
Journal of Materials Chemistry A - pku.edu.cn
10
rsc.li/materials-a Journal of Materials Chemistry A Materials for energy and sustainability ISSN 2050-7488 PAPER Qiang Sun et al. Discovery of a high-pressure phase of rutile-like CoO2 and its potential as a cathode material Volume 6 Number 38 14 October 2018 Pages 18329–18730
Transcript of Journal of Materials Chemistry A - pku.edu.cn

rsc.li/materials-a
Journal of Materials Chemistry AMaterials for energy and sustainability
ISSN 2050-7488
PAPERQiang Sun et al.Discovery of a high-pressure phase of rutile-like CoO2 and its potential as a cathode material
Volume 6 Number 38 14 October 2018 Pages 18329–18730

Journal ofMaterials Chemistry A
PAPER
Publ
ishe
d on
06
Aug
ust 2
018.
Dow
nloa
ded
by P
ekin
g U
nive
rsity
on
8/7/
2019
9:4
6:40
AM
.
View Article OnlineView Journal | View Issue
Discovery of a hi
aDepartment of Materials Science and Eng
University, Beijing 100871, China. E-mail: sbCenter for Applied Physics and Technology,cDepartment of Physics, Virginia Commonwe
† Electronic supplementary informa10.1039/c8ta05840g
Cite this: J. Mater. Chem. A, 2018, 6,18449
Received 19th June 2018Accepted 2nd August 2018
DOI: 10.1039/c8ta05840g
rsc.li/materials-a
This journal is © The Royal Society of C
gh-pressure phase of rutile-likeCoO2 and its potential as a cathode material†
Shuo Wang, a Junyi Liu, a Yu Qie,a Sheng Gong,a Qiang Sun *abc
and Puru Jena c
Due to the structural failure of layered CoO2, there has been considerable effort to improve the reversible
capacity of commercial LiCoO2 cathode materials. Using a global structural search, we have discovered
a rutile-like CoO2 phase as the ground state at 50 GPa. The material is thermally, mechanically and
dynamically stable, even after removing the pressure. We show that, when lithiated, this LiCoO2 material
can serve as an improved cathode material for Li-ion batteries. Its many advantages include good
structural stability against transition metal migration and oxygen loss, one-dimensional channels for Li
ion diffusion with variable energy barriers (0.36–0.86 eV), enhanced ionic mobility, and good intrinsic
electronic conductivity brought about by its metallic band structure. Furthermore, with a voltage
platform of 3.2–3.8 V, it provides better stability against conventional carbonate solvents than
conventional layered cathode materials (>4.0 V). All these features demonstrate that the non-layered
CoO2 phase synthesized at high pressure would be a promising cathode material, providing
a performance beyond that of the currently used layered phase.
Introduction
Layered lithium transition metal (TM) oxides LiMO2 (M ¼ Co,Ni, Mn, etc.)1–3 are the most widely used cathode materials incommercial Li-ion batteries (LiBs). In particular, LiCoO2,introduced by Goodenough and Mizushima4 in the 1980s,shows great electrochemical performance, such as high speciccapacity, high voltage, low over-potential and good revers-ibility.5 But in practical operations only 50% (�140 mA h g�1)6,7
of the theoretical capacity (273 mA h g�1) could be reached inLiCoO2/C Li-ion cells. This is because of the strong repulsion8
between the CoO2 layers and metastable “Li–Co dumbbell”intermediates9 that occurs in the absence of Li atoms. Thisleads to phase transition, oxygen release, and structural failure.Scientists and engineers have made numerous efforts to pushthis limit further via chemical substitution, composite struc-tures and so on, aiming to stabilize the delithiated layeredframework.10–12 As a result, many modied and optimizedcathode materials have been developed by using a layeredstructure with improved capacity (>170 mA h g�1) such as NCA(LiNixCoyAl1�x�yO2),13 NCM (LiNixCoyMn1�x�yO2),14 and[LiCoO2]x/[Li2MnO3]1�x composites.15 However, the funda-mental problem of the instability of the delithiated layered
ineering, College of Engineering, Peking
Peking University, Beijing 100871, China
alth University, Richmond, VA 23284, USA
tion (ESI) available. See DOI:
hemistry 2018
structure has never been solved and a reversible theoreticalcapacity is still not achieved.
To overcome such a limitation, it is necessary to develop newcathode materials to maximize the reversible capacity. Cobaltusually has +2 or +3 valence states in oxides such as Co3O4,Co2O3 and CoO; however, it seldom has a +4 valence state exceptin layered CoO2 obtained by the de-intercalation of LixCoO2
phases. In this paper, we follow a different strategy and explorethe merits of high-pressure synthesis. Note that high pressure isan effective way to change atomic bonding features because itcan overcome the chemical reactivity barrier,16 change inter-atomic distances and bonding patterns,17 reorder the atomicorbitals,18 and induce structural phase transitions.19 Thus,synthesizing materials under high pressure could make theimpossible possible! A classic example is graphite vs. diamond;at high pressure, stable graphite turns into metastable diamonddue to the reversal of the energetic order of the two carbonallotropes. Other examples include H3S with a superconductingtemperature of 203 K at 155 GPa,20 super-hard nano-twinneddiamond with a Vickers hardness of 200 GPa,21 high-energy-density cg-N above 110 GPa (ref. 22) (single-bond cubic form ofnitrogen), etc. In these successful cases, rst-principles calcu-lations and structure predictionmethods have played a key role,providing guidance for experimental explorations.
Encouraged by these advances, we explore the possibility ofachieving a new delithiated cathode material, a CoO2 phase,that is stable at high pressure. Using density functional theorycombined with a global structural search, we discover a rutile-like CoO2 phase to be the ground state at 50 GPa. Equally
J. Mater. Chem. A, 2018, 6, 18449–18457 | 18449

Journal of Materials Chemistry A Paper
Publ
ishe
d on
06
Aug
ust 2
018.
Dow
nloa
ded
by P
ekin
g U
nive
rsity
on
8/7/
2019
9:4
6:40
AM
. View Article Online
important, it remains stable dynamically and thermally, evenaer the pressure is removed. Compared to the layered-struc-ture LiCoO2, the new phase is more stable against oxygenrelease as well as TM displacement. Furthermore, it hasa metallic band structure with a desirable charge/dischargevoltage platform, moderate ionic diffusion barriers andenhanced ionic mobility. In addition, the intrinsic stability andconsiderable ionic diffusivity in the delithiated sample provideimprovement in energy density over traditional cathode mate-rials. The results reinforce the great merit of exploring newcathode materials using high-pressure synthesis techniques.
Methods
Using the CALYPSO (Crystal structure AnaLYsis by ParticleSwarm Optimization) structure prediction package23 anddensity functional theory (DFT) as implemented in the ViennaAb Initio Simulation Package (VASP),24 we performed a globalstructural search at high pressure for stable/metastable struc-tures of a given chemical composition (e.g. CoO2). To simulatethe actual battery operating environment, pressure is removedin subsequent calculations. The projector augmented-wave(PAW) method,25 Perdew–Burke–Ernzerhof (PBE)26 generalized-gradient approximation (GGA) for exchange and correlationpotential, and GGA+U scheme (with a U of 4.91 eV for Co3+/Co4+)27 are used. The structure is relaxed without symmetricrestraint using a conjugated gradient method. The convergencecriteria for total energy and Hellmann–Feynman force are set to1 � 10�4 eV, and 1.0 � 10�2 eV A�1, respectively. The electronicdensity of states is calculated by employing a 10 � 11 � 15Monkhorst–Pack k-point grid.28 A more accurate band structureis also calculated using the Heyd–Scuseria–Ernzerhof (HSE06)29
functional. The dynamical stability is veried by calculating thevibrational spectra using a nite displacement approach asimplemented in the phonopy code.30 The thermodynamicstability is conrmed with ab initio molecular dynamics (AIMD)simulations in the canonical (NVT) ensemble using the Nose–Hoover heat bath scheme (500 K) in a 3 � 3 � 3 supercell. Thenite difference method31 is used to calculate the Hessianmatrix. The mechanical stability is proved using the criteria foran orthorhombic phase.32
Diffusion barriers are calculated using the climbing image-nudged elastic band (CI-NEB) method33 within a 2 � 2 � 4supercell, while lattice vectors are xed to the defect-free relaxedparameters. Furthermore, AIMD simulations lasting for 20 ps attemperatures from 700 K to 2500 K are carried out in xedlattice to study Li-ion transport. Elevated temperatures are usedto speed up the ion-hopping process. The mean squaredisplacement (MSD) is calculated as shown below:34
Dhr.ðtÞ
i2E¼ 1
N
XNi¼1
D½ r.iðtþ t0Þ � r
.iðt0Þ�2
E(1)
where r.ðtÞ is the displacement of the ith Li+ ion at time t and N
is the total number of Li+ ions in the system. The diffusioncoefficient (D) at each temperature is calculated by tting to theMSD according to
18450 | J. Mater. Chem. A, 2018, 6, 18449–18457
D ¼ limt/N
�1
2dt
Dhr.ðtÞ
i2E�; (2)
where d is the dimensionality of diffusion and t is the elapsedsimulation time. From the Nernst–Einstein relation, theconductivity (s) can be calculated as
s ¼ DNe2
kT(3)
N is the number of ion pairs per cm3; other symbols have theircustomary meaning. The activation energy Ea is obtained byusing the Arrhenius model as shown below:
s ¼ A
Texp
�Ea
kT
�(4)
here, A is the constant tting parameter for a specied crystallattice.
In order to nd the most energy-favorable conguration atdifferent Li concentrations, we use the cluster expansion (CE)method to describe the congurational energy. All structuresare fully relaxed in the CE calculation to determine stableintermediate congurations. In our work, LixCoO2 is taken asan alloy system and each possible Li site i is represented by anoccupation variable si, which takes the value 1 if Li resides atthat site and �1 if a vacancy is at that site. The conguration-dependent Hamiltonian can be mapped onto a generalizedIsing Hamiltonian as shown below:
EðsÞ ¼ J0 þXi
Jisi þXj\i
Jijsisj þXk\j\i
Jijksijk þ.
¼ J0 þXa
Ja4a (5)
here the subscripts i, j, and k range over all occupation sites, 4a
is the product of occupation variables si, sj/sk that forma cluster a, which can be a single point, a pair cluster, a triplecluster, etc. Ja is the corresponding effective cluster interactionwhose value converges to zero as the size or distance betweenthe clusters increases. This is obtained by tting them to theenergies of selected congurations calculated from rst-prin-ciples. The tted CE quality is measured using the cross-vali-dation (CV) score, and the tting process is carried out via theAlloy Theoretic Automated Toolkit (ATAT) code.35
Results and discussion1. Structural stability and electronic properties
We perform a global structural search for CoO2 at 50 GPa, whichyielded the ground-state structure to be similar to that of rutileTiO2. However, compared to the standard-rutile structure, wend distortion in the structure of CoO2 with the symmetrychanging from P42/mnm to Pnnm. To conrm the origin of thisdistortion, we calculated the phonon spectra for standard rutileCoO2 under vacuum and at 50 GPa. The results are shown inFig. S1(a),† where the imaginary frequencies exist under bothvacuum and high pressure, suggesting the instability of theregular rutile-type CoO2. The imaginary phonon with lowfrequency comes from the rotation of Co–O octahedra aroundthe octahedral center. Because of the different electronic
This journal is © The Royal Society of Chemistry 2018

Paper Journal of Materials Chemistry A
Publ
ishe
d on
06
Aug
ust 2
018.
Dow
nloa
ded
by P
ekin
g U
nive
rsity
on
8/7/
2019
9:4
6:40
AM
. View Article Online
distribution between Ti (3d24s2) and Co (3d74s2), the octahedrain CoO2 are not regular as those in TiO2. The percentage devi-ation between long and short lattice edges is used as a referenceto measure the level of distortion. Pressure dependence of thedistortion is plotted in Fig. S1(b),† showing that the distortionsrst get enhanced quickly with increasing pressure and thenslow down at high pressure. All these results prove that distor-tion is necessary for the stabilization of rutile-like CoO2 at highpressure. Furthermore, this structure remains intact even aerthe pressure is removed, as shown in Fig. 1(a). Here, Co and Ooccupy 2a and 4g sites, respectively. Details of lattice parame-ters and atomic coordinates are given in Table S1.† To explorethe effect of pressure on materials, pressure dependence ofenthalpy curves with respect to the experimental delithiatedLixCoO2 is calculated (Fig. S2†). We nd the ground-state energyto reverse with increasing pressure to 7 GPa, implying a possiblephase transition. This suggests that it is highly feasible tosynthesize a rutile-like CoO2 phase by compression. This is alsoconrmed by calculating the formation energy of CoO2, asshown below:
Co(s) + O2(g) / CoO2(s) (6)
DH ¼ ECoO2(s)� EO2(g)
� ECo(s) (7)
here, E is the total energy per f.u. Because the accurate energy ofO2 is difficult to calculate directly due to the overestimatedbinding effect of the O2 molecule in the DFT–GGA calculation,36
we obtain the energy of the O2 molecule using the followingequation:
E(O2) ¼ 2EDFT(H2O) � 2EDFT(H2) � 2DEexp(H2O) (8)
Fig. 1 (a) Crystal structure, (b) band structures and partial density offluctuation as a function of time at 500 K, (d) phonon dispersion of rutil
This journal is © The Royal Society of Chemistry 2018
The formation energy is found to be �2.51 eV, which meansthat the chemical reaction can take place. Because oxygenrelease has always been a critical problem during deep deli-thiation, we study oxygen stability by exploring the possibility ofoxygen loss in the following process:
CoO2ðsÞ/CoO2�yðsÞ þ y
2O2ðgÞ (9)
We calculate the resulting change of enthalpy as
DH ¼ CoO2�yðsÞ þ ðy=2ÞEO2� CoO2ðsÞ
y=2(10)
The value is found to be +0.21 eV, which suggests that theoxygen-release reaction is energetically unfavorable, even underfully delithiated conditions. In addition, we simulated theformation of an oxygen dimer between adjacent O atoms ina layered and rutile-like structure, respectively. Aer optimiza-tion, we see from Fig. S3† that Co is displaced and the oxygen-dimer remains in layered LiCoO2. For the rutile-like structure,the displaced O atoms reset themselves and the primaryframework is recovered. Furthermore, the AIMD simulation at1500 K is conducted to compare the relative stability betweenlayered and rutile-like structures; the results are shown inFig. S4.† Oxygen release and Co migration occur in the layeredstructure within 200 fs while the rutile-like framework with-stands thermal disturbance without the formation of theoxygen-dimer and migration of Co, even aer 5000 fs. We thinkthis is because of the combination of the large interspace andbreaking of bonds of O atoms in octahedral coordination in thelayered structure, which provides the possibility for structural
states calculated using PBE and HSE06 functionals, (c) AIMD Energye-like CoO2.
J. Mater. Chem. A, 2018, 6, 18449–18457 | 18451

Journal of Materials Chemistry A Paper
Publ
ishe
d on
06
Aug
ust 2
018.
Dow
nloa
ded
by P
ekin
g U
nive
rsity
on
8/7/
2019
9:4
6:40
AM
. View Article Online
reconstruction. All the above results indicate that the rutile-likestructure has good stability against oxygen loss.
To examine the thermodynamic stability further, ab initiomolecular dynamics (AIMD) simulation is carried out in a 3 � 3� 3 supercell at 500 K using the canonical ensemble (NVT).Total energy uctuation as a function of time is plotted inFig. 1(c). In the inset, we present the geometry aer ten-pico-second relaxation. The average total potential remains nearlyconstant and no structural reconstruction occurs, conrmingthat rutile CoO2 is thermally stable. To verify its dynamicalstability, we calculated the phonon frequencies; the results areshown in Fig. 1(d). Note that there is no imaginary mode in thevibrational spectra in the entire Brillouin zone, which showsthat the rutile CoO2 is dynamically stable. Besides, themechanical stability is also established according to the criteriafor an orthorhombic phase:37
C11 > 0, C22 > 0, C33 > 0, C44 > 0, C55 > 0, C66 > 0,
[C11 + C22 + C33 + 2(C12 + C13 + C23)] > 0,
(C11 + C22 � 2C12) > 0, (C22 + C33 � 2C23) > 0,
(C11 + C33 � 2C13) > 0 (11)
The detailed values of Cxy are listed in Table S2.† Clearly,they satisfy the mechanical stability criteria, and the rutile CoO2
phase is also mechanically stable. Using Voigt–Reuss–Hillapproximation, the bulk modulus (B) and shear modulus (G) ofCoO2 are also calculated. The results are listed in Table 1. Onecan see that the Pnnm CoO2 phase shows comparablemechanical properties with those of standard-rutile TiO2, whichhas been found to have desirable mechanical stability38 whenused as an anode in Li/Na ion batteries.
For a cathode material, a crucial property is electronicconductivity. For example, the practical application of phos-phates (LiFePO4) is held back due to their low intrinsic elec-tronic and ionic conductivities. To further understand theelectronic properties of the studied system, we calculated theband structure and partial density of states (DOS) of rutile CoO2
using the GGA+U (U ¼ 4.91 eV) level of theory. The results aregiven in Fig. 1(b). Different from semi-conducting rutile TiO2,the rutile-like CoO2 is metallic with the O 2p-orbital and Co 3d-orbital crossing the Fermi level. Such a metallic transition wasalso seen in our previous work on a high-pressure Li2MnO3
phase.39 Because pressure usually reduces interatomicdistances and consequently enhances atomic interactions, theenergy bands are broadened at high pressure. The resultingband overlap will oen induce an insulator-to-metal or a semi-conductor-to-metal transition. Such intrinsic metallicity wouldimprove the electronic response during the charge/discharge
Table 1 Bulk modulus (B), shear modulus (G), Young's modulus (E) inGPa and Poisson's ratio (n) of CoO2 and TiO2. Subscript V denotes theVoigt bound, R denotes the Reuss bound and H denotes the Hillaverage
Materials Symmetry BV BR GV GR BH GH E n
CoO2 Pnnm 194 142 130 83 168 107 265 0.23TiO2 P42/mnm 214 204 123 96 209 110 280 0.28
18452 | J. Mater. Chem. A, 2018, 6, 18449–18457
process. More accurate band structure calculations using theHSE06 functional authenticate the metallicity of rutile-likeCoO2 (Fig. 2).
2. Li-ion storage and ionic mobility
Due to the different chemical bonding characteristics betweenCo and Ti, the one-dimensional channels in rutile-like CoO2, asshown in Fig. 4(c), are not rectangular as those in rutile TiO2.The Li atom tends to be at the central position of the one-dimensional channels where the energy is lower and sterichindrance is smaller. Aer all the optimal sites are occupied,the insertion of additional Li atoms would cause positivebinding energy, which means that the half-cell lithiation reac-tion cannot proceed spontaneously because of the strongrepulsive interaction between Li ions. The chemical formulaends up with LiCoO2 aer full lithiation, leading to a theoreticalcapacity of 273 mA h g�1. Aer lithiation, we nd the crystallattice to expand along the x (from 4.41 A to 4.68 A) and y (from4.10 A to 4.67 A) axes, but remains nearly intact along the zdirection. To verify the mechanical stability of the CoO2
frameworks during the lithiation/delithiation process, wecalculated the compressive and tensile stress (s) as a function ofuniaxial strain along the x and y directions and plotted theresults in Fig. 3. The stress shows an approximate linear growthwith increasing compressive and tensile strains along both xand y directions, indicating that the rutile CoO2 framework cansustain the strain caused by the volume change during thecharge/discharge process, thus preventing mechanical failure.
Based on the lithiated conguration, the Li migration energybarriers are calculated using CI-NEB. The corresponding resultsand diagrammatic sketches are shown in Fig. 3. Li ions, shut-tling between the channels, have much higher barrier (>3 eV)than those diffusion along the channels. The diffusion coeffi-cient (D) can be evaluated using eqn (3) and (4). Based on thebarrier difference, the mobility of Li within the one-dimen-sional channel is at least 5.5 � 1034 higher than that of ionicdiffusion between channels at room temperature. Therefore, weexpect that the Li ions should show a one-dimensional mobilebehavior. This expectation is also conrmed in the followingAIMD calculation.
To simulate the Li ion diffusion, a mono-vacancy model isusually used. However, the distribution and concentration ofvacancies can impact ionic motion as found in sodium-iondiffusion in layered P2 and P3 oxides.40 In the actual chargingprocess, more vacancies are introduced which would lead tovacancy clustering because Li ion movements in the one-dimensional diffusion channels may lag behind. To simulatesuch a situation, we adopted a tri-vacancy model. The energybarrier for single atom diffusion is 0.79 eV. The ionic diffusionbarrier is similar (0.86 eV) in the mono-vacancy model butdifferent (0.36 eV) in the tri-vacancy model. Our results arecomparable to those of layered-structure lithium metal oxidessuch as LixCoO2 (0.36–0.76 eV), LixAlO2 (0.65–1.41 eV), LixMnO3
(0.51–0.84 eV), LiNixMnyCo1�x�yO2 (NMC, 0.75–0.90 eV), etc. Fora better understanding of the underlying reasons for differentionic diffusion behaviors in mono-vacancy and tri-vacancy
This journal is © The Royal Society of Chemistry 2018
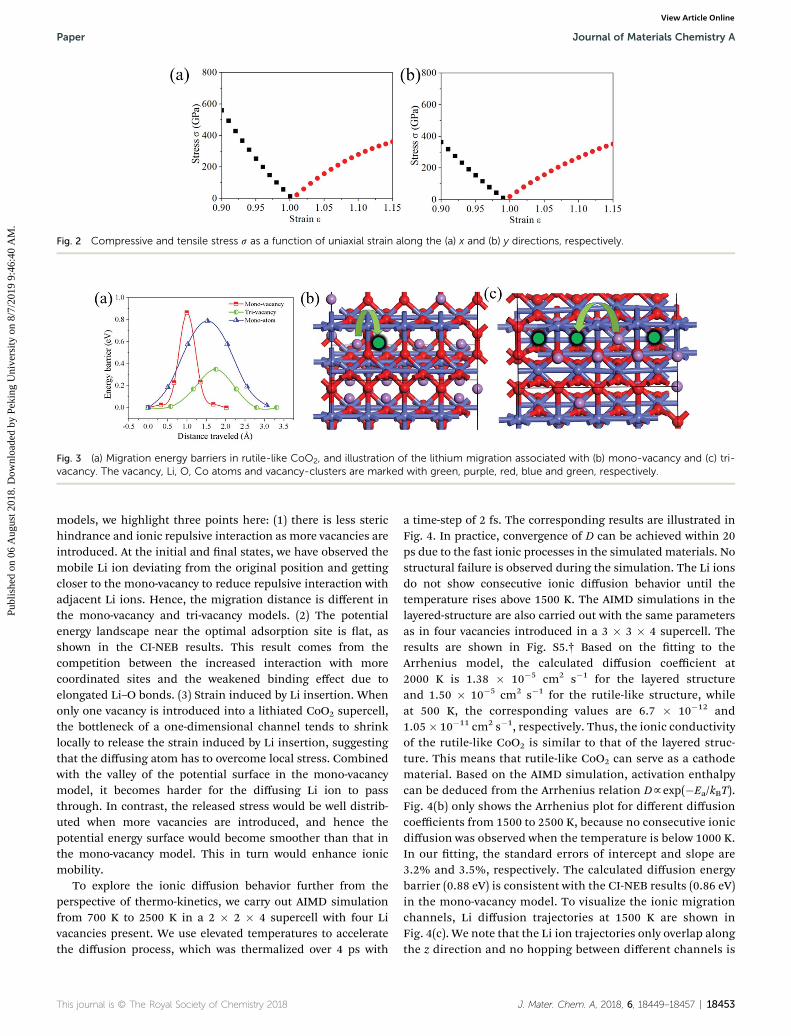
Fig. 2 Compressive and tensile stress s as a function of uniaxial strain along the (a) x and (b) y directions, respectively.
Fig. 3 (a) Migration energy barriers in rutile-like CoO2, and illustration of the lithium migration associated with (b) mono-vacancy and (c) tri-vacancy. The vacancy, Li, O, Co atoms and vacancy-clusters are marked with green, purple, red, blue and green, respectively.
Paper Journal of Materials Chemistry A
Publ
ishe
d on
06
Aug
ust 2
018.
Dow
nloa
ded
by P
ekin
g U
nive
rsity
on
8/7/
2019
9:4
6:40
AM
. View Article Online
models, we highlight three points here: (1) there is less sterichindrance and ionic repulsive interaction as more vacancies areintroduced. At the initial and nal states, we have observed themobile Li ion deviating from the original position and gettingcloser to the mono-vacancy to reduce repulsive interaction withadjacent Li ions. Hence, the migration distance is different inthe mono-vacancy and tri-vacancy models. (2) The potentialenergy landscape near the optimal adsorption site is at, asshown in the CI-NEB results. This result comes from thecompetition between the increased interaction with morecoordinated sites and the weakened binding effect due toelongated Li–O bonds. (3) Strain induced by Li insertion. Whenonly one vacancy is introduced into a lithiated CoO2 supercell,the bottleneck of a one-dimensional channel tends to shrinklocally to release the strain induced by Li insertion, suggestingthat the diffusing atom has to overcome local stress. Combinedwith the valley of the potential surface in the mono-vacancymodel, it becomes harder for the diffusing Li ion to passthrough. In contrast, the released stress would be well distrib-uted when more vacancies are introduced, and hence thepotential energy surface would become smoother than that inthe mono-vacancy model. This in turn would enhance ionicmobility.
To explore the ionic diffusion behavior further from theperspective of thermo-kinetics, we carry out AIMD simulationfrom 700 K to 2500 K in a 2 � 2 � 4 supercell with four Livacancies present. We use elevated temperatures to acceleratethe diffusion process, which was thermalized over 4 ps with
This journal is © The Royal Society of Chemistry 2018
a time-step of 2 fs. The corresponding results are illustrated inFig. 4. In practice, convergence of D can be achieved within 20ps due to the fast ionic processes in the simulated materials. Nostructural failure is observed during the simulation. The Li ionsdo not show consecutive ionic diffusion behavior until thetemperature rises above 1500 K. The AIMD simulations in thelayered-structure are also carried out with the same parametersas in four vacancies introduced in a 3 � 3 � 4 supercell. Theresults are shown in Fig. S5.† Based on the tting to theArrhenius model, the calculated diffusion coefficient at2000 K is 1.38 � 10�5 cm2 s�1 for the layered structureand 1.50 � 10�5 cm2 s�1 for the rutile-like structure, whileat 500 K, the corresponding values are 6.7 � 10�12 and1.05 � 10�11 cm2 s�1, respectively. Thus, the ionic conductivityof the rutile-like CoO2 is similar to that of the layered struc-ture. This means that rutile-like CoO2 can serve as a cathodematerial. Based on the AIMD simulation, activation enthalpycan be deduced from the Arrhenius relation Dfexp(�Ea/kBT).Fig. 4(b) only shows the Arrhenius plot for different diffusioncoefficients from 1500 to 2500 K, because no consecutive ionicdiffusion was observed when the temperature is below 1000 K.In our tting, the standard errors of intercept and slope are3.2% and 3.5%, respectively. The calculated diffusion energybarrier (0.88 eV) is consistent with the CI-NEB results (0.86 eV)in the mono-vacancy model. To visualize the ionic migrationchannels, Li diffusion trajectories at 1500 K are shown inFig. 4(c). We note that the Li ion trajectories only overlap alongthe z direction and no hopping between different channels is
J. Mater. Chem. A, 2018, 6, 18449–18457 | 18453
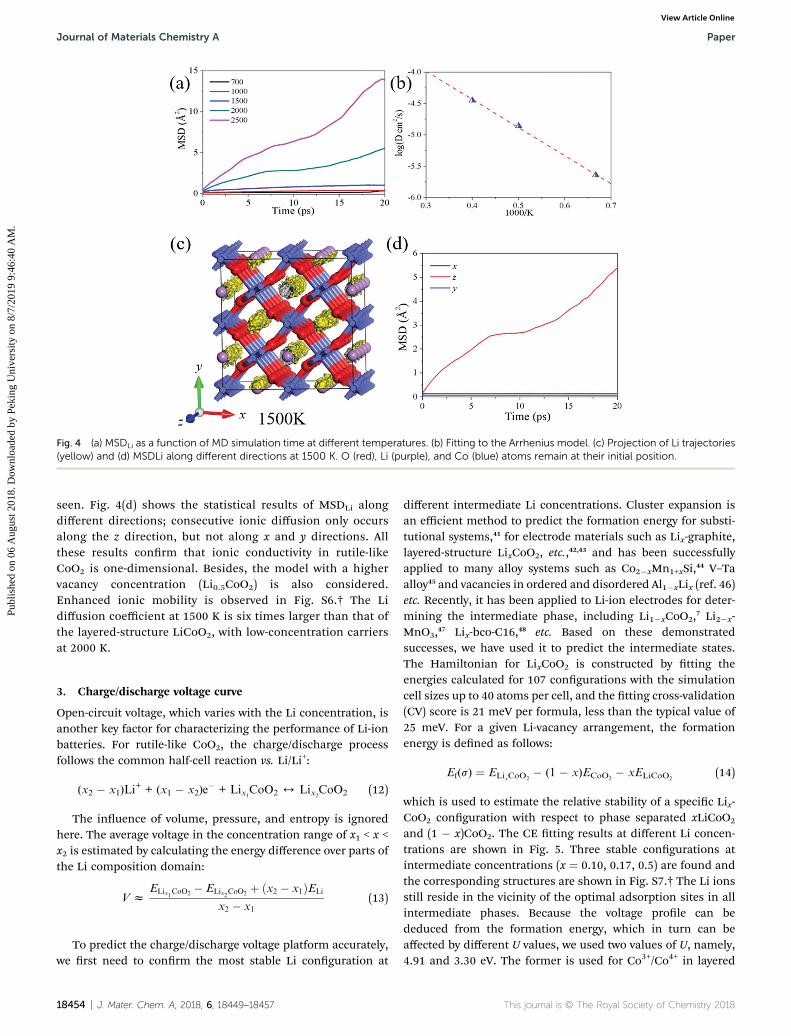
Fig. 4 (a) MSDLi as a function of MD simulation time at different temperatures. (b) Fitting to the Arrhenius model. (c) Projection of Li trajectories(yellow) and (d) MSDLi along different directions at 1500 K. O (red), Li (purple), and Co (blue) atoms remain at their initial position.
Journal of Materials Chemistry A Paper
Publ
ishe
d on
06
Aug
ust 2
018.
Dow
nloa
ded
by P
ekin
g U
nive
rsity
on
8/7/
2019
9:4
6:40
AM
. View Article Online
seen. Fig. 4(d) shows the statistical results of MSDLi alongdifferent directions; consecutive ionic diffusion only occursalong the z direction, but not along x and y directions. Allthese results conrm that ionic conductivity in rutile-likeCoO2 is one-dimensional. Besides, the model with a highervacancy concentration (Li0.5CoO2) is also considered.Enhanced ionic mobility is observed in Fig. S6.† The Lidiffusion coefficient at 1500 K is six times larger than that ofthe layered-structure LiCoO2, with low-concentration carriersat 2000 K.
3. Charge/discharge voltage curve
Open-circuit voltage, which varies with the Li concentration, isanother key factor for characterizing the performance of Li-ionbatteries. For rutile-like CoO2, the charge/discharge processfollows the common half-cell reaction vs. Li/Li+:
(x2 � x1)Li+ + (x1 � x2)e
� + Lix1CoO2 4 Lix2
CoO2 (12)
The inuence of volume, pressure, and entropy is ignoredhere. The average voltage in the concentration range of x1 < x <x2 is estimated by calculating the energy difference over parts ofthe Li composition domain:
V zELix1CoO2
� ELix2CoO2þ ðx2 � x1ÞELi
x2 � x1
(13)
To predict the charge/discharge voltage platform accurately,we rst need to conrm the most stable Li conguration at
18454 | J. Mater. Chem. A, 2018, 6, 18449–18457
different intermediate Li concentrations. Cluster expansion isan efficient method to predict the formation energy for substi-tutional systems,41 for electrode materials such as Lix-graphite,layered-structure LixCoO2, etc.,42,43 and has been successfullyapplied to many alloy systems such as Co2�xMn1+xSi,44 V–Taalloy45 and vacancies in ordered and disordered Al1�xLix (ref. 46)etc. Recently, it has been applied to Li-ion electrodes for deter-mining the intermediate phase, including Li1�xCoO2,7 Li2�x-MnO3,47 Lix-bco-C16,48 etc. Based on these demonstratedsuccesses, we have used it to predict the intermediate states.The Hamiltonian for LixCoO2 is constructed by tting theenergies calculated for 107 congurations with the simulationcell sizes up to 40 atoms per cell, and the tting cross-validation(CV) score is 21 meV per formula, less than the typical value of25 meV. For a given Li-vacancy arrangement, the formationenergy is dened as follows:
Ef(s) ¼ ELixCoO2� (1 � x)ECoO2
� xELiCoO2(14)
which is used to estimate the relative stability of a specic Lix-CoO2 conguration with respect to phase separated xLiCoO2
and (1 � x)CoO2. The CE tting results at different Li concen-trations are shown in Fig. 5. Three stable congurations atintermediate concentrations (x ¼ 0.10, 0.17, 0.5) are found andthe corresponding structures are shown in Fig. S7.† The Li ionsstill reside in the vicinity of the optimal adsorption sites in allintermediate phases. Because the voltage prole can bededuced from the formation energy, which in turn can beaffected by different U values, we used two values of U, namely,4.91 and 3.30 eV. The former is used for Co3+/Co4+ in layered
This journal is © The Royal Society of Chemistry 2018

Fig. 5 (a) Formation energies predicted by the CE method for the 107different Li configurations with three stable intermediate phases and(b) the corresponding voltage profile with different U values (red/bluefor 4.91/3.32) along the minimum energy path.
Paper Journal of Materials Chemistry A
Publ
ishe
d on
06
Aug
ust 2
018.
Dow
nloa
ded
by P
ekin
g U
nive
rsity
on
8/7/
2019
9:4
6:40
AM
. View Article Online
LiCoO2 which agrees well with the experiment49 for the voltageprole. The latter value is used by tting to the experimentaloxidation energies of cobalt oxides.50 The open-circuit voltagecurve is presented in Fig. 5(b). The voltage rises with increasingdelithiation and four voltage regions are displayed. The averagevoltage is 3.7 V for U ¼ 4.91, which is slightly less than thecalculated value (4.0–4.2 V) by Zhou et al.51 using the same U forthe layered-structure. For U¼ 3.30, the maximum voltage is lessthan 3.5 V. The results show that rutile-like CoO2 may providebetter safety due to the stability of conventional carbonatesolvents at low voltage (<4 V).52 Since the energy density isdetermined by the integral of voltage and reversible capacity,when combining intrinsic deep-delithiation stability with anappropriate voltage platform, the rutile-like CoO2 might showimproved energy density as a cathode over the traditionallayered-structure.
Conclusions
In this work, using a global crystal structural search methodcombined with rst-principles calculations, we have identieda stable delithiated high-pressure phase of CoO2. This rutile-likeCoO2 is found to be the ground state at 50 GPa and remainsstable dynamically, thermally and mechanically even aer
This journal is © The Royal Society of Chemistry 2018
pressure is removed. We have studied the potential of this rutileLiCoO2 as a cathode material for Li-ion batteries. We nd it tohave the following advantages over the conventional layeredstructure: (1) better structural stability against oxygen loss andTM displacement than the layered structure, (2) goodmechanical strength to endure volume expansion during thecharge/discharge process, (3) one-dimensional diffusion chan-nels for Li ions with preferred energy barriers (0.36–0.86 eV) andenhanced ionic mobility, (4) a good electronic conductivity dueto the intrinsic metallic band structure, and (5) an improvedsafety against conventional carbonate solvents due to theappropriate average voltage platform (3.2–3.8 V). We hope thatthese encouraging results will stimulate further theoretical andexperimental efforts to develop novel cathode materials usinghigh-pressure synthesis.
Conflicts of interest
The authors declare no competing nancial interest.
Acknowledgements
This work is partially supported by grants from the National KeyResearch and Development Program of China(2016YFB0100200), and from the National Natural ScienceFoundation of China (21573008 and 21773003). P. J. acknowl-edges the support by the U.S DOE, Office of Basic EnergySciences, Division of Material Sciences and Engineering underAward No. DE-FG02-96ER45579. Part of the numerical simula-tions was performed on the High-Performance ComputingPlatform of Peking University and High-performanceComputing Platform of CAPT.
References
1 Y. Wang and G. Cao, Developments in NanostructuredCathode Materials for High-Performance Lithium-IonBatteries, Adv. Mater., 2008, 20(12), 2251–2269.
2 G. Ceder, Y. M. Chiang, D. R. Sadoway, M. K. Aydinol,Y. I. Jang and B. Huang, Identication of cathode materialsfor lithium batteries guided by rst-principles calculations,Nature, 1998, 392(6677), 694–696.
3 J. W. Fergus, Recent developments in cathode materials forlithium ion batteries, J. Power Sources, 2010, 195(4), 939–954.
4 K. Mizushima, P. C. Jones, P. J. Wiseman andJ. B. Goodenough, LixCoO2 (0 < x < �1): A new cathodematerial for batteries of high energy density, Mater. Res.Bull., 1980, 15(6), 783–789.
5 A. Yoshino, Y. Takizawa, A. Koyama, K. Inoue, M. Yamashita,Y. Minato and I. Kuribayashi, in. Secondary battery, US, 1997.
6 S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki,T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara,Electrochemical Na Insertion and Solid ElectrolyteInterphase for Hard-Carbon Electrodes and Application toNa-Ion Batteries, Adv. Funct. Mater., 2011, 21(20), 3859–3867.
7 A. V. D. Ven, M. K. Aydinol, G. Ceder, G. Kresse and J. Hafner,First-principles investigation of phase stability in LixCoO2,
J. Mater. Chem. A, 2018, 6, 18449–18457 | 18455

Journal of Materials Chemistry A Paper
Publ
ishe
d on
06
Aug
ust 2
018.
Dow
nloa
ded
by P
ekin
g U
nive
rsity
on
8/7/
2019
9:4
6:40
AM
. View Article Online
Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58(6), 2975–2987.
8 P. Rozier and J. M. Tarascon, Review—Li-Rich Layered OxideCathodes for Next-Generation Li-Ion Batteries: Chances andChallenges, J. Electrochem. Soc., 2015, 162(14), A2490–A2499.
9 T. Zhang, D. Li, Z. Tao and J. Chen, Understanding electrodematerials of rechargeable lithium batteries via DFTcalculations, Prog. Nat. Sci.: Mater. Int., 2013, 23(3), 256–272.
10 E. J. Wu, P. D. Tepesch and G. Ceder, Size and charge effectson the structural stability of LiMO2 (M ¼ transition metal)compounds, Philos. Mag. B, 1998, 77(4), 1039–1047.
11 H. Yu and H. Zhou, High-Energy Cathode Materials(Li2MnO3�LiMO2) for Lithium-Ion Batteries, J. Phys. Chem.Lett., 2013, 4(8), 1268.
12 L. Jun, P. Qing, W. Weiyang, N. Caiyun, L. Lihong andL. Yadong, Nanoscale Coating of LiMO2 (M ¼ Ni, Co, Mn)Nanobelts with Li+-Conductive Li2TiO3: Toward Better RateCapabilities for Li-Ion Batteries, J. Am. Chem. Soc., 2013,135(5), 1649.
13 G. Jeong, Y. U. Kim, H. Kim, Y. J. Kim and H. J. Sohn,Prospective materials and applications for Li secondarybatteries, Energy Environ. Sci., 2011, 4(6), 1986–2002.
14 N. Nitta, F. Wu, J. T. Lee and G. Yushin, Li-ion batterymaterials: present and future, Mater. Today, 2015, 18(5),252–264.
15 Z. Lu, A. Zhaohui Chen and J. R. Dahn, Lack of CationClustering in Li[NixLi1/3�2x/3Mn2/3�x/3]O2 (0 < x # 1/2)and Li[CrxLi(1�x)/3Mn(2�2x)/3]O2 (0 < x < 1), Chem.Mater., 2003, 15, 3214–3220.
16 E. Zurek, R. Hoffmann, N. W. Ashcro, A. R. Oganov andA. O. Lyakhov, A little bit of lithium does a lot forhydrogen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(42),17640–17643.
17 G. Yang, Y. Wang, F. Peng, A. Bergara and Y. Ma, Gold asa 6p-element in Dense Lithium Aurides, J. Am. Chem. Soc.,2016, 138(12), 4046.
18 Y. Ma, M. Eremets, A. R. Oganov, Y. Xie, I. Trojan,S. Medvedev, A. O. Lyakhov, M. Valle and V. Prakapenka,Transparent dense sodium, Nature, 2009, 458(7235), 182.
19 M. Miao, Cs in high oxidation states and as a p-blockelement, Nat. Chem., 2013, 5(10), 846–852.
20 A. P. Drozdov, M. I. Eremets, I. A. Troyan, V. Ksenofontovand S. I. Shylin, Conventional superconductivity at 203kelvin at high pressures in the sulfur hydride system,Nature, 2015, 525(7567), 73.
21 Q. Huang, D. Yu, B. Xu, W. Hu, Y. Ma, Y. Wang, Z. Zhao,B. Wen, J. He and Z. Liu, Nanotwinned diamond withunprecedented hardness and stability, Nature, 2014,510(7504), 250.
22 C. Mailhiot, L. H. Yang, A. K. Mcmahan and T. W. B. Iii,Polymeric nitrogen, Phys. Rev. B: Condens. Matter Mater.Phys., 1994, 46(22), 14419–14435.
23 Y. Wang, J. Lv, L. Zhu and Y. Ma, Crystal Structure Predictionvia Particle Swarm Optimization, Phys. Rev. B: Condens.Matter Mater. Phys., 2010, 82(9), 7174–7182.
24 G. Kresse and J. Furthmuller, Efficient iterative schemes forab initio total-energy calculations using a plane-wave basis
18456 | J. Mater. Chem. A, 2018, 6, 18449–18457
set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16),11169.
25 P. E. Blochl, Projector augmented-wave method, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50(24), 17953.
26 J. P. Perdew, K. Burke and M. Ernzerhof, Generalizedgradient approximation made simple, Phys. Rev. Lett.,1996, 77(18), 3865.
27 K. Hoang and M. D. Johannes, Defect chemistry in layeredtransition-metal oxides from screened hybrid densityfunctional calculations, J. Mater. Chem. A, 2014, 2(15),5224–5235.
28 H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13(12),5188.
29 J. Heyd, Hybrid functionals based on a screened Coulombpotential, J. Chem. Phys., 2003, 118(18), 8207–8215.
30 A. Togo, F. Oba and I. Tanaka, First-principles calculationsof the ferroelastic transition between rutile-type and CaCl2-type SiO 2 at high pressures, Phys. Rev. B: Condens.Matter Mater. Phys., 2008, 78(13), 134106.
31 Y. L. Page and P. Saxe, Symmetry-general least-squaresextraction of elastic data for strained materials from abinitio calculations of stress, Phys. Rev. B: Condens. MatterMater. Phys., 2002, 65(10), 104104.
32 Z. J. Wu, E. J. Zhao, H. P. Xiang, X. F. Hao, X. J. Liu andJ. Meng, Crystal structures and elastic properties ofsuperhard IrN2 and IrN3 from rst principles, Phys. Rev. B,2007, 76(5), 054115.
33 G. Henkelman, B. P. Uberuaga and H. Jonsson, A climbingimage nudged elastic band method for nding saddlepoints and minimum energy paths, J. Chem. Phys., 2000,113(22), 9901–9904.
34 A. Emly, E. Kioupakis and A. Van der Ven, Phase Stability andTransport Mechanisms in Antiperovskite Li3OCl and Li3OBrSuperionic Conductors, Chem. Mater., 2013, 25(23), 4663–4670.
35 A. van de Walle and G. Ceder, Automating rst-principlesphase diagram calculations, J. Phase Equilib., 2002, 23(4),348–359.
36 R. Xiao, H. Li and L. Chen, Density Functional Investigationon Li2MnO3, Chem. Mater., 2012, 24(21), 4242–4251.
37 J. F. Nye, Physical properties of crystals. Clarendon Press,1985, pp. 391–397.
38 H. Usui, S. Yoshioka, K. Wasada, M. Shimizu andH. Sakaguchi, Nb-doped rutile TiO2: a potential anodematerial for Na-ion battery, ACS Appl. Mater. Interfaces,2015, 7(12), 6567.
39 S. Wang, J. Liu and Q. Sun, New allotropes of Li2MnO3 ascathode materials with better cycling performancepredicted in high pressure synthesis, J. Mater. Chem. A,2017, 5(32), 16936–16943.
40 S. Guo, Y. Sun, J. Yi, K. Zhu, P. Liu, Y. Zhu, G. Zhu, M. Chen,M. Ishida and H. Zhou, Corrigendum: Understandingsodium-ion diffusion in layered P2 and P3 oxides viaexperiments and rst-principles calculations: a bridgebetween crystal structure and electrochemicalperformance, NPG Asia Mater., 2016, 8(4), e266.
This journal is © The Royal Society of Chemistry 2018

Paper Journal of Materials Chemistry A
Publ
ishe
d on
06
Aug
ust 2
018.
Dow
nloa
ded
by P
ekin
g U
nive
rsity
on
8/7/
2019
9:4
6:40
AM
. View Article Online
41 D. B. Laks, L. G. Ferreira, S. Froyen and A. Zunger, Efficientcluster expansion for substitutional systems, Phys. Rev. B:Condens. Matter Mater. Phys., 1992, 46(19), 12587.
42 C. Wolverton and A. Zunger, First-principles prediction ofvacancy order-disorder and intercalation battery voltages inLixCoO2, Phys. Rev. Lett., 1998, 81(3), 606.
43 K. Persson, Y. Hinuma, Y. S. Meng, A. Van der Ven andG. Ceder, Thermodynamic and kinetic properties of the Li-graphite system from rst-principles calculations, Phys.Rev. B: Condens. Matter Mater. Phys., 2010, 82(12), 125416.
44 B. Ouml, R. Uuml and L. Scheffler, Thermodynamics of theHeusler alloy Co2 � xMn1 + xSi: A combined densityfunctional theory and cluster expansion study, Phys. Rev. B:Condens. Matter Mater. Phys., 2009, 79(9), 4407.
45 A. V. D. Walle and M. Asta, Self-driven lattice-model MonteCarlo simulations of alloy thermodynamic, Modell. Simul.Mater. Sci. Eng., 2002, 10(5), 521–538.
46 d. V. A. Van and G. Ceder, Vacancies in ordered anddisordered binary alloys treated with the cluster expansion,Phys. Rev. B, 2005, 71(5), 054102.
47 E. Lee and K. A. Persson, Structural and Chemical Evolutionof the Layered Li-Excess LixMnO3 as a Function of Li Content
This journal is © The Royal Society of Chemistry 2018
from First-Principles Calculations, Adv. Energy Mater., 2014,4(15), 27–32.
48 J. Liu, S. Wang and Q. Sun, All-carbon-based poroustopological semimetal for Li-ion battery anode material,Proc. Natl. Acad. Sci. U. S. A., 2017, 114(4), 651.
49 F. Zhou, M. Cococcioni, C. A. Marianetti, D. Morgan andG. Ceder, First-principles prediction of redox potentials intransition-metal compounds with LDA+U, Phys. Rev. B:Condens. Matter Mater. Phys., 2005, 70(23), 35–40.
50 L. Wang, T. Maxisch and G. Ceder, Oxidation energies oftransition metal oxides within the GGA+U framework,Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73(19),195107.
51 F. Zhou, M. Cococcioni, C. A. Marianetti, D. Morgan andG. Ceder, First-principles prediction of redox potentials intransition-metal compounds with LDA+U, Phys. Rev. B,2004, 70(23), 235121.
52 Q. Li, J. Chen, L. Fan, X. Kong and Y. Lu, Progress inelectrolytes for rechargeable Li-based batteries and beyond,Green Energy & Environment, 2016, 1(1), 18–42.
J. Mater. Chem. A, 2018, 6, 18449–18457 | 18457


















