
hn ghosh_1
9
Interfacial Electron-Transfer Dynamics on TiO 2 and ZrO 2 Nanoparticle Surface Sensitized by New Catec hol Derivati ves of Os(II )-pol ypyrid yl Complexes : Monit oring by Charge-Transfer Emission Sandeep Verma, † Prasenjit Kar, ‡ Amitava Das,* ,‡ Dipak K. Palit, † and Hirendra N. Ghosh* ,† Radiation & Photochemistry Di Vision, Bhabh a Atomi c Resear ch Center Tromb ay, Mumb ai - 400 085, India, and Central Salt and Marine Chemicals Research Institute (CSIR), Bha Vnagar: 364002 , Gujarat , India ReceiVed: October 27, 2007; In Final Form: NoVember 30, 2007 We report synthesis of a new catechol derivative of 2,2 -bipyridyl (L 1 ) and two new Os(II)-polypyridyl complexes, I and II, with pendant catechol functionality (Scheme 1). Both complexes show two strong metal- to-ligand charge transfer (MLCT) absorption bands in the visible region that are attributed to 1 MLCT (spin allowed) and 3 MLCT (spin forbidden) transitions. We have recorded photoluminescence spectra for both complexes at room temperature and at 77 K. We have determined the lifetime for the excited triplet ( 3 MLCT) states using time-resolved emission spectroscopy. Optical absorption studies reveal that both the complexes I and II form charge transfer (CT) complex with TiO 2 and ZrO 2 (higher band gap) nanoparticles. Photoinduced electron injection takes place from the Os(II)-complexes to the conduction band of TiO 2 and surface states of ZrO 2 nanoparticles following excitation of the respective CT complexes. On recombination of these respective charge-separated complexes, CT emission has been detected in the above dye/nanoparticle systems. Monitoring the CT emission, we could determine back electron transfer (BET) rate for the charge recombination process. Introduction Sensitization of nanocrystalline TiO 2 electrodes with molec- ular chromophores forms the basis for efficient solar energy conve rsio n in regen erative photo elect roche mical cells . 1 The study of interfacial electron-transfer dynamics in these solar cells is an area of intense and active investigation. 2-9 In this context Ru(II)-polypyridyl complexes have been used extensively by various researchers as the prospective photosensitizer molecule for the development of dye-sensitized solar cell (DSSC). The fundamental requisite that makes the Ru(II)-polypyridyl com- plexes relevant for these studies is their relatively long-lived lowest lying excited state, which formally attributes as a triplet metal-to-ligand charge transfer ( 3 MLCT) state. 2-22 Among the various polypyridyl complexes, used for design of the photocell, maxi mum photo elect ron conver sion efficien cy of 10.4% is achieved to date using Ru II (dcbp) 2 (NCS) 2 (where, dcbp is 4,4 - dicarboxylic-2,2 -bipyridine). 1,2 However, this sensitizer does not absorb solar photon below 1.77 eV 23 and as a result ∼0.4 V is wasted in the reaction of Ru III (dcbp) 2 (NCS) 2 + and I - to sustain faradic current flow in the cell. 24 Os(II)-polypyridyl complexes, 25-31 owing to their lower energy absorption band in the visible region, may be more suitable for this purpose as it may offer a lower-energy absorption band and also obtain a lower M II/III oxidation potent ial 32 and thereby may of fe r improved performance in photoelectrochemical energy conver- sion . Again thir d-ro w heavy metal -cont aini ng lumi nesce nt comple xes have increa si ngly gained att ent ion due to the ir potential application in electroluminescent devices. 33 The strong spin-orbit coupling effectively promotes an intersystem crossing from singlet excited states to lower triplet emitting states, namely 3 MLCT, 3 ππ *, or a mi xt ur e of the two. 34 Mey er and co- workers 26 have demonstrated ligand-localized electron trapping on TiO 2 nanoparticle surface sensitized by osmium polypyridyl complex. In our earlier studies, we have established that deprotonated catecholate species binds strongly to TiO 2 through the formation of a five-member chelate ring. 13,16,21 This prompted us to design and synthesize two new Os(II)-polypyridyl complexes, having pendant catechol functionality with varying degree of conjuga- tion. This should have enabled us to tune the energy of excited triplet states appropriately, which lie mainly in the ππ * (or nπ *). Further, this would also offers the opportunity to study its effect on the interfacial electron-transfer dynamics between the nano- particul ate semiconductor particle and covalently bound Os(II)- comple xes. Howeve r, to unders tand the dynami cs of the photoinduced processes associated with the interfacial electron- transfer processes, it is very crucial to have a deeper insight in the phot ophy sical proper ties of the se tai lor -ma de Os( II) - polyp yrid yl compl exes and the asso ciate d elect ron-t rans fer kinetics from these photoexcited singlet and triplets states to the semiconductor nanoparticles. To address this specific issue, we have synthesized a new catechol deriv ativ e, 4-[2, 2 ]bipyridinyl-4-yl-benzene-1,2-diol (L 1 ), and have used this ligand to synthesize a new tris-bi pyridyl derivative of Os(II) (I), which has a pendant catechol function- ality to bind the semiconductor surface. Furthermore, we have used a previously reported 35 2,2 -bipyridyl (bpy) derivative, 4-[2- (4 -methyl-[2,2 ]bipyridinyl-4- yl)-vinyl]-benzene- 1,2-diol (L 2 ) for synthesis of another new but analogous Os(II)-complex ( II). Compar is on of the photop hys ica l pro per tie s of the se two complexes is expected to unravel the role of extended conjuga- tion on tuning the excited singlet ( 1 MLCT)/triplet ( 3 MLCT) state * To whom corre spon dence should be addr essed. E-mail: hngh osh@ barc.gov.in (H.N.G.). Fax: 00-91-22-255051 51. [email protected] (A.D.). † Bhabha Atomic Research Center Trombay. ‡ Centra l Salt and Marin e Chemic als Resear ch Instit ute (CSIR). 2918 J. Phys. Chem. C 2008, 112, 2918-2926 10.10 21/jp 71038 25 CCC: $40.75 © 2008 Amer ican Ch emic al Soci ety Published on Web 02/05/2008
Transcript of hn ghosh_1

8/6/2019 hn ghosh_1
http://slidepdf.com/reader/full/hn-ghosh1 1/9
Interfacial Electron-Transfer Dynamics on TiO2 and ZrO2 Nanoparticle Surface Sensitized
by New Catechol Derivatives of Os(II)-polypyridyl Complexes: Monitoring by
Charge-Transfer Emission
Sandeep Verma,† Prasenjit Kar,‡ Amitava Das,*,‡ Dipak K. Palit,† and Hirendra N. Ghosh*,†
Radiation & Photochemistry DiVision, Bhabha Atomic Research Center Trombay, Mumbai - 400 085, India,and Central Salt and Marine Chemicals Research Institute (CSIR), BhaVnagar: 364002, Gujarat, India
ReceiVed: October 27, 2007; In Final Form: NoVember 30, 2007
We report synthesis of a new catechol derivative of 2,2′-bipyridyl (L1) and two new Os(II)-polypyridylcomplexes, I and II, with pendant catechol functionality (Scheme 1). Both complexes show two strong metal-to-ligand charge transfer (MLCT) absorption bands in the visible region that are attributed to 1MLCT (spinallowed) and 3MLCT (spin forbidden) transitions. We have recorded photoluminescence spectra for bothcomplexes at room temperature and at 77 K. We have determined the lifetime for the excited triplet ( 3MLCT)states using time-resolved emission spectroscopy. Optical absorption studies reveal that both the complexesI and II form charge transfer (CT) complex with TiO2 and ZrO2 (higher band gap) nanoparticles. Photoinducedelectron injection takes place from the Os(II)-complexes to the conduction band of TiO2 and surface statesof ZrO2 nanoparticles following excitation of the respective CT complexes. On recombination of these respective
charge-separated complexes, CT emission has been detected in the above dye/nanoparticle systems. Monitoringthe CT emission, we could determine back electron transfer (BET) rate for the charge recombination process.
Introduction
Sensitization of nanocrystalline TiO2 electrodes with molec-ular chromophores forms the basis for efficient solar energyconversion in regenerative photoelectrochemical cells.1 Thestudy of interfacial electron-transfer dynamics in these solar cellsis an area of intense and active investigation.2-9 In this contextRu(II)-polypyridyl complexes have been used extensively byvarious researchers as the prospective photosensitizer moleculefor the development of dye-sensitized solar cell (DSSC). The
fundamental requisite that makes the Ru(II)-polypyridyl com-plexes relevant for these studies is their relatively long-livedlowest lying excited state, which formally attributes as a tripletmetal-to-ligand charge transfer (3MLCT) state.2-22 Among thevarious polypyridyl complexes, used for design of the photocell,maximum photoelectron conversion efficiency of 10.4% isachieved to date using RuII(dcbp)2(NCS)2 (where, dcbp is 4,4′-dicarboxylic-2,2′-bipyridine).1,2 However, this sensitizer doesnot absorb solar photon below 1.77 eV23 and as a result ∼0.4V is wasted in the reaction of RuIII(dcbp)2(NCS)2
+ and I- tosustain faradic current flow in the cell.24 Os(II)-polypyridylcomplexes,25-31 owing to their lower energy absorption bandin the visible region, may be more suitable for this purpose asit may offer a lower-energy absorption band and also obtain a
lower MII/III oxidation potential32 and thereby may offerimproved performance in photoelectrochemical energy conver-sion. Again third-row heavy metal-containing luminescentcomplexes have increasingly gained attention due to theirpotential application in electroluminescent devices.33 The strongspin-orbit coupling effectively promotes an intersystem crossingfrom singlet excited states to lower triplet emitting states, namely
3MLCT, 3ππ *, or a mixture of the two.34 Meyer and co-
workers26 have demonstrated ligand-localized electron trapping
on TiO2 nanoparticle surface sensitized by osmium polypyridyl
complex.
In our earlier studies, we have established that deprotonated
catecholate species binds strongly to TiO2 through the formation
of a five-member chelate ring.13,16,21 This prompted us to design
and synthesize two new Os(II)-polypyridyl complexes, having
pendant catechol functionality with varying degree of conjuga-
tion. This should have enabled us to tune the energy of excited
triplet states appropriately, which lie mainly in the ππ * (or nπ *).
Further, this would also offers the opportunity to study its effect
on the interfacial electron-transfer dynamics between the nano-
particulate semiconductor particle and covalently bound Os(II)-
complexes. However, to understand the dynamics of the
photoinduced processes associated with the interfacial electron-
transfer processes, it is very crucial to have a deeper insight in
the photophysical properties of these tailor-made Os(II)-
polypyridyl complexes and the associated electron-transfer
kinetics from these photoexcited singlet and triplets states to
the semiconductor nanoparticles.
To address this specific issue, we have synthesized a newcatechol derivative, 4-[2,2′]bipyridinyl-4-yl-benzene-1,2-diol
(L1), and have used this ligand to synthesize a new tris-bipyridyl
derivative of Os(II) (I), which has a pendant catechol function-
ality to bind the semiconductor surface. Furthermore, we have
used a previously reported35 2,2′-bipyridyl (bpy) derivative, 4-[2-
(4′-methyl-[2,2′]bipyridinyl-4-yl)-vinyl]-benzene-1,2-diol (L2)
for synthesis of another new but analogous Os(II)-complex (II).
Comparison of the photophysical properties of these two
complexes is expected to unravel the role of extended conjuga-
tion on tuning the excited singlet (1MLCT)/triplet (3MLCT) state
* To whom correspondence should be addressed. E-mail: [email protected] (H.N.G.). Fax: 00-91-22-25505151. [email protected] (A.D.).
† Bhabha Atomic Research Center Trombay.‡ Central Salt and Marine Chemicals Research Institute (CSIR).
2918 J. Phys. Chem. C 2008, 112, 2918-2926
10.1021/jp7103825 CCC: $40.75 © 2008 American Chemical SocietyPublished on Web 02/05/2008

8/6/2019 hn ghosh_1
http://slidepdf.com/reader/full/hn-ghosh1 2/9
energy levels of the respective complexes and thereby theassociated effect on the interfacial electron transfer. We haveprobed the dynamics of the photoinduced processes by opticalsteady state and time-resolved emission techniques. Opticalabsorption spectra of the complexes showed intense metal-to-ligand charge-transfer bands in the visible region of thespectrum. These osmium complexes displayed an additionalband at longer wavelength, which was ascribed to the directexcitation to the triplet states.36,37 We have used time-resolvedemission techniques to study interfacial electron-transfer (IET)
dynamics on the semiconductor surface. Optical absorptionstudies indicate that these complexes interact quite strongly withboth TiO2 and ZrO2 nanoparticles in the ground state. Weintended to study IET dynamics by following time-resolvedemission spectroscopy. In our earlier investigations,11,12,19 wehave observed that back electron-transfer kinetics can bedetermined by monitoring charge transfer (CT) emission. CTemission is basically recombination luminescence, which canbe observed on radiative charge recombination reaction.
2. Experimental Section
(a) Materials and Methods. Materials. Os(bpy)2Cl2, Os-(bpy)3[PF6]2,38 3-(3,4-dimethoxy-phenyl)-propanal (A)39 (Scheme
1), pyridasyl pyridinium iodide salt (B)39
(Scheme 1), and L2
21
were synthesized following reported procedure. [ tBu4N]PF6 wasused as the background electrolyte for electrochemical studiesand was recrystallized from ethanolic solution before use.Diisopropylamine, pyridine, and acetonitrile were dried anddistilled over CaH2 prior to use. All reactions were performedunder argon atmosphere, unless stated otherwise. Water usedwas doubly distilled. All other chemicals and solvents wereobtained locally and were used as such without further purifica-tion.
(b) Synthesis. Synthesis of 4-(3, 4-Dimethoxy-phenyl)-(2,
2′bipyridyl) (Me2 L1). Pyridasyl pyridinium iodide salt (A) 5.2g (0.015 mol) was dissolved in 50 mL of glacial acetic acid ina 250 mL round-bottomed flask. To this solution, ammonium
acetate 12.0 g (0.13 mol) was added, and it was stirred at 100°C. Further, 3.0 g (0.015mol) of 3-(3, 4-dimethoxy-phenyl)-propanal (B) was added to this in three intervals of 1.5 h at 100°C and was kept at that temperature with stirring for 16 h. Then,the reaction mixture was allowed to cool to room temperature,and acetic acid was evaporated completely under vacuum. Tothis, about 50 mL of water was added. pH of this solution wasadjusted to∼8.0, and then the desired compound was extractedwith chloroform. The chloroform layer was dried over anhydrousMgSO4 and evaporated to get the Me2L1 in crude form. Thiswas purified by gravity chromatography using silica as stationaryphase and methanol-chloroform (5:95; v/v) as eluent. Yield:0.80 g (18%). ES-MS (M+): 293 (100%). 1H NMR (CD3OD,ppm): δ 8.72-8.62 (m, 3H, Hpyridyl), 8.44 (d, J ) 8.0 Hz, 1H,
Hpyridyl), 7.84 (t, J ) 8 Hz, 1H, Hpyridyl), 7.50 (d, J ) 8 Hz, 1H,Hpyridyl), 7.29-7.190 (m, 3H, 1Hpyridyl, 2H phenyl), 6.91(d, J ) 8Hz, 1H, Hphenyl), 3.99(S, 3H4-methoxy), 3.946(S, 3H3-methoxy). IR(KBr pellet, cm-1): 1604 (CdC, CdN).
Synthesis of 4-[2,2′]Bipyridyl-4-yl-benzene-1,2-diol (L1). 4-(3,4-Dimethoxy-phenyl)-[2,2′]bipyridyl (0.6 g, 2.05 mmol) wasadded to molten pyridinum chloride (made from 16 mL of drypyridine and 17.5 mL of concentrated HCl at 190 °C) at 160°C ,and then the temperature was raised and maintained at 190
°C for 3 h under nitrogen atmosphere. After cooling it to roomtemperature, the resultant solid mass was dissolved in waterand the pH of the solution was adjusted to approximately 2.0.Undesired organic impurities were removed by extraction withchloroform, and then the pH of this aqueous solution wasadjusted between 6-7 with aqueous KOH. Then, the desiredproduct was extracted into the chloroform layer by solventextraction and was dried over anhydrous MgSO4. Chloroformwas removed under vacuum to isolate the crude product andwas further purified by recrystallization from hot ethanol.Yield: 0.3 g (58%). ES-MS (M+): 265 (100%). 1H NMR (CD3-OD, ppm): δ 8.65-8.46 (m, 3H, Hpyridyl), 8.30 (d, J ) 8 Hz,1H, Hpyridyl), 7.95(t, 1H, Hpyridyl), 7.60 (d, J ) 8 Hz, 1H, Hpyridyl),7.46-7.19 (m, 3H, H
pyridyl, H
phenyl), 6.91(d, 1H, J ) 8.2, H
phenyl).
IR (KBr pellet,cm-1): 1604 (CdC, CdN).
Complex I. [Os(bpy)2Cl2]‚2H2O (0.070 g, 0.114 mmol) andL1 (0.030 g, 0.114 mmol) dissolved in 50 mL of ethanol-watermixture (1:1, v/v) was allowed to reflux for 16 h with constantstirring under inert atmosphere. Then ethanol was removedunder vacuum, and the desired crude complex was precipitatedas a green solid by adding excess of aqueous KPF 6 solution.This was filtered off, washed with cold water, and air-dried.This crude product was further purified by gravity chromatog-raphy using silica as stationary phase and CH3CN-saturatedaqueous KPF6 solution (98:2, v/v) as eluent. Then, acetonitrilewas removed under vacuum, and the desired pure complex wasextracted in dichloromethane layer by solvent extraction.
Dichloromethane was removed under reduced pressure to isolatethe pure compound. Yield: 0.062 g (52%). Elemental analy-sis: calculated, C 40.90, H 2.67, N 7.95; experimental, C 40.72,H 2.58, N 7.65. ES-MS (M+-PF6): 911(85%), (M+-2PF6) 766(60%). 1H NMR (CD3CN, ppm): δ 8.63 (2H, d, J ) 8.4 HzH6,6′ (L1)); 8.48 (4H, d, J ) 8.2 Hz, 2H6,6′ (bpy)); 7.86 (4H, t,
J ) 7.8 Hz, 2H4,4′ (bpy)); 7.72-7.48 (7H, m, 2H5,5′ (bpy), H3,3′,4′
(L1); 7.37-7.27 (8H, m, 2H3,3′ (bpy), 2H5,5′ (L1), H5,6 (phenyl));6.99 (1H, d, J ) 8.2 Hz, H3 (phenyl). IR (KBr pellet, cm-1):3450 (-OH), 1603 (CdC, CdN), 847 (PF6). E 1/2 (V versus Fc/ Fc+, ∆ E (mV)): OsII/III 1.17 V, (105 mV); L1 /L1
•- -1.24 (80mV), bpy/bpy•- -1.48 (100 mV), -1.73 V (110 mV).
Complex II. This was synthesized following the procedure
mentioned above, except L2 was used instead of L1 for thereaction with [Os(bpy)2Cl2]‚2H2O. Os(bpy)2Cl2‚2H2O (0.156 g,0.30 mmol) and 4-[2-(4′-methyl-[2,2′]-bipyridyl-4-yl)-vinyl]-benzene-1,2-diol (L2) (0.110 g, 0.36 mmol) were used for thereaction. Similar workup procedure was adopted for achievingthe complex II in pure form. Yield: 0.19 g (63%). Elementalanalysis: calculated, C 42.70, H 2.91, N 7.66; experimental, C42.98, H 2.78, N 7.82. ES-MS (M+-PF6): 951 (15%), (M+-
2PF6) 806 (5%). 1H NMR (CD3CN, ppm): δ 8.5 (6H, m, 4H6,6′
(bpy), 2H6,6′ (L2)); 8.07 (4H, t, J ) 7.6 Hz, 4H4,4′ (bpy)); 8.01(1H, s, H3 (L2)); 7.82 (1H, d, J ) 5.8 Hz, H3′ (L2)); 7.76-7.56(5H, m, 4H5,5′ (bpy), 1H (ethenyl)); 7.43-7.35 (6H, m, 4H3,3′
(bpy), 2H5,5′ (L2)), 7.23 (1H, d, 8.2 Hz, H5 (phenyl)); 7.03 (1H,d, J ) 8.2 Hz, H6 (phenyl)); 7.13 (1H, d, J ) 16.6 Hz, (ethenyl));
SCHEME 1: Molecular Structure of (bpy)2OsL1 (I) and(bpy)2OsL2 (II)
IET Dynamics on TiO2 and ZrO2 Nanoparticle Surface J. Phys. Chem. C, Vol. 112, No. 8, 2008 2919
8/6/2019 hn ghosh_1
http://slidepdf.com/reader/full/hn-ghosh1 3/9
6.86 (1H, d, J ) 8.2 Hz, H3 (phenyl)); 2.56 (3H, s, -CH3). IR(KBr pellet, cm-1): 3450 (-OH), 1603 (CdC, CdN), 847(PF6). E 1/2 (V vs Fc/Fc+, ∆ E (mV)): OsII/III 1.14 V, (100 mV);L2 /L2
•- -1.21 (85 mV), bpy/bpy•- -1.46 (100 mV), -1.71 V(120 mV).
(c) Synthesis of Nanoparticles. Titanium(IV) isopropoxide(99.9%), zirconium(IV) isopropoxide isopropanol complex Zr-[OCH(CH3)2]4‚(CH3)2CHOH (Aldrich, 99.9%) was used withoutfurther purification. Isopropyl alcohol (Aldrich) was purifiedby distillation. Nanopure water was used for making aqueoussolutions. Nanometer-sized TiO2 and ZrO2 particles wereprepared by controlled hydrolysis of titanium(IV) isopropoxideand zirconium(IV) isopropoxide isopropanol complex, and ithas been described in detail in our earlier work. 11,17
(d) Picosecond Time-Resolved Fluorimeter. Time-resolvedfluorescence measurements were carried out using a diode laserbased spectrofluorometer from IBH (U.K.). The instrumentworks on the principle of time-correlated single-photon counting(TCSPC).40 In the present work, 455 nm (<100 ps, 1 MHz),453 and 589 nm LEDs were used as the excitation light sources,and a TBX4 detection module (IBH) coupled with a specialHamamatsu PMT was used for fluorescence detection.
(e) Cyclic Voltammetry. Electrochemical experiments withOs(II)-complexes (I and II) were carried out with a CH-660A(USA) electrochemical instrument; a conventional three-electrode cell assembly was used. Acetonitrile, dried and distilledprior to the experiment, was used as solvent for electrochemicalstudies. A saturated Ag/AgCl electrode was used as reference,and platinum was used as the working electrode for allmeasurements. Ferrocene was added at the end of eachexperiment as the internal standard, and all potentials are quotedversus the ferrocene/ferrocenium (Fc/Fc+) couple.
3. Results and Discussions
(a) Synthesis. Pyridasyl pyridinium iodide salt (A) and 3-(3,4-
dimethoxy-phenyl)-propanal (B) were allowed to react for thesynthesis of Me2L1 following the basic synthetic methodologyfor the pyridine derivative synthesis (Scheme 2). Then thedesired ligand L1 was obtained by reacting Me2L1 with moltenpyridinium hydrochloride salt at 190 °C. This and L2 were usedfor the reaction with Os(bpy)2Cl2 for the synthesis of the desiredcomplexes I and II, respectively.
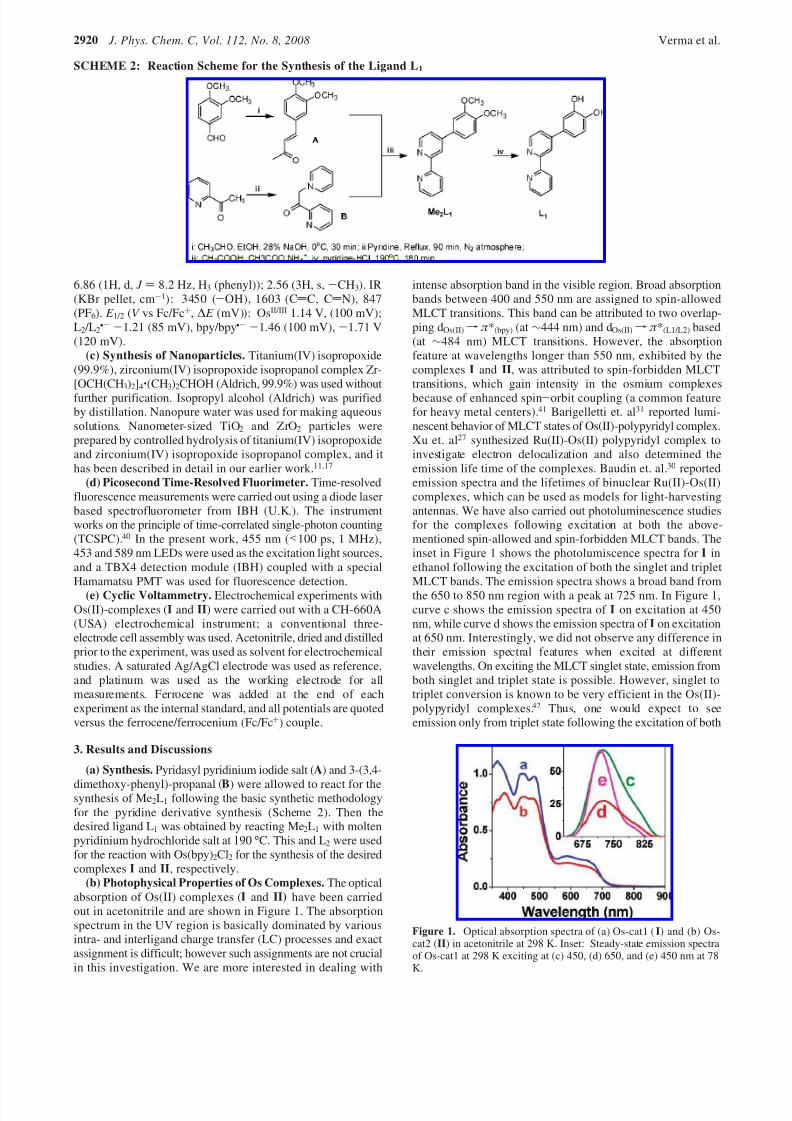
(b) Photophysical Properties of Os Complexes. The opticalabsorption of Os(II) complexes (I and II) have been carriedout in acetonitrile and are shown in Figure 1. The absorptionspectrum in the UV region is basically dominated by variousintra- and interligand charge transfer (LC) processes and exactassignment is difficult; however such assignments are not crucialin this investigation. We are more interested in dealing with
intense absorption band in the visible region. Broad absorptionbands between 400 and 550 nm are assigned to spin-allowedMLCT transitions. This band can be attributed to two overlap-ping dOs(II) f π *(bpy) (at ∼444 nm) and dOs(II) f π *(L1/L2) based(at ∼484 nm) MLCT transitions. However, the absorptionfeature at wavelengths longer than 550 nm, exhibited by thecomplexes I and II, was attributed to spin-forbidden MLCTtransitions, which gain intensity in the osmium complexesbecause of enhanced spin-orbit coupling (a common featurefor heavy metal centers).41 Barigelletti et. al31 reported lumi-nescent behavior of MLCT states of Os(II)-polypyridyl complex.Xu et. al27 synthesized Ru(II)-Os(II) polypyridyl complex toinvestigate electron delocalization and also determined theemission life time of the complexes. Baudin et. al.30 reportedemission spectra and the lifetimes of binuclear Ru(II)-Os(II)complexes, which can be used as models for light-harvestingantennas. We have also carried out photoluminescence studiesfor the complexes following excitation at both the above-mentioned spin-allowed and spin-forbidden MLCT bands. Theinset in Figure 1 shows the photolumiscence spectra for I inethanol following the excitation of both the singlet and tripletMLCT bands. The emission spectra shows a broad band fromthe 650 to 850 nm region with a peak at 725 nm. In Figure 1,curve c shows the emission spectra of I on excitation at 450nm, while curve d shows the emission spectra of I on excitationat 650 nm. Interestingly, we did not observe any difference intheir emission spectral features when excited at differentwavelengths. On exciting the MLCT singlet state, emission fromboth singlet and triplet state is possible. However, singlet totriplet conversion is known to be very efficient in the Os(II)-polypyridyl complexes.42 Thus, one would expect to seeemission only from triplet state following the excitation of both
SCHEME 2: Reaction Scheme for the Synthesis of the Ligand L1
Figure 1. Optical absorption spectra of (a) Os-cat1 (I) and (b) Os-cat2 (II) in acetonitrile at 298 K. Inset: Steady-state emission spectraof Os-cat1 at 298 K exciting at (c) 450, (d) 650, and (e) 450 nm at 78K.
2920 J. Phys. Chem. C, Vol. 112, No. 8, 2008 Verma et al.

8/6/2019 hn ghosh_1
http://slidepdf.com/reader/full/hn-ghosh1 4/9
the MLCT states. To reconfirm the nature of the emissionproperties of the complex, we have carried out time-resolvedemission measurements by exciting at two different MLCTbands, 453 and 589 nm. We have monitored the emission decaytrace at 670 nm on exciting at 453 nm, which primarily excitedthe MLCT singlet state of Os(II) complex in acetonitrilesolution. The emission decay trace could be fitted biexponen-tially with time constants 9.63 ns (27%) and 36.5 ns (73%)(Table 1). However, when we excited the complex at 589 nm,the emission decay trace monitored at 750 nm could be fittedbiexponentially with time constants of 9.2 ns (4%) and 33.0 ns(96%) (Table 1). It was interesting to observe that on excitationat both bands we could get two emission lifetimes. Earlierreports revealed that OsII(bpy)3
2+ complexes generally have very
low quantum yield and a short emission lifetime with a singlecomponent (of the order of 50 ns).43 As the intersystem crossingrate is known to be very fast (<100 fs), we could not attributethe shorter component to the singlet state. To clarify thisconjecture, we have synthesized an [Os(bpy)3]2+ and measuredthe emission lifetime. Experimental data for [Os(bpy)3]2+
revealed that the emission decay trace can be fitted with a singlecomponent with a lifetime of 37.5 ns.44 This led us to presumethat both 9.36 and 36.5 ns components are due to MLCT tripletstate emission. In the present investigation, we have monitoredthe emission lifetime of the two catecholate derivatives (Scheme1), where two dissimilar bipyridyl ligands, bpy and L1 /L2, arecoordinated to the Os(II) center in complex I and II, respec-tively. Thus, for these complexes two different triplet states
could exist in the excited state: one involving bpy ligands andthe other involving the catecholate derivative. For complex I,two lifetimes (9.36 and 36.5 ns) could be attributed to the tripletstate of the osmium complex involving bpy-catecholate ligandand bpy ligands, respectively. Here we could attribute the shortercomponent (9.36 ns) to the triplet state of bpy-catecholateligand. Nonradiative channels will be more active in thecatecholate ligand involving catecholate functionality; as a resultthe excited-state lifetime will be shorter as compared to thenoncatecholate ligand. On the other hand 36.5 ns componentcould be attributed to the triplet state of bpy-ligand in [Os-(bpy)3]2+. Similarly, in complex II we have observed that onexciting at different wavelengths, two lifetime components couldbe detected from the emission decay traces (Table 1). The
shorter component (7.2-
7.4 ns) could be attributed to the MLCTtriplet state involving L2, while the longer component (33-35ns) could be attributed to the MLCT triplet state involving bpy.
(c) Dye Binding with Nanoparticles. The basic aim of thisinvestigation was to study interfacial electron transfer onsemiconductor surface, so it was very important to study bindingproperties between the dye and nanoparticulate semiconductorsystems. The nature of the binding between a sensitizer and asemiconductor is known to influence the excited-state propertiesand interfacial ET behavior.24 A strong binding serves to anchorthe sensitizer in place and control interfacial electronic coupling.This is also known to influence the redox potentials of thesensitizers and the semiconductor nanoparticles. In our earlierstudies,21 we had observed that in Ru(II) complex, analogous
to II, pendant catecholate moiety offered strong binding withTiO2 nanoparticles. To understand the same, optical absorptionmeasurements of Os-catecholate complexes in water and inthe presence of TiO2 colloidal solutions were carried out. Figure2 reveals that the optical absorption spectrum of free I in waterand I adsorbed on TiO2 nanoparticles at different concentrationat pH 2.5. The observed spectral feature of I in water was verysimilar to that in acetonitrile (Figure 1). However, on additionof TiO2 nanoparticles, absorption spectrum of I became broadand shifted to longer wavelength with an appreciable increasein absorbance for both (Osdπ f bpyπ * and Osdπ f L1π *) theMLCT bands. Red shift and broadening of the visible band at465 nm of I in the presence of TiO2 could be attributed to thestrong interaction between the sensitizer and nanoparticle. We
had discussed in our previous reports that the interaction betweenTiO2 nanoparticles and dye molecules such as alizarin,16
triphenyl methane (TPM)-based dye13 (pyrogallol red andbromo-pyrogallol red), and ruthenium catechol21 was verystrong. Interestingly, all the above dyes had catechol moiety asthe pendant functionality, which could interact strongly withTiO2 nanoparticles and form a strong complex with catecholatederivatives. Such a strong complex formation was also proposedby Batista and co-workers through a molecular modelingstudies.45 A similar observation was reported by Moser et. al.46
and Rajh et. al.47 and concluded that the sensitizer moleculeswith pendant catechol functionality formed a CT complex withTiO2 nanoparticles. Further, no such spectral shift was observedwhen [Os(bpy)3]2+ was added to the nanoparticulate dispersion
TABLE 1: Emission Life Times of Os-cat1 (I) and Os-cat2 (II) in Different Media Exciting the Singlet (1MLCT) States at 453nm and Monitoring at 670 nm and Exciting the Triplet ( 3MLCT) States at 589 nm and Monitoring at 750 nm
ZrO2 TiO2
acetonitrile water life time KBET (sec-1) life time KBET (sec-1)
λex ) 453 nm, λem ) 670 nmOs-Cat1 (I)
(10 µM)9.63 ns (27%) 2.2 ns (58%) 0.33 ns (78%) 3 × 109 0.16 ns (99.7%) 6.25 × 109
36.5 ns (73%) 20.5 ns (42%) 9.8 ns (22%) 9.8 ns (0.3%)( χ2 ) 1.18) ( χ2 ) 0.99) ( χ2 ) 1.04) ( χ2 ) 1.11)
Os-
Cat2 (II)(10 µM) 7.45 ns (65%) 2.1 ns (81%) 0.36 ns (84%) 2.7×
10
9
0.18 ns (99.6%) 5.5×
10
9
35.8 ns (35%) 18.7 ns (19%) 7.43 ns (16%) 7.42 ns (0.4%)( χ2 ) 0.97) ( χ2 ) 1.06) ( χ2 ) 1.2) ( χ2 ) 1.2)
λex ) 589 nm, λem ) 750 nmOs-Cat1 (I)
(10 µM)9.2 ns (4%) 2 ns (7%) 0.66 ns (72%) 1.5 × 109 0.16 (65%) 6.25 × 109
33 ns (96%) 21 ns (93%) 19 ns (28%) 0.59 ns (34%) 1.7 × 109
( χ2 ) 1.0) ( χ2 ) 1.03) ( χ2 ) 1.1) >10 ns (1%)( χ2 ) 1.05)
Os-Cat2 (II)(10 µM)
7.2 ns (13%) 1.5 ns (37%) 0.62 ns (87%) 1.6 × 109 0.18 (63%) 5.5× 109
33 ns (87%) 21 ns (63%) 16 ns (13%) 0.6 ns (35.5%) 1.66 × 109
( χ2 ) 1.05) ( χ2 ) 1.15) ( χ2 ) 1.1) >10 ns (1.5%)( χ2 ) 1.1)
IET Dynamics on TiO2 and ZrO2 Nanoparticle Surface J. Phys. Chem. C, Vol. 112, No. 8, 2008 2921

8/6/2019 hn ghosh_1
http://slidepdf.com/reader/full/hn-ghosh1 5/9
of TiO2. To show complex formation more convincingly, wehave shown the optical absorption spectra of I in the presenceof TiO2 particles with increasing concentration (Figure 2). Theformation of a CT complex can be explained by the followingequation:
In the inset of Figure 2, we have shown the Benesi-Hilderbandt(B-H) plot to determine the molar extinction coefficient of theTiO2-I complex, and it is found to be 1.27 × 104 cm-1 M-1.We have also determined the equilibrium constants (at equi-librium condition) for the I /TiO2 system from the B-H plot,and they were found to be 2.5 × 105 M-1.
Thus, based on our observation in the optical spectra we couldconclude that I forms CT complex with TiO2 nanoparticles. Wehave also carried out absorption spectral measurements of I,adsorbed on ZrO2 nanoparticle surface. The use of ZrO2 wouldenable us to study photophysical processes on higher band gapsurface. As the band gap of ZrO2 nanoparticles (∼5.2 eV) andconduction band-edge potential are higher, the photoexcitedcomplex I could not inject an electron into the conduction bandof ZrO2 nanoparticles. Optical absorbance measurements of I
in the presence of ZrO2 nanoparticles were carried out, and thespectra are shown in Figure 3. We have observed that the opticalabsorption of I on ZrO2 was found to increase along with theappearance of extended absorption in 500-600 nm region andin near IR region (700-850 nm). It is evident from the optical
absorbance spectra of I /ZrO2 system that the interaction betweenthe dye and the nanoparticles is not as strong as it was for I /TiO2
system. However, for I /ZrO2 system a weaker charge transferinteraction also exists. Because of the higher band gap energyof ZrO2 nanoparticles, charge transfer interaction can take placethrough surface states of ZrO2 nanoparticles. Similar chargetransfer interaction between ZrO2 nanoparticles and sensitizermolecules has been observed by us17,19,20 in quinizarin/ZrO2
system and also by Rajh et. al.38 in enediol/ZrO2 and alizarin/ ZrO2 system. We have observed that complex formationbetween quinizarin and ZrO2 nanoparticles facilitate electroninjection into the ZrO2 nanoparticles. Similarly, in the presentinvestigation also we can expect electron injection from I intothe surface states ZrO2 nanoparticles.
(d) Steady-State Emission of I on TiO2 and ZrO2 Nano-
particles Surface. In our earlier investigation,11,12,19,20 we hadobserved that emission spectroscopy was a useful technique tostudy the interfacial electron-transfer process in dye/semicon-ductor systems. We have carried out emission spectroscopicmeasurements in the present investigation. Figure 4 shows theemission spectrum of I in water, on TiO2, and on ZrO2
nanoparticle surfaces following excitation at 450 nm. At thispoint, we would like to mention that optical density of allsolutions and the excitation intensity were kept the same to study
the relative emission intensity in different mediums. It wasinteresting to see that emission intensity decreased drasticallyon the TiO2 nanoparticle surface. This decrease in emissionintensity could be attributed to electron injection into theconduction band of TiO2 nanoparticles. In our earlier investiga-tions, we had also observed that emission quantum yield of thesensitizer molecules decreased dramatically on electron injectionto the TiO2 nanoparticle surface. In Scheme 3, we have shownthe energy level diagram of the photoexcited singlet and tripletMLCT states. It is clearly evident that the energy levels of thephotoexcited states are above the conduction band edge of TiO2
nanoparticles. As a result electron injecting into the conductionband of TiO2 nanoparticles from photoexcited state of I waspossible. A gradual decrease in emission intensity was observed
Figure 2. Optical absorption spectra of Os-cat1 (I) in the presence of various TiO2 concentration. Conditions: 25 µM Os-cat1 (I) in 1 cmoptical path length. TiO2 concentrations are (1) 0.0, (2) 0.29, (3) 0.43,(4) 0.59, (5) 0.79, (6) 1.04, (7) 1.5, (8) 2.0, (9) 2.8, (10) 3.7, and (11)5.0 g/L. (12) Normalized spectra of I (0 g/L) is shown and (13) isoptical spectra of TiO2 particles (5 g/L). Inset: Benesi-Hildebrandt plotof the Os-cat1 (I)/TiO2 complex monitored at 530 nm.
TiO2 + I f [TiO2δ-‚‚‚Iδ
+]Complex (1)
Figure 3. Optical absorption spectra of Os-cat1 (I) (a) in water, (b)on ZrO2 nanoparticles (20 g/L), (c) normalized spectra of curve a and(d) ZrO2 nanoparticles (20 g/L). Concentration of Os-cat1 is 50 µM.
Figure 4. Photoluminescence spectra of Os-cat1 (I) (a) in water, (b)on TiO2 nanoparticles (20 g/L), and (c) on ZrO2 nanoparticle (20 g/L)surface. λex is 450 nm and optical density at excitation wavelength is
kept at 0.3 for all the samples. We have also kept the excitation intensitythe same for all three measurements. Inset: Excitation spectra Os-cat1(I) (d) in water ( λem ) 730 nm) and (e) on TiO2 nanoparticles (20 g/L)surface ( λem ) 844 nm).
2922 J. Phys. Chem. C, Vol. 112, No. 8, 2008 Verma et al.

8/6/2019 hn ghosh_1
http://slidepdf.com/reader/full/hn-ghosh1 6/9
with an increase in concentration of the nanoparticulate TiO2.At higher TiO2 nanoparticle concentration (20 g/L), an ap-preciable decrease in the original emission intensity wasobserved due to the electron injection into the nanoparticles;however, a new emission band appears in the red region of thespectra (Figure 4). A similar red-shifted emission band was
observed in coumarin 343/TiO211, xanthene/TiO212, and qui-nizarin(Qz)/ZrO2
19,20 system, which have been attributed to CTemission. On photoexcitation electron injection takes place sothe original emission gets quenched; however, when the injectedelectrons recombine with the parent cation it can emit recom-bination luminescence. We have attributed this recombinationluminescence to CT emission. This process can be explainedby the following equation:
We have also carried out emission spectroscopic measure-ments of I on ZrO2 nanoparticle surface to study the photo-physics of the sensitizer on noninjecting surface. Curve c of Figure 4 shows the emission spectrum of I on ZrO2 nanoparticlesurface. Unlike in I /TiO2 system, where the original emissionintensity of I was found to be appreciably quenched in thepresence of higher concentration of nanoparticulate TiO2 (20g/L), a relatively smaller decrease in emission intensity of I
was observed with nanoparticulate ZrO2. This observation madeus to think about the possibility of electron injection from thephotoexcited molecule I to the ZrO2 nanoparticles. However,Scheme 3 clearly reveals that energy levels of the photoexcitedI lie below the conduction band of ZrO2 nanoparticles. However,
in our earlier investigation17,19,20 we had observed that electroninjection from photoexcited quinizarin molecules into the surfacestates of ZrO2 nanoparticles was possible. This phenomenon ispossible if the sensitizer molecules are strongly coupled withthe ZrO2 nanoparticles and can inject an electron into thenanoparticles. Our optical absorption spectral measurements,discussed in the earlier section, reveal that the sensitizermolecules (I) were strongly coupled with the ZrO2 nanoparticles.The emission spectrum of I on the ZrO2 nanoparticle surface
has a shoulder in the red-shifted side of the spectra that can beattributed due to the CT emission band. As in the earlier Qz/ ZrO2 system, we have observed that the decrease of emissionintensity of I is less as compared to on TiO2 nanoparticle surface,which indicates that not all photoexcited states of I on ZrO2
can inject an electron. However, the appearance of the red-shifted band clearly indicates the presence of recombinationluminescence (CT emission) in I /ZrO2 system.
(e) Time-Resolved Emission Studies in Dye/Semiconductor
System. Time-resolved emission technique can also be goodtool for monitoring electron-transfer dynamics. Electron injectiontime was monitored by Gratzel et. al48 with the help of ultrafastfluorescence up-conversion techniques in coumarin-343/TiO2
system. However, not many reports are available where the time-
resolved emission technique (ET) has been used for determiningback ET dynamics, except for some results reported by us wherewe had determined dynamics of the BET by monitoring CTemission in dye/semiconductor nanoparticles systems. In stronglycoupled systems, the dye-semiconductor nanoparticle forms theCT complex. On excitation of these CT systems, emission of the pure dye molecule decreases drastically due to electroninjection into the nanoparticles. CT emission appears due torecombination of the injected electron and the parent cation.Thus, by monitoring this recombination luminescence (CTemission) lifetime, one can determine the charge recombination(BET) time. In our earlier studies, we reported the CT emissionfrom coumarin 343 (C-343)11 and xanthene12 dye-sensitizedTiO2 nanoparticles and quinizarin sensitized ZrO2 nanopar-ticles.19,20 Later, Hupp and co-workers49 reconfirmed the CTemission in C-343 sensitized TiO2 nanoparticles using stark emission spectroscopy. In our earlier investigations ,we revealedthat it was possible to observe CT emission only when dyemolecules form CT complex with semiconductor nanoparticles.Now, monitoring the lifetime of this recombination lumines-cence one can determine BET dynamics.
In the present investigation, we also observed that I formsCT complex with both TiO2 and ZrO2 nanoparticles. Therefore,we could expect when exciting these CT complexes that onecould achieve electron injection into the nanoparticles. To findout the CT emission lifetimes, we have carried out time-resolvedemission measurements of I and II sensitized TiO2 and ZrO2
nanoparticles. Figure 5 shows the time-resolved emissionmeasurements of I in acetonitrile, water, and on TiO2 and ZrO2
nanoparticle surfaces after excitation at 453 nm laser light. It isinteresting to see that the emission decay trace of I in acetonitrilecould be fitted biexponentially with time constants 9.63 ns (27%)and 36.5 ns (73%), which has been attributed to the triplet stateof the osmium complex involving bpy-catecholate ligand andbpy ligands of I, respectively. As we have carried out sensitiza-tion experiments of I in aqueous solution, we have alsomonitored the emission decay kinetics in water. Figure 5b showsthe kinetics decay trace of I in water. It could be fittedbiexponentially with two time constants 2.2 ns (58%) 20.5 ns(42%), which could again be attributed to the triplet state of the osmium complex involving bpy-catecholate ligand and bpy
SCHEME 3: Mechanistic Scheme Showing Three-LevelModel That Consists of the Ground (So) State, theExcited Triplet (3MLCT) State, and the Excited Singlet(1MLCT) State of Os-cat1 (I) Adsorbed on TiO2 andZrO2 Nanoparticles a
a Excitation of 453 nm inject electrons into the conduction band of TiO2 and shallow surface states of ZrO2 nanoparticles. Excitation of 589 nm inject electron into the conduction band of TiO 2 and deepersurface states of ZrO2 nanoparticles.
[TiO2 - Dye]complex98hν
Electron injection (k ′inj)
[e-
CB(TiO2) + Dye+
]V Recombination
[TiO2 - Dye]complex + hνCT(CT emission)
(2)
IET Dynamics on TiO2 and ZrO2 Nanoparticle Surface J. Phys. Chem. C, Vol. 112, No. 8, 2008 2923

8/6/2019 hn ghosh_1
http://slidepdf.com/reader/full/hn-ghosh1 7/9
ligands of I. It was interesting to see that both the triplet MLCTstate lifetimes were lower in water as compared to those inacetonitrile. This could well be explained on the basis of theformation of the associated H-bonded complex through inter-molecular H-bonding that catecholate functionality of I could
form with the surrounding water molecules. Because of theH-bonding interaction, these photoexcited catecholate moleculesbecame de-excited through nonradiative channel, and conse-quently these photoexcited molecules became short-lived andshowed shorter lifetimes in water as compared to that inacetonitrile. At this point, we would like to discuss the emissionlifetimes observed for the osmium(II) polypyridyl complexeson nanoparticle surfaces at lower pH. As two different emittivespecies were observed, it was very important for us to studywhether the photophysical properties of the complexes changewith a change in the acidity of the solution. Changes in thephotophysical properties with different pH for certain Ru(II)-polypyridyl complexes were reported earlier by Wrighton andco-workers.50 They observed that both optical absorption and
emission bands changed with pH along with the emissionlifetimes. In the present investigation, we have also monitoredoptical properties of the osmium complexes at different pH of the solution. It was clearly seen from our measurements44 thatthe optical spectra of the osmium complex did not change withpH. We have also monitored the steady state and time-resolvedemission of the complexes and did not observe any change withpH; these results are presented in Supporting Information. Figure5c,d shows the emission decay trace of I on the ZrO2
nanoparticle and TiO2 nanoparticle surface. Although theemission quantum yield on both the nanoparticle surfaces aremuch lower as compared to that in neat solvents, we still coulddo the time-resolved emission measurements. The emissiondecay trace on the TiO2 nanoparticle surface could be fitted
biexponentially with time constants 0.16 ns (99.7%) and 9.8 ns(0.3%), while the decay trace on the ZrO 2 nanoparticle surfacecould be fitted biexponentially with time constants 0.33 ns (78%)and 9.8 ns (22%). It is interesting to see that on both nanoparticlesurfaces, the shorter components in the emission decay tracecontributes the maximum. The longer components on both thenanoparticle surfaces could be attributed to triplet MLCT statelifetime on the nanoparticle surface, which does not injectelectron into the nanoparticles. One might attribute the shortercomponent to the emission quenching kinetics due to electroninjection to the semiconductor surfaces. However, our recentultrafast transient absorption studies confirmed that electroninjection in the present systems is single exponential and pulse-width limited (<50 fs). So the shorter time constant cannot be
attributed also to the electron injection process. The shortercomponents observed for both nanoparticulate surfaces wereattributed to CT emission. It is interesting to see that the tripletMLCT lifetime in aqueous colloidal solution of nanoparticlesis higher (9.8 ns) than that in water (2.2 ns). As we weremeasuring the emission decay trace of I on TiO2 and ZrO2
nanoparticles dispersed in water, one could expect the MLCTtriplet state emission lifetime on the nanoparticle surface, whichwere not injecting electron, will be ∼ 2.2 ns. However, we have
observed that the lifetime is much higher (9.8 ns), which wassimilar to the lifetime in acetonitrile. This could be explainedif one considers that I was bound to the nanoparticle surfacethrough catecholate moiety and at that condition most of thephotoexcited states could inject electron. However some of theI do not inject electron; at this condition, the catecholatefunctionality is not available for formation of H-bond withsurrounding water molecule. These noninjected molecules wereattached to the surface of nanoparticles and prevented theintermolecular H-bond formation with the water molecules inthe bulk. At this condition, relaxation due to nonradiativepathway for H-bonding with the solvent did not take place; asa result increase in the triplet state lifetime was observed from2.2 to 9.8 ns.
We have also carried out time-resolved experiments forcomplexes I and II in acetonitrile, water, and in water adsorbedon TiO2 or ZrO2 nanoparticle surface after excitation at 589nm wavelength. This enabled us to monitor the photophysicalproperties after excitation at a different absorption band. Kineticdecay traces were fitted exponentially and are shown in Table1. It is interesting to see that the kinetic decay trace of I inacetonitrile could be best fitted with time constants of 9.2 ns(4%) and 33 ns (96%). It is interesting to see that the timeconstants have not changed on excitation at 589 nm, and onlya change in the contribution due to different MLCT triplet stateswas observed. We have also seen that the lifetimes for both of the components in water have changed to 2 ns (triplet 3MLCTstate lifetime of bpy-catecholate-conjugated Os complex) and21 ns (triplet 3MLCT state lifetime of bpy-conjugated Oscomplex), respectively. Again, the emission decay traces afterexcitation at 589 nm laser light could be fitted with timeconstants 0.16 ns (65%), 0.59 ns (34%), >10 ns (1%) on TiO2
surface, and 0.66 ns (72%), 19 ns (28%) on ZrO 2 surface,respectively (Table 1). Here, we also have attributed the longertime constants to excited MLCT triplet states of Os-catecholateon nanoparticle surfaces and the shorter components to CTemission lifetimes on TiO2 and ZrO2 nanoparticle surfaces.
(f) Electron-Transfer Dynamics from CT Emission. Intime-resolved emission data analysis, we have observed thatthe dye molecules on nanoparticle surfaces show a shorterlifetime component. We also have observed in our earlier
investigations similar shorter components,11,12,19,20
which wasattributed to CT emission. Earlier, we have also confirmed thatthe CT emission can be only observed on excitation of CTcomplex. It has been observed from eq 1 that charge-transfercomplex formation is an equilibrium process. Thus, not all dyemolecules form CT complex, and some free dye molecules alsoexist in the system that do not form CT complex. However,they may be physically adsorbed on the surface. On laserexcitation of the above systems, the molecules that remainedsimply adsorbed on the surface went to the excited-state andcould inject electron into the conduction band of the nanopar-ticle. On the other hand, for the molecules, that form CTcomplex with the nanoparticles, electron injection took placedirectly into the nanoparticle instead via the excited-state of
Figure 5. Single-photon-counting studies of Os-cat1 (I) in differentmedia: (a) in acetonitrile, (b) in water, (c) in ZrO2 colloid (20.0 g/L),(d) in TiO2 colloid (20.0 g/L) after 453 nm excitation. L is the excitationpulse. Emission wavelength was kept for all the measurements at 670nm. Concentration of Os-cat1 was kept at 10 µM.
2924 J. Phys. Chem. C, Vol. 112, No. 8, 2008 Verma et al.

8/6/2019 hn ghosh_1
http://slidepdf.com/reader/full/hn-ghosh1 8/9
the dye after laser excitation. Following electron injection,recombination reaction took place for the charge-separated CTcomplex, and charge-transfer emission was observed during therecombination process.
By monitoring the CT emission, one could determine charge
recombination (BET) time in the dye-nanoparticle systems. Wehave detected CT emission from I /TiO2 and I /ZrO2 systems(Figure 4). Let us first discuss the time-resolved emission dataof I on both TiO2 and ZrO2 nanoparticles. The CT emissiontime constants are 0.16 and 0.33 ns, respectively, after excitationof 453 nm laser light (Table 1). We have drawn a scheme tounderstand the interfacial electron-transfer dynamics from I toTiO2 and ZrO2 nanoparticles (Scheme 3). The redox potentialfor the OsII/III couple in I and II was determined using cyclicvoltametric techniques and was found to be 1.17 and 1.14 V,respectively. The energy levels of excited singlet state (1MLCT)and excited triplet state (3MLCT) were determined from theextrapolation on the wavelength axis ( x-axis) of the opticalabsorption spectrum of MLCT singlet and triplet bands. Scheme
3 reveals that for I, the energy levels for the triplet (3
MLCT)and singlet (1MLCT) states are -0.55 and -1.1 V, respectively.It is evident from the scheme that on photoexcitation with 453nm, it is quite possible to inject electron from the photoexcitedmolecules of I to the conduction band of TiO2 nanoparticles.On the other hand, the photoexcited energy levels of I lie muchbelow the conduction band of ZrO2 nanoparticles. Thus, we donot expect electron injection into the conduction band of ZrO2
nanoparticles (Scheme 3). Photoexcitation of the CT complexcan lead to electron injection into the surface states of ZrO2
nanoparticles. Therefore, the shorter component (0.16 ns) in theI /TiO2 system on excitation at 453 nm can be attributed tocharge recombination (BET) time for the injected electron inthe conduction band and parent cation and 0.33 ns component
in I /ZrO2 system is attributed to charge recombination (BET)time for the injected electron in the surface states of ZrO 2 andparent cation. The data shown in the Table 1 are monitored at670 and 750 nm for different excitation wavelengths. We havealso monitored the emission decay kinetics at 844 nm byexciting 453 nm for the I /TiO2 system where we observed pureCT emission (Figure 4b). In time-resolved analysis data, weobserved the lifetime component of 0.16 ns (100%). Similarobservations also were made for the other systems.
We have also detected CT emission of I-sensitized TiO2 andZrO2 nanoparticles after exciting at 589 nm, which excite mostlythe triplet state band of the sensitizer molecule. It is interestingto note that the lifetime of CT emission for I /TiO2 system for589 nm excitation is 0.16 and 0.59 ns, where as for 453 nm
excitation is 0.16 ns (Table 1). On excitation at 589 nm, electroninjection could take place through triplet state (Scheme 3) of the complex, which has a long tail in a longer wavelengthindicating the involvement of trap state. As a result, excitationat this band may cause partial electron injection in the surfacestates of TiO2 in addition to the conduction band of TiO2. Thus,the shorter component (0.16 ns) observed in the time-resolveddata could be assigned to the charge recombination process of injected electron in the conduction band and parent cation, whilelonger component (0.62 ns) arose due to the recombination of the lower lying surface state electron just below the conductionband edge and parent cation radical. On the other hand CTemission lifetime in I /ZrO2 system on exciting at 589 nm hasbeen observed to be 0.66 ns. This indicates that charge
recombination time for injected electron in the surface states isslower (0.66 ns) for process associated with 589 nm excitationas compared to 453 nm excitation, which was found to be 0.33ns. Scheme 3 shows that both 453 and 589 nm excitation couldcause electron injection to the surface states of ZrO2 nanopar-ticles. However, excitation at 453 nm caused electron injectioninto the surface states just below the conduction band edge,whereas 589 nm excitation caused electron injection in theinterband gap states, which have higher trap depth. As the trap
depths are different for two different kinds of injected electrons,so the charge recombination dynamics are different.
Conclusion
We have used one newly synthesized and one previouslyreported catechol derivative of 2,2′-bipyridyl for synthesizingtwo tailor-made osmium polypyridyl complexes, I and II, tostudy the role of extended conjugation on the photophysicalproperties of the respective complexes and the dynamics of theinterfacial electron transfer on semiconductor nanoparticlesurface. Optical absorption studies featured two strong MLCTabsorption bands at 400-550 and 550-750 nm region, whichare attributed to 1MLCT (spin allowed) and 3MLCT (spinforbidden) transitions. In the present investigation, we haveobserved that unlike analogous ruthenium(II)-polypyridyl com-plex, the triplet states of osmium(II) polypyridyl complexes canbe directly excited for sensitization of semiconductor nanopar-ticles. As a result, the red region of the solar radiation can beused for dye sensitization more effectively as compared to theanalogous ruthenium complex. We have shown the directexcitation of triplet state and electron transfer to the nanopar-ticles. Excited-state lifetimes for both the singlet and triplet stateshave been determined by time-resolved emission studies. Steady-state optical absorption studies confirmed that the osmiumcomplexes form charge-transfer complex with both TiO2 andZrO2 (higher band gap) nanoparticles. On excitation of theseCT complexes, electron injections take place from the Os(II)
complexes to the conduction band of TiO2 and surface statesof ZrO2 nanoparticles. CT emission has been detected in theabove dye/nanoparticle systems when the charge-separatedspecies recombine. By monitoring the CT emission, chargerecombination time could be determined for the BET processfor the electron transfer from the nanoparticle to the parent cationof osmium complexes. We have also shown that back ETdynamics in the dye/nanoparticle system differs on excitationof singlet and triplet MLCT states because on excitation of tripletstates, electron injection can take place in the strongly coupledsurface states in addition to the conduction band.
Acknowledgment. We would like to thank one of thereviewers for their constructive suggestions on the work, which
have significantly improved the quality of the paper. We thank Dr. T. Mukherjee and Dr. S. K. Sarkar of BARC and Dr. P. K.Ghosh of CSMCRI for their constant encouragement. A.D.thanks DST and BRNS while H.N.G. thanks BRNS for financialassistance.
Supporting Information Available: Additional figures andexperimental details. This material is available free of chargevia the Internet at http://pubs.acs.org.
References and Notes
(1) O’Regan, B.; Gratzel, M. Nature 1991, 353, 737.
(2) Nazeeruddin, M. K.; Pechy, P.; Renouard, T.; Zakeeruddin, S. M.;Humphry-Baker, R.; Comte, P.; Liska, P.; Cevey, L.; Costa, E.; Shklover,
e-
(MO2) + Dye+f [MO2 - Dye]Adsorb + hνCT (emission)
(3)
IET Dynamics on TiO2 and ZrO2 Nanoparticle Surface J. Phys. Chem. C, Vol. 112, No. 8, 2008 2925

8/6/2019 hn ghosh_1
http://slidepdf.com/reader/full/hn-ghosh1 9/9
V.; Spiccia, L.; Deacon, G. B.; Bignozzi, C. A.; Gratzel, M. J. Am. Chem.Soc. 2001, 123, 1613.
(3) Nazeeruddin, M. K.; Zakeeruddin, S. M.; Humphry-Baker, R.;Jirousek, M.; Liska, P.; Vlachopoulus, N.; Skhlover, V.; Fischer, C. H.;Gratzel, M. Inorg. Chem. 1999, 38, 6298.
(4) Rice, C. R.; Ward, M. D.; Nazeeruddin, M. K.; Gratzel, M. New J.Chem. 2000, 24, 651.
(5) Hannapel, T.; Burfeindt, B.; Storck, W.; Willig, F. J. Phys. Chem. B 1997, 101, 6799.
(6) Asbury, J. B.; Hao, E.; Wang, Y. Q.; Ghosh, H. N.; Lian, T. J.Phys. Chem. B 2001, 105, 4545.
(7) Sauve, G.; Cass, M. E.; Doig, S. J.; Lauermann, I.; Pomykal, K.;
Lewis, N. S. J. Phys. Chem. B 2000, 104, 3488.(8) Atobello, S.; Argazzi, R.; Caramori, S.; Contado, C.; Da Fre, S.;
Rubino, P.; Chone, C.; Larramona, G.; Bignozzi, C. A. J. Am. Chem. Soc.2005, 127 , 15342.
(9) Tachibana, Y.; Moser, J. E.; Gratzel, M.; Klug, D. R.; Durrant, J.R. J. Phys. Chem. 1996, 100, 20056.
(10) Benko, G.; Kallioinen, J.; Korppi-Tommola, J. E. I.; Yartsev, A.P.; Sundstrom, V. J. Am. Chem. Soc. 2002 , 104, 489.
(11) Ghosh, H. N. J. Phys. Chem. B 1999, 103, 10382.(12) Ramakrishna, G.; Ghosh, H. N. J. Phys. Chem. B 2001, 105, 7000.(13) Ramakrishna, G.; Ghosh, H. N.; Singh, A. K.; Palit, D. K.; Mittal,
J. P. J. Phys. Chem. B 2001, 105, 12786.(14) Ramakrishna, G.; Das A.; Ghosh, H. N. Langmuir 2004, 20, 1430.(15) Ramakrishna, G.; Ghosh, H. N. J. Phys. Chem. A 2002, 106 , 2545.(16) Ramakrishna, G.; Singh, A. K.; Palit, D. K.; Ghosh, H. N. J. Phys.
Chem. B 2004, 108, 1701.(17) Ramakrishna, G.; Singh, A. K.; Palit, D. K.; Ghosh, H. N. J. Phys.
Chem. B 2004, 108, 4775.
(18) Ramakrishna, G.; Singh, A. K.; Palit, D. K.; Ghosh, H. N. J. Phys.Chem. B 2004, 108, 12489.
(19) Ramakrishna, G.; Ghosh, H. N. Langmuir 2004, 20, 7342.(20) Rath, M. C.; Ramakrishna, G.; Mukherjee, T.; Ghosh H. N. J. Phys.
Chem. B 2005 , 109, 20485.(21) Ramakrishna, G.; Jose, D. A.; KrishnaKumar, D.; Das, A.; Palit,
D. K.; Ghosh, H. N. J. Phys. Chem. B 2005, 109, 15445.(22) Ramakrishna, G.; Verma, S.; Jose, D. A.; KrishnaKumar, D.; Das,
A.; Palit, D. K.; Ghosh, H. N. J. Phys. Chem. B 2006, 110, 9012.(23) Gratzel, M.; Kalyansundaram, K. Curr. Sci. 1994, 66 , 706.(24) Hagfeldt, A.; Gratzel, M. Chem. ReV. 1995, 95, 49.(25) Chiorboli, C.; Rodgers, M. A. J.; Scandola, F. J. Am. Chem. Soc.
2003, 125, 483.(26) Heimer, T. A.; Heilweil, E. J.; Bignozzi, C. A.; Meyer, G. J. J.
Phys. Chem. A 2000, 104, 4256.
(27) Xu, D.; Zhang, J. Z.; Hong, B. J. Phys. Chem. A 2001, 105, 7979.(28) Kuciauskas, D.; Monat, J. E.; Villahermosa, R.; Gray, H. B.; Lewis,
N. S.; McCusker, J. K. J. Phys. Chem. B 2002, 106 , 9347.(29) Hoertz, P. G.; Thompson, D. W.; Friedman, L. A.; Meyer, G. J. J.
Am. Chem. Soc. 2002, 124, 9690.(30) Baudin, H. B.; Davidsson, J.; Serroni, S.; Juris, A.; Balzani, V.;
Campagna, S.; Hammarstrom, L. J. Phys. Chem. A 2002, 106 , 4312.(31) Barigelletti, F.; Cola, L. D.; Balzani, V.; Hage, R.; Haasnoot, J.
G.; Reedijk, Vos, J. G. Inorg. Chem. 1991 , 30, 641.(32) Kalanasundaram, K. Photochemistry of Polypyridine and Porphyrin
Complexes; Academic Press: San Diego, 1992; p 626.(33) Baldo, M. A.; O’Brien, D. F.; You, Y.; Shoustikov, A.; Sibley, S.;
Thompson, M. E.; Forrest, S. R. Nature 1998, 395, 151.(34) Liu, Y.; Jiang, S.; Glusac, K.; Powell, D. H.; Anderson, D. F.;
Schanze, K. S. J. Am. Chem. Soc. 2002, 124, 12412.(35) Shukla, A. D.; Whittl, B.; Bajaj, H. C.; Das, A.; Ward, M. D. Inorg.
Chim. Acta. 1999, 285, 89.(36) Carlson, B.; Phelan, G. D.; Kaminsky, W.; Dalton, L.; Jiang, X.;
Liu, S.; Jen, A. K.-Y. J. Am. Chem. Soc. 2002, 124, 14162.(37) Shaw, G. B.; Brown, C. L.; Papanikolas J. M. J. Phys. Chem. A
2002, 106 , 1483.(38) Richter, M. M.; Brewer, K. J. Inorg. Chim. Acta 1991, 180 (1),
125.(39) Kelly-Basetti, Bernadette M.; Cundy, Darren J.; Pereira, Suzanne
M.; Sasse, Wolfgang H. F.; Savage, G. Paul; Simpson, Gregory W. Bioorg. Med. Chem. Letters 1995, 5 (24), 2989.
(40) O’Connor, D. V.; Phillips, D. Time Correlated Single PhotonCounting; Academic Press: New York, 1984.
(41) Kober, E. M.; Caspar, J. V.; Sullivan, B. P.; Meyer, T. J. Inorg.Chem. 1988, 27 , 4587.
(42) Yu, J. K.; Cheng, Y. M.; Hu, Y. H.; Chou, P. T.; Chen, Y. L.; Lee,S. W.; Chi, Y. J. Phys. Chem. B 2004, 108, 19908.
(43) Fetterolf, M. L.; Offen, H, W. J. Phys. Chem. 1985, 89, 3320.(44) Supporting Information.(45) Rego, L. G. C.; Batista, V. S. J. Am. Chem. Soc. 2003, 125, 7989.(46) Moser, J.; Punchihewa, S.; Infelta, P. P.; Gratzel, M. Langmuir
1991, 7 , 3012.(47) Rajh, T.; Chen, L. X.; Lukas, K.; Liu, T.; Thurnauer, M. C.; Teide,
D. M. J. Phys. Chem. B 2002, 106 , 10543.(48) Rehm, J. M.; McLendon, G. L.; Nagasawa, Y.; Yoshihara, K.;
Moser, J.; Gratzel, M. J. Phys. Chem. 1996, 100, 9577.(49) Walters, K. A.; Gaal, D. A.; Hupp, J. T. J. Phys. Chem B 2002,
106 , 5139.(50) Giordano, P. J.; Bock, C. R.; Wrighton, M. S. J. Am. Chem. Soc.
1978, 100, 6960.
2926 J. Phys. Chem. C, Vol. 112, No. 8, 2008 Verma et al.


















