
MECHANICAL AND SWELLING - DiVA portal571374/FULLTEXT01.pdf · MECHANICAL AND SWELLING PROPERTIES OF...
77
MECHANICAL AND SWELLING PROPERTIES OF HYDROGELS Ting Yang AKADEMISK AVHANDLING Som med tillstånd av Kungliga Tekniska högskolan i Stockholm, framlägges till offentlig granskning för avläggande av teknisk doktorsexamen torsdag 6 december 2012, kl. 10:00 i sal F3, Lindstedtsvägen 26, KTH, Stockholm. Avhandlingen försvaras på engelska. Fakultetsopponent: Professor Jöns Hilborn från Uppsala University, Sweden
Transcript of MECHANICAL AND SWELLING - DiVA portal571374/FULLTEXT01.pdf · MECHANICAL AND SWELLING PROPERTIES OF...

MECHANICAL AND SWELLING
PROPERTIES OF HYDROGELS
Ting Yang
AKADEMISK AVHANDLING
Som med tillstånd av Kungliga Tekniska högskolan i Stockholm, framlägges till offentlig
granskning för avläggande av teknisk doktorsexamen torsdag 6 december 2012, kl. 10:00 i
sal F3, Lindstedtsvägen 26, KTH, Stockholm. Avhandlingen försvaras på engelska.
Fakultetsopponent: Professor Jöns Hilborn från Uppsala University, Sweden

Copyright © 2012 Ting Yang
All rights reserved
Paper I © 2011 Wiley Periodicals, Inc.
Paper II © 2012 Wiley Periodicals, Inc.
TRITA-CHE-Report 2012: 63
ISSN 1654-1081
ISBN 978-91-7501-471-5

ABSTRACT
Hydrogels have been used as one of the novel soft materials in many biomedical
applications such as drug delivery and tissue engineering for recent decades.
In the main part of this work, bi-functional poly(ethylene glycol) (PEG) precursors with
either thiols (PEG-SH) or allyls (PEG-Al) , covering molecular weights from 3 kDa to 8
kDa were synthesized and thoroughly characterized by 1H NMR,
13C NMR, FT-Raman
and MALDI-TOF techniques. By combining PEG precursors with complementary
trifunctional crosslinkers, a library of well-defined single-network hydrogels was
efficiently constructed via the robust UV-initiated thiol-ene coupling (TEC) chemistry.
Novel sequential interpenetrating network (seqIPN) hydrogels based on PEG were
fabricated by diffusing and afterwards crosslinking secondary-network precursors within
dense (2 kDa) to loose (8 kDa) primary networks. The impacts of polymer chain length
and diffusion time on the swelling and mechanical properties were assessed for the seqIPN
hydrogels. Additionally, disperse red 13 decorated PEG 2 kDa and 8 kDa were synthesized
and used as probes to monitor the secondary-network precursor diffusion rate by UV/Vis
spectroscopy.
FT-Raman and leaching tests were conducted to evaluate the efficiency of the TEC
reaction for the development of PEG networks and their gel fractions. All gels were fully
crosslinked within 5 minutes and with the gel fraction above 84%. The chain length of
PEG, location of functional groups of PEGs, solvents, solid content were found to have
directly influence on the mechanical and swelling properties of PEG single-network
hydrogels. The utilization of the diffusion time dependent seqIPN strategy enabled further
freedom to control the swelling and mechanical properties of PEG hydrogels, with the
degree of water swelling ranged from 280 – 870% and the tensile modulus ranging from
1135 kPa to 175 kPa.
Furthermore, the seqIPN strategy was utilized for fiber reinforced free radical polymerized
hydrogels. N, N-dimethylacrylamide (DMA) with crosslinker poly(ethylene glycol)
diacrylate were diffused in bacterial cellulose (BC) aerogel thereafter UV crosslinked to
form BC-DMA hydrogels. FT-Raman and leaching tests were conducted to evaluate the
efficiency of the free radical polymerization and the BC-DMA gel fractions. After UV cure
for 10 minutes, robust DMA networks were formed within BC aerogels with over 94% gel
fraction. The high porosity and robust interpenetrating DMA network within BC fibers
were further analysed with FE-SEM. Compression tests showed that fiber reinforced DMA
hydrogels have higher compression modulus than DMA hydrogels, ranging from 4.4 to 8.3
MPa with water content from 78 to 70%.

SAMMANFATTNING
Under de senaste decennierna har hydrogeler studerats som nytt mjukt material och även
tillämpats i biomedicinska applikationer såsom produktion av läkemedel och inom
vävnadsteknik.
I detta arbete har bifunktionell poly(etylenglykol) (PEG) med antingen tioler (PEG-SH 3 kDa till 8 kDa) eller allyler (PEG-Al 3 kDa) syntetiserats och karakteriserats med hjälp av
tekniker såsom 1H-NMR,
13C-NMR, FT-Raman samt MALDI-TOF. Kompletterande
trifunktionella tvärbindare användes för att tvärbinda dessa bifunktionella PEG-system till
väldefinierade nätverk (hydrogeler) med hjälp av robust UV-initierad tiol-en kemi (TEC).
Av PEG-Al-systemet (2 kDa till 8 kDa) tillverkades även sekventiella interpenetrerande
nätverk (seqIPN), där monomanerna till det sekundära nätverket fick diffundera in i ett
tidigare tvärbundet nätverk, varpå det sekundära nätverket också tvärbands. Påverkan av
diffusionstiden hos det sekundära nätverket på seqIPN hydrogelernas egenskaper
utvärderades. Vidare utnyttjades disperse red 13-funktionaliserad PEG 2 kDa och 8 kDa
(PEG-röd) för att utvärdera diffusionshastigheten hos det sekundära nätverket med hjälp av
UV/Vis spektroskopi. Mätningar visade att diffusionshastigheten påverkades av tätheten
hos det primära nätverket, med en lägre diffusionshastighet för tätare tvärbundna nätverk.
Effektiviteten hos TEC-reaktionen utvärderades genom att studera omsättningen och
gelfraktionen av de tillverkade hydrogelerna med FT-Raman och urlakning av oreagerad
monomer. Studien visade att samtliga geler var helt tvärbundna efter 5 minuter med en
gelfraktion på över 84 %. Det visade sig även att längden på PEG-kedjan, placeringen av
funktionella grupper längs PEG-kedjan, vilket lösningsmedel som användes samt andelen
fast material i gelerna hade en direkt inverkan på både svällningsegenskaperna och de
mekaniska egenskaperna hos singelnätverk av PEG. Genom att utnyttja den
diffusionsstyrda seqIPN-strategin erhölls större frihet att kontrollera svällningen och de
mekaniska egenskaperna hos hydrogelerna. Detta resulterade i geler med en svällning i
vatten från 280 till 870 % samt en E-modul mellan 175 och 1135 kPa.
SeqIPN-metoden användes även för att tillverka fiberarmerade friradikalpolymeriserade
hydrogeler. N, N-dimetylakrylamid (DMA) och poly(etylenglykol)diakrylat fick
diffundera in i aerogeler av bakteriell cellulosa (BC), varpå systemet tvärbands genom
exponering för UV-ljus och fiberförstärkta BC-DMA-hydrogeler bildades. Rena
polyDMA-hydrogeler (DMA) tillverkade med samma tvärbindningsdensitet användes som
referens. Omsättningen och gelfraktionen hos nätverk av polyDMA utvärderades med FT-
Raman och urlakning. Efter UV-härdning i 10 minuter erhölls robusta nätverk av DMA i
BC-aerogeler med gelfraktioner på över 94 %. Den höga porositeten hos IPN av DMA i
BC-aerogeler analyserades sedan med FE-SEM. Kompressionstest visade att de
fiberförstärkta nätverken hade bättre kompressionegenskaper än hydrogeler av endast DMA. Mätningar visade att BC-DMA hydrogelerna hade kompressionsmoduler mellan 4,4
och 8,3 MPa samt en vattenhalt mellan 78 och 70 %.

献给爷爷
Dedicated to My Grandfather

LIST OF PAPERS
This thesis is a summary of the following papers:
I. ‘Characterization of Well-Defined Poly(Ethylene Glycol) Hydrogels
Prepared By Thiol-Ene Chemistry’ Yang, T.; Long, H.; Malkoch, M.;
Gamstedt, E. K.; Berglund, L.; Hult, A. Journal of Polymer Science Part A:
Polymer Chemistry 2011, 49, 4044–4054.
II. ‘Sequential Interpenetrating PEG Hydrogels Prepared by UV initiated Thiol-
Ene Coupling Chemistry’ Yang, T.; Malkoch, M.; Hult, A. Journal of
Polymer Science Part A: Polymer Chemistry 2012, published. DOI:
10.1002/pola.26393.
III. ‘The Influence of Diffusion Time on the Properties of Sequential
Interpenetrating PEG Hydrogels’ Yang, T.; Malkoch, M.; Hult, A. Journal of
Polymer Science Part A: Polymer Chemistry 2012, submitted.
IV. ‘Mechanical properties of N, N-dimethylacrylamide hydrogels reinforced
with bacterial cellulose aerogel’ Yang, T.; Hult, A. manuscript.
This thesis contains unpublished results.

ABBREVIATIONS
AEOBA 4-(2-(allyloxy)ethoxy)-4-oxobutanoic acid
Al Allyl
BC Bacterial cellulose
BC-DMA Bacterial cellulose reinforced n, n-dimethyl acrylamide hydrogel
C2,r Mass concentration of polymer in solution before crosslinking Cn Characteristic ratio of the polymer
CDCl3 Deuterated chloroform
Da Daltons
DCC Dicyclohexylcarbodiimide
DCM Dichloromethane
DMA N, N-dimethyl acrylamide
DMAP 4-dimethylaminopyridine
DPTS 4-(dimethylamino)pyridinium 4-toluenesulfonate
E Young’s modulus from tensile test or tensile modulus
Ec Young’s modulus from compression test or compression modulus
EtOH Ethanol
FE-SEM Field emission scanning electron microscopy FRP Free radical polymerization
FT Fourier transform
G Shear modulus
G′ Shear storage modulus
Gf Gel fraction
Gf I Primary network gel fraction
Gf II Secondary network gel fraction
HABA 2-(4-hydroxyphenylazo) benzoic acid
IPN Interpenetrating polymer network
II% Secondary network solid content
Irgacure 651 UV-initiator l Average bond length
Ma Methacrylate
MALDI-TOF MS Matrix-assisted laser desorption/ionization mass spectrometry
Mc Molecular weight between crosslinks
Mc,S Molecular weight between crosslinks estimated from swelling
Mc,T Molecular weight between crosslinks estimated from tensile
modulus
MeOH Methanol
Mn Number molecular weight
Mr Molecular weight of the repeat unit
n Number of repeating units md Weight of dry hydrogel
ms Weight of equilibrium swelling hydrogel
mc Weight of cured hydrogel before dry

NaHSO4 Succinic anhydride, sodium hydrogen sulfate
NMR Nuclear magnetic resonance
PEG Polyethylene glycol
PEG-Al Polyethylene glycol functionalized with allyl group
PEG-Ma Polyethylene glycol functionalized with methacrylate group
PEG-SH Polyethylene glycol functionalized with thiol group
PTSA P-toluene sulfonic acid
Q Volume-swelling ratio
qF After cure weight swelling ratio
qW Equilibrium weight swelling ratio
R Gas constant semiIPN Semi interpenetrating polymer network
seqIPN Sequential interpenetrating polymer network
SH Thiol
T’ Temperature of tensile test
Tdiffusion Secondary network precursors diffusion time
TEC Thiol-ene coupling
THF Tetrahydrofuran
TMP Trimethylolpropane
TTT 1, 3, 5-triallyl-1, 3, 5-triazine-2, 4, 6(1H, 3H, 5H)-triazine
UV Ultra violet
V1 Molar volume of solvent Vis Visible
v2,r Volume fraction of polymer in relaxed gel
v2,s Volume fraction of swollen gel
wt% Weight percent
εB Stress-at-break
ν Poisson’s ratio
ξ Mesh size
ρc Crosslinking density
ρp Polyethylene glycol density
ρsol Solvent density
ρwater Water density
σB Strain-at-break χ Polymer solvent interaction parameter
Specific volume of polymer

TABLE OF CONTENTS
1 PURPOSE OF THE STUDY ................................................................................ 1
2 INTRODUCTION ................................................................................................ 2
2.1 HYDROGELS .................................................................................................... 2 2.2 HYDROGEL CROSSLINKING CHEMISTRY............................................................. 3
2.2.1 Thiol-ene coupling chemistry ................................................................... 3 2.2.1 Functionalized poly(ethylene glycol) ........................................................ 3 2.2.1 Free radical polymerization ..................................................................... 4
2.3 HYDROGEL NETWORKS .................................................................................... 5 2.3.1 Single network ......................................................................................... 5 2.3.2 Interpenetrating polymer networks (IPN) ................................................. 6 2.3.1 Bacterial cellulose (BC)........................................................................... 7 2.3.2 Cellulose-reinforced hydrogels ................................................................ 9
2.4 IMPORTANT PROPERTIES OF HYDROGEL ............................................................ 9 2.4.1 Swelling properties .................................................................................. 9 2.4.2 Mechanical properties ........................................................................... 10
2.5 APPLICATIONS ............................................................................................... 11
3 EXPERIMENTAL .............................................................................................. 13
3.1 MATERIALS ................................................................................................... 13 3.2 INSTRUMENTATION ........................................................................................ 14 3.3 FABRICATION OF PEG HYDROGELS WITH TRIAZINE-BASED CROSSLINKER......... 15 3.4 FABRICATION OF SEQUENTIAL-IPN HYDROGELS WITH TMP-BASED CROSSLINKER18
3.4.1 Preparation of single-network TEC hydrogels ........................................ 18 3.4.2 Preparation of chain length influenced seqIPN TEC hydrogels............... 18 3.4.3 Preparation of diffusion time influenced seqIPN and assessment of the
diffuse rate ........................................................................................................... 20 3.5 PROPERTY ASSESSMENT OF PEG HYDROGELS ................................................. 21
3.5.1 Gel fraction determination by leaching test ............................................ 21 3.5.2 Swelling test .......................................................................................... 22 3.5.3 Structure evaluation .............................................................................. 23
3.5.3.1 Determination of the average molecular weight between crosslinks (Mc,S) and mesh size (ξ) from swelling profile ................................................................................. 24 3.5.3.2 Determination of the average molecular weight between crosslinks (Mc,T) and mesh size (ξ) from the tensile modulus ........................................................................... 24
3.6 FABRICATION OF BACTERIAL-CELLULOSE-REINFORCED HYDROGELS ................ 26 3.6.1 Preparation of BC aerogels ................................................................... 26

3.6.2 Fabrication of DMA and BC-DMA hydrogel .......................................... 26 3.1 PROPERTY ASSESSMENT OF DMA AND BC-DMA HYDROGEL .......................... 28
4 RESULTS AND DISCUSSION .......................................................................... 30
4.1 POLYMER PRECURSORS .................................................................................. 30 4.2 PEG HYDROGELS AND PROPERTY ASSESSMENT ............................................... 30
4.2.1 Influence of solvent and functional group location on hydrogels mechanical
properties ............................................................................................................. 32 4.2.2 Influence of UV-irradiation cure time and PEG chain length on hydrogel
properties ............................................................................................................. 33 4.2.3 PEG-Al and PEG-SH hydrogel structure evaluation............................... 34
4.3 SEQUENTIAL-IPN HYDROGELS PERPARATION AND PROPERTIES ........................ 35 4.3.1 Single-network hydrogels and assessment of their properties .................. 35 4.3.2 PEG chain length influence on seqIPN................................................... 37 4.3.3 Secondary-network diffusion time influence on seqIPN........................... 41
4.4 PROPERTIES OF BACTERIAL-CELLULOSE-REINFORCED DMA HYDROGEL .......... 49
5 CONCLUSIONS ................................................................................................. 56
6 FUTURE WORK ................................................................................................ 58
7 ACKNOWLEDGEMENTS ................................................................................ 59
8 REFERENCES ................................................................................................... 61

Purpose of study
1
1 PURPOSE OF THE STUDY
Hydrogels as novel soft material have been studied for bio-application for over fifty years.
The good biocompatibility of hydrogels emanates from their higher water content (over
70%), but it is also a limitation to the mechanical properties so as the applications. Hereby,
establishing a database relating initial materials, fabrication factors and methods to
swelling and mechanical properties of hydrogels has been the main purpose of this study.
Preparing and assessing well-defined novel hydrogels have been the main part of the study
in which thiol and allyl bi-functionalized poly(ethylene glycol) (PEG) (Mn = 3 kDa to 8k
Da) were used to form hydrogels with the complementary trifunctional crosslinkers via
UV-initiated thiol-ene coupling chemistry. To improve while control the mechanical
properties of PEG hydrogels, sequential interpenetrating networks (seqIPN) were
introduced by diffusing secondary PEG network into dense (2k Da) to loose (8k Da)
crosslinked single networks. To further assess the properties of seqIPN, the influence of
secondary-network diffusion time in dense (2k Da) and loose (8k Da) network hydrogels
was studied. Swelling and tensile tests were conducted to evaluate the swelling and
mechanical properties of PEG hydrogels. Furthermore, PEGs decorated with disperse red
13 were used to investigate the diffusion rate by UV/Vis spectroscopy.
An alternative seqIPN approach was further developed based on fiber reinforced
hydrogels. N, N-dimethylacrylamide (DMA) was used as monomer and combined with
poly(ethylene glycol)diacrylate crosslinker to fabricate hydrogels within the bacterial
cellulose (BC) aerogels via UV-initiated free radical polymerization (BC-DMA).
Compression and rheology tests were conducted to assess the mechanical properties of
BC-DMA hydrogels.

Introduction
2
2 INTRODUCTION
2.1 HYDROGELS
Hydrogels are hydrophilic three-dimensional (3D) networks that are chemically
crosslinked or physically entangled with excellent water swelling capacity.1 On a
molecular level, water in a hydrogel is either bonding to polar hydrophilic groups as ‘bond
water’ or is filling the space between the network chains, pores or voids as ‘free water’.2
As a kind of rapidly developing new material, many scientific reports have been published
since 1870s with focus on the preparation, characterization and applications of hydrogels.2
Hydrogels are characterized as soft material with high water content, which is similar to
soft tissue, so they have good biocompatible properties and have been exploited in many
fields such as food additives, pharmaceuticals, cell culture3 and biomedical implants.4 For
example, polyethylene glycol based hydrogels with water content over 90% are used for
wound healing and N, N-dimethylacrylamide based hydrogels are used for commercial soft
contact lens (Figure 1).
Figure 1. A commercial soft contact lens contains N, N-dimethylacrylamide based
hydrogel.

Introduction
3
2.2 HYDROGEL CROSSLINKING CHEMISTRY
2.2.1 Thiol-ene coupling chemistry
Thiol-ene coupling (TEC) chemistry is an organic reaction between a thiol and an alkene
(Figure 2), which was discovered in 1905.5 The advantages of TEC are high efficiency,
high yielding in the presence of oxygen, tolerance of various solvents and functional
groups.5 In recent years, researchers have considered the TEC reaction to be a "click"
reaction.6 This reaction has been extensively applied in coating,7 nano-printing,8 adhesive
technology9 and in dendrimer chemistry or other well-defined structure syntheses.10 The
TEC addition reaction is typically facilitated by UV irradiation and proceeds through a
thiyl radical species. When TEC utilized in polymer chemistry, the polymerization follows
a free radical, step-growth mechanism.11
Figure 2. Scheme of thiol-ene coupling reaction. R and R’ contains thiol and allyl groups
respectively.
2.2.1 Functionalized poly(ethylene glycol)
Poly(ethylene glycol) (PEG) is a common hydrophilic polymer and characterized as a soft
biocompatible material.3,12,13 The U. S. Food and Drug Administration (FDA) approved
PEG usage in the pharmaceutical, food, and cosmetics industries.14 PEG is a commercial
product made from ethylene oxide with water and ethylene glycol or ethylene glycol
oligomers. PEGs can be functionalized as telechelic polymers for hydrogels preparation.15-
18 Figure 3 shows the chemical structure of PEG and the PEG bi-functionalized by thiol,

Introduction
4
allyl and methacrylate groups, which were used for TEC hydrogels preparation in this
work.
Figure 3. Poly(ethylene glycol) (PEG) and thiol, allyl, methacrylate functionalized PEG.
2.2.1 Free radical polymerization
Free radical polymerization (FRP) is a conventional route to prepare hydrogels.19-21 N, N-
dimethylacrylamide (DMA), which has been studied for biomedical applications,22-24 is a
liquid chemical used as monomer to co-polymerize with crosslinkers into a hydrogel.
Figure 4 shows the chemical structure of a DMA monomer, a homo-polymer of DMA and
a network of DMA after copolymerization with acrylate or methacrylate multifunctional
crosslinkers to form a polymer network. The advantages of FRP are easy preparation, fast
curing and can be performed in an aqueous solution.

Introduction
5
Figure 4. N, N-dimethyl acrylamide (DMA), PolyDMA and a DMA network.
2.3 HYDROGEL NETWORKS
Network is the basic structure of a hydrogel, which is covalently crosslinked or physically
entangled. The network components of hydrogels can be either synthetic or natural, as long
as they absorb and maintain large quantities of water.25
2.3.1 Single network
In the main parts of this work, telechelic polymers based on PEG were used to fabricate
TEC networks. The molecular weight between crosslinks (Mc) is the number average
molecular weight between the crosslinking junctions and is a key component correlated to
diffusion properties of a network.26 The length in between the crosslinking junctions is
mesh size (ξ).27 Figure 5 (a) illustrates an ideal network formed by bi-functional PEG and
complementary tri-functional crosslinkers via TEC, mesh size (ξ) or Mc of this network is
controlled by the polymer chain length.

Introduction
6
Figure 5. (a) TEC network formed by tri-functional crosslinker and bi-functional polymer.
(b) FRP network formed by monomers and bi-functional crosslinker.
Another hydrogel network used in this thesis is FRP network, which is formed by
monomers and bi-functional crosslinkers. Figure 5 (b) illustrates the FRP network that has
a random mesh size or Mc because of the different reactivity between the monomer homo-
polymerization and the monomer and crosslinker copolymerization; besides, the
polymerization can be terminated by different termination mechanisms.28
2.3.2 Interpenetrating polymer networks (IPN)
An interpenetrating polymer networks (IPN) is a system containing multiple physically
interlaced inseparable networks without covalent bonding between them.29 IPN
methodology is an approach to improve hydrogel mechanical properties.30-32 IPNs can be
classified according to two schemes: their synthesis, simultaneous IPN or sequential IPN
(seqIPN), or their structure, full IPN33 and semiIPN.34 Simultaneous IPN is prepared by
forming polymer networks independently in one system via non-interfering reaction
mechanisms,35 which is not included in this work. The seqIPN is prepared by forming the
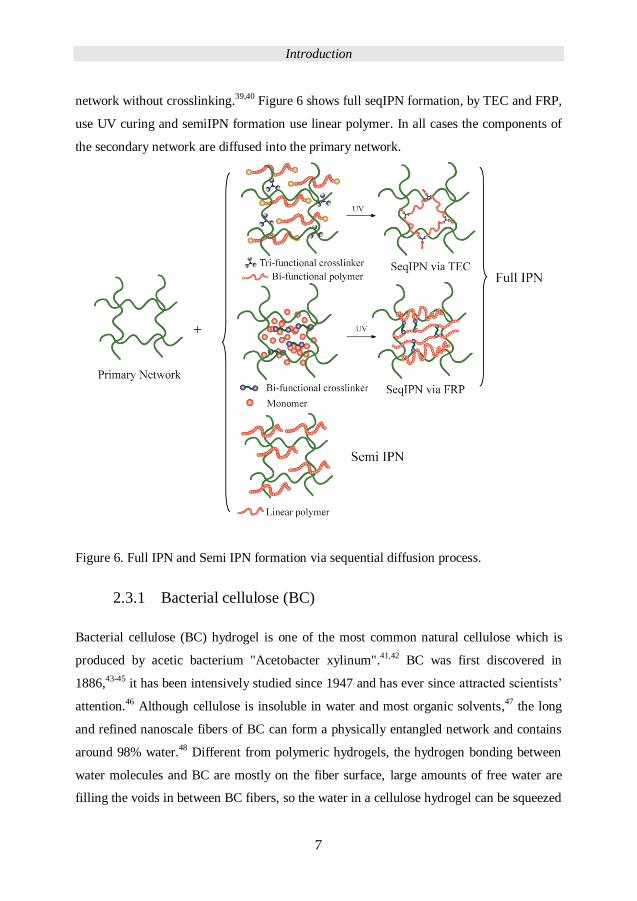
secondary network after the primary network formation by diffusion.36-38
A seqIPN
hydrogel can be prepared by diffusing either polymer precursors or monomers with
crosslinker into a primary network, thereby crosslinking a secondary network within the
primary network. The semiIPN is prepared by diffusing a linear polymer into the primary
Introduction
7
network without crosslinking.39,40 Figure 6 shows full seqIPN formation, by TEC and FRP,
use UV curing and semiIPN formation use linear polymer. In all cases the components of
the secondary network are diffused into the primary network.
Figure 6. Full IPN and Semi IPN formation via sequential diffusion process.
2.3.1 Bacterial cellulose (BC)
Bacterial cellulose (BC) hydrogel is one of the most common natural cellulose which is
produced by acetic bacterium "Acetobacter xylinum".41,42 BC was first discovered in
1886,43-45 it has been intensively studied since 1947 and has ever since attracted scientists’
attention.46 Although cellulose is insoluble in water and most organic solvents,47 the long
and refined nanoscale fibers of BC can form a physically entangled network and contains
around 98% water.48 Different from polymeric hydrogels, the hydrogen bonding between
water molecules and BC are mostly on the fiber surface, large amounts of free water are
filling the voids in between BC fibers, so the water in a cellulose hydrogel can be squeezed
Introduction
8
out easily. The advantages of BC hydrogel are not only green and sustainable49 with high
average molecular weight and crystallinity,50
but also are high dimensional stability after
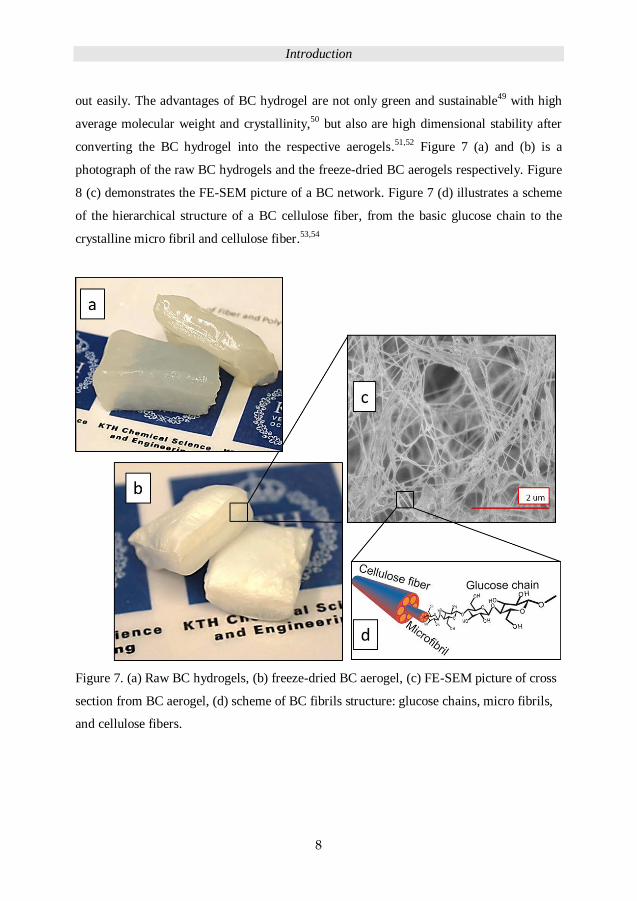
converting the BC hydrogel into the respective aerogels.51,52 Figure 7 (a) and (b) is a
photograph of the raw BC hydrogels and the freeze-dried BC aerogels respectively. Figure
8 (c) demonstrates the FE-SEM picture of a BC network. Figure 7 (d) illustrates a scheme
of the hierarchical structure of a BC cellulose fiber, from the basic glucose chain to the
crystalline micro fibril and cellulose fiber.53,54
Figure 7. (a) Raw BC hydrogels, (b) freeze-dried BC aerogel, (c) FE-SEM picture of cross
section from BC aerogel, (d) scheme of BC fibrils structure: glucose chains, micro fibrils,
and cellulose fibers.

Introduction
9
2.3.2 Cellulose-reinforced hydrogels
Cellulose whiskers have been used as reinforcement to improve the mechanical properties
of hydrogels.55,56 Different from the cellulose whiskers, BC aerogel with the long fiber and
large voids in between the fibers is a suitable scaffold to improve the mechanical
properties of the brittle FRP hydrogels.
2.4 IMPORTANT PROPERTIES OF HYDROGEL
2.4.1 Swelling properties
A crosslinked polymer hydrogels swell but not dissolve when water or a solvent enters it.
The swelling properties, which usually use degree of swelling to define hydrogels, depend
on many factors such as network density, solvent nature, polymer solvent interaction
parameter.1 The properties of water swelling of PEG hydrogels and PEG diffusion in PEG
networks were studied in this work.
Because water acts as a plasticizer in a hydrophilic polymer network system, the swelling
process of the hydrogel can be considered under rubbery state and can be described by the
free energy of mixing ΔGmix from the polymer and solvent interaction and the elastic free
energy ΔGelastic from the crosslinked network:57
ΔGsystem = ΔGmix + ΔGelastic
At the beginning of swelling, the ΔGmix << 0, ΔGelastic > 0, ΔGmix + ΔGelastic < 0, so the
swelling is favoured and the solvent diffuses into the network. During the processing of
swelling, the ΔGmix and ΔGelastic both increased until │ΔGmix │ =│ΔGelastic │ and ΔGsystem =
ΔGmix + ΔGelastic = 0, so that the driving force for swelling is gone: equilibrium swelling is
reached and swelling stops.

Introduction
10
2.4.2 Mechanical properties
Mechanical tests were conducted to assess the hydrogel properties. To establish a library of
mechanical properties of hydrogels is to gather the information of the hydrogel network
and to determine the range of application. 58
Tensile properties were the mainly studied mechanical properties of PEG based hydrogels
in this work. Figure 8 displays a photo of a hydrogel sample in a tensile testing machine.
The hydrogel samples are cut into a dumbbell shape prior to assessment.
Figure 8. Tensile test of a hydrogel sample in an Instron tensile testing machine.
The compression and rheology properties were used to assess the mechanical properties of
fiber-reinforced hydrogels in this work. Rheology is the study of the flow of matter,
measurements being prepared by shearing the sample, as shown in Figure 9.

Introduction
11
Figure 9. Rheology measurements on a hydrogel sample.
2.5 APPLICATIONS
The hydrogels prepared in this work covered a large range of mechanical properties, from
a soft hydrogel such as PEG to a hard hydrogel such as BC-DMA. Because of the high
water content and biocompatibility of hydrogels, many applications are related to
biomedical usage.59,60
Drug delivery is a method of administering a pharmaceutical compound to achieve a
therapeutic effect in or at a certain location in the human body.61,62 Materials used as drug
delivery systems need to have controlled properties, such as absorption and release profile
and no toxicity.63 PEG hydrogels in this work can be used as biodegradable drug delivery
systems, because of the biodegradable ester bond in the structure, the well-controlled
structure and properties;64 most importantly, PEG is a FDA approved polymer for use
inside the human body.65
Because of the low toxicity of its precursors66
and crosslinkers,
PEG can be used to form a hydrogel in situ via TEC. The seqIPN PEG hydrogels can also
change the mechanical properties to suit the applications. Figure 10 pictures a diagram of a
degradable PEG hydrogel as a drug-delivery system.

Introduction
12
Figure 10. Diagram of a drug-delivery system uses a biodegradable PEG hydrogel carrying
drugs and releasing at certain location.
Tissue engineering involves improving or replacing biological functions of the human
body with artificial materials.67,68 Tissue engineering requires certain mechanical and
structural properties for proper functioning for the repair or replacement of portions of the
human body.69 For example, a hydrogel for artificial cartilage needs to have a compression
stress of 0.5 to 10 MPa.70 It needs to be biocompatible and to have interconnected macro-
pores for cell culture.71 Bacterial-cellulose-reinforced DMA hydrogels could be good
candidates for this application.

Experimental
13
3 EXPERIMENTAL
3.1 MATERIALS
PEG (Mn: 2 kDa, 3 kDa, 6 kDa, 8 kDa), trimethylolpropane tris(3-
mercaptopropionate) (TMP-tris-thiol), 2,2-dimethoxy-1,2-diphenylethan-1-one
(IRGACURE 651) and tris[2-(3-mercaptopropionyloxy) ethyl] isocyanurate (TMI),
disperse red 13, 3-mercaptopropionic acid, 99% (MPA), p-toluene sulfonic acid
(PTSA), dicyclohexylcarbodiimide (DCC), 1,3,5-triallyl-1,3,5-triazine-
2,4,6(1H,3H,5H)-triazine (TTT), tris[2-(acryloyloxy)ethyl] isocyanurate13,4-
dimethylaminopyridine, D-glucose, sodium hydroxide, N,N-dimethylacrylamide
(DMA), alpha-ketoglutaric acid, polyethylene glycol diacrylate (Mn=700 Da) were
purchased from Sigma-Aldrich. Chloroform, toluene-4-sulfonic acid monohydrate,
dichloromethane (DCM), diethyl ether, 2-(4-hydroxyphenylazo) benzoic acid
(HABA) were purchased from Fisher Scientific Merck. Ethanol (EtOH, 96% and
100%) was purchased from VWR. Succinic anhydride, sodium hydrogen sulfate
(NaHSO4), 4-dimethylaminopyridine (DMAP), toluene-4-sulfonic acid
monohydrate, diethylether, methanol (MeOH), tetrahydrofuran (THF), toluene and
deuterated chloroform (CDCl3) were purchased from Fisher Scientific Merck. All
the chemicals were used as received. 4-(dimethylamino)pyridinium 4-
toluenesulfonate (DPTS) was prepared as reported elsewhere.72 The synthesis of 4-
(2-(allyloxy)ethoxy)-4-oxobutanoic acid is described in a previous publication.73

Experimental
14
3.2 INSTRUMENTATION
Nuclear Magnetic Resonance (NMR) spectra were recorded with a Bruker Avance
400 MHz instrument using CDCl3 as solvent. The solvent signal was used as
internal standard.
Field Emission Scanning Electron Microscopy (FE-SEM) pictures were collected
from a Hitachi S-4800 scanning electron microscope operated at 1 to 1.5 kV.
MALDI-TOF MS spectra (MALDI-TOF) were conducted on a Bruker UltraFlex
MALDI-TOF MS with SCOUT-MTP Ion Source (Bruker Daltonics, Bremen)
equipped with a N2 laser (337 nm), a gridless ion source and reflector design
FT-Raman spectra were acquired for all samples using a Perkin-Elmer Spectrum
2000 NIR FT-Raman instrument. Each spectrum was based on 128 scans using 800
mW laser powers. The conversion of thiols (2573–2568 cm-1) or vinyl acrylate
(1635 cm-1), allyl (1640-1643 cm-1) was determined using the carbonyl bond (1760-
1763 cm-1) as an internal reference.
UV curing was performed using a Blak-Ray xx-15BlB UV bench lamp for PEG
hydrogels, where the wavelength of the lamp was 365 nm and the intensity of the
UV radiation was 28.5 mW/cm2. Fushion UV cure system model F300 equipped
with Fusion electrodeless bulbs standard type BF9 (Lamp power 300 W/inch, 1800
W total) was used for cuing DMA and BC-DMA hydrogels.
Tensile and compression tests were conducted with a universal testing machine
Instron 5944 with an advanced non-contacting video extensometer (Instron Korea
LLC.) at 23°C and 50% relative humidity using a cross-head speed of 100 mm/min.
For tensile tests, the swollen hydrogels were cut into a dumbbell shape to prevent
unwanted fracture close to the clamping region and to avoid grip slippage.
Compression tests on water swollen hydrogels were performed at 50% relative
humidity and 23°C, using the Miniature materials tester Minimat 2000 with load
cells of 20 N and 200 N.
Rheology tests were performed in TA AR2000 rheometer. An 8 mm sample holder
disk is used for all the samples.

Experimental
15
UV/Vis absorption spectrum was determined by SHIMADZU UV-2550 UV/Vis
spectroscopy. The PEG and PEG-red mixture solution concentration was set to 50
wt% in EtOH. PEG-red-2k (0.05 wt%) and PEG-red-8k (0.01 wt%) as marked
concentration was determined by software UVprobe 2.34.
3.3 FABRICATION OF PEG HYDROGELS WITH TRIAZINE-
BASED CROSSLINKER
To study the formation of the hydrogels using TEC chemistry, PEGs were
functionalized with thiol (PEG-SH) and allyl (PEG-Al) to prepared TEC hydrogels
with the complementary tri-functional triazine based crosslinkers. All the hydrogels
were prepared in 3 to 6 replicas with a thiol to allyl molar ratio set to 1:1 with 3
wt% initiator (Irgacure 651). Solvents and solid content were varied to study the
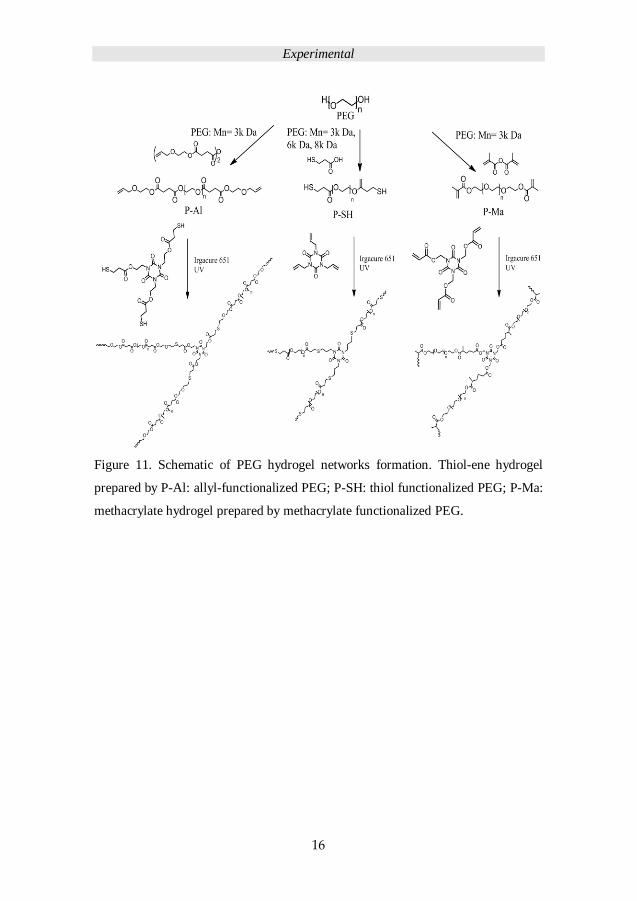
efficient of TEC in different reaction conditions. Figure 11 presents the scheme of
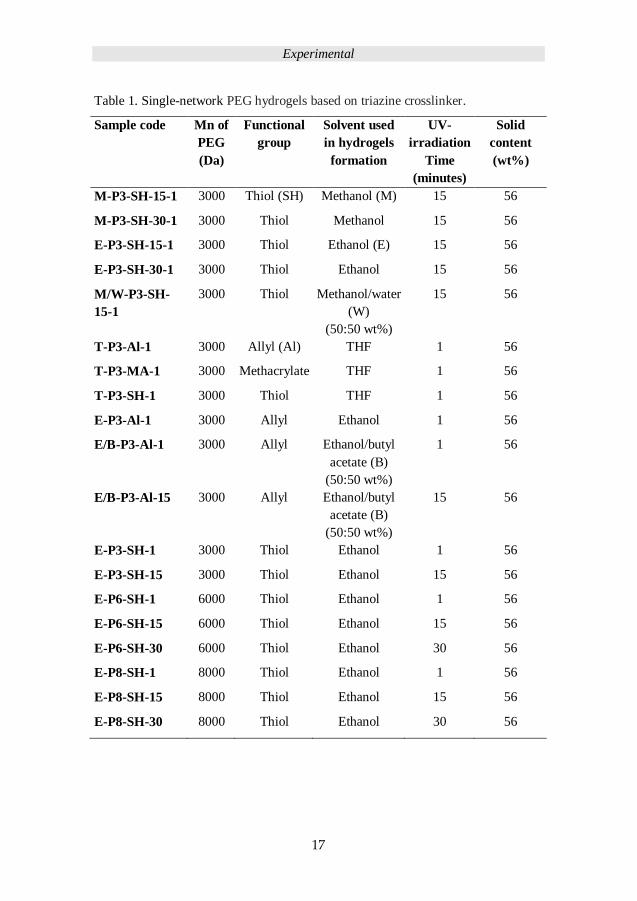
PEG hydrogels fabrication; Table 1 details the sample codes for the single network
hydrogels.
Experimental
16
Figure 11. Schematic of PEG hydrogel networks formation. Thiol-ene hydrogel
prepared by P-Al: allyl-functionalized PEG; P-SH: thiol functionalized PEG; P-Ma:
methacrylate hydrogel prepared by methacrylate functionalized PEG.
Experimental
17
Table 1. Single-network PEG hydrogels based on triazine crosslinker.
Sample code Mn of
PEG
(Da)
Functional
group
Solvent used
in hydrogels
formation
UV-
irradiation
Time
(minutes)
Solid
content
(wt%)
M-P3-SH-15-1 3000 Thiol (SH) Methanol (M) 15 56
M-P3-SH-30-1 3000 Thiol Methanol 15 56
E-P3-SH-15-1 3000 Thiol Ethanol (E) 15 56
E-P3-SH-30-1 3000 Thiol Ethanol 15 56
M/W-P3-SH-
15-1
3000 Thiol Methanol/water
(W)
(50:50 wt%)
15 56
T-P3-Al-1 3000 Allyl (Al) THF 1 56
T-P3-MA-1 3000 Methacrylate THF 1 56
T-P3-SH-1 3000 Thiol THF 1 56
E-P3-Al-1 3000 Allyl Ethanol 1 56
E/B-P3-Al-1 3000 Allyl Ethanol/butyl
acetate (B)
(50:50 wt%)
1 56
E/B-P3-Al-15 3000 Allyl Ethanol/butyl
acetate (B)
(50:50 wt%)
15 56
E-P3-SH-1 3000 Thiol Ethanol 1 56
E-P3-SH-15 3000 Thiol Ethanol 15 56
E-P6-SH-1 6000 Thiol Ethanol 1 56
E-P6-SH-15 6000 Thiol Ethanol 15 56
E-P6-SH-30 6000 Thiol Ethanol 30 56
E-P8-SH-1 8000 Thiol Ethanol 1 56
E-P8-SH-15 8000 Thiol Ethanol 15 56
E-P8-SH-30 8000 Thiol Ethanol 30 56

Experimental
18
3.4 FABRICATION OF SEQUENTIAL-IPN HYDROGELS
WITH TMP-BASED CROSSLINKER
3.4.1 Preparation of single-network TEC hydrogels
Four molecular weights PEGs (2 kDa, 3 kDa, 6 kDa and 8 kDa), equimolar ratio
(1:1) with crosslinker TMP-tris-thiol and Irgacure 651 (3 wt%) were dissolve in
ethanol (EtOH) (50 wt% solid content) respectively to prepare single-network
hydrogels. Using TMP based crosslinker TMP-tris-thiol instead of triazine based
crosslinker was because the better solubility in EtOH for the hydrogel preparation
process. Each mixture was vortexed to homogeneous and poured into a Teflon mold
(thickness: 0.10 cm, width: 1.0 cm) and was covered with a glass slide to prevent
EtOH evaporation. The mixture solution was allowed to gel for 5 minutes under UV
exposure (365 nm, 28.5 mW/cm intensity, Blak-Ray xx-15BlB UV bench lamp) at
room temperature. A small part of the cured hydrogel was cut and dried overnight
for gel fraction tests. The rest of the hydrogels were immersed in deionized water
for 5 hours and immersed in ethanol (100%) for 3 hours to remove residues. The
cleaned hydrogels were dried in air overnight and then placed in a vacuum oven at
40 °C for 1 hour. The single-network hydrogel sample code is S2 prepared from
PEG-Al, Mn = 2 kDa (PEG-Al-2k); hydrogel S3 prepared from PEG-Al, Mn = 3
kDa; hydrogel S6 prepared from PEG-Al, Mn = 6 kDa; and hydrogel S8 prepared
from PEG-Al, Mn = 8 kDa, all hydrogels were prepare in 6 to 10 replicas, the S8
hydrogels were prepared in at least 12 replicas.
3.4.2 Preparation of chain length influenced seqIPN TEC
hydrogels
The dried single-network hydrogels, S2, S3, S6 and S8, were used as primary
network to prepare seqIPN hydrogels. All the primary networks were immersed in
ethanol solutions containing (50 wt% solid content) PEG-Al precursors, crosslinker
TMP-tris-thiol and initiator (3 wt%), the molar ratio of thiol to allyl was set to 1:1.

Experimental
19
The precursors in solutions were allowed to diffuse into the primary networks for 2
hours at 40 °C. The excess of solution on the surface of primary network hydrogels
was removed with tissue paper and was placed in a Teflon mold covered by a glass
slide to prevent solvent evaporation. The secondary networks were allowed to
crosslink for 5 minutes under UV exposure (365nm, 28.5 mW/cm). The crosslinked
seqIPN hydrogels were dried in air overnight and then placed in a vacuum oven at
40 °C for 1 hour. Figure 12 shows the scheme of seqIPN hydrogel preparation.
Figure 12. Preparation of seqIPN, using allyl functionalized PEG (Mn= 2k, 3k, 6k,
8k Da).

Experimental
20
3.4.3 Preparation of diffusion time influenced seqIPN and
assessment of the diffuse rate
Dense and loose single network, 2 kDa (S2) and 8 kDa (S8); hydrogels were used to
assess the diffusion time influence on mechanical properties of seqIPN hydrogels.
S2 and S8 hydrogels were immerged in the ethanol solution of PEG-Al precursors
(2 kDa and 8 kDa) with crosslinker TMP-tris-thiol and initiator (50 wt% solid
content) in an oven at 40°C for 2h, 4h, 20h and 44 hours diffusion time. The
secondary networks were allowed to cure for 5 minutes under UV exposure (365
nm, 28.5 mW/cm2). All seqIPNs were prepared in 12 replicas with a thiol to allyl
molar ratio set to 1:1 with 3 wt% initiator to assess the properties at totally four
diffusion times. The cured seqIPN hydrogels with four different diffusion times
were dried in air overnight and then placed in a vacuum oven at 40°C for 1 hour
respectively.
PEG 2kDa and 8 kDa were dissolved in EtOH to prepare 54.5 mg/mL and 138 mg/l
PEG solution with PEG-red-2k (0.05 wt%) and PEG-red-8k (0.01 wt%)
respectively. S2 and S8 hydrogels were immersed in a 1 mL PEG-red-2k and PEG-
red-8k solution respectively. A total of 24 pieces of samples were placed in an oven
at 40 ° C, the remaining solutions were used to analyse the PEG diffusion after 2, 4,
20, 25, 28 and 44 hours by UV/Vis absorption spectroscopy.
The scheme of diffusion time dependence seqIPN and semiIPN preparation is
demonstrated in Figure 13.

Experimental
21
Figure 13. Preparation of diffusion time dependent IPNs and semi IPN hydrogel
from 2 kDa and 8 kDa PEG.
3.5 PROPERTY ASSESSMENT OF PEG HYDROGELS
3.5.1 Gel fraction determination by leaching test
Leaching tests were conducted to identify any unreacted starting materials and to
determine the gel fraction. The hydrogels were dried in the vacuum oven to remove
the solvent directly after curing, totally three replicas of each hydrogel sample
(thickness: 0.10 cm, length: 1.0 cm, width: 1.0 cm) were weighed (dry weight M)
and immersed in chloroform, one in deuterated chloroform for NMR analysis. All

Experimental
22
samples were swelled for 5 hours with three cycles of solvent exchange. The
leached hydrogels were dried overnight in air and placed in a 40°C vacuum oven
for half hour to remove water and the dried weight was measured as M’. The
leachate in deuterated chloroform was analyzed by NMR to determine remaining
functional groups. The gel fraction of the hydrogel was calculated by the following:
Gel fraction = M’/M ×100% (1)
where M is the mass of the dry hydrogels after cure and M' is the mass of the dry
hydrogels after leaching.
3.5.2 Swelling test
Three or four replicas of each dried hydrogel were swollen in deionized water at
room temperature for 3 days to achieve equilibrium swelling. The degree of
swelling of hydrogels were measured after 5 min, 10 min, 20 min, 0,5 h, 1,5 h, 1
day, 2 days and 3 days. Totally three replicas were measured, the standard
deviations were marked with error bars in the swelling profile charts. The degree of
swelling was calculated as the following:
Degree of swelling = [(Wet weight – Dry weight) / Dry weight] ×100% (2)
The water content of hydrogels were calculated after the equilibrium swelling by
Water content = (Wet weight / Dry weight) ×100% (3)
The molecular weight between cross-links (Mc), effective crosslinking density (ρ),
and mesh size (ξ) were estimated according to the equilibrium swelling result.74
The water-induced volume-swelling ratio75 of hydrogel was calculated as:

Experimental
23
(4)
where v2,r is the volume fraction of polymer in the relaxed gel (hydrogels after cure,
before dry) and v2,s is the volume fraction polymer of swollen gel. These variables
are related to weight fractions by:
,
(5)
[
]
(6)
and [
]
(7)
where is the weight swelling ratio of hydrogels after equilibrium swelling, is
the weight of equilibrium swollen gel, is the weight of dry gel, is the weight
of gel after curing, is the weight ratio after curing. The PEG density is denoted
(1.12 g/cm3), is the density of water (1.00 g/cm3), and is the density
of solvent used in hydrogel cure process (EtOH: 0.79 g/cm3, EtOH/butyl acetate:
0.93 g/cm3).76
3.5.3 Structure evaluation
The structure evaluation is based on swelling and tensile tests result, the average
molecular weight between crosslinks (Mc) and mesh size (ξ) of PEG hydrogel were
calculated. Chain lengths between cross-links of PEG networks were assumed to
follow Gaussian distribution and the equilibrium swelling theory of Flory for
crosslinked polymers.

Experimental
24
3.5.3.1 Determination of the average molecular weight between crosslinks (Mc,S)
and mesh size (ξ) from swelling profile
The Mc,S was calculated with the following equation where ‘S’ indicates swelling77
–
( ) ( ) ( )
( ) (
)
–
(8)
where Mn is the number-average molecular weight of the polymer, is the specific
volume of polymer (0.84 cm3/g for PEG),78 V1 is molar volume of solvent (18
cm3/mol for water) χ is the polymer solvent interaction parameter (0.43 for PEG -
water)76 and is assumed constant in this work.
3.5.3.2 Determination of the average molecular weight between crosslinks (Mc,T)
and mesh size (ξ) from the tensile modulus
Due to the isotropic homogeneous crosslinking of PEG hydrogels, the shear
modulus G can be calculated from the tensile Young’s modulus E according to79:
(9)
where v is Poisson's ratio. When the hydrogel material is fully swollen, its
mechanical behavior is similar to that of an incompressible rubber-like material.
Hence, the Poisson ratio of the hydrogels was assumed to be 0.5 in this work.80
Mc,T, where ‘T’ indicates tensile stiffness,77 was calculated from the equation:
(10)
where C2,r is the mass concentration of polymer in solution before crosslinking (0.9
kg/l in ethanol, 0.86 kg/l in ethanol/butyl acetate mixture), is the gas constant

Experimental
25
(8.31 kPa l/mol K), and T’ is the temperature 298 K at which the tensile testing was
carried.
The crosslinking density ρc was determined 81 by:
(11)
The mesh size ξ was then determined as the root-mean-square of the end-to-end
distance of the polymer chain in the unperturbed state is
(12)
where l is the average bond length (0.146 nm), is the characteristic ratio of the
polymer (typically 4.0 for PEG)82 and n is the number of repeating units in the
crosslink:
(13)
where is the molecular weight of the repeating unit (44 for PEG). The mesh size
ξ can then be expressed as82
(14)
With the above expressions, structural evaluation of the molecular network can be
estimated by tensile measurements and swelling.

Experimental
26
3.6 FABRICATION OF BACTERIAL-CELLULOSE-
REINFORCED HYDROGELS
3.6.1 Preparation of BC aerogels
The bacterial cellulose (BC) was obtained by cultivating the bacterium Acetobacter
in a pre-culture HS medium. This cultivating medium was prepared by pre-
cultivating Acetobacter aceti strain in the Hestrin–Schramm (HS) medium 27 for 7
days at 27 °C. This pre-culture medium (5 mL) was used to inoculate 30 mL of
fresh HS medium for bacterium cultivation. The BC hydrogels were harvested after
7 days of culture at 27 °C under static conditions. They were treated with 0.1 M
NaOH at 80°C for 3 h and washed with de-ionized water. This process was repeated
3 times and the BC hydrogels were finally washed with de-ionized water for several
days until neutrality was reached. After the purified BC has been obtained, scissors
was used to cut the bacterial cellulose into 1x1x1 cm cubic shape and they were
then freeze-dried for 2 days to prepare the BC aerogels.
3.6.2 Fabrication of DMA and BC-DMA hydrogel
N, N-dimethyl acrylamide (DMA) hydrogel was prepared by FRP in aqueous
solution with polyethylene glycol diacrylate (Mn= 700 Da, PEGDA) as crosslinker
and 2-oxo-ketoglutaric acid (5 wt%) as a UV initiator. The DMA monomer (10, 40
and 70 wt%) and crosslinker PEGDA (2 or 4 mol%) were cured in aqueous solution
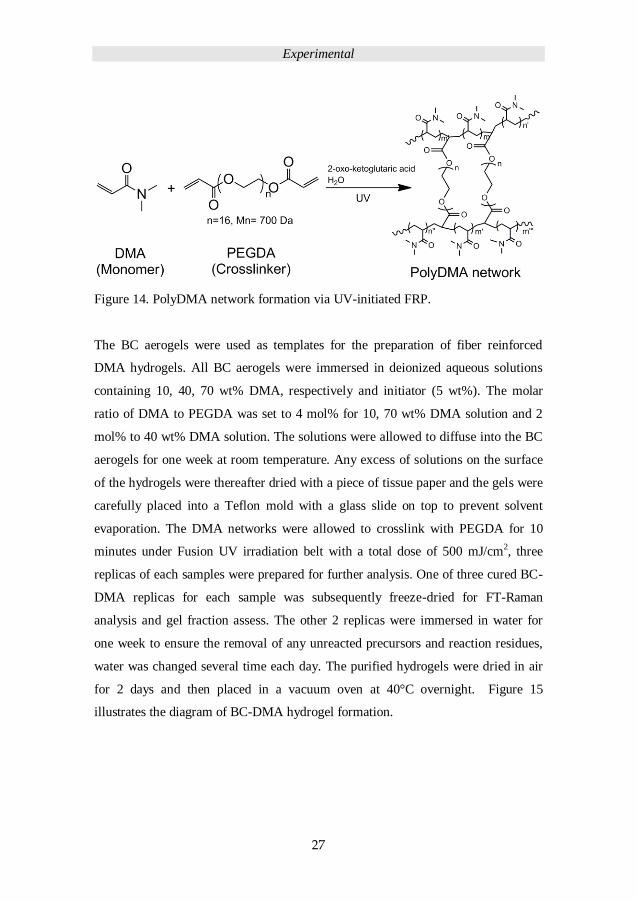
under 10 minutes UV irradiation (Lamp powder 300 W/inch). Figure 14 presents a
diagram of DMA and its network chemical structure.
Experimental
27
Figure 14. PolyDMA network formation via UV-initiated FRP.
The BC aerogels were used as templates for the preparation of fiber reinforced
DMA hydrogels. All BC aerogels were immersed in deionized aqueous solutions
containing 10, 40, 70 wt% DMA, respectively and initiator (5 wt%). The molar
ratio of DMA to PEGDA was set to 4 mol% for 10, 70 wt% DMA solution and 2
mol% to 40 wt% DMA solution. The solutions were allowed to diffuse into the BC
aerogels for one week at room temperature. Any excess of solutions on the surface
of the hydrogels were thereafter dried with a piece of tissue paper and the gels were
carefully placed into a Teflon mold with a glass slide on top to prevent solvent
evaporation. The DMA networks were allowed to crosslink with PEGDA for 10
minutes under Fusion UV irradiation belt with a total dose of 500 mJ/cm2, three
replicas of each samples were prepared for further analysis. One of three cured BC-
DMA replicas for each sample was subsequently freeze-dried for FT-Raman
analysis and gel fraction assess. The other 2 replicas were immersed in water for
one week to ensure the removal of any unreacted precursors and reaction residues,
water was changed several time each day. The purified hydrogels were dried in air
for 2 days and then placed in a vacuum oven at 40°C overnight. Figure 15
illustrates the diagram of BC-DMA hydrogel formation.

Experimental
28
Figure 15. Scheme of BC-DMA hydrogel networks formation.
3.1 PROPERTY ASSESSMENT OF DMA AND BC-DMA
HYDROGEL
To obtain the gel fraction of DMA network a leaching study was conducted. One
replica of 3 samples freeze-dried BC-MDA hydrogel with mass (M) weighed (170
to 230 mg) including any unreacted starting materials was immersed in 200 mL of
deionized water. The deionized water was exchanged every day for one week. All
swollen hydrogels were air dried for 2 days and placed at 50°C in a vacuum oven
overnight. The fully leached and dried sample mass M’ was recorded. The gel
fraction (crosslinking efficiency) of all samples was acquired from mass before and
after leaching using the formula below:
Gel fraction = (M’/M) ×100% (15)
Content of the DMA network within the BC-DMA hydrogel was calculated from
the mass increase after leaching the samples from any unreacted precursors. In this

Experimental
29
case, the dried BC aerogel mass is marked as MBC, the dried BC-DMA mass is
marked as MBC-DMA, enabling the calculation of the secondary-network content:
DMA network content = [(MBC-DMA – MBC) / MBC-DMA] × 100% (16)
Water content of the swollen DMA and BC-DMA hydrogels were collected on
three replicas. The hydrogel samples were dried in air for 2 days and further dried at
50°C in vacuum oven overnight. The drying procedure was completed upon
reaching constant mass loss values. Then after the samples were immersed in
deionized water for one week and the wet weight of each sample was recorded, the
water content was calculated from the formula:
Water content = (Wet weight / Dry weight) ×100% (17)

Results and Discussion
30
4 RESULTS AND DISCUSSION
4.1 POLYMER PRECURSORS
PEGs were functionalized with thiol (3 kDa to 8 kDa) (PEG-SH), allyl (3 kDa) (PEG-Al),
and methacrylate PEG (3 kDa). PEG based precursors were used to study the effect of
chain length, location of functional groups of PEGs, solvents, solid content of TEC
hydrogels. PEG-Al (2 kDa to 8 kDa) was prepared to study the effect of chain length on
single and seqIPN hydrogels. PEG-Al (2 kDa and 8 kDa) was prepared to study the effect
of diffusion time of seqIPN hydrogels. Additionally, PEGs (2 kDa and 8 kDa) were
decorated with disperse red 13 to probe the diffusion rate of PEGs in single-network
hydrogels. 1H NMR and 13C NMR and MALDI-TOF were used to confirm the fully
functionalization of PEG precursors. FT-Raman revealed the S-H stretching peak at 2571
cm-1 and C=C stretching at 1645cm-1. The UV/Vis absorption spectrum of disperse red 13
evidenced a characteristic absorption peak at 503 nm.
4.2 PEG HYDROGELS AND PROPERTY ASSESSMENT
All PEG hydrogels were prepared with a ratio of PEG functional group to crosslinker
functional group of 1:1 with 3wt% UV-initiator. The UV crosslinking of the three systems
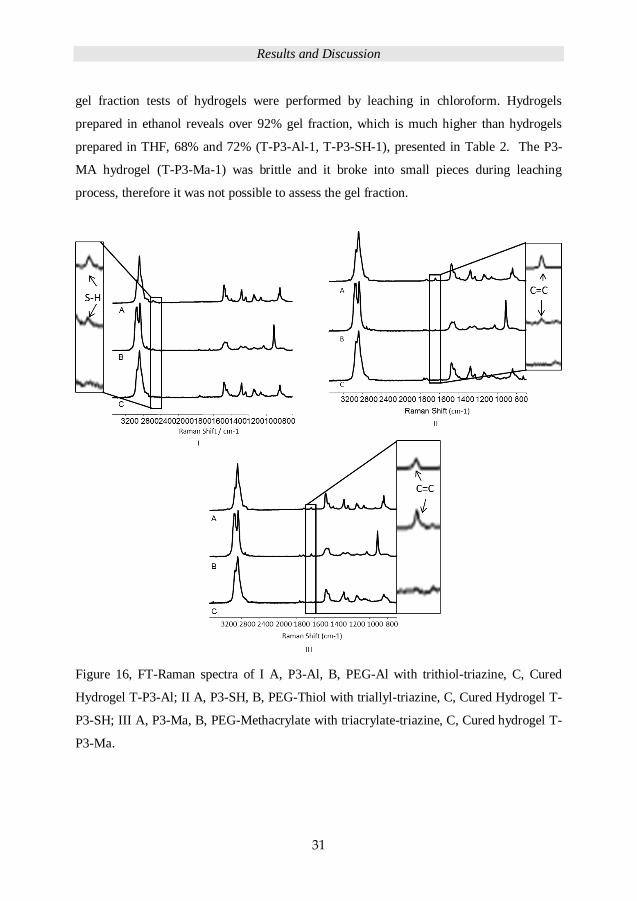
was initially conducted in THF (56 wt%). The FT-Raman spectroscopy was used to assess
the conversion for P3-Al, P3-SH, and P3-MA after 1 minute UV-irradiation time. As
shown in Figure 16(I) there is no trace of the thiol stretching peak at 2570 cm-1 for the
PEG-SH hydrogel and no trace of allyl C=C stretching peak at 1645 cm-1 from PEG-Al
hydrogel, Figure 16 (II) and not for PEG-Ma hydrogel at 1645 cm-1, Figure 16 (III). The
Results and Discussion
31
gel fraction tests of hydrogels were performed by leaching in chloroform. Hydrogels
prepared in ethanol reveals over 92% gel fraction, which is much higher than hydrogels
prepared in THF, 68% and 72% (T-P3-Al-1, T-P3-SH-1), presented in Table 2. The P3-
MA hydrogel (T-P3-Ma-1) was brittle and it broke into small pieces during leaching
process, therefore it was not possible to assess the gel fraction.
Figure 16, FT-Raman spectra of I A, P3-Al, B, PEG-Al with trithiol-triazine, C, Cured
Hydrogel T-P3-Al; II A, P3-SH, B, PEG-Thiol with triallyl-triazine, C, Cured Hydrogel T-
P3-SH; III A, P3-Ma, B, PEG-Methacrylate with triacrylate-triazine, C, Cured hydrogel T-
P3-Ma.

Results and Discussion
32
Table 2. Gel fraction of hydrogels prepared by TEC and FRP. Abbreviation: T:
tetrahydrofuran, E: ethanol, E/B: ethanol/ butyl acetate (50:50 wt%), 1: UV irradiation
time: 1 minute; 15: UV irradiation time: 15 minutes; 30: UV irradiation time: 30 minutes.
Sample
code
T-P3-
Al-1
T-P3-
SH-1
T-P3-
Ma-1
E-P3-
Al-1
E/B-P3-
Al-1
E/B-P3-
Al-15
E-P3-
SH-1
E-P3-
SH-15
E-P6-
SH-1
E-P6-
SH-15
E-P8-
SH-1
E-P8-
SH-15
E-P8-
SH-30
Gel
fraction
(%)
68 72 - 95 92 97 99 99 99 99 99 99 99
4.2.1 Influence of solvent and functional group location on
hydrogels mechanical properties
The influence of solvent and solvent content using during polymerization on the hydrogel
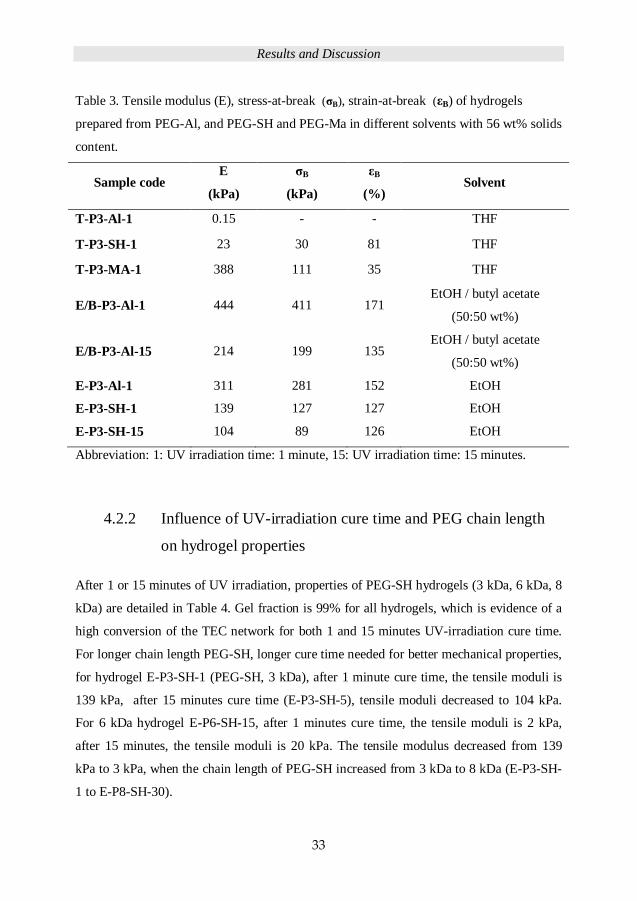
network formation was studied. Table 3 presents the properties of hydrogels prepared with
56 wt% solid content. The hydrogel (E/B-P3-Al-1) prepared in EtOH/ butyl acetate
exhibits a tensile modulus of 444, which is much higher than the hydrogels prepared in
THF (T-P3-Al-1, 0.15 kPa). For hydrogels, PEG-Al system hydrogels E-P3-Al-1 revealed
higher mechanical properties compare to the same chain length PEG-SH hydrogels E-P3-
SH-1, which were assessed in this work. Because thiol-ene reaction has one side reaction
that is thiyl-thiyl radical coupling into disulfide, which was revealed to have more
influence of PEG-SH based hydrogels than PEG-Al based hydrogels. The disulfide
formation between PEG-SH based hydrogel E-P3-SH-1 (tensile moduli 139 kPa)
decreased the network density and demonstrated lower mechanical properties compare to
PEG-Al hydrogel E-P3-Al-1 (tensile moduli 311 kPa).
Results and Discussion
33
Table 3. Tensile modulus (E), stress-at-break (σB), strain-at-break (εB) of hydrogels
prepared from PEG-Al, and PEG-SH and PEG-Ma in different solvents with 56 wt% solids
content.
Sample code E
(kPa)
σB
(kPa)
εB
(%) Solvent
T-P3-Al-1 0.15 - - THF
T-P3-SH-1 23 30 81 THF
T-P3-MA-1 388 111 35 THF
E/B-P3-Al-1 444 411 171 EtOH / butyl acetate
(50:50 wt%)
E/B-P3-Al-15 214 199 135 EtOH / butyl acetate
(50:50 wt%)
E-P3-Al-1 311 281 152 EtOH
E-P3-SH-1 139 127 127 EtOH
E-P3-SH-15 104 89 126 EtOH
Abbreviation: 1: UV irradiation time: 1 minute, 15: UV irradiation time: 15 minutes.
4.2.2 Influence of UV-irradiation cure time and PEG chain length
on hydrogel properties
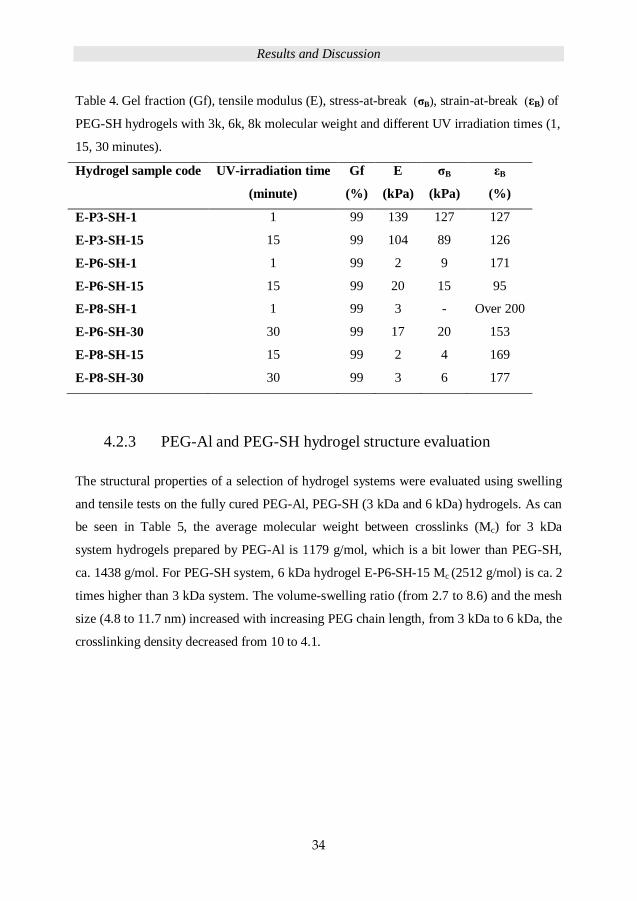
After 1 or 15 minutes of UV irradiation, properties of PEG-SH hydrogels (3 kDa, 6 kDa, 8
kDa) are detailed in Table 4. Gel fraction is 99% for all hydrogels, which is evidence of a
high conversion of the TEC network for both 1 and 15 minutes UV-irradiation cure time.
For longer chain length PEG-SH, longer cure time needed for better mechanical properties,
for hydrogel E-P3-SH-1 (PEG-SH, 3 kDa), after 1 minute cure time, the tensile moduli is
139 kPa, after 15 minutes cure time (E-P3-SH-5), tensile moduli decreased to 104 kPa.
For 6 kDa hydrogel E-P6-SH-15, after 1 minutes cure time, the tensile moduli is 2 kPa,
after 15 minutes, the tensile moduli is 20 kPa. The tensile modulus decreased from 139
kPa to 3 kPa, when the chain length of PEG-SH increased from 3 kDa to 8 kDa (E-P3-SH-
1 to E-P8-SH-30).
Results and Discussion
34
Table 4. Gel fraction (Gf), tensile modulus (E), stress-at-break (σB), strain-at-break (εB) of
PEG-SH hydrogels with 3k, 6k, 8k molecular weight and different UV irradiation times (1,
15, 30 minutes).
Hydrogel sample code UV-irradiation time
(minute)
Gf
(%)
E
(kPa)
σB
(kPa)
εB
(%)
E-P3-SH-1 1 99 139 127 127
E-P3-SH-15 15 99 104 89 126
E-P6-SH-1 1 99 2 9 171
E-P6-SH-15 15 99 20 15 95
E-P8-SH-1 1 99 3 - Over 200
E-P6-SH-30 30 99 17 20 153
E-P8-SH-15 15 99 2 4 169
E-P8-SH-30 30 99 3 6 177
4.2.3 PEG-Al and PEG-SH hydrogel structure evaluation
The structural properties of a selection of hydrogel systems were evaluated using swelling
and tensile tests on the fully cured PEG-Al, PEG-SH (3 kDa and 6 kDa) hydrogels. As can
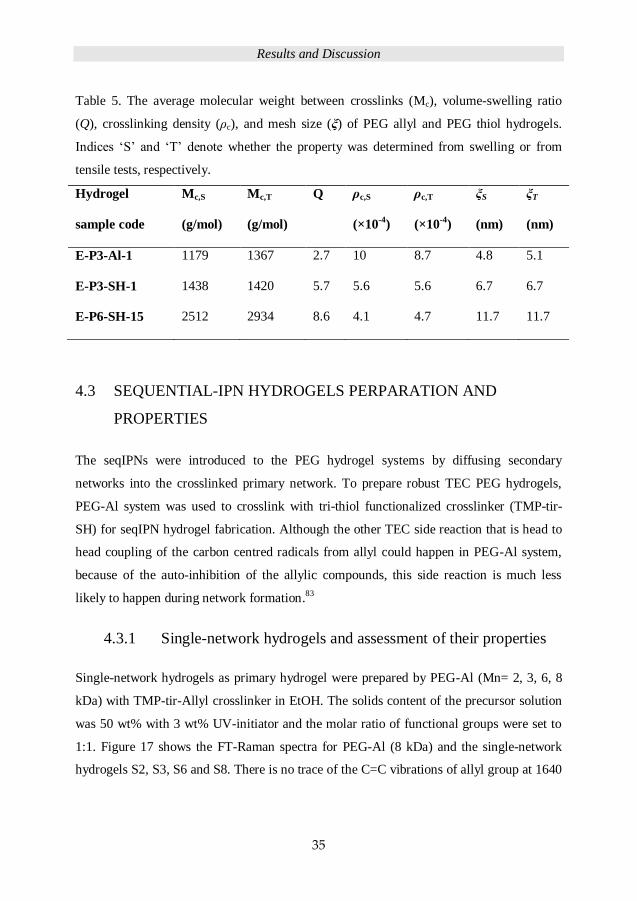
be seen in Table 5, the average molecular weight between crosslinks (Mc) for 3 kDa
system hydrogels prepared by PEG-Al is 1179 g/mol, which is a bit lower than PEG-SH,
ca. 1438 g/mol. For PEG-SH system, 6 kDa hydrogel E-P6-SH-15 Mc (2512 g/mol) is ca. 2
times higher than 3 kDa system. The volume-swelling ratio (from 2.7 to 8.6) and the mesh
size (4.8 to 11.7 nm) increased with increasing PEG chain length, from 3 kDa to 6 kDa, the
crosslinking density decreased from 10 to 4.1.
Results and Discussion
35
Table 5. The average molecular weight between crosslinks (Mc), volume-swelling ratio
(Q), crosslinking density (ρc), and mesh size (ξ) of PEG allyl and PEG thiol hydrogels.
Indices ‘S’ and ‘T’ denote whether the property was determined from swelling or from
tensile tests, respectively.
Hydrogel
sample code
Mc,S
(g/mol)
Mc,T
(g/mol)
Q ρc,S
(×10-4
)
ρc,T
(×10-4
)
ξS
(nm)
ξT
(nm)
E-P3-Al-1 1179 1367 2.7 10 8.7 4.8 5.1
E-P3-SH-1 1438 1420 5.7 5.6 5.6 6.7 6.7
E-P6-SH-15 2512 2934 8.6 4.1 4.7 11.7 11.7
4.3 SEQUENTIAL-IPN HYDROGELS PERPARATION AND
PROPERTIES
The seqIPNs were introduced to the PEG hydrogel systems by diffusing secondary
networks into the crosslinked primary network. To prepare robust TEC PEG hydrogels,
PEG-Al system was used to crosslink with tri-thiol functionalized crosslinker (TMP-tir-
SH) for seqIPN hydrogel fabrication. Although the other TEC side reaction that is head to
head coupling of the carbon centred radicals from allyl could happen in PEG-Al system,
because of the auto-inhibition of the allylic compounds, this side reaction is much less
likely to happen during network formation.83
4.3.1 Single-network hydrogels and assessment of their properties
Single-network hydrogels as primary hydrogel were prepared by PEG-Al (Mn= 2, 3, 6, 8
kDa) with TMP-tir-Allyl crosslinker in EtOH. The solids content of the precursor solution
was 50 wt% with 3 wt% UV-initiator and the molar ratio of functional groups were set to
1:1. Figure 17 shows the FT-Raman spectra for PEG-Al (8 kDa) and the single-network
hydrogels S2, S3, S6 and S8. There is no trace of the C=C vibrations of allyl group at 1640
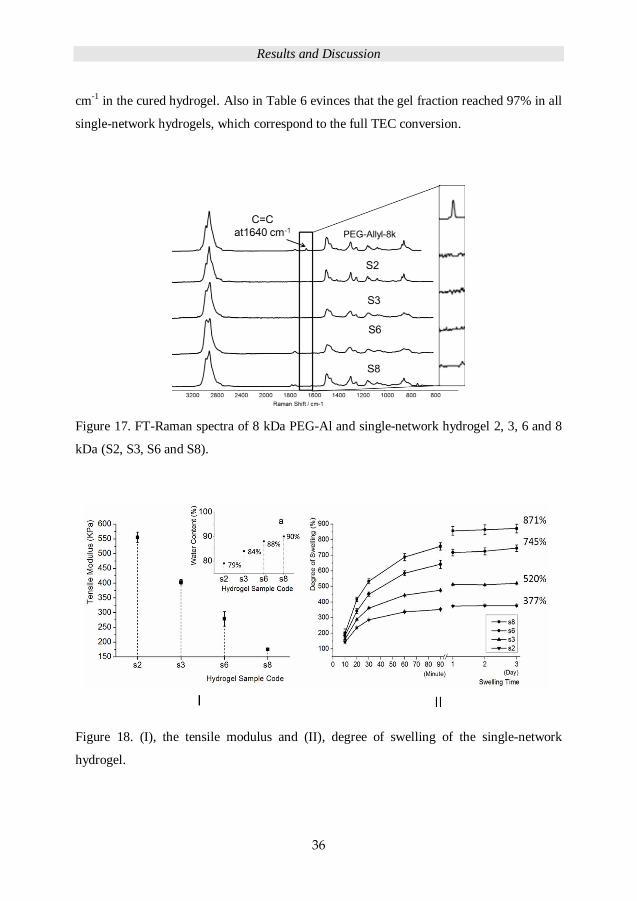
Results and Discussion
36
cm-1 in the cured hydrogel. Also in Table 6 evinces that the gel fraction reached 97% in all
single-network hydrogels, which correspond to the full TEC conversion.
Figure 17. FT-Raman spectra of 8 kDa PEG-Al and single-network hydrogel 2, 3, 6 and 8
kDa (S2, S3, S6 and S8).
Figure 18. (I), the tensile modulus and (II), degree of swelling of the single-network
hydrogel.

Results and Discussion
37
As shown in Figure 18 (I), the tensile modulus decreased from 555 to 175 KPa with
increasing PEG chain length from 2 kDa to 8 kDa, the water content of hydrogels
increased from 79 to 90% as the corresponding degree of swelling increased from 377 to
871%, Figure 18 (II). Table 6 details the relevant structure properties of the hydrogels
including the gel fraction based on the PEG-Al system and the average molecular weight
between crosslinks. As can be seen, the mesh size increased while the crosslinking density
decreased with increasing chain length of PEG precursors.
Table 6. Gel fraction (Gf), tensile modulus (E), stress-at-break (σB), strain-at-break (εB),
average molecular weight between crosslinks (Mc), mesh size (ξ), volume-swelling ratio
(Q), crosslinking density (ρc) and mesh size (ξ) of single-network hydrogel. Indices ‘S’ and
‘T’ indicate values determined from swelling or tensile tests, respectively.
Sample
code
Gf
%
E
(kPa)
σB
(kPa)
εB
(%)
Q
Mc,S
(g/mol)
Mc,T
(g/mol)
ρc,S×10-4
(mol/mL)
ρc,T×10-4
(mol/mL)
ξS
(nm)
ξT
(nm)
S2 95 555±18 226 65 2.3 766 902 15.5 13.2 3.7 4.0
S3 97 403±9 159 67 2.7 1141 1332 10.4 8.9 4.8 5.2
S6 97 279±22 153 75 3.7 2230 2515 5.3 4.7 7.4 7.9
S8 97 175±1 121 102 4.6 3104 3407 3.8 3.5 9.5 9.9
4.3.2 PEG chain length influence on seqIPN
PEG-Al system (Mn = 2, 3, 6, 8 kDa), PEG-Al-2k, PEG-Al-3k, PEG-Al-6k and PEG-Al-
8k were used to fabricate seqIPN hydrogels. All IPNs were prepared by diffusion of
mixtures containing PEG precursors, crosslinker (1:1 molar ratio) and initiator (3 wt%) in
EtOH (50 wt% solid content) for 2 hours at 40 °C, followed by 5 minutes of UV irradiation
at 365 nm to reach a fully cured system.
Results and Discussion
38
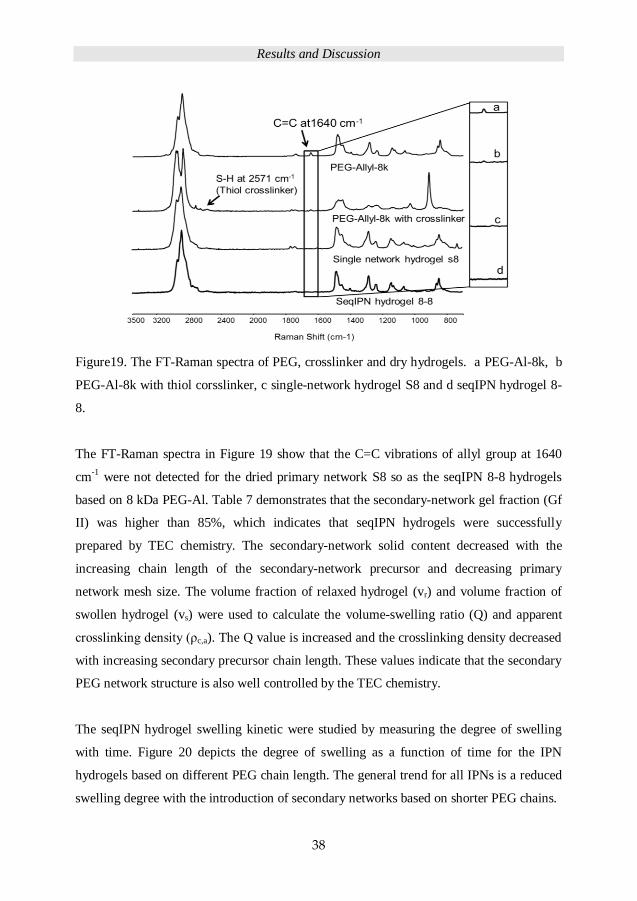
Figure19. The FT-Raman spectra of PEG, crosslinker and dry hydrogels. a PEG-Al-8k, b
PEG-Al-8k with thiol corsslinker, c single-network hydrogel S8 and d seqIPN hydrogel 8-
8.
The FT-Raman spectra in Figure 19 show that the C=C vibrations of allyl group at 1640
cm-1
were not detected for the dried primary network S8 so as the seqIPN 8-8 hydrogels
based on 8 kDa PEG-Al. Table 7 demonstrates that the secondary-network gel fraction (Gf
II) was higher than 85%, which indicates that seqIPN hydrogels were successfully
prepared by TEC chemistry. The secondary-network solid content decreased with the
increasing chain length of the secondary-network precursor and decreasing primary
network mesh size. The volume fraction of relaxed hydrogel (vr) and volume fraction of
swollen hydrogel (vs) were used to calculate the volume-swelling ratio (Q) and apparent
crosslinking density (ρc,a). The Q value is increased and the crosslinking density decreased
with increasing secondary precursor chain length. These values indicate that the secondary
PEG network structure is also well controlled by the TEC chemistry.
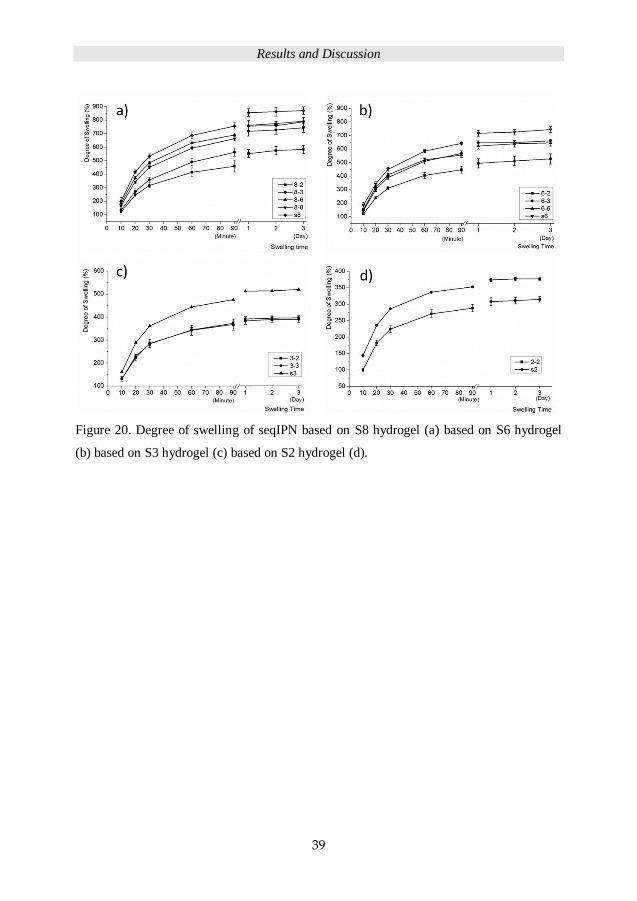
The seqIPN hydrogel swelling kinetic were studied by measuring the degree of swelling
with time. Figure 20 depicts the degree of swelling as a function of time for the IPN
hydrogels based on different PEG chain length. The general trend for all IPNs is a reduced
swelling degree with the introduction of secondary networks based on shorter PEG chains.
Results and Discussion
39
Figure 20. Degree of swelling of seqIPN based on S8 hydrogel (a) based on S6 hydrogel
(b) based on S3 hydrogel (c) based on S2 hydrogel (d).

Results and Discussion
40
Seq
IPN
sam
ple
cod
e
Prim
ary
netw
ork
(Da)
Secon
dary
netw
ork
(Da
)
Gf I
(%)
Gf
II
(%)
II
netw
ork
soli
d
con
ten
t
(%)
Sw
ell
ing
(%)
Wa
ter
con
ten
t
(%)
E
(kP
a)
σB
(kP
a)
ε B
(%)
vr
vs
Q ρ
c,a×
10
-4
(mol/
ml)
8-2
8
00
0
2000
97
95
3
4
58
4±
31
86
3
65
±6
0
12
7±
20
54
±2
0.6
0
.1
4.5
4.3
8-3
8
000
3000
97
94
3
2
74
4±
34
87
2
17
±4
6
72
±1
8
41
±9
0.6
0
.1
5.7
2.6
8-6
8
000
6000
97
92
2
5
79
2 ±
4
89
1
92
±2
1
98
±4
65
±9
0.6
0
.1
5.9
2.4
8-8
8
000
8000
97
85
2
1
81
0±
13
89
2
06
±1
6
13
2±
17
85±
15
0.6
0
.1
5.9
2.4
6-2
6
000
2000
94
95
3
9
52
8±
37
84
3
81
±8
5
11
6±
33
36
±5
0.6
0
.1
4.2
4.1
6-3
6
000
3000
94
92
3
2
66
1±
12
87
3
13
±3
7
10
6±
19
44±
10
0.6
0
.1
4.8
3.5
6-6
6
000
6000
94
91
2
2
64
6±
23
89
3
52
±2
3
17
4±
31
69±
19
0.6
0
.1
5.0
4.0
3-2
3
000
2000
97
93
2
6
39
7±
9
80
5
95
±3
0
28
3±
20
62±
37
0.7
0
.2
3.7
5.5
3-3
3
000
3000
97
97
2
8
39
0±
12
80
5
88
±4
0
26
4±
77
58±
11
0.6
0
.2
3.2
5.8
2-2
2
000
2000
97
94
2
7
31
5 ±
8
76
8
89
±5
0
29
2±
26
38±
5
0.7
0
.2
3.0
7.8
Tab
le 7
. S
eqIP
Ns
hydro
gel
s pri
mar
y n
etw
ork
gel
fra
ctio
n (
Gf
I),
seco
ndar
y g
el f
ract
ion (
Gf
II),
the
seco
ndar
y (
II)
net
work
mas
s
con
ten
t an
d t
he
deg
ree
of
swel
ling,
tensi
le m
od
ulu
s (E
), s
tres
s at
bre
ak (
σB),
str
ain a
t b
reak
(ε B
), v
olu
me
frac
tion o
f re
laxed
hy
dro
gel
(v r
), v
olu
me
frac
tion o
f sw
oll
en h
yd
rog
el (
v s),
vo
lum
e sw
elli
ng
rat
io (
Q),
ap
par
ent
cro
ssli
nk
ing d
ensi
ty (ρ c
,a).
*T
he
app
aren
t cr
oss
linkin
g d
ensi
ty i
s dif
fere
nt
from
th
e cr
oss
lin
kin
g d
ensi
ty c
alcu
late
d f
rom
Mc.
Results and Discussion
41
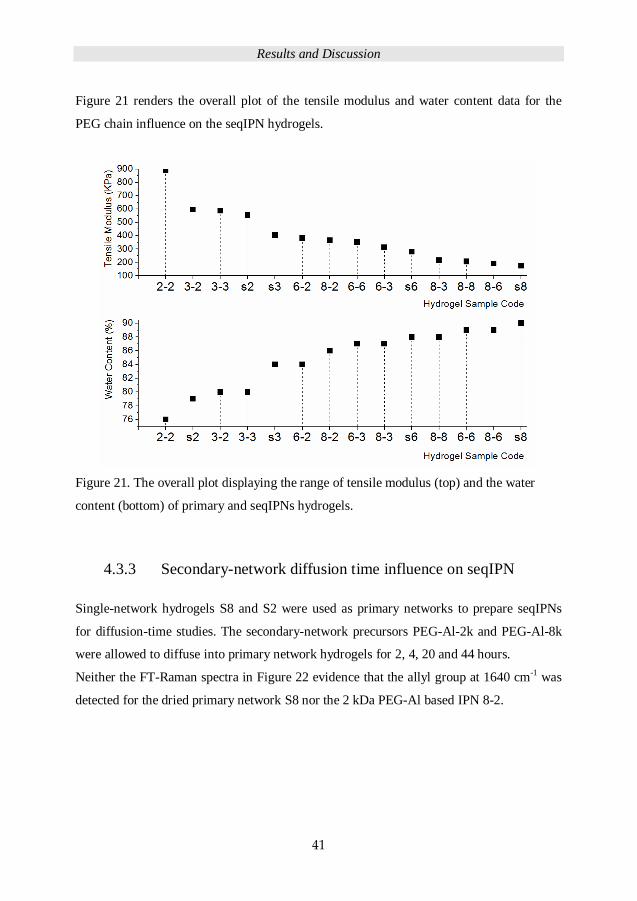
Figure 21 renders the overall plot of the tensile modulus and water content data for the
PEG chain influence on the seqIPN hydrogels.
Figure 21. The overall plot displaying the range of tensile modulus (top) and the water
content (bottom) of primary and seqIPNs hydrogels.
4.3.3 Secondary-network diffusion time influence on seqIPN
Single-network hydrogels S8 and S2 were used as primary networks to prepare seqIPNs
for diffusion-time studies. The secondary-network precursors PEG-Al-2k and PEG-Al-8k
were allowed to diffuse into primary network hydrogels for 2, 4, 20 and 44 hours.
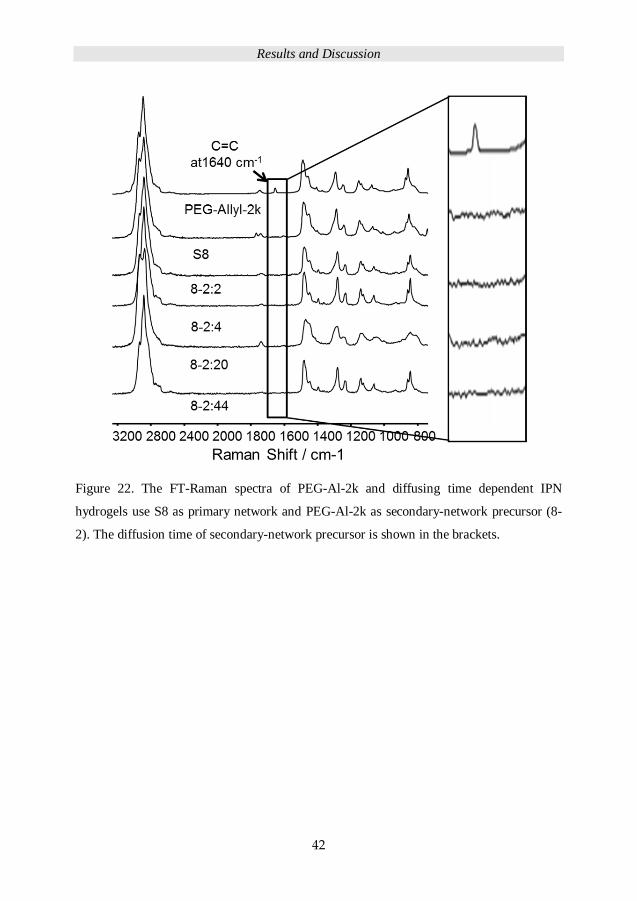
Neither the FT-Raman spectra in Figure 22 evidence that the allyl group at 1640 cm-1 was
detected for the dried primary network S8 nor the 2 kDa PEG-Al based IPN 8-2.
Results and Discussion
42
Figure 22. The FT-Raman spectra of PEG-Al-2k and diffusing time dependent IPN
hydrogels use S8 as primary network and PEG-Al-2k as secondary-network precursor (8-
2). The diffusion time of secondary-network precursor is shown in the brackets.
Results and Discussion
43
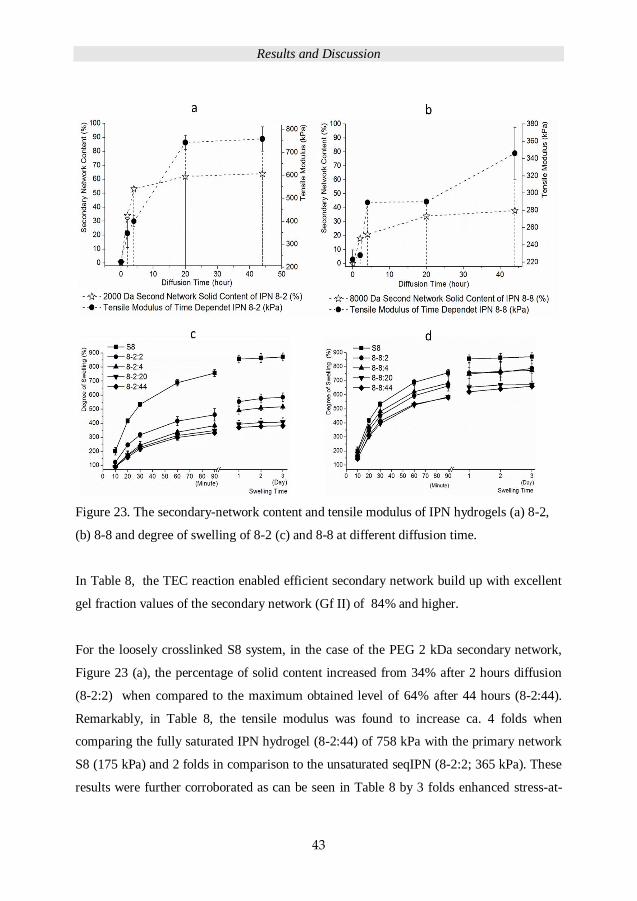
Figure 23. The secondary-network content and tensile modulus of IPN hydrogels (a) 8-2,
(b) 8-8 and degree of swelling of 8-2 (c) and 8-8 at different diffusion time.
In Table 8, the TEC reaction enabled efficient secondary network build up with excellent
gel fraction values of the secondary network (Gf II) of 84% and higher.
For the loosely crosslinked S8 system, in the case of the PEG 2 kDa secondary network,
Figure 23 (a), the percentage of solid content increased from 34% after 2 hours diffusion
(8-2:2) when compared to the maximum obtained level of 64% after 44 hours (8-2:44).
Remarkably, in Table 8, the tensile modulus was found to increase ca. 4 folds when
comparing the fully saturated IPN hydrogel (8-2:44) of 758 kPa with the primary network
S8 (175 kPa) and 2 folds in comparison to the unsaturated seqIPN (8-2:2; 365 kPa). These
results were further corroborated as can be seen in Table 8 by 3 folds enhanced stress-at-

Results and Discussion
44
break (σB = 320 kPa) and 2 folds of strain-at-break values (εB = 106%) for seqIPN 8-2:44
and in relationship to 8-2:2.
Figure 23 (b), the diffusion of 8 kDa secondary-network precursors in S8 scaffolds reached
a maximum solid content of 38% after 44 hours of diffusion (8-8:44). These results
demonstrated a 60% lower solid content of 8 kDa secondary network when compared to
the 2 kDa secondary network. Nonetheless, with the increasing solid content within the
seqIPN 8-2, from 18% to 38% (2 to 44 hours of diffusion), the tensile modules increased
from 206 kPa to 324 kPa (8-2:2 to 8-2:44).
Figure 23 (c) displays the degree of swelling of 8-2 systems supressed by increasing
content of denser secondary network (2 kDa) in 8-2 system, which was the result of
increasing diffusion time. Figure 24 (c) shows that with the same crosslinking density of
primary and secondary network, the increasing secondary solid content still increased the
crosslinking density and decreased the degree of swelling.

Results and Discussion
45
Figure 24. The secondary-network content and tensile modulus of seqIPN hydrogels (a) 2-
2, (b) 2-8 and degree of swelling of 2-2 (c) and 2-8 at different diffusion time.
Similar diffusion behaviour was displayed for the 2 kDa secondary-network precursors
within densely crosslinked S2 primary network, Figure 24 (a) and (b). In Table 8, seqIPN
2-2:2 (with 2 hours diffusion time) exhibited 27% secondary-network solid content and
tensile moduli of 889 kPa. The maximum diffusion was reached after 44 hours and the
fabricated seqIPN 2-2:44 revealed a secondary-network solid content of 49% and a water
swelling capacity of 280%. The seqIPN 2-2:44 displayed the highest tensile modulus of
all fabricated networks with a value of 1135 kPa, which can be compared with the primary
network S2 of 555 kPa. Interestingly, the strain-at-break first decreased from 65% for S2

Results and Discussion
46
to 38% for the seqIPN 2-2:2 and then increased to 78% after 44 hours of diffusion (seqIPN
2-2:44).
For the 2-8 system, Figure 24 (b), is different from the other, since the secondary network
is looser than the primary network. From 2 to 44 hours diffusion time, secondary-network
content of seqIPN 2-8 increased from 8% and reached equilibrium to 22%. However, the
tensile modulus increased from S2, 555 kPa to 829 kPa with 8% secondary network and
dropped to 640 kPa with 22% secondary-network content.
The same suppression of degree of swelling is noticeable in Figure 24 (c) for seqIPN 2-2
system with increasing diffusion time and secondary-network content. For seqIPN 2-8,
Figure 24 (d) the degree of swelling depicts decreased and increased with increased
secondary-network content.
Results and Discussion
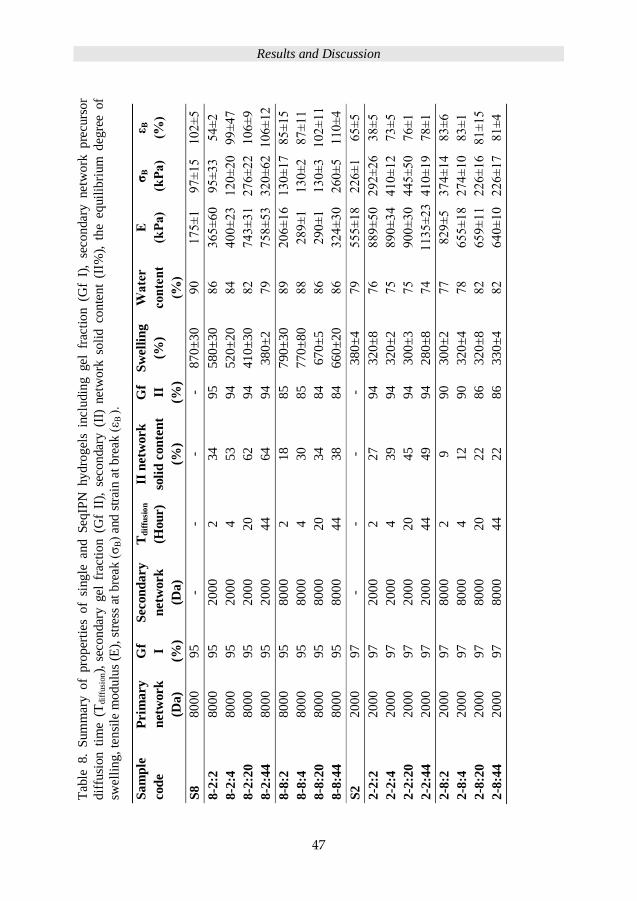
47
Sa
mp
le
cod
e
Prim
ary
netw
ork
(Da)
Gf I
(%)
Secon
dary
netw
ork
(Da)
Td
iffu
sio
n
(Hou
r)
II n
etw
ork
soli
d c
on
ten
t
(%)
Gf
II
(%)
Sw
ell
ing
(%)
Wa
ter
con
ten
t
(%)
E
(kP
a)
σB
(kP
a)
ε B
(%)
S8
8000
95
-
- -
- 8
70
±3
0
90
1
75
±1
97±
15
102±
5
8-2
:2
8000
95
2000
2
34
95
58
0±
30
86
36
5±
60
95
±3
3
54
±2
8-2
:4
8000
95
2000
4
53
9
4
52
0±
20
84
4
00
±2
3
120±
20
99±
47
8-2
:20
8000
95
2000
20
62
9
4
41
0±
30
82
7
43
±3
1
276±
22
106±
9
8-2
:44
8000
95
2000
44
64
9
4
38
0±
2
79
7
58
±5
3
320±
62
106±
12
8-8
:2
8000
95
8000
2
18
85
79
0±
30
89
20
6±
16
130±
17
85
±1
5
8-8
:4
8000
95
8000
4
30
8
5
77
0±
80
88
2
89
±1
130±
2
87±
11
8-8
:20
8000
95
8000
20
34
8
4
67
0±
5
86
2
90
±1
130±
3
102±
11
8-8
:44
8000
95
8000
44
38
8
4
66
0±
20
86
3
24
±3
0
260±
5
110±
4
S2
2000
97
-
- -
- 3
80
±4
79
5
55
±1
8
226±
1
65±
5
2-2
:2
2000
97
2000
2
27
9
4
32
0±
8
76
8
89
±5
0
292
±26
38
±5
2-2
:4
2000
97
2000
4
39
9
4
32
0±
2
75
8
90
±3
4
410±
12
73±
5
2-2
:20
2000
97
2000
20
45
94
30
0±
3
75
90
0±
30
44
5±
50
76
±1
2-2
:44
2000
97
2000
44
49
9
4
28
0±
8
74
1
13
5±
23
410±
19
78±
1
2-8
:2
2000
97
8000
2
9
90
30
0±
2
77
8
29
±5
374±
14
83±
6
2-8
:4
2000
97
8000
4
12
9
0
32
0±
4
78
6
55
±1
8
274±
10
83±
1
2-8
:20
2000
97
8000
20
22
8
6
32
0±
8
82
6
59
±1
1
226±
16
81±
15
2-8
:44
2000
97
8000
44
22
8
6
33
0±
4
82
6
40
±1
0
226±
17
81±
4
Tab
le 8.
Sum
mar
y of
pro
per
ties
of
single
an
d S
eqIP
N hy
dro
gel
s in
clu
din
g gel
fr
acti
on (G
f I)
, se
con
dar
y net
work
pre
curs
or
dif
fusi
on
tim
e (T
dif
fusi
on),
sec
ondar
y g
el f
ract
ion
(G
f II
), s
eco
ndar
y (
II)
net
wo
rk s
oli
d c
on
ten
t (I
I%),
th
e eq
uil
ibri
um
deg
ree
of
swel
lin
g, te
nsi
le m
odulu
s (E
), s
tres
s at
bre
ak (σ
B)
and
str
ain
at
bre
ak (ε B
).
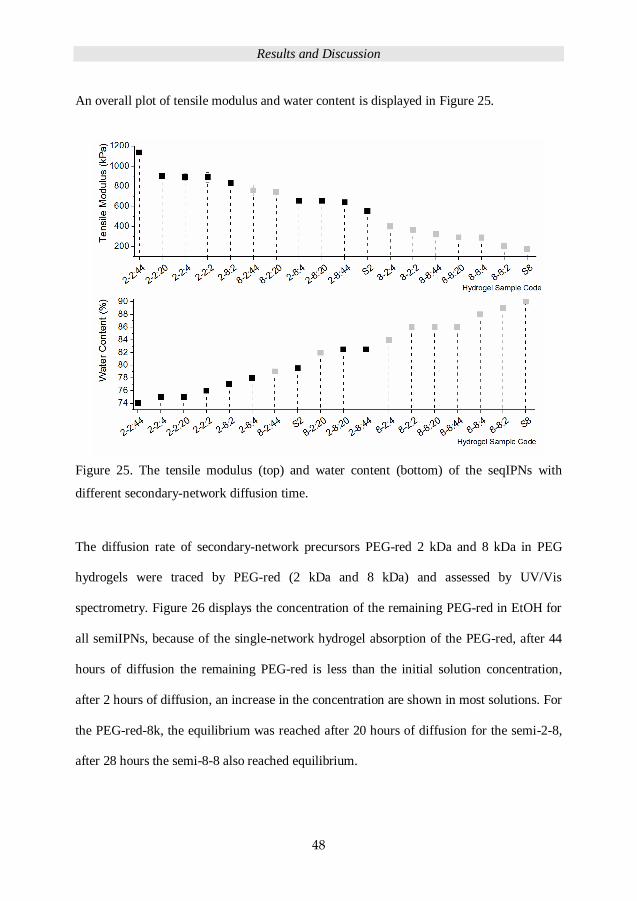
Results and Discussion
48
An overall plot of tensile modulus and water content is displayed in Figure 25.
Figure 25. The tensile modulus (top) and water content (bottom) of the seqIPNs with
different secondary-network diffusion time.
The diffusion rate of secondary-network precursors PEG-red 2 kDa and 8 kDa in PEG
hydrogels were traced by PEG-red (2 kDa and 8 kDa) and assessed by UV/Vis
spectrometry. Figure 26 displays the concentration of the remaining PEG-red in EtOH for
all semiIPNs, because of the single-network hydrogel absorption of the PEG-red, after 44
hours of diffusion the remaining PEG-red is less than the initial solution concentration,
after 2 hours of diffusion, an increase in the concentration are shown in most solutions. For
the PEG-red-8k, the equilibrium was reached after 20 hours of diffusion for the semi-2-8,
after 28 hours the semi-8-8 also reached equilibrium.
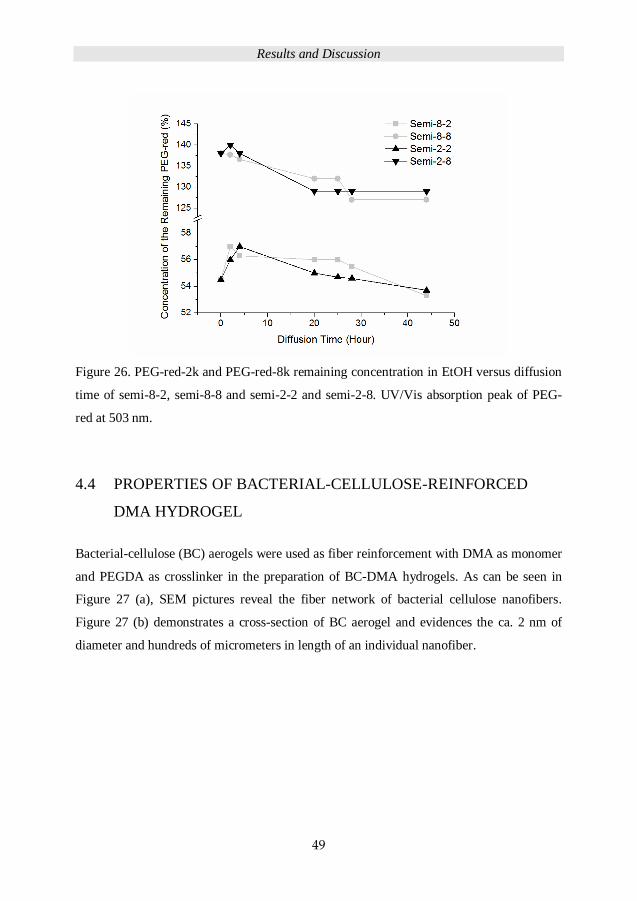
Results and Discussion
49
Figure 26. PEG-red-2k and PEG-red-8k remaining concentration in EtOH versus diffusion
time of semi-8-2, semi-8-8 and semi-2-2 and semi-2-8. UV/Vis absorption peak of PEG-
red at 503 nm.
4.4 PROPERTIES OF BACTERIAL-CELLULOSE-REINFORCED
DMA HYDROGEL
Bacterial-cellulose (BC) aerogels were used as fiber reinforcement with DMA as monomer
and PEGDA as crosslinker in the preparation of BC-DMA hydrogels. As can be seen in
Figure 27 (a), SEM pictures reveal the fiber network of bacterial cellulose nanofibers.
Figure 27 (b) demonstrates a cross-section of BC aerogel and evidences the ca. 2 nm of
diameter and hundreds of micrometers in length of an individual nanofiber.
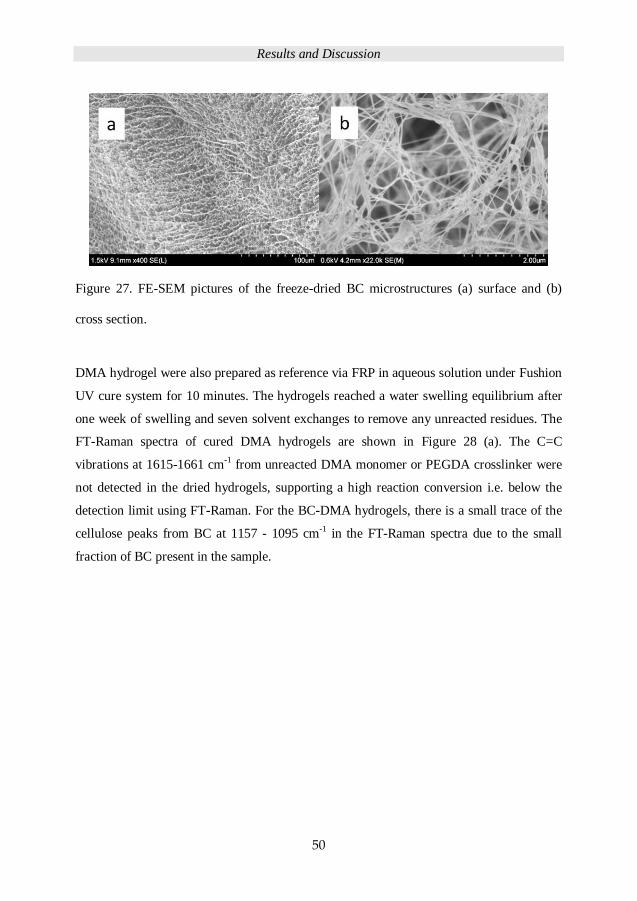
Results and Discussion
50
Figure 27. FE-SEM pictures of the freeze-dried BC microstructures (a) surface and (b)
cross section.
DMA hydrogel were also prepared as reference via FRP in aqueous solution under Fushion
UV cure system for 10 minutes. The hydrogels reached a water swelling equilibrium after
one week of swelling and seven solvent exchanges to remove any unreacted residues. The
FT-Raman spectra of cured DMA hydrogels are shown in Figure 28 (a). The C=C
vibrations at 1615-1661 cm-1 from unreacted DMA monomer or PEGDA crosslinker were
not detected in the dried hydrogels, supporting a high reaction conversion i.e. below the
detection limit using FT-Raman. For the BC-DMA hydrogels, there is a small trace of the
cellulose peaks from BC at 1157 - 1095 cm-1 in the FT-Raman spectra due to the small
fraction of BC present in the sample.
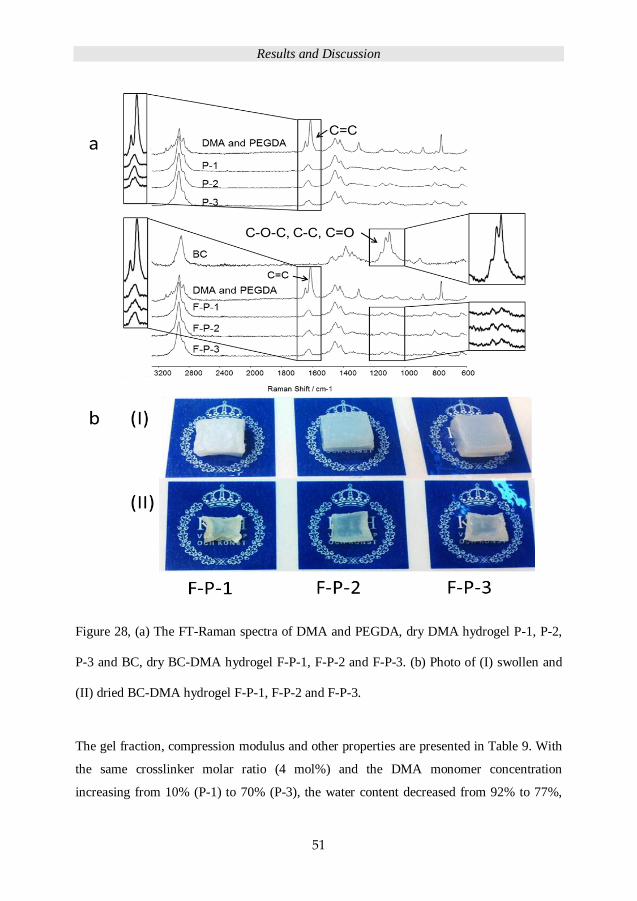
Results and Discussion
51
Figure 28, (a) The FT-Raman spectra of DMA and PEGDA, dry DMA hydrogel P-1, P-2,
P-3 and BC, dry BC-DMA hydrogel F-P-1, F-P-2 and F-P-3. (b) Photo of (I) swollen and
(II) dried BC-DMA hydrogel F-P-1, F-P-2 and F-P-3.
The gel fraction, compression modulus and other properties are presented in Table 9. With
the same crosslinker molar ratio (4 mol%) and the DMA monomer concentration
increasing from 10% (P-1) to 70% (P-3), the water content decreased from 92% to 77%,
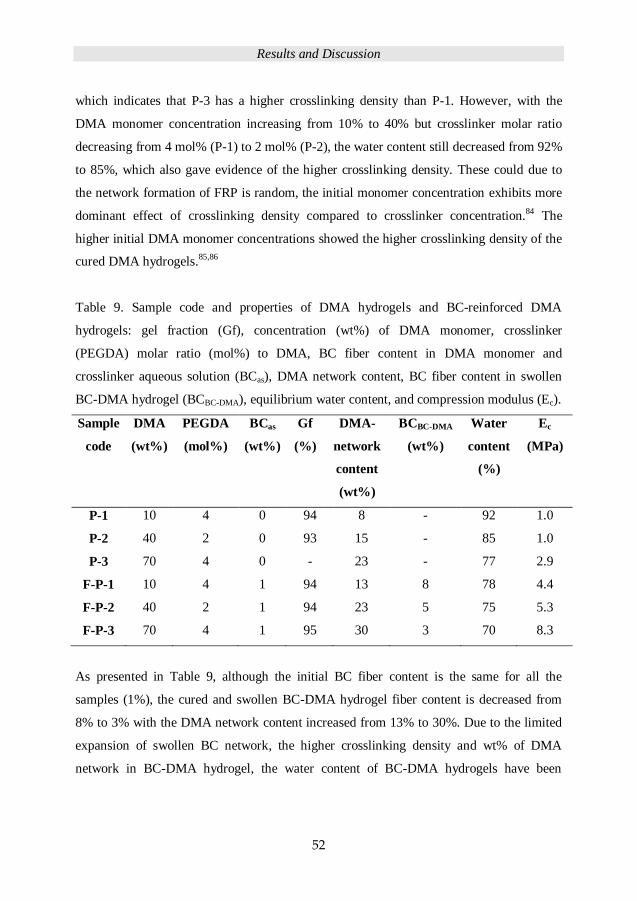
Results and Discussion
52
which indicates that P-3 has a higher crosslinking density than P-1. However, with the
DMA monomer concentration increasing from 10% to 40% but crosslinker molar ratio
decreasing from 4 mol% (P-1) to 2 mol% (P-2), the water content still decreased from 92%
to 85%, which also gave evidence of the higher crosslinking density. These could due to
the network formation of FRP is random, the initial monomer concentration exhibits more
dominant effect of crosslinking density compared to crosslinker concentration.84 The
higher initial DMA monomer concentrations showed the higher crosslinking density of the
cured DMA hydrogels.85,86
Table 9. Sample code and properties of DMA hydrogels and BC-reinforced DMA
hydrogels: gel fraction (Gf), concentration (wt%) of DMA monomer, crosslinker
(PEGDA) molar ratio (mol%) to DMA, BC fiber content in DMA monomer and
crosslinker aqueous solution (BCas), DMA network content, BC fiber content in swollen
BC-DMA hydrogel (BCBC-DMA), equilibrium water content, and compression modulus (Ec).
Sample
code
DMA
(wt%)
PEGDA
(mol%)
BCas
(wt%)
Gf
(%)
DMA-
network
content
(wt%)
BCBC-DMA
(wt%)
Water
content
(%)
Ec
(MPa)
P-1 10 4 0 94 8 - 92 1.0
P-2 40 2 0 93 15 - 85 1.0
P-3 70 4 0 - 23 - 77 2.9
F-P-1 10 4 1 94 13 8 78 4.4
F-P-2 40 2 1 94 23 5 75 5.3
F-P-3 70 4 1 95 30 3 70 8.3
As presented in Table 9, although the initial BC fiber content is the same for all the
samples (1%), the cured and swollen BC-DMA hydrogel fiber content is decreased from
8% to 3% with the DMA network content increased from 13% to 30%. Due to the limited
expansion of swollen BC network, the higher crosslinking density and wt% of DMA
network in BC-DMA hydrogel, the water content of BC-DMA hydrogels have been

Results and Discussion
53
suppressed ca. 10% compared with pure DMA hydrogels (92% to 77%) with the same
polymer-crosslinking density.
In Table 9, while the initial DMA wt% increased from 10 to 70 wt%, the compression
modulus (Ec) of DMA hydrogels increased from 1 to 2.9 MPa without BC. The Ec of BC-
DMA increased from 4.4 to 8.3 MPa with the DMA wt% increased from 10 to 70 wt% (4
mol% PEGDA). Ec of BC-DMA hydrogel increased ca. 3 times compared with the DMA
hydrogel with the same crosslinking density. However, increasing DMA from 10 to 40
wt% and decreasing PEGDA from 4 to 2 mol%, the BC-DMA exhibits an increase Ec from
4.4 to 5.3 MPa. Further presents the increasing initial DMA wt% will increase the
crosslinking density even with less crosslinker mol%.
DMA hydrogels also contributed with improved shear modulus and changed the water
absorption of BC hydrogels. Figure 29, (I) depicts the shear storage modulus G′ of BC-
DMA hydrogels and BC hydrogel. Shear modulus of BC without polymer network is close
to 0 kPa but after curing the DMA network in the BC network, the G′ of the resulting IPN
hydrogel P-F-3 increased up to ca. 350 kPa. The combination of DMA and BC fibers
locked water in the voids of the BC aerogel, as shown in Figure 29 (II) a, with a finger
force twisting the re-swollen BC hydrogel, water can be squeezed out. Water in swollen
BC-DMA hydrogels (Figure 30 (II) b) on the other hand, can only be removed by
evaporation and not by physical force.
Results and Discussion
54
Figure 29. I (a): the photo of BC hydrogel and (b): BC-DMA hydrogel under a finger
shear force. II: the shear storage modulus G′ (kPa) of re-swollen BC hydrogel, BC-DMA
hydrogels F-P-1, F-P-2 and F-P- 3. (ω: 0.1 to 20 Hz, γ: 1%)
The microstructures of BC and DMA interpenetrating networks were observed by FE-
SEM. As can be seen in Figure 30, after DMA network cured in the BC aerogels, the
composites of BC fibers and DMA networks show no trace of individual BC fiber compare
to raw BC aerogel, Figure 27. The FE-SEM pictures reveal that there is no trace of phase
separation between fiber and polymer networks of the BC-DMA hydrogels surface, Figure
30 (a), (b), (c), and cross section, Figure 30 (a’), (b’), (c’); also, the pore size of the
hydrogels decreased from the loose crosslinked BC-DMA hydrogel F-P-1 (10 wt% DMA
and 4 mol% PEGDA) to densely crosslinked BC-DMA hydrogel F-P-3 (70 wt% DMA and
4 mol% PEGDA).
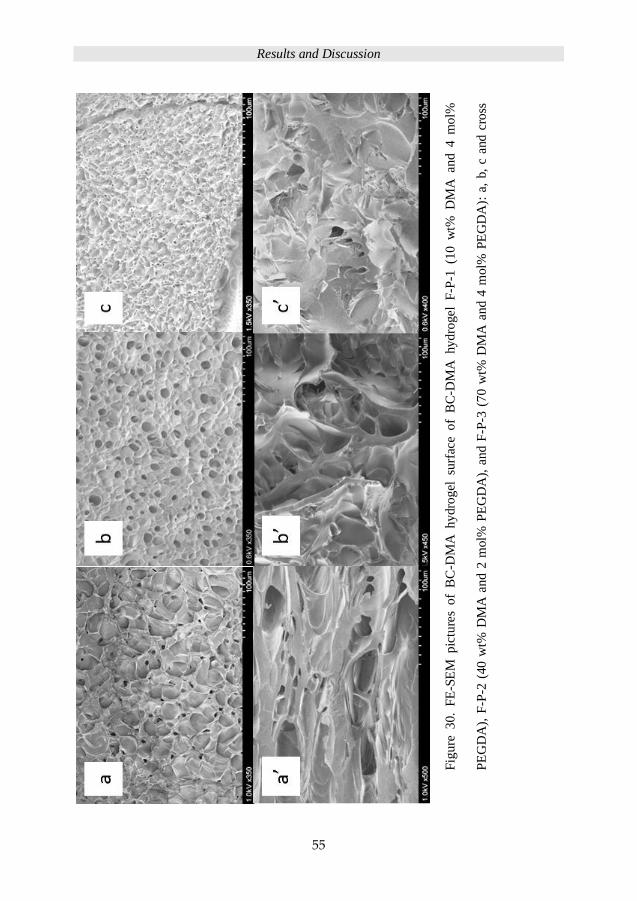
Results and Discussion
55
Fig
ure
30.
FE
-SE
M pic
ture
s of
BC
-DM
A h
ydro
gel
su
rfac
e o
f B
C-D
MA
hy
dro
gel
F
-P-1
(1
0 w
t% D
MA
and 4 m
ol%
PE
GD
A),
F-P
-2 (
40 w
t% D
MA
and 2
mol%
PE
GD
A),
an
d F
-P-3
(7
0 w
t% D
MA
an
d 4
mol%
PE
GD
A):
a,
b,
c an
d c
ross
sect
ion a
’, b
’ an
d c
’.

Conclusions
56
5 CONCLUSIONS
The purpose of this work was to relate and evaluate hydrogel fabrication parameters with
swelling and mechanical properties.
PEG single and sequential interpenetrating polymer network (seqIPN) hydrogels were
successfully prepared and characterized in this work via UV-initiated thiol-ene coupling
(TEC) chemistry.
A library of PEG hydrogels was prepared using PEG-SH or the reversed PEG-Al
precursors in combination with trifunctional crosslinkers. The UV-initiated TEC reaction
proved to be robust for the formation of PEG hydrogels under benign experimental
conditions. The type of solvent, solid content in solution, functional group and PEG chain
length were found to have direct influence on mechanical properties of PEG hydrogels.
Fabricated hydrogels based on PEG-Al displayed stronger mechanical properties than
PEG-SH hydrogels.
Well-defined PEG seqIPN with controllable mechanical properties have also been
prepared and assessed. Properties of a library of seqIPN hydrogel were compiled based on
the information of secondary-network content, gel fraction, swelling kinetic, mechanical
properties and structure evaluations. The seqIPN hydrogels based on the PEG-Al systems
showed 84% and higher gel fraction. The mechanical properties of PEG hydrogel were
increased to reach a tensile modulus over 1 MPa (2-2:44). The range of tensile modulus is
extended from single network, range from 175 kPa to 555 kPa, from seqIPN 365 kPa to
1135 KPa. Water content of PEG hydrogels was supressed by seqIPN from single network,
range 90% to 79% (degree of swelling 870% to 380%), to seqIPN 88% to 74% (degree of
swelling 580% to 280%).

Conclusions
57
The diffusion rate of the disperse red 13 decorated linear PEG with short chain (2 kDa) and
long chain (8 kDa) was evaluated in both a dense (2 kDa) and loose (8 kDa) PEG
crosslinked network.
Finally, BC aerogels were employed as reinforced templates to yield BC-DMA hydrogels
via traditional UV-initiated polymerization. The compression modulus of DMA hydrogel
ranging from 1 MPa - 2.9 MPa increased to 4.4 MPa - 8.3 MPa upon introducing BC
fibers. The water content was supressed by the BC network from DMA hydrogel from
92% to 77% in the pure DMA hydrogels to 78% to 70% in the BC-DMA hydrogels.

Future work
58
6 FUTURE WORK
IPN is an effective way to manipulate mechanical properties of PEG hydrogels. Triple and
multiple networks of PEG IPN will be the next interesting hydrogel to prepare by TEC.
Combining polyDMA with PEG TEC hydrogels could achieve even higher mechanical
properties.
Copper-catalyzed azide–alkyne cycloaddition (CuAAC) could be another interesting
process for preparing hydrogels. FRP with CuAAC in one system could prepare IPN in
one pot.
In order to further explore the potential of TEC chemistry for well-defined hydrogel
preparation, it will be interested to compare dendronized PEG with allyl groups with linear
PEG-Al. The mechanical properties will be expected to improve a lot because of the much
higher crosslinking density.
Dendronized PEG with thiol groups could crosslink without using crosslinker or
dissolution in a solvent.
Nano-febrile cellulose (NFC) could be used to prepare fiber-reinforced hydrogels. The
NFC could be TEMPO-oxidized and surface-treated with polymer to improve the
compatibility with monomers. NFC solid concentrations can be tuned to fit the needs of
the hydrogel applications.

Acknowledgements
59
7 ACKNOWLEDGEMENTS
I would like to thank my supervisors: Prof. Anders Hult is thanked for giving me the
chance to be a PhD student at KTH. Prof. Lars Berglund is thanked for being my co-
supervisor in the first 2 years. Associate prof. Michael Malkoch is thanked for teaching me
from laboratory safety to writing paper and helping me to become a PhD.
All seniors in the department of fiber and polymer technology are thanked for their help.
Thanks to members of biocomposite group. Prof. E. Kristofer Gamstedt and associate prof.
Qi Zhou are thanked for their helps.
The China Scholarship Council (CSC) and the Department of Fiber and Polymer
Technology at KTH are acknowledged for financial support.
Prof. Ling Wang from Chengdu University of Technology is thanked for supporting my
application of CSC scholarship during my master study.
Thanks to members of the coating group: special thanks to Marie, Yvonne for checking my
writings and giving a lot of help when I just arrived in the group. Hui is thanked for
working together with me for my papers and being a nice friend. Kim is thanked for a lot

Acknowledgements
60
of help in the lab. Jan is really thanked for supporting me with the materials to work for the
paper. Oliver, Jonas and Kristina are thanked for helping me with the Swedish abstract
writing. Mats K.G.J is thanked for checking my last paper and being ‘tomten’. Linda is
thanked for being so nice and reviewing my thesis. Camilla is thanked for her nice talks
and warm care. Alireza, Assya and Susanne are thanked for being good office-roommates.
Stacy is thanked for her happy personality. Andreas Fall is thanked for helping me with the
rheometer. Inger is greatly thanked for her help in the department. Big thanks to Eva,
Anna, Emma L, Emma Ö, Bella, Hanna, Sara, Linn, Christian, Carl, Marcus, Martin,
Mauro, Pontus, Carmen, Eric and Petra.
Special thanks to my uncle Jiebing Li and Aunt Yun Yang in Sweden for helping me in
every aspect of my life. Yitong is thanked for her relax outlook on life. Jesper is thanked
for all the fun art things and taping for my niece. Jingyu is thanked for her nice entertain
in Japan and help me a lot in the wedding. Big thanks for Andong Liu, Aihua Pei, Yafang
Yan, Yingzhi Zhu, Wenbin Yu, Xueyu Du, Hongli Zhu, Bi Ran, Lingquan Deng, Yujia
Zhang, Jiayi Yang, Lin Li, Bo Yin, Liming, Zhiqiang Zheng, Chun Ying Shean, Andrew
M., Raquel, Lage, Dany, Pedro and other friends in Sweden, I thank all of you for the
happy moments.
感谢父亲杨竟达和母亲黄果对我的养育之恩。感谢爷爷杨金南对我的栽培和奶奶姜
映南在生活上给我的照顾。感谢外公黄岳飞,外婆王文飞,小姨黄朵,堂哥杨泰宏、
杨经宇,表姐梁晓莉,表哥黄毅对我的关心。
Till familjen, Anders Nilsson (Fluffy) tackas för att vara en omtänksam make; till Rositha,
Leif, Bosse, Elvy, Birgit, Kerstin, Lasse, Ammi, Lisbeth, Michael, Jenny, Stefan, tackas!

References
61
8 REFERENCES
(1) Biomedical Applications of Hydrogels Handbook; Ottenbrite, R. M. P., Kinam;
Okano, Teruo Ed.; Springer, 2010.
(2) Hoffman, A. S.: Hydrogels for biomedical applications. Adv Drug Deliver Rev
2002, 54, 3-12.
(3) Kloxin, A. M.; Kloxin, C. J.; Bowman, C. N.; Anseth, K. S.: Mechanical
Properties of Cellularly Responsive Hydrogels and Their Experimental
Determination. Adv Mater 2010, 22, 3484-3494.
(4) Lin, C. C.; Metters, A. T.: Hydrogels in controlled release formulations:
Network design and mathematical modeling. Adv Drug Deliver Rev 2006, 58, 1379-
1408.
(5) Kade, M. J.; Burke, D. J.; Hawker, C. J.: The Power of Thiol-ene Chemistry. J
Polym Sci Pol Chem 2010, 48, 743-750.
(6) Lowe, A. B.: Thiol-ene “click” reactions and recent applications in polymer
and materials synthesis Polym Chem-Uk 2010, 17-36.
(7) Lundberg, P.; Bruin, A.; Klijnstra, J. W.; Nystrom, A. M.; Johansson, M.;
Malkoch, M.; Hult, A.: Poly(ethylene glycol)-Based Thiol-ene Hydrogel Coatings-
Curing Chemistry, Aqueous Stability, and Potential Marine Antifouling
Applications. Acs Appl Mater Inter 2010, 2, 903-912.
(8) Hagberg, E. C.; Malkoch, M.; Ling, Y.; Hawker, C. J.; Carter, K. R.: Effects of
modulus and surface chemistry of thiol-ene photopolymers in nanoimprinting.
Nano Lett 2007, 7, 233-7.
(9) Nordberg, A.; Antoni, P.; Montanez, M. I.; Hult, A.; Von Holst, H.; Malkoch,
M.: Highly adhesive phenolic compounds as interfacial primers for bone fracture
fixations. ACS Appl Mater Interfaces 2010, 2, 654-7.
(10) Walter, M. V.; Malkoch, M.: Simplifying the synthesis of dendrimers:
accelerated approaches. Chem Soc Rev 2012, 41, 4593-609.
(11) Shipp, D. A.; McQuinn, C. W.; Rutherglen, B. G.; McBath, R. A.: Elastomeric
and degradable polyanhydride network polymers by step-growth thiol-ene
photopolymerization. Chem Commun (Camb) 2009, 6415-6417.

References
62
(12) Zhu, J. M.: Bioactive modification of poly(ethylene glycol) hydrogels for
tissue engineering. Biomaterials 2010, 31, 4639-4656.
(13) Liu, S. Q.; Tay, R.; Khan, M.; Ee, P. L. R.; Hedrick, J. L.; Yang, Y. Y.: Synthetic
hydrogels for controlled stem cell differentiation. Soft Matter 2010, 6, 67-81.
(14) Fuertges, F.; Abuchowski, A.: The Clinical Efficacy of Poly(Ethylene Glycol)-
Modified Proteins. J Control Release 1990, 11, 139-148.
(15) Lin, C. C.; Raza, A.; Shih, H.: PEG hydrogels formed by thiol-ene photo-click
chemistry and their effect on the formation and recovery of insulin-secreting cell
spheroids. Biomaterials 2011, 32, 9685-9695.
(16) Ekblad, T.; Bergstroem, G.; Ederth, T.; Conlan, S. L.; Mutton, R.; Clare, A. S.;
Wang, S.; Liu, Y. L.; Zhao, Q.; D'Souza, F.; Donnelly, G. T.; Willemsen, P. R.;
Pettitt, M. E.; Callow, M. E.; Callow, J. A.; Liedberg, B.: Poly(ethylene glycol)-
Containing Hydrogel Surfaces for Antifouling Applications in Marine and
Freshwater Environments. Biomacromolecules 2008, 9, 2775-2783.
(17) Cha, C.; Kim, E. S.; Kim, I. W.; Kong, H.: Integrative deign of a poly(ethylene
glycol)-poly(propylene glycol)-alginate hydrogel to control three dimensional
biomineralization. Biomaterials 2011, 32, 2695-2703.
(18) Jeong, J. H.; Schmidt, J. J.; Cha, C.; Kong, H.: Tuning responsiveness and
structural integrity of a pH responsive hydrogel using a poly(ethylene glycol)
cross-linker. Soft Matter 2010, 6, 3930-3938.
(19) Nguyen, K. T.; West, J. L.: Photopolymerizable hydrogels for tissue
engineering applications. Biomaterials 2002, 23, 4307-4314.
(20) Rivarola, C. R.; Biasutti, M. A.; Barbero, C. A.: A visible light photoinitiator
system to produce acrylamide based smart hydrogels: Ru(bpy)(3)(+2) as
photopolymerization initiator and molecular probe of hydrogel
microenvironments. Polymer (Guildf) 2009, 50, 3145-3152.
(21) Ruan, W. Q.; Wang, X. G.; Lian, Y. Q.; Huang, Y. L.; Niu, A. J.:
Superabsorbent hydrogel of acrylic acid/potassium acrylate copolymers by
ultraviolet photopolymerization: Synthesis and properties. J Appl Polym Sci 2006,
101, 1181-1187.
(22) Mullarney, M. P.; Seery, T. A. P.; Weiss, R. A.: Drug diffusion in
hydrophobically modified N,N-dimethylacrylamide hydrogels. Polymer (Guildf)
2006, 47, 3845-3855.
(23) Bekiari, V.; Sotiropoulou, M.; Bokias, G.; Lianos, P.: Use of poly(N,N-
dimethylacrylamide-co-sodium acrylate) hydrogel to extract cationic dyes and
metals from water. Colloid Surface A 2008, 312, 214-218.
(24) Caria, G.; Alzari, V.; Monticelli, O.; Nuvoli, D.; Kenny, J. M.; Mariani, A.:
Poly(N,N-dimethylacrylamide) Hydrogels Obtained by Frontal Polymerization. J
Polym Sci Pol Chem 2009, 47, 1422-1428.

References
63
(25) Peppas, N. A.; Klier, J.: Controlled Release by Using Poly (Methacrylic Acid-
G-Ethylene Glycol) Hydrogels. J Control Release 1991, 16, 203-214.
(26) Lin, S.; Sangaj, N.; Razafiarison, T.; Zhang, C.; Varghese, S.: Influence of
Physical Properties of Biomaterials on Cellular Behavior. Pharm Res-Dordr 2011, 28,
1422-1430.
(27) Flory, P. J.: Theory of Elasticity of Polymer Networks - Effect of Local
Constraints on Junctions. J Chem Phys 1977, 66, 5720-5729.
(28) A.M. North, A. M. S.: The free radical polymerization of N,N-
dimethylacrylamide. Polymer (Guildf) 1964, 5, 447-455.
(29) Sperling, L. H.: Interpenetrating Polymer Networks - an Overview. Adv Chem
Ser 1994, 239, 3-38.
(30) Myung, D.; Koh, W. U.; Ko, J. M.; Hu, Y.; Carrasco, M.; Noolandi, J.; Ta, C. N.;
Frank, C. W.: Biomimetic strain hardening in interpenetrating polymer network
hydrogels. Polymer (Guildf) 2007, 48, 5376-5387.
(31) Sun, J.; Xiao, W. Q.; Tang, Y. J.; Li, K. F.; Fan, H. S.: Biomimetic
interpenetrating polymer network hydrogels based on methacrylated alginate and
collagen for 3D pre-osteoblast spreading and osteogenic differentiation. Soft
Matter 2012, 8, 2398-2404.
(32) Lee, S. J.; Kim, S. S.; Lee, Y. M.: Interpenetrating polymer network hydrogels
based on poly(ethylene glycol) macromer and chitosan. Carbohyd Polym 2000, 41,
197-205.
(33) Liu, W. G.; Deng, C.; McLaughlin, C. R.; Fagerholm, P.; Lagali, N. S.; Heyne,
B.; Scaiano, J. C.; Watsky, M. A.; Kato, Y.; Munger, R.; Shinozaki, N.; Li, F. F.;
Griffith, M.: Collagen-phosphorylcholine interpenetrating network hydrogels as
corneal substitutes. Biomaterials 2009, 30, 1551-1559.
(34) Reddy, T. T.; Takahara, A.: Simultaneous and sequential micro-porous semi-
interpenetrating polymer network hydrogel films for drug delivery and wound
dressing applications. Polymer (Guildf) 2009, 50, 3537-3546.
(35) Xu, L. Q.; Yao, F.; Fu, G. D.; Kang, E. T.: Interpenetrating Network Hydrogels
via Simultaneous "Click Chemistry" and Atom Transfer Radical Polymerization.
Biomacromolecules 2010, 11, 1810-1817.
(36) Liu, Y. X.; Chan-Park, M. B.: Hydrogel based on interpenetrating polymer
networks of dextran and gelatin for vascular tissue engineering. Biomaterials 2009,
30, 196-207.
(37) Bae, Y. H.; Kim, S. W.: Hydrogel Delivery Systems Based on Polymer Blends,
Block-Copolymers or Interpenetrating Networks. Adv Drug Deliver Rev 1993, 11,
109-135.
(38) Gong, M.; Zhang, L.; Zuo, Y.; Zou, Q.; Wang, Y. Y.; Wang, L.; Li, Y. B.:
Investigation on the interpenetrating polymer networks (ipns) of polyvinyl

References
64
alcohol and poly(N-vinyl pyrrolidone) hydrogel and its in vitro bioassessment. J
Appl Polym Sci 2012, 125, 2799-2806.
(39) Lira, L. M.; de Torresi, S. I. C.: Conducting polymer-hydrogel composites for
electrochemical release devices: Synthesis and characterization of semi-
interpenetrating polyaniline-polyacrylamide networks. Electrochem Commun 2005,
7, 717-723.
(40) Iwasaki, Y.; Shimakata, K.; Morimoto, N.; Kurita, K.: Hydrogel-like elastic
membrane consisting of semi-interpenetrating polymer networks based on a
phosphorylcholine polymer and a segmented polyurethane. J Polym Sci Pol Chem
2003, 41, 68-75.
(41) Krystynowicz, A.; Czaja, W.; Wiktorowska-Jezierska, A.; Goncalves-
Miskiewicz, M.; Turkiewicz, M.; Bielecki, S.: Factors affecting the yield and
properties of bacterial cellulose. J Ind Microbiol Biot 2002, 29, 189-195.
(42) Tecson-Mendoza, E. M.: Development of functional foods in the Philippines.
Food Sci Technol Res 2007, 13, 179-186.
(43) Eichhorn, S. J.; Baillie, C. A.; Zafeiropoulos, N.; Mwaikambo, L. Y.; Ansell, M.
P.; Dufresne, A.; Entwistle, K. M.; Herrera-Franco, P. J.; Escamilla, G. C.; Groom,
L.; Hughes, M.; Hill, C.; Rials, T. G.; Wild, P. M.: Review: Current international
research into cellulosic fibres and composites. J Mater Sci 2001, 36, 2107-2131.
(44) Somerville, C.: Cellulose synthesis in higher plants. Annu Rev Cell Dev Bi
2006, 22, 53-78.
(45) Siro, I.; Plackett, D.: Microfibrillated cellulose and new nanocomposite
materials: a review. Cellulose 2010, 17, 459-494.
(46) Iguchi, M.; Yamanaka, S.; Budhiono, A.: Bacterial cellulose - a masterpiece of
nature's arts. J Mater Sci 2000, 35, 261-270.
(47) Wach, R. A.; Mitomo, H.; Yoshii, F.; Kume, T.: Hydrogel of biodegradable
cellulose derivatives. II. Effect of some factors on radiation-induced crosslinking of
CMC. J Appl Polym Sci 2001, 81, 3030-3037.
(48) Chang, C. Y.; Zhang, L. N.: Cellulose-based hydrogels: Present status and
application prospects. Carbohyd Polym 2011, 84, 40-53.
(49) Svensson, A.; Nicklasson, E.; Harrah, T.; Panilaitis, B.; Kaplan, D. L.;
Brittberg, M.; Gatenholm, P.: Bacterial cellulose as a potential scaffold for tissue
engineering of cartilage. Biomaterials 2005, 26, 419-431.
(50) Bodin, A.; Backdahl, H.; Fink, H.; Gustafsson, L.; Risberg, B.; Gatenholm, P.:
Influence of cultivation conditions on mechanical and morphological properties of
bacterial cellulose tubes. Biotechnol Bioeng 2007, 97, 425-434.
(51) Czaja, W. K.; Young, D. J.; Kawecki, M.; Brown, R. M.: The future prospects of
microbial cellulose in biomedical applications. Biomacromolecules 2007, 8, 1-12.

References
65
(52) Zhou, Q.; Malm, E.; Nilsson, H.; Larsson, P. T.; Iversen, T.; Berglund, L. A.;
Bulone, V.: Nanostructured biocomposites based on bacterial cellulosic nanofibers
compartmentalized by a soft hydroxyethylcellulose matrix coating. Soft Matter
2009, 5, 4124-4130.
(53) Moon, R. J.; Martini, A.; Nairn, J.; Simonsen, J.; Youngblood, J.: Cellulose
nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev
2011, 40, 3941-3994.
(54) Eichhorn, S. J.; Dufresne, A.; Aranguren, M.; Marcovich, N. E.; Capadona, J.
R.; Rowan, S. J.; Weder, C.; Thielemans, W.; Roman, M.; Renneckar, S.; Gindl, W.;
Veigel, S.; Keckes, J.; Yano, H.; Abe, K.; Nogi, M.; Nakagaito, A. N.; Mangalam, A.;
Simonsen, J.; Benight, A. S.; Bismarck, A.; Berglund, L. A.; Peijs, T.: Review:
current international research into cellulose nanofibres and nanocomposites. J
Mater Sci 2010, 45, 1-33.
(55) Karaaslan, M. A.; Tshabalala, M. A.; Yelle, D. J.; Buschle-Diller, G.:
Nanoreinforced biocompatible hydrogels from wood hemicelluloses and cellulose
whiskers. Carbohyd Polym 2011, 86, 192-201.
(56) Dorkoosh, F. A.; Brussee, J.; Verhoef, J. C.; Borchard, G.; Rafiee-Tehrani, M.;
Junginger, H. E.: Preparation and NMR characterization of superporous hydrogels
(SPH) and SPH composites. Polymer (Guildf) 2000, 41, 8213-8220.
(57) Principles of Polymer Chemistry; Paul.J.Flory, Ed.; Cornell University Press
1953.
(58) Hydrogels: Biological Properties and Applications; Barbucci, R., Ed.; Springer,
2009.
(59) Seliktar, D.: Designing Cell-Compatible Hydrogels for Biomedical
Applications. Science 2012, 336, 1124-1128.
(60) Li, Y. L.; Rodrigues, J.; Tomas, H.: Injectable and biodegradable hydrogels:
gelation, biodegradation and biomedical applications. Chem Soc Rev 2012, 41, 2193-
2221.
(61) Nagai, T.; Machida, Y.: Buccal Delivery Systems Using Hydrogels. Adv Drug
Deliver Rev 1993, 11, 179-191.
(62) Gil, E. S.; Hudson, S. M.: Stimuli-reponsive polymers and their bioconjugates.
Prog Polym Sci 2004, 29, 1173-1222.
(63) Banquy, X.; Suarez, F.; Argaw, A.; Rabanel, J. M.; Grutter, P.; Bouchard, J. F.;
Hildgen, P.; Giasson, S.: Effect of mechanical properties of hydrogel nanoparticles
on macrophage cell uptake. Soft Matter 2009, 5, 3984-3991.
(64) Peppas, N. A.; Keys, K. B.; Torres-Lugo, M.; Lowman, A. M.: Poly(ethylene
glycol)-containing hydrogels in drug delivery. J Control Release 1999, 62, 81-87.
(65) Gong, C. Y.; Dong, P. W.; Shi, S. A.; Fu, S. Z.; Yang, J. L.; Guo, G.; Zhao, X.;
Wei, Y. Q.; Qian, Z. Y.: Thermosensitive PEG-PCL-PEG Hydrogel Controlled Drug

References
66
Delivery System: Sol-Gel-Sol Transition and In Vitro Drug Release Study. J Pharm
Sci-Us 2009, 98, 3707-3717.
(66) Yu, L.; Zhang, Z.; Zhang, H. A.; Ding, J. D.: Biodegradability and
Biocompatibility of Thermoreversible Hydrogels Formed from Mixing a Sol and a
Precipitate of Block Copolymers in Water. Biomacromolecules 2010, 11, 2169-2178.
(67) Khademhosseini, A.; Langer, R.: Microengineered hydrogels for tissue
engineering. Biomaterials 2007, 28, 5087-5092.
(68) Tan, H. P.; Marra, K. G.: Injectable, Biodegradable Hydrogels for Tissue
Engineering Applications. Materials 2010, 3, 1746-1767.
(69) Drury, J. L.; Mooney, D. J.: Hydrogels for tissue engineering: scaffold design
variables and applications. Biomaterials 2003, 24, 4337-4351.
(70) Park, S.; Nicoll, S. B.; Mauck, R. L.; Ateshian, G. A.: Cartilage mechanical
response under dynamic compression at physiological stress levels following
collagenase digestion. Ann Biomed Eng 2008, 36, 425-434.
(71) Yue, Z. L.; Wen, F.; Gao, S. J.; Ang, M. Y.; Pallathadka, P. K.; Liu, L. H.; Yu,
H.: Preparation of three-dimensional interconnected macroporous cellulosic
hydrogels for soft tissue engineering. Biomaterials 2010, 31, 8141-8152.
(72) Moore, J. S.; Stupp, S. I.: Room-Temperature Polyesterification.
Macromolecules 1990, 23, 65-70.
(73) Lundberg, P.; Hawker, C. J.; Hult, A.; Malkoch, M.: Click assisted one-pot
multi-step reactions in polymer science: accelerated synthetic protocols. Macromol.
Rapid Commun. 2008, 29, 998-1015.
(74) Melekaslan, D.; Kasapoglu, F.; Ito, K.; Yagci, Y.; Okay, O.: Swelling and
elasticity of hydrogels based on poly(ethylene oxide) macroinimer. Polym. Int.
2004, 53, 237-242.
(75) Malkoch, M.; Vestberg, R.; Gupta, N.; Mespouille, L.; Dubois, P.; Mason, A.
F.; Hedrick, J. L.; Liao, Q.; Frank, C. W.; Kingsbury, K.; Hawker, C. J.: Synthesis of
well-defined hydrogel networks using Click chemistry. Chem. Commun.
(Cambridge, U. K.) 2006, 2774-2776.
(76) Lee, Y.; Kim, D. N.; Choi, D.; Lee, W.; Park, J.; Koh, W.-G.: Preparation of
interpenetrating polymer network composed of poly(ethylene glycol) and
poly(acrylamide) hydrogels as a support of enzyme immobilization. Polym. Adv.
Technol. 2008, 19, 852-858.
(77) Peppas, N. A.; Merrill, E. W.: Crosslinked poly(vinyl alcohol) hydrogels as
swollen elastic networks. J. Appl. Polym. Sci. 1977, 21, 1763-70.
(78) Cruise, G. M.; Scharp, D. S.; Hubbell, J. A.: Characterization of permeability
and network structure of interfacially photopolymerized poly(ethylene glycol)
diacrylate hydrogels. Biomaterials 1998, 19, 1287-1294.
(79) Polymer Science Dictionary; Alger, M. S. M., Ed.; Chapman & Hall, 1989.

References
67
(80) Ahearne, M.; Yang, Y.; El Haj, A. J.; Then, K. Y.; Liu, K.-K.: Characterizing the
viscoelastic properties of thin hydrogel-based constructs for tissue engineering
applications. J. R. Soc. Interface 2005, 2, 455-463.
(81) Mawad, D.; Odell, R.; Poole-Warren, L. A.: Network structure and
macromolecular drug release from poly(vinyl alcohol) hydrogels fabricated via
two crosslinking strategies. Int. J. Pharm. 2009, 366, 31-37.
(82) Zustiak, S. P.; Leach, J. B.: Hydrolytically Degradable Poly(Ethylene Glycol)
Hydrogel Scaffolds with Tunable Degradation and Mechanical Properties.
Biomacromolecules 2010, 11, 1348-1357.
(83) Koo, S. P. S.; Stamenovic, M. M.; Prasath, R. A.; Inglis, A. J.; Du Prez, F. E.;
Barner-Kowollik, C.; Van Camp, W.; Junkers, T.: Limitations of Radical Thiol-ene
Reactions for Polymer-Polymer Conjugation. J Polym Sci Pol Chem 2010, 48, 1699-
1713.
(84) Bromberg, L.; Grosberg, A. Y.; Matsuo, E. S.; Suzuki, Y.; Tanaka, T.:
Dependency of swelling on the length of subchain in poly(N,N-
dimethylacrylamide)-based gels. J Chem Phys 1997, 106, 2906-2910.
(85) Baker, J. P.; Hong, L. H.; Blanch, H. W.; Prausnitz, J. M.: Effect of Initial Total
Monomer Concentration on the Swelling Behavior of Cationic Acrylamide-Based
Hydrogels. Macromolecules 1994, 27, 1446-1454.
(86) Haraguchi, K.; Farnworth, R.; Ohbayashi, A.; Takehisa, T.: Compositional
effects on mechanical properties of nanocomposite hydrogels composed of
poly(N,N-dimethylacrylamide) and clay. Macromolecules 2003, 36, 5732-5741.


















