
Introduction à la bio-informatique - info.univ-angers.fr · chromosomes et l'identification de...
192
Introduction à la bio-informatique Matthieu Basseur
Transcript of Introduction à la bio-informatique - info.univ-angers.fr · chromosomes et l'identification de...

Introduction à la bio-informatique
Matthieu Basseur

2
Sommaire
� Introduction à la bioinformatique� Bio-informatique?� Notions de biologie moléculaire
� Notions de base� Alignement 2 à 2 de séquences � Alignement multiple de séquences� Phylogénie

3
Bio-informatique?
� Champ de recherche multi-disciplinaire � où travaillent de concert biologistes, informaticiens,
mathématiciens, physiciens, chimistes…� objectif : résoudre un problème scientifique posé par la biologie.
� Décrit également (par abus de langage) toutes les applications informatiques résultant de ces recherches.� l'analyse du génome� modélisation de l'évolution d'une population animale,� modélisation moléculaire, � analyse d'image,� séquençage du génome,
� reconstruction d'arbres phylogénétiques (phylogénie)…
� Cette discipline constitue la « biologie in silico », par analogie avec in vitro ou in vivo.

4
Biologie moléculaire (cf. wikipedia) ?
� La biologie moléculaire est une discipline scientifique au croisement de la génétique, de la biochimie et de la physique, dont l'objet est la compréhension des mécanismes de fonctionnement de la cellule au niveau moléculaire. Le terme « biologie moléculaire », utilisé la première fois en 1938 par Warren Weaver, désigne également l'ensemble des techniques de manipulation d'acides nucléiques (ADN, ARN), appelées aussi techniques de génie génétique.
� La biologie moléculaire est apparue au XXe siècle, à la suite de l'élaboration des lois de la génétique, la découverte des chromosomes et l'identification de l'ADN comme support chimique de l'information génétique.
� Après la découverte de la structure en double hélice de l'ADN en 1953 par James Watson (1928- ), Francis Crick (1916-2004), Maurice Wilkins (1916-2004) et Rosalind Franklin (1920-1958) la biologie moléculaire a connu d'importants développements pour devenir un outil incontournable de la biologie moderne à partir des années 1970.
5
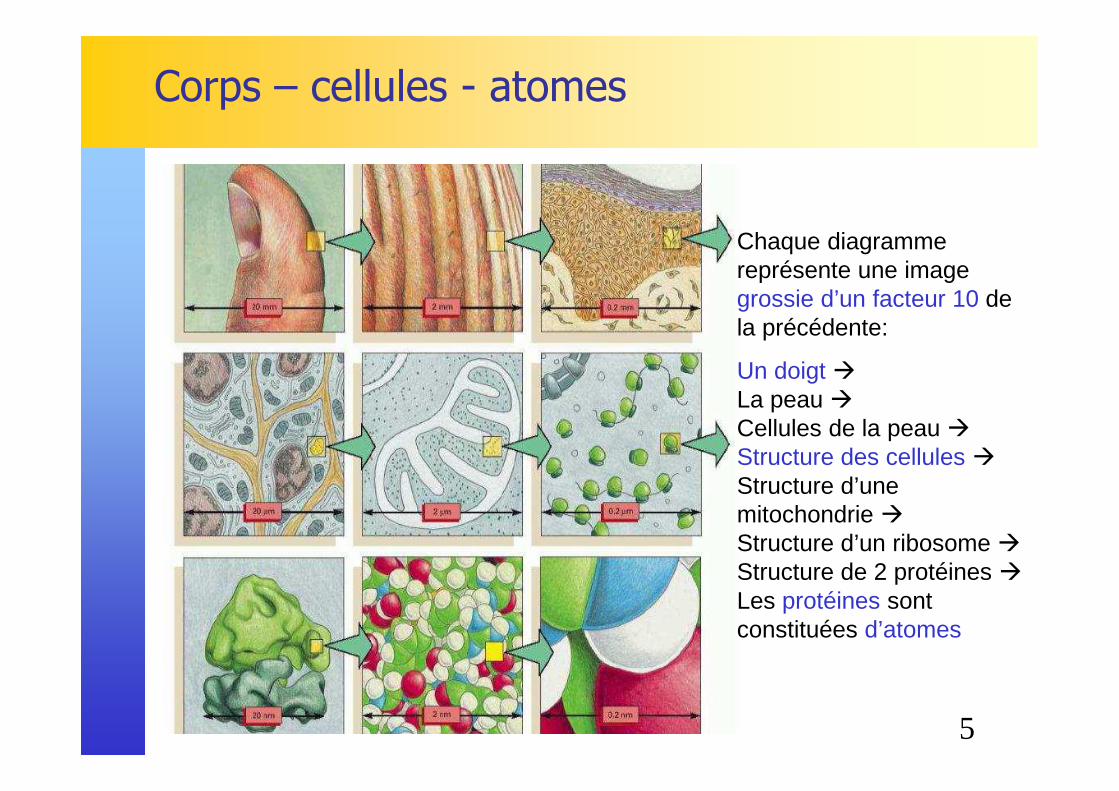
Corps – cellules - atomes
Chaque diagramme représente une image grossie d’un facteur 10 de la précédente:
Un doigt�La peau �Cellules de la peau �Structure des cellules�Structure d’une mitochondrie �Structure d’un ribosome �Structure de 2 protéines �Les protéines sont constituées d’atomes
6
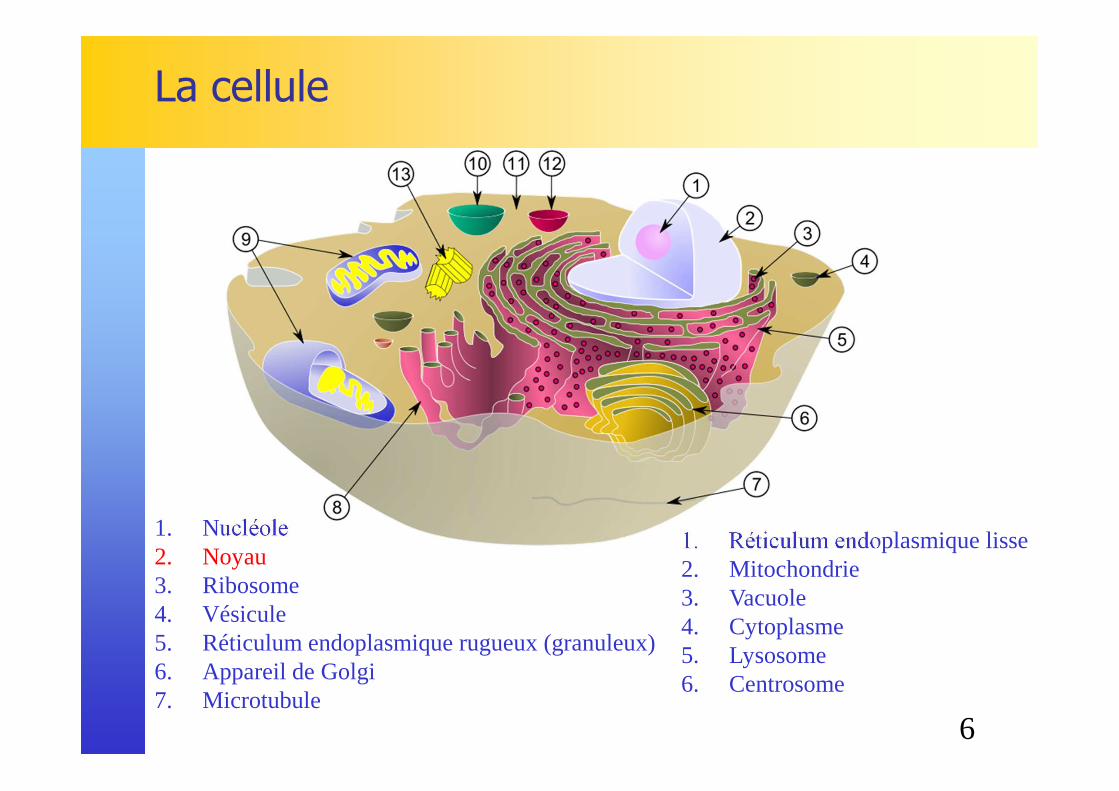
La cellule
1. Nucléole2. Noyau 3. Ribosome4. Vésicule5. Réticulum endoplasmique rugueux (granuleux)6. Appareil de Golgi7. Microtubule
1. Réticulum endoplasmique lisse2. Mitochondrie3. Vacuole4. Cytoplasme5. Lysosome6. Centrosome

7
Les chromosomes
L’ information génétique est contenuedans les chromosomes situés dans lenoyau des cellules*
Chaque cellule d’un être humaincomporte 23 paires de chromosomes
Un chromosome est constitué demolécules d'ADN
* chez les eucariotes seulement. Pour les organismesprocaryotes (organismes unicellulaires), les chromosomesse trouvent dans le cytoplasme.

8
ADN
� ADN est l'abréviation d'acide désoxyribonucléique:� contient sous forme codée toutes les informations relatives à la vie
d'un organisme vivant, du plus simple au plus complexe, animal, végétal, bactérien, viral.
� La fonction de l'ADN est de fabriquer les protéines dont l'organisme a besoin. Les protéines ainsi formées ont différentes fonctions que l'on peut simplifier en les ramenant à deux essentielles: � l'autonomie de l'organisme (sa croissance, sa défense) � sa reproduction
� L'ADN contient donc toutes les informations susceptibles de créer et de faire vivre un organisme. � Si le contenu de la molécule d’ADN humaine était mise sous forme
d'une encyclopédie, il faudrait à peu près 500 volumes de 800 pages chacun.
� Si on étend entièrement l’ADN humain, il mesure + de 1.2 mètre
9
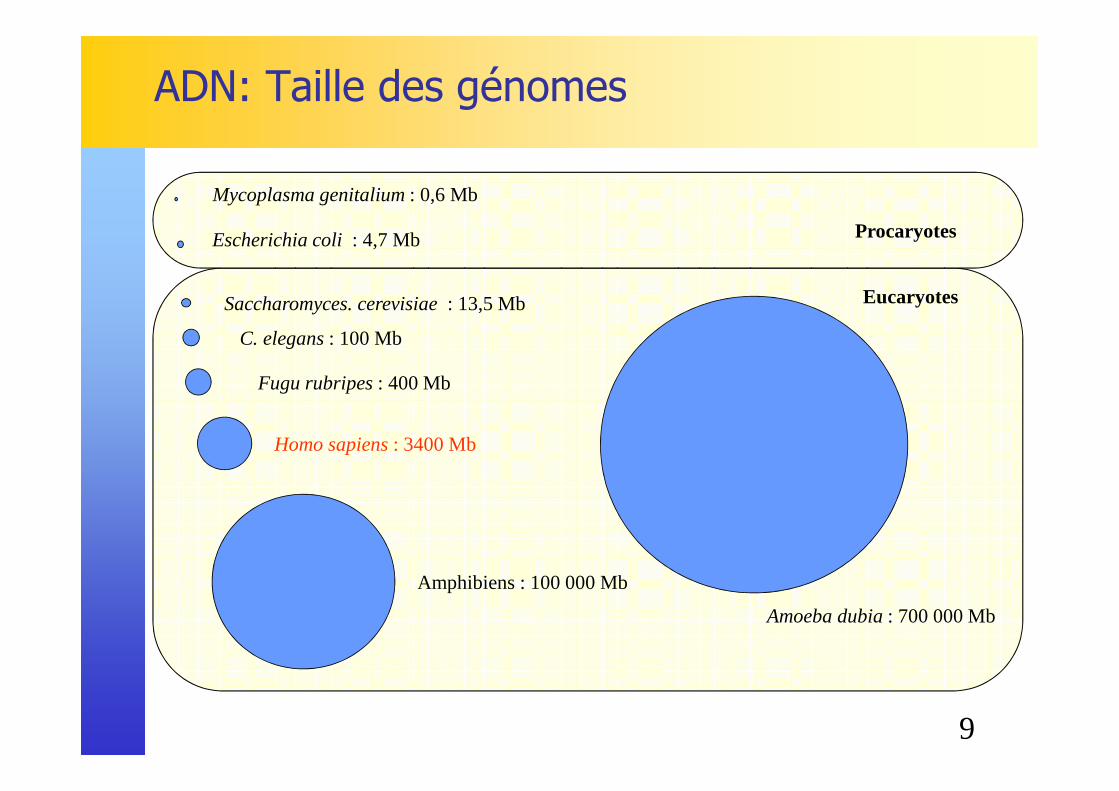
ADN: Taille des génomes
Mycoplasma genitalium : 0,6 Mb
Escherichia coli : 4,7 Mb
Saccharomyces. cerevisiae : 13,5 Mb
C. elegans : 100 Mb
Amoeba dubia : 700 000 Mb
Fugu rubripes : 400 Mb
Homo sapiens : 3400 Mb
Amphibiens : 100 000 Mb
Procaryotes
Eucaryotes

10
ADN
� Une molécule d'ADN se présente sous la forme d'une double hélice enroulée. � macromolécule de millions/milliards d'atomes. C'est un motif
identique tout le temps répété contenant: � des phosphates� des sucres (désoxyribose)� des bases azotées
� Cas du corps humain� Dans l'ensemble des 23 paires de
chromosomes, on compte à peu près trois milliards de bases azotées.
� L’ADN humain est composée de 150 milliards d’atomes.

11
ADN
� Différenciation des motifs : nature de la base azotée. � Le sucre et le phosphate ne sont pas variables.
� 4 bases azotées : � Adénine (A) A C G T
� Cytosine (C)
� Guanine (G)
� Tyrosine(T)
� Propriétés :� Support de l'hérédité (par réplication)� Peut subir des modifications (mutations)
� Naturelles, ou via des facteurs mutagènes (radioactivité, UVs...) � Recombinaisons génétiques (reproduction sexuée, transformation
génétique de bactéries ou artificiellement - OGMs)

12
ADN → Protéines
� Par interaction avec l'environnement, l'ADN se transforme en protéines :� La transcription, transfert de l'ADN vers une autre molécule, l'ARN.� La traduction, transfert depuis l'ARN vers des protéines.
� L'activité des protéines détermine l'activité des cellules� qui vont ensuite déterminer le fonctionnement des organes et de
l'organisme.
� Traduction de l’ADN en protéine:� Les quatre lettres A, C, G et T s'associent en mots de trois lettres
(GGA, CTA...) pour former un codon. Des ribosomes décodent ces codons en acides aminés combinées pour former des protéines.

13
ADN → Protéines

14
ADN → Acides aminés
� 20 Acides aminés :� Acide aspartique� Acide glutamique� Alanine� Arginine� Asparagine� Cystéine� Glutamine� Glycine� Histidine� Isoleucine� Leucine� Lysine� Phénylalanine� Proline� Sérine� Thréonine� Tryptophane� Tyrosine� Valine� Méthionine/Start
� Stop

15
ADN → Acides aminés
� Acides aminés : codes à 1 et 3 lettres� Acide aspartique (D, Asp)
� Acide glutamique (E, Glu)
� Alanine (A, Ala)
� Arginine (R, Arg)
� Asparagine (N,Asn)
� Cystéine (C, Cys)
� Glutamine (Q, Gln)
� Glycine (G, Gly)
� Histidine (H, His)
� Isoleucine (I, Ile)
� Codon Stop : marque la fin de la traduction d'un gène en protéine. Il n'est en général jamais traduit car il n'existe pas d'ARN de transfert correspondant (Il existe 2 acides aminés supplémentaires, la sélénocystéine et la pyrrolysine qui sont insérés lorsqu'un codon STOP particulier est rencontré).
� Leucine (L, Leu)
� Lysine (K, Lys)
� Méthionine (M, Met)
� Phénylalanine (F, Phe)
� Proline (P, Pro)
� Sérine (S, Ser)
� Thréonine (T, Thr)
� Tryptophane (W, Trp)
� Tyrosine (Y, Tyr)
� Valine (V, Val)
16
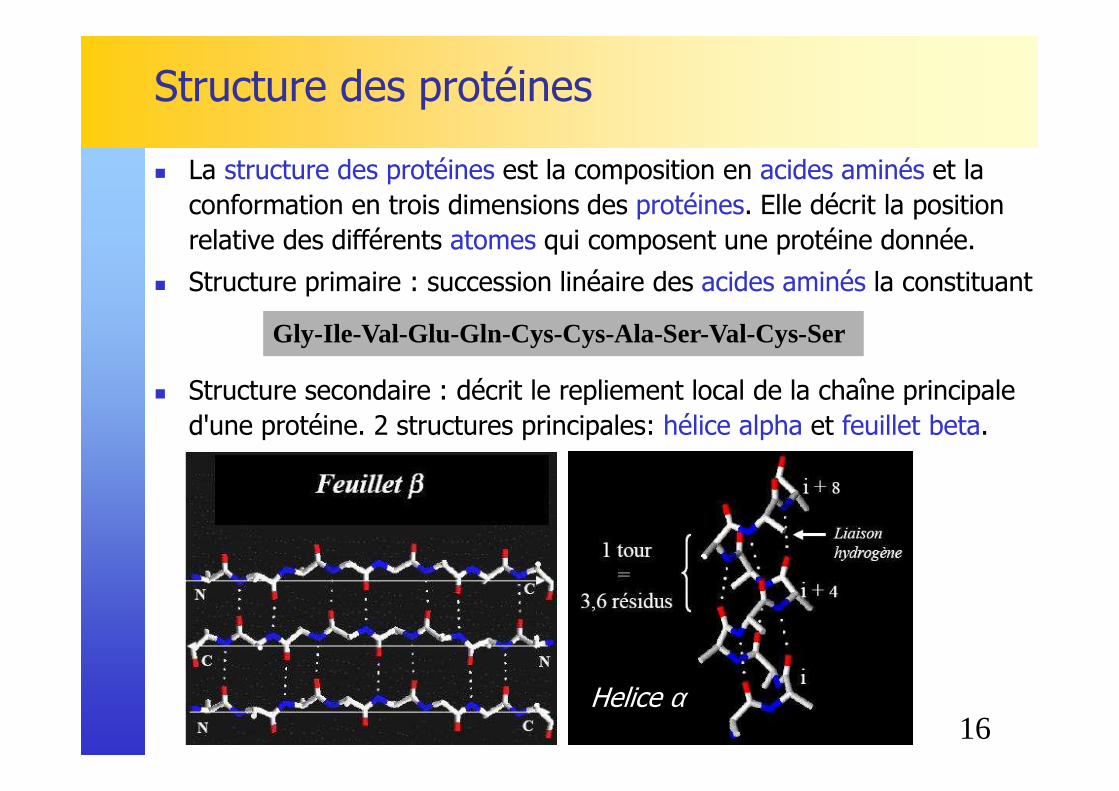
Structure des protéines
� La structure des protéines est la composition en acides aminés et la conformation en trois dimensions des protéines. Elle décrit la position relative des différents atomes qui composent une protéine donnée.
� Structure primaire : succession linéaire des acides aminés la constituant
� Structure secondaire : décrit le repliement local de la chaîne principale d'une protéine. 2 structures principales: hélice alpha et feuillet beta.
Gly-Ile-Val-Glu-Gln-Cys-Cys-Ala-Ser-Val-Cys-Ser
Helice α
17
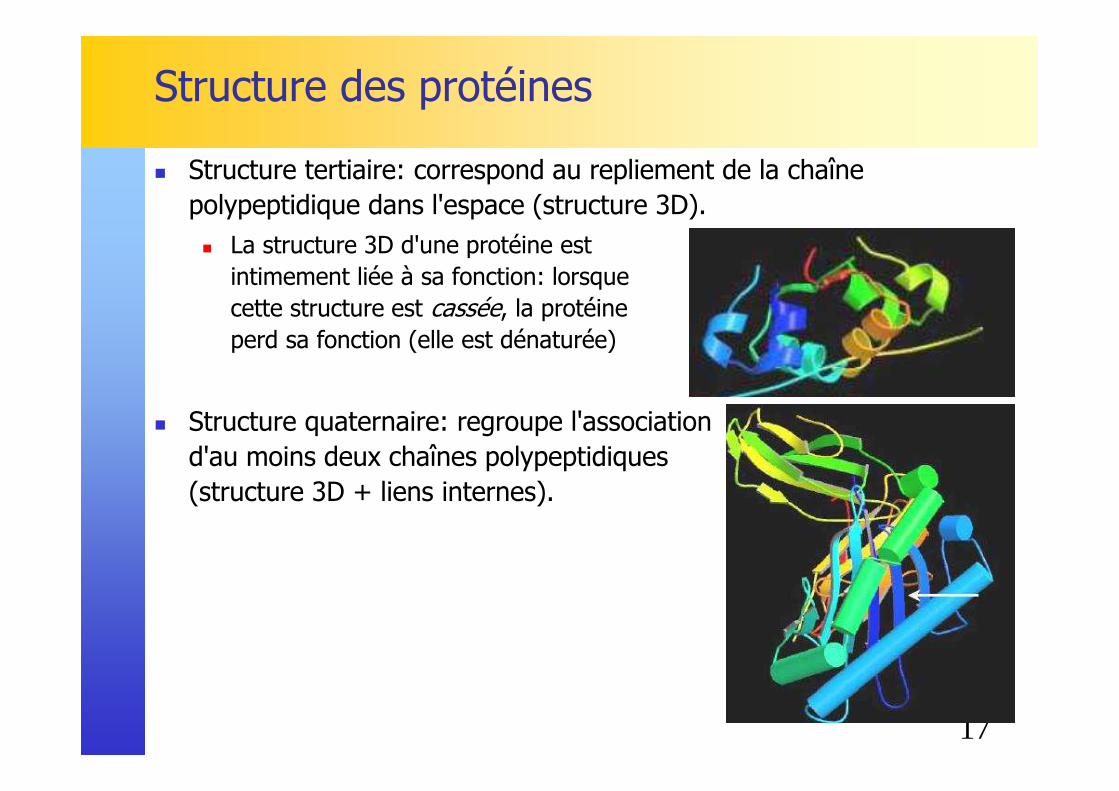
Structure des protéines
� Structure tertiaire: correspond au repliement de la chaîne polypeptidique dans l'espace (structure 3D).� La structure 3D d'une protéine est
intimement liée à sa fonction: lorsque cette structure est cassée, la protéine perd sa fonction (elle est dénaturée)
� Structure quaternaire: regroupe l'association d'au moins deux chaînes polypeptidiques (structure 3D + liens internes).

18
Séquençage de l’ADN
� Séquençage de l’ADN :� Consiste à déterminer l'ordre d'enchaînement des nucléotides
d’un fragment d’ADN donné� Techniques de séquençage apparues fin des années 70
(séquenceurs automatiques � années 90).
� Méthodes de Sanger et de Gilbert � Prix nobel de chimie en 1980� Sanger (UK): Synthèse enzymatique sélective� Gilbert (USA): Dégradation chimique sélective� Méthode de Sanger souvent utilisée actuellement
� méthode de Gilbert : limites de taille, toxicité, difficile à mettre en œuvre
� premier organisme séquencé en 1977: virus bactériophage ϕX174

19
Séquençage de l’ADN
� Séquencage d’un génome complet:� Séquencage de fragments� Réconstitution du génome complet par alignement des séquences
� Séquençage du génome humain:� Décidé en 1980, initié en 1987 avec 400 marqueurs connus, soit
1/10 Mb� Réalisé chromosome par chromosome
� Chromosome 22 publié en 1999� Chromosome 21 publié en 2000� Ébauche du génome humain en Juin 2000� Séquence complète publiée en avril 2003
� Actuellement� Séquençage d’autres espèces (souris, chimpanzé…)� Bactéries, microbes, végétaux etc…

20
Sommaire
� Introduction à la bioinformatique� Notions de base� Alignement 2 à 2 de séquences � Alignement multiple de séquences� Phylogénie

21
Vocabulaire - récapitulatif
� Les êtres humains sont composés de cellules dans le noyau desquelles se trouvent les chromosomes constitués d'ADN. Cet ADN défini des gènes.
� L'information de l'ADN est contenue dans une suite de bases azotées (composée de quatre lettres A, T, C et G)
� un codon est composé de 3 bases azotées.� Un codon peut être traduit en un acide aminé.� En assemblant plusieurs acides aminés, on obtient des
protéines.� Les gènes représente l'ADN qui spécifie une unité
d'information génétique (≠protéines).� Les chromosomes sont constitués de gènes.� L'ensemble des chromosomes d'un individu est le génome.

22
Définitions - Alphabet
� Alphabet� Définition: un alphabet Σ est un ensemble fini de symboles
distincts {a0, a1, …, an}. Dans le cas de séquences d'ADN ou d'acides aminés on définit a0 comme étant le symbole vide ou gapet est représenté par le caractère « - ».
� Alphabet de l’ADN (bases azotées)� L’alphabet des molécules d’ADN est composé de 5 symboles: ΣADN
= {−,A,C,G,T}
� {−,A,C,G,T} représentent respectivement un gap, l’Adénine, la Cytosine, la Guanine et la Thymine.
� Alphabet des Acides aminés� L’alphabet des acides aminés est composé de 21 symboles
ΣAA ={−,A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y} qui représentent les différents acides aminés.

23
Définitions - Séquences
� Séquence : On appelle séquence S une suite ordonnée de caractères S = <x1, x2, . . . , xn> pris dans un alphabet.� On note |S| = n la longueur de la séquence.
� Sous-séquence : Soit S une séquence de longueur n. On appelle sous-séquence de S toute partie de S composée d’un ensemble de caractères consécutifs de S. � On notera S[i..j] avec 1 ≤ i ≤ j ≤ n, la sous-séquence
<xi, xi+1, . . . , xj>. En particulier S[i..i] = S[i] = <xi>.
� Préfixe d’une séquence : Soit S une séquence de longueur n. On appelle préfixe de S toute sous-séquence S[1..p] de longueur p telle que 1 ≤ p < n.

24
Généralités – événement mutationnel
� On part du postulat que l’ensemble des espèces actuelles se sont différenciées au fil du temps grâce à des événements mutationnels.
� 3 événements mutationnels élémentaires� substitution
� insertion
� delétion
� La réalité est sensiblement plus complexe:� Substitution/insertion/delétion par bloc
� ≠ probabilités pour chaque événement mutationnel
� Taux de mutation sensible aux conditions extérieures…
AGACT � AGATT
AGACT � AGACAT
AGACT � AGAT

25
Sommaire
� Introduction à la bioinformatique� Notions de base� Alignement 2 à 2 de séquences
� Introduction / Score d’un alignement� Formulation / Résolution exacte� Alignement global : Needlemann-Wunsch� Alignement local : Smith-Waterman � Modèles de gaps : autres alignements 2 à 2
� Alignement multiple de séquences� Phylogénie

26
Alignement de séquences
� Alignement de séquences d'ADN (ou d’acides aminés):� opération de base en bio-informatique qui a pour but d'identifier
des zones conservées entre séquences.
� Utilité de l'alignement :� identifier des sites fonctionnels
� prédire la ou les fonctions d'une protéine
� prédire la structure secondaire (voire tertiaire ou quaternaire) d'une protéine
� établir une phylogénie (évolution: parenté entre les organismes)
CAGCA-CTTGGATTCT-GG
CAGC---TTG--TACTCGG

27
Alignement de séquences
� On distingue 2 types d'alignements qui diffèrent suivant leur complexité : � l'alignement par paires : consiste à aligner 2 séquences peut être réalisé
grâce à un algorithme de complexité polynomiale. Il est possible de réaliser un alignement : � global, c'est à dire entre les 2 séquences sur toutes leurs longueurs
� local entre une séquence et une partie de l'autre séquence
� l'alignement multiple, qui est un alignement global : consiste à aligner plus de 2 séquences et nécessite un temps de calcul et un espace de stockage exponentiel en fonction de la taille des données.
� Alignement de genres différents:� Alignement de séquences d’ADN
� Alignement de séquences d’acides aminés
CAGCACTTGGATTCT-GG---
CAGC--TTG--TACTCGGATT
RDI--SLVKNA---GIVNADI
RNILVS---DAKNVGIVN-DI

28
Alignement de séquences
� Alignement = Mise en correspondance de deux séquences (ADN ou protéines)
� 3 événements mutationnels élémentaires� substitution
� insertion
� délétion
� Score d'une opération� substitution : score de similarité
� indel : pénalité
� Le score de l'alignement est la somme des scores élémentaires
indel
AGACT � AGATT
AGACT � AGACAT
AGACT � AGAT

29
A C C G A T G A
A C – G C T - A
Somme des paires
� Le score d'un alignement par paires A(S1,S2) est donné par une formule w de somme des paires :
� Exemple (Mismatch: -1, Match: 3, Indel: -2) :
qSSAavecaawSSAw iq
i
i ==∑=
),(),,()),(( 2121
121
A C C G A T G A
A C – G C T - A3 +3 -2 +3 -1 +3 -2 +3 = 10

30
A G T T G T T C
T G – G G T A C
A G T T G T T C
T G – G G T A C-1 +5 -4 -2 +5 +7 -1 +5 = 14
Somme des paires
� Exemple (Mismatch: -1, Match: 2, Indel: -4) :
� Exemple (matrice de substitution) :� Favorise les mutations A�T et G�C� Favorise le match du nucléotide T
A G T T G T T C
T G – G G T A C
A G T T G T T C
T G – G G T A C-1 +2 -4 -1 +2 +2 -1 +2 = 1
- A C G T
- -4 -4 -4 -4
A -4 5 -2 -2 -1
C -4 -2 5 -1 -2
G -4 -2 -1 5 -2
T -4 -1 -2 -2 7

31
Alignement de séquences
� 2 séquences � plusieurs alignements possibles
� Bon/mauvais alignement? matrices de substitutions� Exemple:
� Mismatch: -1� Match: 2� Indel: -2
CAGC----ACTTGGATTCTGG
CAGCTTGTACTCGGATT----
CAGCACTTGGATTCT-GG---
CAGC--TTG--TACTCGGATT
CAGCACTTGGATTCTGG---
CAGC--TTGTACTC-GGATT
7 7
10
- A C G T
- -2 -2 -2 -2
A -2 2 -1 -1 -1
C -2 -1 2 -1 -1
G -2 -1 -1 2 -1
T -2 -1 -1 -1 2

32
Matrices de substitution
� Matrices nucléiques� Il existe peu de matrices pour les acides nucléiques car il n'y a que 5
lettres pour leur alphabet.� La plus fréquemment utilisée est la matrice dite unitaire (ou matrice
identité) où toutes les bases sont considérées comme équivalentes.
� Matrices des acides aminés : beaucoup plus complexe!� Pam [1978], Blosum [1992], Gonnet [1992]…� Basées sur: nombres de mutation nécessaires pour changer d’acide
aminé, propriétés physico-chimiques, évolution…� Page d'Emmanuel Jaspard sur les matrices de substitution
- A C G T
- 0 0 0 0
A 0 1 0 0 0
C 0 0 1 0 0
G 0 0 0 1 0
T 0 0 0 0 1
Match: 1Mismatch: 0
Indel: 0

33
Formulation
■ Définition : Alignement par paire– Soit un alphabet Σ. – Soit S = { S1, S2} 2 séquences de caractères de Σ.– Un alignement de S, noté A(S1, S2) est une matrice 2*q
• Chaque élément au,v de la matrice a est défini dans Σ.• q est plus grand que la plus grande des séquences, et plus
petit que la somme des tailles des séquences.• les séquences {a1,1, a1,2,..., a1,q} et {a2,1, a2,2,..., a2,q} dans
laquelle on supprime les gaps correspondent à S1 et S2
■ Formulation : Problème d'alignement par paire– Soient deux séquences S1 et S2 et une matrice de score w, le
problème d'alignement par paires consiste à déterminer un alignement de coût optimal selon w.

34
Résolution exacte
� Alignement de deux séquences de longueur n:
� Algorithme de Needleman-Wunsch� 1970: A general method applicable to the search for similarities in the
amino acid sequence of two proteins, J Mol Biol. 48(3):443-453� effectue un alignement global de deux séquences, de manière optimale� première application de la programmation dynamique pour la
comparaison de séquences biologiques
CCk
n
n
k
k
knNbAlign •=∑
=+
0
(Énumération exhaustive rapidement impossible)
Longueur des séquences 1 2 3 4 5 6
# alignements 3 13 63 321 1683 8527

35
Programmation dynamique - exemple
� Suite de fibonacci :– La suite de Fibonacci est donnée par la formule récurrente :
• Fib(0) = 0• Fib(1) = 1• Fib(n) = Fib(n-1) + Fib(n-2)
– Pour calculer Fib(n) on peut définir naturellement un algorithme récursif :
Fonction Fib(n : entier) : entier
debut
si n <= 1 alors retour n;
retour Fib1(n-1) + Fib1(n-2);
fin
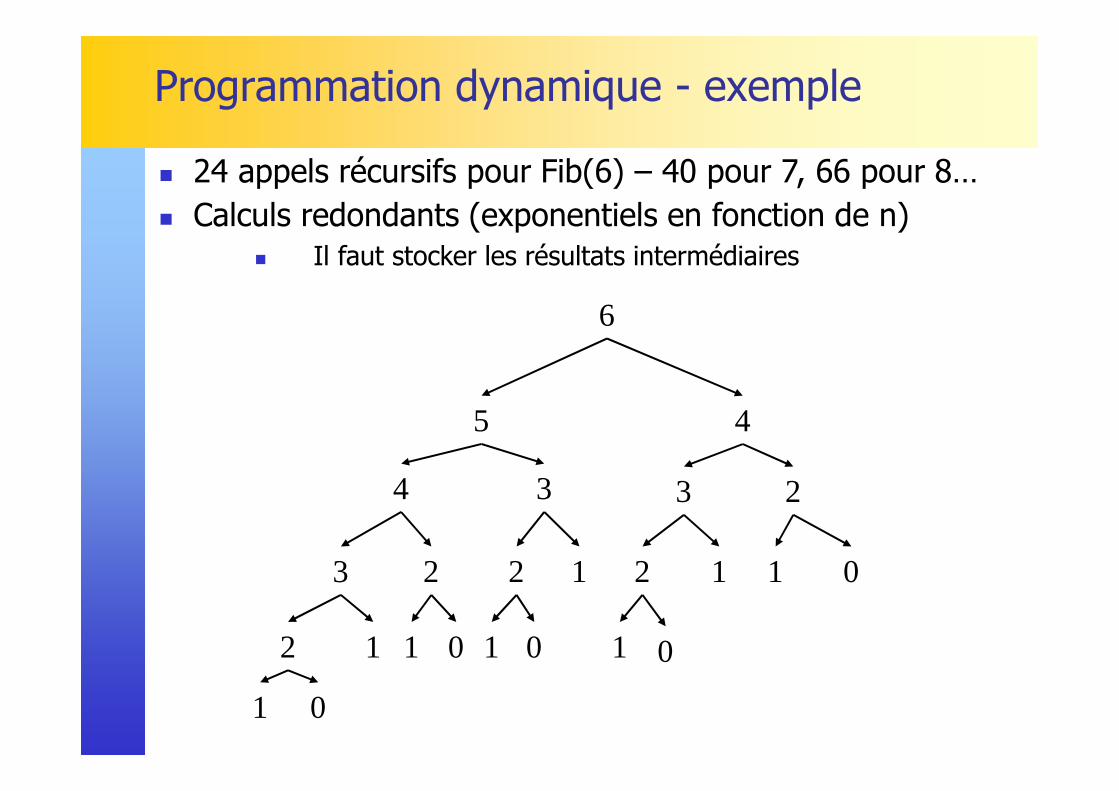
Programmation dynamique - exemple
� 24 appels récursifs pour Fib(6) – 40 pour 7, 66 pour 8… � Calculs redondants (exponentiels en fonction de n)
� Il faut stocker les résultats intermédiaires
6
5 4
4 3 3
3 2
1
022
2
2 11
1
1
1 1
00 0
1 0

37
Programmation dynamique - exemple
� On enregistre les valeurs de Fib(n) une fois calculées– il suffit d'évaluer les Fib(n) dans l'ordre croissant des n
Fonction Fib(n : entier) : entier
debut
tab[0] = 0;
tab[1] = 1;
pour i = 2 à n faire
tab[i] = tab[i-1] + tab[i-2];
fpour
retour tab[n];
Fin
→ Calcul de Fib(n) en temps linéaire !

38
Algorithme de Needleman-Wunsch
� Méthode: Programmation dynamique� Un algorithme de programmation dynamique procède
en réduisant le problème en plusieurs instances plus petites, elle-mêmes résolues par décomposition.
� Les résultats des calculs intermédiaires sont stockés dans une table.
� La solution est ensuite construite à partir de la table, en remontant celle-ci.
� Ici :� calculs intermédiaires = scores d'alignements entre
préfixes des séquences

39
Algorithme de Needleman-Wunsch
� Recherche de Sim(i,j), alignement de score optimal entre les séquences U(1..i) et V(1..j)
� Formule de récurrence :
� Exemple: aligner CAGCTTAavec CGCCTAA
0Sim(0,0)=
Ins(V(j))1)-jSim(0,j)Sim(0, +=
Del(U(i))1,0)-Sim(iSim(i,0) +=
++
+=
Ins(V(j))1)-jSim(i,
Del(U(i))j)1,-Sim(i
V(j))Sub(U(i),1)-j1,-Sim(i
maxj)Sim(i,
CAGC?
C-GC?
CAGCT
C-GCC
CAGCT
C-GC-
CAGC-
C-GCCou ou
40
Algorithme de Needleman-Wunsch
- A C C G A T G A
-
A
C
G
C
T
A
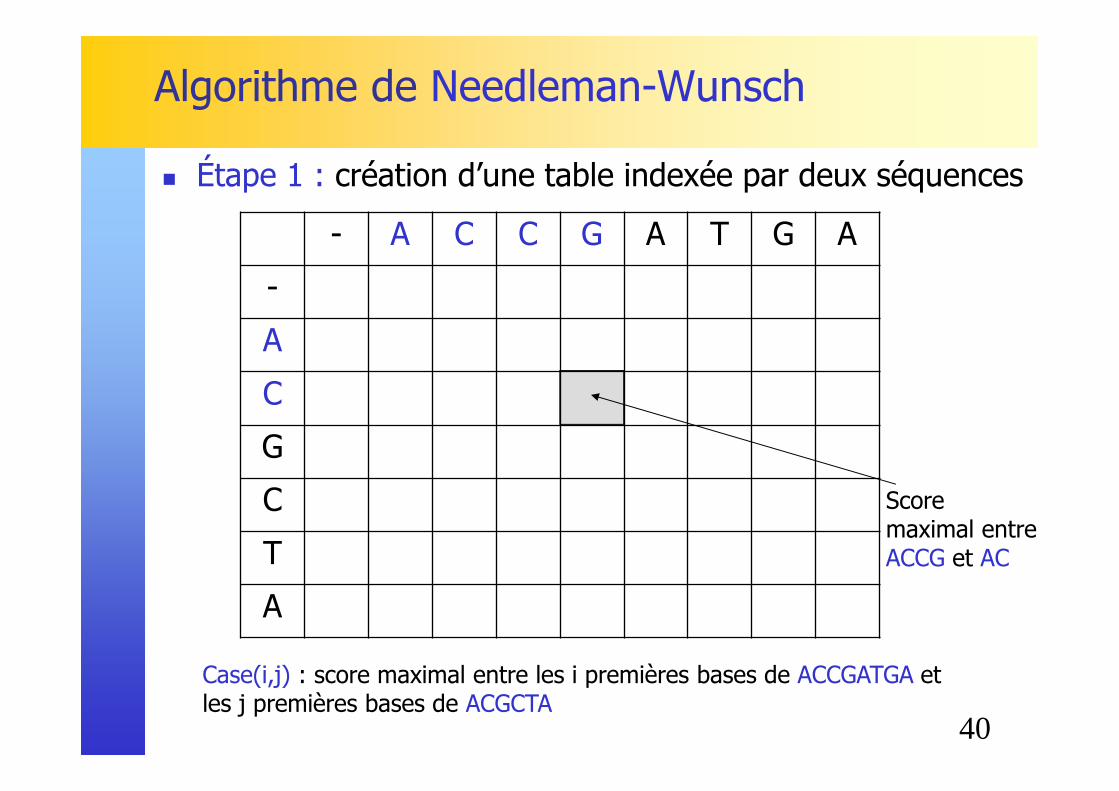
� Étape 1 : création d’une table indexée par deux séquences
Case(i,j) : score maximal entre les i premières bases de ACCGATGA et les j premières bases de ACGCTA
Score maximal entreACCG et AC
41
Algorithme de Needleman-Wunsch
- A C C G A T G A
- 0 0 0 0 0 0 0 0 0
A 0
C 0
G 0
C 0
T 0
A 0
Initialisation (utilisation de la matrice de substitution identité)
� Étape 2 : première ligne / premiere colonne triviales
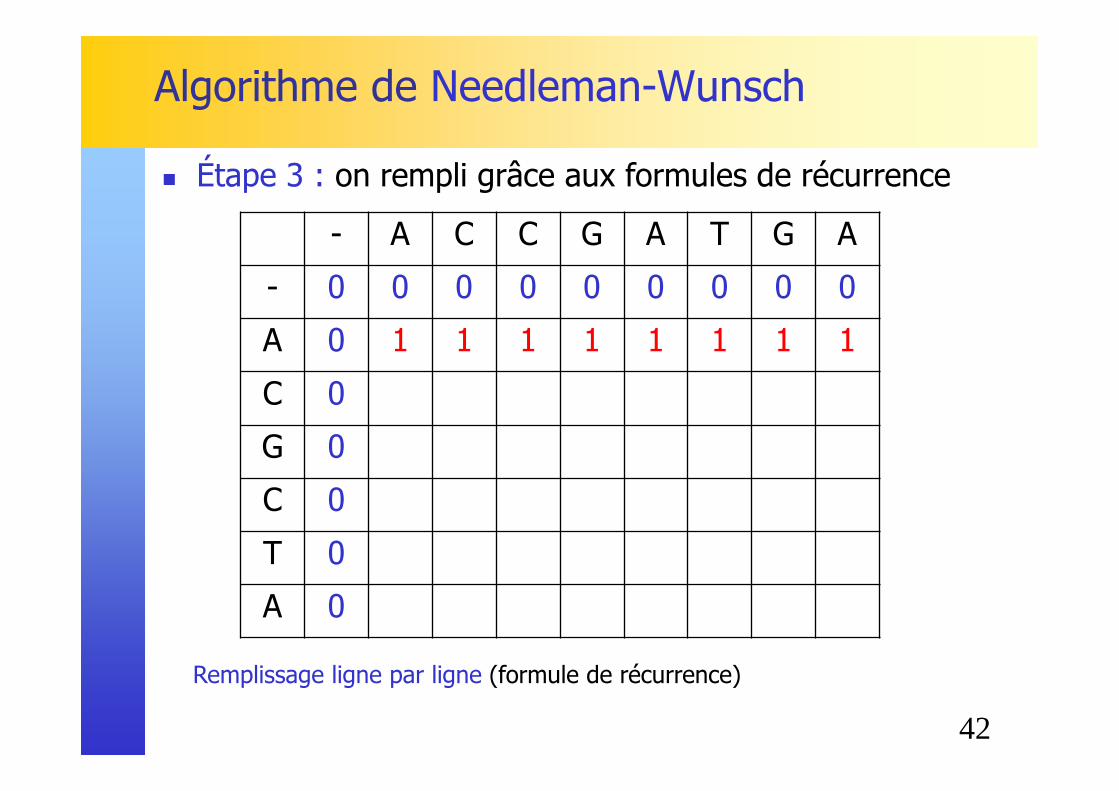
42
Algorithme de Needleman-Wunsch
- A C C G A T G A
- 0 0 0 0 0 0 0 0 0
A 0 1 1 1 1 1 1 1 1
C 0
G 0
C 0
T 0
A 0
Remplissage ligne par ligne (formule de récurrence)
� Étape 3 : on rempli grâce aux formules de récurrence

43
Algorithme de Needleman-Wunsch
- A C C G A T G A
- 0 0 0 0 0 0 0 0 0
A 0 1 1 1 1 1 1 1 1
C 0 1 2 2 2 2 2 2 2
G 0
C 0
T 0
A 0
Remplissage ligne par ligne (formule de récurrence)
� Étape 3 : on rempli grâce aux formules de récurrence
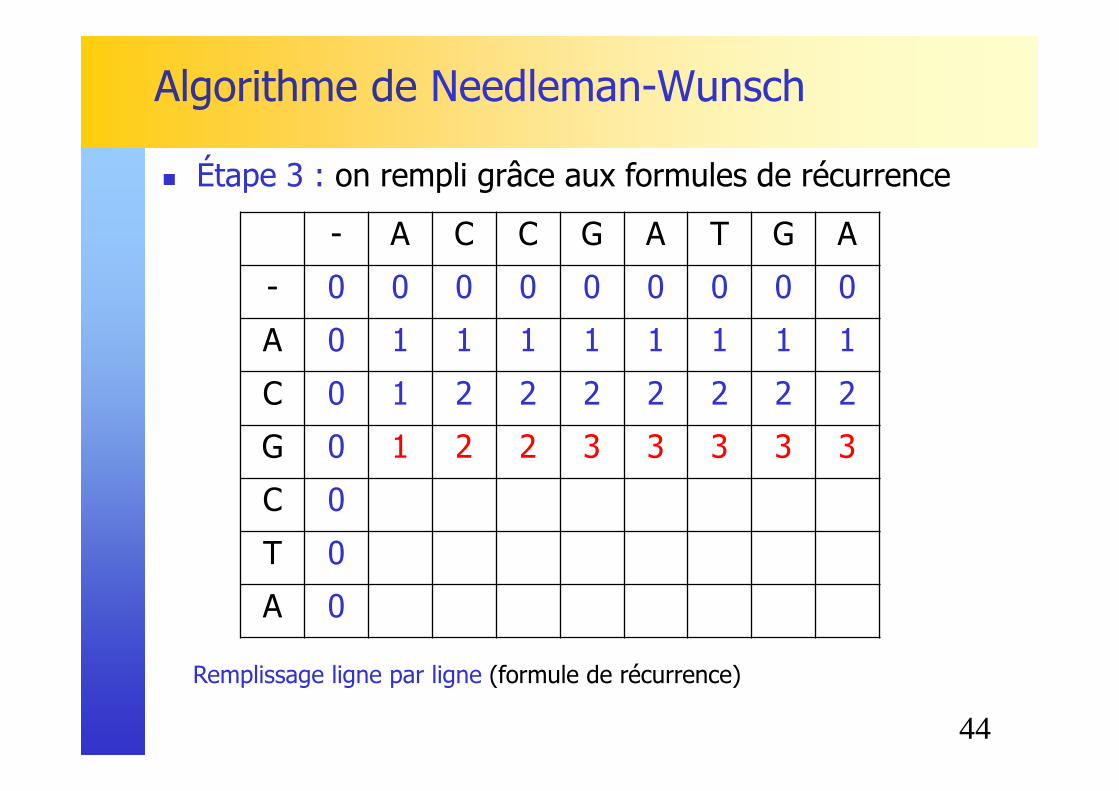
44
Algorithme de Needleman-Wunsch
- A C C G A T G A
- 0 0 0 0 0 0 0 0 0
A 0 1 1 1 1 1 1 1 1
C 0 1 2 2 2 2 2 2 2
G 0 1 2 2 3 3 3 3 3
C 0
T 0
A 0
Remplissage ligne par ligne (formule de récurrence)
� Étape 3 : on rempli grâce aux formules de récurrence
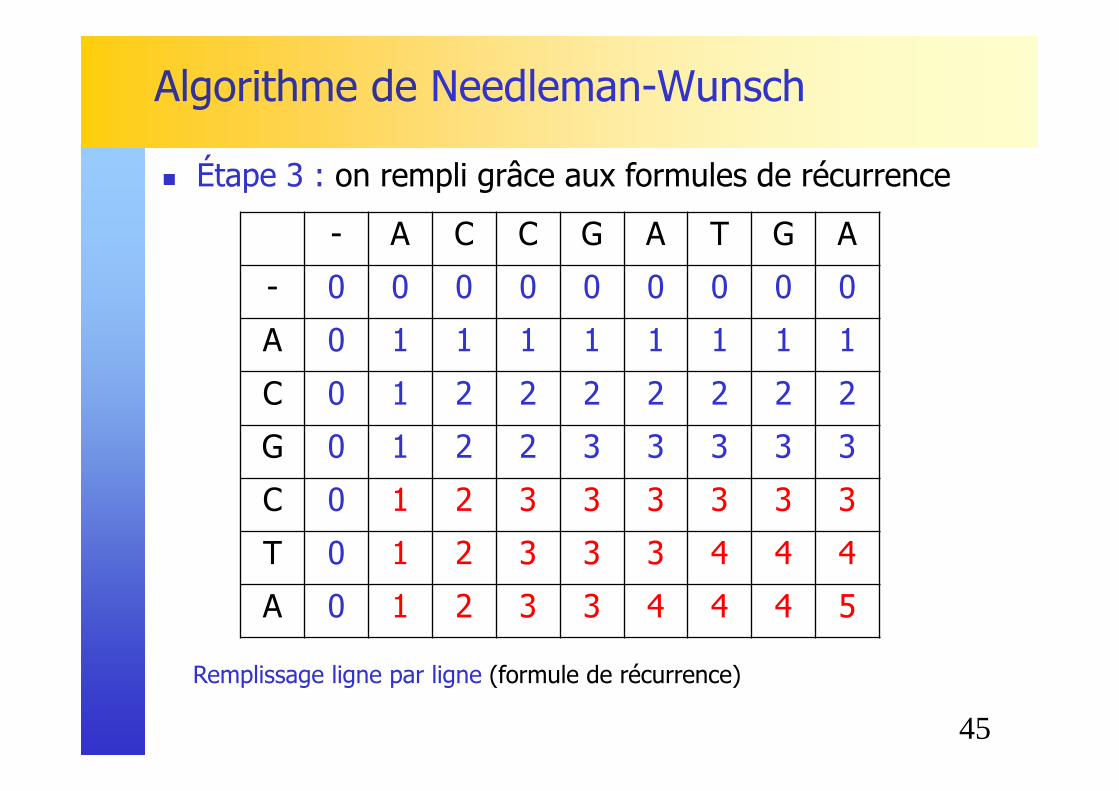
45
Algorithme de Needleman-Wunsch
- A C C G A T G A
- 0 0 0 0 0 0 0 0 0
A 0 1 1 1 1 1 1 1 1
C 0 1 2 2 2 2 2 2 2
G 0 1 2 2 3 3 3 3 3
C 0 1 2 3 3 3 3 3 3
T 0 1 2 3 3 3 4 4 4
A 0 1 2 3 3 4 4 4 5
Remplissage ligne par ligne (formule de récurrence)
� Étape 3 : on rempli grâce aux formules de récurrence
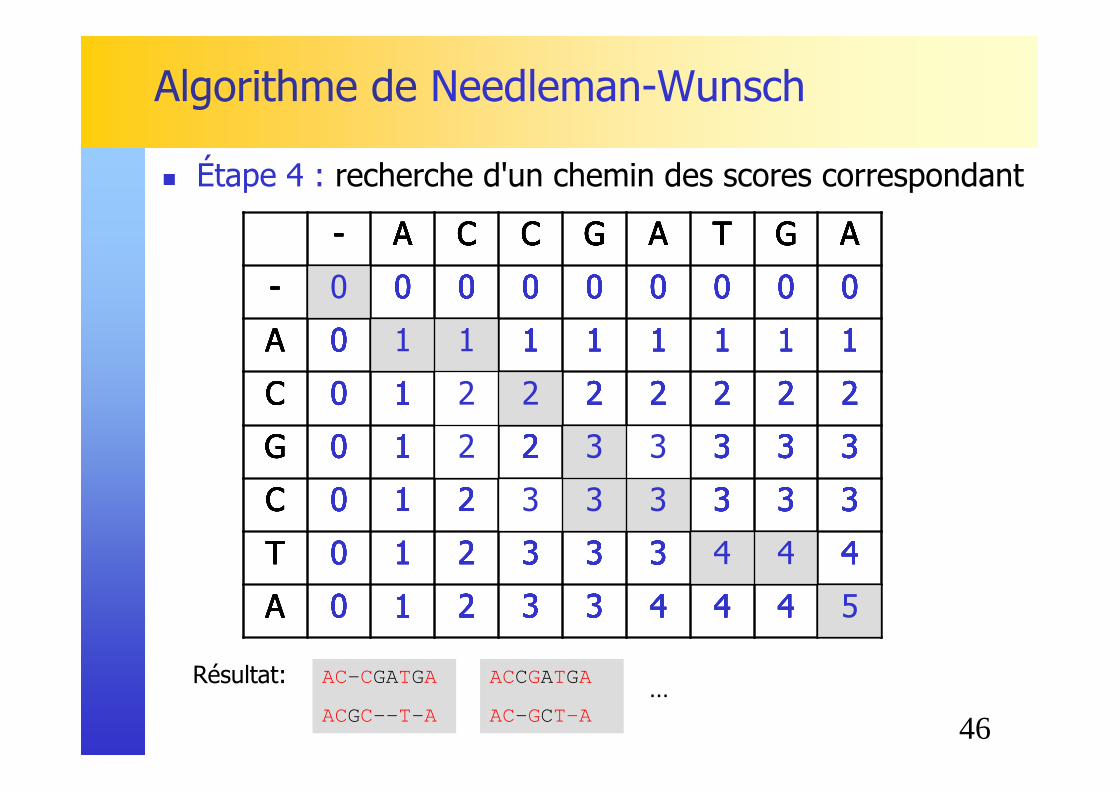
46
Algorithme de Needleman-Wunsch
- A C C G A T G A
- 0 0 0 0 0 0 0 0 0
A 0 1 1 1 1 1 1 1 1
C 0 1 2 2 2 2 2 2 2
G 0 1 2 2 3 3 3 3 3
C 0 1 2 3 3 3 3 3 3
T 0 1 2 3 3 3 4 4 4
A 0 1 2 3 3 4 4 4 5
Résultat:
- A C C G A T G A
- 0 0 0 0 0 0 0 0 0
A 0 1 1 1 1 1 1 1 1
C 0 1 2 2 2 2 2 2 2
G 0 1 2 2 3 3 3 3 3
C 0 1 2 3 3 3 3 3 3
T 0 1 2 3 3 3 4 4 4
A 0 1 2 3 3 4 4 4 5
- A C C G A T G A
- 0 0 0 0 0 0 0 0 0
A 0 1 1 1 1 1 1 1 1
C 0 1 2 2 2 2 2 2 2
G 0 1 2 2 3 3 3 3 3
C 0 1 2 3 3 3 3 3 3
T 0 1 2 3 3 3 4 4 4
A 0 1 2 3 3 4 4 4 5
- A C C G A T G A
- 0 0 0 0 0 0 0 0 0
A 0 1 1 1 1 1 1 1 1
C 0 1 2 2 2 2 2 2 2
G 0 1 2 2 3 3 3 3 3
C 0 1 2 3 3 3 3 3 3
T 0 1 2 3 3 3 4 4 4
A 0 1 2 3 3 4 4 4 5
- A C C G A T G A
- 0 0 0 0 0 0 0 0 0
A 0 1 1 1 1 1 1 1 1
C 0 1 2 2 2 2 2 2 2
G 0 1 2 2 3 3 3 3 3
C 0 1 2 3 3 3 3 3 3
T 0 1 2 3 3 3 4 4 4
A 0 1 2 3 3 4 4 4 5
- A C C G A T G A
- 0 0 0 0 0 0 0 0 0
A 0 1 1 1 1 1 1 1 1
C 0 1 2 2 2 2 2 2 2
G 0 1 2 2 3 3 3 3 3
C 0 1 2 3 3 3 3 3 3
T 0 1 2 3 3 3 4 4 4
A 0 1 2 3 3 4 4 4 5
- A C C G A T G A
- 0 0 0 0 0 0 0 0 0
A 0 1 1 1 1 1 1 1 1
C 0 1 2 2 2 2 2 2 2
G 0 1 2 2 3 3 3 3 3
C 0 1 2 3 3 3 3 3 3
T 0 1 2 3 3 3 4 4 4
A 0 1 2 3 3 4 4 4 5
- A C C G A T G A
- 0 0 0 0 0 0 0 0 0
A 0 1 1 1 1 1 1 1 1
C 0 1 2 2 2 2 2 2 2
G 0 1 2 2 3 3 3 3 3
C 0 1 2 3 3 3 3 3 3
T 0 1 2 3 3 3 4 4 4
A 0 1 2 3 3 4 4 4 5
AC- CGATGA
ACGC-- T- A
ACCGATGA
AC- GCT-A…
� Étape 4 : recherche d'un chemin des scores correspondant
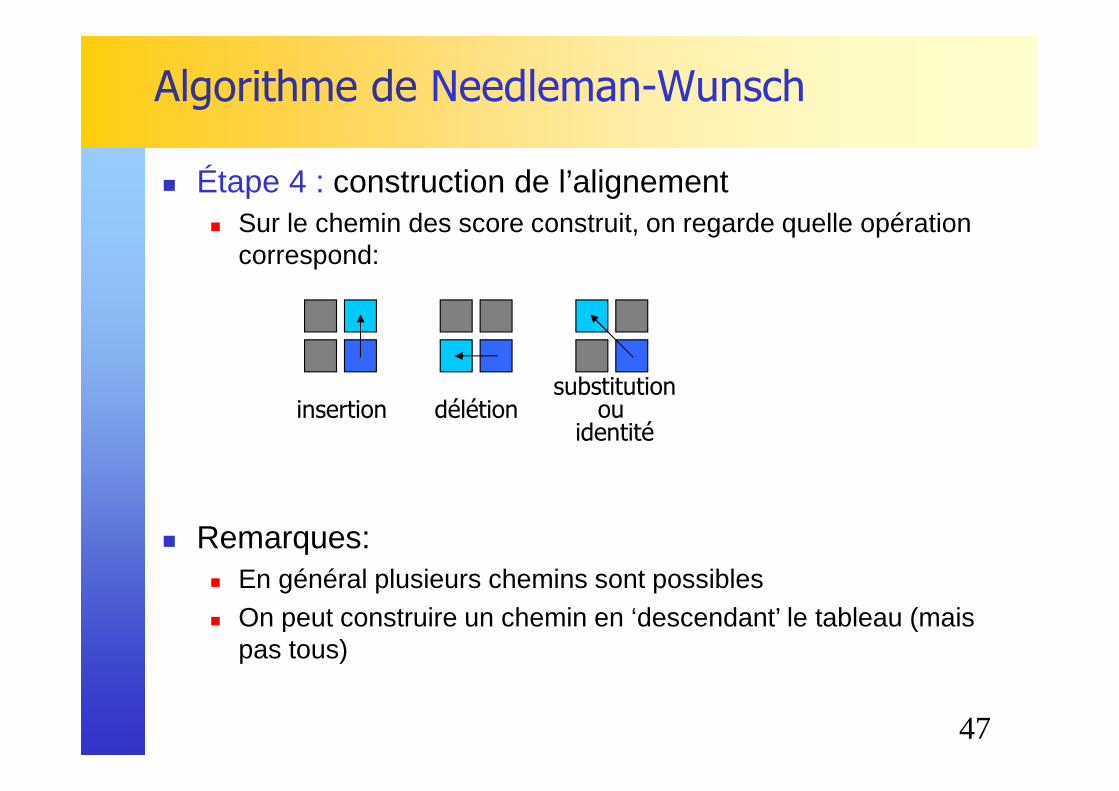
47
Algorithme de Needleman-Wunsch
� Étape 4 : construction de l’alignement� Sur le chemin des score construit, on regarde quelle opération
correspond:
� Remarques:� En général plusieurs chemins sont possibles� On peut construire un chemin en ‘descendant’ le tableau (mais
pas tous)
insertion délétionsubstitution
ou identité

48
Algorithme de Needleman-Wunsch
� Complexité de l’algorithme� Pour le calcul du score d'alignement : (étape 1)
� O(n * m) en temps
� O(min(n,m)) en espace
� Pour la construction de l'alignement : (étapes 1, 2 et 3)
� O(n * m) en temps et en espace(il existe un algorithme pour optimiser la construction de l'alignement, avec espace en O(n). [Myers&Millers – 1988])

49
Algorithme de Needleman-Wunsch
� Sensibilité aux paramètres� Exemple 1 : match 2, mismatch -1, indel –1
� Exemple 2 : match 1, mistmatch -1, indel -2
� L'alignement optimal dépend de la matrice de similarité et des pénalités pour les indels.
ACGGCT- ATC
ACTG- TAATG
ACGGCTATC
ACTGTAATG
alignement optimal
alignement optimal

50
Exemple 2
� Calcul de l’alignement optimal entre la séquence ACCGATGA et la séquence ACGCTA:� Même séquences que pour le premier exemple
� Matrice de substitution, pénalités
� Le fonctionnement de l’algorithme ne change pas!
- A C G T
- -2 -2 -2 -2
A -2 2 -1 -1 -1
C -2 -1 2 -1 -1
G -2 -1 -1 2 -1
T -2 -1 -1 -1 2
Match: 2Mismatch: -1
Indel: -2
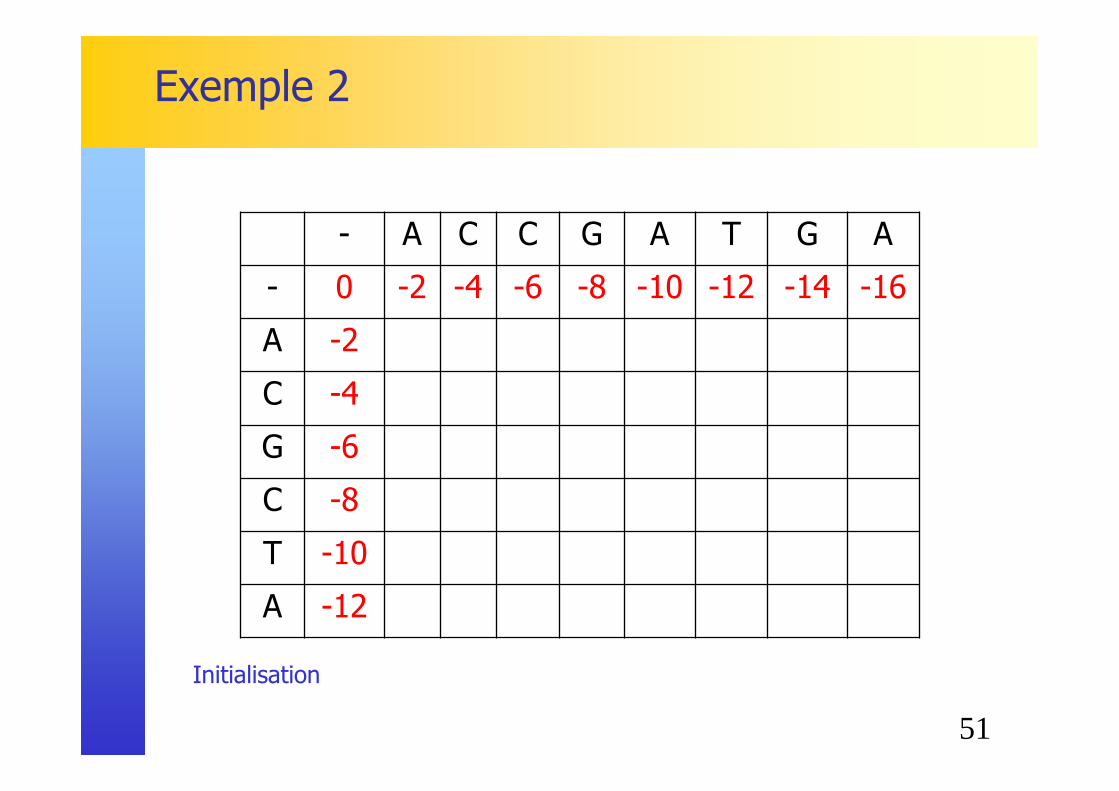
51
Exemple 2
- A C C G A T G A
- 0 -2 -4 -6 -8 -10 -12 -14 -16
A -2
C -4
G -6
C -8
T -10
A -12
Initialisation

52
Exemple 2
Remplissage ligne par ligne (formule de récurrence)
- A C C G A T G A
- 0 -2 -4 -6 -8 -10 -12 -14 -16
A -2 2 0 -2 -4 -6 -8 -10 -12
C -4
G -6
C -8
T -10
A -12
� Étape 3 : on rempli grâce aux formules de récurrence

53
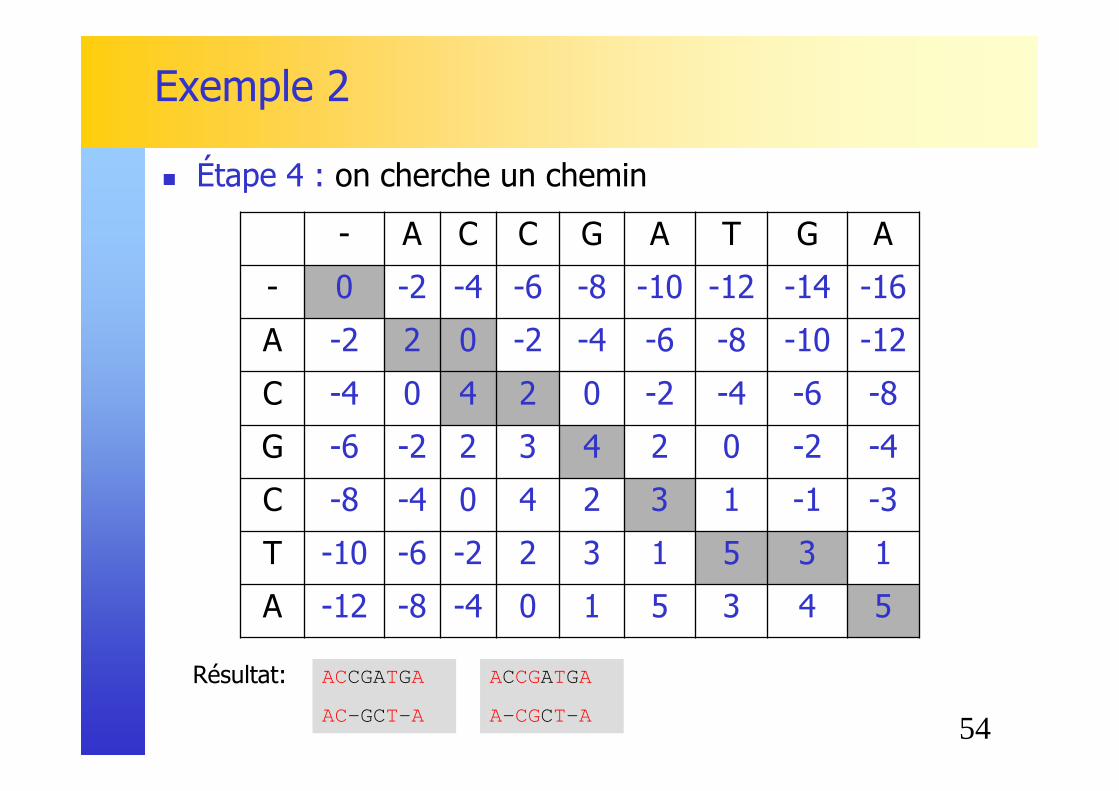
Exemple 2
Remplissage ligne par ligne (formule de récurrence)
- A C C G A T G A
- 0 -2 -4 -6 -8 -10 -12 -14 -16
A -2 2 0 -2 -4 -6 -8 -10 -12
C -4 0 4 2 0 -2 -4 -6 -8
G -6 -2 2 3 4 2 0 -2 -4
C -8 -4 0 4 2 3 1 -1 -3
T -10 -6 -2 2 3 1 5 3 1
A -12 -8 -4 0 1 5 3 4 5
� Étape 3 : on rempli grâce aux formules de récurrence
54
Exemple 2
- A C C G A T G A
- 0 -2 -4 -6 -8 -10 -12 -14 -16
A -2 2 0 -2 -4 -6 -8 -10 -12
C -4 0 4 2 0 -2 -4 -6 -8
G -6 -2 2 3 4 2 0 -2 -4
C -8 -4 0 4 2 3 1 -1 -3
T -10 -6 -2 2 3 1 5 3 1
A -12 -8 -4 0 1 5 3 4 5
Résultat: ACCGATGA
AC-GCT- A
ACCGATGA
A- CGCT- A
� Étape 4 : on cherche un chemin

- A C C G A T G A
-
A
C
G
C
T
A
Exemple 2
� Étape 4 : calcul préalable d’une matrice de direction
Résultat: ACCGATGA
AC-GCT- A
ACCGATGA
A- CGCT- A

56
� Calcul de la matrice des directions Dir est obtenue par les formules suivantes :– Initialisation :
• Dir[0,0] = x• Dir[i,0] = pour tout i de 1 à N• Dir[0,j] = � pour tout j de 1 à P
– Calcul des directions :• Dir[i,j] = Union
– � si M[i,j] = M[i-1,j-1] + w(xi,yj)– � si M[i,j] = M[i-1,j] + w(xi,-)– si M[i,j] = M[i,j-1] + w(-,yj)
Exemple 2

57
Alignement d’acides aminés
� Matrices de substitution des acides aminés� Pam [1978], Blosum [1992]…� Basées sur: nombres de mutation nécéssaires pour changer
d’acide aminé, propriétés physico-chimiques, évolution…� Exemple: BLOSUM62 (indel: -4)

58
Exercice
� Séquences ADN:� Aligner les séquences ADN suivantes:
� ACGGATTACG
� CGCGTATTG
� Match 2, Mismatch –2, Indel -3
� Séquences protéiques:� Aligner les séquences protéiques suivantes:
� STRLPTF
� SRAGDVPY
� Matrice BLOSUM62 (Indel –4)
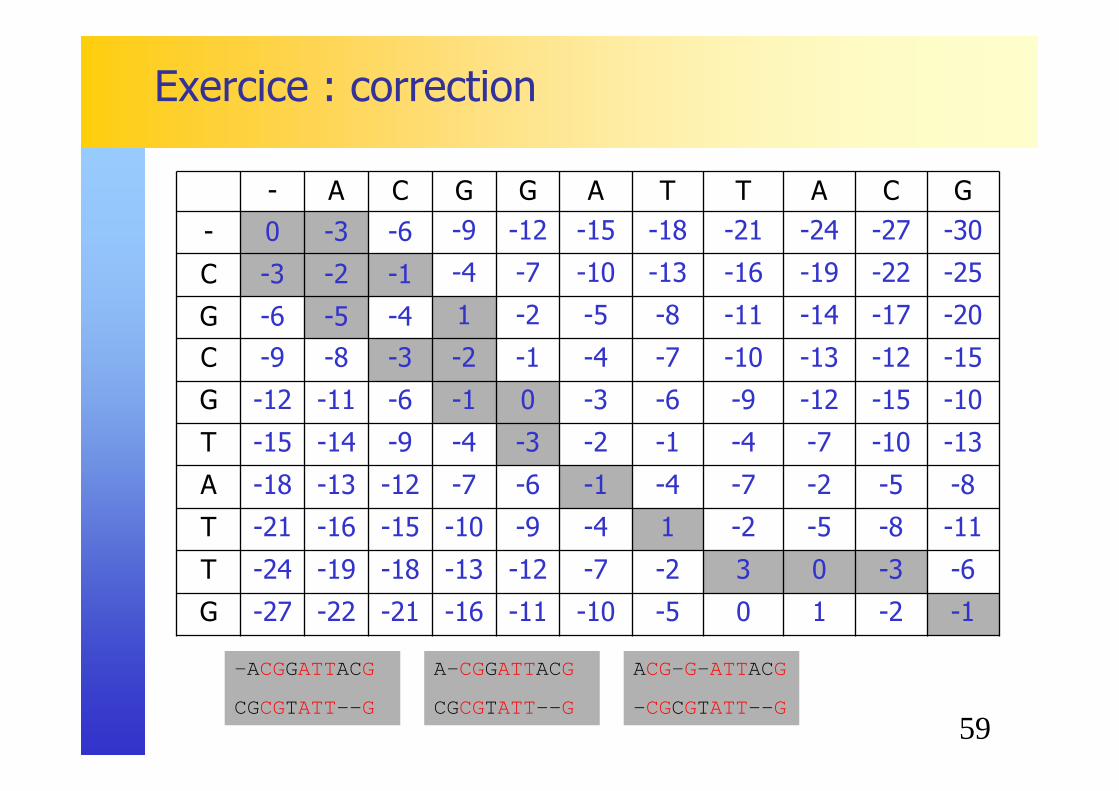
59
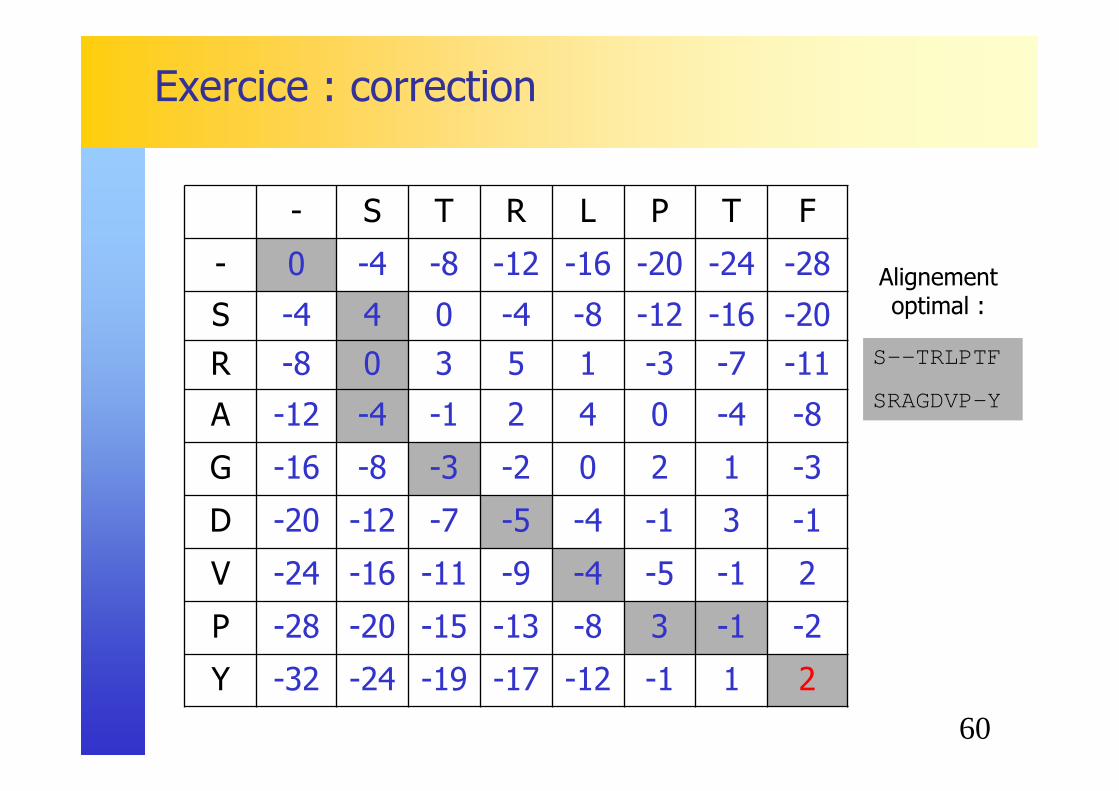
Exercice : correction
- A C G G A T T A C G
- 0 -3 -6 -9 -12 -15 -18 -21 -24 -27 -30
C -3 -2 -1 -4 -7 -10 -13 -16 -19 -22 -25
G -6 -5 -4 1 -2 -5 -8 -11 -14 -17 -20
C -9 -8 -3 -2 -1 -4 -7 -10 -13 -12 -15
G -12 -11 -6 -1 0 -3 -6 -9 -12 -15 -10
T -15 -14 -9 -4 -3 -2 -1 -4 -7 -10 -13
A -18 -13 -12 -7 -6 -1 -4 -7 -2 -5 -8
T -21 -16 -15 -10 -9 -4 1 -2 -5 -8 -11
T -24 -19 -18 -13 -12 -7 -2 3 0 -3 -6
G -27 -22 -21 -16 -11 -10 -5 0 1 -2 -1
-A CGGATTACG
CGCGTATT-- G
A- CGGATTACG
CGCGTATT-- G
ACG- G- ATTACG
- CGCGTATT-- G
60
Exercice : correction
- S T R L P T F
- 0 -4 -8 -12 -16 -20 -24 -28
S -4 4 0 -4 -8 -12 -16 -20
R -8 0 3 5 1 -3 -7 -11
A -12 -4 -1 2 4 0 -4 -8
G -16 -8 -3 -2 0 2 1 -3
D -20 -12 -7 -5 -4 -1 3 -1
V -24 -16 -11 -9 -4 -5 -1 2
P -28 -20 -15 -13 -8 3 -1 -2
Y -32 -24 -19 -17 -12 -1 1 2
S--TRLPTF
SRAGDVP-Y
Alignementoptimal :

61
Alignement global/local
� Les alignements globaux sont plus souvent utilisés quand les séquences mises en jeu sont similaires et de taille égale.
� Alignement global:
� Les alignements locaux sont plus souvent utilisés quand deux séquences dissemblables sont soupçonnées de posséder des motifs semblables malgré l'environnement.
� Alignement local:
� Remarque: Avec des séquences suffisamment identiques, il n'y aucune différence dans les résultats.
FTFTALI LLAVAV
F-- TAL- LLA- AV
FTFTALI LLA- VAV
-- FTAL- LLAAV--

62
Algorithme de Smith et Waterman (1981)
� Cas particulier de l’alignement global : aligner un segment d’une séquence U avec un segment d’une séquence.� Recherche de Sim(i,j), alignement de score optimal entre les séquences
U(a0..a1) et V(b0..b1)� Algorithme presque inchangé! (il suffit de remplacer les scores négatifs
par 0)� Formule de récurrence:
� Reconstruction d’un chemin optimal:� Chercher la valeur maximale (d’indice minimal), puis remonter les
chemins possible jusqu’à aboutir à 0!
[1..b]j [1..a],i0Sim(i,0)j)Sim(0,Sim(0,0) ∈∀∈∀===
++
+
=
0Ins(V(j))1)-jSim(i,
Del(U(i))j)1,-Sim(i
V(j))Sub(U(i),1)-j1,-Sim(i
maxj)Sim(i,

63
- T A T G C A C T A
- 0 0 0 0 0 0 0 0 0 0
T 0 1 1 1 1 1 1 1 1 1
G 0 1 1 1 2 2 2 2 2 2
A 0 1 2 2 2 2 3 3 3 3
- T A T G C A C T A
- X X X X X X X X X X
T X
G X
A X
TATGCACTA
TG-A
Alignement local : exemple
� Matrice de substitution identité :

64
Alignement local : exemple
� Matrice de substitution : match=2, mismatch=-1, indel=-3
- A A T C C A C T G A
- 0 0 0 0 0 0 0 0 0 0 0
A 0 2 2 0 0 0 2 0 0 0 2
C 0 0 1 1 2 2 0 4 1 0 0
G 0 0 0 0 0 1 1 0 3 3 0
A 0 2 2 0 0 0 3 0 0 2 5
C 0 0 1 1 2 2 0 5 2 0 2
C 0 0 0 0 3 4 1 2 4 1 0
G 0 0 0 0 0 2 3 0 1 6 3
T 0 0 0 2 0 0 1 2 2 3 5
T 0 0 0 2 1 0 0 0 4 1 2
AAT CCACTG A
A CGACCG TT

65
Alignement local : variantes
� Beaucoup de variantes possibles:� Alignement préfixes / Alignement suffixes
� Alignement préfixe+suffixe
� Alignement d’une sous séquence
� Algorithmes semblables mais différents pour chaque cas.
-A CGGATTACG
CGCGTATT-- GATTCCTACC
GACCGGCTACCAGGATTACC
TACCAGTATTG-C
ACCCTTCCAGGATTG
GTATTGAGCCTCATAA
ACCCTTCCAGGATTGAGCCTCATAA
GTATTG

66
Alignement par paires : Modèles de gap
� Définition: Un modèle de gap est une application de ℕ�ℝ qui attribue un score généralement négatif (pénalité), à un ensemble de gaps consécutifs.
� 2 modèles traditionnels:� Gap linéaire
� Gap affine
≥⋅=
=1
00)(
nsign
nsing
o
≥⋅−+=
=1)1(
00)(
nsigng
nsing
eo
n: nombre de caractères consécutifs de gap
go<0: pénalité pour l’insertion d’un nouveau gap
go<0: pénalité pour l’introduction d’un nouveau gap
ge<0: pénalité pour l’extension d’un gap existant

67
Alignement par paires : Modèles de gap
� Remarques:� Modèle linéaire: modèle de base, vu précèdemment� Modèle affine: plus proche de la réalité, mais plus
complexe à calculer� Complexité de l’algorithme « naif » en O(n3)� Complexité ramenée en O(n²) en utilisant des matrices
stockant les résultats intermédiaires
� Gap linéaire = cas particulier du gap affine (go=ge)� Gap Affine: ouverture de gap fortement pénalisée
�
� Existance de modèles plus complexes� , algorithme en O(N²log(N)))log()( nng βα +=
oe gg <

68
Exemples de modèles de gap
Gap linéaireGap affine
Gap logarithmique
|gap|
péna
lité

69
Alignement par paires avec gap affine
� En général, considérer que l'insertion d'un gap possède un coût constant ne correspond pas à un modèle réaliste
� On préfère un modèle pour lequel un gap de longueur k est plus probable que k gaps de longueur 1.
� On utilise le modèle de gap affine car il n'augmente pas la complexité du problème d'alignement (O(N²))
� On utilise 4 matrices pour le calcul du meilleur alignement � M la matrice des coûts des meilleurs alignements qui dépend des 3
autres matrices suivantes
� D la matrice des coûts des meilleurs alignements entre xi et yj,
� V la matrice des coûts des meilleurs alignements entre xi et un gap
� H la matrice des coûts des meilleurs alignements entre yj et un gap

70
Alignement par paires avec gap affine
]..1[,)1(],0[],0[
]..1[,)1(]0,[]0,[
0]0,0[]0,0[]0,0[]0,0[
PjgjgjVjD
NigigiHiD
VHDM
eo
eo
∈∀⋅−+==∈∀⋅−+==
====
+=+=
+==
o
o
gHH
gVV
yxsimDD
M
]0,1[]1,1[
]1,0[]1,1[
),(]0,0[]1,1[
max]1,1[11
� Initialisation:
� Calcul de M[1,1]:

71
� Calcul de M[i,1] (pour tout i>1) :
� Calcul de M[1,j] (pour tout j>1) :
Alignement par paires avec gap affine
+=
+−+−+−
=
+−=
=
o
e
o
o
j
gjVjVgjH
gjD
gjV
jH
yxsimjDjD
jM
],0[],1[]1,1[
]1,1[
]1,1[
max],1[
),(]1,0[],1[
max],1[
1
+=
+−+−+−
=
+−=
=
o
o
o
e
i
giHiHgiH
giD
giV
iV
yxsimiDiD
iM
]0,[]1,[]1,1[
]1,1[
]1,1[
max]1,[
),(]0,1[]1,[
max]1,[
1

72
Alignement par paires avec gap affine
+−+−+−
=
+−+−+−
=
+−−+−−+−−
=
=
o
o
e
e
o
o
ji
ji
ji
gjiH
gjiD
gjiV
jiV
gjiH
gjiD
gjiV
jiH
yxsimjiV
yxsimjiH
yxsimjiD
jiD
jiM
],1[
],1[
],1[
max],[
]1,[
]1,[
]1,[
max],[
),(]1,1[
),(]1,1[
),(]1,1[
max],[
max],[
),(]1][1[ ji yxsimjiM +−−=� Calcul de M[i,j] (pour tout i,j>1) :
73
Gap affine : exemple
Matrice D Matrice V
Matrice MMatrice H
- C T G A C A T
-
C
T
A
- C T G A C A T
-
C
T
A
- C T G A C A T
-
C
T
A
- C T G A C A T
-
C
T
A

74
Gap affine: exemple
Matrice D Matrice V
Matrice MMatrice H
- C T G A C A T
- 0
C
T
A
- C T G A C A T
- 0
C
T
A
- C T G A C A T
- 0
C
T
A
- C T G A C A T
- 0
C
T
A

75
Gap affine: exemple
Matrice D Matrice V
Matrice MMatrice H
- C T G A C A T
- 0 X X X X X X X
C X
T X
A X
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C X
T X
A X
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C -3
T -4
A -5
- C T G A C A T
- 0 X X X X X X X
C -3
T -4
A -5
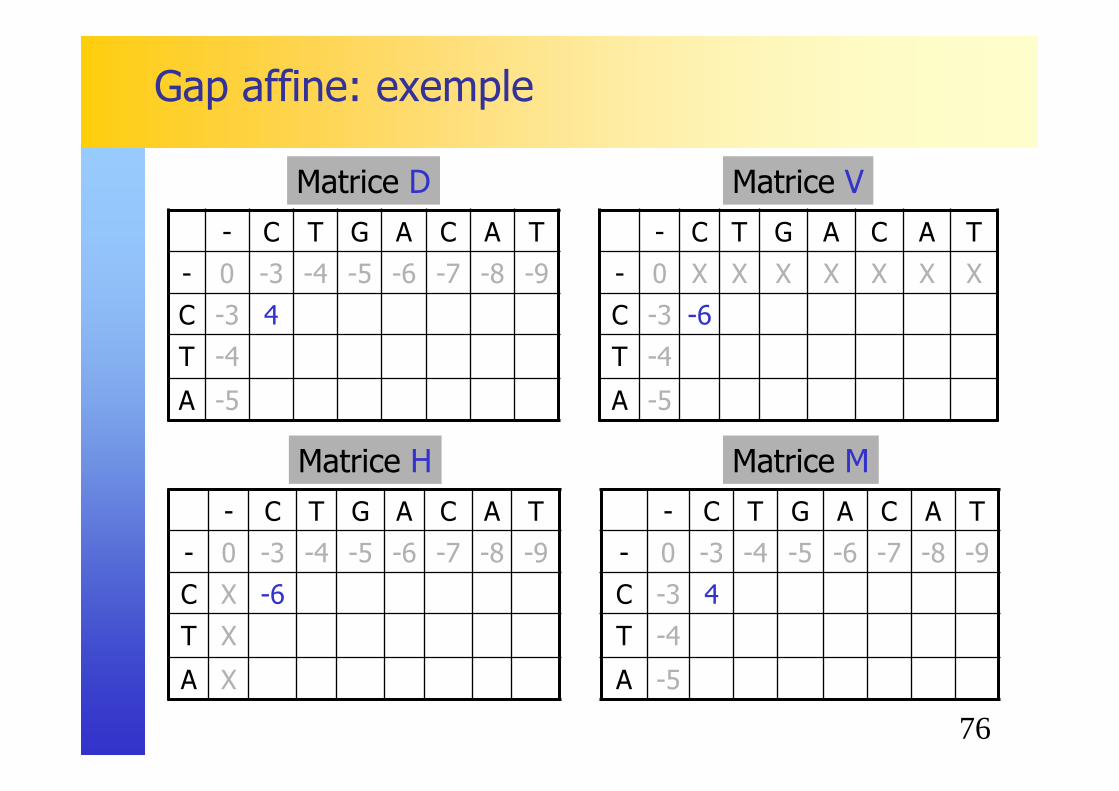
76
Gap affine: exemple
Matrice D Matrice V
Matrice MMatrice H
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C -3 4
T -4
A -5
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C X -6
T X
A X
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C -3 4
T -4
A -5
- C T G A C A T
- 0 X X X X X X X
C -3 -6
T -4
A -5
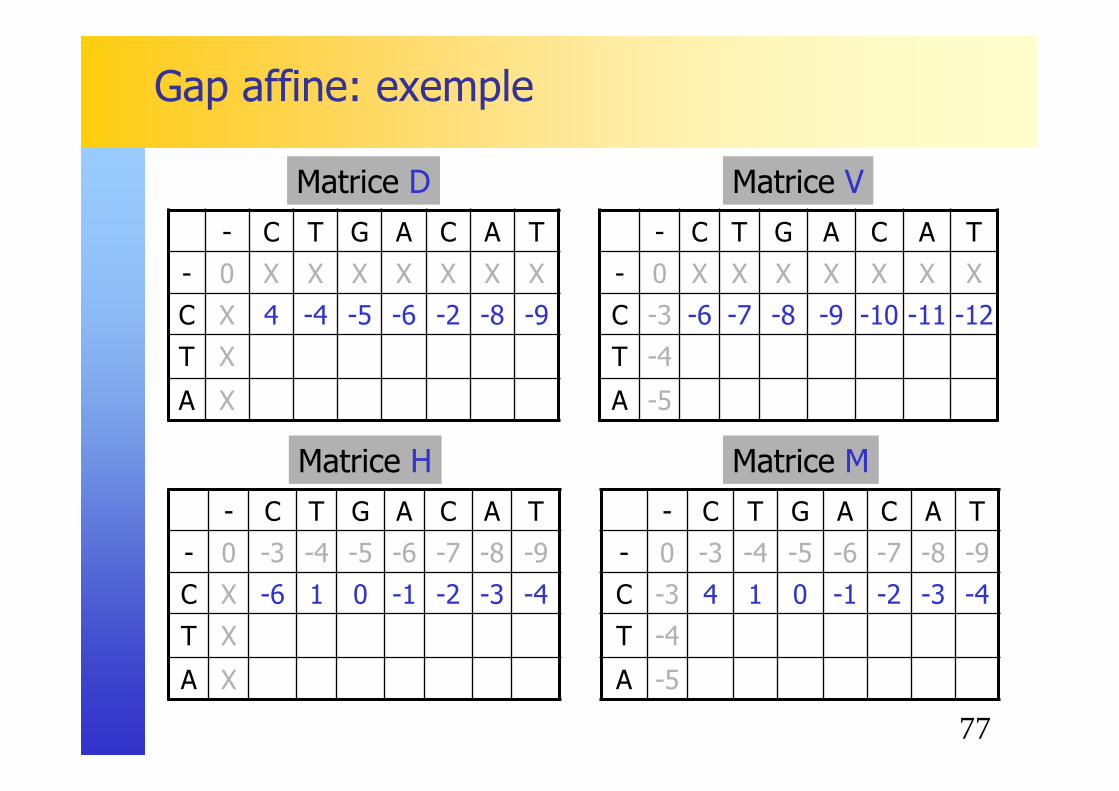
77
Gap affine: exemple
Matrice D Matrice V
Matrice MMatrice H
- C T G A C A T
- 0 X X X X X X X
C X 4 -4 -5 -6 -2 -8 -9
T X
A X
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C X -6 1 0 -1 -2 -3 -4
T X
A X
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C -3 4 1 0 -1 -2 -3 -4
T -4
A -5
- C T G A C A T
- 0 X X X X X X X
C -3 -6 -7 -8 -9 -10 -11 -12
T -4
A -5
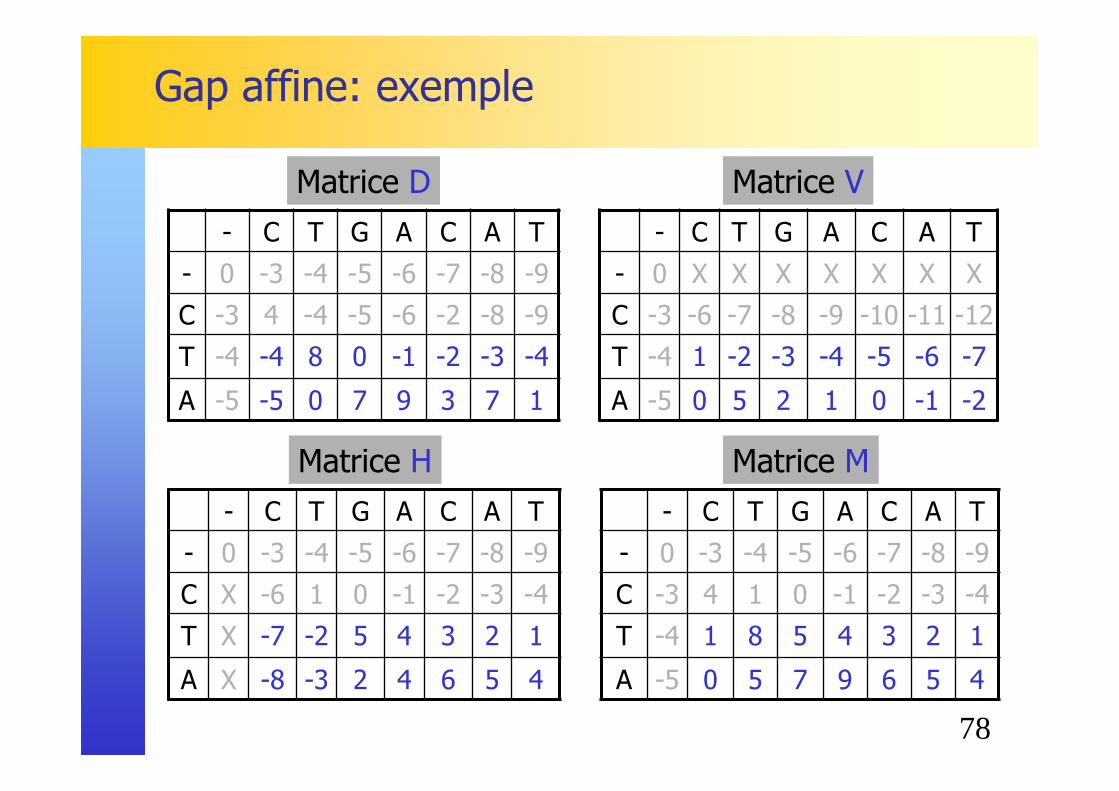
78
Gap affine: exemple
Matrice D Matrice V
Matrice MMatrice H
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C -3 4 -4 -5 -6 -2 -8 -9
T -4 -4 8 0 -1 -2 -3 -4
A -5 -5 0 7 9 3 7 1
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C X -6 1 0 -1 -2 -3 -4
T X -7 -2 5 4 3 2 1
A X -8 -3 2 4 6 5 4
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C -3 4 1 0 -1 -2 -3 -4
T -4 1 8 5 4 3 2 1
A -5 0 5 7 9 6 5 4
- C T G A C A T
- 0 X X X X X X X
C -3 -6 -7 -8 -9 -10 -11 -12
T -4 1 -2 -3 -4 -5 -6 -7
A -5 0 5 2 1 0 -1 -2
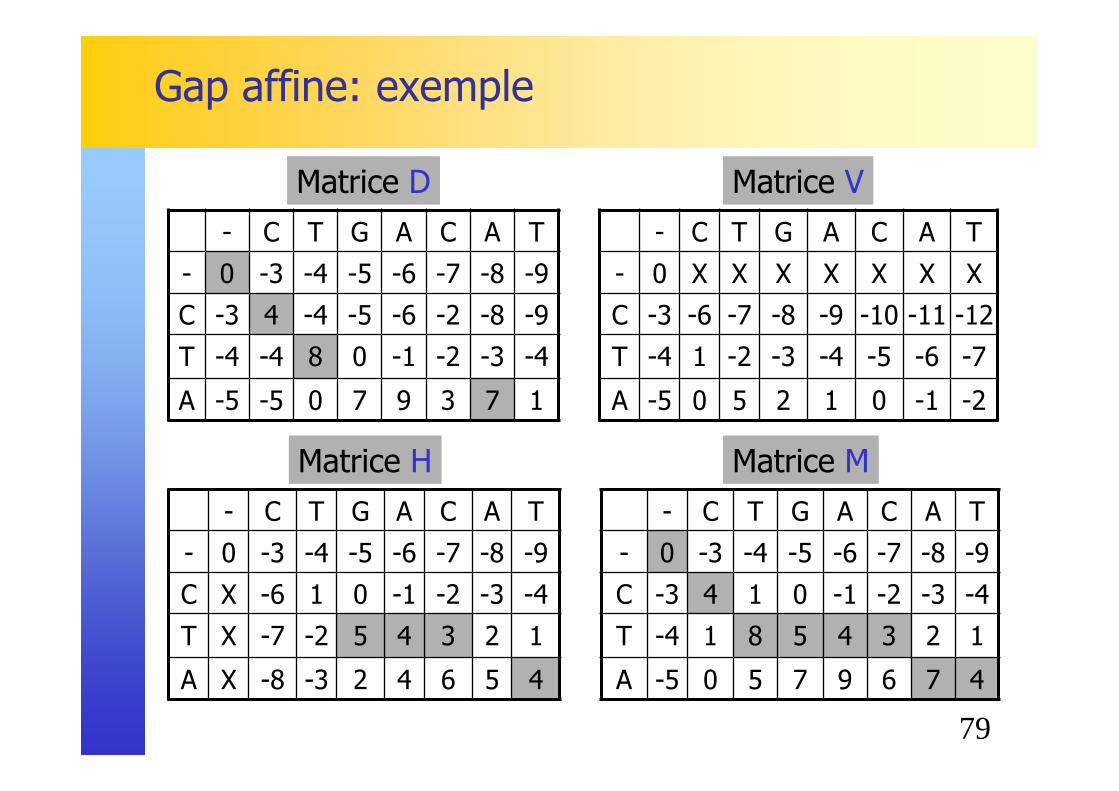
79
Gap affine: exemple
Matrice D Matrice V
Matrice MMatrice H
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C -3 4 -4 -5 -6 -2 -8 -9
T -4 -4 8 0 -1 -2 -3 -4
A -5 -5 0 7 9 3 7 1
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C X -6 1 0 -1 -2 -3 -4
T X -7 -2 5 4 3 2 1
A X -8 -3 2 4 6 5 4
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C -3 4 1 0 -1 -2 -3 -4
T -4 1 8 5 4 3 2 1
A -5 0 5 7 9 6 7 4
- C T G A C A T
- 0 X X X X X X X
C -3 -6 -7 -8 -9 -10 -11 -12
T -4 1 -2 -3 -4 -5 -6 -7
A -5 0 5 2 1 0 -1 -2
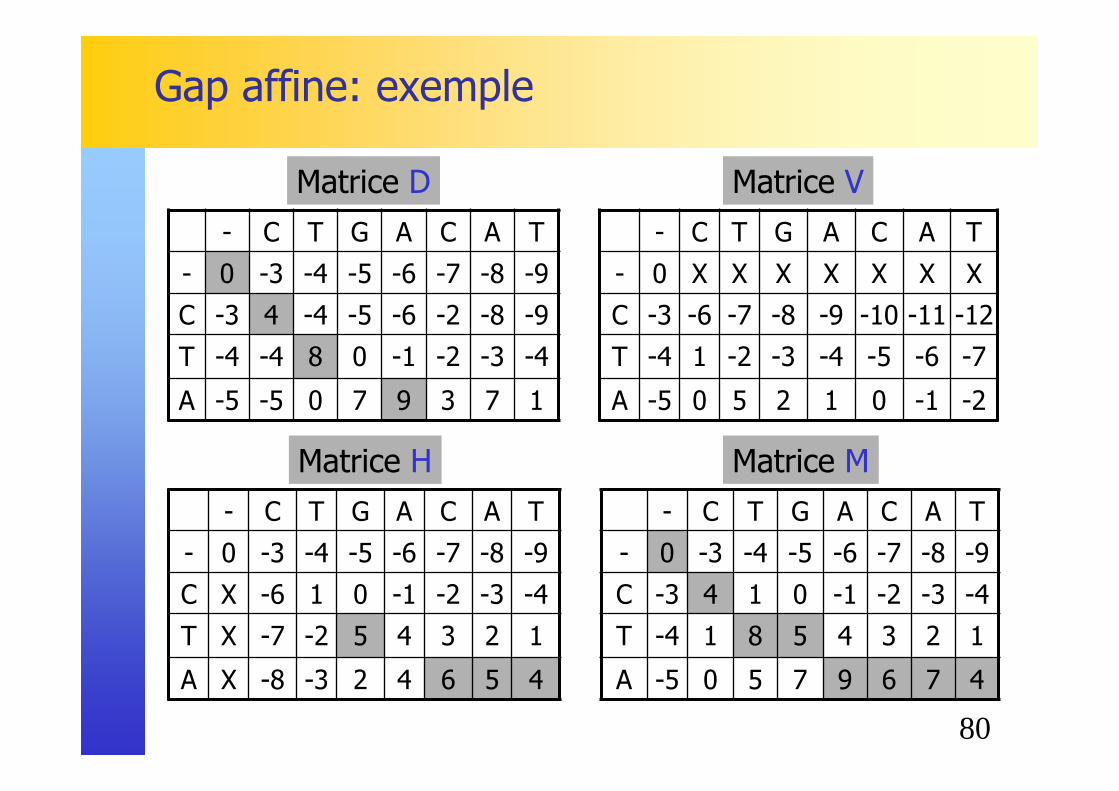
80
Gap affine: exemple
Matrice D Matrice V
Matrice MMatrice H
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C -3 4 -4 -5 -6 -2 -8 -9
T -4 -4 8 0 -1 -2 -3 -4
A -5 -5 0 7 9 3 7 1
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C X -6 1 0 -1 -2 -3 -4
T X -7 -2 5 4 3 2 1
A X -8 -3 2 4 6 5 4
- C T G A C A T
- 0 -3 -4 -5 -6 -7 -8 -9
C -3 4 1 0 -1 -2 -3 -4
T -4 1 8 5 4 3 2 1
A -5 0 5 7 9 6 7 4
- C T G A C A T
- 0 X X X X X X X
C -3 -6 -7 -8 -9 -10 -11 -12
T -4 1 -2 -3 -4 -5 -6 -7
A -5 0 5 2 1 0 -1 -2

81
Gap affine: exemple
� Calculer l’alignement optimal pour les séquences suivantes :� AGCTCGA� TACAGCTTG
� Modèle de gap affine :� Ouverture de gap : -3� Extension de gap : -1� Match : 2� Mismatch : -2

82
- A G C T C G A
- 0 X X X X X X X
T X
A X
C X
A X
G X
C X
T X
T X
G X
- A G C T C G A
- 0 X X X X X X X
T -3
A -4
C -5
A -6
G -7
C -8
T -9
T -10
G -11
- A G C T C G A
- 0 -3 -4 -5 -6 -7 -8 -9
T X
A X
C X
A X
G X
C X
T X
T X
G X
- A G C T C G A
- 0 -3 -4 -5 -6 -7 -8 -9
T -3
A -4
C -5
A -6
G -7
C -8
T -9
T -10
G -11

83
- A G C T C G A
- 0 X X X X X X X
T X -2 -5 -6 -3 -8 -9 -10
A X -1 -4 -7 -8 -5 -8 -5
C X -6 -3 -2 -7 -4 -7 -10
A X -3 -6 -5 -4 -7 -6 -5
G X -8 -1 -8 -7 -6 -5 -8
C X -9 -8 1 -6 -3 -8 -7
T X -10 -9 -6 3 -4 -5 -6
T X -11 -10 -7 0 1 -2 -3
G X -12 -7 -8 -5 -2 3 -4
- A G C T C G A
- 0 -3 -4 -5 -6 -7 -8 -9
T X -6 -5 -6 -7 -6 -7 -8
A X -7 -4 -5 -6 -7 -8 -9
C X -8 -7 -6 -5 -6 -7 -8
A X -9 -6 -7 -8 -7 -8 -9
G X -10 -9 -4 -5 -6 -7 -8
C X -11 -10 -7 -2 -3 -4 -5
T X -12 -11 -8 -5 0 -1 -2
T X -13 -12 -9 -6 -3 -2 -3
G X -14 -13 -10 -7 -4 -5 0
- A G C T C G A
- 0 X X X X X X X
T -3 -6 -7 -8 -9 -10 -11 -12
A -4 -5 -8 -9 -6 -9 -10 -11
C -5 -4 -7 -8 -7 -8 -11 -8
A -6 -5 -6 -5 -8 -7 -10 -9
G -7 -6 -7 -6 -7 -8 -9 -8
C -8 -7 -4 -7 -8 -9 -8 -9
T -9 -8 -5 -2 -5 -6 -7 -8
T -10 -9 -6 -3 0 -3 -4 -5
G -11 -10 -7 -4 -1 -2 -5 -6
- A G C T C G A
- 0 -3 -4 -5 -6 -7 -8 -9
T -3 -2 -5 -6 -3 -6 -7 -8
A -4 -1 -4 -5 -6 -5 -8 -5
C -5 -4 -3 -2 -5 -4 -7 -8
A -6 -3 -6 -5 -4 -7 -6 -5
G -7 -6 -1 -4 -5 -6 -5 -8
C -8 -7 -4 1 -2 -3 -4 -5
T -9 -8 -5 -2 3 0 -1 -2
T -10 -9 -6 -3 0 1 -2 -3
G -11 -10 -7 -4 -1 -2 3 0
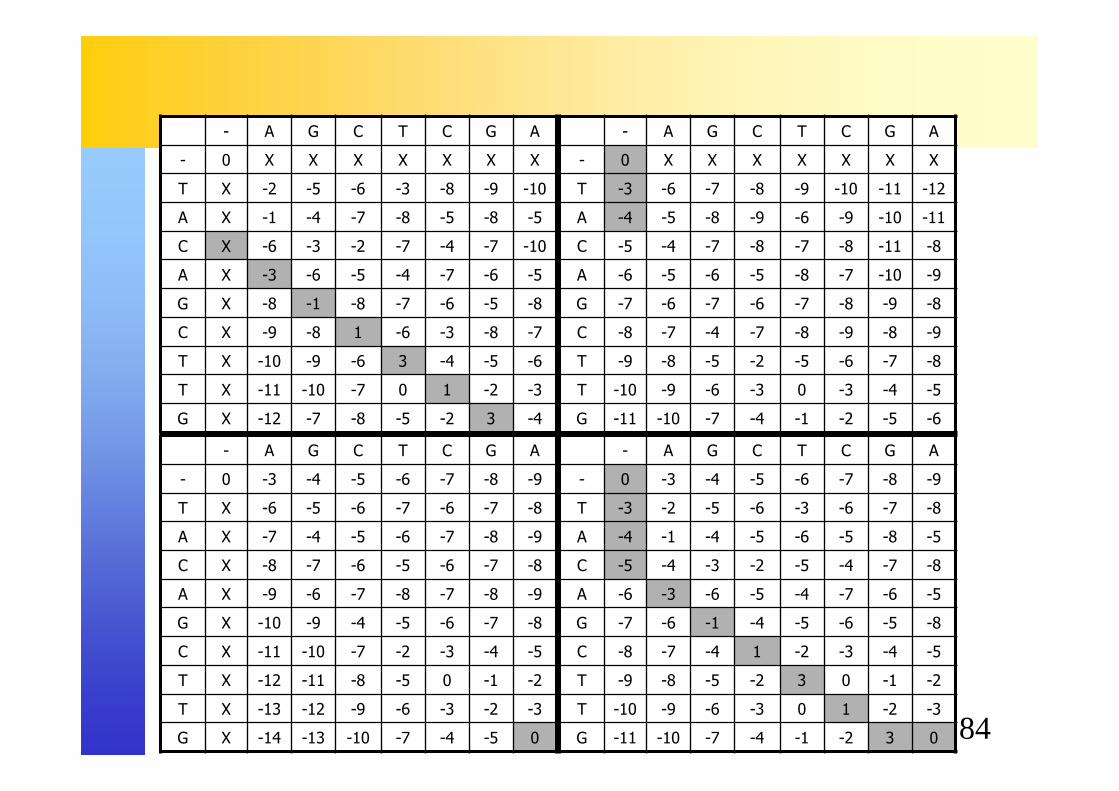
84
- A G C T C G A
- 0 X X X X X X X
T X -2 -5 -6 -3 -8 -9 -10
A X -1 -4 -7 -8 -5 -8 -5
C X -6 -3 -2 -7 -4 -7 -10
A X -3 -6 -5 -4 -7 -6 -5
G X -8 -1 -8 -7 -6 -5 -8
C X -9 -8 1 -6 -3 -8 -7
T X -10 -9 -6 3 -4 -5 -6
T X -11 -10 -7 0 1 -2 -3
G X -12 -7 -8 -5 -2 3 -4
- A G C T C G A
- 0 -3 -4 -5 -6 -7 -8 -9
T X -6 -5 -6 -7 -6 -7 -8
A X -7 -4 -5 -6 -7 -8 -9
C X -8 -7 -6 -5 -6 -7 -8
A X -9 -6 -7 -8 -7 -8 -9
G X -10 -9 -4 -5 -6 -7 -8
C X -11 -10 -7 -2 -3 -4 -5
T X -12 -11 -8 -5 0 -1 -2
T X -13 -12 -9 -6 -3 -2 -3
G X -14 -13 -10 -7 -4 -5 0
- A G C T C G A
- 0 X X X X X X X
T -3 -6 -7 -8 -9 -10 -11 -12
A -4 -5 -8 -9 -6 -9 -10 -11
C -5 -4 -7 -8 -7 -8 -11 -8
A -6 -5 -6 -5 -8 -7 -10 -9
G -7 -6 -7 -6 -7 -8 -9 -8
C -8 -7 -4 -7 -8 -9 -8 -9
T -9 -8 -5 -2 -5 -6 -7 -8
T -10 -9 -6 -3 0 -3 -4 -5
G -11 -10 -7 -4 -1 -2 -5 -6
- A G C T C G A
- 0 -3 -4 -5 -6 -7 -8 -9
T -3 -2 -5 -6 -3 -6 -7 -8
A -4 -1 -4 -5 -6 -5 -8 -5
C -5 -4 -3 -2 -5 -4 -7 -8
A -6 -3 -6 -5 -4 -7 -6 -5
G -7 -6 -1 -4 -5 -6 -5 -8
C -8 -7 -4 1 -2 -3 -4 -5
T -9 -8 -5 -2 3 0 -1 -2
T -10 -9 -6 -3 0 1 -2 -3
G -11 -10 -7 -4 -1 -2 3 0

85
--- AGCTCGATACAGCTTG-
Alignement optimal :(gap affine)
- A G C T C G A
- 0 -3 -4 -5 -6 -7 -8 -9
T -3 -2 -5 -6 -3 -6 -7 -8
A -4 -1 -4 -5 -6 -5 -8 -5
C -5 -4 -3 -2 -5 -4 -7 -8
A -6 -3 -6 -5 -4 -7 -6 -5
G -7 -6 -1 -4 -5 -6 -5 -8
C -8 -7 -4 1 -2 -3 -4 -5
T -9 -8 -5 -2 3 0 -1 -2
T -10 -9 -6 -3 0 1 -2 -3
G -11 -10 -7 -4 -1 -2 3 0

86
Problème lié au gap affine
� Dans certains cas, le résultat trouvé ne semble pas entièrement approprié� exemple:
� Ouverture de gap: -3� Extension de gap: -1� Match: 4� Mismatch: -2
A - - - - - - T G T
A C C T G A T T G T
4-3-1-1-1-1-1+4+4+4=8
- - - - - A - T G T
A C C T G A T T G T
-3-1-1-1-1+4-3+4+4+4=6
A - - - - - - T G T
A C C T G A T T G T
- - - - - A - T G T
A C C T G A T T G T
� Solution: changer le modèle, en ne pénalisant pas les gaps en début de séquence (alignement local affine!)

87
Conclusion
→ L'alignement de séquences par paires est généralement solvable en un temps raisonnable (quadratique)→ De nombreuses variantes selon le résultat recherché
• Matrices de substitution• Modèles de gap• Alignements local/global• …
Remarque : dans certains cas, l'algorithme est trop coûteux• Très grandes séquences (génome)• Recherche de similarité entre une séquence et une banque de
séquences (ex : GenBank)→ Utilisation d'heuristiques (ex : Basic Local Alignment Search Tool)

88
Conclusion
BLAST : • recherche heuristique permettant de trouver les régions similaires entre deux
ou plusieurs séquences de nucléotides ou d'acides aminés. • Permet de retrouver rapidement dans des bases de données, les séquences
ayant des zones de similitude avec une séquence donnée (introduite par l'utilisateur).
• Utilisé pour trouver des relations fonctionnelles ou évolutives entre les séquences et peut aider à identifier les membres d'une même famille de gènes.
• Le terme blast peut être modifié en fonction de la nature de la séquence d'entrée, et de la base de donnée utilisée : blast de nucléotides (séquence nucléotidique vs. BD de séquences nucléotidiques) ; blast de protéines (séqprot. Vs BD prot.) ; blastx (nuc. vs BD prot) ; tblastn (prot. Vs BD nuc.) ; tblastx (nuc.�prot. Vs nuc.� prot.).
• Différentes versions de l'algorithme : BlastN (séquences nucléotidiques, lent donc pas applicable à toute la BD) ; BlastP (protéines, lent) ; Megablast(rapide, séquences similaires) ; PSI-Blast (position-specific iterated BLAST), Blast relancé plusieurs fois par itération. PHI-BLAST (pattern hit initiatedBlast), utilisant un motif utilisé comme point de départ des recherches.

89
Sommaire
� Introduction à la bioinformatique� Notions de base� Alignement 2 à 2 de séquences � Alignement multiple de séquences
� Introduction / Motif protéique� Alignement multiple optimal� Heuristiques pour l’alignement multiple
� Phylogénie

90
Alignement multiple
� Alignement 2 à 2 :� Deux séquences quelconques� Recherche d’une certaine similarité syntaxique� Fonction commune ?
� Alignement multiple :� Famille de séquences partageant une même fonction� Quelle est la conservation syntaxique ?
� Notion de motif protéique

91
� un motif protéïque est une séquence de nucléotides "particulière" qui a une signification biologique.
� Exemple: hormone pancréatique
Motif protéique
PMY_PETMA/1-36 PEE..LSKYMLAVRN YINLIT RQRY
PPY_LOPAM/1-36 PED..WASYQAAVRH YVNLIT RQRY
PAHO_BOVIN/30-65 PEQ..MAQYAAELRR YINMLTRPRY
PAHO_CHICK/26-61 VED..LIRFYNDLQQ YLNVVTRHRY
PAHO_ANSAN/1-36 VED..LRFYYDNLQQ YRLNVFRHRY
NPF_HELAS/4-39 PNE..LRQYLKELNE YYAIMGRTRF
NPF_MONEX/1-39 DNKAALRDYLRQINE YFAIIG RPRF

92
Motif protéique
PMY_PETMA/1-36 PEE..LSKYMLAVRN YINLIT RQRY
PPY_LOPAM/1-36 PED..WASYQAAVRH YVNLIT RQRY
PAHO_BOVIN/30-65 PEQ..MAQYAAELRR YINMLTRPRY
PAHO_CHICK/26-61 VED..LIRFYNDLQQ YLNVVTRHRY
PAHO_ANSAN/1-36 VED..LRFYYDNLQQ YRLNVFRHRY
NPF_HELAS/4-39 PNE..LRQYLKELNE YYAIMGRTRF
NPF_MONEX/1-39 DNKAALRDYLRQINE YFAIIG RPRF
� Exemple : hormone pancréatique
� Expression Prosite� [FY]-x(3)-[LIVM]-x(2)-Y-x(3)-[LIVMFY]-x-R-x-R-[YF]
� Syntaxe� - : séparation des éléments� x : n’importe quel acide aminé� (3,5) : nombre d’occurrences (entre 3 et 5)� [FY] : alternative (F ou Y) – fixer une limite pour le nombre d’alternatives
93
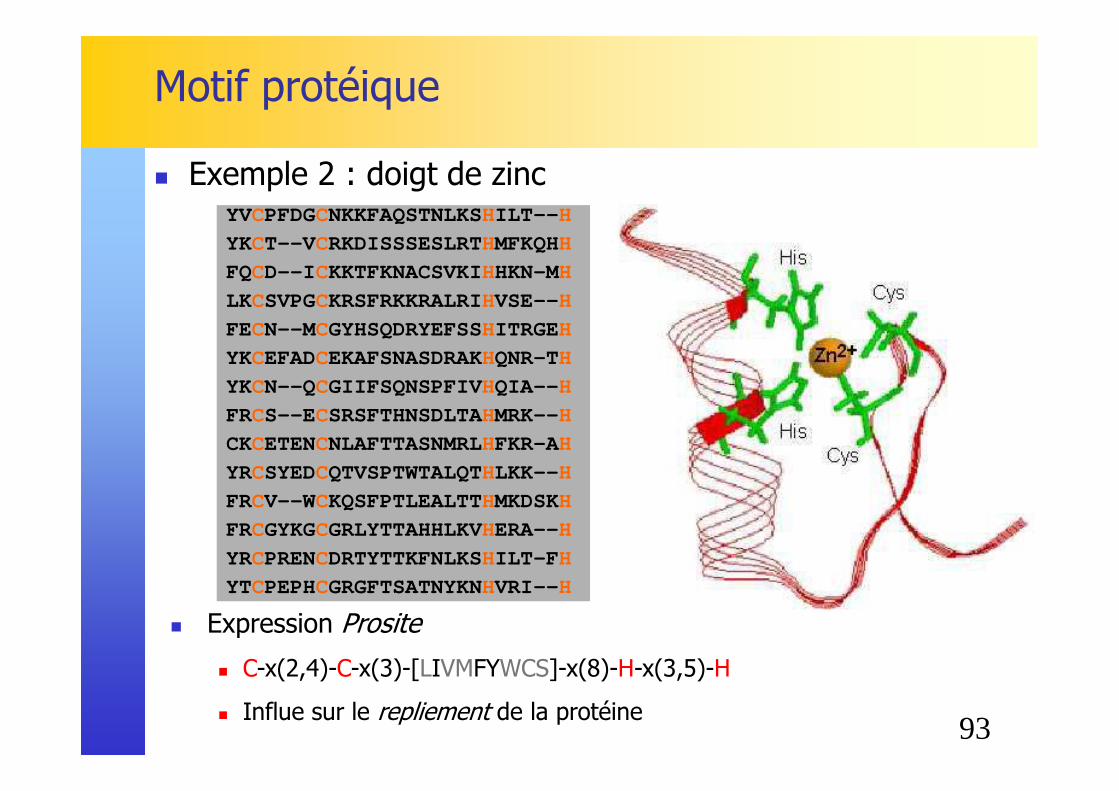
Motif protéique
� Exemple 2 : doigt de zincYVCPFDGCNKKFAQSTNLKSHILT-- H
YKCT--V CRKDISSSESLRTHMFKQHH
FQCD--I CKKTFKNACSVKIHHKN-MH
LKCSVPGCKRSFRKKRALRIHVSE-- H
FECN--M CGYHSQDRYEFSSHITRGEH
YKCEFADCEKAFSNASDRAKHQNR-TH
YKCN--Q CGIIFSQNSPFIV HQIA-- H
FRCS--E CSRSFTHNSDLTAHMRK--H
CKCETENCNLAFTTASNMRLHFKR-AH
YRCSYEDCQTVSPTWTALQTHLKK-- H
FRCV--WCKQSFPTLEALTTHMKDSKH
FRCGYKGCGRLYTTAHHLKVHERA-- H
YRCPRENCDRTYTTKFNLKSHILT-F H
YTCPEPHCGRGFTSATNYKNHVRI-- H
� Expression Prosite
� C-x(2,4)-C-x(3)-[LIVMFYWCS]-x(8)-H-x(3,5)-H
� Influe sur le repliement de la protéine

94
HWGQCGGI---GYSGCKTCTSGTTCQYSNDYYSQCL
HYGQCGGI---GYSGPTVCASGTTCQVLNPYYSQCL
QWGQCGGI---GYTGSTTCASPYTCHVLNPYYSQCY
VWGQCGGQ---NWSGPTCCASGSTCVYSNDYYSQCL
LYGQCGGA---GWTGPTTCQAPGTCKVQNQWYSQCL
IWGQCGGN---GWTGATTCASGLKCEKINDWYYQCV
VWGQCGGN---GWTGPTTCASGSTCVKQNDFYSQCL
DWAQCGGN---GWTGPTTCVSPYTCTKQNDWYSQCL
QWGQCGGQ---NYSGPTTCKSPFTCKKINDFYSQCQ
RWQQCGGI---GFTGPTQCEEPYICTKLNDWYSQCL
HWAQCGGI---GFSGPTTCPEPYTCAKDHDIYSQCV
LYEQCGGI---GFDGVTCCSEGLMCMKMGPYYSQCR
VWAQCGGQ---NWSGTPCCTSGNKCVKLNDFYSQCQ
PYGQCGGM---NYSGKTMCSPGFKCVELNEFFSQCD
AYYQCGGSKSAYPNGNLACATGSKCVKQNEYYSQCV
EYAACGGE---MFMGAKCCKFGLVCYETSGKWSQCR
Motif protéique
HWGQCGGI---GYS GCKTCTSGTTCQYSNDYYSQCL
HYGQCGGI---GYS GPTVCASGTTCQVLNPYYSQCL
QWGQCGGI---GYT GSTTCASPYTCHVLNPYYSQCY
VWGQCGGQ---NWSGPTCCASGSTCVYSNDYYSQCL
LYGQCGGA---GWT GPTTCQAPGTCKVQNQWYSQCL
IWGQCGGN---GWT GATTCASGLKCEKINDWYYQCV
VWGQCGGN---GWT GPTTCASGSTCVKQNDFYSQCL
DWAQCGGN---GWT GPTTCVSPYTCTKQNDWYSQCL
QWGQCGGQ---NYS GPTTCKSPFTCKKINDFYSQCQ
RWQQCGGI---GFT GPTQCEEPYICTKLNDWYSQCL
HWAQCGGI---GFS GPTTCPEPYTCAKDHDIYSQCV
LYEQCGGI---GFD GVTCCSEGLMCMKMGPYYSQCR
VWAQCGGQ---NWSGTPCCTSGNKCVKLNDFYSQCQ
PYGQCGGM---NYS GKTMCSPGFKCVELNEFFSQCD
AYYQCGGSKSAYPNGNLACATGSKCVKQNEYYSQCV
EYAACGGE---MFM GAKCCKFGLVCYETSGKWSQCR
� Exemple 3 : Site de fixation de la cellulose
C-G-G-x(4,7)-G-x(3)-C-x(5)-C-x(3,5)-[NHG]-x-[FYWM]- x(2)-Q-C

95
Alignement multiple
� Entrée : k séquences (nucléiques ou acides aminés)
� Sortie : un tableau contenant les k séquences avec des indels
*******************
*********************
**********************
******************
*********************
*--********---********--**
*******--*********-*****--
****-----*****************
***********--------*******
*--*************---*******

96
Alignement multiple
� Comment scorer un alignement multiple?� Score SP - sums of pairs : somme des scores de ses colonnes
� Comment scorer une colonne ?� adaptable à un nombre quelconque de lignes� indépendant de l'ordre� reflète la similarité
∑≤<≤
=
kjiji
k
ccscore
c
c
scoreSP1
1
),(M
{ } ( ) 0,et,,1 =−−−∪Α∈ scorecc kL

97
Alignement multiple : score
A A C G T A C G A T A
A - C G T A - A A T G
G T C G T A - - T T A
� Exemple :� Identite : +2� Substitution : -1� Indel : -2

98
Alignement multiple : score
A A C G T A C G A T A
A - C G T A - A A T G
G T C G T A - - T T A
� Exemple :� Identite : +2� Substitution : -1� Indel : -2
8

99
Alignement multiple : score
A A C G T A C G A T A
A - C G T A - A A T G
G T C G T A - - T T A
� Exemple :� Identite : +2� Substitution : -1� Indel : -2
85
3

100
Alignement multiple : score
A A C G T A C G A T A
A - C G T A - A A T G
G T C G T A - - T T A
� Exemple :� Identite : +2� Substitution : -1� Indel : -2
Score de l’alignement multiple : 8+5+3=16
85
3

101
Score : Méthode alternative
A A C G T A C G A T A
A - C G T A - A A T G
G T C G T A - - T T A
� Exemple :� Identite : +2� Substitution : -1� Indel : -2

102
Score : Méthode alternative
A A C G T A C G A T A
A - C G T A - A A T G
G T C G T A - - T T A
� Exemple :� Identite : +2� Substitution : -1� Indel : -2
2-1-1=0
-2-1-2=-5
222=6
222=6
222=6
222=6
-2-20=-4
-1-2-2=-5
2-1-1=0
222=6
-12-1=0

103
Score : Méthode alternative
A A C G T A C G A T A
A - C G T A - A A T G
G T C G T A - - T T A
� Exemple :� Identite : +2� Substitution : -1� Indel : -2
Score de l’alignement multiple : 0-5+6+6+6+6-4-5+0+6+0=16
2-1-1=0
-2-1-2=-5
222=6
222=6
222=6
222=6
-2-20=-4
-1-2-2=-5
2-1-1=0
222=6
-12-1=0

104
Score : Visualisation (align. d'acides aminés)
� Notations usuelles :� * → correspondance� : → substitution conservative (acides aminés de même groupe et de
scores ≥ 0)� . → substitution semi-conservative (acides aminés de même groupe)
A L L A L W G P D P AA L L A F W G P D P A A L L A F W G P D P SA L L V L W E P K P SA L L V F S G P G T S
* * * . . * . :

105
Formulation
� Définition : Alignement� Soit un alphabet Σ.� Soit S = { S1, S2, ..., Sk } un ensemble de k séquences.� Un alignement de S, noté A(S1, S2, ..., Sk) est une matrice k*q
� Chaque élément au,v de la matrice a est défini dans Σ.� q est plus grand que la plus grande des séquences, et plus petit
que la somme des tailles des séquences.� Pour tout u tel que q 1≤u≤k, la séquence {au,1, au,2,..., au,q}
dans laquelle on supprime les gaps correspond à Su
� Formulation : Problème d'alignement multiple� Soient k séquences S1,…,Sk et une matrice de score w, le problème
d'alignement multiple consiste à déterminer un alignement de coût optimal selon w.

106
Alignement multiple : approche exacte
� Quelques chiffres concernant le nombre de configurationspour l’alignement multiple. � nombre de configurations différentes pour un alignement de
k séquences de longeur n.
k n
2 3 4 5
2 13 63 321 1683
3 409 16081 699121 3.2e7
4 23917 1.1e7 … …

Alignement multiple : approche exacte
� Problème algorithmique� Trouver l'alignement multiple de score SP maximal.� Approche exacte : programmation dynamique� Alignement deux à deux : chemin dans une matrice de
dimension 2� Alignement multiple : chemin dans une matrice de dimension
supérieure � k séquences à aligner, matrice de dimension k
-G-A-C-GTGAT--G
A
T
G
C G T G
AG

108
Alignement multiple : approche exacte
� Exemple pour trois séquences (U, V et W)� Matrice en dimension trois� Sim(i , j , k) : score optimal entre U(1..i ), V(1..j) et
W(1..k).� Formules de récurrence :

109
Alignement multiple : approche exacte
� Problème de complexité� Explosion combinatoire quand le nombre de séquences
augmente

110
Alignement multiple : approche exacte
� Complexité� s1,…,sk : séquences de taille n� T(i1,…,ik) : score optimal entre les k préfixes
s1(1,…,i1) , … , sk(1,…,ik)
� O(nk2kk2)� Table de taille nk
� Temps de calcul d'une case : dépend de 2k-1 cases précédentes
� Temps de calcul de chaque scoreSP : k(k-1)/2
� Problème de décision NP-Complet

111
Alignement multiple
� Recours à des approches heuristiques� Réduire le temps de calcul � Maximiser le score du résultat� Pas de garantie d’optimalité !
� Quelques exemples d’algorithmes� En étoile (basique)� Clustal (le plus populaire)� Dialign2 (complémentaire à Clustal)� T-coffee, Pima, Multalin, Plasma…� + méthodes heuristiques, métaheuristiques
� Autant d'alignements que de programmes…

112
Alignement multiple progressif
� Les séquences homologues sont reliées d’un point de vue évolutif� Idée : construire progressivement un alignement, à
partir de séries de séquences (ou de groupes de séquences) alignées deux à deux, suivant un ordre de branchement donné par un arbre d’évolution � Alignement des séquences les plus proches d’un point de vue
phylogénétique (évolution)� Intégration progressive des séquences un peu plus éloignées
� Approche suffisamment rapide pour permettre la construction d’alignements contenant un grand nombre de séquences
113
Heuristique en étoile
� Heuristique en étoile� Sélection d'une séquence centrale� Construction de l'alignement multiple, en partant de la
séquence centrale, puis en incorporant une à une les autres séquences
� Exemple :
� indel : -2, substitution : -1, identite : 1
S1 cgatgagtcattgtgactg
S2 cgagccattgtagctactg
S3 cgaccattgtagctacctg
S4 cgatgagtcactgtgactg

114
Heuristique en étoile
� Etape 1 : Alignements globaux de toutes les séquences deux par deux
S1 cgatgagtcattgt-g--actg S2 cgagccattgtagcta-ctg||| | |||||| | |||| ||| |||||||||||| |||
S2 cga-g--ccattgtagctactg S3 cga-ccattgtagctacctg
S1 cgatgagtcattg-tgactg S2 cga-g--ccattgtagctactg||| | | | | | ||| ||| | || ||| | ||||
S3 cgacca-ttgtagctacctg S4 cgatgagtcactgt-g--actg
S1 cgatgagtcattgtgactg S3 cgaccattgtagctacctg|||||||||| |||||||| ||| | | | |||
S4 cgatgagtcactgtgactg S4 cgatgagtcactgtgactg
S1 S2 S3 S4
S1 2 0 17
S2 2 14 0
S3 0 14 -1
S4 17 0 -1
Tableau des scores
k séquences
�
k(k-1)/2 alignements

115
Heuristique en étoile
� Etape 2 : sélection de la séquence centrale à partir du tableau des scores� séquence qui maximise la somme des similarités avec
l'ensemble des autres séquences
� But : Aligner selon la séquence la plus représentative de l’ensemble (centre de gravité)
S1 S2 S3 S4
S1 2 0 17
S2 2 14 0
S3 0 14 -1
S4 17 0 -1
� 19
� 16
� 13
� 16

� Etape 3 : construction de l'alignement multiple par juxtaposition des alignements deux à deux avec la séquence centrale
� L'intégration d'une nouvelle séquence se fait en prenant la séquence centrale comme guide Possible en étirant les gaps de l'alignement multiple courant.
S1 cgatgagtcattgt-g--actg||| | |||||| | ||||
S2 cga-g--ccattgtagctactg
S1 cgatgagtcattg-tgactg||| | | | | | |||
S3 cgacca-ttgtagctacctg
S1 cgatgagtcattgtgactg|||||||||| ||||||||
S4 cgatgagtcactgtgactg
s1 cgatgagtcattgtgactgs4 cgatgagtcactgtgactg�
Alignement multiple
Heuristique en étoile

� Etape 3 : construction de l'alignement multiple par juxtaposition des alignements deux à deux avec la séquence centrale
� L'intégration d'une nouvelle séquence se fait en prenant la séquence centrale comme guide Possible en étirant les gaps de l'alignement multiple courant.
S1 cgatgagtcattgt-g--actg||| | |||||| | ||||
S2 cga-g--ccattgtagctactg
S1 cgatgagtcattg-tgactg||| | | | | | |||
S3 cgacca-ttgtagctacctg
S1 cgatgagtcattgtgactg|||||||||| ||||||||
S4 cgatgagtcactgtgactg
�
Alignement multiple
Heuristique en étoile
s1 cgatgagtcattgtgactgs4 cgatgagtcactgtgactg
s1 cgatgagtcattg - tgactgs3 cgacca-ttgtagctacctgs4 cgatgagtcactg - tgactg
s1 cgatgagtcattg-tgactgs3 cgacca-ttgtagctacctg

� Etape 3 : construction de l'alignement multiple par juxtaposition des alignements deux à deux avec la séquence centrale
� L'intégration d'une nouvelle séquence se fait en prenant la séquence centrale comme guide Possible en étirant les gaps de l'alignement multiple courant.
S1 cgatgagtcattgt-g--actg||| | |||||| | ||||
S2 cga-g--ccattgtagctactg
S1 cgatgagtcattg-tgactg||| | | | | | |||
S3 cgacca-ttgtagctacctg
S1 cgatgagtcattgtgactg|||||||||| ||||||||
S4 cgatgagtcactgtgactg
s1 cgatgagtcattg-t - g-- actgs2 cga-g--ccattg-tagctactgs3 cgacca-ttgtagct - a-- cctgs4 cgatgagtcactg-t - g-- actg
�
Alignement multiple
Heuristique en étoile
s1 cgatgagtcattg-tgactgs3 cgacca-ttgtagctacctgs4 cgatgagtcactg-tgactg
s1 cgatgagtcattgt-g--actgs2 cga-g--ccattgtagctactg

119
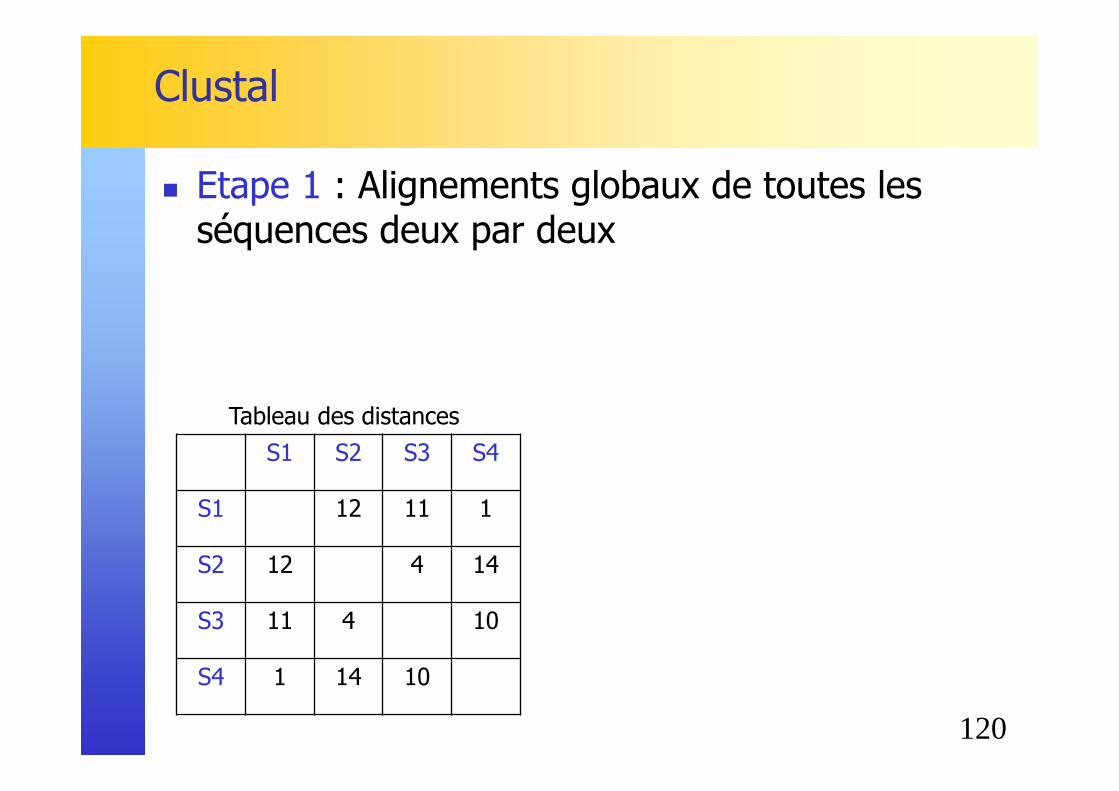
Clustal
� Higgins et Sharp [1988]. CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene, 73, 237-244.
� Clustal = CLUSTer + ALignment� Inspiré par la classification hiérarchique ascendante� Regroupement progressif des séquences� Exemple:
� indel : 2, substitution : 1, identite : 0� Attention, on calcule une distance, d’où les scores utilisés
S1 cgatgagtcattgtgactg
S2 cgagccattgtagctactg
S3 cgaccattgtagctacctg
S4 cgatgagtcactgtgactg
120
Clustal
� Etape 1 : Alignements globaux de toutes les séquences deux par deux
S1 S2 S3 S4
S1 12 11 1
S2 12 4 14
S3 11 4 10
S4 1 14 10
Tableau des distances

121
Clustal
� Etape 1 : Alignements globaux de toutes les séquences deux par deux� Les séquences sont regroupées suivant leur similarité à
partir de la matrice des distances 2 à 2.
S1 S2 S3 S4
S1 12 11 1
S2 12 4 14
S3 11 4 10
S4 1 14 10
Tableau des distances
S1
S4 S3
S2
S1 S4

122
Clustal
� Etape 1 : Alignements globaux de toutes les séquences deux par deux� Les séquences sont regroupées suivant leur similarité à
partir de la matrice des distances 2 à 2.� Nouveaux scores : Neighbour-Joigning (cf. phylogénie)
Tableau des scores
S1
S4 S3
S2
S1 S4
S1/S4 S2 S3
S1/
S412.5 10
S2 12.5 4
S3 10 4

123
Clustal
� Etape 1 : Alignements globaux de toutes les séquences deux par deux� Les séquences sont regroupées suivant leur similarité à
partir de la matrice des distances 2 à 2.� Nouveaux scores : Neighbour-Joigning (cf. phylogénie)
Tableau des scores
S1
S4 S3
S2S1/S4 S2 S3
S1/
S412.5 10
S2 12.5 4
S3 10 4
S1 S4 S2 S3

124
Clustal
� Etape 2 : construction de l'alignement à partir de l'arbre guide� Arbre guide : classication hiérarchique ascendante� Alignement entre deux clusters de séquences :
alignement deux à deux avec le score SP pour le score d'une colonne
� L'alignement est obtenu par extensions successives.� « Once a gap, always a gap »

125
Clustal
S2 cgagccattgtagctac-tg||| ||||||||||||| ||
S3 cga-ccattgtagctacctg
S1 cgatgagtcattgtgactg|||||||||| ||||||||
S4 cgatgagtcactgtgactg
S1 cgatgagtcattgtgactg
S4 cgagccattgtagctactg
S3 cgaccattgtagctacctg
S2 cgatgagtcactgtgactg
S1 cgatgagtcattgt-g--ac-tg
S4 cgatgagtcactgt-g--ac-tg
S2 cga---gccattgtagctac-tgS3 cga----ccattgtagctacctg

126
→ Variante de ClustalThompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequenceweighting, position specific gap penalties and weight matrix choice.Nucleic Acids Res. 22(22):4673-80.
� Modification principale au niveau de la mise à jour de la matrice des distances après regroupement de 2 séquences
� Des poids sont associés selon le nombre de séquences concernées (cf. phylogénie)
Clustal-W

127
Autres algorithmes d’alignement multiple
� Beaucoup d’algorithme dans la littérature !� Les plus classiques/performants :
� clustal omega : nouvelle variante de clustal
� multalin : variante de clustal
� T-coffee : variante de clustal� muscle : fonction de création de profils
� probcons : modèle de Markov
� mafft : transformée de Fourier� dialign : recherche de chemins
� saga : algorithme génétique
� hmmer : modèle de Markov
� Voir: « Multiple sequence alignment », Robert C. Edgarand Serafim Batzoglou, dans « Current Opinion in Structural Biology », 2006, volume 16, pages 368–373.

128
Evaluation des heuristiques d’alignement
� BaliBase (Thompson, Plewniak, Poch 1999)� ensemble d’alignements de référence (considérés comme étant
corrects)� utilisé pour attester de la qualité des logiciels d’alignement multiple
� Base décomposée en 5 sous-ensembles caractéristiques :� set 1 : séquences équidistantes� set 2 : une séquence orpheline� set 3 : familles divergentes� set 4 : longues insertions de gap aux extrémités� set 5 : longues insertions de gap au milieu
� Actuellement :� ProbCons : sets 1, 2 et 3� MAFFT : sets 4 et 5� D’autres candidats à tester ?

129
Sommaire
� Introduction à la bioinformatique� Notions de base� Alignement 2 à 2 de séquences � Alignement multiple de séquences� Phylogénie
� Généralités / Notions sur les arbres� Méthodes de reconstruction phylogénétique
� WPGMA / UPGMA� Neighbourg-joigning� Parcimonie

130
Phylogénie
� La phylogénie est l'étude de la formation et de l'évolution des organismes vivants en vue d'établir leur parenté
� On représente couramment une phylogénie par un arbre phylogénétique. La proximité des branches de cet arbre représente le degré de parenté entre les taxons, les nœuds les ancêtres communs des taxons
� Dans un arbre élaboré par phénétique, la longueur des branches représente la distance génétique entre taxons
Arbre de Haeckel (1866)

131
Phylogénie
� Depuis Darwin, il est communément admis que les êtres vivants descendent tous les uns des autres.
� Jusqu'aux années 1960, les seuls moyens disponibles pour construire des classifications d'espèces étaient:� comparaisons entre les morphologies,
� comparaisons des comportements
� répartition géographiques des espèces…
� La découverte que des protéines homologues (ou acides nucléiques) avaient des séquences en acides aminés (ou en bases) qui variaient d'une espèce à l'autre a fourni un nouveau moyen d'étude : la phylogénie.

132
Phylogénie� Evolution
� L’évolution selon Lamarck (1744-1829)� l'évolution est due à une adaptation continue au
milieu ambiant : un environnement changeant altère les besoins de l'organisme vivant qui s'adapte en modifiant son comportement

133
Phylogénie� Evolution
� L’évolution selon Darwin (1809-1882)� évolution par sélection naturelle : au sein
d'une même lignée, tous les individus sont différents et la nature favorise la multiplication de ceux qui jouissent d'un quelconque avantage

134
� Pour quoi faire ?� Retracer l’histoire évolutive d’une famille de gènes� Reconstruire les relations évolutives entre espèces
� ex : arbre du vivant
� Classer une nouvelle espèce� ex : souche virale
� Comment ?� Aligner correctement les séquences nucléiques ou
protéiques� Appliquer une méthode de génération d’arbres� Évaluer statistiquement la robustesse des arbres
Phylogénie

135
� Deux grands types de méthodes permettant la reconstruction d'arbres phylogénétiques:� Méthodes basées sur les mesures de distances entre séquences
prises deux à deux, c'est à dire le nombre de substitutions de nucléotides ou d'acides aminés entre ces deux séquences.� UPGMA� Neighbour-Joining� …
� Méthodes basées sur les caractères qui s'intéressent au nombre de mutations (substitutions / insertions /délétions) qui affectent chacun des sites (positions) de la séquence. � Parcimonie� Maximum de vraissemblance� …
Phylogénie� Méthodes de reconstruction

136
� Un arbre phylogénétique est caractérisé par :� sa topologie� la longueur de ses branches (éventuellement)
� Nœud : estimation de l’ancêtre commun des éléments appartenant à ce nœud
� Racine (root) : ancêtre commun de tous les éléments de l’arbre.
� Un arbre peut avoir ou non une racine.
Phylogénie� Notions de bases (arbres)
Seq A
Seq C
Seq B Seq ASeq D
Seq B
Seq D
Seq C
nœud internenœud feuille
racine

Phylogénie� Notions de bases (arbres)
� Notation de Newick� Pour stocker un arbre dans un fichier texte, on peut
utiliser la notation suivante � ((A,B),C)
� On peut aussi ajouter la longueur de chaque branche� ((A:1,B:1):2,C:4)
A
B
1
1
C
2
4
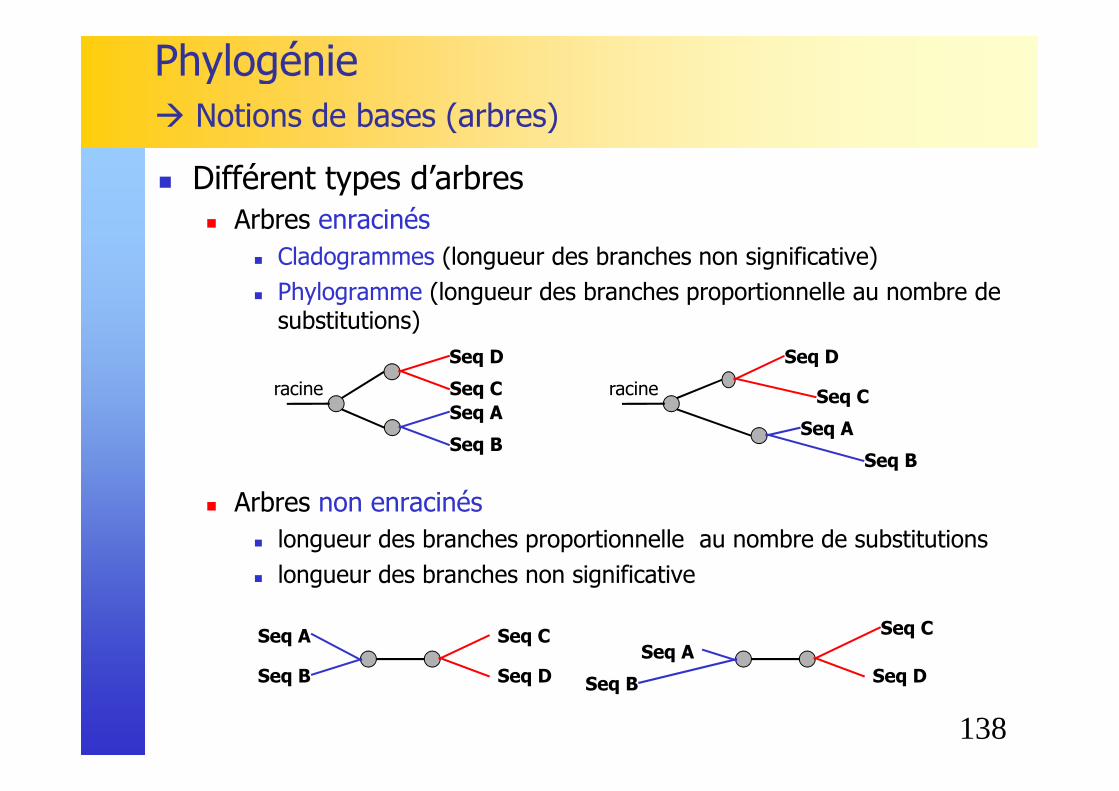
138
� Différent types d’arbres� Arbres enracinés
� Cladogrammes (longueur des branches non significative)� Phylogramme (longueur des branches proportionnelle au nombre de
substitutions)
� Arbres non enracinés� longueur des branches proportionnelle au nombre de substitutions� longueur des branches non significative
Phylogénie� Notions de bases (arbres)
Seq A
Seq B
Seq D
Seq Cracine
Seq A Seq C
Seq B Seq D
Seq A
Seq C
Seq B Seq D
Seq A
Seq B
Seq D
Seq Cracine
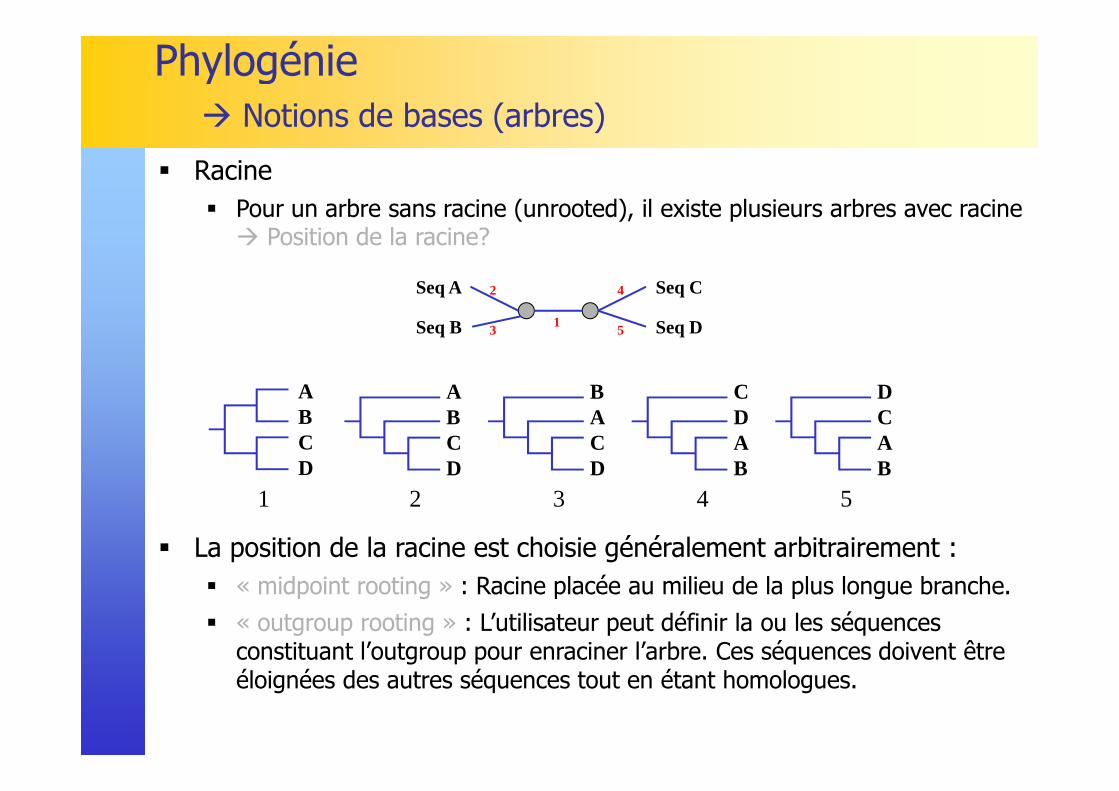
� Racine� Pour un arbre sans racine (unrooted), il existe plusieurs arbres avec racine � Position de la racine?
� La position de la racine est choisie généralement arbitrairement :� « midpoint rooting » : Racine placée au milieu de la plus longue branche.
� « outgroup rooting » : L’utilisateur peut définir la ou les séquences constituant l’outgroup pour enraciner l’arbre. Ces séquences doivent être éloignées des autres séquences tout en étant homologues.
Phylogénie� Notions de bases (arbres)
Seq A Seq C
Seq B Seq D
1
ABCD
ABCD
2
BACD
3
CDAB
4
DCAB
5
1
2
3
4
5

Phylogénie� Notions de bases (arbres)
� Ordre des branches� L’ordre des branches appartenant à un même noeud
n’a aucune importance.� La rotation autour d’un noeud ne change rien à la
topologie de l’arbre.
A B C D I J K L A B C D I J K LE F G H H G F E

141
� Distance d’édition� A partir des opérations nécessaires pour obtenir un alignement, on
peut calculer une distance dite distance d’édition ou de Levenshtein
� Problème de distance d’édition� Consiste à trouver la distance minimum qui permet de transformer
une séquence en une autre séquence en utilisant les opérations d’édition
� Méthode : optimiser l’alignement pour minimiser la distance
Phylogénie� Calcul des distances
( ) ( ) ( ) =
==∑− sinon1
si0,avec,,
1
iiii
q
iiiL
yxyxdyxdVUd
( ) ( )( )VUdVUd LL ,min,* =
cgagccattctagctac-tg||| ||||| ||||||| ||cga-ccattgtagctacctg
� d=3

142
� Correction des distances� Si le temps de divergence entre deux séquences
augmente, la probabilité d’avoir plusieurs substitutionsà un même site augmente
Phylogénie� Calcul des distances
Séq 1 Séq 2 Substitutions observées
Substitutions réelles
Substitution unique C C�A 1 1
Substitutions multiples C C�A�T 1 2
Substitutions coïncidentes C�G C�A 1 2
Substitutions parallèles C�A C�A 0 2
Substitutions convergentes C�A C�T�A 0 3
Substitutions réverses C C�T�C 0 2
� Nombreuses méthodes tentant d’estimer la distance réelle entre séquences

143
� Problème : minimiser la somme des distances d’édition de l’arbre (pour maximiser la vraisemblance de l’arbre)
� WPGMA / UPGMA � « Weighted Pair Group Method with Arithmetic mean »� « Unweighted Pair Group Method with Arithmetic mean »� méthodes utilisées pour reconstruire des arbres phylogénétiques si
les séquences ne sont pas trop divergentes. � algorithmes de clusterisation séquentiel dans lequel les relations
sont identifiées dans l'ordre de leur similarité et la reconstruction de l'arbre se fait pas à pas grâce à cet ordre.
� UPGMA est utilisé pour l’alignement multiple dans l’algo. CLUSTAL
� Principe général� Identification des deux séquences les plus proches� Ces deux séquences sont ensuite traitées comme un tout� On recherche les séquences les plus proches � …et ainsi de suite jusqu'à ce qu'il n'y ait plus que deux groupes
Phylogénie� WPGMA / UPGMA

144
� Hypothèses : � le taux de mutation est le même dans toutes les lignées (horloge
moléculaire)� Pas de mutations multiples
� Méthode� Regroupement des 2 séquences Si et Sj les plus proches� Le noeud Sij est positionné à une distance d de chacune des
séquences (Weighted PGMA)� d = (dist(Si,Sj ))/2
� Calcul de la distance entre le nouveau groupe et les autres séquences� dist((Si,Sj ),Sk) = (dist(Si,Sk)+dist(Sj,Sk))/2
� etc...
Phylogénie� WPGMA
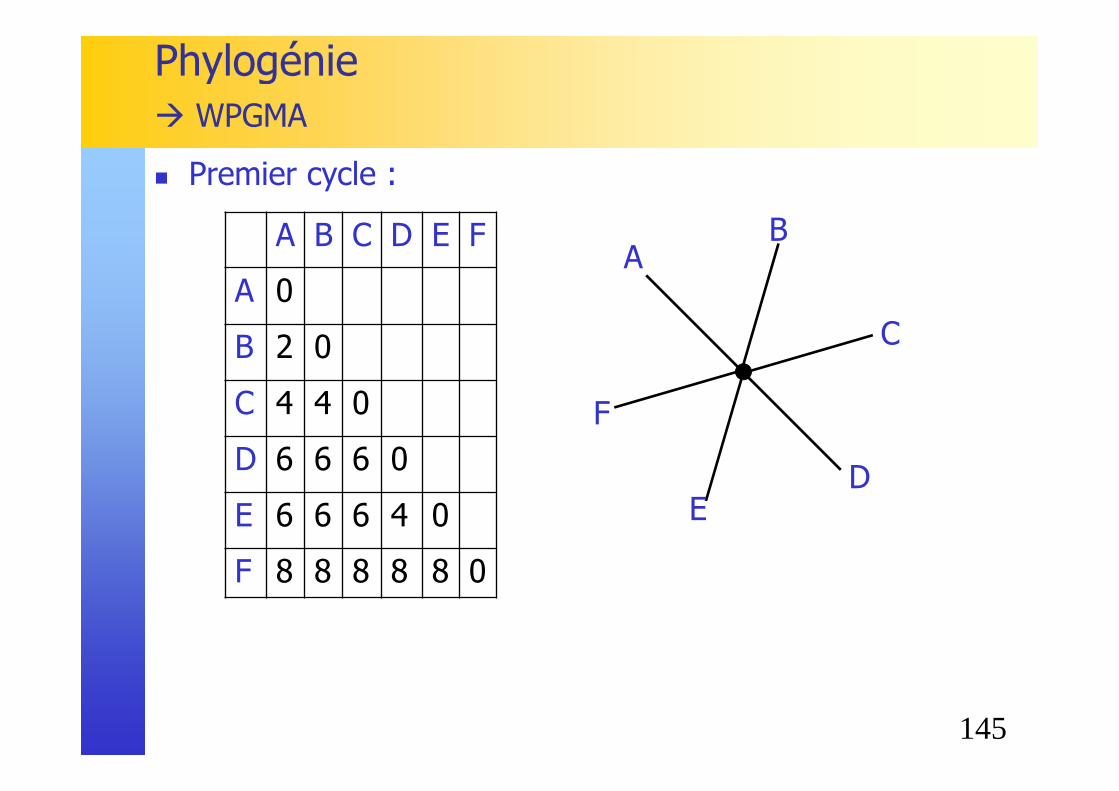
145
A B C D E F
A 0
B 2 0
C 4 4 0
D 6 6 6 0
E 6 6 6 4 0
F 8 8 8 8 8 0
Phylogénie� WPGMA
� Premier cycle :
A
C
F
B
ED

146
dist(A,B),C=(dist AC+dist BC)/2 = 4dist(A,B),D=(dist AD+dist BD)/2 = 6dist(A,B),E=(dist AE+dist BE)/2 = 6dist(A,B),F=(dist AF+dist BF)/2 = 8
A B C D E F
A 0
B 2 0
C 4 4 0
D 6 6 6 0
E 6 6 6 4 0
F 8 8 8 8 8 0
Phylogénie� WPGMA
� Premier cycle :
A
C
F
B
ED
11

147
A,B C D E F
A,B 0
C 4 0
D 6 6 0
E 6 6 4 0
F 8 8 8 8 0
Phylogénie� WPGMA
� Deuxième cycle :
dist(D,E),(A,B)=(dist D(A,B)+dist E(A,B))/2 = 6dist(D,E),C =(dist DC +dist EC )/2 = 6dist(D,E),F =(dist DF +dist EF )/2 = 8
A
C
F
B
ED
11
22
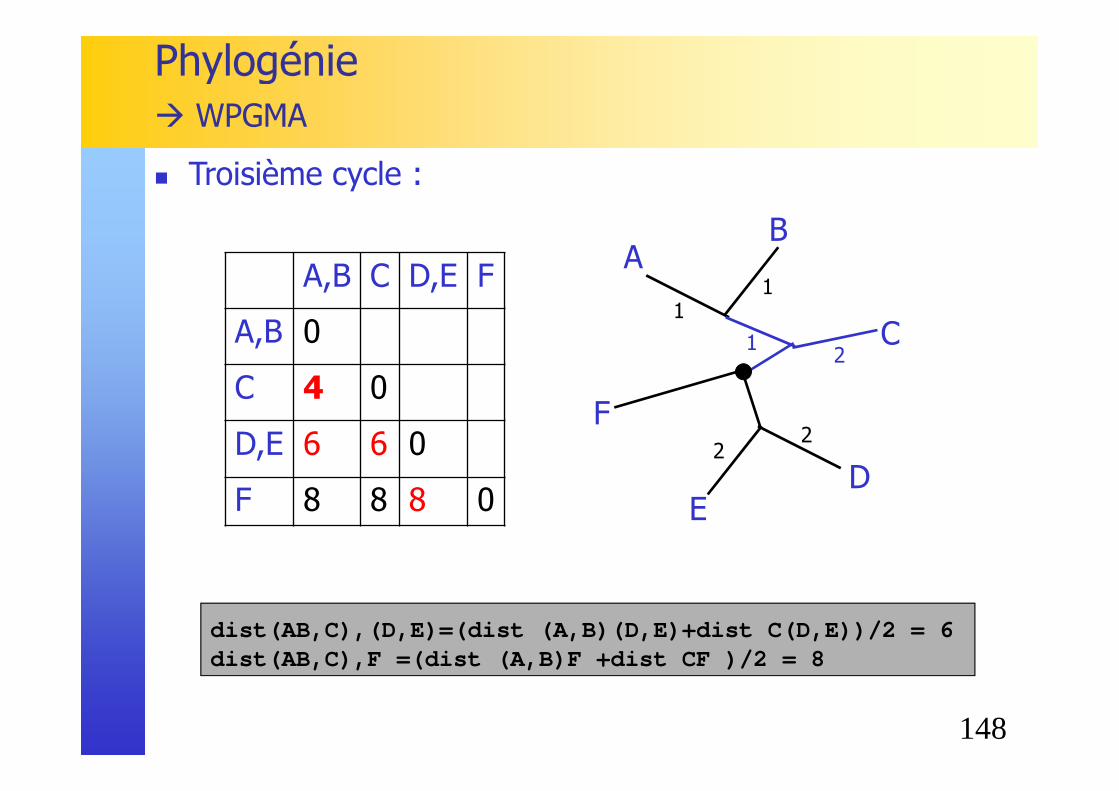
148
A,B C D,E F
A,B 0
C 4 0
D,E 6 6 0
F 8 8 8 0
Phylogénie� WPGMA
� Troisième cycle :
dist(AB,C),(D,E)=(dist (A,B)(D,E)+dist C(D,E))/2 = 6dist(AB,C),F =(dist (A,B)F +dist CF )/2 = 8
A
C
F
B
ED
11
22
21
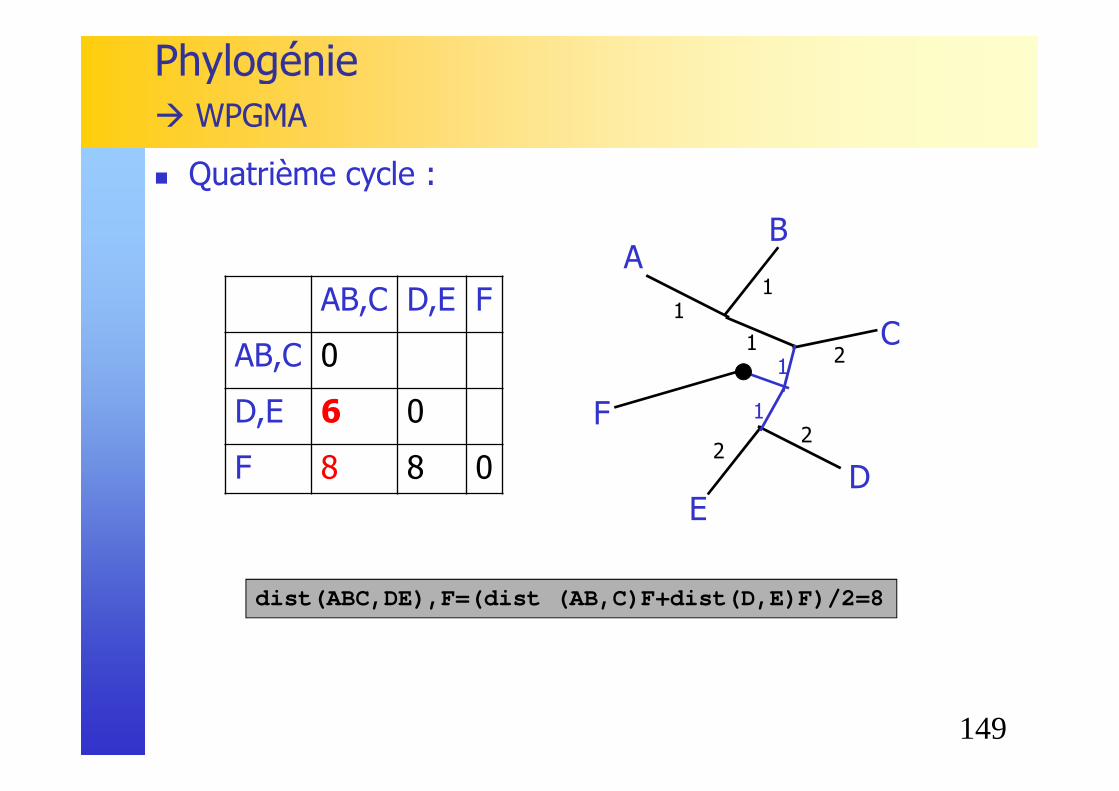
149
dist(ABC,DE),F=(dist (AB,C)F+dist(D,E)F)/2=8
Phylogénie� WPGMA
AB,C D,E F
AB,C 0
D,E 6 0
F 8 8 0
� Quatrième cycle :
A
C
F
B
ED
11
22
211
1

11
1
1
AB
1
1
C
1
2
DE
2
2
1
1
F
1
4
A
C
F
B
ED
11
22
2
4
Phylogénie� WPGMA
� Cinquième cycle :
ABC,DE FABC,DE 0F 8 0

151
� Exercice : étudier la phylogénie d’un gène commun pour 5 organismes, représentés ci-dessous� AGGCCTTACAT� ACCTATAATTGG� ACGATTATCAT� GGCTTACAA� CGATATCCCATT
� Etapes :� Calculer les scores 2 à 2, en recherchant à chaque fois
l’alignement optimal� En déduire la matrice des distances� Choisir le couple des organismes les plus semblables� Reconstruire la matrice des distances, construire l’arbre� Itérer jusqu’à la construction complète de l’arbre
Phylogénie� WPGMA
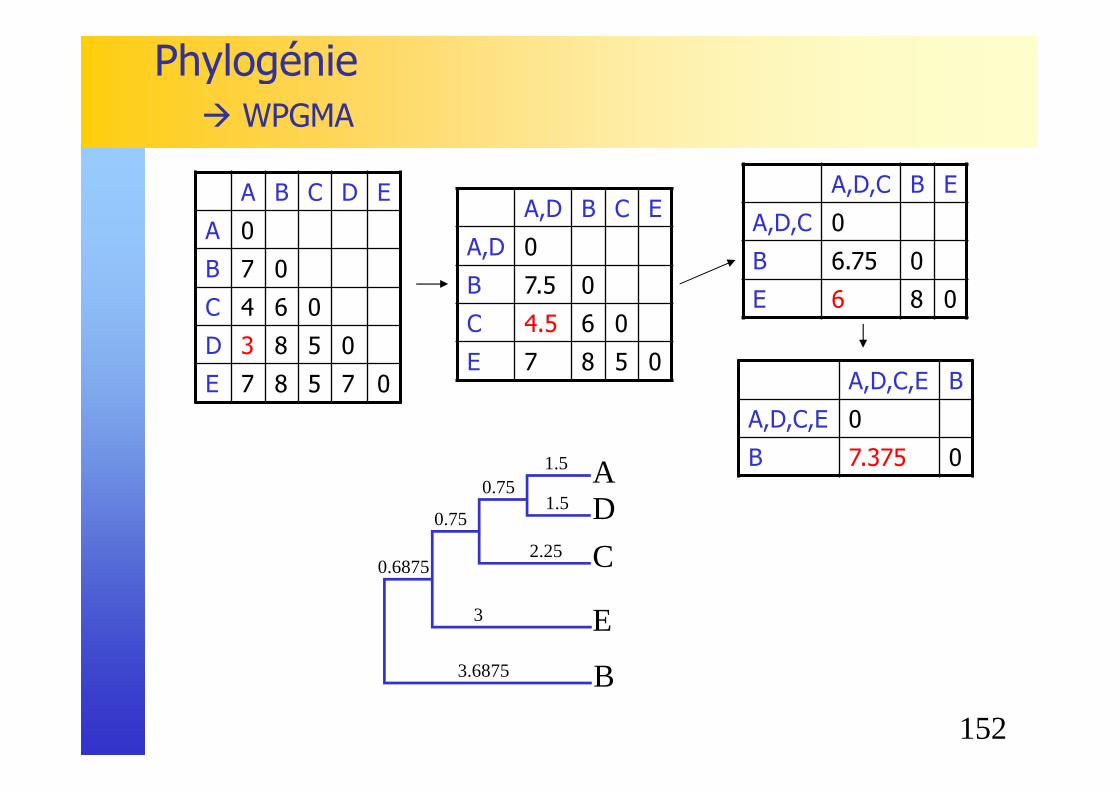
A B C D E
A 0
B 7 0
C 4 6 0
D 3 8 5 0
E 7 8 5 7 0
A,D B C E
A,D 0
B 7.5 0
C 4.5 6 0
E 7 8 5 0
A,D,C B E
A,D,C 0
B 6.75 0
E 6 8 0
A,D,C,E B
A,D,C,E 0
B 7.375 0AD
1.5
1.5
C
0.75
2.25
B
0.6875
3.6875
E
0.75
3
152
Phylogénie� WPGMA

153
� Hypothèses : � le taux de mutation est le même dans toutes les lignées (horloge
moléculaire)� Pas de mutations multiples
� Méthode� Regroupement des 2 séquences Si et Sj les plus proches� Le noeud Sij est positionné à une distance d de chacune des
séquences � d = (dist(Si,Sj ))/2
� Calcul de la distance entre le nouveau groupe et les autres séquences (Unweighted UPGMA)� dist((Si,Sj ),Sk) = (a.dist(Si,Sk)+b.dist(Sj,Sk))/(a+b)
� où a et b sont le nombre de séquences composant Si et Sj
� Etc
→ Exemple
Phylogénie� UPGMA (Unweighted)
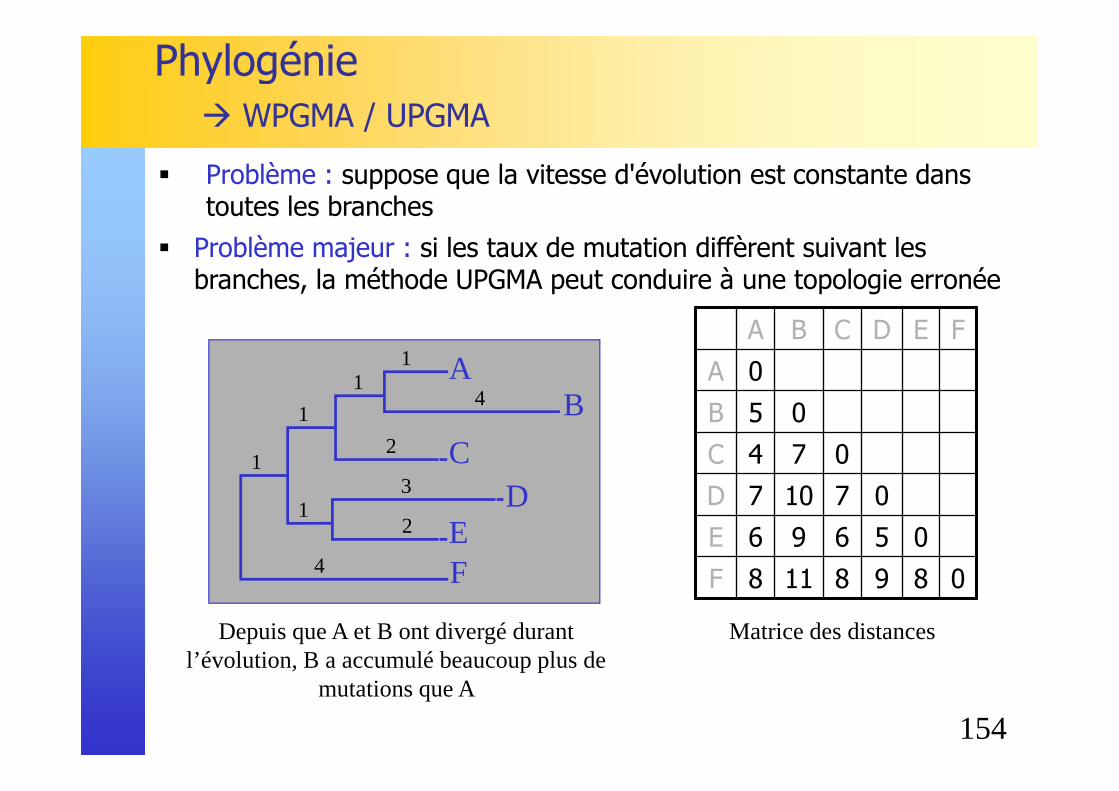
Phylogénie� WPGMA / UPGMA
� Problème : suppose que la vitesse d'évolution est constante dans toutes les branches
� Problème majeur : si les taux de mutation diffèrent suivant les branches, la méthode UPGMA peut conduire à une topologie erronée
AB
1
4
C
1
2
DE
3
2
1
1
F
1
4
A B C D E F
A 0
B 5 0
C 4 7 0
D 7 10 7 0
E 6 9 6 5 0
F 8 11 8 9 8 0
Matrice des distances
154
Depuis que A et B ont divergé durant l’évolution, B a accumulé beaucoup plus de
mutations que A

155
� Développé par Saitou et Nei (1987)
� Tente de corriger la méthode UPGMA afin d'autoriser un
taux de mutation différent sur les branches.
� Même principe que UPGMA :
� Point de départ : matrice des distances 2 à 2.
� Une itération supprime une ligne et une colonne de la matrice (on
lie deux nœuds)
� On itère jusqu’à ce que tout les nœuds soient reliés (matrice vide)
Phylogénie�Neighbor-Joining (NJ)

156
Phylogénie�Neighbor-Joining (NJ)
� Les données initiales permettent de construire une matrice qui donne un arbre en étoile (cf. UPGMA)� Cette matrice de distances est corrigée afin de prendre en compte
la divergence moyenne de chacunes des séquences avec les autres.
� L'arbre est alors reconstruit en reliant les séquences les plus proches dans cette nouvelle matrice (cf. UPGMA)� Lorsque deux séquences sont liées, le noeud représentant leur
ancêtre commun est ajouté à l'arbre tandis que les deux feuilles sont enlevées (cf. UPGMA).
� Ce processus convertit l'ancêtre commun en un noeud terminal dans un arbre de taille réduite (cf. UPGMA).
� L’ancêtre commun est placé de telle sorte que les distances entre les deux feuilles et la reste des séquences reste respecté.

157
Phylogénie�Neighbor-Joining (NJ)
� Exemple
La matrice de distance associée à cet arbre est la suivante :
� Etape 1 : calcul de la divergence de chaque séquence par rapport aux autres� r(A) = 5+4+7+6+8 = 30 � r(B) = 42 � r(C) = 32 � r(D) = 38 � r(E) = 34 � r(F) = 44
A B C D E FA 0 5 4 7 6 8B 0 7 10 9 11C 0 7 6 8D 0 5 9E 0 8F 0

158
Phylogénie�Neighbor-Joining (NJ)
� Etape 2 : cacul de la nouvelle matrice en utilisant la formule M(i,j)= d(ij) -[r(i)+r(j)] / (N-2)� Exemple pour la paire AB :
M(AB)= 5 - [30+42]/4 = -13
� On débute par l’arbre en étoile suivant :
A B C D E F
A 0 -13 -11.5 -10 -10 -10.5
B -13 0 -11.5 -10 -10 -10.5
C -11.5 0 -10.5 -10.5 -11
D -10 0 -13 -11.5
E -10 0 -11.5
F -10.5 0
A
C
F
B
ED

159
Phylogénie�Neighbor-Joining (NJ)
� Etape 3 : Choix des plus proches voisins, c'est à dire des deux séquences ayant le M(i,j) le plus petit� soit A et B soit D et E sur l’exemple.
� On forme un nouveau noeud U avec A et B, et on calcule la longueur de la branche entre U et A ainsi qu'entre U et B: � S (AU) = d (AB) / 2 + [r(A) - r(B)] / 2 (N-2)
= 5/2 + [30-42] /2(6-2) = 1 � S (BU) = d (AB) - S(AU) = 5 - 1 = 4
� On applique à l’arbre en construction:
AC
F
B
ED
1
4
U

160
Phylogénie�Neighbor-Joining (NJ)
� Etape 4 : on définit les nouvelles distances entre U et les autres séquences (ou groupes de séquences) � d (CU) = [d(AC) + d(BC) - d(AB)] /2 = 3� d (DU) = [d(AD) + d(BD) - d(AB)] /2 = 6� d (EU) = [d(AE) + d(BE) - d(AB)] /2 = 5 � d (DU) = [d(AF) + d(BF) - d(AB)] /2 = 7 …
� Création d'une nouvelle matrice :
� La procédure repart de l'étape 1 avec N N-1 = 5.
U C D E FU 0 3 6 5 7
C 0 7 6 8D 0 5 9E 0 8F 0
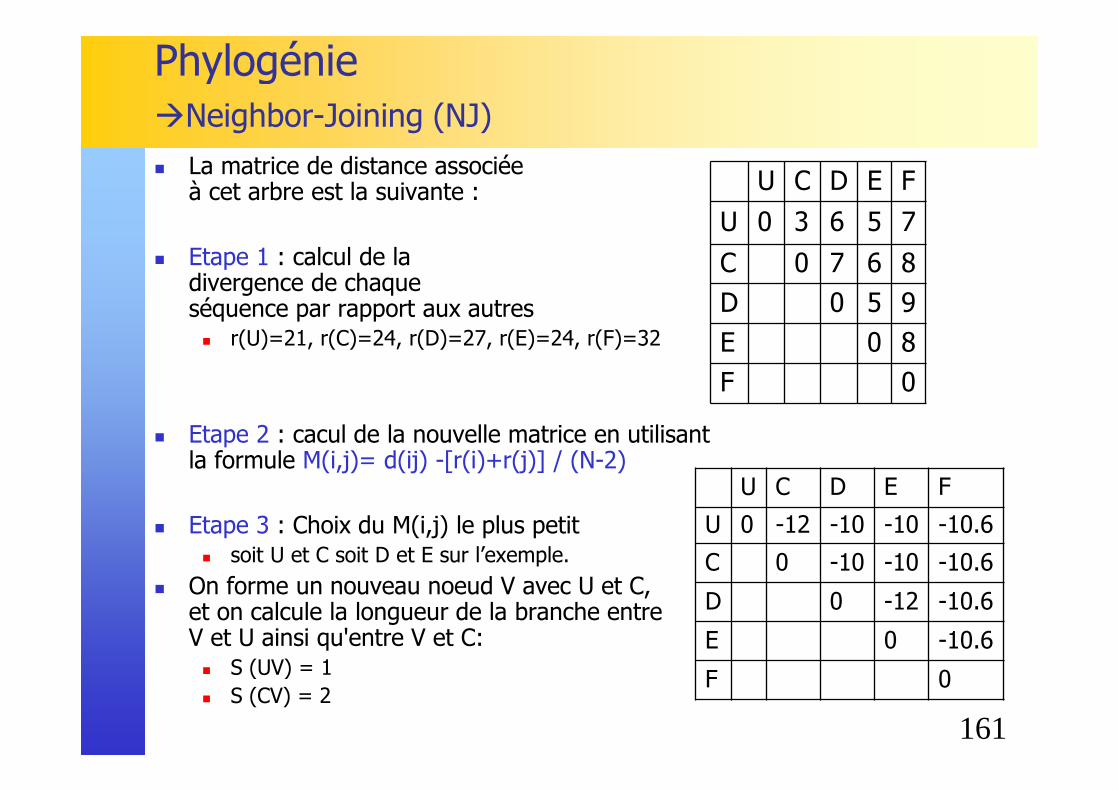
161
Phylogénie�Neighbor-Joining (NJ)� La matrice de distance associée
à cet arbre est la suivante :
� Etape 1 : calcul de la divergence de chaque séquence par rapport aux autres� r(U)=21, r(C)=24, r(D)=27, r(E)=24, r(F)=32
� Etape 2 : cacul de la nouvelle matrice en utilisant la formule M(i,j)= d(ij) -[r(i)+r(j)] / (N-2)
� Etape 3 : Choix du M(i,j) le plus petit� soit U et C soit D et E sur l’exemple.
� On forme un nouveau noeud V avec U et C, et on calcule la longueur de la branche entre V et U ainsi qu'entre V et C: � S (UV) = 1� S (CV) = 2
U C D E F
U 0 3 6 5 7
C 0 7 6 8D 0 5 9E 0 8F 0
U C D E F
U 0 -12 -10 -10 -10.6
C 0 -10 -10 -10.6
D 0 -12 -10.6
E 0 -10.6
F 0

162
Phylogénie�Neighbor-Joining (NJ)
� On applique à l’arbre en construction:
� Etape 4 : on définit les nouvelles distances entre U et les autres séquences (ou groupes de séquences) � d (DV) = 5, d (EV) = 4, d (DV) = 6
� Création d'une nouvelle matrice :
� La procédure repart de l'étape 1 avec N N-1 = 4.
V D E F
V 0 5 4 6
D 0 5 9
E 0 8
F 0
A
CF
B
E D
14
21
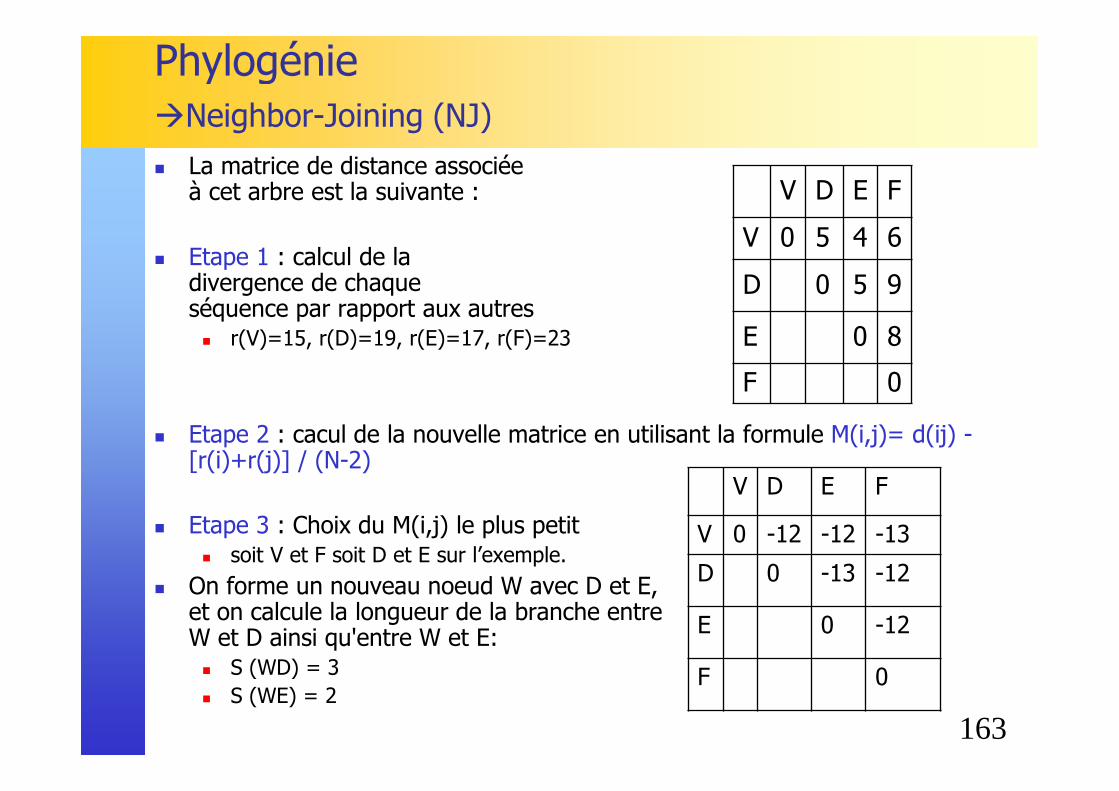
163
Phylogénie�Neighbor-Joining (NJ)� La matrice de distance associée
à cet arbre est la suivante :
� Etape 1 : calcul de la divergence de chaque séquence par rapport aux autres� r(V)=15, r(D)=19, r(E)=17, r(F)=23
� Etape 2 : cacul de la nouvelle matrice en utilisant la formule M(i,j)= d(ij) -[r(i)+r(j)] / (N-2)
� Etape 3 : Choix du M(i,j) le plus petit� soit V et F soit D et E sur l’exemple.
� On forme un nouveau noeud W avec D et E, et on calcule la longueur de la branche entre W et D ainsi qu'entre W et E: � S (WD) = 3� S (WE) = 2
V D E F
V 0 5 4 6
D 0 5 9
E 0 8
F 0
V D E F
V 0 -12 -12 -13
D 0 -13 -12
E 0 -12
F 0

164
Phylogénie�Neighbor-Joining (NJ)
� On applique à l’arbre en construction:
� Etape 4 : on définit les nouvelles distances entre W et les autres séquences (ou groupes de séquences) � d (DW) = 5, d (EW) = 4, d (DW) = 6
� Création d'une nouvelle matrice :
� La procédure repart de l'étape 1 avec N N-1 = 3.
V W F
V 0 2 6
W 0 6
F 0
A
CF
B
E D
14
21
2 3

165
Phylogénie�Neighbor-Joining (NJ)� La matrice de distance associée
à cet arbre est la suivante :
� Etape 1 : calcul de la divergence de chaque séquence par rapport aux autres� r(V)=8, r(W)=8, r(F)=12
� Etape 2 : cacul de la nouvelle matrice en utilisant la formule M(i,j)= d(ij) -[r(i)+r(j)] / (N-2)
� Etape 3 : Choix du M(i,j) le plus petit� V et W sur l’exemple.
� On forme un nouveau noeud X avec V et W, et on calcule la longueur de la branche entre V et X ainsi qu'entre W et X: � S (VX) = 1� S (WX) = 1
V W F
V 0 2 6
W 0 6
F 0
V W F
V 0 -14 -14
W 0 -14
F 0
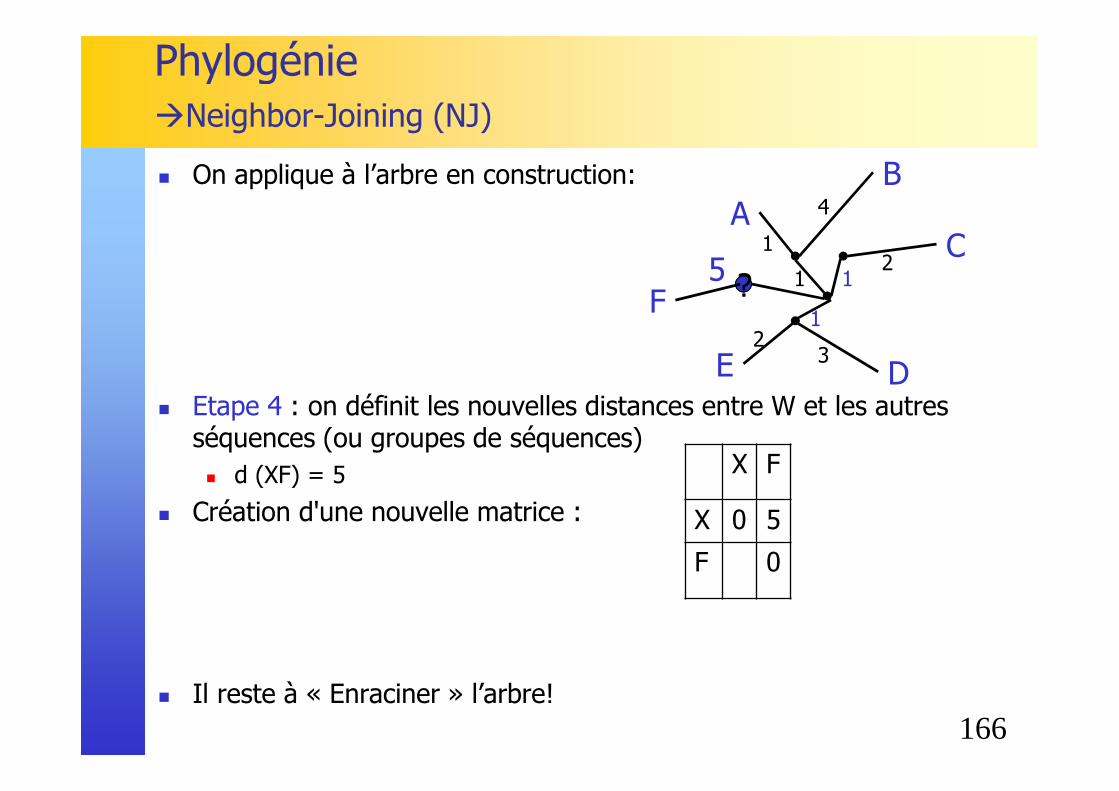
166
Phylogénie�Neighbor-Joining (NJ)
� On applique à l’arbre en construction:
� Etape 4 : on définit les nouvelles distances entre W et les autres séquences (ou groupes de séquences) � d (XF) = 5
� Création d'une nouvelle matrice :
� Il reste à « Enraciner » l’arbre!
X F
X 0 5
F 0
AC
F
B
E D
1
4
21
23
1
1
5 ?

167
Phylogénie�Enraciner un arbre
� Souvent, les méthodes de reconstruction phylogénétiques aboutissent
à des arbres non enracinés :
� Méthode de « l’outgroup » : on ajoute une séquence dont on sait qu'elle
est beaucoup plus ancienne que toutes les autres séquences.
� Si trop éloignée des autres données: peut conduire à des erreurs dans la
topologie de l'arbre.
� Si trop proche des séquences: cela n'est peut-être pas un vrai "outgroup".
� L'utilisation de plus d'un "outgroup" améliore en général l'évaluation de l'arbre.
� En l'absence d'un bon "outgroup", la racine peut être positionnée
approximativement à égale distance de toutes les séquences : on parle
alors de mid-point rooting.
� Si l’état de l’ancêtre commun est connu ou peut être calculé
convenablement, alors enraciner l’arbre en conséquence.

168
Exercice Récapitulatif
� Calculer les alignements 2 à 2 des séquences suivantes:� Homme : AGCCACCGGGTGCA� Gibbon : AGCACCGGATGCA� Gorille : ACCAACGCGGGTGCCA� Chat : AGCATCGTCTGCCGA
� Appliquer UPGMA, NJ� A partir de l’arbre obtenu par NJ, construire un
alignement multiple probable� Appliquer la méthode de Parcimonie à l’alignement
obtenu. En déduire une séquence probable pour l’ancêtre commun.
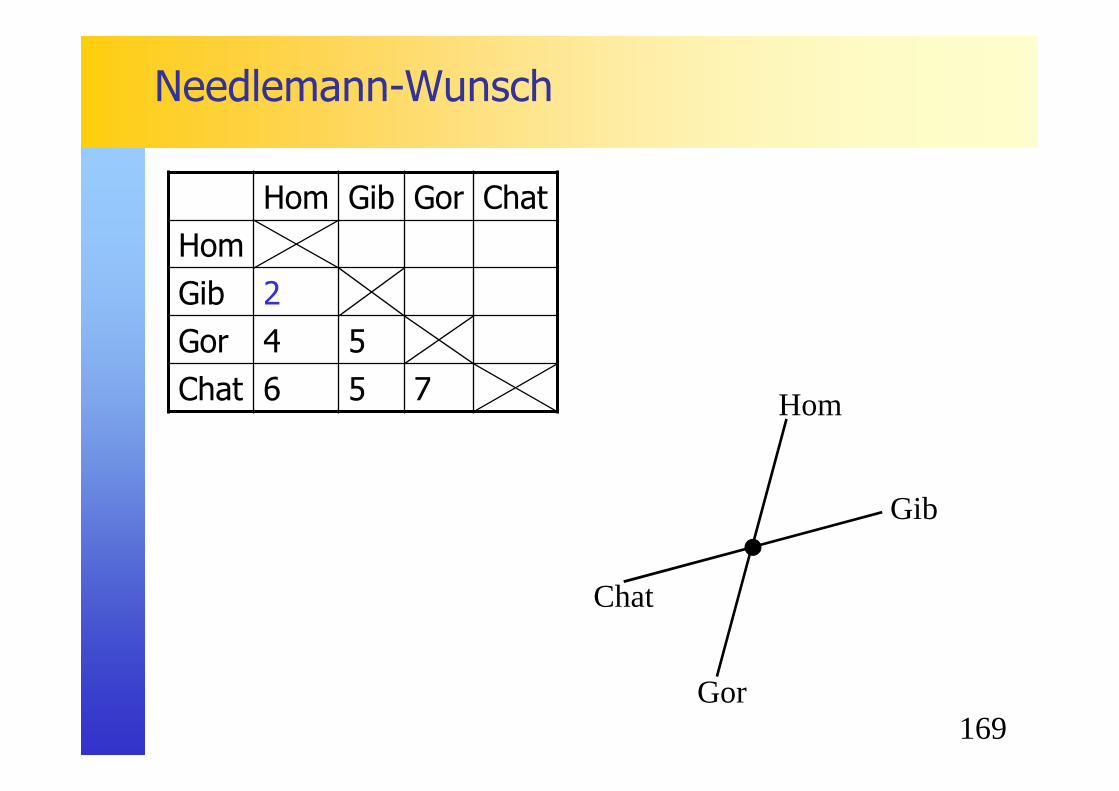
Hom Gib Gor ChatHomGib 2Gor 4 5Chat 6 5 7
Gib
Chat
Hom
Gor169
Needlemann-Wunsch

170
Hom Gib Gor ChatHomGib 2Gor 4 5Chat 6 5 7
X Gor ChatX 4.5 5.5Gor 4.5 7Chat 5.5 7
Gib
Chat
Hom
Gor
1
1X
Gib
Chat
Hom
Gor
UPGMA

171
Y ChatY 6.25Chat 6.25
X Gor ChatX 4.5 5.5Gor 4.5 7Chat 5.5 7
Gib
Chat
Hom
Gor
1
1X
Gib
Chat
Hom
Gor
1
1X
1.25
2.25
UPGMA

172
Y ChatY 6.25Chat 6.25
Gib
Chat
Hom
Gor
1
1X
1.25
2.25
Gib
Chat
Hom
Gor
1
1X
1.25
2.25
3.125
0.875
Hom
Chat
Gib
Gor
1
11.25
2.25
3.125
0.875
UPGMA

173
Arbre en construction:
Etape 4 : on définit les nouvelles distances entre U et les autres séquences (ou groupes de séquences) d (DV) = 5, d (EV) = 4, d (DV) = 6
Création d'une nouvelle matrice :
La procédure repart de l'étape 1 avec N N-1 = 4.
V D E F
V 0 5 4 6
D 0 5 9
E 0 8
F 0
Gib
Chat
Hom
Gor
UPGMA
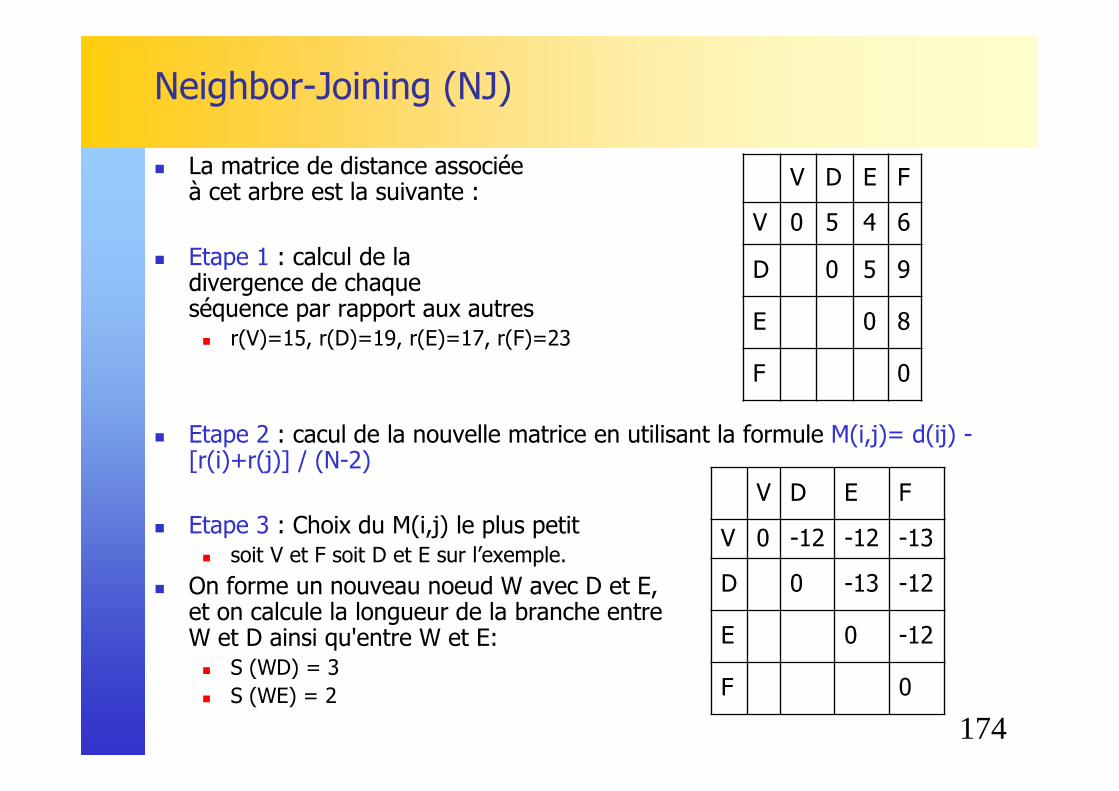
174
Neighbor-Joining (NJ)
� La matrice de distance associée à cet arbre est la suivante :
� Etape 1 : calcul de la divergence de chaque séquence par rapport aux autres� r(V)=15, r(D)=19, r(E)=17, r(F)=23
� Etape 2 : cacul de la nouvelle matrice en utilisant la formule M(i,j)= d(ij) -[r(i)+r(j)] / (N-2)
� Etape 3 : Choix du M(i,j) le plus petit� soit V et F soit D et E sur l’exemple.
� On forme un nouveau noeud W avec D et E, et on calcule la longueur de la branche entre W et D ainsi qu'entre W et E: � S (WD) = 3� S (WE) = 2
V D E F
V 0 5 4 6
D 0 5 9
E 0 8
F 0
V D E F
V 0 -12 -12 -13
D 0 -13 -12
E 0 -12
F 0
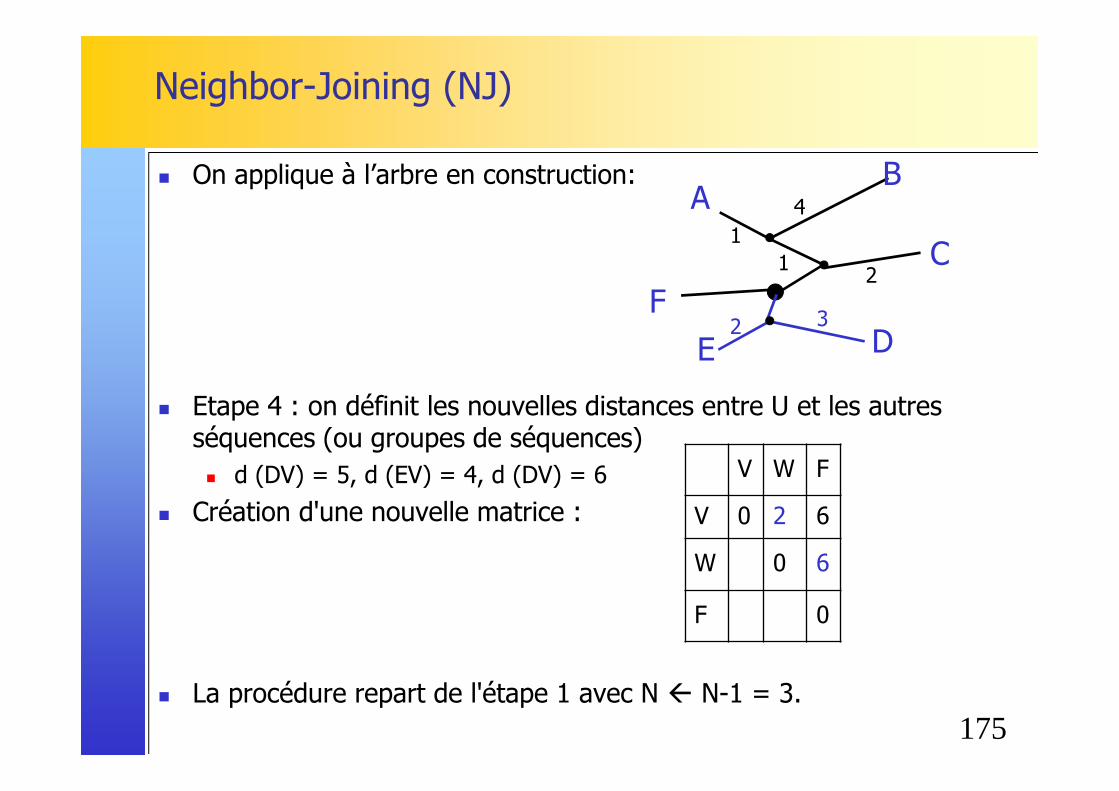
175
Neighbor-Joining (NJ)
� On applique à l’arbre en construction:
� Etape 4 : on définit les nouvelles distances entre U et les autres séquences (ou groupes de séquences) � d (DV) = 5, d (EV) = 4, d (DV) = 6
� Création d'une nouvelle matrice :
� La procédure repart de l'étape 1 avec N N-1 = 3.
V W F
V 0 2 6
W 0 6
F 0
A
CF
B
E D
14
21
2 3

176
Neighbor-Joining (NJ)
� La matrice de distance associée à cet arbre est la suivante :
� Etape 1 : calcul de la divergence de chaque séquence par rapport aux autres� r(V)=8, r(W)=8, r(F)=12
� Etape 2 : cacul de la nouvelle matrice en utilisant la formule M(i,j)= d(ij) -[r(i)+r(j)] / (N-2)
� Etape 3 : Choix du M(i,j) le plus petit� V et W sur l’exemple.
� On forme un nouveau noeud X avec V et W, et on calcule la longueur de la branche entre V et X ainsi qu'entre W et X: � S (VX) = 1� S (WX) = 1
V W F
V 0 2 6
W 0 6
F 0
V W F
V 0 -14 -14
W 0 -14
F 0

177
� Consiste à minimiser le nombre de "pas" (mutations / substitutions) nécessaires pour passer d'une séquence à une autre dans une topologie de l'arbre – hypothèses :� les sites évoluent indépendamment les uns des autres (caractères
non ordonnés)� la vitesse d'évolution est lente et constante au cours du temps
� La méthode de maximum de parcimonie recherche toutes les topologies possibles afin de trouver l'arbre minimal� nombre d'arbres enracinés possibles
pour n séquences = (2n - 3)! / (2exp(n-2))(n-2)! � nombre d'arbres non enracinés possibles
pour n séquences = (2n -5)! / (2exp(n-3))(n-3)!
Phylogénie�Méthode de Parcimonie

Phylogénie�Méthode de Parcimonie
# Séquences Arbres non enracinés
Arbres enracinés
2 1 1
3 1 3
4 3 15
5 15 105
6 105 945
7 945 10395
8 10395 135135
9 135135 2027025
10 2027025 34459425
15 7,906*1012 2,135*1014
20 8,201*1021 3,198*1023
x3
x5
x7
x9
x11
x13
x15
x17

179
� Principe de parcimonie : principe postulant que, pour un groupe d'espèces, la phylogénie la plus vraisemblable est celle qui nécessite le plus petit nombre de changements évolutifs� L'arbre phylogénétique est conçu de manière à impliquer le
minimum d'événements évolutifs. � La longueur de l'arbre L est égale à la somme du nombre de
changements l pour chacun des k sites informatifs.
� Un site est informatif uniquement s'il y a au moins deux types de nucléotides présents dans ce site et si chacun d'eux est représenté dans au moins deux séquences comparées (sinon l’arbre construit n’influe pas sur L)
Phylogénie�Méthode de Parcimonie
∑=
=k
iilL
1

180
� Sites informatifs : quels sont les sites informatifs correspondant à ces séquences? Pourquoi ce résultat?
� Quels sont les sites informatifs correspondant à ces séquences?
Phylogénie�Méthode de Parcimonie – sites informatifs
S1 cgatgagtcattgt-g--ac-tg
S2 cgatgagtcactgt-g--ac-tg
S3 cga---gccattgtagctac-tgS4 cga----ccattgtagctacctgS5 cga---gccattacagctacttgS6 -gatgagtcactgtgg--ac-tg
S1 cgatgagtcattgt-g--ac-tg
S2 cgatg--tcactgt-g--ac-tg
S3 cga---gccattgtagctac-tg

181
� Sites informatifs : quels sont les sites informatifs correspondant à ces séquences? Pourquoi ce résultat?
� Quels sont les sites informatifs correspondant à ces séquences?
Phylogénie�Méthode de Parcimonie – sites informatifs
S1 cgatgagtcattgt-g--ac-tg
S2 cgatgagtcactgt-g--ac-tg
S3 cga---gccattgtagctac-tgS4 cga----ccattgtagctacctgS5 cga---gccattacagctacttgS6 -gatgagtcactgtgg--ac-tg
S1 cgatgagtcattgt-g--ac-tg
S2 cgatg--tcactgt-g--ac-tg
S3 cga---gccattgtagctac-tg

182
� Procédure d'analyse par la méthode de parcimonie :� Données de départ : un alignement multiple des
séquences � Identifier les sites informatifs.� Inférer toutes les topologies d'arbres possibles pour les
séquences données.� Calculer le nombre minimum de substitutions pour
chaque site informatif.� Calculer la somme de changements pour chaque
arbre.� Choisir la topologie de l'arbre qui nécessite le moins de
changements: l’arbre le plus court.
Phylogénie�Méthode de Parcimonie

183
� Algorithme de Fitch (1971)� Données: n taxons, une topologie T à n feuilles et un
caractère c et ses n états pour les n taxons.� Principe
� Associer à chaque noeud interne de T un ensemble d’états menant à une complétion optimale.
� Déduire une complétion optimale du résultat de l’étape 1.
� Exemple� n = 5 et on a CACAG et un arbre T. Déduire l’histoire
évolutionnaire la plus parcimonieuse.
Phylogénie�Méthode de Parcimonie

184{AC}
{ACG}*
{CG}*{AC}*
� Fitch: Mise en œuvre� Étape 1: calcul des états possibles pour chaque caractère à
chaque noeud.� Parcours profondeur postfixé:
� Si le noeud est une feuille alors son état est gardé.� Sinon si l’intersection entre les 2 descendants du nœud est vide alors
l’union de leurs états est prise.� sinon on prend l’intersection.
Phylogénie�Méthode de Parcimonie
{A} {C} {A} {C} {G}
{AC}*
{AC}
{CG}*
{ACG}*
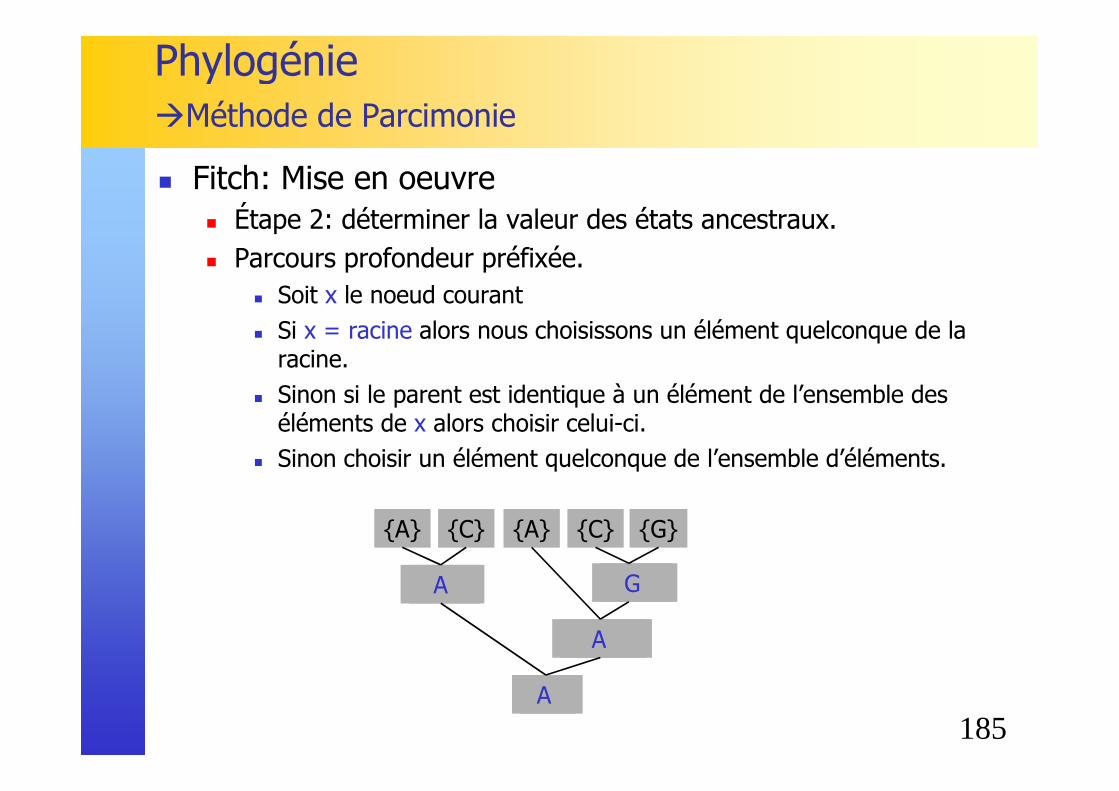
185
� Fitch: Mise en oeuvre� Étape 2: déterminer la valeur des états ancestraux.� Parcours profondeur préfixée.
� Soit x le noeud courant� Si x = racine alors nous choisissons un élément quelconque de la
racine.� Sinon si le parent est identique à un élément de l’ensemble des
éléments de x alors choisir celui-ci.� Sinon choisir un élément quelconque de l’ensemble d’éléments.
Phylogénie�Méthode de Parcimonie
{A} {C} {A} {C} {G}
{AC}*
{AC}
{CG}*
{ACG}*A
A
G A
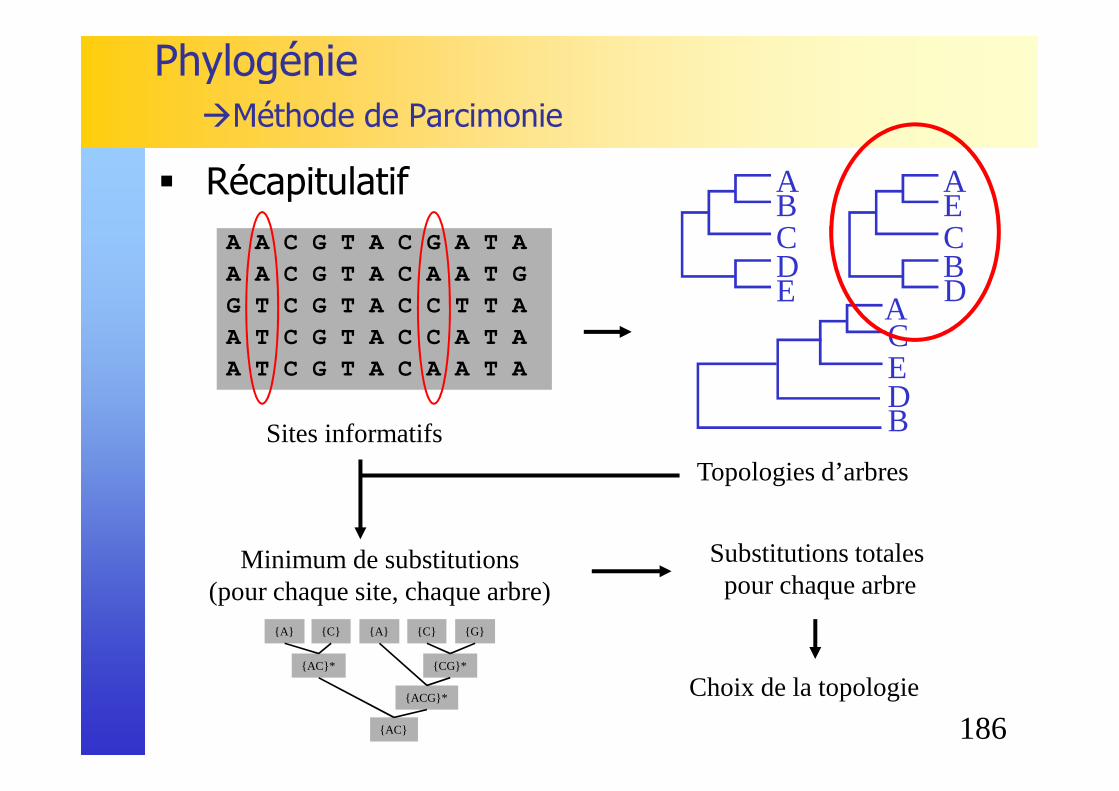
Phylogénie�Méthode de Parcimonie
� RécapitulatifA A C G T A C G A T A
A A C G T A C A A T G
G T C G T A C C T T A
A T C G T A C C A T A
A T C G T A C A A T A
ABCDE
AECBD
ACEDBSites informatifs
Topologies d’arbres
Minimum de substitutions(pour chaque site, chaque arbre)
Substitutions totales pour chaque arbre
Choix de la topologie
{A} {C} {A} {C} {G}
{AC}*
{AC}
{CG}*
{ACG}*
186

187
� Avantages de la parcimonie� Méthode ne réduisant pas la séquence à un simple nombre
� Méthode essayant de donner une information sur les séquences
ancestrales
� Méthode évaluant différents arbres.
� Inconvénients � Méthode très lente par rapport aux méthodes basées sur les distances
� N'utilise pas toute l'information disponible (seuls les sites informatifs sont
pris en compte)
� Pas de corrections pour les substitutions multiples
� Aucune information sur la longueur des branches
Phylogénie�Remarques sur les méthodes de parcimonie

188
� Branch and Bound� Garantie de trouver le meilleur arbre sans évaluer tous les arbres
� Permet de traiter un plus grand nombre de séquences mais reste limitée.
� Recherche heuristique
� Réarrangement des branches à chaque étape
� Ne garantit pas de trouver l'arbre optimal.
� Arbre consensus
� La méthode du maximum de parcimonie peut conduire à trouver
plusieurs arbres équivalents : on crée alors un arbre consensus.
� Arbre consensus construit à partir des noeuds les plus
fréquemment rencontrés sur l'ensemble des arbres possibles.
Phylogénie�Variantes de la méthodes de parcimonie

189
� La topologie des arbres est soumise à de nombreuses variations et
dépendent :
� de la nature des séquences utilisées (acides nucléiques ou protéines),
� de la méthode utilisée (méthodes de distances ou parcimonie),
� de la qualité de l'alignement,
� du nombre de séquences incluses dans l'alignement,
� de l'ordre des séquences dans l'alignement,
� du choix de la racine.
Phylogénie�Remarques générales

190
� On ne garde que les nœuds « fiables »
Phylogénie�Arbre consensus
AB
C
D
E
F
AC
B
D
E
F
AB
C
D
E
F
Arbre consensus :

191
Bootstrap
� Comment évaluer la confiance que l'on peut avoir dans un arbre
� Bootstrap
� Méthode proposée par Bradley Efron (1979) et introduite en phylogénie par
Felsenstein en 1985.
� Méthode divisée en 3 étapes réalisées chacune au moins 100 fois :
� Réalisation d'un pseudo-alignement A' à partir des séquences d'origine en prenant arbitrairement n
colonnes (avec remplacements) de l'alignement d'origine
� Estimation de l'arbre obtenu : T'
� Comparaison des arbres T et T' : pour chaque noeud de T, on regarde s'il est présent dans T'
� On compte ensuite pour chaque noeud le nombre de fois où il est présent dans les
T'. Cette fréquence avec laquelle on retrouve un noeud est la valeur de bootstrap
(plus elle est élevée plus la fiabilité de la branche est importante)
� On supprime alors éventuellement les nœuds à faible fiabilité

192
Bootstrap
AB
C
D
E
F
AB
C
D
E
F
95
100
99
75
95
100
99


















