
“Good Clinical Practices” in Meeting Regulatory Responsibilities Terry VandenBosch, RN, PhD,...
46
“Good Clinical Practices” in Meeting Regulatory Responsibilities Terry VandenBosch, RN, PhD, CIP, CCRP Senior Research Compliance Associate Office of Human Research Compliance Review University of Michigan September 15, 2010
-
date post
19-Dec-2015 -
Category
Documents
-
view
214 -
download
0
Transcript of “Good Clinical Practices” in Meeting Regulatory Responsibilities Terry VandenBosch, RN, PhD,...

“Good Clinical Practices” in Meeting Regulatory
Responsibilities
Terry VandenBosch, RN, PhD, CIP, CCRP
Senior Research Compliance Associate
Office of Human Research Compliance Review
University of Michigan
September 15, 2010

Today’s Discussion
Discuss the growing field of “regulatory science”Describe responsibilities of FDA sponsor-
investigatorsContrast ethical principles and ethical conduct in
research with investigator GCP activitiesIdentify common sense principles for investigators
to implement best practices and GCP in clinical studies

Evidence-based Regulatory Science
Reactions to Research Participation Questionnaire for Adults, Children (RRPQ-C) and the RRPQ for Parents (RRPQ-P)
Comparison of time to enrollment for emergency brain-injured subjects in a study with proxy consent versus exception from consent
Clinical researchers; 1) can address human subjects research compliance issues in
conjunction with answering their main research questions2) should conduct studies that address regulatory compliance issues3) can demonstrate investigator proposed research compliance
procedures are not based on whim or disregard for subject rights and welfare.
Kassam-Adams & Newman, 2002-2006
Wright et al, 2008
Maio, 2008, Excerpts Letter to the Editor, Annals of Emergency Medicine

FDA Sponsor-Investigator
FDA Sponsor defined:“…a person who takes responsibility for and initiates
a clinical investigation (21 CFR 312.3(b)” of a drug, device or biologic.
A sponsor may be an individual, a company, an academic institution, a governmental agency or another organization. (Not the same as a financial sponsor)
FDA Sponsor-Investigator:A sponsor-investigator both initiates and conducts the
clinical investigation

What are FDA Sponsor Responsibilities?
Determine if a study needs an IND/IDE or is exempt (biologics, off-label use & neutraceuticals most difficult)
Submit an IND or IDE application to the FDA
Follow the IND/IDE application approval process and administrative actions
Meet ongoing IND/IDE sponsor responsibilities during study conduct

FDA Sponsor Responsibilities during Study Conduct Assure the investigator conducts the study according to the protocol
and meets investigator responsibilities Maintain an effective IND/IDE with FDA
FDA submissions-amendment updates, adverse events, annual reports, any other FDA communications
Ensure proper monitoring of study data integrity and subject safety Assure test-article control and accountability
Provide for records of disposal of the test-article and control of the test article. Ensure the test article is administered to subjects under the investigator or sub-
investigator’s personal supervision and by investigators or staff authorized to administer it
Keep adequate and accurate sponsor records (Maintenance of sponsor master files/binders)

FDA Monitoring and Ensuring Quality for SI Studies
FDA recommends sponsors build in quality monitoring so problems can be detected and corrected as close to real time as possible
Focus on key parameters of risk to trial integrity and data quality and to subject safety and protection which allows sponsors to set priorities for using resources appropriately

Ethical Principles & GCP
Ethical principles Inform decision-making Basis for federal regulations and guidance
Past abuses of ethical principles stimulated development of GCP Nuremburg Tuskegee syphilis study Willowbrook retarded children hepatitis study
Are these ethical lapses and abuses all in the past? Nicole Wan (healthy volunteer- died), 1996 Jesse Gelsinger (ineligible-died), 1999 Ellen Roche (healthy volunteer-died), 2001 Inadequate monitoring with overdose of pediatric subjects-FDA
warning letter to Pfizer, April 9, 2010

Ethical PrinciplesBelmont Report-1979
Respect for personsAcknowledge autonomy of the individualProtect those with diminished autonomy
BeneficenceDo no harmMaximize possible benefitsMinimize possible harms
JusticeFairness in distribution of burdens and benefits of
research participation

RegulationsRegulations developed in response to
egregious, harmful research conductDeveloped on ethical principlesRegulations are not specificRegulations don’t address everything that is
important in the protection of human subjects No regulations address decision-making
capacity of participantsRegulations MUST be met or sanctions

“GCP” DefinitionA series of key activities that assures ethical principles,
regulations, laws, university policies & procedures are met NIH-”Scientific and ethical standards of human subject
research”FDA-”Standard for the design, conduct, performance,
monitoring, auditing, recording, analyses, and reporting of clinical trials”
Data and reported results are credible, and accurate
Protects rights, welfare and confidentiality of subjects
= Quality Data
= Ethics

Why GCP?
FailuresEthical AtrocitiesPreventable Research Deaths/InjuryScientific Fraud
Subject safetyPublic trust and support of research missionAssure valid data for evidence-based Health CareDrug development trajectory long, arduous,
expensive (GCP assures safety and quality data)Useful products brought to market with known
safety profile and effectiveness

Informed Consent
Obtain Informed Consent

Informed Consent as a Process Interpersonal communication skills assess subject understanding
and motivation to participate Informed consent is freely given and is obtained from each subject
prior to study participation The consent discussion is in language understandable to the
participant or the representative and is done by a qualified person The consent process provides sufficient opportunity for the
participant or the participant’s legally authorized representative to consider whether to participate
The consent process minimizes the possibility of coercion or undue influence (Research is not the same as therapeutic txmt)
The consent discussion is free of exculpatory language The IRB approved document without any changes and with the
elements of informed consent is used Children’s “assent” & “Parental Permission” Adapted from AAHRPP, 2009

Informed Consent ReviewPrivacy respectedVoluntaryConducted as a process by PIProcess follows the IRB approved protocol
Waiver of consent possible
Consent signed prior to any study proceduresCopy of consent given to subjectRe-consent completed and documented as
appropriate100% of consents used correct IRB approved
version and were appropriately signed and dated
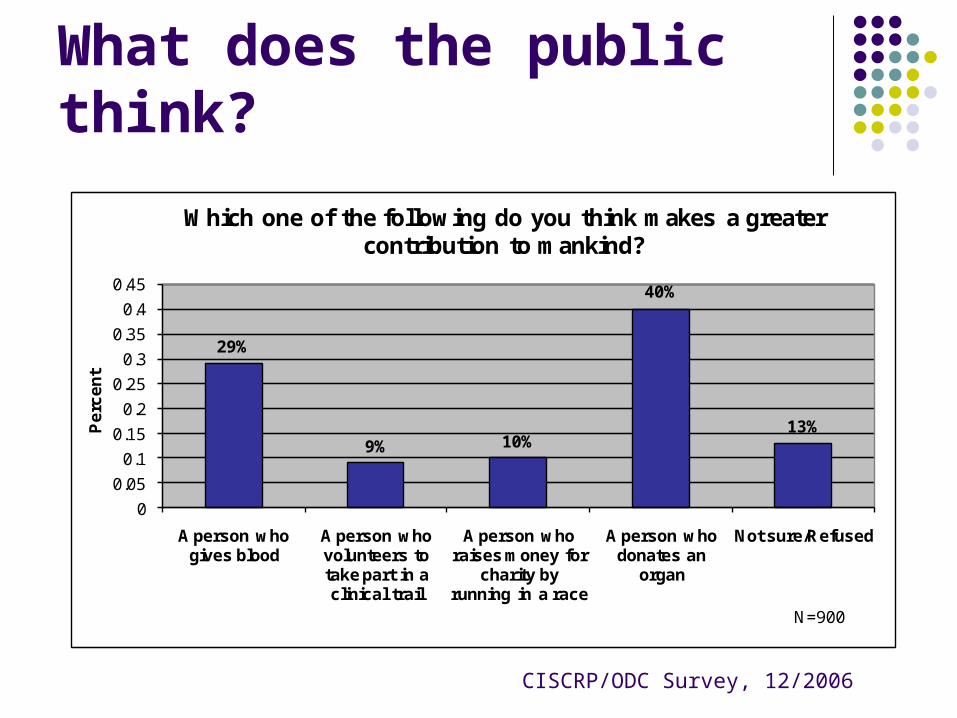
What does the public think?
29%
9% 10%
40%
13%
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
0.45
A person who gives blood
A person who volunteers to take part in a clinical trail
A person who raises money for
charity by running in a race
A person who donates an
organ
Not sure/Refused
Pe
rce
nt
N=900
Which one of the following do you think makes a greater contribution to mankind?
CISCRP/ODC Survey, 12/2006

Subject Safety
Provide for Subject Safety
and Clinical Care

Adverse Events & Harms.
What is an Adverse event (AE)?Prevent, monitor for, identify, provide immediate care for, track,
analyze cause, provide follow up treatment, report to IRB, may submit protocol amendment or changes to consent document & notify Sponsor (FDA)
IRBMED Guidance and timetable for reporting AEs at http://med.umich.edu/irbmed/ae_orio/ae_report.htm
HarmsPhysicalPsychosocialSocialEconomicLegalDignitary

What Can Result in Harms? The protocol/treatment
Side effects of drugs/biologics or adverse device effects NOT following the protocol NOT maintaining up-to-date records NOT maintaining communication with investigator and/or study
sponsor Prevent harm
Qualified person monitors overall study Monitor laboratory results and tests and treat as appropriate May withdraw subject from study Know emergency procedures for breaking a study blind Keep primary care provider in communication as appropriate
AEs are graded by Seriousness Relatedness to the study Expected/Unexpected

The Protocol
Follow the Protocol or Amend it

A Tension in Research
“The principal duty of a physician is to the well-being of the individual.”
“The principal duty of society (social ethics) is to the greatest good for the greatest number of people.”
Research is protocol drivenClinicians often want to adapt the protocol for
an individual

Know and Follow the Protocol Changes to the protocol, “…may not be initiated without IRB review
and approval except when necessary to eliminate apparent immediate hazards to the subject.”
Read it FDA-each person on study team signs it
Protocol Readily Available No mix ups-Clearly label current version
Follow it Prevent and track any protocol deviations
Notes to file-circumstances, CAPA Report to IRB and sponsor as applicable Amend protocol with IRB as needed
Follow randomization procedures If applicable, procedures follow data safety and monitoring plan
(DSMP) submitted to IRB and funding agencies

Confidentiality
Maintain Confidentiality

Data Confidentiality & Security: Outcomes
Data maintained according to IRB approved protocol Access to confidential data is restrictedSafe & secure storage
Don’t share passwords!
UM ITS Safe Computing Polices http://www.safecomputing.umich.edu/faculty&staff.php
Mobile device security for researchers at http://www.safecomputing.umich.edu/MDS
UM Electronic data security Questions to Guide Research Protections at OHRCR website http://www.ohrcr.umich.edu/news/electronicdata.pdf

Record Keeping & Reports
Maintain Accurate, Current, Organized Records and Submit Reports

Study Files
Organized, accurate, up-to-dateDirect/Indirect subject identifiers
Direct-subject identifiers stored with data Indirect-subject identifiers in key & not stored with data
Informed consent-stored with files?FDA-Complete, sign and submit FDA Form 1572Work efficiently
Study schema of subject progress for complex procedures Checklist of forms completed
Maintain records for: FDA- two years after FDA approves NDA NIH- three years after study terminated HIPAA- six years after study terminated

Source Data and Documents (FDA)
All clinical trial information should be recorded, handled, and stored in a way that allows its accurate reporting, interpretation, and verification
“ALCOA” - Attributable, Legible, Contemporaneous, Original, and Accurate (USE “memo to file”; NEVER use white out or back date)
Source Document Definition Original documents, data, and records, (e.g., ALL study records such as
visits, CRF/Data Collection Forms, and, Subjective self-report instruments, hospital, clinical and office charts,
laboratory notes, notes to file, subject diaries, checklists, pharmacy dispensing records, recorded data from automated instruments, X-Rays, digital records…
Source Data Generate and Keep source documents in original records May be using Electronic Data Capture (EDC)

The IRB
Maintain Communication
with the IRB

IRB Communications and SubmissionsInteract with IRB
Ask questions Get to know UM IRB contacts
Initial IRB submission and approvalOngoing oversight
AEs, protocol deviations, unanticipated problems, DSMC reports or safety officer reports, UM OHRCR report, new information that changes risk/benefit of study participation
Continuing review
Terminate a study Don’t let it expire!

Study Oversight & Qualifications

Overall PI/Investigator Responsibilities
Ensure a study is conducted according to the protocol or study plan and applicable regulationsFDA Form 1572
Protect the rights, safety, and welfare of subjects under the investigator’s care
FDA-Control drugs, biological products, and devices under investigation
FDA Guidance for Industry: Investigator Responsibilities, Oct 2009

Study Oversight by the PI/InvestigatorAre individuals who are delegated study
tasks qualified to perform them?Have individuals received training to the
protocol and to the tasks?Is the oversight and involvement in ongoing
conduct of the study appropriate?Where reasonably possible, is there
oversight of 3rd parties?
Guidance for Industry: Investigator Responsibilities, Oct. 2009

Delegating Tasks
The investigator should ensure that any individual to whom a task is delegated is qualified by education, training, and experience (and state licensure where relevant) to perform the delegated task
The level of supervision should be appropriate to the staff, the nature of the trial, and the subject population.

Study Logs & Oversight
Delegation log with study roles, tasks and dates worked on study
Signature log with initials logTrain to protocolTraining logStay up-to-date on subject and overall study
progressRegular staff meetings (FDA-take minutes)

Adequate Resources
Appropriate facilitiesAppropriate equipment
Correct equipment availableCalibratedPreventive maintenanceStudy staff training
Proper laboratory facilities (FDA=CLIA certified)Reference ranges for laboratory testsDetails of analytical methodsQuality assurance information

Investigational Product (FDA)
Accountability for the Investigational Product

Investigational ProductProcess investigational product
Receipt (shipping) and DispensingLabelingAccountability to reconcile records for each tablet,
compounded drugSecure storage of device & device returnReturn/Destroy drug as determined by sponsor Interface with Investigational Drug Services /
Biomedical Engineering staff, as neededInvestigational products should be
manufactured, handled, and stored in accordance with applicable good current manufacturing practice (cGMP)

Essential Documents(FDA)
Maintain Study Binder

Binder Index
1. ALL important study documentation and correspondence from the study sponsor & study monitor
2. Delegation, signature log & monitoring log
3. Signed protocol4. Laboratory Information w nl. Values5. Equipment information6. Blank case report forms (CRFs)7. CVs

Additional Best Practices UMHS HIPAA Training Conflict of Interest Disclosure
Applies to all members of the study team Includes spouses and dependents May have a management plan FDA forms for financial disclosure from sponsor
Maintain all communication with sponsor (FDA) Shipping specimens
UM personnel who ship infectious substances including patient (clinical) specimens, human-derived research materials, infectious micro-organisms, certain genetically-modified organisms, etc. must complete a training program prior to shipping infectious or biological substances.

Summary of “GCP-Best Practices” Key Activities Obtain Informed consent Provide for Subject Safety & Medical Care Follow the IRB approved protocol or submit amendment to IRB Maintain Confidentiality Record keeping-Maintain accurate, current, organized records and submit reports Maintain communication with IRB Provide appropriate oversight of qualified staff FDA-Investigational Product Accountability FDA-Essential Documents Binder Additional areas
Conflict of interest Communication with sponsor (FDA) Shipping regulations
ICH-GCP has no statements on ethical principles such as risks, benefits, selection, privacy, vulnerable populations

Overall
Legal or regulatory is not always adequateA personal commitment to integrity needs to
be coupled with a firm understanding of “GCP”
The public support of research rests on its trust of scientists, scholars and the institutions
Individual actions are important

Ultimate Goal:Responsible Research Practices
“The University of Michigan is committed to the highest standards of ethical behavior by faculty, staff, and students engaged in the conduct and administration of research and other scholarly activity.”
UM Provost Policy Statement on Academic and Research Integrity

Questions?

University of Michigan Policies and ProceduresStandard Practice Guide
Approved by RegentsSection 303, http://spg.umich.edu/section/303
Human Research Protection Program (HRPP) Operations Manualhttp://www.hrpp.umich.edu/om/
IRB Guidance & SOPsSee IRB websites

Resources UM IRBMED & HBHS Workshops Join MICHR research coordinator email network Clinical Trials Network (Duke U)
Forms, education, etc. at https://www.ctnbestpractices.org FDA Device Advice & training
http://www.fda.gov/medicaldevices/deviceregulationandguidance/default.htm Virtual Regulatory Binder
http://www.partners.org/phsqi/vrb/files/index.htm NIH-Office of Human Subject Research
http://ohsr.od.nih.gov/ OHRP Guidance documents
http://www.dhhs.gov/ohrp/policy/ ICH E6
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073122.pdf


















