
Excitation and Deexcitation of...
73
Excitation and Deexcitation of Benzene R. B. CUNDALL Department o f Chemistry and Applied Chemistry University of Salford, United Kingdom and D. A. ROBINSON Department of Chemistry, University of Botswana and L. C. PEREIRA Department of Physics, University of Minho Portugal Lesotho, and Swazi 1 and , Gaborone, Botswana CONTENTS I. Introduction. .................... 148 11. The Nature of the Lower Excited States of Benzene . . 148 111. Transitions Between Lower Energy States ....... 152 A. B. C. Transitions Between Excited Singlet and Excitation from the 'Alg State. ......... 153 Emission to the 'Alg State. ........... 153 Triplet States. ................. 154 IV. V. VI . VII. VIII. IX . X. XI. Excited State Geometry. ............... 154 The Influence of the Environment on Electronic States. ....................... 155 The S1 ++ S Radiative Transition .......... 161 The S1 -++ Triplet Radiationless Transition. ..... 174 The S1 3 So Radiationless Transition ........ 177 The T1 + So Phosphorescence Transition ....... 181 The Tl-+ So Radiationless Transition ........ 189 Transitions from Higher (n > 1) Excited States. ... 192 14 7 Advances in Photochemistry, Volume 10 Edited by ,James N. Pitts Jr., George S. Hammond, Klaus Gollnick Copyright © 1977 John Wiley & Sons, Inc.
Transcript of Excitation and Deexcitation of...

Excitation and Deexcitation of Benzene
R. B. CUNDALL
Department o f Chemistry and Appl ied Chemistry U n i v e r s i t y o f Sal ford, Uni ted Kingdom
and
D. A. ROBINSON
Department o f Chemistry, U n i v e r s i t y o f Botswana
and
L. C . PEREIRA
Department o f Physics, U n i v e r s i t y o f Minho Portugal
Lesotho, and Swazi 1 and , Gaborone, Botswana
CONTENTS
I. Introduction. . . . . . . . . . . . . . . . . . . . . 148
11. The Nature of the Lower Excited States of Benzene . . 148 111. Transitions Between Lower Energy States . . . . . . . 152
A. B. C. Transitions Between Excited Singlet and
Excitation from the 'Alg State. . . . . . . . . . 153 Emission to the 'Alg State. . . . . . . . . . . . 153 Triplet States. . . . . . . . . . . . . . . . . . 154
IV. V.
VI . VII.
VIII.
IX . X. XI.
Excited State Geometry. . . . . . . . . . . . . . . . 154 The Influence of the Environment on Electronic States. . . . . . . . . . . . . . . . . . . . . . . . 155
The S1 ++ S Radiative Transition . . . . . . . . . . 161 The S1 -++ Triplet Radiationless Transition. . . . . . 174 The S1 3 So Radiationless Transition . . . . . . . . 177 The T1 + So Phosphorescence Transition . . . . . . . 181 The Tl-+ So Radiationless Transition . . . . . . . . 189 Transitions from Higher (n > 1) Excited States. . . . 192
14 7
Advances in Photochemistry, Volume 10 Edited by ,James N. Pitts Jr., George S. Hammond, Klaus Gollnick
Copyright © 1977 John Wiley & Sons, Inc.

148 R. B. Cundall, et al.
XII. Relevant Photochemical Reactions of Excited States of Benzene . . . . . . . . . . . . . . . . . . 194
XIII. Benzene Excimer . . . . . . . . . . . . . . . . . . . 196 xIV. Conclusion. . . . . . . . . . . . . . . . . . . . . . 201
Acknowledgements . . . . . . . . . . . . . . . . . . . . . . 201
References . . . . . . . . . . . . . . . . . . . . . . . . . 201
I. INTRODUCTION
A large amount of spectroscopic data are now available for benzene, providing much information on photophysics and photo- chemistry properties of the lowest excited states of this molecule. Nevertheless the nature of the various radiative and nonradiative transitions involved in the deexcitation of the first excited states is far from being completely understood.
of benzene in gas, liquid, and solid phases, with special em- phasis on the experimental advances obtained by spectroscopic techniques in the past few years. Throughout, the symbolism used is that shown in the following reaction scheme:
This work reviews the photophysics and relevant photochemistry
11. THE NATURE OF THE LOWER EXCITED STATES OF BENZENE
The near-ultraviolet spectral characteristics, and photo- physical decay processes of benzene are generally described in terms of transitions between the various molecular orbitals formed by linear combination of the carbon 2p atomic orbitals which have symmetries bZg, eZg(two), elg(two), and aZu within the

Excitation of Benzene 149
D6h point group. of the orbitals are not known precisely, and are, to a degree, dependent upon the complexity of the calculatory method, the sym- metries and ordering of energies generated by the simpler
4 theories remain valid.
Singly excited configurations are produced by promotion of an electron from one of the two degenerate el the degenerate e2 orbitals (1). The predgcted degeneracy of the states so proxuced (B2u, Blu, Elu) is removed by electron interaction. Calculations (2) have shown that the ordering by energy of €he tra sitions in which spin conservation is observed should be Elu > 'B
Experimental oiEervations in the gas phase ( 3 ) , solution ( 4 ) , low-temperature glasses (5), rare gas matrices (6), and the crystal phase (7) have all confirmed that the lowest excited sing- let state has symmetry, B2u, and that a transition from the ground to this state occurs around 260 nm. Band intensity is very low (E at h (max) brationally induced transition. Two more intense bands are ob- served on the high energy side of this 1B2u f Alg absorption; a medium-intensity band (E % 9000 ( 8 ) ) around 205 nm and a high- intensity band (E % 70,000 (8)) around 190 nm.
transition though some controversy is still attached to thls as- signment. Several recent semi-empirical (9-11), and ab initio (11) calculations place an E2 cited 71-71
PIT* excitation; nm band to a lEZg f 'A1 transition. A doubly excited configura- tion may be of comparabfe energy to a singly excited configura- tion due to the sensitivity of the energy of the E2g configura- tion to electron interaction. Contrary evidence comes from another & initio calculation (14) which places the IE state
semi-empirical (15) and ab initio (16) have placed the elusive 'E2 state above 'ElU. that even the most sophisticated computations presently available, incorporating multiple excited configurations and multicenter cor- rections, are unsatisfactory in assigning even relative energies to the doubly excited TI--TI** states. the extreme sensitivity of E state energies to the values chosen for the resonance integrals fb) .
Though the absolute energies and energy spacings
The ground state benzene configuration is (a2u) 2 , (el,) . orbitals to one of
?u* > 'B
250 (8) )* as befits a symmetry forbidden, but vi-
The 205 nm band is widely believed to be the lBlU f lAls
state, arising from a doubly ex- ** transition below ghe Blu state arising from a single su porting an early assignment (13) of the 205
between the lElu and lBlU ~ - - 7 1 * states. Still other cafculations, g
It is clear from this confusing situation
This is apparently due to
*Extinction coefficient units are M-' cm-' throughout the article.

150 k. B. Cundall, et al.
1 f A
transition is consistent with the expected ordering of state energies for those configurations arising from ezu + elg excitation, and with the assigned position of 1Bzu. early inconclusive reports (18), recent evidence from vibrational analyses of low temperature absorption data in rare gas matrices and films (19) and of reflection spectra (20) confirms the lBlU assignment. However, electron impact spectra (21) under high resolution suggests that two electronic transitions occur in the 205 nm region; and this has been confirmed recently by absorption studies on benzene in perfluoro-n-hexane (22) and rare gas matrices (22,23), at low temperature. In these environments the shift in the lEgg -+ 'A1 lBlU lAlg transition %ue to differences in the charge distribu- tion of the configurations and allows separate observation of the two bands. Available experimental evidence favors the energy sequence deduced by Koutecky et al. (9) :
Ig The common assignment of the 205 nm band to the 'B
lu
Despite
transition is greater than that of the
The high-intensity band (E Q, 70,000) around 190 nm is usually as- signed to the lElu * 'Alg symmetry-allowed n - ~ * transition, though some contribution to the intensity may come from the super- position of the first member of one of the Rydberg series. The P-TI assignment has been confirmed (24) from a vibrational anal- ysis of the low-temperature spectrum of benzene in a rare gas matrix. Recent studies using multiphoton ionization spectroscopy have been considered to provide some indications of a lE1 as an alternative to assignments for the elusive lEzK stage (25). This conclusion awaits confirmation or otherwise by urther experiments.
varying extents affected by the molecular environment, as dis- cussed later. Table 1 contai s approximate Franck-Condon maxima for the transitions from the
In addition to transitions in the n manifold discussed above, some attention has been given recently to possible 0--TI
states in the ultraviolet nonradiative transitions from the 1R2u state. Callomon et al. (27) have ostulated the transitions from higher vibrational levels of pB2u to a 3E2u(0-~ ) state to explain theonsetof diffuseness in the high resolution absorption spectrum of vapor phase benzene in the 5.0 eV region. Ab initio (12,28) calculations have predicted the presence of at least one 0 state (e2 ) lying between two filled r-molecular orbitals. Photoionizafion (29,30) experiments, and other evidence (31) ap- pear to agree with the conclusion that a 0-T transition may be
*
state
The relevant energies associated with each state are, to
P Alg state. *
*
*

Excitation of Benzene 151
TABLE 1 . Approximate Franck-Condon maxima for transitions from lAlg ground s ta te t o the lower lying a-electronic s ta tes
1 + Elu
1 + Blu
1 - E 2g
% 7.25 eVa
% 6.75 eV
% 6.08 eV
% 5.9 eV
The location of the second E 2g state, 'E: has been suggested by Birks (26), from an analysis of publisged data on Sx f lBlU absorption experiments.
a
expected in the ultraviolet region conventionally associated with T-a transitions. A recent CNDO -CI*plus SO calculation by Minaev ( 3 2 ) has predicted a 1A2u(a-a ) f 'Alg transition at 5.11 or 5.98 eV depending upon the parameters chosen. Much of the earlier work, theoretical and practical, in the neglected con- sideration of a-electron excitation has been reviewed by Burnelle and Kranepool ( 3 3 ) . While most of the photophysical decay para- meters of benzene*that we shall discuss may be explained in terms of the lowest a-a singlet and triplet states, the mechanism of radiationless S 1 -t So decay may involve the elusive 'Ezg and a-a states discussed above and their participation cannot be d i s - regarded.
Since electron spin may invert during excitation, a-a* tri- plet states, of the same symmetries as the previously described excited singlet states should exist. Again, electron interaction removes the degeneracy of these states, though the precise order- ing of state energies is still a matter of some dispute. The ranking now generally accepted ( 3 4 ) is:
*
*
3 3 B~~ > 3~1u B~~
a sequence for which there is considerable experimental and theoretical justification ( 3 5 ) .

152 R. B. Cundall, et al.
* There is no interference in the lower n--TI manifold from the
3E2g states; both recent calculatory (12,16,17,36) and experimen- tal (37) evidence place the lowest energy 3E2g configuration above the 3B2u state. states are dependent, to a degree, on the molecular environment, and in this case are more difficult to determine experimentally because of the multiplicity as well as symmetry forbidden nature of transitions from the ground state.
The selective data on Franck-Condon maxima for transitions from 'A1 , recorded in Table 2, were obtained from electron im- pact (387 and Tx -+ 3B1u transient absorption studies (39). are considered at this stage to illustrate the relative positions of the triplet states vis-a-vis the singlet states discussed earlier.
Again, the relevant energies of the various
They
ABLE 2. Approximate Franck-Condon maxima for transitions f rom round state to the lower lying triplet 8-electronic excited
3 ~ 2 g
3 ~ 2 u
3 Elu
3 Blu
1 Alg
% 6.55 eV
% 5.60 eV
% 4.75 eV
% 3.95 eV
aData from refs. 38 and 39.
No information is available concerning a-r* transitions, though from the approximate singlet assignments and presuming validity of Hund's Rule a 0-T
to be within the energies of the lower 8-8 teresting to speculate that the radiationless T1 + So decay, ex- plained with such difficulty in theoretical analyses of the ben- zene pro lem, may involve a transition to a previously uncon- sidered (a-a ) state.
* state or states may be expected * manifold. It is in-
9 *
111. TRANSITION BETWEEN THE LOWER ENERGY STATES
Interstate transitions are generally considered to be gov- erned principally by the familiar symmetry and multiplicity

Excitation of Benzene 153
selection rules. The manner excitation and decay routes
A . Excitat
in which these of benzene are
on from the ' A
rules affect the major discussed below.
S t a t e
Within the singlet manifold of the lower energy transitions only the lElu f 'A1 transition to any of the lBlU, lEzg, and 1B2u states can be pro- moted by coupling of the "pure" electronic states with vibrations of symmetry (b2 ezg) (qu, blu, b elu) , and. (:zzii, big) respectively. observed under certain circumstances, although lElu -+ 'A1 expected, the most intense. excitation predict that the modes of blu, bzu, e2u and elu sym- metry should induce the absorption ( 4 0 ) . Studies carried out by Hochstrasser, Sung, and Wessel (111) on a single crystal of benzene at 2'K show that a certain number of vibrations of blu, b2u, elu, and ezu symmetry can be assigned in the two photon absorption spectrum, Gas phase two photon absorption spectra for benzene were reported recently by Wunsch, Neusser, and Schlag ( 4 2 ) , who have found that the most powerful mode is of bZu symmetry which is the extremely weak the v1g mode of elu symmetry. Definitive experiments on the excited states of benzene using this promising technique are awaited.
For excitation into the triplet manifold, the transition 3E1u + 'Alg has f irst-order spin-orbit allowedness , while the 3B1u -+ lAlg transition requires vibronic coupling via ezg, big, or D modes or combination tones. Absorption studies (39) on oxygen free crystalline benzene have revealed a weak T 2 + S 0 tran- sition, but no TI + So transition, confirming the relative allowed- ness of the two transitions. Furthermore, an enhancement of the TI f So absorption induced by high pressures of oxygen was re- cently reported for benzene and for a large number of simple benzene derivatives ( 4 3 ) .
No experimental evidence is available for the 3B2u + lAlS transition which is both spin and symmetry forbidden in the first order, and should occur in the same spectral region as the medium- intensity 1 ~ 1 ~ -+ 1 ~ 1 ~ transition.
excitation is symmetry allowed, though
2; mentioned above, ah!'four transitio s may be is, as
The selection rules for doubfe photon
2g
B. Ern.ission t o t he ' A l g S t a t e
Kasha's Rule ( 4 4 ) requires that emission should occur from the lowest 1B2u and 3B1u states in benzene. This is essentially the case though a predictable very weak emission has been re- ported recently from higher states of oxygen-quenched benzene in gaseous ( 4 5 ) and solution ( 4 6 ) phases. The major fluorescence

154 R. B. Cundall, et al.
and phoaphorescence processes for benzene are thus forbidden, and the radiative decay probabilities may be influenced by the envi- ronment as will be discussed below.
C. T r a n s i t i o n s Between Excited S i n g l e t and Triplet S t a t e s
Intersystem crossing from the 1B2u envelope of vibronic states is thought to occur from the lowest vibrational level, or perhaps the lowest two or three such levels. Given the lower relative energies of 3E1u and 3B1u states, transitions to each of these configurations from 1B2u, are possible. order spin-orbit allowedness in that to the 3B1u state, but vi- bronic coupling is required for the transition to 3E1u. mental evidence for intersystem crossing from higher electronic states is lacking, but there is no reason to neglect this route in the consideration of the decay processes of benzene molecules that have been so excited.
There is first-
Experi-
IV. EXCITED STATE GEOMETRY
The ground state of the benzene molecule is perfectly hexa- gonal and the molecular wave functions may be used as bases for irreducible representations of the D6h point group. Transitions between the molecular energy states are usually excited in terms of the symmetry restrictions of this point group. The forbidden nature of the lower energy electronic transition has led to con- siderable discussion about the geometry of the excited states involved, since even slight deviations from regularity lead to reduction in symmetry, and require consequent modification of the selection rules. Despite early indications (47) that the phos- phorescence spectrum may be explained using regular hexagonal symmetry, and recent evidence ( 4 8 ) the D6h symmetry is maintained for 3B1u benzene in cyclohexane at 77OK, there is much evidence to show that the molecule is distorted when in the 3B1u state. Thirty years ago, Lewls and Kasha (49) presented evidence for a molecular contraction along the 1-4 axis, from analysis of phosphorescence data. oxygen-perturbed absorption spectra (50) led to a D2h assignment of the upper state. van der Waals and co-workers (51-55) have shown by ESR studies that he vibronic coupling in the triplet manifold between 3B1u and 'Elu via the ezg vibrational mode plays the dominant role for the phosphorescence process and that this leads to a non hexagonal phosphorescent state distorted to a quinoidal form. Gwait et al. (56), using phosphorescence micro- wave double resonance, have studied the zero field origin of the
Further results from gas phase 3B1u f 'Alg

E x c i t a t i o n of Benzene 155
0,O band of phosphorescence of benzene-hg i n benzene-dg a t 1.6'K. The r e s u l t s obtained are c o n s i s t e n t w i th a 3 B l u assignment f o r a d i s t o r t e d D2h benzene phosphorescent s ta te and t h e lowest t r i p l e t of toluene is a l s o shown t o have an expanded qu ino ida l ly d i s t o r t e d r i n g by t h e c a l c u l a t i o n s of Haaland and Nieman (57) . Evidence f o r a D2h assignment of t h e lowest t r i p l e t s tate i n s o l i d phase w a s a l s o provided by o t h e r au tho r s ( 5 8 , 5 9 ) .
Due t o t h e h igh ly forbidden na tu re of t h e 3B1u f l A l g ab- s o r p t i o n , obse rva t ions are only poss ib l e i n per turbed systems such as i n t h e presence of oxygen o r i n s o l i d matrices, and i t has been suggested t h a t t h e symmetry d i s t o r t i o n s t h a t have been deduced may arise from t h e in f luence of t h e molecular environment, r a t h e r than a genuine geometric property of t h e i s o l a t e d molecule. While t h i s p o s s i b i l i t y cannot b e overlooked , t h e o r e t i c a l con- s i d e r a t i o n s support t h e view t h a t t h e 3B1u s tate i s i n h e r e n t l y d i s t o r t e d . Davaquet (60) has shown t h a t t h e v i b r a t i o n s t h a t s t a b i l i z e t h e f r e e 3B1v molecules are p r e c i s e l y t h o s e t h a t would cause a s l i g h t d i s t o r t i o n s i m i l a r t o t h a t suggested f o r t h e s o l i d phase t r i p l e t molecule. Fujimura, Yanaguchi, and Nakajima (61) have ca l cu la t ed t h e 3B1u s t a t e t o have qu ino ida l D2h symmetry i n agreement with an earlier t h e o r e t i c a l a n a l y s i s (62). These au tho r s p r e d i c t t h a t t h e l B l u s t a t e w i l l a l s o b e d i s t o r t e d wi th a n t i q u i n o i d a l D2h symmetry, and maintain t h a t t h e lowest e x c i t e d s i n g l e t s ta te 1 ~ 2 ~ w i l l r e t a i n D6h symmetry, a l b e i t w i th a s l i g h t l y enlarged r i n g . This i s n o t i n agreement w i t h conclusions of Nieman and T i n t i (58) who have i n t e r p r e t e d experimental r e s u l t s on t h e abso rp t ion , f l uo rescence , and phosphorescence of s e v e r a l i s o t o p i c benzenes i n terms of a d i s t o r t i o n of both t h e lB2,, and 3~~~ s ta tes .
D e w a r benzene from t h e lBlU s tate (63) and from t h e 3B1u (64,65) i s c o n s i s t e n t w i th t h e qu ino ida l deformations suggested f o r t hese states.
been proposed (66) t o account f o r i n c o n s i s t e n c i e s i n t h e i n t e r - p r e t a t i o n of t h e u l t r a v i o l e t spectrum of pure c r y s t a l l i n e benzene a t low temperatures. Such d i s t o r t i o n s may be t h e r e s u l t of t h e s o l i d medium o r , more probably, t h e s p e c t r a l i n c o n s i s t e n c i e s may be due t o s ta te s p l i t t i n g by t h e environment.
It i s worthy of no te t h a t t h e photochemical product ion of
A d i s t o r t i o n of t h e 1B2u s ta te of benzene t o D3d has a l s o
V. THE INFLUENCE OF THE ENVIRONMENT ON THE ELECTRONIC STATES
The in f luence of d i f f e r e n t environments on molecular s ta tes has been s t u d i e d , t h e o r e t i c a l l y and p r a c t i c a l l y , from changes i n t h e absorpt ion s p e c t r a . Such e f f e c t s as frequency s h i f t s , peak width changes, and v a r i a t i o n of o s c i l l a t o r s t r e n g t h f o r va r ious

156 R. B. Cundall, et al.
transitions are measures of the relative effects of different environments on the state involved.
The first absor tion band of benzene shifts slightly to the red (% 200 - 300 cm- Y ) with the change between vapor phase and solution, and further shifts dependent upon the nature of the solvent occur. The extent of such red shifts may be related qualitatively to medium refractive index and dielectric constant. Several theoretical approaches to the problems of spectral shifts have been attempted (67), but in general these are more useful for polar molecules, and to allowed transitions where shifts are of greater magnitude. Suffice to say that in benzene there is no discontinuity in the trend of shifts with refractive index, through gas, liquid, and solid phases, other than indicating that the forces of interaction between this molecule and the environment are weak. The crystalline phase is exceptional, since the "strict" selection rules for transition are broken down by interaction with the crystal field, which leads to factor group splitting ( 6 8 ) and the appearance of four (69) components of the 0-0 band, all of which are shifted.
Much experimental evidence is available on the small solvent shifts which are measureable but, since little effect on the photophysical decay processes is expected, no review of this work will be attempted; the reader is referred to the work of Eastman and Rehfeld (70) who report data for benzene in the vapor phase and in 37 different solvents.
function of the refractive index of the medium (71), though the effect is much smaller than theoretically expected in the case of benzene. To a first approximation the oscillator strength for the 'BzU + 'Alg transition is independent of molecular environ- ment (70).
cerns the vibrational structure of the spectra. Loss of structure in solution relative to vapor and solid phases is the result of specific solute-solvent interactions and as such, is dependent upon the dielectric properties of the solvent. Lawson, Hirayama, and Lipsky (72) have reported observations on the absorption and fluorescence spectra of benzene in vapor phase and various so l - vents. Their data are consistent with the trend o€ increased broadening of bands and loss of structure with solvent polarity. Perfluorinated solvents exert the least effect, the spectra re- taining much of the fine structure of the vapor phase. Figure 1 shows the data of Lipsky and co-workers (72) for vapor phase ben- zene, and for solutions in hexane and perfluorohexane.
The influence of environmental perturbations on the degree of allowedness of the symmetry-forbidden 0-0 band has been
Changes in oscillator strength are expected since this is a
A typical environmental effect on S1 f-t So transitions con-

E x c i t a t i o n of Benzene 157
Absorption 1.0 -4
Fluorescence
Benzene vapour
0.51A 230 240 250 260 270
Wavelength Imp Wavelength I mp
FIGURE 1 . for benzene in (a ) vapor phase, (b) per- fluorohexane, (c) hexane.
Absorption and emission spectra
(Lawson e t a1 .72) .
thoroughly inves t iga t ed . The f i r s t s tudy on t h e e f f e c t w a s car- r i e d ou t by Ham (73) us ing benzene d i s so lved i n hydrocarbon g l a s s e s with a d d i t i o n of carbon t e t r a c h l o r i d e i n va r ious propor- t i o n s . Bay l i s s and Hulme ( 7 4 ) a l s o found a new abso rp t ion band appearing i n benzene d i s so lved i n carbon t e t r a c h l o r i d e , chloro- form and w a t e r , among o t h e r s o l v e n t s , and a more d e t a i l e d s tudy w a s later c a r r i e d ou t by Kanda (75) and by Bay l i s s and Cant (76). P l a t t (77) i n a review concerned wi th t h e l o c a t i o n of t h e second t r i p l e t s tate of benzene c a l l e d these Ham bands and suggested t h a t they arise from t h e s p i n forbidden 3ElU-+ l A l g t r a n s i t i o n , a view shared by o t h e r au tho r s (78,79). Robinson (80) proposed

158 R. B. Cundall, et al.
that the Ham bands in carbon tetrachloride solid matrices are due to intensity borrowed from the solvent and have nothing to do with the 3E+u f 'Alg transition. was conclusively shown by Leach et al. ( 5 ) , who have resolved the 0-0 band of benzene fluorescence in presence of CCl4 at 77'K, finding a mirror image of the lBZu -+ lA1 including the Ham bands. Further absorption sfudies on Ham bands have been reported by other authors (82-86) and evidence for environmental enhancement of the 0-0 bands of fluorescence and phorphorescence is also available (87-90) .
A detailed investigation of the solvent effects on the in- tensity of the 0-0 bands of CgHg and C6D6 was recently carried out by Cundall and Pereira (91-93) by measuring fully corrected fluorescence and phosphorescence spectra, fluorescence and phosphorescence lifetimes, triplet yields, and electronic absorp- tion spectra at various temperatures in a large variety of sol- vents. It was found that the rate constants for fluorescence and intersystem crossing are only slightly affected by solvent while the rate constant for the nonradiative process from S So is solvent dependent, in agreement with results obtained by Eastman and Rehfeld (70). A good correlation was established between kICM and the extent of solvent-induced 0-0 band inten- sity in both absorption and fluorescence bands (91). A solvent dependence on the rate constant kICM was also found by Luria, Ofran, and Stein (94), who have measured fluorescence lifetimes of benzene in a series of polar and nonpolar solvents and the trend obtained is in good agreement with results of Cundall and Pereira (91). A similar correlation between the solvents en- hancing the fluorescence 0-0 band and the efficiency of the in- ternal conversion from 'Elu to lBzU was reported by Lawson et al. (72). bility of the nonradiative transition from S3 to S1 also increase the nonradiative transition from S1 to So. corrected fluorescence spectrum of benzene solutions in methanol at 77OK and the absorption spectrum at 233OK are represented (91); a good mirror relationship is displayed, including 0-0 bands enhanced bands by solvent perturbation. It was also established by these authors that in the presence of chloroform and carbon tetrachloride the symmetry-forbidden 0-0 bands are enhanced in both fluorescence and phosphorescence, while xenon enhances these bands only in phosphorescence. These effects are illus- trated at 77OK in Figs. 3 and 4, respectively, for benzene-d6 in methylcyclohexane + chloroform and for benzene-hg in ethanol and in ethanol + xenon. The extent of enhancement was given for fluorescence by the ratio Fo/F~ in which Fo represents the peak
The correctness of this view
absorption
1 to
This shows that solvents which give a higher proba-
In Fig. 2 the fully

Exc i t a t ion of Benzene 159
Absorption
J \
Fluorescence
42500 40000 37500 35000 32500 Wavenumber I cm-l
FIGURE 2 . fluorescence spectra o f benzene a t 2330K and 770K, respec ti vely .92
Absorption and ful ly corrected
height of t h e 0-0 band and F1 t h e peak he igh t of t h e v i b r o n i c allowed band a t t h e wavenumber $,-,-606 cm-', and f o r phosphores- cence by t h e r a t i o Po/P2, where Po is t h e peak he igh t of t h e 0-0 band and P2 t h e corresponding peak i n t e n s i t y a t about ;0-0-1500 c m - l , as shown i n Fig. 4 . type of measurement are ind ica t ed i n Tables 3 and 4 . From phos- phorescence l i f e t i m e s , (.cP!, i t w a s shown by t h e above au tho r s t h a t a decrease of T~ and i n c r e a s e of t h e r a t i o of phosphorescence t o f luorescence y i e l d , (x), p a r a l l e l s t h e enhancement of t h e 0-0 phosphorescence band i n most of t h e s o l v e n t s i nves t iga t ed . r e s u l t s w e r e i n t e r p r e t e d i n terms of a d i s t o r t i o n of both t h e
i n t h e 3B1u s t a t e being g r e a t e r than i n t h e l B z U s ta te , i n agree- ment with r e s u l t s reported by Nieman and T i n t i (58).
Some of t h e r e s u l t s obtained from t h i s
The
and 3B1u s ta tes induced by so lven t p e r t u r b a t i o n , d i s t o r t i o n

160 R. B. Cundall, et al.
25000 30000 35000 40000
Wavenumber I cm-1
FIGURE 3. and phosphorescence) o f benzene hexane + 10% chloroform a t 77OK)’ f i r s t - e x c i t a t i o n band.
F u l l y corrected spectra (f luorescence me thy1 cyc l o-
Observations on transitions between S o and the lowest tri- plet state are difficult in nonrigid media and it is not possible to say with certainty that this transition is similarly affected by environment.
Nonradiative processes may occur other than through n-n* configurations. Other types of state may, as a result of their charge distributions, be more susceptible to interaction with the environment than the emitting n,=* states.

Excitation of Benzene 161
PO
Wavenumber I cm -' FIGURE 4. Fully c o r r e c t e d phosphorescence spec t ra o f benzene i n e thano l (----) and i n e thano l t xenon (-) a t 77OK.92
VI. THE S, c-f So RADIATIVE TRANSITION
The lBzU 4- 'Alg absorption transition has been well studied and is discussed in the previous sections. Theory predicts an integrated absorption intensity independent of temperature. Be- low room temperature, Eastman and Rehfeld (70) found a similar absorption spectrum for benzene in 2-methyl-l-propanol at 25OC and at -140°C, that at low temperature being sharper and better resolved. Above room temperature there is a rather more marked effect on peak intensity (95) accompanied by peak broadening and the expected increase in hot band intensity at 260 nm. This is

162 R. B. Cundall, et al.
TABLE 3. phorescence) f o r benzene-h6 i n various so lvents a t 770K (91-93).
I n t e n s i t y o f forbidden bands (f luorescence and phos-
Solvent
Methylcyclohexane 0.050
Methylcyclohexane + 10% CHC13 0.155
Methylcyclohexane + 7% C C l 4 0.094
E t hano 1 0.076
Methanol 0.077
Acetonitrile 0.110
Ethanol + xenon (‘Lo.8M) -
0.074
0.530
0.280
0.070
0.060
-
0.380
TABLE 4. phorescence) f o r benzene-dg i n various so lvents a t 770K (91-93).
I n t e n s i t y o f forbidden bands (f luorescence and phos-
Solvent F o 4 P0/P2
Methylcyclohexane 0.072 0.040
Methylcyclohexane + 10% CHC3 0.234 0.470
Methylcyclohexane + 20% CHC13 0.260 0.440
Methylcyclohexane + 10% Ccl4 0.146 0.260
Ethanol 0.082 0.047
Methanol 0.076 0.060
Acetonitrile 0.100 -
Ethanol + xenon (W.8H) - 0.26
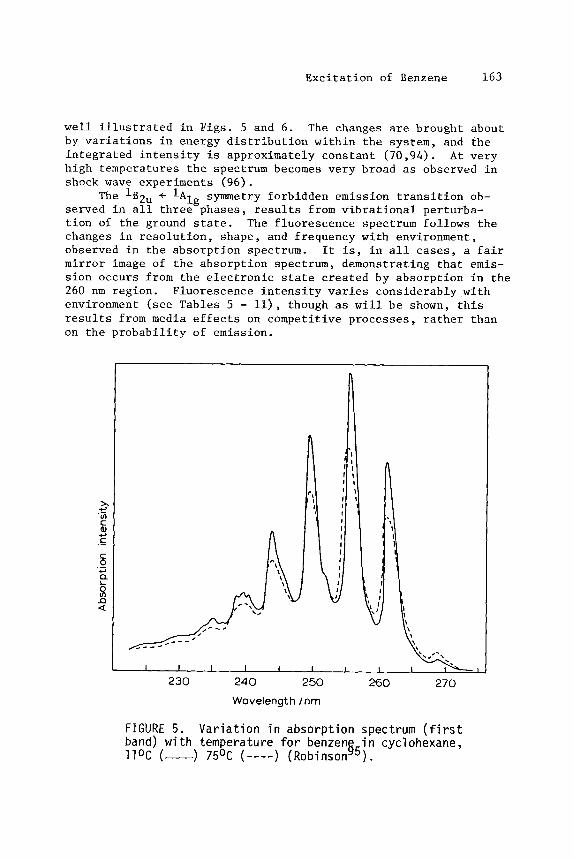
E x c i t a t i o n of Benzene 163
w e l l i l l u s t r a t e d i n F igs . 5 and 6 . by v a r i a t i o n s i n energy d i s t r i b u t i o n w i t h i n t h e system, and t h e i n t e g r a t e d i n t e n s i t y i s approximately cons t an t ( 7 0 , 9 4 ) . A t ve ry h igh tempera tures t h e spectrum becomes v e r y broad as observed i n shock wave experiments (96).
se rved i n a l l t h r e e phases , r e s u l t s from v i b r a t i o n a l per turba- t i o n of t h e ground s ta te . The f luo rescence spectrum follows t h e changes i n r e s o l u t i o n , shape , and frequency wi th environment, observed i n t h e abso rp t ion spectrum. It is , i n a l l cases, a f a i r mi r ro r image of t h e abso rp t ion spectrum, demonstrating t h a t e m i s - s i o n occurs from t h e e l e c t r o n i c s ta te c rea t ed by abso rp t ion i n t h e 260 nm region . environment ( s e e Tables 5 - ll), though as w i l l be shown, t h i s r e s u l t s from media e f f e c t s on compet i t ive p rocesses . r a t h e r than on t h e p r o b a b i l i t y of emission.
The changes are brought about
The l B z U 4 l A l g symmetry forb idden emission t r a n s i t i o n ob-
Fluorescence i n t e n s i t y v a r i e s cons ide rab ly wi th
x .+ m .-
s .+ C
C 0 .+
.-
.-
e 0 m Ll 4
230 24 0 250 260 270
Wavelength I nm
FIGURE 5 . band) w i t h temperature f o r benzen llOC (-) 75OC (----) (RobinsonG5).
V a r i a t i o n i n abso rp t i on spectrum ( f i r s t i n c y c l ohexane ,

164 R. B. Cundall, e t a l .
0.80 - -
0.60 -
-_ - 42500 40000 37500 -_ 45000
Wavenumber Icm-l
FIGURE 6. methanol a t @om temperature (-) and a t
Absorption s p e c t r a o f benzene i n
-4OOC (----) . One of t he m o s t s i gn i f i can t advances i n spectroscopic
techniques i n recent years has given da ta on s ing le vibronic l e v e l f luorescence y i e lds and associated emission l i f e t imes of ind iv idua l v ibronic l eve l s . Deta i l s of t he experimental technique and of t he r e s u l t s achieved have been reviewed by Parmenter (97 ) . Parmenter and Schuyler (98 ,99) succeeded i n recording fluorescence spec t ra from low pressure (<0.2 t o r r ) be zene vapor exci ted with a band pass s u f f i c i e n t l y s m a l l (s 40 cm-') t o ensure tha t s ing le v ib ra t iona l l eve l s of t he l B z U s ta te w e r e created. I n the lower l e v e l s t he spec t ra recorded were sharp and s t ruc tured , as would be expected i n a system i n which c o l l i s i o n a l r ed i s t r ibu t ion of energy p r i o r t o fluorescence does not occur. A t higher energy exc i t a t ion the emission spec t ra become more d i f fuse , and the f luorescence i n t e n s i t y drops t o zero when l eve l s of excess energy

Excitation of Benzene 165
TABLE 5. Monomer f luorescence y i e l d s i n t h e gas phase.
@F Isomer Temp. (OC) - Conditions/Remarks Ref.
0.21
0.32
0.39
0.22
0.27
0.18
0.39
0.18
0.31
0.23
0.53
0.26
0.35
30 2 torrs, 254 nm excitation
30
30
0.077-0.12 torr Xexc 254 nm
0.01-0.4 torr Xexc 254 nm
For zeroth level
For 1st e2 band low-pressure vapor g
25
30
25
30
30
P 2 torr A,,, 254 nm
P > 10 torrs hexc 254 nm
P 10-32 torrs A,,, 264 nm
P 10 torrs Xexc 254 nm
P 4-15 torrs Xexc 264 nm
P 5.2 torrs Xexc 254 nm
Low-pressure-single level fluorescence, zeroth level
> 2500 cm-’ are excited. the various levels were estimated from the established high pressure yield of 0.18. Data are given i n Tables 5 and 6.
times (111-113,115) have also been reported which demonstrate that the radiative transition probability from a vibronic state is determined by the combination of vibrational modes that have been excited. The transition rates that may be of interest in comparing data from all three phases are, of course, those from the lower levels of S1, since relaxation in higher-pressure gases and condensed media will lead to these states. The data of Spears and Rice (111) show that the emission probability from the levels 0 , 6l16l and l1 are 2.2 x 106s-l, 3.4 x 106s-1,
Quantum yields €or the emission from
Further quantum yield measurements (111) and emission life-

166 R. B. Cundall , e t a l .
TABLE 6. Monomer fluorescence l i f e t i m e s i n the gas phase.
T ~ / ~ ~ Isomer Conditions R e € .
60
80
77
80+10
100
79
90
80
72
164
125
92
200
95
72
92
High pressure (5-40 mm) Aexc 230-28Onm (107)
High pressure A,,, 259 nm (108)
A,,, 260, ex t rapola ted t o zero pressure (109)
3-10 t o r r s , Aexc 254 nm (110)
-Zeroth level
1st e 2 level j g
-Zeroth level
-1st e 2 level g
1 low pressure
low pressure
High pressure A,,, 247-266 nm (113)
low pressure Zeroth level
-1st e2 l e v e l g
High pressure l i m i t hexc 247-266 (113)
Zeroth level (113)
High pressure (5-40 t o r r s ) Aexc 230-280 (106)
50 t o r r s indep. of Xexc 245-270 nm (114)
50 t o r r s indep. of Xexc 245-270 nm (114)
3.4 x 106s-l , and 2.5 x 106C1, r e s p e c t i v e l y . parab le t r a n s i t i o n s (106) i n C6D give t h e va lues 1.75 x 10 S- , 2.9 x 106s-l , 3.3 x 106s-l , and 4.0 x 106s-l , respec t ive ly . benzene-d6 t h e sharp drop i n quantum y i e l d w i t h energy i n excess of zero p o i n t level f o r t h e t r a n s i t i o n is noted (106) i n a s i m i l a i r eg ion t o t h a t f o r C6H6, about 3500 cm-l above t h e 0-0 band.
of c o l l i s i o n a l r e d i s t r i b u t i o n of e x c i t a t i o n energy becomes
Data from t h e com- 6 1
I n
With i n c r e a s e i n benzene ( o r added gas) pressure , t h e rate

Excitation of Benzene 167
TABLE 7. benzene-dg, as a f u n c t i o n o f temperature assuming a non- quenchable i n t e r n a l conversion process.
Rate constants f o r f luorescence o f benzene-hg and
T('C) kF x 1O6(s-')
Benzene-h6
25
40
50
60
70
80
100
2.76
2.79
2.84
2.93
3.06
3.24
3.72
25
40
Benzene-d6 50
60
70
80
100
2.85
3.09
3.30
3.51
3.78
4.42
4.98
competitive with the fluorescence rate to an extent which depends upon the total pressure of the system. Quantum yield and emis- sion lifetime measurements reflect the nonequilibrium distribu- t i o n of excited molecules in the medium-pressure range; for example, for excitation of C6H6 at 254 nm, yields of (100) 0.21 (for 2 mm pressure) and a negative dependence upon increasing gas pressure have been reported. A similar dependence upon gas pressure is evident for C6D ; again for excitation at 254 nm, yields of 0.33 (loo), 0.28 ?loo), 0.26 (loo), and 0.23 (116) have been reported for pressures of 1.2 3.0, 5.2, and >10 torrs, re- spectively. A l s o the work of Poole (100) has demonstrated that the yield is a function of excitation wavelength, dropping to zero in both benzene and the per-deutero derivative at excitation wavelengths of 240 nm and below.

168 R. B. Cundall, et al.
TABLE 8 . Monomer fluorescence yields for benzene in solution.
@&I Isomer Temp. Solvent Remarks Ref. (00
25
25
25
25
25
25
25
AMB
20
25
RMT
25
24
RMT
25
25
25
25
25
25
25
25
25
Hexane
Me-cyclohexane
Cyc loh exane
Me-cyclohexane
Hexane
Hexane
Cyclohexane
Hexane
Hexane
Me-cyclohexane
Cy clohexane
Cyclohexane
Me-cyclohexane
Ethanol
E thano 1
Acetonitrile
Methanol
Methanol
Ethane
Acetonitrile
Water
Water
Per-fluoro-n-hexane

Exc i t a t ion of Benzene 169
TABLE 9. solution.
TFM Isomer Temp. Solvent Ref.
Monomer fluorescence emission lifetimes for benzene in
(ns) (OC)
29
24
26
27.5
29.5
32
27.9
45
34
30
33.6
26.6
33
28
30
31
26
20
2 . 2
15
RMT
20
RElT
24
25
20
25
RMT
25
25
25
25
22
25
25
RMT
25
25
25
25
Cyclohexane
Hexane
Cyclohexane
Hexane
Cyclohexane
n-Nonane
Me-cyclohexane
Hexane
Cyclohexane
Me-cyclohexane
Hexane
Cyclohexane
Cyclohexane
Cyclohexane
Me-cyclohexane
Ethanol
Ethanol
Methane
Water
Ace t o n i t r i l e

170 R. B. Cundall, et al.
TABLE 10. Fluorescence yields for benzene i n the solid phase.
0FM Isomer Temp. Solvent Remarks R e f . (OK)
0.21
0.26
0.20
0.22
0.29
0.22
0.19
0.27
0.21
0.18
0.17
0.21
77
77
77
173
77
77
77
77
30
EPA
EPA
Benzene
Crystal
EOA
EPA
EPA
n-Hexane
Methanol
30170 is0 butlisopent
EPA
(148)
2 10% ( 1 4 9 )
(150)
(151)
(152)
Ether/isooctane/ (144) alcohol
(153-155)
(153-155)
(120)
(120)
Limiting low- (120)
(120)
temp. values
TABLE 11. Fluorescence emission lifetimes for benzene i n the solid phase.
TF Temp. Solvent Remarks Ref. (ns) (OK)
a4 173 Benzene No phosphorescence (151) detected
12026 77 Me-cyclohexane ( 93)
115+5 77 Ethanol ( 93)

Excitation of Benzene 17.1
It is considered that pressures of 10 torr or greater are required for complete collisional deactivation to an equilibrium distribution of energy between levels prior to emission. Under these conditions, the fluorescence yield appears independent of further increases in pressure, and achieves a value of 0.18 (100,103,105,116) for all first singlet band excitation wave- lengths in excess of 247 nm.
Fluorescence yields for C H6 and CgD6 decrease (100,105) with increasing temperature; tie data of Dunnicliff (105) are presented in Fig. 7 for 2.5 x 10-4M and 7 . 1 x 10-4M benzene be- tween 20 and 140OC.
Emission lifetimes for benzene with vapor phase are subject to similar variations with pressure and excitation wavelength as are fluorescence yields. Data are collected in Table 6 and can be seen to show considerable variation with source and experi- mental technique. Recent measurements, using a single-proton counting technique (114) have shown the emission lifetimes of high pressure C6H6 and C6D6 to be 77 and 92 ns, respectively, at 25OC. is also shown in Fig. 7.
The temperature dependence of the fluorescence lifetime
20 40 60 80 100 120 140
Temperature1 'C
0.30
OF
020
010
FIGURE 7. The v a r i a t i o n i n flF and f o r vapor- phase benzen as funct ions o f tempergture ( D u n n i c l i f f l g 5 and Lockwoodll4). The filled p o i n t s are f o r CgDg. The open po in ts are f o r C6H6.

172 R. B. Cundall, et al.
At high pressures gaseous system most closely resemble the situation in condensed media, and it is instructive to determine the radiative rate constant for this system, and its variation with temperature. a nonquenchable process which occurs prior to vibrational relaxa- tion from a nonequilibrated state and using results of 0 , and 0 , determined by Cundall and Dunnicliff (105) and values of TF due to Lockwood (114), the calculated kF values are presented in Table 7 for both CgHg and CgDg. for the protonated and deuterated molecules indicates that the large differences in yield and lifetime for the two isomers are the result of an isotope effect on a nonradiative transition.
fluorescence in the collisionally equilibrated gas phase system results from two (117) or three (118) vibrational levels of S1. Whether this be the case, and if emission involves a multistep cascade through vibrational levels, it seems likely from consid- erations of the levels initially populated, the rates of vibra- tional deactivation, and the Boltzmann distribution of levels in the equilibrated system, that emission is dominated by transi- tions from the zeroth level. This is borne out by comparison of high-pressure values of kF with zeroth-le el transition proba- bility discussed previously; 2.8 x 10-6s-' and 2.2 x respectively, for C6H6 at 25OC; though the discrepancy is greater for CgDg for which the values are 2.9 x
cence emission for benzene are dependent upon solvent (see Tables 8 and 9, and more detailed data in reference 70) and temperature (140), but yield has been shown (141-145) to be independent of excitation wavelength within the first absorption band ( A exc > 230 nm). The observed variations arising from different exciting sources and experimental methods, in yield and lifetime for simi- lar and even identical solvents reflect the difficulty experienced in obtaining reliable fluorescence data. Despite this unfortunate limitation, some trends may be discerned. In general, quantum yields and lifetimes tend to be larger in nonpolar solvents (91, 119), although in all cases these quantities are much smaller than in the gas phase. There appears to be no significant .iso- tope effect on either parameter, contrary to expectation from a comparison with gas phase data (107,112).
The oscillator strength for the S1 + So transition is al- most independent of solvent (70,74,94), and it might thus be ex- pected that the effect of solvent on the radiative probability would be slight. Cundall and Pereira (91,119) have reported measurements of k m in polar solvents, nonpolar solvents, and in particular perfluoro-n-hexane (in which solvent the similarity of
Assuming that internal conversion from S1 is
The similarity of rate constants
In the medium pressure region, it has been suggested that
and 1.7(5) x 106s-! In solution, the quantum yields and lifetimes for fluores-

Excitation of Benzene 173
spectral characteristics to those in vapor phase show that the solute-solvent interactions are minimal). It is found that al- though slight variations in k? with solvent occur, the variations are not accounted for by considerations of solvent polarity, and it may well be that km is independent of solvent; lying within the range 1.5 - 1.8 x 1O6s-I. The variation of 0~ and TF with solvent must, therefore, be the result of a medium effect on the nonradiative decay process. The difference between mean monomer transition probability in a range of solvents, and values from high-pressure and zeroth-level gas phase data is significant: 1.5 - 1.8 x 1O6sW1 compared with 2.2 x 1 O 6 s - I and 2.8 x 1O6s-I. Application of the disputed refractive index, n2, correction fur- ther increases the divergence between these data; while the theo- retical value for k m (2.5 x 1O6s-I) derived using the Strickler- Berg equation, neglecting n2 is in close agreement with the gas phase data. The phase effect may be the result of uncertainties in measurement of various parameters, or, more probably inter- actions in the condensed phase which are not explained by the n2 relationship. Reported data on C6D6 are less extensive than for CgH6 and contradictory. The data of Berlman (126) for 0, and T F indicate that k~(C6Db) is larger than that for the perprotonated species. The data used by Sandros (146) and Helman (139) provide conclusions intermediate between those of Berlman (126), and those of Cundall, Olgivie, and Robinson (127), who reported a value for kF(C6D6) lower than that of C6H6. similar trend in radiative lifetimes of the zeroth vibrational level in fluorescence (106,111), and attributed the effect to a greater purity of the electronic levels in perdeuterobenzene, which is also mirrored by changes in oscillator strength with iso- topic substitution (86).
fect on kF in the gas phase. A similar effect is also observed in solution (94 ,119) . Although in nonpolar solvents the effect is relatively small (119,136,147), it is accentuated in more polar environments (119) . The effect is smaller for C6D6.
solid phase are difficult to determine, and accurate data are sparse (see Tables 10 and 11). fluorescence and lifetimes of dilute solutions of benzene increase with decreasing temperature (8,119). In a recent study of similar molecules (156-158), it was shown that the increase in both 0, and T, is smooth, and that both these quantities tend towards a limiting value at low temperature. dependence of the two parameters for benzene in a variety of solvents (119) has indicated that the limiting values for both should be reached at a temperature well above 77'K, since the controlling variable is the radiationless Channel I11 process(es)
Rice and co-workers noted the
It was noted earlier that there was a small temperature ef-
Reliable fluorescence quantum yields and lifetimes in t h e
It is well established that
An analysis of the temperature

174 R. B. Cundall, et al.
which has a solvent dependent activation energy characteristi- cally in excess of 0.2 eV. troduced to describe the process or processes other than fluo- resence or triplet state formation which lead to the disappearance of lBZu state (see Section VIII). thought to provide the limiting low-temperature values for the two fluorescence parameters.
Eastman (120) has reported limiting yields of 0.21, 0.18, 0.17, and 0.21 for benzene in n-hexane, methanol, 30170 isobutanol/ isopentane, and EPA, respectively. The yields are seen to be solvent dependent and determination of kF requires emission life- times in identical media. Such are not available though limiting values of 120 and 115 ns have found (93) for benzene in methyl- cyclohexane and ethanol, respectively, at 77'K. Making the crude assumption that media effects are due to differences in solvent polarity we can calculate radiative rate con tants of 1.75 x 1O6s-I for hydrocarbon solvents and 1.6 x 10's-1 for alcohol solvents.
for room temperature solution work leads to the conclusions that the radiative rate constant is independent of temperature (below ambient) and that the difference between gas phase and condensed phase data is a real effect resulting from some molecular inter- action not accounted for by the n2 correction which conventional theory requires.
The term "Channel 111" has been in-
Measurements at 77OK are
The similarity of these values to those mentioned earlier
VII. THE S1 + TRIPLET RADIATIONLESS TRANSITION
From analysis of fluorescence yields and lifetime measure- ments on emission from single vibronic levels (98,99,111-113,115) "rate constants'' for total radiationless decay may be determined. These are also dependent upon vibrational level involved. Spears and Rice (111) have reported nonradiative lifetimes of 128, 108, 104, 105 ns, respectively, for the levels considered previously. The fluorescence "cut off" region, noted in these reports, sug- gests, in agreement with several other indications discussed in Section VIII, that radiationless S1 + S o decay proceeds via a highly vibrationally energetic level of S1. and co-workers (106) on benzene-dg in this type of experiment shows that the nonradiative transition from S1 is affected by deuteration to a greater extent than the fluorescence transition; the nonradiative lifetimes of the four comparable levels being 565, 345, 300, and 340 ns, respectively.
Triplet quantum yields for benzene vapor in collisionally deactivating systems have been estimated by a number of techniques (Table 12), involving sensitized isomeration, decomposition, or
The report of Rice

Excitation of Benzene 175
TABLE 12. The S1 t r i p l e t t r a n s i t i o n f o r benzene i n t h e gas phase.
dT Isomer Temp. Conditions Technique Ref. (OC)
0.72
0.72
0.71
0.71
0.79
0.63
0.71
0.70
0.68
0.65
0.63
0.61
0.52
0.46
0.37
Room
Room
Room
Room
Room
Room
Room
40
60
80
100
Room
50
70
100
2-19 torrs Aexc254nm
7-26 torrs Aexc252-67nm
10 torrs Aexc252nm
Cyclobutanone
2-19 torrs Xexc254 nm
> 10 torrs Aexc=254 nm
> 10 torrs Xexc=254 nm
> 10 torrs Aexc=254 nm
> 10 torrs Aexc=254 nm
> 10 torrs Xexc=254 nm
> 10 torrs Xexc=254 nm
> 10 torrs Aexc=254 nm
> 10 torrs Aexc=254 nm
> 10 torrs Xexc=254 nm
Butene-2 (159)
Butene-2 (160)
Ketene (161)
Cyclo- (162) butanone
Pyrazine (163)
Butene-2 (159)
Butene-2 (105)
Butene-2 (105)
Butene-2 (105)
Butene-2 (105)
Butene-2 (105)
Butene-2 (105)
Butene-2 (105)
Butene-2 (105)
Butene-2 (105)
luminescence of additives. It is to be expected that the overall intersystem crossing efficiency is dependent upon the extent of collisional deactivation and redistribution of energy following excitation. The presence of appreciable quantities of added gas, which may arise because of the presence of the triplet monitor, and the use of benzene pressures in excess of 10 torrs ensure that the yields of triplets reported for gas phase systems result from intersystem crossing from thermally equilibrated range of levels in S1. Of the many triplet monitors used, it is widely believed that butene-2 effectively quenches only the triplet state.

176 R. B. Cundall, et al.
Dunnicliff (115) has shown that the olefin also quenches singlet S1 benzene, but only with a bimolecular rate constant of 2.2 x lo8 M-ls-l, so at the olefin concentrations commonly used for triplet scavenging, singlet state quenching is negligible compared with that of the triplet state.
Noyes and Harter (160) obtained evidence of a decrease in triplet yield with decrease of excitation wavelength. A limiting high-pressure yield of 0.71 at room temperature after excitation at 254 nm is now well established. The observed isotope effect is large; with corresponding high-pressure limit values of 0.63 (159) and 0.61 (105) reported for CgDg, under the same conditions. Using the emission lifetime measurements of Lockwood (114), inter- system crossing rate constants (for heavy and light benzene) of 7.9 x 106s-1 and 11 x 106s-l are obtained assuming a nonquenchable internal conversion also occurs. For this transition, the crude assumption that the high-pressure rate constant is dominated by the contribution from the zeroth level may not be valid. A s dis- cussed earlier, transition may be to either or both of the 3ElU and 3B1u states. Transition to 3B1u has first-order spin-orbit allowedness, but the rate constant may be reduced by the small Franck-Condon overlap factor resulting from the 1.1 eV energy gap between states. Transition to 3Elp requires vibronic coupling. The positions of the three states In C6D6 relative to the equi- valent states in C6H6 are not accurately known, though spectro- scopic evidence (39,164,165) suggests that the energy spacings are slightly increased (200 cm-l) by deuteration. In addition, deu- terium substitution reduces the energy separation of the vibra- tional levels (166-169). There is some confusion concerning whether deuterium atom substitution affects (170) the radiation- less T1 -+ So transition or otherwise. it is probably unacceptable to seek similarities between the mea- sured rate constant in high-pressure gases and in the nonradiative decay rates of the zeroth vibrational levels of S1 states of C6H6
Because of this uncertainty,
and CgD6. Considerable data are available for triplet yields of ben-
zene in dilute solutions of different solvents (see Table 13). In the main, two techniques have been used; sensitized phosphor- escence of biacetyl, sensitized cis-trans isomerization of butene- 2, octene-2, and stilbene. All yield comparable results, In saturated hydrocarbon solvents at room temperature, the triplet yield for CgH6 is found to be about 0.24 2 0.01. vent dependence of this quantity, the yield dropping to 0.15 in ethanol, 0.13 in methanol, and 0.09 in acetonitrile (91). In determining the effect of environment on the rate constant con- trolling intersystem crossing, values for emission lifetimes in the various systems are needed. These are, as mentioned pre- viously, often unreliable. Cundall and Pereira (91) have reported
There is a sol-

Excitation of Benzene 17 7
consistent data which give the variation in kISCM (= @T/?F) with solvent. It is found that, though differences between solvents occur, there is no consistent trend with solvent polarity, and we can consider kISCM to be independent of solvent within the range of approximately 7(+1) x Further, there appears to be little isotopic effect on kISCM since a rate constant of 8.7 x 1O6S-' has been reported for CgDg (127) in methyl-cyclohexane at room temperature compared with 8.0 x 1O6s-I for the perprotonated solute ( 9 1 ) .
The variation of kISCM with temperature for CgH6 in cyclo- hexane (182) is more marked than that for CgD6 (127) similar to the trends noted in gas phase studies (105). It is erronous to fit this temperature dependence to a simple Arrhenius function, since the yield of phosphorescence at low temperature shows a sub- stantial triplet production in the solid phase. More realisti- cally, the room temperature process may be viewed as the sum of two intersystem crossing steps to the two lowest triplet states. One process being temperature independent (4BlU?) and the other temperature dependent (-t3Elu?).
Estimation of triplet yields from benzene in the solid phase presently requires several approximations, since the available triplet monitoring methods do not function in solid media. Pereira (93) has shown that the quantum deficit l-(flF + @T) may be assigned in benzene although not necessarily to substituted benzene derivatives, to a single process, and that (0F + @T) ap- proaches unity with decreasing temperature. In this way, the deficit (I - OF) may be assigned to 0~ at low temperatures. Using the emission yields and limiting low temperature lifetime data from Tables 10 and 11, together with the solvent effect assum tion noted previo sl and 7.1 x 10Es-?'for intersystem crossing in hydrocarbon and polar media, respectively.
are broadly, that is, within S10 percent, independent of molecular environment. We must conclude that the significant differences noted in fluorescence and triplet yields and liferimes, with phase, must be the result of an effect on the "hidden" Sl j- So decay process.
% we may calculate rate constants of 6.6 x 10
It is seen that the rate constants €or intersystem crossing
VIII. THE S1 -t So RADIATIONLESS TRANSITION
Evidence that decay of the first excited singlet state may occur via a nonradiative process which depends on the vibrational energy and probably does not involve the lower triplet states of the TI manifold comes from various types of experiment:

178 R. B. Cundall, et al.
TABLE 13. T r i p l e t y i e l d s i n t h e s o l u t i o n phase.
OIT Isomer Temp. Conditions Monitor Ref. (OC)
29 Pure benzene Piperylene (172)
25 Pure benzene Bu tene-2 (173)
25 Pure benzene Octene-2 (174)
24 Pure benzene Bu tene- 2 (175)
25 Pure benzene Stilbene (176)
25 Pure benzene Octene (177)
24 0.11 2M cyclohexane Butene-2 (147)
26 6.10-’M Me-cyclohexane Butene-2 (178)
20 Dil. cyclohexane Biace tyl (146)
22 Dil. Me-cyclohexane Octene (177)
25 Dil. cyclohexane Butene-2 ( 91)
25 Dil. Me-cyclohexane Butene-2 ( 91)
25 Dil. ethanol Butene-2 ( 91)
25 Dil. methanol Butene-2 ( 91)
25 Dil. acetonitrile Butene-2 ( 91)
25 Dil. water But ene-2 ( 91)
24 0.IM cyclohexane Butene-2 (175,178)
25 0.3M cyclohexane Flash (179)
25 Dil. cyclohexane S t ilbene (180)
25 Dil. Me-cyclohexane Butene-2 (178)
pho tolysis
20 Dil. cyclohexane Biacetyl (181)
25 Dil. cyclohexane Bu tene-2 (127)
25 Dil. perfluorohexane Butene-2 ( 91)

1.
2.
3.
4.
Excitation of Benzene 179
The decrease in quantum yield for S1 -f So fluorescence with increasing excitation energy has been reported. for gas phase (142), liquid phase (141,143,183), and solid phase (144) benzene systems. The decrease in yield shows that the onset of an additional competitive decay process, occurs at an energy above S1 of the order of 0.3 eV.
Low-pressure, narrow band pass excitation studies have shown (98,99,111-113) that the fluorescence yields from single excited vibrational levels of S 1 drop sharply to zero in re- gion of 0.31 eV above the zeroth level of the 1B2u state. The effect is also noted in the perdeuterated molecule (106).
Callomon and co-workers (27) report the onset of spectral dif- fuseness in the So - S 1 absorption spectrum of benzene vapor at a level approximately 0.31 eV above S 1 .
In the medium-pressure gaseous benzene system, Poole (100) has found that the S1 -+ So fluorescence yield drops to zero for excitation of wavelengths of 240 nm or less, despite effi- cient collisional deactivation.
Such triplet yields, as are available, show that the vibra- tionally enhanced process is not intersystem crossing.
All of these observations are consistent with the existence of a rapid nonradiative route available for the decay of excited benzene molecules with energy approximately 0.30 eV above S1. The variation with temperature of the quantum deficit 01c = 1-(0~i-@T) has been analyzed kinetically in terms of a single pro- cess for benzene-hg (91,147) and benzene-dg (127) in solution. The assumption of a single decay process allows interpretation of the behavior with temperature of the rate constant kICM (= g I C / ~ ~ ) in terms of the Arrhenius equation which yields an activation energy which is solvent dependent (91), typically of the order of 0.25 to 0.37 eV, and is tentatively interpreted as the mean excess energy required by the thermally equilibrated excited benzene molecules, for decay via this hidden route.
creases with temperature in the high-medium-pressure gaseous system. Analysis using the crude model applied to the solution phase data produces an "activation energy'' for Channel 111 decay of 0.14 eV for CgHg and 0.19 eV for CgDg. A kinetic scheme in- volving a nonquenchable radiationless decay of vibrationally ex- cited S 1 states is thought (105) to provide a better fit for the data. Using this approach, the onset of nonradiative deactivation would occur at approximately 0.4 eV above S1.
The experimental evidence proves, without question, the exis- tence of a Channel I11 route; and various attempts have been made
There is also evidence (105) that the quantum deficit in-

180 R. B. Cundall, et al.
to provide some explanation. The theories of radiationless transitions applied with varying success to the Ti -f So step (see section X) have not yet been able to explain satisfactorily the pure singlet decay phenomenon. It has been suggested that photochemical rearrangements could account for the quantum de- ficit. Evidence (184) (reviewed later in Section XII) has shown that the prismane and Dewar isomers would arise from states of Blu symmetry, while benzvalene and fulvene result from the lBzU state. Bryce-Smith and Longuet-Higgins (65,185) and Jan0 (186)have suggested that diradical intermediates consistent with the sym- metries of the 1B2u and 3B1u states may be involved in the photo- isomerization of benzene and its derivatives. Measured radical yields are very low (180), as are the final yields of isomeric products (187). Though rapid re-aromatization of the diradical, o r of benzvalene has been proposed as an explanation of the low isomer yields, it is to be expected that isomerization due to group shifts should occur in the case of benzene derivatives. Examination of the photodecay processes in o-xylene (188) and m-xylene (189) has shown that aromatic isomer products are formed in very small yields, and further, that the activation energy for production of m-xylene from other isomers (0.44 eV) is higher than the activation energy required for the temperature-dependent Channel I11 process (0.21 eV). Birks (190), adopting the isomer theory of Lamola et al. (191) and Noyes and co-workers (192), suggests a "physical" isomer, the most unusual property of which seems to be that, by definition, it is undetectable. It is sug- gested that the potential energy surfaces of this "isomer" over- lap with those of So and S1 benzene, permitting rapid hidden de- cay. It is not clear how this differs from what would be con- ventionally regarded as internal conversion.
Callomon, Leach, and Lopez-Delgado (27) suggested that a O-T* state existed in the region between the 1B2u and 'BlU states (see Section II), and selected 3E2u as the most probable state involved. It has been suggested (156) that a singlet state would be more plausible in view of the very high rate constant for the transition from the lBzU state. newly invoked state remains since it is expected that the transi- tion to ground state would be extremely forbidden, leading con- sequently to a long-lived intermediate. Recent photochemical data (193,194) have shown the existence of a long-lived, non- benzvalene (194) intermediate following 2537 Ao irradiation of benzene. Results obtained by Stein (94,195) in 02-free solu- tions of benzene have indicated the formation of benzvalene with an initial quantum yield of 0.18 after excitation with the 2537 Ao line and that this initial yield is independent of temper- ature in the 9 to 5OoC range.
The problem of decay of this
This result shows that isomer

Excitation of Benzene 181
formation does not account for the thermally activated fluores- cence quenching. Stein proposed that benzvalene arises from a higher vibronic state before vibrational relaxation to the S1 fluorescent state, that is, prior to interaction with the en- vironment. let. Results of Lutz and Stein (195) also show an increase in the efficiency of benzvalene formation at higher temperatures, consistent with a decrease of 0 ~ , as determined by Cundall and co-workers (196). Further results of Ilan and Stein (197) on the fluorescence yields of benzene solutions in the presence of xenon still support the assumption.
A recent tunneling effect model for radiationless transitions (198) has been applied to benzene and other aromatic hydrocarbons. The CH stretching vibrations are considered as dominant for the non-radiative process. Rate constants for the radiationless pro- cess S 1 + So calculated by theory are of the same order of magni- tude as those obtained by experiment. A further possibility for the interpretation of Channel I11 is a delay via a configuration- ally perturbed doubly excited E2g state, though this route seems unlikely following confirmation that the relevant state is of similar energy to 1glu (see Section 11).
Benzvalene is subsequently quenched by benzene trip-
IX. THE Ti -t So PHOSPHORESCENCE TRANSITION
Considering the ease with which phosphorescence emission from solid or glassy phase solutions of benzene is observed, it is perhaps surprising that unambiguous data on the probabil- ities of radiative and radiationless decay of the triplet state are so difficult to obtain and unreliable. The determination of excited state parameters at a particular temperature requires, at least, evaluation of !bp, 0~ and T ~ , and more reliably @p, OF, dT: T?, and T ~ . Direct measurement of op by comparison of the emission of the given compound with that of a standard sample is one method of approach (148,150). However, since it is neces- sary that both sample and standard possess identical optical properties (otherwise corrections for effects of refractive index differences, reflectivity losses, and emission polarization on the measurements must be made), this method is of limited ap- plicability. Kellogg and Bennett (199) estimated phosphorescence yield from measurements of acceptor molecule fluorescence, resulting from triplet energy transfer from the aromatic donor molecule. This method should be reliable, provided solutions are sufficiently dilute to eliminate the effect of triplet-triplet annihilation and provided corrections for anisotropy of emission are made.
Other methods for measuring dP have been reported.

182 R. B. Cundall, et al.
A further method involves a comparison of the luminescence from a glassy sample with the light scattered by a MgO surface (149).
Because of the complexity and difficulty in obtaining re- liable data, some authors (8,200,201) have made the arbitrary as- sumption that kp = 0.030 s-l and is independent of temperature for all unsubstituted hydrocarbons. From this assumption, and measurements of TP, values for k have been estimated. The subsequent theoretical analysis % the radiationless transition probability based on the assumption must , theref ore, be treated with some caution, since it is arbitrary and probably incorrect.
A method for determining kp reported by Cundall and Pereira (91) utilizes data from the parallel studies on parameters con- trolling the spin-allowed emission. In these studies, most ex- haustively pursued for benzene, it is found that both kF and kISC approach limiting low-temperature values, and estimates of their magnitude in the < 100°K region may be given with some certainty. and for 0 /0
With this information, the additional data for -cCp (x) , kp can be determined, since P F
This method does not rely upon any special assumptions, although corrections for photoselection effects (202-205) and for the eigenpolarization of the spectrophotometer must be undertaken. Further studies of fluorescence and phosphorescence at 77’K for simple benzene derivatives using the same spectrophotometer and the same optical arrangement carried out by Cundall and Ogilvie (158) have shown that photoselection and eigenpolari- zation corrections do not alter the results obtained for kp (within 2-3%). The above method assumes reliable data for TP but measurements are subject to uncertainty. Variations in ~p for a specific sample can be found which depend upon the rates of cooling of the sample, and also upon the time which elapses between the freezing process and the measurement operation. The cooling rate may affect the decay if two phases of the solid sol- vent can be formed (206-208). matrices in which cubic matrices are produced by rapid freezing, and monoclinic matrices by slow freezing. Variations in T~ with standing time can be observed if glass relaxation occurs (208- 210) and/or thermal equilibration is achieved only slowly (211). Though the effect of this latter factor may be reduced by using very narrow sample tubes, Martin and Kalantar (212) have shown that T~ varies considerably with time elapsed following freezing, if the solvent utilized undergoes glass transitions in the neigh- borhood of the temperature used. For the particular case of benzene, ‘cP is strongly dependent upon solvent and upon tempera- ture above 60°K.
This is the case for cyclohexane
Solvent effects have been reported by

Excitation of Benzene 183
Leubner (213), Hirayama (214), and Hatch et al. (215), among others (Fig. 8 ) . In some instances, apparent solvent effects may be the result of impurity quenching, particularly in solvents of rela- tively low viscosity. However, this explanation cannot be uni- versally valid and does not assist in understanding the unexpected temperature effect. Leubner (213) could find no correlation be- tween sample viscosity and T ~ , and Ling and Willard (216) re- ported, from measurements on 3-methyl pentane between 77 and 94OK, that the viscosity had a pseudo-activation energy of 15.9 kcal/mol, a factor of 7 greater than the activation energy measured in much the same temperature range for T . A defini- tive examination of the problem has been publisheg recently bv Kilmer and Spangler (217)(Fig. 9). These authors found that the variation in T~ for benzene in cyclohexane (% 10-3M) with tem- perature could be fitted to the relationship of the type
llTp = llTo -I- kl exp(-AEIKT)
For benzene-hg it was found that -to = 11.44s AE/hc = 554 cm-' and kl = 4.2 x lo3,-'. and 218 x lo3,% -rp is, of course, a composite function in- cluding radiative and nonradiative decay processes and there is still speculation concerning which mode of decay is temperature dependent and which not. The weight of evidence seems to favor the nonradiative route as being that which requires thermal acti- vation. Use of the Kilmer-Spangler equation to extrapolate to room temperature produces phosphorescence lifetimes of 3.8 and 5 ms for CgHg and C D6, respectively. not observed in eiteer liquid or gas phases, it is clear that alternative decay routes operate in fluid media.
Particularly of note are the lifetimes in frozen inert gas solu- tions, reflecting the enhancement of T1 + So transition with in- creasing atomic number of the solvent; an effect also noted by other authors. The important results of Haaland and Nieman (222) show a marked solvent effect on phosphorescence lifetimes. Per- fluorocarbons are rhe least perturbing solvents on phosphores- cence, an effect consistent with results obtained in liquid solution (72,91).
of solvent and temperature, shows the importance of the impor- tance of the C-H/C-D modes in promoting one or both of the triplet decay transitions.
Using the values !8F = O.Z'~O, x('@p/@F) = 0.92 (92,219) and -rp = 5.50 s for benzene in methyl cyclohexane, it is possible to calculate values of 0.0417 and 0.401 s-l for kp and kGT.
The values for CgDg are 14.28s, 705 cm-l
Since phosphorescence is
Some emission lifetime data are presented in Table 14.
The effect of deuteration whose influence is independent
The

Temperature ( O K )
FIGURE 8. ethanol ( X ) , EPA (A), 3-methyl entane (O), and MCH-isopentane (+), (Leubner). 91 3
Temperature dependence of T P for CgHg in

Exc i t a t ion of Benzene 185
10 U . .- ? 6
.= 4
2
+ a,
-1
16 I
Temperature 1°K
( b )
16r
-
.- + - Q,
- 1 - .'t 4 -
80 120 160 Temperature 1°K
( a )
FIGURE 9. and C g ~ g i n (a) cyclohexane and ( b ) cyclopentane, (Kilmer and Spangler) .217
Temperature dependence o f T~ f o r CgHg

186 R. B . Cundall, et al.
TABLE 14. Phosphorescence l ifetimes for dilute benzene s o l u t i o n s .
Isomer Solvent Temp. Remarks Ref. T
(S) (OK)
7.1
17.5
29.5
5.75
7.48
4.75
12.02
13.69
14.10
5.50
6.50
5.50
10.50
11.20
9.10
9.83
9.92
10.91
11,19
9 .o
EOA 77
20
20
3-Methylpentane 77
- -
Isopropanol
Cyclohexane
3Mp
Isopropanol
Cyclohexane
Methyl-cyclo- hexane
Ethanol
Methanol
Me thyl-cyclo- hexane
Ethanol
Methanol
Me-cx/Ip
3 -Me th ylp en- tane
EPA
Ethanol
Dioxane
77
77
77
77
77
77
77
77
77
77
77
<60
<60
<60
77
Two sites for decay
This for longest
Relaxed
Frozen quickly
Monoclinic
Relaxed
Frozen quickly
Monoclinic
Frozen quickly
Frozen quickly
Frozen quickly
Frozen quickly
Frozen quickly
Frozen quickly
Independent of
temperature below
60°K but
solvent dependent
-
Excitation of Benzene 187
TABLE 14 (cont.)
5.90
2.0
5.78
11.79
12.11
11.31
13.70
13.40
13.06
9.46
10.32
5.75
8.45
3.04
11.44
14.82
7.46
14.94
29.5
14.5
17.5
0.74
EPA 77
C yclohexane 77
Isopentane 77
3-Methylpentane 77
3-Methylhexane 77
Methyl-cyclo- 77 hexane
Ethanol 77
Iso-propanol 77
EPA 77
C y clohexane 77
Isooc tane 77
3-Methylpentane 77
EPA 77
3-Methylpentane 77 iso-pentane Cyclohexane - Cyclohexane - Cyclopentane -
Cyclopentane -
Argon 20
Argon 20
Argon 20
Krypton 20
Constant (212)

188 R. B. Cundal l , e t a l .
TABLE 14 (cont.)
Xenon 20 0.026
29.5
0.025
15.4
10.9
2 3 . 7
14.3
8.0
11.8
10.2
9 .5.
15.6
12.5
6.3
9 . 4
4.6
Argon 20
Xenon 20
Perfluoromethyl- cyclohexane 65
Methyl-cyclo- 65 hexane
Perf luoro-cyclo- hexane 65
Methyl-cyclo- 65 hexane
EPA 77
Cyclohexane 4.2
Boraz i n e 4.2
C6D6 4.2
Cyclohexane 4.2
Borazine 4.2
EPA 77
EPA 77
EPA 77
phosphorescence rate c o n s t a n t f o r benzene is i n c l o s e agreement w i t h t h a t of Johnson and Ziegl-er (171) who deduced a va lue of 0.041 s-1 f o r ~ 6 ~ 6 and 0.034 s-1 f o r c ~ D ~ . concluded t h a t d e u t e r a t i o n a f f e c t e d t h e v i b r o n i c coupl ing neces- s a r y t o promote t h e r a d i a t i v e t r a n s i t i o n and t h a t t h e deu te r - a t i o n e f f e c t on t h e r a d i a t i o n l e s s t r a n s i t i o n i s s m a l l a t v a r i a n c e w i t h e a r l y t h e o r i e s of r a d i a t i o n l e s s t r a n s i t i o n s (201,223).
From t h e v a r i a t i o n s of ~p w i t h temperature , K i l m e r and Spangler (217) concluded t h a t t h e r a t i o kp kIsC/kF w a s independent
The lat ter a u t h o r s

Excitation of Benzene 189
of temperature and equal to 0.229 ? 0.007 s. Although both kF (134) and kISC (147) may increase with temperature above ambient, by analogy with toluene (156) and other methylated derivatives (224) it is expected that both will be nearly con- stant below room temperature. From a combination of these data it is clear that kp is independent of temperature.
of solute concentration on the phosphorescence emission yield and lifetime is very marked. a decrease in T~ and the x ratio has been reported for EPA (214), EOA (144,152) , methylcyclohexane (219) , and cyclohexane (207) . Phosphorescence is negligible in both pure benzene crystals (225) and polycrystalline powder (144,214). This has been attributed to a rapid triplet-triplet annihilation process, an explanation apparently confirmed by Cundall and Pereira (2191, who detected solution some slight rather diffuse phosphorescence together with delayed fluorescence from pure benzene.
A l l of the above data refer to dilute solutions; the effect
At concentrations of 1M and above,
X. THE Ti -+ So RADIATIONLESS TRANSITION
The benzene triplet has not been observed to phosphoresce in either the gas or liquid phase, as noted previously but in view of the lifetime measured in glassy media at low temperature this is not surprising, since very small amounts of quenching impurities would reduce emission to undetectable levels. Some detailed investigations have shown that there is an intramolecular process which induces radiationless decay of the benzene triplet state, other than impurity quenching.
By competitive methods Ishikawa and Noyes (226) (sensitized biacetyl phosphorescence) and Cundall and Davies (159) (butene-2 isomerization) both estimated the triplet lifetime to be of the order of 10 us. In a reexamination of the butene-2 system, Lee (227) estimated a value of 100 us, a finding confirmed by Cundall and Dunnicliff (105). An examination of the kinetics of the benzene photosensitized composition of cyclopentanone decomposi- tion allowed a value of longer than 3 us to be deduced. This type of experiment is far from satisfactory since photochemical processes can intervene at low pressures, and impurities and the quenching effects of photoproducts can affect the results. These problems can only be overcome by some form of direct measurement. Parmenter and Ring (228) used a flash method in which 20 torrs of benzene and 0.01 torr of biacetyl were submitted to a 20 J, ~
3 us flash of 250 t o 260 nm light. From the time dependence of the biacetyl phosphorescence emission, a lifetime of 26 f 5 p s was deduced. Lee (227) cites a private communication from Parmenter in which the lifetime of the gaseous triplet is given

190 R. B. Cundall, et al.
to be 70 f 20 us for 2 torrs benzene. Burton and Hunziker (229) have developed a modulated transient absorption technique that uses Hg 63P0 atoms to populate the benzene triplet state. The decay of the triplet is observed by an absorption band at 231.5 nm, assigned to the allowed 3E2 -+ 3B1u absorption (36), with f- values of 0.07 and 0.06 for ligit and heavy benzene, respectively. The decay is pseudo-first-order, resulting from bimolecular triplet-triplet interaction. The results are viewed guardedly by other workers because of effects that may arise from the high light absorption which must be used. First order decay rates of 3 x 103d and 2 x lo3,-' were calculated for the presen e of 300 to 800 torrs of nitrogen, needed to promote rhe Hg 6 P1-%i3P0 transition. mixtures at 390°K has given data which can be used to calculate 1.3 x losse1 for the decay constant of the 3B1u state of benzene at a partial pressure of 18 torr (230), from the naphthalene triplet absorption following irradiation.
A new experimental approach to the problem has been used by Hunter and Stock (231) involving measurement of the frequency dependence of the acoustic signal resulting from transformation of electronic excitation from vibrational to translational energy, consequent to excitation of the 1B2u state. This technique was used to measure the decay of the benzene triplet over the range 0 to 6 torrs, and it was found, in agreement with what could he inferred from some of the earlier work, that decay of the triplet depends upon benzene concentration. deca process was found to have a rate constant of (1.7 f 0.1) x 10' s-' and a collision efficiency of 1.6 x deduced for the second order self-quenching process. The authors claim to have discounted the effects of deactivation by traces of oxygen. A strong perturbation of the 3B1u state by ground state molecules seems the most likely mechanism for accounting for the observed concentration dependence; and involvement of the triplet excimer is in accord with the following scheme.
s The pulse radiolysis of naphthalenerbenzene vapor
The pressure-independent
L
k3
M

Excitation of Benzene 1%
The reciprocal of the triplet lifetime -cT-l = k2 + k3(k- +k3) -1 kl[lM]. Analysis of the results shows the k3(k-1 + k3)-I is 1.6 x in other aromatic hydrocarbons in solution where triplet-ground state interaction is proposed to explain similar shortening of measured triplet lifetime (232); in these cases, it would seem that k3 >> k-1.
gas phase system applies also to the liquid phase. nitude agreement between data from the various techniques for measuring ‘IT seems to have been achieved. Cundall and Griffiths (2331, obtained the first indications from studies on the benzene1 butene-2 system, that the triplet state lifetime was very short. The value for kET TT was 21.2~1 at 25OC where kET was the overall rate constant for triplet excitation energy transfer from benzene to butene-2, and TT the lifetime of the benzene triplet. Assum- ing a value of about 5 x 109 ~-1s-1 for kET, TT appears of the order of a few nanoseconds. Although the kinetic analysis may have oversimplified (175), the order of magnitude lifetime is similar to that determined from the work of Kaufman (234) and Hentz and Perkey (176) and also consistent with the results of Dubois (235,236) who failed to detect any evidence for triplet energy transfer to biacetyl in pure benzene. By contrast, Lipsky (237), Sandros (181), and Russian workers (238,239) ob- served triplet energy transfer from benzene to biacetyl for di- lute benzene solutions. This indicates that the triplet life- time in solution is also concentration dependent as is the case for o-xylene (240) where the lifetime increased from a few nano- seconds to microseconds or more, on dilution. This has been con- firmed for benzene (175,189) and further, additional evidence from photochemical (173) and pulse radiolysis (241) systems demonstrate the very short lifetime of the triplet state in pure benzene liquid. It is believed (175) that decay proceeds via the triplet excimer which is only inefficiently quenched by butene-2. Results of electron impact excitation (242) and pulse radiolysis (243) have indicated a triplet lifetime of 500 us for gaseous benzene at low and high pressures.
under these or similar conditions by transient spectroscopy techniques have so far been unsuccessful. Thomas and Mani (244) found a transitory species with an absorption maximum and a decay time of 112 ns in benzene and benzene cyclohexane mixtures. The results from addition of naphthalene, biacetyl, oxygen, and piperylene suggest that this is a product of reaction between triplet and ground state benzenes. Build-in times for this species, which are 3 ns for pure benzene and 20 ns for 10 percent benzene/cyclohexane mixture are temperature independent.
and thus k-1 >> k3. This contrasts with the situation
The self-quenching mechanism which has been invoked for the Order of mag-
Attempts to directly detect the triplet state of benzene
Laser

192 R. B. Cundall, e t a l .
f l a s h photo lys i s (179) r e s u l t s are c o n s i s t e n t wi th a T1 + S o r e a c t i o n f o r t r i p l e t decay. achieved t o d i r e c t observat ion of t h e benzene t r i p l e t i n solu- t i o n a t room temperature.
The l i f e t i m e of t h e t r i p l e t s ta te of benzene i n f l u i d solu- t i o n s ( a t room temperature and above) is determined by t h e , seemingly e f f i c i e n t , i n t e r a c t i o n of t h a t s ta te with ground s t a t e molecules. This is n o t i n c o n s i s t e n t with t h e low temperature behavior from which i t has been deduced (51-55), t h a t a thermally a c t i v a t e d T 1 -f S o t r a n s i t i o n arises from d i s t o r t i o n of t h e ben- zene r i n g which des t roys t h e s t r i c t D6h symmetry. 65OK, i t is apparent t h a t hydrocarbon so lvents are not i n e r t and t h a t perf luorocarbons are t h e least per turb ing of p o s s i b l e s o l v e n t s (222). The concentrat ion quenching mechanism must in- volve some, as y e t unestabl ished, form of chemical o r charge t r a n s f e r i n t e r a c t i o n which reduces t h e forbidden c h a r a c t e r of t h e t r a n s i t i o n . process occurs a t low temperature.
This is t h e most t h a t has been
Even below
There i s no p o s i t i v e evidence t o show t h a t t h i s
XI. TRANSITIONS FROM HIGHER (n > 1)EXCITED STATES
Several years ago, Braun, Kato, and Lipsky (142) showed t h a t i n t h e vapor s o l u t i o n and bulk l i q u i d s , benzene and i t s methyla- ted d e r i v a t i v e s appeared not t o undergo e f f i c i e n t i n t e r n a l con- v e r s i o n o r in te rsys tem cross ing from higher e x c i t e d s i n g l e t s t a t e s t o form e i t h e r S1 o r TI states. Bi rks , Conte, and Walker (143) who pos tu la ted a scheme involving excimers of h igher exc i ted states, t o expla in t h e i r d a t a and n o t e d ' t h a t benzene proved t o be t h e except ion amongst s i m i l a r compounds i n t h a t t h e e f f e c t decreased on d i l u t i o n .
by Voltz and co-workers (145) . Using 2-napthtyl-2-phenyl-5- oxazole-1.3 an e x c i t a t i o n energy acceptor , these au thors ob- ta ined r e s u l t s s i m i l a r t o those of Lipsky (142), but a l s o found t h a t au to ioniza t ion occurred a t wavelengths below 175 nm. There is low e f f i c i e n c y of conversion of Blu and lElu t o t h e lBlU s t a t e , and r e l a t i v e e f f i c i e n c i e s of f luorescence a r e cons tan t bu t d i f - f e r e n t over each of t h e main bands. Voltz ( 1 4 5 ) a l s o claims t h a t energy t r a n s f e r occurs from S2 and S3 states, i n agreement w i t h Laor and Weinreb (245) but a t var iance wi th Lawson, Hirayama, and Lipsky (72,246). E lec t ron scavengers such as 02 and CHC13 which a f f e c t t h e S 3 and S2 states t o only a n e g l i g i b l e e x t e n t , in f luences t h e i o n i z a t i o n e f f e c t s a t higher e x c i t a t i o n energ ies .
This w a s confirmed by
Some more r e c e n t experimental evidence has been presented

E x c i t a t i o n of Benzene 193
Lawson, Hirayama, and Lipsky ( 7 2 , 2 4 6 ) a l s o measured t h e v a r i a t i o n i n quantum e f f i c i e n c i e s f o r i n t e r n a l convers ion from S3 t o S 1 i n s e v e r a l s o l v e n t s and found t h a t t h e v a l u e s va ry ve ry markedly. The e f f i c i e n c i e s measured were 0.45 i n pure benzene, 0.37 t o 0.28 i n a l coho l s and o t h e r p o l a r s o l v e n t s , 0.25 t o 0.22 i n a l i p h a t i c hydrocarbons, 0.04 i n n-C6F14, and about ze ro i n t h e gas phase. The o r d e r of e f f e c t i v e n e s s i n a number of sol- v e n t s p a r a l l e l e d t h e enhancement of t h e 0-0 band i n abso rp t ion . The i m p l i c a t i o n would s e e m t o b e t h a t s o l v e n t p e r t u r b a t i o n pro- duces s ta te mixing and consequent enhancement of i n t e r n a l conver- s i o n Sn -t S 1 .
n a l conversion p rocesses of h ighe r e x c i t e d s i n g l e t s ta tes . Lumb, Braga, and P e r e i r a ( 1 4 4 ) measured t h e f luo rescence and phos- phorescence e x c i t a t i o n s p e c t r a of r i g i d s o l u t i o n s of benzene i n e t h e r / i s o o c t a n e / a l c o h o l glasses a t 77OK. The two e x c i t a t i o n s p e c t r a were i d e n t i c a l , showing t h a t @F and flP va ry i n t h e same manner w i t h Aex . l e v e l s are n o t ze ro , b u t depend upon concen t r a t ion ; a s u r f a c e quenching mechanism may b e involved .
D i r e c t obse rva t ion of f luo rescence from h ighe r s i n g l e t s tates of benzene and some methyl d e r i v a t i v e s has r e c e n t l y been achieved by Hirayama, Gregory, and Lipsky ( 2 4 7 ) . Using ap ara- t u s capable of d e t e c t i n g f luo rescence y i e l d s as low as they recorded t h e emission s p e c t r a from oxygenated s o l u t i o n s of pure benzene and o the r a romat ics e x c i t e d a t 184.9 nm. t i o n of t h e t a i l of t h e r e s i d u a l S emission g i v e s a f luo rescence spectrum wi th A,,, approximately 235 nm and k47 = 8 x benzene. This i s ass igned t o an S2-S0 emiss ion .
could a l s o be d e t e c t e d f o r t h e S3-S0 t r a n s i t i o n . cesses have a l s o been achieved w i t h p-xylene, t o luene , and m e s i - t y l e n e , i n t h e vapor phase, l e a d i n g t o t h e hope t h a t d a t a f o r benzene vapor may soon b e forthcoming.
Some informat ion is a v a i l a b l e on t h e photochemical e f f e c t s t h a t occur a f t e r t h e format ion of h ighe r e x c i t e d s ta tes , which may h e l p , i n p a r t a t least , t o exp la in some of t h e e f f e c t s . Foote, Mallon, and P i t t s ( 2 4 8 ) , and Shindo and Lipsky ( 2 4 9 ) in - v e s t i g a t e d t h e photochemistry of benzene vapor , e x c i t e d i n t o t h e S j state . g i v e a quantum y i e l d of u n i t y for chemical r e a c t i o n . The isomer formed w a s later ( 2 5 0 ) shown t o b e fu lvene , w i t h some polymer format ion . fo l lowing s e c t i o n .
Other t ypes of experiment g ive informat ion about t h e i n t e r -
The e f f i c i e n c i e s of conversion from h i g h e r
Subtrac-
f o r
I n 0 . 1 M s o l u t i o n s of p-xylene and mes i ty l ene , emiss ions S i m i l a r suc-
Both sets of d a t a , when e x t r a p o l a t e d t o ze ro p r e s s u r e ,
This r o u t e f o r energy decay w i l l be examined i n t h e

194 R. B. Cundall, et al.
XI I. RELEVANT PHOTOCHEMICAL REACTIONS OF EXCITED STATES OF BENZENE
As previously stated, molecular isomerization does not pro- vide an explanation for the quantum deficit following excitation of benzene; the first band does not bear close scrutiny. ever, an investigation of the photochemistry of excited benzene is of interest, in view of the variety and complexity of the iso- meric products. An extensive review of conditions under which the formation of isomers of benzene and its derivatives can be found has been compiled by Scott and Jones (250). Interconver- sion of the isomers which would be relevant to photophysical ef- fects are governed by orbital symmetry considerations, and exam- ples of this type of treatment are now appearing (251,252).
In 1966, Bryce-Smith and Longuet-Higgins (65,185) suggested a scheme that seems to provide a generally satisfactory descrip- tion of the facts. Prefulvene and Dewar-like intermediates con- sistent (184) with the symmetry of the 1B2u, and 3B1u excited states which supposedly give rise to them, have been proposed. The existence of these intermediates has not been proven, and for heavily substituted fluoro-benzenes, Dewar isomeric forms arise from excited singlet states in apparent contravention of the sym- metry considerations (253,254). The significance of this is not clear.
No attempt is made here to review all the experimental data, but some investigation relevant to the theme of this article will be cited. Wilzbach, Kaplan, and Harkness (255) showed that benzene-1,3,5,d3 rearranged to the 1,2,4-isomer when excited at 254 nm in the vapor phase, and deduced that this oc- curred through a benzvalene intermediate with a yield of about 3 percent. Subsequently, Kaplan and Wilzbach (256) isolated benzvalene, from benzene excited within the first band, with a yield of 0.02. Addition of added gases increases the yield by stabilization of "hot" benzvalene molecules. The data on the effect of exciting wavelength shows that relaxation from higher vibrational levels of the S1 state is accompanied by a decrease in the yields of both fluorescence and triplet production. The work of Kaplan and Wilzbach provides some evidence for an in- crease in the yield of benzvalene in going from 254 to 237 nm, but no correlation with the sharp increase in nonradiative decay when the vibrational energy exceeds 2500 cm-' has been achieved. An interesting argument against benzvalene formation providing a complete explanation of the nonradiative processes has been advanced by Spears and Rice (111). The conservation of angular momentum must inhibit conversion of benzene into benzvalene, but no correlation of rotational energies with either radiative o r
How-

Excitation of Benzene 195
nonradiative rate constants was found by Parmenter and Schuh (257). A s pointed out in Section V I I I , recent results of Stein (195) have indicated that isomerization cannot explain completely Channel I11 in benzene.
More work on the photochemistry of benzene cannot fail to be informative. A recent example of the type of experiment that shows promise is that reported by Irina and Kurien (194). Phenol and mucondialdehyde are major products found from the photolysis of benzene vapor in the presence of oxygen, and also that phenol alone is formed in the absence of oxygen on interaction with water. The experimental conditions employed a flow of the vapor through a trap, indicating that the products formed must have resulted from the interaction of a long-lived excited inter- mediate with water. This intermediate cannot be benzvalene since it has been established (258) that this reacts with water to give a cyclo pentadiene-1-carboaldehyde, via the following mechanism:
H-C=O I
+ 0 "Internal conversion" in the liquid and solid phases is still obscure. usually of the order of 0.01, and in general are very much less than in the gas phase, presumably due to the rapid collisional stabilization that can occur in condensed phases. Formation of an intermediate of the pre-fulvene type would require several quanta of ring-deforming vibrations, and this could account for the decrease in yields with lowering of temperature.
solution than in the gas phase, and since the controlling para- meters are, to a certain extent, solvent dependent (91,94), the possibility of chemical reactions with solvent by excited state routes cannot be ignored. vented very clear results.
Decomposition and isomerization yields are small,
Since the efficiency of "internal conversion'' is greater in
Experimental difficulties have pre- This is illustrated by the discordant

196 R. B. Cundall, et a l .
d a t a on t h e r e a c t i o n of oxygen with benzene under var ious condi- t i o n s (130,259-261).
It has been known f o r some t i m e t h a t hexa t r ienes can b e formed by r e a c t i o n of exc i ted benzene molecules wi th so lvent i n a f rozen g l a s s (262-264). t h i s process have been deduced by Simons and Smith (90). propose t h a t intermolecular v i b r o n i c i n t e r a c t i o n with so lvent is a necessary b u t no t s u f f i c i e n t requirement f o r production of hexa t r ienes i n v iscous media. The r e a c t i o n i s assoc ia ted with r a d i a t i o n l e s s decay of t h e t r i p l e t s tate r a t h e r than a bimole- c u l a r r e a c t i o n of t r i p l e t benzene wi th neighboring so lvent molecules. Since r a d i a t i o n l e s s decay c o n s t i t u t e s in te rsys tem cross ing , t h e product of t h e r e a c t i o n i s a l s o i n t h e ground s t a t e . It i s s t r e s s e d by t h e au thors t h a t hexa t r ienes a r e not produced i n f l u i d media o r by nonphosphorescent benzene d e r i v a t i v e s i n which t h e r e is a rap id competing decay of t h e t r i p l e t s ta te . The formation of t h e hexa t r iene only occurs when weak i n t e r a c t i o n s occur between t h e re lax ing t r i p l e t and t h e s o l v e n t .
zene t o both i n t e r n a l conversion and t r i p l e t formation by i n t e r - system cross ing r e q u i r e s t h a t t h e t r i p l e t y i e l d measurement tech- niques used should b e r e l i a b l e and t h a t chemical r e a c t i o n s of exc i ted states wi th t r i p l e t monitoring reagents do not occur. A s a r u l e , i n t h e case of o l e f i n s , which have been most used a s t r i p l e t scavengers f o r benzene, i t is reassur ing t o f i n d t h a t f o r t h e types of o l e f i n used a s t r i p l e t monitors a d d i t i o n products a r e u s u a l l y only formed i n very s m a l l y i e l d s (265,268). Consis- tency wi th t r i p l e t y i e l d s obtained by o t h e r methods g ive some f u r t h e r confidence t h a t t h e technique used, i n genera l , provide s e l f - c o n s i s t e n t r e s u l t s .
Examples of t h e type of agreement are given f o r t h e gas phase i n t h e recent review of Noyes and Al-Ani (269) and compari- son of t r i p l e t y i e l d s in s o l u t i o n obtained by Sandros (146,181) using b i a c e t y l as t h e scavenger, and Cundall et a l . (147,156,157) using butene-2.
Some d e t a i l s of t h e mechanism of They
Assignment of nonradia t ive decay from t h e $1 s t a t e of ben-
XIII. BENZENE EXCIMER
The formation of a benzene dimer from a s s o c i a t i o n of a ground s ta te monomer and an exc i ted monomer is a wel l -es tab l i shed observat ion of t h i s s p e c i e s was f i r s t noted by Dammers de Klerk (270) and Ivanova and co-workers (271) i n a s tudy of t h e e f f e c t s of concentrat ion on t h e f luorescence spectrum of benzene. Broad s t r u c t u r e l e s s emission on t h e long wavelength s i d e of monomer emission a t room temperature is c l e a r l y observed a t concent ra t ions

E x c i t a t i o n of Benzene 197
of about l M , and a f u r t h e r i n c r e a s i n g concen t r a t ion becomes t h e dominant component i n t h e spectrum.
The e x i s t e n c e of such a dimeric s p e c i e s , s t a b l e i n t h e ex- c i t e d s ta te , and experiencing d i s s o c i a t i v e r e p u l s i v e i n t e r a c t i o n i n t h e ground s t a t e , i s explained (272) i n terms of configura- t i o n a l i n t e r a c t i o n between e x c i t a t i o n and charge resonance s ta tes of t h e dimer. Its s t r u c t u r e i s g e n e r a l l y be l i eved t o be of an ec l ip sed p a r a l l e l p l a t e form, though Birks and co-workers (273- 275) favor a s l i g h t l y d i sp l aced s t r u c t u r e w i t h one molecule over- lapping t h e o t h e r . The s tudy of Birks and Christophorou (276) has shown t h a t t h e n e t excimer binding energy i s dependent upon in t e rmolecu la r s e p a r a t i o n , and i t seems w e l l e s t a b l i s h e d t h a t t he p a r t i c u l a r r i n g s e p a r a t i o n i n benzene i s 0.31-0.33 nm.
au tho r s (272,277,279) have suggested t h a t emission t o t h e repul- s i v e ground s ta te i s d i p o l e forbidden. It i s p o s s i b l e (272) t h a t a s m a l l r o t a t i o n from t h e strict p a r a l l e l p l a t e conf igu ra t ion i s s u f f i c i e n t t o remove t h i s r e s t r i c t i o n , and Chesnut e t a l . (280) have shown t h a t a r o t a t i o n of 30 degrees decreases t h e excimer binding energy by 0.01 e V . This would suggest t h a t excimer emission p r o b a b i l i t y may be temperature dependent; and such has been found t o be t h e case by Hirayama and Lipsky (125, 281) and Cundall and Robinson ( 1 3 4 ) , b u t n o t by B i rks and co- workers (282).
The parameters t h a t d e s c r i b e t h e p r o p e r t i e s of t h e excimer s ta te may be e x t r a c t e d from k i n e t i c a n a l y s i s of t r a n s i e n t and s t eady s t a t e luminescence da ta . The scheme due t o B i rks , Braga, and Lumb (131,274), a l though c r i t i c i z e d f o r i t s s impli- c i t y (125,283), accounts w e l l f o r experimental decay of t h e s i n g l e t excimer. The observed quantum y i e l d of monomer f luo res - cence flm i s reduced i n t h e presence of excimer from t h e i n t r i n - s ic y i e l d fli~ according t o :
The excimer so descr ibed i s s i n g l e t i n c h a r a c t e r , and many
where l l ch is t h e S t e r n Volmer quenching parameter such t h a t OF, a t c = Ch. A s i m i l a r exp res s ion f o r excimer f luo rescence
i s 0
OFD = 0FD (1 + c h ] c )
Two eva lua t ions O € ch are thus p o s s i b l e f o r a l l temperatures where excimer band emission can be observed. Cundall and Robinson (134) showed t h a t t h e temperature of Ch l e d t o t h e de- duct ion t h a t (Eb -k EICM - E I C ~ D ) = 0.38 e V . r e f e r t o processes descr ibed i n t h e in t roduc to ry s e c t i o n ) . Excimer f luo rescence l i f e t i m e s , -rD can be e x t r a c t e d from know- ledge of t h e measured emission l i f e t i m e of more concentrated
(he re s u b s c r i p t s

198 R. B. Cundall, e t a l .
so lu t ions , and t h e i n t r i n s i c monomer l i f e t i m e T ~ , according t o :
Combination of t h i s da t a wi th t h a t of t he i n t r i n s i c excimer e m i s - s i on y i e ld go probabili ty.FDCundall and Robinson (134) found t h a t t h e ra te con- s t a n t so ca lcu la ted had an assoc ia ted ac t iva t ion energy of 0.15 e V , assigned t o thermal d i s t o r t i o n of t he dimer t o overcome di- po le r e s t r i c t i o n s on the emission. Helman (136,139) has found a concentrat ion dependence f o r km, and he specula tes t h a t t h i s may be due t o the formation of trimeric exci ted complexes.
t he involvement of multimolecule complexes t o explain some ano- malies i n the CCl4 quenching of emission da ta . evidence f o r such species, though they may e x i s t , provided the exciplex is of lower po ten t i a l than the s i n g l e t monomer molecule. The binding energy f o r such a spec ies would be very small. This, together with t h e probable highly forbidden emission , might lead t o d i f f i c u l t i e s i n observation. Proof of t he involvement of t he types of spec ies i n the decay of t h e excimer remains un- ava i lab le .
(136) from t r ans i en t luminescence decay da ta a t l o w temperature (0.26 eV) is a t var iance with the conclusions of o ther authors (125,134) (20.36 eV) using s teady s ta te luminescence da ta . The problem l ies i n determining the temperature dependence, i f any, of t he r a t i o kFD/km, s ince a l l authors agree t h a t t he r a t i o kDM/km has a complete ac t iva t ion energy of approximately 0.25 (t0.03) e V . Theore t ica l e s t i m a t e s of V a l a , H i l l i e r , Rice and Jo r tne r (284) and Chestnut e t a l . (280) p lace the expected binding energy i n the range 0.3 t o 0.5 e V depending upon r i n g separa t ion . The var ious parameters descr ibing t h e s i n g l e t benzene excimer are summarized i n Table 15, compiled wi th reference t o Fig. 10.
The experimental d i f f i c u l t i e s involved i n measurement of f luorescence y i e l d s and l i f e t imes f o r benzene, and o ther systems a l s o , as a funct ion of concentrat ion make i t c e r t a i n t h a t t he var ious thermodynamic q u a n t i t i e s have not ye t been f i rmly estab- l i shed .
t he excimer are s i m i l a r t o those ava i l ab le f o r t he exc i ted monomer. There i s no d i r e c t evidence f o r t he ex is tence of t he t r i p l e t excimer although i ts exis tence has been invoked t o ex- p l a in anomalous t r i p l e t y i e ld da t a (175). H i l l i e r , Glass and
allows computation of km, t he excimer f luorescence
Ludwig and Amata (135) a l s o found it necessary t o pos tu la te
There i s no d i r e c t
The excimer binding energy deduced by Gregory and Helman
It i s reasonable t o presume t h a t t he routes f o r decay of
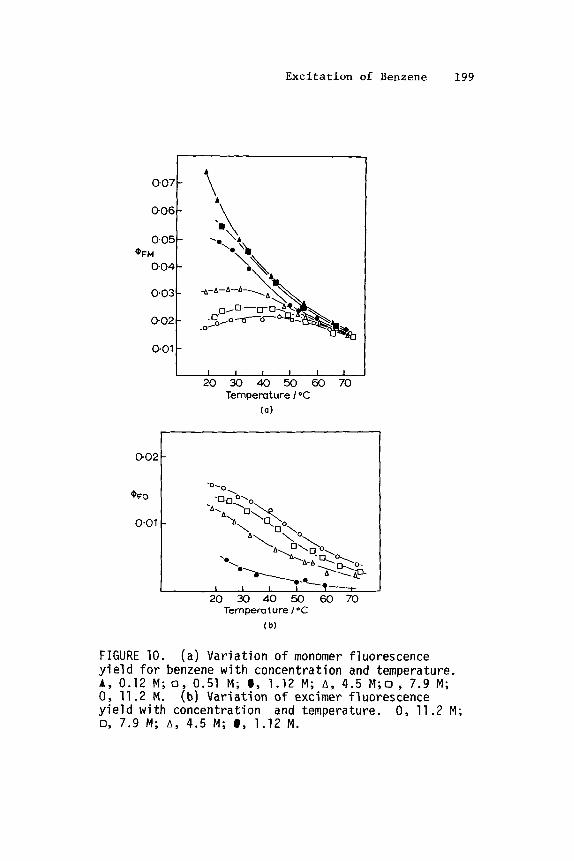
Excitation of Benzene 199
0.07 I \
I I I I I I I
20 30 40 50 60 70 Temperature I OC
( a )
FIGURE 10. ( a ) Variation of monomer fluorescence yield fo r benzene w i t h concentration and temperature. A, 0.12 M ; O , 0.51 M; 0, 1.12 M; A , 4.5 M;O , 7.9 M ; 0 , 11.2 M. (b) Var ia t ion o f excimer fluorescence yield w i t h concentration and temperature. 0 , 11.2 M ; 0, 7.9 M; A , 4.5 M; @, 1.12 M.

200 R. B . Cundall, et al.
TABLE 15.
Parameter Value Temp. Comments Ref.
Decay parameters of the s i n g l e t excimer o f benzene.
(OC)
&D 0.012 20 Hexane (124)
0.016 25 Methylcyclohexane (125)
0.022 20 Cyclohexane, T dependence
Eb 0.26 -3OOC Methyl-cyclohexane (136)
0.22 - Cyclohexane (282)
- >0.36 - Methyl-cyclohexane (125)
0.34 - Cyclohexane (134)
Ch 4.8 24 Hexane (285)
2.1 25 From monomer Me-cyclohexane (125) 5.0 From excimer 5 .O 20 From monomer
16.1 From e-xcimer cyclohexane (283)
5.0 25 From monomer 4.65 From excimer
(very T dependent)
(134)
D 12.3 25 From ionization studies (286)
12. 20 Hexane (282)
30 20 Cyclohexane (134)
T

Exc i t a t ion of Benzene 201
Rice (287) i n f e r r e d i t s e x i s t e n c e from e legan t experiments i n which they examined t h e phosphorescence emission of t h e para- cyclophane compounds. by methylene b r idges i n a sandwich s t r u c t u r e , they observed s h i f t e d phosphorescence s p e c t r a w i t h a s s o c i a t e d loss of s t r u c - t u r e ; t h e most s i g n i f i c a n t e f f e c t s be ing a t a r i n g s e p a r a t i o n of approximately 0.3 nm. P h i l l i p s and Schug (288) i d e n t i f i e d emission a t about 4.30 nm i n 1 MeV e l e c t r o n i r r a d i a t e d benzene a t 1 3 8 O K with t h e t r i p l e t excimer.
a t i c s t u d i e s us ing t r a n s i e n t abso rp t ion techniques. Fu r the r , t h e c h a r a c t e r i s t i c excimer emission h a s n o t been observed i n t h e s o l i d phase, where in t e rmolecu la r d i s t a n c e s are such as t o mini- mize t h e i n t e r a c t i o n and because achievement of t h e necessary o r i e n t a t i o n i s i n h i b i t e d i n t h e f rozen samples.
With t h e two benzene r i n g s he ld toge the r
Conclusive evidence of t h i s e l u s i v e s p e c i e s awaits system-
X I V . CONCLUSION
It i s clear t h a t t h e photophysical behavior of benzene i s complex and t h a t t h e nonrad ia t ive S 1 + So(?) process vaguely descr ibed a t Channel I11 is e s p e c i a l l y important . It is no t p o s s i b l e t o conclude whether benzene is a " typ ica l " aromatic molecule u n t i l comparably d e t a i l e d s t u d i e s of o t h e r systems have been made.
ACKNOWLEDGMENTS
P a r t of t h e work presented h e r e w a s completed during J u l y of 1974 a t t h e Un ive r s i ty of Lourenco Marques, Mozambique. W e wish t o express our g r a t i t u d e t o ULM f o r t h e h o s p i t a l i t y and f o r f i n a n c i a l support f o r one of u s (R.B.C.) and a l s o t o NATO f o r a r e sea rch g ran t . One of u s ( D . A . R . ) g r a t e f u l l y acknowledges a d d i t i o n a l support from t h e Research and P u b l i c a t i o n s Committee, ZnM.
REFERENCES
(1) J. I. S t e i n f e l d , "Molecules and Radiation", Harper and Row, New York, 1974.
(2) For example: (a) M. Goeppart-Mayer and A . L. S k l a r , J. Chem. Phys., 5, 645 (1938); (b) R . C . P a r r , D . P. Craig, and I. C. Ross, J. Chem. Phys. , 18, 1561 (1950).

202 R. B. Cundall, et al .
(3) H. Sponer, G. Nordheim, A. L. Sklar , and E. Teller, J. Chem. Phys. , I , 207 (1939).
(4) J. R. P l a t t and H. B. Klevens, Chem. Rev., 41, 301 (1949).
(5) S. Leach, R. Lopez-Delgado, and F. D e l m a s , J. Mol. Spectrosc. , - 7, 304 (1962).
(6) R. B. Merrithew and C. V. Marusak, J. Mol. Spectrosc. 25, 269 (1968).
(7) H. C. Wolf, "Solid S t a t e Physics" (F. S e i t z and D. Turnbul l , E d s . ) , V o l . 9 , Academic P r e s s , New York, 1959, p . 1.
(8) J. B. Bi rks , "Photophysics of Aromatic Molecules", Wiley, New York, 1970.
(9) J. Koutecky, J. Cizek, J. Dubsky, and K. Hlavaky, Theoret. Chim. Acta, 2, 462 (1964).
(10) J. Koutecky, K. Hlavaky, and P. Hochman, Theor. Chim. Acta, - 3 , 341 (1965).
(11) J. E. Bloor, J. L e e , and S. Gar t s ide , Proc. Chem. Soc., 413, (1960).
(12) R. J. Buenker, J. L. Whitten, and J. D. Pekke, J. Chem. Phys. , 49, 261 (1968).
(13) T . Dunn and C. K. Ingold, Nature, 176, 65 (1965).
(14) S. D. Peyerinholf and R. J. Buenker, Theor. C h i m . Acta, 19, 1 (1970).
(15) J. Karwowski, Chem. Phys. L e t t . , 18, 47 (1973).
(16) P. 5. Hay and I. S h a v i t t , Chem. Phys. L e t t . , 22, 33 (1973).
(17) J. Karwowski, Acta Phys. Polon., A. , 42, 655 (1972).
(18) J. N. Murrel l and T. A . Pople, Proc. Phys. SOC. (London), 6&, 245 (1956).

E x c i t a t i o n of Benzene 203
(20)
(a) For benzene: B. Katz, M. B r i t h , B. Sharf and J. J o r t n e r , J. Chem. Phys., 52, 88 (1970); (b) Confirmed w i t h toluene and xylene: B. Katz, M. B r i t h , B. Sharf and J. J o r t n e r , J. Chem. Phys., s, 3924 (1971); ( c ) E. Pantos and T. D. S. Hamilton, Chem. Phys. L e t t . , 17, 588 (1972).
M. B r i t h , R. Lubert and I. T. S t e inbe rge r , J. Chem. Phys., - 54, 5104 (1971).
E. N . Lassettre, A. Skerbele , M. D i l ton and K. ROSS, J. Chem. Phys., 48, 5066 (1968).
A. M. Taleb, I. H. Munro, and J. B. B i rks , Chem. Phys. L e t t . , 21, 454 (1973).
G. C . Morris and J. G. Angus, J. Mol. Spectrosc. , 45, 2 7 1 (1973).
B. Katz, M. B r i t h , B. Sha r f , and J. J o r t n e r , J. Chem. Phys. , 52, 88 (1970).
P. M. Johnson, J. Chem. Phys., 62, 4562 (1975).
J. B. Birks , Chem. Phys. L e t t . , 3, 567 (1969).
J. H. Callomon, J. E. Pa rk in , and R. Lopez-Delgado, Chem. Phys. L e t t . , 2, 125 (1972).
G. Diesken and J. W. Moskwi tz , J. Chem. Phys., 47, 3491 (1967).
J. Momigny, C . Gof fe r t and L. K ' O r , I n t . J. Mass. Spectrom. Ion. Phys., L, 53 (1968).
A. D. Baker, C . R. Brundle, and D. W. Turner, I n t . J. Mass. Spectrom. Ion. Phys., 1, 443 (1968).
E. Lindholm and B. 0. Johnson, Chem. Phys. L e t t . , 2, 501 (1967).
B. F. Minaev, Isv. Vyssh. Vchet. Zaved. F i z . , 14, 118 (1971).
(33) L. Burnel le and M. J. Kraneppol, J. Mol. Spectrosc. , 37, 383 (1971).

204 R. B. Cundal l , e t a l .
(34)
(35)
(42)
(43)
(44)
(45)
(47)
(48)
(49)
D. Kearns, J. Chem. Phys. , 36, 1608 (1962).
For summary see: S. P. McGlynn, T. Azumi and M. K inosh i t a ,
H a l l , N . J . , 1969. “Molecular Spectroscopy of t h e T r i p l e t S t a t e ” , P ren t i ce -
J. Lec le rq , and J. M. Lec le rq , Chem. Phys. L e t t . , 18, 411 (1973).
R. A s t i e r and Y. H. Meyer, Chem. Phys. L e t t . , 3, 399 (1968).
J. B. Doering, J. Chem. Phys., 51, 2866 (1969).
S. D. Colson and E. R. Be rns t e in , J. Chem. Phys. , 43, 2661 (1965).
R. M. Hochstrasser and J. E. Wessel, Chem. Phys. L e t t . , 24 1 (1974).
R. M. Hochstrasser , H . N . Sung and J. E. Wessel, Chem. Phys. L e t t . , 24, 7 (1974).
L. Wunsch, H. J. Neusser and E. W. Schlag, Chem. Phys. L e t t . , - 31, 433 (1975).
‘J. Metcalfe, M. G. Rockley and D. P h i l l i p s , J. Chem. SOC. Faraday 11, 2, 1660 (1974).
M. Kasha, D i s c . Faraday SOC., 9, 14 (1950).
T. A , Gregory, F. Hirayama and S. Lipsky, J. Chem. Phys. , - 58, 4697 (1973).
See a l s o : R. B. Cundall and T . F. Palmer, Annual R e p o r t s A , Chemical Soc ie ty , London, 1973.
H. S c h u l l , J. Chem. Phys., 17, 295 (1949).
G. S. Kembrovskii, V. P. Bobrovich and A. N . Serchericho, Opt. Spec t rosc . , 24, 213 (1968).
G. N . L e w i s and M. Kasha, J. Am. Chem. SOC., 66, 2100 (1944).
G. W . King and E. H. Pinnington, J. Mol. Spec t rosc . , 15, 394 (1965).

E x c i t a t i o n of Benzene 205
(56)
(57)
(59)
(60)
( 6 1 )
(64)
(65)
M. S . de Groot and J. H. van d e r Waals, Mol. Phys . , 6, 545 (1963) . J. van Egmond and J. H. van d e r Waals, Mol. Phys. , 3, 1147 (1973).
J. van Egmond and J. H. van d e r Waals, Mol. Phys. , 28, 457 (1974).
J. van d e r Waals, A. M. D. Berghuis and M. S. d e Groot, Mol Phys . , 2, 497 (1971).
J. van d e r Waals, A. M. D. Berghuis , and M. S. de Groot, Mol. Phys., 13, 301 (1967).
A . A . Gwaiz, M. A. El-Sayed and D. S . T i n t i , Chem. Phys. L e t t . , 9, 454 (1971).
D. M. Haaland and G. C . Nieman, J. Chem. Phys. , 9, 4435 (19 73) . C. C . Nieman and D. S . T i n t i , J. Chem. Phys. , 46, 1432 (1967).
D. M. Burland, G. Cas t ro and G. W. Robinson, J . Chem. Phys . , 52, 4100 (1970).
A. Devaquet, J. Am. Chem. S O C . , 94, 9013 (1972) .
Y. Fujimura, H. Yanaguchi and T. Nakajima, B u l l . Chem. S O C . Japan , 45, 304 (1972).
A. D. L i e h r , J. Phys. Chem., 67, 389 (1963).
D. Bryce-Smith, A. G i l b e r t and D. A . Robinson, Angew. Chem., - 83, 803 (1971).
E. E. van Tamlen and S . P . Pappas, 3 . Am. Chem. Soc., E, 3297 (1963).
D. Bryce-Smith and H. C . Longuet-Higgins, Chem. Commun., 593 (1966).
(66) G. V. Klimusheva, Teor. Khim. Akad. Nauk. SSSR., 2, 37-43 (1966).

R. B. Cundall , e t a l .
For b r i e f reviews see: (a) B. Linder and S . Abdulnur, J. Chem. Phys., 54, 1807 (1971). (b) J. B. Bi rks , r e f . 8 . pp. 109-119.
For b r i e f review of observable e f f e c t s of Davydoy s p l i t t i n g , see: H. Suzuki, "Elec t ronic Absorption Spectra and Geometry of Organic Molecules", Academic P r e s s , New York, 1967, pp. 112-113.
Three of t h e components are allowed and are e a s i l y observed ( s e e r e f . 68); t h e f o u r t h f a c t o r group i s forbidden b u t has a l s o been observed: S . D. Colson, J. Chem. Phys., 44, 3324 (1968).
J. W. Eastman and S . J. Rehfeld, J. Phys. Chem., 74, 1438 (1970).
For a d e s c r i p t i o n of t h e simple theory and an account of d e v i a t i o n s noted f o r benzene, see r e f . 68, pp. 100-101.
C. W. Lawson, F. Hirayama and S . Lipsky, J. Chem. Phys., - 51, 1590 (1969).
J. S . Ham, J. Chem. Phys., 21, 756 (1953).
N. S . Bayliss and L. Hulme, Aus t ra l . J. Chem., 6, 257 (1953).
Y. Kanda, Y. Gondo and R. Shimada, Spectrochim. Acta, 17, 424 (1961).
N . S. Bayl i ss and N. W. Cant, Spectrochim. A c t a , 18, 1287 (1962) . J. R. P l a t t , J. Mol. Spectrosc. 9, 288 (1962.
R. P a r i s e r , J. Chem. Phys. , 24, 250 (1956).
N. S . Ham and K. Ruedenberg, J. Chem. Phys., 25, 1 (1956).
G. W. Robinson, J. Chem. Phys., 46, 572 (1967).
M. Koyanagi and Y. Kanda, Spectrochim. Acta, 20, 993 (1964).
G. Durocher and C. Sandorfy, J. Mol. Spectrosc. 14, 400 (1964) .

E x c i t a t i o n of Benzene 207
(83) G. Durocher and C. Sandorfy, J. Mol. Spec t rosc . , 20, 410 (1966).
(84) M. Koyanagi, J. Mol. Spec t rosc . , 25, 273 (1968).
(85) N . S. B a y l i s s , J. Mol. Spec t rosc . , 2, 406 (1969).
(86) A. Trombeti and C. Z a u l i , J. Chem. SOC., (B), 1968, 1574- 1577.
(87) Y. Munakami and Y. Kanda, Bu l l . Chem. SOC. Japan 41, 2599 (1968) .
(88) N. C. P e r r i n s , J. P. Simons and A . L. Smith, Trans. Faraday SOC., 67, 3415 (1971).
(89) J. P. Simons and A . L. Smith, J. Chem. SOC. Faraday II., - 70, 53 (1974).
(90) J. P. Simons and A. L. Smith, Chem. Phys. L e t t . , 16, 536 (1972); i b i d . , 25, 97 (1974).
(91) R. B . Cundall and L. C. P e r e i r a , Chem. Phys. L e t t . , 18, 371 (1973).
(92) R. B. Cundall , and L. C. P e r e i r a , submi t ted f o r p u b l i c a t i o n .
(93) L. C . P e r e i r a , Ph.D. T h e s i s , U n i v e r s i t y of Nottingham (1972).
(94) M. L u r i a , M , Ofran and G. S t e i n , J. Phys. Chem., 78, 1904 (1974).
(95) D. A. Robinson, Unpublished r e s u l t s .
(96) R. B. Cundall and A . J. Harrison, To be publ i shed .
(97) C . S. Parmenter, Adv. Chem. Phys. , 22 (1972).
(98) C . S . Parmenter and M. W. Schuyler , J. Chem. Phys., 52, 5366 (1970).
(99) C. S . Parmenter and M. W. Schuyler , Chem. Phys. L e t t . , 5, 339 (1970).
(100) J. A . Poole , J. Phys. Chem., 69, 1343 (1965).

208 R. B. Cundall, et al.
(109)
(111)
(112)
(113)
A. E. Douglas and C. W. Matthews, J. Chem. Phys., 48, 4788 (1968).
E. M. Anderson and G . B. Kistiakowsky, J. Chem. Phys., 48, 4787 (1968).
W. A. Noyes, Jr., W. A. Mulac and D. A. Harter, J. Chem. Phys., 44, 2100 (1966). E. M. Anderson and G . B. Kistiakowsky, J. Chem. Phys., 51, 182 (1969).
K. Dunnicliff, Ph.D. Thesis, University of Nottingham (1971): R. B. Cundall and K. Dunnicliff, To be published.
A. S . Abramson, K. G. Spears and S. A. Rice, J. Chem. Phys., 56, 2291 (1972).
(a) G. M. Breuer and E. K. C . Lee, J. Chem. Phys., 51, 3130 (1969); (b) G. M. Breuer and E. K. C. Lee, J. Chem. Phys., 3, 3615 (1969).
T. Chen and E. W. Schlag, "Molecular Luminescence" (E. C. Lim, Ed.), W. A . Benjamin, New York, 1969, p. 381.
M. Nishikawa and P. K. Ludwig, J. Chem. Phys., 52, 107 (1970).
G . S . Burton and H. E. Hunziker, J. Chem. Phys., 52, 3302 (1970).
K. G. Spears and S . A. Rice, J. Chem. Phys., 55, 5561 (1971).
B. K. Selinger and W. R. Ware, J. Chem. Phys., 53, 3160 (1970).
W. R. Ware, B. K. Selinger, C. S . Parmenter and M. W. Schuyler, Chem. Phys. Lett., 5, 342 (1970).
J. R. Lockwood, Ph.D. Thesis, Nottingham (1974).
B. K. Selinger and W. R. Ware, J. Chem. Phys., 52, 5482 (1970).
R. B. Cundall, K. Dunnicliff and A. J. R. V o s s . , Joint Ann. Mtg. Chem. SOC. and R.I.C., Nottingham, April 1969.

E x c i t a t i o n of Benzene 209
(117) S. J. S t r i c k l e r and R . 3. Watts, J . Chem. Phys . , 44, 5366 (1970) .
(118) G. B . Kis t iakowsky and C . S . Parmenter , J. Chem. Phys . , 42, 2942 (1965) .
(119) R. B. Cundal l and L . C. P e r e i r a , J . Chem. SOC. Faraday 11, - 68, 1152 (1972) .
(120) J. W. Eastman, J. Chem. Phys. , 49, 4617 (1968) .
(121) M. D. Lumb and M. I. Bar radas , L . C . P e r e i r a and C . L. Braga, Rev. F i z . Quin. Eng. U.L.M. , 1, 1 (1969):
(122) G. B . Evans, Ph.D. T h e s i s , Nottingham (1968) .
(123) W. R. Dawson and M. W. Windsor, J. Phys. Chem., 72, 325 (1968) .
(124) M. D. Lumb and D. A. Weyl, J. Mol. S p e c t r o s c . , 3, 365 (1967) .
(125) F. Hirayama and S. L ipsky , J . Chem. Phys . , z, 1939 (1969) .
(126) I . B. Berlman, "Handbook of F luo rescence S p e c t r a of Aro- matic Molecules" , Academic P r e s s , New York, 1971.
(127) R . B. Cundal l , S,. McD. O g i l v i e , and D. A. Robinson, J. Photochem., 1, 417 (1973/1974) .
(128) C. A. P a r k e r , Anal . Chem., 34, 502 (1962) .
(129) M . L u r i a and G. S t e i n , J. Phys. Chem., 3, 165 (1972) .
(130) K. Kaplan, L . A . Wendling and K. E. Wilzbach, J . Am. Chem. Soc . , 93, 3819 (1971) .
(131) J. B. B i r k s , C. L. Braga and M. D. Lumb, P roc . Roy. SOC., A 283, 83 (1965) .
(132) T. V . Ivanova , G. A . Mokeeva and B. Y a . Sveshnikov, Opt. S p e c t r o s c . , 2, 325 (1962); T. V . Ivanova, P. I . Kudryashev, and B. Y a . Sveshnkov, Sov. Phys. Dokl . , 5, 407 (1961).

210 R. B. Cundall, et al.
(144)
(146)
(147)
M. D. Lumb, Private communication, 1971.
R. B. Cundall and D . A . Robinson, J. Chem. Faraday 11, - 68, 1133 (1972).
P. K. Ludwig and C. D. Amata, J. Chem. Phys., 49, 326 (1968).
T. A . Gregory and W. P. Helman, J. Chem. Phys., 56, 377 (19 72) . J. R. Greenleaf and T. A. King, Proc. Int. Conf. Luminescence, Budapest, 212 (1968).
P. M. Froehlich and H. A. Morrison, J. Phys. Chem., 76, 3566 (1972).
W. P. Helman, J. Chem. Phys., 51, 354 (1969). See, for example, ref. 86, 119.
S . Kato, S . Lipsky, and C. L. Braun, J. Chem. Physics, - 37, 190 (1962).
C. L. Braun, S . Kato and S. Lipsky, J. Chem. Phys., 2, 1645 (1963).
J. B. Birks, J. C . Conte and G. Walker, J. Phys. B, 1, 934 (1968).
M.D. Lumb, C. L. Braga and L. C. Pereira, Trans. Faraday SOC., 65, 1992 (1969).
C. Fuchs, F. Heissel and R. Voltz, J. Phys. Chem., 76, 3867 (1972).
K. Sandros, Acta Chem. Scand., 25, 3651 (1971).
R. B. Cundall and D. A. Robinson, J. Chem. S O C . Faraday I1 68, 1145 (1972).
C. A. Parker and C. C. Hatchard, Analyst, 87, 664 (1962).
E. H. Gilmore, G. E. Gibson and D. S . McClure, J. Chem. Phys., 20, 829 (1952) ; ibid, 23, 399 (1955).

E x c i t a t i o n of Benzene 211
(150) E. C . L i m , J. Chem. Phys., 36, 3497 (1962).
(151) M. D. Lumb and A. 0. A 1 Chalab i , Chem. Phys. L e t t . , 2, 365 (1971).
(152) M. D. Lumb and L. C . P e r e i r a , "Organic S c i n t i l l a t o r s and Liquid S c i n t i l l a t i o n Counting" (D. L. Horrocks and C . Peng, Eds.) , Academic P r e s s , New York, 1971, p . 239.
(153) E. C . L i m , P r i v a t e Communication, c i t e d i n D. M. Haaland and G. C . Nieman, J. Chem. Phys. , 2, 1013 (1973).
R. L i and E. C . Lim, J. Chem. Phys. , 57, 605 (1972). (154)
(155) S. F. F i sche r and E. C . Lim, Chem. Phys. L e t t . , 14, 40 (19 72) .
(156) R. B. Cundall , L . C. P e r e i r a and D. A. Robinson, J. Chem. SOC. Faraday 11, 69, 701 (1973).
R. B. Cundall and L. C . P e r e i r a , Chem. Phys. L e t t . , 16, 371 (1972).
(157)
(158) R. B. Cundall and S . M c . O g i l v i e , To b e pub l i shed , 1976.
(159) R. B . Cundall and A. S. Davies, Trans. Faraday SOC., 62, 1151 (1966).
W. A. Noyes, Jr . , and D. A. Harter, J. Chem. Phys., 46, 674 (1967).
(160)
(161) S . Y. Ho and W. A. Noyes, Jr . , J. Am. Chem. SOC., 89, 5091 (1967).
(162) H. 0. Denschlag and E. K . C . Lee, J. Am. Chem. SOC., 90, 3628 (1968).
(163) K. Nakamura, J. Am. Chem. SOC., 93, 3138 (1971).
(164) G. C. Nieman and G. W. Robinson, J. Chem. Phys. , 2, 1298 (1963).
(165) S. D. Colson and G. W. Robinson, J. Chem. Phys. , 48, 2550 (1968).
(166) F. M. Ga r fo r th , G. K. Ingold and H. G . Poole , J. Chem. SOC., 491 (1948).

212 R. B. Cundall , e t a l .
(175)
(179)
E. R .Berns t e in , S . D. Colson, D. S . T i n t i and G. w. Robinson, J. Chem. Phys. , 48 (1968); G. Nonnemacher and R. Mecke, Spectrochim. Acta, 17, 1049 (1961).
F. Langenbucher and R. Mecke, Spectrochim. Acta, 21, 1287 (1965).
A. R. Gee and G. W. Robinson, J. Chem. Phys., 46, 4847 (1967) - J. W. Raba la i s , H. J. Maria and S. P. McGlynn, J. Chem. Phys. , 51, 2259 (1969).
P. M. Johnson and L. Z i e g l e r , J. Chem. Phys., 56, 2169 (1972).
G. S . Hammond and A. A. Lamola, Unpublished, c i t e d i n "Photochemistry", J. G. Ca lve r t and J. N . P i t t s , Jr . , Wiley, New York, 1966.
R. B. Cundall and W. T i p p e t t , Trans. Faraday Soc. , 66, 350 (1970).
R. R. Hentz and H. G. A l t m i l l e r , J . Phys. Chem., 74, 2646 (1970).
R. B. Cundall and D. A. Robinson, J. Chem. SOC. Faraday 11, 68, 1691 (1972).
R. R. Hentz and L. M. Perkey, J. Phys. Chem., 74, 3047 (1970).
R . R. Hentz and R. M. T h i b a u l t , J. Phys. Chem., 77, 1105 (1973).
R. B. Cundall , L . C . P e r e i r a , and D. A. Robinson, Chem. Phys. L e t t . , 13. 253 (1972).
R. V. Bensasson, J. T. Richards, and J. K.. Thomas, Chem. Phys. L e t t . , 2, 13 (1971).
V . M. Be ren fe l ' d and V . A. Krongauz, I z n . Akad. Nauk. SSSR Sen. F i z . , 32 ( 9 ) , 1575 (1968).
K. Sandros, Acta Chem. Scand., 23, 2815 (1969).
D. A. Robinson, Ph.D. Thes i s , Un ive r s i ty of Nottingham (19 71) .

Excitation of Benzene 213
(183) See also J. B. Birks, ref. 8, p. 172.
(184) See, for example: I. Jano and Y. Mori, Chem. Phys. Lett., - 2, 185 (1968).
D. Bryce-Smith, Pure Appl. Chem., 16, 47 (1968). (185)
(186) I. Jano, Chem. Phys. Lett., 2, 205 (1968).
(187) See, for example: (a) K. E. Wilzbach, L. Kaplan and A . L. Harkness, J. Am. Chem. SOC., 90, 1116 (1968); L. Kaplan and K. E. Wilzbach, J. Am. Chem. SOC., 90, 3291 (1968).
(188) R. B. Cundall, D. A . Robinson and A. J. R. Voss, J. Photo- chem., 2, 239 (1973/1974).
(189) A. J. R. Voss, Ph.D. Thesis, Nottingham (1969).
(190) J. B. Birks, Organic Molecular Photophysics, Vol. I, Wiley, New York (1974).
(191) A. A. Lamola, G . S . Hannnond and F. B. Mallory, Photochem. Photobiol., 6, 259 (1965).
(192) D. Phillips, J. Lemaire, C. Burton and W. A. Noyes, Jr., Adv. Photochem., 5, 329 (1968).
(193) G. Stein, Private communication.
(194) J . Irina and K. C. Kurien, Chem. Comun., 1973, 739.
(195) H. Lutz and G. Stein, J. Phys. Chem., 78, 1909 (1974) and references therein.
(196) See, f o r example: R. B. Cundall and S . McD. Ogilvie, "Organic Molecular Photophysics:, Vol. 11, Wiley, New York (1974).
(197) Y. Ilan and G. Stein, Chem. Phys. Lett., 31, 441 (1975).
(198) S. J. Formosinho, J. Chem. SOC. Faraday 11, 2, 605 (1974); S . J. Formosinho and J. Dias da Silva, Mol. Photochem., 6 (41, 409 (1974).
(199) R. E. Kellogg and R. G. Bennett, J. Chem. Phys., 41, 3942 (1964).

214 R. B. Cundall, et al.
(200)
(208)
(210)
W. Siebrand and D. F. Williams, J. Chem. Phys., 46, 403 (1967).
W. Siebrand, "The Triplet State" (A. B. Zahlan, Ed.), Cambridge University Press, 1967.
M. J. Shinitsky, J. Chem. Phys., 56, 5979 (1972). A. H . Kalantar, J. Chem. Phys., 48, 4992 (1968).
M. Almgren, Photochem. Photobiol., 8, 231 (1968).
E. D. Cehelnik, K. D. Mielenz and R. A. Velapoidi, J. Res. NBS, 79A, 1 (1975).
T. E. Martin and A . H . Kalantar, J. Chem. Phys., 50, 1486 (1969).
J. D. Spangler and N.G. Kilmer, J. Chem. Phys., 48, 698 (1968).
T. E. Martin and A. H. Kalantar, J. Chem. Phys., 3, 244 (1968).
T. E. Martin and A. H. Kalantar, J. Phys. Chem., 74, 2030 (1970).
A. Bernas and T. B. Truong, Photochem. Photobiol., 2, 311 (1972).
J. H. Leubner, J . Phys. Chem., 73, 2088 (1969).
T. E. Martin and A . H . Kalantar, J. Phys. Chem., 72, 2265 (1968).
J. H . Leubner, J. Phys. Chem., 74, 77 (1970); J. H. Leubner and J. E. Hodgkins, J. Phys. Chem., 73, 2545 (1969).
(214) F. Hirayama, J. Chem. Phys., 42, 3726 (1965). (215) G. F. Hatch, M. D. Erlitz and G. C. Nieman, J. Chem.
Phys., 3, 3723 (1968); E. C. Lim, Ed., "Molecular Luminescence", W. A . Benjamin, New York, 1969.
(216) A. C . Ling and J. E. Willard, J. Phys. Chem., 72, 1918 (1968).

E x c i t a t i o n of Benzene 215
(217) N . G. K i l m e r and J. D. Spangler , J. Chem. Phys. , 54, 604 (19 71) .
(218) T. E. Mar t in and A. H. Kalantar, J. Chem. Phys. , 48, 4996 (1968).
(219) R. B. Cundall and L. C. P e r e i r a , Chem. Phys. L e t t . , 9, 561 (1974).
(220) P. M. Johnson and M. C. S tudez , Chem. Phys. L e t t . , 18, 371 (1973).
(221) Yei-Ping Hsu and P. M. Johnson, J. Chem. Phys. , 59, 136 (1973).
(222) D. M. Haaland and G. C . Nieman, J. Chem. Phys., 2, 1013 (1973).
(223) G. W. Robinson, J. Chem. Phys. , 47, 1967 (1967), and r e f - e r ences t h e r e i n .
(224) S . McD. Og i lv i e , Ph.D. Thes i s , Un ive r s i ty of Nottingham, (1974).
(225) H. S t e r n l i c h t , G . C. Nieman and G. W. Robinson, J. Chem. Phys., 2, 1326 (1963).
(226) H. Ishikawa and W. A . Noyes J r . , 3. Chem. Phys., 37, 583 (1962).
(227) E . K. C. Lee, H. 0. Denschlag, and G. A . Hanniger, Jr., J. Chem. Phys., 48, 4547 (1968).
C. S . Parmenter and B. L. Ring, J. Chem. Phys. , 46, 1198 (1967).
(228)
(229) C . S . Burton and H. E. Hunziker, Chem. Phys. L e t t . , 6, 352 (1970).
(230) M. Nishikawa and M . C . Sauer, J. Chem. Phys., 51, 1 (1969).
(231) T. F. Hunter and M. G. Stock , Chem. Phys. L e t t . , 22, 368 (1973); T . F. Hunter and M. G. S tock , J. Chem. SOC. Faraday I1 (1974).

216 R. B. Cundall, et al.
(242)
J. Langelaar, G. Jansen, R. P. H. Rettschnick, and G . J. Hoytink, Chem. Phys. Lett., 12, 86 (1971).
R. B. Cundall and P. A. Griffiths, Disc. Faraday SOC., 36, 111 (1963); Trans. Faraday SOC., 61, 1968 (1965).
R. G. Kaufman, Disc. Faraday SOC., 36, 262 (1963).
J. T. Dubois and F. Wilkinson, J. Chem. Phys., 38, 2541 (1963); ibid. , 2, 377 (1963).
J. W. van Loben Sels and J. T. Dubois, J. Chem. Phys., - 45, 1522 (1966).
S. Lipsky, J. Chem. Phys., 2, 2786 (1963).
I. A. Lisovska, M. D. Sakharova and I. C. Kaplan, Optics Spectrosc., 23, 348 (1967).
V. A. Krongauz, Dokl. Akad. Nauk SSSR, 155, 658 (1964).
R. B. Cundall and A . J. R. Voss, Chem. Commun., 1969, 116 (1969); R. B. Cundall. D. A. Robinson and A. J. R. Voss, J. Photochem., 2, 234 (1973/1974).
R. B. Cundall, G . B. Evans and J. P. Keene, J. Phys. Chem. - 72, 3871 (1968).
K. C. Smyth, J. A. Schiavone and R. S. Freund, J. Chem. Phys., 61, 1789 (1974).
Y. Hatano, S. Takao and T. Veno, Chem. Phys. Lett., 2, 429 (1975).
J. K. Thomas and J. Mani, J. Chem. Phys., 2, 1834 (1969).
V. Laor and A. Weinreb, J. Chem. Phys., 43, 1565 (1965).
C. W. Lawson, F. Hirayama and S. Lipsky, in: "Molecular Luminescence", (E. C. Limy Ed.), W. A. Benjamin, New York, 1969, p . 837.
F. Hirayama, T. A. Gregory and S . Lipsky, J. Chem. Phys., - 58, 4886 (1973).

E x c i t a t i o n of Benzene 217
J. K. F o o t e , M. H. Mal lon a n d J. N . P i t t s , J r . , J. Am. Chem. SOC., z, 3698 (1966) .
K. Sh indo and S. L i p s k y , J. Chem. P h y s . , 45, 2292 (1966) .
L . F. S c o t t and M. J o n e s , Jr . , Chem. Rev., 72, 181 (1972) .
P. L e c h t k e n , R. Bres law, A . H. Schmidt , and N . J. T u r r o , J. Am. Chem. SOC., 95, 3025 (1973) .
C . A . Renner, T. J. K a t z , J. P o u l i q u e n , N. J. T u r r o and W. H. Waddel l , J . Am. Chem. SOC. , 97, 2568 ( 1 9 7 5 ) .
G. Camoggi, F. Gozzo and C . C e v i d a l l i , Chem. Commun., 1966, 313.
G. P. Semeluk and R. S. S t e v e n s , Chem. Commun., 1970, 1720.
K.E. Wi lzbach , L. Kaplan and A. L . Harkness , J. Am. Chem. SOC., 90, 1116 (1968) .
L . Kaplan and K, E. Wi lzbach , J. Am. Chem. SOC., 90, 3291 (1968) .
C . S . P a r m e n t e r and M . D. Schuh, Chem. Phys . L e t t . , 13, 120 (1972) .
J. I r i n a and K. C . K u r i e n , Chem. I n d . , 1972, 763.
K. Wei, J. C . Mari and J. N . P i t t s , Jr . , J. Am. Chem. SOC., 89, 4225 (1967) .
C . F a r e n h o r s t , T e t r a h e d r o n L e t t . , 4835 ( 1 9 6 8 ) .
M. L u r i a and G. S t e i n , Chem. Commun., 1970, 1650.
E . M i g i r d i c y a n , J . Chem. P h y s . , 63, 520 (1966) ; i b i d . , - 63, 535 (1966) .
S . Leach, J . Chem. Phys . , 55, 719 (1958) .
E . J. Anderson, H. I. J. C h i l t o n and G. P o r t e r , P r o c . Chem. SOC. , 1960, 352.
K. E. Wi lzbach and L. Kaplan , J. Am. Chem. SOC. , 88, 2066 (1966) .

218 R. B. Cundall, e t al .
(269)
(273)
(274)
(276)
J. Cornel isse , V. Y. Merritt and R. Sr in ivasan , J. Amer. Chem. SOC., 95, 6197 (1973).
W. T i p p e t t , Ph.D. Thesis , Univers i ty of Nottingham (1968).
D. Bryce-Smith, A. G i l b e r t , B. Orger and H. T y r e l l , Chem. Commun., 1974, 334.
W. A. Noyes, Jr., and K. A 1 - h i , Chem. Rev., 74, 29 (1974).
A. Dammers-de-Klerk, Mol. Phys., L, 141 (1958).
T. V. Ivanova, G . A. Mokeeva and B. Y a . Sveshukov, Opt. Spectros . , 12, 325 (1962).
E. Konijnenberg, Ph.D. Thesis , Univers i ty of Amsterdam (1963).
J. B. Birks , A. A. Kazzaz, and T . A. King, Proc. Roy. S O C . , A291, 556 (1966).
J. B. Birks , "Progress i n Reaction Kinet ics" , (G. P o r t e r , Ed.), Vol. 5 , Pergamon, New York, 1970, p. 263.
B. L. Barnes and J. B. Bi rks , Proc. Roy. SOC., E, 570 (1966) . J. B. Birks and L. G. Christophorou, Proc. Roy SOC., A277, 571 (1964).
J. Amuzumi and H. Azumi, Bul l . Chem. SOC. Japan, 2, 2317 (1966).
A. K. Chandra and E. C . L im, J. Chem. Phys., 48, 2589 (1968); i b i d , 9, 5066 (1968).
A. K. Chandra, Chem. Phys. L e t t . , 2, 229 (1970).
D. B. Chestnut, C. J. F r i t c h i e and H. E. Simmons, J. Chem. Phys., 42, 1127 (1965).
F. Hirayama and S. Lipsky, in : "Molecular Luminescence", (E. C. Lim, Ed.), W. A. Benjamin, New York, 1969.
J. R. Greenleaf, M. D. Lumb and J. B. Birks , J. Phys. B . , - 1, 1157 (1968).

E x c i t a t i o n of Benzene 219
(283) Y. Yguerabide, J. Chem. Phys., 49, 1018 (1968).
(284) M. T . Vala, I. H. H i l l i e r , S. A. Rice and J. J o r t n e r , J. Chem. Phys., 49, 326 (1966).
(285) M. D. Lumb, P r i v a t e communication, 1971.
(286) L. G. Chris tophorou , J. G. Carter and M. E. M. Ah-Ze id , J. Chem. Phys. , 49, 3775 (1968).
(287) I. H. Hillier, L. Glass and S . A. R i c e , J. Chem. Phys. , - 45, 3015 (1966).
(288) D. H. P h i l l i p s and J. C. Schug, J. Chem. Phys., 50, 3297 (1969).


















