
NGS Transcriptomic Workflows Hugh Shanahan & Jamie al-Nasir Royal Holloway, University of London.
8
NGS Transcriptomic Workflows Hugh Shanahan & Jamie al-Nasir Royal Holloway, University of London
-
Upload
ronald-clark -
Category
Documents
-
view
215 -
download
0
Transcript of NGS Transcriptomic Workflows Hugh Shanahan & Jamie al-Nasir Royal Holloway, University of London.

NGS Transcriptomic Workflows
Hugh Shanahan & Jamie al-NasirRoyal Holloway, University of London

Transcriptome – total sequence and abundance of RNA generated by a cell
RNA is transcribed from DNA Genome is fixed for a organism Transcriptome is dynamic
Variation between tissues Variation over time
RNA transcripts are 1,000’s-10,000 bases in length
Setting the scene

Interested in How many copies of a particular transcript
are there What is the sequence
- sequence comes from genome but alternative splicing means a transcript may not just be a contiguous block of DNA

Size of transcriptome will vary between species

Fragment transcripts into shorter pieces (reads) 100-300 bases longs
Have many overlapping reads
Amplify (make lots of copies of) the short reads
Can sequence these short reads and then assemble them to reconstruct transcripts.
Size of data set depends on size of transcriptome but also amount of fragmentation (sequencing depth)
Can either assemble with a reference genome or de novo (very hard)
Sequencing steps

NGS Workflow
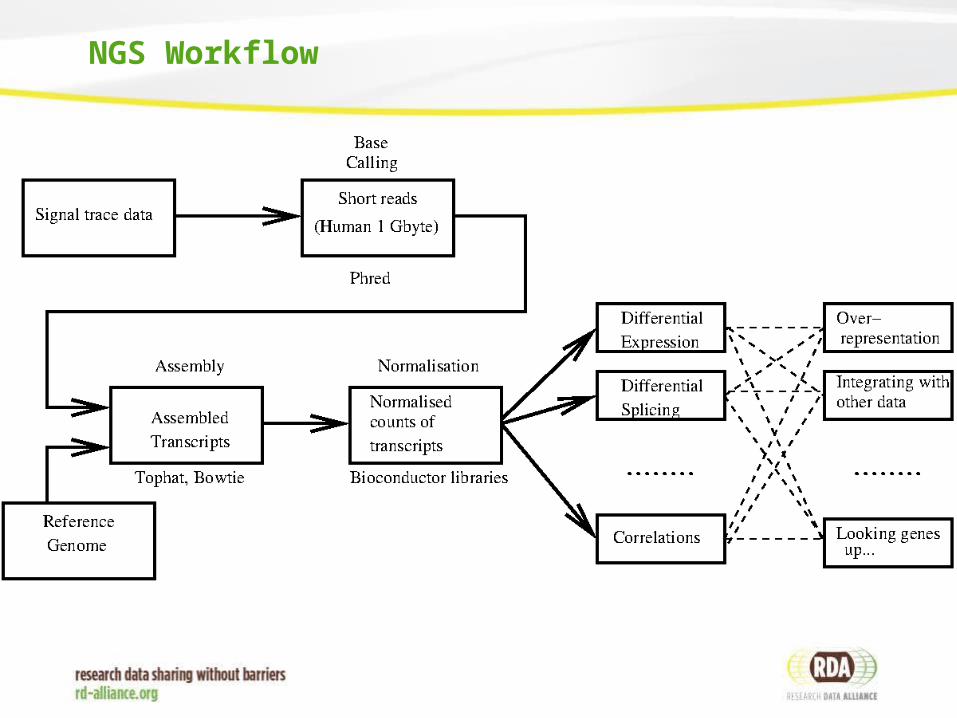
NGS Workflow

File formats have been updated to binary – used to use flat text so sizes were huge (Reference Genome – 39 Gbyte -> 0.8 Gybte)
Raw image data is actually discarded Discussions focusses on assembly and down-stream
analysis Much of this data is deposited in the Sequence Read
Archive (SRA) We’ve papered over everything that happens before
sequencing – i.e. the biochemical steps carried out This is highly variable These steps are not properly annotated
Final points


















