for Legumes
V. alfalfae symptoms on susceptible M.truncatula
7 dpi
12 dpi
A17 (R) F83005.5 (S)
GFP -expressing V31-2 strain (Ben et al. 2013 ; Toueni et al. in preparation.)

Root pathogens enter the plant through root central cylinder
GFP Rs
GUS staining
Ralstonia solanacearum infection
xy
cwhy
* *
cor
xyhy
Verticillium albo-atrum infectionVailleau et al. 2006 Ben et al. 2012

Ancestral genomes may present different levels of resistance to Verticillium wilt
Many eudicot species and cultivars are resistant to the disease and all monocots, gymnosperms and ferns are immune.

Ancestral genomes present different levels of resistance to Verticillium wilt
Linear Model (242 Mt HAPMAP accessions) MSSCorLS ~ K2 + K5 + K7 + K8
r²=0.31, P-value < 2.2 x 10-16

Can phenotype be predicted using GPS?
• Use plant model (Medicago truncatula) to test applicability of admixture framework to predict phenotypes.
• M. truncatula was infected with a fungal pathogen V. alfalfae
• MSS (Maximum symptom score) ranges from 0 (healthy) to 4 (dead), 2 threshold.
GPS was used to predict provenance of 59 unknown samples, and MSS was inferred based on the similarity of admixture profiles to the training set accessions Plants were infected in the lab and symptoms recorded 21/29 samples that are predicted to be resistant were resistant 24/30 samples predicted to be susceptible were susceptible
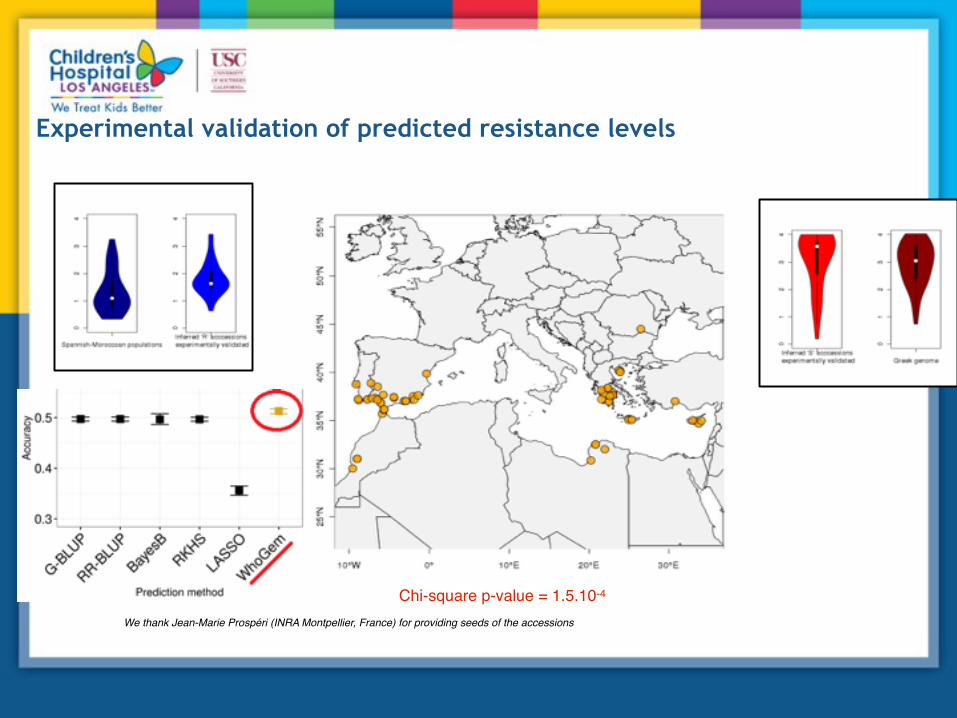
Experimental validation of predicted resistance levels
Chi-square p-value = 1.5.10-4
We thank Jean-Marie Prospéri (INRA Montpellier, France) for providing seeds of the accessions

WhoGem models can be significant predictors of quantitative functional traits in plants
Phenotypic data from: Mazurier et al, in preparation Stanton-Geddes et al., 2013 Bonhomme et al., 2013Gentzbittel et al., pending submission
Comparisons of accuracy of prediction for several quantitative traits of adaptive relevance in M. truncatula using f ive Genomic Selection algorithms and our WhoGEM. Accuracy is computed using 50 runs of five-fold cross-validation for all methods. Phenotypes: (a) Plant height at harvest (b) Number of leaves (c) Nodule number below 5 cm of root growth (d) Nodule number above 5 cm of root (e) Total number of nodules (f) Partial resistance to Aphanomyces euteiches, estimated by root-rot index.

What is missing?Distance between two points should not be just geometric distance. Add: 1. Mode of propagation (e.g. Medicago seeds stick to goats) 2. Soil 3. Climate 4. Geography

This methodology may serve as a basis for analyses in other plant species and for other functional quantitative traits of interest.
Conclusions
• Large proportion of phenotypic variation between individuals may be best explained by population admixture.
• Variation in genome admixture proportion explains most of phenotypic variation for quantitative functional traits.
• We experimentally confirm the prediction of differences in quantitative disease resistance levels in the wild model legume Medicago truncatula.
• Admixture components were found to be significantly related to climate and geography, also positive selection at the species level might not explain current adaptation.
• Phenotypes can be predicted using genome-wide patterns of admixture, when incorporating covariates such as individuals' provenance.
• This insight contributes to the understanding of adaptation, and can accelerate plant and animal breeding, and biomedical research programs.
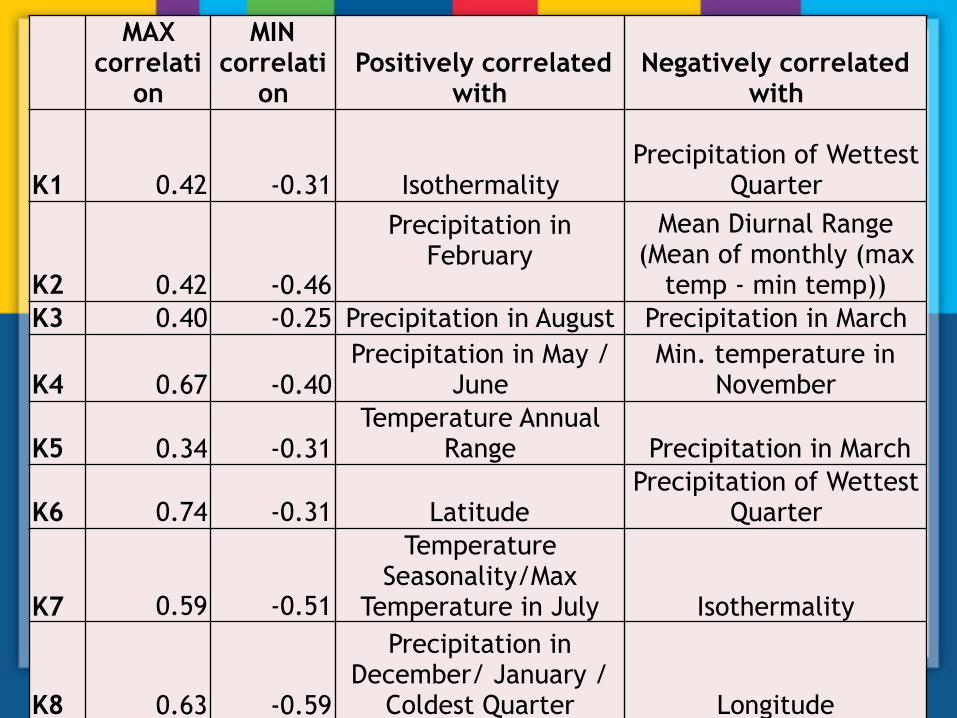
MAX correlati
on
MIN correlati
on Positively correlated
withNegatively correlated
with
K1 0.42 -0.31 Isothermality Precipitation of Wettest
Quarter
K2 0.42 -0.46
Precipitation in February
Mean Diurnal Range (Mean of monthly (max
temp - min temp)) K3 0.40 -0.25 Precipitation in August Precipitation in March
K4 0.67 -0.40Precipitation in May /
June Min. temperature in
November
K5 0.34 -0.31Temperature Annual
Range Precipitation in March
K6 0.74 -0.31 LatitudePrecipitation of Wettest
Quarter
K7 0.59 -0.51
Temperature Seasonality/Max
Temperature in July Isothermality
K8 0.63 -0.59
Precipitation in December/ January /
Coldest Quarter Longitude

Soil/Climate
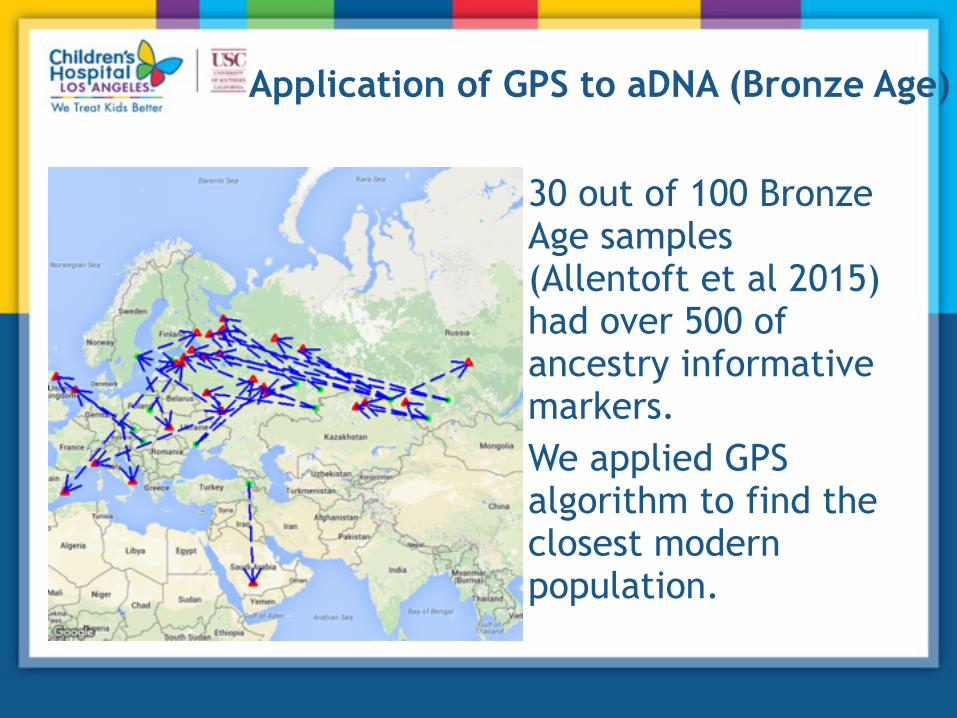
Application of GPS to aDNA (Bronze Age)
30 out of 100 Bronze Age samples (Allentoft et al 2015) had over 500 of ancestry informative markers. We applied GPS algorithm to find the closest modern population.

GPSL
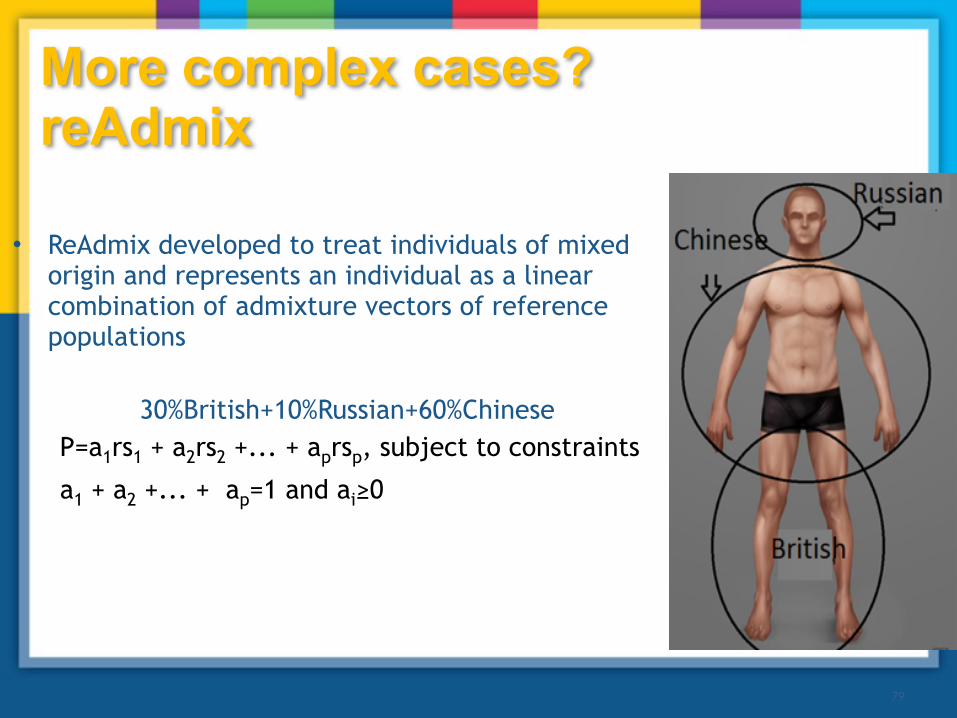
• ReAdmix developed to treat individuals of mixed origin and represents an individual as a linear combination of admixture vectors of reference populations
30%British+10%Russian+60%Chinese P=a1rs1 + a2rs2 +... + aprsp, subject to constraints
a1 + a2 +... + ap=1 and ai≥0
79
More complex cases? reAdmix

How it works
• We assume right away that the given ancient proportions contain error
• Start with a guess population • Add/remove populations to achieve optimal fit • Conditional optimization (such as “I know that there
was a Jewish ancestor somewhere in my pedigree”)
80

31.07.2017
reAdmix approachAim: to find the smallest subset of modern populations whose combined admixture components are similar to those of the individual within a small tolerance margin.
The algorithm consists of three phases:
1. Iteratively build the first candidate solution (LARS) and improve it.2. Generate the predefined number M of additional candidate solutions randomly and apply the Differential Evolution (DEEP).
3. Identify the populations that have stable membership in the solution across the set, that is, are part of solution in at least 75% of cases.
Let R={ri}i=1..I be the set of modern populations where
ri=(ri,1, ..., ri,K) and K is the dimension (K=9).We seek two sets S=(s1,...,sp) and A=(a1,...,ap) where
si are the indices of modern populations ai are the coefficients of modern populationsin the approximation
each
of the test vector T

31.07.2017
Phase 1. Build and improve the initial solution set
S - individual’s ancestry
T – test vector
N - desired size of the solution
R – the set of modern populations
dim(S) < N
j=arg max[F(rj,T)] S = S ∪ {j}
aj=max[α:α·rj<T+ε]×β T = T − ajrj S0 = (S \ si) U {rk}
S = S0
i=0..dim(S) k=0..dim(R)
f(S0,T) < f(S,T)Affinity score
minimizes the loss function
where
Find the population vector with the highest affinity score Append this population to the solution set. Calculate the weight of the population vector
For all populations x in the current solution and for all y outside the solution, replace x with y, if it reduces the error.

31.07.2017
Phase 2. Optimize the solution by global stochastic and local search
2. Local search over all populations close to the preliminary solution
This step selects between related populations (e.g. Belorussian, Russian, and Ukrainian) that could have been misplaced in previous steps
S1,S2,...,SM
are generated randomly or from population groups
Objective function
is the approximation of Twhere
1. Differential Evolution Gmax number of iterations
DE: optimization method used for multidimensional real valued functions. Good for Treatment of noisy problems (Storn and Price, 1997)

31.07.2017
Phase 3. Averaging
S1:(a1,r1)1,(a2,r2)1,..,(ap-1,rp-1)1,(ap,rp)1
S2:(a1,r1)2,(a2,r2)2,..,(ap-1,rp-1)2,(ap,rp)2
SM:(a1,r1)M,(a2,r2)M,..,(ap-1,rp-1)M,(ap,rp)M
...
r belongs to final solution if: r=r1,1=r2,M-1=...=rp,M — is the same modern population
and
is present in L > 75% solutions
and
a=(1/L)(a1,1+a2,M-1+..+ap,M)
Reliable estimate: - the populations that are part of solution in at least 75% of cases,
- their averaged estimates of admixture proportion.
SM-1:(a1,r1)M-1,(a2,r2)M-1,..,(ap-1,rp-1)M-1,(ap,rp)M-1
S1, S2, ... , SM-1, SM — the set of candidate solutions. Final solution:
S=(s1,...,sp)A=(a1,...,ap)
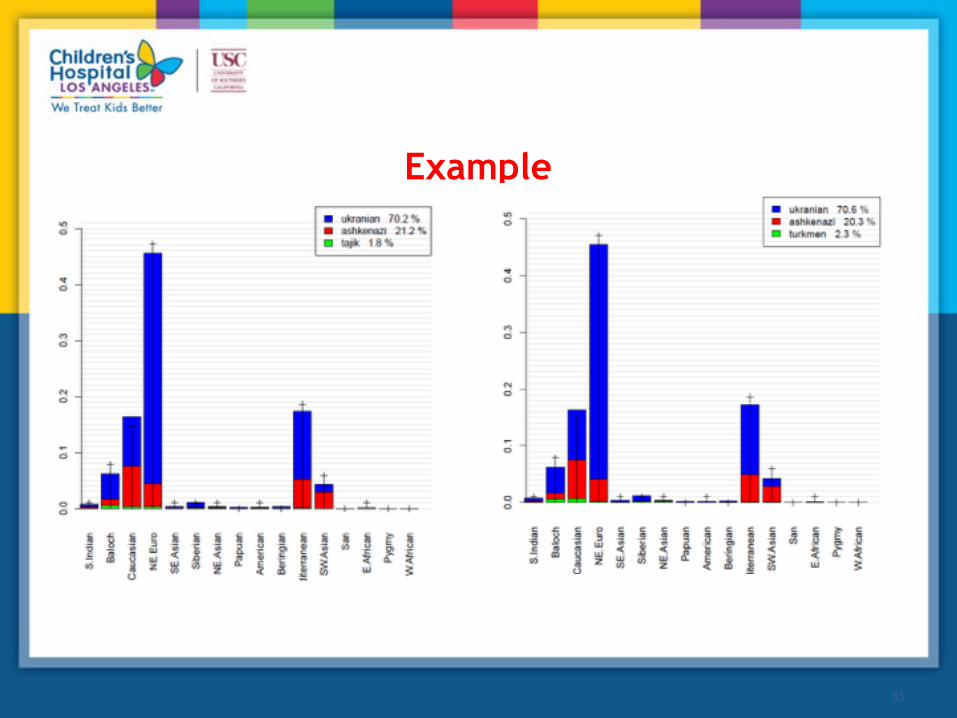
Example
85

Benchmark-1
Take sample that were validated by GPS1
Then we used ReAdmix in (a) unconditional and (b) conditional with incorrect guess mode.
(a) 94% of the samples were mapped to their reported ethnic group, and average distance to the correct location was 54±13 miles.
(b) with randomly chosen incorrect guess, 92% of samples was mapped to the reported ethnic group, with average distance to the correct location equal to 65±17 miles.
In all cases ReAdmix correctly identified the cases as un-mixed
86

Benchmark-2: simulated 500 50:50 mixtures
87
Testing mode Percent of at least one correctly predicted
origin
Percent of completely
correct predictions
Average distance to correct population, miles
Unconditional 78% 56% 324±46
Conditional on the equal weights, populations unknown
91% 74% 176±33
Conditional on one of the populations, weights unknown
NA 71% 104±16

Benchmark-2: simulated 500 50:25:25 mixtures
88
Testing mode Percent of at least one correctly predicted
origin
Percent of completely correct
predictions
Average distance to correct population,
miles
Unconditional 88% 21% 346±34Conditional on the major population, weights unknown
73% 27% 222±26
Conditional on one of the minor populations, weights unknown
78% 33% 234±30

Sohn et (2012) al benchmark• 2 components • 4 components
4-dim space: European, African, Native American and East Asian
Color coding: red-European, green-African, yellow- Native American, blue-East Asian, and white- unassigned


Let’s practice!


















