
HDM2 Regulation by AURKA Promotes Cell Survival in Gastric...
13
Human Cancer Biology HDM2 Regulation by AURKA Promotes Cell Survival in Gastric Cancer Vikas Sehdev 1,5 , Ahmed Katsha 1 , Janet Arras 1 , Dunfa Peng 1,3 , Mohammed Soutto 1,3 , Jeffrey Ecsedy 4 , Alexander Zaika 1,2 , Abbes Belkhiri 1 , and Wael El-Rifai 1,2,3 Abstract Purpose: Suppression of P53 (tumor protein 53) transcriptional function mediates poor therapeutic response in patients with cancer. Aurora kinase A (AURKA) and human double minute 2 (HDM2) are negative regulators of P53. Herein, we examined the role of AURKA in regulating HDM2 and its subsequent effects on P53 apoptotic function in gastric cancer. Experimental Design: Primary tumors and in vitro gastric cancer cell models with overexpression or knockdown of AURKA were used. The role of AURKA in regulating HDM2 and cell survival coupled with P53 expression and activity were investigated. Results: Overexpression of AURKA enhanced the HDM2 protein level; conversely, knockdown of endogenous AURKA decreased expression of HDM2 in AGS and SNU-1 cells. Dual co-immunoprecipitation assay data indicated that AURKA was associated with HDM2 in a protein complex. The in vitro kinase assay using recombinant AURKA and HDM2 proteins followed by co-immunoprecipitation revealed that AURKA directly interacts and phosphorylates HDM2 protein in vitro. The activation of HDM2 by AURKA led to induction of P53 ubiquitination and attenuation of cisplatin-induced activation of P53 in gastric cancer cells. Inhibition of AURKA using an investigational small-molecule specific inhibitor, alisertib, decreased the HDM2 protein level and induced P53 transcriptional activity. These effects markedly decreased cell survival in vitro and xenograft tumor growth in vivo. Notably, analysis of immunohistochemistry on tissue micro- arrays revealed significant overexpression of AURKA and HDM2 in human gastric cancer samples (P < 0.05). Conclusion: Collectively, our novel findings indicate that AURKA promotes tumor growth and cell survival through regulation of HDM2-induced ubiquitination and inhibition of P53. Clin Cancer Res; 20(1); 76–86. Ó2013 AACR. Introduction Gastric cancer exhibits poor patient survival rates due to intrinsic resistance to chemotherapeutic drugs (1, 2). According to current estimates, gastric cancer is a frequently diagnosed disease worldwide with an estimated incidence rate of approximately one million cases and mortality rate of 740,000 cases, respectively (3, 4). Multiple oncogenic signaling mechanisms have been shown to mediate cancer cell survival and drug resistance against several chemother- apeutics in gastric cancers (5–9). Unfortunately, the survival rate for patients with gastric cancer has shown only mar- ginal improvement (10). Therefore, investigations specifi- cally aimed at further understanding of the mechanisms regulating cell death and drug resistance in gastric cancer are essential for the development of novel effective anticancer therapeutic regimens. Aurora kinase A (AURKA), a serine/threonine cell-cycle kinase, has been mapped to the 20q13 chromosomal region, which is frequently amplified in gastric cancer (11). Frequent amplification and/or overexpression of AURKA have been reported in breast, colon, esophageal, gastric, liver, ovarian, and pancreatic cancers (8, 12–18). AURKA plays an important role in facilitating mitosis and its expression is tightly regulated in normal cells. However, deregulated overexpression of AURKA causes genetic insta- bility, dedifferentiated morphology, and oncogenic trans- formation (6, 13). Overexpression of AURKA promotes progrowth and anti-apoptotic signaling pathways resulting in cancer cell proliferation, drug resistance, and poor patient prognosis in gastric cancers (13, 19). The potent Authors' Affiliations: Departments of 1 Surgery and 2 Cancer Biology, Vanderbilt University Medical Center; 3 Department of Veterans Affairs, Tennessee Valley Healthcare System, Nashville, Tennessee; 4 Translational Medicine, Millennium Pharmaceuticals, Inc., Cambridge, Massachusetts; and 5 Department of Pharmacology, Arnold and Marie Schwartz College of Pharmacy and Health Sciences, Long Island University, New York Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/). V. Sehdev and A. Katsha contributed equally to this work. Corresponding Author: Wael El-Rifai, Vanderbilt-Ingram Cancer Center, Vanderbilt University Medical Center, 760 PRB, 2220 Pierce Avenue, Nashville, TN 37232-6308. Phone: 615-322-7934; Fax: 615-322-7852; E-mail: [email protected] doi: 10.1158/1078-0432.CCR-13-1187 Ó2013 American Association for Cancer Research. Clinical Cancer Research Clin Cancer Res; 20(1) January 1, 2014 76 on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187 on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187 on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187
Transcript of HDM2 Regulation by AURKA Promotes Cell Survival in Gastric...

Human Cancer Biology
HDM2 Regulation by AURKA Promotes Cell Survival inGastric Cancer
Vikas Sehdev1,5, Ahmed Katsha1, Janet Arras1, Dunfa Peng1,3, Mohammed Soutto1,3,Jeffrey Ecsedy4, Alexander Zaika1,2, Abbes Belkhiri1, and Wael El-Rifai1,2,3
AbstractPurpose: Suppression of P53 (tumor protein 53) transcriptional function mediates poor therapeutic
response in patients with cancer. Aurora kinase A (AURKA) and human double minute 2 (HDM2) are
negative regulators of P53. Herein, we examined the role of AURKA in regulating HDM2 and its subsequent
effects on P53 apoptotic function in gastric cancer.
Experimental Design: Primary tumors and in vitro gastric cancer cell models with overexpression or
knockdownofAURKAwereused. The role ofAURKA in regulatingHDM2and cell survival coupledwithP53
expression and activity were investigated.
Results: Overexpression of AURKA enhanced the HDM2 protein level; conversely, knockdown of
endogenous AURKA decreased expression of HDM2 in AGS and SNU-1 cells. Dual co-immunoprecipitation
assay data indicated that AURKA was associated with HDM2 in a protein complex. The in vitro kinase assay
using recombinant AURKA andHDM2 proteins followed by co-immunoprecipitation revealed that AURKA
directly interacts and phosphorylates HDM2 protein in vitro. The activation of HDM2 by AURKA led to
inductionof P53ubiquitinationandattenuationof cisplatin-induced activationof P53 in gastric cancer cells.
Inhibition of AURKA using an investigational small-molecule specific inhibitor, alisertib, decreased the
HDM2 protein level and induced P53 transcriptional activity. These effects markedly decreased cell survival
in vitro and xenograft tumor growth in vivo. Notably, analysis of immunohistochemistry on tissue micro-
arrays revealed significant overexpression of AURKA andHDM2 in human gastric cancer samples (P < 0.05).Conclusion: Collectively, our novel findings indicate that AURKA promotes tumor growth and cell
survival through regulation ofHDM2-induced ubiquitination and inhibition of P53.ClinCancer Res; 20(1);
76–86. �2013 AACR.
IntroductionGastric cancer exhibits poor patient survival rates due to
intrinsic resistance to chemotherapeutic drugs (1, 2).According to current estimates, gastric cancer is a frequentlydiagnosed disease worldwide with an estimated incidencerate of approximately one million cases and mortality rateof 740,000 cases, respectively (3, 4). Multiple oncogenic
signaling mechanisms have been shown to mediate cancercell survival and drug resistance against several chemother-apeutics in gastric cancers (5–9).Unfortunately, the survivalrate for patients with gastric cancer has shown only mar-ginal improvement (10). Therefore, investigations specifi-cally aimed at further understanding of the mechanismsregulating cell death and drug resistance in gastric cancer areessential for the development of novel effective anticancertherapeutic regimens.
Aurora kinase A (AURKA), a serine/threonine cell-cyclekinase, has been mapped to the 20q13 chromosomalregion, which is frequently amplified in gastric cancer(11). Frequent amplification and/or overexpression ofAURKA have been reported in breast, colon, esophageal,gastric, liver, ovarian, and pancreatic cancers (8, 12–18).AURKA plays an important role in facilitating mitosis andits expression is tightly regulated in normal cells. However,deregulated overexpression of AURKA causes genetic insta-bility, dedifferentiated morphology, and oncogenic trans-formation (6, 13). Overexpression of AURKA promotesprogrowth and anti-apoptotic signaling pathways resultingin cancer cell proliferation, drug resistance, and poorpatient prognosis in gastric cancers (13, 19). The potent
Authors' Affiliations: Departments of 1Surgery and 2Cancer Biology,Vanderbilt University Medical Center; 3Department of Veterans Affairs,Tennessee ValleyHealthcare System, Nashville, Tennessee; 4TranslationalMedicine, Millennium Pharmaceuticals, Inc., Cambridge, Massachusetts;and 5Department of Pharmacology, Arnold and Marie Schwartz College ofPharmacy and Health Sciences, Long Island University, New York
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
V. Sehdev and A. Katsha contributed equally to this work.
Corresponding Author: Wael El-Rifai, Vanderbilt-Ingram Cancer Center,Vanderbilt University Medical Center, 760 PRB, 2220 Pierce Avenue,Nashville, TN 37232-6308. Phone: 615-322-7934; Fax: 615-322-7852;E-mail: [email protected]
doi: 10.1158/1078-0432.CCR-13-1187
�2013 American Association for Cancer Research.
ClinicalCancer
Research
Clin Cancer Res; 20(1) January 1, 201476
on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187
on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187
on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187

docetaxel, cisplatin, and 5-fluorouracil (DCF) chemother-apy regimen is frequently used against gastric cancers (20,21). Nonetheless, AURKA overexpression has been shownto mediate resistance against taxol and cisplatin and canthereby undermine the therapeutic outcome of the DCFregimen in gastric cancers (22, 23). The efficacy of chemo-therapeutic drugs is dependent on induction of apoptosisby the P53 (tumor protein 53) family of pro-apoptoticproteins. We and others have previously reported that over-expression of AURKA can attenuate both P53 and P73(tumor protein 73) protein expression and function (24,25). These findings indicate that constitutive overexpressionof AURKA can mediate poor response to chemotherapy ingastric cancers. Therefore, further investigation of mecha-nism(s) by which AURKA suppresses P53 expression andfunction can aid in the development of novel and effectivetherapeutic strategies against gastric cancers.Human double minute 2 (HDM2), an E3-ubiquitin
ligase, is one of the critical negative regulators of P53protein function and expression. HDM2 has been shownto inhibit P53 by blocking P53 transcriptional activity,promoting cytosolic translocation of P53 from the nucle-us, and tagging it with ubiquitin for proteasomal degra-dation (26). HDM2 gene amplification has been reportedin various cancers and when overexpressed HDM2becomes a bona fide oncogene that can promote malig-nant transformation and drug resistance (27, 28). Giventhe fact that HDM2 plays a critical role in regulating P53,a function shared with AURKA, we postulated that acti-vation of HDM2 by AURKA regulates P53 in gastric cancercells. In this study, we investigated the role of AURKA inregulating HDM2-mediated P53 inhibition affecting gas-tric tumor growth and cell survival. We also examined therole of an investigational small molecule–specific inhib-
itor of AURKA, alisertib, in suppressing HDM2 and pro-moting cell death in vitro and in vivo.
Materials and MethodsCell culture and pharmacologic reagents
The gastric adenocarcinoma cell lines (AGS, SNU-1, SNU-16, MKN28, MKN45, MKN75, Kato III, and RF-1) wereobtained from American Type Culture Collection (ATCC)and RIKEN BioResource Center. The cell lines were main-tained in F-12 or Dulbecco’s Modified Eagle Medium(DMEM; Gibco) supplemented with 10% (v/v) FBS (Gibco;ref. 29). All cell lines were ascertained to conform to in vitromorphologic characteristics.
Alisertib (Millennium Pharmaceuticals, Inc.) solutionsfor in vitro and in vivo studies were prepared as describedpreviously (8).Nutlin3A (CaymanChemicals) solutionwasprepared in dimethyl sulfoxide (DMSO; Sigma-Aldrich).Cisplatin (CDDP;APPPharmaceuticals, LLC.) solution (3.3mmol/L) was prepared in sterile water. AKT, p-AKT(Ser473), p-AURKA (Thr288), AURKA, HDM2, p-HDM2(Ser166), P53, P21, and b-actin primary antibodies wereobtained from Cell Signaling Technology.
AURKA and HDM2 expression and plasmidsThe AURKA expression plasmid was generated as
described previously (30). The HDM2 expression plasmidwas purchased from Addgene. Transient transfection ofgastric cancer cells was performed using X-tremeGENE HP(Roche Applied Sciences). The recombinant adenovirusexpressing AURKA or control was generated as describedpreviously (31).
Clonogenic cell survival assayGastric cancer cells were seeded at 5,000 cells per well in a
six-well plate and treated with vehicle (DMSO) or alisertib(0.25–5.0 mmol/L) for 24 hours. Next, cells were culturedfor 10 days and colonies were stained and quantified asdescribed previously (8).
Western blot analysisCells were lysed in lysis buffer (50 mmol/L Tris-HCl
buffer, pH 7.4, 150 mmol/L NaCl, 1% Triton X-100, 1%sodium deoxycholate, and 0.1% SDS) supplemented with1�Halt Protease Inhibitor Cocktail (Pierce). Proteins wereanalyzed by Western blot as described previously (32).
Dual immunofluorescenceGastric cancer cells plated in eight-chamber slides (BD
Falcon) were permeabilized and fixed in 2% paraformal-dehyde. Cells were then incubated in a mixture of rabbitAURKA (1:100) and mouse HDM2 (1:100) primary anti-bodies for 3 hours. After washing with PBS, cells werestained with Alexa Fluor 488 anti-rabbit and Alexa Fluor568 anti-mouse secondary antibodies. The cells werewashed and mounted with 40,6-diamidino-2-phenylindole(DAPI) and examined by fluorescence microscopy (Olym-pus America Inc.).
Translational RelevanceGastric cancer is characterized by poor patient survival
and resistance to chemotherapy. Several chemothera-peutic agents used in the treatment of gastric cancerinduce DNA damage and activate P53 (tumor protein53) pro-apoptotic functions. Conversely, Aurora kinaseA (AURKA) and human double minute 2 (HDM2)suppress P53 protein expression and activity. Herein,we report, for the first time, that AURKA and HDM2proteins are frequently co-overexpressed in gastric cancertissues and cell lines. Our data indicate that AURKAregulates the expression and phosphorylation levels ofHDM2, which could be a major mechanism attenuatingtheP53protein function in gastric cancer cells. The use ofAURKA inhibitor, alisertib, reversed these effects, lead-ing to significant suppression of tumor cell growth invitro and in vivo. The fact that AURKA activates HDM2and is frequently overexpressed in gastric cancer stronglyjustifies the use of AURKA inhibitors in the treatment ofgastric cancer.
AURKA Regulates HDM2 E3-Ubiquitin Ligase
www.aacrjournals.org Clin Cancer Res; 20(1) January 1, 2014 77
on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187

ImmunoprecipitationImmunoprecipitation was performed as described previ-
ously (33). Briefly, cells were lysed in lysis buffer andproteins were immunoprecipitated at room temperaturewith primary antibodies previously bound to 50 mL Dyna-beads Protein G (Invitrogen).
In vitro kinase activity assayActive human recombinant AURKA (Cell Sciences) and
HDM2 (Thermo Scientific) proteinswere used for an in vitrokinase assay. Briefly, increasing concentrations of AURKA(0.2–1.0 mg) were added to a fixed concentration of HDM2(0.5 mg) in the assay buffer. The reaction mixtures wereincubated at 37�C for 30 minutes to initiate kinase activity,and the protein samples were subjected to Western blotanalysis.
In vivo tumor xenograftAGS cells (4 � 106) suspended in 200 mL of DMEM
Matrigel mixture (50% DMEM supplemented with 10%FBS and 50%Matrigel) were injected into the flank regionsof female athymic nude-Foxn1 nu/nu mice (Harlan Labo-ratories Inc.). The tumors were allowed to grow until 200mm3 in size before starting the treatmentwith daily alisertib(30 mg/kg, orally) for 21 days. Tumor xenografts weremeasured every 4 days and tumor size was calculatedaccording to the following formula: Tvol ¼ L � W2 � 0.5,in which Tvol is tumor volume, L is tumor length and W istumor width (34). At the end of treatment, the xenografttumors were collected and processed for quantitative real-time PCR (qRT-PCR; PUMA, NOXA, P21, and BAX) orimmunohistochemistry (IHC; P53; SC-126, and HDM2;SMP14; Santa Cruz Biotechnology, Inc.) antibodies asdescribed previously (31).
Tissue microarray and IHCIHCwasperformedon tissuemicroarrays (TMA) contain-
ing 94 deidentified archival cases of gastric cancer and 113cases of esophageal adenocarcinomas (EAC) along with 71normal gastric epithelial tissue samples and 26 normalesophageal tissue samples. All tissue samples were obtainedin accordance with the Institutional Review Board (IRB)–approved protocols at Vanderbilt University. Five-microm-eter thick TMA sections of normal and tumor tissue sampleswere used for IHC staining of AURKA and HDM2 proteins.The intensity and frequency of staining was scored asdescribed previously (35).
AKT and HDM2 silencing by siRNAAGS cells overexpressing AURKA or control vector were
transfected with AKT siRNA, HDM2 siRNA, or controlsiRNA for 48 hours, purchased from Cell Signaling Tech-nology and Integrated DNA technologies Inc., respectively.The cell lysates were analyzed by Western blot.
Statistical analysisData are presented as means � SEM. All in vitro experi-
ments were performed in triplicates. ANOVA with Tukey
post hoc analysis was used to evaluate statistical differencebetween groups. Statistical analyses were carried out usingGraphPad Prism 5 software (GraphPad Software Inc.). Thecorrelation between two parameters was determined by theSpearman correlation and k test. The P value of 0.05 or lesswas considered statistically significant.
ResultsAURKA regulates HDM2 and cell survival in gastricadenocarcinoma cells
We examined AURKA and HDM2 protein expression ingastric adenocarcinoma (gastric cancer) cell lines. TheWestern blot analysis data indicated frequent concomitantoverexpression of AURKAandHDM2proteins in 5/8 gastriccancer cell lines with the RF-1 cell line exhibiting relativelylow expression of AURKA and HDM2 (Fig. 1A). Similarly,the data showed that 4/5 EAC cell lines concomitantlyoverexpressed AURKA and HDM2 proteins, and 3/3untransformed immortalized esophageal cells were nega-tive for AURKA and HDM2 expression (Supplementary Fig.S1). We have previously reported that AURKA suppressesP53 protein function in gastric cancer cells (36). HDM2 isan E3-ubiquitin ligase closely involved in regulating P53protein expression and stability. Because AURKA andHDM2 are frequently overexpressed in gastric cancer celllines, we hypothesized that AURKA regulatesHDM2 expres-sion in gastric cancer. To test this hypothesis, we used P53wild-type AGS and SNU-1 gastric cancer cell in vitromodels. The Western blot analysis indicated that transientoverexpression of AURKA using adenovirus systemenhanced HDM2 protein levels in AGS and SNU-1 cells(Fig. 1B). Following AURKA overexpression, P53 and itsdownstream targets (P21 and BAX) were downregulatedin AGS cells. Conversely, siRNA-mediated knockdown ofendogenous AURKA increased P53, P21, and BAX expres-sion in AGS cells. AURKA overexpression and knockdownexhibited similar effects on HDM2, P53, and P21 in SNU-1 cells (Fig. 1B). However, unlike AGS cells, we did notobserve a change in BAX protein expression in SNU-1cells. To investigate whether AURKA-mediated increasein the HDM2 protein level was dependent on its serine/threonine kinase activity, we used kinase-dead mutantAURKA (D274A). The Western blot analysis revealed that,unlike wild-type AURKA, kinase-dead mutant AURKA(D274A) did not significantly affect HDM2 expressionin the AGS cells (Fig. 1C). To validate the role of AURKAin regulating HDM2 and examine its impact on cellsurvival, we pharmacologically inhibited AURKA withalisertib, an investigational AURKA-specific inhibitor, inAGS and SNU-1 cells. The Western blot analysis dataconfirmed that inhibition of AURKA led to a significantdecrease in the HDM2 protein level coupled with upre-gulation of P53 and P21 expression in both cell lines (Fig.1D). The clonogenic survival assay data indicated thatalisertib-mediated inhibition of AURKA activity signifi-cantly suppressed AGS (P < 0.01) and SNU-1 (P < 0.01)cell survival (Fig. 1E).
Sehdev et al.
Clin Cancer Res; 20(1) January 1, 2014 Clinical Cancer Research78
on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187

AURKA directly interacts and phosphorylates HDM2 ingastric cancerHDM2 localizes to the nucleus, inducing ubiquitination
and degradation of the P53 protein in the proteasome (26).The dual-immunofluorescence analyses results indicatedthat transient overexpression of AURKA significantlyenhanced HDM2 protein expression, confirming the West-ern blot analysis data (Fig. 1B). The immunofluorescencedata showed an overlap of AURKA andHDM2 signals in themerged image, suggesting colocalization of AURKA andHDM2 in both AGS and SNU-1 cells (Fig. 2A and B). Wehave previously reported that AURKA phosphorylates AKT
at Ser473 amino acid residue and active AKT has beenshown to phosphorylate HDM2 at the Ser166 site (36,37). Therefore, it is plausible thatAURKA-mediated increasein HDM2 protein expression could bemediated by an AKT-dependent mechanism. To examine this hypothesis, wetransiently overexpressed AURKA in AGS cells and evaluat-ed HDM2 protein levels after treatment with an AKT inhib-itor (AKTI) or genetic knockdown of AKT with siRNA.Overall, the Western blot analysis data showed that HDM2expression levels were not significantly affected by AKTinhibition or knockdown in AURKA-overexpressing cells(Supplementary Fig. S2). Collectively, the data suggested
Figure 1. AURKA promotes cellsurvival through regulation ofHDM2 and P53 expression ingastric cancer cell lines. A, proteinextracts from a panel of gastriccancer cell lines were subjected toWestern blot analysis of AURKA,P53, and HDM2 proteins. B,Western blot analysis of HDM2,P53, P21, BAX, and AURKAproteins following adenovirus-mediated transient overexpressionof AURKA, control adenovirus,siRNA-mediated knockdown ofAURKA, or control siRNA in AGSand SNU-1 cells is shown.Overexpression of AURKA led toan increase in the protein level ofHDM2 and a decrease in P53.Conversely, inhibition of AURKAreduced the level of HDM2 with anincrease in P53. C, AGS cells weretransiently transfected withpcDNA3.1 empty vector, AURKA,or kinase-dead mutant AURKAD274A. Western blot analysisindicated that AURKA-mediatedincrease in the HDM2 protein levelis dependent on AURKA kinaseactivity. D, AGS and SNU-1 cellswere treated with alisertib (0.5mmol/L) for 48 hours, and celllysates were subjected to Westernblot analysis. The data showed thattreatmentwith alisertib reduced thelevel of HDM2 and increased P53.E, clonogenic cell survival analysisfor AGS and SNU-1 cells wasperformed following treatment withalisertib (0.25–5.0 mmol/L). Thedata indicated a dose-dependentdecrease in cell survival. Ctrl AD,Control Adenovirus; AURKA AD,Aurora kinase A Adenovirus;AURKA Mut, Aurora kinase AD274A Mutant; ��, P < 0.01.
AURKA Regulates HDM2 E3-Ubiquitin Ligase
www.aacrjournals.org Clin Cancer Res; 20(1) January 1, 2014 79
on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187

that AURKA can upregulate HDM2 expression through anAKT-independent mechanism.
P53 suppression by AURKA is dependent on regulationof HDM2 in gastric cancer
On the basis of our results showing that AURKA upre-gulates HDM2 expression, we postulated that AURKA canregulate P53 protein expression and function throughmod-ulation of HDM2 in response to DNA damage in gastriccancer cells. To test this hypothesis, we transiently over-expressed AURKA inAGS and SNU-1 cells, and treated themwith vehicle or cisplatin (CDDP). TheWestern blot analysisdata showed that AURKAoverexpression attenuatedCDDP-induced upregulation of P53 and P21 expression in AGSand SNU-1 cells (Fig. 3A and Supplemental Figure 3A).Because HDM2 regulates P53 through ubiquitination andproteasomal degradation,wehypothesized that AURKA canenhance P53 ubiquitination. To examine this hypothesis,we assessed the effect of AURKA and/or HDM2 expressionon P53 ubiquitination in AGS cells. The immunoprecipi-tation data indicated that transient overexpression ofAURKA or HDM2 significantly enhanced P53 ubiquitina-tion (Fig. 3B). To confirm that inhibition of P53 by AURKAis dependent on HDM2, we transiently overexpressedAURKA or HDM2 and treated with vehicle or Nutlin3A inAGS and SNU-1 cells. Nutlin3A is a HDM2 inhibitor thatprevents HDM2 from binding to P53, thereby preventingubiquitination anddegradation of the P53 protein.Western
blot analysis data indicated that AURKA-mediated down-regulation of P53 and its downstream target P21 was atten-uated by Nutlin3A in AGS cells (Fig. 3C). As a confirmationof the effect of Nutlin3A, the data showed that exogenousHDM2-mediated decrease in P53 and P21 expression waspartially blocked byNutlin3A (Fig. 3C). Similar results wereobtained in SNU-1 cells (Supplementary Fig. S3B). Geneticknockdown ofHDM2by siRNA abrogated AURKA-inducedsuppression of P53 and P21 in AGS cells, confirming theresults obtained by Nutlin3A (Supplementary Fig. S3C).Together, these results indicated that AURKA-induceddownregulation of P53 is dependent on HDM2. In addi-tion, we evaluated resistance to cytotoxic effects of CDDP inAGS cells overexpressing AURKA. The survival data indicatethat transient overexpression of AURKA promotes resis-tance to CDDP in AGS cells (Supplementary Fig. S4).
To ascertain if AURKA interacts with HDM2, we per-formed dual co-immunoprecipitation (Co-IP) followingoverexpression of AURKA in AGS cells. The dual Co-IP dataindicated the presence of endogenous HDM2 protein in animmunocomplex with exogenous AURKA protein (Supple-mentary Fig. S5). To investigate if AURKAdirectly interactedand phosphorylated HDM2 in the presence or absence ofalisertib, we carried out an in vitro kinase assay usingrecombinant AURKA and HDM2 proteins followed byCo-IP assay. The data indicated that AURKA directly inter-acts and phosphorylates HDM2; these effects were abrogat-ed upon inhibition of AURKA with alisertib (Fig. 4A). Tofurther confirmthatAURKAdirectly phosphorylatesHDM2,we performed the in vitro kinase assay with HDM2 andincreasing concentrations of AURKA. The data indicatedthat AURKA protein can directly phosphorylate the HDM2protein at the Ser166 residue in a concentration-dependentmanner (Fig. 4B). Collectively, our data clearly indicatedthat AURKA can directly interact and phosphorylateHDM2.
Pharmacologic inhibition of AURKA induces P53through downregulation of HDM2 in vivo
To establish the role of AURKA in regulating HDM2 andtherebymodulating P53 expression and function in vivo, wetreated AGS mouse xenografts with alisertib and examinedthe tumor growth and expression levels of HDM2 and P53.In addition, we assessed P53 transcriptional activity bymeasuring the mRNA levels of P53 downstream targets,PUMA, NOXA, P21, and BAX. The treatments were initiatedafter the tumor xenografts reached 200 mm3 in size. Theendpoint data for day 21 indicated that treatment withalisertib significantly reduced tumor volume by approxi-mately 55-fold relative to control (P < 0.01; Fig. 5A). TheIHC data revealed that alisertib-mediated inhibition ofAURKA significantly suppressed HDM2 expression (P ¼0.01) and induced P53 expression (P < 0.01) in the AGSxenografts (Fig. 5B and C). The qRT-PCR data showed asignificantly higher induction of P53 downstream targets,P21 (P< 0.01),BAX (P <0.05),NOXA (P<0.05), andPUMA(P ¼ 0.01) in alisertib-treated AGS xenografts than control(Fig. 5D). In accordance with the in vitro data, the xenograft
Figure 2. AURKA expression increases the HDM2 protein level in gastriccancer cell lines. A and B, dual immunofluorescence (IF) analysis forAURKA (green) and HDM2 (red) was performed on AGS and SNU-1 cellstransiently transfected with pcDNA3.1 or AURKA. The nucleus wasstained with DAPI (blue). After merging AURKA, HDM2, and DAPI imagestogether (MERGE), the data showed that overexpression of AURKA leadsto a significant increase in HDM2 protein level. The percentage of cellsthat presented expression of HDM2 was averaged from six differentmicroscopic fields formore than 100 cells. The IF data are represented asthe mean of three different experiments.
Sehdev et al.
Clin Cancer Res; 20(1) January 1, 2014 Clinical Cancer Research80
on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187

results clearly show that AURKA inhibition induces P53through downregulation of HDM2 in vivo.
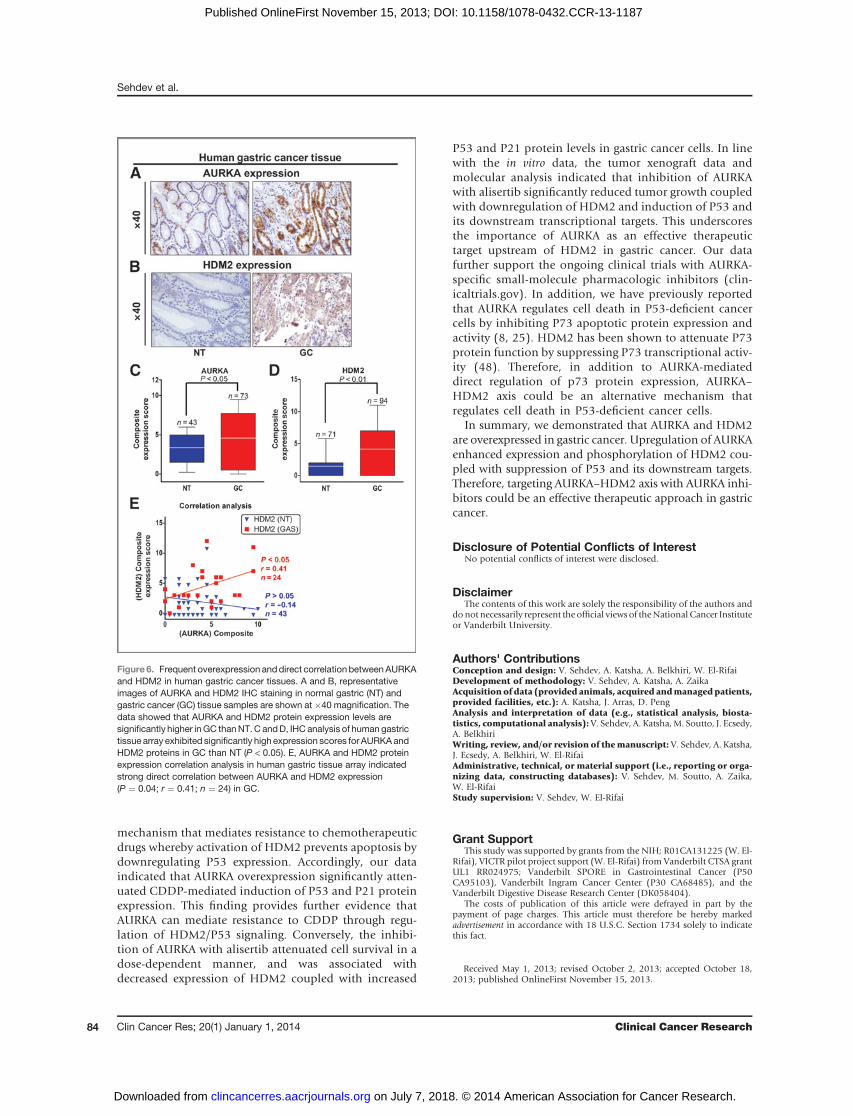
Frequent overexpression of AURKA and HDM2 aredirectly correlated in human gastric cancerWe examined AURKA and HDM2 protein expression in
human gastric TMA containing 94 gastric adenocarcinoma(GC) and71normal gastric (NT) tissue samples by IHC. TheIHC data showed that both AURKA and HDM2 expressionlevels were highly overexpressed in gastric adenocarcinomatissue samples as compared with normal tissue samples(Fig. 6A and B). The data indicated that AURKA waslocalized in both the nucleus and cytoplasm, whereasHDM2 was mainly localized in the nucleus (Fig. 6A andB). The IHC analyses revealed that HDM2 was overex-pressed in approximately 56% (53/94) of gastric adenocar-cinoma tissue samples (Fig. 6D). In addition, AURKA wassimilarly overexpressed in 70.4% (50/73) of gastric adeno-carcinoma tissues (Fig. 6C). The data also indicated asignificant direct correlation between AURKA and HDM2protein expression in gastric adenocarcinoma tissues (r ¼
0.41; P ¼ 0.04; Fig. 6E). Similar results with regard toAURKA and HDM2 overexpression and correlation wereobtained in human esophageal TMA containing 113 EACand 26 normal esophageal (NT) tissue samples by IHCanalysis (Supplementary Fig. S6). Together, these resultsdemonstrate that frequent overexpression of AURKA andHDM2 is directly correlated in human gastric and esoph-ageal adenocarcinoma.
DiscussionGastric cancer is the second most frequently diagnosed
form of cancer accounting for 10% of all cancer-relateddeaths worldwide (4). Despite improvement in chemother-apy, the 5-year survival rate for patients with gastric cancer isa dismal 22% (38, 39). Development of resistance tochemotherapeutic drugs is a major cause for disease recur-rence and low patient survival rates (40–42). Therefore,identification of molecular mechanism(s) mediating che-motherapeutic drug resistance is vital for developing moreeffective therapeutic strategies against gastric cancer. In this
Figure 3. AURKA-mediatedregulation of HDM2 promotes P53ubiquitination and attenuatesCDDP-induced P53 expression ingastric cancer cells. A, AGS cellstransiently expressing AURKA orpcDNA3.1 were treated with CDDP(5.0–10.0 mmol/L) for 12 hours andanalyzed by immunoblotting. Thedata indicated that CDDPtreatment induced P53 and P21expression in control cells. Incontrast, AURKA expressionabrogated these effects inresponse to CDDP. B,immunoprecipitation ofendogenous P53 was done inAGS cells transiently expressingpcDNA3.1, AURKA, or HDM2.Western blot analysis showedthat overexpression of AURKAsignificantly induced P53ubiquitination. C, AGS cellstransiently expressing pcDNA3.1,AURKA, or HDM2 were treatedwith Nutlin3A (4.0 mmol/L) for 12hours. Western blot analysis datasuggested that AURKA-inducedsuppression of P53 and P21expression is mediated by HDM2.IgG, immunoglobulin G; IB,immunoblotting.
AURKA Regulates HDM2 E3-Ubiquitin Ligase
www.aacrjournals.org Clin Cancer Res; 20(1) January 1, 2014 81
on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187

study, we examined the role of AURKA in regulating cellsurvival and apoptosis in gastric cancer. Herein, we reportfor the first time that HDM2 and AURKA are frequentlyoverexpressed in gastric cancer cell lines and in approx-imately half of primary gastric cancer tumors. Similarresults were obtained from EAC tumors and cell lines,suggesting that overexpression of AURKA and HDM2 isa common molecular event in upper gastrointestinalcancers.
We have previously reported that AURKA suppressesP53 transcriptional activity in gastric cancer (36). P53 is avital transcriptional factor that induces expression of pro-apoptotic proteins in response to DNA-damaging chemo-therapeutic agents. AURKA has been reported to inhibitP53 function by directly phosphorylating P53 at Ser315and thereby inducing its proteolytic degradation in H1299human non–small cell lung carcinoma cells (43). Inaddition, AURKA-mediated phosphorylation of P53 atSer215 inhibits P53 transcriptional activity and suppresses
expression of its downstream targets (24). Although wehave previously shown that overexpression of AURKAinhibits P53 expression and activity (36), we did notobserve AURKA/P53 interaction in gastric cancer cellmodels (data not shown). These results indicate that thisprotein interaction may be cell-line dependent, and sug-gest that AURKA-induced suppression of P53 is mediatedby other mechanisms. HDM2, a critical negative regulatorof P53, blocks P53 function by inhibiting its transcrip-tional activity through nuclear export into the cytoplasmand ubiquitin-mediated proteasomal degradation (26).Our results showed that overexpression of AURKA signif-icantly increased the HDM2 protein level coupled withdownregulation of P53 and P21 expression. Conversely,knockdown of endogenous AURKA or pharmacologicinhibition with alisertib attenuated HDM2 protein levelsand upregulated P53 and P21 expression. In addition,mutant AURKA failed to affect expression of HDM2,indicating that AURKA function is dependent on its kinase
Figure 4. AURKA directlyassociates and phosphorylatesHDM2 protein. A, in vitro kinaseassay followed by a subsequentimmunoprecipitation with AURKAantibodywasdoneon recombinantAURKA and HDM2 proteins.Western blot analysis of theimmunoprecipitates showed thatAURKA-mediated interaction ofHDM2 is dependent on AURKAkinase activity. B, Western blotanalysis of in vitro kinase assaywith recombinant AURKA, HDM2,AKT, and GPX7 proteinsindicated that AURKA directlyphosphorylates HDM2 in aconcentration-dependent manner.AKT and GPX7 recombinantproteins were used as positive andnegative controls, respectively.GPX7, glutathione peroxidaseenzyme 7.
Sehdev et al.
Clin Cancer Res; 20(1) January 1, 2014 Clinical Cancer Research82
on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187

activity. These results clearly showed that AURKA is apositive regulator of HDM2 in gastric cancer cells.Phosphorylation of HDM2 (S166) by AKT is known to
enhance HDM2-mediated P53 ubiquitination and degra-dation (26). Of note, we have previously shown thatAURKA can phosphorylate AKT at Ser473 (36). Usingimmunofluorescence, we confirmed the AURKA mediatesan increase in HDM2 protein levels. This is in accordancewith previous reports showing that both AURKA andHDM2 localize to the nucleus, thereby promoting mitosisand suppressing P53 function, respectively (44, 45). The co-IP data confirmed that AURKA and HDM2 were present inthe same immunoprotein complex in gastric cancer cells.The co-IP and in vitro kinase assay data indicated, for the firsttime, that recombinant AURKA directly interacts with andphosphorylates HDM2 recombinant protein. We furthervalidated this novel finding and demonstrated that inhibi-
tion of AURKA with alisertib blocks AURKA/HDM2 inter-action in vitro.
Because HDM2 is a major negative regulator of P53 (46),we examined the effects of AURKA on HDM2-mediatedubiquitination of P53. The immunoprecipitation data indi-cated that AURKA overexpression substantially enhancedHDM2-mediated ubiquitination of P53. To examine ifAURKA-mediated suppression of P53 is dependent onHDM2/P53 interaction,weusedNutlin3A, an investigation-al HDM2-specific inhibitor with antitumor activity (47), todisrupt this protein interaction. Indeed, the data showedthat Nutlin3A treatment can abrogate AURKA-mediatedinhibition of P53, confirming that HDM2/P53 interactionis critical for AURKA-dependent suppression of P53.
Our aforementioned results confirm that AURKA activ-ity is essential for enhancing HDM2-induced ubiquitina-tion and degradation of P53. This could be an important
Figure 5. Alisertib exhibitsantitumor activity, suppressesHDM2, and enhances P53 functionin vivo. AGS xenograft tumorsweretreated with alisertib (30 mg/kg) for21 days and tumor size wasmeasured every 4 days. A, the dataindicated that alisertib hassignificant antitumor activityagainst AGS xenografts. B andC, IHC of AGS xenograft tumorsshowed that inhibition of AURKAsuppressed HDM2 expression andinduced P53 in vivo. D, qRT-PCRanalysis of AGS xenograft tumorsrevealed that blocking AURKAwith alisertib enhanced P53transcriptional activity as indicatedby elevated mRNA levels of P21,BAX, NOXA, and PUMAdownstream target genes.��, P < 0.01.
AURKA Regulates HDM2 E3-Ubiquitin Ligase
www.aacrjournals.org Clin Cancer Res; 20(1) January 1, 2014 83
on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187
mechanism that mediates resistance to chemotherapeuticdrugs whereby activation of HDM2 prevents apoptosis bydownregulating P53 expression. Accordingly, our dataindicated that AURKA overexpression significantly atten-uated CDDP-mediated induction of P53 and P21 proteinexpression. This finding provides further evidence thatAURKA can mediate resistance to CDDP through regu-lation of HDM2/P53 signaling. Conversely, the inhibi-tion of AURKA with alisertib attenuated cell survival in adose-dependent manner, and was associated withdecreased expression of HDM2 coupled with increased
P53 and P21 protein levels in gastric cancer cells. In linewith the in vitro data, the tumor xenograft data andmolecular analysis indicated that inhibition of AURKAwith alisertib significantly reduced tumor growth coupledwith downregulation of HDM2 and induction of P53 andits downstream transcriptional targets. This underscoresthe importance of AURKA as an effective therapeutictarget upstream of HDM2 in gastric cancer. Our datafurther support the ongoing clinical trials with AURKA-specific small-molecule pharmacologic inhibitors (clin-icaltrials.gov). In addition, we have previously reportedthat AURKA regulates cell death in P53-deficient cancercells by inhibiting P73 apoptotic protein expression andactivity (8, 25). HDM2 has been shown to attenuate P73protein function by suppressing P73 transcriptional activ-ity (48). Therefore, in addition to AURKA-mediateddirect regulation of p73 protein expression, AURKA–HDM2 axis could be an alternative mechanism thatregulates cell death in P53-deficient cancer cells.
In summary, we demonstrated that AURKA and HDM2are overexpressed in gastric cancer. Upregulation of AURKAenhanced expression and phosphorylation of HDM2 cou-pled with suppression of P53 and its downstream targets.Therefore, targeting AURKA–HDM2 axis with AURKA inhi-bitors could be an effective therapeutic approach in gastriccancer.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
DisclaimerThe contents of this work are solely the responsibility of the authors and
donot necessarily represent the official views of theNational Cancer Instituteor Vanderbilt University.
Authors' ContributionsConception and design: V. Sehdev, A. Katsha, A. Belkhiri, W. El-RifaiDevelopment of methodology: V. Sehdev, A. Katsha, A. ZaikaAcquisitionofdata (provided animals, acquired andmanagedpatients,provided facilities, etc.): A. Katsha, J. Arras, D. PengAnalysis and interpretation of data (e.g., statistical analysis, biosta-tistics, computational analysis):V. Sehdev, A. Katsha,M. Soutto, J. Ecsedy,A. BelkhiriWriting, review, and/or revision of themanuscript: V. Sehdev, A. Katsha,J. Ecsedy, A. Belkhiri, W. El-RifaiAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): V. Sehdev, M. Soutto, A. Zaika,W. El-RifaiStudy supervision: V. Sehdev, W. El-Rifai
Grant SupportThis study was supported by grants from the NIH; R01CA131225 (W. El-
Rifai), VICTR pilot project support (W. El-Rifai) from Vanderbilt CTSA grantUL1 RR024975; Vanderbilt SPORE in Gastrointestinal Cancer (P50CA95103), Vanderbilt Ingram Cancer Center (P30 CA68485), and theVanderbilt Digestive Disease Research Center (DK058404).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received May 1, 2013; revised October 2, 2013; accepted October 18,2013; published OnlineFirst November 15, 2013.
Figure 6. Frequent overexpression anddirect correlation betweenAURKAand HDM2 in human gastric cancer tissues. A and B, representativeimages of AURKA and HDM2 IHC staining in normal gastric (NT) andgastric cancer (GC) tissue samples are shown at�40 magnification. Thedata showed that AURKA and HDM2 protein expression levels aresignificantly higher inGC thanNT.C andD, IHC analysis of human gastrictissue array exhibited significantly high expression scores for AURKAandHDM2 proteins in GC than NT (P < 0.05). E, AURKA and HDM2 proteinexpression correlation analysis in human gastric tissue array indicatedstrong direct correlation between AURKA and HDM2 expression(P ¼ 0.04; r ¼ 0.41; n ¼ 24) in GC.
Sehdev et al.
Clin Cancer Res; 20(1) January 1, 2014 Clinical Cancer Research84
on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187

References1. Hohenberger P, Gretschel S. Gastric cancer. Lancet 2003;362:
305–15.2. Reim D, Gertler R, Novotny A, Becker K, Buschenfelde CM, Ebert M,
et al. Adenocarcinomasof the esophagogastric junction aremore likelyto respond to preoperative chemotherapy than distal gastric cancer.Ann Surg Oncol 2012;19:2108–18.
3. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimatesofworldwide burden of cancer in 2008:GLOBOCAN2008. Int JCancer2010;127:2893–917.
4. JemalA,BrayF,CenterMM,Ferlay J,WardE, FormanD.Global cancerstatistics. CA Cancer J Clin 2011;61:69–90.
5. Cronin J, McAdam E, Danikas A, Tselepis C, Griffiths P, Baxter J, et al.Epidermal growth factor receptor (EGFR) is overexpressed in high-grade dysplasia and adenocarcinoma of the esophagus and mayrepresent a biomarker of histological progression in Barrett's esoph-agus (BE). Am J Gastroenterol 2011;106:46–56.
6. Rugge M, Fassan M, Zaninotto G, Pizzi M, Giacomelli L, Battaglia G,et al. Aurora kinaseA in Barrett's carcinogenesis. HumPathol 2010;41:1380–6.
7. DengN, Goh LK,WangH, Das K, Tao J, Tan IB, et al. A comprehensivesurvey of genomic alterations in gastric cancer reveals systematicpatterns of molecular exclusivity and co-occurrence among distincttherapeutic targets. Gut 2012;61:673–84.
8. Sehdev V, Peng D, Soutto M, Washington MK, Revetta F, Ecsedy J,et al. The aurora kinase A inhibitor MLN8237 enhances cisplatin-induced cell death in esophageal adenocarcinoma cells. Mol CancerTher 2012;11:763–74.
9. Lord RV, O'Grady R, Sheehan C, Field AF, Ward RL. K-ras codon 12mutations in Barrett's oesophagus and adenocarcinomas of theoesophagus and oesophagogastric junction. J Gastroenterol Hepatol2000;15:730–6.
10. Matuschek C, Bolke E, Peiper M, Knoefel WT, Budach W, Erhardt A,et al. The role of neoadjuvant and adjuvant treatment for adenocar-cinoma of the upper gastrointestinal tract. Eur J Med Res 2011;16:265–74.
11. El-Rifai W, Sarlomo-Rikala M, Andersson LC, Knuutila S, Miettinen M.DNA sequence copy number changes in gastrointestinal stromaltumors: tumor progression and prognostic significance. Cancer Res2000;60:3899–903.
12. Katayama H, Ota T, Jisaki F, Ueda Y, Tanaka T, Odashima S, et al.Mitotic kinase expression and colorectal cancer progression. J NatlCancer Inst 1999;91:1160–2.
13. Sakakura C, Hagiwara A, YasuokaR, Fujita Y, Nakanishi M,Masuda K,et al. Tumour-amplified kinaseBTAK is amplified and overexpressed ingastric cancers with possible involvement in aneuploid formation. Br JCancer 2001;84:824–31.
14. Wang X, Zhou YX, Qiao W, Tominaga Y, Ouchi M, Ouchi T, et al.Overexpression of aurora kinase A in mouse mammary epitheliuminduces genetic instability preceding mammary tumor formation.Oncogene 2006;25:7148–58.
15. Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, et al. Tumouramplified kinase STK15/BTAK induces centrosome amplification,aneuploidy and transformation. Nat Genet 1998;20:189–93.
16. Furukawa T, Sunamura M, Horii A. Molecular mechanisms of pancre-atic carcinogenesis. Cancer Sci 2006;97:1–7.
17. Yen CC, Yeh CN, Cheng CT, Jung SM, Huang SC, Chang TW, et al.Integrating bioinformatics and clinicopathological research of gastro-intestinal stromal tumors: identificationof aurora kinaseAasapoor riskmarker. Ann Surg Oncol 2012;19:3491–9.
18. Fang Z, Xiong Y, Li J, Liu L, Li M, ZhangC, et al. Copy-number increaseof AURKA in gastric cancers in aChinese population: a correlationwithtumor progression. Med Oncol 2011;28:1017–22.
19. Dar AA, Goff LW, Majid S, Berlin J, El-Rifai W. Aurora kinaseinhibitors–rising stars in cancer therapeutics? Mol Cancer Ther2010;9:268–78.
20. Van Cutsem E, Van de Velde C, Roth A, Lordick F, Kohne CH,Cascinu S, et al. Expert opinion on management of gastric andgastro-oesophageal junction adenocarcinoma on behalf of theEuropean Organisation for Research and Treatment of Cancer
(EORTC)-gastrointestinal cancer group. Eur J Cancer 2008;44:182–94.
21. Ilson DH. Docetaxel, cisplatin, and fluorouracil in gastric cancer: doesthe punishment fit the crime? J Clin Oncol 2007;25:3188–90.
22. Sumi K, Tago K, Kasahara T, Funakoshi-Tago M. Aurora kinase Acritically contributes to the resistance to anti-cancer drug cisplatin inJAK2 V617F mutant-induced transformed cells. FEBS Lett 2011;585:1884–90.
23. Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplifica-tion overrides the mitotic spindle assembly checkpoint, inducingresistance to Taxol. Cancer Cell 2003;3:51–62.
24. Liu Q, Kaneko S, Yang L, Feldman RI, Nicosia SV, Chen J, et al.Aurora-A abrogation of p53 DNA binding and transactivation activ-ity by phosphorylation of serine 215. J Biol Chem 2004;279:52175–82.
25. Dar AA, Belkhiri A, Ecsedy J, Zaika A, El-Rifai W. Aurora kinase Ainhibition leads to p73-dependent apoptosis in p53-deficient cancercells. Cancer Res 2008;68:8998–9004.
26. WadeM,WangYV,Wahl GM. Thep53 orchestra:Mdm2andMdmxsetthe tone. Trends Cell Biol 2010;20:299–309.
27. Rayburn E, Zhang R, He J, Wang H. MDM2 and human malignancies:expression, clinical pathology, prognostic markers, and implicationsfor chemotherapy. Curr Cancer Drug Targets 2005;5:27–41.
28. Fakharzadeh SS, Trusko SP, George DL. Tumorigenic potential asso-ciated with enhanced expression of a gene that is amplified in amousetumor cell line. EMBO J 1991;10:1565–9.
29. Soussi T, Asselain B, Hamroun D, Kato S, Ishioka C, ClaustresM, et al.Meta-analysis of the p53 mutation database for mutant p53 biologicalactivity reveals amethodologic bias inmutation detection. Clin CancerRes 2006;12:62–9.
30. DarAA,Belkhiri A, El-RifaiW. Theaurora kinaseA regulatesGSK-3betain gastric cancer cells. Oncogene 2009;28:866–75.
31. Katsha A, SouttoM, Sehdev V, Peng D,WashingtonMK, PiazueloMB,et al. Aurora kinase a promotes inflammation and tumorigenesis inmice and human gastric neoplasia. Gastroenterology 2013;S0016-5085:01270–5.
32. Soutto M, Belkhiri A, Piazuelo MB, Schneider BG, Peng D, Jiang A,et al. Loss of TFF1 is associated with activation of NF-kappaB-mediated inflammation and gastric neoplasia in mice and humans.J Clin Invest 2011;121:1753–67.
33. ZhuS,Hong J, TripathiMK, Sehdev V,Belkhiri A, El-RifaiW.RegulationofCXCR4-mediated invasion byDARPP-32 in gastric cancer cells.MolCancer Res 2013;11:86–94.
34. Gorgun G, Calabrese E, Hideshima T, Ecsedy J, Perrone G, Mani M,et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotox-icity and cell-cycle arrest in multiple myeloma. Blood 2010;115:5202–13.
35. Belkhiri A, Zaika A, Pidkovka N, Knuutila S, Moskaluk C, El-Rifai W.Darpp-32: a novel antiapoptotic gene in upper gastrointestinal carci-nomas. Cancer Res 2005;65:6583–92.
36. Dar AA, Zaika A, Piazuelo MB, Correa P, Koyama T, Belkhiri A, et al.Frequent overexpression of Aurora Kinase A in upper gastrointestinaladenocarcinomas correlateswith potent antiapoptotic functions. Can-cer 2008;112:1688–98.
37. Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathwaypromotes translocation of Mdm2 from the cytoplasm to the nucleus.Proc Natl Acad Sci U S A 2001;98:11598–603.
38. Howlader N, Ries LA, Stinchcomb DG, Edwards BK. The impact ofunderreported Veterans Affairs data on national cancer statistics:analysis using population-based SEER registries. J Natl Cancer Inst2009;101:533–6.
39. Roth AD. Chemotherapy in gastric cancer: a never ending saga. AnnOncol 2003;14:175–7.
40. Ohi S, Takahashi N, NinomiyaK,NakajimaM,HashimotoH, TachibanaT, et al. Establishment and characterization of a cisplatin-resistant cellline (IGSK-1) from a poorly differentiated gastric adenocarcinoma.Hum Cell 2007;20:15–22.
41. Zhao L, Pan Y, Gang Y, Wang H, Jin H, Tie J, et al. Identification ofGAS1asanepirubicin resistance-relatedgene inhumangastric cancer
AURKA Regulates HDM2 E3-Ubiquitin Ligase
www.aacrjournals.org Clin Cancer Res; 20(1) January 1, 2014 85
on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187

cells with a partially randomized small interfering RNA library. J BiolChem 2009;284:26273–85.
42. Huang S, Chen M, Shen Y, Shen W, Guo H, Gao Q, et al. Inhibition ofactivated Stat3 reverses drug resistance to chemotherapeutic agentsin gastric cancer cells. Cancer Lett 2012;315:198–205.
43. Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, et al.Phosphorylation by aurora kinase A induces Mdm2-mediated desta-bilization and inhibition of p53. Nat Genet 2004;36:55–62.
44. Carmena M, Earnshaw WC. The cellular geography of aurora kinases.Nat Rev Mol Cell Biol 2003;4:842–54.
45. Schuster K, Fan L, Harris LC. MDM2 splice variants predominantlylocalize to the nucleoplasm mediated by a COOH-terminal nuclearlocalization signal. Mol Cancer Res 2007;5:403–12.
46. Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res2003;1:1001–8.
47. Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53protein-protein interaction to reactivatep53 function: a novel approachfor cancer therapy. Annu Rev Pharmacol Toxicol 2009;49:223–41.
48. Maisse C, Guerrieri P, Melino G. p73 and p63 protein stability: the wayto regulate function? Biochem Pharmacol 2003;66:1555–61.
Sehdev et al.
Clin Cancer Res; 20(1) January 1, 2014 Clinical Cancer Research86
on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187

Correction
Correction: HDM2 Regulation by AURKAPromotes Cell Survival in Gastric Cancer
In this article (Clin Cancer Res 2014;20:76–86), which was published in the January1, 2014, issue ofClinical Cancer Research (1), an error occurred during the assembly ofthe final Fig. 5A that resulted in the publication of an incorrect representative animalimage. The correct Fig. 5A is shown below. The authors state that this clarificationdoes not change the results, scientific content, interpretations, or conclusions of thearticle. The authors regret this error.
Reference1. Sehdev V, Katsha A, Arras J, PengD, SouttoM, Ecsedy J, et al. HDM2 regulation by AURKApromotes
cell survival in gastric cancer. Clin Cancer Res 2014;20:76–86.
Published online February 2, 2015.doi: 10.1158/1078-0432.CCR-14-3207�2015 American Association for Cancer Research.
AGS xenograft
Alisertib
n = 5n = 5
CtrlA Ctrl
Alisertib
400
300
200
100
0 0 5 10Day(s) of treatment
15 20
Figure 5.A, alisertib exhibits antitumor activity, suppresses HDM2, and enhancesP53 function in vivo. AGS cancer cells were injected in the flanks of 10 femalenude mice. The treatment group received alisertib (30 mg/kg) for21 days. Tumor size was measured every 4 days. The data indicated thatalisertib has significant antitumor activity againstAGS xenografts; �� ,P<0.01.The frozen tissues samples available from the bio-banked tumorxenograft repository of this experiment were used for the experimentsshown in panels B, C, and D.
ClinicalCancerResearch
www.aacrjournals.org 661

2014;20:76-86. Published OnlineFirst November 15, 2013.Clin Cancer Res Vikas Sehdev, Ahmed Katsha, Janet Arras, et al. CancerHDM2 Regulation by AURKA Promotes Cell Survival in Gastric
Updated version
10.1158/1078-0432.CCR-13-1187doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2013/11/15/1078-0432.CCR-13-1187.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/20/1/76.full#ref-list-1
This article cites 48 articles, 15 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/20/1/76.full#related-urls
This article has been cited by 2 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/20/1/76To request permission to re-use all or part of this article, use this link
on July 7, 2018. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 15, 2013; DOI: 10.1158/1078-0432.CCR-13-1187


















