
Pathophysiology of Ischemic Acute Renal Failure Pathophysiology of Ischemic Acute Renal Failure A...
23
14 Pathophysiology of Ischemic Acute Renal Failure A cute renal failure (ARF) is a syndrome characterized by an abrupt and reversible kidney dysfunction. The spectrum of inciting factors is broad: from ischemic and nephrotoxic agents to a variety of endotoxemic states and syndrome of multiple organ failure. The pathophysiology of ARF includes vascular, glomerular and tubular dysfunction which, depending on the actual offending stimulus, vary in the severity and time of appearance. Hemodynamic compromise prevails in cases when noxious stimuli are related to hypotension and septicemia, leading to renal hypoperfusion with sec- ondary tubular changes (described in Chapter 13). Nephrotoxic offenders usually result in primary tubular epithelial cell injury, though endothelial cell dysfunction can also occur, leading to the eventual cessation of glomerular filtration. This latter effect is a con- sequence of the combined action of tubular obstruction and activation of tubuloglomerular feedback mechanism. In the following pages we shall review the existing concepts on the phenomenology of ARF including the mechanisms of decreased renal perfusion and failure of glomerular filtration, vasoconstriction of renal arterioles, how formed elements gain access to the renal parenchyma, and what the sequelae are of such an invasion by primed leukocytes. Michael S. Goligorsky Wilfred Lieberthal CHAPTER
Transcript of Pathophysiology of Ischemic Acute Renal Failure Pathophysiology of Ischemic Acute Renal Failure A...

14
Pathophysiology of Ischemic Acute Renal Failure
Acute renal failure (ARF) is a syndrome characterized by anabrupt and reversible kidney dysfunction. The spectrum ofinciting factors is broad: from ischemic and nephrotoxic agents
to a variety of endotoxemic states and syndrome of multiple organfailure. The pathophysiology of ARF includes vascular, glomerularand tubular dysfunction which, depending on the actual offendingstimulus, vary in the severity and time of appearance. Hemodynamiccompromise prevails in cases when noxious stimuli are related tohypotension and septicemia, leading to renal hypoperfusion with sec-ondary tubular changes (described in Chapter 13). Nephrotoxicoffenders usually result in primary tubular epithelial cell injury,though endothelial cell dysfunction can also occur, leading to theeventual cessation of glomerular filtration. This latter effect is a con-sequence of the combined action of tubular obstruction and activationof tubuloglomerular feedback mechanism. In the following pages weshall review the existing concepts on the phenomenology of ARFincluding the mechanisms of decreased renal perfusion and failure ofglomerular filtration, vasoconstriction of renal arterioles, how formedelements gain access to the renal parenchyma, and what the sequelaeare of such an invasion by primed leukocytes.
Michael S. GoligorskyWilfred Lieberthal
C H A P T E R
14.2 Acute Renal Failure
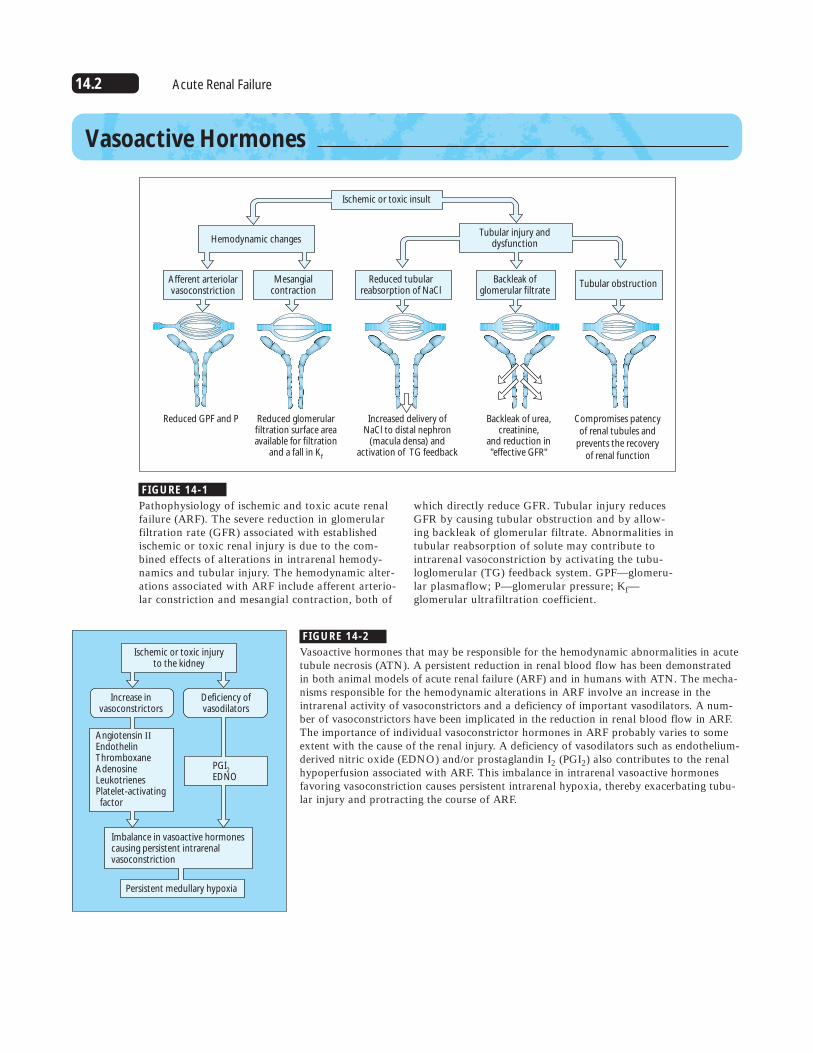
FIGURE 14-1
Pathophysiology of ischemic and toxic acute renalfailure (ARF). The severe reduction in glomerular filtration rate (GFR) associated with establishedischemic or toxic renal injury is due to the com-bined effects of alterations in intrarenal hemody-namics and tubular injury. The hemodynamic alter-ations associated with ARF include afferent arterio-lar constriction and mesangial contraction, both of
Vasoactive Hormones
Reduced GPF and P
Afferent arteriolarvasoconstriction
Mesangialcontraction
Reduced tubularreabsorption of NaCl
Backleak of glomerular filtrate
Tubular obstruction
Reduced glomerularfiltration surface areaavailable for filtration
and a fall in Kf
Increased delivery ofNaCl to distal nephron
(macula densa) andactivation of TG feedback
Backleak of urea, creatinine,
and reduction in"effective GFR"
Compromises patencyof renal tubules and
prevents the recoveryof renal function
Hemodynamic changesTubular injury and
dysfunction
Ischemic or toxic insult
Ischemic or toxic injuryto the kidney
Increase in vasoconstrictors
Deficiency ofvasodilators
Imbalance in vasoactive hormonescausing persistent intrarenalvasoconstriction
Angiotensin IIEndothelinThromboxaneAdenosineLeukotrienesPlatelet-activating factor
PGI2
EDNO
Persistent medullary hypoxia
FIGURE 14-2
Vasoactive hormones that may be responsible for the hemodynamic abnormalities in acutetubule necrosis (ATN). A persistent reduction in renal blood flow has been demonstratedin both animal models of acute renal failure (ARF) and in humans with ATN. The mecha-nisms responsible for the hemodynamic alterations in ARF involve an increase in theintrarenal activity of vasoconstrictors and a deficiency of important vasodilators. A num-ber of vasoconstrictors have been implicated in the reduction in renal blood flow in ARF.The importance of individual vasoconstrictor hormones in ARF probably varies to someextent with the cause of the renal injury. A deficiency of vasodilators such as endothelium-derived nitric oxide (EDNO) and/or prostaglandin I2 (PGI2) also contributes to the renalhypoperfusion associated with ARF. This imbalance in intrarenal vasoactive hormonesfavoring vasoconstriction causes persistent intrarenal hypoxia, thereby exacerbating tubu-lar injury and protracting the course of ARF.
which directly reduce GFR. Tubular injury reducesGFR by causing tubular obstruction and by allow-ing backleak of glomerular filtrate. Abnormalities intubular reabsorption of solute may contribute tointrarenal vasoconstriction by activating the tubu-loglomerular (TG) feedback system. GPF—glomeru-lar plasmaflow; P—glomerular pressure; Kf—glomerular ultrafiltration coefficient.
14.3Pathophysiology of Ischemic Acute Renal Failure
Mesangial cell contractionAngiotensin IIEndothelin–1Thromboxane
Sympathetic nerves
Mesangial cell relaxationProstacyclin
EDNO
Glomerular epithelial cellsM
M
Glomerular capillaryendothelial cells
Glomerular basement membrane
Afferent arteriole
Periportalcell
Macula densacells
Extraglomerularmesangial cells
Glomerus
A
EA
D
AA
PPC
EMC GMCG
MD
ChlorideAdenosinePGE2AngiotensinNitric oxideOsmolarityUnknown?
EA
AA
SMC+GC
PPC
EMC GMCG
MD
C
EA
B
AA
PPC
EMC GMCG
MD
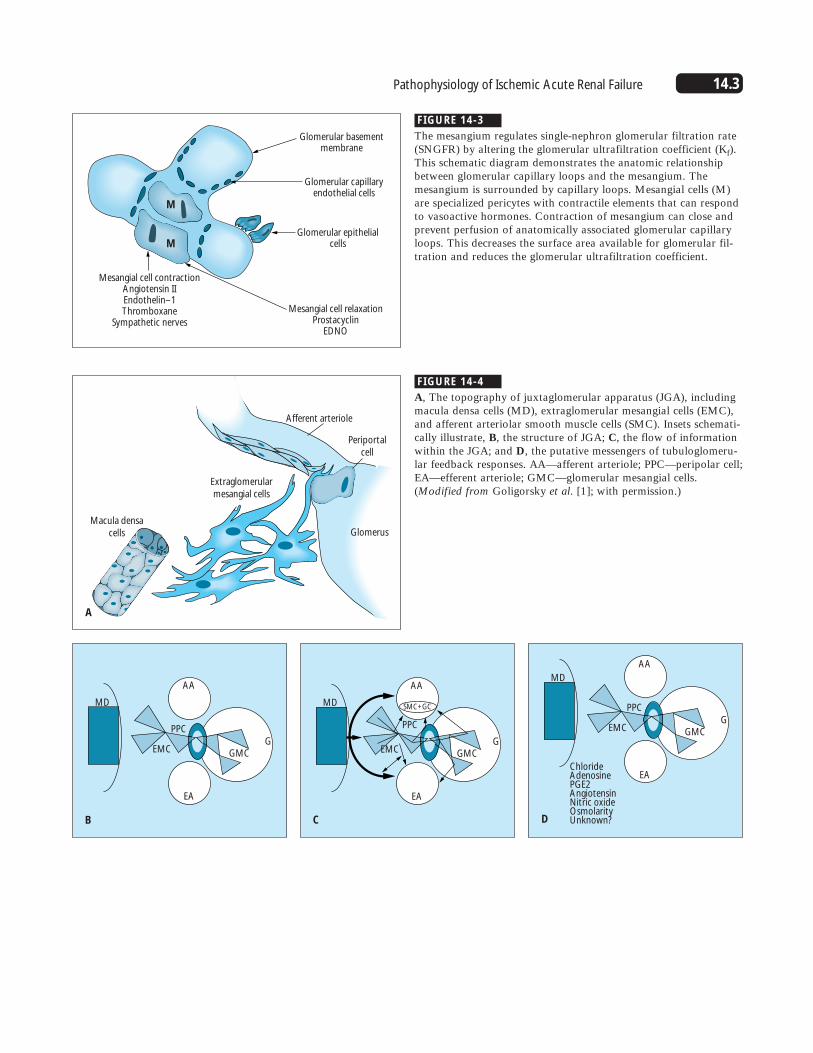
FIGURE 14-3
The mesangium regulates single-nephron glomerular filtration rate(SNGFR) by altering the glomerular ultrafiltration coefficient (Kf).This schematic diagram demonstrates the anatomic relationshipbetween glomerular capillary loops and the mesangium. Themesangium is surrounded by capillary loops. Mesangial cells (M)are specialized pericytes with contractile elements that can respondto vasoactive hormones. Contraction of mesangium can close andprevent perfusion of anatomically associated glomerular capillaryloops. This decreases the surface area available for glomerular fil-tration and reduces the glomerular ultrafiltration coefficient.
FIGURE 14-4
A, The topography of juxtaglomerular apparatus (JGA), includingmacula densa cells (MD), extraglomerular mesangial cells (EMC),and afferent arteriolar smooth muscle cells (SMC). Insets schemati-cally illustrate, B, the structure of JGA; C, the flow of informationwithin the JGA; and D, the putative messengers of tubuloglomeru-lar feedback responses. AA—afferent arteriole; PPC—peripolar cell;EA—efferent arteriole; GMC—glomerular mesangial cells.(Modified from Goligorsky et al. [1]; with permission.)

14.4 Acute Renal Failure
1. SNGFR increasescausing increase in delivery of soluteto the distal nephron.
3. Renin is released from specialized cells of JGA and the intrarenal renin angiotensin system generates release of angiotensin II locally.
2. The composition of filtratepassing the macula densa is altered and stimulates the JGA.
4. Afferent arteriolar and mesangial contraction reduce SNGFR back toward control levels.
The normal tubuloglomerular (TG) feedback mechanism
A
1. Renal epithelial cell injury reduces reabsorptionof NaCl by proximal tubules.
3. Local release of angiotensin IIis stimulated.
2. The composition of filtratepassing the macula densa is altered and stimulates the JGA.
4. Afferent arteriolar and mesangial contraction reduce SNGFR belownormal levels.
Role of TG feedback in ARF
B
FIGURE 14-5
The tubuloglomerular (TG) feedback mech-anism. A, Normal TG feedback. In the nor-mal kidney, the TG feedback mechanism isa sensitive device for the regulation of thesingle nephron glomerular filtration rate(SNGFR). Step 1: An increase in SNGFRincreases the amount of sodium chloride(NaCl) delivered to the juxtaglomerularapparatus (JGA) of the nephron. Step 2:The resultant change in the composition ofthe filtrate is sensed by the macula densacells and initiates activation of the JGA.Step 3: The JGA releases renin, whichresults in the local and systemic generationof angiotensin II. Step 4: Angiotensin IIinduces vasocontriction of the glomerulararterioles and contraction of the mesangialcells. These events return SNGFR backtoward basal levels. B, TG feedback inARF. Step 1: Ischemic or toxic injury torenal tubules leads to impaired reabsorptionof NaCl by injured tubular segments proxi-mal to the JGA. Step 2: The composition ofthe filtrate passing the macula densa isaltered and activates the JGA. Step 3:Angiotensin II is released locally. Step 4:SNGFR is reduced below normal levels. Itis likely that vasoconstrictors other thanangiotensin II, as well as vasodilator hor-mones (such as PGI2 and nitric oxide) arealso involved in modulating TG feedback.Abnormalities in these vasoactive hormonesin ARF may contribute to alterations in TGfeedback in ARF.
14.5Pathophysiology of Ischemic Acute Renal Failure
Osswald's Hypothesis
Increased ATP hydrolysis (increased distal Na+ load)
Increased generation of adenosine
Activation of JGA
Afferent arteriolar vasoconstriction
Nerve endings
Adenosine
ANG II
ANG I
[Na+] Na+
[Cl–]
ATPAdenosine
Renin-containing
cells
Vascularsmoothmuscle
↓ Reninsecretion
↓ GFR
Signal Transmission Mediator(s) EffectsA
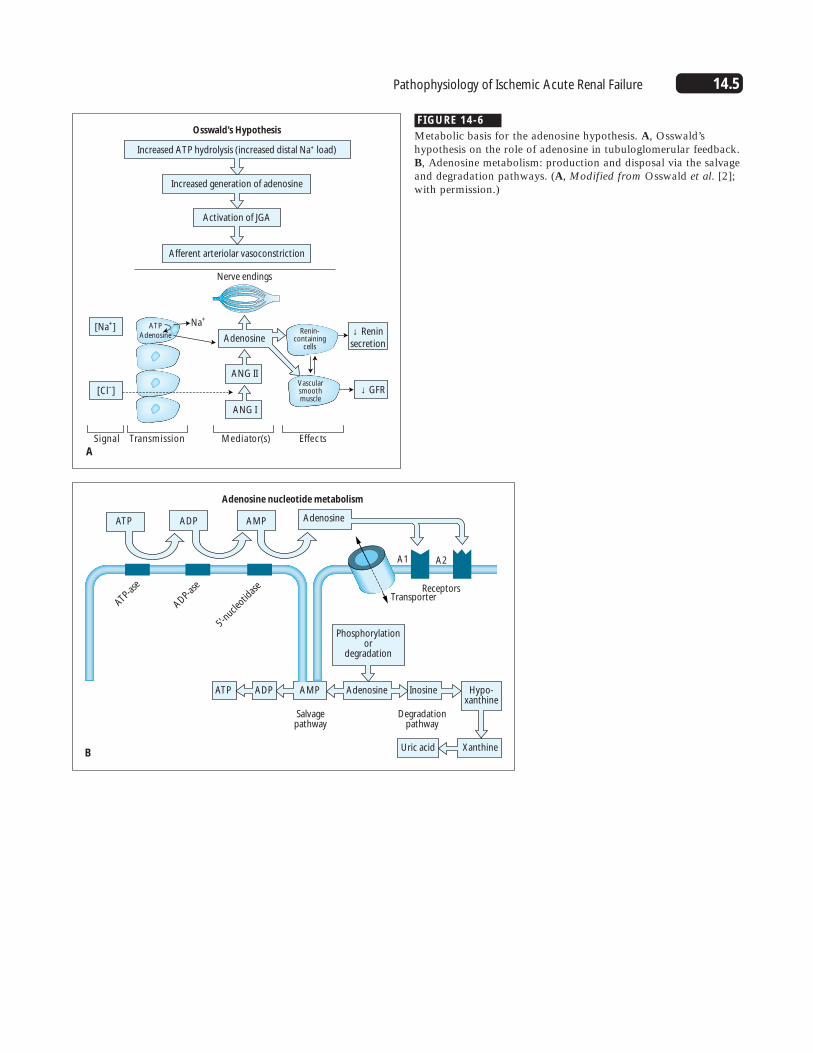
FIGURE 14-6
Metabolic basis for the adenosine hypothesis. A, Osswald’shypothesis on the role of adenosine in tubuloglomerular feedback.B, Adenosine metabolism: production and disposal via the salvageand degradation pathways. (A, Modified from Osswald et al. [2];with permission.)
ATP ADP AMP Adenosine
A1 A2
ATP-ase
ADP-ase
5'-nucle
otidas
e ReceptorsTransporter
Phosphorylationor
degradation
AdenosineADPATP AMP Inosine Hypo-xanthine
XanthineUric acid
Adenosine nucleotide metabolism
B
Degradationpathway
Salvagepathway
14.6 Acute Renal Failure
1
Volume collected, mL
0
0
5
10
Ade
nosi
ne,
nmol
es/m
L 15
20
5
10Inos
ine,
nmol
es/m
L
15
20
25
0
5
10
Hyp
oxan
thin
e,nm
oles
/mL
15
20
25
30
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
FIGURE 14-8
Endothelin (ET) is a potent renal vasoconstrictor. Endothelin (ET)is a 21 amino acid peptide of which three isoforms—ET-1, ET-2and ET-3—have been described, all of which have been shown tobe present in renal tissue. However, only the effects of ET-1 on thekidney have been clearly elucidated. ET-1 is the most potent vaso-constrictor known. Infusion of ET-1 into the kidney induces pro-found and long lasting vasoconstriction of the renal circulation. A,The appearance of the rat kidney during the infusion of ET-1 intothe inferior branch of the main renal artery. The lower pole of thekidney perfused by this vessel is profoundly vasoconstricted andhypoperfused. B, Schematic illustration of function in separatepopulations of glomeruli within the same kidney. The entire kidneyunderwent 25 minutes of ischemia 48 hours before micropuncture.Glomeruli I are nephrons not exposed to endothelin antibody;Glomeruli II are nephrons that received infusion with antibodythrough the inferior branch of the main renal artery. SNGFR—sin-gle nephron glomerular filtration rate; PFR—glomerular renal plas-ma flow rate. (From Kon et al. [4]; with permission.)
A
Glomerul ISNGFR: 17.4±1.7 nL/minPFR: 66.6±5.6 nL/min
Glomeruli IISNGFR: 27.0±3.1 nL/minPFR: 128.7±14.4 nL/min
Post Ischemia
Anti-endothelinB
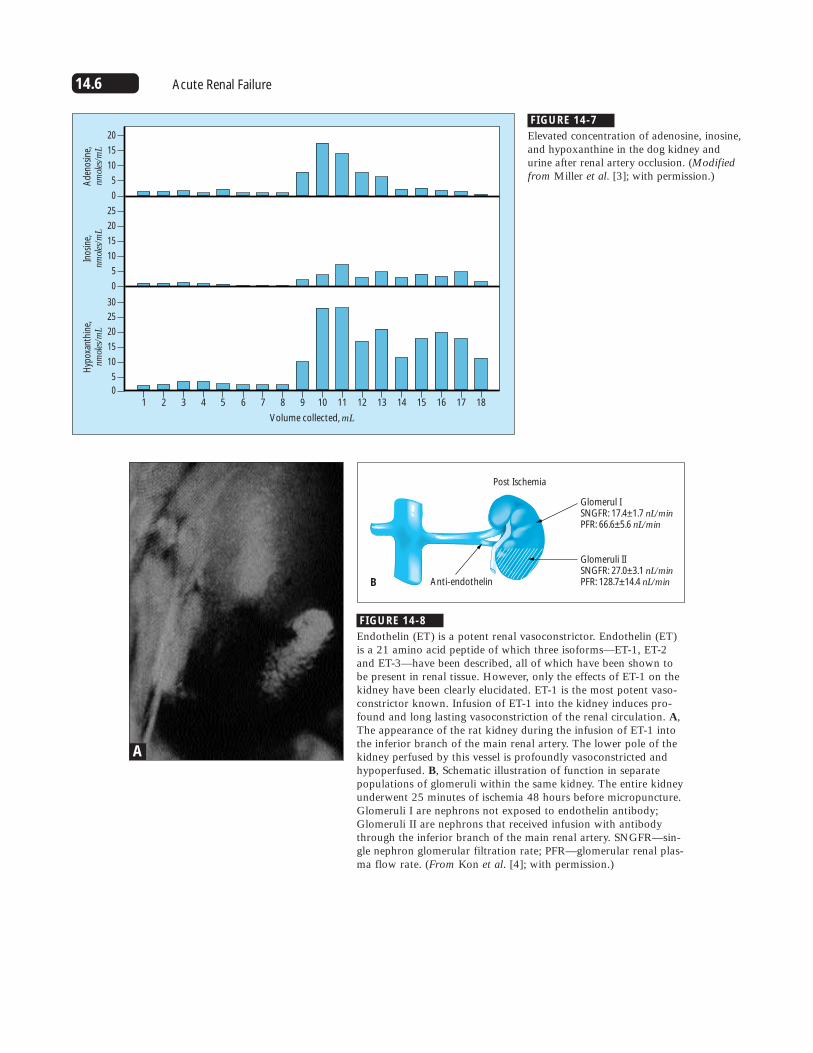
FIGURE 14-7
Elevated concentration of adenosine, inosine,and hypoxanthine in the dog kidney andurine after renal artery occlusion. (Modifiedfrom Miller et al. [3]; with permission.)
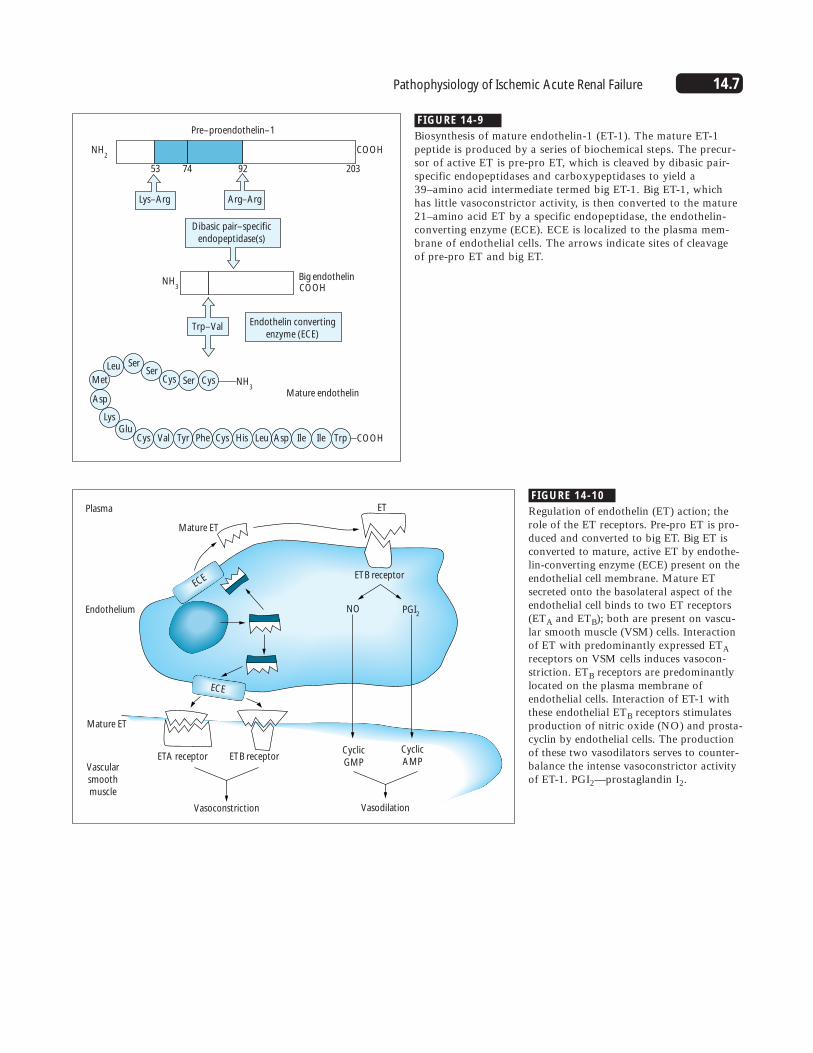
14.7Pathophysiology of Ischemic Acute Renal Failure
FIGURE 14-9
Biosynthesis of mature endothelin-1 (ET-1). The mature ET-1peptide is produced by a series of biochemical steps. The precur-sor of active ET is pre-pro ET, which is cleaved by dibasic pair-specific endopeptidases and carboxypeptidases to yield a39–amino acid intermediate termed big ET-1. Big ET-1, which has little vasoconstrictor activity, is then converted to the mature21–amino acid ET by a specific endopeptidase, the endothelin-converting enzyme (ECE). ECE is localized to the plasma mem-brane of endothelial cells. The arrows indicate sites of cleavage of pre-pro ET and big ET.
Pre–proendothelin–1
53 74 92 203
COOHNH2
Lys–Arg Arg–Arg
Dibasic pair–specificendopeptidase(s)
Big endothelinCOOH
NH3
NH3
Trp–Val Endothelin convertingenzyme (ECE)
CysCys
Lys
Cys Val Tyr Phe Ile Ile TrpHisGlu
Cys Leu Asp
SerSer
SerLeu
Met
Asp
COOH
Mature endothelin
Plasma ET
Endothelium
Mature ET
ETA receptor
ETB receptor
ETB receptorCyclic GMP
Cyclic AMP
Vasoconstriction Vasodilation
Vascular smooth muscle
Mature ET
ECE
ECE
NO PGI2
FIGURE 14-10
Regulation of endothelin (ET) action; therole of the ET receptors. Pre-pro ET is pro-duced and converted to big ET. Big ET isconverted to mature, active ET by endothe-lin-converting enzyme (ECE) present on theendothelial cell membrane. Mature ETsecreted onto the basolateral aspect of theendothelial cell binds to two ET receptors(ETA and ETB); both are present on vascu-lar smooth muscle (VSM) cells. Interactionof ET with predominantly expressed ETAreceptors on VSM cells induces vasocon-striction. ETB receptors are predominantlylocated on the plasma membrane ofendothelial cells. Interaction of ET-1 withthese endothelial ETB receptors stimulatesproduction of nitric oxide (NO) and prosta-cyclin by endothelial cells. The productionof these two vasodilators serves to counter-balance the intense vasoconstrictor activityof ET-1. PGI2—prostaglandin I2.
14.8 Acute Renal Failure
1 2
Nu
mb
er o
f rat
s
Basal
C
A
B
24hcontrol
BQ123(0.1mg/kg • min, for 3h)Ischemia
0
2
8
6
4
10
3 4 5 6 14
1 2
GFR
, mL/
h
Basal 24hcontrol
Ischemia
BQ123(0.1mg/kg • min, for 3h)
0
30
120
90
60
150
3 4 5 6 14
1 2
Pla
sma
K+, m
Eq/L
Basal 24hcontrol
Posttreatment days
IschemiaBQ123(0.1mg/kg • min, for 3h)
0
2
8
6
4
10
3 4 5 6 14
Vehicle
BQ123
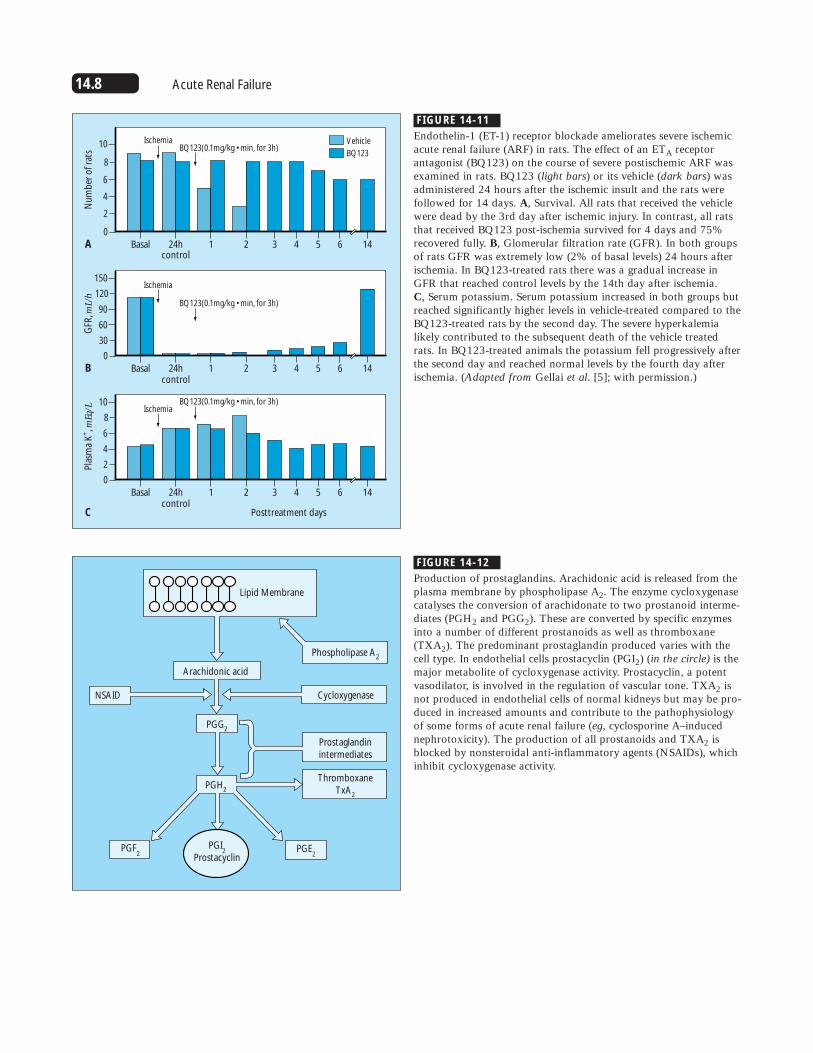
FIGURE 14-11
Endothelin-1 (ET-1) receptor blockade ameliorates severe ischemicacute renal failure (ARF) in rats. The effect of an ETA receptorantagonist (BQ123) on the course of severe postischemic ARF wasexamined in rats. BQ123 (light bars) or its vehicle (dark bars) wasadministered 24 hours after the ischemic insult and the rats werefollowed for 14 days. A, Survival. All rats that received the vehiclewere dead by the 3rd day after ischemic injury. In contrast, all ratsthat received BQ123 post-ischemia survived for 4 days and 75%recovered fully. B, Glomerular filtration rate (GFR). In both groupsof rats GFR was extremely low (2% of basal levels) 24 hours afterischemia. In BQ123-treated rats there was a gradual increase inGFR that reached control levels by the 14th day after ischemia. C, Serum potassium. Serum potassium increased in both groups butreached significantly higher levels in vehicle-treated compared to theBQ123-treated rats by the second day. The severe hyperkalemialikely contributed to the subsequent death of the vehicle treatedrats. In BQ123-treated animals the potassium fell progressively afterthe second day and reached normal levels by the fourth day afterischemia. (Adapted from Gellai et al. [5]; with permission.)
Lipid Membrane
Phospholipase A2
Arachidonic acid
PGG2
PGH2
PGF2
PGE2
CycloxygenaseNSAID
ThromboxaneTxA
2
Prostaglandinintermediates
PGI2
Prostacyclin
FIGURE 14-12
Production of prostaglandins. Arachidonic acid is released from theplasma membrane by phospholipase A2. The enzyme cycloxygenasecatalyses the conversion of arachidonate to two prostanoid interme-diates (PGH2 and PGG2). These are converted by specific enzymesinto a number of different prostanoids as well as thromboxane(TXA2). The predominant prostaglandin produced varies with thecell type. In endothelial cells prostacyclin (PGI2) (in the circle) is themajor metabolite of cycloxygenase activity. Prostacyclin, a potentvasodilator, is involved in the regulation of vascular tone. TXA2 isnot produced in endothelial cells of normal kidneys but may be pro-duced in increased amounts and contribute to the pathophysiologyof some forms of acute renal failure (eg, cyclosporine A–inducednephrotoxicity). The production of all prostanoids and TXA2 isblocked by nonsteroidal anti-inflammatory agents (NSAIDs), whichinhibit cycloxygenase activity.
14.9Pathophysiology of Ischemic Acute Renal Failure
Right renal artery
Left renal artery
Cyclosporine Ain circulation
Aorta
Intra–arterialinfusion of ET
Areceptor antagonist
Right kidney Left kidney
GFR and RPF:near normal
GFR and RPF:Reduced 20-25%
below normal
CSA
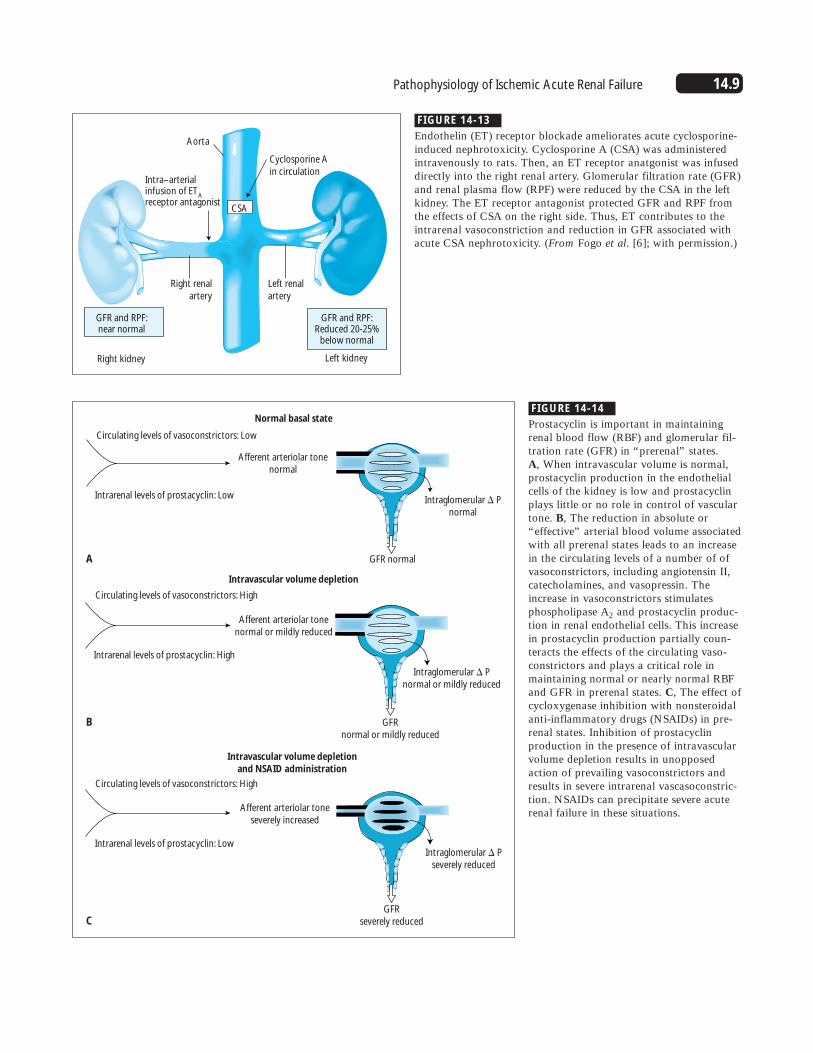
FIGURE 14-13
Endothelin (ET) receptor blockade ameliorates acute cyclosporine-induced nephrotoxicity. Cyclosporine A (CSA) was administeredintravenously to rats. Then, an ET receptor anatgonist was infuseddirectly into the right renal artery. Glomerular filtration rate (GFR)and renal plasma flow (RPF) were reduced by the CSA in the leftkidney. The ET receptor antagonist protected GFR and RPF fromthe effects of CSA on the right side. Thus, ET contributes to theintrarenal vasoconstriction and reduction in GFR associated withacute CSA nephrotoxicity. (From Fogo et al. [6]; with permission.)
GFR normal
Intraglomerular � Pnormal
Afferent arteriolar tonenormal
Circulating levels of vasoconstrictors: Low
Intrarenal levels of prostacyclin: Low
GFR normal or mildly reduced
Intraglomerular � Pnormal or mildly reduced
Afferent arteriolar tonenormal or mildly reduced
Circulating levels of vasoconstrictors: High
Intrarenal levels of prostacyclin: High
GFRseverely reduced
Intraglomerular � Pseverely reduced
Afferent arteriolar toneseverely increased
Circulating levels of vasoconstrictors: High
Intrarenal levels of prostacyclin: Low
Normal basal state
A
B
C
Intravascular volume depletion
Intravascular volume depletionand NSAID administration
FIGURE 14-14
Prostacyclin is important in maintainingrenal blood flow (RBF) and glomerular fil-tration rate (GFR) in “prerenal” states. A, When intravascular volume is normal,prostacyclin production in the endothelialcells of the kidney is low and prostacyclinplays little or no role in control of vasculartone. B, The reduction in absolute or“effective” arterial blood volume associatedwith all prerenal states leads to an increasein the circulating levels of a number of ofvasoconstrictors, including angiotensin II,catecholamines, and vasopressin. Theincrease in vasoconstrictors stimulatesphospholipase A2 and prostacyclin produc-tion in renal endothelial cells. This increasein prostacyclin production partially coun-teracts the effects of the circulating vaso-constrictors and plays a critical role inmaintaining normal or nearly normal RBFand GFR in prerenal states. C, The effect ofcycloxygenase inhibition with nonsteroidalanti-inflammatory drugs (NSAIDs) in pre-renal states. Inhibition of prostacyclin production in the presence of intravascularvolume depletion results in unopposedaction of prevailing vasoconstrictors andresults in severe intrarenal vascasoconstric-tion. NSAIDs can precipitate severe acuterenal failure in these situations.
14.10 Acute Renal Failure
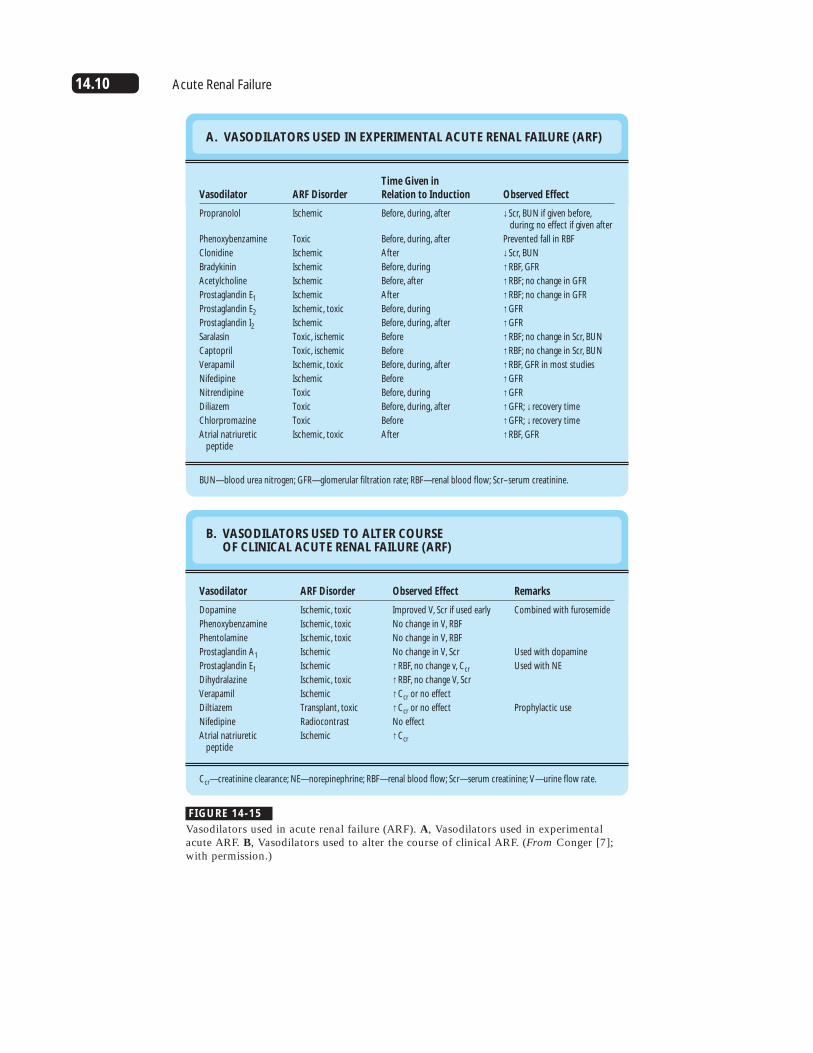
A. VASODILATORS USED IN EXPERIMENTAL ACUTE RENAL FAILURE (ARF)
Vasodilator
Propranolol
Phenoxybenzamine
Clonidine
Bradykinin
Acetylcholine
Prostaglandin E1
Prostaglandin E2
Prostaglandin I2Saralasin
Captopril
Verapamil
Nifedipine
Nitrendipine
Diliazem
Chlorpromazine
Atrial natriuretic peptide
ARF Disorder
Ischemic
Toxic
Ischemic
Ischemic
Ischemic
Ischemic
Ischemic, toxic
Ischemic
Toxic, ischemic
Toxic, ischemic
Ischemic, toxic
Ischemic
Toxic
Toxic
Toxic
Ischemic, toxic
Time Given in Relation to Induction
Before, during, after
Before, during, after
After
Before, during
Before, after
After
Before, during
Before, during, after
Before
Before
Before, during, after
Before
Before, during
Before, during, after
Before
After
Observed Effect
↓Scr, BUN if given before, during; no effect if given after
Prevented fall in RBF
↓Scr, BUN
↑RBF, GFR
↑RBF; no change in GFR
↑RBF; no change in GFR
↑GFR
↑GFR
↑RBF; no change in Scr, BUN
↑RBF; no change in Scr, BUN
↑RBF, GFR in most studies
↑GFR
↑GFR
↑GFR; ↓ recovery time
↑GFR; ↓ recovery time
↑RBF, GFR
BUN—blood urea nitrogen; GFR—glomerular filtration rate; RBF—renal blood flow; Scr–serum creatinine.
B. VASODILATORS USED TO ALTER COURSE OF CLINICAL ACUTE RENAL FAILURE (ARF)
Vasodilator
Dopamine
Phenoxybenzamine
Phentolamine
Prostaglandin A1
Prostaglandin E1
Dihydralazine
Verapamil
Diltiazem
Nifedipine
Atrial natriureticpeptide
ARF Disorder
Ischemic, toxic
Ischemic, toxic
Ischemic, toxic
Ischemic
Ischemic
Ischemic, toxic
Ischemic
Transplant, toxic
Radiocontrast
Ischemic
Observed Effect
Improved V, Scr if used early
No change in V, RBF
No change in V, RBF
No change in V, Scr
↑RBF, no change v, Ccr
↑RBF, no change V, Scr
↑Ccr or no effect
↑Ccr or no effect
No effect
↑Ccr
Remarks
Combined with furosemide
Used with dopamine
Used with NE
Prophylactic use
Ccr—creatinine clearance; NE—norepinephrine; RBF—renal blood flow; Scr—serum creatinine; V—urine flow rate.
FIGURE 14-15
Vasodilators used in acute renal failure (ARF). A, Vasodilators used in experimentalacute ARF. B, Vasodilators used to alter the course of clinical ARF. (From Conger [7];with permission.)
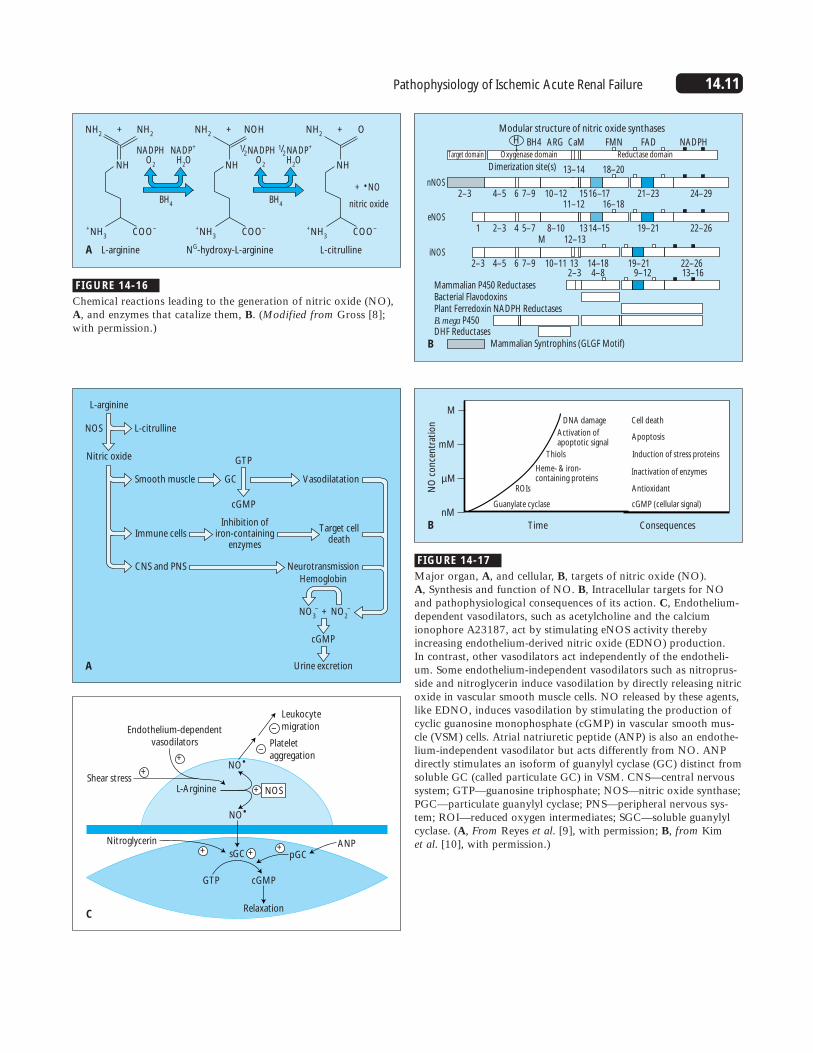
14.11Pathophysiology of Ischemic Acute Renal Failure
A
NH2
+NH3
COO–
NH2
O2
BH4
NADPH NADP+
H2O
+
NH
L-arginine
NH2
+NH3
COO–
NOH
O2
BH4
NADPH NADP+
H2O
+
NH
NG-hydroxy-L-arginine
NH2
+NH3
COO–
O+
NH
L-citrulline
nitric oxide
+ • NO
1/2
1/2
B
Modular structure of nitric oxide synthasesH BH4 ARG CaM FMN FAD NADPH
Target domain Oxygenase domain Reductase domain
13–14
2–3 7–96 10–12 1516–17 21–23 24–294–5
Dimerization site(s)
nNOS
18–20
11–12
1 5–74 8–10 1314–15 19–21 22–262–3
2–3 7–96 10–114–5
eNOS
16–18
12–13M
Mammalian P450 Reductases
13 14–18 19–21 22–26
Bacterial Flavodoxins
DHF ReductasesMammalian Syntrophins (GLGF Motif)
Plant Ferredoxin NADPH Reductases
4–82–3 9–12 13–16
iNOS
B. mega P450
FIGURE 14-16
Chemical reactions leading to the generation of nitric oxide (NO),A, and enzymes that catalize them, B. (Modified from Gross [8];with permission.)
L-arginine
Nitric oxide
Smooth muscle
Immune cellsTarget cell
death
VasodilatationGC
GTP
Inhibition ofiron-containing
enzymes
cGMP
NOS L-citrulline
CNS and PNS Neurotransmission
Hemoglobin
cGMP
Urine excretion
+NO3– NO
2–
A
Endothelium-dependentvasodilators
Leukocytemigration
Plateletaggregation
–
+
+
++ +
+
–
NO•
NO•
sGC
GTP
C
cGMP
Relaxation
pGCANPNitroglycerin
Shear stressL-Arginine NOS
Time Consequences
nM
µM
DNA damage
Thiols
ROIs
Guanylate cyclase
Activation ofapoptotic signal
Heme- & iron-containing proteins
Cell death
Antioxidant
cGMP (cellular signal)
Apoptosis
Inactivation of enzymes
mM
NO
co
nce
ntr
atio
n
M
Induction of stress proteins
B
FIGURE 14-17
Major organ, A, and cellular, B, targets of nitric oxide (NO). A, Synthesis and function of NO. B, Intracellular targets for NOand pathophysiological consequences of its action. C, Endothelium-dependent vasodilators, such as acetylcholine and the calciumionophore A23187, act by stimulating eNOS activity therebyincreasing endothelium-derived nitric oxide (EDNO) production. In contrast, other vasodilators act independently of the endotheli-um. Some endothelium-independent vasodilators such as nitroprus-side and nitroglycerin induce vasodilation by directly releasing nitricoxide in vascular smooth muscle cells. NO released by these agents,like EDNO, induces vasodilation by stimulating the production ofcyclic guanosine monophosphate (cGMP) in vascular smooth mus-cle (VSM) cells. Atrial natriuretic peptide (ANP) is also an endothe-lium-independent vasodilator but acts differently from NO. ANPdirectly stimulates an isoform of guanylyl cyclase (GC) distinct fromsoluble GC (called particulate GC) in VSM. CNS—central nervoussystem; GTP—guanosine triphosphate; NOS—nitric oxide synthase;PGC—particulate guanylyl cyclase; PNS—peripheral nervous sys-tem; ROI—reduced oxygen intermediates; SGC—soluble guanylylcyclase. (A, From Reyes et al. [9], with permission; B, from Kim et al. [10], with permission.)
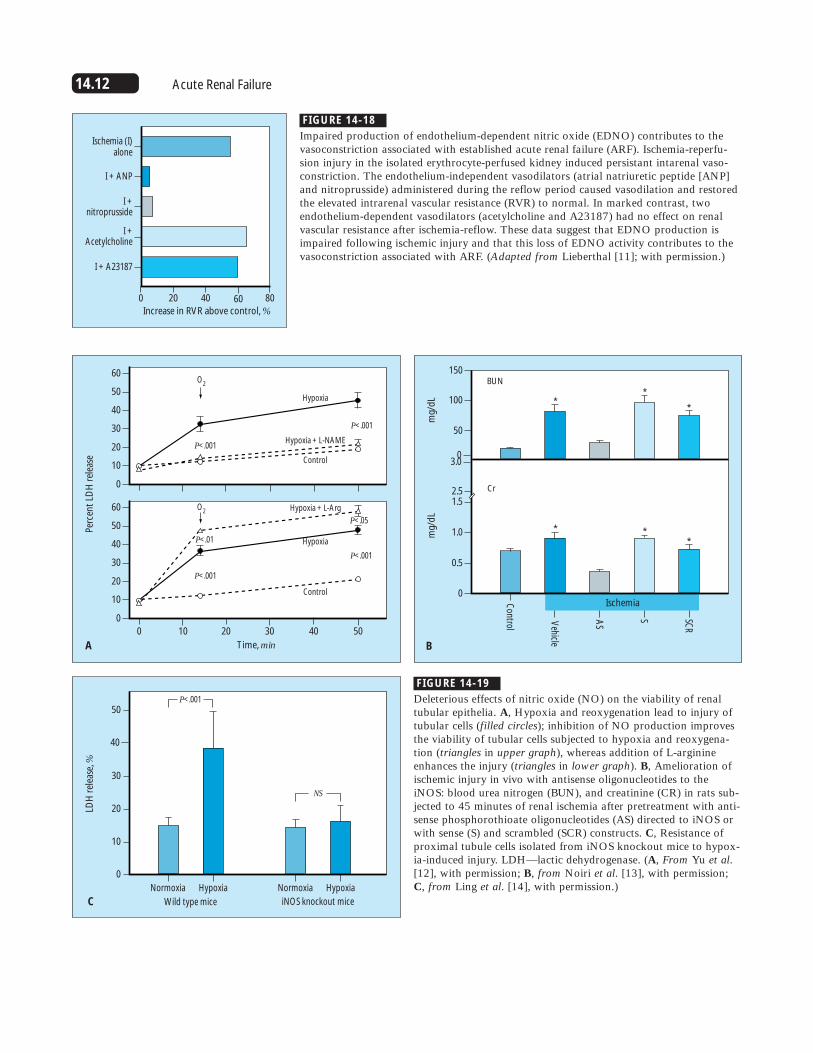
14.12 Acute Renal Failure
0 20 40 60Increase in RVR above control, %
I + A23187
80
I + Acetylcholine
I + nitroprusside
I + ANP
Ischemia (I)alone
FIGURE 14-18
Impaired production of endothelium-dependent nitric oxide (EDNO) contributes to thevasoconstriction associated with established acute renal failure (ARF). Ischemia-reperfu-sion injury in the isolated erythrocyte-perfused kidney induced persistant intarenal vaso-constriction. The endothelium-independent vasodilators (atrial natriuretic peptide [ANP]and nitroprusside) administered during the reflow period caused vasodilation and restoredthe elevated intrarenal vascular resistance (RVR) to normal. In marked contrast, twoendothelium-dependent vasodilators (acetylcholine and A23187) had no effect on renalvascular resistance after ischemia-reflow. These data suggest that EDNO production isimpaired following ischemic injury and that this loss of EDNO activity contributes to thevasoconstriction associated with ARF. (Adapted from Lieberthal [11]; with permission.)
P<.05
A
0
10
20
30
40
50
60
Per
cen
t LD
H r
elea
se
P<.001
P<.001
O2
Hypoxia
Hypoxia + L-NAME
Control
0
10
20
30
40
50
60
0 10 20 30
Time, min
40 50
P<.001
P<.001
P<.01
O2
Hypoxia
Hypoxia + L-Arg
Control
C
Normoxia Hypoxia
Wild type mice
40
50
LDH
rel
ease
, %
0
10
20
30
P<.001
Normoxia Hypoxia
iNOS knockout mice
NS
B
Control
SCR
Vehicle
AS
S
Ischemia
2.5 Cr
BUN
3.00
50
100
150
mg/
dL
mg/
dL
0
0.5
1.0
1.5
* **
**
*
FIGURE 14-19
Deleterious effects of nitric oxide (NO) on the viability of renaltubular epithelia. A, Hypoxia and reoxygenation lead to injury oftubular cells (filled circles); inhibition of NO production improvesthe viability of tubular cells subjected to hypoxia and reoxygena-tion (triangles in upper graph), whereas addition of L-arginineenhances the injury (triangles in lower graph). B, Amelioration ofischemic injury in vivo with antisense oligonucleotides to theiNOS: blood urea nitrogen (BUN), and creatinine (CR) in rats sub-jected to 45 minutes of renal ischemia after pretreatment with anti-sense phosphorothioate oligonucleotides (AS) directed to iNOS orwith sense (S) and scrambled (SCR) constructs. C, Resistance ofproximal tubule cells isolated from iNOS knockout mice to hypox-ia-induced injury. LDH—lactic dehydrogenase. (A, From Yu et al.[12], with permission; B, from Noiri et al. [13], with permission;C, from Ling et al. [14], with permission.)

14.13Pathophysiology of Ischemic Acute Renal Failure
A
0
Iothalamate
Iothalamate
20
No pretreatment
(n = 6)
Minutes MinutesPretreatment with
L-NAME (n = 6)
40
0
50
100
60
150
200
0
50
100
Med
ulla
Cor
tex
Perc
ent
of b
asel
ine
0
Iothalamate
20 40
0
50
100
60
0
50
100
B
Radiocontrast
Mild vasoconstriction
No loss of GFR
Severe vasoconstriction
Acute renal failure
Chronic renalinsufficiency
Compensatory increase inPGI2 and EDNO release
Increasedendothelin
Reduced or absent increase in PGI2 or EDNO
Normal kidneys
FIGURE 14-20
Proposed role of nitric oxide (NO) in radiocontrast-induced acuterenal failure (ARF). A, Administration of iothalamate, a radiocon-trast dye, to rats increases medullary blood flow. Inhibitors ofeither prostaglandin production (such as the NSAID,indomethacin) or inhibitors of NO synthesis (such as L-NAME)abolish the compensatory increase in medullary blood flow thatoccurs in response to radiocontrast administration. Thus, the stim-ulation of prostaglandin and NO production after radiocontrastadministration is important in maintaining medullary perfusionand oxygenation after administration of contrast agents. B,Radiocontrast stimulates the production of vasodilators (such asprostaglandin [PGI2] and endothelium-dependent nitric oxide[EDNO]) as well as endothelin and other vasoconstrictors within
the normal kidney. The vasodilators counteract the effects of thevasoconstrictors so that intrarenal vasoconstriction in response toradiocontrast is usually modest and is associated with little or noloss of renal function. However, in situations when there is pre-existing chronic renal insufficiency (CRF) the vasodilator responseto radiocontrast is impaired, whereas production of endothelin andother vasoconstrictors is not affected or even increased. As a result,radiocontrast administration causes profound intrarenal vasocon-striction and can cause ARF in patients with CRF. This hypothesiswould explain the predisposition of patients with chronic renaldysfunction, and especially diabetic nephropathy, to contrast-induced ARF. (A, Adapted from Agmon and Brezis [15], with per-mission; B, from Agmon et al. [16], with permission.)
FIGURE 14-21
Cellular calcium metabolism and potential targets of the elevatedcytosolic calcium. A, Pathways of calcium mobilization. B, Patho-physiologic mechanisms ignited by the elevation of cytosolic calci-um concentration. (A, Adapted from Goligorsky [17], with permis-sion; B, from Edelstein and Schrier [18], with permission.)
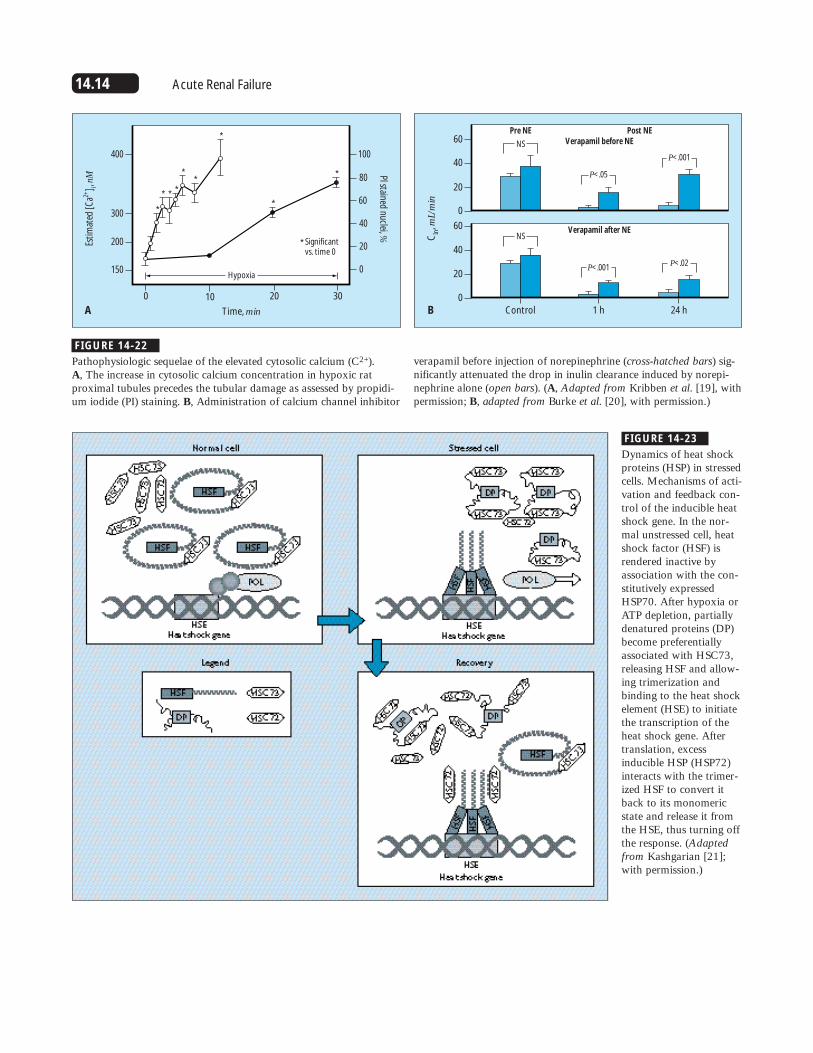
14.14 Acute Renal Failure
A
0
Significantvs. time 0
Hypoxia
3010
Time, min
150
200
20
300
400 100
0
20
40
60
80
Esti
mat
ed [C
a2+] i, n
M Pl stained nuclei, %
*
*
*
*
* *
*
*
*
*
B Control 1 h 24 h
0
20
40
60NS Verapamil before NE
Pre NE Post NE
P<.05
CIn
, mL/
min
P<.001
0
20
40
60NS
Verapamil after NE
P<.001P<.02
FIGURE 14-22
Pathophysiologic sequelae of the elevated cytosolic calcium (C2+). A, The increase in cytosolic calcium concentration in hypoxic ratproximal tubules precedes the tubular damage as assessed by propidi-um iodide (PI) staining. B, Administration of calcium channel inhibitor
verapamil before injection of norepinephrine (cross-hatched bars) sig-nificantly attenuated the drop in inulin clearance induced by norepi-nephrine alone (open bars). (A, Adapted from Kribben et al. [19], withpermission; B, adapted from Burke et al. [20], with permission.)
FIGURE 14-23
Dynamics of heat shockproteins (HSP) in stressedcells. Mechanisms of acti-vation and feedback con-trol of the inducible heatshock gene. In the nor-mal unstressed cell, heatshock factor (HSF) isrendered inactive byassociation with the con-stitutively expressedHSP70. After hypoxia orATP depletion, partiallydenatured proteins (DP)become preferentiallyassociated with HSC73,releasing HSF and allow-ing trimerization andbinding to the heat shockelement (HSE) to initiatethe transcription of theheat shock gene. Aftertranslation, excessinducible HSP (HSP72)interacts with the trimer-ized HSF to convert itback to its monomericstate and release it fromthe HSE, thus turning offthe response. (Adaptedfrom Kashgarian [21];with permission.)

14.15Pathophysiology of Ischemic Acute Renal Failure
O
Innermembrane
Superoxideanion
Mn-SOD(tetramer)
Hydrogen peroxide
Catalase/GPx complex?
Outermembrane Hydroperoxyl
radical
Free Radical Pathways in the Mitochondrion
(From glycolysis/TCA cycle)
Hydrogenperoxide
H2O
2
H2O
2
Hepatocyte(and other cells)
Endoplasmicreticulum
Mitochondrion
Manganesesuperoxidedismatase
(Mn-SOD) mRNA
CatalasemRNA
Catalasesubunit
Copper–zincsuperoxidedismutase
(Cu,Zn–SOD) mRNA
2O2
–
Cu,Zn–SOD(dimer)
Glutathioneperoxidase
(GPx) mRNA
GPxsubunit
Se GPx(tetramer)
Secretory vesicle
Heparinsulfate
proteoglycans
H2O+O2
+GSSGGlutathione
(dimer)
Glutathione(monomer)
Plasmamembranedamaged
(enlarged below)
Plasma EC–SOD
Proteinase?
Tissue EC–SOD
+2GSH
+O2
+2H+
Oxidative enzyme(eg, urate oxidase)
Perxisome reactions
Catalase(tetramer)
Hydrogenperoxide
RH2
+O
2
Lipid
Lipidperoxide
LOH+GSSG+
LOO
Freeradical
Lipidradical
Outsidecell
Lipidchain collpases
(now hydrophilic)
Phospholipidhydroperoxide
glutathioneperoxidase(PHGPx)
H2O
2
2H++
2GSH's + LOOH
Lipid peroxidation of plasma membrane
2H2O+O
2
Heme
HO2
HO2
2O2
–
O2
O2
–
+2H
+
e–
Matrix
+O
2
Vitamin E (a-Tocopherol–)inhibits lipid peroxidation
chain reaction
LOOH
LH
LH
LH
LHLH
LH
R
LOOH
LOO
H
LOH RH
L
L
O
e–
Insidecell
LOOH
Golgicomplex
Peroxisome
Extracellularsuperoxidedismutase(EC-SOD)
mRNAchrom 6
chrom 11chrom 21
chrom 3
Chromosome(chrom) 4
FIGURE 14-24
Cellular sources of reactive oxygen species (ROS) defense systems from free radicals. Superoxide and hydro-gen peroxide are produced during normal cellular metabolism. ROS are constantly being produced by thenormal cell during a number of physiologic reactions. Mitochondrial respiration is an important source ofsuperoxide production under normal conditions and can be increased during ischemia-reflow or gentamycin-induced renal injury. A number of enzymes generate superoxide and hydrogen peroxide during their catalyticcycling. These include cycloxygenases and lipoxygenes that catalyze prostanoid and leukotriene synthesis.Some cells (such as leukocytes, endothelial cells, and vascular smooth muscle cells) have NADH/ or NADPHoxidase enzymes in the plasma membrane that are capable of generating superoxide. Xanthine oxidase, whichconverts hypoxathine to xanthine, has been implicated as an important source of ROS after ischemia-reperfu-sion injury. Cytochrome p450, which is bound to the membrane of the endoplasmic reticulum, can beincreased by the presence of high concentrations of metabolites that are oxidized by this cytochrome or byinjurious events that uncouple the activity of the p450. Finally, the oxidation of small molecules including freeheme, thiols, hydroquinines, catecholamines, flavins, and tetrahydropterins, also contribute to intracellularsuperoxide production. (Adapted from [22]; with permission.)
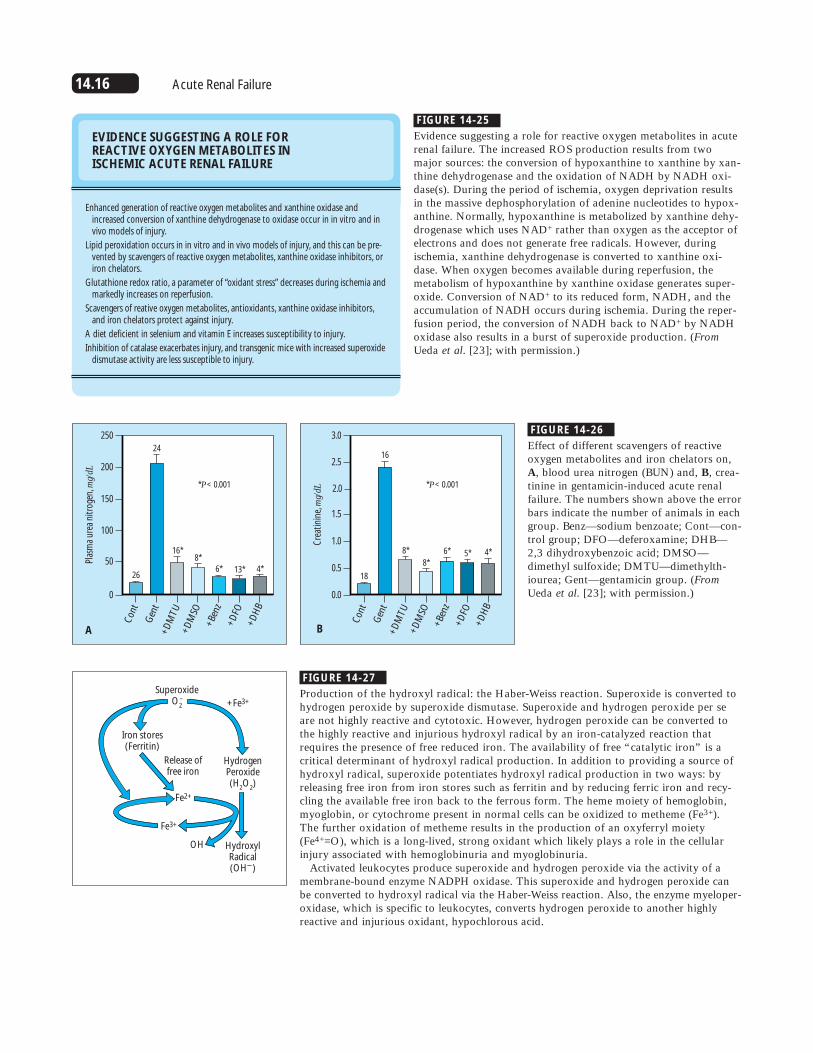
14.16 Acute Renal Failure
FIGURE 14-25
Evidence suggesting a role for reactive oxygen metabolites in acuterenal failure. The increased ROS production results from twomajor sources: the conversion of hypoxanthine to xanthine by xan-thine dehydrogenase and the oxidation of NADH by NADH oxi-dase(s). During the period of ischemia, oxygen deprivation resultsin the massive dephosphorylation of adenine nucleotides to hypox-anthine. Normally, hypoxanthine is metabolized by xanthine dehy-drogenase which uses NAD+ rather than oxygen as the acceptor ofelectrons and does not generate free radicals. However, duringischemia, xanthine dehydrogenase is converted to xanthine oxi-dase. When oxygen becomes available during reperfusion, themetabolism of hypoxanthine by xanthine oxidase generates super-oxide. Conversion of NAD+ to its reduced form, NADH, and theaccumulation of NADH occurs during ischemia. During the reper-fusion period, the conversion of NADH back to NAD+ by NADHoxidase also results in a burst of superoxide production. (FromUeda et al. [23]; with permission.)
EVIDENCE SUGGESTING A ROLE FOR REACTIVE OXYGEN METABOLITES IN ISCHEMIC ACUTE RENAL FAILURE
Enhanced generation of reactive oxygen metabolites and xanthine oxidase andincreased conversion of xanthine dehydrogenase to oxidase occur in in vitro and invivo models of injury.
Lipid peroxidation occurs in in vitro and in vivo models of injury, and this can be pre-vented by scavengers of reactive oxygen metabolites, xanthine oxidase inhibitors, oriron chelators.
Glutathione redox ratio, a parameter of “oxidant stress” decreases during ischemia andmarkedly increases on reperfusion.
Scavengers of reative oxygen metabolites, antioxidants, xanthine oxidase inhibitors,and iron chelators protect against injury.
A diet deficient in selenium and vitamin E increases susceptibility to injury.
Inhibition of catalase exacerbates injury, and transgenic mice with increased superoxidedismutase activity are less susceptible to injury.
Con
t
Gen
t
0
24
*P < 0.001
26
50Plas
ma
urea
nit
roge
n, m
g/dL
100
150
200
250
+D
MTU
16*
+D
MSO
+Be
nz
6*
8*
+D
HB
4*
+D
FO
13*
A B
Con
t
Gen
t
0.0
16
*P < 0.001
180.5
Cre
atin
ine,
mg/
dL
1.0
1.5
2.5
2.0
3.0
+D
MTU
8*
+D
MSO
+Be
nz
6*
8*
+D
HB
4*
+D
FO
5*
FIGURE 14-26
Effect of different scavengers of reactiveoxygen metabolites and iron chelators on,A, blood urea nitrogen (BUN) and, B, crea-tinine in gentamicin-induced acute renalfailure. The numbers shown above the errorbars indicate the number of animals in eachgroup. Benz—sodium benzoate; Cont—con-trol group; DFO—deferoxamine; DHB—2,3 dihydroxybenzoic acid; DMSO—dimethyl sulfoxide; DMTU—dimethylth-iourea; Gent—gentamicin group. (FromUeda et al. [23]; with permission.)
SuperoxideO
2
HydrogenPeroxide(H
2O
2)
HydroxylRadical(OH–)
Iron stores(Ferritin)
Release offree iron
OH
Fe2+
+Fe3+–
Fe3+
FIGURE 14-27
Production of the hydroxyl radical: the Haber-Weiss reaction. Superoxide is converted tohydrogen peroxide by superoxide dismutase. Superoxide and hydrogen peroxide per seare not highly reactive and cytotoxic. However, hydrogen peroxide can be converted tothe highly reactive and injurious hydroxyl radical by an iron-catalyzed reaction thatrequires the presence of free reduced iron. The availability of free “catalytic iron” is acritical determinant of hydroxyl radical production. In addition to providing a source ofhydroxyl radical, superoxide potentiates hydroxyl radical production in two ways: byreleasing free iron from iron stores such as ferritin and by reducing ferric iron and recy-cling the available free iron back to the ferrous form. The heme moiety of hemoglobin,myoglobin, or cytochrome present in normal cells can be oxidized to metheme (Fe3+).The further oxidation of metheme results in the production of an oxyferryl moiety(Fe4+=O), which is a long-lived, strong oxidant which likely plays a role in the cellularinjury associated with hemoglobinuria and myoglobinuria.
Activated leukocytes produce superoxide and hydrogen peroxide via the activity of amembrane-bound enzyme NADPH oxidase. This superoxide and hydrogen peroxide canbe converted to hydroxyl radical via the Haber-Weiss reaction. Also, the enzyme myeloper-oxidase, which is specific to leukocytes, converts hydrogen peroxide to another highlyreactive and injurious oxidant, hypochlorous acid.
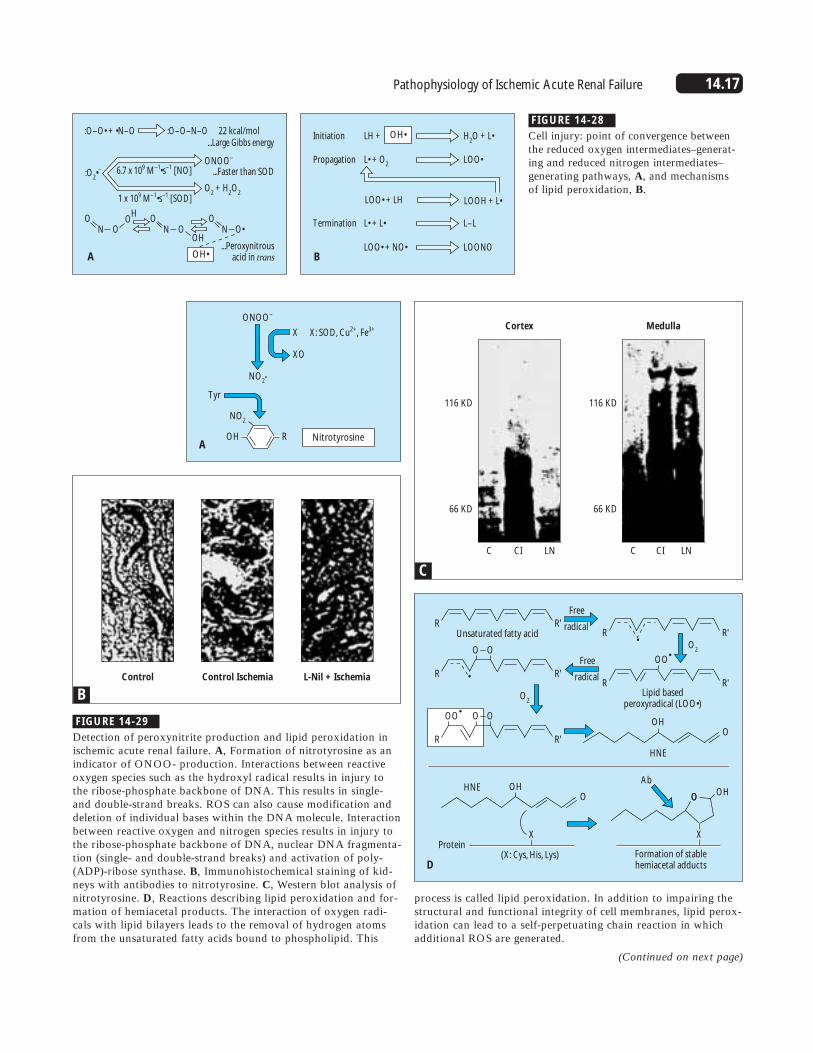
14.17Pathophysiology of Ischemic Acute Renal Failure
:O–O• + •N–O
:O2•–
ON O
ON O
ON O•
OH
OH
:O–O–N–O
ONOO–
O2 + H
2O
2
6.7 x 109 M–1•s–1 [NO]
1 x 109 M–1•s–1 [SOD]
22 kcal/mol...Large Gibbs energy
...Faster than SOD
...Peroxynitrous acid in transA OH•
Initiation LH + H2O + L•
B
OH•
Propagation L• + O2
LOO• + LH LOOH + L•
LOO•
Termination L• + L• L–L
LOO• + NO• LOONO
FIGURE 14-28
Cell injury: point of convergence betweenthe reduced oxygen intermediates–generat-ing and reduced nitrogen intermediates–generating pathways, A, and mechanisms of lipid peroxidation, B.
ONOO–
Tyr
X X: SOD, Cu2+, Fe3+
XO
NO2
•
NO2
ROH NitrotyrosineA
B
Control Control Ischemia L-Nil + Ischemia
FIGURE 14-29
Detection of peroxynitrite production and lipid peroxidation inischemic acute renal failure. A, Formation of nitrotyrosine as anindicator of ONOO- production. Interactions between reactiveoxygen species such as the hydroxyl radical results in injury tothe ribose-phosphate backbone of DNA. This results in single-and double-strand breaks. ROS can also cause modification anddeletion of individual bases within the DNA molecule. Interactionbetween reactive oxygen and nitrogen species results in injury tothe ribose-phosphate backbone of DNA, nuclear DNA fragmenta-tion (single- and double-strand breaks) and activation of poly-(ADP)-ribose synthase. B, Immunohistochemical staining of kid-neys with antibodies to nitrotyrosine. C, Western blot analysis ofnitrotyrosine. D, Reactions describing lipid peroxidation and for-mation of hemiacetal products. The interaction of oxygen radi-cals with lipid bilayers leads to the removal of hydrogen atomsfrom the unsaturated fatty acids bound to phospholipid. This
(Continued on next page)
C
116 KD
66 KD
116 KD
66 KD
Cortex Medulla
C CI LN C CI LN
OH
HNE
HNE
O
(X: Cys, His, Lys)Protein
Ab
O2
R R'
OO•
O2
O O
OO
X
Formation of stablehemiacetal adducts
OH
R R'
OO•
R R'
Free
radical
Lipid basedperoxyradical (LOO•)
Unsaturated fatty acid
O O
OHO
•
R R'R R'
Free
radical
•
X
D
process is called lipid peroxidation. In addition to impairing thestructural and functional integrity of cell membranes, lipid perox-idation can lead to a self-perpetuating chain reaction in whichadditional ROS are generated.

Inactive leukocyte
Activated leukocyte
Selection–mediatedrolling of leukocytes
Firm adhesion of leukocytes
(integrin–mediated)
Diapedesis
Release ofoxidantsproteaseselastases
Tissue injury
A
β2 integrins (LFA
1 or Mac
1)
Selections
ICAMLigand for leukocyte selections
Endothelial adhesion molecules
Leukocyte adhesion moleculesFIGURE 14-30
Role of adhesion molecules in mediatingleukocyte attachment to endothelium. A, The normal inflammatory response ismediated by the release of cytokines thatinduce leukocyte chemotaxis and activation.The initial interaction of leukocytes withendothelium is mediated by the selectins andtheir ligands both of which are present onleukocytes and endothelial cells,
Leukocytes in Acute Renal Failure
14.18 Acute Renal Failure
F
Cortex Medulla
C CI LN C CI LNFIGURE 14-29 (Continued)
E, Immunohistochemical staining of kidneys with antibodies toHNE–modified proteins. F, Western blot analysis of HNE expres-sion. C—control; CI—central ischemia; LN—ischemia with L-Nilpretreatment (Courtesy of E. Noiri, MD.)
E
Control Control Ischemia L-Nil + Ischemia
(Continued on next page)
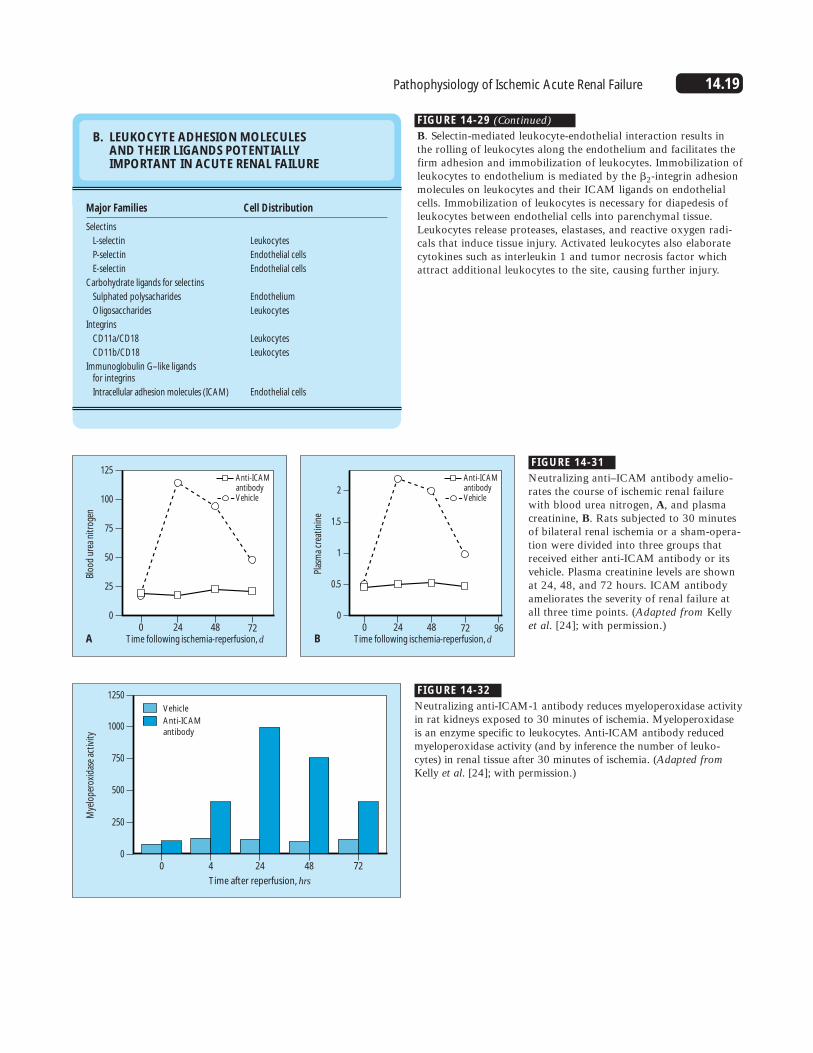
14.19Pathophysiology of Ischemic Acute Renal Failure
B. LEUKOCYTE ADHESION MOLECULES AND THEIR LIGANDS POTENTIALLY IMPORTANT IN ACUTE RENAL FAILURE
Major Families
Selectins
L-selectin
P-selectin
E-selectin
Carbohydrate ligands for selectins
Sulphated polysacharides
Oligosaccharides
Integrins
CD11a/CD18
CD11b/CD18
Immunoglobulin G–like ligands for integrins
Intracellular adhesion molecules (ICAM)
Cell Distribution
Leukocytes
Endothelial cells
Endothelial cells
Endothelium
Leukocytes
Leukocytes
Leukocytes
Endothelial cells
FIGURE 14-29 (Continued)
B. Selectin-mediated leukocyte-endothelial interaction results inthe rolling of leukocytes along the endothelium and facilitates thefirm adhesion and immobilization of leukocytes. Immobilization ofleukocytes to endothelium is mediated by the �2-integrin adhesionmolecules on leukocytes and their ICAM ligands on endothelialcells. Immobilization of leukocytes is necessary for diapedesis ofleukocytes between endothelial cells into parenchymal tissue.Leukocytes release proteases, elastases, and reactive oxygen radi-cals that induce tissue injury. Activated leukocytes also elaboratecytokines such as interleukin 1 and tumor necrosis factor whichattract additional leukocytes to the site, causing further injury.
0 24 48 72Time following ischemia-reperfusion, d
0
25
50
Anti-ICAM antibodyVehicle
75
100
125
Bloo
d ur
ea n
itro
gen
A0 24 48 72 96
Time following ischemia-reperfusion, d
0
0.5
1
Anti-ICAM antibodyVehicle
1.5
2
Plas
ma
crea
tini
ne
B
FIGURE 14-31
Neutralizing anti–ICAM antibody amelio-rates the course of ischemic renal failurewith blood urea nitrogen, A, and plasmacreatinine, B. Rats subjected to 30 minutesof bilateral renal ischemia or a sham-opera-tion were divided into three groups thatreceived either anti-ICAM antibody or itsvehicle. Plasma creatinine levels are shownat 24, 48, and 72 hours. ICAM antibodyameliorates the severity of renal failure atall three time points. (Adapted from Kellyet al. [24]; with permission.)
0 244 48 72
Time after reperfusion, hrs
0
250
500
750
1000
1250
Mye
lope
roxi
dase
act
ivit
y
Vehicle
Anti-ICAM
antibody
FIGURE 14-32
Neutralizing anti-ICAM-1 antibody reduces myeloperoxidase activityin rat kidneys exposed to 30 minutes of ischemia. Myeloperoxidaseis an enzyme specific to leukocytes. Anti-ICAM antibody reducedmyeloperoxidase activity (and by inference the number of leuko-cytes) in renal tissue after 30 minutes of ischemia. (Adapted fromKelly et al. [24]; with permission.)
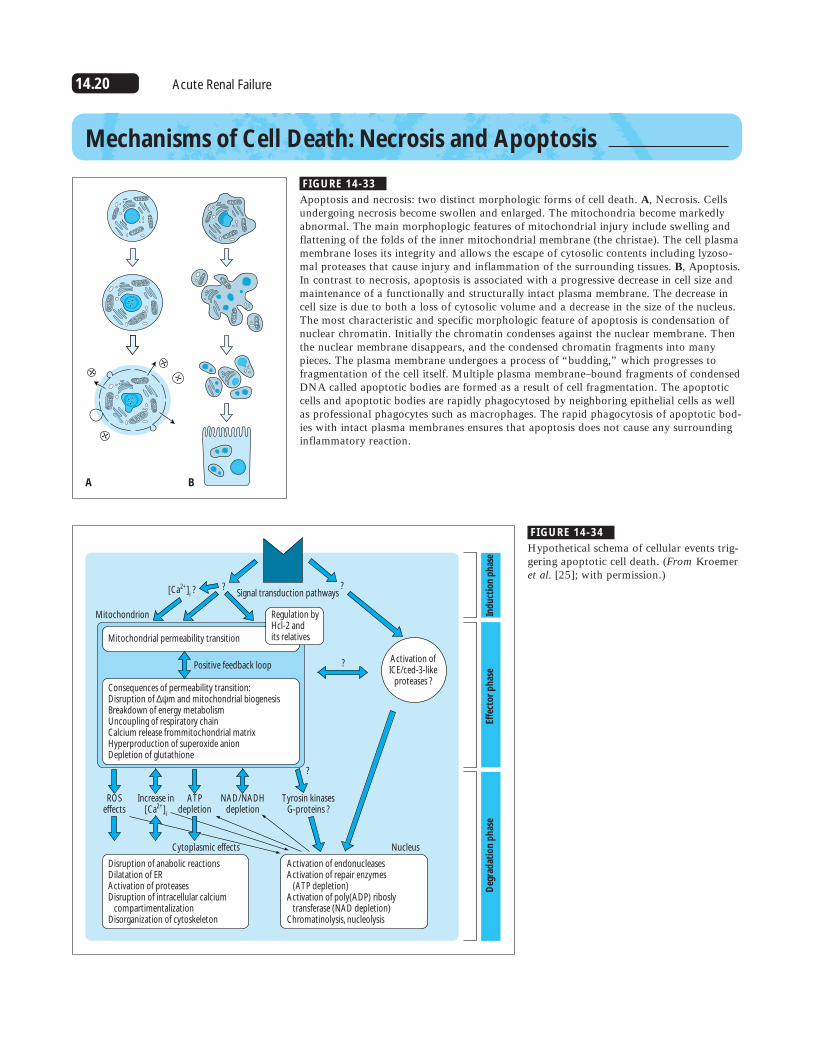
14.20 Acute Renal Failure
Consequences of permeability transition:Disruption of ∆ψm and mitochondrial biogenesisBreakdown of energy metabolismUncoupling of respiratory chainCalcium release frommitochondrial matrixHyperproduction of superoxide anionDepletion of glutathione
Disruption of anabolic reactionsDilatation of ERActivation of proteasesDisruption of intracellular calcium compartimentalizationDisorganization of cytoskeleton
Activation of endonucleasesActivation of repair enzymes (ATP depletion)Activation of poly(ADP) ribosly transferase (NAD depletion)Chromatinolysis, nucleolysis
ROSeffects
Mitochondrial permeability transition
Positive feedback loop
Mitochondrion
Activation ofICE/ced-3-like
proteases ?
Signal transduction pathways[Ca2+]i ?
ATPdepletion
NucleusCytoplasmic effects
?
?
??
Increase in[Ca2+]
i
NAD/NADHdepletion
Tyrosin kinasesG-proteins ?
Regulation byHcl-2 andits relatives
Ind
uct
ion
ph
ase
Effe
cto
r p
has
eD
egra
dat
ion
ph
ase
FIGURE 14-34
Hypothetical schema of cellular events trig-gering apoptotic cell death. (From Kroemeret al. [25]; with permission.)
Mechanisms of Cell Death: Necrosis and Apoptosis
A B
FIGURE 14-33
Apoptosis and necrosis: two distinct morphologic forms of cell death. A, Necrosis. Cellsundergoing necrosis become swollen and enlarged. The mitochondria become markedlyabnormal. The main morphoplogic features of mitochondrial injury include swelling andflattening of the folds of the inner mitochondrial membrane (the christae). The cell plasmamembrane loses its integrity and allows the escape of cytosolic contents including lyzoso-mal proteases that cause injury and inflammation of the surrounding tissues. B, Apoptosis.In contrast to necrosis, apoptosis is associated with a progressive decrease in cell size andmaintenance of a functionally and structurally intact plasma membrane. The decrease incell size is due to both a loss of cytosolic volume and a decrease in the size of the nucleus.The most characteristic and specific morphologic feature of apoptosis is condensation ofnuclear chromatin. Initially the chromatin condenses against the nuclear membrane. Thenthe nuclear membrane disappears, and the condensed chromatin fragments into manypieces. The plasma membrane undergoes a process of “budding,” which progresses tofragmentation of the cell itself. Multiple plasma membrane–bound fragments of condensedDNA called apoptotic bodies are formed as a result of cell fragmentation. The apoptoticcells and apoptotic bodies are rapidly phagocytosed by neighboring epithelial cells as wellas professional phagocytes such as macrophages. The rapid phagocytosis of apoptotic bod-ies with intact plasma membranes ensures that apoptosis does not cause any surroundinginflammatory reaction.

14.21Pathophysiology of Ischemic Acute Renal Failure
FIGURE 14-35
Phagocytosis of an apoptotic body by a renal tubular epithelial cell.Epithelial cells dying by apoptosis are not only phagocytosed bymacrophages and leukocytes but by neighbouring epithelial cells aswell. This electron micrograph shows a normal-looking epithelial cellcontaining an apoptotic body within a lyzosome. The nucleus of anepithelial cell that has ingested the apoptotic body is normal (whitearrow). The wall of the lyzosome containing the apoptotic body (blackarrow) is clearly visible. The apoptotic body consists of condensedchromatin surrounded by plasma membrane (black arrowheads).
DNA fragmentation
Apoptosis Necrosis
800 bp 600 bp 400 bp 200 bp
Nucleosome~200 bp
Internucleosome"Linker" regions
Lossof
histones
Necrotic"smear" pattern
Apoptic"ladder"pattern
DNA electrophoresis
FIGURE 14-36
DNA fragmentation in apoptosis vs necrosis. DNA is made up of nucleosomal units. Eachnucleosome of DNA is about 200 base pairs in size and is surrounded by histones. Betweennucleosomes are small stretches of DNA that are not surrounded by histones and are calledlinker regions. During apoptosis, early activation of endonuclease(s) causes double-strandbreaks in DNA between nucleosomes. No fragmentation occurs in nucleosomes because theDNA is “protected” by the histones. Because of the size of nucleosomes, the DNA is frag-mented during apoptosis into multiples of 200 base pair pieces (eg, 200, 400, 600, 800).When the DNA of apoptotic cells is electrophoresed, a characteristic ladder pattern is found.
In contrast, necrosis is associated with the early release of lyzosomal proteases, whichcause proteolysis of nuclear histones, leaving “naked” stretches of DNA not protected byhistones. Activation of endonucleases during necrosis therefore cause DNA cleavage atmultiple sites into double- and single-stranded DNA fragments of varying size.Electrophoresis of DNA from necrotic cells results in a smear pattern.

14.22 Acute Renal Failure
Execution phase
Caspase activation
Proteolysis of multipleintracellular substrates
Anti-apoptic factors Pro-apoptic factors
BclXLBcl–2 Bax
BAD
Crmap35
Apoptotic Trigger
? Point of no return?
Apoptosis
Commitment phase
FIGURE 14-38
Apoptosis is mediated by a highly coordinated and genetically pro-grammed pathway. The response to an apoptotic stimulus can bedivided into a commitment and execution phases. During the com-mitment phase the balance between a number of proapoptotic andantiapoptotic mechanisms determine whether the cell survives ordies by apoptosis. The BCL-2 family of proteins consists of at least12 isoforms, which play important roles in this commitment phase.Some of the BCL-2 family of proteins (eg, BCL-2 and BCL-XL) pro-tect cells from apoptosis whereas other members of the same family(eg, BAD and Bax) serve proapoptotic functions. Apoptosis is exe-cuted by a final common pathway mediated by a class of cysteineproteases-caspases. Caspases are proteolytic enzymes present in cellsin an inactive form. Once cells are commited to undergo apoptosis,these caspases are activated. Some caspases activate other caspasesin a hierarchical fashion resulting in a cascade of caspase activation.Eventually, caspases that target specific substrates within the cell areactivated. Some substrates for caspases that have been identifiedinclude nuclear membrane components (such as lamin), cytoskeletalelements (such as actin and fodrin) and DNA repair enzymes andtranscription elements. The proteolysis of this diverse array of sub-strates in the cell occurs in a predestined fashion and is responsiblefor the characteristic morphologic features of apoptosis.
POTENTIAL CAUSES OF APOPTOSIS IN ACUTE RENAL FAILURE
Loss of survival factors
Deficiency of renal growth factors (eg, IGF-1, EGF, HGF)
Loss of cell-cell and cell-matrix interactions
Receptor-mediated activators of apoptosis
Tumor necrosis factor
Fas/Fas ligand
Cytotoxic events
Ischemia; hypoxia; anoxia
Oxidant injury
Nitric oxide
Cisplati
FIGURE 14-37
Potential causes of apoptosis in acute renal failure (ARF). Thesame cytotoxic stimuli that induce necrosis cause apoptosis. Themechanism of cell death induced by a specific injury depends inlarge part on the severity of the injury. Because most cells requireconstant external signals, called survival signals, to remain viable,the loss of these survival signals can trigger apoptosis. In ARF, adeficiency of growth factors and loss of cell-substrate adhesion arepotential causes of apoptosis. The death pathways induced byengagement of tumour necrosis factor (TNF) with the TNF recep-tor or Fas with its receptor (Fas ligand) are well known causes ofapoptosis in immune cells. TNF and Fas can also induce apoptosisin epithelial cells and may contribute to cell death in ARF.

14.23Pathophysiology of Ischemic Acute Renal Failure
PMNinfiltration
Hemodynamiccompromise
Loss of tubularintegrity and
function
Stress
Recovery
RBF
GFR andmaintenance
phaseRestoration
of renalhemodynamics
Reparation oftubular integrity
and function
ObstructionBack leak
Kf
• ETR antagonists• Ca channel inhibitors
• Dopamine• ANP• IGF-1
• ICAM-1 antibody• RGD
• Mannitol• Lasix• ANP• RGD
• IGF-1l• T4• HGF
• Avoidance and discontinuation of nephrotoxins• Survival factors (HGF, IGF-1)• ATP-Mg• T4• NOS inhibitors
• Restoration of fluid and electrolyte balance• ETR antagonists• Ca channel inhibitors• ATP-Mg
FIGURE 14-39
Therapeutic approaches, both experimentaland in clinical use, to prevent and manageacute renal failure based on its pathogeneticmechanisms. ETR—ET receptor; GFR—glomerular filtration rate; HGF—hepatocytegrowth factor 1; IGF-1—insulin-like growthfactor 1; Kf—glomerular ultrafiltration coef-ficient; NOS—nitric oxide synthase; PMN—polymorphonuclear leukocytes; RBF—renalblood flow; T4—thyroxine.
References
1. Goligorsky M, Iijima K, Krivenko Y, et al.: Role of mesangial cells inmacula densa-to-afferent arteriole information transfer. Clin ExpPharm Physiol 1997, 24:527–531.
2. Osswald H, Hermes H, Nabakowski G: Role of adenosine in signaltransmission of TGF. Kidney Int 1982, 22(Suppl. 12):S136–S142.
3. Miller W, Thomas R, Berne R, Rubio R: Adenosine production in theischemic kidney. Circ Res 1978, 43(3):390–397.
4. Kon V, et al.: Glomerular actions of endothelin in vivo. J Clin Invest1989, 83:1762–1767.
5. Gellai M, Jugus M, Fletcher T, et al.: Reversal of postischemic acuterenal failure with a selective endothelin A receptor antagonist in therat. J Clin Invest 1994, 93:900–906.
6. Fogo, et al.: Endothelin receptor antagonism is protective in vivo inacute cyclosporine toxicity. Kidney Int 1992, 42:770–774.
7. Conger J: NO in acute renal failure. In: Nitric Oxide and the Kidney.Edited by Goligorsky M, Gross S. New York:Chapman and Hall,1997.
8. Gross S: Nitric oxide synthases and their cofactors. In: Nitric Oxideand the Kidney. Edited by Goligorsky M, Gross S. NewYork:Chapman and Hall, 1997.
9. Reyes A, Karl I, Klahr S: Role of arginine in health and in renal dis-ease. Am J Physiol 1994, 267:F331–F346.
10. Kim Y-M, Tseng E, Billiar TR: Role of NO and nitrogen intermediatesin regulation of cell functions. In: Nitric Oxide and the Kidney. Editedby Goligorsky M, Gross S. New York:Chapman and Hall, 1997.
11. Lieberthal W:Renal ischemia and reperfusion impair endothelium-dependent vascular relaxation. Am J Physiol 1989, 256:F894–F900.
12. Yu L, Gengaro P, Niederberger M, et al.: Nitric oxide: a mediator inrat tubular hypoxia/reoxygenation injury. Proc Natl Acad Sci USA1994, 91:1691–1695.
13. Noiri E, Peresleni T, Miller F, Goligorsky MS: In vivo targeting ofiNOS with oligodeoxynucleotides protects rat kidney againstischemia. J Clin Invest 1996, 97:2377–2383.
14. Ling H, Gengaro P, Edelstein C, et al.: Injurious isoform of NOS inmouse proximal tubular injury. Kidney Int, 1998, 53:1642
15. Agmon Y, et al.: Nitric oxide and prostanoids protect the renal outermedulla from radiocontrast toxicity in the rat. J Clin Invest 1994,94:1069–1075.
16. Agmon Y, Brezis M: NO and the medullary circulation. In: NitricOxide and the Kidney. Edited by Goligorsky M, Gross S. NewYork:Chapman and Hall, 1997.
17. Goligorsky MS: Cell biology of signal transduction. In: Hormones,autacoids, and the kidney. Edited by Goldfarb S, Ziyadeh F. NewYork:Churchill Livingstone, 1991.
18. Edelstein C, Schrier RW: The role of calcium in cell injury. In: AcuteRenal Failure: New Concepts and Therapeutic Strategies. Edited byGoligorsky MS, Stein JH. New York:Churchill Livingstone, 1995.
19. Kribben A, Wetzels J, Wieder E, et al.:Evidence for a role of cytosolicfree calcium in hypoxia-induced proximal tubule injury. J Clin Invest1994, 93:1922.
20. Burke T, Arnold P, Gordon J, Schrier RW: Protective effect ofintrarenal calcium channel blockers before or after renal ischemia. J Clin Invest 1984, 74:1830.
21. Kashgarian M: Stress proteins induced by injury to epithelial cells. In:Acute Renal Failure: New Concepts and therapeutic strategies. Editedby Goligorsky MS, Stein JH. New York:Churchill Livingstone, 1995.
22. J NIH Research
23. Ueda N, Walker P, Shah SV: Oxidant stress in acute renal failure. In: Acute Renal Failure: New Concepts and Therapeutic Strategies.Edited by Goligorsky MS, Stein JH. New York:ChurchillLivingstone, 1995.
24. Kelly KJ, et al.: Antibody to anyi-cellular adhesion molecule-1 pro-tects the kidney against ischemic injury. Proc Natl Acad Sci USA1994, 91:812–816.
25. Kroemer G, Petit P, Zamzami N, et al.: The biochemistry of pro-grammed cell death. FASEB J 1995, 9:1277–1287.


















