
p53 Mutations in Pancreatic Carcinoma and Evidence...
10
[CANCERRESEARCH54, 3025-3033, June1, 19941 ABSTRACT Pancreatic adenocarcinoma is a major cause of cancer death, and yet little is known about its molecular pathogenesis. We identified p53 muta tions in 19 (70%) of 27 primary pancreatic adenocarcinomas. Most were missense point mutations, and the mutations were distributed primarily within the evolutionarily conserved domains. Transitions predominated over transversions, and many of the transitions were at CpG dinucleo tides. Intragenic deletions accounted for 32% of mutations and were associated with decreased survival (P = 0.0016). A review of 1937 pub lishedp53 mutations revealed that the occurrence ofsmall (1—2 base pairs) microdeletions varied among different types of human neoplasms and that pancreatic adenocarcinoma had one ofthe highest frequencies (13% of 47 mutations, P = 0.0036). Many smali deletions occurred in iterations of single bases, but this did not fully account for their pattern of distribution, and there was evidence for the involvement of homocopolymer (poly purine:polypyrimidine) tracts. This may represent a more widespread phenomenon, because microdeletions occur in similar sequence patterns in reports of somatic and germ line mutations among genes other than P53. INTRODUCTION Pancreatic adenocarcinoma is one of the major causes of cancer death in the industrialized world (1). Despite the clinical importance, little is known about its etiology. There is a relationship with smoking, accounting for a 2.5-fold increased risk (2—5).Some studies have indicated an association with diet and possibly other environmental factors, but the implications of this work are unclear (3, 4). Some reports indicate an occasional familial pattern (6). There is otherwise little hint as to what other factors may contribute. Although the high prevalence of K-ras mutations in pancreatic adenocarcinoma has been documented (2, 7), little is yet known about the molecular genetics of these tumors. Knowledge of these events could give us clues to the pathogenesis and possible distinct risk factors. The p53 gene is the most commonly mutated gene in human cancer, and it provides a useful statistical base for analyzing mutational patterns (8—10).Point mutations with amino acid substitutions are the most common changes found in p53, and these are distributed pri marily in four evolutionarily conserved domains. With the increasing numbers of tumors studied, the p53 mutational spectrum has provided clear evidence for carcinogenic â€oefmgerprinting.― This has been seen in (a) aflatoxin-associated mutations at codon 249 in hepatocellular carcinoma (11, 12), (b) CC to U transitions at pyrimidine dimers in UV-induced skin carcinomas (13), and (c) different types of muta tions being favored in different tumors, e.g. , in lung versus colorectal carcinomas, and perhaps relating to the different carcinogenic expo sures of the organs (9). Received 1/11/94; accepted 3/30/94. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. 1 This study was supported in part by Grants RO1-CA56130 and P50-CA62924 from the NIH, by the Medical Research Council of Canada (M. S. R.), and by JNICF Scholarship BD1508/91-ID (L. C.). 2 To whom requests for reprints should be addressed at, 628 Ross Building, The Johns Hopkins School of Medicine, Baltimore, MD 21205-2196. We examined the prevalence and types of p53 mutations in resect able human primary pancreatic adenocarcinoma. We found that p53 mutations were very common, and while amino acid substitutions in conserved domains were the most common changes, intragenic dele tions, and particularly small (1—2base pairs) microdeletions, also accounted for a significant number of the mutations. We reviewed previous reports of such microdeletions to explore a probable and more widespread role for homocopolymer tracts in germ line and somatic deletion events. MATERIALS AND METHODS Patients and Specimens. Specimens were obtainedfrom pancreaticexo crine adenocarcinomas surgically resected at The Johns Hopkins Hospital. Patients met several criteria to be considered eligible for resection, including (a) a preoperative computed tomographic scan in all patients to rule out intraabdominal and hepatic metastases, (b) a preoperative arteriogram in most patients to rule out portal venous occlusion/encasement, and (c) intraoperative evaluation to rule out liver metastases and extrapancreatic lymphatic involve ment. Evidence of regional or distant spread served as exclusion criteria for resection. The immediate peripancreatic lymph nodes are evaluated in the resection specimens and do not serve as exclusion criteria. At the time of surgery, normal and neoplastic tissues were sampled and stored at — 80°C. Forty-three of 51 cases banked from the years 1991—1993were selected for study on the basis of tissue availability. Each case was cryostat microdissected to enrich for neoplastic cells as previously described (14). The threshold of mutation detection by sequencing was established by serial dilution studies using a DNA sample with wild-type p53 and a sample with a homozygousp53 mutation; this threshold was determined to be approximately 10% of alleles. In 24 cases, neoplastic cellularity could be enriched to at least 30% and, thus, could be analyzed by sequencing. Three additional cases with cellularity <30% were also included: one with cellularity of 10—20%, and 2 with cellularity of 5—10%. No other cases with cellularity <30% were selected because of the difficulty in analyzing these cases. A total of 27 cases were examined, including 25 adenocarcinomas, 1 adenosquamous carcinoma, and 1 mucinous cystadenocarcinoma. Twenty-five of these tumors were located in the head of the pancreas and were resected by pancreaticoduodenectomy (Whipple proce dure); 2 were located in the tail of the pancreas and were resected by distal pancreatectomy. There were 23 males and 4 females, and the ages ranged from 44 to 81 years old (mean 64.6 years). Twenty-four were white and 3 were black. This reflects the referral patterns to our hospital for this disease (15). Clinical information was obtained from the patients' charts, from The Johns Hopkins Cancer Registry, from records of the primary physicians, and from the databaseof the ClinicalCorrelatesin PancreaticCancerStudyof The Johns Hopkins Hospital. Tobacco exposure was classified as: heavy, 1 pack/day; moderate, <1 pack/day or quit 5—20 years ago; and light, nonsmoker or quit >20 years ago. Alcohol exposure was classified as: heavy, I 2 oz/week; moderate, <12 ox/week or quit 5—20years ago; and light, nondrinker, quit >20 years ago, or occasional use. Xenografts and Cell Lines. Several primary tumors were implanted into nude mice at the time of surgery. Xenograft samples from tumors grown by this method were available in 5 of the 27 primary tumors included in this study. Nine pancreatic cell lines were obtained from the American Type Culture Collection: AsPcl, BxPc3, CAPAN1, CAPAN2, CFPAC1, Hs766T, MiaPaCa2, Panci, and Su86.86. All cell lines were obtained fresh from the American Type Culture Collection and grown as recommended for three passages in a new laboratory having no other cell lines. 3025 p53 Mutations in Pancreatic Carcinoma and Evidence of Common Involvement of Homocopolymer Tracts in DNA Microdeletions1 Mark S. Redston, Carlos Caldas, Albert B. Seymour, Ralph H. Hruban, Luis da Costa, Charles J. Yeo, and Scott E. Kern2 Departments of Pathology [M. S. R., A. B. S., R. H. H., S. E. K.J, Oncology [C. C., S. E. KJ, and Surgery [C. J. V.], The Johns Hopkins University School of Medicine, and Division of Toxicological Sciences [L. C.], The Johns Hopkins University School of Hygiene and Public Health, Baltimore, Maryland 21205 Research. on September 1, 2018. © 1994 American Association for Cancer cancerres.aacrjournals.org Downloaded from
Transcript of p53 Mutations in Pancreatic Carcinoma and Evidence...

[CANCERRESEARCH54, 3025-3033, June 1, 19941
ABSTRACT
Pancreatic adenocarcinoma is a major cause of cancer death, and yetlittle is known about its molecular pathogenesis. We identified p53 muta
tions in 19 (70%) of 27 primary pancreatic adenocarcinomas. Most weremissense point mutations, and the mutations were distributed primarily
within the evolutionarily conserved domains. Transitions predominatedover transversions, and many of the transitions were at CpG dinucleotides. Intragenic deletions accounted for 32% of mutations and wereassociated with decreased survival (P = 0.0016). A review of 1937 publishedp53 mutations revealed that the occurrence ofsmall (1—2base pairs)microdeletions varied among different types of human neoplasms and thatpancreatic adenocarcinoma had one ofthe highest frequencies (13% of 47mutations, P = 0.0036). Many smali deletions occurred in iterations of
single bases, but this did not fully account for their pattern of distribution,and there was evidence for the involvement of homocopolymer (poly
purine:polypyrimidine) tracts. This may represent a more widespreadphenomenon, because microdeletions occur in similar sequence patternsin reports of somatic and germ line mutations among genes other thanP53.
INTRODUCTION
Pancreatic adenocarcinoma is one of the major causes of cancerdeath in the industrialized world (1). Despite the clinical importance,little is known about its etiology. There is a relationship with smoking,accounting for a 2.5-fold increased risk (2—5).Some studies haveindicated an association with diet and possibly other environmentalfactors, but the implications of this work are unclear (3, 4). Somereports indicate an occasional familial pattern (6). There is otherwiselittle hint as to what other factors may contribute. Although the highprevalence of K-ras mutations in pancreatic adenocarcinoma has beendocumented (2, 7), little is yet known about the molecular genetics ofthese tumors. Knowledge of these events could give us clues to thepathogenesis and possible distinct risk factors.
The p53 gene is the most commonly mutated gene in human cancer,and it provides a useful statistical base for analyzing mutationalpatterns (8—10).Point mutations with amino acid substitutions are themost common changes found in p53, and these are distributed primarily in four evolutionarily conserved domains. With the increasingnumbers of tumors studied, the p53 mutational spectrum has providedclear evidence for carcinogenic “fmgerprinting.―This has been seen in(a) aflatoxin-associated mutations at codon 249 in hepatocellularcarcinoma (11, 12), (b) CC to U transitions at pyrimidine dimers inUV-induced skin carcinomas (13), and (c) different types of mutations being favored in different tumors, e.g. , in lung versus colorectalcarcinomas, and perhaps relating to the different carcinogenic exposures of the organs (9).
Received 1/11/94; accepted 3/30/94.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordance with18 U.S.C. Section 1734 solely to indicate this fact.
1 This study was supported in part by Grants RO1-CA56130 and P50-CA62924 from
the NIH, by the Medical Research Council of Canada (M. S. R.), and by JNICFScholarship BD1508/91-ID (L. C.).
2 To whom requests for reprints should be addressed at, 628 Ross Building, The Johns
Hopkins School of Medicine, Baltimore, MD 21205-2196.
We examined the prevalence and types of p53 mutations in resectable human primary pancreatic adenocarcinoma. We found that p53mutations were very common, and while amino acid substitutions inconserved domains were the most common changes, intragenic deletions, and particularly small (1—2base pairs) microdeletions, alsoaccounted for a significant number of the mutations. We reviewedprevious reports of such microdeletions to explore a probable andmore widespread role for homocopolymer tracts in germ line andsomatic deletion events.
MATERIALS AND METHODS
Patients and Specimens. Specimens were obtainedfrom pancreaticexocrine adenocarcinomas surgically resected at The Johns Hopkins Hospital.
Patients met several criteria to be considered eligible for resection, including(a) a preoperative computed tomographic scan in all patients to rule out
intraabdominal and hepatic metastases, (b) a preoperative arteriogram in mostpatients to rule out portal venous occlusion/encasement, and (c) intraoperative
evaluation to rule out liver metastases and extrapancreatic lymphatic involvement. Evidence of regional or distant spread served as exclusion criteria forresection. The immediate peripancreatic lymph nodes are evaluated in theresection specimens and do not serve as exclusion criteria. At the time ofsurgery, normal and neoplastic tissues were sampled and stored at —80°C.Forty-three of 51 cases banked from the years 1991—1993were selected forstudy on the basis of tissue availability. Each case was cryostat microdissectedto enrich for neoplastic cells as previously described (14). The threshold ofmutation detection by sequencing was established by serial dilution studiesusing a DNA sample with wild-type p53 and a sample with a homozygousp53
mutation; this threshold was determined to be approximately 10% of alleles. In24 cases, neoplastic cellularity could be enriched to at least 30% and, thus,could be analyzed by sequencing. Three additional cases with cellularity <30%were also included: one with cellularity of 10—20%,and 2 with cellularity of5—10%.No other cases with cellularity <30% were selected because of thedifficulty in analyzing these cases. A total of 27 cases were examined,including 25 adenocarcinomas, 1 adenosquamous carcinoma, and 1 mucinouscystadenocarcinoma. Twenty-five of these tumors were located in the head ofthe pancreas and were resected by pancreaticoduodenectomy (Whipple procedure); 2 were located in the tail of the pancreas and were resected by distalpancreatectomy. There were 23 males and 4 females, and the ages ranged from44 to 81 years old (mean 64.6 years). Twenty-four were white and 3 wereblack. This reflects the referral patterns to our hospital for this disease (15).Clinical information was obtained from the patients' charts, from The JohnsHopkins Cancer Registry, from records of the primary physicians, and from thedatabaseof the ClinicalCorrelatesin PancreaticCancerStudyof The JohnsHopkins Hospital. Tobacco exposure was classified as: heavy, 1 pack/day;moderate, <1 pack/day or quit 5—20years ago; and light, nonsmoker or quit>20 years ago. Alcohol exposure was classified as: heavy, I 2 oz/week;moderate, <12 ox/week or quit 5—20years ago; and light, nondrinker, quit >20years ago, or occasional use.
Xenografts and Cell Lines. Several primary tumors were implanted intonude mice at the time of surgery. Xenograft samples from tumors grown bythis method were available in 5 of the 27 primary tumors included in this study.
Nine pancreatic cell lines were obtained from the American Type CultureCollection: AsPcl, BxPc3, CAPAN1, CAPAN2, CFPAC1, Hs766T,MiaPaCa2, Panci, and Su86.86. All cell lines were obtained fresh from theAmerican Type Culture Collection and grown as recommended for three
passages in a new laboratory having no other cell lines.
3025
p53 Mutations in Pancreatic Carcinoma and Evidence of Common Involvement of
Homocopolymer Tracts in DNA Microdeletions1
Mark S. Redston, Carlos Caldas, Albert B. Seymour, Ralph H. Hruban, Luis da Costa, Charles J. Yeo, andScott E. Kern2Departments of Pathology [M. S. R., A. B. S., R. H. H., S. E. K.J, Oncology [C. C., S. E. KJ, and Surgery [C. J. V.], The Johns Hopkins University School of Medicine, andDivision of Toxicological Sciences [L. C.], The Johns Hopkins University School of Hygiene and Public Health, Baltimore, Maryland 21205
Research. on September 1, 2018. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from

Table 1Clinkal characteristics ofpatientswith pancreaticadenocarcinoma from the currentstudyPatientAge
(yr)Tob―EthSiteSize (mm)Grades'Positive nodescFollow-up(days)'@Sta@ePCi58HLHead40M5/6228DODPC570HHHead30M1/4501DODPC672LHHead13M2/8610DODPC868LLHead40P6/12703DODPC948LLHead25M3/3678AWPC1169LLHead35P2/5860AWPCI365LLHead30P3/1467DOCPC1563MLHead45P4/11508AWPC1667LLHead45M6/131015AWPC1760HLHead30M2/377DODPC2367HLHead38M1/8361DOCPC2S64HMHead30P2/17501AWPC2655HLTail40P2/6150DODPC2867MHHead30P4/9140DODPC3O59HHHead30M0/21434AWPC3167HLHead50W4/27467AWPC3367HHHead50M3/10204DODPC3552LLHead15M0/7375DODPC3870HMHead25M2/23283DODPC3968HHHead50P1/575AWPC4O66LLTail15M1/6320AWPC4163HHHead55M0/175DODPC4344LLHead17M0/6275AWPC4469HLHead40M5/18105DODPC4S81LLHead100P4/8160DODPC4780LLHead70W0/12179AWPC6O66LLHead30M0/16137AW
p53 MICRODELETIONS IN PANCREATIC CARCINOMA
a Tob, tobacco; Eth, ethanol; H, heavy; M, moderate; L, light; see text for details.
b Histological grade: W, well differentiated; M, moderately differentiated; P, poorly differentiated.
C Number of nodes with metastases/total number of nodes examined.
d Days after operation.
e Status as of last contact: DOD, dead of disease; AW, alive and well without disease; DOC, dead of other cause without cancer.
P53 Gene Mutations. DNA was preparedfrom microdissected neoplasticand normal tissue as previously described (16). p53 exons 2—4and 5—9wereamplified separately using the polymerase chain reaction as described previously (17, 18). In some cases the PCR3 product could be sequenced directly.In other cases the PCR product was cloned prior to sequencing: the PCRproduct was digested with appropriate restriction enzymes, gel purified, andligated into pBluescript II (Stratagene). DNA pools from at least 200 insertbearing clones were sequenced with primers specific for each exon as described before (17—19).For the 2 cases with cellularity of 5—10%,a total of 60different clones were sequenced for each case, using pools of 5 clones each.p53exons2—4wereexaminedformutationsonlyinthosecaseswhichhadnomutations in exons 5—9and which had a cellularity of >30%. Individual cloneswere sequenced when an ambiguous band was identified in the sequence of thepooled DNA specimen; a mutation was considered to be present when it couldbe found in at least 5 different clones. All mutations were confirmed by asecond PCR and repeat sequencing of the corresponding exon.
,Yzs Gene Mutations. The first exon of the K-ras gene was amplified by thepolymerase chain reaction as described previously (20). In most cases, theresultant 120-base pair product was separated on an acrylamide gel, eluted, andsequenced directly using methods and primers as described in Refs. 20 and 21.In the cases with low cellularity, mutations were identified by a mutationspecific ligation detection assay (22).
lip Allelic Loss. In 7 cases, allelic loss of chromosomal arm Yip wasdetermined by Southern blots as part of a separate study using probes p144D6,pTNH37.3, pYNZ22.1, and pMCT35.1 (23). These cases represented the first7 cases having adequate neoplastic cellularity. Although Southern blots werenot done on the remaining cases, allelic loss could be determined from thesequence gels in those cases with good cellularity and in the xenografts.
Review of Published Gene Mutations. To furtheranalyzethe prevalenceand distribution of small deletions in the p.53gene, the relevant literature wasreviewed. This included all p53 mutational sequences in human tumors,xenografts, and cell lines published to October 1993. In total, 1937 mutationsin the p53 gene were reviewed (English literature).4 Deleted and flankingsequences were then analyzed for relative base frequencies, repeats, andpossible consensus sequences. Cases with incomplete, ambiguous, or conflict
3 The abbreviation used is: PCR, polymerase chain reaction.
4 A complete list of 248 references is available from the authors upon request.
ing sequence data were not included. Reports of intragenic deletions in certaingenes other than p53 were also surveyed in a similar manner, with the factorIX, APC, and NF2 genes being selected for detailed presentation on the basisof the adequacy of their sequence data bases and the dissimilarity of theirdisease associations.
RESULTS
Characteristics of Patient Population. The clinical characteristics of the patients studied are summarized in Table 1. In addition, 9patients had a prior neoplasm: colon adenomas (3 patients), prostatecarcinomas (2 patients), lymphomas (2 patients), breast carcinoma(1 patient), lung carcinoma (1 patient), and kidney neoplasm (1patient). Seven patients had a family history of malignancy: one eachof breast, lung, prostate, head and neck, gastrointestinal, and liver andtwo with unspecffied malignancies.
Mutations of p5.3 Gene. Mutations (Fig. 1 and Table 2) werefound in 19 (70%) of 27 primary neoplasms examined. Eighteen(95%)wereinexons5—9;exons2—4weresequencedin7cases,andone mutation was found. There were 13 point mutations; 11 resultedin amino acid substitutions, while two produced nonsense (stop)codons. Nine of the 13 were transitions, with 7 of the 9 occurring atCpG dinucleotides (78%). Eight of the 11 amino acid substitutionswere located within the evolutionarily conserved domains, while theremaining 3 were at conserved amino acid residues. There were 6intragenic frameshift deletions (32% of all mutations), ranging in sizefrom 1—40base pairs. A number of these were located in homocopolymer (polypurine:polypyrimidine) tracts of 3 or more bases (Table2). Due to the admixture of nonneoplastic cells with neoplastic cellsin the tumors, wild-type signals accompanied the mutant signal in thesequence gels (Fig. 1). The xenografts, on the other hand, werehomozygous for the mutation when present, indicating a lack ofhuman stromal cell contamination as well as indicating allelic loss inthese cases. Sequencing of normal DNA from each patient showedthat none of the mutations were present in the germ line.
3026
Research. on September 1, 2018. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from

p53 MICRODELETIONS IN PANCREATIC CARCINOMA
PC8
Fig. 1.p53 mutations in primary pancreatic adenocarcinomas. A, sequencing gel autoradiograph;samples from 5 tumors are grouped by terminationdideoxynucleotide to ald pattern recognition. Arrow, G to A transition in PC9. This tumor samplehad 30% neoplastic cellularity, and the wild-typesignal present in the 0 lane was reduced in intensity. B, sequencing gel autoradiograph of 3 tumors.Arrow, a one-base pair deletion(del)frameshift(fs)in P€@8.This sample had 50% neoplastic cellularity.bp, base pair.
1 bpdelfs4-.
PC9V V V V
@-
G T•@toA@ ______4
@ @-__
@@ :::::::@—@-@
A GA
C T A C G TB
Mutations were found in 7 of 9 cell lines examined (Table 2). Nomutation was found in cell line Hs766T; however, exons 2—4couldnot be amplified from this cell line. This line was previously shown tohave an internal deletion within p53 (24), suggesting that this deletionoverlapped the PCR primer-annealing site. The results in cell linesBxPc3, CAPAN2, and Panci are in agreement with at least one of theprevious reports (24—26).The mutation we identified in AsPcl differsfrom both of those previously reported (24, 25). CFPAC1, MiaPaCa2,and Su86.86 have not been previously reported.
Mutations of ms Gene. K-ras mutations were identified in 26(96%) of 27 primary tumors (Table 2). In 24 of these cases, themutations were detected by direct sequencing of PCR products. In 2cases (both with 5—10%cellularity) the mutations were detected bythe ligation detection assay. All of the mutations were located incodon 12, and 18 (69%) were transitions. No mutations were found innormal DNA by either assay. There was no association of the type ofras mutation with any clinical or pathological parameters or with p53mutation. Mutations were found in 7 (78%) of 9 cell lines.
Allelic Loss of Chromosomal Arm lip. Presence or absence ofalleic loss of l'7p could be established in 11 cases (Table 2). Therewere 9 cases with allelic loss, all of which had p53 mutations; 4 hadpoint mutations, and 5 had frameshift deletions. All cases with allelicloss showed a marked reduction or complete loss of the wild-typeband on the sequence gel at the site of mutation, indicating that p.53was included in the allelic loss. The two cases without allelic loss bothhad p5.3 mutations, one of which was a point mutation resulting in anonsense codon. The allele status of the remaining 16 cases, including7 caseswith p53 mutations,was not determined.
Clinical Associations with p53 Mutations. A history of moderateto heavy smoking tended to be more common among those patientswhose tumors had a p53 intragenic deletion compared to those with apoint mutation (5 of 6 versus 6 of 13), but this was not statisticallysignificant (P > 0.05, Fisher exact test). There was no difference in theoverall prevalence of p53 sequence mutations in smokers versusnonsmokers (11 of 15 versus 8 of 12). Of those patients with a familyhistory of cancer in a first-degree relative, 7 of 8 had p53 mutationscompared with 12 of 19 of those without a positive family history(P > 0.05,Fisherexacttest).Age, sex, race,alcoholintake,andsecond neoplasms were not associated with the presence or absence ofp53 mutations. p53 mutations tended to be associated with poorerhistological grade: mutations were found in 0 of 2 well-differentiatedtumors, 11 of 16 moderately differentiated tumors, and 8 of 9 poorlydifferentiated tumors (P = 0.044,@ test). No associations of p53mutations with tumor location, size, lymph node status, or duodenalinvasion were identified. A trend toward shorter disease-free survival(mean follow-up in survivors, 420 days) among those patients whose
tumors hadp53 mutations (50% survival, 340 days) compared to thosewithout p53 mutations (50% survival, 580 days) was not statisticallysignificant (P = 0.30, log rank; P = 0.10, Wicoxon-Gehan). However, disease-free survival was significantly reduced in patients whosetumors had a p53 intragenic deletion (50% survival, 140 days) compared (a) to all other patients (50% survival, 610 days, P = 0.0016,log rank test; P = 0.0014, Wilcoxon-Gehan), (b) individually to thosepatients whose tumors had a point mutation (P = 0.0053, log ranktest; P = 0.0062, Wilcoxon-Gehan), or (c) to those patients whosetumors had wild-type p53 (P 0.032, log rank test; P = 0.011,
Wilcoxon-Gehan). No survival differences were seen between thosepatients whose tumors had a p.5.3 point mutation compared to thosewhose tumors had wild-type p53 (P = 0.89, log rank test; P 0.46,Wilcoxon-Gehan).
Reviewof p53 MutationSpectrumin HumanPancreaticAdenocarcinoma. Review of p53 mutations previously reported in pancreatic cancer revealed 28 mutations found within 69 tumors analyzed(prevalence of 41%) (24, 25, 27, 28). Including data from the currentstudy, there are now 47 mutations reported; 37 are point mutations,and of these, 70% are transitions, 48% of which occur at CpGdinucleotides. The majority of the point mutations occur in the evolutionarily conserved domains (Fig. 14). There are 8 frameshift deletions (17% of all mutations) and 2 frameshift insertions.
Literature Review of p5.3 Mutations in Human Neoplasms.Review of sequence data from 1937 published p53 mutations inhuman neoplasms (including current study) revealed a variation in thetypes of mutation (all primary human tumors with at least 10 reportedp53 mutations are compiled in Table 3). Because of the differentmethods of mutation screening utilized in these studies, the denominator was considered indeterminable for providing an accurate assessment of the prevalence rates among the various tumors. Therefore, wecompared the different types of mutation in various neoplasms as afraction of total reported mutations. Point mutations were the mostcommon change in all neoplasms. The majority resulted in amino acidsubstitutions; however, the proportion of nonsense (stop) codonsvaried widely and according to tumor type (P = 0.001, f test). Forexample, 4% of all mutations in sarcomas were nonsense mutations,compared to 18 and 17% for carcinomas of skin and esophagus,respectively. The prevalence of intragenic deletions, and in particularsmall (1—2base pairs) microdeletions, also varied widely, from as lowas 0.8% in colon cancer to as high as 13 and 22% in pancreatic andthyroid carcinoma, respectively. Although the number of mutationsreported for some tumors is small, these differences appear to bestatistically different when comparing all tumor types (Table 3; P0.0036, x2 test for all neoplasms; P = 0.021, Fisher exact test ofpancreatic carcinoma versus all other neoplasms) or when comparing
3027
Research. on September 1, 2018. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from
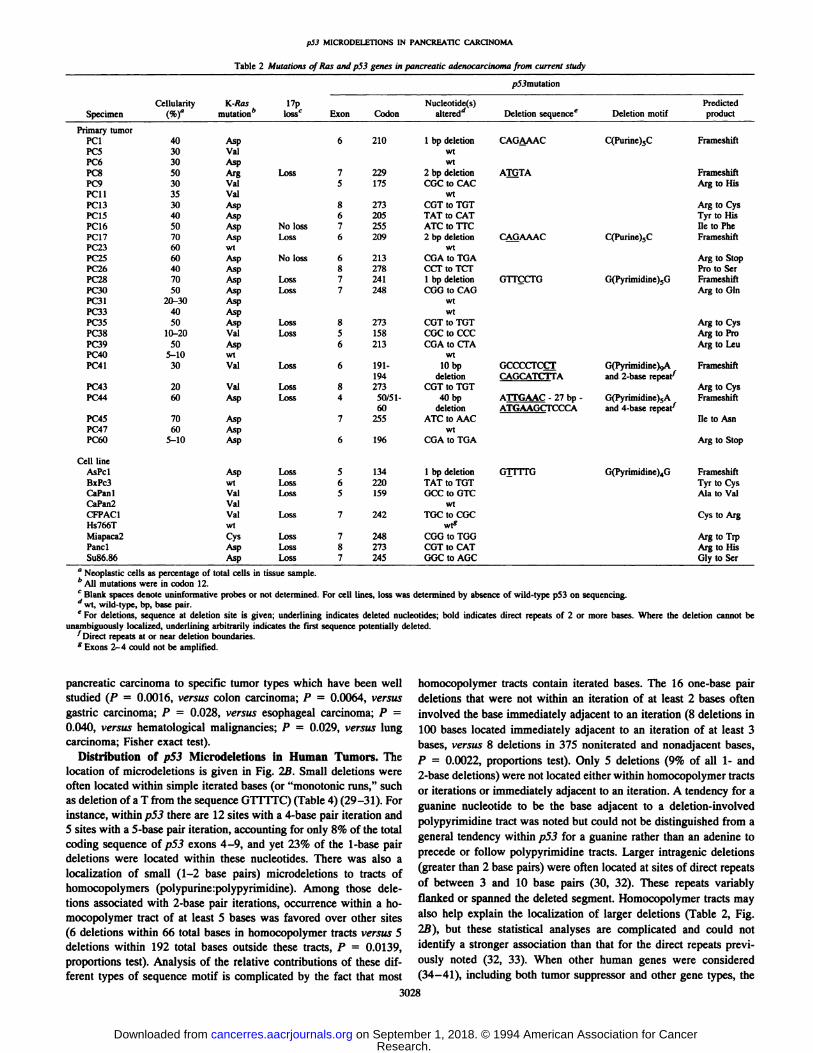
Table 2Mutations ofRas and p53 genes inpancreatic adenocarcinoma from currentstudySpecimenCellularity
(%)aK-Ras mutations'17p losscp53mutationExonCodonNucleotide(s) altered―Deletion sequenceeDeletion motifPredictedproductPrimary
tumorPCi40Asp62101bpdeletionCAG@AACC(Purine)5CFrameshiftPC530ValwtPC630AspwiPc850ArgLoss72292
bpdeletionArnTAFrameshiftPC930Val5175CGCto CACArg toHisPC1135ValwtPC1330Asp8273CUT
to TOTMg toCysPC1540Asp6205TATto CATTyr toHisPC1650AspNoloss7255ATCtoTI'CiletoPhePC1770AspLoss62092
bpdeletionCA@AAACC(Purine),CFrameshiftPC2360wtwtPC2560AspNoloss6213COAtoTOAArgtoStopPC2640Asp8278CCI@
to TCFPro to5crPC2870AspLoss72411bpdeletionOUçCFOG(Pyrimidine)5GFrameshift@c:@o50AspLoss7248COG
to CAGMg toGInP@3120-30Aspwtl't:3340AspwtPC3550AspLoss8273CGTtoTGTArgtoCysP0810-20ValLoss5158CGC
to CCCMg toProP0950Asp6213CGAtoCFAArgtoLeuPC4O5—10wtwtPC4130ValLoss6191-
19410bp
deletionGCCCCFCC@CAGCATC'FTAG(Pyrimidine)@,Aand 2-baserepedFrameshiftPC4320ValLoss8273COTto TOTArg toCysPC4460AspLoss450/51-
6040bp
deletionA1TGAAC- 27 bp -
ATGAAGCI'CCCAG(Pyrimidine)5Aand 4-baserepeat@'FrameshiftPC4570Asp7255ATCIoAACIletoAsnP01760AspwiPC6O5—10Asp6196CGA
to TGAArg toStopCell
lineAsPclAspLoss51341bpdeletionG]ITFGG(Pynmidine)4GFrameshiftBxPc3wtLoss6220TAT
to TOTTyr toCysCaPanlValLoss51590CCto GTCAla toValCaPan2ValwtCFPAC1ValLoss7242TGC
to COCCys toArgHs766TwtMiapaca2CysLoss7248COG
to TOGArg toTrpPanclAspLoss8273COTto CATArg toHisSu86.86AspLoss7245GGCtoAGCGlytoSer
p53 MICRODELETIONS IN PANCREA11C CARCINOMA
a Neoplastic cells as percentage of total cells in tissue sample.
b@ mutations were in codon 12.
C Blank spaces denote uninformative probes or not determined. For cell lines, loss was determined by absence of wild-type p53 on sequencing.
d@ wild-type. bp, base pair.
e For deletions, sequence at deletion site is given; underlining indicates deleted nucleotides; bold indicates direct repeats of 2 or more bases. Where the deletion cannot be
unambiguously localized, underlining arbitrarily indicates the first sequence potentially deleted.‘Directrepeats at or near deletion boundaries.g Exons 2—4could not be amplified.
pancreatic carcinoma to specific tumor types which have been wellstudied (P = 0.0016, versus colon carcinoma; P = 0.0064, versusgastric carcinoma; P = 0.028, versus esophageal carcinoma; P =0.040, versus hematological malignancies; P = 0.029, versus lungcarcinoma; Fisher exact test).
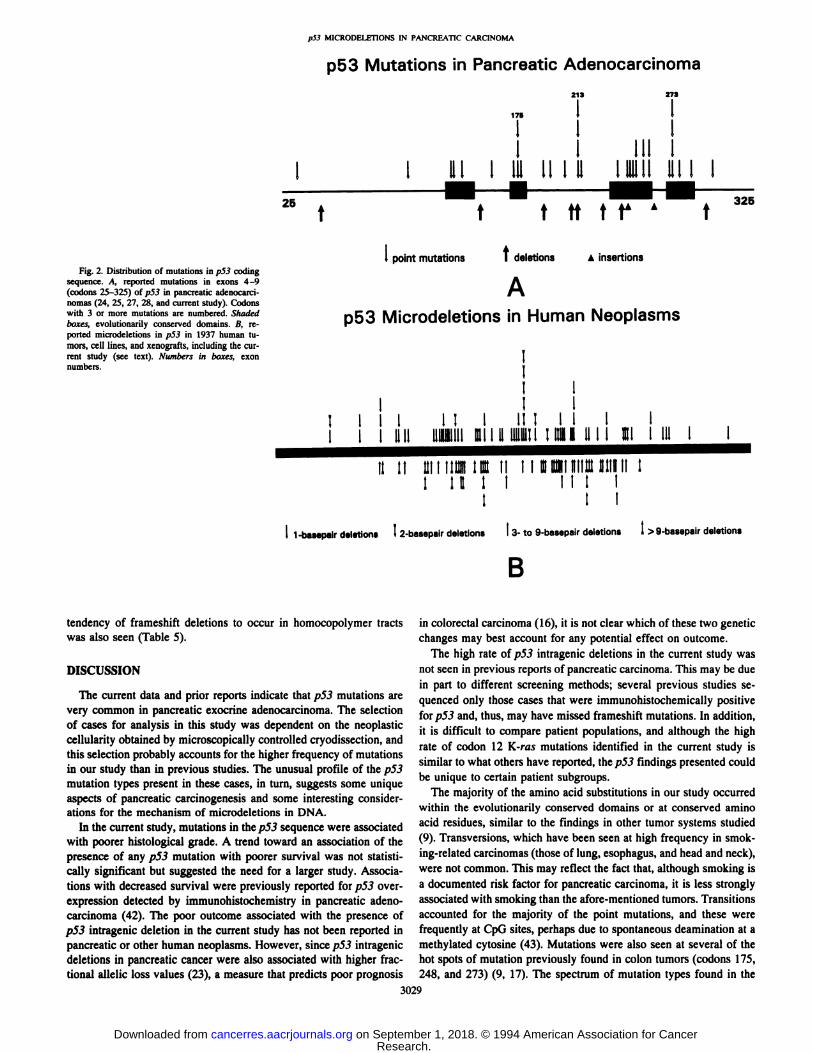
Distribution of p53 Microdeletions in Human Tumors. Thelocation of microdeletions is given in Fig. 2B. Small deletions wereoften located within simple iterated bases (or “monotonicruns,―suchas deletion of a T from the sequence GTITFC) (Table 4)(29—31). Forinstance, within p53 there are 12 sites with a 4-base pair iteration and5 sites with a 5-base pair iteration, accounting for only 8% of the totalcoding sequence of p.53 exons 4—9,and yet 23% of the 1-base pairdeletions were located within these nucleotides. There was also alocalization of small (1—2base pairs) microdeletions to tracts ofhomocopolymers (jlolypurine:polypyrimidine). Among those ddetions associated with 2-base pair iterations, occurrence within a homocopolymer tract of at least 5 bases was favored over other sites(6 deletions within 66 total bases in homocopolymer tracts versus 5deletions within 192 total bases outside these tracts, P = 0.0139,proportions test). Analysis of the relative contributions of these diiferent types of sequence motif is complicated by the fact that most
homocopolymer tracts contain iterated bases. The 16 one-base pairdeletions that were not within an iteration of at least 2 bases ofteninvolved the base immediately adjacent to an iteration (8 deletions in100 bases located immediately adjacent to an iteration of at least 3bases, versus 8 deletions in 375 noniterated and nonadjacent bases,
P = 0.0022,proportionstest).Only5 deletions(9%of all 1- and2-base deletions) were not located either within homocopolymer tractsor iterations or immediately adjacent to an iteration. A tendency for aguanine nucleotide to be the base adjacent to a deletion-involvedpolypyrimidine tract was noted but could not be distinguished from ageneral tendency within p53 for a guanine rather than an adenine toprecede or follow polypyrimidine tracts. Larger intragenic deletions(greater than 2 base pairs) were often located at sites of direct repeatsof between 3 and 10 base pairs (30, 32). These repeats variablyflanked or spanned the deleted segment. Homocopolymer tracts mayalso help explain the localization of larger deletions (Table 2, Fig.2.8), but these statistical analyses are complicated and could notidentify a stronger association than that for the direct repeats previously noted (32, 33). When other human genes were considered(34—41), including both tumor suppressor and other gene types, the
3028
Research. on September 1, 2018. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from
p53 MICRODELETIONS IN PANCREATIC CARCINOMA
I I32525 t
Fig. 2. Distribution of mutations in p53 codingsequence. A, reported mutations in exons 4—9(codons 25-325) ofp53 in pancreatic adenocarcinomas (24, 25, 27, 28, and current study). Codonswith 3 or more mutations are numbered. Shadedboxes; evolutionarily conserved domains. B, reported microdeletions in p53 in 1937 human tumors, cell lines, and xenografts, including the current study (see text). Numbers in boxes, exonnumbers.
f
tendency of frameshift deletions to occur in homocopolymer tractswas also seen (Table 5).
DISCUSSION
The current data and prior reports indicate that p53 mutations arevery common in pancreatic exocrine adenocarcinoma. The selectionof cases for analysis in this study was dependent on the neoplasticcellularity obtained by microscopically controlled cryodissection, andthis selection probably accounts for the higher frequency of mutationsin our study than in previous studies. The unusual profile of the p53mutation types present in these cases, in turn, suggests some uniqueaspects of pancreatic carcinogenesis and some interesting considerations for the mechanism of microdeletions in DNA.
In the current study, mutations in the p53 sequence were associatedwith poorer histological grade. A trend toward an association of thepresence of any p53 mutation with poorer survival was not statistically significant but suggested the need for a larger study. Associations with decreased survival were previously reported for p53 overexpression detected by immunohistochemistry in pancreatic adenocarcinoma (42). The poor outcome associated with the presence ofp53 intragenic deletion in the current study has not been reported inpancreatic or other human neoplasms. However, since p53 intragenicdeletions in pancreatic cancer were also associated with higher fractional allelic loss values (23), a measure that predicts poor prognosis
in colorectal carcinoma (16), it is not clear which of these two geneticchanges may best account for any potential effect on outcome.
The high rate of p5.3 intragenic deletions in the current study wasnot seen in previous reports of pancreatic carcinoma. This may be duein part to different screening methods; several previous studies sequenced only those cases that were immunohistochemically positivefor p53 and, thus, may have missed frameshift mutations. In addition,it is difficult to compare patient populations, and although the highrate of codon 12 K-ras mutations identified in the current study issimilar to what others have reported, the p.53 findings presented couldbe unique to certain patient subgroups.
The majority of the amino acid substitutions in our study occurredwithin the evolutionarily conserved domains or at conserved aminoacid residues, similar to the findings in other tumor systems studied(9). Transversions, which have been seen at high frequency in smoking-related carcinomas (those of lung, esophagus, and head and neck),were not common. This may reflect the fact that, although smoking isa documented risk factor for pancreatic carcinoma, it is less stronglyassociated with smoking than the afore-mentioned tumors. Transitionsaccounted for the majority of the point mutations, and these werefrequently at CpG sites, perhaps due to spontaneous deamination at amethylated cytosine (43). Mutations were also seen at several of thehot spots of mutation previously found in colon tumors (codons 175,248, and 273) (9, 17). The spectrum of mutation types found in the
3029
p53 Mutations in Pancreatic Adenocarcinoma
213 273
‘75 1 1
I I II I III I
III I Ill II I II 1111111III I_I
f ffff@A
11-bempeirdeletionsI2-basepairdeletionsI @-to9-besepairdeletionsI>9-basepairdeletions
B
I pointmutationst deletionsAinsertions
Ap53 Microdeletions in Human Neoplasms
T II I 1
I 1 1 1 11 I III 11 11 1 1 1111 11111111@1I il 1111@T1111111 il I 1 @1I 111
II II 1111IIthIII@Ii I I @@Illh11ll1111111II Ill I I III I
I I I
Research. on September 1, 2018. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from
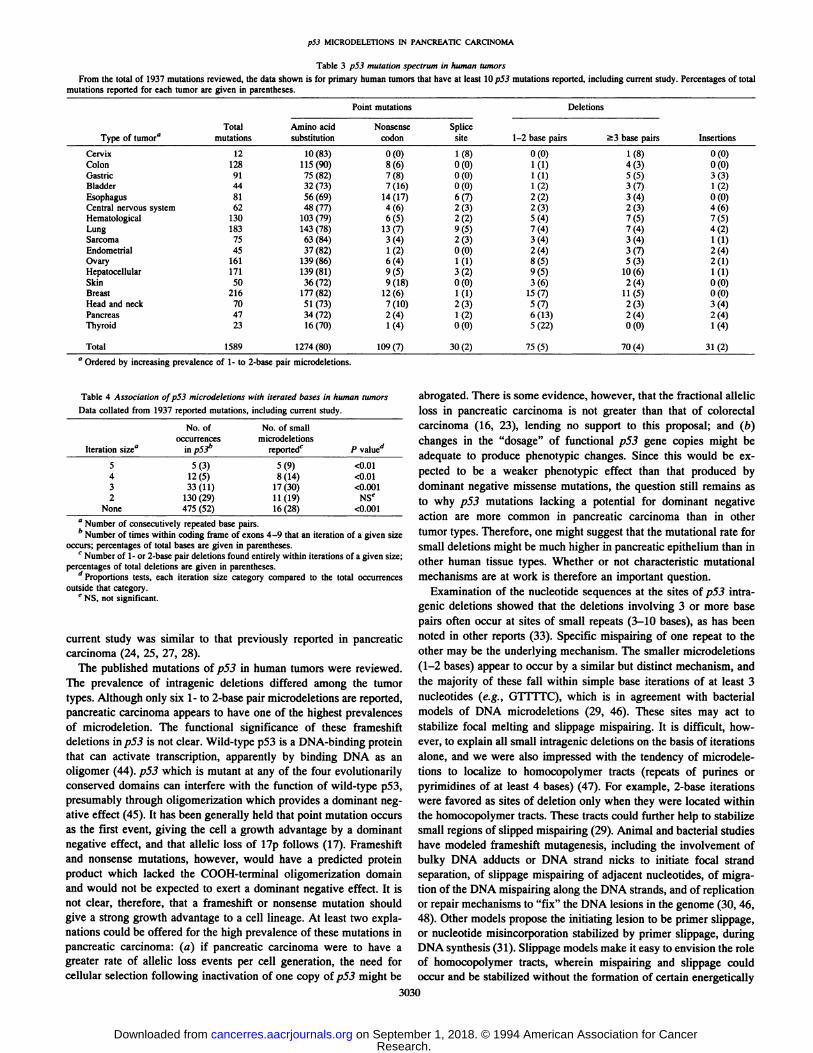
Table 3 p53 mutation spectrum in human tumorsFrom the total of 1937 mutations reviewed, the data shown is for primary human tumors that have at least 10p53 mutations reported, including current study. Percentages of total
mutations reported for each tumor are given in parentheses.
Point mutations Deletions
No. of No. of smalloccurrences microdeletions
Iteration 51@a in p53― reported'@ p valued
5 5(3) 5(9) <0.014 12(5) 8(14) <0.013 33 (11) 17(30) <0.0012 130(29) 11(19) NSe
None 475 (52) 16 (28) <0.001
a Number of consecutively repeated base pairs.
b Number of times within coding frame of exons 4—9 that an iteration of a given size
occurs; percentages of total bases are given in parentheses.C Number of 1- or 2-base pair deletions found entirely within iterations of a given size;
percentages of total deletions are given in parentheses.d Proportions tests, each iteration size category compared to the total occurrences
outside that category.e NS, not significant.
p53MICRODELETIONSINPANCREATICCARCINOMA
Type of tumoraTotal mutationsAminoacid
substitutionNonsense codonSplicesiteCervix1210
(83)0 (0)1(8)Colon128115(90)8 (6)0(0)Gastric9175(82)7(8)0(0)Bladder4432(73)7(16)0(0)Esophagus8156(69)14(17)6(7)Central
nervous system6248 (77)4 (6)2(3)Hematological130103(79)6 (5)2(2)Lung183143(78)13 (7)9(5)Sarcoma7563(84)3 (4)2(3)Endometrial4537(82)1(2)0(0)Ovary161139(86)6 (4)1(1)Hepatocellular171139(81)9 (5)3(2)Skin5036(72)9(18)0(0)Breast216177(82)12 (6)1(1)Head
andneck7051 (73)7(10)2(3)Pancreas4734(72)2 (4)1(2)Thyroid2316(70)1 (4)0(0)30
(2) 75 (5) 70 (4) 31 (2)
1-2 base pairs3 basepairsInsertions0(0)1(8)0(0)1(1)4(3)0(0)1(1)5(5)3(3)1(2)3(7)1(2)2(2)3(4)0(0)2(3)2(3)4(6)5(4)7(5)7(5)7(4)7(4)4(2)3(4)3(4)1(1)2(4)3(7)2(4)8(5)5(3)2(1)9(5)10(6)1(1)3(6)2(4)0(0)15(7)11(5)0(0)5(7)2(3)3(4)6(13)2(4)2(4)5
(22)0 (0)1 (4)
109 (7)
abrogated. There is some evidence, however, that the fractional alleicloss in pancreatic carcinoma is not greater than that of colorectalcarcinoma (16, 23), lending no support to this proposal; and (b)changes in the “dosage―of functional p53 gene copies might beadequate to produce phenotypic changes. Since this would be expected to be a weaker phenotypic effect than that produced bydominant negative missense mutations, the question still remains asto why p53 mutations lacking a potential for dominant negativeaction are more common in pancreatic carcinoma than in othertumor types. Therefore, one might suggest that the mutational rate forsmall deletions might be much higher in pancreatic epithelium than inother human tissue types. Whether or not characteristic mutationalmechanisms are at work is therefore an important question.
Examination of the nucleotide sequences at the sites of p53 intra
genic deletions showed that the deletions involving 3 or more basepairs often occur at sites of small repeats (3—10bases), as has beennoted in other reports (33). Specffic mispairing of one repeat to theother may be the underlying mechanism. The smaller microdeletions(1—2bases) appear to occur by a similar but distinct mechanism, andthe majority of these fall within simple base iterations of at least 3nucleotides (e.g., G1TfTC), which is in agreement with bacterialmodels of DNA microdeletions (29, 46). These sites may act tostabilize focal melting and slippage mispairing. It is difficult, however, to explain all small intragenic deletions on the basis of iterationsalone, and we were also impressed with the tendency of microdeletions to localize to homocopolymer tracts (repeats of purines orpyrimidines of at least 4 bases) (47). For example, 2-base iterationswere favored as sites of deletion only when they were located withinthe homocopolymer tracts. These tracts could further help to stabilizesmall regions of slipped mispairing (29). Animal and bacterial studieshave modeled frameshift mutagenesis, including the involvement ofbulky DNA adducts or DNA strand nicks to initiate focal strandseparation, of slippage mispairing of adjacent nucleotides, of migration of the DNA mispairing along the DNA strands, and of replicationor repair mechanisms to “fix―the DNA lesions in the genome (30, 46,48). Other models propose the initiating lesion to be primer slippage,or nucleotide misincorporation stabilized by primer slippage, duringDNA synthesis (31). Slippage models make it easy to envision the roleof homocopolymer tracts, wherein mispairing and slippage couldoccur and be stabilized without the formation of certain energetically
3030
Total 1589 1274(80)
a Ordered by increasing prevalence of 1- to 2-base pair microdeletions.
Table 4 Association ofps3 microdeletions with iterated bases in human tumors
Data collated from 1937 reported mutations, including current study.
current study was similar to that previously reported in pancreaticcarcinoma (24, 25, 27, 28).
The published mutations of p53 in human tumors were reviewed.The prevalence of intragenic deletions differed among the tumortypes. Although only six 1- to 2-base pair microdeletions are reported,pancreatic carcinoma appears to have one of the highest prevalencesof microdeletion. The functional significance of these frameshiftdeletions in p53 is not clear. Wild-type p53 is a DNA-binding proteinthat can activate transcription, apparently by binding DNA as anoligomer (44). p53 which is mutant at any of the four evolutionarilyconserved domains can interfere with the function of wild-type p53,presumably through oligomerization which provides a dominant negative effect (45). It has been generally held that point mutation occursas the first event, giving the cell a growth advantage by a dominantnegative effect, and that allelic loss of l7p follows (17). Frameshiftand nonsense mutations, however, would have a predicted proteinproduct which lacked the COOH-terminal oligomerization domainand would not be expected to exert a dominant negative effect. It isnot clear, therefore, that a frameshift or nonsense mutation shouldgive a strong growth advantage to a cell lineage. At least two explanations could be offered for the high prevalence of these mutations inpancreatic carcinoma: (a) if pancreatic carcinoma were to have agreater rate of allelic loss events per cell generation, the need forcellular selection following inactivation of one copy ofp53 might be
Research. on September 1, 2018. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from
p53 MICRODELETIONS IN PANCREATIC CARCINOMA
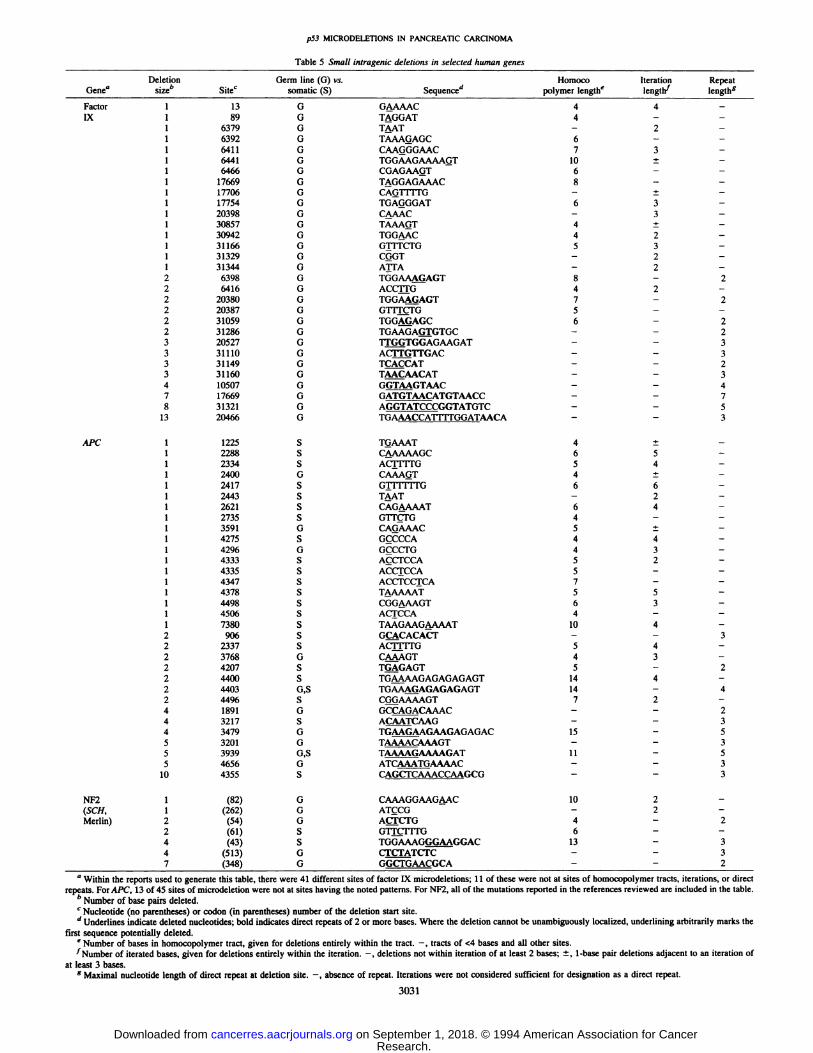
Table 5 Small intragenic deletions in selected human genes
Deletion Germ line (0) vs. Homoco Iteration RepeatGenea size― Site' somatic (5) Sequenced polymer lengthe length lengths
Factor 1 13 0 GAAAAC 4 4 —Ix 1 89 0 TAGGAT 4 - -
1 6379 0 TAAT — 2 —1 6392 0 TAAAGAGC 6 - -1 6411 0 CAA@GGAAC 7 3 -1 6441 0 TGGAAGAAAAGT 10 ± -1 6466 G CGAGAAOT 6 - -1 17669 0 TAGGAGAAAC 8 - -1 17706 0 CAG1TITG - ± -1 17754 G TGAGGGAT 6 3 -1 20398 0 C@AAC — 3 —1 30857 0 TAAA@T 4 ± —1 30942 0 TGGAAC 4 2 -1 31166 0 G1TFCTG 5 3 -1 31329 0 COOT — 2 —1 31344 0 ATFA - 2 -2 6398 G TGGAAAGAGT 8 - 22 6416 G ACCrfG 4 2 -2 20380 0 TGGAAfiAGT 7 - 22 20387 0 GTrT@FG 5 — —2 31059 G TGG@MAGC 6 — 22 31286 0 TGAAGA@GTGC - - 23 20527 0 UGGTGGAGAAGAT - - 33 31110 0 ACITGTrGAC - - 33 31149 G TCACCAT - - 23 31160 0 TAACAACAT - - 34 10507 0 G@MGTAAC - - 47 17669 0 GATGTAACATGTAACC - - 78 31321 0 AGGTATCCCGGTATGTC - - 5
13 20466 0 TGAAACCA1TITGGATAACA - - 3
AFC 1 1225 S T@AAAT 4 ± -1 2288 S C4@AAAAGC 6 5 -1 2334 S ACT1T1'G 5 4 —1 2400 0 CAAAQT 4 ± -1 2417 S GTIT!lIG 6 6 —1 2443 S TAAT - 2 -1 2621 S CAGAAAAT 6 4 —1 2735 5 GTT@TG 4 — —1 3591 0 CAGAAAC 5 ± -1 4275 S GCCCCA 4 4 —1 4296 0 GçCCTG 4 3 -1 4333 5 ACC1'CCA 5 2 —1 4335 S ACCTCCA 5 - -1 4347 S ACCTCCICA 7 - -1 4378 S TAAAAAT 5 5 —1 4498 S CGGAAAGT 6 3 -1 4506 S ACTCCA 4 - -1 7380 S TAAGAAGAAAAT 10 4 -2 906 S GCACACACT - - 32 2337 5 ACITrTG 5 4 —2 3768 0 CAAAGT 4 3 -2 4207 S TGAGAGT 5 — 22 4400 S TGAAAAGAGAGAGAGT 14 4 -2 4403 0,5 TGAAA@AGAGAGAGT 14 - 42 4496 S Cfi@AAAAGT 7 2 -4 1891 0 GC@@CAAAC - - 24 3217 5 A@M3CAAG — — 34 3479 0 TGAAGAAGAAGAGAGAC 15 - 55 3201 0 TAAAACAAAGT - - 35 3939 0,5 TAAAAGAAAAGAT 11 - 55 4656 0 ATCAAATGAAAAC - - 3
10 4355 S CAGCTCAAACCAAGCG - - 3
NF2 1 (82) 0 CAAAGGAAG4AC 10 2 -(SCH, 1 (262) G ATcCcI - 2 -Merlin) 2 (54) 0 ACTCTG 4 — 2
2 (61) 5 GTFC1TI'O 6 — —4 (43) S TGGAAAGGGA@GGAC 13 - 34 (513) 0 CTC'I'@TCTC — — 37 (348) 0 GGCTGAACGCA - - 2
a Within the reports used to generate this table, there were 41 different sites of factor IX microdeletions; 1 1 of these were not at sites of homocopolymer tracts, iterations, or direct
repeats. For APC, 13 of 45 sites of microdeletion were not at sites having the noted patterns. For NF2, all of the mutations reported in the references reviewed are included in the table.b Number of base pairs deleted.
C Nucleotide (no parentheses) or codon (in parentheses) number of the deletion start site.
d Underlines indicate deleted nucleotides; bold indicates direct repeats of 2 or more bases. Where the deletion cannot be unambiguously localized, underlining arbitrarily marks the
first sequence potentially deleted.e Number of bases in homocopolymer tract, given for deletions entirely within the tract. —, tracts of <4 bases and all other sites.
@‘Numberof iterated bases, given for deletions entirely within the iteration. —,deletions not within iteration of at least 2 bases; ±,1-base pair deletions adjacent to an iteration ofat least 3 bases.
g Maximal nucleotide length of direct repeat at deletion site. —,absence of repeat. Iterations were not considered sufficient for designation as a direct repeat.
3031
Research. on September 1, 2018. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from

p53 MICRODELETIONS IN PANCREATIC CARCINOMA
unfavorable mispairs (49) in the transitional alignments (“intermediates of mutation―)which would occur during a slippage event (30).Faulty mismatch repair has been implicated in the genesis of familialcolorectal cancer, producing a mutator phenotype (50) which allowsaccumulation of multiple small deletions and insertions in microsatellite sequences. The human homolog (MSH2) of a bacterial mismatch repair gene (mutS) has been shown to be mutated in some cases(51, 52). However, allelic shifts at microsatellite sequences were notpresent in any of the cases in the current study,4 and there is noevidence to suggest that alterations in the MSH2 gene are involved inthe p53 microdeletions described here.
The tendency of frameshift deletions to occur in homocopolymertracts was also seen among other genes (34—41), including bothsomatic and germ line mutations, and in both tumor suppressor andother gene types (Table 5). A greater appreciation for the sequencepatterns and disease associations of small intragenic deletions mayyield clues to their mechanism of genesis. Especially, there is a needto consider the roles of potential environmental and endogenousmutagens and of the corresponding DNA repair mechanisms.
REFERENCES
1. Boring, C. C., Squires, T. S., and Tong, T. Cancer statistics, 1993.CA-Cancer J. Clin.,43: 7—26,1993.
2. Hruban, R. H., van Mansfeld, A. 0. M., Offerhaus, 0. J. A., van Weering, D. H. J.,Allison, D. C., Goodman, S. N., Kensler, T. W., Bose, K. K., Cameron, J. L, andBoa, J. L. K-rat oncogene activation in adenocarcinomas of the human pancreas: astudy of 82 carcinomas using a combination of mutant-enriched polymerase chainreaction analysis and allele-specific oligonucleotide hybridization. Am. J. Pathol.,143:545—554,1993.
3. Fraumeni, J. F. Cancers of the pancreas and biiary tract: epidemiological considerations. Cancer Res., 35: 3437—3446,1975.
4. Liu, R. S., and Kessler, I. I. A multifactorial model for pancreatic cancer in man.J.Am. Med.Assoc.,245:147—152,1981.
5. Falk, R. T., Williams Pickle, L, Fontham, E. T., Correa, P., and Fraumem, J. F.Life-style risk factors for pancreatic cancer in Louisiana: a case-control study. Am. J.Epidemiol., 128: 324—336, 1988.
6. Lynch, H. T., Fitzsimmons, M. L., Smyrk, T. C., Lanspa, S. J., Watson, P., McClellan,J., and Lynch, J. F. Familial pancreatic cancer: clinicopathologic study of 18 nuclearfamilies. Am. J. Gastroenterol., 85: 54—60, 1990.
7. Almoguera, C., Shibata, D., Forrester, K., Martin, J., Arnheim, N., and Perucho, M.Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell,53: 549—554,1988.
8. Nigro, J. M., Baker, S. J., Preisinger, A. C., et aL p53 gene mutations occur in diversehuman tumour types. Nature (Lond.), 342: 705—708,1989.
9. Hollstein, M., Sidransky, D., Vogelstein, B., and Harris, C. C. p53 mutations inhuman cancers. Science (Washington DC), 253: 49—53,1991.
10. de Fromentel, C. C., and Soussi, J. TP53 tumor suppressor gene: a model forinvestigating human mutagenesis. Genes Chromosomes Cancer, 4: 1—15,1992.
11. Bressac B., Kew, M., Wands, J., and Ozturk, M. Selective 0 to T mutations ofp53 gene in hepatocellular carcinoma from southern Africa. Nature (Lond.), 350:429—431,1991.
12. Hsu, I. C., Metcalf, R. A., Sun, T., Welsh, J. A., Wang, N. J., and Harris, C. C.Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature(Lond.), 350: 427—428,1991.
13. Brash, D. E., Rudolf, J. A., Simon, J. A., Lin, A., McKenna, 0. J., Baden, H. P.,Halperin, A. J., and Pontén,J. A role for sunlight in skin cancer: UV-induced p53mutations in squamous cell carcinoma. Proc. Natl. Acad. Sci. USA, 88: 10124—10128,1991
14. Bos, J. L., Fearon, E. R., Hamilton, S. R., et aL Prevalence of ras gene mutations inhuman colorectal cancers. Nature (Lond.), 327: 293—297,1987.
15. Cameron, J. L., Crist, D. W., Sitzman, J. V., Hruban, R. H., Boitnott, J. K., Seidler,A. J., and Coleman, J. Factors influencing survival after pancreaticoduodenectomy forpancreatic cancer. Am. J. Surg., 161: 120—125,1991.
16. Vogelstein, B., Fearon, E. R., Kern, S. E., Hamnilton, S. R., Preisinger, A. C.,Nakamura, Y., and White, R. Allelotype of colorectal carcinomas. Science (Washington DC), 244: 207—211, 1989
17. Baker, S. J., Preisinger, A. C., Jessup, J. M., Paraskeva, C., Markowitz, S., Willson,J. K. V., Hamilton, S. R., and Vogelstein, B.p.53gene mutations occur in combinationwith lip allelic deletions as late events in colorectal tumorigenesis. Cancer Res., 50:7717—7720,1990.
18. Runnebaum, I. B., Nagarajan, M., Bowman, M., Soto, D., and Sukumar, S. Mutationsin p53 as potential molecular markers for human breast cancer. Proc. Natl. Acad. Sci.USA, 88: 10657—10661,1991.
19. Leach, F. S., Tokino, T., Meltzer, P., Burrell, M., Oliner, J. D., Smith, S., Hill, D. E.,Sidransky, D., Kinzler, K. W., and Vogeistein, B. p53 mutation and MDM2 amplification in human soft tissue sarcomas. Cancer Res., 53: 2231—2234,1993.
20. Sidransky, D., Tokino, T., Hamilton, S. R., Kinzler, K. W., Levin, B., Frost, P., andVogelstein, B. Identification of ras oncogene mutations in the stool of patients withcurable colorectal tumors. Science (Washington DC), 256: 102—105,1992.
21. Hollstein, M. C., Metcalf, R. A., Welsh, J. A., Montesano, R., and Harris, C. C.Frequent mutation of the p53 gene in human esophageal cancer. Proc. Nail. Aced. Sci.USA, 87: 9958—9961,1990.
22. Landegren, U., Kaiser, R., Sanders, J., and Hood, L. Aligase-mediated gene detectiontechnique. Science (Washington DC), 241: 1077—1080,1988.
23. Seymour, A. B., Hruban, R. H., Redston, M., Caldas, C., Powell, S. M., Kinzler,K.W.,Yeo,C. J., andKern,S. E. Allelotypeof pancreaticadenocarcinoma.CancerRca., 54: 2761—2764,1994.
24. Ruggeri, B., Zhang, S-Y., Caamano, J., DiRado, M., Flynn, S. D., and Klein-Szanto,A. J. P. Human pancreatic carcinomas and cell lines reveal frequent and multiplealterations in the p53 and Rb-i tumor-supressor genes. Oncogene, 7: 1503—1511,1992.
25. Barton, C. M., Staddon, S. L, Hughes, C. M., Hall, P. A., O'Sullivan, C., Kloppel,0., Theis,B.,Russell,R.C.0., Neoptolemos,J., Williamson,R.C. N.,Land,D. P.,and Lemoine, N. R. Abnormalities of the p53 tumour suppressor gene in humanpancreatic cancer. Br. J. Cancer, 64: 1076—1082, 1991.
26. Kalthoff, H., Schmiegel, W., Roeder, C., Kasche, D., Schmidt, A., Lauer, 0., Thiele,H-G., Honold, 0., Pantel, K., Riethmuller, 0., Scherer, E., Maner, J., and Deppert, W.p53 and K-RAS alterations in pancreatic epithelial cell lesions. Oncogene, 8:289—298, 1993.
27. Scarpa, A., Capeili, P., MUkai, K, Zamboni, G., Ode, T., lacono, C., and Hirohashi,S. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am. J. PathoL,142: 1534—1543,1993.
28. Casey, 0., Yamanka, Y., Freiss, H., Kobrin, M. S., Lopez, M. E., Buchler, M., Beger,H. 0., and Korc, M. p53 mutations are common in pancreatic carcinoma and areabsent in chronic pancreatitis. Cancer Left., 69: 151—160,1993.
29. Streisinger, 0., and (Emrich) Owen, J. Mechanisms of spontaneous and inducedframeshiftmutationsin bacteriophageT4. Genetics,109:633—659,1985.
30. Ripley, L. S. Frameshift mutation: determinants of specificity. Annu. Rev. Genet., 24:189, 1990.
31. Kunkel, T. A. Misalignment-mediated DNA synthesis errors. Biochemistry, 29:8003-8011, 1990.
32. Krawczak, M., and Cooper, D. C. Gene deletions causing human genetic disease:mechanisms of mutagenesis and the role of the local DNA sequence environment.Hum. Genet., 86: 425-441, 1991.
33. Jego, N., Thomas, 0., and Hamelin, R. Short direct repeats flanking deletions andduplicating insertions in p53 gene in human tumors. Oncogene, 8: 209—213, 1993.
34. Powell, S. M., Zilz, N., Brazer-Barclay, Y., Bryan, 1. M., Hamilton, S. R.,Thibodeau, S. N., Vogelstein, B., and Kinzler, K. W. APC mutations occur earlyduring colorectal tumorigenesis. Nature (Land.), 359: 235—237,1992.
35. Nakatsuru, S., Yanagisawa, A., Ichii, S., Tahara, E., Kato, Y., Nakamura, Y., andHoril, A. Somatic mutations of the APC gene in gastric cancer: frequent mutations invery well differentiated adenocarcinoma and signet-ring cell carcinoma. Hum. MoLGenet., 1: 559—563,1992.
36. Miyoshi, Y., Nagase, H., Ando, H., Horii, A., Ichii, S., Nakatsuru, S., Aoki, T., Miki,Y.,Mon,T.,andNakamura,Y.SomaticmutationsofAPCgeneincolorectaltumors:mutation cluster region of the APC gene. Hum. Mol. Genet., 1: 229—233, 1992.
37. Miyoshi, Y., Ando, H., Nagase, H., Nishisho, I., Horn, A., Mild, Y., Mori, T.,Utsunomiya, J., Baba, S., Peterson, 0., Hamilton, S. R., Kinzler, K. W., Vogelstein,B. V., and Nakamura,Y. Germ-linemutationsof the APC gene in 53 familialadenomatous polyposis patients. Proc. Natl. Acad. Sci. USA, 89: 4452—4456,1992.
38. Olschwang, S., Laurent-Puig, P., Groden, J., White, R., and Thomas, 0. Germ-linemutations in the first 14 exons of the adenomatous polyposis coli (APC) gene. Am.J. Genet., 52: 273—279,1993.
39. Giannelli, F., Green, P. M., High, K. A., Sommer, S., Poon, M-C., Ludwig, M.,Schwaab,R., Reitsma,P. H., Goossens,M., Yoshioka,A., and Brownlee,0. 0.Haemophilia B: database of point mutations and short additions and deletions—fourthedition, 1993. Nucleic Acids Res., 21: 3075—3087, 1993.
40. Rouleau, 0. A., Merel, P., Lutchman, M., Sanson, M., Zucman, J., Marineau, C.,Hoang-Xuan, K., Demczuk, S., Desmaze, C., Plougastel, B., Pulst, S. M., Lenoir, 0.,Bijlsma, E., Fushold, R., Dumanski, J., de Jong, P., Parry, D., Eldrige, R., Aurias, A.,Delattre, 0., and Thomas, 0. Alterations in a gene encoding a putative membraneorganizing protein causes neuro-fibromatosis type 2. Nature (Lond.), 363: 515—521,1993.
41. Trofatter, J. A., MacCoffin, M. M., Rutter, J. L., Murrell, J. R., Duyao, M. P., Parry,D. M., Eldridge, R., Kley, N., Menon, A. 0., Pulaski, K., Haase, V. H., Ambrose,C. M., Munroe,D., Bove,C., Haines,J. L, Martuza,R. L, MacDonald,M. E.,Seizinger, B. R., Short, M. P., Buckler, A. J., and Gusella, J. F. A novel moesin-,ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor.Cell, 72: 791—800, 1993.
42. DiGiuseppe, J. A., Hruban, R. H., Goodman, S. N., Allison, D. A., Cameron, J. L,and Offerhaus, J. 0. A. Overexpression of the p53 tumor suppressor protein inpancreatic adenocarcinoma. Am. J. Gin. Pathol., 101: 684—688,1994.
43. Rideout, W. M., Ill, Coetzee, 0. A., Olumi, A. F., and Jones, P. A. 5-Methylcytosineas an endogenous mutagen in the human LDL receptor and p53 genes. Science(Washington DC), 249: 1288—1290.
44. Kraiss, S., Qualser, A., Oren, M., and Montenarh, M. Oligomerization of oncoproteinp53. J. Virol., 62: 4737—4744,1988.
45. Kern, S. E., Pietenpol, J. A., Thiagalingam, S., Seymour, A., Kinzler, K. W., andVogelstein, B. Oncogenic forms of p53 inhibit p53-regulated gene expression.Science (Washington DC), 256: 827—830, 1992.
46. Roth, J. R. Frameshift mutations. Annu. Rev. Genet., 8: 319—346, 1974.47. Hentschel, C. C. Homocopolymer sequences in the spacer of a sea urchin histone gene
3032
Research. on September 1, 2018. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from

p53 MICRODELETIONSIN PANCREA11C CARCINOMA
repeatare sensitiveto Si nuclease.Nature(Land.),295: 714—716,1982.48. Streisinger, 0., Okada, Y., Emrich, J., Tsugita, A., Terzaghi, E., and Inouye, M.
Frameshift mutations and the genetic code. Cold Spring Harbor Symp. Quant. BioL,
31: 77—84,1966.49. Ke,S-H.,andWartdll,R. M.Influenceof nearestneighborsequenceon thestability
of basepairmismatchesin long DNA:determinationby temperature-gradientgelclectrophoresis. Nucleic Acids Res., 21: 5137—5143,1993.
50. Parsons, R@,Li, G-M., Langley, M. J., Fang, W., Papadopoulos, N., Jen, J., de laChapelle, A., Kinzler, K. W., Vogelstein, B., and Modrich, P. Hypermutability andmismatch repair deficiency in RER@ tumor cells. Cell, 75: 1227—1236,1993.
51. Fishel, R., Lescoc, M. K., Rao, M. R. S., COpCland,N. 0., Jenkins, N. A., Gerber, J.,
Kane, M., and Kolodner, R. The human mutator gene homolog MSH2 andits association with hereditary nonpolyposis colon cancer. Cell, 75: 1027—1038,1993.
52. Leach, F. S., Nicolaides, N. C., Papadopoulos, N., Liu, B., Jen, J., Parsons, R.,PeltomAki, P., Sistonen, P., Aaltonene, L A., Nystrom-Lahti, M., Guan, X-Y., Zhang,J., Meltzer,P. S., Yu,J.W.,Kao,F. 1., Chen,D. J., Cerosaletti,K. M.,Fournier,R.E. K.,Todd,S., Lewis,T., Leach,R.J., Naylor,S. L, Weissenbach,J., Mecklin,i-P., JSrvinen,H., Peterson,0. M.,Hamilton,S. R.,Green,J., lass, I., Watson,P.,Lynch,H. 1., Trent,I. M.,de Ia Chapelle,A., Kinzler,K. W., and Vogelslein,B.Mutations of a matS homolog in hereditary nonpolyposis colorectal cancer. Cell, 75:1215-1235,1993.
3033
Research. on September 1, 2018. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from

1994;54:3025-3033. Cancer Res Mark S. Redston, Carlos Caldas, Albert B. Seymour, et al. MicrodeletionsCommon Involvement of Homocopolymer Tracts in DNA
Mutations in Pancreatic Carcinoma and Evidence ofp53
Updated version
http://cancerres.aacrjournals.org/content/54/11/3025
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/54/11/3025To request permission to re-use all or part of this article, use this link
Research. on September 1, 2018. © 1994 American Association for Cancercancerres.aacrjournals.org Downloaded from


















