
pp 1-1转曲 - Scientific Research Publishing · Pharmacology & Pharmacy Journal Information...
44
Transcript of pp 1-1转曲 - Scientific Research Publishing · Pharmacology & Pharmacy Journal Information...



Pharmacology & Pharmacy, 2010, 1, 1-38 Published Online July 2010 in SciRes (http://www.SciRP.org/journal/pp/)
Copyright © 2010 SciRes. PP
TABLE OF CONTENTS
Volume 1 Number 1 July 2010
Anti-Amnesic Activity of Vitex negundo in Scopolamine Induced Amnesia in Rats
A. Kanwal, J. Mehla, M. Kuncha, V. G. M. Naidu, Y. K. Gupta, R. Sistla…………………………………………………………1
Development and Evaluation of a New Interpenetrating Network Bead of Sodium
Carboxymethyl Xanthan and Sodium Alginate for Ibuprofen Release
R. Ray, S. Maity, S. Mandal, T. K. Chatterjee, B. Sa…………………………………………………………………………………9
Preparation and Evaluation of Rapidly Disintegrating Fast Release Tablet of
Diazepam-Hydroxypropyl-β-Cyclodextrin Inclusion Complex
T. K. Giri, B. Sa…………………………………………………………………………………………………………………18
The Porcine Pulmonary Surfactant Protein A (pSP-A) Immunogenicity
Evaluation in the Murine Model
S. de Cássia Dias, F. L. dos Santos, D. Sakauchi, D. Iourtov, I. Raw, F. S. Kubrusly……………………………………………27
New Design of Biopharmaceuticals through the Use of Microalgae Addressed to Global
Geopolitical and Economic Changes. Are You Ready for New Development in Biopharma?
A. B. Avagyan……………………………………………………………………………………………………………………33

Pharmacology & Pharmacy
Journal Information
SUBSCRIPTIONS
The Pharmacology & Pharmacy (Online at Scientific Research Publishing, www.SciRP.org) is published quarterly by Scientific
Research Publishing, Inc., USA.
Subscription rates: Print: $50 per copy.
To subscribe, please contact Journals Subscriptions Department, E-mail: [email protected]
SERVICES
Advertisements
Advertisement Sales Department, E-mail: [email protected]
Reprints (minimum quantity 100 copies)
Reprints Co-ordinator, Scientific Research Publishing, Inc., USA.
E-mail: [email protected]
COPYRIGHT
Copyright© 2010 Scientific Research Publishing, Inc.
All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by
any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except as described below, without the
permission in writing of the Publisher.
Copying of articles is not permitted except for personal and internal use, to the extent permitted by national copyright law, or under
the terms of a license issued by the national Reproduction Rights Organization.
Requests for permission for other kinds of copying, such as copying for general distribution, for advertising or promotional purposes,
for creating new collective works or for resale, and other enquiries should be addressed to the Publisher.
Statements and opinions expressed in the articles and communications are those of the individual contributors and not the statements
and opinion of Scientific Research Publishing, Inc. We assumes no responsibility or liability for any damage or injury to persons or
property arising out of the use of any materials, instructions, methods or ideas contained herein. We expressly disclaim any implied
warranties of merchantability or fitness for a particular purpose. If expert assistance is required, the services of a competent
professional person should be sought.
PRODUCTION INFORMATION
For manuscripts that have been accepted for publication, please contact:
E-mail: [email protected]

Pharmacology & Pharmacy, 2010, 1, 1-8 doi:10.4236/pp.2010.11001 Published Online July 2010 (http://www.SciRP.org/journal/pp)
Copyright © 2010 SciRes. PP
1
Anti-Amnesic Activity of Vitex negundo in Scopolamine Induced Amnesia in Rats
Abhinav Kanwall,2, Jogender Mehla3, Madhusudana Kunchal, Vegi Ganga Modi Naidul, Yogendra Kumar Gupta3, Ramakrishna Sistla1*
1Division of Pharmacology, Indian Institute of Chemical Technology (IICT), Hyderabad, India; 2National Institute of Pharmaceutical Education and Research (NIPER), Hyderabad, India; 3Department of Pharmacology, All India Institute of Medical Sciences (AIIMS), New Delhi, India. Email: [email protected] Received June 8th, 2010; accepted July 12th, 2010.
ABSTRACT
In the present study we investigated the anti-amnesic activity of Vitex negundo in scopolamine induced amnesia in rats. Wistar rats (180-200 g) were trained on active avoidance task. Each animal received session of 15 trials with inter trial duration of 15 s for 5 days. Scopolamine (3 mg/kg, i.p) was administered at different time periods on the basis of stages of memory i.e acquisition, consolidation and retention in different groups (n = 6). Effect of Vitex negundo extract was evaluated and compared to a standard drug, Donepezil. Significant (p < 0.05) increase in the avoidance response on the 5th session has been observed as compared to 1st session in control group. Scopolamine treatment significantly (p < 0.05) reduced the avoidance response compared to control. Extract treated groups shown significant (p < 0.05) in-crease in number of avoidance responses as compared to scopolamine treated groups. Increased oxidative stress in brain after scopolamine treatment, as observed by increase in MDA & decrease in GSH & SOD, was lowered in the groups treated with extracts. AChE activity was also improved after V. negundo treatment. Results of the study have shown that V. negundo treated groups decrease the phenomenon of amnesia by increasing learning of memory through antioxidant effect and decreasing AChE activity. Keywords: Vitex negundo, Amnesia, Acetylcholinestrase, Scopolamine, Learning and Memory, Oxidative Stress
1. Introduction
The Memory is the most important function of the brain. Memory is the process by which organisms are able to record their experiences and use this information to adapt their responses to the environment. Hence it is vital for survival [1]. Central cholinergic system is considered as the most important neurotransmitter involved in regula-tion of cognitive functions [2]. Impaired cognitive func-tions are the major features of Alzheimer disease (AD) [3]. Presence of acetylcholine within the neocortex is sufficient to ameliorate learning deficits and restore memory [4]. The prevalence of AD increases with the age (65 yrs) from 2% to 30-45% in those over 85 yrs [5]. AD and stroke together rank as the third most common causes of death [6]. The incidence of AD for those aged 65yrs and older was 3.24 per 1000 individuals in a year [7]. One study in India showed that, the median survival time determined to be 3.3 yrs for patients with dementia and 2.7 yrs for patients with AD [8]. Scopolamine, a
nonselective muscarinic cholinergic antagonist, is a well- known centrally acting cholinergic probe, which causes impairment in learning [9]. In addition, scopolamine also causes increase in cognitive impairment in healthy eld-erly subjects compared to young adults [10]. The treat-ment with AChE inhibitors and muscarinic receptors agonists which increases cholinergic neurotransmission causes an improvement in cognitive deficits in AD [11]. Besides reducing cholinergic activity, oxidative stress plays an important role and is one of the major causes for memory loss in AD [12,13]
Extensive research is going on different plants all around the world as plant extracts have a relatively higher therapeutic window, lesser side effects and are economical. Plant extracts may also provide a source of new compound as many synthetic drugs have been originated from herbal sources. Vitex negundo, a de-ciduous shrub belonging to family Verbenaceae that comprises 75 genera and nearly 2500 species, chiefly occurs in Pakistan, India and Srilanka. Though almost all

Anti-Amnesic Activity of Vitex negundo in Scopolamine Induced Amnesia in Rats
Copyright © 2010 SciRes. PP
2
parts of the plant are used, the extract from leaves and the roots is the most important in the field of phytomedi-cine and is sold as drugs. The leaf extract is used in Ay-urvedic and Unani system of medicine [14]. Water ex-tract of mature fresh leaves exhibited anti-inflammatory, analgesic and antihistamine properties [15]. Literature survey of V. negundo revealed the presence of volatile oil, triterpenes, diterpenes, sesquiterpenes, lignan, flavonoids, flavones glycosides, iridoid glycosides, and stilbene de-rivative [14]. Lignans, one class of natural compounds present in V. negundo, showed anti-cholinesterase activ-ity in in-vitro [14]. However no studies were conducted to explore the effect of V. negundo extract against mem-ory impairment in in-vivo.
In the process of learning and memory, three important stages have been suggested viz., acquisition, consolida-tion and recall of the learned task [16]. The Scopolamine hydrobromide is an anticholinergic drug, which produces amnesia by reducing the levels of acetylcholine, which is considered to be an important neurotransmitter for the learning and memory. Therefore, the present study was aimed to investigate the anti-amnesic effect of V. ne-gundo aqueous extract on scopolamine administered at different stages of active avoidance learning in rats.
2. Materials and Methods
2.1 Materials
Aqueous extract of the plant Vitex negundo was obtained from Amruta herbals Pvt Limited, Indore (M.P), India, (Batch no. AHVN/556.) along with the copy of certifi-cate of analysis. Scopolamine hydrobromide, Thiobarbi-turic acid (TBA), Glutathione, DTNB, Acetylthiocholine all were purchased from Sigma-Aldrich (Bangalore, In-dia). SOD kit was purchased from Fluka. Other chemical and reagents are of analytical grade.
2.2 Animals
Male Wistar rats weighing between 180-200 g were ob-tained from National Institute of Nutrition, Hyderabad. The animals were housed in an animal facility of Indian Institute of Chemical Technology (IICT). The animal house maintained at 20 ± 2°C and 50-60% relative humidity. A 12-hour dark/light cycle was maintained throughout the study. Air changes were maintained with 5µ HEPA filter. Rats had free access to food (pellet diet supplied from M/s Petcare India Ltd., Bangalore) and water ad libitum. This study protocol was approved by the Institutional Animal Ethics Committee of Indian In-stitute of Chemical Technology, Hyderabad.
2.3 Behavioral Test
2.3.1 Two-Way Active Avoidance with Negative (Punishment) Reinforcement
The animals were trained on Active Avoidance Task in
an automatic reflex conditioner with two-way shuttle box (Ugo Basile, Italy). The rats were treated orally with the standard drug through an intragastric feeding tube. Simi-larly the plant extract were administered for 14 days. For this purpose each rat is placed in a compartment sepa-rated from the other one by a guillotine door in the shut-tle box. Exploration period of 2 min is given initially. There after, the trial start. In each trial the animal is sub-jected to a light for 30 s followed by a sound stimulus for 10s. Immediately after the sound stimulus, the rat re-ceives a single low intensity foot shock (0.5 mA; 3 s) from 10th day to 14th through the floor grid if it does not transfer to the other shock free compartment. Infrared sensors monitor the transfer time from one compartment to another, which is recorded as avoid (after the stimulus of either light alone or both light and sound) and escape (after the foot shock) response. Each animal received a daily session of 15 trials with an inter-trial duration of 15 s for 5 days i.e., a maximum of 75 trials. The rats were evaluated on the basis of their performance in the last session i.e., in the 5th session for their decrease in amne-sic activity and increased learning and memory. The cri-terion for improved cognitive activity was taken as sig-nificant increase in the avoidance response on 5th session (retention) compared to 1st session.
2.4 Scopolamine Induced Loss of Memory in Rat
Acquisition: scopolamine was administered 5 min prior to 1st Trial on 1st session.
Consolidation: scopolamine was administered 5 min after the 15th (i.e., last) trial on 1st session (Training ses-sion).
Retention: scopolamine was administered 5 min prior to the 1st trial on the last session i.e., 5th session (Training session).
Dementia effect of scopolamine was evaluated on the basis of significant decrease in number of avoidance re-sponse in the treated groups as compared to that of con-trol group in the last session i.e., 5th session.
2.5 Treatment Schedule
The animals were divided into eight different groups (n = 6). Scopolamine (3 mg/kg, i.p) was administered at dif-ferent time periods in the three groups (GR-2, GR-3, and GR-4) as follows:
Group I (GR-1)–Saline (control). Group II (GR-2)–scopolamine was administered 5 min
prior to 1st Trial on 1st session (Training session). Group III (GR-3)–scopolamine was administered 5
min after the 15th (i.e., last) trial on 1st session (Training session).
Group IV (GR-4)–scopolamine was administered 5 min prior to the 1st trial on the last session i.e., 5th ses-sion.
Group V (GR-5)–Standard drug, Donepezil (5 mg/kg)

Anti-Amnesic Activity of Vitex negundo in Scopolamine Induced Amnesia in Rats
Copyright © 2010 SciRes. PP
3
is given to GR-2 rats prior to 1 hour of 1st trial. Group VI (GR-6)–Similar to GR-2 but rats were
pre-treated with plant extract for 14 days. Group VII (GR-7)–Similar to GR-3 but rats were
pre-treated with plant extract for 14 days. Group VIII (GR-8)–Similar to GR-4 but rats were pre-
treated with plant extract for 14 days.
2.6 Biochemical Estimation of Markers of Oxidative Stress
On day 14th following the behavioral testing, animals were sacrificed and the brain tissues were quickly re-moved, cleaned with ice-cold saline and stored at –80°C for biochemical estimation.
2.6.1 Preparation of Brain Homogenate Brain-tissue samples were thawed and homogenized with 10 times (w/v) ice-cold 0.1 M phosphate buffer (pH 7.4). Aliquots of homogenates from the rat brains were sepa-rated and used to measure protein, lipid peroxidation and glutathione. The remaining homogenates were centri-fuged at 10,000 rpm for 15 min and the supernatant was then used for enzyme assay. Superoxide dismutase was determined within 24 h.
2.6.2 Estimation of Malondialdehyde (MDA) Aliquotes of 0.5 ml distilled water and 1.0 ml 10% TCA were added to a volume of 0.5 ml brain tissue homoge-nate, mixed well and centrifuged at 3000 rpm for 10 min. To 0.2 ml supernatant, 0.1 ml thiobarbituric acid (TBA) (0.375%) was added. The total solution was placed in a water bath at 80ºC for 40 min and then cooled to room temperature. The absorbance of the clear supernatant was measured at 532 nm in spectrophotometer [17].
2.6.3 Estimation of Superoxide Dismutase (SOD) The SOD activity of the brain tissue was analyzed by using the SOD Assay kit (Fluka). For the assay, 200 µl of working solution, 20 µl of dilution buffer and 20 µl of enzyme working solution was added. Incubate the plate at 37°C for 20 min. Absorbance was read at 450 nm us-ing a microplate reader.
2.6.4 Measurement of Glutathione Pipette out 100 µl of the brain supernatant and add 50 µl of O-ophthaldehyde (100 µl/ml). Incubate at room tem-perature for 15 min. The flouroscent complex formed was read at an excitation wavelength of 350 nm and emission wavelength of 420 nm [18].
2.6.5 Estimation of Cholinergic Status in the Rat Brain The cholinergic marker, acetylcholinesterase was esti-mated in the whole brain according to the method of [19]. Briefly, the brains of the rats were removed over ice and the brain was separated using fine forceps. The tissue was then homogenized in 100 mM phosphate buffer. 0.1
ml of this homogenate was incubated for 5 min with 2.7 ml of phosphate buffer and 0.1 ml of DTNB. Then, 0.1 ml of freshly prepared acetylthiocholine iodide, pH 8 was added and the absorbance was read at 412 nm for 3 min at 30, 60, 90, 120, 150 and 180 sec.
2.7 Statistical Analysis
All data were expressed as mean ± SD. The significance of difference among the values of control, scopolamine treated, standard drug and extract treated groups for each session was determined by ANNOVA (one-way) fol-lowed by Dunnett’s test. The difference between values on 1st session and 5th session of the same group was ana-lyzed by student’s t-test.
3. Results
3.1 Selection of the Dose
One single dose (300 mg/kg) of the herbal extract has been selected after the initial pilot study. This pilot study was done by taking limited number of Wistar rats. In the pilot study three different doses (100, 300 & 900 mg/kg) were taken. Based on initial data (data not shown) from active avoidance test 300 mg/kg was selected for the main study. It was also seen that animals with higher dose (900 mg/kg) tolerated the shock and remained at one place, which is not acceptable for the avoidance test. However, with lower dose (100 mg/kg) there was no significant difference in the number of avoidances be-tween different groups of animals.
3.2 Automatic Reflex Conditioner
There was a significant (p < 0.05) increase in avoidance response on 5th session (6.4 ± 1.67) as compared to 1st session (3.0 ± 1.00) in the control group (Table 1). All groups except GR-3 have shown significant (p < 0.05) increase in avoidance response compared to their first session data. Significant (p < 0.05) reduction of avoid-ance response was observed in scopolamine treated group (GR-2) compared to control group (GR-1). How-ever standard drug (donepezil) treatment and extract feeding (GR-5 and GR-6) significantly (p < 0.05) in-creased the avoidance response in their first session compared to their corresponding scopolamine treated group (GR-2). This reflects the effectiveness of donepe-zil as well as aqueous extract during scopolamine in-duced memory loss. However donepezil group (GR-5) showed improved response compared to extract treated group (GR-6) (5.8 ± 0.83 vs 4.8 ± 0.44) at the end of 5th session. While extract treatment showed significant (p < 0.05) improvement of avoidance response at the end of 5th session in GR-7, no improvement was observed in GR-8.
Anti-Amnesic Activity of Vitex negundo in Scopolamine Induced Amnesia in Rats
Copyright © 2010 SciRes. PP
4
Table 1. The number of avoidance responses in control (GR-1), scopolamine (GR-2, 3, 4) and drug treated (GR-6, 7, 8) groups (Mean ± SD, n = 6)
S.NO. GROUPS DAY1 DAY2 DAY3 DAY4 DAY5
1 GR-1 03 ± 1.00 3.6 ± 1.14 4.2 ± 0.83 05 ± 0.70 6.4 ± 1.67**
2 GR-2 2.4 ± 0.55 03 ± 1.00 3.4 ± 1.14 3.8 ± 0.83 4.2 ± 0.83**,†
3 GR-3 3.2 ± 1.09 2.6 ± 0.54 3.4 ± 0.54 3.4 ± 0.89 3.2 ± 0.44
4 GR-4 3.4 ± 0.54 3.6 ± 0.89 4.2 ± 0.83 4.6 ± 0.54 4.8 ± 0.44*
5 GR-5 03 ± 0.70 04 ± 100 4.4 ± 0.54 5.4 ± 0.89 5.8 ± 0.83**, Ψ
6 GR-6 3.4 ± 0.54 3.8 ± 0.83 4.2 ± 0.83 4.8 ± 0.83 4.8 ± 0.44**,†, Ψ
7 GR-7 3.6 ± 0.54 3.6 ± 0.54 3.8 ± 0.44 4.4 ± 1.52 4.6 ± 0.54*, Ψ
8 GR-8 3.6 ± 0.89 3.8 ± 0.44 4.4 ± 0.54 4.8 ± 0.44 5.4 ± 0.54**
*p < 0.05 vs. Day 1; **p < 0.01 vs. Day 1; †p < 0.05 vs. Standard drug (GR-6); Ψp < 0.05 vs. corresponding scopolamine treated group
3.3 Markers of Oxidative Stress in Rat Brain
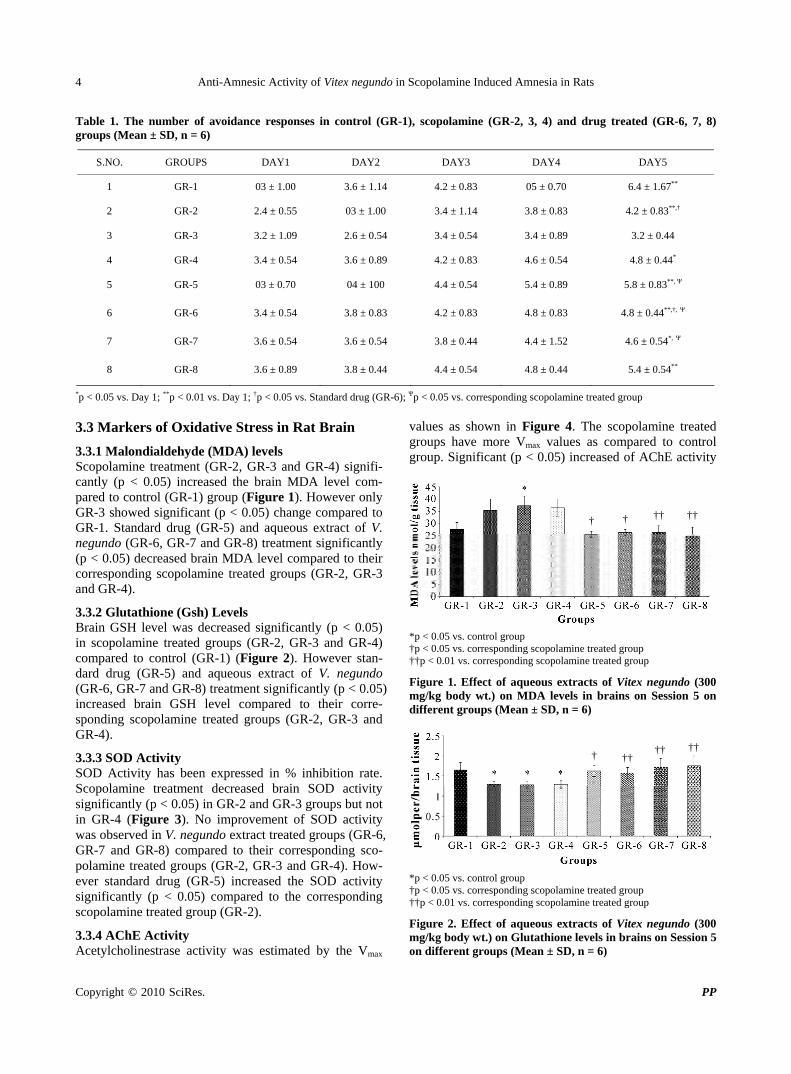
3.3.1 Malondialdehyde (MDA) levels Scopolamine treatment (GR-2, GR-3 and GR-4) signifi-cantly (p < 0.05) increased the brain MDA level com-pared to control (GR-1) group (Figure 1). However only GR-3 showed significant (p < 0.05) change compared to GR-1. Standard drug (GR-5) and aqueous extract of V. negundo (GR-6, GR-7 and GR-8) treatment significantly (p < 0.05) decreased brain MDA level compared to their corresponding scopolamine treated groups (GR-2, GR-3 and GR-4).
3.3.2 Glutathione (Gsh) Levels Brain GSH level was decreased significantly (p < 0.05) in scopolamine treated groups (GR-2, GR-3 and GR-4) compared to control (GR-1) (Figure 2). However stan-dard drug (GR-5) and aqueous extract of V. negundo (GR-6, GR-7 and GR-8) treatment significantly (p < 0.05) increased brain GSH level compared to their corre-sponding scopolamine treated groups (GR-2, GR-3 and GR-4).
3.3.3 SOD Activity SOD Activity has been expressed in % inhibition rate. Scopolamine treatment decreased brain SOD activity significantly (p < 0.05) in GR-2 and GR-3 groups but not in GR-4 (Figure 3). No improvement of SOD activity was observed in V. negundo extract treated groups (GR-6, GR-7 and GR-8) compared to their corresponding sco-polamine treated groups (GR-2, GR-3 and GR-4). How-ever standard drug (GR-5) increased the SOD activity significantly (p < 0.05) compared to the corresponding scopolamine treated group (GR-2).
3.3.4 AChE Activity Acetylcholinestrase activity was estimated by the Vmax
values as shown in Figure 4. The scopolamine treated groups have more Vmax values as compared to control group. Significant (p < 0.05) increased of AChE activity
† † ††††
*p < 0.05 vs. control group †p < 0.05 vs. corresponding scopolamine treated group ††p < 0.01 vs. corresponding scopolamine treated group
Figure 1. Effect of aqueous extracts of Vitex negundo (300 mg/kg body wt.) on MDA levels in brains on Session 5 on different groups (Mean ± SD, n = 6)
† †† †† ††
*p < 0.05 vs. control group †p < 0.05 vs. corresponding scopolamine treated group ††p < 0.01 vs. corresponding scopolamine treated group
Figure 2. Effect of aqueous extracts of Vitex negundo (300 mg/kg body wt.) on Glutathione levels in brains on Session 5 on different groups (Mean ± SD, n = 6)

Anti-Amnesic Activity of Vitex negundo in Scopolamine Induced Amnesia in Rats
Copyright © 2010 SciRes. PP
5
††
*p < 0.05 vs. control group †p < 0.05 vs. corresponding scopolamine treated group ††p < 0.01 vs. corresponding scopolamine treated group
Figure 3. Effect of aqueous extracts of Vitex negundo (300 mg/kg body wt.) on SOD levels in brains on Session 5 on different groups (Mean ± SD, n = 6)
††††
††
*p < 0.05 vs. control group †p < 0.05 vs. corresponding scopolamine treated group ††p < 0.01 vs. corresponding scopolamine treated group
Figure 4. Effect of aqueous extracts of Vitex negundo (300 mg/kg body wt.) on AChE levels in brains on Session 5 on different groups (Mean ± SD, n = 6) was observed in scopolamine treated groups (GR-2 and GR-3, not in GR-4) compared to control (GR-1) (Figure 4). However standard drug (GR-5) and aqueous extract of V. negundo (GR-6 and GR-7, not in GR-8) treatment significantly (p < 0.01) decreased brain AChE activity compared to their corresponding scopolamine treated groups (GR-2, GR-3 and GR-4).
4. Discussion
V. negundo possesses many medicinal properties. Leaves of V. negundo have been investigated for its anti-in- flammatory activity [15,20]. Telang et al. first noticed non-steroidal anti-inflammatory (NSAID) activity of V. negundo. Similarly, fresh leaves of V. negundo have been suggested to possess anti-inflammatory and pain sup-pressing activities. Antinociceptive activity study of ethanolic leaf extract of V. negundo showed that it pos-sesses both central and peripheral analgesic activity [21]. V. negundo has been also used in adjuvant therapy to standard anti-inflammatory drugs [22]. Literature survey of V. negundo also revealed the presence of lignans de-rivative, which is responsible for anti-cholinesterase ac-
tivity in in-vitro [14]. LD50 dose of V. negundo leaf ex-tract is 7.58 g/kg which practically falls in the non- toxic dose range [23].
The administration of the antimuscarinic agent sco-polamine produces transient memory deficit. Also, sco-polamine has been shown to impair memory retention when given to rat shortly before training in an avoidance task. The ability of a range of different cholinergic ago-nist drugs to reverse the amnesic affects of scopolamine is now well documented in animals and human volun-teers [24]. The scopolamine amnesia test is widely used as primary screening test for so called anti-Alzheimer drugs [24]. Here, scopolamine is given at different time of training sessions and trials. Such protocol is adapted to distinguish between the three different stages of memory i.e. acquisition, consolidation and retention.
In the preliminary screening of the present study showed that the improvement in learning and memory tasks in the shuttle-box was only observed at a dose of 300 mg/kg body wt. Therefore, the aqueous extract with 300 mg/kg was evaluated in more details. The avoid-ance responses shown by the animals were due to their ability to learn the task, which reflects the cognitive function. The task was investigated by using the sco-polamine-induced dementia with the aqueous herbal ex-tract of V. negundo. The animals were treated with sco-polamine at different time intervals of trial in the sessions according to the different stages of the memory. Sco-polamine administered 5 min prior to 1st trial on 1st ses-sion was for the acquisition whereas scopolamine ad-ministered 5 min after the 15th (i.e., last) trial on 1st ses-sion was for the consolidation stage of the memory. Similarly for the requisition (recall) the scopolamine was administered 5 min prior to the 1st trial on the last session i.e., 5th session. To evaluate the effect of the herbal aqueous extract of V. negundo, scopolamine was admin-istered in the similar pattern in the pretreated herbal ex-tract animals. The efficacy and potency of the extract was compared with the vehicle control and standard drug (Donepezil) group.
From the behavioral test i.e. two way shuttle box ac-tive avoidance test, it is clearly seen that there was a general decrease in the performance in the active avoid-ance in the scopolamine treated groups. The memory loss effect of scopolamine is more prominent compared to the control group. The aqueous herbal extract of V. negundo improved the memory loss effect of scopola-mine in all three events like acquisition, consolidation and retention. As scopolamine-induced memory loss was more prominent in acquisition period, we administered standard drug, donepezil with this group. In comparison with Donepezil, the extract treated group had almost equal avoidance responses which indicates therapeutic efficacy of V. negundo against memory loss.
The present study therefore demonstrates the probable

Anti-Amnesic Activity of Vitex negundo in Scopolamine Induced Amnesia in Rats
Copyright © 2010 SciRes. PP
6
mechanism by which V. negundo enhanced the anti-am- nesic activity by increasing the performance of learning and memory. It had been suggested that the varying de-grees of behavioral impairments are associated with ag-ing and age associated neurodegenerative diseases. Oxi-dative stress due to free radicals generation is responsible for producing the neuronal changes mediating these be-havioral deficits [25]. Oxidative stress in brain generates oxygen radicals like superoxide anion, hydroxyl radical, and hydrogen peroxide, which act on polyunsaturated fatty acids in brain, thereby propagating the lipid peroxi-dation [26]. The major antioxidant and oxidative free- radical scavenging enzymes like glutathione, SOD and catalase plays an important role to reduce oxidation stress in brain. In the present study rats after scopolamine treatment showed a significant increase in the brain lev-els of malondialdehyde, which is the measure of lipid peroxidation and free radical generation. At the same time there was a significant reduction in levels of glu-tathione, a tripeptide found in all cells, which reacts with free radicals to protect cells from superoxide radical, hydroxyl radical and singlet oxygen [27]. Pre-treatment of V. negundo reduced the MDA levels and increased GSH content in brain after scopolamine treatment. Sco-polamine reduced the SOD activity in brain. SOD is the only enzyme that uses the superoxide anions as the sub-strate and produces hydrogen peroxide as a metabolite. Super oxide anion is more toxic than H2O2 and has to be removed. Pretreatment with V. negundo significantly prevented the reduction of SOD activity in brain during scopolamine treatment. Our results also suggest that the aqueous extract of V. negundo reduced oxidative stress by reducing lipid peroxidation and increasing the en-dogenous antioxidant enzymes in brain. Other important activity has been shown by the extract is that it has ace-tylcholinetrase (AChE) inhibiting activity. This activity tends to allow the more retention of acetylcholine in the brain, which is important for the cognitive functions, learning and memory.
In conclusion, the present study demonstrates that aqueous V. negundo extract has potential therapeutic effects on improving the anti-amnesic activity in rats through inhibiting lipid peroxidation, augmenting en-dogenous antioxidant enzymes and decreasing acetylcho- linestrase (AChE) activity in brain. Further study is war-ranted to find its potential use in humans.
5. Aknowledgements
Authors are very thankful to the Project Director, Na-tional Institute of Pharmaceutical Education and Re-search, Hyderabad and Director Indian Institute of Chemical Technology, Hyderabad for supporting this work. We are also thankful to Amruta Herbals Pvt Lim-ited, Indore (M.P), India for providing the plant material.
REFERENCES [1] J. Dunning and J. D. Matthew, “Molecular Mechanisms
of Learning and Memory,” Expert Reviews in Molecular Medicine, Vol. 5, No. 25, 2003, pp. 1-11.
[2] A. Blockland, “Acetylcholine: A Neurotransmitter for Learning and Memory?” Brain Research Review, Vol. 21, No. 3, 1996, pp. 285-300.
[3] M. F. Siddiqui and A. I. Levey, “Cholinergic Therapies in Alzheimer’s Disease,” Drugs of the Future, Vol. 24, No. 4, 1999, pp. 417-444.
[4] J. Winkler, S. Suhr and F. Gage, “Essential Role of Neo-cortical Acetylcholine in Spatial Memory,” Nature, Vol. 375, No. 6531, 1995, pp. 484-487.
[5] T. F. Wernicke and F. M. Reischies, “Prevalence of De-mentia in Old Age: Clinical Diagnoses in Subjects Aged 95 and Older,” Neurology, Vol. 44, No. 2, l994, pp. 250- 253.
[6] D. C. Ewbank, “Deaths Attributable to Alzheimer’s Dis-ease in the United States,” American Journal of Public Health, Vol. 89, No. 1, 1991, pp. 90-92.
[7] V. Chandra, R. Pandav and H. H. Dodge, “Incidence of Alzheimer’s Disease in a Rural Community in India: The Indo-US Study,” Neurology, Vol. 57, No. 6, 2001, pp. 985-989.
[8] V. Chandra, M. Ganguli and R. Pandav, “Prevalence of Alzheimer’s Disease and Other Dementias in Rural India: The Indo-US Study,” Neurology, Vol. 51, No. 4, 1998, pp. 1000-1008.
[9] T. Sunderland, P. N. Tariot, R. M. Cohen, H. Weingarbier, E. A. Mueller and D. L. Murphy, “Anticholinergic Sensi-tivity in Patients with Dementia of the Alzheimer’s Type and Age Matched Controls,” Archives of General Psy-chiatry, Vol. 44, No. 5, l987, pp. 418-426.
[10] C. Flicker, S. H. Ferris and M. Serby, “Hypersensitivity to Scopolamine in the Elderly,” Psychopharmacology, Vol. 107, No. 2-3, 1992, pp. 437-441.
[11] E. Giacobini, “The Cholinergic System in Alzheimer's Disease,” In: S. M. Aquilonius and P. G. Gillberg, (Eds.), Brain Research. Cholinergic Neurotransmission: Func-tional and Clinical Aspects, Elsevier, Amsterdam, 1990, pp. 321-332.
[12] W. R. Markesbery, “Oxidative Stress Hypothesis in Alz-heimer’s Disease,” Free Radical Biology and Medicine, Vol. 23, No. 1, 1997, pp. 134-147.
[13] M. A. Lovell, W. D. Ehmann, S. M. Butler and W. R. Markesberg, “Elevated Thiobarbituric Acid Reactive Substances and Antioxidant Enzyme Activity in the Brain in Alzheimer’s Disease,” Neurology, Vol. 45, No. 8, 1995, pp. 1594-1608.
[14] U. H. Azhar and M. Abdul, “Enzymes Inhibiting Lignans from Vitex negundo,” Chemical and Pharmaceutical Bul-letin, Vol. 52, No. 11, 2004, pp. 1269-1272.
[15] M. G. Dharmasiri, J. R. Jayakodym, G. Galhenam, S. S. Liyanagem and W. D. Ratnasooriyam, “Anti Inflamma-tory and Analgesic Activities of Mature Fresh Leaves of Vitex negundo,” Jounal of Ethnopharmacology, Vol. 87,

Anti-Amnesic Activity of Vitex negundo in Scopolamine Induced Amnesia in Rats
Copyright © 2010 SciRes. PP
7
No. 2-3, 2003, pp. 199-206.
[16] A. C. Guyton and J. E. Hall, “Textbook of Medical Phy-siology,” Harcourt Asia Pte Ltd, Singapore, 1999.
[17] H. Ohkawa, N. Ohishi and K. Yagi, “Assay for Lipid Peroxides in Animal Tissues by Thiobarbituric Acid Re-action,” Analytical Biochemistry, Vol. 95, No. 2, 1979, pp. 351- 358.
[18] P. J. Hissin and R. Hilf, “A Fluorometric Method for Determination of Oxidized and Reduced Glutathione in Tissues,” Analytical Biochemistry, Vol. 74, No. 1, 1976, pp. 214-226.
[19] G. L. Ellman, D. K. Courtney, V. Andres and R. M. Fea-therstone, “A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity,” Biochemical Pharma-cology, Vol.7, No. 2, 1961, pp. 88-95.
[20] R. S. Telang, S. Chatterjee and C. Varshneya, “Studies on Analgesic and Anti-Inflammatory Activities of Vitex ne-gundo Linn,” Indian Journal of Pharmacology, Vol. 31, No. 5, 1999, pp. 363-366.
[21] R. K. Gupta and V. R. Tandon, “Antinociceptive Activity of Vitex-Negundo Linn Leaf Extract,” Indian Journal of Pharmacology, Vol. 49, No. 2, 2005, pp. 163-170.
[22] R. K. Gupta and V. R. Tandon, “Vitex negundo Linn (VN) Leaf Extract as an Adjuvant Therapy to Standard Anti- Inflammatory Drugs,” The Indian Journal of Medical Re- search, Vol. 124, No. 4, 2006, pp. 447-450.
[23] M. N. Ghosh, “Fundamentals of Experimental Pharma-cology,” Scientific Book Agency, Calcutta, 1984.
[24] M. H. V. Kumar and Y. K. Gupta, “Antioxidant Property of Celastrus Paniculatus Willd: A Possible Mechanism in Enhancing Cognition,” Phytomedicine, Vol. 9, No. 4, 2002, pp. 302-311.
[25] C. I. Cantuti, B. Shukitt-Hale and J. A. Joseph, “Neuro-behavioural Aspects of Antioxidants in Aging,” Interna-tional Journal of Developmental Neuroscience, Vol. 18, No. 4-5, 2000, pp. 367-381.
[26] T. Coyle and P. Puttfarcven, “Oxidative Stress, Glutamate and Neurodegenerative Disorder,” Science, Vol. 262, No. 5134, 1993, pp. 89-695.
[27] J. B. Schulz, J. Linderau and J. Dichgans, “Glutathione, Oxidative Stress and Neurodegeneration,” European Jour-nal of Biochemistry, Vol. 267, No. 16, 2000, pp. 4904- 4911.

Anti-Amnesic Activity of Vitex negundo in Scopolamine Induced Amnesia in Rats
Copyright © 2010 SciRes. PP
8
Graphical Abstract

Pharmacology & Pharmacy, 2010, 1, 9-17 doi:10.4236/pp.2010.11002 Published Online July 2010 (http://www.SciRP.org/journal/pp)
Copyright © 2010 SciRes. PP
9
Development and Evaluation of a New Interpenetrating Network Bead of Sodium Carboxymethyl Xanthan and Sodium Alginate for Ibuprofen Release
Rajat Ray, Siddhartha Maity, Sanchita Mandal, Tapan K. Chatterjee, Biswanath Sa
The Division of Pharmaceutics, Department of Pharmaceutical Technology, Jadavpur University, Kolkata, India. Email: [email protected] Received June 6th, 2010; accepted July 8th, 2010.
ABSTRACT
Interpenetrating network (IPN) beads of sodium carboxymethyl xanthan (SCMX) and sodium alginate (SAL) were pre-pared by ionotropic gelation process using AlCl3 as a cross-linking agent. The effect of different formulation variables like total polymer concentration, gelation time, concentration of cross-linking agent, and drug load on the extent of release of ibuprofen (IBP), a non steroidal anti-inflammatory drug, was examined. The formation of IPN structure was examined using Fourier Transform Infra-red (FTIR) analysis and the compatibility of the drug in the bead was evalu-ated through FTIR, X-ray diffraction (XRD) and Differential Scanning Calorimetry (DSC) analyses. While increase in the concentration of total polymer, gelation time, and drug load decreased the drug release in both acidic (pH-1.2) and phosphate buffer (PB) solution (pH-6.8), increase in the concentration of cross-linking agent tended to increase the drug release. However, from all the formulations, the drug release in acidic medium was considerably slow and a maximum 14% of the loaded drug was released in 2 h. Complete drug release was achieved in PB solution within 210 to 330 min depending upon the formulation variables. The release of the drug followed non-Fickian transport process in acidic medium and case-II transport mechanism in PB solution and these release behaviour correlated well with the kinetics of dynamic swelling of IPN beads. The study indicated that the IPN beads of SCMX and SAL could be a suit-able dosage form to minimize the drug release in acidic solution and to control the drug release in PB solution depend-ing upon the need. Keywords: IPN Bead, Ibuprofen, Drug Release, Kinetics, Swelling
1. Introduction
Among the most abundant natural polymers, polysaccha-rides are widely used in pharmaceutical dosage forms as excipients like suspending agents, emulsifying agents, tablet binders, gelling agents. With the advent of mac-romolecular chemistry, the use of polysaccharides has been extended towards new applications in pharmaceuti-cal, biomedical, and agricultural fields. Although natu-rally available polysaccharides exhibit certain limitations in terms of their reactivity and processibility, these can be overcome by modification of the polysaccharides through either physical or chemical cross-linking, graft-ing with other materials and developing hydrogels or interpenetrating network (IPN) structures.
Since the homopolymers alone can not meet divergent
demand in terms of properties and performances, devel-opment of IPN appears to be a better approach [1] and one of the easiest ways for modification of the properties of polysaccharides. IPN consists of two polymers, each in network form, which can be cross-linked in the pres-ence of each other to give a three dimensional network structure [2] and hence, combine the properties of two cross-linked polymers in a network form [3]. IPNs are thus emerging as a rapidly developing branch of polymer blended technology and are finding applications in artifi-cial implants, dialysis, membranes, drug delivery systems [4], and in agricultural field [5].
Sodium alginate (SAL), a hydrophilic biopolymer ob-tained from brown sea weeds, is a polysaccharide com-posed of varying proportions of D-mannuronic acid (M) and L-guluronic acid (G) residues which are arranged in
Development and Evaluation of a New Interpenetrating Network bead of Sodium Carboxymethyl Xanthan and Sodium Alginate for Ibuprofen Release
Copyright © 2010 SciRes. PP
10
MM or GG blocks interspersed with MG blocks [6]. Its unique property of forming water insoluble calcium al-ginate gel through ionotropic gelation with Ca2+ ions in a simple and mild condition has made possible to encapsu-late both macromolecular agents [7] and low molecular weight therapeutic agents [8,9] in calcium alginate beads. However in physiological environment, calcium alginate beads tend to have poor mechanical stability [10]. To overcome this limitation, IPN beads of SAL with gelatin or egg albumin [2], polyvinyl alcohol-graftedpolyacry- lamide [11], N,O-Carboxymethyl chitosan [12] for con-trolled drug delivery, and with gelatin [5] for controlled release of pesticides have been developed.
Although xanthan gum, a polysaccharide obtained from Xanthomonas campestris, can not form gel beads, its Na-salt of carboxymethyl derivative is able to form gel beads through ionotropic gelation with Al3+ ions [13]. Sodium carboxymethyl xanthan (SCMX) beads have been found capable of encapsulating albumin [14] and diltiazem hydrochloride [15]. However hitherto there are no reports on IPN beads of SCMX with SAL for drug release study.
The objective of the present work was to develop a new IPN bead composed of SCMX and SAL and to eva-luate the beads for encapsulation and release behavior of ibuprofen (IBP).
2. Experimental
2.1 Materials
Ibuprofen (Indian Pharmacopoeia) and xanthan gum were obtained as gift samples from respectively M/S Albert David Limited and M/S Deys Medical Stores (Mfg). Pvt. Limited, Kolkata, India. Sodium alginate (Mol. wt. 240kDa), AlCl3·2H2O (SD Fine Chem Pvt. Ltd, Mumbai, India), Monochloro acetic acid (Loba Chemie Pvt. Ltd, Mumbai, India) and all other analytical grade reagents were obtained commercially and used as re-ceived.
2.2 Preparation of Sodium Carboxymethyl Xanthan (SCMX)
Xanthan gum was derivatised to SCMX having O-car- boxymethyl substitution of 0.8 following the method reported previously [13]. In brief, required amount of xanthan gum was dispersed in ice cold solution of 45% w/v sodium hydroxide. The dispersion was kept at 5-8°C with continuous stirring for 1h. Monochloroacetic acid solution (75% w/v) was added with stirring in the reac-tion mixture and the temperature was raised slowly to 15-18°C. After 30 min, the temperature was raised to 75°C and maintained for additional 30 min. The reaction mixture was, then cooled to room temperature, cut into small pieces and dried at 50°C. The dried product was
milled, washed with 80% v/v methanol and again dried.
2.3 Preparation of Interpenetrating Network (IPN) Bead
Required amount of ibuprofen (IBP) was homogenously dispersed in an aqueous solution of SCMX and SAL. The resulting dispersion was extruded through 21 G flat-tip hypodermic needle into AlCl3 solution. Gelation of the beads was carried out for different periods of time. The beads were, then collected by filtration, washed with deionized water, dried at 45°C in a hot air oven to con-stant weight and kept in a dessicator until used. The beads were prepared using the following variables:
1) Keeping the drug load constant at 50% w/w of total polymer and the concentration of AlCl3 constant at 2% w/v, the total polymer concentration was varied from 2-4% w/v (SCMX to SAL weight ratio 1:1) and the gela-tion time was varied from 0.5 to 2 hour.
2) Keeping the drug load constant at 50% w/w of total polymer, the gelation time at 0.5 hour and total polymer concentration 3% w/v, concentration of AlCl3 was varied from 2-8% w/v.
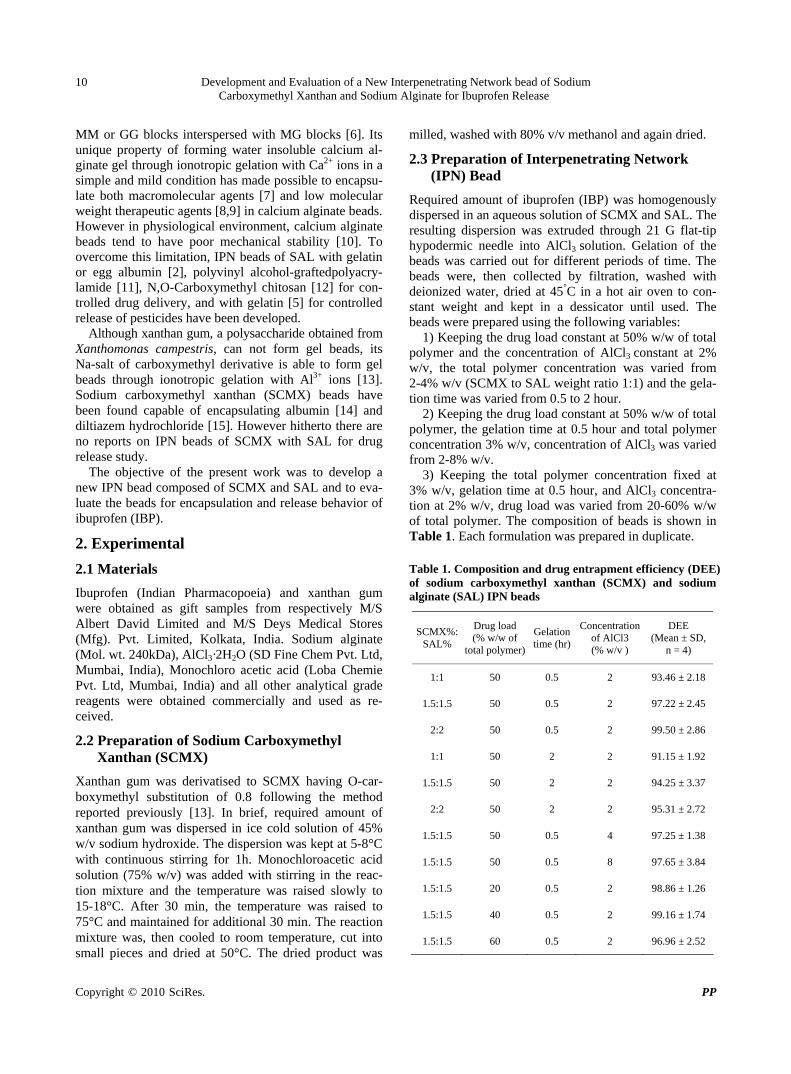
3) Keeping the total polymer concentration fixed at 3% w/v, gelation time at 0.5 hour, and AlCl3 concentra-tion at 2% w/v, drug load was varied from 20-60% w/w of total polymer. The composition of beads is shown in Table 1. Each formulation was prepared in duplicate. Table 1. Composition and drug entrapment efficiency (DEE) of sodium carboxymethyl xanthan (SCMX) and sodium alginate (SAL) IPN beads
SCMX%: SAL%
Drug load (% w/w of
total polymer)
Gelation time (hr)
Concentration of AlCl3 (% w/v )
DEE (Mean ± SD,
n = 4)
1:1 50 0.5 2 93.46 ± 2.18
1.5:1.5 50 0.5 2 97.22 ± 2.45
2:2 50 0.5 2 99.50 ± 2.86
1:1 50 2 2 91.15 ± 1.92
1.5:1.5 50 2 2 94.25 ± 3.37
2:2 50 2 2 95.31 ± 2.72
1.5:1.5 50 0.5 4 97.25 ± 1.38
1.5:1.5 50 0.5 8 97.65 ± 3.84
1.5:1.5 20 0.5 2 98.86 ± 1.26
1.5:1.5 40 0.5 2 99.16 ± 1.74
1.5:1.5 60 0.5 2 96.96 ± 2.52

Development and Evaluation of a New Interpenetrating Network bead of Sodium Carboxymethyl Xanthan and Sodium Alginate for Ibuprofen Release
Copyright © 2010 SciRes. PP
11
2.4 Fourier Transform Infrared (FTIR) Analysis
FTIR spectra of SCMX, SAL and drug free IPN bead were recorded in a FTIR spectrophotometer (Perkin- El-mer, model Spectrum RX-1, UK). Each sample was mixed with KBr and converted into disc at 100 kg pres-sure using a hydraulic press. The spectra were recorded within 4000-400 cm-1 wave numbers. Similarly, the FTIR spectra of IBP and drug loaded IPN beads were recorded.
2.5 Powder X-Ray Diffraction (XRD) Analysis
Qualitative XRD studies were performed using an X-ray diffractometer (Bruker D8 advanced powder diffracto-meter, USA). Pure IBP and powdered beads were scanned from 5° to 55° diffraction angle (2θ) range under the following conditions:
Source, Ni-filtered Cu-Kα (λ = 1.54) radiation; voltage, 40 kV; Current, 40 mA; scan speed, 16°/min
2.6 Differential Scanning Calorimetry (DSC) Study
DSC thermograms of IBP and powdered beads were ob-tained in the following way:
A weighed amount (about 6 mg) of sample was kept in a hermetically sealed aluminium pan and heated at a scan speed of 10°C /min over a temperature range of 35°C- 310°C in a Differential Scanning Calorimeter (Perkin- Elmer, model Pyris Diamond TG/DTA, UK ) which was calibrated against indium. A nitrogen purge (20 ml/min) was used throughout the runs.
2.7 Photomicrograph
Photomicrograph of IPN beads were taken at 4X magni-fication with an optical microscope (Leica DM 2500P) fitted with a camera (Cannon Power Shot S-80, Japan).
2.8 Drug Entrapment Efficiency
IPN beads (20 mg) were accurately weighed in an elec-tronic balance (Precisa XB 600 MC, Precisa Instrument Ltd; Switzerland), immersed in 250 ml USP phosphate buffer (PB) solution (pH 6.8), and shaken for 2h on a mechanical shaker. The beads were crushed and further shaken for 1h. The solution was filtered and an aliquot following suitable dilution was analyzed at 222 nm in a UV-Visible spectrophotometer (model Cary-50 Bio-sp- ectrophotometer, VARIAN, Australia)) and the content of the beads was determined using a calibration curve constructed using PB solution of pH 6.8. The reliability of the above analytical method was judged by conducting recovery analysis at three levels of spiked drug solution in the presence or absence of the polymers for three con-secutive days. The recovery averaged 98.45 ± 2.68%. DEE was determined using the following relation:
DEE (%) = (Determined drug content/Theoretical drug
content) × 100.
2.9 In-Vitro Drug Release Study
In-vitro drug release study was carried out in acidic solu-tion 0.1 (N) HCl (pH 1.2) and in USP PB solution (pH 6.8) using USP-II dissolution rate test apparatus (model TDP-06P Electro Lab, Mumbai, India). 20 mg beads were placed in 500 ml acidic solution or 500 ml PB solu-tion (37 ± 1°C) and rotated with paddle at 75 rpm. Ali-quot was withdrawn at different times and replenished immediately with the same volume of fresh solution. Undiluted or suitably diluted withdrawn samples were analyzed spectrophotometrically at 220 nm for acidic solution and 222 nm for PB solution. The amount of drug released in acidic solution and PB solution were calcu-lated from the calibration curves drawn respectively, in 0.1 (N) HCl and PB solution (pH 6.8). Each release study was conducted four times.
2.10 Swelling Study
Dried drug-free IPN beads (50 mg) were immersed in 25ml acidic solution (pH 1.2) at 37°C. The beads were removed at different times by filtration and blotted care-fully to remove excess surface water. The swollen beads were weighed. The swelling ratio of the beads were de-termined using the following formula:
Swelling ratio = (weight of swollen beads-weight of dry beads)/weight of dry beads
Swelling ratio of the beads in PB solution (pH 6.8) was determined in a similar way.
2.11 Statistical Analysis
Each formulation was prepared in duplicate, and each analysis was duplicated. Effect of formulation variables on drug release was tested for significance level by using analysis of variance (ANOVA: single factor and two factor) with the aid of Microsoft® Excel 2003. Differ-ence was considered significant when p < 0.05.
3. Results & Discussion
3.1 Formation of IPN
IPN beads composed of SCMX and SAL were prepared by inotropic gelation process using AlCl3 as a common cross-linking agent for both the polymers. Formulation of IPN structure was verified by FTIR analysis (Figure 1). FTIR spectrum of SCMX showed the presence of bands corresponding to asymmetric and symmetric carboxylate anions at respectively 1605 cm-1 and 1419 cm-1, a broad band at 3419 cm-1 corresponding to stretching vibration of hydroxyl group, a peak at 1327 cm-1 corresponding to C = O stretching of carboxymethyl group. These results are similar to the findings reported earlier [14]. The spectrum of SAL showed the bands characteristics of
Development and Evaluation of a New Interpenetrating Network bead of Sodium Carboxymethyl Xanthan and Sodium Alginate for Ibuprofen Release
Copyright © 2010 SciRes. PP
12
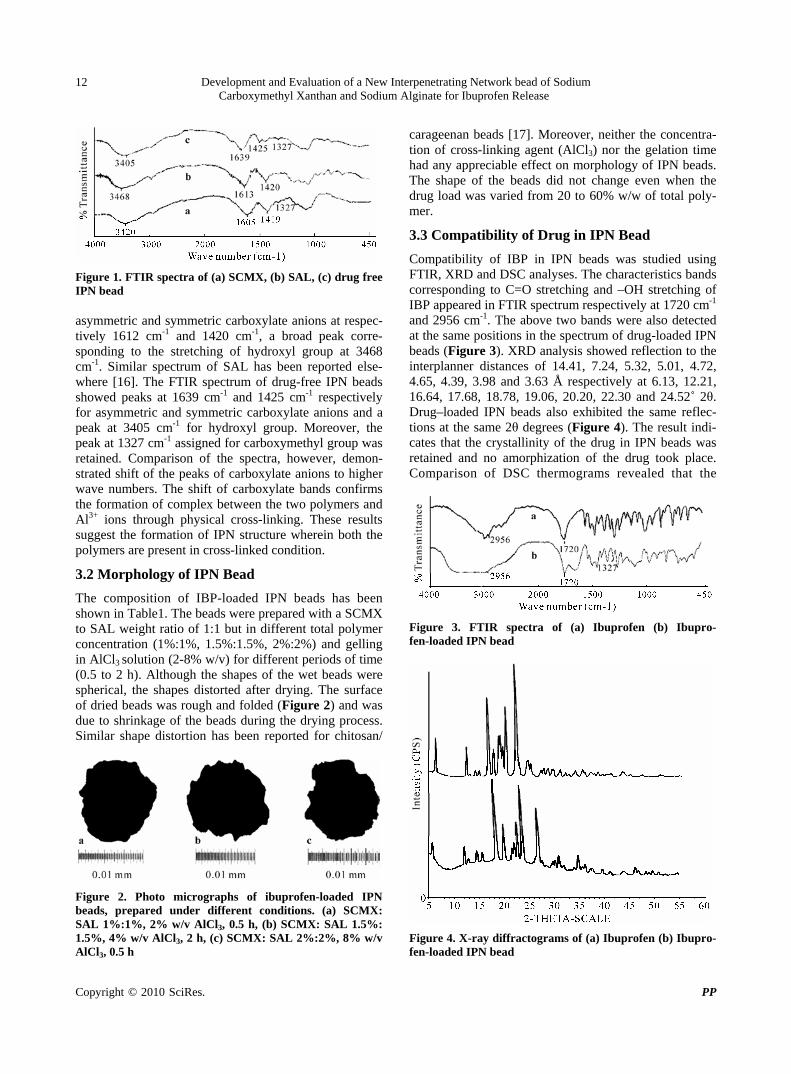
Figure 1. FTIR spectra of (a) SCMX, (b) SAL, (c) drug free IPN bead asymmetric and symmetric carboxylate anions at respec-tively 1612 cm-1 and 1420 cm-1, a broad peak corre-sponding to the stretching of hydroxyl group at 3468 cm-1. Similar spectrum of SAL has been reported else-where [16]. The FTIR spectrum of drug-free IPN beads showed peaks at 1639 cm-1 and 1425 cm-1 respectively for asymmetric and symmetric carboxylate anions and a peak at 3405 cm-1 for hydroxyl group. Moreover, the peak at 1327 cm-1 assigned for carboxymethyl group was retained. Comparison of the spectra, however, demon-strated shift of the peaks of carboxylate anions to higher wave numbers. The shift of carboxylate bands confirms the formation of complex between the two polymers and Al3+ ions through physical cross-linking. These results suggest the formation of IPN structure wherein both the polymers are present in cross-linked condition.
3.2 Morphology of IPN Bead
The composition of IBP-loaded IPN beads has been shown in Table1. The beads were prepared with a SCMX to SAL weight ratio of 1:1 but in different total polymer concentration (1%:1%, 1.5%:1.5%, 2%:2%) and gelling in AlCl3 solution (2-8% w/v) for different periods of time (0.5 to 2 h). Although the shapes of the wet beads were spherical, the shapes distorted after drying. The surface of dried beads was rough and folded (Figure 2) and was due to shrinkage of the beads during the drying process. Similar shape distortion has been reported for chitosan/
Figure 2. Photo micrographs of ibuprofen-loaded IPN beads, prepared under different conditions. (a) SCMX: SAL 1%:1%, 2% w/v AlCl3, 0.5 h, (b) SCMX: SAL 1.5%: 1.5%, 4% w/v AlCl3, 2 h, (c) SCMX: SAL 2%:2%, 8% w/v AlCl3, 0.5 h
carageenan beads [17]. Moreover, neither the concentra-tion of cross-linking agent (AlCl3) nor the gelation time had any appreciable effect on morphology of IPN beads. The shape of the beads did not change even when the drug load was varied from 20 to 60% w/w of total poly-mer.
3.3 Compatibility of Drug in IPN Bead
Compatibility of IBP in IPN beads was studied using FTIR, XRD and DSC analyses. The characteristics bands corresponding to C=O stretching and –OH stretching of IBP appeared in FTIR spectrum respectively at 1720 cm-1 and 2956 cm-1. The above two bands were also detected at the same positions in the spectrum of drug-loaded IPN beads (Figure 3). XRD analysis showed reflection to the interplanner distances of 14.41, 7.24, 5.32, 5.01, 4.72, 4.65, 4.39, 3.98 and 3.63 Å respectively at 6.13, 12.21, 16.64, 17.68, 18.78, 19.06, 20.20, 22.30 and 24.52˚ 2θ. Drug–loaded IPN beads also exhibited the same reflec-tions at the same 2θ degrees (Figure 4). The result indi-cates that the crystallinity of the drug in IPN beads was retained and no amorphization of the drug took place. Comparison of DSC thermograms revealed that the
Figure 3. FTIR spectra of (a) Ibuprofen (b) Ibupro-fen-loaded IPN bead
Figure 4. X-ray diffractograms of (a) Ibuprofen (b) Ibupro-fen-loaded IPN bead
Development and Evaluation of a New Interpenetrating Network bead of Sodium Carboxymethyl Xanthan and Sodium Alginate for Ibuprofen Release
Copyright © 2010 SciRes. PP
13
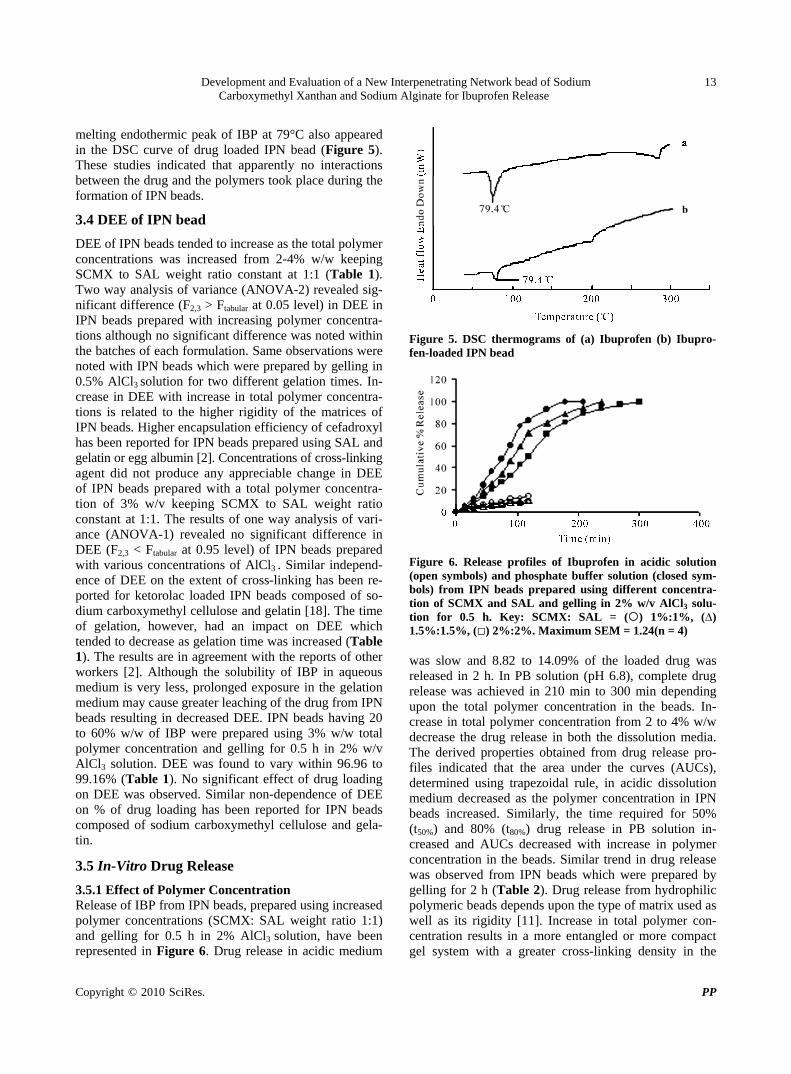
melting endothermic peak of IBP at 79°C also appeared in the DSC curve of drug loaded IPN bead (Figure 5). These studies indicated that apparently no interactions between the drug and the polymers took place during the formation of IPN beads.
3.4 DEE of IPN bead
DEE of IPN beads tended to increase as the total polymer concentrations was increased from 2-4% w/w keeping SCMX to SAL weight ratio constant at 1:1 (Table 1). Two way analysis of variance (ANOVA-2) revealed sig-nificant difference (F2,3 > Ftabular at 0.05 level) in DEE in IPN beads prepared with increasing polymer concentra-tions although no significant difference was noted within the batches of each formulation. Same observations were noted with IPN beads which were prepared by gelling in 0.5% AlCl3 solution for two different gelation times. In-crease in DEE with increase in total polymer concentra-tions is related to the higher rigidity of the matrices of IPN beads. Higher encapsulation efficiency of cefadroxyl has been reported for IPN beads prepared using SAL and gelatin or egg albumin [2]. Concentrations of cross-linking agent did not produce any appreciable change in DEE of IPN beads prepared with a total polymer concentra-tion of 3% w/v keeping SCMX to SAL weight ratio constant at 1:1. The results of one way analysis of vari-ance (ANOVA-1) revealed no significant difference in DEE (F2,3 < Ftabular at 0.95 level) of IPN beads prepared with various concentrations of AlCl3 . Similar independ-ence of DEE on the extent of cross-linking has been re-ported for ketorolac loaded IPN beads composed of so-dium carboxymethyl cellulose and gelatin [18]. The time of gelation, however, had an impact on DEE which tended to decrease as gelation time was increased (Table 1). The results are in agreement with the reports of other workers [2]. Although the solubility of IBP in aqueous medium is very less, prolonged exposure in the gelation medium may cause greater leaching of the drug from IPN beads resulting in decreased DEE. IPN beads having 20 to 60% w/w of IBP were prepared using 3% w/w total polymer concentration and gelling for 0.5 h in 2% w/v AlCl3 solution. DEE was found to vary within 96.96 to 99.16% (Table 1). No significant effect of drug loading on DEE was observed. Similar non-dependence of DEE on % of drug loading has been reported for IPN beads composed of sodium carboxymethyl cellulose and gela-tin.
3.5 In-Vitro Drug Release
3.5.1 Effect of Polymer Concentration Release of IBP from IPN beads, prepared using increased polymer concentrations (SCMX: SAL weight ratio 1:1) and gelling for 0.5 h in 2% AlCl3 solution, have been represented in Figure 6. Drug release in acidic medium
Figure 5. DSC thermograms of (a) Ibuprofen (b) Ibupro-fen-loaded IPN bead
Figure 6. Release profiles of Ibuprofen in acidic solution (open symbols) and phosphate buffer solution (closed sym-bols) from IPN beads prepared using different concentra-tion of SCMX and SAL and gelling in 2% w/v AlCl3 solu-tion for 0.5 h. Key: SCMX: SAL = () 1%:1%, (∆) 1.5%:1.5%, () 2%:2%. Maximum SEM = 1.24(n = 4) was slow and 8.82 to 14.09% of the loaded drug was released in 2 h. In PB solution (pH 6.8), complete drug release was achieved in 210 min to 300 min depending upon the total polymer concentration in the beads. In-crease in total polymer concentration from 2 to 4% w/w decrease the drug release in both the dissolution media. The derived properties obtained from drug release pro-files indicated that the area under the curves (AUCs), determined using trapezoidal rule, in acidic dissolution medium decreased as the polymer concentration in IPN beads increased. Similarly, the time required for 50% (t50%) and 80% (t80%) drug release in PB solution in-creased and AUCs decreased with increase in polymer concentration in the beads. Similar trend in drug release was observed from IPN beads which were prepared by gelling for 2 h (Table 2). Drug release from hydrophilic polymeric beads depends upon the type of matrix used as well as its rigidity [11]. Increase in total polymer con-centration results in a more entangled or more compact gel system with a greater cross-linking density in the
Development and Evaluation of a New Interpenetrating Network bead of Sodium Carboxymethyl Xanthan and Sodium Alginate for Ibuprofen Release
Copyright © 2010 SciRes. PP
14
Table 2. Derived properties of drug release in different dissolution media from IPN beads prepared using different polymer concentrations and gelling for 2 h in 2% w/v AlCl3 solution
In acidic solution In phosphate buffer solution SCMX%:
SAL% AUC (% mg · min/ml) (Mean ± SD, n = 4)
t50% (min) (Mean ± SD, n = 4)
t80% (min) (Mean ± SD, n = 4)
AUC (% mg·min/ml) (Mean ± SD, n = 4)
1:1 885.6 ± 56.31 94.8 ± 5.20 143.5 ± 15.21 14336.1 ± 25.63
1.5:1.5 729.2 ± 35.50 108.5 ± 8.75 174.1 ± 10.11 12553.9 ± 27.75
2:2 574.5 ± 46.21 132.4 ± 6.31 209.6 ± 8.65 10564.5 ± 14.02
matrix [12]. As a result, the rigidity of gel matrix in-creases and free volume of the matrix decreases [19]. This hinders easy transport of drug molecules through the matrix and reduces drug release from the matrix.
3.5.2 Effect of Swelling of IPN Bead The release of a drug from a polymeric matrix is con-trolled by the swelling behaviour of the polymer. To study the effect of swelling of IPN beads on drug release, swelling ratio of beads was measured in terms of water uptake at selected time intervals and the results have been represented in Figure 7. While the swelling ratio of the IPN beads was very low in acidic solution, the same property increased considerably in PB solution (pH 6.8). The main functional group present in both the polymers that undergoes cross-linking with Al3+ ions is –COOH group. In acidic solution, –COOH group remains proto-nated and exerts insignificant electrostatic repulsive force. As a result, the beads swell to very less extent. At higher pH value of PB solution, –COOH group undergoes ioni-zation which exerts electrostatic repulsion between the ionized groups, and results in higher swelling. Moreover, upon ionization, the counter ion concentration inside the polymeric network increases, and an osmotic pressure dif- ference exists between the internal and external solutions of the beads. The increased osmotic pressure is balanced by the swelling of the beads [20]. The higher the swelling of the polymers, the higher is the drug release from the IPN beads. Thus the slower release of IBP in acidic solu-tion and faster release in PB solution are related to the swelling behaviour of IPN beads in the respective disso-lution media. It was further observed that increase in total polymer concentration from 2 to 4% w/v decreased the swelling of IPN beads in both the media. At low po-lymer concentration, the polymeric network is loose with a greater hydrodynamic free volume which allows more of the liquid to be absorbed and produces higher swelling. This, in turn, facilitates transport of the drug molecule through the matrix and causes higher drug release [21]. On the other hand, at higher polymeric concentration, opposite phenomenon takes place resulting in slower release of drug.
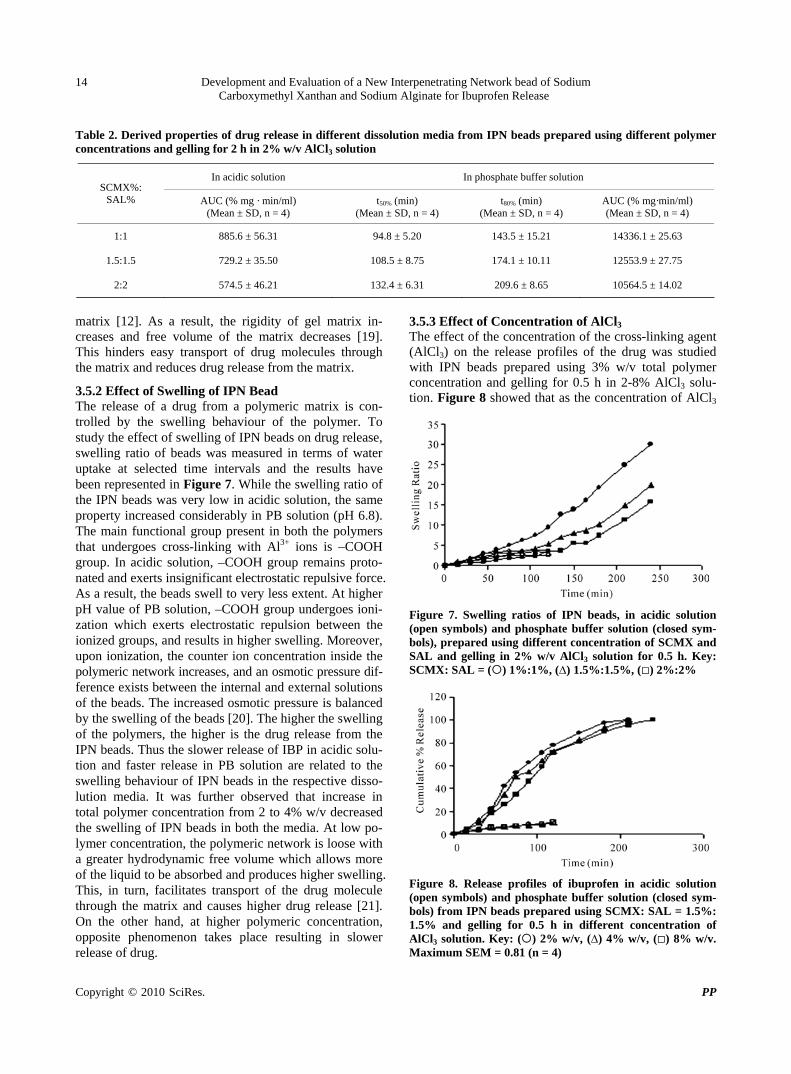
3.5.3 Effect of Concentration of AlCl3 The effect of the concentration of the cross-linking agent (AlCl3) on the release profiles of the drug was studied with IPN beads prepared using 3% w/v total polymer concentration and gelling for 0.5 h in 2-8% AlCl3 solu-tion. Figure 8 showed that as the concentration of AlCl3
Figure 7. Swelling ratios of IPN beads, in acidic solution (open symbols) and phosphate buffer solution (closed sym-bols), prepared using different concentration of SCMX and SAL and gelling in 2% w/v AlCl3 solution for 0.5 h. Key: SCMX: SAL = () 1%:1%, (∆) 1.5%:1.5%, () 2%:2%
Figure 8. Release profiles of ibuprofen in acidic solution (open symbols) and phosphate buffer solution (closed sym-bols) from IPN beads prepared using SCMX: SAL = 1.5%: 1.5% and gelling for 0.5 h in different concentration of AlCl3 solution. Key: () 2% w/v, (∆) 4% w/v, () 8% w/v. Maximum SEM = 0.81 (n = 4)

Development and Evaluation of a New Interpenetrating Network bead of Sodium Carboxymethyl Xanthan and Sodium Alginate for Ibuprofen Release
Copyright © 2010 SciRes. PP
15
was increased during the preparation of beads, the release of drug increased in both the dissolution media. Statisti-cal analysis in terms of ANOVA-1 also confirmed this phenomenon as the AUCs in acidic medium increased and t50%, t80% decreased and AUCs increased in PB solu-tion significantly. This unusual release behaviour could be explained in the following way. When the IPN beads were prepared with higher concentration of AlCl3, a thick outer gel layer might have been formed along the periph-ery of the beads. The thicker outer gel layer provided higher diffusional resistance to further influx of Al3+ ions resulting in the formation of inhomogeneous gel beads and less densely cross-linked matrix in the core of the beads. During dissolution study, once the outer thick gel layer swelled, quick drug release occurred from the beads. At lower concentration of AlCl3, Al3+ ions diffuse more uniformly into the beads and form homogenous gel beads resulting in slow drug release.
3.5.4 Effect of Gelation Time The derived properties obtained from drug release pro-files (Table 3) indicated that increase in gelation time decreased the drug release appreciably. The higher the gelation time, the greater is the cross-linking density and rigidity of the matrix which resulted in a fall in drug re-lease.
3.5.5 Effect of Drug Load The effect of drug load on the release dynamics of IBP was studied using IPN beads prepared using 3% w/v total polymer concentration (SCMX:SAL in a weight ratio 1:1) and gelling for 0.5 h in 2% w/v AlCl3 solution, and the results are shown in Figure 9. Increase in drug load from 20 to 60% w/w of total polymer decreased the drug re-lease in both the dissolution media. Generally, higher drug load provides higher concentration gradient be-tween the drug in the dosage form and the external dis-solution medium and results in faster drug release. The release of a drug is governed not only by drug diffusion
through the polymeric network but also by the relaxa- tional process of the polymer on solvent penetration. Low drug load in IPN beads forms larger pore fraction resulting in higher swelling and consequently faster drug release. On the other hand, at higher drug load, larger crystalline domain of drug is formed in the beads. This causes reduction as well as shrinkage of pores of the ma-trix and results in fall in drug release. Decrease in drug release with increase in drug load from various IPN beads have been reported [5,18,22-23].
3.5.6 Release Kinetics Drug release from a swellable matrix primarily depends on the degree of gelation, hydration, chain relaxation, and erosion of polymer. To understand the mode of drug transport through the IPN beads, the release data were fitted to the classical power law expression [24]
Mt/Mα = Ktn
where Mt and Mα are, respectively, the amount of drug released at time t and at infinite time, K represents a con-stant incorporating structural and geometrical character-istics of the dosage forms, n denotes the diffusion expo-nent indicative of the mechanism of drug release. Values of n ranging from 0.45 to 0.5 indicate Fickian or diffu-sion controlled release, values of n ranging from 0.5 to 0.89 indicate non-Fickian or anomalous release, and val-ues of n ranging from 0.89 to 1.0 indicate Case-II trans-port mechanism. By applying least squares method to release data, the values of n were estimated and have been shown in Table 4 along with the correlation co-efficient (r2). The results indicate that drug release in acidic medium followed non-Fickian mechanism and in PB solution drug release occured following case-II tran- sport mechanism. When the swelling data of drug-free IPN beads were fitted to the above power law expression, it was found that swelling in acidic medium took place following the non-Fickian mechanism and that in PB solu-tion followed Case-II transport mechanism (Table 4).
Table 3. Effect of gelation time on derived properties of drug release in different dissolution media from IPN beads prepared using different polymer concentration and gelling in 2% w/v AlCl3 solution for different periods of time
In acidic solution In phosphate buffer solution
Gelation time 0.5 h Gelation time 2 h Gelation time 0.5 h Gelation time 2 h SCMX%:
SAL% AUC (% mg · min/ml)
(Mean ± SD, n = 4)
AUC (% mg · min/ml)
(Mean ± SD, n = 4)
t50% (min) (Mean ± SD,
n = 4)
t80% (min) (Mean ± SD,
n = 4)
AUC (% mg · min/ml)
(Mean ± SD, n = 4)
t50% (min) (Mean ± SD,
n = 4)
t80% (min) (Mean ± SD,
n = 4)
AUC (% mg · min/ml)
(Mean ± SD, n = 4)
1%:1% 970.1 ± 22.63 885.4 ± 56.31 77.5 ± 7.86 111.1 ± 11.53 13151.5 ± 31.46 94.8 ± 5.20 143.5 ± 15.21 14336.1 ± 25.63
1.5%:1.5% 702.2 ± 27.15 729.2 ± 35.50 94.3 ± 10.45 148.1 ± 9.45 11117.7 ± 20.46 108.5 ± 8.75 174.1 ± 10.11 12553.9 ± 27.75
2%:2% 565.5 ± 13.36 574.5 ± 46.21 119.7 ± 12.61 176.4 ± 14.63 9230.4 ± 26.81 132.4 ± 6.31 209.6 ± 8.65 10564.5 ± 14.02

Development and Evaluation of a New Interpenetrating Network bead of Sodium Carboxymethyl Xanthan and Sodium Alginate for Ibuprofen Release
Copyright © 2010 SciRes. PP
16
Table 4. Kinetic data (n) and correlation coefficient (r2) of (A) drug release and (B) swelling of IPN beads prepared by gelling in 2% w/v AlCl3 solution for 0.5 h
In acidic solution In phosphate buffer solutionSCMX:SAL
n r2 n r2
1%:1% 0.73 0.997 1.26 0.989
1.5%: 1.5% 0.69 0.996 1.28 0.994 A
2%:2% 0.87 0.993 1.30 0.989
1%:1% 0.69 0.877 1.40 0.996
1.5%:1.5% 0.64 0.935 1.33 0.982 B
2%:2% 0.80 0.907 1.23 0.923
Figure 9. Effect of drug load on release of ibuprofen in acidic solution (open symbols) and phosphate buffer solu-tion (closed symbols) from IPN beads prepared using SCMX:SAL = 1.5%:1.5% and gelling for 0.5 h in 2% AlCl3 solution. Key: drug load, () 20% w/v, (∆) 40% w/v, () 60%w/v of total polymer. Maximum SEM = 1.27 (n = 4)
4. Conclusions
SCMX-SAL interpenetrating network beads were pre-pared by inotropic gelation method using Al3+ ions as cross-linking agent for both the polymers. Formation of IPN structure was verified by FTIR analysis and the ab-sence of drug-polymer interaction in IPN beads was con-firmed by FTIR, XRD, and DSC analysises. DEE of IPN beads were found to be reasonably high (91.15 to 99.50%) and was not affected by formulation variables except the gelation time, the increase of which tended to decrease DEE. While the release of IBP decreased in both acidic (pH 1.2) and PB solution (pH 6.8) with increase in total polymer concentration, gelation time, and drug load, the drug release increased in both the media with increase in the concentration of AlCl3. However, all the formulations showed considerably low release in acidic medium and the release followed non-Fickian transport mechanism
due to poor swelling of the beads. Complete drug release was achieved in PB solution (pH 6.8) at different periods of time depending on the formulation variables and the release followed case II transport process due to swelling and erosion of the beads. The results of the study indicate that high drug-loaded IPN beads can be prepared using SCMX and SAL by ionotropic gelation process and could be used to minimize the release of IBP in acidic medium and to modulate the drug release in PB solution (pH 6.8).
REFERENCES [1] M. Changez, K. Burugapalli, V. Koul and V. Chowdary,
“The Effect of Composition of Poly (Acrylic Ac-id)-Gelatin Hydrogel on Gentamycin Sulphate Release in Vitro,” Biomaterials, Vol. 24, No. 4, 2003, pp. 527-536.
[2] A. R. Kulkarni, K. M. Soppimath, T. M. Aminabhavi and W. E. Rudzinski, “In-Vitro Release Kinetics of Ce-fadroxyl Loaded Sodium Alginate Interpenetrating Net-work Beads,” European Journal of Pharmaceutics and Biopharmaceutics, Vol. 51, No. 2, 2001, pp. 127-133.
[3] T. T. Hsieh, K. H. Hsieh, G. P. Simon and C. Tiu, “Inter-penetrating Polymer Networks of 2-Hydroxylethyl Meth- acrylate Terminated Polyurethanes and Urethanes,” Po-lymer, Vol. 40, No. 11, 1999, pp. 3153-3163.
[4] A. K. Bajpai, J. Bajpai and S. Shukla, “Water Sorption through Semi-Interpenetrating Polymer Network with Hydrophilic and Hydrophobic Chains,” Reactive and Functional Polymers, Vol. 50, No. 1, 2002, pp. 9-21.
[5] A. Roy, A. K. Bajpai and J. Bajpai, “Designing Swellable Beads of Alginate and Gelatin for Controlled Release of Pesticide (Cypermethrin),” Journal of Macromolecular Science, Part A, Vol. 46, No. 9, 2009, pp. 847-859.
[6] P. Aslani and R. A. Kennedy, “Studies on Diffusion in Alginate Gels 1. Effect of Cross-Linking with Calcium or Zinc Ions on Diffusion of Acetaminophen,” Journal of Controlled Release, Vol. 42, No. 1, 1996, pp. 75-82.
[7] T. L. Bowersock, H. HogenEsch, M. Suckow, P. Gui-mond, S.Martin, D. Borie, S.Torregrosa, H. Park and K.Park, “Oral Vaccination of Animals with Antigens En-capsulated in Alginate Microspheres,” Vaccine, Vol. 17, No. 13-14, 1999, pp. 1804-1811.
[8] M. L. Gonzalez-Rodriguez, M. A. Holgado, C. San-chez-Lafuente, A. M. Rabasco and A. Finni, “Alginate/ Chitosan Particulate Systems for Sodium Diclofenac Re-lease,” International Journal of Pharmaceutics, Vol. 232, No. 1-2, 2002, pp. 225-234.
[9] A. Halder, S. Maiti and B. Sa, “Entrapment Efficiency and Release Characteristics of Polyethyleneimine-Treated or Untreated Calcium Alginate Beads Loaded with Pro-pranolol,” International Journal of Pharmaceutics, Vol. 302, No. 1-2, 2005, pp. 84-94.
[10] N. P. Desai, A. Sojomihardjo, Z. Yao, N. Ron and P. Soon-Shiong, “Interpenetrating Polymer Networks of Al-ginate and Polyethylene Glycol for Encapsulation of Is-

Development and Evaluation of a New Interpenetrating Network bead of Sodium Carboxymethyl Xanthan and Sodium Alginate for Ibuprofen Release
Copyright © 2010 SciRes. PP
17
lets of Langerhans,” Journal of Microencapsulation, Vol. 17, No. 6, 2000, pp. 677-690.
[11] S. G. Kumber and T. M. Aminabhavi, “Preparation and Characterisation of Interpenetrating Network Beads of Poly (Vinyl Alcohol)-Grafted-Poly (Acrylamide) with Sodium Alginate and their Controlled Release Character-istics of Cypermethrin Pesticides,” Journal of Applied Polymer Science, Vol. 84, No. 3, 2002, pp. 552-560.
[12] Y. H. Liu, H.-F. Lian, C.-K. Chung, M.-C. Chen and H. -W. Sung, “Physically Cross-Linked Alginate/N,O-Carbo- xymethyl Chitosan Hydrogels with Calcium for Oral De-livery of Protein Drugs,” Biomaterials, Vol. 26, No. 14, 2005, pp. 2105-2213.
[13] B. Sa and M. Setty, “Novel Gel Microbeads Based on Natural Polysaccharides,” Indian Patent, No. 224992, 31 October 2008.
[14] S. Maiti, S. Roy, B. Mondal, S. Sarkar and B. Sa, “Car-boxymethyl Xanthan Microparticles as a Carrier for Pro-tein Delivery,” Journal of Microencapsulation, Vol. 24, No. 8, 2007, pp. 743-756.
[15] S. Ray, S. Maiti and B. Sa, “Preliminary Investigation on the Development of Diltiazem Resin Complex Loaded Carboxymethyl Xanthan Beads,” AAPS PharmSciTech, Vol. 9, No. 1, 2008, pp. 295-301.
[16] C. M. Setty, S. S. Sahoo and B. Sa, “Alginate-Coated Alginate-Polyethyleneimine Beads for Prolonged Release of Furosemide in Simulated Intestinal Fluid,” Drug De-velopment and Industrial Pharmacy, Vol. 31, No. 4-5, 2005, pp. 435-446.
[17] P. Piyakulawat, N. Praphairaksit, N. Chantarasiri and N. Muangsin, “Preparation and Evaluation of Chitosan/Car- Rageenan Beads for Controlled Release of Sodium Di-clofenac,” AAPS PharmSciTech, Vol. 8, No. 4, 2007, pp. 1-10.
[18] A. P. Rokhade, S. A. Agnihotri, S. A. Patil, N. N. Mal-
likarjuna, P. V. Kulkarni and T. M. Aminabhavi, “Semi- Interpenetrating Polymer Network Microspheres of Gela-tin and Sodium Carboxymethyl Cellulose for Controlled Release of Ketorolac Tromethamine,” Carbohydrate Po-lymers, Vol. 65, No. 3, 2006, pp. 243-252.
[19] S. A. Agnihotri and T. M. Aminabhavi, “Development of Novel Interpenetrating Network Gellan Gum-Poly (Vinyl Alcohol) Hydrogel Microspheres for the Controlled Re-lease of Carvedilol,” Drug Development and Industrial Pharmacy, Vol. 31, No. 6, 2005, pp. 491-503.
[20] K. S. Soppimath, A. R. Kulkarni and T. M. Aminabhavi, “Chemically Modified Polyacrylamide-G-Guar Gum Based Crosslinked Anionic Microgels as PH Sensitive Drug Delivery Systems: Preparation and Characteriza-tion,” Journal of Controlled Release, Vol. 75, No. 3, 2001, pp. 331-345.
[21] R. V. Kulkarni and B. Sa, “Novel PH-Sensitive Inter-penetrating Network Hydrogel Beads of Carboxymethyl Cellulose-(Polyacryl Amide-Grafted-Alginate) for Con-trolled Release of Ibuprofen: Preparation and Characteri-zation,” Current Drug Delivery, Vol. 5, No. 4, 2008, pp. 256-264.
[22] S. Benita, A. Barkai and Y. U. Pathak, “Effect of Drug Loading Extent on the in Vitro Release Kinetic Behaviour of Nifedipine from Polyacrylate Microspheres,” Journal of Controlled Release, Vol. 12, No. 3, 1990, pp. 213-222.
[23] K. S. Soppimath, A. R. Kulkarni and T. M. Aminabhavi, “Controlled Release of Antihypertensive Drug from the Interpenetrating Network Poly (Vinyl Alcohol)-Guar Gum Hydrogel Microspheres,” Journal of Biomaterials Science Polymer Edition, Vol. 11, No. 1, 2000, pp. 27-43.
[24] P. L. Ritger and N. A. Peppas, “A Simple Equation for Description of Solute Release. II Fickian and Anomalous Release from Swellable Devices,” Journal of Controlled Release, Vol. 5, No. 1, 1987, pp. 37-42.

Pharmacology & Pharmacy, 2010, 1, 18-26 doi:10.4236/pp.2010.11003 Published Online July 2010 (http://www.SciRP.org/journal/pp)
Copyright © 2010 SciRes. PP
Preparation and Evaluation of Rapidly Disintegrating Fast Release Tablet of Diazepam-Hydroxypropyl-β-Cyclodextrin Inclusion Complex
——Rapidly Disintegrating Fast Release Tablet
Tapan Kumar Giri, Biswanath Sa*
Centre for Advanced Research in Pharmaceutical Sciences, Department of Pharmaceutical Technology, Jadavpur University, Kolkata, India. Email: [email protected] Received June 1st, 2010; accepted July 8th, 2010.
ABSTRACT
This study was undertaken to develop tablets of diazepam-hydroxypropyl-β-cyclodextrin inclusion complex that disinte-grate within 3 minutes and release 85% of drug within 30 minutes to provide rapid action of the drug through oro-mucosal route. Formation of inclusion complex was verified using X-ray diffraction and differential scanning calo-rimetric studies. Enhanced of aqueous solubility, as evident from phase solubility study, and dissolution of the drug were related with the formation of inclusion complex. Among the various formulations, tablet containing inclusion complex of drug/hydroxypropyl-β-cyclodextrin in a molar ratio of 1:2, and a combination of microcrystalline cellu-lose/lactose in a ratio of 4:1 disintegrated in 13 seconds and released 85% drug within 9 minutes. Addition of 10% w/w polyvinyl pyrrolidone in the tablet formulation further enhanced the drug release. Accelerated stability study indicated that mean dissolution time of the drug from the tablet did not change significantly within 6 months. Keywords: X-Ray Diffraction, Phase Solubity, Dissolution Efficiency, Mean Dissolution Time, Stability
1. Introduction
Though conventional oral and parenteral routes are used widely to achieve systemic action of drugs, various mu-cosae are being explored as possible alternative routes for drug delivery. Since the invention of nitroglycerin sublingual tablets, the oral mucosal route is drawing at-tention of both academia and industries as a substitute drug delivery approach. Several constraints like difficulty in swallowing experienced by many paediatrics and geri-atrics [1], and in chewing by edentulous [2]; nausea and vomiting experienced with certain drugs when released in stomach [3]; degradation and metabolism of suscepti-ble drugs in gastrointestinal tract [4]; tissue necrosis and irritation from repeated administration of parenterals [5], high expenses due to sterile manufacturing [6] are avoided through oromucosal delivery of drugs. In certain diseases like epilepsy, rapid onset of drug action is nec-
essary to suppress convulsion and terminate seizures. Thus early termination of seizures by initiating therapy as soon as possible, preferably at home, has been empha-sized as a key to minimize morbidity of these seizures [7-9].
Benzodiazepines are used for the acute management of severe seizures and have a rapid onset of action once delivered into the central nervous system and are safe. [10] Diazepam, a benzodiazepine, is included in the “WHO Essential Drug list” for the treatment of convul-sion and epileptic seizure [11-14]. Although intravenous therapy is the most rapid way to suppress epileptic con-vulsion, it may produce toxic manifestation due to exces-sive drug concentration [15,16], requires great care and caution to avoid thrombophlebitis and irritation [17] and may not be feasible where adequate medical facilities are not available in the immediate vicinity. While absorption of diazepam from intramuscular route is poor and erratic

Preparation and Evaluation of Rapidly Disintegrating Fast Release Tablet of Diazepam-Hydroxypropyl-β-Cyclodextrin Inclusion Complex
Copyright © 2010 SciRes. PP
19
[18], the time to reach peak plasma concentration fol-lowing oral administration is 1-2 hours [19] and is ac-companied with acid hydrolysis and extensive liver me-tabolism [10].
If diazepam is formulated in a rapidly disintegrating fast dissolving tablet dosage form, the high vascularity and rich blood supply of oral mucosa [20] may provide rapid absorption and faster onset of action [21] and could enable a patient for self medication even without the aid of water in a situation where onset of convulsion is ap-prehended.
Two principle criteria appear to be important for de-veloping rapidly disintegrating fast dissolving tablets: 1) disintegration time preferably < 3 minutes [22] and 2) rapid drug dissolution: time required for 85% dissolution (t85%) less than 30 minutes [23]. Valuable research re-ports for formulation of rapidly disintegrating tablets are available [24]; also, various technologies for improving dissolution property of poorly water soluble drugs have been documented to enhance bioavailability following oral absorption [25]. Among the various strategies, for-mulation of solid dispersions with hydrophilic carriers especially polyethylene glycols (PEGS) have been suc-cessfully used for enhancing dissolution of poorly water soluble drugs [26-29]. However, development of dosage forms like tablet and capsule using the solid dispersion encounters problems in pulverization/sifting of the solid dispersion which are usually soft and tacky and exhibits poor flow properties. In recent years, inclusion com-plexes of poorly water soluble drugs with cyclodextrins especially hydroxypropyl-β-cyclodextrins (HPβCD) have become popular to enhance the solubility and bioavail-ability of drugs.
Loftsson [30] reported that the solubility of diazepam and various poorly water soluble drugs improved in solu-tion with the natural cyclodextrins and their derivatives. Subsequent studies also shows that the incorporation of hydrophilic polymer such as sodium carboxymethyl cel-lulose, hydroxypropyl methyl cellulose and polyvinyl pyrrolidone increase the solubilizing effect of the cyclo-dextrins and reduce the amount of cyclodextrin required.
The objective of this study was to develop a rapidly disintegrating fast dissolving tablet of diazepam that can disintegrate in less than 3 minutes and release/dissolve 85% of the drug within 30 minutes in the oral cavity. The initial part of this work involved preparation of diaze-pam-HPβCD inclusion complex by kneading method and characterization of the complex using X-ray diffrac-tion(XRD) and differential scanning calorimetric (DSC) studies. The subsequent phase involved the preparation of tablets of diazepam-HPβCD complex by direct com-pression method to meet the specified time limits of dis-integration and drug dissolution. Finally the optimized tablets were subjected to accelerated stability study.
2. Experimental
2.1 Materials
Diazepam (East India Pharmaceutical Works Ltd., Kol-kata, India), Saccharin-Na, Crosscarmellose sodium (Ac- Disol), Microcrystalline Cellulose (Avicel, PH-102) [Dey’s Medical Stores (Mfg.) Ltd., Kolkata, India], Hydroxypro-pyl beta cyclodextrin (HPβCD) [Dr Reddy’s Lab, Hy-derabad, India] were obtained as gift samples. Mannitol, lactose monohydrate and sorbitol (Merc, India), PVP- K30 (Qualigens, Mumbai, India), magnesium stearate and all other ingredients were obtained commercially and used as received.
2.2 Preparation of Solid Complexes
Solid inclusion complexes of diazepam-HPβCD were prepared in 1:1 and 1:2 ratio by kneading method with and without the addition of polyvinyl pyrrolidone (PVP). PVP was added at a concentration of 10% (w/w) of the solid complex. Mixture was triturated for one hour in a mortar with a small volume of water to obtain a homo-geneous paste. During the process, the water content of the paste was empirically adjusted to maintain the con-sistency of the paste. The paste was dried at 45°C for 48 hours, pulverized and passed through sieve # 100.
2.3 Thermal Analysis
Differential thermal analysis of diazepam, HPβCD, di-azepam/HPβCD physical mixture, and inclusion complex were carried out using Perkin-Elmer instrument (Pyris Diamond TG/DTA, Singapore) equipped with a liquid nitrogen subambient accessory. About 4 mg samples were kept in aluminum pan, hermetically sealed and scanned at a rate of 5°/min between 30-210°C under ni-trogen atmosphere.
2.4 X-Ray Diffraction Study
X-ray powder diffraction patterns of diazepam, HPβCD, diazepam/HPβCD physical mixture, and inclusion com-plex were conducted with a X-ray powder diffractometer (Rigaku-MiniFlax, Tokyo, Japan.) using a copper Kα target with a nickel filter at 30 kV voltage, 15 mA cur-rent and at scanning speed of 1°/min over a 2θ range of 5°-60°.
2.5 Phase Solubility Study
Phase solubility studies were carried out by adding ex-cess amounts of drug to 10 ml of USP phosphate buffer solution (pH 5.8) containing various concentrations of HPβCD (3-15 mM) in stoppered conical flasks. The flasks were shaken at 50 revolutions per minute in a shaking incubator (Model KMC 8480 SL, Vision Scien-tific Company, Ltd., Seoul, South Korea) at 37 ± 0.5°C

Preparation and Evaluation of Rapidly Disintegrating Fast Release Tablet of Diazepam-Hydroxypropyl-β-Cyclodextrin Inclusion Complex
Copyright © 2010 SciRes. PP
20
until equilibrium (about 90hours) was reached. The re-sulting mixtures were filtered and aliquots, following suitable dilutions, were analyzed using a spectropho-tometer (Genesyis, 10 UV, Thermo Electron Corporation, Wisconsin, USA.) at 230 nm against blanks prepared in the same concentration of HPβCD in USP phosphate buffer solution (pH 5.8). Phase solubility studies were conducted with and without the addition of PVP. The PVP was added at a concentration of 0.5% w/v to the solution containing HPβCD. The solubility experiments were conducted in triplicate.
2.6 Preparation of Placebo Tablet by Direct Compression Method
Microcrystalline cellulose (MCC), lactose or mannitol or sorbitol and crosscarmellose sodium were mixed without drug for 10 minutes. The resulting mixture was lubri-cated with magnesium stearate and mixed for 5 minutes. The final powder mixture was then compressed into tab-let using concave punches (approx 9.5 mm diameter) in a 10 station Minipress tablet machine (RIMEK, Karnavati Engineering Ltd, Gujarat, India).
2.7 Preparation of Tablet with Drug by Direct Compression Method
MCC, lactose or mannitol or sorbitol and crosscarmel-lose sodium were mixed with drug (as such or solid in-clusion complexes) for 10 minutes. The resulting mixture was lubricated with magnesium stearate and mixed for 5 minutes. The final powder mixture was then compressed into tablet using concave punches (approx 9.5 mm di-ameter) in a 10 station Minipress tablet machine (RIMEK, Karnavati Engineering Ltd, Gujarat, India).
2.8 Measurement of Disintegration Time
Disintegration times were measured using a modified disintegration test method. [31] To determine disintegra-tion time, 10ml of USP phosphate buffer solution (pH 5.8) was taken in a petridish (10 cm diameter) and a tablet was carefully placed in the centre and agitated mildly. Time for the tablet to completely disintegrate into fine particles was noted using a stop watch.
2.9 Measurement of in Vitro Drug Release
Dissolution profiles of the diazepam tablets were deter-mined using USP II dissolution rate test apparatus (mod-el TDP-06P, Electrolab, Mumbai, India).The dissolution medium was 500ml of USP phosphate buffer solution (pH 5.8) maintained at 37 ± 0.5°C and stirring speed was 50 rpm. At appropriate time intervals, 10ml samples were withdrawn, suitably diluted, and analyzed spectro-photometrically for diazepam content at 230 nm. The initial volume of dissolution medium was maintained by
adding 10ml of fresh medium. The amount of drug re-leased was determined from the calibration curve. The reliability of the above analytical method was judged by conducting recovery analysis in the presence or absence of the excipients. Low, middle, and high concentrations of drug solution were spiked and recovery was found to vary from 99.05 to 100.83%.
2.10 Stability Study
The stability of the drug in the optimized tablet was as-sessed by keeping the tablets in a sealed glass bottle and subsequently placing the bottle in a Stability Test Cham-ber (Humidity Cabinet, Testing instruments manufactur-ing company, Kolkata) at 40°C and 75% RH for different periods of time. The tablets were analyzed immediately (0 month), and after 1, 3 and 6 months for appearance, hardness, friability, drug content, disintegration time and dissolution profiles of the drug.
3. Results and Discussion
Diazepam-HPβCD inclusion complexes were prepared by kneading method. The resulting complex was ana-lyzed by XRD, DSC, and phase solubility studies.
Thermal behavior of diazepam, HPβCD, diazepam/ HPβCD physical mixture, and diazepam/HPβCD inclu-sion complex are shown in Figure 1. The DSC thermo-gram of diazepam shows one sharp endothermic peak at 133°C which corresponds to the melting point of the drug. The DSC trace of HPβCD did not show any endothermic peak. The thermal event of the physical mixture demon-strated the appearance of a sharp peak having diminished intensity at 133°C which was assigned to the melting point of the drug. The DSC thermogram of the inclusion
Figure 1. Differential scanning calorimetric thermograms of a) diazepam, b) HPβCD, c) physical mixture, d) inclusion complex

Preparation and Evaluation of Rapidly Disintegrating Fast Release Tablet of Diazepam-Hydroxypropyl-β-Cyclodextrin Inclusion Complex
Copyright © 2010 SciRes. PP
21
complex revealed that although the intensity of the melt-ing endotherm of the drug at 133°C decreased to a great extent, it was not abolished completely. The decrease in endothermic peak of the drug was due to entrapment of the crystalline drug within the cavity of HPβCD. This indicates the formation of an inclusion complex of di-azepam with HPβCD. During the preparation of solid dispersion using melting method [32] solvent/co-evap- oration method [33], the crystalline drugs are completely converted into amorphous form and consequently, no thermal event is evident in the DSC scan. On the other hand the endothermic melting peak of a crystalline drug may not be abolished, although may be diminished to a great extent, when a drug forms solid complex with HPβCD by kneading method [32].
The XRD patterns of drug, HPβCD, physical mixture, and inclusion complex are shown in Figure 2. The dif-fractogram of the drug exhibited a series of intense peaks due to its crystalline structure. The XRD pattern of HPβCD was an amorphous halo. The diffractogram of the physical mixture appeared to represent the superim-position of each components spectrum although the drug crystallinity reduced considerably. A no. of sharp peaks, although of reduced intensity, were still present in the diffractogram of the solid complex .Close examination of the diffractogram revealed that many peaks of the drug disappeared(2θ of 26.12°, 26.69°, 27.56°, 28.28°) and many new peaks emerged (2θ of 25.67° and 26.99°). Similar to the DSC results, the XRD analysis does not show diffraction pattern of drugs when the solid disper-sions are prepared by melting method [32] or solvent/ co-evaporation method [33]. However, the inclusion complexes prepared by kneading process still show peaks on the diffractogram [32]. These results indicate the for-mation of inclusion complex.
Phase solubility diagram (Figure 3) demonstrated a linear increase in the aqueous solubility of diazepam with the concentration of HPβCD. The improved solubility of various poorly soluble drugs through complexation with HPβCD is well documented in scientific journals [34,35]. The complexation of diazepam with HPβCD was type AL
[36] and as the slope of the straight line of concentration of diazepam verses concentration of HPβCD plots was < 1, the complexation took place in 1:1 molar ratio. The apparent stability constant (KC) was calculated from the slope of the linear plot of the phase solubility diagram following the equation KC = Slope/So (1-Slope), where So is the solubility of the drug in the absence of HPβCD. The value of KC (Table 1) indicated that the complex was quite stable.
Tablets formulated using MCC and low-substituted hydroxypropyl cellulose has been reported to disintegrate and dissolve rapidly in the saliva of humans [37]. How-ever, such tablets provide a gritty mouth feel due to the
Figure 2. X-ray diffraction pattern of a) diazepam, b) HPβCD, c) physical mixture, d) inclusion complex
Figure 3. Phase solubility diagrams of diazepam-hydroxy- propyl β-cyclodextrin complex in the presence (■) and absence (◆) of PVP Table 1. Effect of PVP on stability constant (KC) and solubi-lizing efficiency of diazepam-HPβCD complexes†
Sample KC (M-1) Solubilizing Efficiency .٭
D-HPβCD 499.087 3.880
D-HPβCD-PVP 515.094 4.271
†HPβCD indicates hydroxypropyl β-cyclodextrin; PVP, polyvinyl pyrrolidone; and D, diazepam. Ratio of drug solubility in USP buffer solution (pH 5.8) (15 mM) of٭cyclodextrin (with or without PVP) to drug solubility in buffer. presence of insoluble crystalline cellulose [38].
To formulate tablets that can disintegrate rapidly and reduce gritty mouth feel characteristics, several water soluble diluents like sorbitol, manitol and lactose were selected, and 9 placebo tablets containing of MCC and
Preparation and Evaluation of Rapidly Disintegrating Fast Release Tablet of Diazepam-Hydroxypropyl-β-Cyclodextrin Inclusion Complex
Copyright © 2010 SciRes. PP
22
either sorbitol, manitol or lactose in the ratios of 1:1, 2:1, and 4:1 were prepared by direct compression method. The compositions of the placebo tablets are shown in Table 2.
The blended powder of each formulation exhibited good flowability as evident from the measurement of angle of repose (measured by conventional method) that varied from about 31.3° to 34° (Table 3). Angle of repose below 40° is an indication of good flowability of pow-der/granules [39]. The compression force during tablet-ting was adjusted in such a way that the hardness (meas-ured using Monsanto type hardness tester) of the tablets, each weighing 300 mg, was 2 Kg-F. The friability of the tablets (determined using Friabilator, Veego instrument, Mumbai, India) was found between 0.03 to 0.29% that was below 1% indicating sufficient mechanical integrity and strength of the placebo tablets.
The disintegration time of tablets (P1) which were prepared using MCC and sorbitol in 1:1 ratio was 42.85 seconds. The tablets P2 and P3 which consisted of MCC and sorbitol in a ratio of 2:1 and 4:1 respectively disinte-grated in 29.03 and 27.34 seconds. The result indicates that increase in MCC/sorbitol ratio decreased the disin-
tegration time of the tablets and this decrease was found significant at 95% confidence limit (p < 0.05). MCC is considered as one of the most versatile excipients in tab-let manufacturing. In addition to its performance as dilu-ents and dry binder, it is also regarded as an excellent disintegrant for tablets prepared by direct compression method. This property is related to its wicking action due to which water penetrates into the tablet and the devel-oped hydrostatic pressure causes break down of the tablet. The rate and extent of water penetration is related to the porosity (determined using laboratory pycnometer) it provides in the tablets. The greater the amount of MCC, the greater will be the porosity in the tablet matrix. Table 3 shows that as the ratio of MCC/sorbitol increased, the porosity of the tablets increased significantly (p < 0.05) that provided faster penetration of water. Determination of wetting time (determined by the method described by Bi et al. [40]) demonstrated that the wetting time of the tablets decreased significantly (p < 0.05) with increase in the amount of MCC (Table 3) indicating faster penetra-tion of water into the tablets. Similar observations were noted for the tablets prepared using MCC/manitol (tab-lets P4, P5, P6) and MCC/lactose (tablets P7, P8, P9).
Table 2. Composition of placebo tablets (without drug) prepared by direct compression method
Ingredients(mg/tablet) P1 P2 P3 P4 P5 P6 P7 P8 P9
MCC 144.25 193 230.8 144.25 193 230.8 144.25 193 230.8
Lactose - - - - - - 144.25 96.5 57.7
Mannitol - - - 144.25 96.5 57.7 - - -
Sorbitol 144.25 96.5 57.7 - - - - - -
Crosscarmellose-Na 7.5 7.5 7.5 7.5 7.5 7.5 7.5 7.5 7.5
Mg-St. 1 1 1 1 1 1 1 1 1
Saccharin-Na 3 3 3 3 3 3 3 3 3
Total 300 300 300 300 300 300 300 300 300
Table 3. Physical characteristics of placebo tablets (without drug) prepared by direct compression method
P1 P2 P3 P4 P5 P6 P7 P8 P9
Angle of Repose 34.03 (0.33)
33.56 (0.39)
31.47 (1.4)
33.99 (0.29)
32.66 (0.69)
32.49 (1.13)
35.38 (0.8)
32.37 (1.02)
31.29 (0.96)
Porosity 9.99
(0.13) 15.21 (0.21)
22.39 (0.21)
17.26 (0.14)
21.28 (0.16)
26.17 (0.28)
19.31 (0.19)
24.34 (0.42)
31.87 (0.7)
Disintegration time, seconds 42.85 (1.89)
29.03 (0.67)
27.34 (0.62)
43.29 (0.77)
18.76 (0.25)
15.3 (0.9)
26.49 (1.36)
17.25 (0.17)
13.23 (0.46)
Wetting time, seconds 119.92 (1.63)
92.69 (2.1)
84.78 (1.85)
82.56 (1.86)
52.69 (2.81)
42.53 (1.7)
80.67 (1.78)
50.3 (1.07)
35.43 (1.62)
Figures in parentheses indicate ± SD; n = 6 for disintegration time and ± SD, n = 3 for Angle of Repose, porosity and wetting time
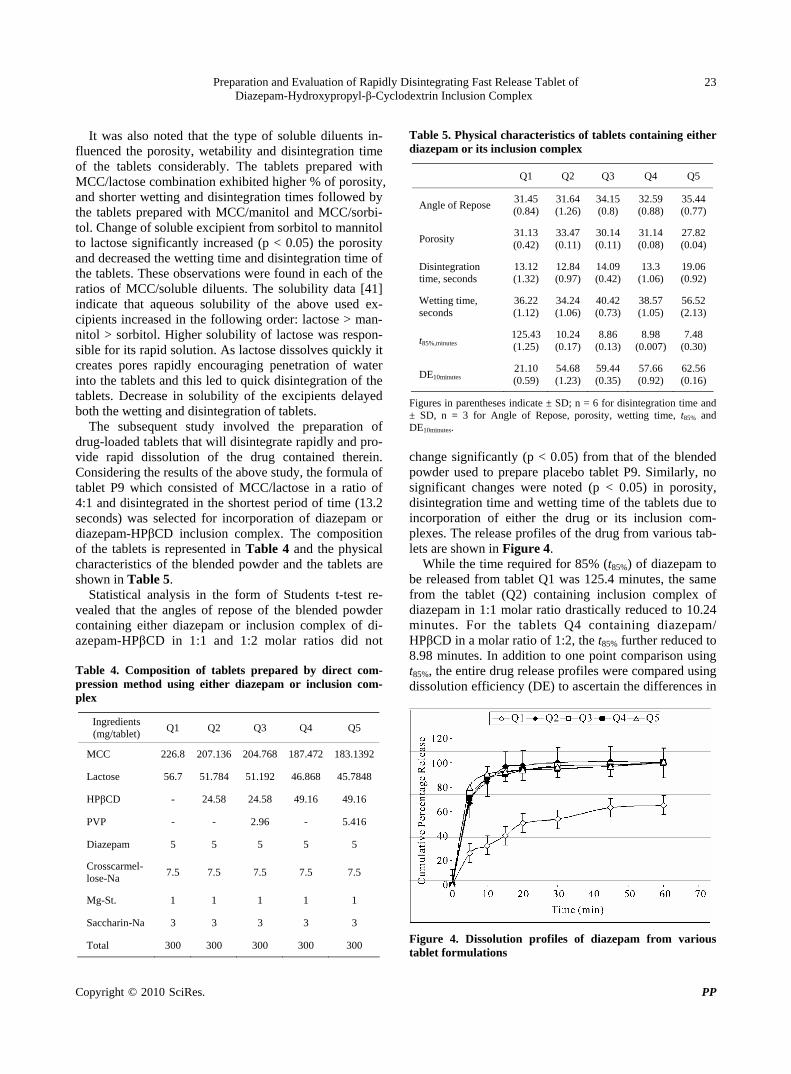
Preparation and Evaluation of Rapidly Disintegrating Fast Release Tablet of Diazepam-Hydroxypropyl-β-Cyclodextrin Inclusion Complex
Copyright © 2010 SciRes. PP
23
It was also noted that the type of soluble diluents in-
fluenced the porosity, wetability and disintegration time of the tablets considerably. The tablets prepared with MCC/lactose combination exhibited higher % of porosity, and shorter wetting and disintegration times followed by the tablets prepared with MCC/manitol and MCC/sorbi- tol. Change of soluble excipient from sorbitol to mannitol to lactose significantly increased (p < 0.05) the porosity and decreased the wetting time and disintegration time of the tablets. These observations were found in each of the ratios of MCC/soluble diluents. The solubility data [41] indicate that aqueous solubility of the above used ex-cipients increased in the following order: lactose > man-nitol > sorbitol. Higher solubility of lactose was respon-sible for its rapid solution. As lactose dissolves quickly it creates pores rapidly encouraging penetration of water into the tablets and this led to quick disintegration of the tablets. Decrease in solubility of the excipients delayed both the wetting and disintegration of tablets.
The subsequent study involved the preparation of drug-loaded tablets that will disintegrate rapidly and pro-vide rapid dissolution of the drug contained therein. Considering the results of the above study, the formula of tablet P9 which consisted of MCC/lactose in a ratio of 4:1 and disintegrated in the shortest period of time (13.2 seconds) was selected for incorporation of diazepam or diazepam-HPβCD inclusion complex. The composition of the tablets is represented in Table 4 and the physical characteristics of the blended powder and the tablets are shown in Table 5.
Statistical analysis in the form of Students t-test re-vealed that the angles of repose of the blended powder containing either diazepam or inclusion complex of di-azepam-HPβCD in 1:1 and 1:2 molar ratios did not Table 4. Composition of tablets prepared by direct com-pression method using either diazepam or inclusion com-plex
Ingredients (mg/tablet)
Q1 Q2 Q3 Q4 Q5
MCC 226.8 207.136 204.768 187.472 183.1392
Lactose 56.7 51.784 51.192 46.868 45.7848
HPβCD - 24.58 24.58 49.16 49.16
PVP - - 2.96 - 5.416
Diazepam 5 5 5 5 5
Crosscarmel-lose-Na
7.5 7.5 7.5 7.5 7.5
Mg-St. 1 1 1 1 1
Saccharin-Na 3 3 3 3 3
Total 300 300 300 300 300
Table 5. Physical characteristics of tablets containing either diazepam or its inclusion complex
Q1 Q2 Q3 Q4 Q5
Angle of Repose 31.45(0.84)
31.64 (1.26)
34.15 (0.8)
32.59 (0.88)
35.44(0.77)
Porosity 31.13(0.42)
33.47 (0.11)
30.14 (0.11)
31.14 (0.08)
27.82(0.04)
Disintegration time, seconds
13.12(1.32)
12.84 (0.97)
14.09 (0.42)
13.3 (1.06)
19.06(0.92)
Wetting time, seconds
36.22(1.12)
34.24 (1.06)
40.42 (0.73)
38.57 (1.05)
56.52(2.13)
t85%,minutes 125.43(1.25)
10.24 (0.17)
8.86 (0.13)
8.98 (0.007)
7.48 (0.30)
DE10minutes 21.10(0.59)
54.68 (1.23)
59.44 (0.35)
57.66 (0.92)
62.56(0.16)
Figures in parentheses indicate ± SD; n = 6 for disintegration time and ± SD, n = 3 for Angle of Repose, porosity, wetting time, t85% and DE10minutes.
change significantly (p < 0.05) from that of the blended powder used to prepare placebo tablet P9. Similarly, no significant changes were noted (p < 0.05) in porosity, disintegration time and wetting time of the tablets due to incorporation of either the drug or its inclusion com-plexes. The release profiles of the drug from various tab-lets are shown in Figure 4.
While the time required for 85% (t85%) of diazepam to be released from tablet Q1 was 125.4 minutes, the same from the tablet (Q2) containing inclusion complex of diazepam in 1:1 molar ratio drastically reduced to 10.24 minutes. For the tablets Q4 containing diazepam/ HPβCD in a molar ratio of 1:2, the t85% further reduced to 8.98 minutes. In addition to one point comparison using t85%, the entire drug release profiles were compared using dissolution efficiency (DE) to ascertain the differences in
Figure 4. Dissolution profiles of diazepam from various tablet formulations

Preparation and Evaluation of Rapidly Disintegrating Fast Release Tablet of Diazepam-Hydroxypropyl-β-Cyclodextrin Inclusion Complex
Copyright © 2010 SciRes. PP
24
the release of the drug from various tablets. The DE is defined as the area under the dissolution curve upto a certain time, t, expressed as a percentage of the area of the rectangle described by 100% dissolution in the same time [42].
0
100
.
D.E 100%.
t
y dt
y t
where y is the drug percent dissolved in time t. DE can have a range of values depending on the time
interval chosen. However, while comparing a set of data, a constant time interval should be selected. In the present study, DE10minutes (dissolution efficiency upto 10 minutes) were calculated from the dissolution profile of each tab-let and used for comparison. It was found that in com-parison to DE10minutes obtained from tablet containing diazepam, a 2.59 and 2.73 fold increase in DE10minutes were obtained from the tablets prepared using diazepam/ HPβCD inclusion complex in molar ratios of 1:1 and 1:2 respectively. These increase in DE10minutes were found to be statistically significant (p < 0.05). Faster release of diazepam from the tablets prepared using its inclusion complexes was related to the enhanced solubility of the drug because of the formation of complex with HPβCD.
In another two batches of tablets namely Q3 and Q5 containing respectively 1:1 and 1:2 diazepam-HPβCD complex, PVP, a hydrophilic polymer was added at a concentration of 10% w/w of the solid complexes (Table 4) to investigate its effect on the various physical char-acteristics which are shown in Table 5. Incorporation of PVP in the formula increased the angle of repose of the blended powders and decreased the porosity of the tab-lets significantly (P < 0.05). Moreover, PVP increased both the disintegration time and wetting time of the tab-lets significantly (p < 0.05). The larger the amount of
PVP, the higher the disintegration time and the wetting time. PVP which acts as a binder densified the powder resulting in decreased flowability of the powder blend and reduced the porosity of the tablets. Reduced porosity, in turn, protracted the wetting and the disintegration of the tablets. Addition of PVP in the tablet formulations, however, further reduced t85% and increased DE indicat-ing that drug dissolution took place faster than that from the tablets without containing PVP. Addition of PVP in the complex also increased the solubility linearly having a slope < 1 as evident from phase solubility diagram (Figure 3). At each point of determination, the solubility of the drug was higher than that produced by the com-plex in absence of PVP. The stability constant was also found to be higher (Table 1). The potentiated solubility of diazepam in complex form in the presence of PVP was evaluated by determining the solubilization efficiency, which is defined as ratio of solubility of drug in USP phosphate buffer solution (pH 5.8) of 15 mM HPβCD (with or without PVP) and the solubility of the drug in buffer solution. Table 1 show that while HPβCD without PVP produced 3.88 fold increases, the same in presence of PVP produced 4.27 fold increases in the solubility of diazepam. It, therefore, appears that presence of hydro-philic polymer like PVP enhances the solubilizing effi-ciency of HPβCD.
Accelerated stability test on the tablets (Q5) was con-ducted at 40°C and 75% RH in accelerated stability test chamber (Humidity Cabinet, Testing instruments manu-facturing company, Kolkata) and the physical character-istics of the tablets observed after different periods of time are shown in Table 6.
The physical characteristics such as appearance, fri-ability and drug content did not change and were con-fined within the specified limits upto 6 months of time. The disintegration time and t85% of the tablets upto one month storage did not change significantly when com-pared to those of the fresh tablets. However, marginal
Table 6. Various characteristics of Diazepam tablets stored at 40°C and 70% RH
Storage
Time (month) Conditions Appearance Friability (%) Content (mg) DT (seconds) t85% (minutes) MDT (minutes)
Specifications White,round-flat tablet < 1% 95-105% <3 minutes < 30 minutes -
Initial Complies 0.140
(0.010) 4.96
(0.015) 19.53
(0.395) 7.56
(0.15) 2.85
(0.045)
1 Complies 0.147
(0.012) 4.98
(0.015) 20.26
(0.352) 7.90
(0.17) 2.848
(0.049)
3 Complies 0.177
(0.012) 4.94
(0.006) 20.84 (0.40)
8.22 (0.16)
2.847 (0.050)
6
40°C /75% RH
Complies 0.157
(0.025) 4.97
(0.006) 21.71
(0.262) 8.21
(0.11) 2.677 (0.12)

Preparation and Evaluation of Rapidly Disintegrating Fast Release Tablet of Diazepam-Hydroxypropyl-β-Cyclodextrin Inclusion Complex
Copyright © 2010 SciRes. PP
25
changes in the two characteristics were observed after 3 and 6 months storage. Although, the changes were statis-tically significant, the tablets complied with the specified limits even after 6 months. To further investigate the effect of storage conditions on drug dissolution, mean dissolution time (MDT) were calculated from the release profiles of the tablets kept under stressed condition for different periods of time and were compared with that from freshly prepared tablet. MDT was calculated from the following equation:
1
1
n
midin
i
t MMDT
M
where tmid is the time at the midpoint between i and i-1, and M is the additional amount of drug dissolved be-tween i and i-1.
Table 6 shows that MDT values of the tablets did not differ significantly (p < 0.05). This indicates that the drug dissolution from the tablets (Q5) which were kept under stressed condition were similar to that from the freshly prepared tablets upto 6 months.
4. Conclusions
Tablets of diazepam were prepared by direct compres-sion method that disintegrated quickly (in 13.3 seconds) and released 85% drug rapidly (in 8.98 minutes). Rapid disintegration was achieved using MCC and lactose as excipients in a ratio of 4:1. Rapid drug dissolution was obtained through the formulation of inclusion complex of the drug with HPβCD. Inclusion of PVP further in-creased the drug dissolution. Such tablets seem to be suitable for achieving rapid onset of action of the drug through oro-mucosal route.
REFERENCES [1] J. S. Schindler and J. H. Kelly, “Swallowing Disorders in
the Elderly,” Laryngoscope, Vol. 112, No. 4, 2002, pp. 589-602.
[2] J. C. Ofstehage and K. Magilvy, “Oral Health and Ag-ing,” Geriatric Nursing, Vol. 81, No. 7, 1986, pp. 238- 241.
[3] G. Motola, F. Russo, F. Mazzeo, B. Rinaldi, A. Capuano, F. Rossi and A. Filipelli, “Over-the-Counter Oral Non-steroidal Anti-Inflammatory Drugs: A Pharmacoepidemi-ologic Study in Southern Italy,” Advances in Therapy, Vol. 18, No. 5, 2001, pp. 216-222.
[4] L. Yang, S. C. James and A. Joseph, “Colon-Specific Drug Delivery: New Approaches and in Vitro/in Vivo Evaluation,” International Journal of Pharmaceutics, Vol. 235, No. 1-2, 2002, pp. 1-15.
[5] K. D. Tripathi, “Essentials of Medical Pharmacology,” 5th Edition, Jaypee Brothers Medical Publishers (P) LTD,
New Delhi, 2003.
[6] L. Li, S. Gorukanti, Y. M. Choi and K. H. Kim, “Rap-id-Onset Intranasal Delivery of Anticonvulsants: Phar-macokinetic and Pharmacodynamic Evaluation in Rab-bits,” International Journal of Pharmaceutics, Vol. 199, No. 1, 2000, pp. 65-76.
[7] B. Alldredge, A. Gelb, S. Isaacs, M. Corry, F. Allen, S. Ulrich, M. Gottwald, N. O’Neil, J. Neuhaus, M. Segal and D. A. Lowenstein, “Comparison of Lorazepam, DZ, and Placebo for the Treatment of Out-of-Hospital Status Epilepticus,” New England Journal of Medicine, Vol. 345, No. 9, 2001, pp. 631-637.
[8] C. O’Dell, “What Do We Tell Parents of a Child with Simple or Complex Febrile Seizures?” In: T. Z. Baram, and S. Shinnar, Eds., Febrile Seizures, San Diego, CAS
Academic Press, 2002, pp. 305-316.
[9] C. O’Dell, S. Shinnar, K. Ballaban-Gil, M. Hornick, M. Sigalova, H. Kang and S. Moshe, “Rectal DZ Gel in the Home Management of Seizures in Children,” Pediatric Neurology, Vol. 33, No. 3, 2005, pp. 166-172.
[10] G. Abdelbary and R. H. Fahmy, “Diazepam-Loaded Solid lipid Nanoparticles: Design and Characterization,” AAPS PharmSciTech, Vol. 10, No. 1, 2009, pp. 211-219.
[11] E. Bechgaard, S. Gizurarason and R. K. Hjortkjaer, “Pharmacokinetic and Pharmacodynamic Response after Intranasal Administration of Diazepam to Rabbits,” Journal of Pharmacy Pharmacology, Vol. 49, No. 8, 1997, pp. 747-750.
[12] S. Gizurarson, F. K. Gudbrandsson, H. Jonsson and E. Bechgaard, “Intranasal Administration of Diazepam Aiming at the Treatment of Acute Seizures: Clinical Tri-als in Healthy Volunteers,” Biological and Pharmaceuti-cal Bulletin, Vol. 22, No. 4, 1999, pp. 425-427.
[13] E. Rey, J. M. Treluyer and G. Pons, “Pharmacokinetic Optimization of Benzodiazepine Therapy for Acute Sei-zures. Focus on Delivery Routes,” Clinical Pharmacoki-netics, Vol. 36, No. 6, 1999, pp. 409-424.
[14] F. U. Knudsen, “Rectal Administration of Diazepam in Solution in the Acute Treatment of Convulsions in Infants and Children,” Archives of Disease in Childhood, Vol. 54, No. 11, 1979, pp. 855-857.
[15] E. Norris, O. Marzouk, A. Nunn, J. Mclntyre and I. Choonara, “Respiratory Depression in Children Receiving Diazepam for Acute Seizures: A Prospective Study,” De-velopmental Medicine and Child Neurology, Vol. 41, No. 5, 1999, pp. 340-343.
[16] B. R. Ogutu, C. R. J. C. Newton, J. Crawley, S. N. Mu-chohi, G. O. Otieno, G. Edwards, K. Marsh and G. O. Kokwaro, “Pharmacokinetics and Anticonvulsant Effects of Diazepam in Children with Severe Falciparum Malaria and Convulsions,” British Journal of Clinical Pharma-cology, Vol. 53, No. 1, 2002, pp.49-57.
[17] C. W. Graham, R. R. Pagano and J. T. Conner, “Pain and Clinical Thrombophlebitis Following Intravenous Diaze-pam and Lorazepam,” Anaesthesia, Vol. 33, No. 2, 1978, 188-191.

Preparation and Evaluation of Rapidly Disintegrating Fast Release Tablet of Diazepam-Hydroxypropyl-β-Cyclodextrin Inclusion Complex
Copyright © 2010 SciRes. PP
26
[18] C. Lehmann and G. L. Wannarka, “Bioavailability and Dose Proportionality of Intramuscular Diazepam Admin-istered by Autoinjector,” Journal of Clinical Pharmacol-ogy, Vol. 48, No. 4, 2008, pp. 436-444.
[19] J. G. Hardman, L. E. Limbard and A. G. Gilman, “The Pharmacological Basis of Therapeutics,” 10th Edition, McGraw-Hill Medical Publishing Division, New Delhi, 2001.
[20] A. J. Hoogstrate, J. C. Verhoef, B. Tuk and A. Pijpers, “In-Vivo Buccal Delivery of Fluorescin Isothiocyanate- Dextran 4400 with Glycodeoxycholate as an Absorption Enhancer in Pig,” Journal of Pharmaceutical Sciences, Vol. 85, No. 5, 1996, pp. 457-460.
[21] T. Keiko, O. Yasuko, N. Tsuneji, L. Thorseinn and T. Kozo, “Buccal Absorption of Ergotamine Tartrate Using the Bioadhesive Tablet System in Guinea-Pigs,” Interna-tional Journal of Pharmaceutics, Vol. 238, No. 1-2, 2002, pp. 161-170.
[22] B. S. Kuchekar, S. B. Bhise and V. Arumugam, “Design of Fast Dissolving Tablet,” Indian Journal of Pharma-ceutical Education, Vol. 35, No. 4, 2001, pp. 150-152.
[23] T. Loftsson, “Cyclodextrins and the Biopharmaceutics Classification System of Drugs,” Journal of Inclusion Phenomenon, Vol. 44, No. 1-4, 2002, pp. 63-67.
[24] M. Sugimoto, S. Narisawa, K. Matsubara, H. Yoshino, M. Nakano and T. Handa, “Effect of Formulated Ingredients on Rapidly Disintegrating Oral Tablets Prepared by the Crystalline Transition Method,” Chemical and Pharma-ceutical Bulletin, Vol. 54, No. 2, 2006, pp. 175-180.
[25] H. N. Joshi, R. W. Tejwani, M. Davidovich, V. P. Sahas-rabudhe, M. Jemal, M. S. Bathala, S. A. Varia and A. T. M. Serajuddin, “Bioavailability Enhancement of a Poorly Water-Soluble Drug by Solid Dispersion in Polyethylene Glycol-Polysorbate 80 Mixture,” International Journal of Pharmaceutics, Vol. 269, No. 1, 2004, pp. 251-258.
[26] P. C. Sheen, V. K. Khetarpal, C. M. Cariola and C. E. Rowlings, “Formulation Studies of a Poorly Water-Solu- ble Drug in Solid Dispersions to Improve Bioavailabil-ity,” International Journal of Pharmaceutics, Vol. 118, No. 2, 1995, pp. 221-227.
[27] P. Mura, A. Manderioli, G. Bramanti and L. Ceccarelli, “The Properties of Solid Dispersions of Naproxen in Various Polyethylene Glycols,” Drug Development and Industrial Pharmacy, Vol. 22, No. 9-10, 1996, pp. 909- 916.
[28] T. Kai, Y. Akiama, S. Nomura and M. Sato, “Oral Ab-sorption Improvement of Poorly Soluble Drug Using Solid Dispersion Technique,” Chemical and Pharmaceu-tical Bulletin, Vol. 44, No. 3, 1996, pp. 568-571.
[29] C. Leuner and J. Dressman, “Improving Drug Solubility for Oral Delivery Using Solid Dispersions,” European Journal of Pharmaceutical Sciences, Vol. 50, No. 1, 2000, pp. 47-60.
[30] T. Loftsson, “Cyclodextrin Complexation,” US Patent, 1995, 5472954.
[31] M. Gohel, M. Patel, A. Amin, R. Agarwal, R. Dave and N. Bariya, “Formulation Design and Optimization of Mouth Dissolve Tablets of Nimesulide Using Vacuum Drying Technique,” AAPS PharmSciTech, Vol. 5, No. 3, 2004, Article 36.
[32] S. Chutimaworapan, G. C. Ritthidej, E. Yonemochi, T. Oguchi and K. Yamamoto, “Effect of Water-Soluble Car-riers on Dissolution Characteristics of Nifedipine Solid Dispersions,” Drug Development and Industrial Phar-macy, Vol. 26, No. 11, 2000, pp. 1141-1150.
[33] H. G. Choi, K. M. Kim, H. W. Jun, B. K. Yoo and C. S. Yong, “Improvement of Dissolution and Bioavailability of Nifedipine by Inclusion in Hydroxypropyl-β-Cyclo- dextrin,” Drug Development and Industrial Pharmacy, Vol. 29, No. 10, 2003, pp. 1085-1094.
[34] H. J. Ahn, K. M. Kim, S. J. Choi and C. K. Kim, “Effects of Cyclodextrin Derivatives on Bioavailability of Keto-profen,” Drug Development and Industrial Pharmacy, Vol. 23, No. 4, 1997, pp. 397-401.
[35] A. Latrofa, G. Trapani, M. Franco, M. Serra, M. Muggi-roni, F. P. Franizzi, A. Cutrignelli and G. Liso, “Com-plexation of Phenytoin with Some Hydrophilic Cyclodex-trins: Effect on Aqueous Solubility, Dissolution Rate and Anti-Convulsant Activity in Mice,” European Journal of Pharmaceutics and Biopharmaceutics, Vol. 52, No. 1, 2001, pp. 65-73.
[36] T. Higuchi and K. A. Connors, “Phase-Solubility Tech-niques,” Advances in Analytical Chemistry and Instru-mentation, Vol. 53, No. 4, 1965, pp. 117-212.
[37] Y. Watanable, K. I. Koizumi, Y. Zama, M. Kiriyama and M. Matsumoto, “New Compressed Tablet Rapidly Disin-tegrating in Saliva in the Mouth Using Crystalline Cellu-lose and a Disintegrant,” Biological and Pharmaceutical Bulletin, Vol. 18, No. 9, 1995, pp. 1308-1310.
[38] K. I. Koizumi, Y. Watanable, K. Morita, N. Utoguchi and M.Matsumoto, “New Method for Preparing High-Poros-ity Rapidly Saliva Soluble Compressed Tablets Using Mannitol with Camphor, a Subliming Material,” Interna-tional Journal of Pharmaceutics, Vol. 152, No. 1, 1997, pp. 127-131.
[39] H. A. Lieberman, L. Lachman and J. B. Schwartz, “Phar- maceutical Dosage Forms: Tablets Vol. 2,” 2nd Edition, Marcel Dekker, New York, 1990, pp. 1-71.
[40] Y. Bi, H. Sunada, Y. Yonezawa, K. Danjo, A. Otsuka and K. Iida, “Preparation and Evaluation of a Compressed Tablet Rapidly Disintegrating in the Oral Cavity,” Chem-ical and Pharmaceutical Bulletin, Vol. 44, No. 11, 1996, pp. 2121-2127.
[41] R. C. Rowe, P. J. Sheskey and P. J. Weller, “Handbook of Pharmaceutical Excipients,” 4th Edition, Pharmaceutical Society of Great Britain, London, 2003.
[42] K. A. Khan, “The Concept of Dissolution Efficiency,” Journal of Pharmacy and Pharmacology, Vol. 27, No. 1, 1975, pp. 48-49.

Pharmacology & Pharmacy, 2010, 1, 27-32 doi:10.4236/pp.2010.11004 Published Online July 2010 (http://www.SciRP.org/journal/pp)
Copyright © 2010 SciRes. PP
27
The Porcine Pulmonary Surfactant Protein A (pSP-A) Immunogenicity Evaluation in the Murine Model
Sandra de Cássia Dias1,3, Fernanda Lucio dos Santos2, Dirce Sakauchi2, Dmitri Iourtov2, Isaias Raw3, Flavia Saldanha Kubrusly3
1Engenharia de Bioprocessos-Campus Alto do Paraopeba, Universidade Federal de São João Del-Rei, Minas Gerais, Brazil; 2Divisão Bioindustrial, Instituto Butantan, Sao Paulo, Brazil; 3Centro de Biotecnologia, Instituto Butantan, Sao Paulo, Brazil. Email: [email protected] Received June 8th, 2010; accepted July 9th, 2010.
ABSTRACT
This paper investigated the porcine surfactant protein A (pSP-A) immunogenicity in murine model. Many elegant stud-ies about SP-A therapeutic applications are available however specific studies about its exogenous immunogenicity were not easily assumed. Therefore, we investigated the immunogenicity of this porcine protein in mice. The mice re-ceived pSP-A subcutaneously on days 0 and 7. The animals were observed during 90 days and the blood was collected on days 30, 60 and 90 for assessment the immunogenic potential of pSP-A. Some animals showed circulating antibodies above the screening cut point, which was calculated based on control mice sera signals. However, those antibodies were considered false positive read-outs by the performed competitive inhibition assay. Also no neutralizing antibodies were detected able to avoid the porcine protein ability to promote lipid aggregation. So far in this model, porcine sur-factant protein-A could be considered not immunogenic. Keywords: Immunogenicity, Porcine Surfactant Protein A, Anti-Drug Antibody, Murine Model
1. Introduction
Alveolar type II cells produce surfactant protein A, SP-A. This protein belongs to a group of soluble humoral pat-tern recognition receptors, called collectins, which mod-ulate the immune response to microorganisms [1]. The primary unit of a collectin contains an amino-terminal collagen like domain and a carboxyl terminal lectin or carbohydrate recognition domain (CRD) united by a more hydrophobic neck. SP-A is assembled as hexamers of trimers. This super structure can bind, agglutinate, opsonize and neutralize many different pathogens and can also modulate the uptake of these microorganisms by phagocytic cells as well as the inflammatory and the adaptive immune responses. Recent data have also high-lighted their involvement in clearance of apoptotic cells, hypersensitivity and a number of lung diseases [2-4].
Because of its ability to protect from infection by a wide variety of microorganisms and its capacity to regu-late the inflammatory response SP-A might be used as a paradigm to develop drugs to prevent or treat lung infec-tions [3]. Despite of a huge number of reports deals with
the role of SP-A for pulmonary pathology and its proba-bly use in lung disease treatment, very few reports men-tioned the immunogenicity of SP-A after exogenous ad-ministration [5]. The SP-A high molecular mass, oli-gomeric structures and glycosylation can contribute for immunogenicity development, but theses properties are important to SP-A activity in vivo and should be main-tained for therapeutics applications of this molecule.
Regulatory discussions about immunogenicity of the-rapeutic proteins represent today a central issue of bio-pharmaceuticals both by developers and by regulators cause an unwanted immunogenicity may lead to a loss of product efficacy besides severe side effects. These ef-fects could develop more deleterious consequences to the patient (allergy, anaphylaxis, serum sickness, neutraliza-tion of the drug or native protein) [6,7].
Since we have been studying porcine lung as a raw material for the development of potential lung disease medicines, we purified porcine SP-A (pSP-A) as by-product of lung surfactant production. The reduced number of reports about immunogenicity of exogenous SP-A administration prompted us to investigate the im-

The Porcine Pulmonary Surfactant Protein A (pSP-A) Immunogenicity Evaluation in the Murine Model
Copyright © 2010 SciRes. PP
28
munogenicity of pSP-A in the murine model.
2. Materials and Methods
2.1 Purification of Porcine Surfactant Protein A (pSP-A)
The pSP-A was purified from a lung extract previously clarified. This extract is a waste from the main produc-tion. The short protocol involved an acid precipitation of the rejected extract before affinity chromatography (pat-ent pending). LPS free pSP-A was obtained after the en-dotoxin removal by polymixin B-agarose (Pierce).
2.2 Gel Filtration Chromatography
Oligomerization was assessed by size exclusion chroma-tography. The pSP-A was loaded onto Superdex 200 HR column (10/30) (Ge Healthcare) in 0.1 M ammonium acetate- acetic acid buffer (pH 8.0). The flow rate was 0.3 mL/min and the peaks were manually collected. The chromatography was calibrated using the molecular mass standards (Thyroglobulin-669 kDa, aldolase-158 kDa, albumin-66 kDa, ovalbumin-43 kDa, chymotrypsinogen- 25 kDa).
2.3 Protein Assay
Protein concentration was determined using a bicin-choninic acid protein assay Kit (BCA; Pierce) using bo-vine serum albumin (BSA) as a standard.
2.4 Gel Electrophoresis
Purity of pSP-A were determined by 12.5% SDS-PAGE under reducing (β-mercaptoethanol) and non-reducing conditions. Relative molecular masses of pSP-A were estimated using molecular mass standards run in parallel.
2.5 Western Blot
The pSP-A was resolved by SDS-PAGE (12.5%) under reducing conditions and electroblotted from gel to the polyvinylidene difluoride membrane. After blocking, the membrane was incubated with rabbit anti-human SP-A polyclonal antibody from patients with rheumatoid ar-thritis or with mouse anti-human SP-A monoclonal anti-body (HYB 238-04-S BIOPORTO) or with rabbit an-ti-porcine SP-A polyclonal antibody.
2.6 Subcutaneous Administration of pSP-A
Forty female Swiss mice (20-22 g) were housed in cages at room temperature (22 ± 1°C) and 12 h light-dark cycle. The Instituto Butantan Committee for Research and Animal Ethics approved the experimental protocol. Mice were separated in two experimental groups: control and treatment group. Treatment group-Twenty mice were injected subcutaneously (sc) with 100 µl of pSP-A at dose of 5 mg/kg on days 0 and 7. Control group-Twenty
mice were divided in two subgroups. Ten animals were sc injected with 100 µl of saline on days 0 and 7 and the other ten were not injected. All animals were bled through the ophthalmic plexus on days 30, 60 and 90. The serum was separated and individually stored at –20°C.
2.7 Screening for Circulating Antibodies against pSP-A
The presence of anti-pSP-A polyclonal antibody in mice serum was investigated using an indirect ELISA assay. Wells were coated with 100 ng of pSP-A in coating buf-fer (carbonate-bicarbonate pH 9.6) overnight at 4°C fol-lowed by blockage with 10% fetal calf serum in PBS-T. Control or treated mice sera diluted 10, 100 and 1000 times were added to search circulating antibodies against the protein. After that incubation, the second antibody (goat anti-mouse IgG peroxidase-conjugate from Sigma) was added. The optical density was read at 492 nm using a microtiter plate spectrophotometer (Multiskam). The screening cut point was calculated using the individual control sera results in triplicate obtained from the three bleedings and from the three used dilutions. ANOVA estimated the mean and the standard deviation results to be used in the parametric method: mean + 1.645SD [8].
2.8 Competitive Immunoassay to Confirm the Positive Read-Outs against pSP-A
A competitive immunoassay was performed to dis- cri-minate false positive to the actual positive read-outs. The same indirect ELISA procedure described above was used with exception that potential positive samples di-luted 1:10 were previously incubated or not with pSP-A 25 μg/mL, overnight at 4°C. The assay was standardized using the rabbit polyclonal anti-pSP-A produced with Freund complete adjuvant (FCA) as the positive control diluted 1:2500 versus different pSP-A concentrations to built a calibration curve to establish the necessary amount of pSP-A to assure the specific inhibition or the specificity cut point. The chosen pSP-A concentration exceeded forty times the necessary amount. The per-centage of signal inhibition is the ratio of pSP-A inhib-ited sample by uninhibited sample calculated by the for-mula:
100 1
percentage of the signal inhibition
study dtug inhibited sample
unhibited sample
2.9 Screening for Neutralizing Antibodies (NAbs) against pSP-A
The presence of NAbs was investigated in potential posi-tive mice sera. The proposed assay verified the pSP-A dependent of Ca+2 ability to aggregate phospholipids
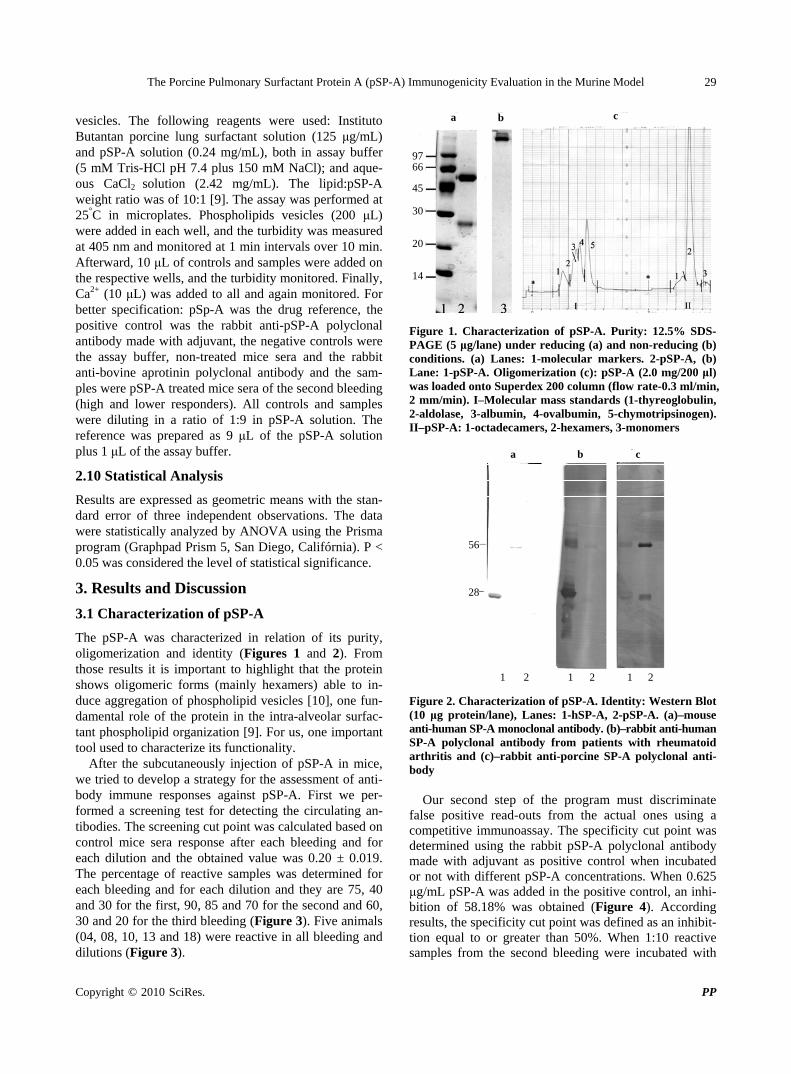
The Porcine Pulmonary Surfactant Protein A (pSP-A) Immunogenicity Evaluation in the Murine Model
Copyright © 2010 SciRes. PP
29
vesicles. The following reagents were used: Instituto Butantan porcine lung surfactant solution (125 μg/mL) and pSP-A solution (0.24 mg/mL), both in assay buffer (5 mM Tris-HCl pH 7.4 plus 150 mM NaCl); and aque-ous CaCl2 solution (2.42 mg/mL). The lipid:pSP-A weight ratio was of 10:1 [9]. The assay was performed at 25°C in microplates. Phospholipids vesicles (200 μL) were added in each well, and the turbidity was measured at 405 nm and monitored at 1 min intervals over 10 min. Afterward, 10 μL of controls and samples were added on the respective wells, and the turbidity monitored. Finally, Ca2+ (10 μL) was added to all and again monitored. For better specification: pSp-A was the drug reference, the positive control was the rabbit anti-pSP-A polyclonal antibody made with adjuvant, the negative controls were the assay buffer, non-treated mice sera and the rabbit anti-bovine aprotinin polyclonal antibody and the sam-ples were pSP-A treated mice sera of the second bleeding (high and lower responders). All controls and samples were diluting in a ratio of 1:9 in pSP-A solution. The reference was prepared as 9 μL of the pSP-A solution plus 1 μL of the assay buffer.
2.10 Statistical Analysis
Results are expressed as geometric means with the stan- dard error of three independent observations. The data were statistically analyzed by ANOVA using the Prisma program (Graphpad Prism 5, San Diego, Califórnia). P < 0.05 was considered the level of statistical significance.
3. Results and Discussion
3.1 Characterization of pSP-A
The pSP-A was characterized in relation of its purity, oligomerization and identity (Figures 1 and 2). From those results it is important to highlight that the protein shows oligomeric forms (mainly hexamers) able to in-duce aggregation of phospholipid vesicles [10], one fun-damental role of the protein in the intra-alveolar surfac-tant phospholipid organization [9]. For us, one important tool used to characterize its functionality.
After the subcutaneously injection of pSP-A in mice, we tried to develop a strategy for the assessment of anti-body immune responses against pSP-A. First we per-formed a screening test for detecting the circulating an-tibodies. The screening cut point was calculated based on control mice sera response after each bleeding and for each dilution and the obtained value was 0.20 ± 0.019. The percentage of reactive samples was determined for each bleeding and for each dilution and they are 75, 40 and 30 for the first, 90, 85 and 70 for the second and 60, 30 and 20 for the third bleeding (Figure 3). Five animals (04, 08, 10, 13 and 18) were reactive in all bleeding and dilutions (Figure 3).
a b c
9766
45
30
20
14
Figure 1. Characterization of pSP-A. Purity: 12.5% SDS- PAGE (5 μg/lane) under reducing (a) and non-reducing (b) conditions. (a) Lanes: 1-molecular markers. 2-pSP-A, (b) Lane: 1-pSP-A. Oligomerization (c): pSP-A (2.0 mg/200 μl) was loaded onto Superdex 200 column (flow rate-0.3 ml/min, 2 mm/min). I–Molecular mass standards (1-thyreoglobulin, 2-aldolase, 3-albumin, 4-ovalbumin, 5-chymotripsinogen). II–pSP-A: 1-octadecamers, 2-hexamers, 3-monomers
a c b
28
56
21 2 1 2 1
Figure 2. Characterization of pSP-A. Identity: Western Blot (10 μg protein/lane), Lanes: 1-hSP-A, 2-pSP-A. (a)–mouse anti-human SP-A monoclonal antibody. (b)–rabbit anti-human SP-A polyclonal antibody from patients with rheumatoid arthritis and (c)–rabbit anti-porcine SP-A polyclonal anti-body
Our second step of the program must discriminate false positive read-outs from the actual ones using a competitive immunoassay. The specificity cut point was determined using the rabbit pSP-A polyclonal antibody made with adjuvant as positive control when incubated or not with different pSP-A concentrations. When 0.625 μg/mL pSP-A was added in the positive control, an inhi-bition of 58.18% was obtained (Figure 4). According results, the specificity cut point was defined as an inhibit- tion equal to or greater than 50%. When 1:10 reactive samples from the second bleeding were incubated with

The Porcine Pulmonary Surfactant Protein A (pSP-A) Immunogenicity Evaluation in the Murine Model
Copyright © 2010 SciRes. PP
30
Figure 3. Humoral immune response to porcine SP-A administered subcutaneous rout. The animals were bled and their indi-vidual serum used for detection of anti-pSP-A circulating antibodies. The screening cut point (SCP) (0.20 ± 0.019) was ob-tained using the control group sera response
Figure 4. Competitive inhibition curve of rabbit anti-pSP-A antibody made with adjuvant. The antibody was incubated with different concentrations of pSP-A, and the inhibition quantified by Elisa. The specificity binding cut point was defined as inhibition equal to or greater than 50% pSP-A 25 μg/mL (forty times greater to have drug ex- cess), they showed different inhibitions, but they were less than 50% for all of them (Figure 5). Thus, pSP-A treated mice did not develop specific antibodies against this protein.
Exogenous SP-A administration routes and treatment regime are still unknown. In this study we used highly immunogenic conditions, subcutaneous rout and two doses on days 0 and 7 with physiologic protein concen- tration. However, in the last bleeding we observed a de- crease of the supposed anti-pSP-A antibodies. Theses results appointed to an antibody transient production and probably do not do have clinical relevance.
One last step was performed to conclude our valida- tion program, we screened for NAbs in potential positive samples. For that purpose we used the pSP-A ability to
Figure 5. Competitive inhibition assay. Mice second bleed- ing individual serum (1:10) were incubated with pSP-A (25 μg/mL). The protein concentration was forty times greater than used in the inhibition curve. Elisa quantified the inhi- bition aggregate phospholipid vesicles [11]. Ruano et al. (1996) showed that after Ca2+ addition the light absorbance of SP-A/lipid aggregates increases 20-25% [9]. We had similar results (Figure 6). Moreover, any mice treated sera did not affect the p-SP-A ability to phospholipids aggregation, which means we did not detected NAbs against p-SP-A (Figure 6). All reactive samples are false positives.
Our pSP-A immunogenicity evaluation led us to con- clude that at least for the murine model the porcine pro- tein has no immunogenic potential.
Are those results enough to propose p-SP-A as one safety and efficacy drug for human lung disease treatment? Or we must consider the predictive value of animal mod- els for the evaluation of immunogenicity in humans to be usually low [8]. So, what to do?

The Porcine Pulmonary Surfactant Protein A (pSP-A) Immunogenicity Evaluation in the Murine Model
Copyright © 2010 SciRes. PP
31
Figure 6. Assay for detect neutralizing antibodies (Nabs) in serum of mice treated with pSP-A. The Nabs must destroy pro- tein ability to lipid aggregation. Samples and references were filled with 125 μg/mL phospholipids. After 10 min, buffer, sam- ples, positive and negative controls were added in respective wells. Next, Ca2+ (1 mM final concentration) was added in all wells

The Porcine Pulmonary Surfactant Protein A (pSP-A) Immunogenicity Evaluation in the Murine Model
Copyright © 2010 SciRes. PP
32
4. Acknowledgements
Thanks to Dr. RMF Piazza for kindly donation of the reagents human SP-A and human SP-A antiserum.
Support: FAPESP, PRONEX, SADIA, Fundação Bu-tantan.
REFERENCES [1] P. Waters, M. Vaid, U. Kishore and T. Madan, “Lung
Surfactant Proteins A and D as a Pattern Recognition Proteins,” Advances in Experimental Medicine Biology, Vol. 653, December 2009, pp. 74-97.
[2] N. Palaniyar, J. Nadesalingam, H. Clark, M. J. Shih, A. W. Dodds and K. B. Reid, “Nucleic Acid is a Novel Li-gand for Innate, Immune Pattern Recognition Collectins Surfactant Proteins A and D and Mannose-Binding Lec-tin,” The Journal of Biological Chemistry, Vol. 279, No. 31, May 2004, pp. 32728-32736.
[3] A. Haczku, “Protective Role of the Lung Collectins Sur-factant Protein A and Surfactant Protein D in Airway In-flammation,” The Journal of Allergy and Clinical Immu-nology, Vol. 122, No. 5, November 2008, pp. 861-879.
[4] M. E. Famuyide, J. D. Hasday, H. C. Carter, K. L. Chesko, J. R. He and R. M. Viscardi, “Surfactant Pro-tein-A Limits Ureaplasma-Mediated Lung Inflammation in a Murine Pneumonia Model,” Pediatric Research, Vol. 66, No. 2, August 2009, pp. 162-167.
[5] S. G. Kremlev and D. S. Phelps, “Surfactant Protein A Stimulation of Inflammatory Cytokine and Immu-noglobulin Production,” American Journal Physiology,
Vol. 267, No. 6, December 1994, pp. L712-L719.
[6] G. Schernthaner, “Immunogenicity and Allergenic Poten-tial of Animal and Human Insulins,” Diabetes Care, Vol. 16, Suppl. 3, December 1993, pp. 155-165.
[7] E-V. Jahn, C. K. Schneider, “How to Systematically Evaluate Immunogenicity of Therapeutic Proteins— Regulatory Considerations,” New Biotechnology, Vol. 25, No. 5, June 2009, pp. 280-286.
[8] G. Shankar, V. Devanarayan, L. Amaravadi, Y. C. Barrett, R. Bowsher, D. Finco-Kent, M. Fiscella, B. Gorovits, S. Kirschner, M. Moxness, T. Parish, V. Quarmby, H. Smith, W. Smith, L. A. Zuckerman and E. Koren, “Recommen-dations for the Validation of Immunoassays Used for De-tection of Host Antibodies against Biotechnology Prod-ucts,” Journal of Pharmaceutical and Biomedical Analy-sis, Vol. 48, No. 5, December 2008, pp. 1267-1281.
[9] M. L. F. Ruano, E. Miguel, J. Perez-Gil and C. Casals, “Comparison of Lipid Aggregation and Self-Aggregation Activities of Pulmonary Surfactant Associated Protein A,” Biochemical Journal, Vol. 313, Part 2, January 1986, pp. 683-689.
[10] F. Sánchez-Barbero, J. Strassner, R. García-Canero, W. Steinhilber and C. Casals, “Role of the Degree of Oli-gomerization in the Structure and Function of Human Surfactant Protein,” The Journal of Biological Chemistry, Vol. 280, No. 9, March 2005, pp. 7659-7670.
[11] H. P. Haagsman, A. Hogenkamp, M. van Eijk and E. J. Veldhuizen, “Surfactant Collectins and Innate Immunity,” Neonatology, Vol. 93, No. 4, June 2008, pp. 288-294.

Pharmacology & Pharmacy, 2010, 1, 33-38 doi:10.4236/pp.2010.11005 Published Online July 2010 (http://www.SciRP.org/journal/pp)
Copyright © 2010 SciRes. PP
33
New Design of Biopharmaceuticals through the Use of Microalgae Addressed to Global Geopolitical and Economic Changes. Are You Ready for New Development in Biopharma?
Armen B. Avagyan
Research & Industry Center of Photosynthesizing Organisms, Feed Additives and Physiologically Active Compounds, Yerevan, Armenia. Email: [email protected] Received May 6th, 2010; accepted July 8th, 2010.
ABSTRACT
The Biopharma industry is enduring sweeping change in response to the financial crisis, but one aspect of the industry that emerged relatively unscathed and that perhaps has directly benefited from the crisis is new revolutionary innova- tion solution. Identifying opportunities in the next wave of technologies for Biopharma, along with other policy initia- tives including financial crisis actions and climate policy, will affect on manufacturing biopharmaceutical products today and in the future in cost effective manner, and will be our adequate answer addressing to global geopolitical, economic and climate changes. It also underscores the search for new approach, evidenced by trends around new for- mulations to serve lower income patients. Microalgae biomass comes in many strains, and can be used by means of variety product developments. In the last years the key task of our R & D was to find a solution for these tasks. The bio- fuel market development dynamics include significant opportunity of microalgae raw material and microalgae proc- essing biomass rest of biodiesel manufacturing for Biopharma global growth in cost effective manner. Second new source of microalgae raw material for Biopharma include microalgae production through waste and wastewater cleaning. This should provide the opportunity to see the future in a new vision, where technology can serve as a revela- tion of the truth and where every endeavor is governed by reflection on and appreciation of the environment and thus leading to resolution of global tasks facing the world community and inclusion of microalgae in production and bio cycles open new cost effective ways for Biopharma companies and conservation of nature. A truly coherent microalgae raw material and Biopharma production policy has to find ways to bring these two traces closer for cost effective manufacturing, well being Biopharma economy and human health. Keywords: Biopharma Development, Microalgae, Biofuel, Wastewater Cleaning
1. Biopharma Market Development Policy
Biopharma development progressive policy can be cate- gorized in many different ways. The Obama administra- tions policies will affect the pharmacy industry going forward [1,2]. One is cost-containment track. This track has been built by legislative initiatives and key health-care post nominations. The second is science-innovation track, powered by the economic stimulus package ($10 billions for National Institute of Health and with pres- timuus budget of more than $30 billions) [2]. Those 10 years also witnessed the first clear signs emerging of a fundamental shift in the balance of economic and politi- cal power from West to East, as the big emerging mar-
kets of Asia—and beyond—began to challenge the long- standing dominance of the U.S. and Europe. The issues identified, reimbursement challenges in emerging mar- kets dominate the list, with potentially game-changing initiatives underway in Korea and China, and also in Ja-pan [3]. Growth in the emerging markets is predicted 11-14% from 2008 to 2013. However, at last years, the trend has been for large pharmaceutical manufacturers to shed their consumer health divisions to narrow their business focus to a highly profitable core. As a result the last decade sales of biopharmaceutical products have grown dramatically and relatively steadily from less than $25 billion in 2000 to nearly $90 billion in 2008. At the same time, technology has created an environment for

New Design of Biopharmaceuticals through the Use of Microalgae Addressed to Global Geopolitical and Economic Changes. Are You Ready for New Development in Biopharma?
Copyright © 2010 SciRes. PP
34
tremendous short- and long-term currency volatility as legions of investors can pile into or out of a currency in milliseconds. While the forces sharping the world of busi-ness are much the same—technology, population changes, natural resources, regulation, environmental concerns, political movies and social pressure—the effects of these forces are a constant state of flux, as companies learn new ways to interact with their customers. Today, proc-ess innovation is becoming as important as product in-novation, because without finding better ways to promote access to needed medicines, industry’s best scientific assets can never be fully leveraged to support real gains in health outcomes. The determination to find a solution may have been fueled in part by desperation—and the fact that the world had just been dealt a sobering slap in the face by the financial crisis. It also underscores the search for new approach, evidenced by trends around new formulations to serve lower income patients. What will happen to the innovative category of drugs and raw materials that have been eligible for sale at a competitive low price? What is certain is that the shotgun approach belongs to the past, while precision focus and selectivity are taking hold across the industry. But many leading pharma manufacturers have been shaken out of their in- ertia when it comes to the existing R & D focus and the sales of biopharmaceutical product faced with a triple hammy of declining R & D productivity and increasing pricing forward looking companies must embracing new outsourcing strategies to achieve and maintain profitable growth [2,4,5]. Technology must been key factor in new changes. Nowadays we need a series of new R & D solu- tions for Biopharma, which may be well advised to view portfolios of R & D-based manufacturers from a new perspective. The world could actually enhance economic output and welfare by pursuing a path of mitigation cost through profitable innovation. This creates both new op- portunities and new headaches. Brainstorming is a useful way of generating radical solutions to problems and its effective strategies is prime the pump and improve abi- lity to generate bright ideas. It is particularly useful when you want to break out of stale, established patterns of thinking, so that you can develop new ways of looking at things. This article present situtional analyses and our approch in the framework of our concept addressed to global sustainable development through including micro- algae and its biomass in Production (such as wastewater treatment and biofuel manufacturing) and Bio Cycles (such as biopharmaceuticals, feed, perfumery etc. pro- ductions) [6,7].
2. Biopharma Project-Thinking to the Future
The Biopharma industry is enduring sweeping change in response to the financial crisis, but one aspect of the in-
dustry that emerged relatively unscathed and that perhaps has directly benefited from the crisis is new revolution-ary innovation solution. It is already clear that the world’s infrastructure will change dramatically in the decade ahead as a result of trends already in place. In analyzing supply and demand for bio-manufacturing ca- pacity, we able to identify and track industry-wide trends in production of biopharmaceutical products and to fore- cast future directions for this highly dynamic field. The level of economic development directions and the policy choices are important factors determining the nature of the problems faced Biopharma and the ways in which they are solving. Despite several decades of experience in monitoring bioreactors and refining cell culture opera- tions, the production of safe, pure and potent biologics remains a tricly business [1-5].
Rising energy and transportation costs and impending rules on carbon emission are promoting companies to reassess their sourcing strategies. The last year ended with United Nations conference of Copenhagen in De- cember, with many hoping for a Kyoto-style consensus to shape globally environmental policy. The countries involved failed come to a binding agreement, but instead signed the Copenhagen Accord, which pledged $30 bil- lion a year to a fund for poor countries to adapt to cli- mate change from 2010-2012, and $100 billion a year by 2020. The fuel economy improvements and introduction of bioetanol and biodisiel add more opportunities for Biopharma developing. Biofuel manufacturing is ex- pected to be a new rapidly growing global market for algae biomass and increase volume of its products [8]. Our vision infers suggests that algae have emerged as one of the most promising sources especially for Bio- pharma development. The validity of this approach in- creases and confirms in the face of Biofuels Digest up- dated Advanced Biofuels tracking database, based on announced projects and updated company guidance tracking 56 companies with advanced biofuels projects in 13 countries [9]. According this database algal biodiesel volume projected to reach 421 million gallons per year in 2013. The biofuel market development dynamics include significant opportunity of microalgae raw material for Biopharma global growth in cost effective manner and new technological innovation leading to the ability to develop a roll-out platform.
Second new source of microalgae raw material for Biopharma. The EU Landfill Directive has forced waste management policies across the member states of the EU to reduce the amount of waste sent for disposal in landfill. The directive requires that progressively increasing quantities of biologically active waste are diverted away from landfill. May be the criteria of the Landfill Direc- tive can become the main drivers for the use microalgae aimed to cleaning their wastewaters in Biopharma? It is

New Design of Biopharmaceuticals through the Use of Microalgae Addressed to Global Geopolitical and Economic Changes. Are You Ready for New Development in Biopharma?
Copyright © 2010 SciRes. PP
35
known that the biological method is considered the most effective and economically efficient method for the puri-fication of industrial wastewater by using of the microbio- logical active slime or alga. However, bacteria of the active slime have low stability to high concentration of organic and mineral components, thus considering big water flow volumes [7,10]. This method also requires further destruction of superfluous quantity of active slime, which contains also pathogenic microorganisms. Micro- algae have higher stability, which enables working in more concentrated and toxic environments. Chlorella actively utilizes mineral elements, spirits, sugar, and amino acids and as compared with active slime enables higher purification rate (up to 96-98% for organic and 80% for mineral components, accordingly). In the last years the key task of our R & D was to find a solution for this problem, because microalgae possess higher stability, which enables their use in more concentrated and toxic environments, and our Center strategy believes also that the cost saving of raw material with the use of waste- waters through their biological cleaning will help raise the availability of microalgae biomass for biofuel, bio-pharmaceuticals, food, agriculture producers, thus lead-ing to resolution of global tasks facing the world com-munity. The Center carried out researchers for develop-ment technologies of microalgae cultivation in some wastewaters of industrial plants [8]. As a result the Cen-ter developed a cost-effective technology applying new innovative approaches in various stages of microalgae production and this technology for microalgae produc-tion may be applied all around the world. The exhaust steam and effluent gas (including greenhouse emissions) may be used for heating microalgae suspension in bio-technological pools so the biomass manufacture not will be available year-around. Simultaneously the high norms of wastewater purification from organic and mineral compounds were achieved and in parallel to this it was accompanied by sharp reduction of the bacteria contents in strongly microbiological infected biotechnilogical wastewater. Therefore receiving microalgae biomass by waste and wastewater cleaning and inclusion it in bio cycles open new cost effective way for Biopharma com-panies and conservation of nature. This should provide the opportunity to see the future in a new vision, where technology can serve as a revelation of the truth and where every endeavor is governed by reflection on and appreciation of the environment and thus leading to reso-lution of global tasks facing the world community. Thus, manufacturing microalgae through the use and purifica- tion of wastewaters, as well as microalgae processing biomass rest of biodiesel manufacturing must be ways for manufacturing biopharmaceutical in cost effective manner and an additional source of profit.
3. Which Value Can Obtain Biopharma from Microalgae Biomass?
Microalgae are a diverse group of microscopic plants with the wide range of physiological and biochemical characteristics and contain up to 50-70% protein (up to 50% in meat, and 15-17% in wheat), 30% lipids, over 40% glycerol, up to 8-14% carotene and a fairly high concentration of vitamins B1, B2, B3, B6, B12, E, K, D, etc., compared with other plants or animals [11]. More- over, microalgae are meant to be an important raw mate- rial for amino acids, vitamins and productions of other pharmateuticals. The cultivation of microalgae is known to be the most profitable business in the biotechnological industry. It is a wasteless, ecologically pure, energy and resource saving process. They are also harvested very quickly; dramatically speeding up production process with small water consumption. Additionally algae can adsorb up to 450 tons of CO2 per acre when grown com- mercially.
The potential of microalgae biomass for big Pharma practical uses is certainly great. The first use of microal- gae by humans dates back 2000 years to the Chinese, who used Nostoc to survive during famine. At present around 110 commercial producers of microalgae are in the Asia-Pacific region, with annual production capa- city ranging from 3 to 500 tones [12]. The commercially cultivated microalgae include Chlorella, Spirulina, Du- naliella, Nannochloris, Nitzschia, Crypthecodinium, Sc- hizochytrium, Tetraselmis, Skeletonema etc. The market survey shows that being developed in the last 20-30 years, the microalgae production volume increased excessively [10]. In fact, the former USSR was the first to become a large scale manufacturing of microalgae, in the frame- work of producing high quality feed additives [11]. In 1980 more than 500 Chlorella manufacturings were in farms of Uzbekistan (mainly for sheep, adding 1 liter microalgae suspension in the daily diet of sheep in- creased their weight gain by 15-20%) as well as ad- diitional facielties in other Soviet republics. Chlorella powder raised the poultry the average daily weight by 13%, egg-laying quality by 10-30% (paste–by 26-30%), quantity of vitamins in liver increased 2-3 times and rate of poultry mortality decreased up to 7-23%. Chlorella protein digestibility reached up to 85 percent. The aver- age daily weight gain of pigs increased twice due to the use of Chlorella paste. Chlorella protein digestibility for pigs was 52-72%. In our Industrial test ccombined feed with 1% of Chlorella powder (produced through cleaning of biotechnological wastewater of crystallic lysine) used on 25200 species of fishes (average weight-12.7 g). Dur- ing the first 20 days the daily average weight gain in- creased by 20% (0.97 g in tested and 0.73 g in the control group), and the mortality reduced by 48% (0.77% and

New Design of Biopharmaceuticals through the Use of Microalgae Addressed to Global Geopolitical and Economic Changes. Are You Ready for New Development in Biopharma?
Copyright © 2010 SciRes. PP
36
1.25%, respectively) [11]. However, the disintegration of the USSR has caused interruption of all these manufac- turing.
Second-generation microalgae large scale manufac-turing volume sharply increased due to significant influ- ence of food, high-quality perfumery additives related industry development (the U.S. (Sun Wellness Inc., Cya- notech. Corp., etc), Japan (Yaeyama Factory, etc)). Most of the commercially produced algal biomass is being marketed as health food, in the forms of tablets and cap-sules. Algae and their extract are also included in noodles, wine, beverages, breakfast cereals and cosmetics. So, currently over 75% of pharmaceutical product develop-ment is generated by the food suppliment production comprising also microalgae. About 61% of Americans (spending $6 billion yearly) and 43% of Europeans use food additives.
4. Which Role Can Microalgae and its Processing Raw Materials of the Biofuel Production and Wastewater Cleaning Take in the Production Patterns?
Microalgae biomass comes in many strains, and can be used by means of variety product developments. Blue-green algae are a group of prokaryotes which history goes back to 2700 million years. Blue-green cyanophytes are not true algae. They have no nucleus, the structure that en- closes the DNA, and no chloroplast, the structure that encloses the photosynthetic membranes, the structures that are evident in photosynthetic true algae. It was set at a level considered to be safe for human consumption. In China, Taiwan and Japan, several cyanophytes are served as a side dish and are considered a delicacy [12]. The very potent toxins produced by many solitary, filamen-tous or colonial aquatic cyanophytes, are responsible for an increasing number of water-related poisonings of both wildlife and people [7]. The World Health Organization has a limit on the toxin Microcystin at one part per bil-lion (ppb). In 1998, the U. S. Environmental Protection Agency (EPA) included freshwater cyanobacteria and their toxins on the first Candidate Contaminant List (CCL) (Federal Register 1998). For this reason, the use of cyanophytes blue-green must be allow only from large producer-companies with good developed toxin control.
American Cyanotech. Corp. and Russian Convercia produce vitamin-mineral complex (pills of Spirullina) as a food additive and milk substitute [9]. It effectively re- moves slag’s (heavy metals, radio nucleotides and leu- kocytes) from organism, reduces influence of irradiation, sugar and cholesterol in blood, cardiovascular diseases as well as increases immunity and improves skin condition. It is also useful for liver diseases, arthritis, asthenia and insomnia, regulates normal pregnancy and lactation and
prevents accumulation of excessive weight. Natural products of microalgae are leading sources of novel molecules that have been used in the pharmaceutical and nutraceutical industries since their inception. So, product list of Cyanotech. Corp. included also natural astaxanthin BioAstin for humans, produced from Spirullina, which reviewed by the U.S. Food and Drug Administration and have up to 550 times the antioxidant activity of vitamin E and 10 times the antioxidant activity of beta-carotene, and surpasses many of the antioxidant benefits of vitamin C and other carotenoids [13].
Green algae evolved from prokaryotes between 2500 and 1000 million years ago. Chlorella is a microscopic, green, single cell and has not toxins. During 12 hours Chlorella cell provides fourfold reproduction of cells in optimum conditions. Compared to the traditional plants, the water consumption of manufacturing is over 10 times as low. The biomass yield per sq. are 5 times higher. Other main Chlorella biological priorities are as follow- ing [11]:
1) High concentration of protein (50%) and amino ac-ids,
2) High concentration of chlorophyll (5-10 times as much, compared to Spirullina or Lucerne), the molecule of which is identical to hemoglobin molecule structure. Therefore, if getting in blood flow, it sates blood with oxygen and is transformed to hemoglobin. The chloro- phyll is an effective means for the treatment of anemia, a pancreatitis, skin ulcer, diabetes, recovery of peristalsis affect and normalization of digestive juice excretion. It owns anticancer, anti-inflammatory, antiseptic and regen- erating properties,
3) Unique properties of a cell wall which consists of three layers (the largest-a middle part consists of cellu- lose, and the outer layer is formed of polymeric carotene which is capable of adsorbing toxic elements and re- moving them from organisms),
4) High contents of vitamins, especially, pro-vitamin A–carotene, which not only plays an important role dur- ing the growth process, but destructs cancer cells as well in initial stages and improves the generation of macro- bacteriophage in immune system,
5) Ability to intensively synthesize high concentration of nucleonic acids with a combination of high contents of fibers, peptides, amino acids, other vitamins, sugars and trace elements. This not only promotes super fast repro-duction of Chlorella, but as a growth factor also provides favorable conditions for the use of Chlorella in other organisms.
6) Chlorella has also organic acids, which prevent the growth of pathogenic microorganisms in wastewater and feed. While antibiotics were proven to be effective in improving agricultural production, their use came under pressure as an increasing number of consumers feared

New Design of Biopharmaceuticals through the Use of Microalgae Addressed to Global Geopolitical and Economic Changes. Are You Ready for New Development in Biopharma?
Copyright © 2010 SciRes. PP
37
that their inclusion in animal feed rations would lead to antibiotic resistant bacteria that are pathogenic to humans. In 2005, the EU removed the last antibiotic growth pro- moters from pig and poultry diets. As consensus begins to develop among the scientific community on this sub- ject, a few approaches stand out in terms of efficacy, technological and economical feasibility, particularly in terms of organic acids and the use of essential or botani- cal oils [7,8]. Organic acids provide a natural alternative, reducing production of toxic components by bacteria and causing a change in the morphology of the intestinal wall that reduces colonization of pathogens, thus preventing damage to the epithelial cells. Anions of organic acids deactivate the RNA transferase enzyme, which damage the nucleic acid multiplication process and eventually result in death of the organisms. But the manufacturing of organic acids and essential oils for the feed industry are potentially a source of other problems: corrosion, worker safety, handling, vitamin stability in pre-mixes, environmental concerns, and the stability of products [8,11]. With all this in mind, the use of microalgae Chlorella could become the best solution, since microal- gae contain natural organic acids (hexadecatetraenoic (up to 7-8% of total fatty acid quantity) and octadecatet- raenoic acids, as well as oxy, aldehyde and keto acids, which increase spectrum of antibacterial action) that re- duce colonization of pathogens. Therefore, thanks to this feature Chlorella is also used for feed conservation, and the reduction of microbiological pollution of wastewa- ters.
Chlorella studies have shown the plant’s cells are pro- active in stimulating T-cells, and largely improving the immune system’s ability to ward off the formation of diseases like cancer, hypoglycemia, and bacteria [14]. Chlorella’s high concentration of chlorophyll has been cited to eliminate halitosis in a matter of just days and after reversing constipation, Chlorella can improve the stink of heavily accented stools.
In the end of 1990 in former USSR clinical tests ex- amined Lipid concentrate of Chlorella (for the treatment of colitis, cervical erosion and burn) and Chlorella hy- drolysate (for the improvement of working capasity). According to all-Union State Standards of the CIS coun- tries this products can use for shampoos, creams, tooth pastes, lotions etc. as bioactive additive.
The yellow microalgae can be used as sourse of ara- chidonic eicosapentaenoic acids aimed to manufacturing of drugs.
The growing worldwide market value of carotenoids is projected to reach over US$1,000 million and there the use of microalgae open possibility aimed to reduction in production costs [15]. The Israel and Australia compa- nies specialized in carotene production from unicellular green Dunaliella microalgae (up to 8-14% carotene, over
40% of glycerol). Omega-3-fatty aids can not be constructed within our
bodies. Therefore they must be obtained from the diet, making outside source. DHA is found at low levels in fish and at high levels in certain microalgae oils. Con- centrated omega-3 DHA dietary supplements made from microalgae oils or extracted from fish oils currently are the best way to get medical dosages of DHA at levels above 1 gram daily [16]. They extracted from algae with organic solvents from protein while the fish oil is pressed out of cooked fish mass during fishmeal production. In California, BioCentric Energy Holdings announced the commercialization of its closed-loop Photo-Bioreactor system for the mass production and subsequent comer- cialization of algae products for Omega-3 (EPA) oil and algal biomass [17].
Recitals of the experience gained on microalgae use may continue, but the above discussion demonstrates an increasing need to the use of microalgae in Biopharma. Identifying opportunities in the next wave of technolo- gies for Biopharma, along with other policy initiatives including financial crisis actions and climate policy, will affect on manufacturing biopharmaceutical products to- day and in the future in cost effective manner, and will be our adequate answer addressing to global geopolitical, economic and climate changes. A truly coherent micro- algae raw material and Biopharma production policy has to find ways to bring these two traces closer for cost ef- fective manufacturing, well being Biopharma economy and human health. Further isolation and identification of novel metabolites from microalgae will help to feed the pipeline of the biopharmaceutical industries for the development of new therapeutic agents, but also open a door for nutraceutical and functional food industries.
REFERENCES [1] J. Wechdier, “Issue and Opportunities Challenge Biotech
Manufacturers,” Biopharm International, Vol. 23, No. 1, 2010, pp. 16-17.
[2] M. Hoffman, “Parallel Track. Obama Cost-Containment and Science Innovation Initiatives Need to Overlap,” Pharmateuticals Technology, Vol. 33, No. 4, April, 2009, p. 12.
[3] E. C. Langer and M. Pavicant, “Global Economic Crunch. Benefits Biomanufacturing Outsourcing to Asia,” Inside Outsoursing, November 2009, pp. 24-28.
[4] D. C. Esola, “An Introduction from the Publisher,” Inside Outsoursing, November 2009, p. 8. http://www.nxtbook. com/nxtbooks/advanstar/insideoutsourcing_200911/#/10/ OnePage.
[5] A. C. Cacich, R. Bjella, M. J. Cosko, et al., “8 Key Play-ers. On The Future of Global Outsourcing, and Forming Alliances with CRO’s/CMO’s,” Inside Outsoursing, No-vember 2009, pp. 32-37.

New Design of Biopharmaceuticals through the Use of Microalgae Addressed to Global Geopolitical and Economic Changes. Are You Ready for New Development in Biopharma?
Copyright © 2010 SciRes. PP
38
http://www.nxtbook.com/nxt-books/advanstar/insideoutsourcing_200911/#/32/OnePage.
[6] A. B. Avagyan, “A Contribution to Global Sustainable Development: Inclusion of Microalgae and their Biomass in Production and Bio Cycles,” Clean Technologies and Environmental Policy, Vol. 10, No. 4, 2008, pp. 313-317.
[7] A. B. Avagyan, “New Design & Build Biological System through the Use of Microalgae Addressed to Sustainable Development,” Journal of Environmental Protection, Vol. 1, No. 2, 2010, pp. 183-200.
[8] A. B. Avagyan, “Global Prospects for Microalgae Pro- duction for Biofuels and for the Preservation of Nature,” Global Fuel Magazine, February 2008, pp. 22-27. http:// www.propubs.com/global-fuels/eGF_Feb08_LowRes.pdf
[9] J. Lane, “Advanced Biofuels Tracking Database,” Biofuels Digest, March 2010. http://www.biofuelsdigest.com/bdi- gest/2010/03/23/advanced-biofuels-tracking-database/
[10] A. B. Avagyan, “Microalgae Production Development Glo- bal Prospects and Profitable Technology Wasterwater Pu-rification by the Use Microalgae,” Water and Wastewater International, 2008. http://ww.pennnet.com/articles/article_ display.cfm?article_id=340236&dcmp=WaterWorldEnl
[11] A. B. Avagyan, “Microalgae: Big Feed Potential in a Small Package,” Feed International, March 2008, pp. 16-18.
http://www.fi-digital.com/fi/200803/data/feedinternational200803-win32.zip
[12] Y.-K. Lee, “Commercial Production of Microalgae in the Asia-Pacific Rim,” Journal of Applied Phycology, Vol. 9, No. 5, October 1997, pp. 403-411.
[13] Cyanotech Corp.,“BioAstin.”’ http://www.cyanotech.com/bioastin.html
[14] The Colon Cleansing & Constipation Resource Center, “How Chlorella Relates to Constipation,” 2007. http:// www.articlesbase.com/alternative-medicine-articles/how-chlorella-relates-to-constipation-89878.html
[15] J. A. Del Campo, M. García-González, M. G. Guerrero, “Outdoor Cultivation of Microalgae for Carotenoid Pro-duction: Current State and Perspectives,” Applied Micro-biology and Biotechnology, Vol. 74, No. 6, 2007, pp. 1163- 1174.
[16] H. Chapell, “PhD Presents Omega-3 Replacements for Medical Fish Oils,” February 2009. http://www.chiroeco. com/chiropractic/news/7480/856/PhD-presents-Omega-3- replacements-for-medical-fish-oils/
[17] J. Lane, “BioCentric to Deploy Five Algae Photobioreactor Units in Client Trial,” Biofuels Digest, April 2010. http:// biofuelsdigest.com/bdigest/2010/04/23/biocentric-to-deploy- five-algae-photobioreactor-units-in-client-trial/




















