Lim Domain Binding 2: A Key Driver of Transendothelial ...
11
Björkegren Tom Michoel, Eric E. Schadt, Christer Betsholtz, Josefin Skogsberg and Johan L.M. Karin Leander, Ulf de Faire, Anders Hamsten, Arno Ruusalepp, Olle Melander, Torbjörn Ivert, Asl, Rajeev K. Jain, Aranzazu Rossignoli, Cecilia Cedergren, Angela Silveira, Bruna Gigante, Ming-Mei Shang, Husain A. Talukdar, Jennifer J. Hofmann, Colin Niaudet, Hassan Foroughi Atherosclerosis Lim Domain Binding 2: A Key Driver of Transendothelial Migration of Leukocytes and Print ISSN: 1079-5642. Online ISSN: 1524-4636 Copyright © 2014 American Heart Association, Inc. All rights reserved. Greenville Avenue, Dallas, TX 75231 is published by the American Heart Association, 7272 Arteriosclerosis, Thrombosis, and Vascular Biology doi: 10.1161/ATVBAHA.113.302709 2014;34:2068-2077; originally published online June 12, 2014; Arterioscler Thromb Vasc Biol. http://atvb.ahajournals.org/content/34/9/2068 World Wide Web at: The online version of this article, along with updated information and services, is located on the http://atvb.ahajournals.org/content/suppl/2014/06/12/ATVBAHA.113.302709.DC1.html Data Supplement (unedited) at: http://atvb.ahajournals.org//subscriptions/ at: is online Arteriosclerosis, Thrombosis, and Vascular Biology Information about subscribing to Subscriptions: http://www.lww.com/reprints Information about reprints can be found online at: Reprints: document. Question and Answer Permissions and Rights page under Services. Further information about this process is available in the which permission is being requested is located, click Request Permissions in the middle column of the Web Copyright Clearance Center, not the Editorial Office. Once the online version of the published article for can be obtained via RightsLink, a service of the Arteriosclerosis, Thrombosis, and Vascular Biology in Requests for permissions to reproduce figures, tables, or portions of articles originally published Permissions: at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014 http://atvb.ahajournals.org/ Downloaded from
Transcript of Lim Domain Binding 2: A Key Driver of Transendothelial ...

BjörkegrenTom Michoel, Eric E. Schadt, Christer Betsholtz, Josefin Skogsberg and Johan L.M. Karin Leander, Ulf de Faire, Anders Hamsten, Arno Ruusalepp, Olle Melander, Torbjörn Ivert,Asl, Rajeev K. Jain, Aranzazu Rossignoli, Cecilia Cedergren, Angela Silveira, Bruna Gigante, Ming-Mei Shang, Husain A. Talukdar, Jennifer J. Hofmann, Colin Niaudet, Hassan Foroughi
AtherosclerosisLim Domain Binding 2: A Key Driver of Transendothelial Migration of Leukocytes and
Print ISSN: 1079-5642. Online ISSN: 1524-4636 Copyright © 2014 American Heart Association, Inc. All rights reserved.
Greenville Avenue, Dallas, TX 75231is published by the American Heart Association, 7272Arteriosclerosis, Thrombosis, and Vascular Biology
doi: 10.1161/ATVBAHA.113.3027092014;34:2068-2077; originally published online June 12, 2014;Arterioscler Thromb Vasc Biol.
http://atvb.ahajournals.org/content/34/9/2068World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://atvb.ahajournals.org/content/suppl/2014/06/12/ATVBAHA.113.302709.DC1.htmlData Supplement (unedited) at:
http://atvb.ahajournals.org//subscriptions/
at: is onlineArteriosclerosis, Thrombosis, and Vascular Biology Information about subscribing to Subscriptions:
http://www.lww.com/reprints
Information about reprints can be found online at: Reprints:
document. Question and AnswerPermissions and Rightspage under Services. Further information about this process is available in the
which permission is being requested is located, click Request Permissions in the middle column of the WebCopyright Clearance Center, not the Editorial Office. Once the online version of the published article for
can be obtained via RightsLink, a service of theArteriosclerosis, Thrombosis, and Vascular Biologyin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from

2068
Despite in-depth knowledge about several key pathways in atherosclerosis, such as the transendothelial migration of
leukocyte (TEML) pathway,1 no approved drug therapies act directly against coronary and carotid artery disease (CAD). Rather, therapies for CAD act indirectly by modifying CAD risk factors, such as low-density lipoprotein cholesterol. Why targeting candidate genes acting within atherosclerotic lesions
has failed thus far to generate novel CAD therapies remains unclear. One possible explanation is that most known can-didate genes within atherosclerotic lesions are effectors (ie, nonregulatory genes) that are typically found in the periphery of molecular networks and have few interactions (ie, edges)
© 2014 American Heart Association, Inc.
Arterioscler Thromb Vasc Biol is available at http://atvb.ahajournals.org DOI: 10.1161/ATVBAHA.113.302709
Objective—Using a multi-tissue, genome-wide gene expression approach, we recently identified a gene module linked to the extent of human atherosclerosis. This atherosclerosis module was enriched with inherited risk for coronary and carotid artery disease (CAD) and overlapped with genes in the transendothelial migration of leukocyte (TEML) pathway. Among the atherosclerosis module genes, the transcription cofactor Lim domain binding 2 (LDB2) was the most connected in a CAD vascular wall regulatory gene network. Here, we used human genomics and atherosclerosis-prone mice to evaluate the possible role of LDB2 in TEML and atherosclerosis.
Approach and Results—mRNA profiles generated from blood macrophages in patients with CAD were used to infer transcription factor regulatory gene networks; Ldlr–/–Apob100/100 mice were used to study the effects of Ldb2 deficiency on TEML activity and atherogenesis. LDB2 was the most connected gene in a transcription factor regulatory network inferred from TEML and atherosclerosis module genes in CAD macrophages. In Ldlr–/–Apob100/100 mice, loss of Ldb2 increased atherosclerotic lesion size ≈2-fold and decreased plaque stability. The exacerbated atherosclerosis was caused by increased TEML activity, as demonstrated in air-pouch and retinal vasculature models in vivo, by ex vivo perfusion of primary leukocytes, and by leukocyte migration in vitro. In THP1 cells, migration was increased by overexpression and decreased by small interfering RNA inhibition of LDB2. A functional LDB2 variant (rs10939673) was associated with the risk and extent of CAD across several cohorts.
Conclusions—As a key driver of the TEML pathway in CAD macrophages, LDB2 is a novel candidate to target CAD by inhibiting the overall activity of TEML. (Arterioscler Thromb Vasc Biol. 2014;34:2068-2077.)
Key Words: atherosclerosis ◼ coronary artery disease ◼ gene regulatory networks
Received on: October 15, 2013; final version accepted on: May 28, 2014.From the Division of Cardiovascular Genomics (M.M.S., H.A.T., H.F.A., A.R., C.C., J.S., J.L.M.B.), Division of Vascular Biology, Department of
Medical Biochemistry and Biophysics (M.M.S., H.A.T., J.J.H., C.N., H.F.A., A.R., C.C., C.B., J.S., J.L.M.B.), Computational Medicine Unit, Department of Medicine Solna, Center of Molecular Medicine (M.M.S.), and Department of Environmental Medicine (B.G., K.L., U.d.F.), Karolinska Institutet, Solna, Sweden; Clinical Gene Networks AB, Karolinska Science Park, Solna, Sweden (M.M.S., A.R., J.L.M.B.); Division of Cardiovascular Genomics, Department of Pathological Anatomy and Forensic Medicine, University of Tartu, Tartu, Estonia (R.K.J., A.R., J.L.M.B.); Cardiovascular Genetics and Genomics, Department of Medicine Solna, Karolinska Institutet, Solna, Sweden (A.S., A.H.); Department of Cardiac Surgery, Tartu University Hospital, Tartu, Estonia (A.R.); Department of Clinical Sciences, Hypertension and Cardiovascular Disease, Clinical Research Center, Skåne University Hospital, Malmö, Sweden (O.M.); Department of Cardiothoracic Surgery and Anesthesiology and Department of Molecular Medicine and Surgery, Karolinska University Hospital Solna, Karolinska Institutet, Sweden (T.I.); School of Life Sciences–LifeNet, Freiburg Institute for Advanced Studies, University of Freiburg, Freiburg im Breisgau, Germany (T.M.); The Roslin Institute, The University of Edinburgh, Easter Bush, Midlothian, United Kingdom (T.M.); and Institute for Genomics and Multi-Scale Biology, Mount Sinai School of Medicine, New York, NY (E.E.S., J.L.M.B.).
*These authors share last authorship.The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.113.302709/-/DC1.Correspondence to Johan L.M. Björkegren, MD, PhD, Cardiovascular Genomics Group, Department of Medical Biochemistry and Biophysics,
Karolinska Institutet, Scheeleväg 2, 171 77 Stockholm, Sweden. E-mail [email protected]
Lim Domain Binding 2A Key Driver of Transendothelial Migration of Leukocytes
and Atherosclerosis
Ming-Mei Shang, Husain A. Talukdar, Jennifer J. Hofmann, Colin Niaudet, Hassan Foroughi Asl, Rajeev K. Jain, Aranzazu Rossignoli, Cecilia Cedergren, Angela Silveira,
Bruna Gigante, Karin Leander, Ulf de Faire, Anders Hamsten, Arno Ruusalepp, Olle Melander, Torbjörn Ivert, Tom Michoel, Eric E. Schadt, Christer Betsholtz,
Josefin Skogsberg,* Johan L.M. Björkegren*
See accompanying editorial on page 1809
Translational Sciences
at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from
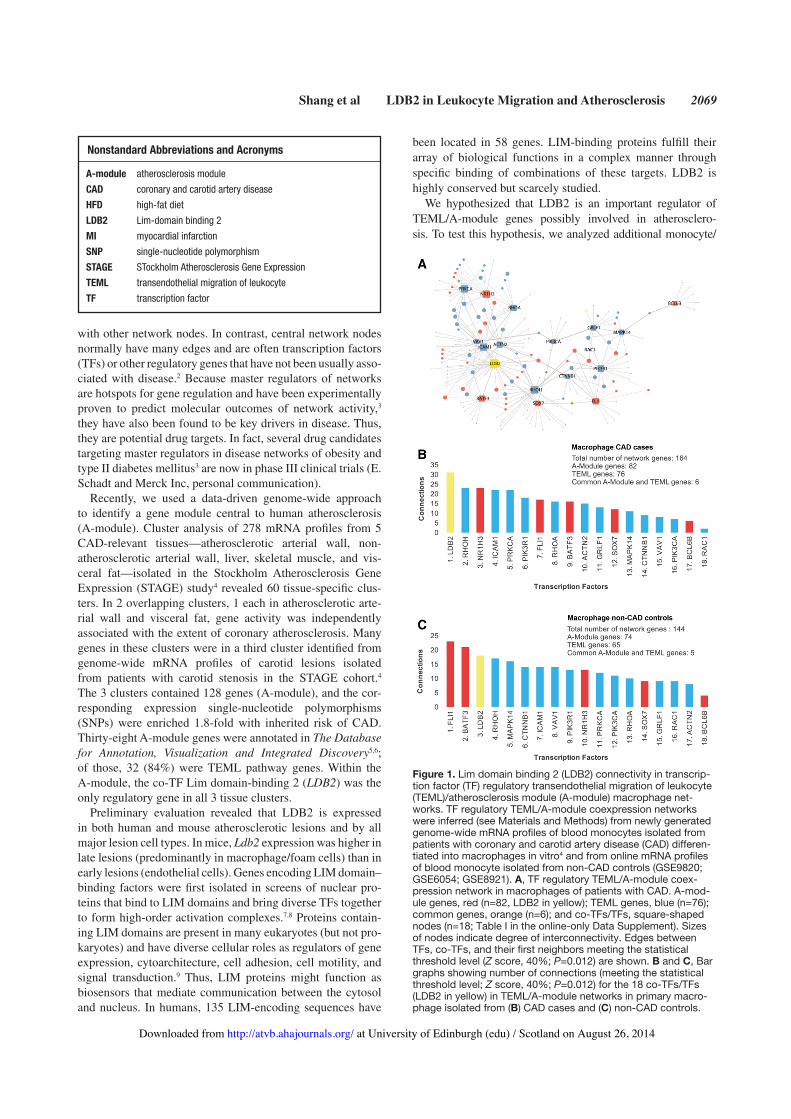
Shang et al LDB2 in Leukocyte Migration and Atherosclerosis 2069
with other network nodes. In contrast, central network nodes normally have many edges and are often transcription factors (TFs) or other regulatory genes that have not been usually asso-ciated with disease.2 Because master regulators of networks are hotspots for gene regulation and have been experimentally proven to predict molecular outcomes of network activity,3 they have also been found to be key drivers in disease. Thus, they are potential drug targets. In fact, several drug candidates targeting master regulators in disease networks of obesity and type II diabetes mellitus3 are now in phase III clinical trials (E. Schadt and Merck Inc, personal communication).
Recently, we used a data-driven genome-wide approach to identify a gene module central to human atherosclerosis (A-module). Cluster analysis of 278 mRNA profiles from 5 CAD-relevant tissues—atherosclerotic arterial wall, non-atherosclerotic arterial wall, liver, skeletal muscle, and vis-ceral fat—isolated in the Stockholm Atherosclerosis Gene Expression (STAGE) study4 revealed 60 tissue-specific clus-ters. In 2 overlapping clusters, 1 each in atherosclerotic arte-rial wall and visceral fat, gene activity was independently associated with the extent of coronary atherosclerosis. Many genes in these clusters were in a third cluster identified from genome-wide mRNA profiles of carotid lesions isolated from patients with carotid stenosis in the STAGE cohort.4 The 3 clusters contained 128 genes (A-module), and the cor-responding expression single-nucleotide polymorphisms (SNPs) were enriched 1.8-fold with inherited risk of CAD. Thirty-eight A-module genes were annotated in The Database for Annotation, Visualization and Integrated Discovery5,6; of those, 32 (84%) were TEML pathway genes. Within the A-module, the co-TF Lim domain-binding 2 (LDB2) was the only regulatory gene in all 3 tissue clusters.
Preliminary evaluation revealed that LDB2 is expressed in both human and mouse atherosclerotic lesions and by all major lesion cell types. In mice, Ldb2 expression was higher in late lesions (predominantly in macrophage/foam cells) than in early lesions (endothelial cells). Genes encoding LIM domain–binding factors were first isolated in screens of nuclear pro-teins that bind to LIM domains and bring diverse TFs together to form high-order activation complexes.7,8 Proteins contain-ing LIM domains are present in many eukaryotes (but not pro-karyotes) and have diverse cellular roles as regulators of gene expression, cytoarchitecture, cell adhesion, cell motility, and signal transduction.9 Thus, LIM proteins might function as biosensors that mediate communication between the cytosol and nucleus. In humans, 135 LIM-encoding sequences have
been located in 58 genes. LIM-binding proteins fulfill their array of biological functions in a complex manner through specific binding of combinations of these targets. LDB2 is highly conserved but scarcely studied.
We hypothesized that LDB2 is an important regulator of TEML/A-module genes possibly involved in atherosclero-sis. To test this hypothesis, we analyzed additional monocyte/
Nonstandard Abbreviations and Acronyms
A-module atherosclerosis moduleCAD coronary and carotid artery diseaseHFD high-fat dietLDB2 Lim-domain binding 2MI myocardial infarctionSNP single-nucleotide polymorphismSTAGE STockholm Atherosclerosis Gene ExpressionTEML transendothelial migration of leukocyteTF transcription factor
Figure 1. Lim domain binding 2 (LDB2) connectivity in transcrip-tion factor (TF) regulatory transendothelial migration of leukocyte (TEML)/atherosclerosis module (A-module) macrophage net-works. TF regulatory TEML/A-module coexpression networks were inferred (see Materials and Methods) from newly generated genome-wide mRNA profiles of blood monocytes isolated from patients with coronary and carotid artery disease (CAD) differen-tiated into macrophages in vitro4 and from online mRNA profiles of blood monocyte isolated from non-CAD controls (GSE9820; GSE6054; GSE8921). A, TF regulatory TEML/A-module coex-pression network in macrophages of patients with CAD. A-mod-ule genes, red (n=82, LDB2 in yellow); TEML genes, blue (n=76); common genes, orange (n=6); and co-TFs/TFs, square-shaped nodes (n=18; Table I in the online-only Data Supplement). Sizes of nodes indicate degree of interconnectivity. Edges between TFs, co-TFs, and their first neighbors meeting the statistical threshold level (Z score, 40%; P=0.012) are shown. B and C, Bar graphs showing number of connections (meeting the statistical threshold level; Z score, 40%; P=0.012) for the 18 co-TFs/TFs (LDB2 in yellow) in TEML/A-module networks in primary macro-phage isolated from (B) CAD cases and (C) non-CAD controls.
at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from

2070 Arterioscler Thromb Vasc Biol September 2014
macrophage-specific genome-wide RNA data from patients with CAD and non-CAD controls and assessed atherosclerosis devel-opment and TEML activity in Ldb2-deficient mice bred onto an atherosclerosis-prone background (Ldb2–/–Ldlr–/–Apob100/100).
Materials and MethodsMaterials and Methods are available in the online-only Supplement.
ResultsA Master Regulator of TEML/A-Module Network Genes in CAD MacrophagesPreviously, we used a data-driven whole-genome approach to identify LDB2 as a master regulator (ie, a key TF or co-TF in molecular processes) of A-module genes in the arterial wall.4
Here, we first determined the extent to which LDB2 is a mas-ter regulator of A-module genes (n=128) and all other known TEML genes (n=113). For these studies, we used mRNA pro-files isolated from primary monocytes in the blood of STAGE patients (Table 1).4 Before RNA isolation, monocytes were dif-ferentiated into macrophages in vitro (mimicking macrophage differentiation in TEML). TF regulatory network inference of all 233 genes (Table I in the online-only Data Supplement), of which 18 encoded TFs or co-TFs, was performed at Z score of 40% (P=0.012). Of the 233 genes, 164 (76 TEML, 82 A-module, and 6 in common) were part of a highly interconnected TF regu-latory network in CAD blood macrophages (Figure 1A; Figure I and Table I in the online-only Data Supplement). The most connected genes were LDB2 (31 connections) and RHOH and NR1H3 (23 connections each; Figure 1B).
Next, we examined the role of LDB2 in TEML/A-module networks inferred from 5 sets of mRNA profiles isolated from primary blood monocytes/macrophages in non-CAD controls (3 GSE982 data sets, GSE6054, GSE8921). In TEML/A-module networks inferred from 2 of the GSE9820 data sets of differenti-ated macrophages (Figure 1C; Table II in the online-only Data Supplement) and lipopolysaccharide-stimulated monocytes, LDB2 had 18 connections, making it the third most connected TF (of 18). In 3 data sets (GSE6054, GSE8921, and 1 GSE9820) of mRNA profiles from nonstimulated primary monocytes, LDB2 had 10 connections in 2 networks and 5 connections in 1, representing the 13th or 14th most connected TF (of 18).
To examine disease causalities of the TF regulatory network genes from CAD macrophages, we determined their enrich-ment in CAD/myocardial infarction (MI) risk (see Materials and Methods), using the genome-wide association CAD database from the Wellcome Trust Case-Control Consortium (WTCCC).10 SNPs in network genes had a 4.5-fold greater enrichment in CAD/MI risk (P=1×10−156) than 5000 randomly selected, equal-sized sets of SNPs, strongly indicating causal-ity for the network genes.
Exacerbated Atherosclerosis in Ldb2–/– MiceThe role of LDB2 as a TEML master regulator in CAD mac-rophages suggests that targeting Ldb2 might affect athero-sclerosis development. To test this hypothesis, we obtained Ldb2–/– mice from Mutant Mouse Regional Resource Center. Ldb2 was targeted by replacing the first exon (of 9) by homologous recombination. We bred the Ldb2–/– mice with Ldlr–/–Apob100/100 mice and backcrossed to ensure >99% C57/BL6 strain background. Ldlr–/–Apob100/100 mice have a human-like low-density lipoprotein cholesterol profile and are prone to atherosclerosis.11 Ldb2 deficiency did not affect plasma
Table 1. Basic Characteristics of Carotid Patients of the Stockholm Atherosclerosis Gene Expression Cohort4
Characteristics
No. of patients (men/women) 38 (25/13)
Age, y 69±10
Body mass index, kg/m2 25±3.2
Waist-to-hip ratio 0.91±0.07
Cholesterol, mmol/L
Total 4.56±1.09
VLDL 0.27±0.19
LDL 2.45±0.85
HDL 1.70±0.42
Triglycerides, mmol/L
Total 1.30±0.49
VLDL 0.85±0.41
LDL 0.30±0.09
HDL 0.20±0.07
IMT, mm (mean±SD) 1.20±0.16
Diagnoses and therapies
Hyperlipidemia 7 (18%)
Statins 25 (66%)
Hypertension 16 (42%)
ACE inhibitors 12 (32%)
β-Blocker 20 (53%)
Calcium channel blockers 7 (18%)
Loop diuretics 4 (11%)
Values are mean±SD or number (%) of patients. ACE indicates angiotensin-converting enzyme; HDL, high-density lipoprotein; IMT, intima-media thickness; LDL, low-density lipoprotein; and VLDL, very low-density lipoprotein
Table 2. Weight and Plasma Lipid and Glucose Levels in the Study Mice
Age, wk
Cholesterol, mg/dL Triglycerides, mg/dL Glucose, mg/dL Weight, g
Ldb2+/+ Ldb2–/– Ldb2+/+ Ldb2–/– Ldb2+/+ Ldb2–/– Ldb2+/+ Ldb2–/–
10 358±35 334±57 101±23 98±14 159±51 157±42 … …
20 377±53 381±68 106±24 93±14 198±60 174±32 … …
30 447±60 430±141 124±40 128±39 300±86 311±72 31.4±5.0 30.8±5.3
Ldb2–/– (Ldb2–/–Ldlr–/–Apob100/100), n=10; Ldb2+/+ (Ldb2+/+Ldlr–/–Apob100/100), n=13. P>0.05 at each age for all Ldb2–/– and Ldb2+/+ comparisons.
at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from

Shang et al LDB2 in Leukocyte Migration and Atherosclerosis 2071
cholesterol, triglyceride, or glucose levels over time or weight at euthanization (Table 2). However, after 30 weeks, chow-fed Ldb2–/–Ldlr–/–Apob100/100 mice had ≈2-fold more aortic ath-erosclerosis compared with littermate Ldb2+/+Ldlr–/–Apob100/100
controls, as judged by lesion surface areas in pinned-out aortas (Figure 2A, upper panel). A similar difference in atherosclero-sis was observed at 25 weeks of age in mice fed a high-fat diet (HFD; 24% of energy; Figure 2A, lower panel).
Figure 2. Atherosclerotic lesion size and immunohistochemical characteristics in Ldb2–/–mice. A, Bar graph of average lesion area as a percentage of the total area in pinned-out aortas stained with Sudan IV (left; n=9 mice per group) and representative images (×10; right) in 30-week-old chow-fed mice (top) and 25-week-old mice fed a high-fat diet (bottom). B to E, Average percentage (%) color or immu-nohistochemical staining in frozen cross-sections relative to the entire aortic root cross-section area (left) and representative images (×50; top right; ×630, right bottom) from 30-week-old chow-fed mice. B, Oil-Red-O (n=8–9 mice per group in chow-fed mice; n=4 mice per group in high-fat diet–fed mice). C, CD68 (n=9 mice per group in chow-fed mice; n=4 mice per group in high-fat diet–fed mice). D, SM22α (n=3 mice per group in chow-fed mice; n=4 mice per group in high-fat diet–fed mice). E, Collagen staining (n=3 mice per group in chow-fed mice; n=4 mice per group in high-fat diet–fed mice). F, Average number of hematoxylin-stained nuclei in areas (≈100×100 μm) of lesion stained with Oil-Red-O (ie, foam cell number, 1 area/section in 5–6 areas per section per mouse; n=3 mice per group; left) and representative examples of such areas (right). Arrows indicate nuclei of foam cells (hematoxylin-stained endothelial cell nucleus). G, Plaque stability scores (calculated as [collagen+SM22α]/[Oil-Red-O+CD68] % areas). All bar graphs show mean±SEM. *P<0.05; &P<0.01; #P<0.001; scale bars, 100 μm.
at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from

2072 Arterioscler Thromb Vasc Biol September 2014
Composition of Atherosclerotic Lesions in Ldb2–/– MiceNext, we assessed the composition of atherosclerotic lesions in aortic root cross-sections from chow- and HFD-fed Ldb2–/– Ldlr–/–Apob100/100 mice and littermate Ldb2+/+Ldlr–/–Apob100/100 controls at 30 and 25 weeks, respectively. Unlike Sudan IV lipid staining of pinned-out aortas, which shows lesion surface area, Oil-Red-O staining of cross-sections of the aortic root shows lesion thickness. Relative to the entire area of each aor-tic root cross-section, Ldb2-/–Ldlr–/–Apob100/100 mice had 50% more Oil-Red-O staining and 67% more staining for the mac-rophage marker CD68 compared with controls (Figure 2B and 2C). In contrast, the area of staining for the smooth muscle cell marker SM22α was smaller in Ldb2–/–Ldlr–/–Apob100/100 mice (Figure 2D); the staining of lesion collagen was largely unaf-fected (Figure 2E).
The increase in staining in Ldb2-/–Ldlr–/–Apob100/100 mice could reflect a greater number of lesion foam cells (ie, CD68-positive cells) or increased lipid accumulation and expression of cell surface CD68 in larger foam cells. To assess these pos-sibilities, we counterstained Oil-Red-O–positive lesion areas (≈200×100 μm) in aortic root cross-sections with hematoxylin
and counted nuclei (indicating number of foam cells). In mul-tiple cross-sections of aortic root in 3 Ldb2–/–Ldlr–/–Apob100/100 and 3 littermate controls, lesion foam cells were ≈3-fold more abundant in Ldb2–/–mice (Figure 2F).
Next, we used the immunohistochemical characteristics of atherosclerotic lesions to determine plaque stability scores, calculated as (SM22α+collagen)/(CD68+Oil-Red-O)% areas12 of Ldb2–/–Ldlr–/–Apob100/100 and littermate Ldb2+/+Ldlr–/– Apob100/100 controls (Figure 2B–2E). Ldb2–/– plaques were ≈50% less stable (Figure 2G). The compositional differences in the atherosclerotic lesions of Ldb2–/– and wild-type mice in the chow-fed group were also reflected in the aortic root lesions and in the plaque stability scores of 25-week-old HFD-fed mice (Figure 2B–2E and 2G). Ldb2–/– plaques from HFD-fed mice were ≈42% less stable than those in HFD-fed controls (Figure 2G).
Adhesion Molecule Activity in Ldb2–/– Vascular Wall and Blood LeukocytesAt 30 weeks, the exacerbated atherosclerosis and accumula-tion of foam cells led to more unstable plaques in Ldb2–/– Ldlr–/–Apob100/100 mice than in controls, consistent with
Figure 3. Leukocyte and vascular wall characteristics of Ldb2–/– mice. White blood cell (WBC) count and vascular and leukocyte expres-sion patterns were examined in Ldb2–/–Ldlr–/–Apob100/100 mice and littermate Ldb2+/+Ldlr–/–Apob100/100 control mice. A, WBC and lympho-cyte, monocyte, and granulocyte WBC subclasses determined in 6- to 8-week-old mice with the scil Vet abc Animal Blood Counter (n=5 and 6 mice per group). B, Expression levels of Vcam1 in the aortic arch of 6- to 8-week-old mice determined by reverse transcription polymerase chain reaction (RT-PCR; n=6 mice per group; left) and immunofluorescently stained Vcam1 (white) in representative aortic root sections; 6- to 8-week-old (top right) and 30-week-old mice also doublestained for CD68 lesion macrophages (red; bottom right). C, Expression levels of Icam1 in the aortic arch of 6- to 8-week-old mice determined by RT-PCR (n=6 mice per group; left), and repre-sentative white immunofluorescently stained aortic root sections (right). D and E, Expression levels of Vcam1 (D) and Icam1 (E) in 6- to 8-week-old study mice in primary blood leukocytes stimulated with tumor necrosis factor α in vitro before RNA isolation (n=4 mice per group). All bar graphs show mean±SEM. *P<0.05; scale bars, 100 μm.
at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from

Shang et al LDB2 in Leukocyte Migration and Atherosclerosis 2073
increased TEML activity. To determine whether increased TEML activity exacerbated atherosclerosis development, we examined Ldb2–/–Ldlr–/–Apob100/100 and littermate controls at 6 to 10 weeks of age, before atherosclerotic lesions formed. In these mice, any differences in molecular phenotypes should be primarily caused by Ldb2 deficiency, whereas in 30-week-old mice, such differences would largely reflect the 2-fold difference in the extent of atherosclerosis. The 2 groups had equally elevated plasma cholesterol levels (Table 2) and no difference in circulating leukocyte levels (Figure 3A).
Genome-wide expression analysis of RNA from the aor-tic arch of 6 Ldb2–/–Ldlr–/–Apob100/100 mice and 6 littermate controls at 6 to 10 weeks of age showed that 40 genes were
differentially expressed (false discovery rate <0.05; Table II in the online-only Data Supplement). The pathways most significantly enriched with these genes—antigen processing and presentation (P=0.0005), viral myocarditis (P=0.001), and cell adhesion (P=0.005)—were all highly enriched with TEML genes. The vascular wall cell adhesion activity at 6 to 10 weeks was supported by an increase in Vcam1 expression (Figure 3B, left panel) and mirrored by Vcam1 protein expres-sion in aortic root cross-sections (Figure 3B, upper right panels). Icam1 expression was not significantly increased (Figure 3C). In primary leukocytes from 6- to 10-week-old mice, Vcam1 expression was also significantly higher in Ldb2–/– mice than in wild-type controls (Figure 3D); Icam1 expres-sion was not significantly increased (Figure 3E). Of note, in
Figure 4. Transendothelial migration of leukocyte in Ldb2–/– mice. A, Leukocyte subtypes assessed by fluorescence-activated cell sorting in exudate, collected from subcutaneous dorsal air pouches (4 mL of sterile air injected twice) 24 h after tumor necrosis factor α injections (50 ng per mouse), from 6- to 8-week-old study mice (n=6 mice per group); leukocytes were identified as CD11B+F4/80+, macorphages as CD11B+F4/80+MHCII+, monocytes as CD11B+F4/80+MHCII− and neutrophils as CD11B+F4/80−GR1+. B, Representative images of the retinal vasculature (green, isolectin staining) and monocyte/macrophages (white; F4/80 staining) from 6- to 8-week-old study mice. White and red arrows indicate F4/80+ cells within and migrating across the vessel wall, respectively. Scale bar, 50 μm. C, Mean F4/80 staining as a percentage of total vessel area (n=2 retinas per mouse; 3 mice per group). D, Expression of Lim domain binding 2 (LDB2) in THP1 mono-cytes and in primary leukocytes from 6- to 8-week-old study mice (left) and littermate controls and its effect on migration of these cells in vitro (right) assessed with the Chemicon QCM cell migration assay using moncyte chemotactic protein 1 as chemoattractant. The mRNA expression levels and migration rates are shown as relative to their respective control. E, Arterial recruitment of leukocytes investigated by ex vivo in situ perfusion. Representative images of yellow gold latex beads labeled primary leukocyte from 6- to 8-week-old Ldb2–/–Ldlr–/– Apob100/100 mice and littermate controls (n=3 mice per group) adherence on isolated aortas from 6- to 8-week-old Ldb2+/+Ldlr–/–Apob100/100 mice (right), and quantification of the yellow gold labeled area is showing as percentage of the total aortic arch (left). F, Gene expression levels of Ki67 in tumor necrosis factor α–stimulated primary leukocytes isolated from 6- to 8-week-old study mice (left). Immunofluores-cence of representative aortic root sections from 30-week-old mice demonstrating Ki67 staining (green) and colocalization (yellow) with CD68 lesion macrophages (red; bottom right). Top right panels show nuclei staining with DAPI (blue). Scale bar, 100 μm. All bar graphs show mean±SEM. *P<0.05; &P<0.01; #P<0.001 as compared with its control.
at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from

2074 Arterioscler Thromb Vasc Biol September 2014
30-week-old mice, Vcam1 expression was to a large extent colocalized with macrophages within the aortic root lesions in Ldb2–/–Ldlr–/–Apob100/100 mice (Figure 3B, lower right panels).
TEML Activity in Ldb2–/– MiceThe early adhesion activity in vascular wall and circulat-ing monocytes in 6- to 10-week-old mice should facilitate TEML. To examine this possibility, we created dorsal air pouches in 6- to 10-week-old Ldb2–/–Ldlr–/–Apob100/100 mice and littermate controls by subcutaneous injections of sterile air and 50 ng tumor necrosis factor α. After 24 hours, exu-dates were extracted and examined by fluorescence-activated cell sorting.13 Leukocytes in air pouches, especially macro-phages and monocytes, were markedly increased in Ldb2–/–
Ldlr–/–Apob100/100 mice (Figure 4A). Next, the migration of leukocytes was examined in the retinal vasculature of 6- to 10-week-old mice by confocal microscopy. In littermate con-trols, several small leukocytes were observed inside arterioles/venules (Figure 4B, upper panels), consistent with reports14,15 of slightly activated but circulating monocytes in hypercho-lesterolemic mouse models. These intraluminal small leuko-cytes were largely absent in equally hypercholesterolemic Ldb2–/–Ldlr–/–Apob100/100 mice (Figure 4B, lower panels). Instead, we saw a few larger leukocytes (eg, macrophages) migrating through the vessel wall (Figure 4B, lower right panels). Owing to the absence of the intraluminal cells, F4/80 staining was significantly reduced in the retinal vasculature of Ldb2-/–Ldlr–/–Apob100/100 mice (Figure 4C), despite similar levels of circulating leukocytes (Figure 3A). These findings support the notion that TEML activity is increased in Ldb2–/–
Ldlr–/–Apob100/100 mice, consistent with the results of air-pouch studies (Figure 4A).
Effects of Perturbing Physiological LDB2 Expression Levels on Leukocyte Migration In VitroTo further investigate the role of LDB2 in TEML activity, we examined the migration of primary blood leukocytes from Ldb2–/–Ldlr–/–Apob100/100 and littermate controls in vitro after stimulation with monocyte chemotactic protein 1. Consistent with findings in the air-pouch and the retinal vascular models, migration of Ldb2–/– leukocytes was significantly increased (Figure 4D). Thus, in the complete absence of Ldb2, the migra-tory properties of primary leukocytes and TEML activity
clearly increased. However, the effects of complete and life-long absence of a key regulator, such as LDB2, can differ from those of subtle deviations from physiological expression levels. To examine how changes in physiological LDB2 expression levels affect monocyte migration, we measured the migra-tion of human THP1 monocytes in vitro after small interfer-ing RNA inhibition or overexpression of LDB2. Migration was increased by overexpression and decreased by lower expression (Figure 4D). Thus, like complete deficiency, LDB2 overex-pression increases the migration of human THP1 monocytes, whereas decreased LDB2 expression levels decrease migration.
Arterial Recruitment of Leukocytes in Ldb2–/– MiceTo determine whether increased TEML activity triggered by LDB2 deficiency is caused by an activated endothelium or activated/migration-prone leukocytes, we performed ex vivo perfusion studies. Latex bead–labeled primary leukocytes from 6- to 10-week-old Ldb2-/–Ldlr–/-Apob100/100 mice and lit-termate controls were perfused in situ into the aortic arches of littermate controls. After 2 hours, adhesion of primary leuko-cytes from Ldb2-/–Ldlr–/–Apob100/100 mice was ≈120% greater than that of control leukocytes, indicating that the leukocyte phenotype is the primary reason for the increased TEML activity (Figure 4E). To investigate whether the primary phe-notype in leukocytes is unrelated to modification in the circu-lation (making them more adherent), we investigated the in vitro migration of primary spleen leukocytes, which have not been exposed to the circulation,16 isolated from the spleen of 6- to 10-week-old Ldb2–/–Ldlr–/–Apob100/100 mice and littermate controls. Again, the Ldb2–/– leukocytes had greater migration compared with control leukocytes isolated from littermate controls (Figure 4D). Thus, Ldb2 deficiency in leukocytes seems to be the primary cause of their increased migration in Ldb2–/–Ldlr–/–Apob100/100 mice.
Leukocyte Proliferation in Ldb2–/– MiceRecently, local proliferation within plaques, rather than de novo influx, of macrophages was shown to promote athero-sclerosis development.17 To investigate a possible contribu-tion of macrophage proliferation to the observed increase in atherosclerosis burden in Ldb2-deficient mice, we mea-sured gene and protein expression levels of the prolifera-tion marker Ki67. The proliferation rate as indicated by
Figure 5. Manhattan plot showing association between single-nucleotide polymorphisms (SNPs) and Lim domain binding 2 (LDB2) expression in arterial wall in the Stockholm Atherosclerosis Gene Expression cohort. y axis shows the association P value (−log10) between each SNP and LDB2 expres-sion in arterial wall. x axis represents the position of each SNP and LDB2 in chromosome 4; the box underneath shows the LDB2 transcript with exons marked as vertical blue lines. Triangles represent genotyped SNPs (n=102) in LDB2. The solid black triangle shows the SNP with the highest association to LDB2 expression (rs10939673). The horizontal line shows the threshold of P<0.05.
at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from

Shang et al LDB2 in Leukocyte Migration and Atherosclerosis 2075
Ki67 mRNA levels in tumor necrosis factor α–stimulated primary leukocytes isolated from Ldb2-deficient mice was found to be 2-fold higher than in stimulated leuko-cytes from littermate controls (Figure 4F, left panel). The increase in mRNA levels was also found to be mirrored by Ki67 protein expression colocalizing with the CD68 mac-rophage marker in aortic root cross-sections of 30-week-old Ldb2–/–Ldlr–/–Apob100/100 mice (Figure 4F, right panels). Apparently in parallel to increased TEML activity, Ldb2 deficiency also seems to increase proliferation of lesion macrophages/foam cells that may contribute to exacerbat-ing atherosclerosis.
Functional LDB2 Variant, Risk of CAD/MI, and Extent of Clinical and Subclinical AtherosclerosisBecause LDB2 is a key driver of TEML and atherosclerosis, subtle alterations in its protein levels mediated by functional DNA variants in LDB2 may affect the extent of atherosclero-sis and possibly risk for CAD/MI. To test this hypothesis, we identified 5 SNPs distributed in LD-blocks of LDB2 and exam-ined their functionality by reverse transcription polymerase chain reaction analysis of tissues from the STAGE study.4 For 1 SNP, rs10939673, carriers of the T-allele had lower LDB2 mRNA levels in arterial wall (P=0.004) and mediastinal fat (P=0.001) compared with carriers of the C-allele. To con-firm that this SNP had the strongest association with LDB2 expression, we examined associations between all 102 SNPs in LDB2 that were genotyped in the STAGE study4 (repre-senting all haploblocks) and arterial wall expression of LDB2. rs10939673 had by far the strongest association with LDB2 expression levels (Figure 5).
Consistent with our findings of LDB2 expression levels in human monocytes (Figure 4D), examination of the allele distribution of rs10939673 in 2 CAD case/control studies18,19 showed that carriers of the minor T-allele were significantly under-represented among CAD cases versus controls in both cohorts (Table 3). In 1926 CAD cases and 2938 population controls in the WTCCC study,20 rs10939673 was not geno-typed, but 1 SNP (rs1496747) genotyped in WTCCC was in LD with rs10939673 (r2=0.62). The minor G-allele of rs1496747 was also under-represented among CAD cases (P<0.041).
Next, we examined whether the T-allele also was associ-ated with the extent of atherosclerotic lesions in 2 cohorts in which coronary18 and carotid21 lesions had been evaluated by quantitative coronary angiography and B-mode ultrasound, respectively. The rare allele (T) of the functional rs10939673 SNP (associated with lower LDB2 expression) was associated with a lower stenosis score (P=0.040) and a smaller plaque area (P=0.024) in 375 MI cases and with a thinner intima-media (P=0.004) at the carotid bulb in 3118 healthy subjects (Table 4). The latter association was independent of other established CAD/MI risk factors, including age, sex, smok-ing, systolic/diastolic blood pressures, body mass index, and plasma levels of triglycerides, high-density lipoprotein, low-density lipoprotein cholesterol, and C-reactive protein (P<0.005).
DiscussionThe idea of inhibiting TEML to treat atherosclerosis has not yet been fully examined.1 Targeting TEML in atheroscle-rosis is attractive, because leukocyte migration across the
Table 3. Association Analyses of rs10939673 in 2 Case/Control Studies of MI
Genotype Distribution Allele Frequencies
CC CT TT P Value C T MAF P Value
Genotype and allele frequencies for rs10939673 in 375 MI cases and controls
MI patients 160 170 45 0.047 490 260 0.347 0.034
MI controls 130 200 53 460 306 0.399
Genotype and allele frequencies for rs10939673 in SHEEP 929 MI cases and controls
MI patients 351 440 138 0.175 1142 716 0.385 0.031
MI controls 440 626 217 1506 1060 0.413
Genotype and allele frequencies for rs10939673 in the 2 studies combined
MI patients 511 610 183 0.015 1632 976 0.374 0.005
MI controls 570 826 270 1966 1366 0.410
MAF indicates minor allele frequency; MI, myocardial infarction; and SHEEP, Stockholm Heart Epidemiology Program.
Table 4. Association Analyses of rs10939673 and Atherosclerosis Measurements in MI Cases and Healthy Subjects
CC CT TT P Value
Associations between rs10939673 and atherosclerosis measurements in 375 MI cases
Stenosis score 8.7±2.9 7.7±3.0 7.5±3.9 0.040
Plaque area 68±30 62±20 53±27 0.024
Associations between rs10939673 and IMT in the carotid bulb in 3118 healthy subjects
IMT, mm 1.4±0.6 1.4±0.6 1.3±0.6 0.004
Values are mean±SD. IMT indicates intima-media thickness; and MI, myocardial infarction.
at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from

2076 Arterioscler Thromb Vasc Biol September 2014
endothelium is an early inflammatory event occurring before the differentiation and activation of leukocytes and subsequent downstream cascades of immune and inflammatory reactions. TEML is important in both early and late stages of atheroscle-rosis,1 but the TEML pathway is also essential for many, if not all, inflammatory and immune reactions22 and is particularly crucial as the first defense against pathogens. In fact, impaired TEML induced by targeting nonregulatory pathway genes has caused severe opportunistic infections.23,24 Therefore, strate-gies to target TEML should not disrupt isolated steps in the TEML pathway but should generally downregulate overall TEML activity.
From this perspective, our finding that LDB2 is a key net-work driver of TEML in CAD macrophages and regulates many TEML genes that are causally related to CAD (ie, enriched with inherited CAD risk) is potentially important. Master network regulators are increasingly recognized as key drivers in disease.25 However, a master regulatory role in a gene network is not proof of a corresponding key driver role in disease, which must be experimentally validated. Our find-ings in atherosclerosis-prone mice provide such a validation and strongly support the notion that LDB2 is a key driver of TEML, atherosclerosis development, and plaque stability, pos-sibly also promoting proliferation of lesion macrophages.17 This notion is further supported by our finding of a functional LDB2 variant showing that subtle changes in LDB2 expression levels are relevant to CAD/MI risk and the extent of atheroscle-rosis. In summary, our study provides strong support for LDB2 as a new candidate target gene for atherosclerosis and CAD.
Apart from our current and previous findings relating to LDB2 expression in the atherosclerotic arterial wall,4 noth-ing is known about LDB2 in CAD. Interestingly, however, targeting LDB2 increases fibroblast migration in vitro.26,27 Specifically, LDB2 seems to anchor STE20-like kinase, pre-venting STE20-like kinase–mediated phosphorylation (activa-tion) of the actin–tubulin system, which controls cell migration and motility. It is not known whether regulation of the TEML pathway by LDB2 in monocytes involves STE20-like kinase. In fibroblasts, however, LDB2 seemed to be required in a steady state to maintain normal migration, whereas major deviations (in either direction) leads to increased migra-tion.26,27 Thus, it was not surprising that both the complete lack and high overexpression of LDB2 increased leukocyte migra-tion, whereas decreased LDB2 expression inhibited migration (Figure 4D). A similar pattern was reported for another key driver in disease, peroxisome proliferator–activated receptor δ,28 in controlling the inflammatory status of macrophages/foam cells in atherosclerotic lesions. Briefly, when key drivers regulating many downstream genes are completely depleted (ie, knocked out), the underlying network genes lose their main regulation, resulting in increased activity of the regu-lated network. But when these key drivers are stimulated or inhibited at more physiological conditions, the effects can be the reverse (ie, inhibition leads to less network activity and stimulation to more), as in our study (Figures 2A and 4D).
We used the Ldlr–/–Apob100/100 mouse model to study Ldb2 deficiency. These mice have a plasma lipid profile similar to that of patients with CAD29,30 with high low-density lipoprotein
cholesterol and apolipoprotein B100 levels. Atherosclerosis development in these mice is aggressive and occurs on a chow diet.11 Because the hypercholesterolemia causes leukocytosis in these mice,14,15 they are a convenient model to study TEML during atherogenesis. Because LDB2 is a master regulator of TEML genes in a CAD macrophage regulatory network, we examined TEML activity in a range of in vivo, ex vivo, and in vitro models in these mice. In vivo, we examined how leuko-cytes migrate into an air-pouch during a longer time period. In this model, Ldb2 deficiency increased migration of leukocytes, macrophages, and monocytes. To visualize migration in vivo, we isolated retinas, which again indicated increased migra-tion of Ldb2–/– leukocytes. Ex vivo perfusion studies helped to establish that the main Ldb2 phenotype is within the leu-kocytes, not in the arterial wall. An in vitro migration assay confirmed the primary role of Ldb2 in spleen leukocytes. In addition, we found that Vcam1, a key gene for adhesion in the TEML pathway, is upregulated both in Ldb2–/– leukocytes and the aortic arterial wall. Thus, we cannot rule out that LDB2 also is affecting migration in the arterial wall (by increasing mono-cyte adhesion) besides a direct role in leukocytes. The role of Vcam1 expression in the arterial wall is well established,31,32 whereas the significance of the increased expression in LDB2-deficient leukocytes and lesion macrophages is unknown.
In conclusion, through its regulatory effects on the TEML pathway, LDB2 is a novel therapeutic target for atherosclero-sis and plaque stability. Future studies sorting out the molecu-lar mechanism in leukocytes and the arterial wall by which LDB2 regulates TEML should include STE20-like kinase.
AcknowledgmentsWe thank Lennart Lindbom and Ellinor Kenne (Karolinska Institutet) and Vinay Choubey (University of Tartu) for advice on experiments, and Stephen Ordway for editorial assistance. This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk.
Sources of FundingThis work was supported by Clinical Gene Networks AB, PROCARDIS (PRecOcious Coronary ARtery DISease) in the Sixth EU-framework program (LSHM-CT-2007–037273, www.procardis.org), the Swedish Heart-Lung Foundation (to J.L.M. Björkegren and U.d. Faire), the Swedish Research Council (J.L.M. Björkegren, U.d. Faire, J. Skogsberg), the King Gustaf V and Queen Victoria’s Foundation of Freemasons (J.L.M. Björkegren, 2008–2010; J. Skogsberg, 2011–2013), the Swedish Society of Medicine (to J.L.M. Björkegren, J. Skogsberg). This work was also supported by grant from University of Tartu (SP1GVARENG, to J.L.M. Björkegren) and the Estonian Research Council (to J.L.M. Björkegren; #ETF8853).
DisclosuresJ.L.M. Björkegren is founder, main shareholder, and chairman of the board for Clinical Gene Networks AB (CGN). A. Ruusalepp and E. Schadt are board members and shareholders. T. Michoel is share-holder. CGN has an invested interest in microarray data from carotid artery disease macrophages. The other authors report no conflicts.
References 1. Braunersreuther V, Mach F. Leukocyte recruitment in atherosclero-
sis: potential targets for therapeutic approaches? Cell Mol Life Sci. 2006;63:2079–2088.
at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from

Shang et al LDB2 in Leukocyte Migration and Atherosclerosis 2077
2. Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12:56–68.
3. Chen Y, Zhu J, Lum PY, et al. Variations in DNA elucidate molecular net-works that cause disease. Nature. 2008;452:429–435.
4. Hägg S, Skogsberg J, Lundström J, et al. Multi-organ expression profiling uncov-ers a gene module in coronary artery disease involving transendothelial migra-tion of leukocytes and LIM domain binding 2: the Stockholm Atherosclerosis Gene Expression (STAGE) study. PLoS Genet. 2009;5:e1000754.
5. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative anal-ysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
6. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13
7. Agulnick AD, Taira M, Breen JJ, Tanaka T, Dawid IB, Westphal H. Interactions of the LIM-domain-binding factor Ldb1 with LIM homeodo-main proteins. Nature. 1996;384:270–272.
8. Jurata LW, Gill GN. Functional analysis of the nuclear LIM domain inter-actor NLI. Mol Cell Biol. 1997;17:5688–5698.
9. Kadrmas JL, Beckerle MC. The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol. 2004;5:920–931.
10. The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared con-trols. Nature. 2007;447:661–678
11. Skogsberg J, Lundström J, Kovacs A, Nilsson R, Noori P, Maleki S, Köhler M, Hamsten A, Tegnér J, Björkegren J. Transcriptional profiling uncovers a network of cholesterol-responsive atherosclerosis target genes. PLoS Genet. 2008;4:e1000036.
12. Ni W, Egashira K, Kitamoto S, Kataoka C, Koyanagi M, Inoue S, Imaizumi K, Akiyama C, Nishida KI, Takeshita A. New anti-monocyte chemoattractant protein-1 gene therapy attenuates atherosclerosis in apo-lipoprotein E-knockout mice. Circulation. 2001;103:2096–2101.
13. Soehnlein O, Zernecke A, Eriksson EE, Rothfuchs AG, Pham CT, Herwald H, Bidzhekov K, Rottenberg ME, Weber C, Lindbom L. Neutrophil secretion products pave the way for inflammatory monocytes. Blood. 2008;112:1461–1471.
14. Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205.
15. Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo CL, Wang M, Sanson M, Abramowicz S, Welch C, Bochem AE, Kuivenhoven JA, Yvan-Charvet L, Tall AR. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest. 2011;121:4138–4149.
16. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616.
17. Robbins CS, Hilgendorf I, Weber GF, et al. Local proliferation domi-nates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–1172.
18. Samnegård A, Silveira A, Lundman P, Boquist S, Odeberg J, Hulthe J, McPheat W, Tornvall P, Bergstrand L, Ericsson CG, Hamsten A, Eriksson P. Serum matrix metalloproteinase-3 concentration is influenced by MMP-3 -1612 5A/6A promoter genotype and associated with myocardial infarction. J Intern Med. 2005;258:411–419.
19. Gigante B, Bennet AM, Leander K, Vikström M, de Faire U. The interaction between coagulation factor 2 receptor and interleukin 6 haplotypes increases the risk of myocardial infarction in men. PLoS One. 2010;5:e11300.
20. Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453.
21. Hamrefors V, Hedblad B, Engström G, Almgren P, Sjögren M, Melander O. A myocardial infarction genetic risk score is associated with markers of carotid atherosclerosis. J Intern Med. 2012;271:271–281.
22. Fischer A. Leukocyte adhesion. Clin Exp Rheumatol. 1993;11(suppl 9):S23–S24.
23. Lindå H, von Heijne A, Major EO, Ryschkewitsch C, Berg J, Olsson T, Martin C. Progressive multifocal leukoencephalopathy after natalizumab monotherapy. N Engl J Med. 2009;361:1081–1087.
24. Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, Verbeeck J, Geboes K, Robberecht W, Rutgeerts P. Progressive multifo-cal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N Engl J Med. 2005;353:362–368.
25. Albert R, Jeong H, Barabasi AL. Error and attack tolerance of complex networks. Nature. 2000;406:378–382.
26. Wagner SM, Sabourin LA. A novel role for the Ste20 kinase SLK in adhe-sion signaling and cell migration. Cell Adh Migr. 2009;3:182–184.
27. Storbeck CJ, Wagner S, O’Reilly P, McKay M, Parks RJ, Westphal H, Sabourin LA. The Ldb1 and Ldb2 transcriptional cofactors interact with the Ste20-like kinase SLK and regulate cell migration. Mol Biol Cell. 2009;20:4174–4182.
28. Lee CH, Chawla A, Urbiztondo N, Liao D, Boisvert WA, Evans RM, Curtiss LK. Transcriptional repression of atherogenic inflammation: mod-ulation by PPARdelta. Science. 2003;302:453–457.
29. Grundy SM. Cholesterol and coronary heart disease. A new era. JAMA. 1986;256:2849–2858.
30. Lieu HD, Withycombe SK, Walker Q, Rong JX, Walzem RL, Wong JS, Hamilton RL, Fisher EA, Young SG. Eliminating atherogen-esis in mice by switching off hepatic lipoprotein secretion. Circulation. 2003;107:1315–1321.
31. Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–1262.
32. Park JG, Ryu SY, Jung IH, Lee YH, Kang KJ, Lee MR, Lee MN, Sonn SK, Lee JH, Lee H, Oh GT, Moon K, Shim H. Evaluation of VCAM-1 antibod-ies as therapeutic agent for atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 2013;226:356–363.
The transendothelial migration of leukocyte (TEML) pathway is important in inflammatory diseases, including atherosclerosis. Several genes in this pathway have been targeted in mice with substantial effects on atherosclerosis development. At least 1 drug (natalizumab) has been developed against the key TEML step of leukocyte adherence. Although natalizumab was effective against targeted diseases (multiple scle-rosis and Crohns disease), it can have severe side effects (opportunistic brain infections), highlighting the physiological importance of TEML as the first barrier against viral and bacterial infections. However, this role does not disqualify TEML as a suitable target for inflammatory diseases. Instead, the risk of severe side effects underscores the importance of controlling the overall TEML activity as opposed to hamper-ing key steps in TEML. From this perspective, our finding of a regulatory gene with overall effects on TEML activity is significant. Lim domain binding 2 and possibly its coregulator SLK (STE-20 like kinase) merit further attention as novel targets to lower TEML activity to prevent coronary and carotid artery disease and possibly other inflammatory diseases.
Significance
at University of Edinburgh (edu) / Scotland on August 26, 2014http://atvb.ahajournals.org/Downloaded from