
In Copyright - Non-Commercial Use Permitted Rights ...31720/eth-31720-02.pdf · sen, Am.Soc. 7_^,...
67
Research Collection Doctoral Thesis Zur Kenntnis des Zehnrings Author(s): Schenker, Karl Publication Date: 1953 Permanent Link: https://doi.org/10.3929/ethz-a-000087608 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection . For more information please consult the Terms of use . ETH Library
Transcript of In Copyright - Non-Commercial Use Permitted Rights ...31720/eth-31720-02.pdf · sen, Am.Soc. 7_^,...

Research Collection
Doctoral Thesis
Zur Kenntnis des Zehnrings
Author(s): Schenker, Karl
Publication Date: 1953
Permanent Link: https://doi.org/10.3929/ethz-a-000087608
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Prom. Nr. 2214
Zur Kenntnis des Zehnrings
Von der
Eidgenössischen Technischen
Hochschule in Zürich
zur Erlangung
der Würde eines Doktors der
Naturwissenschaften
genehmigte
PROMOTIONSARBEIT
vorgelegt von
KARL SCHENKER
dipl. Naturwissenschafter
von Däniken (Kanton Solothurn)
Referent : Herr Prof. Dr. V. PrelogKorreferent: Herr Prof. Dr. L. Ruzicka
Juris-Verlag Zürich
1953

Leer - Vide - Empty

DEM ANDENKEN
MEINER LIEBEN ELTERN
IN DANKBARKEIT

Leer - Vide - Empty

Meinem hochverehrten Lehrer,
Herrn Prof. Dr. L. Ruzicka,
bin ich für sein Wohlwollen und die
Unterstützung dieser Arbeit zu grossem
Dank verpflichtet.
Herrn Prof. Dr. V. Prelog,
unter dessen Anleitung ich die vorlie¬
gende Promotionsarbeit ausführte, danke
ich herzlich für die vielen wertvollen
Ratschläge und die unermüdliche Hilfe,
die er mir stets zuteil werden Hess.

Leer - Vide - Empty

- 7 -
Inhaltsübersicht.
Theoretischer Teil.
Einleitung 9
eis- und trans-Cyclodecen 12
IE.-Absorptionsspektren 20
Die Oxydation der Cyclodecene mit Perameisen¬
saure, eine transanulare Reaktion 28
Die Oxydation von cis-Cyclodecen
mit Perameisensaure 29
Die Oxydation von trans-Cyclodecen
mit Perameisensaure 31
Experimenteller Teil.
Zur Herstellung des Cyclodecanol-(l)-ons-(2) 41
Cyclodecm 43
cis-Cyclodecen 45
trans-Cyclodecen 47
cis-Cyclodecandiol-(l,2) 48
trans-Cyclodecandiol-(l,2) 50
Herstellung von Cyclodecenen durch
Dehydratisierung von Cyclodecanol 51
Isomerisierungsversuche mit Cyclodecenen 53
cis-l,2-0xido-cyclodecan 53
trans-1,2-0xido-cyclodecan 54
Die Reaktionsgeschwindigkeiten der Oxydation von
stereoisomeren Cyclodecenen mit Benzoepersaurc .... 54
Die Reaktionsgeschwindigkeiten der Oxydation
von stereoisomeren Cyclodecandiolen-(l,2)mit Blei(IV)-acetat 55
Oxydation von cis-Cyclodecen mit Perameisensaure .. 55
Oxydation von trans-Cyclodecen mit Perameisensaure 56
Einzelne isolierte Verbindungen 58
Zusammenfassung 64

Leer - Vide - Empty

THEORETISCHER TEIL
I. Einleitung
Im Jahre 1924 entdeckte L. Ruzicka ) bei der
Konstitutionsaufklärung der natürlichen Riechstoffe
Muscon und Zibeton die ersten Beispiele für vielglied-
rige Ringverbindungen.
In der Folge wurden von ihm und seinen Mitarbei¬
tern homocyclische Ringe mit 9 bis 14 Kohlenstoffato¬
men hergestellt und ihre chemischen und physikalischen
Eigenschaften, soweit es das in beschränktem Masse
vorhandene Material erlaubte, untersucht. Die Ergeb¬
nisse dieser Untersuchungen und besonders ihre Aus¬
wirkung auf die "Baeyer'sehe Spannungstheorie" sind
heute allgemein bekannt und haben Eingang in alle neue-
ren Lehrbücher der organischen Chemie gefunden ).
In den auf die Entdeckung der vielgliedrigen Rin¬
ge folgenden Jahren arbeitete man trotz der mannig¬
faltigen Forachungsmöglichkeiten nur in wenigen Labo¬
ratorien auf diesem neuen Gebiete der Chemie5). Der
Grund dazu lag in präparativen Schwierigkeiten. Zwar
konnten K. Ziegler, H. Eberle und H. Ohlinger ) im
Jahre 1933 die Ausbeute im Bereich der grossen Ringe )
stark verbessern, doch waren damit die vielgliedrigen
Ringverbindungen nicht wesentlich leichter zugänglich
h Helv. 2., 230 (1926)) Vgl. z.B. G.W.Wheland, Advanced Organic Chemistry,
,2. Auflage (1951) S. 371 ff.
?) Es seien neben der Arbeitsgruppe L.Ruzicka und der
mit ihr verbundenen von M.Stoll noch angeführt:W.H.Carothers, R.Adams, A.T.Blomquist in USA und
K.Ziegler, A.Lüttringhaus, H.Hunsdiecker in Deutsch-
.land.
*) A. 521, 94 (1933)') Vgl. die von H.C.Brown, R.S.Fletcher ft R.B.Johanne-
sen, Am.Soc. 7_^, 212 (1951) vorgeschlagene Klassi¬
fikation der alicyclischen Verbindungen.

- 10 -
geworden wegen der experimentell mühsamen Technik
des Verdünnungsprinzips. Die Methode von Ziegler
ergab zudem, gleich wie diejenige von Ruzicka, für
mittlere Ringe ausserordentlich schlechte Ausbeuten.
Erst die Einfuhrung und Verbesserung des Acyloin-
Ringschlussverfahrens ) ermöglichte auch die Herstellung
von Ringen mit 8 bis 12 Rmggliedern in befriedigender
Ausbeute und grosseren Mengen. Damit war der Weg zur
genaueren Untersuchung der vollständigen rmghomolo-
gen Verbindungsreihen frei.
Im Verlaufe dieser Untersuchungen zeigte sich
eine eigentumliche und unerwartete Abhängigkeit so¬
wohl der physikalischen als auch der chemischen Ei-
genschaften von der Rmggrosse )« Die mittleren Ringe,
vor allem der Zehnring, nehmen dabei eine besondere
Stellung ein, die sich offensichtlich auf Konstellations-
effekte bei diesen Verbindungen zurückfuhren lässt ).
H.C. Brown, S.S. Fletcher und R.B. Johannesen )
erklaren das aussergewohnliche Verhalten der mittle¬
ren Ringe durch das Vorhandensein eines sogenannten
"I-Strains", d.h. ausschliesslich durch die Aenderung
der nichtklassischen ( = fitzer1sehen )) Spannung der
mittleren Ringe im Verlaufe einer chemischen Reaktion.
Als "I-Strain" definieren diese Autoren im Falle
der mittleren Ringe die Aenderung der Spannung im Ring,
die aus der Aenderung der Koordinationszahl des an ei¬
ner Reaktion beteiligten Ring-Kohlenstoffatoms resul¬
tiert. Reaktionen mit einem Wechsel der Koordinations¬
zahl von 4 nach 5 (Sj,l-Reaktionen) vermindern bei mitt¬
leren Ringen die Ringspannung ("I-Strain" negativ) und
erfolgen daher rasch. Ebenso sind Reaktionen mit einem
1) V.Prelog, L.Frenkiel. M.Kobelt 4 P.Barman,Helv. 20, 1741 (1947)M.Stoll & J.Hulstkamp, Helv. 29., 1815 (1947)
3M.Stoll 4 A.Rouvé, Helv. 29., 1822 (1947)
"z) V.Prelog, Soc. 1929, 420l) Am.Soc. 22, 212 (1951)4) K.S.Pitzer, Nature 191, 672 (1945)

- 11 -
Wechsel der Koordinationszahl von 4 nach 5 (Sjj2-Reaktionen) begünstigt. Reaktionen hingegen, die eine
Erhöhung der Koordinationszahl von 3 auf 4 zur Folge
haben, vergrössern die Ringspannung ("I-Strain po¬
sitiv) und verlaufen daher langsam.
Die Zürcher Schule fasst den Begriff des Kon¬
stellationseffekts weiter. Neben der unbestreitbaren
Bedeutung der nichtklassischen Spannung für das ab¬
norme Verhalten der mittleren Ringe zieht sie zusätz¬
lich Wechselwirkungen der Atome quer über den Ring
in Betracht, die bei mittleren Ringen besonders gross
sind und als transanulare Effekte bezeichnet werden ).
Im Zusammenhang mit diesen Ueberlegungen schien
es uns lohnenswert, weitere Untersuchungen in der
Cyclodecan-Reihe vorzunehmen, um die Brauchbarkeit
der beiden Anschauungen zu überprüfen und wenn mög¬
lich Beweise für die Existenz von transanularen
Effekten beim Zehnring zu finden.
) Vgl. Abstracts of Papers 120 Meeting Am.Chem.Soc. New York, September 3-7, 1951.

- 12 -
II. cia- und trans-Cyclodecen"1-)
Durch das Acyloin-Ringschlussverfahren sind die
Verbindungen mit einem Zehnring so leicht zugänglich
geworden, dass es uns naheliegend schien, verschiedene
bisher nicht behandelte Probleme der Chemie des Zehn-
rings anzugreifen ).
Das als Ausgangsmaterial für unsere Versuche die¬
nende Cyclodecanol-(l)-on-(2) (I) liess sich aus Seba-
cinsäure-dimethylester nach Einführung kleiner techni¬
scher Verbesserungen mit einer Ausbeute von 53 ^ ge¬
winnen.
Von V. Prelog und Mitarbeitern ) wurde die Herstel¬
lung des Cyclodecanons (II) aus dem Cyclodecanol-(l)-
on-(2) durch Reduktion mit Zink und Salzsäure in Eis¬
essig beschrieben. Inzwischen konnte festgestellt wer¬
den, dass die Ausbeuten bei dieser Reaktion stark von
Verunreinigungen der verwendeten Reagentien abhängig
sind. Die besten Ausbeuten wurden in unserem Laborato¬
rium bei Anwendung reiner Zink-Wolle ) und reinster
konz. Salzsäure gewonnen. Durch Zusatz von kleinen Men¬
gen Eisen-, Nickel- und Platin-Salzen lässt sich die
Reduktion vollständig hemmen, Kupfer-Salze sind weniger
schädlich, während Blei-Salze kaum hemmend wirken.
In Anwesenheit von Quecksilber-Salzen entsteht statt
des Ketons der gesättigte cyclische Kohlenwasserstoff.
Die Reduktion des Gyclodecanol-(l)-ons-(2) nach Clemmen-
1) V.Prelog, K.Schenker Sc, Hs.H.Günthard, Helv. 35, 1598
2(1952)
) Nach Abschluss dieser Arbeit haben auch A.T.Blom-
quist. R.E.Bürge jr. & A.C.Sucsy, Am.Soc. 2À.> 3636(1952) Untersuchungen über die stereoisomeren Cyclo-
,decene veröffentlicht.
->) Vgl. Anmerkung 1 S.10
4) H.C.Brown & M.Borkowski, Am.Soc. 2!» 1901 (1952)konnten mit sehr reinem Zink die Reaktion nicht
durchführen, erhielten dagegen gute Ergebnissemit techn. Zinkstaub (Mallinckrodt Chemical Works)

- 15 -
sen stellt den einfachsten Weg zur Herstellung von
Cyclodecan (III) dar, welches von uns auf diesem Wege
in grösseren Mengen für physikalische Messungen be¬
reitet wurde ).
Das Gyclodecanol (IV), welches früher aus Cyclo-
decanon durch Hydrierung mit Raney-Nickel hergestellt
worden war, ist neuerdings sehr bequem auch durch Re¬
duktion des Cyclodecanons mit Lithiumaluminiumhydrid
zugänglich geworden. Durch Dehydratisierung des Cyclo-
decanols an Aluminiumoxyd bei 400 entsteht, wie aus
späteren Versuchen folgt, ein Gemisch von annähernd
gleichen Mengen der stereoisomeren Cyclodecene.
Das reine, einheitliche cis-Cyclodecen (VIII)
konnte aus Cyclodecin (VII) durch partielle kataly-
tische Hydrierung mit einem vor kurzem von H. Lindlar )
beschriebenen Palladium-Blei-Calciumcarbonat-Kataly-
sator in methanolischer Lösung hergestellt werden.
Die letztere Verbindung haben A.T. Blomquist und Mit-
arbeiter ) in einer vorläufigen Mitteilung beschrie¬
ben. Wir haben das Cyclodecin nach demselben Verfah¬
ren in leicht modifizierter Form hergestellt. Durch
Oxydation von Cyclodecanol-(l)-on-(2) mit Chrom-(VI)-
oxyd wurde das Cyclodecandion-(l,2) (V) bereitet ).
Das Dihydrazon VI der letzteren Verbindung gab durch
Oxydation mit Quecksilber-(II)-oxyd in siedendem Toluol
das Cyclodecin.
) Das Cyclodecan wurde Prof. F.D.Rossini, CarnegieInstitute of Technologie, Pittsburg, Pa. zur Ver¬
fügung gestellt, der damit die Bestimmung der iäntro-
pie, der Verdampfungsw^rme und Verbrennungswnrme durch¬
führen wird. Dadurch hofft man eine bessere Einsicht
„in die Spannungsverhältnisse beim Zehnring zu erhalten.
) Helv. 21, 446 (1952). In der Arbeit sind auch Bei-
~ spiele für die cis-Anlagerung gegeben.) A.T.Blomquist, R.E.Bürge jr., L.Huang Li, J.C.Bohrer,A.C.Sucsy & J.Kleis, Am.Soc. J2, 5510 (1951). Die
ausführliche Veröffentlichung erfolgte nach Abschluss
. unserer Arbeiten. Vgl. Anm. 2, S. 12.
*) V.Prelog, L.Frenkiel, M.Kobelt & P.Barman, Helv.
20, 1746 (1947).

- 14
Das reine, einheitliche trans-Gyclodecen (XI) wurde
in Anlehnung an die von K. Ziegler und H. Wilms )
beschriebene Herstellung des trans-Cyclooctens durch
den Hofmann'sehen Abbau aus Cyclodecyl-trimethyl-
ammoniumhydroxyd (X) gewonnen. Die quaternäre Ammo¬
nium-Base liess sich vorteilhaft durch Methylierung
von Cyclodecyl-methylamin (IX) erhalten, das durch ka-
talytische Hydrierung von Cyclodecanon mit Raney-Nickel
in Gegenwart von Methylamin zugänglich ist.
Die Einheitlichkeit der so hergestellten stereo¬
isomeren Cyclodecene folgt besonders aus ihrem Verhal¬
ten gegenüber Osmium(VIII)-oxyd. Aus cis-Cyclodecen
(VIII) entsteht dabei einheitlich ein Gyclodecandiol-(l,2)
(XII) vom Smp. 140°, welches früher aus Cyclodecanol-(l)-
on-(2) durch katalytische Hydrierung mit Platinoxyd-2
Katalysator bereitet wurde ). Das trane-Cyclodcen lie¬
fert dagegen in ausgezeichneter Ausbeute ein bisher
nicht beschriebenes Cyclodecandiol-(l,2) (XIII) vom
Smp. 54 ). Dieses letztere liess sich auch neben dem
Gyclodecandiol-(l,2) vom Smp. 140° durch Hydrierung
von Cyclodecanol-(l)-on-(2) mit Raney-Hickel in Fein¬
sprit gewinnen. Die beiden stereoisomeren Diole lassen
sich sehr gut durch Chromatographieren an Aluminium¬
oxyd trennen. Die Oxydation mit Osmium(VIII)-oxyd und
o) A. 567, 37 (1950)\\ VglTHelv. 2S, 1746 (1947)?) Das gleiche Cyclodecandiol-(l,2 ) entsteht neben dem
höher schmelzenden Isomeren auch bei der Reduktion
von Cyclodecanol-(l)-on-(2) mit Lithiumaluminium¬
hydrid.D.D.Coffman, A.P. 2 212 151. führte die Hydrierungin Toluol mit Nickel bei 110° und 200 atm. durch und
erhielt in 50-prozentiger Ausbeute das höher schmel¬
zende Diol.
A.T.Blomquist & Mitarb., Am.Soc. 2i, 3636 (1952)erhielten das bei 140 schmelzende Cyclodecandiol-(l,2)auf drei verschiedenen Wegen aus Cyclodecanol-(l)-on-(2):a) durch Reduktion nach Meerwein-Ponndorf in 47 ?>,b) durch Reduktion mit Lithiumaluminiumhydrid in 52 ji,c) durch katalytische Hydrierung mit Platioxyd-
Katalysator in 75 i» Ausbeute. Das schwer kristal¬
lisierende, bei 54° schmelzende Diol scheint bei¬
den Autoren entgangen zu sein.

- 15 -
nachfolgende chromatographische Trennung aes Diol-Gre-
misches eignet sich deshalb zur annähernd quantitativen
Bestimmung der stereoisomeren Cycloaecene in Gemischen.
Nach diesem Verfahren wurde auch festgestellt, dass das
Gremisch der Cyclodecene, welches sich durch Behydrati—
sierung von Gyclodecanol an Aluminiumoxyd bei 400° bil¬
det, aus etwa gleichen Mengen von eis— und trans-Cyclo-
decen besteht.
Die Konfiguration der Cyclodecene folgt sowohl aus
ihrer Entstehungsweise als auch aus den IR.^Absorp¬
tionsspektren, die in einem besonderen Abschnitt be¬
sprochen werden.
Bekanntlich sind die Cycloolefme mit einer trans-
Konfiguration der Doppelbindung, ebenso wie die Cyclo-
alkme, erst von einer bestimmten Ringgrosse an mög¬
lich. Zuerst wurden zwei cis-trans-isomere Cycloole¬
fme bei Zibeton beschrieben ). Spater haben K. Zieg¬
ler und H. Wilms gezeigt, dass das trans-Isomere
schon beim Achtring existenzfähig ist, wahrend nach
N.A. Dommm ) auch das Cyclooctm herstellbar sein soll.
Jedenfalls scheint das Cyclononin schon recht bestän¬
dig zu sein.
Zum Unterschied vom trans-Cycloocten, welches
leicht polymerisiert, scheint das trans-Cyclodecen
bestandig zu sein. Durch Erhitzen mit p-Toluolsulfo-
saure auf 170° entsteht aus beiden reinen, stereoiso¬
meren Cyclodecenen das gleiche Gemisch, in welchem
cis-Cyclodecen stark überwiegt. Dies folgt sowohl
aus dem Ergebnis der Oxydation mit Osmium(VIII)—oxyd
als auch aus den IR.-Absorptionsspektren.
-1) L.Ruzicka, rielv. £, 230 (1926). H.Hunsdiecker,Naturw. 20, 587 (1942), B. 2k, 142 (1943),B. 21, 135 (1944). M. Stoll, J.hulstkamp *
A.Rouvé, Helv. 21, 543 (1948).
9 A.T.ßlomquist, Am.Soc. 22, 34 (1948)eL) C. 1939. II, 1470.

- 16 -
Aus der Konfiguration der Cyclodecene lässt
sich die Konfiguration der daraus mit Osmium(VIII)-
oxyd erhaltenen Cyclodecandiole-(l,2) ableiten. Das
höher schmelzende Stereoisomer besitzt wohl die cis-
Konfiguration XII und das niedrig schmelzende die
trans-Konfiguration XIII. Es war interessant zu prüfen,
wie sich die Stereoisomeren bei der Oxydation mit
Blei(IV)-acetat verhalten. Bekanntlich haben R. Crie¬
gee und Mitarbeiter1) gefunden, dass in der Cyclopentan-
und in der Cyclohexan-Reihe das cis-Isomere mit grös¬
serer Geschwindigkeit oxydiert wird als das trans¬
isomere. Die Cyclodecandiole-(l,2) zeigen nun ein
umgekehrtes Verhalten. In Tabelle 1 sind die von uns
in Eisessig bei 20° gemessenen Geschwindigkeitskon¬
stanten in (mol"1.1.min"1) der "Glykolspaltung" von
stereoisomeren Cyclodecandiolen-(1,2) und Cyclohexan-
diolen-(l,2) mit den von H. Criegee und Mitarbeitern
an Cyclohexandiolen-(l,2) und Cyclopentandiolen-(l,2)
erhaltenen zusammengestellt.
Tabelle 1
Verbindung k20 Konstellation
Cyclodecandiol-(l,2)
eis-, Smp 140 ....
trans-, Smp. 54 . . .
2,61
100
F
H
Cyclohexandiol-(l,2)
eis-, Smp. 98 ... .
trans-, Smp. 104 . .
4,53 5,042)0,222 0,2242
C
D (E)
Cyclopentandiol-(1,2)40'0005)
12,8 3)A
B
1) R.Criegee, R.Kraft & B.Rank, A. 507, 159 (1933)„ H. Criegee & E.Büchner B. 21, 563, 571 (1940)i) A. 501, 184 (1933)*) B. 22, 572 (1940)

CM
H-P
PCD
bflPI
a«a!
CJ
Ma
«I
XI
<*~"S
«^^S.
Fh
cdFh
•H
P.
•H
CJ
•H
CD
HCJ
•H
co
*XI
O-P
CD
CD
•H
OCD
Hfi>
WO
Scd
+>
X-p
1fi
H-p
•3
bO
>5-rj
CD
CD
PL|U
>>
PP
cdCO
CD
oCD
rH
ÜCJ
1F-i
-p
XI
3CD
PH
cdfi
fiM
HO
CD
CD
fi•H
co
H<«!
Ha
•SrHXl -p
O3
^T3
§•H
CSI
CD
03
Üfi
CJ
XI
NP.
Fh
CM
bflO
:3
CD
>»
CD
w•
p.
cm
1^
co
T3
!x|CD
XI
XI
cd-p
T3
CJ
•H
HfirHpq
0J
O-f»
bfl00
MP.
CD
CD
3o
CD
H3
fiX
•»
CD
Fh>
CD
•H
MCJ
sFh
•H
CD
>!
•cd
-p
ficd
N•H
QU
T3
<H
XI
>>
03
HPP
cdfi
•
'U
=P
3 oCD
03
XI
Sco
:cdCD
fiS
-P
oCJ
XI
CQ
-P
S1-3
-H
•1
r-iCD
t3
3r4
h1-a
aCD
CDbO
•fi-H
CJ
CJ
Fh
sCQ
CD
OH
uPp
03
"M
3>J
x!
T3
OX
00
-P
fiS
CD
O<
CO
ao
o-p
cdCJ
CD
cdH
>CD
CM
CD
-p
P.
CJ
O•H
-<1>
HXI
MCD
CQ
>s
•3
U^•-k
1cd
CD
CD
•H
-P
co
bflCD
O-P
3CD
OH
fi-p
co
CD
3CD
•H
T3
3+>
•H
P+>
O>5
CD
-P
a)o
T3
Mco
CD
3Fh
CD
cdCD
•H
O«.
HS
co
CD
S-3
fiCJ
-p
CJ
mh
co
HS
CD
co
fifi
•CD
•H
CD
SXI
oO
-a
1•
hFh
CJ
SFh
CD
CD
OPP
aXI
cd«
X!
1fi
H•H
CD
CD
bO
mfi
S«
-PClH
CD
bflO
co
HU
P.
0J
gCD
bO
•H
fiH
>a
CO
1-3
CD
-P
bO
•O
4«!O
CD
Xi-P
4-'
0CJ
P.
H3
fiCD
cd>>
fico
•H
rH
efl•s
Sa
•H
HO
o-*|
CCICD
Ohü)
PÜ
OH
CM
bO
O-3
fiÖ
Hg
"r<
:rfCD
XI
TJ
+J
!>>•H
r-i-P
OQ
CJ-
XI
CD
CD
3"?
>CJ
Ocd
T3
rH
T3
CD
LT»
CO
>l
-P
fiCJ
3M
Or-i
rH
CO
<D
•H
CD
fiir\
oco
»Cg
bO
•H
fiO
1CO
uCJ
OCD
«\
Si
ri
PP
Co
T3
%«^
CD
XI
fi00
CJ
bflCD
T3
00O
CD
CD
Hfi
OS
•—
fiX)
HÖ
•H
Ö1
fiÜ
•»
>j
bO
TJ
Xi
H-P
XI
•H
OCJ
-p
CD
CM
0Q
OCD
•H
QCTi
M•H
-P
firH
*«!>s
co
fi*-^
•H
HXI
Ofi
CM
Pbfl
bfl1
HÜ
p.
CD
oO
CD
HCO
CD
3^-*
3H
U•H
XI
X)
fi<D
+»
HS
-P
•H
3i-H
aO
0U
fiCD
1CD
afi
bfla»
H-3
ÜCD
co
HO
^^
r-i3
-3
CD
CM
fiCD
XI
cdCJ
ocd
•H
HH
ficd
XI
fi*—v
•H
CJ
CD
bfl>i
*fi
CD
Mcd
bO
co
00
3cd
fi•H
Üo
1CM
+»
co
Hcd
sc
•O
CD
H«
-p
*—V
•H
3X
OCD
U•H
3CD
uo
HCD
MM
"*—*»
fiCD
co
•H
Sfi
LT\
M>>
Xfi
OXI
§•H
00
CM
oM
"3
M >Si 9
CD
CO 1
-P
fi
Oa
bO
•H
w>5
•H
Hco
CO
CJ
-p
X>
oH
•P
a*
Oo
•CD
:cdT3
CO
FhFh
CD
a•ai
CD
CD
CJ
•H
3S
§VI
«3 CD
T3
>M*
ci
-p
gH
>y
Fh
OM
Ocd
HM
gP.
CJ
iM
•3
•H
X)
^_^
*-^
CO
HI
-3
Oa
MO
CD
a:o
CM
«u
CD
hfl
.»*"""*
>5
Fh
r-ia
US
CD
rH
>CD
••—k
o
oa
ri
XI
aCD
CQ
-p
•»
CQ
1-P CD
•3
aCO
Hran
cdp.
fiH
CO
CD
•P
1CO
m•H
bO
SCD
*Fh
CJ
CO
Ocd
H
T3
§3 co
O 3NI
•H
**
CD
CD
CD
-p
Ü•H
T3
UUi
cdS
MO
00
S>3
OC
co
CD
ri
fiCD
1O
CO
-P
•a
OH
-P
CD
cd>>
CD
CD
HCO
O1
Hr-i
rH
V_ri*
rH
cd-P
OH
cd
-P
Fh3
•t
-P
•H
co
CM
•H
oS
O."s
ofi
CD
CD
fi1
fiCO 1
>T3
Xi
«t
T3
p-3s;
Oca
cdo
•H
CD
i•H
cdcd
>»
•i-lfi
fiCD
-p
T3
3sa
bO
fiO
fiSP
u3
MCD
HT>
co
-p
cdO
-3
CD
fiCO
•H
su
AÜ
W•H
Uco
T3
ow
CD
XI
CD
ca
M-P
S>CO
O•H
sCO
§h
-p
3CD
o•H
XI
CO
aCO
M3
CO
<H
fifi
CO
CD
(U
X!
1•H
CD
MXI
a*
ficd
OCD
bflH
Hë
-a
ta
+»
•ri
a>>
XI
CD
CD
UCD
cdO
OXI
T3
Ocd
cfi
•H
CD
M3
3•H
XI
cdCD
Hh
ofi
fiCD
o-P
+»
HO
Fha
Oca
Su
äH
•H
*-p
•3
•H
•H
XI
-P
NCiJ
co
cdCO
bO
CD
CD
-p
CD
p"rH
-p
3DO
3m
CJ
Fh
CD
fi>»
Oco
cd
-p
a•H
CD
>u
XI
M«H
•*
cCP
ÜI3
o*
*:o
o=H
3fi
yXI
T3
SI
uîcB
CD
oH
cd3
o•H
>XI
,5
bXI
co
-p
cdâ
«fi
Xi
O•H
CO
-p
10
a.
om
CO
Om
•H
HCO
XI
+»
co
-p
cd
aCO
MÖ
-P
a>
1Ü
cd#»
fico
Hco
fiCO
0)
(h*—*
XI
H•
fip
fiXI
T3
-P
HH
CD
-p
T3
•t
3CD
COH
oM
fiCÖ
Öxo
•H
CO
i-Hcd
<H
CM
co
CQ
mbO
SÜ
H-P
<H
*—.
MCJ
CQ
^s
3XI
cd•H
CO
oX!
OH
Û(0
-P
Os
XI
fiH
SXI
•H
aa
•H
fiXI
CD
co
•H
CD
co
OH
a)o
CO
(S-P
-p
CD
ö^
oXI
•H
+5
fiCD
hU
CD
co
T3
XI
CD
CJ
•H
CO
<H
-
:cdH
co
cdcd
XI
co
Ö<H
CJ
r-iCD
3Pf
CJ
«co
o0)
*XI
cdfi
00
XI
co
GXI
T>
•H
-P
-p
co
-P
cdCO
s<D
H•H
MT3
3CD
Mh
bO
heu
ri
oCd
CD
+»
-P
co
CI)•
co
Üfi
fifi
a•H
HN
CO
oXI
3•H
*o
cdCO
Fhn
xi
ci
«.
X-P
co
fi-P
H?
fio
CO
O+»
Fhco
Ho
aCD
T3
OH
obO
>X!
5P«
•H
§cd
CD
CD
co
co
co
fia
•H
ofi
o09
HU
fico
fico
-p
00
uCO
HCD
••
CJ
+>
CO
••H
a«
1fi
XI
«H
CD
3*
^-»»
Ht)0
CQ
•H
•CD
CO •
fiCO

- 18 -
H
HOT^OH
C5 trans
C1Q eis C1Q trans C/0 trans
Pig. 1
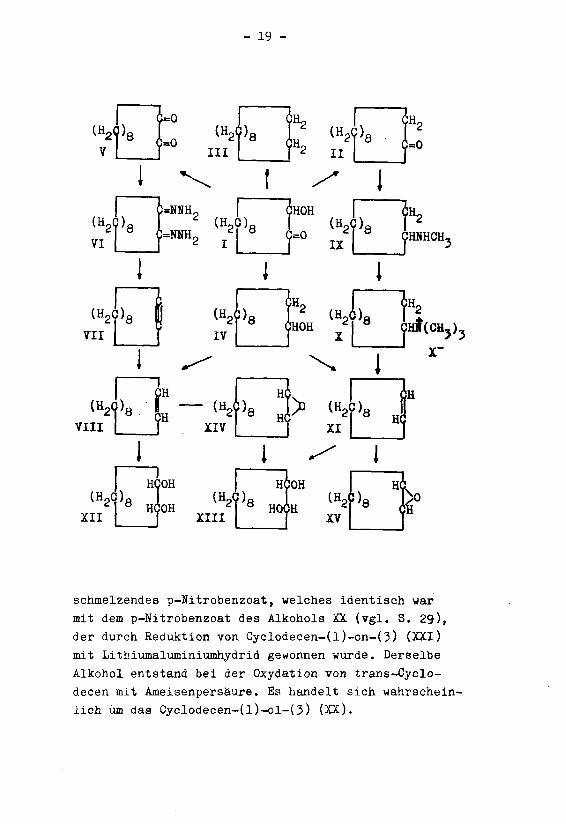
- 19 -
(H2Ç)8VII
(H2a) I (h2c)8 P (h2ç)„ T 2
v! î=0 ni! K --' i=0
IV
o)Q f2 (hJ)8 n2,8
CHÎ(CH3)3JHOHX
\ i
(H2VIII
I > I 1 1 ]I QtJ TjA I /
Ve |H — <Wa J> (H2f>8 JI j XIV I j xi I j
| HÇOH
^' öHCOH
2
XII
(H9Ç)HÇOH
XIII
(H0Ç)S8
HOÇH 2f8
OH
schmelzendes p-Nitrobenzoat, welches identisch war
mit dem p-Nitrobenzoat des Alkohols XX (vgl. S. 29),
der durch Reduktion von Cyclodecen-(l)-on-(3) (XXI)
mit Lithiumaluminiunihydrid gewonnen wurde. Derselbe
Alkohol entstand bei der Oxydation von trans-Cyclo-
decen mit Ameisenpersaure. Es handelt sich wahrschein¬
lich um das Cyclodecen-(l)-ol-(3) (XX).

- 20 -
Ein ahnliches Verhalten zeigen nach K. Ziegler und H.
Wilms die beiden stereoisomeren 1,2-Oxido-cyclooctane.
Die Geschwindigkeit, mit welcher die stereoisomeren
Cyclodecene mit ßenzoepersaure reagieren,ist ziemlich
stark verschieden. Bei 0° wurden m Chloroform folgen¬
de Geschwindigkeitskonstanten k (mol- .l.min" ) ge¬
funden:
trans-Cyclodecen 9,40
cis-Cyclodecen 1,20
Cyclohexen 1|15
Wahrend die beiden cis-Cycloolefme fast gleich
schnell reagieren, zeigt das trans-Cyclodecen eine
beträchtlich höhere Reaktionsgeschwindigkeit. Da sich
die eis- und trans-Stereoisomeren m der aliphatischen
Reihe gegenüber Persauren nicht stark verschieden ver¬
halten ),ist die grossere Reaktionsfähigkeit des trans-
Cyclodecens wohl auf eine "Spannung" in dieser letz¬
teren Verbindung zurückzuführen, auf die man auch aus
der geringen Stabilität bei der Isomerisierung mit
Sauren schliessen kann.
IR. -Absorptionsspektren
pDie IR.-Absorptionsspektren ) der m dieser Ar¬
beit untersuchten Verbindungen waren in mehrfacher Hin¬
sicht von Interesse. In bezug auf die Kohlenwasserstoffe
*) Vgl. z.B. D.Swern, Chem. Reviews ü, 49 (1949)) Aufgenommen auf einem Baird-Zweistrahl-Spektrogra-phen mit NaCl-Optik m Schichtdicken von 0,02 - 0,10mm bei Zimmertemperatur. Die Spektren der festen Ver¬
bindungen wurden an der Suspension in Nujol aufge¬nommen. Die Eichung des Gerätes wurde dauernd durch
Vergleich mit NH,-Banden überprüft und durfte m der
Nahe von 3u. und ou auf 0,02 bzw. 0,01*«. genau sein.
Herrn Prof. Dr. Hs.H. Gunthard danke ich bestens fur
die Aufnahme, Ausmessung und Interpretation der Spektren.

- 21 -
war die Frage nach dem Verhalten der rf(CH„)-Banden,
der Bestimmung der Konfiguration an der Doppelbindung
und der Möglichkeit des Nachweises der dreifachen
Bindung von Bedeutung. Bei dem Gyclodecanon stand die
Prüfung des Einflusses der Rmggrosse auf die Carbonyl-
frequenz im Vordergrund, und schliesslich war bei den
Epoxyden, Diolen und deren Acetonylacetalen die Deutung
des Zusammenhangs der Konfiguration mit den Spektren
praktisch wichtig.
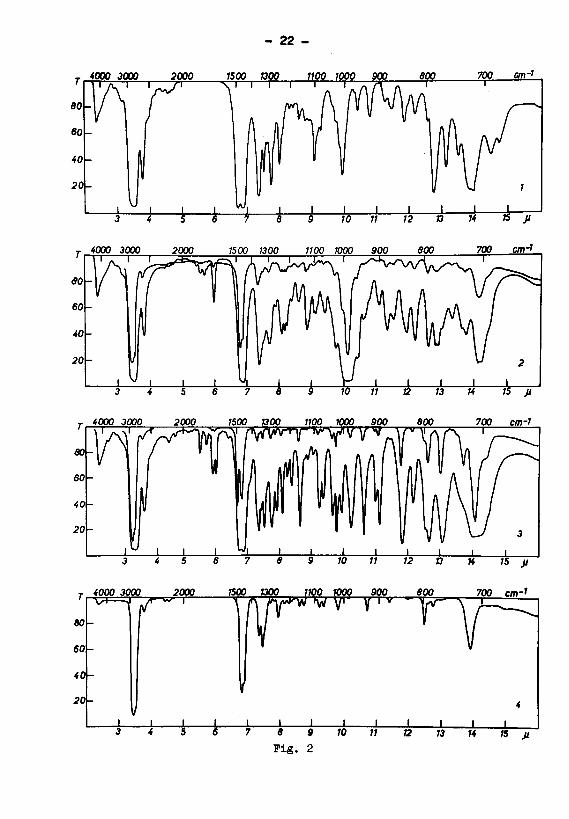
Kohlenwasserstoffe (vgl. Fig.2). Die in der 3—4m -
und 6,5-8u-Region beobachteten Banden ) des Cyclode-
cans (Kurve 1), der beiden Cyclodecene iKurve 3 cis-
und Kurve 2 trans-) und des Cyclodecms (Kurve 4) wei¬
sen folgende Frequenzen auf:
V(CH)2) ?«WJ) ^(CHg)
Cyclodecan . . . 2910 vs — 1488 va, 1462 va
(Kurve 1) 2680 w ~ 1451 va, 1385 s
1361 s,1357 s
eis—Cyclodecen . 2915 vs 1715 w 1484 vs, 1462 vs
(Kurve 3) 2680 w 1684 m 1451 vs, 1357 s
1656 m 1346 w,1330 s
trans-Cyclodecen 2915 vs 1667 m 1468 vs, 1463 vs
(Kurve 2) 2675 w — 1449 vs, 1356 s
1328 S
Cyclodecin 2915 vs - 1464 vs, 1449 vs
(Kurve 4) 2680 w " 1441 Ss, 1355 s
1337 s,1323 S
Die V(CH)-Frequenzen der Verbindungen sind - soweit
sich dies mittels Kochsalzoptik entscheiden lasst - von
) Es bedeuten vs, s, m, w, vw, resp. sehr stark, stark,mittel, schwach, sehr schwach. S = Schulter. Die No¬
menklatur fur die Normalschwingungen ist die von
R. Mecke, Leipziger Vortrage 1931, S. 23 {f. bm-
2 dungsstreckende, 6- bindungsbiegende Schwingung).) Die Frequenzen beziehen sich auf die Mitte der
CH-Banden.
- 22 -
OOP 3000 2000 7500 7300 7700 7000 900 eoo 700 cm-i
80
60
40
20
_L _L _L J_3 4 5 6 7 fl 9 10 11 12 73
4000 3000 2000 7500 7300 7700 7000 900 800
14 IS p
700 cm-1
J I L3 ^ 5 6 7 5 510 TT
4000 3000 2000 7500 7300 7700 7000 900
13 14 IS JJ
800 700 cm-1
3 4 5 6 7 8 9 10 11 12 O K IS jt
4000 3000 2000 1500 1300 1100 1000 900 800 700 cm-1
Fig. 2

- 25 -
gleicher Grösse wie bei den offenkettigen Kohlenwasser¬
stoffen. Alle Spektren weisen im Vergleich zu denen des
n-Decans ) und des n-1-Decens ) eine kompliziertere
é(CH„)-Region auf. Insbesondere treten - neben einem
—1regelmässig wiederkehrenden Triplett in dem 1450 cm -
Gebiet - zwischen 1320 und 1380 cm" mindestens zwei
Banden auf, die man bei Kohlenwasserstoffen meist einer
symmetrischen Deformationsschwingung der Methylgruppe
zuschreibt. In unseren Spektren dürften diese Banden
von £(CH2)-Schwingungen herrühren.
Die beiden Cycloolefine zeigen merkliche Unterschie¬
de in der 1>(C=C)-Region. So weist das cis-Cyclodecen
ein Dublett (1656, 1684 cm~ ) auf, dessen kurzwelliger
Zweig wohl als Kombinationston anzusehen ist.
Die ^)(C=C)-Bande des trans-Isomeren hat eine auffal-
lend hohe Frequenz ). Die Figerprint-Gebiete der bei¬
den stereoisomeren Cyclodecene sind völlig verschieden
und eignen sich für die analytische Bestimmung.
Darüber hinaus erlauben gewisse Merkmale der é(CH)-
Banden eine Zuteilung der Konfiguration, indem beim
cis-Cyclodecen die Banden 977 m und 710 vs und beim
trans-Cyclodecen die Banden 986 vs und 707 m vorkom-•7.
men ).
) American Petroleum Research Institute, Research
Project 44, Carnegie Institute of Technologie (CIT).Catalogue of Infrared Spectral Data. Ser.Nr. 390,
2n-Decane.
) So ist vergleichsweise 1>(C=C) bei Cyclohexen und
Cyclohepten 1652 bzw. 1651 cm". Siehe z.B. K.W.F.
Kohlrausch, Hand- u. Jahrbuch der Chemischen Physik,
,Bd. 9, VI, S. 349.
?) Wegen der Erkennbarkeit der eis- und transconfigu¬ration bei Olefinen siehe z.B. G.B.M.Sutherland & Cow.
Soc.1950. 915 und R.N.Jones, Am.Soc. 22, 5322 (1950).Der Vergleich mit Spektren des API, Res. Project 44,CIT, Catalogue of Infrared Spectral Data, z.B. Ser.
Nr. 21, cis-2-Butene, Ser. Nr. 22, trans-2-Butene, Ser.
Nr. 357, cis-2-Pentene, Ser. Nr. 727, cis-2-Hexene,Ser. Nr. 728, trans-2-Hexene & Ser. Nr. 621, trans-3-
Hexene, stützt diese Zuordnung. Vgl. auch die Raman-
spektren der Cyclooctene bei Y.Goubeau, A. 567. 214
(1950).

- 24 -
Das IH.-Spektrum des Cyclodecms ) weist in der
V(C5C)-ßegion zwei Banden auf (2208 und 2267 cm-1),deren Intensität nahezu gleich gross scheint. Die Deu-
tung dieses Dubletts kann Vorlaufig nicht gegeben werden ).
Es durfte sich aber bei dem einen der beiden Zweige um
einen Kombinationston handeln, der durch Fermi-Reso-
nanz mit der V(CSC)-Schwingung verstärkt wird.
Cvclodecanonr Cyclodecanol. Cvclodecandiole-(1.2)
und ihre Acetale (vgl. Fig. 3 und 4). Die nachstehende
Tabelle enthalt die (mutmasslichen) charakteristischen
Gruppenfrequenzen dieser Verbindungen.
V(0H) v(c=o) J(CH3) J(C0)3) J(0H)4)
Cyclodecanon(Kurve 5)
1701
Cyclodecanol(Kurve 6)
3380 s 1012 s
cis-Cyclode-candiol-(l,2)(Kurve 9)
3405 Ss
3305 s
1021 s
1050 s
trans-Cyclode-candiol-(l,2)(Kurve 10)
3495 Ss
3390 s
1052 s
cis-Acetal
(Kurve 11)1381 s, 1064 vs
1372 s, 1047 vs
trans-Acetal
(Kurve 12)1381 s, 1064 vs
1374 s, 1060 vs
Die Spektren der stereoisomeren Diole und ihrer
Acetale sind im Fmgerpnntgebiet stark verschieden,
zeigen aber trotzdem eine Parallele bei den entsprechen-
},) Vgl. A.T.Blomquist à Mitarb., Am.Soc. 21, 5511 (1951); Im allgemeinen sind die Intensitäten von i>(C=C)-Bandenin nahezu symmetrisch disubstituierten Acetylenen ver¬
hältnismässig klein. In dem von A.T.Blomquist veröffent¬lichten Spektrum, welches an dickeren Schichten gemes¬sen wurde, scheint der kurzerwellige Zweig des Dubletts
, weniger intensiv zu sein.
) Analog zu diesen beiden, beim trans- wenig und beim cis-
stark aufgespaltenen Dubletts, tritt in jedem Spektrumnoch ein zweites mit ganz ähnlichen Eigenschaften auf:
. trans- 1250 vs, 1240 vs, eis- 1250 vs, 1224 vs cm".
) Diese Zuordnung wurde nicht durch Deuterierung gesichert.

- 25 -
4000 3000"I ' '
2000 1500 7300 7700 7000 900 800 700 cm-*
â0-
60-
40-
20-
W
J I L J L
~5 6 7 S 5 70 77 12 13 14 15 p
2000 1500 1300 1100 1000 900 600 700 Cirri4000 3000
3 4 5
4000 3000 2000
7 â 9 10 77 12 W
1500 1300 1100 7000 900 800
14 iSjl
700 cm-1
3 4 5 6 7 6 9 10 11 12 13 14 75 Jl
4000 3000 2000 1500 1300 1100 1000 900 000 700 cm-1
Fig. 3

- 26 -
4000 300C 2000 7500 1300 1100 1000 900 âOO 700 cm-1
60
1 1 1 1 I I 1 [ 1 1
l\Al
1
V\A/N
60 "
/I (1/ tM,/lf
a/V v vv\/\
\s^~_^-~^-40
7/ I/!/"
\rT-
20lk1 1
\1
If1 1 i 1 1 i i 1
9
13 4
4000 3000
5
2000
6 7 6
1500 1300
9 10 11 12 13
1100 1000 900 600
75 Jl
700 an-1
4000 3000
40
20
8
Pig. 4
70 12 13 IS p

- 27 -
den Stereoisomeren. Bei beiden cis-Verbindungen weisen
die den <J(QH)- bzw. v(C°)-Schwingungen vermutlich zu¬
zuschreibenden Banden eine kräftige Aufspaltung in ein
Dublett auf, die den Spektren der trans-Verbindungen
zu fehlen scheint. Ob diese Unterschiede z.B. auf die
Symmetrie der die beiden OH-Oszillatoren verknüpfenden
Bindungen oder auf durch verschiedene Geometrie beding¬
te Unterschiede in den mechanischen Kopplungsgraden
zurückgeht, konnte nicht aufgeklärt werden ).
Das Vorhandensein der beiden geminalen Methyl¬
gruppen in den Acetalen kennzeichnet sich durch Dubletts1 2
sehr grosser Intensität bei 1381 und 1372 cm ).
) Modellbetrachtungen zeigen, dass Konstellationender trans-Diole existieren, bei denen die nähere
Umgebung der 1,2-Dioxygruppierung angenähert ein
Symmetriezentrum aufweist (siehe Fig. 1, D und G).Dann wäre wegen der g,u-Hegel eine Zuordnung möglich.Das trans-Cyclohexandiol-(l,2) besitzt ein ebenes
O-vq^Cvq -Gerüst und ein den OH-Gruppen wahrschein¬
lich zuzuordnendesDublett bei 1080 und 1045 cm
(Aufspaltung 35 cm ). Das Spektrum des cis-Iso-
meren weist zwei starke Banden bei 1080 und 990cm~ auf, die ©(OH)-Schwingungen zugeordnet werdenkönnen und deren Aufspaltung 90 cm~l beträgt.Auch in der Cyclodecan-Heihe entspricht das Spek¬trum mit den grösseren Aufspaltungen dem cis-Iso-
meren. Das chemische Verhalten zeigt jedoch, dass
im trans-Cyclodecandiol-(l,2) die Konstellation G
wohl nicht vorliegt (rasche Bildung des Acetals, usw.).) Diese Banden sind deutlich intensiver als die
^(CHp)-Banden der methylgruppenfreien Verbindungen.Der Hachweis der Methylgruppen in organischen Verbin¬
dungen mittels der <J(CH.,)-Bande ist daher nicht
immer einwandfrei durchführbar.

- 28 -
Die Carbonylfrequenz (1701 cm" ) von Gyclodecanon
ist nur wenig vom beim Cyclohexanon gemessenen Wert
(1706 cm~ ) verschieden. Auch hier weist das IR.-
Spektrum ê{CH„)-Banden in zwei Gruppen (1479, 1462,
1447 vs, 1423 s und 1370, 1362, 1350 s) auf.
lf2-0xido-cyclodecane (vgl. Fig. 3, Kurve 7 (eis)
und Kurve 8 (trans)). In den IR.-Absorptionsspektren
dieser Verbindungen scheinen einige Zusammenhänge zu
existieren: gemeinsame Banden treten im Fingerprint¬
gebiet auf bei 1362 w, 965 vs, 887 s und 708 s cm-1.
Von diesen können eventuell die Banden bei 1362, 887
und eine Bandengruppe bei 800-810 cm- mit dem Aethy-
lenoxydring in Zusammenhang gebracht werden.
III. Die Oxydation der Cyclo¬
decene mit Perameisensäure,
eine transanulare Reaktion )
Im vorhergehenden Abschnitt dieser Arbeit be¬
schrieben wir die Herstellung der beiden stereoiso¬
meren Cyclodecene. Erwartungsgemäss bildet sich durch
Oxydation mit Osmium(VIII)-oxyd aus cis-Cyclodecen das
cis-Cyclodecandiol-(l,2) und aus trans-Cyclodecen das
trans-Cyclodecandiol-(l,2). Zu unerwarteten Ergebnis¬
sen führte dagegen die Oxydation der Cyclodecene mit
Perameisensäure. Bekanntlich liefert die Oxydation
von Cyclopenten und Cyclohexen durch Persäuren in sau—o
rer Lösung trans-l,2-Diole ).
h V.Prelog 4 K.Schenker, Helv. 22., 2044 (1952)) Vgl. D.Swern, Chem. Reviews 4_5_, 1 (1949)

- 29 -
1. Die Oxydation von cis-Çyclodecen
mit Perameisensäure
Aus cis-Cyclodecen (XVIII) entsteht durch Oxy¬
dation mit Perameisensäure ein kompliziertes Reaktions-
Gemisch, aus dem durch Chromatographie an Aluminium¬
oxyd bisher folgende Verbindungen isoliert werden konn¬
ten:
a) ein flüssiger Alkohol C-joHtqO,
b) ein Diol G10ü20°2 vom Smp. 144°,
c) eine Verbindung C-jqHtqOp vom Smp. 98 und
d) eine Verbindung C-|0H,802 vom Smp. 74°.
a) Der Alkohol C-,qH,qQ ist ungesättigt und dem¬
nach monocyclisch. Zu seiner Charakterisierung eignet
sich besonders das schön kristallisierende p-Nitro-
benzoat, welches identisch ist mit dem p—Nitrobenzoat
eines Alkohols, der durch Reduktion von Cyclodecen-(l)-
on-(3) (XXI) mit Lithiumaluminiumhydrid gewonnen wur¬
de. Derselbe Alkohol entstand auch bei der Behandlung
des trans-l,2-Oxido-cyclodecans (XXII) ) mit Aluminium¬
oxyd. Es handelt sich demnach wohl um das Cyclodecen-
ol-(l)-ol-(3) (XX).
b) Das Diol ^iqH20^2 i3t gesättigt, monocyclisch
und nicht identisch mit den beiden früher hergestellten
stereoisomeren Cyclodecandiolen-(l,2). Der Vergleich
mit authentischen Präparaten zeigte, dass es sich um
eines der beiden stereoisomeren Cvclodecandiole-(lr6)(XXIII) handelt, das zuerst von ¥. Hückel und Mitar-
beitern ) aus Cyclodecandion-(l,6) durch katalytische
Hydrierung mit Platinmohr gewonnen wurde. Später erhiel-
) A. 474_,!9-
138 (1929)

- 50 -
ten die gleiche Verbindung PI. A. Plattner und J. Hulst-
kamp ) durch katalytische Reduktion von Cyclodecandion-
(1,6) mit Raney-Nickel und R. Criegee durch Reduktion
von Cyclodecanol-(l)-on-(6) mit Natriumamalgam ). Die
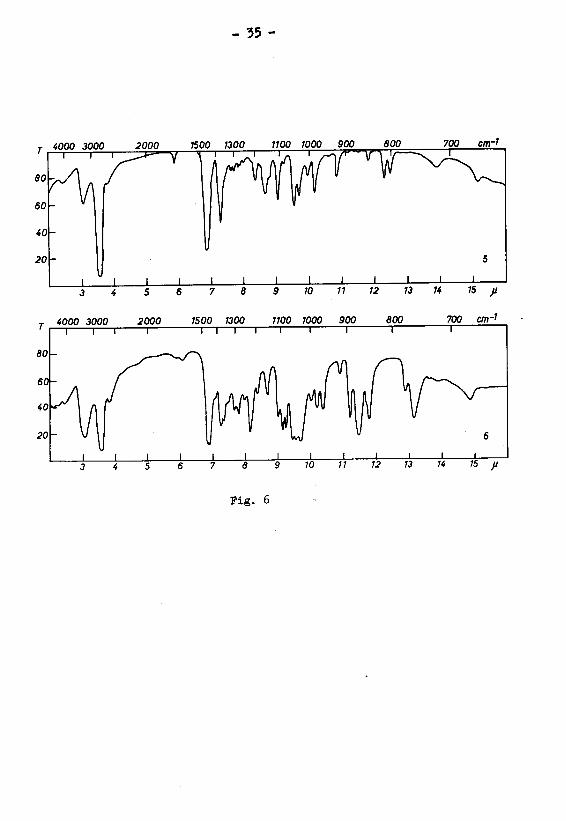
in Fig. 5 (Kurven 1 und 2) dargestellten IR.-Absorptions¬
spektren des freien Diols und seines Diacetates aus
Cyclodecen einerseits und aus Cyclodecandion-(l,6)
andrerseits waren identisch.
c) Die Verbindung gtohi8°2 vom ^mp* ^®° ist eea&^~
tigt und demnach bicyclisch. Sie gibt ein Mono-p-nitro-
benzoat vom Smp. 142 und ein Mono-acetat vom Smp. 33 •
Die Bestimmung des aktiven Wasserstoffs nach Zerewi-
tinoff zeigt, dass die Verbindung nur eine Hydroxyl¬
gruppe besitzt. In Uebereinstimmung damit führt die
Oxydation mit Chrom(VI)-oxyd in Eisessig zu einem ge¬
sättigten Mono-keton O-jQ^gOp, dessen 2,4-Dinitrophe-
nylhydrazon bei 159 schmilzt.
Die IR.-Absorptionsspektren des Mono-acetats
(Fig. 7, Kurve 8) und des Mono-ketons (Fig. 7, Kurve 9)
weisen in der <?(0H)-Reglon keine Absorptionsbande auf,
was auf Abwesenheit einer Hydroxylgruppe hinweist, da¬
gegen findet sich, gleich wie bei der Verbindung
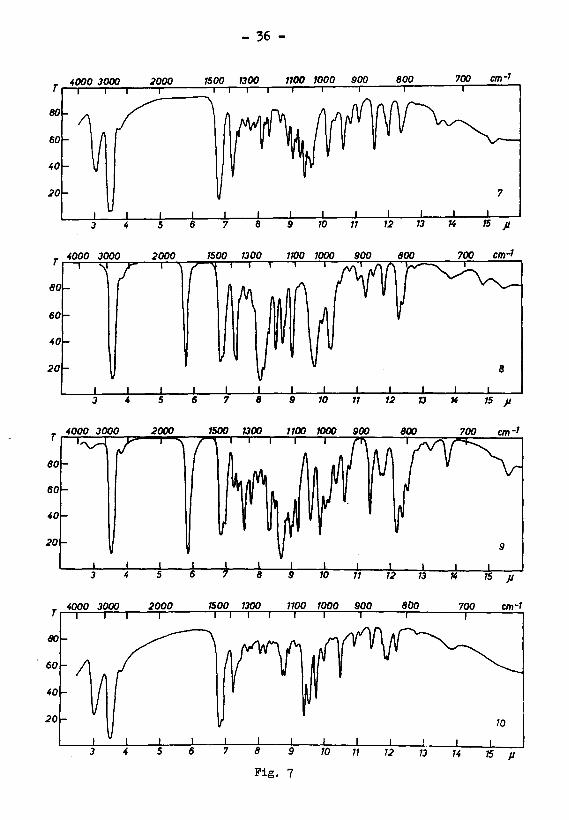
C10H18°2 selbs* (Fiß- 6» Kurve 5), eine charakter¬
istische Bande im Bereich von 1100 cm-,welche ver¬
mutlich einer Aethergruppierung zugeordnet werden kann.
Auf Grund dieser Beobachtungen ziehen wir für die Ver¬
bindung C-joH-igOp vom Smp. 98 die Konstitution eines
bicyclischen Oxy-äthers der Formel XVI oder XVII in
Betracht.
* * * *
H—&H0H ,
CH—GHOH
(H
yn—'
.c:rcMCH2)4
XVII
Sie muss durch Einwirkung von 2 Mol Perameisensàure auf
ein Mol Kohlenwasserstoff entstanden sein.
hHelv_. 21,218
(1944)
,ne±v. t±, c±o \,±wij
d) B. 21, 723, 725 (1944)

- 31 -
d) Die Verbindung G10H1802 vom SmP* 74° UH.-
Absorptionsspektrum Fig. 6, Kurve 6) scheint mit der
isomeren Verbindung vom Smp. 98 nahe verwandt zu sein.
Ihr Mono-p-nitrobenzoat schmilzt bei 129°. Die Bestim¬
mung des aktiven Wasserstoffs lässt auf nur eine Hydro¬
xylgruppe schliessen. Die Oxydation mit Chrom(VI)-oxyd
in Eisessig liefert ein Mono-keton, dessen 2,4-ßinitro-
phenylhydrazon bei 154 - 155° schmilzt und mit dem 2,4-
Dinitro-phenylhydrazon des Oxydationsproduktes aus der
isomeren Verbindung eine beträchtliche Schmelzpunkts—
depression ergibt.
Die beiden Isomeren müssen demnach entweder stel¬
lungsisomer sein in bezug auf die eine Haftstelle der
Aetherbrücke (vgl. Formeln XVI und XVII, Kohlenstoffatom 5
oder 6) oder an den asymmetrischen Stellen (Kohlenstoff¬
atom 5 bzw. 6 und 10) verschiedene Konfiguration aufwei¬
sen oder beides. Für weitere Untersuchungen reichte das
vorhandene Material nicht aus.
2. Die Oxydation von trans-Cvclodecen
mit Perameisensäure
Aus dem Reaktions-Gemisch, das durch Einwirkung
der Perameisensäure auf trans—Cyclodecen entsteht, lies-
sen sich durch chromatographische Analyse an Aluminium¬
oxyd folgende Verbindungen abtrennen:
a) Cyclodecanon (XXVI),
b) ein Diol C1QH2002 vom Smp. 152°,c) ein Alkohol C1C)Hl60 vom Smp. 63°,d) eine Verbindung G10Hi8°2 vom Smp 98° und
e) eine Verbindung üiqH1802 vom Smp* 6^°*

- 32 -
b) Das gesattigte Diol C10H20Û2 vom Smp* 152°
war verschieden sowohl von den beiden Cyclodecan-
diölen-(1,2) als auch vom Cyclodecandiol-(l,6) vom
Smp. 144 .Es erwies sich dagegen als identisch mit
dem zweiten stereoisomeren Cyclodecandiol-(1.6) (XXVII),
das zuerst von El.A. Plattner und J. Hulstkamp ) durch
katalytische Reduktion von Cyclodecandion-(l,6) mit
Raney-Nickel und dann von R. Criegee ) durch Reduktion
von Gyclodecanol-(l)-on-(6) bereitet worden war. So¬
wohl die freien Diole als auch ihre Diacetate gaben
miteinander keine Schmelzpunktserniedrigung und ihre
IR.-Absorptionsspektren, die in Fig. 5 (Kurven 3 und 4)
dargestellt sind, waren gleich.
c) Der gesattigte Alkohol C10Hi8° vom Smp" ^°
liess sich durch den Vergleich mit einem authentischen
Präparat^) mit dem bekannten trans-Dekalol-(1) (XXVIII)
identifizieren. Es handelt sich um das Stereoisomere,
dem nach ¥. Hucke1 ) wahrscheinlich die Konfiguration
XXVIIIa zukommt. Sein IR.-Absorptionsspektrum ist in
Fig. 7 (Kurve 10) abgebildet.
d) Die Verbindung C-]0H1802 vom SmP« 98° is"t
identisch mit der Verbindung C-jahi802 vom gleichen
Smp., die durch Oxydation von cis-Cyclodecen entstand.
e) Die dritte Verbindung C-.QH-.gQp vom SmP- 63°
(IR.-Absorptionsspektrum Fig. 7, Kurve 7) gab mit al¬
len bisher isolierten Isomeren eine starke Schmelz-
punktsemiedrigung und besitzt, gleich wie jene, nur
eine Hydroxylgruppe. Auch ihr kommt vermutlich die
Konstitution eines bicyclischen Oxy-athers zu. Da die
Verbindung nur in geringen Mengen isoliert wurde, konn¬
te sie bis jetzt nicht weiter untersucht werden.
i) Helv. 27, 218 (1944)5 B. 21, 723, 725 (1944)I) W.Huckel, A. Ml. 33 (1925)°t) B 62A, 134 (1934)

XXVIII
CHg(H2C)5
CHOH(H2C)5
XXVI
0=(H2Ç)7
XXVII
~°H2t(H2C>
HOCHHOCH—
1*2(H2?)3
XXV
CHp)•?CHo
HCiï-K&>
hc:
XXIV
:h2ç)
-HCC)„>lCHi
(H2Ô)7
XXIIIXIX
ÔHj(H^
HCOHHOCH•
ÇHg(HoÇ),
CIL,(H^Ofcj
CHHÖH''"
XXIIXX
XVIII
ÇH(H2Ç)7
XXI
CH„'I
?Ï
-CH
E»HÇ(H0Ç),CH(H2Ç)7
^CH
'
IH2C—
CH
C)7CH
(H2Ç)7
-ÇHOH-0=0
-33-

- 34 -
4000 3000 2000 1500 1300 1100 7000 900 800 700 cm-1
Fig. 5
- 35 -
T4000 3000 2000 7500 7300 7700 7000 900 «00 700 cm-'
1 1 i^i y ] ,y\. '/\ '/-V^MaT"S A*
BOy\/\ / I1/ w W
60 .
V'
40 -
J
20
1 1 1 1 1 1 1 1 1 1 1 ,
5
1
3 4 5 6 7 8 9 70 77 12 13 14 75 p
T4000 3000 2000 1500 1300 1100 1000 900 800 700 an'1
Fig. 6
- 36 -
4000 3000 2000 1500 1300 1100 1000 900 800 700 cm-1
S 6 7 8 9 10 11 12 13
2000 1500 1300 1100 1000 900 800
H 15 ß
700 cm-1
80
60
40
20
3 4 S 6 7 8 9 10 11 12 13
4000 3000 2000 1500 1300 1100 1000 900 800
3 4 5 6 7 8 9 10 ~fî 12 W
4000 3000 2000 1500 1300 1100 1000 900 800
15 ji
700 cm-'
« 15 ji
700 cm-'

- 37 -
Diskussion.
Die Entstehung der bisher aufgeklarten Produkte
der Oxydation der Cyclodecene mit Perameisensaure lasst
sich unserer Ansicht nach plausibel erklaren, wenn man
annimmt, dass sich zuerst die instabilen stereoisomeren,
ionischen Zwischenprodukte XIX und XXV bilden ). Bei
gewohnlichen klassischen Ringen ) reagieren die analo¬
gen Zwischenprodukte direkt mit Ameisensaure, welche
dabei die Rolle einer Base (Elektronendonators) spielt,
unter Entstehung von 1,2-Diol-Denvaten ). Die Stereo-
spezifitat diese Vorganges, der ausschliesslich zu
tran8-l,2-Diolen fuhrt, spricht dafür, dass es sich
um eine Sj,2-Reaktion handelt. Wie man aus den Modell¬
betrachtungen leicht ableiten kann, ist der besondere
räumliche Bau der Zwischenprodukte XIX und XXV in der
Zehnring-Reihe fur eine normale S„2- Reaktion ungun¬
stig. Ihre Stabilisierung wird deshalb auf andere
Weise erreicht:
a) Durch Abspaltung eines Protons an den Kohlen¬
stoffatomen 1 bzw. 5 entstehen die Enol-Form des Cvclo-
decanons (XXVI) bzw. das Cyclodecen-(l)-ol-(3) (XX).
) Durch das Symbol ^Q-=H soll die Gesamtheit
aller mesomeren und valenztautomeren Formenr also
sowohl das Oxonium-Ion als auch die Carbemum-
Ionen dargestellt werden.
) Ueber die Bezeichnung des 5-, 6- und 7-Ringes als
gewohnliche klassische Ringe (common classic rings)vgl. H.C.Brown, R.S.Fletcher & R.B.Johannesen,Am.Soc. 22., 212 (1951).
) Vgl. S.Winstein & R.B.Henderson in R.C.Elderfield,Heterocyclic Compounds, Vol. I, New York, London 1950.

. - 58 -
b) Besondere bemerkenswert ist die Entstehung
der beiden Cyclodecandiole-(l,6). Wir nehmen
an, das in der Cyclodecan-Reihe eine abnormale S~2-
Reaktion stattfinden kann, indem die Sauerstoff-Base
nicht am Kohlenstoffatom 2 des Kations, sondern an
dem gegenüberliegenden Kohlenstoffatom 6 angreift.
Gleichzeitig wandert ein Wasserstoff mit seinen bei¬
den Valenzelektronen von diesem letzteren zum Kohlen¬
stoffatom 2. Solche neuartige Reaktionen wollen wir
transanulare Reaktionenen nennen, )).
Zum besseren Verständnis der sterischen Verhältnisse
sind in Pig. 8 und 9 die Kalotten-Modelle der stereo¬
isomeren Cyclodecene abgebildet. Durch Pfeile sind die
Angriffsstellen des Oxydationsmittels (0) und der
Base (B) sowie des wandernden Wasserstoffs (H) bezeich¬
net. Man kann daraus ersehen, dass die sterischen Ver¬
hältnisse bei den Cyclodecenen für eine transanulare
Reaktion ausserordentlich günstig sind. Es sei noch
besonders hervorgehoben, dass es sich bei der Entstehung
von Cyclodecandiolen-(l,6) aus den Cyclodecenen um
stereospezifische Vorgänge handelt. Aus sterisch ver¬
schiedenen Cyclodecenen entstehen sterisch verschie¬
dene Cyclodecandiole-(l,6).
Wenn man die plausible Annahme macht, dass der
transanulare Angriff der Sauerstoff-Base (B) am Koh¬lenstoffatom 6 wie bei normalen Sjf2-Reaktionen unter
Konfigurationsumkehrung stattfindet, so würde daraus
folgen, dass dem Cyclodecandiol-(l,o) aus cis-Cyclo-decen die cis-Konfiguration und dem stereoisomeren
Cyclodecandiol-(l,6; aus trans-Cyclodecen demnach die
trans-Konfiguration zukommt.
) Von trans=hinüber, quer, jenseits und anulus=Klng.) Wir danken Herrn Prof. A.C.Cope, M.I.T. Cambridge,Mass., für die Mitteilung, dasa er eine ähnliche
transanulare Reaktion in der Cyclooctan-Reihebeobachtet hat.

- 39 - .,
L il *-<xo'd eâ
r"a -'CV W , J -an-ctC
*j- fiUO-S j Q'JJJT.l
ai ? (ler1 Co J H8^>
1t3 a£l t ^
i
«1 "i
É^) 15- '
'
#"i,a5: y<•
Kr>/ ?> v
r l
Pig. 8
ois-Cyclod#e#n
Pig. t-
trans-Cyclodeoea

- 40 -
Es bleibt noch abzuklären, ob die transanulare
Oxydation vom beschriebenen Typus auch bei anderen
Ringen beobachtet werden kann ) und ob auch andere
Reaktionen transanular verlaufen können.
c) Die Entstehung von trans-Dekalol-(l) (XXVIII)
vom Smp. 63° bei der Oxydation von trans-Gyclodecen
mit Perameisensäure bildet eine weitere Stütze für
die hier entwickelten Anschauungen. Es handelt sich
um eine weitere transanulare Reaktion. Eines der bei¬
den formell möglichen Carbenium-Ionen im Zwischen¬
produkt XXV scheint quer durch den Ring mit den Koh¬
lenetoffatomen 6 bzw. 7 unter Abspaltung eines Protons
zu reagieren. Im Einklang mit einer aolchen Annahme
steht die Stereochemie des resultierenden trans-üeka-
lols-(l) (XXVIIIa). Wenn der Ringschluss unter Konfi-
gurationsumkehrung am Carbenium-Ion-Kohlenstoff statt¬
findet, so ist zu erwarten, dass die Wasserstoffatome
1 und 9 des Dekalin-Gerüstes in trans-Stellung stehen.
Zur Erklärung des aussergewöhnlichen Verhaltens
verschiedener Ringverbindungen mit mittlerer Ringglie-
derzahl (8-13) hat V. Prelog ) schon früher relativ
starke Wechselwirkungen quer über den Ring angenommen,
die er als transanulare Effekte bezeichnete. Die in
der vorliegenden Arbeit beschriebenen transanularen
Reaktionen dürfen als starke Stütze für die Annahme
der Existenz solcher transanularer Effekte betrach¬
tet werden. Damit scheint auch bewiesen zu sein, dass
die Sonderstellung der Verbindungen mit einem mittle¬
ren Ring im Hinblick auf ihre chemischen und physika¬
lischen Eigenschaften durch die "I-Strain" - Theorie
von H. C. Brown und Mitarbeitern nicht ausreichend
erklärt werden kann.
h Vgl. Anm. 2, S. 38.
*) Abstracto of Papers 120"1 Meeting Am. Chem. Soc.
New York, Sept. 3-7, 1951.

EXPERIMENTELLER TEIL1)
Zur Herstellung des Cyclodecanol-(l)-ons-(2) (I).
Die fur die vorliegende Arbeit benotigten gros¬
seren Mengen von Cyclodecanol-(l)-on-(2) wurden in
einem zylindrischen Reaktionsgefass mit halbkugelfor-
migem Boden aus nichtrostendem V2A-Stahl von 10 1 In¬
halt hergestellt. Der Boden des Reaktionsgefasses war
mit elektrischen Heizkörpern versehen, welche durch
ein Kontakt-Thermometer gesteuert wurden. Der auf¬
schraubbare und mit einer Kunstharz-Abdichtung versehe¬
ne Deckel aus V2A-Stahl trug neben einer Thermometer-
hulse fur das Kontakt-Thermometer und drei Stutzen
(fur die Zuleitung des Esters, fur die Zuleitung des
Stickstoffs und fur den Ruckflusskuhler) eine Fuhrung
fur die Achse eines überdimensionierten Yibro-Mischers ).
Dieser erlaubte eine feine Verteilung des Natriums und
hatte nicht die vielen Nachteile, welche die schnell¬
laufenden Rotationsruhrer aufweisen. Die Zugabe des
Esters erfolgte nach dem von K. Ziegler und Mitarbei-
tern ) empfohlenen Einpress-Verfahren mittels Queck¬
silber.
In das Reaktionsgefass wurden 5 1 absolutes Xylol
vorgelegt, die Apparatur wurde mit sorgfaltig vorge-
reinigtem Stickstoff gespult, worauf man 500 com Xylol
abdestillierte. Man liess dann die Temperatur auf etwa
150° fallen und fugte 170 g blankea Natrium zu, welches
in kurzer Zeit vom Vibrator pulverisiert wurde. Darauf
presste man 410 g Sebacinsaure-methylester in 200 ccm
absolutem Xylol em (Dauer 28 Std., Ruhren mit Vibra¬
tor). Nach der Zugabe des Esters rührte man weitere
) Alle Schmelzpunkte sind korrigiert.
,) von der A.G. fur Chemie-Apparatebau, Zurich.
>) A. 52£, 125 (1935).

- 42 -
2 Std., worauf das überschüssige Natrium durch Zugabe
von 200 ccm Methanol zerstört wurde. Has Reaktions¬
gemisch versetzte man mit 40-prozentiger Schwefelsaure
bis zu stark saurer Reaktion und trennte die Jiylol-
schicht im Scheidetrichter ab. Nach Waschen mit Natnum-
carbonat-Losung und Wasser wurde das Xylol durch eine
kurze Kolonne abdestilliert und der Rückstand in Stick¬
stoff-Atmosphäre m einem Hickman-Kolben destilliert.
Bei 0,1 mm Druck und 70 - 88° gingen 161 g (53 $ d. Th.)
Acylom über, welches nach kurzem Stehen kristallin
erstarrte.
Cyclodecanon (II).
Bei Anwendung reiner Zink-Wolle ) und analytisch
reiner Salzsaure konnte nach der veröffentlichten
Vorschrift von verschiedenen Experimentatoren das Cyclo¬decanon in reproduzierbar guten Ausbeuten erhalten
werden. So gaben 20 g rohes Cyclodecanol-(l)-on-(2)
19 g (77# d. Th.) reines, aus Methanol umkristalli-
siertes Cyclodecanon-semicarbazon vom Smp. 203—205°.Daraus liessen sich mit 60 g Phtalsaureanhydrid im
Wasserdampfström 12,3 g (66?4 d. Th.) reines Cyclode¬
canon vom Sdp.12 nm102-104° gewinnen.
IR.-Absorptionsspektrum Fig. 3, Kurve 5.
Die zeitraubende Reinigung über das Semicar-
bazon kann, wie sich spater herausgestellt hat, unter¬
lassen werden. So erhielt man aus 23 g Cyclodecanol-
(l)-on-(2) 19,3 g (93> d. Th.) durch zweimalige Fraktio¬
nierung an einer Vigreux-Kolonne gereinigtes, reines
Cyclodecanon voa Sdp.g,- 112-114°.
Cyclodecan (III)2).
300 g reine Zink-TSolle wurden mit 30 g Quecksil¬
ber (II)-chlorid, 500 ccm Wasser und 30 ccm konzentrier¬
ter, reiner Salzsaure wlhrend 5 Min. in einem 3 1 Rund¬
kolben stehen gelassen. Man dekantierte die Flüssigkeit
2) Bezogen von Dr.Bender 4 Dr.Hobein, Zurich.) Dieser Versuch wurde von Dr.M.Zimmermann ausgeführt.

- 43 -
vom amalgamierten Zink und setzte 50 g Cyclodecanol-
(l)-on-(2) in 300 ccm Eisessig und 200 ccm konzentrier¬
ter, reiner Salzsäure hinzu. Zum Reaktionsgemisch,
welches in heftigem Sieden gehalten wurde, gab man in
6-stündigen Intervallen je 50 ccm, insgesamt 830 ccm,
konz. Salzsäure zu. Nach 88 Std. wurde die Lösung vom
überschüssigen Zink abgegossen, mit dem gleichen Volu¬
men Wasser verdünnt und mit Aether ausgeschüttelt. Die
ätherische Schicht wurde mit Natronlauge und Wasser ge¬
waschen, mit Natriumsulfat getrocknet und destilliert.
Aus 150 g Cyclodecanol-(l)-on-(2) wurden so 108 g
Cyclodecan vom Sdp.pQ 82-83° erhalten.
IR.-Absorptionsspektrum Fig. 2, Kurve 1.
Cyclodecanol (IV).
In einem 500 ccm Schliffkolben wurden zu einer
Lösung von 1 g Lithiumaluminiumhydrid in 100 ccm abs.
Aether unter Kühlen mit Wasser und Schütteln 8,0 g
Cyclodecanon in 200 ccm abs. Aether langsam zugetropft.
Nach Abschluss der Reaktion erwärmte man das Reaktions¬
gemisch und zersetzte es dann mit Wasser und verd.
Schwefelsäure. Die übliche Aufarbeitung ergab 7,40 g
Cyclodecanol vom Sdp.,2 __ 123*-126°, welches aus Petrol-
Sther umkristallisiert, bei 42° schmolz.
3,656 mg Subst. gaben 9,076 mg C02 und 4,231 mg RgO
G10H20° Ber- C 76»86 * H 12»9° >
Gef. C 76,88 fr H 12,95 fr
IR.-Absorptionsspektrum in Nujol Fig. Kurve 6.
Cyclodecin (VII).
70 g frisch destilliertes Gyclodecanol-(l)-on-(2)
gaben durch Oxydation mit Chrom(VI)-oxyd nach der frü-

- 44 -
her veröffentlichten Vorschrift 49,5 g (71,55t d. Th.)
Cyclodecandion-(1.2) (V) vom Sdp.18 120-125° X).
40 g Cyclodecandion-(l,2) wurden unter Kühlung mit
50 g wasserfreiem Hydrazin gemischt und dann 15 Std.
im Einschlussrohr auf 100° erhitzt. Beim Abkühlen er¬
starrte das Reaktionsprodukt. Die nadeligen Kristalle
wurden mit Benzol aufgeschwemmt und abgesaugt. Durch
Umlosen aus Benzol konnten insgesamt 52,5 g (69> à. Th.)
reines Dihydrazon (VI) gewonnen werden, das zur Ana¬
lyse noch zweimal aus Benzol umkristallisiert wurde.
Smp. 126-150° (Zers.).
4,056 mg Subst. gaben 9,076 mg C02 Und 5,669 mg HgO5,570 mg Subst. gaben 0,954 ccm N2 (22°, 718 mm)
C10H20N4 Ber* G 61'18 * H 10'27 * N 2Ö»55 >
Gef. C. 61,57 > H 10,17 5* N 28,60 -/>
In einem Dreihalskolben mit Ruhrwerk und Ruckfluss-
kuhler, der mit einem Wasserabscheider versehen war,
wurden 25 g Quecksilber(II)-oxyd, 1 g fein gemahlenes
Kaliumhydroxyd und 20 g wasserfreies Natriumsulfat in
100 ccm Toluol zu schwachem Ruckfluss erhitzt ).
) Die Oxydation des Cyclodecanol-(l)-ons-(2) nach dem
von A.T.Blomquist, L.Huang lau <5c J.C.Bohrer, Am.Soc.
2i, 5645 (1952) beim Cyclononanol-(l)-on-(2) ange¬
wendeten Verfahren (Kupferacetat m wassngem Eis-
2essig) fuhrt zu Ausbeuten von über 90>6 an Diketon.
) A.T.Blomquist. R.E.Bürge jr., A.C.Sucsy, Am.Soc.
74. 5656 (195*) verwendeten absolutes Benzol als
Losungsmittel. Nach 70-stundiger Reaktion erhielten
sie das Cyclodecm m einer Ausbeute von 56> d. Th..
Verwendung von Toluol als Losungsmittel und Ent¬
fernung des bei der Oxydation sich bildenden «as¬
sers mittels Wasserabscheider erhöht die Ausbeute
bei wesentlich kürzerer Reaktionsdauer bedeutend.
Das wasserfreie Natriumsulfat dient dabei lediglichals Tragersubstanz fur das frei werdende wuecksil-ber und erleichtert die nachfolgende Filtration.
Zugleich kann anhand des am Wasserabscheider abge¬trennten Wassers der Verlauf der Reaktion kontrol¬
liert werden.

- 45 -
Zu der Suspension gab man unter starkem Ruhren im Ver¬
laufe von 30 Mm. 11,0 g feinst zerriebenes Cyelodecan-
dion-(l,2)-dihydrazon in 6 Portionen. Der Stickstoff
entwich dabei unter Aufschäumen, und der Kolbeninhalt
färbte sich durch das gebildete Quecksilber schwarz.
Nach zweistündigem Kochen war die Stickstoff-Entwick¬
lung beendet, worauf die filtrierte, gelbe Toluol-
Losung durch eine Saule von 10 g Aluminiumoxyd (Akt.
II bis III) filtriert wurde. Das nun farblose Filtrat
gab bei der Destillation 5,12 g (67^ d. Th.) eines
unangenehm riechenden, farblosen Oels.
Nach sorgfaltiger Fraktionierung mit einer Vig-
reux-Kolonne zeigte das Cyclodecin folgende Eigen¬
schaften: Sdp.12 ^ 78,5°, d420= 0,8973, nD20= 1,4950.
MD ber. 44,18 gef. 44,22.
3,528 mg Subst. gaben 11,406 mg C02 und 3,780 mg Ho0
C10H16 Ber. C 88,16 £ H 11,84 5*
Gef. C 88,23 > H 11,99 y
IR.-Absorptionsspektrum Fig. 2, Kurve 4.
cis-Cyclodecen (VIII).
3,05 g Cyclodecin wurden m 30 ccm abs. Methanol
mit 1,0g Palladium-Blei-Calciumcarbonat-Katalysator
nach Lmdlar katalytisch hydriert. Nach Aufnahme von
1,016 Mol Wasserstoff, was 195 Min. erforderte, stand
die Hydrierung still. Der Katalysator wurde abfiltriert,
das Filtrat m Wasser gegossen und dreimal mit Petrol-
ather ausgeschüttelt. Nach Abdampfen des Losungsmittels
und Destillation wurden 2,95 g cis-Cyclodecen erhalten,
welche zur Analyse über Natrium in einem Vigreux-Kol-
ben destilliert wurden. Sdp.1(- mm
73°, d.,20= 0,8743,
9nr
15 mm' 4 ' '
n^ = 1,4885. MD ber. 45,65 gef. 45,52.
3,717 mg Subst. gaben 11,810 mg C02 und 4,356 mg H20
G10H18 Ber* G 86»87 * H 13'1:5 *
Gef. C 86,71 > H 13,11 i»
IR.-Absorptionsspektrum Fig. 2, Kurve 3.

- 46 -
Neben Methanol hat sich auch Feinsprit für die
partielle Hydrierung des Cyclodecins bewährt. In
Petroläther und Benzol verlief die Hydrierung sehr
langsam, und auch der sonst empfohlene Zusatz von
Chinolin zeigte in keinem der vier versuchten Lösungs¬
mittel eine günstige Wirkung. cis-Cyclodecen ist -
gleich wie trans-Cyclodecen und Cyclodecin — mit Fein¬
sprit und Methanol leicht flüchtig und muss deshalb
über den Umweg der Extraktion mit Petroläther isoliert
werden.
Cyclodecyl-methvlamin (IX).
28,2 g über Semicarbazon gereinigtes Cyclodecanon
wurden mit 40 g wasserfreiem Methylamin in 120 ccm abs.
Methanol und 3 g Raney-Nickel bei 80 atü Wasserstoff
und 100 im Hochdruckautoklaven 20°Std. geschüttelt.
Der Katalysator wurde darauf abfiltriert und das
Reaktionsgemisch zur Entfernung des Methylamins unter
Rückfluss gekocht. Nach Zugabe von 15 ccm konz. Salz¬
säure dampfte man im Vakuum ein. Der Rückstand wurde
in Wasser gelöst und mit Aether ausgeschüttelt, wobei
9,0 g neutrale Anteile entfernt werden konnten. Aus
der wässerigen salzsauren Lösung wurden die Basen mit
konz. Natronlauge in Freiheit gesetzt und in Aether
aufgenommen. Aus den mit Kaliumhydroxyd getrockneten
ätherischen Auszügen konnten durch Destillation 17,05 g
Cyclodecyl-methylamin vom Sdp.,p „,»105-108° gewonnen
werden. pKA = 9,56 in 80-proz. Methyl-cellosolve.
3,956 mg Subst. gaben 11,330 mg C02 und 4,766 mg RgO2,834 mg Subst. gaben 0,208 com Ng (22°, 723 mm)
C11H23N Ber* C 78>05 * H 15»69 * K 8»27 *
Gef. C 78,16 fL H 13,68 ji N 8,08 i>

- 47 -
Cyclodecyl-trimethyl-ammonium.iodid (X).
Zu 16,8 g Cyclodecyl-methylamin in 20 ccm abs.
Methanol tropfte man bei 0° unter Rühren eine Lösung
von 14,1 g Methyljodid in 15 ccm abs. Methanol. Nach
kurzem Stehen versetzte man ebenfalls bei 0° nochmals
mit der gleichen Menge Methyljodid und fügte gleich¬
zeitig eine Lösung von 5,55 g Kaliumhydroxyd in 50 ccm
abs. Methanol hinzu. Nach 2 Std. wiederholte man die
Zugabe von Methyljodid und Kaliumhydroxyd, lieas 3 Std."
bei Zimmertemperatur stehen und erhitzte darauf noch
1 Std. auf dem Wasserbad, bis das Reaktionsgemisch
neutral reagierte. Nach dem Abdampfen des Lösungsmit¬
tels blieb ein weisser Kuchen zurück, der in 100 ccm
Wasser gelöst und in 100 ccm kalte 10-prozentige Natron¬
lauge gegossen wurde. Das Cyclodecyl-trimethyl-ammonium-
jodid fiel als dichter, farbloser Niederschlag aus.
Ausbeute 30,3 g. Zur Analyse löste man aus Aceton um.
Das quaternäre Ammoniumsalz kristallisierte in farblosen
Schuppen vom Smp. 265-268° (Zers.).
3,806 mg Subst. gaben 6,662 mg G02 und 2,810 mg H20
C13H28NJ Ber# G 48»00 S* H 8»68 &
Gef. C 47,78 $4 H 8,26 5*
trans-Gyclodecen (XI).
30 g Cyclodecyl-trimethyl-ammoniumjodid wurden in
100 ccm abs. Methanol mit 13 g Silberoxyd 2 Std. ge¬
schüttelt. Der Niederschlag wurde abgenutscht und gründ¬
lich mit Methanol gewaschen. Das Filtrat wurde eingedampft
und der Rückstand, ein gelbes Oel, in einem Hickman-
Kolben bei 150 mm Druck langsam erhitzt. Die Zersetzung
der quaternären Base begann bei einer Badtemperatur von
etwa 100°. Das Destillat nahm man in Aether auf, schüt¬
telte es mit verd. Salzsäure, verd. Natriumcarbonat-
Lösung und Wasser aus und destillierte durch eine kur-

- 48 -
ze Vigreux-Kolonne.
Man erhielt so 6,75 g {bOjb d. Th. ) reines trans-
Cyclodecen vom Sdp.,0 78-80 . Zur Analyse wurde* IB 01
?Qüber Natrium destilliert. Sdp.,a 78 ,
d. = 0,8681,
or» 1° mm * 4 ' '
nD = 1,4846. MD ber. 45,65 gef. 45,53.
3,568 mg Subst. gaben 11,362 mg C02 und 4,204 mg RgO
G10H18 Ber* C 86'87 * H 15>15 *
Gef. C 86,90 i> H 13,18 i>
IH.-Absorptionsspektrum Fig. 2, Kurve 2.
Durch Hydrierung mit Platinoxyd-Katalysator in
Eisessig nahm das trans-Cyclodecen langsam 1 Mol Was¬
serstoff auf unter Entstehung von Cyclodecan.
cis-Cyclodecandiol-(1.2) (XII).
a) Aus cis-Cyclodecen.
125 mg cis-Cyclodecen gaben mit 190 mg 0smium(VIII)-
oxyd und 10 Tropfen Pyridin 378 mg hellbraunes, kristal¬
lines Addukt, welches in 10 ccm Methylenchlorid, 400 mg
Mannit und 400 mg Kaliumhydroxyd in 5 ccm Wasser so lan¬
ge geschüttelt wurde, bis das Methylenchlorid farblos
war. Nach dem Eindampfen der Methylenchlorid-Lösung
erhielt man 130 mg eines Produktes vom Smp. 132-34°,welches nach Umlösen aus Alkohol-Benzol 108 mg reines
cis-Cyclodecandiol-(l,2) vom Smp. 139-40° gab.
b) Aus Cyclodecanol-(l)-on-(2).
35 g frisch destilliertes Gyclodecanol-(l)-on-(2)
wurden in 100 ccm Feinsprit mit Raney-Nickel bei 100°
und 100 atü 10 Std. katalytisch hydriert. Nach dem Ab—
filtrieren des Katalysators und dem Abdestillieren des
Losungsmittels blieb ein Gemisch der stereoisomeren
üiole zurück, aus welchem sich die Hauptmenge des cis-
Gyclodecandiols-(l,2) durch Kristallisieren aus Alko¬
hol-Benzol, bzw. Petrolather erhalten liess. Ausbeute
16,85 g (47,5> d. Th.).

- 49 -
Zur Analyse wurde aus Alkohol-Benzol umkristallisiert,
Smp. 140°.
3,671 mg Subst. gaben 9,353 mg C02 und 3,840 mg HgO
C10H20°2 Ber' G 69'72 * H llt7° *
Gef. C 69,53 + H 11,70 1>
IH.-Absorptionsspektrum in Nujol Fig. 4, Kurve 9-
Diacetat.
1 g des Diols wurde mit 5 ccm Acetanhydrid und
I ccm Pyridin 3 Std. am Rückfluss acetyliert. Das auf
übliche Weise gewonnene ölige Diacetat wurde schliess¬
lich durch Chromatographieren an Aluminiumoxyd (Akt.
II bis III) gereinigt und im Hochvakuum destilliert,
Sdp.n ,
„„
100°.r
0,1 mm
3,937 mg Subst. gaben 9,486 mg C02 und 3,355 mg HgO
C14H24°4 Ber' ° 65'59 * H 9>44 *
Gef. C 65,75 % H 9,54 ft
Acetonyl-acetal.
0,5 g des Diols liess man mit 5 ccm wasserfreiem
Aceton und zehn Tropfen konz. Schwefelsäure über Nacht
stehen. Das Reaktionsgemisch wurde hierauf in Wasser
gegossen, in Petroläther aufgenommen, mit Natriumcar-
bonat-Lösung gewaschen und schliesslich an Aluminium¬
oxyd (Akt. II bis III) chromatographiert. Die Destil¬
lation der Petroläther-Eluate lieferte 470 mg farb¬
loses Oel, d420 = 0,9773, nD20= 1,4765.
3,764 mg Subst. gaben 10,147 mg C02 und 3,831 mg HgO
C13H24°2 Ber# ° 73»55 * H 11'39 *
Gef.. C 73,53 $ H 11,39 Ï
IR.-Absorptionsspektrum Fig. 4, Kurve 11.

- 50 -
tranB-Cyclodecandiol-(1.2) (XIII).
a) Aus trans-Cyclodecen.
100 mg trans-Cyclodecen gaben mit 184 mg
Osmium(VIIl)-oxyd 310 mg Additionsverbindung, aus
welcher 84 mg trana-Cyclodecandiol-(l,2) vom Smp.
48-50° erhalten werden konnten. Einmaliges Umlösen aus
Petroläther ergab 59 mg eines Produktes vom Smp. 52-
53°.
b) Aus Cyclodecanol-(l)-on-(2).
Die Mutterlauge des Hydrierungsproduktes von
Cyclodecanol-(l)-on-(2) mit Raney-Nickel, aus der die
Hauptmenge von cis-Cyclodecandiol-(l,2) durch Kristal¬
lisation abgetrennt worden war, destillierte man im
Hochvakuum, wobei neben 3,33 g Vorlauf vom Sdp.,c' » " r
j.5 mm
80-105, 14,45 g (413t d. Th.) eines farblosen Oels
vom Sdp.«,c 105-113 erhalten wurden. Diese*wur-
0,15 mm
de an 400 g Aluminiumoxyd (Akt. II bis III) chromato-
graphiert. Aus den Aether-Eluaten konnten insgesamt
10,10 g kristallines trans-Cyclodecandiol-(l,2) gewon¬
nen werden. Zur Analyse wurde aus Petroläther umkri¬
stallisiert, Smp. 54°.
3,762 mg Subst. gaben 9,600 mg C02 und 3,978 mg HgO
C10H20°2 Ber' C 69>72 * H 11t70 3*
Gef. C 69,63 % H 11,83 £
IR.-Absorptionsspektrum in Nujol Fig. 4, Kurve 10.
Diacetat.
Der auf gleiche Weise wie beim cis-Isomeren
hergestellte Ester kristallisierte aus den Benzol-
Petroläther-Eluaten des Ohromatogramms und schmolz nach
Umlösen aus Petroläther bei 37°.
3,894 mg Subst. gaben 9,368 mg G02 und 3,238 mg HgO
C14H24°4 Ber* C 65»59 ?6 H 9,44 *
Gef. C 65,65 % H 9,30 £

- 51 -
Acetonyl-acetal.
Aus 578 mg Diol erhielt man 650 mg eines farb-
losen Oels: d^u = 0,9761, n^ = 1,4740.
3,680 mg Subst. gaben 9,940 mg C02 und 3,771 mg 1^0
C13H24°2 Ber* C 75»53 * H n'59 *
Gef. C 73,71 # H 11,47 t
IE.-Absorptionsspektrum Fig 4, Kurve 12.
Herstellung von Cyclodecenen durch Dehydra-
tisierung von Cyclodecanol.
7,0 g Cyclodecanol wurden im iasserstrahlVakuum
durch ein Rohr von 10 cm Lange und 1 cm Veite, das mit
feingekorntem Aluminiumoxyd ("Alcoa", activated Alumi¬
na, Grade F-l, Mesh 8-14) gefüllt war, destilliert.
Das Rohr, welches elektrisch auf 400°± 10° erhitzt
wurde, war mit einer eisgekühlten Vorlage verbunden,
der eine zweite, auf - 50° gekühlte Vorlage folgte, die
an das Wasserstrahlvakuum angeschlossen war. Die De¬
stillation dauerte 40 Min. und lieferte ein Gemisch
von Cyclodecenen und Wasser, aus dem 5,47 g (88?4 d. Th.)
Cyclodecene vom Sdp.,2 74-76° gewonnen werden
konnten.
Zur Analyse wurde über Natrium destilliert:
d420 « 0,8712, nD20 = 1,4867.
3,470 mg Subst. gaben 11,069 mg C02 und 4,077 mg H20
G10H18 Ber* G 86»87 * H 13»13 *
Gef. C 87,05 1» H 13,15 *
Aus 60 mg des Gemisches von Stereoisomeren konn¬
ten durch Oxydation mit Qsmium(VIII)-oxyd und nach¬
folgende chromatographische Trennung an 3 g Aluminium—
oxyd (Akt. II bis III) 3o mg cis-Cyclodecandiol-(l,2)und 26 mg trans-Cyclodecandiol-(l,2) gewonnen werden.

- 52
14,15 g eines auf gleiche Weise gewonnenen Cyclo-
decen-Gemisches wurden mit einer hochwirksamen Kolonne
(Podbielniak Semi-automatic High Temperature Micro Di¬
stillation Analyzer mit Heli-Grid-Packung) destilliert.
Als Schiebeflüssigkeit wurden 4 ccm 1-Methyl-naphtalinverwendet. Bei 14,7 mm Druck und einem Rücklauf-Destil¬
latverhältnis von 100:1 wurden 14 Fraktionen zu je 1 g
aufgefangen.
Fr. 3 vom Sdp.14 ? ^ 72,2-72,7°, zeigte folgen¬
de Eigenschaften:
d420 = 0,8684, nD20 = 1,4858.
Gef. C 86,62 $> H 13,05 Jt.
101,5 mg der Fraktion 3 gaben mit 187 mg Osmium(VIII)-
oxyd 67 mg farbloses Oel, aus welchem durch einmali¬
ges Umlosen aus Petrolather 56 mg trans-Cyclodecan-
diol-(l,2) vom Smp 52-53° erhalten werden konnten.
Die Anwesenheit des hoch schmelzenden eis—Cyclodecan—
diols-(l,2) liess sich nicht nachweisen.
Fr. 11 vom Sdp.14 7 ^73,5° zeigte folgende
Eigenschaften :
d420 - 0,8738, nD20 - 1,4881.
öef. C 86,61 i> H 13,44 $•
Aus 107 mg der Fraktion 11 und 197 mg Osmium(VIII)-
oxyd Hessen sich 68 mg rohes kristallines cis-
Cyclodecandiol-(l,2) vom Smp. 125-28° isolieren,welche nach Umlosen aus Alkohol-Benzol 38 mg reines
cis-Cyclodecandiol-(l,2) vom Smp. 137-38° liefer¬
ten.
Die IR.-Absorptionsspektren zeigten, in Ueber-
einstimmung mit diesen Ergebnissen des oxydativen Ab¬
baus, dass die Fraktion 3 vorwiegend aus trans-Cyclode-cen besteht. In der Fraktion 11 war cis-Cyclodecen stark
angereichert.

- 53 -
Isomensierungsversuche mit Cvclodecenen.
Je 300 mg der reinen, stereoisomeren ungesättig¬
ten Kohlenwasserstoffe wurden im Einschlussrohr mit
25 mg p-Toluolsulfosaure 3 Std. auf 170 erhitzt. Die
durch Destillation über Natrium erhaltenen Kohlenwas¬
serstoffgemische zeigten identische IR.-Absorptions¬
spektren, welche auf ein Gemisch von viel cis-Cyclo—
decen mit wenig trans-Cyclodecen hinwiesen. Im Ein¬
klang damit erhielt man aus 70 mg des Reaktionsgemi¬
sches mit 130 mg 0smium(VIII)-oxyd 60 mg cis-Cyclodecan-
diol-(l,2) vom Smp. 137°.
cis-1.2-0xido-cylodecan (XIV).
2,0 g cis-Cyclodecen wurden mit 38 ccm einer
0,446-molaren Losung von Benzoepersaure in Chloro¬
form 15 Std. bei 0° stehengelassen. Die Aufarbeitung
ergab 1,94 g eines gelblichen Oels vom Sdp.,,
100-105°.
Um unverändertes Ausgangsmaterial zu entfernen,
chromatographierte man an 60 g Aluminiumoxyd lAkt. I).
Mit Petrolather wurden zuerst 75 mg Cyclodecen zu¬
rückerhalten, darauf liessen sich mit Petrolather-
Benzol 1:1 227 mg reines, öliges cis-lr2-0xido-cyclo-
decan eluieren, welches mit Tetranitromethan keine
Gelbfärbung gab und folgende Eigenschaften aufwies:
Sdp'll mm105°' d42° - °.9619, nD20 = 1,4847.
5,488 mg Subst. gaben 9,949 mg C02 und 3,641 mg HpO
C10H18° üer* C 77'86 * H Utl6 *
Üef. C 77,93 £ H 11,68 y.
IR-Absorptionsspektrum Fig. 3 Kurve 7.
Der Rest der Oxido-Verbindung wurde beim Chro¬
matographieren am Aluminiumoxyd hydrolytisch gespalten
und konnte erst mit Methanol eluiert werden. Man er-

- 54 -
hielt 878 mg eines hochviskosen Oels, welches nach
einmaligem Umlösen aus Petroläther bei 52-53 schmolz
und mit trans-Cyclodecandiol-(l,2) keine Schmelzpunkts-
erniedrigung gab.
trans-lf2—Qxido-cyclodecan (XV).
Auf analoge Weise wie das cis-Isomere wurde aus
1,5 g trans-Cyclodecen und 32 ccm einer 0,362-molaren
Benzoepersäure-Lösung das trans-Oxido-cyclodecan her¬
gestellt. Durch Chromatographieche Trennung an Alu¬
miniumoxyd (Akt. I) wurden 265 mg trans-Cyclodecen
zurückerhalten, worauf 575 mg reines, öliges trans-
1.2-0xido-cyclodecan folgten, welches folgende Eigen¬
schaften besass: Sdp.,,„,„,
105°, d.20 = 0,9614,
n^ - 1,4840.
3,522 mg Subst. gaben 10,056 mg C02 und 3,719 mg H20
C10H18° Ber* C 77'86 * H 11'76 *
Gef. C 77,93 i> H 11,82 $>
IR.-Absorptionsspektrum Fig. 3t Kurve 8.
Die Eluierung mit Methanol ergab 514 mg eines
viskosen Oels vom Sdp.,n,
125°, das mit Tetranitro-r 11 mm
'
methan eine deutliche Gelbfärbung zeigte. Das p-Nitro-
benzoat schmolz nach dreimaligem Umkristallisieren
aus Essigester bei 152 und gab mit dem p-Nitroben-
zoat des durch Reduktion von Cyclodecen-(l)-on-(3)
mit Lithiumaluminiumhydrid, bzw. durch Behandlung
von trans-Cyclodecen mit Perameisensäure erhaltenen
Cyolodecen-(l)-ola-(3) keine Schmelzpunktserniedri¬
gung.
Die Reaktionsgeschwindigkeiten der Oxydation von
von stereoisomeren Cyclodecenen mit Benzoepersäure.
Aequimolekulare Mengen von Cycloolefin und Benzoe¬
persäure in Chloroform wurden bei 0° vermischt. In be-

- 55 -
stimmten Zeitabstanden, die in Vorversuchen ermittelt
worden waren, nahm man aliquote Teile der Reaktions¬
losung heraus und bestimmte die überschüssige Persaure,
indem man die entnommene Probe möglichst rasch zu
1 com einer Losung von 8,3 g Kaliumjodid in 50 ccm Al¬
kohol, 40 ccm Wasser und 10 ccm Eisessig zugab. Das
ausgeschiedene Jod wurde darauf mit 0,1-n Natrium-
thiosulfat titriert. Die Reaktionsgeschwindigkeits¬
konstanten k wurden aus der Formel
, o 1 a ,—1 , —1k =
t*
aIi=IT mo1 •1-min
berechnet. Die Ergebnisse sind im theoretischen Teil
auf S.20 zusammengestellt.
Reaktionsgeschwindigkeiten der Oxydation von
stereoisomeren Cyclodecandiolen-(1.2) mit
Blei(IV)-acetat.
Die Messungen wurden nach der von R. Criegee
und Mitarbeitern angegebenen Vorschrift durchgeführt.
Als Losungsmittel wurde reiner Eisessig vom Smp. 15,4
verwendet.
Die Ergebnisse sind in Tabelle 1 auf S. 16 zusammen¬
gestellt.
Oxydation von cis-Cyclodecen mit Perameisensaure.
3,0 g cis-Cyclodecen wurden mit 40 ccm Ameisen¬
saure und 5 g 30—prozentigem Wasserstoffperoxyd kraf¬
tig geschüttelt. Nach einigen Hin. erwärmte sich das
Reaktionsgemisch von selbst zum Sieden, und der Koh¬
lenwasserstoff ging m Losung. Nach dreistündigem Er¬
hitzen auf dem Wasserbad wurde der Hauptanteil der
Ameisensaure im Vakuum abde3tilliert. Den Ruckstand

- 56 -
verdünnte man mit 100 ccm Wasser, worauf mit Petrol-
äther extrahiert wurde. Die mit Natriumhydrogencarbo-
nat-Lösung und Wasser gewaschenen Auszüge hinterliessen
beim Eindampfen 5,05 g eines gelblichen, beweglichen
Oels. Dieses wurde 1 Std. mit 1 g Kaliumhydroxyd in
50 ccm 50-prozentigem Methanol am Rückfluss verseift.
Durch Extrahieren mit Methylenchlorid Hessen sich
daraus 2,54 g eines dickflüssigen Oels erhalten, wel¬
ches an 80 g Aluminiumoxyd (Akt. II bis III) Chromate—
graphiert wurde. Es wurden dabei Fraktionen von je
80 ccm aufgefangen.
Fr. Eluierungsmittel Eluat, mg
1-5 Petroläther 242 ölig
4-8 Petroläther-Benzol
25:1 bis 9:1
10 ölig
9-11 Petroläther-Benzol 1:1 240 krist.
12-16 Benzol 586 ölig
17-22 Benzol-Aether 1:1 950 krist.
25 Aether-Aceton 1:1 178 krist.
Aus den Petroläther-Benzol-Eluaten (9-11) wurde
die Verbindung CjQHjßOg YOm Smp» 74° erhalten, die
Benzol-Aether-Eluate (17-21) lieferten die isomere Ver¬
bindung CipH-in0" Yom ^mp» ^8° und aus dem Ae'ther-A06*011-
Eluat (25) wurde schliesslich das Cvclodecandiol-(1T6)vom Smp 144° isoliert.
Ueber einen unter milderen Bedingungen durchgeführt
ten Oxydâtionsversuch siehe unter Cvclodecen-(l)-ol-(5).
Oxydation von trans-Cvclodecen
mit Perameisensäure.
5,0 g trans-Cyclodecen wurden unter denselben Be¬
dingungen wie das cis-Stereoisomere mit Perameisensäu-

- 57 -
re oxydiert. Nach analoger Aufarbeitung erhielt man
2,58 g eines teilweise kristallisierenden dickflüssi¬
gen Oels. Die chromatographische Trennung an 80 g
Aluminiumoxyd (Akt. II bis III) ergab folgende Fraktio¬
nen:
Fr. Eluierungsmittel Eluat, mg
1-2 Petrolather 46 ölig
3-5 Petrolather-Benzol 9:1 253 ölig
6-8 Benzol 392 krist.
9 Benzol 108 ölig
10-12 Benzol 331 krist.
13-16 Aether 779 krist.
17-18 Aether-Aceton 1:1 237 krist.
19 Aether-Aceton 1:1 112 ölig
Die öligen Petrolather-Benzol-Eluate (3-5) gaben
ein 2,4-Dinitrophenylhydrazon vom Smp 165,welches mit
Cyclodecanon-2.4-dinitrophenylhvdrazon keine Smp.-Ernied¬
rigung gab. Aus den ersten Benzol-Eluaten (6-8) liess
sich das trans-Pekalol-(l) vom Sap. 63 isolieren.
Die spateren Benzol-Eluate (10-12) lieferten die Ver¬
bindung C10H1002 vom Smp> 63°« Mlt Aetner (13-16) kam
die isomere Verbindung ^in^inQr; voa Smo. 98 aus der
Saule, und schliesslich gaben die Aether-Aceton-Eluate
(17-18) das Gvclodecandiol-(1.6) vom Smp. 152°.

- 58 -
Einzelne isolierte Verbindungen.
trans-Dekalol-(l) Smp 63° (XXVIIIa).
Die spateren Benzol-Eluate des OxydâtionsPro¬
duktes aus trans-Cyclodecen schmolzen nach Sublimation
im Hochvakuum bei 61-62 und gaben mit einem authen¬
tischen trans-Dekalol-(l) vom Smp. 63° keine Smp.-Er¬
niedrigung. Das I.R.-Absorptionsspektrum war mit dem¬
jenigen des Vergleichspraparates identisch.
IR.-Absorptionsspektrum Fig. 7, Kurve 10.
Das aus Methanol umkristallisierte p-Nitrobenzoat.
das bei 87° schmolz, gab ebenfalls mit einem authen¬
tischen Vergleichspraparat keine Smp.-Erniedrigung.
4,409 mg Subst. gaben 10,907 mg COg und 2,749 mg HgO
C17H21°4N Ber* C 67>51 * H 6'98 *
Gef. C 67,51 ft H 6,98 fi
Cyclodecandiol-(1.6) Smp. 144° (XXIII).
Die Aether-Aceton-Eluate des Oxydationsproduktes
aus cis-Cyclodecen gaben nach viermaligem Umlosen aus
EBSigester-Methanol farblose Nadeln vom Smp. 144°, die
mit einem Vergleichspraparat keine Smp.-Erniedrigung
zeigten.
3,770/3,860 mg Subst. gaben 9,609/9,873 mg C02und 3,921/4,040 mg HgO
C10H20°2 Ber* C 69'72 * H H»70 ^
Oef. C 69,57 ft H 11,64 ft
C 69,80 ft H 11,70 fi
Der Vergleich des IR.-Absorptionsspektrums mit
demjenigen des Vergleichspraparates zeigte, dass es
sich um das Cyclodecandiol-(1,6 ) Smp. 144 handelt.
Fig. 5, Kurve 1.

- 59 -
Das Diacetat. welches aus 12 mg Diol hergestellt
wurde, schmolz nach chromatographischer Reinigung an
Aluminiumoxyd bei 56-59 und gab mit einem Vergleichs¬
präparat vom Smp. 63 keine Smp.-Erniedrigung. Auch
hier waren die IR.-Absorptionsspektren gleich: Fig. 5,
Kurve 2.
Zur Charakterisierung des niedriger schmelzenden
Cyelodecandiols-(l,6) eignet sich sein Di-p-nitro-
benzoat: Kristalle aus Essigester-Methanol vom Smp.
176°, keine Smp.-Erniedrigung mit dem Vergleichs¬
präparat .
4,484 mg Subst. gaben 10,086 mg C02 und 2,187 mg HgO
C24H26°8N2 Ber* ° 61'27 * H 5'57 *
Gef. C 61,38 <f> H 5,46 <jL
Cyclodecandiol-(1.6) Smp. 152° (XXYII).
Die farblosen Prismen, die aus den Aether—Aceton—
Eluaten des Oxydationsproduktes von trans-Cyclodecen
durch Umkristallisieren aus Chloroform—Petroläther er¬
halten wurden, schmolzen bei 152 und gaben mit einem
Vergleichspräparat keine Smp.-Erniedrigung.
3,980 mg Subst. gaben 10,175 mg C02 und 4,187 mg HgO
G10H20°2 Ber* C 69»72 * H 11'1° *
Gef. C 69,77 # H 11,77 *
Das IR.-Absorptionsspektrum, Fig. 5, Kurve 3, war
mit demjenigen des Vergleichspräparates identisch.
Das auf die übliche Weise hergestellte Diacetat
(IR.-Absorptionsspektrum Fig. 5, Kurve 4) schmolz bei
104,5 und zeigte mit dem Vergleichspräparat ebenfalls
keine Smp.-Erniedrigung.
Verbindung C10H10°2 Smp 98°'
Die Benzol-Aether-Eluate des Oxydationsproduktes
aus cis-Cyclodecen gaben nach dreimaligem Umlösen aus

- 60 -
Benzol-Petroläther feine Nadeln vom Smp. 98°.
3,470/3,921 mg Subst. gaben 8,968/10,147
mg C02 und 3,346/3,772 mg HgO
C10H18°2 Ber* G 7°»54 * H 10'66 *
Gef. C 70,53 1> H 10,78 Jt
C 70,62 i> H 10,76 +
4,385 mg Subst. gaben 0,651 ccm CH. (25°/728 mm)
Ber. 1 "H" 0,59 > Gef. 0,59 *
IR.-Absorptionsspektrum Fig. 6, Kurve 5.
Dieselbe Verbindung liess sich auch aus den
Aether-Eluaten des Oxydationsproduktes von trans-Cyclo-
decen erhalten.
Das Mono-p-nltrobenzoat schmolz nach zweimaligem
Umkristallisieren aus Essigester-Feinsprit bei 142°.
4,371 mg Subst. gaben 10,222 mg C02 und 2,591 mg HgO
°17H21°5N Ber* G 65'93 * H 6'63 *
Gef. C 63,82 1» H 6,63 >
Mono-acetat der Verbindung C-, qhiqQ2 Smp» 98°*
160 mg der Verbindung üioH18°2 wurden mi't 5 ccm
Acetanhydrid und 10 Tropfen Pyridin während 2 Std. zum
Sieden erhitzt. Mach Abdampfen des überschüssigen Acet-
anhydrids am Vakuum chromatographierte man den Rück¬
stand an 3 g Aluminiumoxyd (Akt. II bis III). Die Elu-
ation mit Petroläther lieferte 147 mg plattige Prismen-
die nach Sublimation am Hochvakuum bei 33 schmolzen.
4,074/3,957 mg Subst. gaben 10,176/9,830
mg C02 und 3,346/3,772 mg H^O
C12H20°3 Ber* ° 67»89 * H 9,50 >•
Gef. C 68.16 >• H 9,53 >
0 67,79 v» H 9,51 >
IK.-Absorptionsspektrum Fig. 7, Kurve 8.

- 61 -
Oxydation der Verbindung CipH-ir^ ^mp. qQ°
mit Chrom(VI)-oxvd.
Zu 500 mg der Verbindung c1qH20°2 vrurden in essig¬
saurer Losung bei 0° unter Ruhren 140 mg Chrom(VI)-
oxyd in 5 com Eisessig im Verlaufe einer Stunde zuge¬
tropft. Nach Stehen über Nacht wurde der Eisessig im
Vakuum abdestilliert, der Ruckstand mit Wasser verdünnt
und dreimal mit Petrolather extrahiert. Die vereinigten
Auszuge wurden mit Natnumcarbonat-Losung und Wasser
gewaschen, mit Natriumsulfat getrocknet und hmterlies-
sen nach Abdampfen des Losungsmittels 152 mg Oel, das
in Petrolather gelost und durch eine kurze Saule von
Aluminiumoxyd (Akt. II bis III) filtriert wurde. Das
isolierte farblose Oel (123 mg) destillierte man im
Kragenkolbchen zur Analyse, Sdp.,2 120°.
3,811 mg Subst. gaben 9,988 mg G02 und 3,304 mg H20
C10H16°2 Ber' G 71,39 9è H 9,59 >
Gef. C 71,52 j. H 9,70 ft
IR.-Absorptionsspektrum Fig. 7, Kurve 9.
Das 2.4-Dinitrophenyl-hvdrazon schmolz nach drei¬
maligem Umkristallisieren aus Essigester-Methanol bei
159°.
4,530 mg Subst. gaben 9,116 mg C02 und 2,383 mg HgO3,426 mg Subst. gaben 0,500 ccm Ng (24°/729 mm)
°16H20°5N4 Ber. C 55,16 ji H 5,79 > N 16,08 *
Gef. C 54,92 # H 5,89 1> N 16,10 >
Verbindung C^H^On smP. 74°.
Die aus den Petrolather-ßenzol-Eluaten des Oxy¬
dationsproduktes von cis-Cyclodecen erhaltene Verbin¬
dung war in allen Losungsmitteln leicht loslich und
konnte zur Analyse nur durch Sublimieren im Hochvakuum
gereinigt werden.

- 62 -
3,836 mg Subst. gaben 9,848 mg G02 und 3,627 mg H20
4,150 mg Subst. gaben 0,637 can CH4 (22°/722 mm)
C10H18°2 Ber* C 7°»54 > H 10»66 >
üef. C 70,06 y H 10,58 #
1 H aktiv Ber. 0,59 u>
üef. 0,69 >
IR.—Absorptionsspektrum Fig. 6, Kurve 6.
Das Mono-p-nitrobenzoat wurde dreimal aus Methanol
umkristallisiert: Smp. 129°.
3,680 mg Subst. gaben 8,631 mg C02 und 2,218 mg HgO
C^Hg^N Ber. C 63,93 > H 6,63 1»
Gef. C 64,01 jk H 6,74 %
Oxydation der Verbindung C-iQHin02 Smp» 74°
mit Chrom(VI)-oxyd.
30 mg der Verbindung C10H18°2 Smp* ^4° wurden
mit 13 mg Chrom(VI)-oxyd auf gleiche Weise wie das
Isomere oxydiert. Das nach dem Chromatographieren
erhaltene Keton gab ein orangegelbes 2.4-Dinitro-
phenyl-hydrazon. das nach Umkristallisieren aus Essig-
e8ter-Methanol bei 154-155° schmolz und mit dem 2,4-
Dinitrophenyl-hydrazon des Oxydationsproduktes aus der
isomeren Verbindung eine Smp.-Depression von 15° zeigte.
2,926 mg Subst. gaben 0,423 com N2 (23°/723 mm)
G16H20°5N4 Ber* N l6>08 * Gef« N 15,86 f
Verbindung C10H1002 Smp. 63°.
Aus den späteren Benzol-Eluaten des Oxydations¬
produktes von trans-Cyclodecen Hessen sich durch Sub¬
limation im Hochvakuum Nadeln vom Smp. 63° erhalten,
die mit den isomeren Verbindungen ctoH18°2 starke
Smp.-Depressionen gaben.

- 63 -
3,592 mg Subst. gaben 9,250 mg COg und 3,471 mg H20
5,552 mg Subst. gaben 0,807 ccm GH. (22°/727 mm)
C10H18°2 Ber# G 70»54 * K 10'66 *
Gef. G 70,27 £ H 10,81 j>
1 H aktiv Ber. 0,59 £ Gef. 0,58 jS
IR.—Absorptionsspektrum Fig. 7, Kurve 1.
Das Mono-p-nitrobenzoat. welches nach Umkristalli¬
sieren aus Methanol bei 132° schmolz, gab mit den Mono-
p-nitrobenzoaten der Isomeren starke Smp.—Erniedrigun¬
gen.
3,650 mg Subst. gaben 8,515 mg COg und 2,114 mg HgO
C17H21°5N Ber# C 63»93 * H 6»63 *
Gef. G 63,67 1> H 6,48 fk
Cyclodecen-(l)-ol-(3) (XX).
Zur Isolierung von Cyclodecen-(l)-ol-(3) wurde das
Cyclodecen unter milderen Bedingungen oxydiert. 0,75 g
cis-Cyclodecen wurden 3 Std. mit einem Gemisch von 0,70 g
30—proz. Wasserstoffperoxyd, 20 ccm Ameisensäure und
10 ccm Methylenchlorid auf 40 erwärmt. Die Aufarbeitung
und Verseifung ergab 0,64 g eines öligen Oxydationspro¬
duktes, aus dem durch Chromatographie an 20 g Alumi—
niumoxyd (Akt. II bis III) mit Petroläther-Benzol 1:1
288 mg eines öligen Produktes erhalten wurden. Dieses
überführte man auf übliche Weise in sein p-Nitrobenzoat.
das nach dreimaligem Umlösen aus Essigester bei 152°
schmolz.
3,754 mg Subst. gaben 9,250 mg C02 und 2,323 mg H20
3,638 mg Subst. gaben 0,160 ccm N2 (27°/726 mm)
G17H21°4N Ber* G 67»51 ^ H 6'98 * N 4'62 *
Gef. C 67,24 ^ H 6,92 ji N 4,78 ^

- 64 -
Das p-Nitrobenzoat gab mit dem p-Nitrobenzoat
des durch Reduktion von Cyclodecen-(l)-on-(3) mit
Lithiumaluminiumhydrid bzw. durch Behandlung von
trans-1,2—Oxido-cyclodecan mit Aluminiumoxyd erhal¬
tenen Gyclodecen-(l)-ols-(5) keine Smp.-Erniedrigung.
Neben Cyclodecen-(l)-ol-(3) wurden aus den spä¬
teren Eluaten des Ghromatogramms die gleichen Produk¬
te erhalten wie bei energischerer Oxydation.
Die Analysen wurden im mikroanalytischen Labo¬
ratorium des Organisch-Chemischen Institutes der
Eidg. Technischen Hochschule unter der Leitung von
Herrn W. Manser ausgeführt, dem ich an dieser Stelle
meinen besten Dank ausspreche.
Ebenso danke ich Herrn Prof. Dr. Hs.H. Günthard
für die Diskussion und Interpretation und Herrn A.
Hübscher für die Aufnahme der IR.-Absorptionsspektren.
Zusammenfassung .
Ausgehend von Cyclodecanol-(l)-on—(2) wurden
verschiedene Verbindungen mit einem Zehnring her¬
gestellt, unter anderem: das Oyclodecin und die
beiden cis-trans-etereoisomeren Cyclodecene, Cyclo-
decandiole-(l,2) und 1,2-Oxido-cyclodecane.
Auf Grund ihrer Entstehungßweise und ihrer IR.-
Ab8orption8spektren wurden den hergestellten Stereo-
isomeren bestimmte Konfigurationen zugeteilt.

- 65 -
Die beiden stereoisomeren Gyclodecandiole-(l,2)lassen sich leicht in ihre Acetonyl-acetale überfüh¬
ren, und das trans-Cyclodecandiol-(l,2) reagiert viel
schneller mit Blei(IV)-acetat als das cis-lsomere.
Dieses Verhalten, welches verschieden ist von dem
Verhalten der Cyclopentan- und Cyclohexandiole-(l,2)mit analoger Konfiguration, lässt sich auf verschie¬
dene Konstellation der Ringhomologen zurückführen.
Die IR.-Absorptionsspektren der hergestellten
Zehnring-Verbindungen wurden aufgenommen und kurz
interpretiert.
Die reinen, stereoisomeren Cyclodecene wurden
mit Perameisensäure oxydiert.
Die Oxydation des cis-Cyclodecens mit Perameisen¬
säure liefert: a) Cyclodecen-(l)-ol-(3), b) Cyclode-
candiol-(lf6) vom Smp. 144,
c) und d) zwei noch nicht
völlig aufgeklärte, isomere Verbindungen ciq%q^2 vom
Smp. 98° bzw. 74°.
Die gleiche Reaktion mit trans-Cyclodecen ergab:
a) Cyclodecanon, b) Cyclodecandiol—(1,6) vom Smp. 152°,c) trans-Dekalol-(l) vom Smp. 63°, d) und e) zwei noch
nicht aufgeklärte Verbindungen C-,0H,g02 vom Smp. 98
bzw. 63°, von welchem die erste mit dem Isomeren vom
gleichen Smp. identisch ist, das sich bei der Oxydation
von cis-Cyclodecen bildet.
Ein Entstehungsmechanismus für die aufgeklärten
Oxydationsprodukte wird vorgeschlagen. Es wird dabei
angenommen, dass sich die beiden Cyclodecandiole-(l,6)
und das trans-Dekalol-(l) aus den Cyclodecenen durch
eine transanulare Reaktion bilden.

CURRICULUM VITAE
Am 16. Januar 1927 wurde ich in meiner Heimat¬
gemeinde Däniken (Kt. Solothurn) geboren. Nach dem
Besuch der dortigen Primarschule und der Bezirks¬
schule in Schönenwerd trat ich 1943 in die Oberreal-
Abteilung der Aargauischen Kantonsschule ein, wo ich
im Herbst 1946 die Maturitätsprüfung (Typus C) ab¬
legte .
Noch im gleichen Jahre immatrikulierte ich mich
an der Abteilung für Naturwissenschaften der Eidge¬
nössischen Technischen Hochschule in Zürich, wo ich
im Frühjahr 1950 die Diplomprüfung als Naturwissen¬
schafter bestand. Nach kurzer Unterrichtstätigkeit
an der Aargauischen Kantonsschule führte ich im
Organisch-Chemischen Laboratorium der E.T.H. unter
der Leitung von Herrn Prof. Dr. L. Ruzicka und Herrn
Prof. Dr. V. Prelog die vorliegende Promotionsarbeit
aus.
Zürich, im Dezember 1952 Karl Schenker


















