
lysosomal membrane stability and cathepsins in cell death
160
Linköping University Medical Dissertations No. 1325 LYSOSOMAL MEMBRANE STABILITY AND CATHEPSINS IN CELL DEATH HANNA APPELQVIST Department of Clinical and Experimental Medicine, Faculty of Health Sciences, Linköping University, Sweden Linköping 2012
-
Upload
truongdieu -
Category
Documents
-
view
221 -
download
3
Transcript of lysosomal membrane stability and cathepsins in cell death

Linköping University Medical Dissertations No. 1325
LYSOSOMAL MEMBRANE STABILITY
AND CATHEPSINS IN CELL DEATH
HANNA APPELQVIST
Department of Clinical and Experimental Medicine, Faculty of Health Sciences, Linköping University, Sweden
Linköping 2012

© Hanna Appelqvist 2012 Cover picture: Filipin staining of cholesterol in human fibroblasts treated with quinacrine. Published papers have been reprinted with the permission of the respective copyright holder Paper I © 2012 Association of Clinical Scientists Paper II © 2011 Elsevier Printed in Sweden by LiU-tryck, Linköping, Sweden 2012 ISBN: 978-91-7519-803-3 ISSN: 0345-0082

Till min familj
Undret är inte att flyga i luften eller gå på vattnet,
utan att vandra på jorden
Kinesiskt ordspråk

SUPERVISOR Karin Öllinger, Professor Experimental Pathology, Division of Cell Biology, Department of Clinical and Experimental Medicine, Faculty of Health Sciences, Linköping University
CO-SUPERVISORS Katarina Kågedal, Assistant Professor Experimental Pathology, Division of Cell Biology, Department of Clinical and Experimental Medicine, Faculty of Health Sciences, Linköping University
Petra Wäster, PhD Division of Dermatology and Venereology, Department of Clinical and Experimental Medicine, Faculty of Health Sciences, Linköping University
FACULTY OPPONENT Maria Shoshan, Associate Professor Department of Oncology-Pathology, Karolinska Institutet
COMMITTEE BOARD Ebo de Muinck, Professor Division of Cardiovascular Medicine, Department of Medical and Health Sciences, Faculty of Health Sciences, Linköping University
Marianne Farnebo, Associate Professor Department of Oncology-Pathology, Karolinska Institutet
Peter Strålfors, Professor Division of Cell Biology, Department of Clinical and Experimental Medicine, Faculty of Health Sciences, Linköping University

ABSTRACT
Lysosomes are acidic organelles that are critically involved in a number of physiological processes, including macromolecule degradation, endocytosis, autophagy, exocytosis and cholesterol homeostasis. Several pathological conditions, such as cancer, neurodegenerative disorders and lysosomal storage diseases, involve lysosomal disturbances, indicating the importance of the organelle for correct cellular function. The aim of this thesis was to investigate the role of lysosomes in cell death signaling.
Previous studies have shown that permeabilization of the lysosomal membrane and release of hydrolytic enzymes such as cathepsin D to the cytosol occurs during apoptosis. We identified Bid and 14-3-3 proteins as cytosolic targets of cathepsin D in human fibroblasts. Truncated Bid, generated by cathepsin D proteolytic cleavage, stimulates Bax-mediated release of pro-apoptotic factors from the mitochondria, thereby engaging the intrinsic pathway to apoptosis.
Since the presence of cathepsins in the cytosol is sufficient to induce apoptosis, the permeability of the lysosomal membrane influences the fate of the cell. In this thesis, we demonstrated that the stability of the lysosomal membrane can be manipulated by altering the lysosomal cholesterol content. Cells with high lysosomal cholesterol content were less prone to undergo apoptosis when challenged with stimuli known to induce lysosome-mediated cell death. In addition, cholesterol accumulation was associated with increased expression of lysosome-associated membrane proteins and storage of other lipids; however, these factors did not contribute to lysosomal stabilization.
Lysosomal membrane permeabilization and cathepsins contribute to ultraviolet (UV) irradiation-induced apoptosis. We demonstrate plasma membrane damage induced by UVA irradiation to be rapidly repaired by lysosomal exocytosis in human keratinocytes. Despite efficient plasma membrane resealing, the cells

underwent apoptosis, which was dependent on early activation of caspase-8. The activation of caspase-8 was lysosome-dependent and occurred in vesicles positive for lysosomal markers.
This thesis demonstrates the importance of lysosomal stability for apoptosis regulation and that this stability can be influenced by drug intervention. Modulation of the lysosomal membrane permeability may have potential for use as a therapeutic strategy in conditions associated with accelerated or repressed apoptosis.

SAMMANFATTNING
Varje dygn bildas mer än tusen miljarder nya celler i en människokropp, och lika många celler dör. Celldöd som sker enligt ett förbestämt program kallas apoptos, eller programmerad celldöd. Om obalans mellan cellbildning och celldöd uppstår kan det bidra till allvarliga sjukdomar. Vid cancer sker för lite celldöd och skadade celler tillåts överleva. Neurodegenerativa sjukdomar, som till exempel Alzheimers sjukdom, orsakas istället av en alltför omfattande nervcellsdöd. Lysosomer fungerar normalt som cellens motsvarighet till återvinningsstationer där cellkomponenter bryts ned till sina beståndsdelar som kan återanvändas av cellen. Denna avhandling syftar till att undersöka lysosomernas roll vid celldöd.
Lysosomer innehåller många enzymer, däribland cathepsiner. Normallt befinner sig cathepsinerna inuti lysosomen och skiljs från resten av cellen av ett skyddande hölje, lysosommembranet. Vid apoptos går lysosomens membran sönder och cathepsinerna läcker ut och påverkar övriga delar av cellen. Denna avhandling visar att cathepsin D bidrar till celldöd genom att aktivera proteinet Bid. Eftersom läckage av cathepsiner från lysosomen är tillräckligt för att en cell ska dö är det viktigt att lysosommembranets genomsläpplighet regleras noga. Det är dock till stor del oklart vilka faktorer som påverkar membranet och hur. Avhandlingen visar att kolesterol är en sådan faktor. Ett högt kolesterolinnehåll i lysosommmebranet gör det mindre genomsläppligt och bidrar till att cellen blir mer motståndskraftig mot celldöd.
När vi är ute i solsken träffas vår hud av ultraviolett strålning som skadar cellens yttre hölje. Reparation av denna skada är viktig för cellens överlevnad och vi visar att lagningen sker med hjälp av lysosomer. Våra resultat visar att trots att lagningen är effektiv kommer vissa celler att dö på grund av skador orsakade av strålningen. Denna celldöd är viktig för att förhindra uppkomsten av hudcancer.

Genom att studera hur apoptos regleras och kartlägga orsakerna till felreglerad celldöd kan man utveckla nya sätt att behandla sjukdomar som har ett samband med ökad eller minskad celldöd.

TABLE OF CONTENTS
LIST OF PAPERS ............................................................................... 13
ABBREVIATIONS ............................................................................. 15
INTRODUCTION .............................................................................. 17
APOPTOSIS ................................................................................................................................. 17
Caspases .............................................................................................................................................. 19
Apoptotic signaling pathways ................................................................................................................ 20
The Bcl-2 protein family ...................................................................................................................... 23
Bax ................................................................................................................................................................. 25
Consequences of mitochondrial outer membrane permeabilization ........................................................................... 29
Dysregulated apoptosis in disease .......................................................................................................... 31
LYSOSOMES - MULTIFUNCTIONAL ORGANELLES ........................................................................... 33
The lysosomal membrane ..................................................................................................................... 33
Bis(monoacylglycero)-phosphate (BMP) ................................................................................................................ 34
Acid hydrolases and lysosomal membrane proteins ................................................................................ 34
Lysosome-associated membrane proteins (LAMPs) .................................................................................................. 35
Cathepsins ........................................................................................................................................................ 36
Functions of the lysosomal compartment .............................................................................................. 38
Degradation of macromolecules ........................................................................................................................... 39
Endocytosis ....................................................................................................................................................... 39
Autophagy ........................................................................................................................................................ 42
Membrane repair by lysosomal exocytosis .............................................................................................................. 43
Cholesterol homeostasis ....................................................................................................................................... 45
Lysosomal participation in cell death signaling ...................................................................................... 48
Mechanisms of lysosomal membrane permeabilization ............................................................................................ 49
Functions of cathepsins in the cytosol.................................................................................................................... 55
Lysosomes in disease............................................................................................................................. 57
Lysosomal storage disorders ................................................................................................................................. 57
Adult neurodegenerative disorders ....................................................................................................................... 59
Cancer ............................................................................................................................................................. 60

AIMS ............................................................................................. 63
MATERIALS AND METHODS ............................................................. 65
CELLS .......................................................................................................................................... 65
APOPTOSIS INDUCERS ................................................................................................................. 68
INHIBITORS ................................................................................................................................. 73
MODULATION OF LYSOSOMAL CHOLESTEROL CONTENT .............................................................. 76
DETECTION OF CELL DEATH .......................................................................................................... 79
METHODS FOR THE ANALYSIS OF LYSOSOMES AND THEIR STABILITY ............................................ 83
WESTERN BLOT ANALYSIS ............................................................................................................ 86
IMMUNOCYTOCHEMISTRY .......................................................................................................... 86
MEASUREMENT OF CYTOSOLIC PH ............................................................................................... 87
CELL-FREE EXPERIMENTS ............................................................................................................. 89
MICROINJECTION ........................................................................................................................ 90
ANALYSIS OF LIPIDS ..................................................................................................................... 91
STATISTICAL ANALYSIS ................................................................................................................ 92
ETHICAL CONSIDERATIONS .......................................................................................................... 93
RESULTS ........................................................................................ 95
PAPER I: Cathepsin D-specific processing of Bid at Phe24, Trp48 and Phe183 ................................ 95
PAPER II: Attenuation of the lysosomal death pathway by lysosomal cholesterol accumulation .... 98
PAPER III: Modulation of lysosomal cholesterol content influences lysosome-dependent cell death sensitivity .......................................................................................................................... 99
PAPER IV: Lysosomal exocytosis repairs the plasma membrane after UVA and is followed by caspase-8 induced apoptosis ..................................................................................................... 102
DISCUSSION ................................................................................. 105
THE ROLE OF CATHEPSIN D IN APOPTOSIS SIGNALING ................................................................ 105
Pro-apoptotic Bid processing ...............................................................................................................108
The proteolytic activity of cathepsin D is influenced by pH ..................................................................110
Cathepsin D vs. cysteine cathepsins .....................................................................................................111
THE EFFECT OF CHOLESTEROL ON LYSOSOMES ........................................................................... 112
Cholesterol modulates lysosomal membrane stability ............................................................................113
Cholesterol accumulation induces alterations of the lysosomal compartment .........................................115

LYSOSOMAL EXOCYTOSIS .......................................................................................................... 116
CASPASE-8 ACTIVATION ............................................................................................................ 118
CONCLUSIONS............................................................................... 123
CLINICAL IMPLICATIONS ................................................................ 125
FUTURE PERSPECTIVES .................................................................. 127
TACK ...........................................................................................129
REFERENCES ................................................................................. 133


13
LIST OF PAPERS
This thesis is based on the following papers, which will be referred to in the text by Roman numerals:
I. Cathepsin D-specific processing of Bid at Phe24, Trp48, and Phe183. Hanna Appelqvist, Ann-Charlotte Johansson, Emma Linderoth, Uno Johansson, Bruno Antonsson, Robert Steinfeld, Katarina Kågedal and Karin Öllinger Annals of Clinical and Laboratory Science 42(3): 231-242, 2012
II. Attenuation of the lysosomal death pathway by lysosomal cholesterol accumulation. Hanna Appelqvist, Cathrine Nilsson, Brett Garner, Andrew J Brown, Katarina Kågedal and Karin Öllinger American Journal of Pathology 178(2):629-39, 2011
III. Modulation of lysosomal cholesterol content influences lysosome-dependent cell death sensitivity. Hanna Appelqvist, Linnea Sandin, Karin Björnström, Paul Saftig, Brett Garner, Karin Öllinger and Katarina Kågedal Accepted for publication in PLoS One
IV. Lysosomal exocytosis repairs the plasma membrane after UVA and is followed by caspase-8 induced apoptosis. Hanna Appelqvist, Petra Wäster, Ida Eriksson, Inger Rosdahl and Karin Öllinger Manuscript


15
ABBREVIATIONS
3-MA 3-methyladenine
25-HC 25-hydroxycholesterol
ACAT acyl CoA:cholesterol acyltransferase
AIF apoptosis inducing factor
AO acridine orange
Apaf-1 apoptosis protein activating factor-1
aSMase acid sphingomyelinase
BCECF 2',7'-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein
BH Bcl-2 homology
BMP bis(monoacylglycero)-phosphate
CAD cationic amphiphilic drug
CARD caspase activation and recruitment domain
CE cholesterol ester
CHO Chinese hamster ovary
CLEAR coordinated lysosomal expression and regulation
DAPI 4’,6-diamidino-2-phenylindole
DD death domain
DED death effector domain
DISC death-inducing signaling complex
DMSO dimethyl sulfoxide
EEA1 early endosomal antigen 1
ER endoplasmic reticulum
FADD Fas associated death domain
FC free cholesterol
GAPDH glyceraldehyde-3-phosphate dehydrogenase
Hsp heat shock protein
IAP inhibitor of apoptosis
ILV intraluminal vesicle
LAMP lysosome-associated membrane protein
LDH lactate dehydrogenase
LDL low density lipoprotein
LMP lysosomal membrane permeabilization
M6P mannose-6-phosphate
MβCD methyl-β-cyclodextrin

16
MEF mouse embryonic fibroblast
MOMP mitochondrial outer membrane permeabilization
MSDH O-methyl-serine dodecylamine hydrochloride
MTT 3-(4,5-dimethylthiazol-2-yl)-,5-diphenyltetrazolium bromide
NAG N-acetyl-β-glucosaminidase
NPC Niemann-Pick disease type C
Pep A pepstatin A
PLA2 phospholipase A2
ROS reactive oxygen species
SNARE soluble N-ethylmaleimide-sensitive factor attachment protein receptors
STS staurosporine
Syt VII synaptotagmin VII
TFEB transcription factor EB
TGN trans-Golgi network
TNF tumor necrosis factor
TRADD TNF receptor 1-associated death domain
TRAIL TNF-related apoptosis-inducing ligand
tBid truncated Bid
UV ultraviolet
wt wild type
XIAP X-linked inhibitor of apoptosis

INTRODUCTION
17
INTRODUCTION
APOPTOSIS Programmed cell death, or apoptosis, is a fundamental process that is evolutionary conserved. Apoptosis is essential for normal development because it removes unnecessary or excessive cells during tissue formation, but it is also important later in life for the precise regulation of cell numbers to maintain tissue homeostasis (Penaloza et al., 2006). In addition, cell death is also crucial as a defense mechanism to remove damaged or potentially dangerous cells, such as malignant cells, virus-infected cells and self-reactive lymphocytes (Moffitt et al., 2010). Traditionally, cell death has mainly been categorized as apoptotic or necrotic based on morphological changes.
The term apoptosis was coined in 1972 by John Kerr, Andrew Wyllie and Alastair Currie and is used to describe the specific morphological changes associated with this type of cell death (Kerr et al., 1972). The apoptotic cell death process is divided into three phases: the initiation phase, which involves the activation of heterogeneous signaling pathways; the commitment phase, during which the cell becomes irreversibly committed to death; and the execution phase, during which the morphological changes characterizing apoptosis occur. As illustrated in Figure 1, these alterations include a reduction of cellular volume, retraction of pseudopods, chromatin condensation, nuclear fragmentation, plasma membrane blebbing and disassembly of the cell into apoptotic bodies (Häcker, 2000). In vivo, this process culminates in the engulfment of the apoptotic bodies by other cells, preventing the release of cellular content into the extracellular space. By contrast, necrosis is generally considered an acute and uncontrolled mode of cell death that is associated with cell swelling and lysis, resulting in inflammation in the tissue. However, increasing evidence suggests that the execution of necrotic cell death may also be finely regulated (Festjens et al., 2006; Galluzzi et al., 2011; McCall, 2010).

INTRODUCTION
18
Figure 1. Morphological changes associated with apoptosis and necrosis. Necrotic morphology is characterized by increased cellular volume and loss of plasma membrane integrity, resulting in the release of cellular content to the surroundings. Apoptosis is associated with a reduction in cell volume, condensation and nuclear fragmentation. During this process, the plasma membrane remains intact, and the formation of apoptotic bodies, which are engulfed by neighboring cells, prevents inflammation.
Although cell death has traditionally been categorized based on morphology, a new molecular classification of cell death modalities has recently been proposed (Galluzzi et al., 2012a). It has become evident that the morphology of cell death can be similar even though the lethal signaling cascade may vary. Therefore, definitions based on biochemical rather than morphological criteria may be more appropriate. According to the new classification, cell death is divided into extrinsic apoptosis, caspase-dependent or caspase-independent intrinsic apoptosis, regulated necrosis, mitotic catastrophe and autophagic cell death (Galluzzi et al., 2012a). Although multiple death modalities exist, the majority of described cell death processes are mediated by a caspase-dependent apoptotic mechanism. The apoptotic program involves a highly sophisticated and well-regulated machinery to efficiently eliminate cells. There are several pathways through which the apoptotic machinery can be activated, which frequently converge in the activation of caspases, which are responsible for the dismantling of the cell.

INTRODUCTION
19
Caspases Caspases are a structurally related family of proteases. They are cysteine-dependent aspartate-specific proteases, hence their name. In humans, 12 caspases have been identified, seven of which are involved in apoptotic signaling (Würstle et al., 2012). The other caspases participate in cytokine activation as part of immune responses. The apoptotic caspases are classified into two categories; initiation caspases (caspase-2, -8, -9 and -10) and effector caspases (caspase-3, -6 and -7) (Pop and Salvesen, 2009). As shown in Figure 2, caspases are synthesized and exist in healthy cells as inactive zymogens containing a pro-domain, a large subunit and a small subunit (Würstle et al., 2012). Effector caspases possess short prodomains, while initiator caspases have long prodomains harboring specific motifs essential for their activation. Initiation caspases are activated through specific and highly regulated mechanisms that generally involve the formation of multimeric protein complexes. As illustrated in Figure 2, initiator caspases activate effector caspases via proteolytic removal of the prodomain, thus generating a proteolytic cascade (Pop and Salvesen, 2009). Dimerization is required for caspase activity, as it allows the formation of the active site and the substrate pocket (van Raam and Salvesen, 2012). In contrast to executor caspases which exist as inactive dimers, initiator procaspases are monomers, which dimerize during activation.
Figure 2. Caspases are synthesized as inactive zymogens, which are activated during apoptosis. Caspase zymogens consist of a prodomain, a large subunit and a small subunit. Effector caspases, including caspase-3, have a short prodomain, while initiator caspases (for example caspase-8 and -9) have long prodomains with death effector domains (DEDs) or caspase recruitment domains (CARDs), which enable their activation in protein complexes. Activation of an effector caspase occurs through proteolytic processing at internal aspartic residues, resulting in removal of the prodomain. Two caspases dimerize, allowing the formation of the active site () and thus active caspases are found as dimers in the cell.

INTRODUCTION
20
Effector caspases have a large number of cytosolic substrates, and their proteolysis results in the biochemical and morphological changes associated with apoptosis, such as fragmentation of DNA, chromatin condensation and plasma membrane blebbing (Häcker, 2000). Caspase activity can be controlled upstream, by the regulation of signals that lead to zymogen activation, or downstream, by inhibitors that prevent caspases from interacting with their substrates (Pop and Salvesen, 2009). Inhibitor of apoptosis proteins (IAPs) are a family of proteins that can inhibit caspases. X-linked IAP (XIAP) is the only mammalian IAP that directly functions as a caspase inhibitor, and it effectively inhibits caspase-3, -7 and -9 (Gyrd-Hansen and Meier, 2010). In addition, XIAP and other members of this family, including cIAP1 and cIAP2, can indirectly inhibit the activity of caspases by ubiquitination, which may lead to proteasomal degradation (Gyrd-Hansen and Meier, 2010). Although caspases are normally key mediators of apoptosis, caspase activation is not an absolute requirement for apoptosis to occur (Galluzzi et al., 2012a).
Apoptotic signaling pathways There are two classical signaling pathways leading to the activation of the caspase cascade: the intrinsic and the extrinsic pathways (Figure 3). The apoptotic demise of cells can be triggered by a number of intracellular stress conditions, including DNA damage and oxidative stress. Although the signaling that initiates intrinsic apoptosis is heterogeneous, mitochondrial participation is unifying for the intrinsic pathway. Normally, a number of signaling cascades (both pro- and anti-apoptotic) converge at the level of mitochondria. When the pro-apoptotic signals dominate, the integrity of the mitochondrial membrane is lost in a process known as mitochondrial outer membrane permeabilization (MOMP) (Martinou and Youle, 2011). MOMP results in the release of pro-apoptotic factors, including cytochrome c, from the intermembrane space of mitochondria to the cytosol. As shown in Figure 3, cytosolic cytochrome c acts as a cofactor for the assembly of the apoptosome, a protein complex in which caspase-9 is activated (Würstle et al., 2012).
The extrinsic pathway to apoptosis is dependent on death receptors belonging to the tumor necrosis factor (TNF) receptor family. These include the Fas receptor

INTRODUCTION
21
(APO-1/CD95), TNF receptor 1 and TNF-related apoptosis-inducing ligand (TRAIL) receptors 1 and 2 (Dickens et al., 2012). The extrinsic pathway is activated by the ligation of death receptors found on the cell surface by their respective ligands (Fas ligand, TNF-α and TRAIL). In the case of Fas receptor signaling, Fas spontaneously trimerizes at the plasma membrane. Ligand binding stabilizes these trimers and induces a conformational change that permits the assembly of a multimeric protein complex at the cytosolic part of the receptors. The adaptor protein Fas associated death domain (FADD) is recruited and interacts with the death receptor via their respective death domains (DDs). FADD, in turn, recruits procaspase-8 via a homotypic interaction between their respective death effector domains (DEDs) (Dickens et al., 2012). Lethal signaling by TNF receptor 1 requires the participation of additional adaptor proteins, including tumor necrosis factor receptor type 1-associated death domain (TRADD) (Cabal-Hierro and Lazo, 2012). The assembled protein complex is known as the death inducing signaling complex (DISC), which serves as a platform for dimerization of caspase-8 (Figure 3). The caspase-8 dimer is active in the absence of proteolytic processing, but after dimerization proteolytic cleavage serves to stabilize the active conformation and increase the activity (van Raam and Salvesen, 2012). The active caspase-8 dimer can be released from the DISC-complex by proteolytic cleavage between the DED and the large subunit.
In type I cells, active caspase-8 directly activates caspase-3, thereby triggering the execution phase of apoptosis in a mitochondria-independent manner (Figure 3). However, in most cells (type II cells), amplification of the death signal by mitochondrial engagement is required for efficient activation of the caspase cascade (Kantari and Walczak, 2011). The difference between type I and type II cells is the expression of XIAP (Jost et al., 2009). High expression of XIAP results in the inability of caspase-8 to efficiently activate caspase-3, and thus mitochondrial amplification of the death signal is required. Mitochondrial engagement in type II cells is achieved by caspase-8-mediated proteolytic processing of the Bcl-2 protein Bid, which, in its active truncated form, promotes MOMP (Kantari and Walczak, 2011).
Caspase-10, like caspase-8, contains a DED, indicating a role in death receptor signaling. Indeed, caspase-10 is activated in a FADD-dependent manner in the

INTRODUCTION
22
DISC, but it is not required for apoptosis and cannot substitute for caspase-8 deficiency (Sprick et al., 2002). Interestingly, signaling by death receptors appears to be regulated by their intracellular localization, and endocytosis of the receptor complexes modulates the apoptotic signaling (Akazawa et al., 2009; Lee et al., 2006; Schneider-Brachert et al., 2004).
Figure 3. The extrinsic and intrinsic pathways to apoptosis. The two classical apoptosis pathways are characterized by the activation of death receptors and the permeabilization of the mitochondrial outer membrane, respectively. These signaling pathways are interconnected by the protein Bid, and both ultimately result in the activation of the caspase cascade. Abbreviations: Apaf-1, apoptosis protein activating factor-1; Cyt c, cytochrome c; DD, death domain; DED, death effector domain; DISC, death inducing signaling complex; FADD, Fas associated death domain; MOMP, mitochondrial outer membrane permeabilization; and tBid, truncated Bid.
In addition to the well-known role of DISC in caspase-8 activation, alternative modes of caspase-8 activation have been described. For example, activation of caspase-8 and the subsequent proteolytic processing of Bid have been demonstrated

INTRODUCTION
23
to be due to caspase-8 insertion, homodimerization and autoactivation at the mitochondrial outer membrane (Gonzalvez et al., 2008).
The machinery for apoptosis is present in essentially all mammalian cells at all times. The danger of such a suicide program is obvious, and therefore a complex regulatory network has evolved. The Bcl-2 protein and its homologs are key elements in this regulatory network.
The Bcl-2 protein family The Bcl-2 protein family consists of more than 20 proteins that share homology in at least one of four conserved motifs known as Bcl-2 homology domains (BH1-BH4) (Shamas-Din et al., 2011). As illustrated in Figure 4, the Bcl-2 protein family is divided into three functional groups based on the expression of these BH-domains: i) anti-apoptotic Bcl-2 proteins (Bcl-2, Bcl-XL, Bcl-W, Mcl-1 and A1), which possess all four BH-domains; ii) pro-apoptotic Bcl-2 proteins, which express multiple BH-domains (Bax and Bak); and iii) pro-apoptotic Bcl-2 proteins, which only contain the BH3 domain and are therefore referred to as BH3-only proteins (including Bad, Bim, Bid, Bik, Noxa and Puma) (Chipuk et al., 2010). The pro-apoptotic multidomain proteins Bax and Bak were originally described to possess BH1-BH3 but have since been discovered to contain a conserved BH4 motif, as well (Kvansakul et al., 2008). In addition, a third member of this subclass, Bok, has been described, but its action is not well characterized (Hsu et al., 1997a; Ke et al., 2012).
Figure 4. The three subgroups of the mammalian Bcl-2 protein family. The presence of the Bcl-2 homology (BH) domains is used to classify the Bcl-2 protein family into three functional groups. The anti-apoptotic Bcl-2 proteins express all four BH domains, while their pro-apoptotic relatives are either multi domain proteins or express only the BH3 domain. Many of the proteins of the Bcl-2 family also contain a transmembrane domain (TM).

INTRODUCTION
24
The integrity of the mitochondrial outer membrane is tightly controlled by the Bcl-2 proteins. Pro-apoptotic members cooperate to induce MOMP, while the anti-apoptotic members preserve mitochondrial integrity. Because the Bcl-2 family acts at the mitochondria and, thus, usually upstream of irreversible cellular damage, these proteins play a pivotal role in whether a cell will live or die. This fate is determined by the level of pro- versus anti-apoptotic Bcl-2 family members present in the cell.
There are several proposed models for how Bcl-2 family members interact with each other (Shamas-Din et al., 2011). The lipid composition and the structural organization of the membrane in which these interactions generally occur significantly influences binding and protein conformation, thus leading to the proposal of the “embedded together theory” of Bcl-2 interactions (Bogner et al., 2010). In most cases, a Bcl-2 protein interaction is dependent on the BH3 domain of one protein and a hydrophobic groove on its partner (Westphal et al., 2011). Anti-apoptotic members block apoptosis by sequestering both BH3-only proteins and Bax/Bak (Llambi et al., 2011). BH3-only proteins can be classified either as sensitizers (Bad, Bik and Noxa) or activators (Bim, tBid and possibly Puma) (Letai et al., 2002). Activators directly bind to and activate Bax and Bak, while sensitizers promote MOMP indirectly (Figure 5). In healthy cells, BH3-only proteins are either inactive or sequestered by anti-apoptotic proteins. BH3-only proteins act as pathway-specific sensors in the cell, and in response to an apoptotic signal, activation can be fulfilled by several mechanisms, such as transcriptional induction, phosphorylation or cleavage (Shamas-Din et al., 2011). In addition, activator BH3-only proteins can be released from anti-apoptotic Bcl-2 proteins by sensitizer BH3-only proteins that bind to the anti-apoptotic relatives with higher affinity (Shamas-Din et al., 2011). After their activation or release, the activator BH3-only proteins transmit the death signal to the multidomain Bcl-2 family members, Bax and Bak, which are the executioners of MOMP (Wei et al., 2001). These two proteins seem to be functionally redundant, and inactivation of both is required to fully impair apoptosis in most tissues (Lindsten et al., 2000).

INTRODUCTION
25
Figure 5. Proteins of the Bcl-2 protein family regulates apoptosis. A) The ratio between pro- and anti-apoptotic Bcl-2 family proteins determines the fate of the cell. Excessive anti-apoptotic proteins results in cell survival, but when the pro-apoptotic proteins prevail apoptosis is induced. B) There are several proposed models for the interactions between the different subgroups of Bcl-2 proteins, but to date, the exact mechanisms remain obscure. BH3-only proteins promote the activation of Bax and/or Bak either directly or indirectly. BH3-only proteins capable of directly activating Bax/Bak are called activators; tBid, Bim and Puma belong to this group. Sensitizing BH3-only (Bad, Bik and Noxa) proteins promote Bax/Bak activation by binding to and neutralizing anti-apoptotic Bcl-2 proteins such as Bcl-2 and Bcl-XL. Anti-apoptotic Bcl-2 proteins inhibit mitochondrial outer membrane permeabilization (MOMP) by interacting with both BH3-only proteins and Bax/Bak.
Bax Bax was the first pro-apoptotic protein of the Bcl-2 family to be isolated and was given the name Bcl-2 associated protein X because it co-immunoprecipitates with Bcl-2 and blocks Bcl-2´s prosurvival activity when co-expressed (Oltvai et al., 1993). In contrast to Bak, which is normally present in the mitochondrial membrane, Bax is localized mainly to the cytoplasm but redistributes to mitochondria in response to stress stimuli (Hsu et al., 1997b). It is widely accepted that Bak and Bax are critical pro-apoptotic effectors that increase the permeability of the outer mitochondrial membrane, but the exact mechanism remains a subject of debate.
Sequestration of Bax by 14-3-3 proteins The mechanisms underlying Bax translocation and insertion into the mitochondrial membrane are not fully understood, but it has been suggested that there is a need for a signal that releases Bax from inhibitory interactions with sequestering proteins (Sawada et al., 2003; Won et al., 2003). A number of proteins have been shown to negatively regulate apoptosis by binding to Bax and
BH3-only protein BH3- only protein
Anti-apoptoticBcl-2 protein Bax/Bak
Sensitizer Direct activator
Death stimulus
MOMPMOMP
Death stimulus
Cell survival Cell deathCell deathsurvival
Pro-apoptoticproteins
Anti-apoptoticproteins
Checkpoint
A B

INTRODUCTION
26
sequestering it from the mitochondria, including Ku70, humanin, 14-3-3 and apoptosis repressor with caspase recruitment domain (ARC) (Guo et al., 2003; Gustafsson et al., 2004; Nomura et al., 2003; Sawada et al., 2003). However, the presence and biological significance of these inhibitory interactions remains unclear (Vogel et al., 2012).
14-3-3 proteins are a family of highly expressed regulatory proteins. Seven isoforms are found in mammals and they are known for their great ability to bind other proteins (Obsil and Obsilova, 2011). Through their interactions with key signaling molecules, 14-3-3 proteins regulate central signaling events such as metabolism, signal transduction, stress response and progression through the cell cycle (Gardino and Yaffe, 2011). 14-3-3 proteins control the induction of apoptosis at multiple levels, including interaction with Bcl-2 family proteins (Gardino and Yaffe, 2011). The 14-3-3 proteins were first discovered to interact with Bad (Subramanian et al., 2001; Zha et al., 1996) but were later shown to bind to Bax. When overexpressed, 14-3-3 proteins selectively inhibit Bax-mediated apoptosis (Nomura et al., 2003; Samuel et al., 2001). Bax is thought to be held in an inactive conformation by the 14-3-3 proteins, and this interaction is supposed to be disrupted upon the induction of apoptosis (Nomura et al., 2003; Samuel et al., 2001). However, contradictory evidence indicates that the interaction persists during apoptosis (Sutheesophon et al., 2006). Bax liberation is partly dependent on caspase-mediated cleavage of the 14-3-3 proteins, which reduces the binding affinity and results in Bax release and redistribution to the mitochondria (Nomura et al., 2003; Won et al., 2003). In one study apoptosis-associated cleavage of the 14-3-3θ (also called τ) isoform was almost unaffected by the addition of a broad caspase inhibitor; thus, the authors proposed that non-caspase proteases contribute to 14-3-3 processing (Kuzelova et al., 2009). In addition, translocation of Bax to the mitochondria occurs upstream of caspase activation in a number of experimental systems, suggesting the existence of other mechanisms responsible for the dissociation of Bax from 14-3-3 proteins.

INTRODUCTION
27
Activation of Bax by Bid The requirement for the release of Bax from interactions in the cytosol is controversial, while the need for an activating signal that unleashes its pro-apoptotic potential is widely recognized. Such a signal can be provided by BH3-only proteins, which are activated in response to various apoptotic stimuli (Shamas-Din et al., 2011).
Bid (BH3 interacting domain death agonist) was identified as a death agonist capable of interacting with both Bax and Bcl-2 (Wang et al., 1996). It belongs to the large group of BH3-only proteins but has a unique function because it connects the extrinsic and intrinsic apoptotic pathways. Under normal conditions, Bid is found in the cytosol in an inactive form, and its pro-apoptotic function is activated by proteolytic processing (Kantari and Walczak, 2011). Caspase-8 was the first protease shown to cleave Bid, but other caspases, calpains, granzymes and cysteine cathepsins have been demonstrated to activate Bid as well (Barry et al., 2000; Chen et al., 2001; Cirman et al., 2004; Li et al., 1998; Mandic et al., 2002; Milhas et al., 2005; Slee et al., 2000). Proteolytic processing of Bid by caspases yields a 15 kDa truncated form known as tBid (Li et al., 1998; Luo et al., 1998). Cleavage removes the N-terminus of Bid, which has an inhibitory effect on its pro-apoptotic activity (Tan et al., 1999). Moreover, removal of this domain results in exposure of the BH3 domain, thus enabling membrane insertion and facilitating protein interactions (McDonnell et al., 1999). In most cases, truncation of Bid seems crucial for its pro-apoptotic function, although some reports have demonstrated that full-length Bid can induce apoptosis as well (Maas et al., 2011; Sarig et al., 2003).
The Bax protein consists of nine α-helices and adopts a locked globular structure under normal conditions. It is mainly cytosolic, but can to a minor extent be found attached to the membranes of the mitochondria and endoplasmic reticulum (ER) (Westphal et al., 2011). The C-terminal helix (α9), which may act as a membrane anchor, is buried in the hydrophobic groove, which may explain why Bax is predominantly a cytosolic protein (Suzuki et al., 2000). As illustrated in Figure 6, Bax undergoes major conformational changes during apoptosis, from an inert monomer to a pore-forming oligomer, and translocates from the cytosol to the mitochondria (Hsu et al., 1997b; Wolter et al., 1997). Bid is a direct activator of

INTRODUCTION
28
Bax and is thus able to induce the conformational changes that ultimately result in MOMP (Desagher et al., 1999; Eskes et al., 2000). After proteolytic processing, tBid translocates to the mitochondria, where its membrane-bound form stimulates translocation and membrane insertion of Bax (Eskes et al., 2000; Gross et al., 1999).
Figure 6. Activation of Bax, dimer formation and membrane insertion. A proposed sequence of Bax structural rearrangement involved in the conversion from an inert cytosolic monomer to a membrane-inserted oligomer. Exposure of the N-terminus results in opening of the Bax structure and exposure of the BH3-domain. This domain is only transiently exposed because it participates in symmetric BH3:groove dimer formation by binding the hydrophobic groove of another Bax protein. Membrane insertion involves the C-terminal α9 helix, as well as the α5 and α6 (black) helices. Dimers can be linked via the α6-helices to form oligomers (Westphal et al., 2011). Oligomerization is necessary and sufficient for membrane permeabilization.
Bax-mediated permeabilization of the mitochondrial membrane Bax and Bak are absolutely required for MOMP and cells from Bax and Bak double knockout mice are resistant to a number of apoptotic stimuli that induce apoptosis via activation of the intrinsic pathway (Wei et al., 2001). However, the exact mechanism by which Bax permeabilizes membranes to allow the release of proteins remains unclear. It has been suggested that Bax could modulate the opening of an existing channel, such as the permeability transition pore or the voltage dependent anion channel. However, genetic studies have demonstrated that it is unlikely that any of these channels participate in Bax-induced MOMP (Tait and Green, 2010). Instead, the prevalent hypothesis is that Bax, alone or in combination with other factors, forms a channel in the mitochondrial membrane. This was first suggested based on the structural similarities between Bcl-2 family proteins and bacterial pore-forming toxins (Muchmore et al., 1996). Indeed, Bax has been shown to form channels in liposomes and phospholipid bilayers, and

INTRODUCTION
29
purified Bax can release cytochrome c from isolated mitochondria (Narita et al., 1998; Schlesinger et al., 1997). However, it is not clear whether Bax induces the formation of a proteinaceous or a lipidic pore (Figure 7). Bax pores formed in planar lipid bilayers and liposomes have the characteristics of a lipidic pore, but a protein pore was observed in mitochondria isolated from apoptotic cells (Martinez-Caballero et al., 2009). This protein pore was named the mitochondrial apoptosis-induced channel (MAC). This channel formed de novo during apoptosis, and it has been suggested to contain roughly ten Bax monomers (Martinez-Caballero et al., 2009). It is not known if MAC formation is the only mechanism for the release of apoptotic factors from the mitochondria.
Figure 7. Two possible mechanisms by which Bax might porate the mitochondrial membrane. In the proteinaceous pore, part of the Bax protein aligns to form a barrel-like pore that spans the membrane. Bax could also form a lipidic pore by inserting into the membrane and inducing membrane curvature such that the outer and inner leaflet are continuous, thus allowing cytochrome c (Cyt c) release.
Consequences of mitochondrial outer membrane permeabilization Irreversible MOMP has a number of lethal consequences for the cell: i) disruption of the mitochondrial membrane potential, which interrupts mitochondrial ATP synthesis; ii) release of pro-apoptotic proteins from the intermembrane space to the cytosol; and iii) inhibition of the respiratory chain, resulting in overproduction of reactive oxygen species (Tait and Green, 2010). The most well-known of the proteins released from the mitochondria during apoptosis is cytochrome c, but a number of other proteins that normally reside within mitochondria assist in the lethal signaling upon release to the cytosol (Figure 8). Apoptosis inducing factor (AIF) and endonuclease G translocate to the nucleus, where they mediate DNA

INTRODUCTION
30
fragmentation independent of caspases (Li et al., 2001; Susin et al., 1996). Second mitochondrial activator of caspases/direct inhibitor of apoptosis-binding protein with low pI (Smac/Diablo) and the mammalian homolog of the bacterial high temperature requirement protein (HtrA2/OMI) both function by inhibiting the anti-apoptotic function of XIAP, thereby derepressing caspase activation (Du et al., 2000; Verhagen et al., 2002).
Figure 8. Release of pro-apoptotic factors during mitochondrial outer membrane permeabilization (MOMP). Cytosolic cytochrome c (Cyt c) participates in the formation of the apoptosome, a cytosolic protein complex in which caspase-9 is activated. In addition to cytochrome c, several proteins with pro-apoptotic properties are released from the mitochondria to the cytosol during MOMP. Apoptosis inducing factor (AIF) and endonuclease G (Endo G) mediate DNA fragmentation, while second mitochondrial activator of caspases/direct inhibitor of apoptosis-binding protein with low pI (Smac/Diablo) and the mammalian homolog of the bacterial high temperature requirement protein (HtrA2/OMI) facilitate caspase activation by repressing the activity of X-linked inhibitor of apoptosis (XIAP).

INTRODUCTION
31
Dysregulated apoptosis in disease The enormous importance of the apoptotic process in the normal function of multicellular organisms is indicated by the number of diseases for which dysregulated apoptosis is a causative or contributing factor (Figure 9). Inappropriate apoptosis is implicated in neurodegenerative diseases, stroke, ischemic injury following myocardial infarction, acquired immunodeficiency syndrome (AIDS), sustained viral infections, autoimmune disorders and cancer (Moffitt et al., 2010).
Figure 9. Deregulated apoptosis contributes to many diseases. Homeostasis is achieved when cell proliferation is perfectly balanced with cell death. Neurodegenerative disorders, acquired immunodeficiency syndrome (AIDS) and ischemic injuries are examples of diseases involving excessive apoptosis. There are also conditions in which apoptosis is inadequate, resulting in autoimmune diseases or the growth of malignant cells.
Normally, the cell death machinery assures the elimination of cells with DNA damage. However, evasion of cell death is a characteristic feature of cancer, and tumor cells have a variety of mechanisms to protect against apoptotic elimination. Mutations in the tumor suppressor p53, the normal function of which is to induce cell cycle arrest and apoptosis in response to DNA damage, are found in more than 50% of human carcinomas (Reinhardt and Schumacher, 2012). In addition, tumors frequently display altered expression of IAPs and Bcl-2 family proteins (Fulda and Vucic, 2012; Moffitt et al., 2010). Radiotherapy and many

INTRODUCTION
32
chemotherapeutic drugs have been shown to induce apoptosis. The efficacy of these treatments depends on their ability to induce substantial cellular damage, as well as the ability of the cells to respond to damage. Thus, in addition to permitting the survival of genetically modified cells, alterations in key components of the apoptosis signaling network may lead to treatment resistance. Modifying the ratio of pro- versus anti-apoptotic proteins to reactivate the cell death program in cancer cells is a promising strategy to overcome resistance to treatment, which is a major unsolved problem in clinical oncology. Indeed, several apoptosis-based cancer therapeutics have entered clinical trials for the treatment of several malignancies (Fulda, 2009). These novel treatments include TRAIL receptor agonists and compounds designed to inhibit or decrease the expression of the anti-apoptotic Bcl-2 family proteins and IAPs (Fulda and Vucic, 2012; Leber et al., 2010). Interestingly, approximately 80% of human cancer cell lines are sensitive to TRAIL, whereas most normal cells are completely resistant (Nicholson, 2000). Due to the differential sensitivity toward TRAIL-mediated apoptosis, TRAIL receptor activation has attracted considerable interest as a mechanism for inducing apoptosis specifically in cancer cells.
The therapeutic interventions described above aim to induce cell death; however, preventing apoptosis may be beneficial in pathological conditions associated with excessive death. During embryonic development, cells of the central nervous system are overproduced, and the large quantity of superfluous cells must be eliminated by apoptosis. However, post-development excessive apoptosis in the central nervous system is deleterious. Neurons do not divide, meaning that dead cells cannot be replaced; therefore, neuronal cell loss is associated with deprivation of vital functions. Loss of specific subsets of neurons in selective parts of the brain characterize neurodegenerative disorders, such as Alzheimer’s, Huntington´s and Parkinson´s diseases (Moffitt et al., 2010). For pathological conditions for which prevention of apoptosis would be desirable, there are two important questions to consider. First, will the cells survive, or will death occur through other mechanisms? Second, if the cells survive, will they be functional? Blocking key apoptotic signaling molecules, for example caspases, could prevent apoptosis, but the therapeutic benefit is not achieved if the cells die nevertheless. The functionality of cells after apoptosis inhibition is probably influenced by the cell

INTRODUCTION
33
type, the context and the degree of damage. Apoptosis associated with acute injuries, such as cerebral stroke, trauma-induced neurodegeneration, cardiac ischemia-reperfusion injury, transplantation, acute liver injury and sepsis, are more likely to be successfully treated than chronic stress situations (Nicholson, 2000).
LYSOSOMES - MULTIFUNCTIONAL ORGANELLES Lysosomes are membrane-bound cytoplasmic organelles with an acidic interior and are found in virtually all eukaryotic cells (Saftig and Klumperman, 2009). Lysosomes were originally described in the 1950s by Christian de Duve (Appelmans et al., 1955; de Duve, 1959), a finding that yielded de Duve the Nobel Prize. Lysosomes were long regarded as simple waste bags, but are now known as advanced organelles that are involved in many cellular processes and are considered crucial regulators of cell homeostasis.
The lysosomal membrane Lysosomes are limited by a single 7-10 nm phospholipid-bilayer (Saftig et al., 2010). A unique feature of the lysosomal membrane is its high carbohydrate content. Lysosomal membrane proteins are generally heavily glycosylated at their luminal domain and form a glycocalyx, which is suggested to protect the membrane from the action of the hydrolytic enzymes contained within this organelle (Granger et al., 1990). One crucial role of the membrane limiting lysosomes is to separate the potent activities of lysosomal acid hydrolases from other cellular constituents, thereby preventing uncontrolled proteolytic damage (Saftig et al., 2010). The lysosomal membrane also facilitates interaction and fusion with other cellular compartments, including endosomes, autophagosomes and the plasma membrane (Schröder et al., 2010). In addition to the limiting lysosomal membrane lysosomes have intralysosomal membranes, which represent the main site of membrane degradation within this organelle (Schulze et al., 2009).

INTRODUCTION
34
Bis(monoacylglycero)-phosphate (BMP) Bis(monoacylglycero)-phosphate (BMP), also known as lyso-bis-phosphatidic acid (LBPA), is an unusual phospholipid that is found mainly in the inner membrane of lysosomes and late endosomes (Hullin-Matsuda et al., 2009; Möbius et al., 2003). The unusual stereo conformation of BMP results in higher resistance to the action of phospholipases compared to other phospholipids (Matsuzawa and Hostetler, 1979). In the endolysosomal system, hydrophobic lipids and membranes are digested by hydrophilic enzymes, a process in which BMP serves as an important factor. BMP is negatively charged at the acidic pH of lysosomes, and these negative charges facilitate the adhesion of the soluble positively charged hydrolases, thus allowing the hydrolases to degrade lipids at the interface of the inner membranes of lysosomes (Gallala and Sandhoff, 2011; Kolter and Sandhoff, 2005). In addition, evidence suggests that BMP regulates the dynamics of the internal membranes of late endosomes, is involved in protein- and lipid-sorting and plays a critical role in endo/lysosomal cholesterol trafficking (Chevallier et al., 2008; Hullin-Matsuda et al., 2009; Kobayashi et al., 2002).
Acid hydrolases and lysosomal membrane proteins Two categories of proteins are essential for the correct function of lysosomes: integral membrane proteins and soluble hydrolytic enzymes. The approximately 60 resident hydrolases have different target substrates, and their collective action permits the degradation of all types of macromolecules (Lübke et al., 2009).
Lysosomal proteins are synthesized at the rough ER, transferred to the Golgi apparatus and targeted to the lysosome by specific sorting mechanisms. Targeting of newly synthesized lysosomal proteins can be direct, from the trans-Golgi network (TGN) to the endosomal system, or indirect, involving transport to the plasma membrane and subsequent endocytosis (Saftig and Klumperman, 2009). The best characterized route is the direct pathway, which is dependent on the mannose-6-phosphate (M6P) receptor, through which the majority of lysosomal hydrolases end up in lysosomes (Coutinho et al., 2012). After synthesis, proteins move to the cis-Golgi network, where they are covalently modified by the addition of M6P residues (Coutinho et al., 2012). The M6P-tagged lysosomal hydrolases are recognized and bound by M6P receptors in the TGN and sorted into transport

INTRODUCTION
35
vesicles, which bud off from the TGN and fuse with late endosomes. At the low pH of the late endosome, the hydrolases dissociate from the M6P receptors, and the empty receptors are recycled to the Golgi apparatus for further transport (Coutinho et al., 2012).
Approximately 25 lysosomal membrane proteins have been identified, which reside primarily in the limiting lysosomal membrane (Lübke et al., 2009; Saftig and Klumperman, 2009). Proteins residing in the lysosomal membrane are usually highly glycosylated transmembrane proteins, which mediate a number of essential functions for the organelle, including acidification of the lysosomal lumen, import of protein from the cytosol and transport of degradation end products out of the lysosome. The characteristic acidic pH of lysosomes is a result of the action of the vacuolar H+-ATPase, a transmembrane multimeric protein complex (Mindell, 2012). The vacuolar H+-ATPase uses energy from ATP hydrolysis to pump protons from the cytosol against their electrochemical gradient into the lysosomal lumen (Mindell, 2012). Other lysosomal membrane proteins are involved in interactions and fusion with other cell components, including endosomes, phagosomes and the plasma membrane. The most abundant lysosomal membrane proteins are lysosome-associated membrane protein (LAMP)-1 and -2, lysosomal integral membrane protein (LIMP)-2 and CD63 (Saftig and Klumperman, 2009).
Lysosome-associated membrane proteins (LAMPs) LAMP-1 and -2 have been estimated to constitute 50% of lysosomal membrane proteins (Saftig et al., 2010). LAMPs are transmembrane proteins with a large luminal domain, a transmembrane domain and a short C-terminal cytoplasmic tail (Fukuda, 1991). They are heavily glycosylated, as indicated by the increase in the mass of the polypeptide from approximately 40 kDa to 120 kDa after glycosylation (Carlsson et al., 1988).
Mice deficient in LAMP-1 are viable and demonstrate a mild phenotype with normal lysosomal morphology and function (Andrejewski et al., 1999). Deficiency of LAMP-2 induces a more severe phenotype with extensive accumulation of autophagic vacuoles in many tissues, and degradation of long-lived proteins is severely impaired (Tanaka et al., 2000). These findings indicate that LAMP-2 is critical for autophagy (described later), which is further substantiated by the

INTRODUCTION
36
finding that LAMP-2 deficiency in humans causes Danon's disease. This disease is a lysosomal glycogen storage disease that is associated with the accumulation of autophagic material in striated myocytes, resulting in a pathological condition associated with cardiomyopathy, myopathy and variable mental retardation (Danon et al., 1981; Nishino et al., 2000).
Cathepsins Among the lysosomal hydrolases, the best known are the cathepsin family of proteases. A number of human cathepsins have been identified and are categorized into three distinct groups based on the amino acid found in the active site; serine (A and G), cysteine (B, C, F, H, K, L, O, S, V, X and W) and aspartic cathepsins (D and E) (Turk et al., 2012). The aspartic cathepsin D and some of the cysteine cathepsins, including cathepsins B, L, C and H, are ubiquitous and among the most abundant lysosomal proteases (Rossi et al., 2004). By contrast, the expression of some cathepsins, such as cathepsins S and K, is tissue- and cell type-specific. Cathepsins participate in the bulk degradation of proteins within the lysosomes, but many cathepsins have also been shown to be critically involved in distinct physiological processes, including bone remodeling, proprotein processing, antigen presentation, degradation of extracellular matrix and initiation of cell death (Reiser et al., 2010; Turk et al., 2012). Cathepsins are predominantly endopeptidases and relatively nonspecific enzymes. However, cathepsins B and H can act as both endo- and exopeptidases, while cathepsins A, C and X are exopeptidases only (Rawlings et al., 2012).
Cathepsin D
Cathepsin D is synthesized in the ER, and during transport to lysosomes, the 52 kDa human pro-cathepsin D is proteolytically processed to form a 48 kDa single chain active intermediate (Figure 10). Further processing yields a mature active lysosomal protease, which is composed of a heavy 34 kDa chain and a light 14 kDa chain (Metcalf and Fusek, 1993). The catalytic site of cathepsin D involves two critical aspartic acid residues located on the different chains (Faust et al., 1985).

INTRODUCTION
37
Figure 10. Trafficking and maturation of cathepsin D. 1) Cathepsin D is synthesized as a pre-proenzyme at the endoplasmic reticulum (ER). The pre-proenzyme includes a N-terminal signal peptide (S) and a propeptide (P). 2) The signal peptide is removed, and sugars are attached to two glycosylation sites. 3) In the Golgi complex, the sugars are modified, and their mannose residues are phosphorylated, generating mannose-6-phosphate (M6P) tags. These tags are bound by M6P receptors in the trans-Golgi network, and transport of cathepsin D to the lysosomal compartment is, to a great extent, dependent on this recognition. 4) In the acidic pH environment of the endosomes, cathepsin D dissociates from the M6P-receptors and the phosphate groups are removed. 5) The propeptide is cleaved from cathepsin D, generating a single-chain active form (48 kDa). The catalytic aspartic residues (amino acids 97 and 293) are indicated by a star. 6) The intermediate form is further processed into the mature two-chain form of cathepsin D, which is composed of an N-terminal light chain and a C-terminal heavy chain.
Cathepsin D is of general importance for cell growth and tissues homeostasis, as indicated by gene knockout studies (Saftig et al., 1995). Cathepsin B, C, L and S knockout mice are viable, fertile and display no gross developmental defects (Gocheva et al., 2006). By contrast, mice deficient for cathepsin D die at postnatal day 26. Cathepsin D deficiency is associated with cell death in several tissues, including the brain, intestine, lymphoid organs and retina (Koike et al., 2003; Saftig et al., 1995; Tyynelä et al., 2000), and cathepsin D has thus been considered essential for survival. However, bulk proteolysis within lysosomes was maintained, suggesting that the vital functions of cathepsin D are exerted by limited proteolysis of proteins regulating cell growth and tissue homeostasis. In support of this, high expression of cathepsin D stimulates cancer cell proliferation (Masson et al., 2010).
Interestingly, there are a number of indications that cathepsin D is involved in intracellular lipid homeostasis and trafficking (Getty and Pearce, 2011). Cathepsin D modulation of the ATP-binding cassette protein A1 results in increased phospholipid and cholesterol efflux from the cell (Haidar et al., 2006). Moreover, cathepsin D deficiency causes lysosomal accumulation of cholesterol and glycosphingolipids (Haidar et al., 2006; Jabs et al., 2008). The underlying

INTRODUCTION
38
mechanisms are unknown but could include defective sphingolipid catabolism because cathepsin D is involved in the proteolytic conversion of prosaposin into active saposins, which are required for lysosomal hydrolysis of sphingolipids (Hiraiwa et al., 1997). An additional link between cathepsin D and lipids is the ceramide-induced activation of cathepsin D (Heinrich et al., 1999).
Functions of the lysosomal compartment Lysosomes are responsible for the disposal and recycling of old and damaged macromolecules and organelles, as well as the digestion of extracellular and foreign material. As illustrated in Figure 11, lysosomes are involved in various physiological processes, including plasma membrane repair, cholesterol homeostasis and cell death (Saftig and Klumperman, 2009). These functions make the lysosome a central and dynamic organelle and just not the final destination in degradation processes.
Figure 11. Major functions of the lysosomal compartment. Lysosomes are cellular organelles that are critically involved in many cellular processes, including endocytosis, autophagy, plasma membrane repair, cell death and cholesterol homeostasis. LAMP, lysosome-associated membrane proteins; NPC, Niemann-Pick type C protein.

INTRODUCTION
39
Degradation of macromolecules The Greek word lysosome means digestive body, which is a suitable name because the main function of lysosomes is degradation of macromolecules. Intracellular components intended for lysosomal degradation reach the lysosomes by different forms of autophagy, while exogenous material ingested by endocytosis passes through the endocytic compartment before reaching the lysosomes. To successfully degrade all macromolecules, lysosomes are equipped with a plethora of hydrolytic enzymes, including proteases, peptidases, phosphatases, nucleases, glycosidases, sulfatases and lipases (Bainton, 1981). The acidic environment of the lysosomal lumen (pH 4.5-5.0) aids the degradation process by loosening the structures of macromolecules and is optimal for the activity of lysosomal hydrolases (Mindell, 2012). Together, the hydrolases are able to decompose proteins, polysaccharides, lipids and nucleic acids into their monomeric constituents. The end products of lysosomal digestion are reused by the cell after diffusion or carrier-mediated transport through the lysosomal membrane (Schröder et al., 2010).
Endocytosis Endocytosis is a process by which cells internalize the plasma membrane along with cell surface receptors and soluble molecules. Cells have multiple mechanisms for endocytosis, including clathrin-dependent and -independent routes (Andersson, 2012). Lysosomes represent the terminal station for the degradative endocytic pathway, which starts at the plasma membrane (Figure 12). The cargo first arrives at the early endosome, which is the main sorting station in the endocytic pathway. The majority of cargos, including most receptors, are returned to the plasma membrane via the recycling endosomes (Huotari and Helenius, 2011). It has been estimated that 50% of the surface area of the plasma membrane is cycled in and out of a typical mammalian cell every hour (Steinman et al., 1983). Cargos destined for degradation are retained in the early endosome, which, through a process involving exchange of material and multiple fusion events, transforms into a late endosome. Late endosomes are round and contain lysosomal membrane proteins (such as LAMP) and acid hydrolases. In late endosomes, cargos undergo further sorting and are transported to other organelles such as TGN (Figure 12). Trafficking between TGN and endosomes is a continuously ongoing process that is responsible for the removal of endosomal components and the delivery of

INTRODUCTION
40
lysosomal components (Huotari and Helenius, 2011). Newly synthesized enzymes are delivered to the appropriate endolysosomal compartment from the TGN network, and components are returned to the TGN for reuse. Lysosomes receive cargo from late endosomes; in addition, new lysosomal hydrolases and membrane proteins from the TGN are also transferred to the lysosomes. The influx of new components is essential, and without incoming endosomal traffic, lysosomes lose their intactness, acidity and perinuclear localization (Bucci et al., 2000).
Figure 12. The endocytic pathway. Lysosomes represent the end station for the degradative endocytic pathway, which starts with early endosomes budding from the plasma membrane. In early endosomes, cargos are either sorted into a recycling pathway back to the cell surface via a recycling endosome or are retained in the endosome. Through gradual maturation, the early endosome is transformed into a late endosome and eventually a lysosome, where cargo is degraded. One of the characteristic features of endosomes is the accumulation of internal membranes within the lumen of the organelle (Kobayashi et al., 1998). Intraluminal vesicles (ILVs) are detached membranous structures that form from the limiting membrane in the endocytic pathway, and their presence is essential for efficient cargo sorting (Woodman and Futter, 2008). In the early endosomes, the formation of ILVs begins, and in the late endosomes, proteins are sorted between

INTRODUCTION
41
the limiting membranes and ILVs. Due to the high content of ILVs, late endosomes are sometimes referred to as multivesicular bodies (Huotari and Helenius, 2011). Via the generation of ILVs, lipids and membranes are delivered to lysosomes in a form that is easily accessible to lysosomal hydrolases. In contrast to the limiting membrane, the membrane of ILVs has no protective coat of glycosylated proteins (Huotari and Helenius, 2011). The lipid composition of the ILV membrane is also different and contains more cholesterol, sphingolipids and BMP (Huotari and Helenius, 2011). As presented in Figure 12, lysosomes contain fewer ILVs, and luminal lipids are observed as multilamellar membrane whirls (Huotari and Helenius, 2011).
The maturation process from early endosome to lysosome takes approximately 40 minutes. During this time, the vesicle has undergone a multitude of changes, including exchange of membrane components, movement to the perinuclear area, formation of ILVs, a decrease in luminal pH, acquisition of lysosomal components and changes in morphology by which the tubular extensions of early endosomes are lost (Huotari and Helenius, 2011). The low pH within lysosomes provides a better milieu for the acid hydrolases but is also essential for membrane trafficking and sorting of cargo.To summarize, on its way to the lysosome, endocytosed cargo passes through several endosomal intermediates that are distinguished by their cargo, molecular composition, morphology and pH (Table I).
Table I. Characteristics of intermediates in the endocytic pathway.
Early endosomes
Late endosomes
Lysosomes
Endocytosed proteins destined for recycling to the plasma membrane
+++ + -
Endocytosed protein destined for degradation
+++ +++ +++
Proteins that recycle to the TGN ++ ++ ++ Lysosomal proteins + ++ +++ Intraluminal vesicles (ILVs) Contain 0-8
ILVs Contain >9
ILVs Some ILVs
can be present pH ~6 5-6 4.5-5 Modified from Saftig and Klumperman (Saftig and Klumperman, 2009). ILV; intraluminal vesicle, TGN; trans-Golgi network. The abundance of proteins is expressed on a gradient scale: absent (-), low (+), intermediate (++) and high (+++).

INTRODUCTION
42
The widely used distinction between early endosomes, late endosomes and lysosomes simplifies the complexity of the endocytic pathway. There is a continuous exchange of content between the intermediates in the endocytic pathway, and therefore it is difficult to identify markers that specifically label a single organelle. However, early endosomal antigen 1 (EEA1) and Rab5 are widely used as markers of early endosomes (Mu et al., 1995). Late endosomes and lysosomes have an overlapping molecular complement, including LAMPs and acid hydrolases. However, lysosomes can be distinguished from late endosomes by their lack of M6P receptors (Sachse et al., 2002).
Autophagy During autophagy, cytoplasmic components, damaged proteins and entire organelles are degraded and recycled to generate building blocks for anabolic processes (Mizushima and Komatsu, 2011). Depending on the pathways to deliver the cargo, autophagy in mammalian cells can be divided into macroautophagy, microautophagy, and chaperone-mediated autophagy (Mizushima and Komatsu, 2011). Chaperone-mediated autophagy is a process by which cytosolic proteins harboring specific recognition motifs are delivered to the lysosomes via the action of a chaperone and the proposed lysosomal receptor LAMP-2A (Arias and Cuervo, 2011). Microautophagy involves direct engulfment of cytoplasmic cargo at the limiting lysosomal membrane (Li et al., 2012). During macroautophagy, sequestration of a small portion of the cytoplasm, including soluble materials and organelles, within a newly generated double membrane called the isolation membrane (or phagophore) results in the formation of an autophagosome (Burman and Ktistakis, 2010). Autophagosomes fuse with lysosomes for the degradation and recycling of their contents (Figure 13). This secures the supply of building blocks in the cell during starvation and permits the disposal of unneeded or non-functional organelles (Mizushima and Komatsu, 2011). Macroautophagy is thought to be the major type of autophagy, and it has been more extensively studied than microautophagy and chaperone-mediated autophagy.

INTRODUCTION
43
Figure 13 Macroautophagy - the formation and fate of an autophagosome. During macroautophagy, the part of the cytosol containing the material to be degraded is surrounded by a double membrane known as the isolation membrane. The isolation membrane elongates and seals itself to form the autophagosome. During the formation of the autophagolysosome, the outer autophagosomal membrane fuses with the lysosomal membrane, and the inner vesicle is released into the interior. The vesicle and its macromolecular cargo are degraded and returned to the cytosol for reutilization. During autophagy, the soluble form of microtubule-associated protein 1A/1B-light chain 3 (LC3-I) is converted into LC3-II, which is recruited to the autophagosomal membranes (Tanida et al., 2008). The presence of LC3-II is used as a marker for autophagy.
There is normally a basal rate of autophagy in cells to maintain homeostasis, but there is a strong induction of autophagy to protect cells under various physiological stresses, such as nutrient depletion and the presence of aggregated proteins (Mizushima and Komatsu, 2011). In mammalian cells, cell death is often associated with autophagic vacuolization, leading to the conclusion that autophagic cell death was a third cell death modality (Bursch et al., 2000). However, the presence of autophagic vesicles does not necessarily indicate that cell death is mediated by autophagy. Accumulating evidence suggests that autophagic cell death is usually an attempt of the damaged cell to adapt to stress rather than a mechanism to execute cell death (Shen et al., 2012).
Membrane repair by lysosomal exocytosis Lysosomes are involved in a secretory pathway known as lysosomal exocytosis. Initially, lysosomal exocytosis was thought to be limited to specialized secretory cells, but this process seems to occur in all cell types (Andrews, 2000; Rodriguez et al., 1997). Lysosomal exocytosis is a two-step process. First, lysosomes relocate from their perinuclear localization to the close vicinity of the plasma membrane
Autophagosome
Lysosome
AutophagolysosomeLC3
Isolation membrane
LC3
LC3
LC3
LC3 LC3
LC3
LC3
LC3
LC3
LC3
LC3

INTRODUCTION
44
(Jaiswal et al., 2002), where they fuse with each other. This process is followed by lysosomal fusion with the plasma membrane, which occurs in response to an increased intracellular concentration of calcium (Andrews, 2000; Jaiswal et al., 2002). Lysosomal exocytosis plays a major role in important processes such as immune responses, bone resorption and plasma membrane repair (Andrews, 2000; Andrews, 2005).
During plasma membrane damage, restoration of plasma membrane integrity is essential for the survival of the cell. The resealing is a rapid process because integrity is restored within seconds up to a minute, depending on the damage (McNeil and Steinhardt, 1997). As shown in Figure 14, the plasma membrane damage can be repaired by translocation and fusion of lysosomes (Reddy et al., 2001). The formation of a lysosomal patch that eventually fuses with the plasma membrane restores plasma membrane integrity (McNeil, 2002). As a direct consequence of lysosomal exocytosis, lysosomal enzymes are released extracellularly, and the luminal part of LAMP-1 appears at the plasma membrane (Rodriguez et al., 1997). The presence of LAMP-1 at the cell surface is commonly used as a marker of lysosomal exocytosis (Medina et al., 2011; Reddy et al., 2001; Rodriguez et al., 1997). The lesion formed in the plasma membrane is removed by endocytosis to promote wound resealing (Idone et al., 2008). The endocytosis process is dependent on the action of acid sphingomyelinase (aSMase), which is released extracellularly during lysosomal exocytosis (Tam et al., 2010). aSMase processes sphingomyelin to generate ceramide, which is believed to play an important role in the stress response. A high membrane ceramide content results in an inward bending of the membrane, which facilitates endocytosis (Holopainen et al., 2000). Moreover, ceramide-enriched rafts form signal transduction platforms that are involved in processes such as apoptosis signaling (Corre et al., 2010).

INTRODUCTION
45
Figure 14. Plasma membrane repair by lysosomal exocytosis is followed by endocytosis. Damage to the plasma membrane results in calcium influx into the cell (1), which triggers a repair process involving lysosomal exocytosis. This process involves the translocation of lysosomes to the periphery, where they form a patch that fuses with the plasma membrane in a calcium-dependent manner (2). Exocytosis results in the extracellular release of lysosomal enzymes, including acid sphingomyelinase (aSMase). aSMase converts sphingomyelin at the outer leaflet of the plasma membrane into ceramide (3), thus facilitating membrane bending and endocytosis (4).
Cholesterol homeostasis Cholesterol is an essential structural element of cellular membranes as well as a precursor for the synthesis of steroid hormones, bile acids and lipoproteins (Simons and Ikonen, 2000). There are marked asymmetries in cholesterol concentration among intracellular membranes. Recycling endosomes, Golgi membranes and the membranes of ILVs are cholesterol rich, whereas the ER, late endosomes and lysosomes contain less cholesterol (Maxfield and Wustner, 2002; Möbius et al., 2003). The majority of cholesterol (up to 80%) is found in the plasma membrane, where it constitutes approximately 40% of total lipids (Liscum and Munn, 1999; Maxfield and Wustner, 2002). Cholesterol participates in the formation of lipid rafts, which provide a special platform for proteins engaged in important cellular processes, such as signal transduction or vesicular transport (Pike, 2009). In healthy cells, lipids rafts are present at the plasma membrane, early endosomes and TGN, while membranes of late endosomes and lysosomes are relatively deprived of such cholesterol-containing microdomains, most likely due to the presence of BMP and hydrolytic enzymes (Simons and Gruenberg, 2000).

INTRODUCTION
46
In addition to de novo cholesterol synthesis in the ER, the uptake of low density lipoprotein (LDL) via receptor-mediated endocytosis is an important route for cholesterol entry into the cell (Brown and Goldstein, 1986). LDL-derived cholesterol esters are transported to the lysosomes, where the action of acid lipase liberates free unesterified cholesterol. After being released from lysosomes, free cholesterol is transported to other cellular sites such as the Golgi, plasma membrane and ER. When the free cholesterol level is too high, acyl-coenzyme A: cholesterol acyltransferase (ACAT), an ER resident enzyme, converts it into cholesterol esters, which are stored in cytoplasmic lipid droplets (Chang et al., 2009). The intracellular cholesterol transport is summarized in Figure 15.
Figure 15. Intracellular transport of LDL-derived cholesterol. After its internalization, the cholesterol esters (CE) of low density lipoprotein (LDL) particles are hydrolyzed by an acid lipase, generating free cholesterol (FC). FC is then distributed to the plasma membrane and endoplasmic reticulum (ER). In the ER, FC is esterified by acyl-coenzyme A: cholesterol-acyltransferase (ACAT), and the CE generated can be stored as cytosolic lipid droplets.
Cholesterol synthesis, uptake and efflux are tightly regulated processes that ensure that there is sufficient cholesterol for the various needs of the cell while preventing overaccumulation of cholesterol, which is toxic and contributes to disease (Brown and Goldstein, 1997). Cellular free cholesterol levels are kept within a tight range in normal cells by transcriptional control of cholesterol biosynthetic enzymes and the LDL receptor, as well as by activation of ACAT (Brown and Goldstein, 1999; Chang et al., 2009).
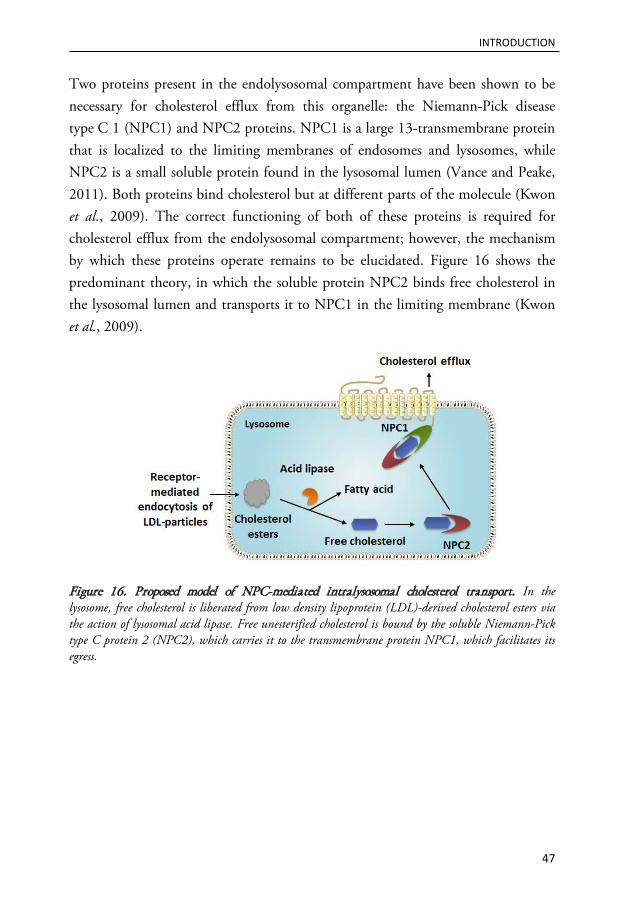
INTRODUCTION
47
Two proteins present in the endolysosomal compartment have been shown to be necessary for cholesterol efflux from this organelle: the Niemann-Pick disease type C 1 (NPC1) and NPC2 proteins. NPC1 is a large 13-transmembrane protein that is localized to the limiting membranes of endosomes and lysosomes, while NPC2 is a small soluble protein found in the lysosomal lumen (Vance and Peake, 2011). Both proteins bind cholesterol but at different parts of the molecule (Kwon et al., 2009). The correct functioning of both of these proteins is required for cholesterol efflux from the endolysosomal compartment; however, the mechanism by which these proteins operate remains to be elucidated. Figure 16 shows the predominant theory, in which the soluble protein NPC2 binds free cholesterol in the lysosomal lumen and transports it to NPC1 in the limiting membrane (Kwon et al., 2009).
Figure 16. Proposed model of NPC-mediated intralysosomal cholesterol transport. In the lysosome, free cholesterol is liberated from low density lipoprotein (LDL)-derived cholesterol esters via the action of lysosomal acid lipase. Free unesterified cholesterol is bound by the soluble Niemann-Pick type C protein 2 (NPC2), which carries it to the transmembrane protein NPC1, which facilitates its egress.

INTRODUCTION
48
Lysosomal participation in cell death signaling
Due to their high hydrolase content, lysosomes are potentially harmful to the cell. Damage to the lysosomal membrane results in leakage of lysosomal content to the cytosol. Lysosomes were referred to as “suicide bags” by Christian de Duve because partial permeabilization of the membrane induces apoptosis and massive lysosomal rupture induces necrosis (de Duve, 1959; Turk and Turk, 2009). The executors of lysosome-mediated apoptosis are not the lysosomes themselves but their hydrolases, more specifically, the cathepsins. A critical step in the mediation of apoptotic signaling by cathepsins is the release of cathepsins to the cytosol, a process known as lysosomal membrane permeabilization (LMP). The importance of the cytosolic location of cathepsins for their pro-apoptotic function is highlighted by studies in which microinjection of cathepsins into the cytosol was sufficient to induce apoptosis (Bivik et al., 2006; Roberg et al., 2002; Schestkowa et al., 2007). Depending on the lethal stimuli and the cell type, LMP can be a triggering event that is critical for activation of the signaling cascade, or it can occur later in the apoptotic process and contribute to amplification of the death signal. Signaling after LMP often involves activation of the caspase cascade, usually via the intrinsic pathway; however, cathepsins can also mediate cell death in a caspase-independent manner (Broker et al., 2004). Cathepsin B induces typical apoptosis-associated changes, including chromatin condensation, DNA fragmentation, phosphatidylserine exposure and plasma membrane blebbing (Foghsgaard et al., 2001; Vancompernolle et al., 1998). Thus, cathepsins have the ability to participate in both the initiation and execution phases of apoptosis.
Evidence supporting the role of lysosomes as cell death mediators comes from studies of compounds that directly target the lysosomes and affect the integrity of their membranes, such as lysosomotropic detergents. However, LMP is also an active contributor to apoptosis signaling induced by more classical apoptosis stimuli, such as ligation of death receptors, p53 activation, ultraviolet (UV) irradiation, growth factor deprivation and oxidative stress (Bivik et al., 2006; Brunk and Svensson, 1999; Guicciardi et al., 2000; Roberg and Öllinger, 1998; Yuan et al., 2002). Although experimental data obtained in recent decades robustly illustrate the important and active contribution of lysosomes to cell death induced by a wide variety of stimuli, in vivo evidence for lysosome-mediated cell death has

INTRODUCTION
49
been sparse. Thus, critics have suggested that the lysosomal death pathway only occurs in vitro or under pathological conditions. However, a lysosome-mediated cell death pathway was recently shown to be active in the regression of the mammary gland after lactation (Kreuzaler et al., 2011). In addition, cathepsin D expression is increased and has been functionally implicated in physiological cell death during embryonic development (Zuzarte-Luis et al., 2007). Thus, lysosomes and cathepsins are also active in cell death signaling under physiological conditions. In yeasts, the cathepsin D orthologue Pep4p is released from the vacuole, an analog of the lysosome, to the cytosol as a response to apoptotic stimuli, similar to the release of cathepsin D in mammalian cells (Sousa et al., 2011). These results suggest that the release of proteases from membrane-bound acidic organelles is an evolutionarily conserved cell death mechanism, indicating the crucial function of lysosomes for cell death signaling.
The mechanism by which lysosomal content reaches the cytosol is unknown. The simultaneous release of cathepsins, protons and lysosomal dyes suggest a nonspecific release mechanism, such as pore formation or limited membrane damage. The release of lysosomal content has been suggested to be size-selective, as small (up to 40 kDa) but not large (over 70 kDa) FITC-dextran complexes are released from lysosomes during apoptosis (Bidére et al., 2003). However, the size selectivity does not apply in all cell death scenarios because a 150 kDa lysosomal hydrolase has been shown to be released during LMP under certain experimental conditions (Blomgran et al., 2007; Kågedal et al., 2005; Nylandsted et al., 2004).
Mechanisms of lysosomal membrane permeabilization The mechanism by which cathepsins escape the lysosomal compartment during LMP remains elusive, but several mechanism have been proposed (Guicciardi et al., 2004; Johansson et al., 2010). Multiple mechanisms may exist that are employed differentially, depending on the inducing stimuli and cell type (Figure 17). Obviously, the constituents of the lysosomal membrane are essential for maintaining the integrity of the organelle. Damage to the membrane components or alterations in membrane structure and fluidity can affect lysosomal stability. There are reports suggesting that lysosomes differ in their vulnerability to LMP. For example, increased size and high lysosomal iron content can make lysosomes

INTRODUCTION
50
more prone to losing their integrity (Kurz et al., 2011; Ono et al., 2003). The most studied factors promoting or inhibiting LMP will be discussed here; for a comprehensive description of potential mediators, I refer to our review article, “Regulation of apoptosis-associated lysosomal membrane permeabilization” (Johansson et al., 2010).
Figure 17. Factors involved in the regulation of lysosomal membrane permeabilization (LMP). The relative contributions of the regulatory factors probably depend on the cell type and the apoptotic stimuli. Changes in membrane lipid composition include membrane destabilizing factors, such as phospholipase A2 and sphingosine, as well as protecting factors, including cholesterol and sphingomyelin. A regulated and partial destabilization of the lysosomal membrane results in apoptotic signaling, while total lysosomal rupture induces necrosis. ROS, reactive oxygen species; Hsp70, heat shock protein 70; LAMP, lysosome-associated membrane protein.
The Bcl-2 protein family Similar to MOMP, LMP appears to be regulated by proteins belonging to the Bcl-2 family. In support of this hypothesis, Bax is located at the lysosomal membrane during apoptosis initiated by different stimuli (Castino et al., 2009; Feldstein et al., 2006; Kågedal et al., 2005; Werneburg et al., 2007). Down regulation of Bax expression prevents LMP, indicating an active function at this intracellular site. In addition, Bax inserts into the membrane of isolated lysosomes and induces leakage, similar to its function at the mitochondrial membrane

INTRODUCTION
51
(Kågedal et al., 2005). There are no reports of Bak-mediated LMP, but Bak is also localized to the lysosomal membrane (Feldstein et al., 2004). Further supporting a role for Bcl-2 family proteins in the regulation of LMP, Bim localizes with Bax at lysosomes during apoptosis, and down regulation of either Bim or Bax prevents LMP (Werneburg et al., 2007). Similarly, Bid is found at lysosomes, and the release of cathepsin B is absent in Bid-/- cells (Werneburg et al., 2004). In strong support of BH3-only mediated activation of Bax as a LMP-inducing mechanism, a BH3 domain peptide can induce release from lysosomes in the presence of Bax (Werneburg et al., 2007). However, as LMP occurs in Bax/Bak double-knockout cells, other mechanisms for LMP must exist (Boya et al., 2003).
p53 Destabilization of the lysosomal membrane occurs early during p53-induced apoptosis, and ectopic p53 expression is sufficient to trigger LMP (Li et al., 2007; Yuan et al., 2002). In a number of models, p53 inhibition or down regulation inhibits LMP. p53 localizes to lysosomes and triggers LMP in a transcription-independent manner (Li et al., 2007). p53 may be recruited to the lysosomal membrane due to its interactions with the lysosome-associated apoptosis-inducing protein containing the pleckstrin homology and FYVE domain (LAPF). The presence of LAPF is essential for p53-induced LMP because LAPF down regulation prevents lysosomal release (Li et al., 2007). Interestingly, p53 translocates to the mitochondrial membrane and cooperates with Bcl-2 family proteins to induce MOMP during apoptosis (Mihara et al., 2003). A similar mechanism may govern p53-induced LMP. p53 also promotes apoptosis, by increasing the transcription and subsequent expression of the BH3-only proteins Noxa and Puma (Reinhardt and Schumacher, 2012); thus, p53 could induce LMP or MOMP without translocating to the lysosome or mitochondria, respectively. Indeed, UVB induces p53-dependent LMP without the appearance of p53 at the lysosomes but with an increased expression of Puma and Noxa (Wäster and Öllinger, 2009). Thus, p53 may link a number of apoptosis stimuli, e.g., DNA damage, to LMP.

INTRODUCTION
52
Proteases Caspases have been suggested to be involved in the induction of LMP; caspase-2, -8 and -9 have been indicated upstream of LMP (Guicciardi et al., 2000; Gyrd-Hansen et al., 2006; Huang et al., 2009). Both caspase-2 and -8 are demonstrated to induce LMP in isolated lysosomes (Guicciardi et al., 2000). Caspase-8-mediated lysosomal destabilization is enhanced by the addition of cytosol, suggesting that the effect of caspase-8 is at least partly dependent on additional agents, possibly Bid (Guicciardi et al., 2000; Werneburg et al., 2004). However, in another study, caspase-8 and active Bid failed to induce cathepsin release from isolated lysosomes, suggesting that the effect is dependent on other factors as well (Kågedal et al., 2005).
A number of studies have suggested that calpains mediate LMP during neuronal death (Yamashima et al., 1996; Yap et al., 2006). Calpains are proteases activated by elevation in cytosolic calcium. The involvement of calpains in LMP is supported by their localization to lysosomes prior the release of cathepsins and the fact that their inhibition abrogates LMP (Guicciardi et al., 2000; Yamashima et al., 1996; Yap et al., 2006). Moreover, calpain can induce cathepsin release from isolated lysosomes (Guicciardi et al., 2000). Cathepsins have also been suggested to be involved in their own release; in particular, a role for cathepsin B has been demonstrated. LMP fails in cathepsin B-deficient cells, and inhibition of cathepsin B reduces the extent of LMP in other experimental settings (Eriksson et al., 2012; Feldstein et al., 2004; Guicciardi et al., 2007; Werneburg et al., 2002). However, because cathepsin B is constitutively active inside lysosomes, cathepsin B activity cannot be sufficient for LMP.
Oxidative stress Oxidative stress, in which the production of reactive oxygen species is increased or the cellular detoxifying system decreased, can induce LMP by causing peroxidation of membrane lipids. Due to the degradation of iron-containing proteins, lysosomes contain free iron (Kurz et al., 2011). Fenton-like chemistry, by which iron reacts with hydrogen peroxide, generates highly reactive hydroxyl radicals, which likely destabilizes the lysosomal membrane (for further details see Figure 23 on page 72). In support of this theory, iron chelators inhibit lysosomal damage (Persson et al., 2003).

INTRODUCTION
53
Phospholipase A2 Phospholipase A2 (PLA2) is an enzyme that regulates membrane lipid composition. Activation of PLA2 occurs during apoptosis and is suggested to destabilize membranes by degrading membrane phospholipids (Jäättelä et al., 1995; Zhao et al., 2001). PLA2 induces lysosomal leakage in isolated lysosomes and has been implicated in lysosomal membrane destabilization in several different experimental systems (Johansson et al., 2010; Wang et al., 2006; Zhao et al., 2001; Zhao et al., 2003b).
Sphingosine Sphingosine is a sphingolipid implicated in LMP and apoptosis (Kågedal et al., 2001b; Ullio et al., 2012; Werneburg et al., 2002). Sphingosine is proposed to accumulate inside lysosomes and to permeabilize the membrane in a detergent-like fashion. Sphingosine is generated in the lysosome by the sequential action of aSMase and acid ceramidase, which convert sphingomyelin into ceramide and, subsequently, sphingosine (Figure 18). In contrast to sphingosine, sphingomyelin has been shown to protect against lysosomal destabilization (Caruso et al., 2005). Thus, the conversion of sphingomyelin to sphingosine has dual negative effects on lysosomal stability.
Figure 18. Conversion of sphingomyelin into sphingosine. Sphingomyelin is converted into ceramide, a reaction catalyzed by sphingomyelinase. By the action of ceramidase, ceramide is transformed into sphingosine. Sphingosine can accumulate inside lysosomes and induce lysosomal membrane permeabilization in a detergent-like fashion. Sphingosine can also be converted to sphingosine-1-phosphate, which is considered anti-apoptotic.
The addition of sphingosine to isolated lysosomes induces membrane destabilization, which is dependent on the presence of cathepsin B (Werneburg et al., 2002). In apoptotic cells, the levels of sphingosine may increase due to the enhanced activity of aSMase and cathepsin B-mediated degradation of sphingosine
Sphingomyelin Ceramide SphingosineSphingomyelinase Ceramidase
Sphingosine-1-phosphate
Sphingosine kinase-1
Sphingomyelin synthase Ceramide synthase
Sphingosine-1-phosphate phosphatase

INTRODUCTION
54
kinase-1 (Taha et al., 2005), which normally converts pro-apoptotic sphingosine into anti-apoptotic sphingosine-1-phosphate. This effect may explain, at least in part, the dependence of sphingosine on cathepsin B for efficient LMP-induction.
Safeguards of lysosomal integrity Some proteins have been shown to function as stabilizers of the lysosomal membrane through either genetic or pharmacological inhibition, e.g., heat shock protein 70 (Hsp70) and LAMPs. Anti-apoptotic Bcl-2 proteins are also considered lysosomal safeguards, due to their inhibitory effects on their pro-apoptotic relatives. Indeed, the abundance of Bcl-2, Bcl-XL or Mcl-1 influences cell sensitivity to LMP; overexpression decreases the sensitivity, while downregulation makes cells more prone to leakage (Feldstein et al., 2006; Paris et al., 2007; Werneburg et al., 2007; Zhao et al., 2000). LAMPs, which are the major protein constituents of the lysosomal membrane, have been shown to modulate the sensitivity to lysosomal cell death (Fehrenbacher et al., 2008). In addition, lysosomal destabilization can be inhibited by antioxidants, which scavenge reactive oxygen species. For example, α-tocopherol (vitamin E), a lipid soluble antioxidant, protects from lipid peroxidation and inhibits lysosomal destabilization (Roberg and Öllinger, 1998; Yoshida et al., 2007).
Hsp70 fulfills essential survival functions as a molecular chaperone in the cytosol. Hsp70 assists the folding of newly synthesized and stress-denatured proteins, as well as the import of proteins into organelles and the dissociation of aggregated proteins. Interestingly, Hsp70 is found at the plasma membrane and in lysosomes in many tumor and stressed cells and has been shown to prevent LMP (Bivik et al., 2007; Nylandsted et al., 2004). Overexpression of Hsp70 may be a defense mechanism to ensure tumor cell survival because down regulation of Hsp70 results in LMP and cathepsin-mediated cell death in the absence of other stimuli (Dudeja et al., 2009; Nylandsted et al., 2004). Several mechanisms by which Hsp70 can confer protection from LMP have been proposed. Hsp70 can bind intralysosomal free iron and thereby prevent the generation of the hydroxyl radical and permeabilization of the lysosomal membrane (Doulias et al., 2007; Kurz and Brunk, 2009). In addition, Hsp70 binds to Bax and prevents its translocation to the mitochondria (Gotoh et al., 2004; Stankiewicz et al., 2005), and a similar mechanism could prevent Bax-mediated LMP. Moreover, Hsp70 interacts with

INTRODUCTION
55
BMP within the internal lysosomal membranes, thereby facilitating aSMase activity (Kirkegaard et al., 2010). Inhibition of this interaction or inhibition of aSMase activity abolishes the lysosomal stabilizing effect of Hsp70, indicating that the major protective effect of Hsp70 is due to its facilitation of lysosomal sphingolipid catabolism (Kirkegaard et al., 2010).
Functions of cathepsins in the cytosol The involvement of lysosomal cathepsins in apoptosis has been demonstrated in various cellular models by both genetic manipulation and pharmacological inhibitors. The presence of endogenous cysteine cathepsin inhibitors in the cytosol can suppress the activity of accidentally released cathepsins. However, no endogenous inhibitors of cathepsin D have been identified (Kirkegaard and Jäättelä, 2009). Instead, the main regulatory mechanism seems to be pH-dependent proteolysis. Cathepsins are highly active at acidic pH but are inactivated at the neutral pH of the cytosol, due to irreversible unfolding (cysteine cathepsins) or reversible deprotonation of the active site aspartates (cathepsin D) (Turk et al., 1995; Turk and Turk, 2009). However, cathepsins can be stabilized by substrate binding, and their proteolytic activity can be preserved by the cytosolic acidification reported to be associated with apoptosis (Gottlieb et al., 1995; Nilsson et al., 2006). The abundant cysteine cathepsins B and L and the aspartate cathepsin D have been most studied with respect to their roles in apoptosis signaling.
In contrast to caspases, which require activation to promote apoptosis, cathepsins are already active when released from the lysosome following LMP. In the cytosol, cathepsins process other proteins to promote death signaling. In contrast to caspases, which have hundreds of identified substrates, only a small number of apoptosis-associated cathepsin substrates have been identified (Figure 19). The most studied cathepsin substrate is Bid (Repnik et al., 2012). Multiple cysteine cathepsins (including cathepsin B, H, K, L and S) activates Bid, but data regarding cathepsin D-dependent Bid cleavage are inconclusive (Caruso et al., 2006; Cirman et al., 2004; Heinrich et al., 2004). In cells lacking the endogenous cysteine cathepsin inhibitor, stefin B, neuronal apoptosis cannot be prevented by ablation of Bid, which suggested the existence of other cathepsin substrates (Houseweart et al.,

INTRODUCTION
56
2003). This result led to the discovery of anti-apoptotic Bcl-2 proteins as cathepsin substrates (Droga-Mazovec et al., 2008). Engagement of the mitochondrial pathway is a common event downstream of LMP, as evidenced by the finding that cell death is absent in Bax/Bak double-deficient mice after LMP (Boya et al., 2003). Moreover, mimicking LMP by microinjection of cathepsin D directly into the cytosol induces cytochrome c release (Roberg et al., 2002). XIAP is degraded by cysteine cathepsins, indicating that cathepsins also participate in apoptosis signaling downstream of mitochondria (Droga-Mazovec et al., 2008). One report suggests that membrane-associated guanylate kinases, which are involved in cell-cell contacts, are cathepsin substrates (Ivanova et al., 2011). Another target of cathepsins is sphingosine kinase-1, the normal activity of which promotes proliferation and survival (Taha et al., 2005). Direct cleavage and activation of caspase-3 by lysosomal proteases has been suggested (Ishisaka et al., 1998; Katunuma et al., 2001), but in general, caspases are poor substrates for most cathepsins (Cirman et al., 2004). However, caspase-8 has been demonstrated to be activated by cathepsin D during neutrophil apoptosis (Conus et al., 2008).
Figure 19. Proposed cathepsin substrates during apoptosis. After release from lysosomes following lysosomal membrane permeabilization, cathepsins perform their pro-apoptotic function in the cytosol. Many of the known cathepsin substrates results in the engagement of the mitochondrial pathway to apoptosis. However, the cathepsin-mediated cleavage of X-linked inhibitor of apoptosis (XIAP) suggests that cathepsins also participate in apoptosis signaling downstream of mitochondrial outer membrane permeabilization.

INTRODUCTION
57
Lysosomes in disease As many cellular functions involve the lysosomal compartment, lysosomal disturbance has a profound impact on homeostasis. Therefore, it is not unexpected that lysosomal dysfunction causes and contributes to many diseases. Lysosomes have a central role in lysosomal storage disorders, but increasing evidence indicates that lysosomes are involved also in more widespread diseases, such as cancer and Alzheimer’s disease (Kirkegaard and Jäättelä, 2009; Nixon et al., 2008). Due to the essential role of lysosomes in autophagy, lysosomal dysfunction impairs this process, thereby contributing to disease (Levine and Kroemer, 2008).
Lysosomal storage disorders Lysosomal storage diseases represent a class of inborn pathologies characterized by the accumulation of material in lysosomes. These conditions are caused by the absence or reduced activity of lysosomal proteins, which results in the lysosomal accumulation of substances dependent on these particular proteins (Bellettato and Scarpa, 2010). Often, this material will be stored because digestion is impaired due to enzyme deficiency, but disease can also arise when transport out of the lysosomal compartment is compromised. Over 50 human lysosomal storage conditions have been recognized, and although individually rare, their combined prevalence is approximately 1 in 8 000 births (Filocamo and Morrone, 2011). The massive accumulation of substances affects the function of lysosomes and other organelles, resulting in secondary changes, such as impairment of autophagy, mitochondrial dysfunction and inflammation (Figure 20). Lysosomal storage disorders frequently involve the central nervous system, where neuronal dysfunction or loss results in mental retardation, progressive motor degeneration and premature death.

INTRODUCTION
58
Figure 20. Lysosomal dysfunction contributes to neurodegeneration. Improper function of lysosomal proteins results in accumulation of substrates within the lysosomal compartment. The storage is associated with expansion of the compartment, inefficient lysosomal degradation and impairment of autophagy. The defective degradation results in increase amount of aged organelles and toxic proteins, further exacerbating the dysfunction of the cell. The accumulation of toxic protein and protein aggregates, aged organelles and autophagosomes might contribute in neurodegeneration.
Niemann-Pick disease type C In contrast to Niemann-Pick disease type A and B, which are both lysosomal storage disorders caused by aSMase deficiency (Schuchman, 2009), the type C (NPC) is caused by a deficiency in the cholesterol handling proteins NPC1 and NPC2. Mutations within these proteins result in a fatal neurodegenerative storage disorder characterized by the accumulation of abnormal amounts of cholesterol and other lipids in the late endosomes and lysosomes of cells (Vanier, 2010). The majority of cases (95%) are due to mutations in the NPC1 protein, while 5% are due to mutations in NPC2 (Carstea et al., 1997; Naureckiene et al., 2000; Vanier et al., 1996). NPC is a rare disease with an incidence of approximately 1:120 000 (Vanier, 2010), but due to its similarities with Alzheimer’s disease, NPC is relatively well-studied (Nixon, 2004). The severity and progression varies, but generally, the disease presents with symptoms in the first years of life. The disease is characterized by progressive neurodegeneration and hepatosplenomegaly and is usually fatal before adolescence. An effective cure for the disease has yet to be discovered.
Neuronal ceroid lipofuscinoses The neuronal ceroid lipofuscinoses (NCLs) are severe neurodegenerative lysosomal storage disorders and are the most common cause of pediatric neurodegenerative disease (Haltia, 2003). These disorders are caused by mutations in different recessively inherited genes that encode soluble and membrane proteins, the majority of which are resident in endosomes and lysosomes (Jalanko and Braulke,
Incresed level of agedorganelles and toxic proteinsLysosomal expansion
and secondary storage Defective autophagy
Substrate accumulation NeurodegenerationAltered expression,
trafficking or functionof lysosomal proteins

INTRODUCTION
59
2009). Despite being a genetically heterogeneous group, with ten disease causing genes identified, the NCLs share histopathological and clinical characteristics, including degeneration of nerve cells and accumulation of autofluorescent ceroid lipopigments (Jalanko and Braulke, 2009). In contrast to most lysosomal storage disorders, the stored material is not a disease-specific substrate, but the deposits consist mainly of subunit c of mitochondrial ATP synthase or sphingolipid activator proteins (saposins) A and D (Jalanko and Braulke, 2009). NCL patients suffer a clinical course with progressive decline in congenital and motor functions, with severe visual deficits and seizures being common symptoms. At present, there is no cure for NCL, and treatment is limited to palliation of symptoms.
One of the subtypes of NCLs is ceriod lipofuscinosis neuronal 10 (CLN10), which is caused by cathepsin D deficiency. Patients devoid of proteolytic activity of cathepsin D present a severe form of NCL, with disease onset before or shortly after birth and death occurring within a few weeks (Siintola et al., 2006). There is also a CLN10 form with a slower progression in which the patients have a degree of residual cathepsin D activity (Steinfeld et al., 2006). NCL pathologies due to mutations or deletions of cathepsin D homologs have also been identified in flies, mice, sheep and dogs (Awano et al., 2006; Koike et al., 2000; Myllykangas et al., 2005; Saftig et al., 1995; Tyynelä et al., 2000).
Adult neurodegenerative disorders Many of the secondary pathological changes (e.g., brain inflammation, alteration of intracellular trafficking and impairment of autophagy) linked to lysosomal storage disorders are also observed in adult neurodegenerative disorders, such as Alzheimer´s, Parkinson’s and Huntington’s diseases (Bellettato and Scarpa, 2010). Even if lysosomal disturbances are not a direct cause of the disease, neurodegeneration associated with lysosomal dysfunction and defective autophagy is well-documented (Nixon et al., 2008). Correct autophagic function is essential for cells, particularly for neurons, which rely on autophagy for survival, and the inactivation of crucial autophagy genes in mice results in severe neurodegeneration (Bellettato and Scarpa, 2010). Alterations to the lysosomal compartment are particularly evident and closely linked to Alzheimer’s pathology (Nixon et al., 2008; Nixon and Yang, 2011). Current evidence strongly indicates disruption of

INTRODUCTION
60
substrate proteolysis within autophagolysosomes as the principal mechanism underlying autophagy failure in Alzheimer´s disease (Lee et al., 2010; Nixon and Yang, 2011).
Independent of the primary defect, disturbances in lysosomal function seem to have more severe consequences in the central nervous system than in other parts of the body. The reason for the selective vulnerability of neurons is unknown but is suggested to be due to i) their limited regenerative potential, ii) their status as postmitotic cells, which are unable to dilute undigested material by cell division, iii) the lack of compensatory metabolic pathways and iv) their dependence on efficient intracellular trafficking to connect the distal parts with the cell body (Bellettato and Scarpa, 2010).
Because lysosomal dysfunction is associated with many degenerative disorders, therapeutic interventions aiming at restoring lysosomal function may be useful for the treatment of diseases such as Alzheimer’s and Parkinson’s. Indeed, the restoration of lysosomal proteolysis and autophagy efficiency in mouse models of Alzheimer´s disease has yielded promising therapeutic effects on neuronal function and cognitive performance (Nixon and Yang, 2011).
Cancer Rapidly dividing cells, such as cancer cells, are highly dependent on effective lysosomal function, and dramatic changes in lysosomal volume, composition and subcellular localization occur during transformation and cancer progression (Kallunki et al., 2012; Kirkegaard and Jäättelä, 2009). Several members of the cathepsin family have been implicated in cancer due to their increased expression, activity and mislocalization in various tumors (Palermo and Joyce, 2008). Increased expression and activity of lysosomal hydrolases often correlates with malignant progression, a high risk of recurrence and poor prognosis (Kallunki et al., 2012; Palermo and Joyce, 2008). The pro-tumorigenic effect of cysteine cathepsins are considered to be due to proteolytic activity after secretion to the extracellular space, where they stimulate tumor angiogenesis, tumor growth and tumor invasion (Gocheva et al., 2006). The cancer-promoting effects of cathepsin D are partly independent of its proteolytic activity, and the pro-form acts as a mitogen by binding to a cell surface receptor (Masson et al., 2010).

INTRODUCTION
61
Tumorigenesis is associated with several changes that alter cellular susceptibility to programmed cell death. The majority of changes are anti-apoptotic; however, alterations within the lysosomal compartment can be considered pro-apoptotic (Vasiljeva and Turk, 2008). Oncogene-driven transformation is associated with increased cathepsin expression and higher sensitivity toward lysosome-mediated cell death (Fehrenbacher et al., 2004). Cathepsin-mediated degradation of LAMPs may contribute to increased lysosomal instability (Fehrenbacher et al., 2008). Inhibition of aSMase also destabilizes the lysosomal membrane (Groth-Pedersen and Jäättelä, 2010). Interestingly, the expression of aSMase is significantly reduced in multiple carcinoma, possibly contributing to lysosomal destabilization (Kallunki et al., 2012). However, increased lysosomal Hsp70 in tumor cells counteracts this destabilizing effect. Thus, transformation-associated changes in the lysosomal compartment have opposing effects in cancer cells because they increase the tumorigenic potential and simultaneously sensitize the cells to lysosome destabilization and cell death (Figure 21).
Figure 21. Transformation-induced alterations within the lysosomal compartment. The overexpression of cathepsins promotes tumor progression via their extracellular action; however, in the lysosome, cathepsins degrade lysosome-associated membrane proteins (LAMPs), thereby destabilizing the lysosomal membrane. Tumor cells often display decreased expression of acid sphingomyelinase (aSMase), potentially reducing lysosomal stability. However, the increased expression of heat shock protein 70 (Hsp70) counteracts this effect by enhancing the activity of aSMase.
The significant pro-neoplastic properties of cathepsins make them interesting targets for cancer therapy (Groth-Pedersen and Jäättelä, 2010; Kallunki et al., 2012). Cathepsin inhibition could be beneficial in cancer treatment; however, multiple cathepsins must be inhibited, due to their redundant function. The
Increased cathepsin expression
Transformation
Tumor growth, invasion and angiogenesis
Lysosomal destabilization
Increasedlysosomal Hsp70
Decrease in LAMPs
IncreasedaSMase activity
Transformation Lysosomal destabilization
DecreasedaSMase
expression

INTRODUCTION
62
inhibition of lysosomal exocytosis may be a better strategy because it would decrease the content of extracellular cancer-promoting cathepsins and simultaneously increase the intracellular content of cathepsins, which could compromise the survival of the cancer cell by lysosomal destabilization.
Cancer cells often display defects in apoptotic pathways; fortunately, cell death can still occur through the release of lysosomal enzymes (Kirkegaard and Jäättelä, 2009). p53 is normally a key player in apoptosis induction, and the high frequency of p53 mutations is believed to contribute to resistance to commonly used chemotherapeutic agents. Interestingly, a small-molecule screen revealed that a majority of compounds capable of inducing cell death independent of p53 did so by triggering LMP and cathepsin-mediated killing of tumor cells (Erdal et al., 2005). This finding suggests that cancer cells that are insensitive to traditional therapies may be killed by agents that trigger the lysosomal cell death pathway.

AIMS
63
AIMS
The general objective of the studies in this thesis was to investigate the role of lysosomes in cell death signaling.
SPECIFIC AIMS OF PAPER I • To determine if the proteolytic activity of cathepsin D, when released to the
cytosol, is important for its pro-apoptotic function. • To identify cytosolic target(s) of cathepsin D. • To examine the cytosolic pH of apoptotic cells and its influence on
cathepsin activity.
SPECIFIC AIMS OF PAPERS II AND III • To determine if lysosomal cholesterol accumulation affects cellular
sensitivity to apoptosis. • To determine if sensitivity to lysosomal membrane permeabilization can be
modulated by altering the lysosomal cholesterol content. • To identify the component that protects NPC1 mutated cells and cells
exposed to U18666A, an inhibitor of lysosomal cholesterol efflux, from permeabilization of the lysosomal membrane and apoptosis.
SPECIFIC AIMS OF PAPER IV • To study the repair of plasma membrane damage induced by UV irradiation
in normal human keratinocytes. • To examine lysosomal participation in apoptosis signaling after UVA
irradiation.


MATERIALS AND METHODS
65
MATERIALS AND METHODS
This section will present the theoretical background, principles, additional information and personal reflections on the methods used in the papers I-IV. For a description of the execution of the methods, please see the materials and methods sections of respective paper.
CELLS
Human fibroblasts Papers I, II and III are primarily based on studies using human fibroblasts. These cells were chosen due to their human origin and untransformed phenotype because it is known that transformation and oncogenic progression results in significant changes in the lysosomal compartment (Kallunki et al., 2012) and alters important components of cell death signaling (Kelly and Strasser, 2011). Compared to transformed cells and cancer cells, normal fibroblasts have a lower proliferation rate and can only be cultivated for a limited number of passages before senescence occurs. Therefore, fibroblasts in passages 12-24 were used in the experiments.
In papers I and II, the apparently normal human fibroblasts AG01518, which are derived from foreskin explants, were used. In paper III, NPC1 mutated human fibroblasts (GM18436) were used to investigate the effect of cholesterol accumulation in the lysosomal system. The donor subject is heterozygous at the NPC1 gene locus; allele 1 includes one missense mutation at codon 612, and allele 2 harbors a deletion at nucleotide 16281, resulting in inappropriate protein expression and function. The apparently normal fibroblasts GM05659 were chosen
1 http://ccr.coriell.org/Sections/Search/Sample_Detail.aspx?Ref=GM18436&PgId=166

MATERIALS AND METHODS
66
as a wild type (wt) control because the donor matched the GM18436 donor with respect to age, gender and ethnicity.
Bid deficient fibroblasts In paper I, mouse embryonic fibroblasts (MEFs) lacking Bid expression, a generous gift from Stanley Korsmeyer (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, USA), were used. These cells are derived from wt and Bid knock out (Bid-/-) mice and were immortalized by simian virus 40 (SV40) T antigen-transformation as previously described (Yin et al., 1999). Due to the expression of the T antigen, which interacts with the p53 and Rb proteins, the cells can be propagated in culture indefinitely (Ahuja et al., 2005).
Cathepsin D deficient fibroblasts To investigate the pro-apoptotic function of cathepsin D, cathepsin D deficient cells were employed in paper I. MEFs that were derived from a cathepsin D knockout background and had been transfected with an empty vector, human wt cathepsin D or a mutated form of cathepsin D were compared. The mutated form of cathepsin D, W383C, was originally identified in a patient with a neurodegenerative disorder (Steinfeld et al., 2006). This was the first reported case of human neuronal ceroid lipofuscinosis caused by cathepsin D deficiency, and genetic analysis revealed that the patient had two missense mutations in the gene for cathepsin D, resulting in normal RNA levels but only minor catalytic activity (less than 7% of normal) (Steinfeld et al., 2006).
CHO cells In paper II, Chinese hamster ovary (CHO) cells were used as a genetically manipulated model to verify the results of drug-induced cholesterol accumulation. CHO 2-2 cells were obtained by treating immortalized wt cells (CHO-K1) with the mutagenic ethyl methanesulfonate (Dahl et al., 1992). In CHO 2-2 cells cholesterol accumulates in lysosomes, which is in concordance with a NPC phenotype (Dahl et al., 1992). Genetic analysis demonstrated that the NPC1 gene in CHO 2-2 cells contained an insertion that caused a frame shift and early termination of translation, resulting in a reduced amount of NPC1 mRNA and protein (Wojtanik and Liscum, 2003).

MATERIALS AND METHODS
67
Neurons In paper III, because NPC is a neurodegenerative disorder, a neuronal cell model was employed to investigate the effect of cholesterol accumulation in a more physiologically relevant experimental setting. Primary neuronal cultures were obtained from the cortexes of newborn rats according to an established method (Hansson and Rönnbäck, 1989). The cultures consisted of a mix of neurons and glia cells; the majority of the cells had a neuron-like morphology. Cultures established by this method were analyzed using neuron-specific enolase as a marker, and the cultures were estimated to consist of approximately 75% neurons (Hansson and Rönnbäck, 1989). However, when a more recently identified neuronal marker (microtubule-associated protein 2) was used for analysis, the percentage of neurons was substantially lower (Björklund et al., 2010).
LAMP deficient fibroblasts To investigate the role of LAMP proteins in lysosomal membrane stability in paper III, LAMP deficient MEFs were used. In contrast to a single deficiency of either LAMP-1 or -2, double deficiency is embryonically lethal in mice (Eskelinen et al., 2004). The MEFs used in our study were established from wt, single (LAMP-1-/- and LAMP-2-/-), or double deficient (LAMPnull) mice and were thereafter transformed. The phenotype caused by the simultaneous disruption of both LAMPs was characterized by the accumulation of autophagic vacuoles and altered lysosomal distribution and appearance. In addition, unesterified cholesterol accumulated in the endo-lyosomal compartment of LAMPnull cells, similar to NPC cells (Eskelinen et al., 2004).
Keratinocytes Normal human skin keratinocytes were used in paper IV. Human skin is composed of two layers, the epidermis and the dermis. The epidermis consists of a stratified squamous epithelium and is the outermost layer of the skin, functioning as a protective barrier (Lippens et al., 2009). The majority of the cells (90%) of the epidermis are keratinocytes, but other cells, including melanocytes, are also present. The epidermis undergoes continuous self-renewal due to the proliferation of the cells in the basal layer. During their movement through the epidermis, the

MATERIALS AND METHODS
68
keratinocytes undergo a differentiation program called cornification, and the terminally differentiated cells in the outermost layer of epidermis are ultimately shed from the skin (Lippens et al., 2009).
Primary cultures of keratinocytes were established from biopsies obtained from fair-skin donors (0-2 years of age) via foreskin circumcisions and cells were isolated according to an established protocol (Andersson et al., 2001; Larsson et al., 2005). The procedure includes incubation of the surgical specimen in dispase, followed by separation of the epidermal layer from the dermis and incubation in trypsin/EDTA solution to dissociate the cells. Pure keratinocyte cultures, without contaminating melanocytes, were obtained by removal of melanocytes by differential trypsinization. Experiments were conducted at passages 2-7.
APOPTOSIS INDUCERS Apoptosis was induced when cell cultures were sub-confluent, and all cell cultures to be compared were seeded to reach equal confluence at the time of apoptosis induction. The dose or concentration of the apoptosis inducers was optimized to induce substantial apoptosis but limited necrotic cell death, as determined by morphological evaluation of cell cultures.
Staurosporine Staurosporine (STS), which was used in papers I and II, was first discovered in 1977 and is a natural product isolated from the bacterium Streptomyces staurosporeus (Omura et al., 1977). STS functions as a broad spectrum protein kinase inhibitor by blocking the binding of ATP to kinases, thereby inhibiting their catalytic activity (Prade et al., 1997). STS binds to many kinases with high affinity, and this lack of specificity has precluded its clinical use. However, STS is still a valuable research tool and has been demonstrated to potently induce apoptosis in a variety of cells. Although it is frequently used as a pro-apoptotic stimulus, the targets and mechanisms involved in apoptosis signaling after exposure to STS are poorly understood.

MATERIALS AND METHODS
69
A previous publication from our group have established that apoptosis signaling in human fibroblasts exposed to STS is initiated by the early permeabilization of the lysosomal membrane with ensuing release of cathepsins to the cytosol, followed by mitochondrial engagement and activation of the caspase cascade (Johansson et al., 2003). Cell death signaling in the fibroblast model is highly dependent on cathepsin D; cytochrome c release, caspase activation and apoptosis-associated morphological changes were prevented when cathepsin D was inhibited (Johansson et al., 2003). Although simultaneously released from lysosomes (Kågedal et al., 2001a), the cysteine cathepsins did not exhibit the same pro-apoptotic action as cathepsin D in the human fibroblast model, since microinjection of cathepsin D but not cathepsin B induced apoptosis (Roberg et al., 2002), and inhibition of cathepsin B failed to inhibit caspase activation.
MSDH O-methyl-serine dodecylamine hydrochloride (MSDH), a lysosomotropic detergent, was used in papers I, II and III to study cell death signaling induced by the enforced release of lysosomal content. MSDH was kindly provided by Gene M. Dubowchik (Bristol-Myers Squibb, Wallingford, CT, USA). The term lysosomotropic was coined by de Duve to describe various weak amines that are subject to considerable accumulation (often hundredfold) within lysosomes (de Duve et al., 1974). Lysosomotropic amines contain a moderately basic amino group with a pKa of 5-9, and at the neutral pH of the cytosol, they remain predominantly in their unprotonated form and are able to passively diffuse across cell membranes. However, as illustrated in Figure 22, in the acidic milieu of lysosomes, they become protonated and are no longer able to penetrate the membrane; thus the amines are trapped within the lysosome (Firestone et al., 1979). If the compound also has a long hydrophobic chain, it will become a lysosomotropic detergent when protonated, possibly acting with its hydrophobic tail buried in the membrane and the hydrophilic protonated head group facing the aqueous interior of the lysosomes (Firestone et al., 1979). The amine will accumulate inside the lysosomes until the concentration is sufficient to dissipate the lysosomal membrane (Dubowchik et al., 1995).

MATERIALS AND METHODS
70
Figure 22. A lysosomotropic agent accumulates in lysosomes due to ion trapping. A weakly basic amine (B) will, in its unprotonated form, passively diffuse through cell membranes. In the acidic interior of lysosomes, the amine will become charged and can no longer pass through the lysosomal membrane, thus accumulating in the lysosomal compartment.
In contrast to other detergents that kill cells by acting at the plasma membrane, lysosomotropic detergents primarily act from within the lysosomes. Consequently, red blood cells, which lack lysosomes, are not lysed by lysosomotropic detergents (Firestone et al., 1979), and agents that raise the lyso somal pH confer protection from cell death induced by lysosomotropic detergents (Ghosh et al., 2011; Miller et al., 1983).
MSDH fulfills the criteria of a lysosomotropic detergent; it is an amine with a long hydrophobic chain and a pKa of 5.9 (Dubowchik et al., 1995). In the acidic interior of lysosomes, MSDH becomes protonated and acquires detergent properties, thereby inducing the release of lysosomal content and induces cell death (Ghosh et al., 2011; Li et al., 2000; Nilsson et al., 2006; Terman et al., 2002; van Nierop et al., 2006; Zhao et al., 2003a).
UV irradiation Exposure to UV irradiation was used to induce apoptosis in cathepsin D deficient MEFs (paper I) and keratinocytes (paper IV). UV radiation is composed of electromagnetic rays of wavelengths shorter than that of visible light. The UV spectrum of sunlight is divided into three categories based on wavelength; UVA (320-400 nm), UVB (280-320 nm) and UVC (200-280 nm). UVC is biologically irrelevant because it is almost completely absorbed by the ozone layer. However, both UVA and UVB radiation penetrates to the earth and have deleterious effects on living organisms.

MATERIALS AND METHODS
71
Malignant transformation of keratinocytes and melanocytes can give rise to skin cancer, and UV radiation is considered to be the most important environmental carcinogen in the development of skin cancer (Armstrong and Kricker, 2001). The mutagenic and carcinogenic properties of sunlight have been mostly attributed to UVB, although there is no doubt that the damaging effect of UVA also contributes to skin cancer (Agar et al., 2004; Bennett, 2008). UVA and UVB differ in their biological effects and in their depth of penetration through the skin layers. UVB can directly induce DNA damage, while the effect of UVA is indirect and is due to the generation of free radicals and reactive oxygen species in the cell (Marrot and Meunier, 2008). Severe damage induced by UV exposure normally activates the apoptotic elimination of the damaged cells, which may carry mutations that can aid malignant transformation. Efficient removal of irreparable damaged cells is thus an essential defense mechanism against skin carcinogenesis.
The signaling following UV irradiation is complex, but most evidence suggests that the intrinsic pathway is the main apoptotic route (Pustisek and Situm, 2011), although there are some reports indicating activation of the extrinsic pathway by ligand-independent clustering of death receptors (Aragane et al., 1998; Sheikh et al., 1998). In addition, LMP and cathepsins have a significant role in apoptotic signaling in melanocytes after both UVA and UVB irradiation (Bivik et al., 2006).
Cisplatin In paper II, the platinum-containing drug cisplatin was used as an apoptosis inducer. Cisplatin, one of the most potent chemotherapeutic drugs, has been used during the past 30 years as a treatment for several types of solid tumors (Siddik, 2003). It binds to DNA and induces crosslinking, which blocks the transcription process (Todd and Lippard, 2009). At clinically relevant concentrations, the acute apoptotic response of cisplatin is most likely of minor importance, and the efficacy of the drug is primarily due to growth arrest (Fayad et al., 2009; Havelka et al., 2007). The cytotoxic effect of cisplatin is generally believed to be due to DNA damage, however, in cultured cells, high concentrations of cisplatin induce an apoptotic response that is independent of its DNA-damaging properties (Berndtsson et al., 2007; Mandic et al., 2003). Alternative targets of cisplatin include the ER, mitochondria and lysosomes (Mandic et al., 2003; Santin et al.,

MATERIALS AND METHODS
72
2012). In cultured head and neck squamous cell carcinoma cells, exposure to cisplatin was associated with LMP, and inhibition of cathepsins partly prevented cell death, indicating lysosomal participation in cisplatin-induced cell death signaling (Nilsson et al., 2010).
Oxidative stress Oxidative stress is caused by an imbalance between the production of reactive oxygen species and the reducing capacity of the cellular antioxidant defense system. Disturbances in the redox state can damage cellular components such as protein, lipids and DNA via the generation of free radicals and peroxides. Destabilization of the lysosomal membrane has been detected early during cell damage induced by oxidative stress (Roberg et al., 1999; Zdolsek et al., 1993; Öllinger and Brunk, 1995). In cell culture, oxidative stress is commonly induced by adding hydrogen peroxide, which is completely membrane permeable and can reach lysosomes. As shown in Figure 23, the acidic pH and high iron content of lysosomes facilitates a Fenton-type reaction that results in the intralysosomal production of reactive oxygen species, which in turn can destabilize the lysosomal membrane via the peroxidation of membrane lipids (Repnik et al., 2012).
Figure 23. Destabilization of the lysosomal membrane by oxidative stress. During oxidative stress, hydrogen peroxide (H2O2) diffuses into the lysosome. In the lysosome, the acidic pH and the presence of iron, which is derived from the degradation of iron-containing proteins, promotes the reduction of iron and the generation of hydroxyl radicals (HO•) via Fenton chemistry. Peroxidation of the membrane lipids, induced by the hydroxyl radical, results in the loss of lysosomal membrane integrity and leakage of the lysosomal content into the cytosol.

MATERIALS AND METHODS
73
In paper III, oxidative stress-induced apoptosis was studied by exposing LAMP-deficient MEFs and neurons to hydrogen peroxide or glucose oxidase. The direct addition of hydrogen peroxide to cell cultures yields a short duration of oxidative stress because such a bolus dose of hydrogen peroxide is consumed within minutes (Antunes and Cadenas, 2000). By contrast, when glucose oxidase, which is an oxido-reductase that catalyzes the oxidation of glucose generating hydrogen peroxide, is added, there is a continuous generation of hydrogen peroxide during exposure, resulting in prolonged oxidative stress (Antunes and Cadenas, 2001). This technique likely produces oxidative stress conditions that are more physiologically relevant than those induced by the conventional bolus-addition method (Antunes and Cadenas, 2001).
INHIBITORS
Pepstatin A We used the aspartic protease inhibitor pepstatin A to investigate the role of cathepsin D in cell death processes. Pepstatin A is a pentapeptide produced and secreted by Streptomyces species (Morishima et al., 1970; Umezawa et al., 1970). It contains the rare amino acid statin, which reacts with the active site residues of aspartic proteases and reversibly inhibits their proteolytic activity. Pepstatin A is not a specific inhibitor of cathepsin D but also inhibits cathepsin E and the two extracellular proteases pepsin and renin (Kunimoto et al., 1974). However, only intracellular proteases are relevant in our experimental model, and cathepsin D is more abundant than cathepsin E, which is mainly expressed in immune cells. Thus, we propose that the effect of pepstatin A in our experimental model is due to its inhibition of cathepsin D.
Pepstatin A efficiently inhibits the proteolytic activity of cathepsin D, but due to its poor solubility (Umezawa et al., 1970) and negligible cell permeability, a rather high pepstatin A concentration (100 μM) was used in cell culture experiments. Because pepstatin A is not cell permeable, cells were pretreated overnight to allow pepstatin A to be pinocytosed and reach lysosomes (Dean, 1983) before the induction of apoptosis.

MATERIALS AND METHODS
74
E64d In paper IV, E64d was employed. E64d is an irreversible inhibitor of cysteine proteases, including cathepsin B and L (Tamai et al., 1986). In addition to its inhibitory effect on cysteine cathepsins, E64d also inhibits calpains (McGowan et al., 1989). E64d is taken up by pinocytosis (Wilcox and Mason, 1992) and eventually enters the lysosomal compartment, indicating a primary effect of E64d within this compartment.
Caspase inhibitors Synthetic caspase inhibitors have been developed both as research tools and with the hope that they may eventually be used clinically to prevent the cell loss that is associated with some disorders. These inhibitors are based on the peptide sequence preceding the cleavage site of known caspase substrates (Callus and Vaux, 2007). The peptide sequence is conjugated to various functional groups to improve efficiency, stability and cell permeability. z-VAD-fmk is a potent inhibitor that suppresses the activity of a broad spectrum of caspases (Garcia-Calvo et al., 1998) by binding to the catalytic site and acting as a pseudo-substrate (Callus and Vaux, 2007). Notably, z-VAD-fmk does not inhibit all caspases with equal efficiency, and at higher concentrations (>10 μM), it also reduces the proteolytic activity of non-caspase proteases, including calpains and cysteine cathepsins (Bizat et al., 2005; Kroemer et al., 2009; Waterhouse et al., 1998), which is of importance when interpreting the results obtained with a high concentration of the inhibitor. Caspase-8 activity was inhibited with z-IETD-fmk.
3-Methyladenine In paper II, 3-methyladenine (3-MA), a commonly used inhibitor of autophagy, was employed. It is capable of blocking autophagy due to its inhibitory effect on phosphoinositide 3-kinase (PI3K) activity (Blommaart et al., 1997), which is known to be essential for the induction of autophagy (Seglen and Gordon, 1982).

MATERIALS AND METHODS
75
Myriocin In paper III, myriocin was used to investigate the effect of cholesterol accumulation relative to the accumulation of other lipids. Myriocin, also known as ISP-1, is an atypical amino acid and an antibiotic derived from certain thermophilic fungi. It is a potent inhibitor of serine palmitoyltransferase, which catalyzes the first step in sphingosine biosynthesis (Miyake et al., 1995). Thus, myriocin is used in biochemical research as a tool for depleting cells of sphingolipids, including ganglioside, ceramide and sphingomyelin (Figure 24). To verify the effect of myriocin on sphingolipid biosynthesis in human fibroblasts, the cellular content of sphingomyelin was measured using a previously described method, which involves a four-step enzymatic process (Glaros et al., 2010).
Figure 24. Sphingolipid biosynthesis. De novo sphingolipid synthesis begins with formation of 3-ketosphinganine by serine palmitoyltransferase. In a series of reactions ceramide is formed. Ceramide can be converted into sphingosine, sphingomyelin or the glycosphingolipid glycosylceramide (GlcCer), which can be further processed into lactocylceramide (LacCer). LacCer serves as a precursor for a large number of glycosphingolipids, including the gangliosides (with the exception of GM4) (Kolter et al., 2002). Enzyme names are italicized, and framed lettering represents inhibitors.
Desipramine In paper IV, desipramine was used to inhibit the activity of acid sphingomyelinase (aSMase). aSMase is a lysosomal hydrolase that is responsible for the conversion of sphingomyelin to phosphocholine and ceramide (Figure 24). Desipramine induces

MATERIALS AND METHODS
76
a rapid decrease in cellular aSMase activity when added to cells (Hurwitz et al., 1994). Desipramine disrupts the binding of aSMase to the inner membrane of lysosomes and aSMase is then subjected to enhanced proteolytic degradation by lysosomal proteases (Kolzer et al., 2004).
MODULATION OF LYSOSOMAL CHOLESTEROL CONTENT
U18666A In papers II and III, hydrophobic amines, that disturb intracellular cholesterol homeostasis and induce cholesterol accumulation in the endolysosomal compartment, were used. The synthesis of the amphipathic steroid 3-β-[2-(diethylamino)-ethoxy]androst-5-en-17-one (U18666A) was first described in 1951 (Cavallini and Massarani, 1951). It blocks endogenous cholesterol biosynthesis by inhibiting several enzymes involved in sterol synthesis (Cenedella, 2009). The discovery that U18666A also inhibits the egress of cholesterol from the lysosomal compartment (Liscum and Faust, 1989) was the basis for the use of U18666A in cellular models of NPC disease. In the presence of U18666A, LDL-derived free cholesterol is trapped in lysosomes and is unable to induce the normal regulatory responses, such as the stimulation of cholesterol esterification and suppression of endogenous cholesterol synthesis. The exact mechanism of U18666A action is unknown. A direct interaction between U18666A and NPC1 has been suggested, but the effect can also be secondary due to alterations of the membrane in which the NPC1 protein resides (Cenedella et al., 2004; Cenedella, 2009; Underwood et al., 1996). Hence there are three actions of U18666A that may influence its cellular effect; i) its inhibitory effect on sterol synthesis, ii) its interference in intracellular trafficking of cholesterol and iii) its ability to intercalate into membranes and alter their order.
There are reports suggesting that U18666A treatment induces apoptosis of cultured lens epithelial cells, melanoma cells and cortical neurons (Cenedella et al., 2004; Cheung et al., 2004; Di Stasi et al., 2005), but in our experiments, U18666A treatment did not induce any cytotoxicity in fibroblasts, melanoma cells or cortical neurons.

MATERIALS AND METHODS
77
Quinacrine In addition to U18666A, a number of cationic amphiphilic compounds with similar properties have been identified (Klingenstein et al., 2006; Liscum, 1990). These are small lysosomotropic structures commonly used as therapeutic drugs and include antidepressants and antiarrhythmics. The ability of such drugs to inhibit lysosomal lipid catabolism and induce lipid accumulation is closely associated with a subgroup of lysosomotropic amines known as cationic amphiphilic drugs (CADs), to which quinacrine belongs. CADs trapped in the acidic compartment will concentrate at the negatively charged inner membranes and neutralize the charge provided by BMP (Gallala and Sandhoff, 2011). This neutralization loosens the binding of lysosomal hydrolases and displaces the enzymes from their membrane-bound substrate, impairing degradation and causing lipid accumulation, similar to the phenotype observed in lysosomal storage disorders.
Quinacrine is a synthetic compound that was discovered in the 1920´s and has been used for decades worldwide for a number of different indications (Wallace, 1989). It was initially approved as an antimalarial drug but has also been used to treat other parasitic infections and autoimmune disorders, such as rheumatoid arthritis. In addition, it has been suggested that quinacrine could be used to treat prion diseases and tumors (Gurova, 2009; Klingenstein et al., 2006).
After an initial screen of five compounds (progesterone, imipramine, amiodarone, desipramine and quinacrine) demonstrated to induce lipidosis, quinacrine was chosen for use in paper III due to its high efficacy in inducing cholesterol accumulation in wt fibroblasts.
Methyl-β-cyclodextrin Cyclodextrins are naturally occurring amylose-derived oligomers that were identified in 1891. The three naturally occurring cyclodextrins (α, β and γ) are composed of a number of glucose units (six, seven and eight, respectively) that form a ring structure. Their ability to solubilize drugs has made them valuable in the pharmaceutical industry, and the use of cyclodextrins as carriers has made it possible to increase the delivery of hydrophobic medical agents into biological systems. This property is related to the cone-like structure of the cyclodextrin

MATERIALS AND METHODS
78
molecule, which includes a hydrophilic exterior and a hydrophobic interior into which hydrophobic molecules such as cholesterol, steroids and vitamins may enter. β-cyclodextrin has long been used to deliver poorly soluble drugs, but in 2009, β-cyclodextrin itself was shown to have powerful pharmacological properties (Abi-Mosleh et al., 2009). β-cyclodextrin significantly reduced intracellular cholesterol levels in NPC1 and NPC2 mutant cells (Abi-Mosleh et al., 2009; Rosenbaum et al., 2010), and it has been proposed to act as a cholesterol-chelating agent and shuttle cholesterol to membranes in a similar manner as the NPC2 protein (McCauliff et al., 2011). The ameliorating effect of β-cyclodextrin was also observed in vivo, where it substantially reduced neurodegeneration and increased the lifespan of NPC1-/- mice (Davidson et al., 2009).
Recently, it was demonstrated that methyl-β-cyclodextrin (MβCD) is more potent than hydroxypropyl-β-cyclodextrin (Rosenbaum et al., 2010), and MβCD was therefore employed in paper III. However, most in vivo studies have used hydroxypropyl-β-cyclodextrin due to its improved water solubility and presumed lower toxicity (Gould and Scott, 2005). In paper III, cells were treated with MβCD for 1 h and then incubated for 24 h prior to analysis or apoptosis induction. This procedure was shown to deplete cholesterol from the endolysosomal compartment rather than the plasma membrane (Rosenbaum et al., 2010).
25-hydroxysterol The hydroxylated derivatives of cholesterol, such as the oxysterols, play important roles in lipid metabolism. The search for the physiological role of 25-hydroxysterol (25-HC) began when it was shown to suppress cholesterol synthesis in cultured cells (Brown and Goldstein, 1974). 25-HC also reverts cholesterol accumulation caused by mutations in the NPC proteins or due to U18666A treatment (Frolov et al., 2003; Lange et al., 1998). There are several possible mechanisms for the actions of 25-HC because oxysterols has been demonstrated to act at multiple sites in cholesterol homeostasis, for example, by increasing the expression of cholesterol efflux genes and decreasing cholesterol uptake and synthesis (Brown and Goldstein, 2009; Gill et al., 2008). Interestingly, 25-HC is also reported to bind the luminal

MATERIALS AND METHODS
79
loop of the NPC1 protein (Infante et al., 2008), but whether oxysterol binding regulates the activity of NPC1 remains to be established.
Several other substances also have the ability to decrease lysosomal cholesterol, including orlistat, an inhibitor of lysosomal acid lipase, and lovastatin, an inhibitor of enzymes involved in cholesterol biosynthesis (Reid et al., 2008). However, in our cell model using NPC1 mutated fibroblasts, these compounds were less efficient than MβCD and 25-HC in reducing cholesterol load.
DETECTION OF CELL DEATH A single biochemical readout cannot be used as an unequivocal indicator of a precise death modality. Therefore, the Nomenclature Committee on Cell Death highly recommends the use of more than one method to assess ongoing cell death to reduce the probability of artifacts (Galluzzi et al., 2012a; Kroemer et al., 2009). Therefore, several methods were used in papers I-IV to assess cell death.
Cellular morphology The morphological changes characterizing apoptosis, such as reduced cellular volume, condensation of the nuclei and formation of apoptotic bodies, are easily detected by light microscopic inspection of cell cultures (Figure 25). In all of the experiments upon which this thesis is based, cell cultures were inspected by light microscopy before analysis. Representative images of cell morphology are often presented in parallel with viability data in papers.
Figure 25. Morphology of control and apoptotic fibroblasts. Typical morphology of healthy and apoptotic human fibroblasts, as observed by light microscopy. Images were acquired 8 h after exposure to 0.2 μM staurosporine.
Control Apoptosis

MATERIALS AND METHODS
80
Analysis of plasma membrane integrity After toxic insult, the integrity of the plasma membrane can be assessed by the addition of a cell impermeable dye. Cells with an intact plasma membrane exclude the dye, while damaged cells are stained. In paper IV, propidium iodide was used to assess plasma membrane damage after UV irradiation.
Assessment of viability using the MTT reduction assay In papers II and III, cell culture viability was analyzed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay. Since its first description as a viability assay for cultured cells (Mosmann, 1983), the MTT assay has been widely used to assess cytotoxicity and cell viability and in proliferation studies. The method relies on the conversion of MTT in metabolically active cells to a water-insoluble violet-blue formazan product, which can be extracted with an organic solvent and quantified spectrophotometrically. Although MTT was once assumed to be reduced by the mitochondrial enzyme succinate dehydrogenase, nicotinamide coenzymes appear to be the principal factor involved in MTT reduction, although other reducing agents may assist (Stockert et al., 2012). Thus, in contrast to previous assumptions, the MTT assay is not appropriate for assessing mitochondrial function.
Generally, the amount of MTT formazan product is directly proportional to the number of living cells, but there are reports showing that the direct reducing effect of some drugs and antioxidants may interfere with the assay (Stockert et al., 2012). In addition, variations in the MTT formazan product as a function of lipid content have been demonstrated; the formazan product was shown to localize and accumulate in lipid droplets but not in lysosomes (Stockert et al., 2012). These phenomena are important to be aware of, particularly when working with models involving lipid accumulation. Lipid droplets are the storage site for excess cholesterol after esterification in the ER. In NPC-deficient cells, this esterification is hindered, and therefore the amount of lipid droplets in these cells would be expected to be lower than normal. Accordingly, the amount of lipid droplets in LAMPnull cells is lower than that in wt cells (Eskelinen et al., 2004). It is not known if the formazan product also accumulates in cholesterol-loaded lysosomes. Thus, there are some concerns with the method. However, the use of multiple methods

MATERIALS AND METHODS
81
for analyzing cell death sensitivity excludes the possibility that the higher viability of cholesterol-loaded fibroblasts, as measured by the MTT reduction assay, is caused by excessive accumulation of the formazan product due to increased lipid load.
Crystal violet staining of cells Staining of cells with crystal violet is a simple method for obtaining quantitative information about the relative density of cells. This assay is based upon the binding of the dye to DNA. Upon solubilization, the amount of dye can be measured spectrophotometrically. While this method can be used to determine the presence of cells, it does not provide any information about their condition, dead or alive. Therefore, this method is suitable for use during late-stage apoptosis, when dying cells have detached.
Viability assessment using the MTT assay or crystal violet staining is not a pure indicator of apoptotic processes; rather, the results of these assays are correlated with the number of cells in the culture. A reduction in the number of cells may be due to inhibition of proliferation or cell death processes (apoptosis or other cell death modalities). However, by using additional methods, we have demonstrated that the reduced viability measured by these two methods (in papers II and III) is substantially due to apoptotic cell death.
Caspase activity measurement Activation of the caspase cascade is generally believed to be an indicator of apoptotic cell death. In all of the papers, the proteolytic activity of caspase-3 was measured with the fluorescence conjugated synthetic tetrapeptide substrate Ac-DEVD-AMC, and in paper IV, the activity of the initiator caspase-8 was analyzed with Ac-IETD-AFC. Both of these synthetic substrates have a short amino acid sequence containing a cleavage site for the caspase. Proteolytic cleavage at the aspartic amino acid (D) liberates the formerly quenched fluorogenic group (AMC/AFC), which can be quantified spectrophotometrically. Because the caspase family includes a number of members with similar substrate specificities, it is likely that other caspases, including caspase-7, which has a very similar specificity profile as caspase-3 (Thornberry et al., 1997), contribute to substrate processing. Because

MATERIALS AND METHODS
82
caspase-3 measurement is used to confirm the general engagement of the caspase cascade, this lack of specificity is not a problem, but for correctness, the activity is expressed as caspase-3-like activity in papers I-III. The Ac-IETD-AFC substrate is highly specific for caspase-8, according to the manufacturer.
Caspase activity must be interpreted with caution. Caspase activation does not necessarily mean that the cell is undergoing apoptosis because the presence of active caspases can be linked to non-lethal processes (Galluzzi et al., 2008; Galluzzi et al., 2012b; Garrido and Kroemer, 2004). Conversely, a cell can undergo apoptosis without activation of the caspase cascade during caspase-independent apoptosis (Galluzzi et al., 2012a). In combination with other methods, caspase-3 activity measurement is a valuable technique for the determination of cell death in a cell population, even though it does not provide any information about the frequency of cells with activated caspases or where the active caspases are located.
Visualization of nuclear morphology During apoptosis, the nucleus is subjected to a number of changes, including condensation, shrinkage, deformation and, finally, fragmentation. These changes can be visualized using 4’,6-diamidino-2-phenylindole (DAPI), which is a fluorescent stain that binds to the minor groove of DNA (Kubota et al., 2000). Nuclear morphology was analyzed by fluorescence microscopy, and for quantification, hundreds of randomly selected nuclei were counted in each sample. In the control cultures, most of the nuclei were round with homogenous fluorescence, while the nuclei of apoptotic cells were characterized by condensed chromatin and an irregular shape (Figure 26).
Figure 26. Nuclear morphology as assessed by DAPI staining. Examples of the nuclear morphology of control and apoptotic human fibroblasts after staining with 4’,6-diamidino-2-phenylindole (DAPI). Images were acquired 6 h after exposure to 0.2 μM staurosporine. Note the condensed appearance and the irregular shape of the apoptotic nuclei.

MATERIALS AND METHODS
83
During late-stage apoptosis, the number of cells with apoptotic nuclear morphology may be underestimated by this method because dead cells detach and are lost. If cells have detached, the cell culture medium can be collected. Cells pelleted by centrifugation and cytospinned onto a cover slip before fixation and staining can give a more correct frequency of cells with apoptotic nuclear morphology.
METHODS FOR THE ANALYSIS OF LYSOSOMES AND THEIR STABILITY
There are a number of methods available for the assessment of lysosomal membrane permeabilization. In the papers in this thesis, the release of cathepsin D from lysosomes was studied visually using immunocytochemistry and biochemically using digitonin extraction of cytosol followed by Western blot analysis. The stability of the lysosomes was investigated using the lysosomotropic and metachromatic dye acridine orange and by digitonin extraction.
Analysis of Lysotracker fluorescence Lysotracker probes consist of a fluorophore linked to a weak base and are therefore lysosomotropic and can be used to track lysosomes in live cells. In papers II and III, flow cytometric quantification of Lysotracker fluorescence was used to study alterations to the lysosomal compartment induced by lipid accumulation. The analysis of cells by flow cytometry is ideal for the rapid analysis of a large number of cells (usually 10 000/sample), but every cell is also analyzed individually (Haynes, 1988). In paper IV, microscopic analysis of Lysotracker fluorescence was used to monitor lysosomal translocation to the periphery.
Determination of lysosomal membrane stability using acridine orange In paper III, acridine orange (AO) was employed to investigate the stability of the lysosomal membrane in living cells. AO is a lysosomotropic weak base, metachromatic fluorophore and a photosensitizer. At low concentrations, AO is

MATERIALS AND METHODS
84
present in a monomeric form that emits green light upon excitation with blue light, but at higher concentrations, AO has a stacked conformation, which emits red light (Bradley and Wolf, 1959). AO accumulates in lysosomes due to proton trapping, and thus viable cells loaded with AO exhibit mainly red lysosomal fluorescence, due to the high concentration of AO within this compartment. When released into the cytosol, upon permeabilization of the lysosomal membrane, AO fluorescence shifts to green cytosolic staining.
Due to the photosensitizing properties of AO, there is a rapid redistribution of the dye from the lysosomes during intense blue laser exposure, as a consequence of peroxidative damage to the lysosomal membrane (Abok et al., 1983; Olsson et al., 1989). As illustrated in Figure 27, the relocalization of AO can be seen either as decreased red fluorescence or increased green fluorescence (Zdolsek et al., 1990).
Figure 27. Relocalization of acridine orange following blue light exposure. Human fibroblasts were loaded with acridine orange (AO) and excited with blue light. Images were acquired every third second from the start of laser exposure. The images demonstrate a time-dependent decrease in red lysosomal AO fluorescence and an increase in green cytosolic fluorescence, indicative of permeabilization of the lysosomal membrane.
In paper III, the stability of the lysosomal membrane in fibroblasts was studied by the AO relocalization method, and the lag time, i.e., the time between the beginning of laser irradiation and start of the increase in green fluorescence, was used to compare the stabilities of the lysosomes. A longer lag time indicates increased stability of the lysosomal membrane. A major advantage of analyzing lysosomal stability by the photooxidation of AO is the ability to study LMP in single cells in real time.
0 s 3 s 6 s 9 s 12 s 15 s 18 s 21 s 24 s
27 s 30 s 33 s 36 s 39 s 42 s 45 s 48 s 51 s
54 s 57 s 60 s 63 s 66 s 70 s 73 s 76 s 79 s

MATERIALS AND METHODS
85
Digitonin extraction of cytosol Digitonin is a detergent that effectively solubilizes cholesterol. Due to the asymmetric distribution of cholesterol in biological membranes, digitonin, when used at an optimized concentration, effectively permeabilizes the plasma membrane, which has high cholesterol content, but leaves intracellular membranes with lower cholesterol content intact (Werneburg et al., 2002). Thus, proteins found in the cytosol can be extracted, precipitated and analyzed by Western blotting. To determine the optimal digitonin concentration, the activities of a cytosolic and a lysosomal marker enzyme were analyzed simultaneously. The activity of the cytosolic enzyme lactate dehydrogenase (LDH) was measured to verify that a substantial amount of cytosol was released in all samples, while the activity of the lysosomal enzyme β-N-acetylglucosaminidase (NAG) was assessed to confirm that lysosomes were intact (Figure 28). The digitonin concentration was optimized for individual cell types, and the procedure was precisely timed for all samples to obtain a pure cytosolic fraction.
Figure 28. Extraction of cytosol with the cholesterol-solubilizing detergent digitonin. An optimal digitonin concentration results in maximal release of cytosolic content and leaves lysosomes intact. Lactate dehydrogenase (LDH) was used as a marker enzyme for cytosolic content, while the activity of the lysosomal enzyme β-N-acetylglucosaminidase (NAG) was used as a lysosomal marker. In this example, the preferred digitonin concentration is 20 μg/ml because it results in >85% release of LDH activity but less than 3% of NAG activity.
0 10 20 30 400
20
40
60
80
100
0
10
20
30
40
50
•
•
•
• • • •
•
Digitonin concentration (µg/ml)
LDH
rel
ease
(% o
f max
) NA
G release (%
of max)

MATERIALS AND METHODS
86
WESTERN BLOT ANALYSIS The expression of proteins in cell extracts was determined by Western blotting, in which denatured proteins are separated based on molecular mass by polyacrylamide gel electrophoresis and then transferred onto a membrane. The protein of interest is then detected with a primary antibody, followed by horseradish peroxidase (HRP)-conjugated secondary antibodies. Upon the addition of a chemiluminescent substrate (luminol), HRP catalyzes a reaction that produces light, which can be detected on a photographic film (Figure 29). The presence of protein is detected as dark bands whose intensity can be measured by densitometric analysis. The Western blot method is considered semi-quantitative and can be used to compare the expression of proteins in different cells or after drug exposure. For precise quantification of protein expression in a large number of samples, other methods are usually more appropriate, such as enzyme-linked immunosorbent assay (ELISA).
Figure 29. Schematic illustration of the principle for protein detection by Western blot analysis. After separation by electrophoresis, the proteins are transferred to a membrane, where they are visualized by incubation with primary antibodies followed by horseradish peroxidase (HRP)-conjugated antibodies. In the presence of a luminol detection reagent, HRP catalyzes a reaction that produces light, which is detected as dark bands on a photographic film.
IMMUNOCYTOCHEMISTRY A common feature of proteins during apoptosis is a change in localization; this applies, for example, to cathepsins, which change their localization from lysosomal to cytosolic, and the pro-apoptotic Bcl-2 proteins Bid and Bax, which are redistributed to the mitochondria during the execution of apoptosis. The subcellular localization of proteins can be visualized by immunocytochemistry. Immunocytochemistry is an antibody-based staining method for the visualization of antigens in cells. A two-step immunocytochemistry method was used in which

MATERIALS AND METHODS
87
the antigen of interest was labeled by a primary unlabeled antibody directed against the antigen, followed by a secondary fluorescence-conjugated antibody directed against the primary antibody. The two-step method is more sensitive than using directly conjugated antibodies because the signal is enhanced by the binding of multiple secondary antibodies to the primary antibody. Before staining of intracellular antigens, the cells are fixed in paraformaldehyde, which cross-links proteins and preserves the morphology of the cell. To visualize cathepsins, an additional fixation in methanol was employed. It is worth noting that visualization of BMP and filipin staining of cholesterol (see below) are not compatible with methanol fixation. To detect LAMP-1 on the extracellular leaflet of the plasma membrane, primary antibodies were added to unfixed cells. To minimize endocytosis of bound antibodies, cells were preincubated in serum and kept on ice.
For improved visualization of the compartment in which a protein resides, colocalization of the protein with marker proteins or an organelle dye can be investigated. Optimally, these markers should not change localization during apoptosis so that redistribution of the protein of interest can be observed as an increased or decreased colocalization with an organelle marker. For example, in paper I, cathepsin D was costained with the lysosomal membrane protein LAMP-2, and Bax redistribution was studied by staining with the fluorescent dye Mitotracker Red before fixation.
MEASUREMENT OF CYTOSOLIC PH Since its introduction in 1982 (Rink et al., 1982), 2′,7′‑bis‑(2‑carboxyethyl)‑5‑ (and‑6)‑carboxyfluorescein (BCECF) has been the most widely used fluorescent indicator for intracellular pH. Two advantages of BCECF are its pKa of 7, which is ideally matched to the normal range of cytoplasmic pH, and its pH-dependent excitation (Figure 30A).
In paper I, the cytosolic pH of BCECF-loaded control and apoptotic fibroblasts was measured with a microscopic ratiometric technique, which was modified from a previously published method for measuring cytosolic pH using flow cytometry (Nilsson et al., 2003). The acetoxymethyl (AM) ester of BCECF was used because esterification renders the molecule uncharged and hence enables penetration of cell
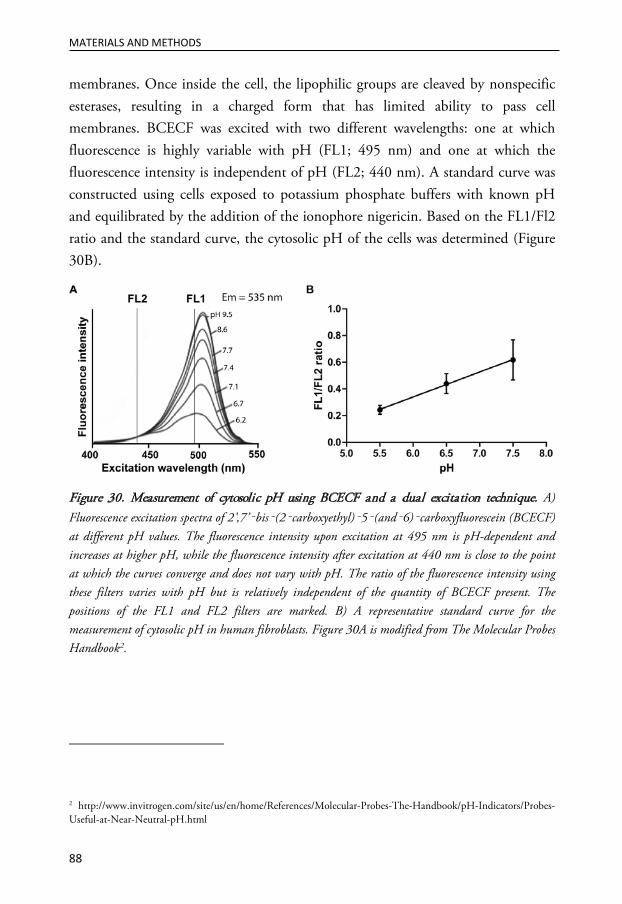
MATERIALS AND METHODS
88
membranes. Once inside the cell, the lipophilic groups are cleaved by nonspecific esterases, resulting in a charged form that has limited ability to pass cell membranes. BCECF was excited with two different wavelengths: one at which fluorescence is highly variable with pH (FL1; 495 nm) and one at which the fluorescence intensity is independent of pH (FL2; 440 nm). A standard curve was constructed using cells exposed to potassium phosphate buffers with known pH and equilibrated by the addition of the ionophore nigericin. Based on the FL1/Fl2 ratio and the standard curve, the cytosolic pH of the cells was determined (Figure 30B).
Figure 30. Measurement of cytosolic pH using BCECF and a dual excitation technique. A) Fluorescence excitation spectra of 2’,7’‑bis‑(2‑carboxyethyl)‑5‑(and‑6)‑carboxyfluorescein (BCECF) at different pH values. The fluorescence intensity upon excitation at 495 nm is pH-dependent and increases at higher pH, while the fluorescence intensity after excitation at 440 nm is close to the point at which the curves converge and does not vary with pH. The ratio of the fluorescence intensity using these filters varies with pH but is relatively independent of the quantity of BCECF present. The positions of the FL1 and FL2 filters are marked. B) A representative standard curve for the measurement of cytosolic pH in human fibroblasts. Figure 30A is modified from The Molecular Probes Handbook2.
2 http://www.invitrogen.com/site/us/en/home/References/Molecular-Probes-The-Handbook/pH-Indicators/Probes-Useful-at-Near-Neutral-pH.html

MATERIALS AND METHODS
89
CELL-FREE EXPERIMENTS One of the major benefits of cell-free experiments is the ability to investigate a specific process without the involvement of too many unidentified factors. These experiments can answer simple questions; however, care must be taken when extrapolating the results to in vivo situations.
Isolation of cytosol and mitochondria from rat liver The cytosolic and mitochondrial fractions of rat hepatocytes were isolated for use in cell-free experiments. The isolation method used was simple; differential centrifugation was used to obtain the different fractions. This method has the advantage of a relatively short preparation time. The more extensive protocols initially tested resulted in damage to the mitochondria and problems with keeping mitochondria intact until experiments. The drawback to the simple differential centrifugation method is that the mitochondrial fraction may be contaminated by other organelles. The mitochondrial fraction was intended for use in studying the release of cytochrome c, which is exclusively found in mitochondria; therefore, obtaining intact mitochondria was more important than obtaining a pure mitochondrial fraction.
Proteolytic processing in test tubes In paper I, the capability of cathepsin D to proteolytically process substrates and the pH-dependence of this proteolytic activity were investigated in cell free conditions. It is difficult to determine the proportion of enzyme and substrate; the concentrations are likely in excess of those found cellularly. However, these concentrations are often necessary to detect the proteins by relatively insensitive methods such as Western blotting.
If possible, a positive control should be included to ensure that the conditions used are relevant. For Bid processing, the addition of caspase-8 served as a positive control because it is well known that caspase-8 can proteolytically process Bid. We used 200 nM Bid, which is substantially less than the 20 μM reported previously in an analogous experiment (Cirman et al., 2004). We used 1 μM cathepsin D for test tube processing experiments and cathepsins have been used in a concentration

MATERIALS AND METHODS
90
range of 1 nM to 3 μM in similar experimental settings (Cirman et al., 2004; Stoka et al., 2001).
The processing of 14-3-3 by cathepsin D was studied in cytosol isolated from rat liver because recombinant 14-3-3 proteins were not commercially available. Processing was also performed in the presence of the caspase inhibitor z-VAD-fmk in order to exclude the possibility that cathepsin-mediated processing was indirect and due to activation of caspases found in the cytosol.
The reactions were performed in sodium acetate buffers of different pH as previously described for cathepsin D processing in test tubes (Heinrich et al., 2004). The pH of the buffers was chosen by matching a single initial experiment measuring the cytosolic pH of cells. In this experiment, the cytosolic pH was 7.3, 6.5 and 5.7 in control, STS- and MSDH-exposed fibroblasts, respectively.
Release of cytochrome c from isolated mitochondria Proteolytically processed Bid and monomeric Bax were incubated together with isolated mitochondria. The mitochondria were then pelleted by centrifugation, and the supernatant was analyzed for the release of cytochrome c.
MICROINJECTION Release of lysosomal content during apoptosis can be mimicked by microinjection of cathepsins directly into the cytosol of cells. Microinjection of cathepsin D into the cytosol of human fibroblasts induces classical apoptosis, with ensuing relocalization of cytochrome c, activation of the caspase cascade and typical alteration of cellular morphology (Roberg et al., 2002). In paper I, active cathepsin D was microinjected into the cytosol of human fibroblasts in order to study the downstream signaling resulting in mitochondrial engagement. Cathepsin D was microinjected simultaneously with Alexa Fluor 488-conjugated dextran to keep track of those cells that had been microinjected. Because the plasma membrane is damaged by a needle during microinjection, this treatment alone has the capacity to induce persistent damage to the cells. Therefore, negative controls, i.e., microinjection without cathepsin D, are critical for comparisons. A

MATERIALS AND METHODS
91
drawback of this method is that it is time consuming and technically advanced. Each cell must be injected individually, and therefore microinjection is not compatible with analysis methods requiring a large number of cells.
ANALYSIS OF LIPIDS
Cholesterol visualization using filipin Filipin, a polyene antibiotic and antifungal agent, is commonly used to visualize the intracellular distribution of cholesterol because it binds specifically to free 3-β-hydroxysterols, including unesterified cholesterol (Severs, 1982). Filipin III, which is one of the predominant and most frequently used of the four isomers isolated from Streptomyces filipinensis, was employed in papers II and III to visualize cholesterol and determine its intracellular localization. Due to the cytotoxic effect of filipin, staining demands prior fixation of cells. Whether filipin staining reflects the correct distribution of cholesterol, particularly at intracellular sites that are not easily accessible or are prone to artifacts during fixation, has been a subject of concern (Pelletier and Vitale, 1994; Steer et al., 1984). However, methods for the visualization of cholesterol in living cells are not readily available, and filipin staining is therefore still commonly used to visualize and quantify unesterified cholesterol in cells.
Cholesterol measurement The levels of unesterified cholesterol in cell lysates were measured by a fluorometric method in papers II and III. Upon the addition of cholesterol oxidase, cholesterol is oxidized into a ketone product, generating hydrogen peroxide. The hydrogen peroxide is detected with the reagent Amplex Red, which is a stable and sensitive probe. In the presence of HRP, Amplex Red reacts with hydrogen peroxide to produce the fluorescent product resorufin, which can be quantified.
Cholesterol levels in CHO cells were measured at Prince of Wales Medical Research Institute, Sydney, Australia, according to their established method for cholesterol quantification by high-performance liquid chromatography.

MATERIALS AND METHODS
92
Detection of lipid rafts The membranes of eukaryotic cells are not homogenous; rather, they contain microdomains that are often referred to as lipid rafts. Lipid rafts are detergent-insoluble membrane microdomains that are enriched in sphingolipids and cholesterol (Reeves et al., 2012). Signaling proteins and receptors are also parts of the lipids rafts, and the lipid raft assemblies can regulate the signaling by these proteins. Lipid rafts have been demonstrated to play a role in various cellular processes, including apoptosis signaling and trafficking of membrane proteins and lipids (George and Wu, 2012).
Cholera toxin, an oligomeric protein complex secreted by the bacterium Vibrio cholerae, is a major agent causing severe diarrheal disease (Vanden Broeck et al., 2007). The cholera toxin complex consists of six subunits: a single copy of the catalytic toxic A subunit and five copies of the receptor binding B subunit (Sanchez and Holmgren, 2011). In paper IV, cholera toxin subunit B was used to stain lipid rafts in cells. The B subunit conjugate is produced from recombinant cholera toxin and is completely free of the toxic subunit A, thereby eliminating any concern for toxicity in cells. Cholera toxin subunit B has strong affinity for its receptor, the membrane ganglioside GM1 (Critchley et al., 1982; Fishman, 1982), and can therefore be used as a marker for lipid rafts, which are enriched in ganglioside GM1 (Fra et al., 1994; Parton, 1994). After incubating cells with fluorescent conjugated cholera toxin subunit B, an antibody that specifically recognizes cholera toxin subunit B is then used to crosslink the labeled lipid rafts into distinct patches on the plasma membrane, which can be visualized by fluorescence microscopy.
STATISTICAL ANALYSIS The results are presented as the means and standard deviations of independent samples. Data were statistically evaluated using the nonparametric Kruskal-Wallis analysis of variance, followed by the Mann-Whitney U test for comparison of two samples. P values ≤0.05 were considered significant and are marked with an asterisk in figures.

MATERIALS AND METHODS
93
ETHICAL CONSIDERATIONS The isolation of mitochondria and cytosol from rat liver (paper I) and the preparation of neuronal cultures from rat brain (paper III) were approved by the Ethics Committee for Animal Research at Linköping University (permit numbers 57-05,106-10 and 101-08). The use of residual skin after surgery for cell culture studies of UV effects on keratinocytes (paper IV) was approved by the Regional Ethical Review Board at Linköping University (permit number 98-309).


RESULTS
95
RESULTS
PAPER I: Cathepsin D-specific processing of Bid at Phe24, Trp48 and Phe183
Previous studies indicate that apoptotic signaling after LMP and the release of cathepsin D to the cytosol of human fibroblasts proceeds by engaging the mitochondrial pathway. This study was performed to investigate apoptotic signaling upstream of cytochrome c release in human fibroblasts and identify cytosolic targets of cathepsin D. More specifically, we aimed to investigate if the proteolytic activity of cathepsin D is necessary for its pro-apoptotic function and to investigate the cytosolic pH during apoptosis.
Exposure of human fibroblasts to STS or MSDH resulted in an early (within 1 h) redistribution of cathepsin D from the lysosome to the cytosol, which was followed by caspase-dependent apoptosis. Both agents induced the translocation of Bax to mitochondria, which could be prevented by pretreatment with the cathepsin D inhibitor pepstatin A. Furthermore, mimicking LMP by microinjecting cathepsin D directly into the cytosol also resulted in Bax translocation, indicating that cytosolic cathepsin D is sufficient for Bax redistribution to occur.
Interestingly, MEFs lacking cathepsin D or expressing an inactive form were less prone to undergo apoptosis, arguing that the pro-apoptotic effect of cathepsin D is dependent on its catalytic activity. Therefore, we sought to identify possible substrates of cathepsin D during apoptosis and investigated the hypothesis that cathepsin D stimulates Bax translocation by interfering with the interaction of Bax with inhibitory 14-3-3 proteins. Of the seven isoforms, 14-3-3ε and 14-3-3θ are known to be regulated by proteolytic cleavage (Kuzelova et al., 2009; Nomura et al., 2003; Won et al., 2003). Indeed, both 14-3-3 isoforms decreased in abundance during apoptosis. Inhibition of cathepsin D prevented this decrease, and cell free

RESULTS
96
experiments indicated that cathepsin D can proteolytically process both 14-3-3ɛ and 14-3-3θ, although only at acidic pH (pH 5.7). However, processing of 14-3-3 is a relatively late event in the apoptotic process (observed after 16 h) compared to Bax translocation (significant after 2 h), and we therefore suggest that cathepsin D-mediated 14-3-3 processing is indispensable for the initial apoptotic signaling.
Once truncated, Bid can activate Bax. Bid has been suggested to be proteolytically truncated by a number of proteases, including cysteine cathepsins, but data regarding cathepsin D-mediated activation are ambiguous. We demonstrated that Bid-deficient MEFs are less sensitive to apoptosis initiated by the enforced release of lysosomal content induced by MSDH. Notably, there was a similar extent of cathepsin D release in these cells compared to wt cells. This finding indicates that Bid is a key mediator of apoptotic signaling downstream of LMP. Accordingly, a time-dependent decrease in the full-length form of Bid was observed in human fibroblasts after STS exposure, and two truncated forms of Bid with approximate masses of 15 and 19 kDa appeared. The Bid processing was cathepsin D-dependent and could be imitated by MSDH treatment.
Test tube experiments verified that Bid was directly processed by cathepsin D and that the cleavage yielded two fragments with apparent molecular weights of 15 and 19 kDa, similar to the fragments generated within cells. Bid cleaved by cathepsin D was pro-apoptotic because it, in combination with monomeric Bax, induced cytochrome c release from isolated mitochondria. Sequence analysis identified three cleavage sites located at Phe24, Trp48 and Phe183 in the mouse Bid sequence. Thus, we proposed cleavage at Phe24 to generate the 19 kDa Bid fragment, and sequential cleavage at TrP48 and Phe183 to yield the 15 kDa fragment (Figure 31). In contrast to 14-3-3 processing, which occurs only at acidic pH (5.7), Bid processing was evident even at a neutral pH. The processing was, however, more efficient at acidic pH in agreement with the function of cathepsin D as an acid hydrolase. Moreover, apoptosis induced by STS and MSDH was associated with cytosolic acidification, which favors the proteolytic activity of cathepsin D in the cytosol.

RESULTS
97
Figure 31. Cathepsin D-mediated proteolytic processing of Bid. Cathepsin D-mediated processing of Bid in human fibroblasts as well as in test tube experiments generates two Bid fragments of approximately 15 and 19 kDa. Sequence analysis revealed three cathepsin D-specific cleavage sites at Phe24, Trp48 and Phe183.
In human fibroblasts exposed to STS or MSDH, we demonstrated that apoptotic signaling is initiated by LMP and the release of cathepsin D to the cytosol. At this cellular site, cathepsin D cleaves Bid, resulting in two fragments. Activated Bid and Bax co-operate to induce MOMP and cytochrome c release, thus engaging the intrinsic pathway to apoptosis (Figure 32). At a later stage, cathepsin D-mediated processing of 14-3-3 may contribute to amplification of the death signal.
Figure 32. Cathepsin D-mediated Bax activation. Lysosome-mediated apoptosis is initiated by the release of cathepsins from the lysosome. In the cytosol, cathepsin D proteolytically processes Bid, thereby unleashing its pro-apoptotic potential. Truncated Bid activates Bax, which translocates to the mitochondria and induces cytochrome c release. Theoretically, cathepsin D could stimulate Bax translocation to the mitochondria either by activating Bid or by releasing Bax from inhibitory interactions by degrading 14-3-3 proteins. However, the degradation of 14-3-3 proteins is a late event and likely does not contribute to Bax translocation.

RESULTS
98
PAPER II: Attenuation of the lysosomal death pathway by lysosomal cholesterol accumulation
This study was conducted to investigate the role of cholesterol in lysosomal stability and cell death sensitivity. Treatment of wt human fibroblasts with U18666A resulted in a NPC-like phenotype characterized by the accumulation of cholesterol and BMP in the endolysosomal compartment. In addition, the volume of the lysosomal compartment increased, as did the expression of both soluble (cathepsin D) and membrane-associated (LAMP-2) lysosomal proteins. Together, these results suggest that U18666A induces major changes in the lysosomal compartment without affecting cell viability.
Lysosomal cholesterol accumulation decreased the cellular sensitivity to apoptosis initiated by enforced release of lysosomal content induced by MSDH. U18666A also exhibited a protective effect when apoptosis was induced by STS or cisplatin, indicating that U18666A does not exclusively protect cells from cell death induced by lysosomotropic detergents. Moreover, similar results were obtained in CHO cells in which the NPC phenotype was induced by genetic manipulation of the NPC1 gene.
The apoptosis-inhibitory effect was demonstrated to occur at the lysosomes because pretreatment with U18666A prevented the redistribution of cathepsin D from lysosomes after MSDH-treatment. The magnitude of protection was dependent on the amount of cholesterol stored in the lysosomes, demonstrating a correlation between lysosomal cholesterol content and the stability of lysosomes. Thus, U18666A-treatment induces changes that affect the permeability of the lysosomal membrane.
Cholesterol accumulation and the protective effect of U18666A were abolished by co-treatment with 25-HC, indicating that the effect of U18666A is due to its cholesterol accumulating properties and not due to, for example, its inhibitory effect on enzymes involved in sterol synthesis. Changes induced by U18666A treatment other than increased cholesterol content may contribute to lysosomal stabilization. However, the expression and localization of lysosomal membrane stabilizers (anti-apoptotic Bcl-2 proteins and Hsp70) did not change. Induction of

RESULTS
99
autophagy was also ruled out as a protective mechanism (Figure 33). The increased expression of LAMPs and accumulated sphingolipids may contribute to the enhanced lysosomal stability and reduced sensitivity to cell death.
Figure 33. Possible mechanisms for U18666A-induced protection from lysosomal membrane permeabilization and apoptosis. U18666A-treatment of human fibroblasts induces the accumulation of cholesterol in the lysosomal system, and these cells are protected from lysosome-mediated cell death. Neither increased expression nor altered localization of anti-apoptotic Bcl-2 proteins or heat shock protein 70 (Hsp70) contribute to this protection, and no induction of autophagy occurs. However, an increased expression of LAMPs or storage of multiple sphingolipids could contribute to the stabilization of the lysosomal membrane after U18666A treatment.
In summary, U18666A-induced cholesterol accumulation decreases cellular sensitivity to LMP and apoptosis. The stability of the lysosomal membrane correlates with the lysosomal cholesterol content.
PAPER III: Modulation of lysosomal cholesterol content influences lysosome-dependent cell death sensitivity
This project was conducted to explore further the mechanism of protection associated with cholesterol accumulation. Human fibroblasts derived from a patient with NPC disease were employed, and wt cells treated with cholesterol-accumulating drugs were used for comparison (Figure 34). Thus, the model includes both genetic- and drug-induced cholesterol accumulation. Treatment of

RESULTS
100
wt fibroblasts with U18666A and quinacrine did, as expected, result in cholesterol accumulation, which was associated with an expansion of the endolysosomal system. Similar cholesterol storage was found in NPC1 mutated fibroblasts, and this phenotype could be reverted by treating cells with 25-HC or MβCD (Figure 34).
Figure 34. Modulation of lysosomal cholesterol content in human fibroblasts. Lysosomal cholesterol accumulation was evident in NPC1 mutated cells, as well as in wild-type cells, after treatment with U18666A or quinacrine. Cholesterol storage was reverted in NPC1 mutated fibroblasts by treatment with methyl-β-cyclodextrin (MβCD) or 25-hydroxysterol (25-HC).
Supporting the hypothesis that lysosomal cholesterol content determines the sensitivity toward LMP and apoptosis, wt cells were less prone to MSDH-induced apoptosis after U18666A or quinacrine treatment. Similarly, NPC1-mutated fibroblasts could be sensitized by reducing the cholesterol content with 25-HC or MβCD. The cell death-protecting effect of U18666A was also demonstrated under clinically relevant conditions in which primary neuronal cultures were exposed to oxidative stress.
By directly measuring the stability of the lysosomal membrane by photo-oxidation of AO and comparing the lag time until LMP, the lysosomes of NPC1mutated cells were shown to be more stable than those of wt cells. U18666A treatment increased the stability of wt cells. Similarly, MβCD treatment increased the sensitivity of NPC1 mutated cells to lysosomal destabilization.
To investigate the contribution of other accumulating lipids in lysosomal stabilization, cells were treated with myriocin. Myriocin treatment decreased the

RESULTS
101
sphingolipid content (measured as the cellular sphingomyelin content) but did not influence cholesterol content or localization. Importantly, myriocin treatment did not influence cellular sensitivity to MSDH, indicating that the accumulation of sphingolipids does not play a major role in the protective mechanism.
In paper II, U18666A-induced cholesterol accumulation was shown to be associated with an increased expression of LAMP-2. Similarly, NPC1 mutated cells exhibited increased expression of LAMP-2 compared to wt cells. Therefore, we investigated if the increased expression of LAMP-2 contributes to lysosomal stabilization. In these experiments, apoptosis was induced by oxidative stress because LAMP expression is more likely to influence lysosomal sensitivity to LMP in a more physiologically relevant experimental setting in which lysosomal release is not induced by membrane perturbation caused by a lysosomotropic detergent. MEFs deficient for either LAMP-1 or LAMP-2 did not display an increased sensitivity to apoptosis compared with wt MEFs. Interestingly, LAMPnull MEFs displayed prominent cholesterol accumulation and were less sensitive to apoptosis than wt cells. U18666A-treatment did not alter the cholesterol load or apoptosis sensitivity of LAMPnull MEFs, but MβCD-treatment sensitized these cells toward lethal stress. Thus, LAMPnull MEFs are less sensitive to cell death than are wt cells, despite their total lack of LAMPs. This decreased sensitivity is probably due to the inherently high lysosomal cholesterol content of the mutant MEFs.
In summary, lysosomal cholesterol accumulation results in lysosomal membrane stabilization and decreased cellular sensitivity to lysosome-mediated cell death. Sphingolipid storage and the increased LAMP expression are of minor importance for the protective against LMP (Figure 35). Modulation of the lysosomal cholesterol content may thus represent a new therapeutic target, for example, to protect cells during acute stress.

RESULTS
102
Figure 35. The NPC phenotype confers protection against cell death mainly due to cholesterol accumulation within the endolysosomal system. Lysosomal cholesterol accumulation, induced by U18666A treatment or NPC1 deficiency, protects from lysosome-dependent cell death. The protective effect of the accompanying sphingolipids was excluded by the use of myriocin, and LAMP deficiency did not sensitize cells to cell death.
PAPER IV: Lysosomal exocytosis repairs the plasma membrane after UVA and is followed by caspase-8 induced apoptosis
The aim of this study was to investigate lysosomal participation in plasma membrane repair and apoptosis signaling due to the damaging effect of UV irradiation.
UVA irradiation induced plasma membrane damage, but the integrity of the plasma membrane was rapidly restored in the presence of calcium. UVA-induced plasma membrane damage is repaired by lysosomal exocytosis as evident by i) redistribution of lysosomes from their normal perinuclear location to a peripheral localization, ii) detection of the luminal portion of LAMP-1 at the plasma membrane, and iii) extracellular release of lysosomal content (cathepsin D was used as a marker). Although the plasma membrane was efficiently resealed, the apoptotic program was initiated in the cells and this initiation was associated with an early activation of caspase-8. Interestingly, although UVB also induces an apoptotic response, detected as nuclear fragmentation, UVB was not associated with plasma membrane damage, lysosomal exocytosis or caspase-8 activation.

RESULTS
103
Investigation of the apoptotic signaling induced by UVA irradiation revealed that caspase-8 is an important mediator of the signaling cascade because caspase-8 inhibition reduces nuclear fragmentation. We excluded the possibility that the early caspase-8 activation was due to UVA-induced TNF-α production and identified oxidative events as important mediators because pretreatment with the antioxidant α-tocopherol inhibited caspase-8 activation and apoptosis.
UVA irradiation was associated with increased aSMase activity. Lipid rafts were detected at the plasma membrane immediately after UVA irradiation, and their presence was dependent on the activity of aSMase since desipramine treatment reduced their presence. However, caspase-8 activation was not affected by desipramine treatment. By contrast, caspase-8 activation was dependent on lysosomal function, because treatment with either cathepsin inhibitors or ammonium chloride, which increases the lysosomal pH, decreased caspase-8 activation after UVA. Perturbation of cathepsin activities also resulted in decreased cell death, indicating a significant role of cathepsins in apoptosis signaling.
We used an antibody that specifically recognizes the active form of caspase-8 and after UVA irradiation active caspase-8 was localized in vesicles positive for LAMP-1 and cathepsin D. Caspase-8 also co-localized with the early endosomal markers EEA1 and Rab5A, although to a lesser extent. Inhibition of endocytosis partially decreased caspase-8 activation.
In summary, UVA-induced plasma membrane damage in human keratinocytes was rapidly repaired by lysosomal exocytosis (Figure 36). The subsequent UVA-induced apoptosis is characterized by an early activation of caspase-8, which occurs within vesicles positive for LAMP-1 and cathepsin D. The activation of caspase-8 is dependent on an acidic pH and the activity of lysosomal cathepsins.

RESULTS
104
Figure 36. Lysosomal participation in the repair of UVA-induced plasma membrane damage and apoptosis. UVA irradiation induces plasma membrane damage, which is rapidly repaired by lysosomal exocytosis. During this process lysosomes redistribute to the periphery and eventually fuse with the plasma membrane in a calcium-dependent manner. Lysosomal exocytosis is detected by the appearance of lysosome-associated membrane protein (LAMP)-1 at the plasma membrane and the extracellular release of lysosomal content. Acid sphingomyelinase (aSMase) generates ceramide thereby inducing the formation of lipid rafts, which are important signaling platforms. Caspase-8 is activated in vesicles positive for LAMP-1 and cathepsin D. The activation is partly dependent on endocytosis, acidic pH and cathepsin activity.

DISCUSSION
105
DISCUSSION
THE ROLE OF CATHEPSIN D IN APOPTOSIS SIGNALING The first indication of a pro-apoptotic role of cathepsin D was obtained from a screen for mediators of interferon-γ-induced apoptosis (Deiss et al., 1996). Since then, cathepsin D has been implicated in cell death in a number of experimental settings. However, the role of cathepsin D in apoptosis is not consistent because it has been shown to be both dispensable and indispensable for cell death. When an active role is demonstrated, cathepsin D can act in both an anti- and a pro-apoptotic manner, increasing the complexity of the situation (Benes et al., 2008).
In paper I, we conclude that cathepsin D has a proapoptotic role in fibroblasts that is dependent on its catalytic activity. This conclusion is based on the observation that cells expressing an inactive form of cathepsin D are less sensitive to apoptosis induction than wt fibroblasts. In addition, inhibition of the proteolytic activity of cathepsin D suppresses the pro-apoptotic effect. Numerous lines of evidence in the literature support a pro-apoptotic action of cathepsin D: i) pepstatin A inhibits cell death induced by a variety of stimuli (Bivik et al., 2006; Johansson et al., 2003; Roberg et al., 1999), ii) ectopic expression of cathepsin D kills cells in the absence of an external stimulus (Deiss et al., 1996), iii) microinjection of cathepsin D into the cytosol induces apoptosis (Roberg et al., 2002; Schestkowa et al., 2007), iv) down regulation of cathepsin D inhibits cell death (Deiss et al., 1996; Emert-Sedlak et al., 2005; Trincheri et al., 2007), v) fibroblasts from cathepsin D-deficient mice are more resistant to apoptosis induction (Fehrenbacher et al., 2004); and vi) mice with a cathepsin D-/- hematopoietic system demonstrate reduced apoptosis in vivo (Bewley et al., 2011). Although the pro-apoptotic function of cathepsin D has been shown to be dependent on its catalytic activity in several studies, alternative mechanisms exist because microinjection of the catalytically inactive proenzyme also provokes an apoptosis-promoting effect

DISCUSSION
106
(Schestkowa et al., 2007). Likewise, proteolytic inactive cathepsin D can increase apoptosis induced by chemotherapy in cancer cells (Beaujouin et al., 2006). These results suggest that cathepsin D stimulates apoptosis in at least two different ways, dependent on and independent of its catalytic activity.
There are also reports that cathepsin D can prevent cell death. For example, overexpression of cathepsin D increases the resistance of neuroblastoma cells to apoptosis, while the opposite is true for cathepsin D down regulation or inhibition (Sagulenko et al., 2008). In support of an anti-apoptotic function dependent on the catalytic activity of cathepsin D, overexpression of wt but not proteolytic inactive cathepsin D inhibits tumor cell apoptosis in xenografts (Berchem et al., 2002). Similarly, overexpression of proteolytically active cathepsin D inhibits oxidative stress-induced cell death in HeLa cells (Hah et al., 2012). Interestingly, one report demonstrate that high cathepsin D expression is associated with activation of autophagy, and indicates that cathepsin D can function as an anti-apoptotic mediator by inducing autophagy under cellular stress (Hah et al., 2012). The anti-apoptotic versus the pro-apoptotic function of cathepsin D can potentially be determined by the concentration of cathepsin D in the cytosol. My unpublished results demonstrate that a sub-lethal concentration of MSDH induces cellular vacuolization, which could be a result of limited lysosomal release and induction of autophagy, thus rescuing cells from cell death. By contrast, as shown in papers I, II and III, a higher MSDH concentration induces extensive release of lysosomal content and irreparable damage to the cell, resulting in cell death.
Cathepsin D has also been proposed to inhibit cell death by activating the protein Aven (Melzer et al., 2012). Aven is an anti-apoptotic protein that is suggested to interfere with Apaf-1 assembly during apoptosome formation (Melzer et al., 2012). Cathepsin D-mediated proteolysis of Aven removes its N-terminal inhibitory domain and thereby unleashes its cytoprotective function (Melzer et al., 2012). Nuclear translocation of cathepsin D has also been described during cell death, but the function of cathepsin D at this cellular localization remains to be explored (Zhao et al., 2010). Thus, cathepsin D has multiple functions at different cellular sites and seems to be important for both the life and death of the cell (Figure 37). Of note, in our study that demonstrates a pro-apoptotic role of cathepsin D (paper I), non-transformed fibroblasts were used. By contrast, the studies cited

DISCUSSION
107
above that suggested an apoptosis-preventing effect of cathepsin D all used transformed cells or cancer cell lines and often overexpressed cathepsin D. Thus, the anti-apoptotic effect of cathepsin D in normal cells remains to be proven.
Figure 37. The many functions of cathepsin D. After synthesis in the endoplasmic reticulum (ER), cathepsin D is transported via the Golgi apparatus in a mannose-6-phosphate (M6P)-dependent manner to the endolyosomal compartment, where it is activated. In the lysosome, cathepsin D participates in the proteolysis of cargo entering the lysosomes. Cathepsin D is also involved in the regulation of lipid trafficking and efflux. In response to apoptotic stimuli, cathepsin D can be released to the cytosol, a process known as lysosomal membrane permeabilization (LMP). Cathepsin D can either inhibit or promote apoptosis through mechanisms that are dependent on or independent of its catalytic activity. In addition, cathepsin D is present in the nucleus, but its nuclear function is unknown. Pro-cathepsin D is secreted to the extracellular space, particularly in cancer cells, in which it stimulates proliferation and acts as a mitogenic factor by binding to a cell surface receptor. The presence of proteolytically active cathepsin D outside the cell has been proposed to be a result of extracellular activation of secreted pro-cathepsin D but could also be a result of lysosomal exocytosis. Extracellular cathepsin D has been proposed to promote cancer progression by stimulating proliferation, invasion and angiogenesis.

DISCUSSION
108
Pro-apoptotic Bid processing Bid activation by caspase-8 and granzyme B is well-known, and Bid processing by cysteine proteases is also fairly well characterized. By contrast, the role of cathepsin D as a Bid activator had, when we initiated our investigations, not been convincingly demonstrated. The first indication that cathepsin D might activate Bid was revealed in an experiment using lysosomal extract, where pepstatin A partially inhibited Bid processing (Cirman et al., 2004). The ability of cathepsin D to activate Bid was studied in more detail, with conflicting results (Blomgran et al., 2007; Caruso et al., 2006; Cirman et al., 2004; Heinrich et al., 2004). In paper I, Bid wass verified as a cathepsin D substrate during apoptosis, and for the first time, the cathepsin D cleavage sites were identified. One of the cleavage sites is located within the so called bait loop, the flexible loop connecting the second and the third alpha helices of Bid. This loop has been demonstrated to be an excellent target for proteolytic activation of Bid (Repnik et al., 2012). We identify cathepsin D-mediated processing of Bid to occur at three sites. Cysteine cathepsins have also been reported to cleave Bid at multiple sites, with major cleavage sites being Arg65 and Arg71 (Cirman et al., 2004). Interestingly, the exact cleavage site seems to be of minor importance for the pro-apoptotic function of truncated Bid because Bid variants truncated at Tyr47, Gln57, Arg65 and Arg71 were all pro-apoptotic (Cirman et al., 2004). It should be noted that published results regarding test tube processing of Bid by cathepsins, including those in our study, were performed on mouse Bid. A sequence comparison between mouse and human Bid shows that the cleavage sites for caspase-8 and granzyme B are conserved, although this is not true for the majority of cysteine cathepsin cleavage sites (Repnik et al., 2012). For cathepsin D, only the most N-terminal cleavage site is conserved (Figure 38). Nevertheless, Bid fragments of approximately the same size are generated in test tube experiments and in human fibroblasts, suggesting that processing by cathepsin D is similar in human and mouse Bid.

DISCUSSION
109
Figure 38. Proteolytic processing of Bid. Both the human and mouse Bid protein consist of 195 amino acids and have eight α-helices. The number over each helix represents the starting and ending amino acid. The flexible loop that connects the second and third helices contains the majority of known cleavage sites. The cleavage sites for caspase-8 (Asp59) and granzyme B (Asp75), the major cleavage site for calpain (Gly70), one of the cleavage sites of cysteine cathepsins (Arg71), and one cleavage site of cathepsin D (Phe24) are conserved between mouse and man. JNK; c-Jun N-terminal kinase.
Although the majority of reported cleavage sites are located within the flexible loop, cysteine cathepsins have been reported to cleave Bid at multiple sites, and some are located outside this region (Ser6 and Gly12) (Cirman et al., 2004). Similarly, our results demonstrate that cathepsin D cleaves Bid at two residues located at other sites. TNF-α exposure was reported to result in Bid-activation in HeLa cells (Deng et al., 2003). The processing was caspase-8 independent, and Bid was instead cleaved by an unidentified protease at Leu25, generating a jBid fragment (Deng et al., 2003). Interestingly, jBid was capable of inducing the release of Smac/Diablo but not cytochrome c from the mitochondria (Deng et al., 2003). Thus, removal of the extreme N-terminal region is sufficient for the redistribution of Bid to the mitochondria and the induction of the release of pro-apoptotic factors. In paper I, cell-free experiments demonstrate that Bid cleavage by cathepsin D is pro-apoptotic because cleaved Bid in combination with Bax is able to induce cytochrome c release from isolated mitochondria. However, it is not clear if both Bid fragments have this ability. Because all reported Bid fragments generated by cleavage within the flexible loop are pro-apoptotic, it is reasonable to believe that the 15 kDa fragment generated by cathepsin D-mediated cleavage in the flexible loop would possess this property as well. The similarity of the

DISCUSSION
110
cathepsin D-generated 19 kDa fragment with jBid suggests that it translocates to the mitochondria but is incapable of inducing cytochrome c release; however, this possibility was not investigated.
Several studies have demonstrated that cathepsin D engages the intrinsic pathway to apoptosis by promoting the translocation of Bax to the mitochondria (Bidére et al., 2003; Bivik et al., 2006; Castino et al., 2009; Gan et al., 2008; Laforge et al., 2007; Trincheri et al., 2007). Using human fibroblasts, we demonstrate that this is a result of the proteolytic activation of Bid in the cytosol (paper I). However, cathepsin D-mediated Bax translocation can also occur in a Bid-independent manner (Bidére et al., 2003). Under these circumstances, cathepsin D-mediated processing of 14-3-3 proteins might play a more prominent role than in human fibroblasts, in which we propose cathepsin D-mediated processing of 14-3-3 proteins to be of minor importance for Bax translocation. The results presented in paper I, nevertheless, demonstrate that 14-3-3 proteins can be added to the short list of cathepsin D substrates during apoptosis. Cathepsin D can also act in a pro-apoptotic manner by reducing the expression of the anti-apoptotic Bcl-2 family protein Mcl-1 during apoptosis. Loss of Mcl-1 is prevented by pepstatin A; however, no evidence of Mcl-1 as a direct cathepsin D substrate was found (Bewley et al., 2011), indicating an indirect effect.
The proteolytic activity of cathepsin D is influenced by pH Because cathepsins are lysosomal hydrolases, they are optimally active at a pH of approximately 4.5; thus, the capability of cathepsins to be proteolytically active after release to the cytosol has been debated (Liaudet-Coopman et al., 2006). In paper I, we demonstrate that cathepsin D processes 14-3-3 in test tube experiments but only at a low pH. By contrast, Bid processing mediated by cathepsin D occurs at neutral pH, although the processing is more efficient at acidic pH. The occurrence of cathepsin D-mediated Bid processing at neutral pH has not been reported previously; by contrast, other reports have suggested that cathepsin D is unable to cleave Bid at neutral pH (performed at pH 7.2) (Cirman et al., 2004) or that cathepsin D-mediated Bid activation only occurs at pH values below 6.2 (Heinrich et al., 2004).

DISCUSSION
111
Cathepsins can process cytosolic substrates, suggesting that proteolytic activity is preserved after their release from lysosomes. Of note, some cathepsins can retain their proteolytic activity at neutral pH for several hours, allowing transient activity in the cytosol (Kirschke et al., 1989). Another explanation for the preserved proteolytic action of cathepsins is cytosolic acidification. Indeed, in paper I, we demonstrate that lysosome-mediated apoptosis in human fibroblasts is associated with a significant decrease in cytosolic pH. In the literature, both alkalization and acidification of the cytosol has been associated with apoptosis (Gottlieb et al., 1995; Khaled et al., 1999; Matsuyama et al., 2000; Nilsson et al., 2006; Tafani et al., 2002), and changes in intracellular pH might influence cell death signaling. In vitro, changes in pH regulate the activation and activity of caspases as well as translocation, dimerization and pore-formation by Bcl-2 family proteins (Matsuyama and Reed, 2000). We suggest that apoptosis-associated cytosolic acidification allows cathepsins released to the cytosol to maintain their proteolytic activity.
Cathepsin D vs. cysteine cathepsins The majority of studies investigating the role of cathepsins in cell death have been focusing on analyzing the participation of cysteine cathepsins. Cysteine cathepsins represent the largest group of cathepsins, and studies on theses proteases have been facilitated by the existence of cell-permeable inhibitors. The relative contribution of cysteine cathepsins and cathepsin D varies depending on the experimental setup. In a series of investigations, our group have investigated apoptotic signaling in human fibroblasts and demonstrated that, after LMP, cathepsin D but not cysteine cathepsins is required for the propagation of apoptotic signaling (Johansson et al., 2003; Kågedal et al., 2001a; Roberg et al., 2002). Similarly, MEFs deficient for cathepsin D but not cathepsins B and L are highly resistant to STS-induced apoptosis (Fehrenbacher et al., 2004). However, the opposite is true when these cells are treated with TNF-α, indicating a stimuli-specific engagement. This is not the sole explanation because microinjection of cathepsin B induces apoptosis in melanocytes but not in fibroblasts (Bivik et al., 2006; Roberg et al., 2002). A possible explanation for these differences is variable expression of the individual cathepsins and cathepsin inhibitors in different cell types. Human fibroblasts may

DISCUSSION
112
have a high expression of cystatins or stefins, which inhibit the action of cysteine cathepsins in the cytosol. Taken together, these findings demonstrate that cathepsins are involved in apoptosis signaling in a stimulus- and cell type-dependent manner. Interestingly, tumor cells have been shown to be highly dependent on cathepsins for apoptosis, while their role in primary cells is less pronounced (Foghsgaard et al., 2001). However, cathepsins have been demonstrated to be important cell-death mediators in a number of primary cells, including melanocytes, keratinocytes and fibroblasts (Bivik et al., 2006; Roberg, 2001). It is possible that the different results obtained in different model systems are partly due to changes in the endolysosomal system caused by transformation or oncogenic progression (Kallunki et al., 2012; Kirkegaard and Jäättelä, 2009).
THE EFFECT OF CHOLESTEROL ON LYSOSOMES After an extensive investigation of the proteins involved in apoptosis, the attention now turns to lipids. In recent years, it has become evident that lipids are important components of diverse cellular processes, including cell death signaling. Well known events during apoptosis have been demonstrated to be significantly influenced by a variety of lipids. For example, sphingolipids were recently shown to serve as specific cofactors for Bax/Bak activation that lower the threshold for apoptosis-associated cytochrome c release (Chipuk et al., 2012). In addition, the mitochondria-specific phospholipid cardiolipin acts as a targeting signal for pro-apoptotic proteins of the Bcl-2 family, and similarly, phosphatidic acid, one of the major acidic phospholipids found in the lysosomal membrane, facilitates Bid insertion and is essential for tBid-induced LMP (Lutter et al., 2000; Zhao et al., 2012). In papers II and III, we studied the influence of cholesterol on the lysosomal compartment.

DISCUSSION
113
Cholesterol modulates lysosomal membrane stability Cholesterol is known to regulate the fluidity of lipid bilayers. Because of its specific structure, cholesterol intercalates between the saturated hydrocarbon chains of phospholipids and condenses the packing of sphingolipids (Ikonen, 2008). Therefore, alterations of the cholesterol content may alter the physiochemical properties of the membrane (von Arnim et al., 2008). In papers II and III, we demonstrate that high lysosomal cholesterol content reduces the sensitivity of cells to LMP and apoptosis (Figure 39). The decreased sensitivity of cholesterol-loaded lysosomes was demonstrated by two different methods: photooxidation of AO and digitonin-extraction. In addition, reversal of cholesterol storage sensitizes NPC mutated cells to LMP and apoptosis. Our results are in good agreement with previous data showing that the addition of cholesterol to isolated lysosomes reduces their permeability (Fouchier et al., 1983). In addition, cholesterol depletion by MβCD treatment increases the permeability of the lysosomal membrane and thus destabilizes the organelle (Deng et al., 2009; Hao et al., 2008; Jadot et al., 2001).
Figure 39. Cholesterol accumulation is associated with increased lysosomal stability and decreased sensitivity to lysosome-mediated cell death. Mutations in the NPC1 protein or treatment with U18666A block the efflux of cholesterol from the endolysosomal compartment and result in the accumulation of cholesterol in the lysosomal membrane. Lysosomal cholesterol accumulation correlates with lysosomal stability, and thus cells with high lysosomal cholesterol content are protected from lysosomal membrane permeabilization (LMP) and lysosome-dependent apoptosis.

DISCUSSION
114
It is assumed that the majority of cholesterol accumulates in the internal membranes of late endosomes and lysosomes. Changes in the lipid composition of inner lysosomal membranes have a direct and potent influence on the stability of the entire lysosome (Petersen et al., 2010), but it is not known if this is due to a direct effect on the lipid composition of the outer membrane. In paper II, we demonstrate that cholesterol accumulation affects the sensitivity of the lysosomal membrane to digitonin-permeabilization, arguing that cholesterol also increases in the limiting membrane of lysosomes. Similarly, cholesterol accumulation in LAMPnull MEFs was observed in both the limiting and internal membranes of endosomes and lysosomes (Eskelinen et al., 2004), indicating that unesterified cholesterol increases at both locations.
Cholesterol is increased in most cancer cells and contributes to their resistance to cytotoxic stress (Lucken-Ardjomande, 2012). More specifically, cholesterol inhibits Bax oligomerization and thereby reduces the release of pro-apoptotic factors from the mitochondria (Lucken-Ardjomande et al., 2008). The authors suggested that the protective effect of cholesterol is exerted at the mitochondria, but it is tempting to speculate that lysosomal cholesterol accumulation might also contribute to the protective effect. Reducing intracellular cholesterol levels might represent an interesting novel approach to sensitize cancer cells to chemotherapeutic agents.
The distinct loss of cells associated with some lysosomal storage disorders could be a consequence of decreased lysosomal stability. According to such a theory, the accumulation of substances might cause instability or rupture of the lysosomal membrane and result in leakage of lysosomal hydrolases to the cytosol; however, there is little evidence supporting such a basis for the neurodegeneration associated with these disorders (Futerman and van Meer, 2004). Our results from papers II and III rather demonstrate that, at least in NPC disease, lysosomes exhibit increased stability and are less prone to LMP. Thus, cell death seems to be a consequence of disturbed cellular function due to lysosomal dysfunction, rather than a result of lysosomal membrane instability.

DISCUSSION
115
Cholesterol accumulation induces alterations of the lysosomal compartment The capacity to mobilize cholesterol and transfer it outside of the inner lysosomal membrane is crucial for the ability of hydrolases to degrade lipids of the inner membrane. The presence of cholesterols in a membrane affects its packaging; therefore, membrane degradation decreases as its cholesterol content increases (Gallala and Sandhoff, 2011). This effect of cholesterol might explain the accumulation of other lipids that occurs secondary to cholesterol accumulation. As a result of lipid storage, the membrane fluidity of late endosomes and lysosomes is reduced, which affects the distribution and activity of proteins associated with the membranes, resulting in lysosomal dysfunction.
In papers II and III, we demonstrate that cholesterol accumulation is associated with an increased size of the lysosomal compartment. In line with this, NPC1 mutations increase the size of the endolysosomal compartment up to three fold (Sobo et al., 2007). Treatment with the amphiphilic compund imipramine similarly induces the expansion of the lysosomal volume, which can be reduced by treatment with hydroxylpropyl-β-cyclodextrin (Funk and Krise, 2012). These data indicate that expansion of the lysosomal volume occurs as a response to an increase in lysosomal cholesterol content and might represent a way for the cell to store undigested material.
In paper II, we demonstrate that lysosomal cholesterol accumulation is associated with an increased expression of both a lysosomal membrane protein (LAMP-2) and a soluble hydrolase (cathepsin D). These findings could suggest that these proteins are upregulated by a common mechanism. Indeed, such a regulatory network has been identified in recent years and is currently being explored. Many genes encoding lysosomal proteins, including NPC1, cathepsin D and LAMP-1, harbor a short sequence near the transcription start site called a coordinated lysosomal expression and regulation (CLEAR) element (Sardiello et al., 2009; Schultz et al., 2011). The transcription factor EB (TFEB) can enter the nucleus and bind to the CLEAR elements, thereby inducing the transcription of these genes. There are 471 direct targets of TFEB, including genes involved in lysosomal biogenesis and autophagy (Palmieri et al., 2011; Settembre et al., 2011). Thus, TFEB can be

DISCUSSION
116
considered a master regulator of lysosomal biogenesis and autophagy. Interestingly, the TFEB pathway is activated under lysosomal storage conditions and might be responsible for the increased size of the lysosomal compartment (Sardiello et al., 2009). In addition, enhanced clearance of stored material was observed as a consequence of TFEB overexpression, suggesting that activation of the TFEB/CLEAR network could restore the cellular defect in many lysosomal storage disorders (Sardiello et al., 2009; Schultz et al., 2011).
LYSOSOMAL EXOCYTOSIS Plasma membrane injury is a frequent event in mammalian cells. In paper IV, we demonstrate that lysosomal exocytosis is involved in the repair of UVA irradiation-induced plasma membrane damage. In addition to UVA-induced plasma membrane damage, disruption of the plasma membrane is common in cells that operate under conditions of mechanical stress, such as in muscles and skin (McNeil, 2002). The permeability barrier can also be breached, for example, by pathogens gaining access to host cells by secreting pore-forming toxins. If disruptions of the plasma membrane are not resealed, calcium will flood into the cytosol of the wounded cell, and cytoplasmic constituents can escape. Therefore, cell survival depends on the rapid restoration of plasma membrane integrity after injury. As illustrated in paper IV, resealing of the plasma membrane by lysosomal exocytosis is associated with the extracellular release of lysosomal content. The release of aSMase is essential for the formation of lipid rafts, which are known to influence various signaling processes, including death receptor signaling (Corre et al., 2010).
Originally, it was assumed that cell membranes would spontaneously reseal if broken, but it is now known that plasma membrane repair is the outcome of a dynamic and complex mechanism involving lysosomal exocytosis (McNeil, 2002). Lysosomal exocytosis is dependent on the presence of synaptotagmin VII (Syt VII), which is located at the lysosomal membrane (Reddy et al., 2001). Synaptotagmins are a family of proteins that are thought to function as transducers of calcium signaling in membrane fusion events, for example, during neurotransmitter release in brain synapses (Andrews and Chakrabarti, 2005). The importance of Syt VII for

DISCUSSION
117
efficient plasma membrane repair was highlighted in experiments in which Syt VII inhibition prevented membrane resealing. Of note, impaired resealing is also observed in cells microinjected with antibodies against the cytosolic tail of LAMP-1 [35], suggesting an unidentified function of LAMP-1 during lysosomal exocytosis. During exocytosis, calcium binds to Syt VII at the lysosomal membrane and facilitates its interaction with soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs; Figure 40), which are essential for membrane fusion.
Figure 40. Synaptotagmin VII (Syt VII) and soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) cooperate in the fusion of lysosomes and the plasma membrane. Calcium binds the calcium-sensing protein Syt VII, which is located at the lysosomal membrane, to facilitate the interaction with SNAREs. Syt VII specifically interacts with the vesicle (v)-SNARE VAMP7 found on lysosomes and with the target (t)SNAREs syntaxin 4 and SNAP-23 at the plasma membrane (Rao et al., 2004). SNARE complexes are involved in the formation of a fusion pore between the lysosome and the plasma membrane (Andrews and Chakrabarti, 2005).
Extensive evidence demonstrates that UV radiation is a major skin carcinogen. The harmful effects of UV are mostly attributed to direct and indirect DNA damage (Pfeifer and Besaratinia, 2012). However, it is likely that UV irradiation influences the cells of the skin in a number of ways. The results presented in paper IV demonstrate that UVA-induced lysosomal exocytosis results in the release of lysosomal content to the extracellular space. Thus, chronic UV exposure might

DISCUSSION
118
contribute to cancer progression by providing a continuous source of extracellular active cathepsin. Cathepsins promote tumorigenesis by acting in both an autocrine and a paracrine manner to stimulate proliferation, enhance angiogenesis and promote invasion by degrading the extracellular matrix. Melanoma is indeed one of the most invasive cancers (Zaidi et al., 2012).
CASPASE-8 ACTIVATION Human fibroblasts show a strong dependence on cathepsin D during apoptosis, while cysteine cathepsins are dispensable. However, inhibition of both cathepsin D and cysteine cathepsins protects melanocytes (Bivik et al., 2006) and keratinocytes from UV-induced apoptosis (paper IV). In human melanocytes, UV irradiation triggers intrinsic apoptosis and cell death signaling that is not associated with early activation of caspase-8 (Bivik et al., 2006). By contrast, in paper IV, we demonstrate that UVA irradiation of human keratinocytes results in early caspase-8 activation, indicating a difference in apoptotic signaling in keratinocytes compared to melanocytes. Early caspase-8 activation was observed in keratinocytes exposed to UVA but not UVB. Similarly, UVA irradiation but not UVB irradiation induced plasma membrane damage and lysosomal exocytosis, suggesting a possible link between these processes.
Caspase-8 is classically activated in the DISC complex as part of extrinsic apoptotic signaling. However, other mechanisms for caspase-8 activation have been demonstrated, including activation at the mitochondrial membrane (Gonzalvez et al., 2008). The involvement of death receptor signaling in keratinocytes exposed to UVA irradiation is supported by a study showing ligand-independent Fas receptor aggregation after UV irradiation (Aragane et al., 1998; Rehemtulla et al., 1997; Sheikh et al., 1998). In addition, keratinocytes have been shown to secrete TNF-α as a response to UVB irradiation (Kock et al., 1990). However, under our experimental conditions, there is no release of TNF-α before the activation of caspase-8, eliminating this alternative as the triggering signal. aSMase has been shown to translocate from lysosomes to the plasma membrane during apoptosis, and this redistribution resulted in sphingomyelin breakdown and ceramide production in the outer leaflet of the plasma membrane (Grassme et al., 2001).

DISCUSSION
119
Ceramide enhances early Fas receptor signaling events, including its translocation into lipid rafts. Therefore, it is not surprising that impaired ceramide production due to aSMase deficiency confers resistance to Fas receptor-mediated cell death (Segui and Legembre, 2010). In paper IV we demonstrate UVA irradiation to be associated with formation of lipid rafts, suggesting that death receptor recruitment and aggregation could occur in these membrane domains. Inhibition of aSMase activity with desipramine decreases lipid raft formation but do not influence caspase-8 activation. These results suggest that lipid rafts are not involved in UVA-induced caspase-8 activation in keratinocytes.
Death receptor signaling, including assembly of the DISC complex and activation of caspase-8, was initially suggested to occur at the cytosolic leaflet of the plasma membrane. However, it is now clear that the death receptors require endocytosis to activate apoptotic signaling, and caspase-8 activation is thought to occur in intracellular vesicles sometimes referred to as receptosomes (Tchikov et al., 2011). A requirement for endocytosis has been described for the TNF-, TRAIL- and Fas-receptors (Akazawa et al., 2009; Lee et al., 2006; Schneider-Brachert et al., 2004) and in a series of investigations, Schütze and co-workers characterized the signaling that occurs downstream of TNF receptor-1 activation (Tchikov et al., 2011). As presented in Figure 41, in this experimental model TNF-α binding to the TNF receptor, results in endocytosis of the receptor. DISC components are recruited to the endosome, where activation of caspase-8 occurs (Schneider-Brachert et al., 2004). Because receptor-containing vesicles are positive for a marker of the TGN, the receptosomes have been proposed to fuse with TGN-derived vesicles containing pro-aSMase and pro-cathepsin D (Schneider-Brachert et al., 2004). The receptors are then sorted into ILVs of multivesicular bodies, where the lethal signal is amplified by caspase-8-mediated activation of caspase-7, which in turn activates aSMase (Edelmann et al., 2011). Ceramide, generated by aSMAse, activates cathepsin D, which, after its release to the cytosol, processes Bid (Heinrich et al., 1999; Heinrich et al., 2004).

DISCUSSION
120
Figure 41. A proposed model for signaling by endocytosed death receptors. Binding of tumor necrosis factor (TNF)-α to TNF receptor 1 initiates endocytosis and recruitment of the death inducing signaling complex (DISC) components: Fas associated death domain (FADD) and procaspase-8. Within the receptosome, the DISC complex serves as a platform for the dimerization and activation of caspase-8. The receptosome fuses with a vesicle derived from the trans-Golgi network, containing pro-cathepsin D and pro-acid sphingomyelinase (aSMase), to form a multivesicular body. In the fusion organelle, caspase-8 activates caspase-7, which in turn activates aSMase. Activation of aSMase results in ceramide production, which activates cathepsin D. Cathepsin D is released to the cytosol, where it activates Bid, thereby amplifying the death signal by mitochondrial engagement.
The results presented in paper IV indicate that a signaling pathway similar to the one described above can possibly be operating in UVA-irradiated keratinocytes; however, there are some divergences. In keratinocytes, caspase-8 activation is lysosome-dependent, placing lysosomes upstream rather than downstream of caspase-8. Likewise, aSMase activity is increased after UVA but independent of caspase-8, arguing against caspases as activators of newly synthesized aSMase. In paper IV, we demonstrate that the early activation of caspase-8 occurs in intracellular vesicles positive for LAMP-1 and cathepsin D and is dependent on lysosomal function. We have not yet determined if any death receptor is involved

DISCUSSION
121
in the activation of caspase-8 after UVA irradiation. An alternative caspase-8 activation mechanism is direct proteolytic processing by cathepsin D, as described in neutrophils (Conus et al., 2008). Under these circumstances, both proteolysis and dimerization of caspase-8 are required for robust activation of caspase-8 (Conus et al., 2012). Other proteases, including granzyme B and caspase-6, are also capable of activating caspase-8 (van Raam and Salvesen, 2012). Although the exact mechanism remains to be revealed, we conclude that caspase-8 is activated in a lysosome-dependent manner within intracellular vesicles positive for LAMP-1 and cathepsin D and is partly dependent on endocytosis.


CONCUSIONS
123
CONCLUSIONS
From the results presented in this thesis, the following conclusions can be drawn:
• In fibroblasts, the pro-apoptotic activity of cathepsin D is dependent on its proteolytic activity, which is favored by apoptosis-associated cytosolic acidification.
• After lysosomal release, cathepsin D engages the intrinsic pathway to apoptosis by proteolytic activation of Bid, which is followed by Bax translocation to the mitochondria.
• Cell-free experiments verify that cathepsin D proteolytically processes Bid, generating a truncated Bid with pro-apoptotic properties. This cleavage occurs at neutral pH but is more efficient at acidic pH.
• Drugs or mutations resulting in lysosomal cholesterol accumulation increase the stability of lysosomes and render cells less sensitive to apoptotic induction. Secondary changes, including the accumulation of sphingolipids and increased expression of LAMPs, are non-essential for the increased lysosomal stability.
• Cellular sensitivity to lysosome-mediated apoptosis can be modulated by alteration of the lysosomal cholesterol content.
• Plasma membrane damage induced by UVA irradiation is repaired by lysosomal exocytosis.
• UVA irradiation-induced apoptosis in human keratinocytes is characterized by an early activation of caspase-8. Caspase-8 activation is lysosome-dependent.


CLINICAL IMPLICATIONS
125
CLINICAL IMPLICATIONS
Future targeting of lysosomes as therapeutic intervention?
Understanding physiological processes in biochemical and molecular detail not only offers insight into disease pathogenesis but also permits the development of new diagnostic and prognostic tools, as well as the design of novel therapeutic compounds. The role of lysosomes as key components of many cellular processes make them attractive therapeutic targets.
The acidic pH of the lysosomal compartment is essential for the correct functioning of this organelle. Cargo release, hydrolase maturation, degradation, autophagy and intracellular trafficking are all dependent on a low pH (Schultz et al., 2011). An elegant study demonstrated that the Alzheimer’s disease related protein presenelin-1 regulates the trafficking of the vacuolar H+-ATPase to the lysosomes. Mutations within presenilin-1, which is one of the major risk factors for familial Alzheimer´s disease, disrupts trafficking of the H+-ATPase and results in aberrant lysosomal pH, defective cathepsin activation and faulty degradation (Lee et al., 2010). Some lysosomal storage disorders have also been associated with lysosomal alkalization (Schultz et al., 2011). This is, however, not a general characteristic of lysosomal storage disorders because the lysosomal pH is not elevated in NPC disease (Elrick et al., 2012; Lloyd-Evans et al., 2008). In disorders with an aberrant lysosomal pH, restoration of lysosomal acidification by therapeutic intervention could represent an efficient way to promote the degradation and clearance of accumulating agents.
The search for therapies that reduce lysosomal storage is a continuous ongoing project. The cholesterol depleting effect of cyclodextrin derivatives is known and generally believed to be due to the solubilization of cholesterol within the interior of the cyclodextrin molecules. Unexpectedly, a variant of cyclodextrin incapable of solubilizing cholesterol was as effective at decreasing cholesterol load as its normal

CLINICAL IMPLICATIONS
126
counterpart (Ramirez et al., 2011). A possible explanation for this effect is the finding that hydroxypropyl-β-cyclodextrin reduces cholesterol storage in NPC1-/- mice by inducing lysosomal exocytosis (Chen et al., 2010). Reversion of pathologic lysosomal storage by lysosomal exocytosis was also demonstrated in vivo and in vitro by TFEB overexpression (Medina et al., 2011). In support of such clearance mechanisms operating in vivo, patients with lysosomal storage disorder secrete the storage products into the urine (Schultz et al., 2011). However, inducing exocytosis as a therapeutic mechanism is a major concern because the potentially toxic stored contents might not be adequately cleared after release to the extracellular space in the brain (Schultz et al., 2011).
Another way to induce the clearance of accumulated material is the induction of autophagy, which has proven beneficial as a therapeutic strategy in neurodegenerative disorders associated with protein aggregation (Harris and Rubinsztein, 2012). A prerequisite for a favorable effect of autophagic induction is a functional lysosomal compartment capable of cargo degradation. In NPC, in which accumulation of autophagosomes is already a prominent feature, autophagy induction would most likely have a limited beneficial effect. In support of such a conclusion, it was recently shown that defective autophagosome clearance in NPC cells is due to inhibition of lysosomal cathepsins by the stored lipids (Elrick et al., 2012). Furthermore, autophagy of lipid droplets contributes to lipid storage (Elrick et al., 2012). These data suggest that promotion of autophagy in NPC disease would further impair cellular function rather than meliorate the disease phenotype.
An opposite intervention in the autophagic process, impairment of autophagy, is being investigated as a therapeutic approach to sensitize cancer cells to apoptosis-inducing agents. In general, autophagy functions as a protective mechanism that counteracts apoptosis by providing the cell with energy and building blocks as well as disposal of damaged and non-functional mitochondria, which are a source of reactive oxygen species (Lee et al., 2012). Destabilization of the lysosomes by lysosomotropic detergents could be a promising strategy because destabilization would not only impair autophagy but also promote tumor cell apoptosis through the release of cathepsins, thereby activating the lysosomal cell death pathway (Repnik et al., 2012).

FUTURE PERSPECTIVES
127
FUTURE PERSPECTIVES
The results from paper II and III suggest that cellular sensitivity to cell death can be modulated by changing the lysosomal cholesterol content. It would be highly interesting to investigate this in vivo; for example, can short pretreatment with a drug that induces lysosomal lipid accumulation decrease the damage caused by an acute toxic insult such as ischemia? It would also be of interest to investigate the lysosomal cholesterol content of tumor cells for comparison with their normal counterparts, to determine if lysosomal cholesterol content, similar to mitochondrial cholesterol content, is higher in malignant cells. In addition, new approaches to improve lysosomal function in vitro that could promote clearance of storage and restore cellular function in vivo would be of great interest.
Moreover, the similar cellular phenotype, with lysosomal accumulation of cholesterol and glycosphingolipids, resulting from cathepsin D, LAMP or NPC protein deficiency is intriguing. Currently, the cause of lipid accumulation in cathepsin D deficient cells is unknown, and investigation of the causative mechanism would add new valuable information.
Further studies of the signaling upstream of caspase-8 activation in human keratinocytes after UVA irradiation is necessary to reveal if caspase-8 activation is dependent on death receptors or if a novel mechanism is engaged. The involvement of UV irradiation-induced lysosomal exocytosis in skin cancer progression is another exploitable research area.


TACK
129
TACK
Detta är min avhandling, men den hade inte funnits idag utan många personers medverkan. Jag är omgiven av fantastiska människor och jag vill passa på att tacka alla som hjälpt och stöttat mig under arbetet, inklusive:
Karin - som under min tid som student, stipendiat och doktorand har handlett mig i med- och motgång. Stort tack för att du generöst delar med dig av din stora erfarenhet och kunskap, samtidigt som du låter mig arbeta självständigt och ta eget ansvar. Att alltid kunna knacka på din dörr för att ställa en fråga eller be om råd är guld värt. Tack också för att jag fått chansen att presentera vår forskning runt om i världen.
Katarina - du är en inspiration och en person med sociala förmågor utöver det vanliga. Tack för ditt engagemang och omtanke, och för att du har förmåga att se när jag behöver lätta mitt hjärta. Jag beundrar din förmåga att tänka i andra banor och komma på nya idéer som kan förvandlas till spännande forskningsprojekt. Tack för att du såg till att jag kan skriva forskningsvistelse i Australien på mitt CV.
Petra - din effektivitet och simultanförmåga är otrolig. Tack för att du jobbar så hårt med arbete IV, som aldrig hade blivit något utan dig. Jag värdesätter att du läst och kommenterat det jag skrivit och det snabbt som ögat. Det är trevligt att vi nu till slut lyckats befinna oss Patologen samtidigt.
Lotta - det är tack vare dig som mitt intresse för apoptos väcktes i en föreläsningssal för 10 år sedan och sen dess är jag fast. Det har varit roligt att arbeta med dig, tänk att Bax II blev publicerat till sist. Tack för att du tog dig tid att läsa min kappa trots en fulltecknad kalender. Jag uppskattar dina ärliga åsikter och konstruktiva kritik.

TACK
130
Cathrine - du var en sann förebild, med ditt skarpa intellekt, otroliga argumentationsförmåga och enorma medkänsla. Helt enkelt en fenomenal doktorandkollega, reskamrat och vän. Saknar dig.
Linnea - din positiva inställning till allt, litet som smått, är beundransvärd. Jag uppskattar dina uppmuntrande hejarop under avhandlingsarbetet. Tack också för ditt tålamod med alla dessa utsådder med mängder av celltätheter. Du är drottning över LAMP MEFarna och en riktig glädjespridare.
Brett Garner - thanks for your highly valuable scientific input and for the invitation to come and visit you and your lab in Australia. I will always remember the close encounter with the kangaroos at the South coast.
Britt-Marie – tack för att du lärde mig att cellodla och hjälpte mig med så mycket min första tid på Patologen. Jag uppskattade ditt sällskap tidiga morgnar. Nu för tiden är det alltid lika trevligt att träffas över en lunch eller middag.
Uno - tack för hjälp med pH-mätningar och krånglande apparatur.
Ida och Linda - tack för praktisk hjälp under åren som gått.
Tack till alla medförfattare för ett givande samarbete. Especially thanks to Karin Björnström, Andrew J. Brown, Paul Saftig and Robert Steinfeld for providing cells. Tack till Mats Söderström för hjälp med plasmider och till Cecilia Bivik för trevliga diskussioner om forskning och annat. Thanks to Kajsa Holmgren Peterson, Sangeeta Nath and Bengt-Arne Fredriksson for help with microscopy. Jag vill också passa på att tacka Camilla Höglund för administrativ hjälp.
Alla nuvarande och före detta kollegor på Patologen plan 09, tack för den trevliga arbetsplats som ni skapar. Ett extra tack till Lotta Agholme, Fredrik Jerhammar och övriga invånare i doktorandrummet, där det alltid finns någon att fråga när man behöver hjälp. Ett speciellt tack också till de kollegor som jag haft nöjet att uppleva Bergen, Budapest, Heidelberg, Spetses, Portoroz, Shanghai, Sydney, Honolulu och Rom tillsammans med.

TACK
131
Tack till vänner och släktingar för all trevlig samvaro. En promenad, en lunch, ett telefonsamtal eller lite skvaller över en kopp te är mycket värt och får mig att ladda batterierna. Tack också till alla er som intresserat er för mitt arbete och frågat om min forskning för att sedan fundersamt rynka pannan när jag försökt förklara.
Mamma och Pappa som alltid finns där för att hjälpa och stötta, och min stora lillebror Niklas. Vad hade jag varit utan er och all möjlig tänkbar service som ni erbjuder? Oavsett om det gäller att vara resesällskap, sitta barnvakt, rulla gräsmatta, installera takfotsbelysning, skjutsa mig eller utföra diverse sy-uppdrag känns det tryckt att veta att ”Mild, Mild & Mild” ställer upp. Tack också för den stående inbjudan att komma till Lillstugan, för att koppla av i trevligt sällskap.
Christer - ända sen första gången vi sågs har det varit vi två och det är jag så lycklig för. Det var precis som Per Håkan beskrev det ”Om du kommer ihåg”. Tack för att du påminner mig om att det inte finns några bekymmer som inte känns bättre (åtminstone lite) efter fika och ansiktsmassage. Du finns för mig, du kommer så fort du bara kan. Du lyssnar på mig, håller om mig och erbjuder en hand. För mig är Ingenting är värt någonting alls, utan nån som du. Jag älskar dig!
Sist och minst (men bara längdmässigt); Felix - du gör mig stolt och mitt liv så mycket roligare och som har förmåga att på ett ögonblick få mig tänka på något helt annat än mitt arbete. Christer & Felix ni är mina absoluta favoriter och det mest värdefulla jag har!
Jag vill avsluta med att tacka de generösa bidragsgivare som stött mitt arbete genom stipendier och anslag; Anna Cederbergs stiftelse, Cancerfonden, European Cell Death Organisation, Fonden för Familjerna Carl och Albert Molins i Motala minne, Knut och Alice Wallenbergs stiftelse, Kommittén för Medicinsk FoU inom Östergötlands läns landsting, Lions forskningsfond mot folksjukdomar, Signhild Engkvist stiftelse, Sven och Dagmar Saléns stiftelse och Svenska Sällskapet för Medicinsk Forskning. Studierna har även kunnat genomföras med hjälp av projektanslag från Cancerfonden och Vetenskapsrådet.


REFERENCES
133
REFERENCES
Abi-Mosleh L, Infante RE, Radhakrishnan A, Goldstein JL, Brown MS (2009) Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc Natl Acad Sci U S A; 106(46): 19316-19321.
Abok K, Hirth T, Ericsson JL, Brunk U (1983) Effect of iron on the stability of macrophage lysosomes. Virchows Arch B Cell Pathol Incl Mol Pathol; 43(2): 85-101.
Agar NS, Halliday GM, Barnetson RS, Ananthaswamy HN, Wheeler M, Jones AM (2004) The basal layer in human squamous tumors harbors more UVA than UVB fingerprint mutations: a role for UVA in human skin carcinogenesis. Proc Natl Acad Sci U S A; 101(14): 4954-4959.
Ahuja D, Saenz-Robles MT, Pipas JM (2005) SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene; 24(52): 7729-7745.
Akazawa Y, Mott JL, Bronk SF, Werneburg NW, Kahraman A, Guicciardi ME et al. (2009) Death receptor 5 internalization is required for lysosomal permeabilization by TRAIL in malignant liver cell lines. Gastroenterology; 136(7): 2365-2376.
Andersson E, Vahlquist A, Rosdahl I (2001) Beta-carotene uptake and bioconversion to retinol differ between human melanocytes and keratinocytes. Nutr Cancer; 39(2): 300-306.
Andersson ER (2012) The role of endocytosis in activating and regulating signal transduction. Cell Mol Life Sci; 69(11): 1755-1771.
Andrejewski N, Punnonen EL, Guhde G, Tanaka Y, Lullmann-Rauch R, Hartmann D et al. (1999) Normal lysosomal morphology and function in LAMP-1-deficient mice. J Biol Chem; 274(18): 12692-12701.
Andrews NW (2000) Regulated secretion of conventional lysosomes. Trends Cell Biol; 10(8): 316-321.
Andrews NW (2005) Membrane repair and immunological danger. EMBO Rep; 6(9): 826-830.
Andrews NW, Chakrabarti S (2005) There's more to life than neurotransmission: the regulation of exocytosis by synaptotagmin VII. Trends Cell Biol; 15(11): 626-631.
Antunes F, Cadenas E (2000) Estimation of H2O2 gradients across biomembranes. FEBS Lett; 475(2): 121-126.
Antunes F, Cadenas E (2001) Cellular titration of apoptosis with steady state concentrations of H(2)O(2): submicromolar levels of H(2)O(2) induce apoptosis through Fenton chemistry independent of the cellular thiol state. Free Radic Biol Med; 30(9): 1008-1018.

REFERENCES
134
Appelmans F, Wattiaux R, De Duve C (1955) Tissue fractionation studies. 5. The association of acid phosphatase with a special class of cytoplasmic granules in rat liver. Biochem J; 59(3): 438-445.
Aragane Y, Kulms D, Metze D, Wilkes G, Poppelmann B, Luger TA et al. (1998) Ultraviolet light induces apoptosis via direct activation of CD95 (Fas/APO-1) independently of its ligand CD95L. J Cell Biol; 140(1): 171-182.
Arias E, Cuervo AM (2011) Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol; 23(2): 184-189.
Armstrong BK, Kricker A (2001) The epidemiology of UV induced skin cancer. J Photochem Photobiol B; 63(1-3): 8-18.
Awano T, Katz ML, O'Brien DP, Taylor JF, Evans J, Khan S et al. (2006) A mutation in the cathepsin D gene (CTSD) in American Bulldogs with neuronal ceroid lipofuscinosis. Mol Genet Metab; 87(4): 341-348.
Bainton DF (1981) The discovery of lysosomes. J Cell Biol; 91(3 Pt 2): 66s-76s.
Barry M, Heibein JA, Pinkoski MJ, Lee SF, Moyer RW, Green DR et al. (2000) Granzyme B short-circuits the need for caspase 8 activity during granule-mediated cytotoxic T-lymphocyte killing by directly cleaving Bid. Mol Cell Biol; 20(11): 3781-3794.
Beaujouin M, Baghdiguian S, Glondu-Lassis M, Berchem G, Liaudet-Coopman E (2006) Overexpression of both catalytically active and -inactive cathepsin D by cancer cells enhances apoptosis-dependent chemo-sensitivity. Oncogene; 25(13): 1967-1973.
Bellettato CM, Scarpa M (2010) Pathophysiology of neuropathic lysosomal storage disorders. J Inherit Metab Dis; 33(4): 347-362.
Benes P, Vetvicka V, Fusek M (2008) Cathepsin D-many functions of one aspartic protease. Crit Rev Oncol Hematol; 68(1): 12-28.
Bennett DC (2008) Ultraviolet wavebands and melanoma initiation. Pigment Cell Melanoma Res; 21(5): 520-524.
Berchem G, Glondu M, Gleizes M, Brouillet JP, Vignon F, Garcia M et al. (2002) Cathepsin-D affects multiple tumor progression steps in vivo: proliferation, angiogenesis and apoptosis. Oncogene; 21(38): 5951-5955.
Berndtsson M, Hägg M, Panaretakis T, Havelka AM, Shoshan MC, Linder S (2007) Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int J Cancer; 120(1): 175-180.
Bewley MA, Marriott HM, Tulone C, Francis SE, Mitchell TJ, Read RC et al. (2011) A cardinal role for cathepsin d in co-ordinating the host-mediated apoptosis of macrophages and killing of pneumococci. PLoS Pathog; 7(1): e1001262.
Bidére N, Lorenzo HK, Carmona S, Laforge M, Harper F, Dumont C et al. (2003) Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem; 278(33): 31401-31411.

REFERENCES
135
Bivik C, Larsson P, Kågedal K, Rosdahl I, Öllinger K (2006) UVA/B-induced apoptosis in human melanocytes involves translocation of cathepsins and Bcl-2 family members. J Invest Dermatol; 126(5): 1119-1127.
Bivik C, Rosdahl I, Öllinger K (2007) Hsp70 protects against UVB induced apoptosis by preventing release of cathepsins and cytochrome c in human melanocytes. Carcinogenesis; 28(3): 537-544.
Bizat N, Galas MC, Jacquard C, Boyer F, Hermel JM, Schiffmann SN et al. (2005) Neuroprotective effect of zVAD against the neurotoxin 3-nitropropionic acid involves inhibition of calpain. Neuropharmacology; 49(5): 695-702.
Björklund U, Persson M, Rönnback L, Hansson E (2010) Primary cultures from cerebral cortex and hippocampus enriched in glutamatergic and GABAergic neurons. Neurochem Res; 35(11): 1733-1742.
Blomgran R, Zheng L, Stendahl O (2007) Cathepsin-cleaved Bid promotes apoptosis in human neutrophils via oxidative stress-induced lysosomal membrane permeabilization. J Leukoc Biol; 81(5): 1213-1223.
Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ (1997) The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem; 243(1-2): 240-246.
Bogner C, Leber B, Andrews DW (2010) Apoptosis: embedded in membranes. Curr Opin Cell Biol; 22(6): 845-851.
Boya P, Andreau K, Poncet D, Zamzami N, Perfettini JL, Metivier D et al. (2003) Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J Exp Med; 197(10): 1323-1334.
Bradley DF, Wolf MK (1959) Aggregation of Dyes Bound to Polyanions. Proc Natl Acad Sci U S A; 45(7): 944-952.
Broker LE, Huisman C, Span SW, Rodriguez JA, Kruyt FA, Giaccone G (2004) Cathepsin B mediates caspase-independent cell death induced by microtubule stabilizing agents in non-small cell lung cancer cells. Cancer Res; 64(1): 27-30.
Brown MS, Goldstein JL (1974) Suppression of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity and inhibition of growth of human fibroblasts by 7-ketocholesterol. J Biol Chem; 249(22): 7306-7314.
Brown MS, Goldstein JL (1986) A receptor-mediated pathway for cholesterol homeostasis. Science; 232(4746): 34-47.
Brown MS, Goldstein JL (1997) The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell; 89(3): 331-340.
Brown MS, Goldstein JL (1999) A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci U S A; 96(20): 11041-11048.
Brown MS, Goldstein JL (2009) Cholesterol feedback: from Schoenheimer's bottle to Scap's MELADL. J Lipid Res; 50 Suppl: S15-27.

REFERENCES
136
Brunk UT, Svensson I (1999) Oxidative stress, growth factor starvation and Fas activation may all cause apoptosis through lysosomal leak. Redox Report; 4(1-2): 3-11.
Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B (2000) Rab7: a key to lysosome biogenesis. Mol Biol Cell; 11(2): 467-480.
Burman C, Ktistakis NT (2010) Autophagosome formation in mammalian cells. Semin Immunopathol; 32(4): 397-413.
Bursch W, Ellinger A, Gerner C, Frohwein U, Schulte-Hermann R (2000) Programmed cell death (PCD). Apoptosis, autophagic PCD, or others? Ann N Y Acad Sci; 926: 1-12.
Cabal-Hierro L, Lazo PS (2012) Signal transduction by tumor necrosis factor receptors. Cell Signal; 24(6): 1297-1305.
Callus BA, Vaux DL (2007) Caspase inhibitors: viral, cellular and chemical. Cell Death Differ; 14(1): 73-78.
Carlsson SR, Roth J, Piller F, Fukuda M (1988) Isolation and characterization of human lysosomal membrane glycoproteins, h-lamp-1 and h-lamp-2. Major sialoglycoproteins carrying polylactosaminoglycan. J Biol Chem; 263(35): 18911-18919.
Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C et al. (1997) Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science; 277(5323): 228-231.
Caruso JA, Mathieu PA, Reiners JJ, Jr. (2005) Sphingomyelins suppress the targeted disruption of lysosomes/endosomes by the photosensitizer NPe6 during photodynamic therapy. Biochem J; 392(Pt 2): 325-334.
Caruso JA, Mathieu PA, Joiakim A, Zhang H, Reiners JJ, Jr. (2006) Aryl hydrocarbon receptor modulation of tumor necrosis factor-alpha-induced apoptosis and lysosomal disruption in a hepatoma model that is caspase-8-independent. J Biol Chem; 281(16): 10954-10967.
Castino R, Peracchio C, Salini A, Nicotra G, Trincheri NF, Demoz M et al. (2009) Chemotherapy drug response in ovarian cancer cells strictly depends on a cathepsin D-Bax activation loop. J Cell Mol Med; 13(6): 1096-1109.
Cavallini G, Massarani E (1951) [Relation between chemical constitution and biological activity; new vasodilators and curare-simulants derived from sex hormones]. Farmaco; 6(3): 291-299.
Cenedella RJ, Jacob R, Borchman D, Tang D, Neely AR, Samadi A et al. (2004) Direct perturbation of lens membrane structure may contribute to cataracts caused by U18666A, an oxidosqualene cyclase inhibitor. J Lipid Res; 45(7): 1232-1241.
Cenedella RJ (2009) Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids; 44(6): 477-487.
Chang TY, Li BL, Chang CC, Urano Y (2009) Acyl-coenzyme A:cholesterol acyltransferases. Am J Physiol Endocrinol Metab; 297(1): E1-9.
Chen FW, Li C, Ioannou YA (2010) Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS One; 5(11): e15054.

REFERENCES
137
Chen M, He H, Zhan S, Krajewski S, Reed JC, Gottlieb RA (2001) Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J Biol Chem; 276(33): 30724-30728.
Cheung NS, Koh CH, Bay BH, Qi RZ, Choy MS, Li QT et al. (2004) Chronic exposure to U18666A induces apoptosis in cultured murine cortical neurons. Biochem Biophys Res Commun; 315(2): 408-417.
Chevallier J, Chamoun Z, Jiang G, Prestwich G, Sakai N, Matile S et al. (2008) Lysobisphosphatidic acid controls endosomal cholesterol levels. J Biol Chem; 283(41): 27871-27880.
Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR (2010) The BCL-2 family reunion. Mol Cell; 37(3): 299-310.
Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ et al. (2012) Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell; 148(5): 988-1000.
Cirman T, Oresic K, Mazovec GD, Turk V, Reed JC, Myers RM et al. (2004) Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J Biol Chem; 279(5): 3578-3587.
Conus S, Perozzo R, Reinheckel T, Peters C, Scapozza L, Yousefi S et al. (2008) Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J Exp Med; 205(3): 685-698.
Conus S, Pop C, Snipas SJ, Salvesen GS, Simon HU (2012) Cathepsin D primes caspase-8 activation by multiple intra-chain proteolysis. J Biol Chem; 287(25): 21142-21151.
Corre I, Niaudet C, Paris F (2010) Plasma membrane signaling induced by ionizing radiation. Mutat Res; 704(1-3): 61-67.
Coutinho MF, Prata MJ, Alves S (2012) Mannose-6-phosphate pathway: a review on its role in lysosomal function and dysfunction. Mol Genet Metab; 105(4): 542-550.
Critchley DR, Streuli CH, Kellie S, Ansell S, Patel B (1982) Characterization of the cholera toxin receptor on Balb/c 3T3 cells as a ganglioside similar to, or identical with, ganglioside GM1. No evidence for galactoproteins with receptor activity. Biochem J; 204(1): 209-219.
Dahl NK, Reed KL, Daunais MA, Faust JR, Liscum L (1992) Isolation and characterization of Chinese hamster ovary cells defective in the intracellular metabolism of low density lipoprotein-derived cholesterol. J Biol Chem; 267(7): 4889-4896.
Danon MJ, Oh SJ, DiMauro S, Manaligod JR, Eastwood A, Naidu S et al. (1981) Lysosomal glycogen storage disease with normal acid maltase. Neurology; 31(1): 51-57.
Davidson CD, Ali NF, Micsenyi MC, Stephney G, Renault S, Dobrenis K et al. (2009) Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One; 4(9): e6951.

REFERENCES
138
de Duve C (1959). Lysosomes, a new group of cytoplasmic particles. Subcellular Particles. H. T. New York, The Ronald Press Co.: 128-159.
de Duve C, de Barsy T, Poole B, Trouet A, Tulkens P, Van Hoof F (1974) Commentary. Lysosomotropic agents. Biochem Pharmacol; 23(18): 2495-2531.
Dean RT (1983) The aspartic proteinase inhibitor pepstatin, enters mouse peritoneal macrophages by adsorptive pinocytosis. Cell Biol Int Rep; 7(6): 405.
Deiss LP, Galinka H, Berissi H, Cohen O, Kimchi A (1996) Cathepsin D protease mediates programmed cell death induced by interferon-gamma, Fas/APO-1 and TNF-alpha. EMBO J; 15(15): 3861-3870.
Deng D, Jiang N, Hao SJ, Sun H, Zhang GJ (2009) Loss of membrane cholesterol influences lysosomal permeability to potassium ions and protons. Biochim Biophys Acta; 1788(2): 470-476.
Deng Y, Ren X, Yang L, Lin Y, Wu X (2003) A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell; 115(1): 61-70.
Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S et al. (1999) Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol; 144(5): 891-901.
Di Stasi D, Vallacchi V, Campi V, Ranzani T, Daniotti M, Chiodini E et al. (2005) DHCR24 gene expression is upregulated in melanoma metastases and associated to resistance to oxidative stress-induced apoptosis. Int J Cancer; 115(2): 224-230.
Dickens LS, Powley IR, Hughes MA, MacFarlane M (2012) The 'complexities' of life and death: death receptor signalling platforms. Exp Cell Res; 318(11): 1269-1277.
Doulias PT, Kotoglou P, Tenopoulou M, Keramisanou D, Tzavaras T, Brunk U et al. (2007) Involvement of heat shock protein-70 in the mechanism of hydrogen peroxide-induced DNA damage: the role of lysosomes and iron. Free Radic Biol Med; 42(4): 567-577.
Droga-Mazovec G, Bojic L, Petelin A, Ivanova S, Romih R, Repnik U et al. (2008) Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J Biol Chem; 283(27): 19140-19150.
Du C, Fang M, Li Y, Li L, Wang X (2000) Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell; 102(1): 33-42.
Dubowchik GM, Gawlak SL, Firestone RA (1995) The in vitro effect of three lysosomotropic detergents against three human tumor cell lines. Bioorg Med Chem Lett; 5(8): 893-898.
Dudeja V, Mujumdar N, Phillips P, Chugh R, Borja-Cacho D, Dawra RK et al. (2009) Heat shock protein 70 inhibits apoptosis in cancer cells through simultaneous and independent mechanisms. Gastroenterology; 136(5): 1772-1782.
Edelmann B, Bertsch U, Tchikov V, Winoto-Morbach S, Perrotta C, Jakob M et al. (2011) Caspase-8 and caspase-7 sequentially mediate proteolytic activation of acid sphingomyelinase in TNF-R1 receptosomes. EMBO J; 30(2): 379-394.

REFERENCES
139
Elrick MJ, Yu T, Chung C, Lieberman AP (2012) Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease. Hum Mol Genet; Epub ahead of print
Emert-Sedlak L, Shangary S, Rabinovitz A, Miranda MB, Delach SM, Johnson DE (2005) Involvement of cathepsin D in chemotherapy-induced cytochrome c release, caspase activation, and cell death. Mol Cancer Ther; 4(5): 733-742.
Erdal H, Berndtsson M, Castro J, Brunk U, Shoshan MC, Linder S (2005) Induction of lysosomal membrane permeabilization by compounds that activate p53-independent apoptosis. Proc Natl Acad Sci U S A; 102(1): 192-197.
Eriksson I, Joosten M, Roberg K, Öllinger K (2012) The histone deacetylase inhibitor trichostatin A reduces lysosomal pH and enhances cisplatin-induced apoptosis. Exp Cell Res; Epub ahead of print
Eskelinen EL, Schmidt CK, Neu S, Willenborg M, Fuertes G, Salvador N et al. (2004) Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol Biol Cell; 15(7): 3132-3145.
Eskes R, Desagher S, Antonsson B, Martinou JC (2000) Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol; 20(3): 929-935.
Faust PL, Kornfeld S, Chirgwin JM (1985) Cloning and sequence analysis of cDNA for human cathepsin D. Proc Natl Acad Sci U S A; 82(15): 4910-4914.
Fayad W, Brnjic S, Berglind D, Blixt S, Shoshan MC, Berndtsson M et al. (2009) Restriction of cisplatin induction of acute apoptosis to a subpopulation of cells in a three-dimensional carcinoma culture model. Int J Cancer; 125(10): 2450-2455.
Fehrenbacher N, Gyrd-Hansen M, Poulsen B, Felbor U, Kallunki T, Boes M et al. (2004) Sensitization to the lysosomal cell death pathway upon immortalization and transformation. Cancer Res; 64(15): 5301-5310.
Fehrenbacher N, Bastholm L, Kirkegaard-Sorensen T, Rafn B, Bottzauw T, Nielsen C et al. (2008) Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res; 68(16): 6623-6633.
Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R et al. (2004) Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology; 40(1): 185-194.
Feldstein AE, Werneburg NW, Li Z, Bronk SF, Gores GJ (2006) Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol Gastrointest Liver Physiol; 290(6): G1339-1346.
Festjens N, Vanden Berghe T, Vandenabeele P (2006) Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim Biophys Acta; 1757(9-10): 1371-1387.
Filocamo M, Morrone A (2011) Lysosomal storage disorders: molecular basis and laboratory testing. Hum Genomics; 5(3): 156-169.

REFERENCES
140
Firestone RA, Pisano JM, Bonney RJ (1979) Lysosomotropic agents. 1. Synthesis and cytotoxic action of lysosomotropic detergents. J Med Chem; 22(9): 1130-1133.
Fishman PH (1982) Role of membrane gangliosides in the binding and action of bacterial toxins. J Membr Biol; 69(2): 85-97.
Foghsgaard L, Wissing D, Mauch D, Lademann U, Bastholm L, Boes M et al. (2001) Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J Cell Biol; 153(5): 999-1010.
Fouchier F, Mego JL, Dang J, Simon C (1983) Thyroid lysosomes: the stability of the lysosomal membrane. Eur J Cell Biol; 30(2): 272-278.
Fra AM, Williamson E, Simons K, Parton RG (1994) Detergent-insoluble glycolipid microdomains in lymphocytes in the absence of caveolae. J Biol Chem; 269(49): 30745-30748.
Frolov A, Zielinski SE, Crowley JR, Dudley-Rucker N, Schaffer JE, Ory DS (2003) NPC1 and NPC2 regulate cellular cholesterol homeostasis through generation of low density lipoprotein cholesterol-derived oxysterols. J Biol Chem; 278(28): 25517-25525.
Fukuda M (1991) Lysosomal membrane glycoproteins. Structure, biosynthesis, and intracellular trafficking. J Biol Chem; 266(32): 21327-21330.
Fulda S (2009) Therapeutic opportunities for counteracting apoptosis resistance in childhood leukaemia. Br J Haematol; 145(4): 441-454.
Fulda S, Vucic D (2012) Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov; 11(2): 109-124.
Funk RS, Krise JP (2012) Cationic amphiphilic drugs cause a marked expansion of apparent lysosomal volume: implications for an intracellular distribution-based drug interaction. Mol Pharm; 9(5): 1384-1395.
Futerman AH, van Meer G (2004) The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol; 5(7): 554-565.
Gallala HD, Sandhoff K (2011) Biological function of the cellular lipid BMP-BMP as a key activator for cholesterol sorting and membrane digestion. Neurochem Res; 36(9): 1594-1600.
Galluzzi L, Joza N, Tasdemir E, Maiuri MC, Hengartner M, Abrams JM et al. (2008) No death without life: vital functions of apoptotic effectors. Cell Death Differ; 15(7): 1113-1123.
Galluzzi L, Vanden Berghe T, Vanlangenakker N, Buettner S, Eisenberg T, Vandenabeele P et al. (2011) Programmed necrosis from molecules to health and disease. Int Rev Cell Mol Biol; 289: 1-35.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al. (2012a) Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ; 19(1): 107-120.
Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G (2012b) Non-apoptotic functions of apoptosis-regulatory proteins. EMBO Rep; 13(4): 322-330.

REFERENCES
141
Gan Y, Zhao X, Hu J, Wang ZG, Zhao XT (2008) HCCS1 overexpression induces apoptosis via cathepsin D and intracellular calcium, and HCCS1 disruption in mice causes placental abnormality. Cell Death Differ; 15(9): 1481-1490.
Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA (1998) Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem; 273(49): 32608-32613.
Gardino AK, Yaffe MB (2011) 14-3-3 proteins as signaling integration points for cell cycle control and apoptosis. Semin Cell Dev Biol; 22(7): 688-695.
Garrido C, Kroemer G (2004) Life's smile, death's grin: vital functions of apoptosis-executing proteins. Curr Opin Cell Biol; 16(6): 639-646.
George KS, Wu S (2012) Lipid raft: A floating island of death or survival. Toxicol Appl Pharmacol; 259(3): 311-319.
Getty AL, Pearce DA (2011) Interactions of the proteins of neuronal ceroid lipofuscinosis: clues to function. Cell Mol Life Sci; 68(3): 453-474.
Ghosh M, Carlsson F, Laskar A, Yuan XM, Li W (2011) Lysosomal membrane permeabilization causes oxidative stress and ferritin induction in macrophages. FEBS Lett; 585(4): 623-629.
Gill S, Chow R, Brown AJ (2008) Sterol regulators of cholesterol homeostasis and beyond: the oxysterol hypothesis revisited and revised. Prog Lipid Res; 47(6): 391-404.
Glaros EN, Kim WS, Garner B (2010) Myriocin-mediated up-regulation of hepatocyte apoA-I synthesis is associated with ERK inhibition. Clin Sci (Lond); 118(12): 727-736.
Gocheva V, Zeng W, Ke D, Klimstra D, Reinheckel T, Peters C et al. (2006) Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev; 20(5): 543-556.
Gonzalvez F, Schug ZT, Houtkooper RH, MacKenzie ED, Brooks DG, Wanders RJ et al. (2008) Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol; 183(4): 681-696.
Gotoh T, Terada K, Oyadomari S, Mori M (2004) hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ; 11(4): 390-402.
Gottlieb RA, Giesing HA, Zhu JY, Engler RL, Babior BM (1995) Cell acidification in apoptosis: granulocyte colony-stimulating factor delays programmed cell death in neutrophils by up-regulating the vacuolar H(+)-ATPase. Proc Natl Acad Sci U S A; 92(13): 5965-5968.
Gould S, Scott RC (2005) 2-Hydroxypropyl-beta-cyclodextrin (HP-beta-CD): a toxicology review. Food Chem Toxicol; 43(10): 1451-1459.
Granger BL, Green SA, Gabel CA, Howe CL, Mellman I, Helenius A (1990) Characterization and cloning of lgp110, a lysosomal membrane glycoprotein from mouse and rat cells. J Biol Chem; 265(20): 12036-12043.
Grassme H, Jekle A, Riehle A, Schwarz H, Berger J, Sandhoff K et al. (2001) CD95 signaling via ceramide-rich membrane rafts. J Biol Chem; 276(23): 20589-20596.

REFERENCES
142
Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C et al. (1999) Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem; 274(2): 1156-1163.
Groth-Pedersen L, Jäättelä M (2010) Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer Lett; Epub ahead of print
Guicciardi ME, Deussing J, Miyoshi H, Bronk SF, Svingen PA, Peters C et al. (2000) Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest; 106(9): 1127-1137.
Guicciardi ME, Leist M, Gores GJ (2004) Lysosomes in cell death. Oncogene; 23(16): 2881-2890.
Guicciardi ME, Bronk SF, Werneburg NW, Gores GJ (2007) cFLIPL prevents TRAIL-induced apoptosis of hepatocellular carcinoma cells by inhibiting the lysosomal pathway of apoptosis. Am J Physiol Gastrointest Liver Physiol; 292(5): G1337-1346.
Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC et al. (2003) Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature; 423(6938): 456-461.
Gurova K (2009) New hopes from old drugs: revisiting DNA-binding small molecules as anticancer agents. Future Oncol; 5(10): 1685-1704.
Gustafsson AB, Tsai JG, Logue SE, Crow MT, Gottlieb RA (2004) Apoptosis repressor with caspase recruitment domain protects against cell death by interfering with Bax activation. J Biol Chem; 279(20): 21233-21238.
Gyrd-Hansen M, Farkas T, Fehrenbacher N, Bastholm L, Hoyer-Hansen M, Elling F et al. (2006) Apoptosome-independent activation of the lysosomal cell death pathway by caspase-9. Mol Cell Biol; 26(21): 7880-7891.
Gyrd-Hansen M, Meier P (2010) IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer; 10(8): 561-574.
Hah YS, Noh HS, Ha JH, Ahn JS, Hahm JR, Cho HY et al. (2012) Cathepsin D inhibits oxidative stress-induced cell death via activation of autophagy in cancer cells. Cancer Lett; 323(2): 208-214.
Haidar B, Kiss RS, Sarov-Blat L, Brunet R, Harder C, McPherson R et al. (2006) Cathepsin D, a lysosomal protease, regulates ABCA1-mediated lipid efflux. J Biol Chem; 281(52): 39971-39981.
Haltia M (2003) The neuronal ceroid-lipofuscinoses. J Neuropathol Exp Neurol; 62(1): 1-13.
Hansson E, Rönnbäck L (1989). Primary cultures of astroglia and neurons from different brain regions. A dissection and tissue culture manual of nervous system. d. Shahar A, A. Vernadakis and B. Haber. New York, Liss: 92-104.
Hao SJ, Hou JF, Jiang N, Zhang GJ (2008) Loss of membrane cholesterol affects lysosomal osmotic stability. Gen Physiol Biophys; 27(4): 278-283.

REFERENCES
143
Harris H, Rubinsztein DC (2012) Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol; 8(2): 108-117.
Havelka AM, Berndtsson M, Olofsson MH, Shoshan MC, Linder S (2007) Mechanisms of action of DNA-damaging anticancer drugs in treatment of carcinomas: is acute apoptosis an "off-target" effect? Mini Rev Med Chem; 7(10): 1035-1039.
Haynes JL (1988) Principles of flow cytometry. Cytometry Suppl; 3: 7-17.
Heinrich M, Wickel M, Schneider-Brachert W, Sandberg C, Gahr J, Schwandner R et al. (1999) Cathepsin D targeted by acid sphingomyelinase-derived ceramide. EMBO J; 18(19): 5252-5263.
Heinrich M, Neumeyer J, Jakob M, Hallas C, Tchikov V, Winoto-Morbach S et al. (2004) Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ; 11(5): 550-563.
Hiraiwa M, Martin BM, Kishimoto Y, Conner GE, Tsuji S, O'Brien JS (1997) Lysosomal proteolysis of prosaposin, the precursor of saposins (sphingolipid activator proteins): its mechanism and inhibition by ganglioside. Arch Biochem Biophys; 341(1): 17-24.
Holopainen JM, Angelova MI, Kinnunen PK (2000) Vectorial budding of vesicles by asymmetrical enzymatic formation of ceramide in giant liposomes. Biophys J; 78(2): 830-838.
Houseweart MK, Vilaythong A, Yin XM, Turk B, Noebels JL, Myers RM (2003) Apoptosis caused by cathepsins does not require Bid signaling in an in vivo model of progressive myoclonus epilepsy (EPM1). Cell Death Differ; 10(12): 1329-1335.
Hsu SY, Kaipia A, McGee E, Lomeli M, Hsueh AJ (1997a) Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members. Proc Natl Acad Sci U S A; 94(23): 12401-12406.
Hsu YT, Wolter KG, Youle RJ (1997b) Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci U S A; 94(8): 3668-3672.
Huang WC, Lin YS, Chen CL, Wang CY, Chiu WH, Lin CF (2009) Glycogen synthase kinase-3beta mediates endoplasmic reticulum stress-induced lysosomal apoptosis in leukemia. J Pharmacol Exp Ther; 329(2): 524-531.
Hullin-Matsuda F, Luquain-Costaz C, Bouvier J, Delton-Vandenbroucke I (2009) Bis(monoacylglycero)phosphate, a peculiar phospholipid to control the fate of cholesterol: Implications in pathology. Prostaglandins Leukot Essent Fatty Acids; 81(5-6): 313-324.
Huotari J, Helenius A (2011) Endosome maturation. EMBO J; 30(17): 3481-3500.
Hurwitz R, Ferlinz K, Sandhoff K (1994) The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Biol Chem Hoppe Seyler; 375(7): 447-450.
Häcker G (2000) The morphology of apoptosis. Cell Tissue Res; 301(1): 5-17.

REFERENCES
144
Idone V, Tam C, Goss JW, Toomre D, Pypaert M, Andrews NW (2008) Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J Cell Biol; 180(5): 905-914.
Ikonen E (2008) Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol; 9(2): 125-138.
Infante RE, Abi-Mosleh L, Radhakrishnan A, Dale JD, Brown MS, Goldstein JL (2008) Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J Biol Chem; 283(2): 1052-1063.
Ishisaka R, Utsumi T, Yabuki M, Kanno T, Furuno T, Inoue M et al. (1998) Activation of caspase-3-like protease by digitonin-treated lysosomes. FEBS Lett; 435(2-3): 233-236.
Ivanova S, Gregorc U, Vidergar N, Javier R, Bredt DS, Vandenabeele P et al. (2011) MAGUKs, scaffolding proteins at cell junctions, are substrates of different proteases during apoptosis. Cell Death Dis; 2: e116.
Jabs S, Quitsch A, Kakela R, Koch B, Tyynela J, Brade H et al. (2008) Accumulation of bis(monoacylglycero)phosphate and gangliosides in mouse models of neuronal ceroid lipofuscinosis. J Neurochem; 106(3): 1415-1425.
Jadot M, Andrianaivo F, Dubois F, Wattiaux R (2001) Effects of methylcyclodextrin on lysosomes. Eur J Biochem; 268(5): 1392-1399.
Jaiswal JK, Andrews NW, Simon SM (2002) Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J Cell Biol; 159(4): 625-635.
Jalanko A, Braulke T (2009) Neuronal ceroid lipofuscinoses. Biochim Biophys Acta; 1793(4): 697-709.
Johansson AC, Steen H, Öllinger K, Roberg K (2003) Cathepsin D mediates cytochrome c release and caspase activation in human fibroblast apoptosis induced by staurosporine. Cell Death Differ; 10(11): 1253-1259.
Johansson AC, Appelqvist H, Nilsson C, Kågedal K, Roberg K, Öllinger K (2010) Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis; 15(5): 527-540.
Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DC et al. (2009) XIAP discriminates between type I and type II FAS-induced apoptosis. Nature; 460(7258): 1035-1039.
Jäättelä M, Benedict M, Tewari M, Shayman JA, Dixit VM (1995) Bcl-x and Bcl-2 inhibit TNF and Fas-induced apoptosis and activation of phospholipase A2 in breast carcinoma cells. Oncogene; 10(12): 2297-2305.
Kallunki T, Olsen OD, Jäättelä M (2012) Cancer-associated lysosomal changes: friends or foes? Oncogene; Epub ahead of print
Kantari C, Walczak H (2011) Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochim Biophys Acta; 1813(4): 558-563.

REFERENCES
145
Katunuma N, Matsui A, Le QT, Utsumi K, Salvesen G, Ohashi A (2001) Novel procaspase-3 activating cascade mediated by lysoapoptases and its biological significances in apoptosis. Adv Enzyme Regul; 41: 237-250.
Ke F, Voss A, Kerr JB, O'Reilly LA, Tai L, Echeverry N et al. (2012) BCL-2 family member BOK is widely expressed but its loss has only minimal impact in mice. Cell Death Differ; 19(6): 915-925.
Kelly GL, Strasser A (2011) The essential role of evasion from cell death in cancer. Adv Cancer Res; 111: 39-96.
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer; 26(4): 239-257.
Khaled AR, Kim K, Hofmeister R, Muegge K, Durum SK (1999) Withdrawal of IL-7 induces Bax translocation from cytosol to mitochondria through a rise in intracellular pH. Proc Natl Acad Sci U S A; 96(25): 14476-14481.
Kirkegaard T, Jäättelä M (2009) Lysosomal involvement in cell death and cancer. Biochim Biophys Acta; 1793(4): 746-754.
Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I et al. (2010) Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature; 463(7280): 549-553.
Kirschke H, Wiederanders B, Bromme D, Rinne A (1989) Cathepsin S from bovine spleen. Purification, distribution, intracellular localization and action on proteins. Biochem J; 264(2): 467-473.
Klingenstein R, Lober S, Kujala P, Godsave S, Leliveld SR, Gmeiner P et al. (2006) Tricyclic antidepressants, quinacrine and a novel, synthetic chimera thereof clear prions by destabilizing detergent-resistant membrane compartments. J Neurochem; 98(3): 748-759.
Kobayashi T, Stang E, Fang KS, de Moerloose P, Parton RG, Gruenberg J (1998) A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature; 392(6672): 193-197.
Kobayashi T, Beuchat MH, Chevallier J, Makino A, Mayran N, Escola JM et al. (2002) Separation and characterization of late endosomal membrane domains. J Biol Chem; 277(35): 32157-32164.
Kock A, Schwarz T, Kirnbauer R, Urbanski A, Perry P, Ansel JC et al. (1990) Human keratinocytes are a source for tumor necrosis factor alpha: evidence for synthesis and release upon stimulation with endotoxin or ultraviolet light. J Exp Med; 172(6): 1609-1614.
Koike M, Nakanishi H, Saftig P, Ezaki J, Isahara K, Ohsawa Y et al. (2000) Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J Neurosci; 20(18): 6898-6906.
Koike M, Shibata M, Ohsawa Y, Nakanishi H, Koga T, Kametaka S et al. (2003) Involvement of two different cell death pathways in retinal atrophy of cathepsin D-deficient mice. Mol Cell Neurosci; 22(2): 146-161.

REFERENCES
146
Kolter T, Proia RL, Sandhoff K (2002) Combinatorial ganglioside biosynthesis. J Biol Chem; 277(29): 25859-25862.
Kolter T, Sandhoff K (2005) Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu Rev Cell Dev Biol; 21: 81-103.
Kolzer M, Werth N, Sandhoff K (2004) Interactions of acid sphingomyelinase and lipid bilayers in the presence of the tricyclic antidepressant desipramine. FEBS Lett; 559(1-3): 96-98.
Kreuzaler PA, Staniszewska AD, Li W, Omidvar N, Kedjouar B, Turkson J et al. (2011) Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol; 13(3): 303-309.
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH et al. (2009) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ; 16(1): 3-11.
Kubota Y, Kubota K, Tani S (2000) DNA binding properties of DAPI (4',6-diamidino-2-phenylindole) analogs having an imidazoline ring or a tetrahydropyrimidine ring: groove-binding and intercalation. Nucleic Acids Symp Ser; 44): 53-54.
Kunimoto S, Aoyagi T, Nishizawa R, Komai T, Takeuchi T (1974) Mechanism of inhibition of pepsin by pepstatin. II. J Antibiot (Tokyo); 27(6): 413-418.
Kurz T, Brunk UT (2009) Autophagy of HSP70 and chelation of lysosomal iron in a non-redox-active form. Autophagy; 5(1): 93-95.
Kurz T, Eaton JW, Brunk UT (2011) The role of lysosomes in iron metabolism and recycling. Int J Biochem Cell Biol; 43(12): 1686-1697.
Kuzelova K, Grebenova D, Pluskalova M, Kavan D, Halada P, Hrkal Z (2009) Isoform-specific cleavage of 14-3-3 proteins in apoptotic JURL-MK1 cells. J Cell Biochem; 106(4): 673-681.
Kvansakul M, Yang H, Fairlie WD, Czabotar PE, Fischer SF, Perugini MA et al. (2008) Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ; 15(10): 1564-1571.
Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS et al. (2009) Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell; 137(7): 1213-1224.
Kågedal K, Johansson U, Öllinger K (2001a) The lysosomal protease cathepsin D mediates apoptosis induced by oxidative stress. FASEB J; 15(9): 1592-1594.
Kågedal K, Zhao M, Svensson I, Brunk UT (2001b) Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem J; 359(Pt 2): 335-343.
Kågedal K, Johansson AC, Johansson U, Heimlich G, Roberg K, Wang NS et al. (2005) Lysosomal membrane permeabilization during apoptosis - involvement of Bax? Int J Exp Pathol; 86(5): 309-321.

REFERENCES
147
Laforge M, Petit F, Estaquier J, Senik A (2007) Commitment to apoptosis in CD4(+) T lymphocytes productively infected with human immunodeficiency virus type 1 is initiated by lysosomal membrane permeabilization, itself induced by the isolated expression of the viral protein Nef. J Virol; 81(20): 11426-11440.
Lange Y, Ye J, Steck TL (1998) Circulation of cholesterol between lysosomes and the plasma membrane. J Biol Chem; 273(30): 18915-18922.
Larsson P, Andersson E, Johansson U, Öllinger K, Rosdahl I (2005) Ultraviolet A and B affect human melanocytes and keratinocytes differently. A study of oxidative alterations and apoptosis. Exp Dermatol; 14(2): 117-123.
Leber B, Geng F, Kale J, Andrews DW (2010) Drugs targeting Bcl-2 family members as an emerging strategy in cancer. Expert Rev Mol Med; 12:e28.
Lee J, Giordano S, Zhang J (2012) Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J; 441(2): 523-540.
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM et al. (2010) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell; 141(7): 1146-1158.
Lee KH, Feig C, Tchikov V, Schickel R, Hallas C, Schütze S et al. (2006) The role of receptor internalization in CD95 signaling. EMBO J; 25(5): 1009-1023.
Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell; 2(3): 183-192.
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell; 132(1): 27-42.
Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell; 94(4): 491-501.
Li LY, Luo X, Wang X (2001) Endonuclease G is an apoptotic DNase when released from mitochondria. Nature; 412(6842): 95-99.
Li N, Zheng Y, Chen W, Wang C, Liu X, He W et al. (2007) Adaptor protein LAPF recruits phosphorylated p53 to lysosomes and triggers lysosomal destabilization in apoptosis. Cancer Res; 67(23): 11176-11185.
Li W, Yuan X, Nordgren G, Dalen H, Dubowchik GM, Firestone RA et al. (2000) Induction of cell death by the lysosomotropic detergent MSDH. FEBS Lett; 470(1): 35-39.
Li WW, Li J, Bao JK (2012) Microautophagy: lesser-known self-eating. Cell Mol Life Sci; 69(7): 1125-1136.
Liaudet-Coopman E, Beaujouin M, Derocq D, Garcia M, Glondu-Lassis M, Laurent-Matha V et al. (2006) Cathepsin D: newly discovered functions of a long-standing aspartic protease in cancer and apoptosis. Cancer Lett; 237(2): 167-179.
Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA et al. (2000) The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell; 6(6): 1389-1399.

REFERENCES
148
Lippens S, Hoste E, Vandenabeele P, Agostinis P, Declercq W (2009) Cell death in the skin. Apoptosis; 14(4): 549-569.
Liscum L, Faust JR (1989) The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-beta-[2-(diethylamino)ethoxy]androst-5-en-17-one. J Biol Chem; 264(20): 11796-11806.
Liscum L (1990) Pharmacological inhibition of the intracellular transport of low-density lipoprotein-derived cholesterol in Chinese hamster ovary cells. Biochim Biophys Acta; 1045(1): 40-48.
Liscum L, Munn NJ (1999) Intracellular cholesterol transport. Biochim Biophys Acta; 1438(1): 19-37.
Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL et al. (2011) A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell; 44(4): 517-531.
Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ et al. (2008) Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med; 14(11): 1247-1255.
Lucken-Ardjomande S, Montessuit S, Martinou JC (2008) Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death Differ; 15(3): 484-493.
Lucken-Ardjomande S (2012) Cholesterol, cardiolipin, and mitochondria permeabilisation. Anticancer Agents Med Chem; 12(4): 329-339.
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell; 94(4): 481-490.
Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X (2000) Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol; 2(10): 754-761.
Lübke T, Lobel P, Sleat DE (2009) Proteomics of the lysosome. Biochim Biophys Acta; 1793(4): 625-635.
Maas C, de Vries E, Tait SW, Borst J (2011) Bid can mediate a pro-apoptotic response to etoposide and ionizing radiation without cleavage in its unstructured loop and in the absence of p53. Oncogene; 30(33): 3636-3647.
Mandic A, Viktorsson K, Strandberg L, Heiden T, Hansson J, Linder S et al. (2002) Calpain-mediated Bid cleavage and calpain-independent Bak modulation: two separate pathways in cisplatin-induced apoptosis. Mol Cell Biol; 22(9): 3003-3013.
Mandic A, Hansson J, Linder S, Shoshan MC (2003) Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem; 278(11): 9100-9106.
Marrot L, Meunier JR (2008) Skin DNA photodamage and its biological consequences. J Am Acad Dermatol; 58(5 Suppl 2): S139-148.

REFERENCES
149
Martinez-Caballero S, Dejean LM, Kinnally MS, Oh KJ, Mannella CA, Kinnally KW (2009) Assembly of the mitochondrial apoptosis-induced channel, MAC. J Biol Chem; 284(18): 12235-12245.
Martinou JC, Youle RJ (2011) Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell; 21(1): 92-101.
Masson O, Bach AS, Derocq D, Prebois C, Laurent-Matha V, Pattingre S et al. (2010) Pathophysiological functions of cathepsin D: Targeting its catalytic activity versus its protein binding activity? Biochimie; 92(11): 1635-1643.
Matsuyama S, Llopis J, Deveraux QL, Tsien RY, Reed JC (2000) Changes in intramitochondrial and cytosolic pH: early events that modulate caspase activation during apoptosis. Nat Cell Biol; 2(6): 318-325.
Matsuyama S, Reed JC (2000) Mitochondria-dependent apoptosis and cellular pH regulation. Cell Death Differ; 7(12): 1155-1165.
Matsuzawa Y, Hostetler KY (1979) Degradation of bis(monoacylglycero)phosphate by an acid phosphodiesterase in rat liver lysosomes. J Biol Chem; 254(13): 5997-6001.
Maxfield FR, Wustner D (2002) Intracellular cholesterol transport. J Clin Invest; 110(7): 891-898.
McCall K (2010) Genetic control of necrosis - another type of programmed cell death. Curr Opin Cell Biol; 22(6): 882-888.
McCauliff LA, Xu Z, Storch J (2011) Sterol transfer between cyclodextrin and membranes: similar but not identical mechanism to NPC2-mediated cholesterol transfer. Biochemistry (Mosc); 50(34): 7341-7349.
McDonnell JM, Fushman D, Milliman CL, Korsmeyer SJ, Cowburn D (1999) Solution structure of the proapoptotic molecule BID: a structural basis for apoptotic agonists and antagonists. Cell; 96(5): 625-634.
McGowan EB, Becker E, Detwiler TC (1989) Inhibition of calpain in intact platelets by the thiol protease inhibitor E-64d. Biochem Biophys Res Commun; 158(2): 432-435.
McNeil PL, Steinhardt RA (1997) Loss, restoration, and maintenance of plasma membrane integrity. J Cell Biol; 137(1): 1-4.
McNeil PL (2002) Repairing a torn cell surface: make way, lysosomes to the rescue. J Cell Sci; 115(Pt 5): 873-879.
Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C et al. (2011) Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell; 21(3): 421-430.
Melzer IM, Fernandez SB, Bosser S, Lohrig K, Lewandrowski U, Wolters D et al. (2012) The Apaf-1-binding protein Aven is cleaved by Cathepsin D to unleash its anti-apoptotic potential. Cell Death Differ; 19(9): 1435-1445.
Metcalf P, Fusek M (1993) Two crystal structures for cathepsin D: the lysosomal targeting signal and active site. EMBO J; 12(4): 1293-1302.

REFERENCES
150
Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P et al. (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell; 11(3): 577-590.
Milhas D, Cuvillier O, Therville N, Clave P, Thomsen M, Levade T et al. (2005) Caspase-10 triggers Bid cleavage and caspase cascade activation in FasL-induced apoptosis. J Biol Chem; 280(20): 19836-19842.
Miller DK, Griffiths E, Lenard J, Firestone RA (1983) Cell killing by lysosomotropic detergents. J Cell Biol; 97(6): 1841-1851.
Mindell JA (2012) Lysosomal acidification mechanisms. Annu Rev Physiol; 74: 69-86.
Miyake Y, Kozutsumi Y, Nakamura S, Fujita T, Kawasaki T (1995) Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochem Biophys Res Commun; 211(2): 396-403.
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell; 147(4): 728-741.
Moffitt KL, Martin SL, Walker B (2010) From sentencing to execution--the processes of apoptosis. J Pharm Pharmacol; 62(5): 547-562.
Morishima H, Takita T, Aoyagi T, Takeuchi T, Umezawa H (1970) The structure of pepstatin. J Antibiot (Tokyo); 23(5): 263-265.
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods; 65(1-2): 55-63.
Mu FT, Callaghan JM, Steele-Mortimer O, Stenmark H, Parton RG, Campbell PL et al. (1995) EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine "fingers" and contains a calmodulin-binding IQ motif. J Biol Chem; 270(22): 13503-13511.
Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS et al. (1996) X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature; 381(6580): 335-341.
Myllykangas L, Tyynelä J, Page-McCaw A, Rubin GM, Haltia MJ, Feany MB (2005) Cathepsin D-deficient Drosophila recapitulate the key features of neuronal ceroid lipofuscinoses. Neurobiol Dis; 19(1-2): 194-199.
Möbius W, van Donselaar E, Ohno-Iwashita Y, Shimada Y, Heijnen HF, Slot JW et al. (2003) Recycling compartments and the internal vesicles of multivesicular bodies harbor most of the cholesterol found in the endocytic pathway. Traffic; 4(4): 222-231.
Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H et al. (1998) Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc Natl Acad Sci U S A; 95(25): 14681-14686.
Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R et al. (2000) Identification of HE1 as the second gene of Niemann-Pick C disease. Science; 290(5500): 2298-2301.

REFERENCES
151
Nicholson DW (2000) From bench to clinic with apoptosis-based therapeutic agents. Nature; 407(6805): 810-816.
Nilsson C, Kågedal K, Johansson U, Öllinger K (2003) Analysis of cytosolic and lysosomal pH in apoptotic cells by flow cytometry. Methods Cell Sci; 25(3-4): 185-194.
Nilsson C, Johansson U, Johansson AC, Kågedal K, Öllinger K (2006) Cytosolic acidification and lysosomal alkalinization during TNF-alpha induced apoptosis in U937 cells. Apoptosis; 11(7): 1149-1159.
Nilsson C, Roberg K, Grafström RC, Öllinger K (2010) Intrinsic differences in cisplatin sensitivity of head and neck cancer cell lines: Correlation to lysosomal pH. Head Neck; 32(9): 1185-1194.
Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T et al. (2000) Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature; 406(6798): 906-910.
Nixon RA (2004) Niemann-Pick Type C disease and Alzheimer's disease: the APP-endosome connection fattens up. Am J Pathol; 164(3): 757-761.
Nixon RA, Yang DS, Lee JH (2008) Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy; 4(5): 590-599.
Nixon RA, Yang DS (2011) Autophagy failure in Alzheimer's disease--locating the primary defect. Neurobiol Dis; 43(1): 38-45.
Nomura M, Shimizu S, Sugiyama T, Narita M, Ito T, Matsuda H et al. (2003) 14-3-3 Interacts directly with and negatively regulates pro-apoptotic Bax. J Biol Chem; 278(3): 2058-2065.
Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Hoyer-Hansen M et al. (2004) Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med; 200(4): 425-435.
Obsil T, Obsilova V (2011) Structural basis of 14-3-3 protein functions. Semin Cell Dev Biol; 22(7): 663-672.
Olsson GM, Rungby J, Rundquist I, Brunk UT (1989) Evaluation of lysosomal stability in living cultured macrophages by cytofluorometry. Effect of silver lactate and hypotonic conditions. Virchows Arch B Cell Pathol Incl Mol Pathol; 56(4): 263-269.
Oltvai ZN, Milliman CL, Korsmeyer SJ (1993) Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell; 74(4): 609-619.
Omura S, Iwai Y, Hirano A, Nakagawa A, Awaya J, Tsuchya H et al. (1977) A new alkaloid AM-2282 OF Streptomyces origin. Taxonomy, fermentation, isolation and preliminary characterization. J Antibiot (Tokyo); 30(4): 275-282.
Ono K, Kim SO, Han J (2003) Susceptibility of lysosomes to rupture is a determinant for plasma membrane disruption in tumor necrosis factor alpha-induced cell death. Mol Cell Biol; 23(2): 665-676.

REFERENCES
152
Palermo C, Joyce JA (2008) Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol Sci; 29(1): 22-28.
Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M et al. (2011) Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet; 20(19): 3852-3866.
Paris C, Bertoglio J, Breard J (2007) Lysosomal and mitochondrial pathways in miltefosine-induced apoptosis in U937 cells. Apoptosis; 12(7): 1257-1267.
Parton RG (1994) Ultrastructural localization of gangliosides; GM1 is concentrated in caveolae. J Histochem Cytochem; 42(2): 155-166.
Pelletier RM, Vitale ML (1994) Filipin vs enzymatic localization of cholesterol in guinea pig, mink, and mallard duck testicular cells. J Histochem Cytochem; 42(12): 1539-1554.
Penaloza C, Lin L, Lockshin RA, Zakeri Z (2006) Cell death in development: shaping the embryo. Histochem Cell Biol; 126(2): 149-158.
Persson HL, Yu Z, Tirosh O, Eaton JW, Brunk UT (2003) Prevention of oxidant-induced cell death by lysosomotropic iron chelators. Free Radic Biol Med; 34(10): 1295-1305.
Petersen NH, Kirkegaard T, Olsen OD, Jäättelä M (2010) Connecting Hsp70, sphingolipid metabolism and lysosomal stability. Cell Cycle; 9(12): 2305-2309.
Pfeifer GP, Besaratinia A (2012) UV wavelength-dependent DNA damage and human non-melanoma and melanoma skin cancer. Photochem Photobiol Sci; 11(1): 90-97.
Pike LJ (2009) The challenge of lipid rafts. J Lipid Res; 50 Suppl: S323-328.
Pop C, Salvesen GS (2009) Human caspases: activation, specificity, and regulation. J Biol Chem; 284(33): 21777-21781.
Prade L, Engh RA, Girod A, Kinzel V, Huber R, Bossemeyer D (1997) Staurosporine-induced conformational changes of cAMP-dependent protein kinase catalytic subunit explain inhibitory potential. Structure; 5(12): 1627-1637.
Pustisek N, Situm M (2011) UV-radiation, apoptosis and skin. Coll Antropol; 35 Suppl 2: 339-341.
Ramirez CM, Liu B, Aqul A, Taylor AM, Repa JJ, Turley SD et al. (2011) Quantitative role of LAL, NPC2, and NPC1 in lysosomal cholesterol processing defined by genetic and pharmacological manipulations. J Lipid Res; 52(4): 688-698.
Rao SK, Huynh C, Proux-Gillardeaux V, Galli T, Andrews NW (2004) Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J Biol Chem; 279(19): 20471-20479.
Rawlings ND, Barrett AJ, Bateman A (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res; 40(Database issue): D343-350.
Reddy A, Caler EV, Andrews NW (2001) Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell; 106(2): 157-169.
Reeves VL, Thomas CM, Smart EJ (2012) Lipid rafts, caveolae and GPI-linked proteins. Adv Exp Med Biol; 729: 3-13.

REFERENCES
153
Rehemtulla A, Hamilton CA, Chinnaiyan AM, Dixit VM (1997) Ultraviolet radiation-induced apoptosis is mediated by activation of CD-95 (Fas/APO-1). J Biol Chem; 272(41): 25783-25786.
Reid PC, Lin S, Vanier MT, Ohno-Iwashita Y, Harwood HJ, Jr., Hickey WF et al. (2008) Partial blockage of sterol biosynthesis with a squalene synthase inhibitor in early postnatal Niemann-Pick type C npcnih null mice brains reduces neuronal cholesterol accumulation, abrogates astrogliosis, but may inhibit myelin maturation. J Neurosci Methods; 168(1): 15-25.
Reinhardt HC, Schumacher B (2012) The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet; 28(3): 128-136.
Reiser J, Adair B, Reinheckel T (2010) Specialized roles for cysteine cathepsins in health and disease. J Clin Invest; 120(10): 3421-3431.
Repnik U, Stoka V, Turk V, Turk B (2012) Lysosomes and lysosomal cathepsins in cell death. Biochim Biophys Acta; 1824(1): 22-33.
Rink TJ, Tsien RY, Pozzan T (1982) Cytoplasmic pH and free Mg2+ in lymphocytes. J Cell Biol; 95(1): 189-196.
Roberg K, Öllinger K (1998) Oxidative stress causes relocation of the lysosomal enzyme cathepsin D with ensuing apoptosis in neonatal rat cardiomyocytes. Am J Pathol; 152(5): 1151-1156.
Roberg K, Johansson U, Öllinger K (1999) Lysosomal release of cathepsin D precedes relocation of cytochrome c and loss of mitochondrial transmembrane potential during apoptosis induced by oxidative stress. Free Radic Biol Med; 27(11-12): 1228-1237.
Roberg K (2001) Relocalization of cathepsin D and cytochrome c early in apoptosis revealed by immunoelectron microscopy. Lab Invest; 81(2): 149-158.
Roberg K, Kågedal K, Öllinger K (2002) Microinjection of cathepsin D induces caspase-dependent apoptosis in fibroblasts. Am J Pathol; 161(1): 89-96.
Rodriguez A, Webster P, Ortego J, Andrews NW (1997) Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol; 137(1): 93-104.
Rosenbaum AI, Zhang G, Warren JD, Maxfield FR (2010) Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc Natl Acad Sci U S A; 107(12): 5477-5482.
Rossi A, Deveraux Q, Turk B, Sali A (2004) Comprehensive search for cysteine cathepsins in the human genome. Biol Chem; 385(5): 363-372.
Sachse M, Ramm G, Strous G, Klumperman J (2002) Endosomes: multipurpose designs for integrating housekeeping and specialized tasks. Histochem Cell Biol; 117(2): 91-104.
Saftig P, Hetman M, Schmahl W, Weber K, Heine L, Mossmann H et al. (1995) Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO J; 14(15): 3599-3608.

REFERENCES
154
Saftig P, Klumperman J (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol; 10(9): 623-635.
Saftig P, Schröder B, Blanz J (2010) Lysosomal membrane proteins: life between acid and neutral conditions. Biochem Soc Trans; 38(6): 1420-1423.
Sagulenko V, Muth D, Sagulenko E, Paffhausen T, Schwab M, Westermann F (2008) Cathepsin D protects human neuroblastoma cells from doxorubicin-induced cell death. Carcinogenesis; 29(10): 1869-1877.
Samuel T, Weber HO, Rauch P, Verdoodt B, Eppel JT, McShea A et al. (2001) The G2/M regulator 14-3-3sigma prevents apoptosis through sequestration of Bax. J Biol Chem; 276(48): 45201-45206.
Sanchez J, Holmgren J (2011) Cholera toxin - a foe & a friend. Indian J Med Res; 133: 153-163.
Santin G, Scietti L, Veneroni P, Barni S, Bernocchi G, Bottone MG (2012) Effects of Cisplatin in neuroblastoma rat cells: damage to cellular organelles. Int J Cell Biol; Epub ahead of print
Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA et al. (2009) A gene network regulating lysosomal biogenesis and function. Science; 325(5939): 473-477.
Sarig R, Zaltsman Y, Marcellus RC, Flavell R, Mak TW, Gross A (2003) BID-D59A is a potent inducer of apoptosis in primary embryonic fibroblasts. J Biol Chem; 278(12): 10707-10715.
Sawada M, Sun W, Hayes P, Leskov K, Boothman DA, Matsuyama S (2003) Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat Cell Biol; 5(4): 320-329.
Schestkowa O, Geisel D, Jacob R, Hasilik A (2007) The catalytically inactive precursor of cathepsin D induces apoptosis in human fibroblasts and HeLa cells. J Cell Biochem; 101(6): 1558-1566.
Schlesinger PH, Gross A, Yin XM, Yamamoto K, Saito M, Waksman G et al. (1997) Comparison of the ion channel characteristics of proapoptotic BAX and antiapoptotic BCL-2. Proc Natl Acad Sci U S A; 94(21): 11357-11362.
Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J et al. (2004) Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity; 21(3): 415-428.
Schröder BA, Wrocklage C, Hasilik A, Saftig P (2010) The proteome of lysosomes. Proteomics; 10(22): 4053-4076.
Schuchman EH (2009) The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. Int J Clin Pharmacol Ther; 47 Suppl 1: S48-57.
Schultz ML, Tecedor L, Chang M, Davidson BL (2011) Clarifying lysosomal storage diseases. Trends Neurosci; 34(8): 401-410.
Schulze H, Kolter T, Sandhoff K (2009) Principles of lysosomal membrane degradation: Cellular topology and biochemistry of lysosomal lipid degradation. Biochim Biophys Acta; 1793(4): 674-683.

REFERENCES
155
Seglen PO, Gordon PB (1982) 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A; 79(6): 1889-1892.
Segui B, Legembre P (2010) Redistribution of CD95 into the lipid rafts to treat cancer cells? Recent Pat Anticancer Drug Discov; 5(1): 22-28.
Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S et al. (2011) TFEB links autophagy to lysosomal biogenesis. Science; 332(6036): 1429-1433.
Severs NJ (1982) Cholesterol distribution and structural differentiation in the sarcoplasmic reticulum of rat cardiac muscle cells. A freeze-fracture cytochemical investigation. Cell Tissue Res; 224(3): 613-624.
Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW (2011) BH3-only proteins: Orchestrators of apoptosis. Biochim Biophys Acta; 1813(4): 508-520.
Sheikh MS, Antinore MJ, Huang Y, Fornace AJ, Jr. (1998) Ultraviolet-irradiation-induced apoptosis is mediated via ligand independent activation of tumor necrosis factor receptor 1. Oncogene; 17(20): 2555-2563.
Shen S, Kepp O, Kroemer G (2012) The end of autophagic cell death? Autophagy; 8(1): 1-3.
Siddik ZH (2003) Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene; 22(47): 7265-7279.
Siintola E, Partanen S, Stromme P, Haapanen A, Haltia M, Maehlen J et al. (2006) Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain; 129(Pt 6): 1438-1445.
Simons K, Ikonen E (2000) How cells handle cholesterol. Science; 290(5497): 1721-1726.
Simons K, Gruenberg J (2000) Jamming the endosomal system: lipid rafts and lysosomal storage diseases. Trends Cell Biol; 10(11): 459-462.
Slee EA, Keogh SA, Martin SJ (2000) Cleavage of BID during cytotoxic drug and UV radiation-induced apoptosis occurs downstream of the point of Bcl-2 action and is catalysed by caspase-3: a potential feedback loop for amplification of apoptosis-associated mitochondrial cytochrome c release. Cell Death Differ; 7(6): 556-565.
Sobo K, Le Blanc I, Luyet PP, Fivaz M, Ferguson C, Parton RG et al. (2007) Late endosomal cholesterol accumulation leads to impaired intra-endosomal trafficking. PLoS One; 2(9): e851.
Sousa MJ, Azevedo F, Pedras A, Marques C, Coutinho OP, Preto A et al. (2011) Vacuole-mitochondrial cross-talk during apoptosis in yeast: a model for understanding lysosome-mitochondria-mediated apoptosis in mammals. Biochem Soc Trans; 39(5): 1533-1537.
Sprick MR, Rieser E, Stahl H, Grosse-Wilde A, Weigand MA, Walczak H (2002) Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signalling complexes in a FADD-dependent manner but can not functionally substitute caspase-8. EMBO J; 21(17): 4520-4530.

REFERENCES
156
Stankiewicz AR, Lachapelle G, Foo CP, Radicioni SM, Mosser DD (2005) Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem; 280(46): 38729-38739.
Steer CJ, Bisher M, Blumenthal R, Steven AC (1984) Detection of membrane cholesterol by filipin in isolated rat liver coated vesicles is dependent upon removal of the clathrin coat. J Cell Biol; 99(1 Pt 1): 315-319.
Steinfeld R, Reinhardt K, Schreiber K, Hillebrand M, Kraetzner R, Bruck W et al. (2006) Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am J Hum Genet; 78(6): 988-998.
Steinman RM, Mellman IS, Muller WA, Cohn ZA (1983) Endocytosis and the recycling of plasma membrane. J Cell Biol; 96(1): 1-27.
Stockert JC, Blazquez-Castro A, Canete M, Horobin RW, Villanueva A (2012) MTT assay for cell viability: Intracellular localization of the formazan product is in lipid droplets. Acta Histochem; 114(8): 785-796.
Stoka V, Turk B, Schendel SL, Kim TH, Cirman T, Snipas SJ et al. (2001) Lysosomal protease pathways to apoptosis. Cleavage of bid, not pro-caspases, is the most likely route. J Biol Chem; 276(5): 3149-3157.
Subramanian RR, Masters SC, Zhang H, Fu H (2001) Functional conservation of 14-3-3 isoforms in inhibiting bad-induced apoptosis. Exp Cell Res; 271(1): 142-151.
Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A et al. (1996) Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med; 184(4): 1331-1341.
Sutheesophon K, Kobayashi Y, Takatoku MA, Ozawa K, Kano Y, Ishii H et al. (2006) Histone deacetylase inhibitor depsipeptide (FK228) induces apoptosis in leukemic cells by facilitating mitochondrial translocation of Bax, which is enhanced by the proteasome inhibitor bortezomib. Acta Haematol; 115(1-2): 78-90.
Suzuki M, Youle RJ, Tjandra N (2000) Structure of Bax: coregulation of dimer formation and intracellular localization. Cell; 103(4): 645-654.
Tafani M, Cohn JA, Karpinich NO, Rothman RJ, Russo MA, Farber JL (2002) Regulation of intracellular pH mediates Bax activation in HeLa cells treated with staurosporine or tumor necrosis factor-alpha. J Biol Chem; 277(51): 49569-49576.
Taha TA, Kitatani K, Bielawski J, Cho W, Hannun YA, Obeid LM (2005) Tumor Necrosis Factor Induces the Loss of Sphingosine Kinase-1 by a Cathepsin B-dependent Mechanism. J Biol Chem; 280(17): 17196-17202.
Tait SW, Green DR (2010) Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol; 11(9): 621-632.
Tam C, Idone V, Devlin C, Fernandes MC, Flannery A, He X et al. (2010) Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J Cell Biol; 189(6): 1027-1038.

REFERENCES
157
Tamai M, Matsumoto K, Omura S, Koyama I, Ozawa Y, Hanada K (1986) In vitro and in vivo inhibition of cysteine proteinases by EST, a new analog of E-64. J Pharmacobiodyn; 9(8): 672-677.
Tan KO, Tan KM, Yu VC (1999) A novel BH3-like domain in BID is required for intramolecular interaction and autoinhibition of pro-apoptotic activity. J Biol Chem; 274(34): 23687-23690.
Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R et al. (2000) Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature; 406(6798): 902-906.
Tanida I, Ueno T, Kominami E (2008) LC3 and Autophagy. Methods Mol Biol; 445: 77-88.
Tchikov V, Bertsch U, Fritsch J, Edelmann B, Schütze S (2011) Subcellular compartmentalization of TNF receptor-1 and CD95 signaling pathways. Eur J Cell Biol; 90(6-7): 467-475.
Terman A, Neuzil J, Kågedal K, Öllinger K, Brunk UT (2002) Decreased apoptotic response of inclusion-cell disease fibroblasts: a consequence of lysosomal enzyme missorting? Exp Cell Res; 274(1): 9-15.
Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M et al. (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem; 272(29): 17907-17911.
Todd RC, Lippard SJ (2009) Inhibition of transcription by platinum antitumor compounds. Metallomics; 1(4): 280-291.
Trincheri NF, Nicotra G, Follo C, Castino R, Isidoro C (2007) Resveratrol induces cell death in colorectal cancer cells by a novel pathway involving lysosomal cathepsin D. Carcinogenesis; 28(5): 922-931.
Turk B, Bieth JG, Bjork I, Dolenc I, Turk D, Cimerman N et al. (1995) Regulation of the activity of lysosomal cysteine proteinases by pH-induced inactivation and/or endogenous protein inhibitors, cystatins. Biol Chem Hoppe Seyler; 376(4): 225-230.
Turk B, Turk V (2009) Lysosomes as "suicide bags" in cell death: myth or reality? J Biol Chem; 284(33): 21783-21787.
Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B et al. (2012) Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim Biophys Acta; 1824(1): 68-88.
Tyynelä J, Sohar I, Sleat DE, Gin RM, Donnelly RJ, Baumann M et al. (2000) A mutation in the ovine cathepsin D gene causes a congenital lysosomal storage disease with profound neurodegeneration. EMBO J; 19(12): 2786-2792.
Ullio C, Casas J, Brunk UT, Sala G, Fabrias G, Ghidoni R et al. (2012) Sphingosine mediates TNFalpha-induced lysosomal membrane permeabilization and ensuing programmed cell death in hepatoma cells. J Lipid Res; 53(6): 1134-1143.

REFERENCES
158
Umezawa H, Aoyagi T, Morishima H, Matsuzaki M, Hamada M (1970) Pepstatin, a new pepsin inhibitor produced by Actinomycetes. J Antibiot (Tokyo); 23(5): 259-262.
Underwood KW, Andemariam B, McWilliams GL, Liscum L (1996) Quantitative analysis of hydrophobic amine inhibition of intracellular cholesterol transport. J Lipid Res; 37(7): 1556-1568.
Wallace DJ (1989) The use of quinacrine (Atabrine) in rheumatic diseases: a reexamination. Semin Arthritis Rheum; 18(4): 282-296.
van Nierop K, Muller FJ, Stap J, Van Noorden CJ, van Eijk M, de Groot C (2006) Lysosomal destabilization contributes to apoptosis of germinal center B-lymphocytes. J Histochem Cytochem; 54(12): 1425-1435.
van Raam BJ, Salvesen GS (2012) Proliferative versus apoptotic functions of caspase-8 Hetero or homo: the caspase-8 dimer controls cell fate. Biochim Biophys Acta; 1824(1): 113-122.
Vance JE, Peake KB (2011) Function of the Niemann-Pick type C proteins and their bypass by cyclodextrin. Curr Opin Lipidol; 22(3): 204-209.
Vancompernolle K, Van Herreweghe F, Pynaert G, Van de Craen M, De Vos K, Totty N et al. (1998) Atractyloside-induced release of cathepsin B, a protease with caspase-processing activity. FEBS Lett; 438(3): 150-158.
Vanden Broeck D, Horvath C, De Wolf MJ (2007) Vibrio cholerae: cholera toxin. Int J Biochem Cell Biol; 39(10): 1771-1775.
Wang JW, Sun L, Hu JS, Li YB, Zhang GJ (2006) Effects of phospholipase A2 on the lysosomal ion permeability and osmotic sensitivity. Chem Phys Lipids; 144(2): 117-126.
Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ (1996) BID: a novel BH3 domain-only death agonist. Genes Dev; 10(22): 2859-2869.
Vanier MT, Duthel S, Rodriguez-Lafrasse C, Pentchev P, Carstea ED (1996) Genetic heterogeneity in Niemann-Pick C disease: a study using somatic cell hybridization and linkage analysis. Am J Hum Genet; 58(1): 118-125.
Vanier MT (2010) Niemann-Pick disease type C. Orphanet J Rare Dis; 5(16): 1-18.
Vasiljeva O, Turk B (2008) Dual contrasting roles of cysteine cathepsins in cancer progression: apoptosis versus tumour invasion. Biochimie; 90(2): 380-386.
Waterhouse NJ, Finucane DM, Green DR, Elce JS, Kumar S, Alnemri ES et al. (1998) Calpain activation is upstream of caspases in radiation-induced apoptosis. Cell Death Differ; 5(12): 1051-1061.
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ et al. (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science; 292(5517): 727-730.
Verhagen AM, Silke J, Ekert PG, Pakusch M, Kaufmann H, Connolly LM et al. (2002) HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J Biol Chem; 277(1): 445-454.

REFERENCES
159
Werneburg NW, Guicciardi ME, Bronk SF, Gores GJ (2002) Tumor necrosis factor-alpha-associated lysosomal permeabilization is cathepsin B dependent. Am J Physiol Gastrointest Liver Physiol; 283(4): G947-956.
Werneburg NW, Guicciardi ME, Yin XM, Gores GJ (2004) TNF-alpha-mediated lysosomal permeabilization is FAN and caspase 8/Bid dependent. Am J Physiol Gastrointest Liver Physiol; 287(2): G436-443.
Werneburg NW, Guicciardi ME, Bronk SF, Kaufmann SH, Gores GJ (2007) Tumor necrosis factor-related apoptosis-inducing ligand activates a lysosomal pathway of apoptosis that is regulated by Bcl-2 proteins. J Biol Chem; 282(39): 28960-28970.
Westphal D, Dewson G, Czabotar PE, Kluck RM (2011) Molecular biology of Bax and Bak activation and action. Biochim Biophys Acta; 1813(4): 521-531.
Vogel S, Raulf N, Bregenhorn S, Biniossek ML, Maurer U, Czabotar P et al. (2012) Cytosolic Bax: does it require binding proteins to keep its pro-apoptotic activity in check? J Biol Chem; 287(12): 9112-9127.
Wojtanik KM, Liscum L (2003) The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J Biol Chem; 278(17): 14850-14856.
Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ (1997) Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol; 139(5): 1281-1292.
von Arnim CA, von Einem B, Weber P, Wagner M, Schwanzar D, Spoelgen R et al. (2008) Impact of cholesterol level upon APP and BACE proximity and APP cleavage. Biochem Biophys Res Commun; 370(2): 207-212.
Won J, Kim DY, La M, Kim D, Meadows GG, Joe CO (2003) Cleavage of 14-3-3 protein by caspase-3 facilitates bad interaction with Bcl-x(L) during apoptosis. J Biol Chem; 278(21): 19347-19351.
Woodman PG, Futter CE (2008) Multivesicular bodies: co-ordinated progression to maturity. Curr Opin Cell Biol; 20(4): 408-414.
Würstle ML, Laussmann MA, Rehm M (2012) The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Exp Cell Res; 318(11): 1213-1220.
Wäster PK, Öllinger KM (2009) Redox-dependent translocation of p53 to mitochondria or nucleus in human melanocytes after UVA- and UVB-induced apoptosis. J Invest Dermatol; 129(7): 1769-1781.
Yamashima T, Saido TC, Takita M, Miyazawa A, Yamano J, Miyakawa A et al. (1996) Transient brain ischaemia provokes Ca2+, PIP2 and calpain responses prior to delayed neuronal death in monkeys. Eur J Neurosci; 8(9): 1932-1944.
Yap YW, Whiteman M, Bay BH, Li Y, Sheu FS, Qi RZ et al. (2006) Hypochlorous acid induces apoptosis of cultured cortical neurons through activation of calpains and rupture of lysosomes. J Neurochem; 98(5): 1597-1609.
Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B et al. (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature; 400(6747): 886-891.

REFERENCES
160
Yoshida Y, Saito Y, Jones LS, Shigeri Y (2007) Chemical reactivities and physical effects in comparison between tocopherols and tocotrienols: physiological significance and prospects as antioxidants. J Biosci Bioeng; 104(6): 439-445.
Yuan XM, Li W, Dalen H, Lotem J, Kama R, Sachs L et al. (2002) Lysosomal destabilization in p53-induced apoptosis. Proc Natl Acad Sci U S A; 99(9): 6286-6291.
Zaidi MR, De Fabo EC, Noonan FP, Merlino G (2012) Shedding light on melanocyte pathobiology in vivo. Cancer Res; 72(7): 1591-1595.
Zdolsek J, Olsson G, Brunk U (1990) Photooxidative damage to lysosomes of cultured macrophages by acridine orange. Photochem Photobiol; 51(1): 67-76.
Zdolsek J, Zhang H, Roberg K, Brunk U (1993) H2O2-mediated damage to lysosomal membranes of J-774 cells. Free Radic Res Commun; 18(2): 71-85.
Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ (1996) Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell; 87(4): 619-628.
Zhao K, Zhou H, Zhao X, Wolff DW, Tu Y, Liu H et al. (2012) Phosphatidic acid mediates the targeting of tBid to induce lysosomal membrane permeabilization and apoptosis. J Lipid Res; 53(10): 2102-2114.
Zhao M, Eaton JW, Brunk UT (2000) Protection against oxidant-mediated lysosomal rupture: a new anti-apoptotic activity of Bcl-2? FEBS Lett; 485(2-3): 104-108.
Zhao M, Brunk UT, Eaton JW (2001) Delayed oxidant-induced cell death involves activation of phospholipase A2. FEBS Lett; 509(3): 399-404.
Zhao M, Antunes F, Eaton JW, Brunk UT (2003a) Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis. Eur J Biochem; 270(18): 3778-3786.
Zhao M, Antunes F, Eaton JW, Brunk UT (2003b) Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis. Eur J Biochem; 270(18): 3778-3786.
Zhao S, Aviles ER, Jr., Fujikawa DG (2010) Nuclear translocation of mitochondrial cytochrome c, lysosomal cathepsins B and D, and three other death-promoting proteins within the first 60 minutes of generalized seizures. J Neurosci Res; 88(8): 1727-1737.
Zuzarte-Luis V, Montero JA, Kawakami Y, Izpisua-Belmonte JC, Hurle JM (2007) Lysosomal cathepsins in embryonic programmed cell death. Dev Biol; 301(1): 205-217.
Öllinger K, Brunk UT (1995) Cellular injury induced by oxidative stress is mediated through lysosomal damage. Free Radic Biol Med; 19(5): 565-574.


















