
EVALUATION OF THE REGULATORY REVIEW … · time for all pharmaceutical company products, ... List...
259
I EVALUATION OF THE REGULATORY REVIEW PROCESS OF THE GCC CENTRALISED PROCEDURE: DEVELOPMENT OF A MODEL FOR IMPROVING THE APPROVAL PROCESS A thesis submitted in accordance with the condition governing candidates for the degree of DOCTOR OF PHILOSOPHY in CARDIFF UNIVERSITY Presented by MOHAMMED HAMDAN AL-RUBAIE 2013
Transcript of EVALUATION OF THE REGULATORY REVIEW … · time for all pharmaceutical company products, ... List...

I
EVALUATION OF THE REGULATORY REVIEW PROCESS OF
THE GCC CENTRALISED PROCEDURE: DEVELOPMENT OF
A MODEL FOR IMPROVING THE APPROVAL PROCESS
A thesis submitted in accordance with the condition
governing candidates for the degree of
DOCTOR OF PHILOSOPHY
in
CARDIFF UNIVERSITY
Presented by
MOHAMMED HAMDAN AL-RUBAIE
2013

II
DECLARATION
This work has not been submitted in substance for any other degree or award at this or any
other university or place of learning, nor is being submitted concurrently in candidature for any
degree or other award.
Signed (candidate) Date 27-11-2013
STATEMENT 1
This thesis is being submitted in partial fulfillment of the requirements for the degree of PhD
(insert MCh, MD, MPhil, PhD etc, as appropriate)
Signed (candidate) Date 27-11-2013
STATEMENT 2
This thesis is the result of my own independent work/investigation, except where otherwise
stated.
Other sources are acknowledged by explicit references. The views expressed are my own.
Signed (candidate) Date 27-11-2013
STATEMENT 3
I hereby give consent for my thesis, if accepted, to be available for photocopying and for inter-
library loan, and for the title and summary to be made available to outside organisations.
Signed (candidate) Date 27-11-2013
STATEMENT 4: PREVIOUSLY APPROVED BAR ON ACCESS
I hereby give consent for my thesis, if accepted, to be available for photocopying and for inter-
library loans after expiry of a bar on access previously approved by the Academic
Standards & Quality Committee.
Signed (candidate) Date 27-11-2013

III

IV
ACKNOWLEDGEMENTS
I am greatly indebted to my supervisors, Prof Sam Salek and Prof Stuart Walker for
their outstanding supervision and expert advice and guidance throughout, and for their
continuous encouragement and ultimate support throughout the preparation of this
thesis.
Thanks also go to all the staff at the School of Pharmacy and pharmaceutical sciences,
especially Helen Harron and Justine Jenkins who assisted me throughout the study.
I would also like to express my gratitude and appreciation to my colleagues in the
Health authorities in the Gulf States and in the Executive Board of the Health Ministers
Council for GCC States, without whose input in terms of data provision, it would not
have been possible to conduct this research.
I dedicate my thesis to my family, especially my parents, my loving wife, my kids,
,brothers and sisters, I would like to thank them for their encouragement, support and
love. Their belief in me gave me the strength to pursue my research successfully.

V
ABSTRACT
The aim of this study was to evaluate seven GCC regulatory authorities and pharmaceutical
companies active in the region in order to identify the strengths and weakness of the current
GCC centralised procedure. The GCC regulatory authorities and the pharmaceutical companies
who had registered their products through the GCC centralized registration procedure and the
national registration systems were recruited into the study and asked to complete the
questionnaires specifically designed for this study.
The regulatory review process in Oman was evaluated to identify areas for improvement in the
system. Information on the total application numbers and approval dates were obtained directly
from the Oman Ministry of Health archives. Another study was conducted to evaluate the
regulatory review process and approval times of the remaining six GCC countries (Bahrain,
Kuwait, Qatar, Saudi Arabia, UAE and Yemen) and the GCC central registration, with respect to
review time for new and existing substances, to identify the strengths and weaknesses of the
process and to propose strategies that could help the policy maker in the GCC to enhance the
review process.
The results of the Omani regulatory system showed no significant increase (p>0.05) in the total
number of registered pharmaceutical products from 2006 to 2010. The approval time in Oman
showed that there was a significant increase in approval times for pharmaceutical products from
2006 to 2010 (p<0.001). The findings show that although there was an increase in the approval
time for all pharmaceutical company products, the median approval time for the five year period
was 117 days. This was within the time limit (4 months) fixed by the health authority for the
overall registration time. The comparative study of the GCC States showed a downward trend in
the median approval time for most of the GCC States, during 2008 to 2010. However, the
approval time for all approved products in the GCC States during this period varied from 60
days in Qatar and Oman (2009 and 2010) to 609 days in Saudi Arabia (2008). The main
reasons for the decrease in approval time in the Gulf States were due to the positive effect of
the Gulf Central Registration, the rise in the number of reviewers in some GCC drug authorities,
and the parallel procedure used in the regulatory approval review process. The study of the
regulatory review process of the GCC central registration showed that a total of 413 products
(96 NASs and 317 EASs) were approved during the period 2006-2010 with an overall significant
increase in the EASs (p<0.001). The approval times increased from 107 calendar days in 2006
to 265 in 2010 (p<0.001). The lowest approval time was for EASs submitted by the Gulf
companies (134 days) and the longest for NASs submitted by international companies (346
days) (p<0.001).

VI
Both the regulatory authorities and the pharmaceutical companies agreed that the centralised
procedure is an effective system for authorising medicinal products in all seven GCC countries
in one procedure and is the way forward in the future but there is room for improvement in the
procedure and the follow ups. They also agreed that clear guidelines, transparency of
procedures, effective interactions between authorities and companies, increase in the number
of the committee meetings per year, use of electronic on-line submissions will improve approval
time for registration of new medicines, enhance the quality of review practice and encourage
the pharmaceutical companies to use the GCC central registration system. This research has
enabled development of a new model of the GCC central registration procedure to be proposed
for the GCC Health Authorities which could improve patient access to medicines in the GCC
states.

VII
TABLE OF CONTENTS
Acknowledgements …………………………………………………………………… IV
Abstract ………………………………………………………………………………. V
Table of contents …………………………………………………………………….. VII
Glossary of abbreviations …………………………………………………………… IX
Glossary of terms ……………………………………………………………………. XI
List of figures ………………………………………………………………………… XVI
List of tables ………………………………………………………………………….. XIX
Chapter one……………………………………………………………………………. 1
Background………………………………………………………………………….. 2
Regulatory Reviews Process, Approval Times and Patients' Access to Medicines 2
Quality of the Review Process …………………………………………………… 5
Benefit-risk assessment and decision-making …………………………………… 7
Role of harmonisation ……………………………………………………………. 8
The Gulf Cooperation Council (GCC) States ……………………………………. 9
GCC Drug Regulatory Authorities ………………………………………………. 10
Evolution of GCC-DR …………………………………………………………… 11
The quality of the review in the GCC …………………………………………… 15
Global drug development and its influence on the regulatory environment in the Gulf
Region ……………………………………………………………………………. 16
Aims and objectives of the study ………………………………………………….. 18
Chapter two ……………………………………………………………………….... 19
Study rationale & methodological framework
Rational of the study …………………………………………………………….... 20 Methodology framework …………………………………………………………. 21
Development of the Study Plan ………………………………………………..... 31
Development of the Study Instruments ………………………………………….... 33
Data processing and analysis …………………………………………………... … 35
Summary ………………………………………………………………………….. 40
Chapter three ……………………………………………………………………… 41
Evaluation of the Pharmaceutical Regulatory Review Process in Oman
Introduction ……………………………………………………………………… 42 Objectives ……………………………………………………………………….. 43
Methods …………………………………………………………………………. 43 Results …………………………………………………………………………... 44 Discussion ………………………………………………………………………. 61 Conclusion ……………………………………………………………………… 65 Summary ………………………………………………………………………… 66
Chapter four ………………………………………………………………………. 67
Evaluation of Regulatory Review Times in the Gulf States
Introduction ……………………………………………………………………… 68 Objectives ……………………………………………………………………….. 69
Methods …………………………………………………………………………. 69 Results …………………………………………………………………………… 70

VIII
Discussion ………………………………………………………………………….. 90
Summary …………………………………………………………………………… 95 Chapter five …………………………………………………………………………... 96
An Evaluation of the GCC Centralised Regulatory Review Process
Introduction ………………………………………………………………………… 97
Objectives ………………………………………………………………………….. 98 Methods ……………………………………………………………………………. 99
Results ……………………………………………………………………………... 101
Discussion …………………………………………………………………………. 112
Summary …………………………………………………………………………... 115
Chapter six …………………………………………………………………………... 117
An Evaluation of the Gulf States Views and Experiences with GCC Centralised Procedure
Introduction ………………………………………………………………………… 118
Objectives ………………………………………………………………………….. 119
Methods ……………………………………………………………………………. 119
Results ……………………………………………………………………………... 119
Discussion …………………………………………………………………………. 147
Summary …………………………………………………………………………... 150
Chapter seven ………………………………………………………………………… 152
An Evaluation of the Pharmaceutical Companies Experiences with the GCC Centralised
Procedure
Introduction ………………………………………………………………………... 153
Objectives ………………………………………………………………………….. 154
Methods ……………………………………………………………………………. 154
Results ……………………………………………………………………………... 155
Discussion ………………………………………………………………………………... 183
Summary …………………………………………………………………………………. 187
Chapter eight ………………………………………………………………………... 188
The Gulf Centralised Procedure- A Comparison between the Experiences of Regulatory
Authorities and Pharmaceutical Companies, a proposed new model
Introduction ……………………………………………………………………….. 189
Objectives ………………………………………………………………………… 190
Methods …………………………………………………………………………… 190
Results &Discussion …………………………………………………………………… 191
Summary ………………………………………………………………………………… 209 Chapter nine ………………………………………………………………………... 210
General Discussion
Limitation of the study …………………………………………………………… 219 Recommendations ……………………………………………………………..... 220 Future work ……………………………………………………………………… 223 Conclusion ……………………………………………………………………..... 224
References …………………………………………………………………………. 225
Publications ……………………………………………………………………….. 237
Appendix……………………………………………………………………………. 239

IX
LIST OF ABBREVIATIONS
AMRH: African Regulatory Harmonization Initiative
APEC: Asia Pacific Economic Cooperation
ADEC: Australian Drug Evaluation Committee.
ATC: Anatomical Therapeutic and Chemical code.
CACP: Companies Assessment of Central Procedure
CATI: Computer assisted telephone interviewing
CBER: Centre for Biologics Evaluation and Research.
CIF: Cost, Insurance and Freight
CIRS: Centre for Innovation in Regulatory Science
CDER: Centre for Drug Evaluation and Research.
CLADF: Central Laboratory for Analysis of Drugs and Food.
CTD: Centralised Technical Document
CHMP: Committee for Medicinal Products for Human Use.
CMR: Centre for Medicines Research
CPP: Certificate of Pharmaceutical Product.
DGPA&DC: Directorate General of Pharmaceutical Affairs and Drug Control
EMA: European Medicines Agency.
EU: European Union.
FDA: Food and Drug Administration.
GACP: Gulf Assessment of Centralised Procedure
GCC: Gulf Cooperation Council
GCC-CP: Gulf Cooperation Council Centralised Procedure
GCC-DR: Gulf Cooperation Council Drug Registration committee.
GCP: Good Clinical Practice.
GRP: Good Review Practice.
HAS: Health Sciences Authority of Singapore.
ICH: International Conference on Harmonisation.
IND: Investigational New Drug.
IT: Information Technology.
KSA: Kingdom of Saudi Arabia.
MA: Marketing Authorisation.
MCA: Medicines Control Agency.

X
MEB: Medicines Evaluation Board.
MERC: Middle East Regulatory Conference.
MERCOSUR: Mercado Comundel Sur
MHLW: Ministry of Health, Labour and Welfare.
MHW: Ministry of Health and Welfare.
MPA: Medical Products Agency.
NAS: New Active Substance.
NCE: New Chemical Entity.
NDA: New Drug Application.
NME: New Molecular Entity.
NGT:Nominal Group Technique
OCD: Office Centre Director.
PD: Pharmacodynamics.
PDUFA: Prescription Drug User Fee Act.
PANDRH:Pan American Network for Drug Regulatory Harmonisation
PK: Pharmacokinetics.
PMS: Post Marketing Surveillance.
PPRS: Pharmaceutical Pricing Regulation Scheme.
QA: Quality Assurance.
QCL: Quality Control Laboratory
R&D: Research and Development.
RC: Registration Committee.
RMS: Reference Member State.
SADC: Southern African Development Community
SOP: Standard Operating Procedure.
TCR&P: Technical Committee for Registration of Pharmaceutical Manufacturers & Their
Products & Pricing
TGA: Therapeutics Goods Administration.
TRIPS: Trade-Related Aspects of Intellectual Property Rights.
UAE: United Arab Emirates.
WHO: World Health Organisation.
WTO: World Trade Organisation.

XI
GLOSSARY OF TERMS
Adverse event: Any untoward medical occurrence in a patient or clinical investigation of a subject
administered a pharmaceutical product and which does not necessarily have a causal relationship
with this treatment.
Africa, the Southern African Development Community (SADC): Angola, Botswana, Democratic
Republic of Congo, Lesotho, Malawi, Mauritius, Mozambique, Namibia, Seychelles, South Africa,
Swaziland, Tanzania, Zambia and Zimbabwe.
Approval: The active substance has been approved for licence by a regulatory authority in one or
more markets (when the authority grants a licence and subject to pricing/ reimbursement issues the
product can be legally marketed).
Arab Central Registration (ACR): Jordan, UAE, Bahrain, Tunisia, Algeria, Djibouti, KSA, Sudan,
Syria, Somalia, Iraq, Oman, Palestine, Mauritania, Yemen, Qatar, Comoros, Kuwait, Lebanon,
Libya, Egypt and Morocco.
Asia-Pacific Economic Cooperation (APEC): Australia, Brunei Darussalam, Canada, Chile, China,
Hong Kong, Indonesia, Japan, South Korea, Malaysia, Mexico, New Zealand, Papua New Guinea,
Peru, Philippines, Russia, Singapore, Taiwan, Thailand, USA and Vietnam.
Association of Southeast Asian Nations (ASEAN): Brunei, India, Thailand, Philippines, Indonesia,
Malaysia, Singapore, Myanmar and Cambodia.
Benefit/risk ratio: The benefit to risk ratio relates the potential or actual benefit that a patient
derives from a treatment to the risks incurred by the patient through using the treatment. The benefit
to risk ratio can be described in qualitative terms (high, low, equal, very low etc).
Binding: Advice provided by a regulatory authority prior to submission that is acknowledged and
considered a regulatory agreement.
Biological: A substance isolated from animal tissues e.g. vaccines, hormones, antigens.
Biotech product: A naturally occurring or modified polypeptide, protein, DNA or RNA product
(produced by recombinant DNA or hybridoma technology and expressed in cell lines, transgenic
animals or transgenic plants) for therapeutic, prophylactic or in vivo diagnostic use in humans. The
only types of vaccines included in the biotech category are recombinant vaccines.

XII
Centralised procedure: The Centralised procedure is used when marketing authorization
covering the entire EU region is applied for, for example, for new biotechnological medicinal
products and new innovative medicinal products. The applications for marketing authorization
are then submitted to the European Agency for the Evaluation of Medicinal Products (EMEA).
Clinical trial: Any investigation in human subjects intended to discover or verify the clinical,
pharmacological and/or other pharmacodynamic effects of an investigational product, and/or to
identify any adverse reactions to an investigational product, and/or to study the absorption,
distribution, metabolism and excretion of an investigational product, with the objective of
ascertaining its safety and/or efficacy.
Collaborative or sponsored research: The active substance is discovered as a result of research
carried out in collaboration with, or sponsored by, another company, a university, government
agency or an individual.
Co-operation Council of the Arab Gulf States (GCC): Bahrain, Kuwait, Oman, Qatar, United
Arab Emirates and the Kingdom of Saudi Arabia.
Drug product: A finished formulation, for example, a tablet or capsule that contains the active
substance, generally in association with one or more other ingredients.
Drug Regulatory Authority (DRA): The agency that develops and implements most of the
legislation and regulation of medicines.
European Union Member States (EU): Austria, Belgium, Cyprus, Czech Republic, Denmark,
Estonia, Finland, France, Germany, Greece, Hungary, Ireland, Italy, Latvia, Lithuania,
Luxembourg, Malta, Poland, Portugal, Slovakia, Slovenia, Spain, Sweden The Netherlands and
the United Kingdom.
Extrapolation of foreign clinical data: The generalisation and application of the safety,
efficacy and dose response data generated in a population of a foreign region to the population of
the new region.
Foreign region: Any region outside the new region where product authorisation is being sought.
ICH Regions: European Union, Japan and USA.
ICH: International Conference on Harmonisation.

XIII
Indication: The specific indication for which the active substance for the project is designed.
This may represent the cure, alleviation, treatment, prevention or diagnosis of disease in humans.
Investigational New Drug (IND): An application that a drug sponsor must submit to FDA
before beginning tests of a new drug in humans. The IND contains the plan for the study and is
supposed to give a complete picture of the drug, including structural formula, animal test results,
and manufacturing information.
Launched: The active substance is made available for sale as an ethical prescription only
medicine in the first market in any country.
Line extension: NASs which are already marketed but are being further developed for new
indications, formulations, dosages, routes of administration, or novel drug delivery systems.
Local study: A study conducted in a single country with the primary aim of providing local
experience with a compound.
Marketing Authorisation (MA): Legal approval granted to a company by a national (or
regional) authority to market a medicinal product in that particular country (or region).
Marketing Authorisation Application (MAA): An application by a company for a marketing
authorisation to be submitted to each country (or region) in which marketing approval is sought.
Mutual Recognition procedure: The Mutual Recognition (MR) procedure utilizes the
marketing authorization granted for an active substance by another EU Member State, Norway,
or Iceland. The Member State whose assessment is recognized as a basis for marketing
authorization is called the Reference Member State (RMS).
National procedure: The national procedure is mainly used in cases where marketing
authorisation is being applied for in a single member state within the.
New Active Substance (NAS): A chemical, biological or radiopharmaceutical substance not
previously authorised as a medicinal product. The term NAS also includes: an isomer, mixture of
isomers, a complex or derivative or salt of a chemical substance previously authorised as a
medicinal product but differing in properties with regard to safety and efficacy from that
substance previously authorised; a biological substance previously authorised as a medicinal product,
but differing in molecular structure, nature of source material or manufacturing process; a
radiopharmaceutical substance that is a radionuclide or a ligand not previously authorised as a

XIV
medicinal product. Alternatively, the coupling mechanism linking the molecule and the radionuclide
that has not been previously authorised.
New Chemical Entity (NCE): An entity produced by chemical synthesis.
New Drug Application (NDA): An application requesting regulatory approval to commercially
market a new drug for human use.
New Molecular Entity (NME): A product (including new chemical entities, biological products,
vaccines and products of biotechnology) that has not been previously available for therapeutic use in
man and is destined to be made available as a „prescription-only medicine‟, to be used for the cure,
alleviation, treatment, prevention or in vivo diagnosis of diseases in man. New salts, pro drugs and
esters of existing products and certain biological compounds (e.g. antigens) are excluded.
Combination products are also excluded unless one or more of the active constituents has never been
previously marketed.
Pan-America Network for Drug Regulatory Harmonisation (PANDRA): Mexico, Costa Rica,
Trinidad, Argentina and Colombia.
Pharmacodynamics (PD): Involves the study of the effect that a drug has on the body. This will
include looking at what effect the amount of drug has, called a dose response, and will also look at
the site of action of the drug, such as the kinetics of how a drug interacts with a receptor.
Pharmacogenetics: is the study of interindividual variations in DNA sequence related to drug
response.
Pharmacogenomics: is the study of the variability of the expression of individual genes relevant to
disease susceptibility as well as drug response at cellular, tissue, individual or population level. The
term is broadly applicable to drug design, discovery, and clinical development.
Pharmacokinetics (PK): Involves the study of the manner in which the body handles a drug. It
looks at the rate at which drug concentrations change in the body, and is involved with analysing the
kinetics of absorption, distribution, metabolism and excretion of a drug.
Phase I: Initial safety trials on a new medicine, usually conducted in normal male volunteers. An
attempt is made to establish the dose range tolerated by volunteers for single and multiple doses.
Phase I trials are sometimes conducted in severely ill patients (e.g. in the field of cancer) or in less ill
patients (e.g. metabolism of a new antiepileptic medicine in stable epileptic patients whose
microsomal liver enzymes have been induced by other epileptic medicines). Pharmacokinetic trials

XV
are usually considered Phase I trials regardless of when they are conducted during the development
of medicines.
Phase II: Clinical trials (pilot and well-controlled) to evaluate efficacy (and safety) in patients with
the disease or condition to be treated, diagnosed, or prevented. Objectives may focus on dose-
response, type of patient, frequency of dosing, or numerous other characteristics of safety and
efficacy.
Phase III: Clinical trials conducted in patient populations for which the medicine is eventually
intended. These generate additional data on both safety and efficacy in relatively large numbers of
patients, in both controlled and uncontrolled trials. Clinical trials are also conducted in special
groups of patients (e.g. renal failure patients) or under special conditions dictated by the nature of the
medicine and disease. These trials often provide much of the information needed for the package
insert and labelling of the medicine and also include trials that are conducted after the regulatory
submission of a new drug application or other dossier, but prior to the medicine‟s approval and
launch.
Phase IV: Phase IV clinical trials are (other than routine surveillance) performed after drug approval
and related to the approved indication. They are not considered necessary for approval but are often
important for optimising the drug‟s use. They may be of any type but should have valid scientific
objectives. Commonly conducted studies include additional drug-drug interaction, dose-response, or
safety studies and studies designed to support an extended claim under the approved indication, e.g.,
mortality/morbidity studies.
Pivotal study: Well-controlled trial to evaluate efficacy (and safety) in patients with the disease or
condition to be treated, diagnosed, or prevented. These clinical trials usually represent the most
rigorous demonstration of a medicine‟s efficacy.
Preclinical: In vivo and in vitro studies to support administration to man.
Pre-submission: The last patient visit for the last pivotal study to be included in the regulatory
dossier is complete and the dossier is being prepared but has not yet been submitted to a regulatory
authority.
Reference Member State (RMS): The EU member state that is chosen by a company to conduct the
first review in the EU for marketing authorisation through the Mutual Recognition procedure.

XVI
LIST OF FIGURES
Figure 1.1
Regulatory approval times from date of submission to date of approval for
New Active Substances (NASs) approved 2007-2011
4
Figure 1.2 Number of NASs approved by ICH agencies (EMA, FDA, PMDA)
by approval year (2003-2012)
4
Figure 1.3 Median approval time of NASs approved by ICH agencies (2003-2012) 5
Figure 1.4 The Eight Step of the Benefit Risk Framework 8
Figure 1.5 Map of the seven Gulf Cooperation Council (GCC) States 10
Figure 2.1 Adoption of the Dolphi approach to be used in this study 31
Figure 3.1 Process map for sultanate of Oman 46
Figure 3.2 Total number of pharmaceutical products approved in Oman (2006-2010) 49
Figure 3.3 Number of EASs approved in Oman for the period (2006-2010) 50
Figure 3.4 Number of NASs approved in Oman for the period (2006-2010) 51
Figure 3.5 Approved pharmaceutical products (EASs & NASs) for GCC, international,
non-GCC Arab and Asian companies (2006-2010)
52
Figure 3.6 Median approval time for all pharmaceutical products in Oman (2006-2010) 53
Figure 3.7 Median approval time for Existing Active Substance (EASs) in Oman
(2006-2010)
53
Figure 3.8 Median approval time for NASs in Oman (2006-2010) 54
Figure 3.9 Median approval time for GCC, international, non-GCC Arab, and Asian
companies Existing Active Substances (EASs) in Oman (2006-2010).
55
Figure 3.10 Median approval time for different dosage forms for EASs and NASs in
Oman, (2006-2010)
56
Figure 3.11 Median approval times for different therapeutic classes for EASs and NASs
in Oman (2006-2010) 57
Figure 4.1 Process map for Bahrain 72
Figure 4.2 Process map for Kuwait 73
Figure 4.3 Process map for Oman 74
Figure 4.4 Process map for Qatar 75
Figure 4.5 Process map for Kingdom of Saudi Arabia 76
Figure 4.6 Process map for United Arab Emirates 77
Figure 4.7 Process map for Yemen 78
Figure 4.8 Number of approved products by the five regulatory authorities (2008-2010) 83
Figure 4.9 Median approval times for all products (2008-2010) 84

XVII
Figure 4.10 Median approval time for Gulf companies products (2008-2010) 86
Figure 4.11 Median approval time for Arab non-Gulf companies products from
(2008-2010)
87
Figure 4.12 Median approval time for International companies‟ products
(2008-2010)
88
Figure 4.13 Median approval time for Asian companies‟ products (2008-2010) 89
Figure 5.1 Process map for GCC centralised registration 100
Figure 5.2 Total number of pharmaceutical products approved in the Gulf
Centralised Procedure (2006-2010)
102
Figure 5.3 Number of EASs approved in the Gulf Centralised Procedure (2006-2010) 105
Figure 5.4 Number of NASs approved in the Gulf Centralised Procedure
(2006-2010)
106
Figure 5.5 Approved pharmaceutical products (EASs & NASs) from GCC
international, Arab non-GCC and Asian companies (2006-2010)
106
Figure 5.6 Median approval time for all pharmaceutical products in the Gulf
Centralised Procedure (2006-2010)
107
Figure 5.7 Median approval time for EASs in the Gulf Centralised Procedure
(2006-2010)
108
Figure 5.8 Median approval time for NASs in the Gulf Centralised Procedure (2006-
2010)
110
Figure 5.9 Median approval time for different dosage forms for EASs and NASs in the
Gulf Centralised Procedure (2006-2010)
110
Figure 5.10 Median approval time for different therapeutic class for EASs and NASs in
the Gulf Centralised Procedure (2006-2010)
112
Figure 6.1 An evaluation of the Gulf states views and experience with Assessment of
GCC Central registration process (Instrument assessment)
122
Figure 6.2 An evaluation of the Gulf states views and experience with Assessment of
GCC Central registration process (Questionnaire)
123
Figure 6.3 The products which could potentially be registered under the Centralised
Procedure
131
Figure 6.4 Proposed approval timeline for products in Centralised procedure 132
Figure 6.5 Responses for the GCC authorities with regard to improving the GCC
Centralised Procedure
133
Figure 6.6 Member states assessment of the Centralised Procedure 135
Figure 6.7 Level of interaction between GCC-DR Secretariat and the Committee 136
Figure 6.8 Number of GMP Inspections conducted by GCC (2008 - 2010) 137
Figure 6.9 Number of Products evaluated by Oman, Qatar and Saudi Arabia (2008 -
2010)
138
Figure 6.10 Factors that affect the sustainability of the Centralised Procedure 140
Figure 6.11 The value of professional functions to the GCC-DR system by the Gulf
States
142
Figure 6.12 A comparison between the centralised and the national systems 145
Figure 7.1 An evaluation of the Pharmaceutical Companies experience with GCC
Central registration process (Instrument assessment)
157

XVIII
Figure 7.2
An evaluation of the Pharmaceutical Companies experience with GCC
Central registration process (Questionnaire)
159
Figure 7.3 Number of companies and their preferred pharmaceutical registration
system
165
Figure 7.4 Reasons for companies preference in using the Centralised
Procedure for product registration
166
Figure 7.5 Reasons why companies did not prefer the Centralised Procedure 167
Figure 7.6 Number of products submitted and approved through the Centralised
Procedure
168
Figure 7.7 The quality of decision-making of the Centralised Procedure 169
Figure 7.8 Companies‟ suggestions on how certain aspects of the registration
procedure could be simplified
170
Figure 7.9 Companies‟ view on the Centralised Procedure guidelines 172
Figure 7.10 Companies‟ views on the quality of the GCC-DR scientific advice,
efficiency and effectiveness of the centralised pharmacovigilance
procedures and the transparency of the Centralised Procedure
172
Figure 7.11 Companies‟ view of the level of interaction on the quality of information
received, timing of the registration procedure and their relationship with the
Registration Department of the GCC-DR
175
Figure 7.12 Companies‟ views on some of the ways to improve the relationship between
the companies and the GCC-DR
176
Figure 7.13 A comparison of the companies‟ views of the national versus the centralised
system with respect to scientific opinion and consistency of evaluations
179
Figure 7.14 Companies‟ views as to why a standardised pricing is not appropriate 179
Figure 7.15 Summary of responses on the questionnaire 182
Figure 8.1 Key issues to be consider in the new proposed GCC model 196
Figure 8.2 The Current GCC-CP registration system 199
Figure 8.3
Figure8.18
The proposed model for GCC-CP 200

XIX
LIST OF TABLES
Table 1.1 The demographic structure of the gulf cooperation council (GCC) states 11
Table 1.2 The structure, responsibilities and scope of activities within each of the
seven GCC regulatory authorities
12
Table 2.1 Data source for the seven Gulf authorities 26
Table 2.2 Features of consensus methods
29
Table 4.1 Models of assessment and characteristics in the Gulf States
79
Table 4.2 Number of approved products for Gulf Cooperation Council
(GCC) States in 2008, 2009 and 2010
82
Table 5.1 Number of EASs Approved in the Gulf Centralised Procedure from various
regions, dosage forms and therapeutic classes
103
Table 5.2 Number of NASs Approved in the Gulf Centralised Procedure from various
regions, dosage forms and therapeutic classes
104
Table 6.1 List of the seven participating authorities in the Gulf Cooperation Council
(GCC) States
130
Table 6.2 Number of internal and external personnel in different areas of expertise
within the Gulf States
141
Table 6.3 Relationships between capacity and workload for each Gulf State
(2008, 2009 & 2010)
142
Table 6.4 Summary of the major advantages
146
Table 6.5 Summary of the major disadvantages
147
Table 7.1 Companies‟ opinion regarding regulatory expertise within the GCC-DR
178
Table 8.1 Registration fees in the GCC States 208

1
CHAPTER 1
General Introduction

2
BACKGROUND
Modern medicines regulations are the foundation of any country‟s drug policy. This ensures a
viable pharmaceutical industry as well as a high standard for the drug approval process.
Throughout history, governments have regulated food and drug products and in general, the
focus of this regulation has been safeguarding the quality, safety and efficacy of these
products. Drug regulation is almost universal in the industrialised nations of the world and is
being progressively developed in third world countries. It is increasingly based on the
hypothesis that regulation is needed to ensure that new medicines will be comparatively safe
and effective, labelled accurately and marketed responsibly. In short, potential benefits and
harms of modern medicines are too important to be left to the market place and the
unregulated functioning of the pharmaceutical industry (Philip et al 1986).
Today, pharmaceutical products are highly regulated in most countries and need to comply
with strict legislative requirements before being authorised for sale. Therefore, individual
pharmaceutical regulatory bodies in each country are responsible for ensuring the safety and
effectiveness of human and veterinary medicines in their jurisdiction, in compliance with
local pharmaceutical legislation.
Regulatory Reviews Process, Approval Times and Patients' Access to
Medicines
Anderson (2004) commented on the importance of timeliness for national regulatory
authorities in order to approve new medicines, which may influence directly the stakeholders
and patients during the process. A lengthy approval process results in a delay in marketing
new approved medicines for patients. Normally, the length of the review process depends on
the type of products being registered, the requirements of the approval process and the type of
review carried out.
Governments are obligated to provide their citizens with access to new medicines and
therapies when they are needed. In other words, conducting safety and efficacy reviews of
marketing authorisation applications for new medicines and biotherapy products in a timely
manner should be a major priority for safeguarding and enhancing public health, which is
part of the core mission of health authorities. However, expediting the review process for new

3
medicines and biological products to ensure it is completed within predefined deadlines
should never mean that regulatory standards can be compromised as they are essential to
proving the safety, efficacy and quality of new medicines.
During the process of review and approval of new applications, the most important hurdles to
the pharmaceutical industry is the length of the review time (CMR, 2001). Therefore, efforts
have been made by several national authorities to allow patients‟ access to medicinal
products in a timely manner by reviewing their strategies to monitor the efficiency of the
review process, as well as the performance of the regulatory authorities (Mallia-Milanes,
2010).
Hill and Johnson (2004) suggested that in order to exploit the differences in registration
requirements of different countries for manufacturers they should synchronize and streamline
the registration process. Ultimately these unifying processes reduce the time lag for
registering new products, as well as reinforcing the 'assurance of quality' of medicines.
Furthermore, it is possible to complete and monitor the whole review process transparently
within a reasonable time frame satisfying both the manufacturers and regulatory authorities.
An effective review process is perceived through the overall approval times from submission
to patients‟ access to new medicines (Rawson, 2000; Anderson, 2004).
Many countries have legislated maximum times allowed for the review of dossiers. For
example, the target time-frame for completing the review process in the European Union
centralised system is 210 days. This authorisation period has two points known as „clock-
stops‟- at 120th
day and 180th
day. A time-scale of three months and one month, respectively,
will now be mandated for applicants to respond and these periods may be doubled upon
request (EMA, 2009). The longest review time usually occurs when the benefits of the
product are not evident.
A study reported by the Centre for Innovation in Regulatory Science (CIRS, 2013) on New
Active Substances (NASs) approved in the emerging markets between 2007 and 2011
indicated that there were considerable differences among countries in the time taken to
review medicinal products with median approval times ranging from one to three years
(Figure1.1 ).

4
Figure 1.1 Regulatory approval times from date of submission to date of approval
for New Active Substances (NASs) approved 2007-2011
Data are shown for NASs that were approved between 01/01/2007 and 31/12/2011. (n1) = number of drug applications, (n2)
= number of companies providing data. Box: 25th and 75th percentiles. Whiskers 5th and 95th percentiles =median
These study data (CIRS, 2013) for the median times for patients‟ access to NASs approved
by the three major drug regulatory authorities namely, FDA, PMDA, and EMA from 2003 to
2012 showed that the median time for patients access achieved by the US FDA was the
shortest among these three authorities (Figures 1.2 and 1.3).
Figure.1.2.Number of NASs approved by ICH agencies (EMA, FDA, PMDA)
by approval year (2003-2012)

5
Figure.1.3. Median approval time of NASs approved by ICH agencies (2003-2012)
Quality of the Review Process
It is a recognised fact that the efficiency of the drug regulatory review process does not
depend only on the speed of the review. In reality, the effectiveness of the regulatory process
should be measured in terms of the approval time and the quality of decision-making. Both
pharmaceutical companies and patients require a rapid registration process, but they should
understand that this must not be at the expense of the quality of the review (Jefferys,
1997;Lumpkin, 2000).
In the case of new drug applications (Lumpkin, 2000) the quality of the review depends
mainly on (i) the competence, consistency and experience of the reviewer and (ii) the
scientific soundness in communication, transparency, consistency and adequacy of the
process and feedback to the industry. Ultimately, the regulatory authority‟s primary
responsibility to the public is to expedite the availability of safe and effective new therapies,
however, pressure is also exerted on the authorities to constantly improve the efficiency and
quality of their review processes. On the other hand, dossier quality, as well as the robustness
and quality of the data submitted, are critical factors for achieving successful registration and
expediting market access (McAuslane et al, 2004). Furthermore, companies and regulatory
authorities have established open and frequent dialogue as regulatory factors that may

6
invariably result in enhancing the review process and review time (Cocchetto et al, 2000).
Normally, the drug review process (Jefferys, 1997) depends on the following parameters:
The review input: In order to ensure the availability of safe and effective medicines in the
market, governments usually have both legal requirements and guidelines. The assessment of
quality involves identifying and defining the role of legal requirements and how they can be
uniformly met with consistency. The clarity of information and communication between the
regulatory agency and the pharmaceutical companies about the expected quality will
influence the efficiency of the review process.
Clarity of roles: The credibility of the review process depends largely on the experts
involved although conflicts of interest undermine the process. The scientific review must be
independent of conflicts with suppliers of technology and they must also be protected from
political influence. An independent scientific review process will hold both suppliers and
purchasers accountable for decisions in the light of the scientific data.
Quality of data: Setting high standards of evidence are essential to the integrity of the drug
review process. However, it is often uncertain whether all of the data from the available trials
are being provided for the review. Requiring public registration of clinical trials and protocols
will enable agencies to know which findings are not being reported by manufacturers.
The scientific assessment: It is the responsibility of the reviewing personnel to establish the
consistency of the review and to maintain the quality and rigour of the scientific analysis. The
scientific basis on which a decision is made must be underpinned on up to date scientific
knowledge, procedures and expertise.
Transparency of rationale: Accountability in drug coverage decision-making requires that
all stakeholders understand the reasons for decisions. One of the major obstacles to such
transparency is currently the commercial confidentiality imposed by manufacturers (Morgan
et al 2006).
The output: The review process should help to ensure greater accountability of both
decision-makers and manufacturers by requiring full disclosure of all reasonable evidence
relevant to a medicine‟s potential coverage. The final decision and its communication to the

7
pharmaceutical company generally and the wider public must be rational, well-documented,
well-organised and transparent (Lumpkin, 2000). The quality of the review (Moss, 2004) will
finally depend on the methodical assessment of the reviewers and the reliable evidence
embedded in the data.
Benefit-risk assessment and Decision-making
Benefit-risk assessment is the basis of any regulatory authority‟s decisions in the pre-market
and post-market review process (EMA, 2012). A framework for medicines' benefit-risk
decision-making that summarises the relevant facts, uncertainties, and key areas of judgment,
clearly explains how these factors influence a regulatory decision and can greatly inform and
clarify the regulatory discussion. A universal eight step framework has been developed for
the Benefit-Risk assessment of medicines (Figure 1.4). This consists of the decision context
(step one), building the value tree (step two), refining the value tree (step three), assessing the
relative importance of Benefits and Risks (step four), evaluating the options (step five),
evaluating uncertainty (step six), a concise presentation and visualisation of results (step
seven) and expert judgment and communication (step eight). Such a framework can provide
transparency regarding the basis of conflicting recommendations made by different agencies
using the same information. Benefits and risks or harms assessment provide a summary of the
submitted evidence regarding the drug under review (Coplan et al, 2011). Key considerations
of benefits include the results of the pivotal clinical trials as well as appropriate analyses of
subpopulations for efficacy. Also the key considerations of risks include the robustness of the
safety database, the severity and reversibility of adverse events, and the sub-optimal
management of the post-market surveillance studies. In assessing benefits and risks,
consideration is also given to some factors that may be significant for a particular drug
review, including nonclinical pharmacology and toxicology data, pharmacodynamics and
pharmacokinetics, chemistry, manufacturing and controls and clinical microbiology. Risk
Management or ideally benefit risk management provides a summary and assessment of any
efforts that could help to lessen the identified safety concerns, or in other words ensure that
the medicine is prescribe for those patients for whom the risk or harm is considered
acceptable in the light of the benefits. Measures to reform the drug approval process have
focused increasingly on the decision bias that minimises unsafe or ineffective medicines but
neglects the costs of denying safe and effective drugs.

8
Figure 1.4 The Eight Step of the Benefit Risk Framework
Role of harmonisation
Harmonisation involves the formation of effective networks between regulatory authorities
(nationally, regionally and internationally) to facilitate the sharing of best practices, making
the best use of scarce resources and eliminating duplication of effort. Such networks are an
important element in building regulatory capacity and trust between different regulatory
systems (WHO, 2008). In recent years, considerable efforts have been made to harmonise
regulatory requirements in terms of scientific content and presentation of data in the
registration dossier, across various regions and countries. The meaning of „harmonisation‟ in
terms of the regulatory point of view is the standardisation of technical requirements for
medicines regulation. The policies of global standardisation (Trouiller, 2002) of regulatory
requirements have been set up, with the most notable example being the initiation of the
International Conference on Harmonisation of Technical Requirements for Registration of
Pharmaceuticals for Human Use (ICH). This initiative has disseminated a common set of
technical requirements relating to quality control (validation of procedures), drug safety and
efficacy as well as certain aspects of manufacturing practice for active ingredients.
Since medicines are essential to healthcare they should be available to the population of every
country. Medicines regulations remain an important component of promotion and protection
Framing the
decision
Step 1: Decision Context
Step 2: Building the Value Tree
Identifying benefits and risks
Step 3: Refining the Value Tree
Step 4: Relative
Importance of Benefit and
Risks
Step 5: Evaluating the
Options
Step 6: Evaluating Uncertaint
y
Step 7: Concise Presentation of Results Visualisation
Step 8: Expert
Judgement and Communication
Interpretation and recommendations

9
of public health because they help to ensure that patients have access to high quality, safe,
and effective medicines. Investing in the regulatory harmonisation initiative provides an
opportunity for strengthening regulatory capacity and the cost-effective use of limited
financial and human resources. Today regulatory authorities are thinking in terms of
regionalisation (Molzon.2010, 2011) through the efforts made by the International
Conference on Harmonisation.
The Global Cooperation Group (GCG) now includes the European Medicine Agency
(EMA), the African Regulatory Harmonisation Initiative (AMRH), the United States FDA,
the Association of Southeast Asian Nations (ASEAN), the Pan American Network for Drug
Regulatory Harmonisation (PANDRH), the Asia Pacific Economic Cooperation (APEC),
Southern African Development Community (SADC) and Japan. The Mercado Comundel Sur
(MERCOSUR) is the South American “Common Market” consisting of Brazil, Bolivia,
Argentina, Chile, Paraguay and Uruguay. This organisation is highly structured among the
Central and South American trade groups for regulatory harmonisation of pharmaceuticals
(Marcelo et al, 1998).
The structures of medicines regulation that exist today which include the drug laws, drug
regulatory agencies, the drug evaluation boards, quality control (QC) laboratories, drug
information centres, have evolved over time. Regulation of medicines encompasses a variety
of functions including licensing, inspection of manufacturing facilities and distribution
channels, product assessment and registration, adverse drug reactions (ADR) monitoring,
laboratory testing for the quality control of promotion and advertising and control of clinical
trials (Sauwakon et al, 2002). Each of these functions target a different aspect of
pharmaceutical activity and act in concert for effective consumer protection.
The Gulf Cooperation Council (GCC) States
The Gulf Cooperation Council (GCC) was established by an agreement concluded on the 25
th
May 1981 in Abu Dhabi (UAE) among Bahrain, Kuwait, Oman, Qatar, Saudi Arabia and
UAE in view of their special relationship, geographic proximity, similar political systems
based on Islamic beliefs, joint destiny and common objectives (Figure 1.5). The GCC States
are located in the Arabian Peninsula southwest of Asia, bordered by Iraq and Jordan in the
North, the Republic of Yemen and the Arabian Sea in the South, the Arabian Gulf in the East,

11
and the Red Sea in the West. The total population of the six member states is 44,223,488
inhabitants in a total area of 2,572,954 square kilometres. Yemen is currently in negotiations
for GCC membership and hopes to join by 2016. The GCC has already approved Yemen‟s
accession to some areas such as the GCC Council of Health Ministers and the GCC Council
of Labour and Social Affairs Ministers. The demographic characteristics of the GCC are
shown in the table 1.1.
Figure 1.5 Map of the Seven Gulf Cooperation Council (GCC) States
GCC Drug Regulatory Authorities
The GCC authorities official names are as follows, Bahrain National Health Regulatory
Authority; Kuwait Drug and Food Control; The Directorate General of Pharmaceutical
Affairs & Drug Control Oman; The Pharmacy & Drug Control Department Qatar, Saudi
Food & Drug Authority; The Registration & Drug Control Department UAE; and the
Supreme Board of Drugs & Medical Appliances Yemen. These seven authorities form the
membership of the GCC Centralised Procedure system (GCC-CP). Five authorities are under
their respective Health Ministries and fully funded by their respective governments. Saudi
Arabia and Yemen, however, are independent stand-alone authorities that rely on registration
fees as the major source of their funding.

11
Table 1.1 The demographic structure of the Gulf Cooperation Council (GCC) States
Ref. The Cooperation Council for the Arab States of Gulf Secretariat General, Statistical Bulletin, Information Sector, Statistical
Department (Volume Twenty, 2012).
The seven GCC authorities regulate pharmaceutical products for human use with their main
activities such as marketing authorisation, post-marketing surveillance and quality control of
pharmaceutical and health products (Al-Essa, 2011). They also have a variety of other
responsibilities depending on the size and resources available for each regulatory authority
(Table 1.2).
Evolution of GCC-DR
The harmonisation of regulatory processes in the GCC States was initiated following the
consent of the GCC Health Ministers‟ Council Decree No. 8 in 1976 regarding the formation
of a study group to report on how a centralised registration system should be set up to
monitor medicines and establish common guidelines for the participating authorities (Hashan,
2005). In 1997, the State of Bahrain submitted a proposal for the formation of a Central
Committee of Gulf States to register pharmaceutical companies and their products to ensure
basic standards for the manufacture of good quality medicines and also to unify the
regulations relating to export of medicines from the Council States.
Country Area / sq
Km
Population Median
age (years)
Life expectancy
at birth (years)
GDP ($) GDP per
capita ($)
Bahrain 760 1,234,571 30.4 75.4 28.27 billion 17763
Kuwait 17,818 2,672,926 26.4 77.89 137.7 billion 46509
Oman 309,500 2,773,479 23.9 73.97 72.78 billion 21355
Qatar 11,586 1,715,010 30.8 75.51 100.8 billion 74839
Saudi
Arabia
2,149,690 27,563,432 24.9 73.87 590.9 billion 16355
UAE 83,600 8,264,070 30.2 76.32 191.9 billion 36017
Yemen 527,968 23,495,361 17.9 63.36 57.95billion 2,500
Total 3,100,922 67,718,849 - - - -
Average - - 26.4 73.76 168.47 30,762.57

12
Table 1.2 The structure, responsibilities and scope of activities within each of the seven GCC regulatory authorities*
*Adopted from Al-Essa, 2011
Country Bahrain Kuwait Oman Qatar Saudi Arabia UAE Yemen
Name of Authority National Health
Regulatory
Authority
Kuwait Drug and
Food Control
The Directorate
General of
Pharmaceutical
Affairs & Drug
Control
The Pharmacy &
Drug Control
Department
Saudi Food &
Drug Authority
The Registration
& Drug Control
Department
Supreme Board of
Drugs & Medical
Appliances
Independent stand-alone authority X X X X √ X √
Budget / GBP NA 2million NA NA 85million 1.6million 2million
Fees / GBP 9 230 130 None >5000 NA 470
Scope of
registration
responsibilities
Medicines for
human use
√ √ √ √ √ √ √
Veterinary
medicines
X √ X X √ √ √
Medical devices
and in-vitro
diagnostics
√ √ √ √ √ √ X
Cosmetic
products
X √ X X X X X
Food
supplements
X √ X X X X X
Herbal medicines X √ X X X X X
Scope of
activities
Marketing
authorisation
√ √ √ √ √ √ √
Post-marketing
surveillance
√ √ √ √ √ √ √
Sample analysis √ √ √ √ √ √ √
Advertising
control
X √ √ X √ √ X
Price regulation √ √ √ √ √ √ √
GMP inspection √ √ X √ X X
Clinical trial
authorisation
√ X X X √ √ X

13
The Gulf Central Committee for Drug Registration (GCC-DR) was formed in May 1999 and
includes Bahrain, Kuwait, Oman, Qatar, Saudi Arabia, UAE, and Yemen. The GCC Drug
Registration Committee, composed of two members from each of the seven countries, advanced
in three stages. In September 2005, an intra-GCC Free Trade Area Agreement, came into effect
by which all GCC national products including pharmaceuticals were exempted from custom
duties within the GCC and any single consignment of foreign goods were charged import duty
only at the port of entry into the GCC area. The formation of effective networks among national
regulatory authorities participating in various harmonisation initiatives has several advantages. It
facilitates the sharing of limited resources; eliminates duplication of activities; saves capital
expenditure for all; supports cooperation and collaboration and international understanding;
facilitates the building of an efficient regulatory capacity; and enhances public trust in the
regulatory efforts (Jamel, 1998). The first stage of this initiative was a two year period during
which all pharmaceutical and research-based companies in the Gulf region were registered; in
the second stage the GCC-DR systems were evaluated and generic medicines from local
manufacturers registered and the third stage involved the appraisal and registration of all
complementary pharmaceutical products such as cosmetics and herbal compounds.
The Ministers of Health in 1999 agreed the two-phase proposal submitted by the Executive
Office of the GCC Ministers of Health for Central Registry and approved the registry
regulations. During phase one (1999-2001) new pharmaceutical products were assigned to
register with both the local authority and the new Central Registration authority and included the
registration of pharmaceutical companies. This also applied to new chemical entities and
biological products. Phase two of this process started in 2002, mainly targeted to assess the
whole system of the registration process and the inspection of pharmaceutical manufacturing
plants. In fact, it was recognised that if the knowledge and experience of different authorities are
shared for a specific task or responsibility, the best practice from each system could be combined
for the advantage of all member states.
Since the GCC States constitute one regional community in its religion, language, population,
similarity in geography, values, history, traditions, economic resources, social and cultural
circumstances, they had to unify their efforts in different fields of life to face the rapid changes
and the overall development requirements. The procurement of safe and effective pharmaceutical

14
products, hospital sundries and equipment of high quality at appropriate prices are organised
through a central group purchasing program. Furthermore, the mission of GCC–DR is to
establish a unified Gulf Drug Registration System that ensures effectiveness, safety and quality
of medications as well as good manufacturing practices (GMP) according to international
standards and post marketing surveillance after registration. The GCC-DR also emphasizes the
use of highly effective and safe medications to protect public health in the Gulf Countries. The
government control over medicines has grown in the last hundred years and now
pharmaceuticals are among the most-regulated products in the world. At present, about 20% of
countries have fully operational medicines regulations, 30% have either none or very limited
drug regulation and a few have regulations of varying capacity (WHO 2006). In reality, many
developing countries cannot ensure the safety, efficacy and quality of medicines available in
their markets because they are resource constrained in terms of staffing, standard systems and
training (WHO, 2008). However, the globalisation of pharmaceutical markets and production has
also increased the spread and prevalence of unsafe medicines.
Protection of public health is the primary aim of any drug regulatory authority and is the
foundation of any country‟s national drug policy that ensures a viable pharmaceutical industry as
well as a high standard drug approval process. However, regulation is perceived as an obstacle
to the availability of medicines in national or regional markets and has placed a significant
demand on regulators to accelerate reviews and evaluations to approve new medicines in the
shortest possible time (Hill, 2004)
The study by Hashan (2005) emphasized the differences between authorities in the GCC States in
the areas which include structures, procedures, available resources, measures of quality &
performance, degree of harmonisation within and between processes and approval times. The
study provided a comprehensive understanding of the medicine regulatory processes in the Gulf
States. It also elaborated the need for greater standardisation in requirements, performance,
procedures and guidelines in the Gulf and other Arab countries which may lead to one regulatory
body in the Gulf region.
The study by Al-Essa (2011) demonstrated the added value of the a harmonised strategic
planning process in the GCC region. This research explored the differences in the Gulf States

15
which included structures, procedures, resources, quality measures and strategic parameters
which facilitate an effective drug approval process for pharmaceutical companies in the GCC
region.
The quality of the review in the GCC
The regulatory review process consists of the scientific evaluation of all data generated during
product development which are described in the registration dossier and when data are
considered satisfactory a marketing approval is granted. The key advantage of the centralised
system in the Gulf is the availability of external expertise to complement the internal evaluators.
This enhances the quality of the reports and more information is focused on the registration to
ensure that product evaluation is carried out according to the required standards. In this manner,
the quality of the product registration is assured across the GCC States.
Apart from obtaining an improved scientific evaluation from more than one state following an
evaluation of the application dossiers, the CP allows further scope for the exchange of
knowledge, ideas and experiences among the National Drug Regulatory Authorities in the GCC.
This results in better coordination and quality reviews within the GCC.
The scientific evidence developed by a pharmaceutical company is evaluated to ensure that the
product can be made available for use or prescribed to patients. Regulators must balance between
the speed of access of the medicine to patients and ensuring that its benefits outweigh any risks.
According to Dureja el al (2010) a well-funded, consistent, robust and transparent regulatory
review system is indispensable to protect the public health and build confidence in the marketed
medicines. Gradual strengthening of the regulatory authorities in the GCC region is fundamental
so that they have the expertise and tools for evaluating new medicines efficiently. Also it is very
important to analyse and scrutinise the differences and uniformity among the regulatory review
processes carried out in all the GCC authorities.
Mallia-Milanes et al (2010) reported on the influence of the type of assessment carried out by
different authorities which may affect the regulatory approval times. The study pointed out the
existence of three types of review process offered for pharmaceuticals in Singapore which
includes full, abridged and verification reviews.

16
A full review relates to products that have not yet received approval elsewhere and takes around
270 working days in Singapore to be completed and supported by external reviewers. The
abridged review usually takes 180 working days, which is used for applications that have been
approved by a recognized regulatory authority, (eg. US FDA, EMA, TGA, etc), before
submission in Singapore. The verification review can be effected if the medicine has been
approved by at least two benchmark authorities and takes a minimum of 120 working days.
McAuslane and Walker (2007) reported in a study of ten agencies, that Argentina alone utilizes
the verification review system for pharmaceuticals and biologicals while Mexico, Egypt, Saudi
Arabia, Malaysia and Singapore make use of the abridged assessment for most of their
applications. Walker et al., (2005a, 2005b and 2005c) stressed the significance of the pattern of
the review process carried out by the regulatory authorities for pharmaceuticals in a timely
manner.
The primary issue to be addressed is the understanding of the type of model being used to carry
out dossier assessment in each GCC country and to quantify the extent of the scientific review
approved in each member state. Karlton et al (1997) pointed out the importance of the quality of
the dossiers in the realisation of rapid regulatory reviews and approval of new applications.
Mediocre documents complicate the review process and may impact negatively upon the
confidence in quality of the medicinal product and its manufacturer (Zellerhoff, 2001). It is the
duty of the manufacturing companies to submit high quality dossiers for improving the
efficiency of the approval process. Valuable capacity development and an effective application
tracking system, assessment and decision-making, appropriate use of information technology,
effective working, accountability and transparency of the medicine registration related decisions
finally result in the overall competence of a unified registration process.
Global drug development and its influence on the regulatory environment in
the Gulf Region
The World Trade Organisation (WTO) established a global patent protection act named TRIPS
(Trade-Related Aspects of Intellectual Property Rights) (WHO, 2003a) in regards to New
Chemical Entities (NCEs) in 1994. Patents can relate directly to active ingredients, formulations,

17
pharmaceutical salts, isomers, polymorphs, combinations, manufacturing processes and also the
patent holder has the complete freedom to set the price. The seven Gulf States, with the
exception of Yemen, recognised the TRIPS agreements. However, the TRIPS agreement
eventually results in a price increase for new medicinal products which is unaffordable to most
people living in developing countries (WHO, 1999,2004).
The world pharmaceutical market in 2014 is estimated to be around one thousand billon US$.
The Middle East and North Africa (MENA) in 2007 comprise around 2% of the global
pharmaceutical market, with an average annual growth of 10.4%, the Arab market (22 Arab
countries) comprises 1.5%, valued at around US$6.2 billion, of which the largest market is the
Saudi Arabia, valued at around US$1.2 billion (Veronica, 2011; Nixon et al, 2013). The
increasing number of conferences and studies on the central drug registration process reflects the
need for greater harmonisation in medicine regulation within the Middle East and it is with this
background that this research was initiated.

18
AIM AND OBJECTIVES OF THE STUDY
Aim
The aim of this study is to evaluate the GCC centralised procedure and the approval times since
it inception in 1999 to 2010.
Objectives
Evaluate the regulatory review process in Oman and identify the milestones and timelines
(2006-2010)
Identify the strengths and weaknesses of the Oman regulatory review process and propose
strategies for improvement.
Determine the similarities and differences in the regulatory reviews in each of the seven GCC
states together with the characteristics of the review process.
Examine the trends in the submission, registration and the associated approval timelines for
patients‟ access to medicines in the GCC states.
Appraise the GCC central registration regulatory review process.
Examine the views and experiences of the GCC states and the pharmaceutical companies
with respect to the GCC registration procedure.
Identify the strengths and weaknesses of the centralised review process and propose
strategies for improvement
Develop a new model for the GCC-DR based on the evaluation and outcome of the
centralised procedure since its inception.

19
CHAPTER 2
Study Rationale and Methodological
framework

21
RATIONALE OF THE STUDY
The decision to centralise Drug registration within the GCC was a political one. Many were
skeptical as to whether such centralisation would improve the time line in registering new
medicines and thus facilitate earlier availability of new drugs to the population. Furthermore,
individual countries were experienced in registration processes and believed that they were
efficient enough. A number of such officials indicated that the individual countries were being
deprived of their sovereignty and equality, they were therefore skeptical about the success of
such initiative. Pharmaceutical companies were also doubtful about the GCC-DR system as to
whether it would improve the review process and the approval timelines in the GCC region.
Many countries had adopted joint drug registration procedures and it is assumed that the process
works and delivers efficiency. As it is now more than 10 years (1999) since the centralised drug
registration system was established in the GCC, it was timely for an assessment of the GCC-DR
timelines and the number of registered products to be evaluated. In addition it was decided to
conduct a comparative study between the GCC-DR and the national approval timelines to
complete the picture for both the individual authorities and the pharmaceutical companies.
It is intended to conduct studies involving the major stakeholders in the registration process.
Current officials responsible for registration in the respective country, pharmaceutical
manufacturers and those within the GCC-DR secretariat will be requested to participate in these
studies. The questionnaire technique will be used for data collection to determine the success and
impediment to the intended progress of the GCC-DR.
In the light of the GCC Central Drug Registration system, the seven regulatory authorities have
had a long experience in joining their efforts to centrally register pharmaceutical products in the
Gulf region. They were apprehensive about the possibility of changing their entire procedures
and practices.
These concerns led to the conceptualisation of this study in order to examine the views of the
seven GCC authorities about the current status of the GCC-DR system and its impact on their
individual processes. Also the current research has focused on evaluating the pharmaceutical

21
industry‟s views about the advantages and disadvantages of the GCC-DR system and how this
has impacted the speed of marketing authorisation for their products in each Gulf State.
METHODOLOGICAL FRAMEWORK
Development of the Study design
The present study employs the principles of quantitative, exploratory and constructive research
methodologies with the primary aim of evaluating the GCC central drug registration procedure.
Background and review of the Study design
The Quantitative data collection methods rely on random sampling and structured data collection
instruments that fit diverse experiences into programmed response categories. These methods
produce results that are easy to summarise, compare, and generalise. Depending on the research
questions, participants may be randomly assigned to different treatments and data collected on
participants relevant to the outcome being measured. (Kidder et al,1987).
A cross sectional design will be adopted for data collection to gather specific data pertaining to
all the seven Gulf States at different phases of the study. There are several advantages of the
cross-sectional design, such as requiring less resources and being able to collect a large volume
of data in the shortest possible time. The questionnaires, as assessment tools are important
aspects of any research of such nature and usually reflect the perception of individual members
of the target population about a particular issue. Furthermore, questionnaires techniques are used
in studies to elicit reports of facts, attitudes, and other subjective states. A questionnaire is
usually composed of a set of questions, answered by potential clients or the target population.
The results can be presented as a table or a graph. A well-structured study using questionnaire
techniques includes a clear and comprehensive statement of the study goals, data requirements,
and the analysis plan (Dillman & Don et al, 1978; Bradburn et al, 1982).
Several different types of studies are utilized depending on the approach used and the overall
study objectives expected. A one-off data collection is selected for the present study which can

22
also impart the advantages of baseline and comparative assessment points (Salant et al, 1994;
Anon, 2000; Passmore et al, 2002).
Th e ma jo r s t reng th s and w ea kn ess e s o f questionnaire techniques are as follows:
S t reng th s
1. Are particularly useful in describing the characteristics of a large population.
2. Can be self-administered making it feasible to recruit large samples.
3. Are flexible and the least costly method of data collection.
4. Results can be generalised.
W eakn es s es
1. Often appear superficial in their coverage of complex topics.
2. There is no way of telling how much thought a respondent has put in to the responses.
3. There is no way to tell how truthful a respondent is being.
4. Lacks validity.
5. People may read differently into each question and therefore reply based on their own
interpretation of the question.
6. Questionnaires also invite people to lie and answer the questions very vaguely which they
would not do in an interview.
Data Collection
Development of Data Collection Techniques
In deicing an appropriate data collection technique for this study questionnaires were reviewed
alongside interviews. The cardinal difference between the two is that the questionnaires can
produce either quantitative or qualitative data whereas interviews produce qualitative data. The
attributes and different types of the two techniques are presented here.
Questionnaires
Questionnaires are usually an inexpensive and efficient method where no special conditions or
equipment are required. It can also be compiled anywhere and distributed easily and the
information can be collected from a large group of people in a timely manner. In a questionnaire

23
everyone answers the same questions therefore it can be compared easily. Learning how to
design and use structured interviews, questionnaires and observation instruments is an important
skill for researchers. Interviews have certain advantages over self-completion questionnaires.
The interviewer can explain questions that the respondent has not understood and can ask for
further elaboration of replies. However, interviews are more time consuming and it may also be
open to interviewer bias, where the interviewer influences the replies by revealing their own
opinions. Such biases can be avoided by using self-completion questionnaires.
Interviews
The interview is a more flexible form than the questionnaire technique and, if wisely used, can
generally be employed to gather information of greater depth and be more sensitive to contextual
variations in meaning. Interviews, however, can be non-scheduled, though still partly
standardised. This is sometimes called a semi-structured interview. Here, the interviewer works
from a list of topics (checklist) that need to be covered with each respondent, but the order and
exact wording of questions is not important. Generally, such interviews gather qualitative data,
although this can be coded into categories to be made amenable to statistical analysis.
The main advantages of the interview technique include (McNamara, 1999; Bourque and
Fielder, 2003a):
The presence of an interviewer allows for complex questions to be explained, if
necessary, to the interviewee.
Interviews can generally be longer than when self-completion techniques are used as
interviewees are less likely to be put off by the length or to give up halfway through.
There is more scope to ask open questions since respondents do not have to write in their
answer and the interviewer can pick up on non-verbal clues that indicate what is relevant
to the interviewees and how they are responding to different questions.
Visual aids can also be used in the face-to-face situation.
The major limitations of the face to face interview are discussed below.
The cost associated with face-to-face interviews can limit the size and geographical coverage of
the study. Interviewers can introduce bias, which will affect the reliability of responses. Such
bias might emerge from the way in which questions are asked, or in the personal characteristics

24
of the interviewer, or in respondents‟ wish to give socially desirable responses. For instance,
there tends to be an over-reporting of voting activity and of participation in voluntary activities
in data gathered through interviews.
The studies using self-completion questionnaires have some distinct advantages over face-to-
face interviews:
They are inexpensive to administer. The only costs are those associated with printing or
designing the questionnaires, their postage or electronic distribution.
They allow for a greater geographical coverage than face-to-face interviews without incurring
the additional costs of time and travel. Thus, they are particularly useful when carrying out
research with geographically dispersed populations. Using self-completion questionnaires
reduces biasing error caused by the characteristics of the interviewer and the variability in
interviewers‟ skills. The absence of an interviewer provides greater anonymity for the
respondent. When the topic of the research is sensitive or personal it can increase the reliability
of responses.
Telephone interviews
Telephone interviews using interview schedules are becoming increasingly efficient with
sophisticated developments in computer technology. Computer assisted telephone interviewing
(CATI) systems are available and these provide clear instructions for the interviewer, display the
interview schedule and allow electronic recording of responses as they are given. This omits the
data entry part of the research because responses are recorded directly onto the computer.
The major advantages associated with telephone interviews are as follows:
The researcher does not have to travel; interviews can take place over a wider
geographical area.
There are fewer interviewer effects ie. The personal characteristics of the researcher will
be less obvious than in face-to face situations and is therefore less intrusive.
The physical safety of the interviewer is not an issue.
Telephone interviews are subject to greater levels of monitoring because supervisors can
unobtrusively listen in to interviews to ensure that they are carried out correctly

25
Different Types of Questionnaire Techniques Used in Data Collection
Several methods are available for collecting data using the questionnaire technique and also
depend on many factors for selecting the most appropriate method. These include the type and
size of population being studied, timelines, budget, resources and purpose of the study (Diem,
2002a).
Paper or electronic mail-delivered
This method utilizes minimum resources to prepare; enables privacy of responses and it is
relatively inexpensive or free of charge, still it may take time and requires follow-up to obtain
representative responses.
Group-administered
This approach involves gathering a group of responders together administering the
questionnaires and asking the group to complete them individually.
The method ensures a high response rate from those who receive the questionnaire and enables a
full explanation of the study with the opportunity for questioning. The disadvantages of this
method are the limited time availability for the responders to formulate their answers and the
slow turnaround time. (Trochim, 2006).
Telephone-administered
This method utilizes pre-arranged telephone calls with the study participants and by recording
the calls; the investigator can generate a digital data base with the interview texts. The time
zones and languages among respondents may be a hurdle of this method leading to no contacts
(Diem, 2002a; Trochim, 2006). It may also be possible to utilize an automated system where the
study participants record their responses via a touch-tone telephone to a computer-based
interview system. Text messages, to remind the respondents, together with cell phone facility
may be utilised in this method.
Web-based method
In this method, the questionnaires may be displayed and posted on a web site which is to be
completed by respondents in a fixed time period remotely. Web-based methods enable a quick
and easy response and are inexpensive (Diem, 2002b; Bourque et al, 2003a ).

26
Data Source
Information will be sought from the seven regulatory authorities who are members of the Gulf
Cooperation Council (GCC) States (Table 2.1).
Table 2.1 Data source for the seven Gulf authorities
Inclusion Criteria
For the Oman study and the evaluation of the review process and timeline in the individual GCC
countries, all products from local, Gulf, Arab non-Gulf and international manufacturers, which
have been approved for human use between 2006 and 2010 and 2008-2010 respectively, will be
included. This includes: New Active Substances (NASs) and Existing Active Substances (EASs).
The products which have complete date information e.g. submission date, regulatory evaluation
time, companies‟ response time to the regulatory queries and approval dates will be included in
the total approval time. In the GCC Centralised Procedure and GCC States studies, all products
from local, Gulf, Arab not-Gulf and international manufacturers approved for human use
between 2008 and 2010 will be included.
Position Agency Country
Director, Pharmacy & Drug Control Ministry of Health Bahrain
Drug Registration and Release Superintendent Drug and Food Control -
Ministry of Health
Kuwait
Director General of Pharmaceutical Affairs &
Drug Control
Directorate General of
Pharmaceutical Affairs and
Drug Control- Ministry of
Health
Oman
Head of Drug Registration Section Drug Control Department-
Supreme Council of Health
Qatar
Executive Director of Licensing Department -
Drug Sector
Saudi Food and Drug
Authority
Kingdom of Saudi
Arabia (KSA)
Head of Registration and Pricing Department
Drug Control Department -
Ministry of Health
UAE
Member of the board council Supreme board of drugs and
medical appliances
Yemen

27
The opinions and suggestions of all the seven GCC states from the respective regulatory
authorities as well as the pharmaceutical companies on their experiences with GCC central
registration procedure will be collected by means of questionnaires.
Exclusion Criteria
Products which have incomplete data such as missing submission date or approval date will be
excluded from the final analysis.
Data Collection Procedure
Confidential procedures will be used for all parts of the study. All the collected data may be
aggregated to avoid identification of individual responders. The electronic or interviewer
delivered questionnaires will be used to collect data which may allow the confidentiality and/or
anonymity of the procedure ((Crow and Wiles, 2008). A questionnaire is useful when there is a
need to collect information from the study participants in a reasonable time period. It is a
structured technique for collecting primary data in a study using a series of structured questions
for which the respondent provides answers. (Diem, 2002b)
Data Collection Monitoring and Timeline
A face-to-face meeting will take place to follow-up the data to be obtained from the Oman drug
authority and to closely observe the regulatory review processes and the approval timelines in
Oman. In addition, a face-to-face meeting with all the Directors and General Directors of
Regulatory Affairs in the Ministry of Health in the GCC States will take place during the GCC-
DR committee meetings. All the participants will be asked to provide the registration data for
products approved in 2008, 2009 and 2010 based on the milestones in each authority.
Clarification (validation) regarding the data set will be obtained from the GCC authorities
through telephonic conferences and electronic mails.
The comparisons of the Gulf States and pharmaceutical companies' perspective on the
effectiveness of the GCC centralized procedure require telephone-interview and email delivered
questionnaire in order to:
Identify the similarities and differences between the experiences of regulatory authorities
and pharmaceutical companies with the Gulf Centralised Procedures

28
Provide recommendations to improve the Gulf Centralised Registration Procedure
Propose the strategies to maximise the benefits of a centralised registration system
Collect feedback about the GCC centralized procedure questionnaire from the GCC
regulatory authorities and pharmaceutical companies. Attempts will be made to clarify
areas of the questionnaire which maybe unclear to the participants
Development of Questionnaires
There are different types of self-completed questionnaires such as mail-delivered, web-based or
email-delivered. The email-delivered and web-based questionnaires will be adopted for this
study . Following a series of consultations with experts and key regulators, it will be possible to
test the applicability of the questions to evaluate views and experiences of the Gulf States with
respect to the Centralised Procedure by conducting a pilot study involving two randomly selected
GCC authorities.
Another questionnaire will be developed to evaluate the experiences and views of
pharmaceutical companies with respect to the central registration review process. A pilot study
will be conducted involving five pharmaceutical companies (two innovative and three generic) to
examine the practicality and applicability of the industry questionnaire.
Chapters three, four, five, six, seven and eight will aim to provide evidence to establish
consensus opinions on topics that do not have adequate information currently available. The
studies in these chapters will focus on the collection of both qualitative and quantitative
information. In situations where there is a need to define levels of agreement on controversial
subjects but there is no unanimity of opinion because little evidence exists or available evidence
is contradictory, consensus methods can be used. These methods attempt to assess the extent of
agreement (consensus measurement) and resolve disagreement (consensus development) (Jones
et al., 2000).
The most commonly used consensus methods are the Delphi process, the nominal group
technique (also known as the expert panel) and the consensus development conference. The aim

29
of consensus methods is to determine the extent to which experts agree about a given issue (Jones
et al., 1995). The features of consensus methods are described in table 2.2.
Table 2.2 Features of Consensus Methods (Jones and Hunter, 1995)
Anonymity To avoid dominance; achieved by use of a questionnaire in Delphi
and private ranking in nominal group
Iteration Processes occur in "rounds", allowing individuals to change their
opinions
Controlled feedback Showing the distribution of the group's response (indicating to each
individual their own previous response in Delphi)
Statistical group response Expressing judgement using summary measures of the full group
response, giving more information that just a consensus statement
Consensus Development Conference (CDC) method
Consensus Development Conference is a formal method developed by the US National Institutes
of Health in 1977, for gaining feedback facilitated through face-to-face contact. A key feature of
this method is the appointment of a carefully selected panel of people thought to be without
vested interest, to listen to the evidence presented at a CDC meeting and prepare a report on the
topic under discussion with recommendations (Fink et al, 1984).
Nominal Group Technique
The nominal group technique (NGT) is a group problem solving process involving problem
identification, solution generation and decision-making. The NGT is a structured method for
group brainstorming that encourages contributions from everyone.
This approach was developed in the USA in the 1960s which includes a highly structured
meeting organized to collect information from appropriate experts about a given topic or issue. It
involves two rounds in which panelists rate, discuss and then re-rate a series of items or
questions. This technique is most commonly used in healthcare to examine the appropriateness
of clinical interventions and has some features in common with focus groups (Jones et al, 2000).
This method focuses on a single goal, e.g. the definition of criteria to assess the appropriateness

31
of a gene therapy intervention, rather than eliciting a range of ideas and it will therefore not be
appropriate for the studies considered for this thesis.
Delphi approach
It is a forecasting method based on the results of questionnaires sent to a panel of experts.
Several rounds of questionnaires are sent out, and the anonymous responses are aggregated and
shared with the group after each round. The experts are allowed to adjust their answers in
subsequent rounds.
Because multiple rounds of questions are asked and each member of the panel is told what the
group thinks as a whole, the Delphi Method seeks to reach the "correct" response through
consensus. The Delphi technique was developed in the 1950s by RAND Corporation in Santa
Monica, California (a non-profit institution that helps improve policy and decision-making
through research and analysis) (Kaplan et al., 1950; Woudenberg, 1991). Since then the method
has been adopted and interpreted widely in health services research (Mead et al, 2001) to obtain
judgement from expert panels by systematically collecting and aggregating informed opinions
from a group on specific issues.
The Delphi method seeks to aggregate opinions from a diverse set of experts, and can be done
without having to bring everyone together for a physical meeting. This anonymity promotes a
sense of freedom to express opinions without negative repercussion (Annells et al, 2003). The
Panel members are encouraged to revise previous responses in subsequent iterations after
reviewing new information submitted by other experts.
The Delphi approach can be a quick, inexpensive and a relatively efficient way of combining the
knowledge and abilities of an expert group on a particular issue. It is viewed as a useful
communication tool for generating debate as opposed to reaching conclusions. For the purpose
of this study, the essence of the Delphi approach will be used to develop consensus of opinions
in each of the previously defined topic areas as well as to collect information that can be used as
scientific evidence (Figure 2.1). It is, therefore, imperative that detailed information be given
about the proposed method of data collection and if questionnaires are used, their development
should be described. Having defined the key problem in each of the research areas and identified

31
the appropriate individuals from regulatory authorities in the area, a pilot study will be conducted
in the first round. Comments from the experts will then be incorporated and used to refine the
questionnaire that will be mailed to all participants. Then the results from the questionnaire will
be aggregated and analyzed before being reported back to participants.
Figure 2.1 Adoption of the Delphi Approach to be used in this study (Mead et al., 2001)
Development of the Study Plan
A comprehensive review and critical analysis of the literature reported in chapter one,
demonstrated a significant gap in the area of the drug regulatory process for the Gulf countries
(i.e. Bahrain, Kuwait, Oman, Qatar, Saudi Arabia, UAE and Yemen) and the GCC Centralised
Procedure. In order to close this gap and improve the regulatory performance, it is vital to
understand the regulatory review practices and evaluate the Gulf States and pharmaceutical
companies' views and experiences with GCC Centralised Procedure to achieve an effective and
improved Gulf Centralised regulatory system. Therefore, the following study plan was developed
to capture data on the regulatory environment in the Gulf. These were then subjected to a
significant scrutiny to fulfill the objectives of the study.

32
The research project will consist of six smaller studies each forming a thesis chapter that
explores the core concepts of GCC drug regulatory field, namely,
Evaluation of Oman Pharmaceutical Regulatory Review Process (Chapter three)
Evaluation of the Regulatory Review Times in the GCC States (Chapter four)
A comparative study of the centralised regulatory review processes in the seven
GCC States. (Chapter five)
An Evaluation of the Gulf States Views and Experiences with GCC Centralised
Procedure. (Chapter six)
An Evaluation of the Pharmaceutical Companies Experiences with the GCC
Centralised Procedure. (Chapter seven)
The Gulf Centralised Procedure- A Comparison between the Experiences of
Regulatory Authorities and Pharmaceutical Companies (Chapter eight)
The outcomes of the research will be combined to derive the major areas that require further
improvement to achieve a standardized and efficient regulatory review process for the GCC
centralised system. The following methods will be employed to collect the required data:
1. To obtain a list of the key regulatory personnel in each drug regulatory authority in the
GCC region.
2. A review of the literature and/publications on the subject of regulatory review process in
the GCC States their centralised system.
3. To consult with regulatory experts and senior managers in the GCC regulatory
authorities.
4. To develop two questionnaires with which to evaluate the Gulf States and pharmaceutical
companies' views and experiences with GCC Centralised Procedure.
5. To produce two reports as a result of the assessment of the GCC centralised regulatory
review process and the Comparison between the experiences of regulatory authorities and
pharmaceutical companies with the GCC Centralised Procedure.
6. The reports will form the basis of the relevant chapters in this thesis.

33
Development of the Study Instruments
The sequence of events to be carried out to achieve the aim of the study will start with an
evaluation of the regulatory approval timelines in Oman. This is a lengthy process which
involves collecting data on the registration dates for 2006, 2007, 2008, 2009 and 2010 from the
saved documents in the Oman MOH archives.
During the course of collecting data on the approval timelines in Oman, two questionnaires to
examine the views and experiences of the Gulf Cooperation Council states and pharmaceutical
companies, with respect to the Gulf central registration procedure will be prepared. The first
questionnaire is planned to be distributed among the seven regulatory authorities in the GCC
states and the second is intended for completion by pharmaceutical companies, already registered
or yet to register their products through GCC centralised registration procedure. A pilot study is
planned to be carried out by coordinating two GCC states and five pharmaceutical companies to
assess the practicality, feasibility, applicability and relevance of the study instrument for
assessing the Gulf Centralised Procedure.
The review times for New Active Substances (NASs) and Existing Active Substances (EASs)
submitted to the GCC Centralised Registration will be obtained directly from the Executive
Board of the Health Ministers Council for GCC States. Since the GCC registration office do not
maintain a robust and complete electronic system of data capture, the data collection will have to
be carried out manually.
Psychometric evaluation of the survey questionnaires
Evaluation process
Questionnaires should undergo evaluation of their psychometric properties before they are relied
on for making decisions. Psychometric validation is used in various fields including education,
psychology, and health research to define the properties of the scales which are included in such
questionnaires. Psychometric principles such as applicability and acceptability, validity,
reliability, consistency and responsiveness are considered in the present study (Mallia-Melanes,
2010).

34
Applicability and acceptability for use
Usually the applicability of a study instrument (Stewart, et al 1990) guarantees the
appropriateness of its content to the purpose of the research being conducted. Furthermore,
applicability describes the suitability of an instrument for its intended use in terms of wording,
clarity and simplicity of language (Higginson and Carr, 2001). The willingness to respond to the
questions by the study participants is another critical aspect in the acceptability of the study
instrument. It also considers the time required from the participants to complete the
questionnaire and whether the questions are clear, concise and easy to understand (McLeod et al,
2008).
Practicality
Practicality issues include the participant‟s comfort, cost and mode of administration of the study
instrument (e.g. interviews or self-administered), convenience and ease of understanding of the
questions (Ware et al, 1980a, 1981). The participants‟ lack of engagement can result in a low
response rate, and high refusal rate, missing responses and longer administration time.
Validity
According to Kaiser and Smith (2001) validity is the most fundamental consideration in
developing and evaluating tests. The most commonly used validity methods for assessing study
instrument are content (face) validity and criterion validity. The concept refers to the degree to
which evidence and theory support the interpretations of test scores demanded by proposed uses
of tests". Ultimately the concept of validity measures what the method claims to measure and
investigate sensitive issues of such method.
Content Validity
This method assesses the extent to which questions in a study serve to encompass the important
facets of the notion the indicator is supposed to represent in a balanced way. The weighting of
the results are also reviewed with the set of indicators (Anon, 2001).
Criterion Validity
The aim of the criterion validity is to correlate a new indicator with reference to a previously
well established indicator (Anon, 2001) and assesses how the observed values of the indicator
compare with another related measure. For example piloting the questionnaires by assessesing

35
the practicality and applicability of questions will ensure that they are clear, feasible and
unambiguous before using the final version for the main study participants
Construct Validity
Guyatt et al., (1993) describe this method as the most rigorous approach to establish validity.
Construct validity examines the logical relationship that exists between a measure and the
characteristics of the system being studied and also involves comparisons between the measures.
Sub-types of construct validity include convergent validity (positive correlation with a related
measure) and discriminate validity (low correlation coefficient) (Mallia-Milanes, 2010 ; Saw,
2001).
Reliability
The consistency and reproducibility of an instrument are the main parameters to be examined for
testing its reliability (Pinar, 2002, McLeod et al., 2008). This quality of the instrument is always
demonstrated by using internal consistency and test-retest reliability.
Sensitivity
The sensitivity of an instrument lies in its ability to detect small changes in parameters such as
approval times, milestones, quality measures and strategic goals (Mallia-Milanes, 2010;
Higginson and Carr, 2001 and McLeod et al, 2008).
DATA PROCESSING AND ANALYSIS
Data will be collected using electronic mail, postal mail, fax, interviewer delivered, computer
assisted telephone interviews and semi-structured interviews. Irrespective of the delivery
method, data will be entered into Microsoft Excel and developed specifically for each study.
Data processing and analysis will be carried out using Microsoft excel and the Statistical
Product and Service Solutions (SPSS). The sample mean will be used extensively for estimating
the population means (the 'middle' value), as was the median (the value halfway through the
ordered data set, below and above which there lies an equal number of data values).Where
applicable ranges around the median and average will be shown to indicate the spread of the
data. Scatter plot will be used to summarise data sets that are bivariate in order to give a good
visual picture of the relationship between the two variables. Box and Whisker plot will be used
on data sets measured on an interval scale to show the shape of the distribution, the central value

36
and variability. If the data is quantitative, descriptive statistics such as mean, median and range
will be used and if the data is qualitative, content analysis will be employed to generate themes
and sub-themes.
Quantitative Content
Hypothesis testing
Hypothesis testing refers to the process of choosing between competing hypotheses about a
probability distribution, based on observed data from the distribution. There are two types of
statistical hypotheses.
Null hypothesis: The null hypothesis, denoted by H0, is usually the hypothesis that sample
observations result purely from chance.
Alternative hypothesis: The alternative hypothesis, denoted by H1 or Ha, is the hypothesis that
sample observations are influenced by some non-random cause.
To perform hypothesis testing a theory needs to be presented that is believed to be true or is to be
used as a basis for an argument, but has not been proved. An example of a hypothesis could
be the claim that a new drug is better than the current drug for treatment of the same symptoms
(Easton and McColl. 2001). In each case two contrasting hypotheses are assessed: the null
hypothesis, denoted by H0 and alternative hypothesis denoted by H1. These two hypotheses are
not treated on an equal basis as special consideration is given to the null hypothesis. This is due
to the fact that the null hypothesis relates to the statement being tested, whereas alterative
hypothesis relates to the statement to be accepted if or when the null hypothesis is rejected
(Easton and McColl, 2001). Plausible arguments will be generated to test a series of hypotheses
arising from the aims and objectives of the study.
Hypothesis testing will be used to:
Evaluate the approval time in Oman
Evaluate the approval time in the GCC Centralised Procedure.
Evidence will be generated to test a series of hypotheses for each of these studies.

37
Choice of Statistical testing procedures
A statistical test provides a mechanism for making quantitative decisions about a process or
processes. The intent is to determine whether there is enough evidence to reject a conjecture or
hypothesis about the process. Therefore, statistical inferences make use of information from a
sample to draw conclusions (inferences) about the population from which the sample was taken
(Easton et al., 2001). The sample mean will be used extensively for estimating the population
means as will the median. Where applicable, ranges around the median and mean will be shown
to indicate the spread of the data. Scatter plots will be used to summarise data sets that are
bivariate (two variables) to give a good visual picture of the relationship between the two
variables. Box and whisker plots will be used on data sets measured on an interval scale to show
the shape of the distribution, the central value, and variability. The figure produced consists of
the most extreme values in the data set (5th
and 95th
percentile values), the lower and upper
quartiles (25th
and 75th
percentile values), and the median. Occasionally, pie charts will be used
for sets of categorical data, each segment representing a particular category and the area of each
segment being proportional to the number of cases in that category (Easton et al., 2001). Several
statistical tests will be used in the present study, depending on the research question asked, the
distribution of the data and the number and characteristics of the data set. For each analysis, the
test used will be specified.
Regression
In statistics, regression analysis is a statistical technique for estimating the relationships among
variables. It includes many techniques for modeling and analysing several variables, when the
focus is on the relationship between a dependent variable and one or more independent
variables. Regression analysis will be used to express the relationship between two (or more)
variables algebraically. The regression equation indicates the nature of the relationship between
two (or more) variables. In particular, it indicates the extent to which some variables can be
predicted by knowing others, or the extent to which some are associated with others. A linear
regression equation is usually written as Y = a + bX + e, where „Y‟ is the dependent variable, „a‟
is the intercept, „b‟ is the slope or regression coefficient, „X‟ is the independent variable (or
covariate) and „e‟ is the error term (Easton et al., 2001).

38
Correlation
Correlation analyses can be used to show the strength of a relationship between two variables. A
correlation coefficient, statistically referred to as r, is a number between -1 and 1 which measures
the degree to which two variables are linearly related. If there is a perfect linear relationship with
a positive slope between the two variables, the correlation coefficient equals +1. There is a
positive correlation whenever one variable has a high value and so does the other, or vice versa.
If there is a perfect linear relationship with a negative slope between the two variables, the
correlation coefficient equals -1. A correlation coefficient of 0 indicates that there is no linear
relationship between the variables (Easton et al., 2001).
Mann-Whitney U-test
The Mann-Whitney U-test is a non-parametric test used to test the difference between the
medians of two independent samples (Crichton, 2000a; Easton et al, 2001),for example males
and females.
Wilcoxon Signed Rank test
The Wilcoxon Signed Rank test is a non-parametric test used to test the median difference in
paired data (related samples). This test is the non-parametric equivalent of the paired t-test
(Crichton, 2000b; Easton et al., 2001).
Kruskal-Wallis test
The Kruskal-Wallis one-way analysis of variance (ANOVA) is a nonparametric test used to
compare three or more independent samples. It is used to test the null hypothesis that all
populations have identical distribution functions against the alternative hypothesis that at least
two of the samples differ only with respect to location. The outcome of this test is not conclusive
as to which of the samples differ. The Kruskal-Wallis test is a logical extension of the Mann-
Whitney U-Test (Easton et al, 2001).
Qualitative Content
The content analysis is a widely used qualitative research technique. Rather than being a single
method, current applications of content analysis show three distinct approaches: conventional,
directed, or summative. All three approaches are used to interpret meaning from the content of

39
text data and, hence, adhere to the naturalistic archetype. Content analysis does not proceed in a
linear fashion and is more complex and difficult than quantitative analysis because it is less
standardized and formulaic (Polit &Beck, 2004). Content analysis is a method of analysing
written, verbal or visual communication messages (Cole, 1988). It is also known as a method of
analysing documents allowing the researcher to test theoretical issues to enhance understanding
of the data.
This method also serves for making replicable and valid inferences from data to their context,
with the purpose of providing knowledge, new insights, a representation of facts and a practical
guide to action (Krippendorff 1980; Sandelowski, 1995). Miles & Huberman (1994) presents the
following three parallel flows of activity to explain the analysis.
Data Reduction: the process of selecting, focusing, simplifying, abstracting and
transforming the data. The purpose is to organize the data so that the final conclusion can
be drawn and verified.
Data display: taking the reduced data and displaying it in an organized compressed way
so that the conclusions can be more easily drawn.
Conclusion drawing/verification: deciding what things mean, noting regularities,
patterns, explanations, possible configurations, causl flows, and propositions.
Miles and Huberman (1994) further describe pattern coding as a way to present data for a
qualitative analysis, pattern coding is important since it reduces large amounts of data into a
smaller number of analytic units. Codes are tags or labels for assigning units of meaning to the
descriptive or inferential information compiled during a study. Codes are attached to “lumps” of
varying size – words, phrases, sentences, or whole paragraphs, connected or unconnected to a
specific setting‟ . This allows the researcher for a better focused analysis and helps to elaborate a
cognitive map in order to understand local incidents and interactions. Codes are used to retrieve
and organize the data which includes descriptive codes, interpretive codes, pattern code etc.
In this study, a set of steps is followed in order to analyze the generated data. The questionnaires
prepared are decided to be piloted with two GCC regulatory authorities before the
appropriateness of the final questionnaires is determined. Then they are e- mailed to the targeted

41
key regulators in the rest of the authorities for completion at pre-scheduled dates. The data
present in the completed questionnaires will then be analysed and reduced where the required
data is abstracted according to pre-set targeted information in individual GCC regulatory agency.
Furthermore, the data will be displayed in a report format where the respondent‟s answers will
be compared to one another in a clear organised manner. At the end the conclusions from the
analyses will be drawn based on patterns of similarities and dissimilarities which are revealed in
the data reduction and data display processes.
SUMMARY
The chapter describes the rationale for carrying out the study to evaluate the GCC
Centralised Procedure for the last ten years since its inception.
The various methodologies, techniques and instruments that will be used in analysing the
data obtained from the seven GCC regulatory authorities and the pharmaceutical
companies have been described.
A detailed description of the developmental technique of the two questionnaires was also
provided and how the information obtained from these questionnaires are reduced,
analysed and displayed in an organized manner.
Methodological choices related to database management, data processing and data
analyses were evaluated.
The data to be collected from the GCC States revolve around three major areas, namely,
the regulatory review processes and milestones, the registration timelines and the
stakeholders views‟ and experiences with GCC Centralised Procedur

41
CHAPTER 3
Evaluation of the Pharmaceutical Regulatory
Review Process in Oman

42
INTRODUCTION
The regulation of pharmaceuticals is mandated such that the government takes on responsibility
as the gatekeeper of the public‟s health and safety and it does this by regulating the behaviour of
the private sector for public purposes (Ratanawijitrasin 2002). Ensuring the safety, efficacy and
quality of medicines available to the public is the main aim of drug regulation. If regulatory
goals are to be achieved, appropriate structures must be established and the activities carried out
to achieve the desired goals.
Ideally, a government should endeavour to have a drug regulatory authority that is created by
comprehensive and up-to-date laws, is maintained as a unified but independent organisation,
contains competent human resources, is free from political and commercial influence, is
maintained by adequate and sustainable financial resources, is guided by transparent standards
and procedures, is driven by outcome-oriented implementation and is monitored and evaluated
systematically (Ratanawijitrasin 2002).
The time required by drug regulatory authorities to approve a new medicine may affect
healthcare professionals and patients and unnecessarily long approval times delay access to new
medicines that may improve the health status of patients (Rawson 2000). Oman had the
advantage of being the last country in the GCC to start adopting the registration system and as a
result the MOH review times decreased soon after the implementation of the new registration
guideline (Ministerial Decision (86/2000). Currently the registration decision must be within
four months from the date of first submission of the registration file. However, many factors still
affect the review process and delay product approvals e.g. lack of experts and resources.
This chapter evaluates the regulatory review process of new medicines in Oman and examines
the regulatory aspirations, barriers, problems and priorities, related to the review of new
medicines that could have an impact on their availability to patients. It assesses the current
regulatory situation in Oman in order to identify the key issues for improving review practices
and patients‟ access to new medicines.

43
OBJECTIVES
The objectives of this study were to:
1. Evaluate the regulatory review process in Oman and identify the milestones.
2. Review the regulatory timelines for pharmaceutical products in Oman between January
2006 and December 2010
3. Identify the strengths and weaknesses of the Oman regulatory review process.
4. Propose strategies that can improve the regulatory review process
METHODS
Data source
Information on the total application numbers and approval dates from January 2006 to December
2010 were obtained directly from the Oman Ministry of Health. This includes New Active
Substances (NASs), Existing Active Substances (EASs), biological products and vaccines. The
data also includes the application submission date, the registration date and the overall review
time. The products were classified into four groups (regions) according to the location of the
manufacturing companies (GCC, non-GCC Arab, international and Asian). A questionnaire for
studying the regulatory review process in the emerging markets was used in this research
(McAuslane et al. 2009). This questionnaire consisted of four parts namely, a review of the
regulatory review process, the requirements for submission of new drug applications, good
review practices, and the timeline for the review process.
Hypotheses
This study examined the following hypotheses:
1. There was an increase in the number of pharmaceutical products approved in Oman
between 2006 and 2010.
2. The largest number of products approved in Oman originated from international
companies.
3. There was a significant increase in the total approval time in Oman between 2006 and
2010.
4. The time of approval for products from the Gulf pharmaceutical companies was shorter
than for other companies.

44
5. There was a significant increase in New Active Substances (NASs) approval times in
Oman between 2006 and 2010.
6. There is no difference in approval times between major therapeutic groups for NASs.
DATA PROCESSING AND ANALYSIS
The data were processed using Excel and SPSS for windows. The statistical analysis for Existing
Active Substances (EASs) and New Active Substances (NASs) covered: the total number of
registered products between 2006 and 2010; and regulatory approval time in Oman between
2006 and 2010 for different cohorts of pharmaceutical products (GCC, non-GCC Arab,
international and Asian); different dosage forms (i.e. solids, semi-solids, liquids, injectables);
and different therapeutic indications (i.e. Cardiovascular system (CVS), Central nervous system
(CNS), Gastro-intestinal system, and Anti-Infectives). Non parametric tests were applied for the
analysis of the data presented in this chapter.
RESULTS
For the purpose of clarity the results will be presented in three parts:
Part I: Review process map and milestones.
Part II: Products reviewed and their characteristics.
Part III: Good review practices in the assessment and registration process.
Part I: Review process map and milestones
The review process map (figure 3.1) reflects the current situation of the Directorate General of
Pharmaceutical Affairs and Drug Control (DGPA&DC) and is a simplified representation of the
main steps being followed for the review of New Active Substances (NASs) and Existing Active
Substances (EASs). The map represents the review and authorisation of a product that is
approved on the first cycle and does not include a second cycle for products approved subject to
submission of additional data. The map also does not include the steps involved in the case of a
refusal of an application (such as hearing, appeal etc.)
The Drug Control and Quality Control Laboratory Departments in DGPA&DC are responsible
for reviewing the marketing authorisation applications. The two departments currently have a
staff of 43 people, 22 of whom are involved in the process of review. There is no formal „fast

45
track‟ procedure, however, life saving medicines are not queued but are selected for review as a
priority, although, the data requirements and review process are the same. The process map and
milestones for Oman is shown in figure 3.1. The review process milestones in the review and
approval are as follows;
a) The receipt and validation stage: This includes administrative registration (reference
number) and validation of legal requirements, the status of the company, details of the
local agent and manufacturer as well as a „checklist‟ validation of the application content
(e.g. technical sections, CPP status). The product applications received are duly recorded
electronically which then generate a unique tracking number. After receiving the product
file, the company must pay the registration fee within one week. The file is rejected if the
information is not complete and a new application must be made after completing the
missing data. The time required for the receipt and validation procedures is only one day.
b) Queuing for review: Once the file is accepted, it will be entered into a queue for review
which takes around one week. The product dossier is divided into two parts; the safety
and efficacy part which are reviewed by the Drug Control Department and quality by the
Quality Control Laboratory. Administrative time is a measure of the „backlog‟ time while
valid applications queue for commencement of their review.
c) Scientific review: Scientific review and analysis takes three months to complete for the
Drug Control Department and Quality Control Laboratory, although both proceed in
parallel. The start and finish of the scientific assessment is formally recorded. The QC
laboratory sends the quality assessment report to the Drug Control Department where the
reviewer, after completing a scientific product report form, submits the file to the
Technical Committee for Registration of Pharmaceutical Manufacturers & Their Products
& Pricing (TCR&P) for a decision.

46
Figure 3.1 PROCESS MAP FOR SULTANATE OF OMAN
(A) RECEIVING OF APPLICATION (B) ACCEPTED FOR REVIEW
(C) SCIENTIFIC REVIEW STARTS (D) Start of Registration Committee Procedure (E) QUESTIONS TO COMPANY REGISTRATION REJECTION
Receipt & Validation Procedures Validation Time: 1 day
Queuing for review Administration Time: 2 weeks
Safety Efficacy Sample
analysis
Quality
Assessment Time: 3 months
Drug Control Department Quality Control Department
<<< Parallel review >>>
Discussion in Technical Committee for Registration of Pharmaceutical
Manufacturers & their Products and Pricing of those products Meets
every
2
weeks
Assessment Time: 1 month
Questions Processed by Company
Reply from Company
TCR&P
REGISTRATION
Company Time: 1 -6 months (Clock stop)
Assessment time: 1 month
Overall registratio
n tim
e 4
mo
nth
s

47
d) Registration committee procedure: The registration committee meets routinely every
two weeks. The TCR&P, established by the Minister of Health, consists of members
from two different directorates of the Ministry of Health, six members from DGPA&DC
and two members from the Directorate General Medical Supplies. All of the members
are Pharmacists and there are no external members. The company can meet with the
internal directorate staff to discuss questions and queries that arise during the assessment,
but they are only permitted to meet the Director and/or Section Heads. If the committee is
satisfied with the safety profile of the product, it will be registered, otherwise rejected. If
registered, a Product Registration Certificate signed by the Chairperson of TCR&P is
issued within two weeks from the date of registration. In the case of a rejection, the
company can appeal within two months from the date of receiving the Committee's
rejection decision.
e) Questions to the companies: In the case of any queries raised by the Committee, they
will be conveyed by the Committee Secretary to the company to respond within a
stipulated deadline which is normally given from 1 to 6 months. Once the response from
the company is received within this stipulated deadline, it is discussed in TCR&P within
one month from the date of its receipt and accordingly a final decision is taken. The issue
of product authorisation also depends on the pricing agreement and discussion of pricing
is separate from the technical review but does not delay the approval of products. The
pricing negotiation starts after the end of the scientific review.
Models of regulatory review:
Many authorities apply different levels of data assessment to different applications, according to
the type of product and/or its regulatory status with other authorities. Three basic types of
review models for the scientific assessment have been identified as a result of discussions with
regulatory authorities and workshop reports from CMR International Institute for Regulatory
Science (McAuslane, 2006), namely,
1. Verification model (type I assessment)
2. Abridged model (type II assessment)
3. Full review model (type III assessment)

48
Verification model (type I assessment)
This model is used to reduce duplication of effort by agreeing that the importing country will
allow certain products to be marketed locally once they have been authorized by two or more
recognized reference agencies, elsewhere. The main responsibility of the authority in the
importing country is to „verify‟ that the product intended for local sale has been duly registered
as declared in the application and that the product characteristics (formulation, composition) and
the prescribing information (use, dosage, precautions) for local marketing conforms to that
agreed in the reference authorization(s).
Abridged model (type II assessment)
This model conserves resources, by not re-assessing scientific supporting data that has been
reviewed and accepted elsewhere but includes an „abridged‟ independent review of the product
in terms of its use under local conditions. This might include a review of the Chemical and
Manufacturing Control (CMC) data in relation to climatic conditions and distribution
infrastructure and a benefit-risk assessment in relation to use in the local ethnic population,
medical practice/culture and patterns of disease and nutrition. Approval by a recognized agency
elsewhere is a pre-requisite before the local authorization can be granted but the initial
application need not necessarily be delayed until formal documentation such as a Certificate of a
Pharmaceutical Product (CPP) is available.
Full review model (type III assessment)
In this model the authority has suitable resources, including access to appropriate internal and
external experts, to carry out a „full‟ review and evaluation of the supporting scientific data
(quality, pre-clinical, clinical) for a major application. A Type III assessment could be carried
out on a new application that has not been approved elsewhere but in practice legal requirements
may dictate that the product must be authorized by a reference authority before the local
authorization can be finalized. The module used by Oman in the assessment of all major
application is an Abridged Assessment. Oman MOH is capable to conduct an independent
review of product applications. However, Oman carries out a unique practice whereby the New
Active Substances (NASs), being considered for approval, must be marketed in the product
country of origin for at least 24 months before it can be registered in Oman. This requirement
may be waived, depending on the product type, if evidence of registration in recognized

49
international reference agencies such United States Food and Drug Administration (US FDA),
European Medicines Agency (EMA) and/or GCC Centralised Drug Registration (GCC-DR)
System.
Part II: Products considered and their characteristics
Approval of all products in Oman (2006 - 2010)
A total of 807 pharmaceutical products for human use were successfully registered in Oman
between 2006 and 2010 (Figure 3.2). This number included EASs and NASs with a similar
portion for the years 2006, 2008, 2009 and 2010 (176; 22%; 171; 21%; 172; 21%; and 178; 22%,
respectively), but there was dip in 2007 (110; 15%).
Figure 3.2: Total number of pharmaceutical products approved in Oman (2006-2010)
Statistical analysis of these data using a simple linear regression showed no significant increase in the total number
of registered pharmaceutical products over the five year period (p>0.05).
This was possibly due to the development of new regulations for the approval of health products
and alternative medicines. These new regulations resulted in an overload for the Quality Control
Laboratory, increasing the analysis time required for pharmaceutical products. The results
showed no significant increase (p>0.05) in the total number of registered pharmaceutical
products from 2006 to 2010 (Figure 3.2).
0
20
40
60
80
100
120
140
160
180
200
2006 2007 2008 2009 2010
Nu
mb
er
of
pro
du
cts
Registration Year
Number of product Line of best fit

51
Approval of Existing Active Substances (EASs) and New Active Substances
(NASs) in Oman (2006 – 2010)
The total number of products registered in Oman between 2006 and 2010 was 807 and this
included 573 EASs and 234 NASs. The highest number of EASs registered in Oman was seen in
year 2010 (137) followed by year 2008 (123), year 2009 (116) year 2006 (112) and year 2007
(87) (Figure 3.3). Statistical analysis using a simple linear regression showed a significant
increase in EASs registered over the five year period (p<0.001).
Figure 3.3: Number of EASs approved in Oman for the period (2006-2010)
Statistical analysis using a simple linear regression showed a significant increase in EASs registered over the five
year period, (p<0.001).
The highest number of NASs was registred in 2006 (64; 36%), while the lowest number was in
year 2007 (25; 23%) (Figure 3.4). From these 234 NASs, 226 (97%) were found to be from
international companies. Statistical analysis using a simple linear regression showed a significant
increase in the number of registered EASs with a significant decrease in the number of registered
NASs (p<0.001).
0
20
40
60
80
100
120
140
160
Num
ber o
f Pro
duct
s
Registration Year
EASs Line of best fit

51
Figure 3.4: Number of NASs approved in Oman for the period (2006-2010)
Statistical analysis using a simple linear regression showed a significant decrease in the number of registered
NASs over the five year period (p<0.001).
The number of pharmaceutical products registered for international, GCC,
non-GCC Arab and Asian companies (2006-2010)
The number of products registered during the five years period varied for different cohorts of
companies based on their geographical location, for example the highest number was for the
international companies products (393; 49%), followed by the GCC Companies products (962;
33%), non-GCC Arab Companies products (95; 12%) and Asian Companies products (50; 6%)
(Figure 3.5).
The Existing Active Substances (EASs) approved for GCC, international non-
GCC Arab and Asian companies in Oman (2006-2010)
A total of 573 Existing Active Substances (EASs) were registered in Oman between 2006 and
2010. When examined by individual regions 267(47%) medicines were identified as being
approved from GCC, 167(29%) from international companies, 94 (16%) from non-GCC Arab,
and 45 (8%) from Asian companies. Statistical analysis of the data using Kruskal-Wallis Test
(K-Test) over the five-year period showed that there was a significant increase in the number of
registered Existing Active Substances (EASs) between 2006 and 2010 (p<0.001).
0
10
20
30
40
50
60
70
Num
ber o
f Pro
duct
s
Registration Year
NASs Line of best fit

52
Figure 3.5: Approved pharmaceutical products (EASs & NASs) for GCC, international, non-GCC
Arab and Asian companies (2006-2010)
Approval times of all pharmaceutical products in Oman (2006 – 2010)
The total approval times for pharmaceutical products in Oman for the period of the study (2006 -
2010) was calculated for all of the 807 Existing Active Substances (EASs) and New Active
Substances (NASs) from different pharmaceutical companies. The median approval time varied
from 55 days in 2006 to 207 in 2008 while in 2010 it was 138 days showing a 33% decrease in
the approval time(Figure 3.6). Statistical analysis using Kruskal-Wallis Test (K-Test) over the
five-year period showed that there was a significant increase in approval times for
pharmaceutical products from 2006 to 2010 (p<0.001).
Approval time for Existing Active Substances (EASs) in Oman (2006 - 2010)
Approval time for 573 Existing Active Substances (EASs) registered in Oman varied over the
period (2006-2010) (Figure 3.7). The lowest median in this period was seen for 2006 (39 days)
and the highest median for 2009 (176 days). Statistical analysis using Kruskal-Wallis Test (K-
Test) over the five-year period showed that there was a significant upward trend in the median
approval time (39, 93, 149, 176 and 138 days respectively) for EASs between 2006 to 2010
(p<0.001).

53
Figure 3.6: Median approval time for all pharmaceutical products in Oman (2006-2010)
Statistical analysis using Kruskal-Wallis Test (K-Test) showed that there was a significant increase in approval
times for pharmaceutical products from 2006 to 2010 (p<0.001).
Figure 3.7: Median approval time for Existing Active Substance (EASs) in Oman (2006-2010)
Statistical analysis of the data using non-parametric K- test shows a significant upward trend in the median
approval time over the five year period (P<0.001).

54
Approval time for New Active Substances (NASs) in Oman (2006 - 2010)
The highest number of products (64) was registered in the year 2006 with the median approval
time of 72 days followed by 133 days for 25 products in 2007 and 292 days for 48 products in
2008. In year 2009, 56 products were registered with a median approval time of 147 days and
159 days for 41products in 2010. The drop in the number of registered products in year 2007
was due to the development of the new regulations for approval of health and alternative
products. These new regulations created an overload for the Quality Control Laboratory,
increasing the time of analysis of pharmaceutical products. The results obtained from performing
analysis of variance test showed that there was a significant upward trend in the median approval
time for NASs products between 2006 and 2010 (p<0.05). (Figure 3.8)
Figure 3.8: Median approval time for NASs in Oman (2006-2010)
The results of variance test analysis showed that there was a significant upward trend in the median approval time
between 2006 and 2010 (p<0.05).
Approval time for Existing Active Substances (EASs) submitted by GCC,
international, non-GCC Arab and Asian companies (2006-2010)
The median approval time for different cohorts of companies varied from 80 for the GCC
companies to 254 for the Asian companies (Figures 3.9). The rapid approval time for the GCC
States is due to their efforts; particularly Oman, Saudi Arabia, UAE, and Kuwait, to improve

55
their local manufacturing capabilities and the production capacity for the local population. EASs
from Asian companies had the longest median approval time of 254 days. Statistical analysis of
the data using non-parametric K- test showed that there was a significant difference in approval
times between different cohorts of companies (P<0.001).
Figure 3.9: Median approval time for GCC, international, non-GCC Arab, and Asian companies
Existing Active Substances (EASs) in Oman (2006-2010).
Statistical analysis of the data using non-parametric K- test showed a significant difference in approval time
between different cohorts of companies (P<0.001).
Approval time for different dosage forms for all Existing Active Substances
(EASs) and New Active Substances (NASs) in Oman, (2006-2010)
During the five years, 573 EASs were registered in Oman with the lowest number represented by
the semi-solid dosage forms (48) followed by injectables, liquid dosage forms and the highest
number was represented by solid dosage forms (364). Out of 234 NASs registered, the lowest
number is represented by the semi-solid dosage forms (3) with a median approval times of 73
days and the highest by the solid dosage forms both in term of numbers (121) and median
approval time (145 days) (Figure 3.10).
0
50
100
150
200
250
300
Gulf International Arab Asian
Tota
l Med
ian
App
rova
l Tim
e (D
ays)
Company Type
Median Time

56
Figure 3.10 Median approval time for different dosage forms for EASs and NASs in Oman,
(2006-2010)
Approval times for Existing Active Substances (EASs) and New Active
Substances (NASs) by therapeutic class, (2006-2010)
An examination of EASs by therapeutic class showed that the lowest number was represented by
the immunological preparations (1 preparation) and the highest number by the injectables (117
preparations). The analysis of the approval time of EASs registered between 2006 and 2010
involved the four major therapeutic groups which dominate the pharmaceutical market in Oman,
namely,
The cardiovascular system (CVS)
The central nervous system (CNS)
The gastro-intestinal system
Anti-Infectives
The range of median approval times for EASs analysed by therapeutic class varied from 141
calendar days for CNS products to 57 for gastro-intestinal products. The differing patterns of
approval times for EASs from different therapeutic groups were attributed to the authority‟s
assessment requirements which may or may not include the need for clinical and bioequivalent
studies. This can be a lengthy process and has an impact on the overall approval timeline.

57
Another reason for such differences is the sponsor‟s response time to the authority‟s request and
the level of communication between the two parties. During the period of study, 234 NASs were
approved in Oman, with the lowest number represented by the ear, nose and oro-larynx
preparations (1) and the highest number by cardiovascular preparations (40). Figure 3.11 shows
the range of approval times for NASs analysed by therapeutic class and these varied from 216
days for anti-infectives to 49 for gastro-intestinal products.
Figure 3.11: Median approval times for different therapeutic classes for EASs and NASs in Oman
(2006-2010)
There were several factors leading to the variation in approval times for different therapeutic
groups. In some cases, consultations with different therapeutic committees within the Ministry of
Health, for example Central Drug Committee (CDC) and National Antibiotic committee, which
do not have regular meetings, may have caused delays in the approval time. Some medicines
have longer pricing times due to a protracted period of negotiation with the companies.
Part III: Good review practice in the assessment and registration process
The ultimate measure of success in regulatory performance is the quality reviews and decisions,
as well as the quality of the dossiers (Kendle, 2009; Karlton and Johnson, 1997 and McAuslane
and Cone, 2006). The regulatory authority, industry and patients benefit from having a high
quality review process that is well managed (Hynes et al., 2001 and Booz Allen Hamilton, 2008).

58
An efficient review allows the regulatory authorities to fulfill their public health mission to
ensure that safe and effective medicines are made available to patients in a timely manner and
allows for efficient use of resources. Patients benefit from the timely access to safe and effective
therapies while pharmaceutical companies are able to market the product sooner and generate
revenues (Booz Allen Hamilton, 2008). The regulatory challenge is to allow access to the safest
and most effective pharmaceutical products in the shortest possible time with a highest degree of
certainty (Alder, 2001).The results showed that the Oman regulatory authority, the Directorate
General of Pharmaceutical Affairs & Drug Control (DGPA&DC), intends to implement a
number of quality measures in the review and authorisation of medicinal products. It includes a
well defined internal quality policy, good review practices, standard operating procedures,
assessment templates and a peer review process.
General measures used to achieve quality
The DGPA&DC currently does not have an internal quality policy which defines the intentions
and directions of the organisation related to quality as formally expressed by top management.
But it is likely to be established in the near future. Similarly, good review practices are not
formally documented at present but will be established soon. A well defined code about the
process and documentation of review procedures would standardize and improve the overall
documentation and ensure timeliness, predictability, consistency and high quality reviews.
Standard operating procedures were implemented in the DGPA&DC for certain procedures
while for the remaining processes, SOPs will soon be established. Assessment templates would
set out the content and format of written reports on scientific reviews however they are not in use
currently but are likely to be established within the next few years. A peer review procedure is
currently used for vaccines as there are two committees reviewing the vaccines marketing
authorisation applications. Peer review, defined as an additional evaluation of the original
assessment that is carried out by an independent person or committee, is unlikely to be
established in the next few years.
Quality management
The three most important measures which the DGPA&DC identified for establishing quality
measures were (1) to be most efficient, (2) to minimise errors and (3) to increase transparency.

59
To improve quality, the following activities were undertaken to bring about continuous
improvement in the assessment and authorisation process:
Reviewing stakeholders‟ feedback (e.g. through complaints, meetings or workshops) and
taking necessary action.
Carrying out internal quality audits (e.g. self assessments) and using the findings to
improve the system.
Having external quality audits by an accredited certification body to improve the system
(e.g. WHO)
The DGPA&DC does not have a dedicated department for assessing and/or ensuring quality in
the assessment and registration process for new medicines, however, there are plans to set up
such a department in the future.
Quality in the review and assessment process
The guidelines issued under the Ministerial Decision No. 86/2000 serve as an official document
to assist the industry in the registration of medicinal products in Oman. This Ministerial Decision
is also available on the Ministry of Health website. Pre-application scientific advice is not
available to applicants, but can be obtained during the file submission day. The applicants are
allowed to make formal contacts with DGPA&DC staff to discuss an application during its
review process.
There is a registration committee called “The Technical Committee for Registration of
Pharmaceutical Companies and their Products and Pricing”. The committee consists of eight
members and meets once every two weeks. New Active Substances (NASs) applications are
reviewed by this committee while Major Line Extensions (MLE) are reviewed by the Drug
Control Department in the DGPA&DC and not forwarded to the registration committee. The
committee receives assessment reports from the Drug Control and Quality Control Laboratory
staff that review the applications. Shared/Joint reviews are undertaken when products are
submitted through the GCC Central Registration Procedure.

61
Training and continuing education
To maintain or improve the quality of the work, it is essential that reviewers follow training or
refresher courses from time to time. These may concern general training about new
developments in the regulatory science field or specialized training in carrying out a quality
review process. The DGPA&DC does not currently have any formal training programmes for
assessors. Training is mainly carried out through external courses, post-graduate degrees,
participation in international workshops/conferences, placements and secondments in other
regulatory agencies and inviting speakers to the authority. After training or attending a course,
the reviewer should report and convey his/her experience or knowledge to colleagues and top
managers and make proposals for any change of existing procedures or adoption of new
practices to improve the overall performance of the reviewing staff.
Transparency of the review procedure
The DGPA&DC assigns a high priority to transparency of the review procedure. It has identified
the following three top incentives for assigning resources to activities that enhance the openness
of the regulatory system namely: political will, the need to increase confidence in the system and
to provide assurances to the pharmaceutical industry. Information such as product approval and
the price of medicines is routinely provided to the general public. The information can be
accessed from the ministry‟s official website or provided on submission of a formal request. As
far as transparency is concerned, the companies can follow up the progress of their application
either through phone contact, email or meetings with the officials with prior appointment. There
is an electronic system for registration and tracking of applications, which also can be used to:
Track applications that are under review and identify the stage of the process
Signal that target review dates have been exceeded
Record the terms of the authorisation once granted
Archive information on applications in a way that can be retrieved.
Drivers and barriers facing the review process in Oman
Understanding the drivers and barriers to achieving a quality regulatory review process can
promote innovative and creative ideas and practices to help reviewers assess new products and
underline the importance of monitoring the quality of the review process.

61
There are several motivating factors identified by DGPA&DC that contribute to accomplishing
the desired effectiveness and efficiency of the authority‟s review procedures and decision-
making for new applications, namely:
Good tracking system
Utilization of an assessment template.
Good Management Plan
Well qualified, trained and experienced reviewers
However, the DGPA&DC is facing several obstacles that are hindering its ability to ensure that
new medicines are available in a timely manner, i.e:
Shortage of experience personnel
Lack of an independent budget
Lack of an internal quality policy
DISCUSSION
This study has evaluated the pattern of total regulatory approval time in Oman between 2006 and
2010. It has also addressed the various factors which may have a positive or negative effect on
the total approval time. It has been shown that the total number of registered Pharmaceutical
Products in Oman (2006 - 2010) is 807 and this number includes Existing Active Substances
(EASs) and New Active Substances (NASs).
Six hypotheses were generated for this study, as outlined previously and the findings are
discussed below.
1) There was an increase in the number of pharmaceutical products
approved in Oman between 2006 and 2010.
The largest number of registered medicines was found in 2006. In this year, there was no
overload on the quality control laboratory since the actual approval of health products, using the
recent guidelines and regulations (Circular 64/2005) did not start until 2005. However, the
number of the registered medicines was similar in the years 2006, 2008 & 2009 with the
exception of 2007 when there was a fall. This was largely due to the development of new
regulations for approval of health products. These new regulations resulted in an overload on the

62
Quality Control Laboratory, increasing the time of analysis of pharmaceutical products. This
hypothesis has therefore rejected.
2) The largest number of the products approved in Oman originated from
international companies.
The number of products registered during the five year period (2006-2010) varied for different
companies based on their geographical location. The largest number was for the international
companies products, followed by the GCC companies, non-GCC Arab, and finally the Asian
companies. International companies were the largest group registering products every year in
Oman. This was due to the fact that the pharmaceutical companies must submit a full registration
application when the manufacturing site has been changed for the same registered company.
Thus, there was an increase in the number of changes in product manufacturing sources
applications from the international companies during this five year period. Furthermore,
international companies increased the number of their submissions to the Omani authority to
obtain registration approval as evidence that would support their GCC-DR approval. The pricing
process in Oman is the final step and price negotiations occur at the end of the scientific
assessment. The Registration Committee makes the decision on the product registration and
pricing. Oman has a robust pricing control system and this is one of the main reasons that
pharmaceutical companies register their products in Oman prior to other GCC states. Using a
price reference system, the pricing committee considers the manufacturer's wholesale and retail
prices in the country of origin, export Cost-Insurance-Freight (CIF) prices to Oman as well as
CIF prices in other GCC states. This hypothesis was accepted.
3) There was a significant increase in the total approval time for medicines in
Oman between 2006 and 2010.
During the five years, the median approval time varied from 55 days in 2006 to 119 days in
2010. There was a significant increase in the median approval time between 2006 and 2008
which was due to the new regulations that were implemented by Oman MOH in 2006 for the
registration of human health products. This was due to the increase in the number of local
registration applications and the applications received from GCC-DR for Oman MOH
evaluations. Registration for the pharmaceutical companies and their products must be renewed
every five years as required by the Oman registration system. In 2006 Oman MOH requested

63
pharmaceutical companies to re-register their products. This re-registration was for the first time
and led to an increase in the number of applications and an overload for the Drug Control
Department and Quality Control Laboratory.
The WHO has clearly stated that the shortage of qualified personnel is the greatest problem
faced by drug regulatory authorities (WHO, 2003a), and is one of the factors affecting patients‟
access to new medicines in Oman. All staff are paid according to established salary bands for the
entire public service. Consequently, Oman MOH is unable to use salaries to attract and retain
some of the skills that would strengthen the regulatory role. This hypothesis was therefore
accepted.
4) The time of approval for products from Gulf pharmaceutical companies
was shorter than other companies.
Median approval time varied for different companies from 80 to 254 days for GCC and Asian
companies respectively. The rapid approval time is mainly due to the GCC States making efforts,
particularly Oman, Saudi Arabia, UAE, and Kuwait, to improve their local manufacturing
capabilities and the production capacity for the local population (Al-Essa, 2011). This led to a
positive outcome in approval times, giving a median time of 80 days. There are other factors
which have a positive effect on the approval time for local and Gulf companies but have a
negative effect on the international, non-GCC Arab and Asian companies. For example, in
decision-making, the geographical proximity of the company to the authority leads to an
effective interaction and thus a faster response to any questions from the authority. Additionally,
most locally manufactured products are EASs and do not need very much consultation during the
scientific review compared to NASs. Furthermore, an understanding of the culture of the
regulatory authority where there is no language barrier has a positive effect on the review
process. However, a priority procedure is a common practice in many authorities, but often in
relation to the importance of the medicine rather than geographic location. In Canada and
Australia, pharmaceutical companies can apply to receive a priority review for their medicines
and the regulatory authority then decides which medicines will be reviewed in this way. The
FDA classifies all new medicine applications to receive either a priority or a standard review
(Rawson et al., 2000a). In this study, it was found that the 31% of the products, which received
priority approval, were manufactured by local companies or were for serious life-threatening

64
medical groups, e.g. cancer medicines. For the 5-year study period, products from Asian
companies had the longest median approval time of 254 days. Oman requests limited clinical
data in the form of bioequivalence studies to demonstrate that the EAS is bioequivalent to the
original product. Bioequivalence presents the first challenge for the EAS registration because
many manufacturers find it difficult and costly to perform high standard controlled trials that
compare the proposed EAS with the original product. Meanwhile, for EAS‟s the laboratory
sample analysis is carried out in parallel with the scientific review but the analytical step can be
waived if the product is registered in Saudi Arabia, UAE, and/or Kuwait, which have recognized
laboratories in the region. From these results this hypothesis has confirmed.
5) There was a significant increase in New Active Substances (NASs)
approval time in Oman between 2006 and 2010.
The pricing process can markedly affect the registration time in Oman. The pricing time for
NASs is longer than for EASs due to the period of negotiation. The registration committee sets
the lowest possible price for the original brand product, which is usually the export price to the
Saudi market. Saudi Arabia has much better negotiating power and hence the Omani agency
wants to use this power to reduce the medicine prices. Pricing is the final step and no product
will be issued a registration certificate before it is priced, but that proposed by the company is
not necessarily agreed, although the company is given an opportunity to appeal before the final
price is decided. Pricing negotiations start after the granting of registration approval of the
medicine. This approach prevents the pricing step from being integrated into the review process
and this delays the final approval time. Therefore, it would be preferable for the pricing process
to be carried out in parallel with the review process rather than after granting registration
approval. The analytical step is one of the approval requirements and takes up to almost 50% of
the total approval time for all companies. Unlike Oman, none of the international authorities
analyse the products pre-registration. Therefore, it is recommended that this step be carried out
post-approval with product samples being selected from the market for analysis. This would
reduce the registration time. Another inappropriate milestone is the submission of a Certificate of
pharmaceutical Product and the price certificate which has to be legalized by the relevant
authority and Oman embassy in the country of origin. Such regulations lead to registration delays
and increase the burden for the companies and it is not a WHO requirement.

65
The findings from this study indicate that the company time for answering questions from the
authority when companies submit analytical data are longer than the authority time for all
cohorts of companies. No doubt this issue also causes a further delay in the registration of
products, which affects patients‟ access to medicines submitted by international companies.
Furthermore, the delay caused by companies leads to a waste of authority time and resources. In
order to reduce the company time, it is recommended to implement an official clock-stop system
by the Omani health authority while the company respondes. This hypothesis was accepted.
6) There is no difference in approval time between major therapeutic groups
for the NASs.
The results of this study shows that there are significant differences in the approval time between
therapeutic groups, for example the approval time for the anti-infectives is three times longer
than those for gastrointestinal products. There are several factors leading to these differences in
approval times. In some cases, consultations with different therapeutic committees within the
Ministry of Health for example the Central Drug Committee (CDC) and the National Antibiotic
committee may cause delays in the approval time. In addition, the nature of the products
evaluated, e.g. biological products may require a more detailed review than others. Some
medicines have longer pricing time due to periods of protracted negotiations. Using a price
reference system, the pricing committee considers the manufacturers wholesale and retail prices
in the country of origin, export CIF (Cost, Insurance, and Freight) prices to Oman and CIF prices
in other GCC states. These results do not support this hypothesis.
CONCLUSIONS
This study has assessed the trend in the regulatory approval times in Oman for the period 2006 to
2010. The findings show that although there is an increase in the approval time for all
pharmaceutical company products, the median approval time for the five year period was 117
calendar days. This is within the time limit (4 months) which is fixed by the health authority for
the overall registration time. It was found that products in the different cohorts of companies,
which include local, GCC, non-GCC Arab and international companies have varying approval
times which may be due to an internal unpublished policy of prioritization within the authority.
However, there are many factors affecting the approval time, which include the quality of the

66
dossier, the geographical location of the company, communication within companies and various
cultural factors. This study enabled the regulatory authority in Oman to be made aware of its
own review time and the timing of each step within the process. Furthermore, since this study
was based on regulatory authority data, a reliable and accurate source, it may enable
pharmaceutical companies to better understand the factors that influence the review process and
the overall review time. This, in turn, may help to accelerate the registration procedure and save
time and resources by allowing a more rapid access to innovative medicines. The companies
have the responsibility for most of the delay of their own product registration in Oman because
they take a long time to respond to questions asked by the authority or to submit the analytical
data.
SUMMARY
The review time for the registration process during the period 2006 to 2010 is within the
time limit fixed by the Oman health authority, but there has been an upward trend in
regulatory approval times since 9206.
The total approval time varies according to the geographical location of the company,
and it was found that the shortest approval time was for products from local and Gulf
companies.
In addition to the factors already mentioned, the absence of separate sections for health
products and herbal preparations in the Drug Control and Quality Control Laboratory
Departments and the large number of samples to be analysed by the QCL from
Directorate of medical supplies has a significant impact on total approval time.
The pricing process for the NASs, the pre-registration analysis, lack of official clock-
stop system and the delay in replying to regulatory queries, all have a delaying impact on
approval time.
The fees for the registration of the companies and their products are very low in Oman.
This has led to a high number of applications and an increase in the median time of
registration.

67
CHAPTER 4
Evaluation of Regulatory Review Times
in the Gulf States

68
INTRODUCTION
Long regulatory approval timelines for new medicines delay their access to the market that may
have the potential to improve patients' health status and of alleviating suffering. Variation in the
availability of medicines in different countries has been studied since the early 1970s (Rawson,
2000), and some marked differences have been identified. Regulatory approval time is a key
metric that is used to evaluate the performance of regulatory agencies (Hirako et al., 2007). A
study was previously made to evaluate the milestones and timelines involved in the regulatory
review processes for different authorities (Hirako et al., 2007). A specific study of the Gulf
regulatory authorities (Hashan, 2005) highlighted important aspects of the drug approval
procedures in each of the seven member states. However, the study provided limited information
about the approval timelines and the duration of the milestones and stages involved in the review
process because the authorities did not have electronic tracking systems to monitor such
activities. This made it difficult, if not impossible, to allow cohorts of pharmaceutical products to
be monitored retrospectively (Al-Essa, 2011).
Currently, within the GCC States, there are two registration systems, the national and the
centralised procedure. In order to benchmark and make true comparisons between the different
authorities, it is essential to identify first the common processes that occur during registration.
However, there are no major differences in the legislation, regulations or requirements for
marketing pharmaceutical products in the GCC States (Bahrain, Kuwait Oman, Qatar, Saudi
Arabia, UAE and Yemen). All the national regulatory departments in the GCC States have
regulations that govern and regulate most aspects of the pharmaceutical services by effective
laws (Hashan, 1998). Apart from the registration process of medicinal products, some countries
have price control also, in their remit.
It is recognized that individual authorities have different experiences and knowledge that could
be of value to other member states through comparison of various systems and sharing of best
practices to the benefit of all. To this effect, this study was conducted to explore the timelines of
the approval processes in the GCC States. However, it is important to have a basic understanding
of the regulatory review process in these countries in order to place the approval time in an
appropriate context.

69
OBJECTIVES
The major objectives of this study are to:
1. Compare the number of registration applications submitted for pharmaceutical products over
the three-year period from January 2008 to December 2010 in the GCC States.
2. Compare the regulatory approval times for registered products for Gulf, Arab non-Gulf,
Asian and International pharmaceutical companies in the GCC States.
3. Assess the trends in approval times over the period 2008-2010.
4. Identify areas of the approval process in each of the seven Gulf States which might lead to
delays in the registration of new medicines.
5. Provide possible solutions for addressing the delays in the registration timelines in the Gulf
States.
METHODS
Participants
The participants were the authorities responsible for the regulation of pharmaceutical products in
the GCC States (i.e. (Bahrain, Kuwait Oman, Qatar and Saudi Arabia) Yemen was excluded
from this study because they could not provide data due to the unstable political situation in the
country during the period of this study. The United Arab Emirates was also excluded from this
study, as they are unable to furnish the full data due to changes in the drug regulatory
organization that took place during the time this study was conducted. Additionally, they were
also unable to extract some of the data as it was manually stored making retrieval of the required
information very difficult.
Data collection
A face-to-face meeting with all the Directors and General Directors of Regulatory Affairs in the
Ministry of Health in the GCC States took place during the GCC-DR meetings in 2011and 2012.
They provided the data on approval timelines as an Excel spreadsheet including the registration
data for products approved in 2008, 2009 and 2010, based on the milestones in each authority.
Clarification regarding the data set was obtained from the five authorities during 2012 through
telephonic conferences and electronic mails.

71
The data consisting of all products from Gulf, Arab non-Gulf, Asian and International
manufacturers, approved for human use in 2008, 2009 and 2010 in the GCC States, were
included in this study. Products reviewed included both New Active Substances (NASs) and
Existing Active Substances (EASs) and only those with a complete data set e.g. submission date
and approval date were included in the analyses. In addition, relevant information regarding the
regulatory review process of the five Gulf countries was collected by literature search. The
overall data includes milestone and tables reflecting the specific characteristic of each system
(Al-Essa et al, 2012).
Models of regulatory review:
Three GCC authorities utilize an abridged assessment (Bahrain, Oman and Qatar) as describe in
chapter three. This is a critical practice to ensure the appropriateness of the product under local
conditions. Bahrain uses a verification review for biological and biotech products because they
have to be registered in other reference authorities to be accepted for review in Bahrain, and an
abridged review for other major applications. Saudi Arabia is the only country that performs a
full review on all types of applications, while Kuwait carry out a simple verification review on
the registration dossiers.
DATA PROCESSING AND ANALYSIS
The Regulatory approval times were calculated as the time from submission of the product
dossier to the regulatory authority until the final approval. The data sets processed include: the
total number of registered products between 2008 and 2010 and regulatory approval time in
GCC States between 2008 and 2010 for different groups of pharmaceutical products (GCC, non-
GCC Arabs, International and Asian). Descriptive statistics including frequencies was used to
present the results.
RESULTS
The results of this study are presented in two parts,
Part 1: Includes the process maps for the regulatory reviews in each of the five states together
with characteristics of the review process.
Part 2: The results are presented under three headings:

71
I: Total number of product approved in each Gulf states.
II: Median approval times for the Gulf States by year 2008-2010
III: Median approval times for the different pharmaceutical companies.
Part 1: The regulatory process maps for the Gulf States:
All the GCC authorities share similar goals and obligations to safeguard public health when
assessing the safety, quality and efficacy of medicines before they are authorized for marketing.
They also have a similar structure when reviewing pharmaceutical product dossiers, but the
position of each milestone in the review process differs from one state to another.
The first study in the GCC was initiated by Hashan (2005) to assess the regulatory environment
in the six GCC states (Bahrain, Kuwait, Oman, Qatar, Saudi Arabia and UAE) using
comparative data on a country and regional level, in order to identify the key issues for
improving regulatory practices and ensuring new medicines were available in an efficient and
timely manner. The second study by Al-Essa (2011) was to map the key milestones and
associated activities for each agency in the GCC region and to determine the quality measures
employed by the agencies in their different procedures. Figures 4.1- 4.7 illustrate the process
maps for each of the countries participating in this study as well as the characteristics of the
process.
This part describes the comparison between the regulatory review processes in the seven GCC
regulatory authorities.
Validation process
The validation process is the first contact between the sponsors and the regulatory authorities.
Validation is an important checking point to ensure the correctness and completeness of the
submitted data before entering the scientific assessment stage. The seven authorities record the
date of receiving the registration dossier and carrying out a validation process to ensure that the
documents submitted for registration are complete before they can be accepted for review. After
examining the validation process, a considerable difference was observed in the time taken to
validate the registration dossier from one country to another, with Oman performing the
validation process immediately after submission while Bahrain takes 14 days to complete it.

72
Figure 4.1 PROCESS MAP FOR BAHRAIN

73
Figure 4.2 PROCESS MAP FOR KUWAIT

74
Figure 4.3 PROCESS MAP FOR OMAN

75
Figure 4.4 PROCESS MAP FOR QATAR

76
Figure 4.5 PROCESS MAP FOR KINGDOM OF SAUDI ARABIA
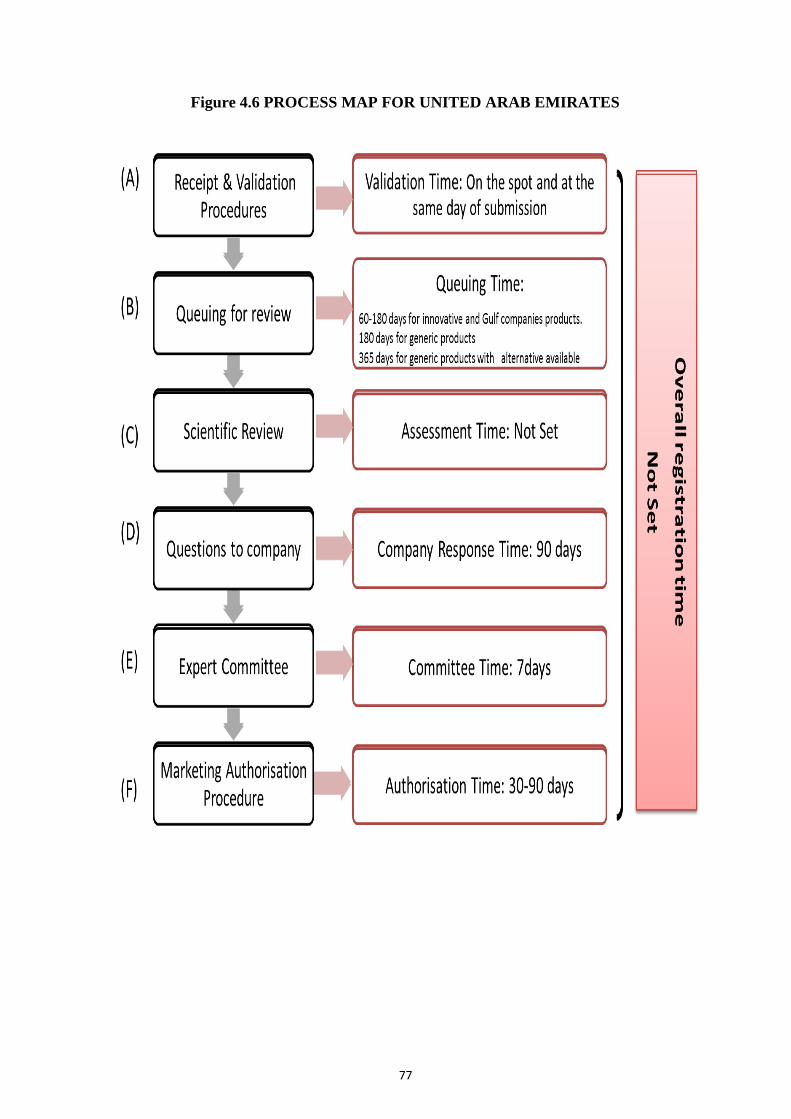
77
Figure 4.6 PROCESS MAP FOR UNITED ARAB EMIRATES

78
Figure 4.7 PROCESS MAP FOR YEMEN
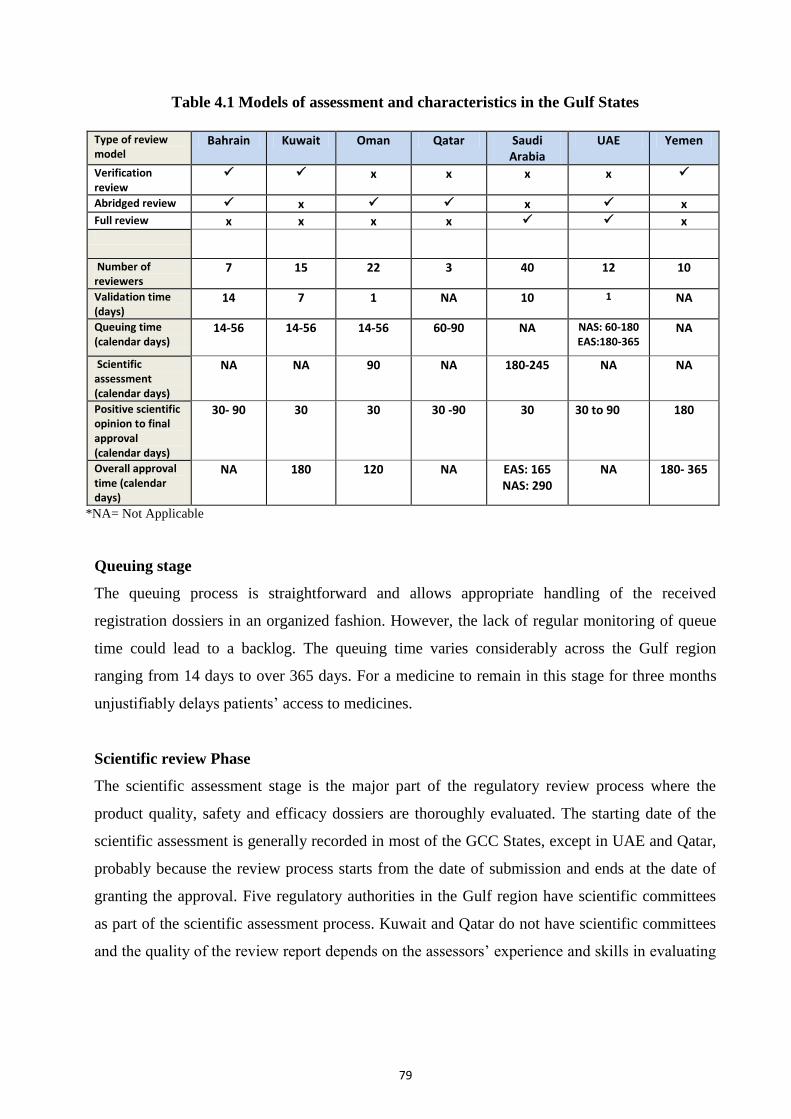
79
Table 4.1 Models of assessment and characteristics in the Gulf States
Type of review model
Bahrain Kuwait Oman Qatar Saudi Arabia
UAE Yemen
Verification review
x x x x
Abridged review x x x Full review x x x x x
Number of reviewers
7 15 22 3 40 12 10
Validation time (days)
14 7 1 NA 10 1 NA
Queuing time (calendar days)
14-56 14-56 14-56 60-90 NA NAS: 60-180 EAS:180-365
NA
Scientific assessment (calendar days)
NA NA 90 NA 180-245 NA NA
Positive scientific opinion to final approval (calendar days)
30- 90 30 30 30 -90 30 30 to 90 180
Overall approval time (calendar days)
NA 180 120 NA EAS: 165 NAS: 290
NA 180- 365
*NA= Not Applicable
Queuing stage
The queuing process is straightforward and allows appropriate handling of the received
registration dossiers in an organized fashion. However, the lack of regular monitoring of queue
time could lead to a backlog. The queuing time varies considerably across the Gulf region
ranging from 14 days to over 365 days. For a medicine to remain in this stage for three months
unjustifiably delays patients‟ access to medicines.
Scientific review Phase
The scientific assessment stage is the major part of the regulatory review process where the
product quality, safety and efficacy dossiers are thoroughly evaluated. The starting date of the
scientific assessment is generally recorded in most of the GCC States, except in UAE and Qatar,
probably because the review process starts from the date of submission and ends at the date of
granting the approval. Five regulatory authorities in the Gulf region have scientific committees
as part of the scientific assessment process. Kuwait and Qatar do not have scientific committees
and the quality of the review report depends on the assessors‟ experience and skills in evaluating

81
the registration dossier. The scientific assessment is not a target time in five GCC authorities
while Oman and Saudi Arabia specified a time limit of 90 days and 180 to 245 days respectively.
Queries to companies
In this phase all the communications occur between the sponsor and the authority with regards to
the registration of a new medicine in each GCC State. Questions and queries arising during the
scientific assessment and quality control analysis stages are collected into a single batch to be
sent to the sponsor by each of the seven GCC authorities. The sponsor‟s response time varies
significantly between the seven GCC States with the shortest time limit being 30 days enforced
by the Saudi Food and Drug Authority (SFDA) and the longest is approximately 365 days in
Qatar. The clock-stop concept is perceived differently across the region. In Kuwait, for example,
the authority does not enforce a limit for the sponsor‟s response time but if the sponsor fails to
respond in two years, the authority ceases the registration process and returns the dossiers to the
local agent and a new application is officially requested should the sponsor be still considering
the product registration in Kuwait Bahrain and UAE, however, do not have a clock-stop system
but they target a specified limit for the sponsors response time.
Overall target time for registration
While Bahrain Qatar and UAE were not setting a target approval time, the other four authorities
have slight differences in their overall target approval times with the shortest one in Oman being
120 days.
Part 2: Total number of product approved in each Gulf states, median approval times for
the Gulf States by year 2008-2010 and the median approval times for the different
pharmaceutical companies
In this study, three major areas were examined, for the period 2008-2010 namely, (1) Trends in
submission and registration of new pharmaceutical products; (2) Changes in the approval
timelines; (3) Approval times for Gulf, Arab non-Gulf, Asian and International pharmaceutical
products in the GCC States. The number of approved products in any regulatory authority is
affected by many factors. For example, the number of dossiers submitted to the authority and the
number of reviewers in the review section (Hashan, 2005). The GCC States are no exception and
like many other authorities the number of approved products varies from year to year influenced
by these and other factors.

81
Total number of products approved in each Gulf state
In general, the countries reviewed a similar number of products, but there was an increase in the
number of approved products in 2010 in most GCC States compared to 2008 and 2009 with the
exception of Saudi Arabia (Table 4.2). This may reflect an increased interest by the industry in
the Gulf market or the authorities in the region were able to cope with an increased work load,
translating into an increased number of products approved. The GCC regulatory authorities share
common regulatory review practices that are critical in the establishment of a standardized
review process for the GCC region. The differences identified in the five review processes were
mainly due to the order of the steps carried out or the time spent in carrying out a certain
procedure. As per Al-Essa (2011) the approval timelines in the GCC States depend on the type of
products being registered, the quality of the submitted data and the level of interaction between
the sponsor and the authority.
In 2010, Kuwait registered the highest number of products among all the GCC countries with a
total of 210 products registered which is almost twice that of Saudi Arabia (92 products) (Table
4.2). Bahrain had the lowest number of products registered in total for 2008, 2009 and 2010 at
87, 149 and 174 products respectively. The analysis for each country is provided in the following
paragraphs. Oman is the only country in the GCC with a more or less constant number of
products registered between 2008 to 2010. Bahrain saw a steady rise in the number of registered
medicines from 87 approved products in 2008, 149 in 2009 and 174 in 2010 (Figure 4.8). This
steady increase in the registration of medicines in Bahrain was due to the decision from the
Ministry of Health (MOH) in Bahrain to facilitate the procedures for the drug registration if the
product is approved by advanced regulatory authorities such as EMA and USFDA or by one of
the reference countries in the Gulf.
Kuwait, on the other hand, saw a decrease in the number of registered medicines in 2009 and
2010 of 198 and 210 respectively as compared to 416 in 2008. According to the Kuwait
regulatory authority, the high number of approvals in 2008 was attributed to the pending files
with minimum requirement which were finalised in 2008, Hence, an increase in total number of
registered products in that year.

82
Table 4.2 Number of approved products for Gulf Cooperation Council
(GCC) States in 2008, 2009 and 2010
Countries Type of company 2008 2009 2010
Approved
products
Approved
products
Approved products
Bahrain
Gulf 5 5 51
Arab non-gulf 25 37 42
Asian 7 2 19
International 50 105 62
Total 87 149 174
Kuwait
Gulf 81 51 43
Arab non-gulf 94 38 46
Asian 17 3 5
International 224 106 116
Total 416 198 210
Oman
Gulf 52 37 56
Arab non-gulf 15 25 29
Asian 15 5 12
International 89 105 81
Total 171 172 178
Qatar
Gulf 56 57 134
Arab non-gulf 39 13 11
Asian 1 0 0
International 107 30 62
Total 203 100 207
Saudi
Arabia
Gulf 150 91 16
Arab non-gulf 25 52 19
Asian 0 0 0
International 134 132 57
Total 309 275 92
In Saudi Arabia, the decrease in the number of registered medicines in 2010 as compared to
2008 and 2009 was attributed to the transfer of the tasks from the Ministry of Health to the
Saudi Food and Drug Administration in July 2010. During the transition period, the ministry
refrained from accepting new registrations until the transfer was completed, hence the
decline in the number of products registered. In Qatar, the decrease in the number of
registered medicines in 2009 as compared with 2008 and 2010 was attributed to the limited
number of registration committee meetings in 2009. Since the number of meetings was
reduced, the number of products registered was also affected.

83
Figure 4.8 Number of approved products by the five regulatory authorities
(2008-2010)
Approval times in Gulf States
There are differences in the milestones within the approval process among the GCC States.
For example, the analysis step starts with the scientific assessment in Oman, Saudi Arabia,
and Qatar (parallel procedure), but in Kuwait and Bahrain the analytical step comes after the
scientific assessment (sequential procedure) and this could negatively impact the approval
time. The GCC authorities also waive the analytical stage for products registered in Kuwait,
Saudi Arabia, Oman, the GCC central drug registration (GCC-DR), and/or in countries with
advanced regulatory systems such as the United States Food and Drug Administration or the
European Medicines Agency (EMA).
The median approval time for all approved products in the GCC States ranged from 69 days
in Oman and Qatar in 2010, to 587 days in Saudi Arabia in 2008 (Figure 4.9). The Saudi
Arabia median approval time was two to three times the regulatory approval time in other
GCC States. This may be attributable to some products remaining in the approval process for
a longer time, or because some steps in the regulatory approval required more time to
complete or because they carry out a full review. However, upon verification with the Saudi
Arabia authority, the decrease in registration period for the three years (from 587 Day in
2008, 515 in 2009 and 439 day in 2010) was due mainly to the decrease in the number of
registration applications received by the MOH. With fewer files, the evaluation process and
decision-making were carried out in a timely manner.

84
Figure 4.9 Median approval times for all products (2008-2010)
Bahrain, recorded a decline in the registration period between 2008 and 2010. This decline in
the registration period in the year 2010 (from 264 days in 2008 to 166 days in 2010) was
attributed to the increase in the number of registration committee meetings and the increased
number of reviewers. This expedited the approval process and reduced the registration
process by 98 days.
Qatar also experienced a decrease in the registration period for the three years; 143 days in
2008, 87 Days in 2009 and 69 days in 2010. This was attributed to the increased number of
reviewers, hence expediting the approval process. By taking into account products that had
been approved by regulatory authorities in advanced countries such as the EMA and
USFDA, the local approval process becomes more streamlined and shortened. Kuwait
recorded a decline in the registration period for the same three years ranging from 346 days
in 2008 to 300 days in 2010. Oman also recorded a decline in the median approval time from
143 days in 2008 to 69 in 2010. The increase in the approval time in 2008 was due to the
development of the new regulations for approval of health and alternative products. These
new regulations created an overload for the Quality Control Laboratory, increasing the time
for the analysis of Pharmaceutical products.
Approval time for Gulf, Arab non-Gulf, Asian and International pharmaceutical
products in the GCC States
The approval times in the Gulf States varied based on the geographical location of the
manufacturer. For local companies it is easier to contact the authority and faster to reply to

85
questions asked by the authority. International company products need a longer time for
approval for many reasons, including priority given to local companies and cultural
influences. Therefore, the following four figures examine the difference in the approval time
for different types of companies in the GCC States.
Gulf companies products
The median approval time for the products from Gulf companies varied from about 70 days
in Oman and Qatar in 2008, 2009, 2010 to more than 600 days in Saudi Arabia. The longest
approval time was for Saudi Arabia at 609 days in 2008, as shown in (Figure 4.10). The
regulatory review process for medicines in Saudi Arabia is carried out in the newly
established autonomous “Saudi Food and Drug Authority” which commenced its activities in
2008. The review process in Saudi Arabia comprises thirteen steps which are critical to the
whole process (Al-Essa, 2012). As a result, the approval time decreased significantly over
the period of this study. The Saudi Arabia authority ascertained that the reduction in approval
time was partly due to fewer product registration applications received in 2008, 2009 and
2010 from the GCC companies as well as the rapid responses from these companies
regarding any clarifications. The improvement in the approval time in Oman and Qatar in
2010 was due to the positive effects of the Gulf Centralised Registration system. Qatar MOH
for example requested the Gulf companies to register their products in the GCC Centralised
system before they submit their products to the local procedure and this led to a reduction in
the time needed to register the products. In Bahrain, the increase in the number of reviewers
had a positive effect on approval times. But in Kuwait, the health authority thought the slight
increase in approval times in 2008 and 2010 was mainly attributable to the delays in the
companies‟ response to the authority‟s queries which affects the overall approval time.
Bahrain, on the other hand, recorded an increase in the duration of medicine registration from
Gulf pharmaceutical companies in 2008 (520 days), a decrease in duration recorded in 2010
and no registration of any products from Gulf companies in 2009.The decline in the
registration period in 2010 was due to the increase in number of the registration committee
meetings and in the number of reviewers. On verification, there were no specific reasons
recorded as to why there was no registration for any products from Gulf pharmaceutical
companies in 2009. One of the probable reasons might have been that the Bahraini market is
very small in size and hence not attractive for companies to register their products locally.

86
Figure 4.10 Median approval time for Gulf companies products (2008-2010)
Arab non-Gulf companies products
The median approval time varied from 91 days in Oman in 2008 to 567 days in Saudi Arabia
in 2010. The approval time for Arab non-Gulf products in Saudi Arabia was four times that
in the other GCC States (Figure 4.11). The increase in the median approval time to more than
560 days in 2009 and 2010, respectively, was due to an accumulated backlog for the
registration applications during the transition period for the newly established authority
SFDA. Furthermore, priority was also given to products from local and Gulf companies at
the expense of Arab non-Gulf companies. Saudi Arabia also cited that the slow responses
from Arab companies further delayed the approval process. In Kuwait, the median approval
time was 559, 437 and 450 days in 2008, 2009 and 2010 respectively and the increase in
2008 was mainly due to lack of follow-up from the company to submit more documents or
answer questions asked by the authority. The reduction by 90 days between 2008 and 2010
was mainly due to an increase in the number of reviewers in the registration section. The
approval time in Bahrain, Oman and Qatar varied from 90 days to 531 (Figure 4.11). Qatar
recorded a reduction in the duration of registration of medicines from Arab non-Gulf
pharmaceutical companies and the median approval time is more constant over the 3 year
period. This is attributed to an initiative by the Qatari MOH requesting the companies to
register their products in the GCC Centralised system before they submit their products to the
local system which led to the reduction in the approval time.

87
Figure 4.11 Median approval time for Arab non-Gulf companies products from
(2008-2010)
The increase in the approval time in Oman in 2009 and 2010 was mainly attributable to the
delays of the company responses to the authority queries which affects the overall approval
time. A significant drop in the registration timelines in Bahrain for the products from Arab
non-Gulf companies was due to the decision from the Ministry of Health (MOH) to facilitate
the procedures for drug registration for products approved by any reference authorities such
as USFDA, EMA or SFDA.
International companies products
The median approval time in 2008 for products submitted by International companies varied
from 143 days in Qatar to 587 days in Saudi Arabia (Figure 4.12). The approval time for
Saudi Arabia is two to three times that of the other countries, with 587, 473 and 434 days in
2008, 2009 and 2010, respectively, which may reflect the fact that they carry out a full
review. The mean fluctuated with an increase in 2008, mainly due to priority given to local
and Gulf companies, which had a negative effect on the approval time for International
company products. However, between 2009 and 2010, there was an increase in the median
approval time, mainly due to products remaining in the approval process for a longer time.
The median approval time for other GCC States varied from 70 days to 301 days, but it
decreased with 143, 110 and 96 days in 2008, 2009 and 2010 in Qatar, due to the fact that the
International companies submit complete dossiers. Furthermore, in Kuwait the reduced
approval time was mainly because the NASs do not enter the quality control analysis stage as

88
long as the sponsor has provided complete pharmaceutical documents to ensure that this
product is of the desired quality. EASs, however, are sent to the Quality Control laboratory
for sample analysis and take the companies some time to follow up the queries.
Figure 4.12 Median approval time for International companies’ products
(2008-2010)
In Bahrain, there was an increase in the duration of the registration time for medicines from
the Europe and the U.S. in the year 2009 (264 days) as compared to 164 days in 2008 and
127 days in 2010. The Bahraini authority stated that this was due to some changes in the
administration process which led to a delay in drug registrations in 2009. In Oman, there was
a decrease in the median approval time for products from International companies from 217
days in 2008 to 182 days in 2010. The reduction in approval time is due to the fact that more
products from the International companies had been registered in GCC Centralised system
which reduces the time needed to register the products locally.
Asian companies’ products
The median approval time for products submitted in 2008 by Asian companies varied from
127 days in Qatar to 864 days in Oman (Figure 4.13). Saudi Arabia did not register any
products from Asian companies during this period because these companies were unable to
fulfill some of the registration requirements such as their product must first be registered
with an advanced regulatory body such as EMA and USFDA. Similarly Qatar also did not

89
register any products from Asian companies in 2009 and 2010. Thus, only Oman, Kuwait
and Bahrain registered products from Asian countries during these periods.
Figure 4.13 Median approval time for Asian companies’ products (2008-2010)
Bahrain registered an increase in the timelines of registration for Asian pharmaceutical
companies in 2009 (664 days) as compared to 203 days in 2010. The Bahraini authority
stated that the increase was attributed to several reasons, for example the low demand for
Asian pharmaceutical products in Bahrain, the products were not registered with reference
authorities or the products did not fulfill all the registration requirements such as bio-
equivalence studies.
The approval time for Oman was 697, 864 and 216 days in 2008, 2009 and 2010,
respectively. The mean fluctuated with an increase in 2008 and 2009 mainly due to the
development of new regulations for the approval of health products and alternative medicines
and the delay in the company responses to the authority queries. While Oman and Bahrain
registered an increased in the median approval time for Asian companies, Kuwait recorded a
decline in 2009 from 380 days in 2008 to 181 days in 2009. The Kuwaiti authority stated that
this was due to the increased number of reviewers. However, in 2010 the approval time had
doubled from that in 2009.

91
DISCUSSION
The regulatory environment has changed in recent years, largely as a consequence of internal
reviews designed to increase the efficiency and quality of the approval process. At the same
time relationships have become more open both those between different national regulatory
authorities and those between industry and the agencies perhaps due to the impact of
restructuring and performance improvement initiatives.
In order to make a consistent, timely and high-quality review decision the drug registration
agency has to acquire the following key components in their quality assurance system: 1.
Standard operating procedures; 2. Guidelines for the reviewers‟; 3. Review templates; 4.
Training programs; and 5. Management review of individual files.
This study investigated the pattern of the total regulatory approval time in GCC States in
2008, 2009 and 2010 for pharmaceutical products from Gulf, Arab (non-Gulf), Asian and
International companies. It revealed that the shortest median approval time was for the
products from Gulf companies. In contrast, products submitted by Arab non-Gulf, Asian and
International companies had the longest median approval times. The median regulatory
approval time varied according to the geographical situation of the pharmaceutical
companies. Several factors led to an improvement in the median regulatory approval time in
the GCC States during the study period, such as the Gulf Central Registration which had a
positive impact on submissions from the Gulf companies.
1. Trends in the overall approval times in the Gulf States
The study data showed a downward trend in the median approval time for most of the GCC
States, during 2008 to 2010. However, the approval time for all approved products in the
GCC States during this period varied from about 60 days in Qatar and Oman (2009 and
2010), and to about 609 days in Saudi Arabia (2008).
There are many factors which can affect and explain the differences in approval time in the
GCC States. The main factor is the difference in the positions of milestones within the
approval process, for example, the analytical step starts with the scientific assessment in
Oman, Saudi Arabia, and Qatar (parallel procedure), but in Kuwait and Bahrain the
analytical step comes after the scientific assessment (sequential procedure). The study

91
identified the negative influence of the sequential procedure on the products approval times.
The speed and uniformity of the sample analysis can be achieved by fixing a time limit,
which may improve the handling of the analytical procedure along with the required quality
control tests in order to meet the target time. Furthermore, carrying out the quality control
analysis in parallel with, rather than after the scientific assessment, would be a rational
decision to avoid any impediment to patients‟ access to medicines.
Bahrain and Qatar do not specify target approval times as there are several factors involved
in their decisions. The major influencing factors are, the types of products being registered
(i.e. whether they are NASs, EASs, or therapeutically important or life saving products), the
quality of the submitted dossiers and the level of follow-up and interaction between the
pharmaceutical company and the authority. The other three authorities showed slight
differences in their overall target approval times with the shortest one in Oman being 120
days. Regulatory approval times have also been influenced by the type of assessment carried
out by different authorities. Bahrain and Kuwait conduct a verification reviews for all types
of products registered in countries with competent authorities. Saudi Arabia performs a full
review on all types of application while Oman and Qatar perform an abridged review for all
products. It must be noted that all the GCC regulatory authorities require the submission of
the Certificate of Pharmaceutical Product at one point during the registration process as this
is the most important requirement for successful completion of the approval process in the
five member states. The GCC states could take the Singapore system as an example to reduce
the overall approval time by conducting verification review for all types of medicines which
are previously authorized by at least two reference authorities, except for biological and
biotechnology products.
GCC states should seek to increase the level of funding to bring about the required expertise
and resources to conduct a more extensive review of important medicines, including
biological and biotechnology products. A clock stop is another important approach which is
not fully enforced in the GCC authorities to control the overall approval time. EMA is
obliged by the regulation to reach a decision within 210 days, although the clock is stopped if
it is necessary to ask the applicant for clarification or further supporting data (EMA, 2009).
In any case, the clock stop is an important practice that has several advantages for the review
process, namely,
1. It controls the approval time

92
2. It keeps the sponsor alert to the time limit and the consequences of delays of their
responses to the authority.
3. It improves the interaction and follow-up practices between the sponsor and the authority
4. It minimizes the backlog problem
2. The review times in the Gulf States
The review of new drug applications by regulatory agencies around the world is an
immensely complex process and the factors influencing regulatory behaviour are
numerous. According to the Centre for Drug Evaluation and Research (CDER) of FDA, the
staffing and disease politics (media coverage of diseases) play an important role in the drug
approval process. An appropriate number of staff has been the dominant influence on
review times for New Active Substances. It is essential to have a target time for the
scientific assessment and to monitor the assessment time in order to prevent delays that
may impact the final approval time. In the US FDA reviewers are under constant pressure
to meet time goals. They do not only review new drug applications (NDAs), but also other
key documents submitted by sponsors, some of which also have time goals attached. At the
same time, reviewers must provide advice to sponsors and stay abreast of the latest
scientific advances in their fields. The results of a study on the US FDA‟s review process
for NDAs, revealed that the allotted six months priority review and ten months standard
reviews were found to be inadequate due to concerns of time pressure on the FDA
reviewers which ultimately required the agency to hire close to an additional 300
employees within five years with funds from user fees (Rehnquist, 2003).
The main factors (Barrocliffes et al. 1994, Donnelly et al 1996) which may impact on
review times are the harmonisation of technical requirements, increase in dialogue between
industry and authorities, restructuring and reorganization of authorities, introduction by
authorities of user fees and target review times. This suggests that differences in review
times between competent authorities may be related to the way in which the review is
managed. The time taken from reaching a positive opinion by the scientific committee to
the final approval varies considerably across the region taking less than 30 days in Kuwait
and Oman and less than 90 days in Bahrain, Qatar and Saudi Arabia. This is the time
period to complete the final administrative procedures before granting the registration
approval in each country. It is important for the authorities to consider improving their

93
internal bureaucratic procedures that cause unnecessary delays in the authorization time
without any justifiable reason related to the quality of the submitted registration dossiers.
Most regulatory authorities have procedures for accelerated reviews like the fast track
procedure in Europe, based solely on potential gain time from conditional licensing or from
authorisation under exceptional circumstances. Hence the need for the product does not seem
to be a determinant of the speed of the review. The utilisation of abridged reviews, mutual
recognition and assessment review sharing among regulatory authorities may enhance the
efficiency of scientific reviews. Initiatives such as increased dialogue, production of early
guidelines and training of regulators may enable increased efficiency. The speed of review
may depend on the need for the product, the quality of the dossier and the efficiency of the
regulatory process. Still there is little evidence to show that the need for the product is a
determinant of the review speed or the time to bring the product to the market. Accelerated
reviews, fast track procedures and priority assessments constitute the efficiency of the
regulatory agency (Lumley et al 1996). Facilities for accelerating the development of certain
products for licensing under exceptional circumstances also are added measures in the
regulatory process. Sometimes a fast track procedure means that under certain very
specialized circumstances the regulatory authorities will assess a dossier ahead of time. The
criteria should be solely those of a significant potential gain for public health. Adherence to
strict time limits has been one of the quantitative measures for the success of a regulatory
authority. This has been achieved partly through the introduction of a pre-submission phase
and clear dialogue (sound scientific advice) with the applicant. Performance indicators like
the Application Tracking Systems (ATS), monthly publication of decision tables, quality
management systems, questionnaires, may accelerate the overall assessment and the drug
registration process in the GCC states.
3. The influence of the type of Companies on approval times
Analysis of the 2008 data showed that, the approval time for the products of Gulf companies
to be the shortest followed by Arab non-Gulf and International companies in most of the Gulf
countries. The main reason for this is the priority given to local manufacturers in the Gulf
States. In addition, the rapid responses from these companies also help to expedite the
approval processes. The local manufacturers have an unwritten priority in all regulatory
review processes, as a part of governmental support to encourage local industry. Such

94
priority treatment reflects positively on approval times. The approval time for products from
the Gulf companies was the shortest, compared to other types of companies. The median
approval time in some authorities in 2010 was similar e.g. in Qatar there was no significant
difference between types of companies nevertheless the approval time for local products still
showed continuous improvement during 2008, 2009 and 2010 at 102 days, 86 days and 69
days, respectively. The longest approval time was for products from Asian countries in Oman
which was 864 days in 2009 and 395 days in Kuwait in 2010. An unwritten review priority
has always been given not only to products from the Gulf companies but also to products
indicated for serious, life-threatening conditions in the GCC states. This is in line with the
Canadian priority review for medicines and medical device applications intended for the
treatment, prevention or diagnosis of serious, life threatening or severely debilitating
illnesses or conditions. Their priority review is specifically applicable where no product is
currently marketed in Canada and/or where a new product represents a significant increase in
efficacy and/or significant decrease in risk such that the overall benefit-risk profile is better
than that of existing therapies (Canadian Review process, 2006). The longer approval time
for the products from local companies in Saudi Arabia, compared to other GCC States, may
be due to the sequential approval process followed in Saudi Arabia. However, the median
approval time for products from Gulf companies in 2010 in Oman was about 79 days and
Qatar it was 70. These shorter approval times may be due to the parallel approval procedure
used in Oman and Qatar. It is important to note that the scientific assessments in Saudi
Arabia are carried out by independent specialist academic staff which could lead to more
difficult questions and consequently require more time to respond on the part of
pharmaceutical companies, while in the other GCC States, the scientific assessments are
carried out by internal pharmacists.
In addition to the priority procedure, there are many other factors which affect the approval
time of local companies positively, like the ease of establishing contact with key individuals
in the authority and a rapid response to queries raised by the authority. Overall the median
approval time is reduced radically for all products from all regions. The main reasons for
this decrease in approval time between 2008 and 2010 in the Gulf States are due to the
positive effect of the Gulf Central Registration, a rise in the number of reviewers in some
GCC drug authorities and the parallel procedure used in the regulatory approval review
process.

95
SUMMARY
The findings from this study showed a downward trend in the median approval time
for most of the GCC States between 2008 and 2010.
The main reasons for the decrease in approval time in the Gulf States are due to the
positive effect of the Gulf Central Registration, the rise in the number of reviewers in
some GCC drug authorities and the parallel procedure used in the regulatory approval
review process.
Regulatory approval times in the GCC states have been influenced by the type of
assessment carried out by different authorities.
Clock stop is not fully enforced in the GCC authorities to control the overall approval
time.
The GCC states should seek to increase the level of funding to bring about the
required expertise and resources to conduct a more extensive review of important
medicines, biological and biotechnology products.
Performance indicators such as Application Tracking Systems, monthly publication
of decisions and quality management systems, may accelerate the overall assessment
and drug registration process in the GCC states.

96
CHAPTER 5
An Evaluation of the GCC Centralised
Regulatory Review Process

97
INTRODUCTION
The approval of medicines in different countries across the world has been and still is
performed by regulatory agencies according to their national regulations, although, attempts
to harmonise the regulations have been made for several years within the GCC region. The
harmonisation of the regulatory review processes in the GCC States was initiated following
the issuance of the GCC Health Ministers‟ Council Decree No. 8 in 1976 regarding the
formation of a study group to report on how a centralised registration review system should
be established to monitor the marketing of medicines and develop common guidelines for the
participating authorities (Al-Essa, 2011).
This was followed by a series of GCC Ministerial Decrees relating to the establishment of a
centralised registration system which was not approved until the Kingdom of Bahrain
submitted a proposal for the formation of a “Central Committee for the Gulf States” to
register pharmaceutical companies and their products. This committee ensures that the
pharmaceutical companies apply satisfactory standards to guarantee manufacturing of high
quality, safe and effective medicines and to standardise their regulations with regards to
medicines importation practices in the Gulf States.
The Centralised Procedure for the registration of pharmaceutical companies and medicinal
products has been established under the Executive Board of the Health Ministers Council for
GCC States. The Executive Board is chaired by the Director General, who is responsible for
supervising the work of the board and following up the resolutions and recommendations of
the Health Ministers‟ Council, assisted by technical, administrative and financial
departments.
The Gulf Centralised Committee for Drug Registration (GCC-DR) was formed in May 1999
and included Bahrain, Kuwait, Oman, Qatar, Saudi Arabia, the United Arab Emirates with
Yemen (joining in 2003). The GCC-DR consists of two representatives from each member
state. The main role of the committee is to register pharmaceutical companies and their
products through the joint coordination of the safety, efficacy and quality of medicinal
products.

98
In order to adopt the Central Registration System, the following two phases were proposed.
Phase 1: In the year 2000, pharmaceutical companies in the Gulf States had to register their
products using the local national system while at the same time it was mandated that they
also use the centralised system. During a period of two years the following was required:
The registration of research companies by the GCC-DR in accordance with the
centralised registration.
The registration of new chemical entities, biological and new technology products.
A priority was given to the Gulf States companies to register centrally.
The registration of products which have not been through the centralised procedure but
have been selected for the GCC international tender.
Phase II: The second phase took effect in 2002 following an evaluation of phase I and
during this phase all the companies could submit their products through the GCC procedure.
During the eleven years (1999-2010) the GCC centralised system received 1,824 medicinal
product applications and approved 1,165. This study has highlighted the different steps in the
review process in the GCC-DR system and the way in which these influenced the overall
timelines. Information was obtained to identify the practices that accelerate or delay the
marketing authorisation. Therefore this study mapped the key milestones and associated
activities and evaluated the quality measures employed by the GCC-DR process.
OBJECTIVES
The objectives of this study were to:
Appraise the regulatory review process in the GCC Central Registration operation.
Evaluate the review times for New Active Substances (NASs) and Existing Active
Substances (EASs) submitted to the GCC Centralised Registration (January 2006 to
December 2010).
Identify the strengths and weaknesses of the centralised review process.
Propose strategies that could help the GCC-DR enhance the drug review process
leading to improved patient access.

99
METHODS
Information on the total number of applications and approvals together with the timelines
from January 2006 to December 2010 were obtained directly from the Executive Board of
the Health Ministers Council for GCC States. The data included the application submission
date, the registration date and the overall review time. The products were classified into four
groups according to the location of the manufacturing companies (GCC, non-GCC Arab,
international and Asian). The data also included information on different dosage forms
(solids, semi-solids, liquids, injectables) and the therapeutic indications.
THE GULF CENTRALISED PROCEDURE REVIEW PROCESS MAP AND
MILESTONES
Currently the Executive Board is responsible for receiving the registration files and the
prescribed fee for the registration of pharmaceutical manufacturing companies and their
products. After receiving the files, the Executive Board forwards the files to all the member
states for their evaluation. Every member state should review the files and provide their
recommendations to the GCC-DR. The registration of the company is decided based on the
recommendation of the inspection team. If the company has been recommended to be
registered, the GCC-DR issues the registration certificate; otherwise the company will be
notified accordingly. The inspection team consists of three members from three different
member States. The selection of members is carried out based on certain criteria where every
member state would have an equal number of visits. Upon completion of the visit, the head
of the inspection team will prepare a visit report and it is signed by other members of the
team and then the report is submitted to the GCC-DR. Similarly, the GCC-DR forwards the
registration files of the products to each individual Member State for evaluation. The
samples are sent for analysis to one of the four laboratories (Saudi Arabia, UAE, Kuwait and
Oman) which are accredited by GCC-DR. The registration of products is determined on the
basis of the analysis reports. There are basically five steps for the regulatory authority in the
Gulf Central Registration System. Some of these steps are critical and constitute a substantial
part of the review process (Figure 5.1). The review process map indicates the main steps in
the review and approval process and identifies the key milestones for monitoring and
analysing timelines. Receipt and validation include administrative registration (reference
number) and checks on legal requirements, the status of the company, manufacturer etc. as
well as a „checklist‟ validation of the application content (e.g. technical sections, CPP status).
The pharmaceutical

111
Figure 5.1: PROCESS MAP FOR GCC CENTRALISED REGISTRATION
(A) RECEIPT OF APPLICATION BY EXECUTIVE BOARD
(B) ACCEPTED FOR REVIEW
(C) SCIENTIFIC REVIEW STARTS
(D) START OF GCC-DR PROCEDURE
(E) QUESTIONS TO COMPANY REGISTRATION REJECTION
Receipt & Validation Procedures Validation Time
7-10 days
Queuing for review Administration Time
1-2 weeks
Safety Efficacy Sample analysis
Registration Dept. in GCC Countries Quality Control Laboratory
<<< Parallel review, >>>
Questions processed by Company
Reply from Company
GCC-DR
REGISTRATION
Quality
<<2 Months>> <<4-5 months>>
Meets every 3 months
Registration Certificate
Overall registratio
n tim
e 6
-8 m
on
ths

111
companies are required to submit 16 samples of their product in addition to eight copies of
the product dossier and one original copy which would remain with the Gulf Central
Registration Office. While two separate GCC States would be chosen through a computer
program alphabetically according to the countries name, to act as rapporteurs, the other GCC
States would keep the dossiers for documentation purposes. The rapporteurs are not chosen
because of their expertise in a certain area of regulatory science, but to give equal
opportunities to all the members states in a systematic manner of selection. Similarly, the
reference laboratory chosen, to carry out the analysis, is based on the equity of the number of
dossiers distributed between these laboratories and not on the ability of these laboratories to
perform the analysis.
Hypotheses
This study examined the following hypotheses,
1. There was a significant increase in the number of products registered from 2006-
2010.
2. The highest number of products approved by GCC-DR is linked to those originating
from the companies in the GCC region.
3. There was a significant increase in the approval time between 2006-2010 in the GCC-
DR.
4. The approval time for products from GCC pharmaceutical companies was shorter
than for other companies.
RESULTS
Products considered and their characteristics
During the five year period (2006 - 2010), the number of pharmaceutical products for human
use successfully registered through the GCC Centralised Registration procedure was 413. For
the purpose of clarity the results will be presented in four parts as follows:
Part I - Number of products approved in the Gulf Centralised Procedure.
Part II - Approval time in the Gulf Centralised Procedure.
Part III - Approval time for different dosage forms
Part IV - Approval time for different therapeutic groups.
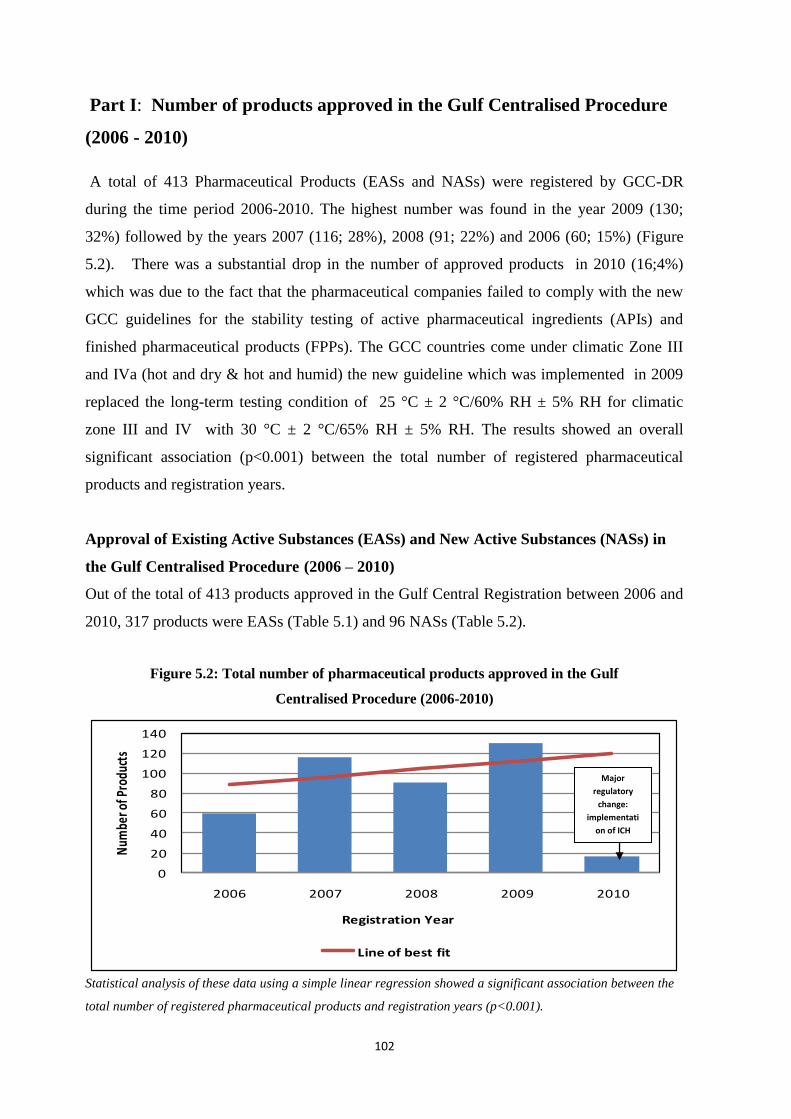
112
Part I: Number of products approved in the Gulf Centralised Procedure
(2006 - 2010)
A total of 413 Pharmaceutical Products (EASs and NASs) were registered by GCC-DR
during the time period 2006-2010. The highest number was found in the year 2009 (130;
32%) followed by the years 2007 (116; 28%), 2008 (91; 22%) and 2006 (60; 15%) (Figure
5.2). There was a substantial drop in the number of approved products in 2010 (16;4%)
which was due to the fact that the pharmaceutical companies failed to comply with the new
GCC guidelines for the stability testing of active pharmaceutical ingredients (APIs) and
finished pharmaceutical products (FPPs). The GCC countries come under climatic Zone III
and IVa (hot and dry & hot and humid) the new guideline which was implemented in 2009
replaced the long-term testing condition of 25 °C ± 2 °C/60% RH ± 5% RH for climatic
zone III and IV with 30 °C ± 2 °C/65% RH ± 5% RH. The results showed an overall
significant association (p<0.001) between the total number of registered pharmaceutical
products and registration years.
Approval of Existing Active Substances (EASs) and New Active Substances (NASs) in
the Gulf Centralised Procedure (2006 – 2010)
Out of the total of 413 products approved in the Gulf Central Registration between 2006 and
2010, 317 products were EASs (Table 5.1) and 96 NASs (Table 5.2).
Figure 5.2: Total number of pharmaceutical products approved in the Gulf
Centralised Procedure (2006-2010)
Statistical analysis of these data using a simple linear regression showed a significant association between the
total number of registered pharmaceutical products and registration years (p<0.001).
0
20
40
60
80
100
120
140
2006 2007 2008 2009 2010
Num
ber o
f Pro
duct
s
Registration Year
Line of best fit
Major
regulatory
change:
implementati
on of ICH
stability
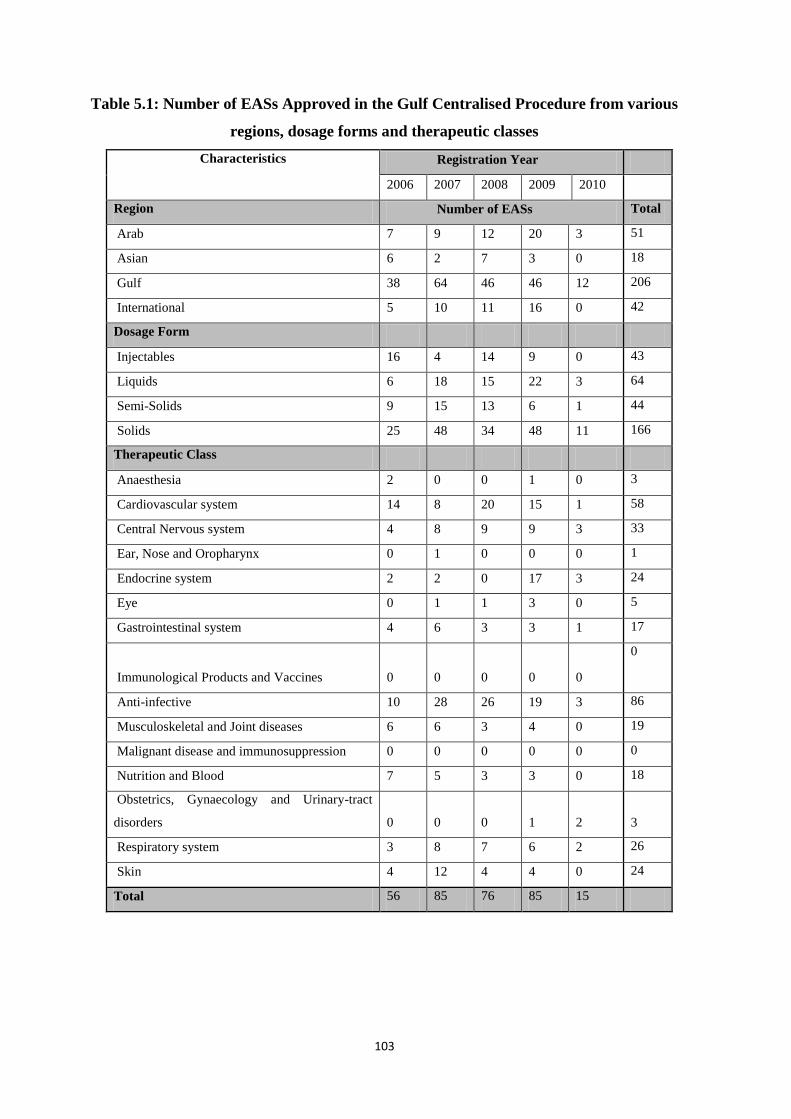
113
Table 5.1: Number of EASs Approved in the Gulf Centralised Procedure from various
regions, dosage forms and therapeutic classes
Characteristics Registration Year
2006 2007 2008 2009 2010
Region Number of EASs Total
Arab 7 9 12 20 3 51
Asian 6 2 7 3 0 18
Gulf 38 64 46 46 12 206
International 5 10 11 16 0 42
Dosage Form
Injectables 16 4 14 9 0 43
Liquids 6 18 15 22 3 64
Semi-Solids 9 15 13 6 1 44
Solids 25 48 34 48 11 166
Therapeutic Class
Anaesthesia 2 0 0 1 0 3
Cardiovascular system 14 8 20 15 1 58
Central Nervous system 4 8 9 9 3 33
Ear, Nose and Oropharynx 0 1 0 0 0 1
Endocrine system 2 2 0 17 3 24
Eye 0 1 1 3 0 5
Gastrointestinal system 4 6 3 3 1 17
Immunological Products and Vaccines 0 0 0 0 0
0
Anti-infective 10 28 26 19 3 86
Musculoskeletal and Joint diseases 6 6 3 4 0 19
Malignant disease and immunosuppression 0 0 0 0 0 0
Nutrition and Blood 7 5 3 3 0 18
Obstetrics, Gynaecology and Urinary-tract
disorders 0 0 0 1 2
3
Respiratory system 3 8 7 6 2 26
Skin 4 12 4 4 0 24
Total 56 85 76 85 15
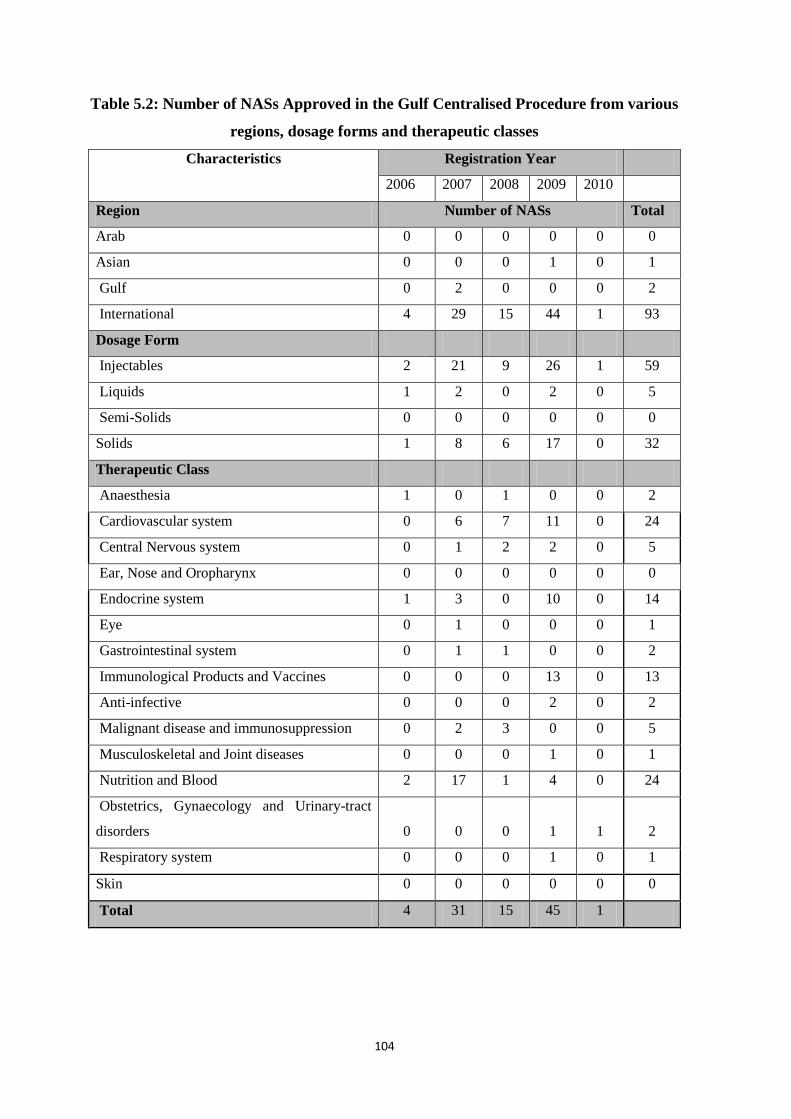
114
Table 5.2: Number of NASs Approved in the Gulf Centralised Procedure from various
regions, dosage forms and therapeutic classes
Characteristics Registration Year
2006 2007 2008 2009 2010
Region Number of NASs Total
Arab 0 0 0 0 0 0
Asian 0 0 0 1 0 1
Gulf 0 2 0 0 0 2
International 4 29 15 44 1 93
Dosage Form
Injectables 2 21 9 26 1 59
Liquids 1 2 0 2 0 5
Semi-Solids 0 0 0 0 0 0
Solids 1 8 6 17 0 32
Therapeutic Class
Anaesthesia 1 0 1 0 0 2
Cardiovascular system 0 6 7 11 0 24
Central Nervous system 0 1 2 2 0 5
Ear, Nose and Oropharynx 0 0 0 0 0 0
Endocrine system 1 3 0 10 0 14
Eye 0 1 0 0 0 1
Gastrointestinal system 0 1 1 0 0 2
Immunological Products and Vaccines 0 0 0 13 0 13
Anti-infective 0 0 0 2 0 2
Malignant disease and immunosuppression 0 2 3 0 0 5
Musculoskeletal and Joint diseases 0 0 0 1 0 1
Nutrition and Blood 2 17 1 4 0 24
Obstetrics, Gynaecology and Urinary-tract
disorders 0 0 0 1 1
2
Respiratory system 0 0 0 1 0 1
Skin 0 0 0 0 0 0
Total 4 31 15 45 1

115
There were a similar number of EASs approved centrally in 2007 and 2009 (85; 27%)
followed by 2008 (76; 24%), 2006 (56; 18%) and 2010 (15; 5%) (Figure5.3). Statistical
analysis using a simple linear regression analysis showed that the increase in the number of
registered EASs was statistically significant (p<0.001).
The highest number of NASs was registered in 2009 (45), whilst the lowest was in 2010 (1)
(Figure 5.4). Out of the 96 NASs, 93 products (97%) were found to be from international
companies. Statistical analysis using a simple linear regression showed a significant increase
in the number of registered NASs (p<0.001).
By individual regions, 208 (50%) medicines were identified as being approved from Gulf, 51
(12%) from Arab non-GCC, 19 (5%) from Asian and 135 (33%) from international
companies (Figure 5.5). Out of the total of 317 EASs approved in the Gulf Central
Registration between 2006 and 2010, by individual regions, 206 (65%) drugs were identified
as being approved from Gulf, 51 (16%) from Arab non-GCC, 18 (6%) from Asian and 42
(13%) from international companies.
Figure 5.3: Number of EASs approved in the Gulf Centralised Procedure
(2006-2010)
Statistical analysis using a simple linear regression showed a significant increase in the number of registered
EASs (p<0.001).
0
20
40
60
80
100
Num
ber o
f Pro
duct
s
Rigistration Year
Line of best fit
Major regulatory
change:
implementation
of ICH stability
guideline

116
Figure 5.4: Number of NASs approved in the Gulf Centralised Procedure
(2006-2010)
Statistical analysis using a simple linear regression showed a significant increase in the number of registered
NASs (p<0.001).
Figure 5.5: Approved pharmaceutical products (EASs & NASs) from GCC international, Arab
non-GCC and Asian companies (2006-2010)
0
50
100
150
200
250
Gulf International Arab Asian
Nu
mb
er
of
Pro
du
cts
Company type
New Active Substances Existing Active Substances

117
Part II: Approval times in the Gulf Centralised Procedure (2006 – 2010)
The total approval time for the period studied (2006 - 2010) includes all the 413 EASs and
NASs from different companies. During the five years, the median approval time ranged
from 107 days in 2006 to 265 days in 2010. There was a significant increase in the median
approval time between 2006 and 2010 which was due to several factors. These included the
limited number of meetings by the GCC-DR. although the seven member states met four to
five times a year to discuss the product review reports, on the other hand there was a steady
increase in the number of registration applications. Another reason for the delay in
registration was the assessment review process where two authorities are selected
alphabetically to review a registration dossier. There was also a lack of a standard evaluation
template for product assessment which leads to an increase in the correspondence which is
sent to the sponsor in several batches and at different times. Statistical analysis using the non
parametric test (Kruskal-Wallis test) across the five-year period showed that there was a
significant increase in the approval time for pharmaceutical products (p<0.001) (Figure 5.6)
Figure 5.6: Median approval time for all pharmaceutical products in the Gulf
Centralised Procedure (2006-2010)
0
50
100
150
200
250
300
2006 2007 2008 2009 2010
App
rova
l Tim
e(Ca
lend
er d
ays)
Registration Year
Line of best fit
Statistical analysis using non parametric test (Kruskal-Wallis test) over the five-year period showed that there
was a significant increase in the approval times for pharmaceutical products (p<0.001).

118
Approval time of Existing Active Substances (EASs) in the Gulf Centralised Procedure
(2006 - 2010)
Approval time for the 317 EASs approved in the Gulf Centralised Procedure between 2006
and 2010 varied from one year to the next. The lowest median approval time was for 2006
(114 days) and the highest median for 2010 (265 days) (Figure 5.7). Statistical analysis using
the non parametric test (Kruskal-Wallis test) across the five-year period showed that there
was a significant upward trend in the median approval time for EASs (p<0.05). In general for
the years 2006 to 2009 there is a wide range because in the centalised procedure there is no
defined target time within which the company time is also not specified (no clock stop).
However, in 2010 the range is significantly reduced because there were very few products
approved as a result of the implementation of ICH stability guideline.
Figure 5.7: Median approval time for EASs in the Gulf Centralised Procedure
(2006-2010)
Statistical analysis using the non parametric test (Kruskal-Wallis test) over the five-year period showed that
there was a significant difference in the median approval time for EASs (p<0.05)
Approval time for Existing Active Substances (EASs) by GCC, international, Arab non-
GCC and Asian companies (2006-2010)
The median approval time for EASs varied for different cohorts of companies based on
location, for example the shortest approval time was for Gulf company products (134 days)
and the longest for those of international companies (346 days). The shortest approval time

119
for Gulf company products was due to the waiver of analysis requirements. A product is
waived from analysis if it is registered in any one of the 4 GCC countries (Oman, Saudi
Arabia, United Arab Emirates or Kuwait) where the Quality Control Laboratory for these
States has the accreditation from the GCC. The median approval time for Arab companies
was 227 and 332 calendar days for Asian companies. Statistical analysis using the non
parametric test (Mann-Whitney U test) showed that there was a significant difference of
approval time between international companies (median approval time=346 days ) and Gulf
companies (median approval time=134 days) (p<0.001).
Approval time for New Active Substances (NASs) in the Gulf Centralised Procedure
(2006 - 2010)
In the year 2009, the highest number of NASs was registered (45 products) with a median of
288 days. In 2006, only four products were registered with median of 31 days. In 2007, 31
NASs were registered with a median of 138 days. In 2008, 15 products were registered with
median of 140 days. In 2010, the lowest number of products was registered (1 product only)
with a registration time of 131 days. The results obtained from performing analysis of
Kruskal-Wallis test showed that there was a highly significant upward trend in the median
approval time for NASs products between 2006 and 2010 (p<0.001). This was due to several
factors, including a limited number of meetings by the GCC-DR (only four to five times a
year) and during this period there was a steady increase in the number of registration
applications (Figure 5.8). The reason for the variation in approval times is similar to that
expressed for EASs. This variation is not acceptable and could be mitigated by introducing
target times.
Part III: Approval time for different dosage forms for all Existing Active
Substances (EASs) and New Active Substances (NASs) in the Gulf
Centralised Procedure (2006-2010)
From the 317 EASs registered with the Gulf Centralised Procedure between 2006-2010, the
lowest number constituted the injectable forms (14%) followed by the semi-solid dosage
forms ( 20% ) with the highest represented by solid dosage forms (52%). Approval times

111
Figure 5.8: Median approval time for NASs in the Gulf Centralised Procedure
(2006-2010)
Statistical analysis using the non parametric test (Kruskal-Wallis test) showed that there was a significant
difference in the median approval time for NASs between 2006 and 2010 (p<0.001).
ranged from 138 days for liquids to 350 days for injectables (Figure 5.9). In general, when
the dosage form is more complex, additional time is needed to approve the product. Special
studies and more steps are needed to analyse some products such as solutions for IV infusion
which need additional tests such as sterility and pyrogen testing.
Figure 5.9: Median approval time for different dosage forms for EASs and NASs in the Gulf
Centralised Procedure (2006-2010)
0
50
100
150
200
250
300
350
400
Injectables Liquids Solid Semi solid
App
rova
l Tim
e(D
ays)
Dosage Form
Existing Active Substances New Active Substances

111
In terms of dosage forms, out of the 96 NASs registered in the Gulf Centralised Procedure
(2006-2010), the lowest number registered was represented by the liquid dosage forms (5)
with a median approval time 128 days. The highest number registered was represented by the
injectable forms (59) with a median approval time 288 days.
Part IV: Approval times for Existing Active Substances (EASs) and New
Active Substances (NASs) by therapeutic class, (2006-2010)
The therapeutic class of 314 EASs was assessed and the lowest number was represented by
ear, nose and oropharynx preparations (1 preparation) and the highest number represented by
anti-Infective (86 preparations). The analysis of the approval time of EASs registered
between 2006 and 2010 involved the four largest therapeutic groups, namely,
The cardiovascular system (CVS)
The central nervous system (CNS)
The gastro-intestinal system
Anti-infective
The range of approval times for EASs analysed by therapeutic class varied from 206 days for
central nervous system products to 162 for anti-infective products (Figure 5.10). The trend in
approval times for EASs from different therapeutic groups was attributed to the GCC-DR
assessment requirements depending on the nature of the products which may include the
need for both clinical and bioequivalent studies and the prolongation of the analytical time.
Ninety-six NASs were approved between 2006-2010 and the lowest number was represented
by the eye, musculoskeletal and joint disease and respiratory system preparations (one
preparation) whereas, the highest number was represented by cardiovascular and nutrition
and blood preparations (24 preparations). The median approval times for NASs analysed by
the main therapeutic classes ranged from 274 days for anti-infective products to 92 for
gastro-intestinal products. The main factor for the variation in the approval time for different
therapeutic groups was the nature of the products evaluated.

112
Figure 5.10: Median approval time for different therapeutic class for EASs and NASs in the
Gulf Centralised Procedure (2006-2010)
DISCUSSION
This study investigated the pattern of total regulatory approval times in the Gulf Centralised
Procedure over the period from 2006 to 2010. It also addressed the various factors which
may have a positive or negative effect on the total approval times. It showed that the total
number approved in the Gulf Centralised Procedure was 413 including both Existing Active
Substances (EASs) and New Active Substances (NASs). The outcomes revealed that the
largest number of medicines submitted for review and approved by GCC-DR occurred in
2009. The study examined four hypotheses and their respective findings are discussed below.
Hypothesis 1: There was a significant increase in the number of products registered
from 2006 to 2010.
The highest percentage of approved products was in the year 2009. There was a considerable
decrease in the number of approved products in 2010. This was due to the fact that the
pharmaceutical companies failed to comply with the new GCC guideline for stability testing
of active pharmaceutical ingredients (APIs) and finished pharmaceutical products (FPPs).
The new guideline was a major challenge to most of the pharmaceutical companies leading to
the rejection of those which failed to meet the new stability study specifications. Other
factors influencing the number of approved products in 2010 were that the Saudi FDA began
to evaluate all the products in addition to the two rapporteurs from the Gulf States designated
0
50
100
150
200
250
300
Cardiovascular system
Central Nervous system
Gastrointestinal system
Anti-infective
Appr
oval
Tim
e (D
ays)
Therapeutic Class
Existing Active Substances(EASs) New Active Substances (NASs)

113
to review the products. This resulted in a large number of queries raised from the Saudi Food
and Drug administration (SFDA) which substantially delayed the review process and there
was a significant decrease in the number of products approved in 2010. However, despite this
change in 2010, this hypothesis was accepted.
Hypothesis 2: The highest number of products approved by GCC-DR is linked to those
originating from the companies in the GCC region.
The number of products approved during the period of the study varied for different
companies based on their geographical location. The highest percentage of products
approved was from the Gulf companies, followed by Arab companies, international
companies and finally the Asian companies. However, the highest number of products
approved from GCC companies could be due to several factors including; that the GCC
manufacturers increased their efforts to obtain the GCC-DR approval in order to facilitate
entry into the international tender which is the common purchase route by GCC states. This
has an impact on the number of submissions and approvals in GCC-DR. Other factors that
might have also contributed to the increased number of products approved by GCC-DR from
GCC companies was the decision to waive the requirement to submit full time stability
studies during the first submission. The GCC manufacturers need to submit only one year
full time stability data during the first submission. However, non-GCC manufacturing
companies need to submit full time stability studies for the whole shelf life. This is because
the GCC States are making efforts to support and improve their local manufacturing
capabilities and the production capacity for the local population. The lower number of
products approved by GCC-DR was from non-GCC manufacturing companies due to the
strict GCC regulations that require evidence of registration in countries with developed
regulatory systems. Therefore this hypothesis was accepted.
Hypothesis 3: There was a significant increase in the approval time between 2006-2010
in the GCC-DR.
During the five year period of the study (2006 – 2010), the median approval time
significantly increased due to several factors which included the limited number of meetings
by the GCC-DR and the increased number of applications for registration. Other factors that
delayed the registration were the assessment review process where two authorities are

114
selected alphabetically to review the registration dossier. In addition, the lack of a standard
evaluation template for product assessment leads to an increase in the correspondence which
is sent to the sponsor in several batches and at different times. It is recommended to have one
standard assessment template and rather than selecting the reviewing authorities
alphabetically, the dossier should be sent to all countries and all assessments combined into
one final report on which the final decision in the GCC-DR could be based. A clock-stop
system is not enforced in the Gulf Centralised Procedure and this delays the company
response to queries from the GCC Central registration committee. The implementation of a
clock-stop will ensure the sponsor meets the deadline and would allow more time for the
reviewers to complete the assessment of the submitted data. The sample analysis stage is an
essential part of the review process that impacts on the overall approval time. It is completed
after the scientific assessment as the outcome of the sample analysis affects the final approval
decision. Carrying out the quality control analysis in parallel, rather than after the scientific
assessment, would avoid the impact in the overall registration time. The queuing process is
straightforward and allows appropriate handling of the registration dossiers in an organised
manner. However, the lack of regular monitoring of queue time could lead to a backlog.
Managing the priority review is another important issue that needs recognition by GCC-DR
and should be dealt with according to set guidelines and SOPs that clearly specify the
conditions under which products can be taken out of the queue for such review. Therefore,
adequate resources and electronic handling of the queuing process should be provided to
support accurate follow-up of the pending dossiers, priority reviews and fast-track products.
On the basis of these finding this hypothesis has been accepted.
Hypothesis 4: The approval time for products from GCC pharmaceutical companies
was shorter than for other companies.
The approval time for pharmaceutical products varied for different cohorts of companies
based on their location, for example the shortest approval time was for the Gulf company
products and the longest for international companies and this was due to the waiver of
analysis requirement. The analysis is waived for a product if it is registered in any one of the
four GCC countries (Oman Saudi Arabia, United Arab Emirates or Kuwait) where the
Quality Control Laboratory has the accreditation from the GCC. There are other factors
which have a positive effect on the approval time for Gulf companies but have a negative
effect on the international, Arab non-GCC and Asian companies.

115
This includes the geographical proximity of the company to the authority which leads to an
effective interaction and thus a faster response to any question asked by the authority.
Additionally, most locally manufactured products are EASs and do not need very much
consultation during the scientific review compared to NASs.
However, a priority procedure is a common practice in many authorities, the rapid approval
time is also due to the GCC States making efforts; particularly (Oman, Saudi Arabia, UAE,
and Kuwait) to improve their local manufacturing capabilities and the production capacity for
the local population (Al-Essa, 2011). The data supported this hypothesis and therefore it was
accepted.
For the first time this study has examined the centralised regulatory review process in the
Gulf region and identified which steps within the process take the most time. In addition, it
has highlighted the factors which may improve or delay the approval process resulting in
accelerating patients' access to medicines. Furthermore these results may improve the
registration process thereby saving time and resources.
SUMMARY
There was a significant upward trend in the regulatory approval times in the Gulf
Centralised Registration process between 2006 and 2010.
The limited number of meetings per year led to a delay in the feedback for the
submitted files to GCC-DR by the companies.
Further correspondence after registration should be dealt with by individual local
health authorities rather than centrally.
The reference laboratory chosen to carry out the analysis is based on equity of the
number of dossiers distributed between these laboratories, not on the ability of these
laboratories to perform the analysis and that can lead to an increase in the analysis
period.
There was no clock-stop system and this delayed the company response to queries
from the GCC Central registration committee.
The re-registration (renewal) of pharmaceutical companies and their products led to a
reduction in the number of new product applications reviewed by GCC-DR in 2010.

116
The lack of a standard assessment template for product evaluation leads to a number
of requests for additional data at different stages of registration. A standard template
could possibly avoid such delays and lead to more consistent outcomes.
Using information technology and e-mails could speed up the registration process
rather than the manual exchange of the product registration files between the
Executive Office and the member states.

117
CHAPTER 6
An Evaluation of the Gulf States Views and
Experiences with GCC Centralised Procedure

118
INTRODUCTION
The historical implementation of the central registration system was subject to several
criticisms with challenges from both the pharmaceutical industry as well as government. The
GCC-DR received 1,824 medicinal product applications out of which 1,165 (64%)
applications were approved during the eleven years from 1999 to 2010. The pharmaceutical
industry was apprehensive about whether the GCC-DR system would be a hindrance to the
timely approval of medicines in the region while the government officials were concerned
about losing sovereignty to the centralised authority (Hashan, 2005). The collaborative
efforts between the member countries however proved to be effective and substantiated the
support of the GCC-DR system for each GCC country improving the regulatory approval
processes and operation efficiencies at the national level.
Two members from each of the seven GCC countries represent the GCC Central Drug
Registration (GCC-DR) Committee with the current procedure in force and two member
countries are chosen to review a registration dossier. The selection of those countries is
unbiased and purely based on alphabetical order and all the GCC countries are equally
responsible for evaluating the quality, safety and efficacy of medicines. Thus, all seven GCC
countries consisting of Bahrain, Kuwait, Oman, Qatar, Saudi Arabia, UAE and Yemen are
provided with copies of the product registration dossier for their individual assessments. The
member countries meet four to five times a year to discuss the product review reports issued
by the reviewers from each country and an approval decision is made by common agreement.
The only studies that have addressed the GCC region was the one reported by Hashan in
2005 and later by Al-Essa in 2011. They reported that there are three common phases in the
GCC regulatory review process: submission, evaluation, and authorisation. The similarities
and differences in the key milestones and activities in the GCC states made it possible to
propose a new process, which could underpin the degree of standardisation required for a
strong regulatory body that facilitates an effective drug approval process for the region (Al-
Essa,2011). These are the key components essential for the development of a strategic plan
for strengthening drug regulations in the Gulf region. This chapter evaluates the experiences
and opinions of all seven Gulf States regulatory authorities with the GCC Central
Registration Process.

119
OBJECTIVES
The main objectives of this study were to:
Evaluate the regulatory views and experiences of all seven Gulf States with respect to the
Centralised Procedure.
Appraise the barriers and opportunities to the full implementation of the Gulf Centralised
Procedure.
Make recommendations to improve the Gulf Centralised system as well as to identify its
future strategy based on the views of the national regulatory authorities.
METHODS
There is a lack of information and research on the GCC Centralised Registration Procedure
and therefore, with this limited information, there is a need to develop suitable measurement
tools in order to obtain the relevant data required for this study. The development of the
assessment instrument was carried out over 5 phases;
Phase 1 – Identify target population and item generation
Phase 2 – Development of themes and item reduction
Phase 3 – Development of the first version of the instrument, Gulf Assessment of
Centralised Procedure (GACP)
Phase 4 – Pilot study
Phase 5 – Development of the final version of the GACP
This final version of the study instrument was sent to the seven Gulf States and followed up
after one month.
RESULTS
This section presents the results from the development of the GACP and the views of the
seven GCC regulatory authorities. For the purpose of clarity the results will presented in two
parts: Part I - The development of the study instrument (GACP); and Part II - The views of
seven Gulf states about the Centralised Procedure.

121
Part I: The Development of the Study Instrument (GACP)
Phase 1 – Target population and item generation
An informal interaction and dialogue was held with the members of the GCC-DR which
consist of two individuals (The head of the agency the head of the registration department)
from each of the seven member states in the Gulf region. This allowed the generation of
items for the development of the GACP. In addition, the relevant literature was also reviewed
to inform this phase of the development.
Phase 2 – Development of themes and item reduction
There were a total of five themes emerging from the item generation phase based on
the objectives of the study. The first version of the questionnaire comprised 5 sets of
questions pertaining to the following headings;
Theme 1- Value of centralised procedure consisting of seventeen sub-themes.
Theme 2- Utilization of resources consisting of five sub-themes;
Theme 3- Regulatory expertise consisting of eight sub-themes;
Theme 4- Importance of CP consisting of five sub-themes;
Theme 5- General observations consisting of seven sub-themes.
Phase 3 – Development of the first version of the instrument: Gulf Assessment
of Central Procedure (GACP)
Any item that was mentioned by only one individual was not included in the themes and sub-
themes unless these were country or regulatory authority specific items. Having selected the
themes and sub-themes, these were put in an appropriate structured questionnaire. These
were then reviewed by an expert panel using four appropriate parameters; namely; language
clarity, completeness of the statement, appropriate relevance and scaling. Account was also
taken with regard to the length of the items and their readability.
Phase 4 - Pilot study
The purpose of the pilot study was to assess the practicality, feasibility, applicability and
relevance of the study instrument to assessing the Gulf Centralised Procedure. Having
developed a structured questionnaire it was sent to the head of agency in two GCC States;

121
namely Oman and the UAE. Feedback on the first version of the questionnaire was obtained
from Oman and UAE Drug Control Authorities. The pilot study clearly indicated that such a
study into the GCC Central Registration Procedure was essential to bring about
standardisation and harmonisation of procedures. This would inevitably improve the
registration processes for both the applicants as well as the approving authorities.
Based on the feedback from the pilot study, the following item was deleted.
Part 1 – Question 1.1.7: Do you think your agency has the necessary expertise for the
Centralised Procedure and are you willing to contribute?
The following item was added in Part 1 of the questionnaire.
Part 1 – Question 1.1.8: Is your agency willing to contribute to the revision of these
guidelines?
Finally at the end of the questionnaire, six simple questions were added to enable feedback
and opinions about the questionnaire that was used to obtain the information from each of the
Drug Regulatory Authorities in the GCC. This feedback was critical in order to design more
effective assessment instruments for future studies (Figure 6.1).
Phase 5 – Development of the final version of the GACP
Following the pilot study and implementation of the changes suggested by the participating
regulatory authorities, the development of GACP was completed. Thus, the final version of
the instrument consists of 42 items grouped into five major categories. In addition it includes
an introductory page collecting the respondents' personal details (Figure 6.2). The final
version of the questionnaire was sent to all seven Gulf States with a clear guideline on the
aims and objectives of the study and a confidentiality note that would ascertain the
confidential procedure for the collection of data to prevent the identification of individual
respondents. Generally the structure of the questionnaire did not differ from that used in the
pilot study. Only minor adjustments and refinements were carried out to make the final
questionnaire more user friendly and easily understood by the respondents.

122
Figure 6.1

123
Figure 6.2

124

125

126

127

128

129

131
The final version of the questionnaire was well received by all the Drug Regulatory
Authorities in the region. They successfully completed the questionnaire and submitted the
completed documents in good time. Their full co-operation was significant for the success of
this study.
Part II: The views of seven Gulf States about the Centralised Procedure
The regulatory authorities who are responsible for the regulation of pharmaceutical products
in the seven Gulf States participated in the survey and a 100% response rating was achieved.
(Table 6.1).
Table 6.1 - List of the seven participating authorities in the Gulf Cooperation Council (GCC)
States
Position Agency Country
Director, Pharmacy & Drug Control Ministry of Health Bahrain
Drug Registration and Release Superintendent Drug and Food Control -
Ministry of Health
Kuwait
Director General of Pharmaceutical Affairs &
Drug Control
Directorate General of
Pharmaceutical Affairs and
Drug Control- Ministry of
Health
Oman
Head of Drug Registration Section Drug Control Department-
Supreme Council of Health
Qatar
Executive Director of Licensing Department -
Drug Sector
Saudi Food and Drug
Authority
Kingdom of Saudi
Arabia (KSA)
Head of Registration and Pricing Department
Drug Control Department -
Ministry of Health
UAE
Member of the board council Supreme board of drugs and
medical appliances
Yemen
Section 1 - Value of the Centralised Procedure (CP)
The complete responses from all GCC member states indicated that the Centralised
Procedure (CP) had indeed contributed to an effective system for authorising medicinal
products for the GCC. This was ascertained from their views that the GCC-DR achieved its
main objective, by providing the best possible scientific opinion for the CP. They also agreed
that the CP has appropriate guidelines for regulatory review in the GCC-DR and that the

131
quality of the CP decision-making is satisfactory to good as compared to the national system.
The CP which has been put in place since 1999 is clearly still functioning well as six of the
member countries indicated that the quality of the CP Decision-Making process is good.
Only Saudi Arabia rated the quality of processes as satisfactory.
This signifies that all GCC member countries support the centralised procedure in registering
pharmaceutical products. With the strong support for the CP, four member countries
(Kuwait, Saudi Arabia, UAE and Yemen) indicated that the CP should be extended to other
products to include herbal products, health products and medical devices while Bahrain,
Oman, and Qatar reported that CP procedure should not be extended to other products
(Figure 6.3).
Figure 6.3 The products which could potentially be registered under the Centralised Procedure
The suggested range for the approval timeline for products reviewed in the Centralised
Procedure should be between 6-12 months as indicated by four of the member countries
(Oman, Saudi Arabia, UAE, and Yemen) while Bahrain, Kuwait and Qatar indicated that it
should be 3-6 months (Figure 6.4). The latest data available (2010) indicated that the median
approval time for the Centralised Procedure was nine months (chapter 5).

132
Figure 6.4 Proposed approval timeline for products in Centralised procedure
Responses for GCC authorities with regard to improving the CP
Registration process and timelines
The long approval time could be the main obstacle in advocating the CP as the main route for
registration of products in the region. As a result, five of the member countries (Bahrain,
Kuwait, Oman, Saudi Arabia and UAE) agreed that the CP process could be shortened,
although two (Yemen and Qatar) indicated otherwise (Figure 6.5). Bahrain and UAE
indicated that the CP approval time can be reduced by following the exact rules and
regulations in the guidelines by all sectors. In addition, if the verifications were carried out at
the time when the applications were submitted, the time line could be reduced. UAE also
added that another way would be for the reviewing country to send the applications with
preliminary assessment before the committee meets for their discussion as the committee
sees the assessment for the first time at their meeting. To reduce the timeline further, Oman
stated that the member states should not demand more requirements in order to avoid
delaying the whole process; but they should encourage the sponsors to respond expeditiously
to any such requests and by increasing the number of meetings. Kuwait stated that adopting
mutual recognition from mature regulatory authorities would shorten the approval time.
Saudi Arabia, on the other hand, advocated parallel national approvals in the Gulf could also
expedite the process.

133
Figure 6.5 – Responses for the GCC authorities with regard to improving the GCC Centralised Procedure

134
Bahrain, Oman, Saudi Arabia and UAE, agreed that certain aspects of the registration
procedure could be simplified to reduce the duration of the CP process. Bahrain stated
that this could be done by developing a written policy for fast track registration, while
Saudi Arabia and UAE advocated the implementation of electronic communication along
with an online submission. Oman stated that the countries must consolidate all the
requests for additional information to be sent to the sponsors as one single batch instead
of in several batches and at different times.
Centralised Procedure guidelines
Bahrain, Kuwait, Oman, Qatar, and Yemen agreed that the CP has appropriate guidelines
for the regulatory review in GCC-DR while UAE and Saudi Arabia report that currently
the guidelines are not adequate. Bahrain, Kuwait, Qatar, Saudi Arabia, and UAE,
indicated that some current guidelines need to be revised or updated. The areas
recommended include GMP inspections, generics registration, contract manufacturing
and biological / biosimilar registration. These guidelines should also be revised on a
regular basis according to international requirements. All the member countries with the
exception of Yemen indicated that they would be willing to contribute to the revisions of
CP guidelines.
GMP Inspections
All member countries indicated their concerns about GMP inspections. Six of the member
countries reported on the quality of expertise for GMP inspectors as well as the quality of
the reports. UAE cited other causes which included the inconsistency of the inspections
and decision-making with regard to classification of defects and time given for corrective
actions and their follow-up.
The effectiveness and efficiency of the GCC-DR
Only Qatar and Oman indicated that the organisation and administration of the GCC-DR
are effective and efficient. Bahrain, Kuwait, Saudi Arabia, UAE, and Yemen reported that
the organisation and administration of the GCC-DR is not effective and efficient. Bahrain,
Saudi Arabia, UAE and Yemen cited the voting system as the contributing factor to the
ineffectiveness which in turn affects the efficiency of the GCC-DR system. Kuwait cited
that choosing the countries to conduct the evaluation affected the effectiveness and

135
efficiency. In addition, Kuwait indicated that there is political pressure in some Gulf
States for special consideration to be given to local manufacturers. Bahrain also added
that the SOP for the voting system, which requires total agreement between all member
states, should be written into the policy document.
Contribution to GCC-DR
Four member states (Bahrain, Kuwait, Qatar and Yemen) considered that every agency
made an adequate and appropriate contribution to GCC-DR while the remaining three
member states (Oman, Saudi Arabia and UAE) believe that all agencies did not make an
adequate contribution.
Assessment of Centralised Procedure
In general the GCC Drug Regulatory Authorities are satisfied with the GCC-DR
assessment procedure, implementation of the regulatory guidelines and the availability of
the scientific and technical expertise as is shown in Figure 6.6.
Figure 6.6 – Member states assessment of the Centralised Procedure
However, only two member countries (UAE and Yemen) stated that the quality of the CP
decision-making is better that of their national system (Figure 6.6) while four members
(Bahrain, Kuwait, Oman and Qatar) indicated that it is about the same as their national
system. On the other hand, Saudi Arabia indicated that the quality of the CP decision-making

136
is worse than that of their national system and this is certainly a cause of concern that needs
to be further investigated.
GCC-DR Secretariat activities
The GCC-DR secretariat activities were also evaluated to ascertain the quality and
deficiency, if any, within the organisation. Five member countries (Bahrain, Kuwait, Oman,
Qatar and UAE) indicated that the level of interactions between the GCC-DR Secretariat and
the GCC-DR Committee was good to excellent. Only two member countries (Saudi Arabia
and Yemen) stated that it was only satisfactory. However, all member states, with the
exception of Qatar, indicated that the GCC-DR Secretariat‟s support to the GCC-DR
Committee should be increased (Figure 6.7).
Figure 6.7– Level of interaction between GCC-DR Secretariat and the Committee
The member states added that the Secretariat‟s support could be enhanced by increasing the
number of staff and improving the follow up system between the members. They also
recommended that there should be expert pharmacists at the Secretariat‟s office to validate
the submitted files and this would ensure that the approval process is streamlined. This view
was also reiterated by another member country who stated that the Secretariat needed to
include one or two pharmacists who have enough experience in drug registration and GMP
inspections. Furthermore, since it is an administrative issue, more scientific staff are needed
to organise the dossiers distribution, follow up the status of the companies, and communicate
with other GCC countries members.

137
Section 2 – Utilization of Resources
The key issues explored here centred on the allocation of resources to various activities
carried out at the national or centralised level. All member states indicated that limited
resources did pose moderate problems for their agency in relation to their activities with the
Centralised Procedure. However, only Kuwait, Qatar, Saudi Arabia and UAE indicated that
they did not lack the expertise required for the assessment of the Centralised Procedure
applications. Bahrain, Oman and Yemen stated that they did lack the necessary expertise and
that they would be able to overcome this issue by increasing their budget and engaging the
resources required.
Further information was gathered on how the GCC States evaluated their allocation of
resources depending on the various CP activities. Only Oman, Saudi Arabia and UAE
conducted GMP inspections at both national and centralised level (Figure 6.8). Generally
there is an increasing trend for the conduct of GMP inspections at a centralised level.
Figure 6.8 – Number of GMP Inspections conducted by GCC (2008 - 2010)
Based on the data collected from Oman, Qatar and Saudi Arabia (Figure 6.9), it could be
ascertained that the number of product evaluations at a national level is more than those
carried out under the CP. This indicates that most pharmaceutical companies prefer to receive
approval at a national level rather than through the CP. This is further reinforced by the fact
that half of the states indicated that the current CP evaluation system would not be
sustainable due to the lack of resources, together with the current expertise and the heavy
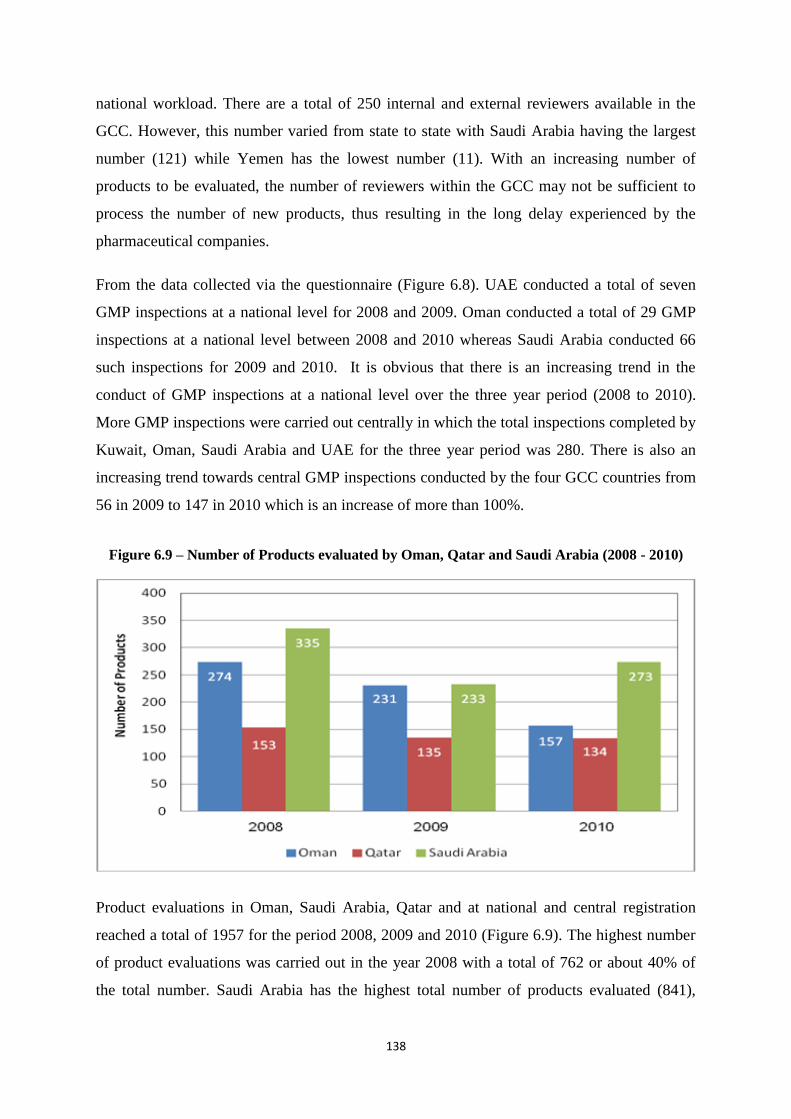
138
national workload. There are a total of 250 internal and external reviewers available in the
GCC. However, this number varied from state to state with Saudi Arabia having the largest
number (121) while Yemen has the lowest number (11). With an increasing number of
products to be evaluated, the number of reviewers within the GCC may not be sufficient to
process the number of new products, thus resulting in the long delay experienced by the
pharmaceutical companies.
From the data collected via the questionnaire (Figure 6.8). UAE conducted a total of seven
GMP inspections at a national level for 2008 and 2009. Oman conducted a total of 29 GMP
inspections at a national level between 2008 and 2010 whereas Saudi Arabia conducted 66
such inspections for 2009 and 2010. It is obvious that there is an increasing trend in the
conduct of GMP inspections at a national level over the three year period (2008 to 2010).
More GMP inspections were carried out centrally in which the total inspections completed by
Kuwait, Oman, Saudi Arabia and UAE for the three year period was 280. There is also an
increasing trend towards central GMP inspections conducted by the four GCC countries from
56 in 2009 to 147 in 2010 which is an increase of more than 100%.
Figure 6.9 – Number of Products evaluated by Oman, Qatar and Saudi Arabia (2008 - 2010)
Product evaluations in Oman, Saudi Arabia, Qatar and at national and central registration
reached a total of 1957 for the period 2008, 2009 and 2010 (Figure 6.9). The highest number
of product evaluations was carried out in the year 2008 with a total of 762 or about 40% of
the total number. Saudi Arabia has the highest total number of products evaluated (841),

139
followed by Oman (662) and Qatar (422). This survey also indicated that four member states
did not provide the data for products evaluated by them between 2008 to 2010 for the central
registration. The data for Saudi Arabia was combined with that of the national data as it was
not differentiated when the data was first collected. There is no data available from Bahrain,
Kuwait, UAE and Yemen for centralised product evaluation. In general, it could be
ascertained that the number of products evaluated at a national level is more than those
conducted within the Centralised proceed.
Limitation of resources for product evaluation
All member countries indicated that limited resources posed moderate problems for their
agency in relation to the activities within the Centralised Procedure. However, Kuwait, Qatar,
Saudi Arabia and UAE indicated that they did not lack the expertise required for the
assessment of the Centralised Procedure submissions although Bahrain, Oman and Yemen
indicated that they did lack the necessary expertise. The member states which indicated that
the lack of expertise would be a problem in the future stated that they would seek additional
budget and utilise external reviewers. All member states also indicated that they have a unit
or person within their authority that coordinates CP activities. The entity varies from state to
state depending on the size of the organisation and the related activities.Bahrain, Kuwait,
Oman and Yemen indicated that the current system is not sustainable while the remaining
Qatar, Saudi Arabia and UAE indicated that it is. The countries which indicated that the
current system is not sustainable cited the lack of resources as the main factor. The lack of
expertise is another key concern as well as the national workload as the third factor (Figure
6.10).
Part III: Regulatory Expertise
This section of the report examines the regulatory expertise residing within the national
agencies that would enable the evaluation of different parts of the registration applications.
Table 6.2 provides a summary of all the data pertaining to the internal and external expertise
available in each agency.

141
Figure 6.10 – Factors that affect the sustainability of the Centralised Procedure
Generally local internal specialised reviewers are available within each agency and these
account for almost three times the number of external experts. The highest number of internal
experts are involved with the quality data review (30%) while the lowest number is involved
in the review of the biological data (4%) (Table 6.2). Saudi Arabia has the largest number of
internal reviewers totalling 77 while Yemen has the lowest number (11). Oman, UAE and
Yemen rely entirely on internal reviewers. However, there is a direct correlation between the
number of experts, the number of products evaluated and GMP inspections carried out by
these member states (Table 6.3).
Saudi Arabia reported the highest number of evaluators totaling 121 (internal and external)
and they managed to complete 841 product evaluations at national and central levels (2008,
2009 & 2010) as shown in Table 6.3. With their reviewers, Saudi Arabia was able to carry
out 65% of the total GMP inspections carried out at national level. Product evaluations in
Oman, Qatar and Saudi Arabia at national and central registration reached a total of 1,957 for
the period 2008, 2009 and 2010.
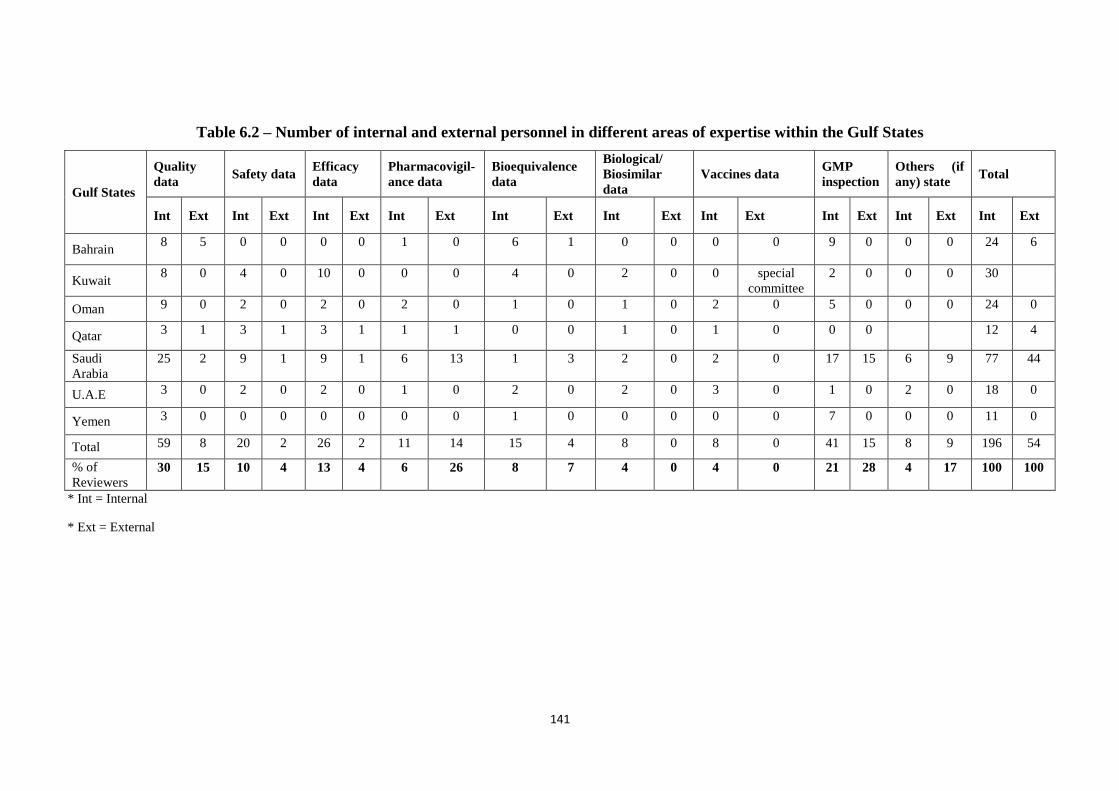
141
Table 6.2 – Number of internal and external personnel in different areas of expertise within the Gulf States
Gulf States
Quality
data Safety data
Efficacy
data
Pharmacovigil-
ance data
Bioequivalence
data
Biological/
Biosimilar
data
Vaccines data GMP
inspection
Others (if
any) state Total
Int Ext Int Ext Int Ext Int Ext Int Ext Int Ext Int Ext Int Ext Int Ext Int Ext
Bahrain 8 5 0 0 0 0 1 0 6 1 0 0 0 0 9 0 0 0 24 6
Kuwait 8 0 4 0 10 0 0 0 4 0 2 0 0 special
committee
2 0 0 0 30
Oman 9 0 2 0 2 0 2 0 1 0 1 0 2 0 5 0 0 0 24 0
Qatar 3 1 3 1 3 1 1 1 0 0 1 0 1 0 0 0 12 4
Saudi
Arabia
25 2 9 1 9 1 6 13 1 3 2 0 2 0 17 15 6 9 77 44
U.A.E 3 0 2 0 2 0 1 0 2 0 2 0 3 0 1 0 2 0 18 0
Yemen 3 0 0 0 0 0 0 0 1 0 0 0 0 0 7 0 0 0 11 0
Total 59 8 20 2 26 2 11 14 15 4 8 0 8 0 41 15 8 9 196 54
% of
Reviewers 30 15 10 4 13 4 6 26 8 7 4 0 4 0 21 28 4 17 100 100
* Int = Internal
* Ext = External

142
Table 6.3 – Relationships between capacity and workload for each Gulf State
(2008, 2009 & 2010)
Gulf States
Internal evaluators
External evaluators
Total Percentage
%
Total Product
Evaluation at National & CP Level
Percentage %
Total GMP Inspections
done at National
Level
Percentage %
Bahrain 24 6 30 12 NA NA NA NA
Kuwait 30 NA 30 12 NA NA NA NA
Oman 24 0 24 10 662 34 29 28
Qatar 12 4 16 6 422 22 NA NA
Saudi
Arabia
77 44 121 48 841 44 66 65
U.A.E 18 0 18 7 NA NA 7 7
Yemen 11 0 11 4 N A NA NA NA
Total 196 54 250 100 1925 100 102 100
NA – Not Available
Section 4 – Importance of GCC-DR
Despite the large number of products being evaluated at a national level, the GCC
member states agreed that the GCC-DR system provides salient advantages to them in
terms of the availability of specialised expertise, training, sharing of
pharmacovigilance data and harmonisation of guidelines. The most valued service is
the collaboration with other national agencies and the provision of guidelines for
harmonisation of the central and national processes. (Figure 6.11).
Figure 6.11 – The value of professional functions to the GCC-DR system by the
Gulf States

143
Generally, Bahrain, Qatar and UAE indicated that all the services rendered by the
GCC-DR system are of high value to their respective agencies while Kuwait, Oman
and Yemen indicated that they are of moderate value. Only Saudi Arabia indicated
that the services provided by the GCC-DR system are of little value. The most valued
service is the collaboration with other national agencies and the harmonisation of the
guidelines.
Expertise
In addition, the member states added that by continuously engaging with the GCC-DR
evaluators through discussions and debates, national evaluators would be challenged
to acquire more information and knowledge about the subject of debate and in so
doing expand their experience. These active intellectual exchanges are essential since
those applications which would be evaluated and registered centrally would be then
registered automatically by the individual GCC States.
Training
With regards to training, the respondents stated that such training could be of higher
value if the frequency for shared workshops is increased and becomes an essential
part of each GCC-DR committee meeting attended by reviewers, evaluators and
committee members. Many shared workshops at a GCC level enriched the expertise
of the evaluators who attended. In addition, through such workshops there is a great
potential for the companies to participate at GCC level and opens up the opportunity
to share knowledge and expertise for all participants. To facilitate the training efforts,
it was suggested that training plans be prepared periodically at GCC-DR level.
Collaborations with other agencies
The member states indicated that the exchange of information, experience and plans
should be initiated between all national agencies in the region on a regular basis. This
is clearly beneficial and natural when the committee members are meeting on a
regular basis and have the opportunity to share information. Networking among
member states becomes easier with better established teamwork and this facilitates
communication and enhances further collaborations.

144
Sharing of Pharmacovigilance Data
The member states indicated that the sharing of pharmacovigilance data is most
important because at least three member states do not have reviewers in this area.
They added that the potential benefit of the GCC-CP is not fully exploited. If it was,
then the CP will be of very high value to all member countries. It needs more
resources and a specialised unit at the central level equipped with the right software
and supported by unified systems at the country levels. In addition, the respondents
suggested that there should be a system for sharing scientific data between all national
agencies
Guidelines Harmonisation
The member states indicated that the GCC-CP is a great opportunity to harmonise
guidelines and requirements. The GCC States recognised the importance of expediting
these guidelines as eventually the GCC registered products will have the option to be
marketed in their country. If guidelines are not harmonised this would mean
inconsistency in the product quality and the guidelines would also avoid duplication
of effort. All members reiterated that similar registration requirements and
circumstances permit the adoption of guideline harmonisation. In addition, they stated
that the regular updating of guidelines is essential.
Section 5 – General Observations
All the national agencies in the GCC indicated that the integrity of the evaluation
results and better scientific opinion are the key to the success of the centralised
evaluation system (Figure 6.12). Hence, it can be ascertained that the CP has achieved
its mandate to provide a better and efficient registration system in comparison to the
national system. The GCC agencies provided details of the main advantages and
disadvantages of the CP system in comparison to the national system. Their
recommendations to improve the CP system included streamlining and harmonising
the approval process.

145
Figure 6.12 – A comparison between the centralised and the national systems
Kuwait, Oman, Qatar, UAE and Yemen indicated that the centralised evaluation
system mostly provided a better scientific opinion than the national system. Bahrain,
on the other hand indicated that the centralised evaluation system always provides a
better scientific opinion than the national system while Saudi Arabia indicated that it
was rarely better (Figure 6.12).
All the GCC States with the exception of Saudi Arabia indicated that the current
centralised system mainly provides consistent evaluations in comparison with national
evaluation. Hence, this is clear evidence that the integrity of the evaluation results and
better scientific opinions are the key to the success of the centralised evaluation
system.
All GCC member countries agreed on having a standard price for specific medicines
throughout the GCC States as shown. However, Qatar was against this, citing the
reason that there would be differences in the proportions of sales and consumption of
medicines between the states and this leads to different profit margins. All GCC
member countries agreed to support the continuity of national registration in parallel
to the centralised procedure while Yemen did not agree to support such a motion due
to limited financial capabilities in comparison to other six member states and for this
reason the Centralised Procedure is a cost-effective option for Yemen.

146
Major advantages of the CP system compared to the national system
The major advantages of the centralised procedure as compared with the national
system are shown in table 6.4. Generally, respondents agreed that the first key
advantage of the CP system is the availability of external expertise to complement the
internal reviewers. This enhances the quality of the report and more information is
focused on the registration to ensure that product evaluation is carried our according
to the standards. In this manner, the quality of the product registration is assured
across the GCC States.
Major disadvantages of the CP system compared to the national system
The main disadvantage of the system is the long approval process together with the
differences in recommendations between the national and the centralised procedure.
The inflexibility of the system and the limited number of meetings per year further
delays the whole process thus making the CP system not as attractive as national
registration for companies (Table 6.5).
Practicality and Applicability of the GACP
An evaluation of the GCC states experiences in participating and completing the
questionnaire for this study was carried out. Generally all the participating states
found the questionnaire easy to complete and understand and they found the
questionnaire relevant and clear. Six states found the length of the questionnaire about
right while only Kuwait indicated it was too long. Three states (Oman, Qatar and
Yemen) indicated that they took 30 minutes to complete the questionnaire, Bahrain
took only 20 minutes while Kuwait and UAE took about 120 minutes. Saudi Arabia
could not ascertain the amount of time taken to complete the questionnaires.
Table 6.4 – Summary of the major advantages
1. Better quality scientific evaluation since more than one state is studying the file
2. More scope for exchange of knowledge, ideas and experiences
3. Drug regulatory coordination
4. The burden and workload is divided
5. Better GMP Inspections
6. Guidelines consensus and harmonisation
7. Exchange of Pharmacovigilance reports

147
Table 6.5 – Summary of the major disadvantages
1. The delay in registration process in comparison to the national system
2. Conflict of some of the recommendations and/or designs with national system
3. The lack of ability to contact the applicant directly to verify or clarify issues
4. The inability to track the full history of the application as the process of review and
follow up is fragmented
5. Lack of a standardised assessment template
6. The limited number of meetings
7. Increase in the number of applications lead to rushed evaluation by the countries
8. Difficulty in activating some of the recommendations
DISCUSSION
This study was carried out to examine the regulatory views and experiences of all
seven Gulf States with respect to the Central Registration Procedure as well as
identifying the barriers and opportunities to improve the CP. Several challenges face
the GCC health authorities to successfully operate the GCC-DR system. The seven
states identified the need for regulatory reforms during the last 12 years, and this has
enabled the improvement of their national system which has enhanced patients‟ access
to high quality medicines throughout the region. The central registration system has
faced several criticisms both from the pharmaceutical industry who were
apprehensive about whether the GCC-DR system would be an obstacle to the timely
approval of medicines in the region as well as government officials who were
concerned about losing sovereignty to the centralised authority (Hashan, 2005).
However, the effective collaborative efforts between the member states substantiated
the support of the GCC-DR system from each GCC authority with an improvement to
regulatory approval processes and operational efficiencies at the national level.
The most significant finding from this study is that the National Drug Regulatory
Authorities in the GCC believe that the Centralised Procedure (CP) has contributed to
an effective system for authorising medicinal products in the GCC. This is ascertained
from their views that the GCC-DR CP achieved its main objective of providing the
best possible scientific opinion for the CP. They also agreed that the CP has
appropriate guidelines for the regulatory review in the GCC-DR and that the quality
of the CP decision-making is satisfactory to good as compared to the national
systems. Apart from obtaining better quality scientific evaluations, since more than

148
one state is studying the application dossiers, the CP allows more scope for the
exchange of knowledge, ideas and experiences among the National Drug Regulatory
Authorities in the GCC. This results in better drug regulatory coordination within the
GCC. As a result of this study it could be ascertained that product evaluation at a
national level is more frequently carried out than at the CP as most pharmaceutical
companies prefer to have their products reviewed at a national level. As an outcome
of this more than three of the states indicated that the current CP evaluation system
would not be sustainable due to the lack of resources and the heavy national
workload.
This lack of resources presents moderate problems for the GCC States in relation to
their activities within the Centralised Procedure. The GCC States recognise the
importance of resources, both human and financial, for the development of strong
regulatory systems. However, the lack of resources can be compensated to some
extent by effective collaboration among countries and information sharing (WHO,
2008). Furthermore, the GCC authorities are able to allocate financial resources due to
their strong economic status, but the availability of human resources and expertise
remains a challenge for the development of a robust drug regulatory system.
There are a total of 250 internal and external professional evaluators available in the
GCC. However, this number of evaluators varied from country to country with Saudi
Arabia having the largest number (121) and Yemen having the lowest with only 11.
With the increasing number of products to be evaluated, the number of specialised
experts within the GCC may not be sufficient to process product registration, which
will result in further delays for pharmaceutical companies. Increasing the number of
external experts in the GCC States may be the best solution for this problem to reduce
the workload on the internal reviewers but this may have a possible impact on the
quality of the review.
Pharmaceutical companies have mixed feelings about whether they can gain faster
marketing authorisation through the national regulatory systems or the regional
centralised system. The goal of any pharmaceutical company is to complete the
registration requirements and gain access to the national GCC markets in the shortest
possible time. In the end, two approval routes, national and centralised exist side-by-
side in the GCC region. For the centralised system to dominate, member states should
seek ways to increase their collaborative efforts to bring their systems closer towards

149
standardisation that would facilitate the regional registration process and maintain
patients‟ access to safe and effective medicines within a reasonable time frame. The
long approval timeline in the CP approval process is a major obstacle to advocating a
central approval procedure. The majority of GCC States agreed that the CP process
should be shortened and they indicated that the timeline for the Centralised Procedure
could be reduced by:
All member states following the GCC guidelines.
The verifications of the registration documents being carried out at the time of
submission.
The reviewing agency sending the applications with preliminary
remarks/deficiencies before the committee meets for their discussions.
The member states not demanding additional data to avoid delaying the whole
process.
Increasing the number of GCC-DR meetings.
Taking in to account the prior approval by mature regulatory authorities.
One way forward is to consider the verification model for the assessment of a new
registration dossier. This model can be used for applications for products that are
registered in a reference country with established mature regulatory authorities
(McAuslane et al., 2009). If not, a more specialised review can be considered for
products which are not registered elsewhere. Singapore, for example, conducts a
verification review for all types of medicines which are previously authorized by at
least two reference authorities, except for biological and biotechnology products.
The member States also agreed that certain aspects of the registration procedure could
be simplified to reduce the duration of the CP process, these aspects are to:
Implement electronic communication together with online submissions.
Send all the requests for further information or clarification questions to the
companies at one time instead of in several batches.
Have a written policy for fast track registration such as life-saving anti-cancer
and anti-HIV drugs.

151
Despite the large number of products being evaluated at a national level, the GCC
member states agreed that the GCC-DR system provides significant advantages in
terms of the availability of specialised expertise, training, sharing of
pharmacovigilance data and harmonisation of guidelines. The most valued service is
the collaboration with other national agencies and the provision of guidelines for
harmonisation of the central and national processes.
The GCC agencies agreed that the key advantage of the CP system is the exchange of
knowledge and information between the member States. This enhances the quality of
the report and more attention is focused on the registration to ensure that the product
evaluation is carried out according to set standards and thus the quality of the product
registration is assured across the GCC States. In addition, the inflexibility of the
system and the limited number of meetings per year further delays the whole process
thus making the CP system less attractive for companies to obtain their product
approval.
SUMMARY
This study has demonstrated that the GCC-DR Centralised Procedure achieved
its main objective by providing the best possible scientific opinion. Appropriate
guidelines for the regulatory review in GCC-DR was of considerable value while
the quality of the CP decision-making ranged from satisfactory to good as
compared to the national system.
All member countries indicated that limited resources did present moderate
problems for their agency in relation to their activities with the Centralised
Procedure.
There are more products being registered at a national level than at the CP.
Despite the preference to evaluate products at a national level, these agencies
agreed that the Centralised Procedure (CP) had indeed contributed to an effective
system of authorising medicinal products for the GCC by providing the best
possible scientific opinion for the quality of the CP decision-making as compared
to the national system.

151
The main disadvantage of the system is the long process to obtain the approval.
The inflexibility of the system and the limited number of meetings per year
further delays the whole process thus, making the CP system less attractive for
companies.
The member countries recommended that the GCC-DR streamlines and
harmonises the CP approval process with national systems in order to improve
the effectiveness and efficiency of the Centralised Procedure.

152
CHAPTER 7
An Evaluation of the Pharmaceutical
Companies Experiences with the GCC
Centralised Procedure

153
INTRODUCTION
The primary aim of drug regulation is the protection of public health. Hill (2004)
suggested that for some the balance between controlling pharmaceuticals in the
interests of ensuring public health and encouraging the development of
pharmaceutical products has shifted in favour of the innovative industry. However,
others perceive regulation as an obstacle to the availability of medicines in national or
regional markets and this has placed a significant demand on regulators to expedite
reviews and evaluations to approve medicines (Hill, 2004). Nevertheless, medicines
regulation is the foundation of any country‟s national drug policy that ensures a viable
pharmaceutical industry as well as a high standard drug approval process.
The establishment of the International Conference on Harmonization (ICH) between
the United States, Europe and Japan in 1990 reflected a need felt by the research
based industry and certain governments to streamline the approval process for the
registration of new medicines (Mallia-Milanes, 2010). The term harmonisation refers
to the standardisation of technical requirements for medicines regulation, of which the
requirements relate to quality, safety and efficacy of medicines and this can differ
from one country to the other. Since its inception, ICH has evolved, through its
Global Cooperation Group (GCG) to respond to the increasingly global phase of drug
development. The GCG was originally formed as a subcommittee in 1999, in response
to the growing interest in ICH guidelines beyond the ICH regions and one such
harmonisation was that of GCC, the Gulf cooperation council. This collaborative
mechanism ensures ensure a more transparent and streamlined process for the
marketing authorization of pharmaceutical products in the GCC region.
The Gulf centralised registration process is facing several challenges which lead to
delays in patients‟ access to medicines with the need to re-register products in
individual countries after central registration. In addition there is a requirement to
register the manufacturer prior to submitting the product file to the Central
Registration Office. Currently there is the need to establish effective methods for
sharing the workload amongst the regulatory authorities in the GCC states. A paucity
of studies at the GCC level, probing into the views and experiences of individual
pharmaceutical companies with the GCC central registration system, has made its
improvement a constant challenge. Hence, this study is envisaged to explore the
importance of the system and identify challenges and critical success factors to

154
enhance the effectiveness and efficiency of the central registration procedure.
Patients benefit from the timely access to safe and effective therapies while
pharmaceutical companies are able to market the product sooner and generate
revenues for further research and development.
OBJECTIVES
The objectives of this research were to:
Evaluate the experiences and views of pharmaceutical companies with respect to
the central registration review process.
Appraise the barriers and opportunities to the implementation of the Gulf
Centralised Procedure.
Make recommendations to improve the Gulf Centralised system as well as to
identify its future strategy based on the views of the pharmaceutical companies.
METHODS
With limited information and the lack of data on the GCC Central Registration
Procedure, there is a need to develop suitable measurement tools in order to obtain the
relevant information required in this study. In Chapter 6, an assessment tool for the
evaluation of the Centralised Procedure by the Drug Authorities in the GCC was
developed and the findings reported. In this Chapter, the emphasis will be on the
pharmaceutical companies and evaluating their experiences in obtaining approval for
their products. The development of the assessment instrument was completed over 5
phases;
Phase 1 – Identify target population and item generation
Phase 2 – Development of themes and item reduction
Phase 3 – Development of the first version of the instrument, Companies Assessment
of Centralised Procedure (CACP)
Phase 4 – Pilot study
Phase 5 – Development of the final version of the CACP
This final version of the study instrument was sent to 100 pharmaceutical companies
(50 generic and 50 innovative) and followed up after one month.

155
RESULTS
This section presents the results from the development of the CACP and the views of
pharmaceutical companies. For the purpose of clarity the results will be presented in
two parts: Part I - The development of the study instrument (CACP); and Part II - The
views of the pharmaceutical companies about the Centralised Procedure
Part I: The Development of the Study Instrument (CACP)
Phase 1 – Target population and item generation
An informal interaction and dialogue was held with a number of pharmaceutical
companies at a consultation meeting organised by the GCC-DR in the spring of 2010.
This allowed the generation of items for the development of the CACP. In addition
these items were cross-referenced with the Gulf States instrument in order to ensure
that relevant items were also retained for the CACP.
Phase 2 – Development of themes and item reduction
There were total of five themes emerging from the item generation phase based on the
objectives of the study. The first version of the questionnaire comprised five sets of
questions pertaining to the following headings;
Theme 1- General Introduction consisting of eight sub-themes;
Theme 2- Centralised Registration Procedure consisting of eight sub-themes;
Theme 3- Interaction with GCC-DR consisting of six sub-themes;
Theme 4- Scientific Opinion consisting of nine sub-themes;
Theme 5- General observations consisting of eight sub-themes.
The General Information included their preference for using the system, national vs.
centralised, data on the number of products submitted and registered through the
centralised procedure, along with their experience and opinion of the system itself.
Theme 2 deals with the guidelines, quality of scientific opinions, appeal procedure,
implementation of the common technical document submission format and
harmonisation of the procedure. Interaction of the company with GCC–DR with
respect to the quality and timing of information received, their relationship with
individual member countries, influence of the pharmaceutical companies on the

156
GCC–DR formed Theme 3 Scientific opinion in terms of regulatory expertise was
included in Theme 4, and Theme 5 contains general observations from pricing to
recommendations to improve the procedure, their views on the present system and
future strategies.
Phase 3 – Development of the first version of the instrument, Companies
Assessment of Central Procedure (CACP)
The selected themes and sub-themes were put into an appropriate structured
questionnaire. These were then reviewed by a social science expert using four
appropriate parameters; namely; language clarity, completeness of the statement,
appropriate relevance and scaling. Account was also taken with regard to the length of
the items and their readability.
Phase 4 – Pilot study
The purpose of the pilot study was to assess the practicality, feasibility, applicability
and relevance of the study instrument to assessing the Gulf Centralised Procedure
from the companies‟ perspective. The structured questionnaire was sent to five
targeted companies (two innovative and three generic).
Feedback from the first questionnaire was obtained from these companies. This pilot
study clearly indicated that such a project into GCC Central Registration Procedure
from the pharmaceutical companies‟ perspective was essential to bring about
standardisation and harmonisation of the approval process. This would inevitably
improve the registration system for both the applicants as well as the approving
authorities. Based on the respondents feedback minor changes were made to the
questionnaire. These changes included adding refinement to questions 2.1 and 2.2 in
Part 2 and repositioning questions 3.3 and 3.4 in Part 3 of the questionnaire. Finally,
six simple questions were added to the questionnaire to obtain the views of the
respondents about the practicability, applicability and relevance of the CACP. This
feedback was critical in order to design a more effective assessment instrument for
future studies (Figure 7.1).

157
Figure 7.1

158
Phase 5 – Development of the final version of the CACP
Following the pilot study and implementation of the changes suggested by the
participating companies the development of CACP was completed. Thus, the final
version of the instrument consists of 39 items grouped into five major categories. In
addition it includes an introductory page collecting the respondents' personal details
(Figure 7.2). The final version of the questionnaire was sent to 100 companies who
had registered their products through the either centralised registration procedure
and/or national registration systems. The questionnaire was sent with a clear guideline
regarding the aims and objectives of the study with a statement assuring them of the
confidentiality of the study.
Part II: The views of the pharmaceutical companies about the
Centralised Procedure
An analysis of the target audience was carried out by listing all pharmaceutical
companies who had interactions with the national registration system and/or the CP in
the GCC region. This list of the companies registered with the CP was obtained from
the GCC-DR office in Saudi Arabia. The profile of each company was analysed to
ascertain their suitability and their willingness to provide information for the study.
Fifty generic and 50 innovative companies were selected for the study from the list of
registered companies, half of them with experience registering their products through
the CP while the other half had the experience of registering their products through
the national system.
Characteristics of the study participants:
The CACP was distributed to 100 pharmaceutical companies out of which 30
responses were received (30%) (Appendix 1). The remaining, who did not respond,
could be categorised into four groups:
1- Unwilling to participate
2- Lack of experience
3- Lack of response, and
4- Sensitivity of the information required

159
Figure 7.2

161

161

162

163

164

165
The CACP scores:
In order to answer the main evaluation questions covering the experience of these
companies with GCC-DR Central Registration Procedure the results are presented
under five sections.
Section 1 – Companies general views about the Centralised procedure
In this section, the companies‟ experiences in registering their products through the
Centralised Procedure were examined. Their preference for using this system, national
versus centralised was determined. Data on the number of products submitted and
registered through the centralised procedure, along with their experience and opinion
of the system itself were also recorded. Generally, more than half of the companies
who responded preferred the national registration procedure to the Centralised
Procedure (Figure 7.3) The majority of the pharmaceutical companies preferred to
register their products through the national system than the centralised system. Eleven
innovative companies preferred the national system rather than the CP while six
generic companies choose the national system instead of the CP. Nineteen companies
had submitted their products for registration under the centralised system, while more
generic companies (12) had submitted their products to the CP than the innovative
companies (Figure 7.3).
Figure 7.3 – Number of companies and their preferred pharmaceutical registration
system

166
Only the 19 companies who had the experience of registering their products through
the CP were able to provide critical information on the strengths and weaknesses of
the CP registration system. The main reasons for choosing the centralised registration
system over the national system are illustrated in Figure 7.4. This shows that 14
pharmaceutical companies were able to meet the requirements to GCC tender
specification, convenience (12 companies) and timeliness of registration,
cost/resources required and ease of subsequent CP registration. One of the
respondents indicated that the central registration team is more accessible, easier to
communicate with, flexible and understanding about the companies‟ issues. It is also
faster in approval than the national authority. Another respondent indicated their
choice of product registration system would be dependent on their company‟s strategy
and the nature of the product.
Figure 7.4 – Reasons for companies preference in using the Centralised
Procedure for product registration
*Other Factors
1. Central registration team more accessible, easier to communicate with flexible and
understanding about the companies’ issues. Faster in approval than the national and is a
respectable authority.
2. Depending on the company strategy and the nature of the product
Figure 7.5 shows the reasons why pharmaceutical companies did not prefer the
centralised system. Eight of the companies stated that their product strategy was
aligned to the registration in selected GCC states, hence the preference for national
registration. The timeline for registration and the experience with the national systems
registrations resulted in a total of seven responses. The rest cited the cost and resource
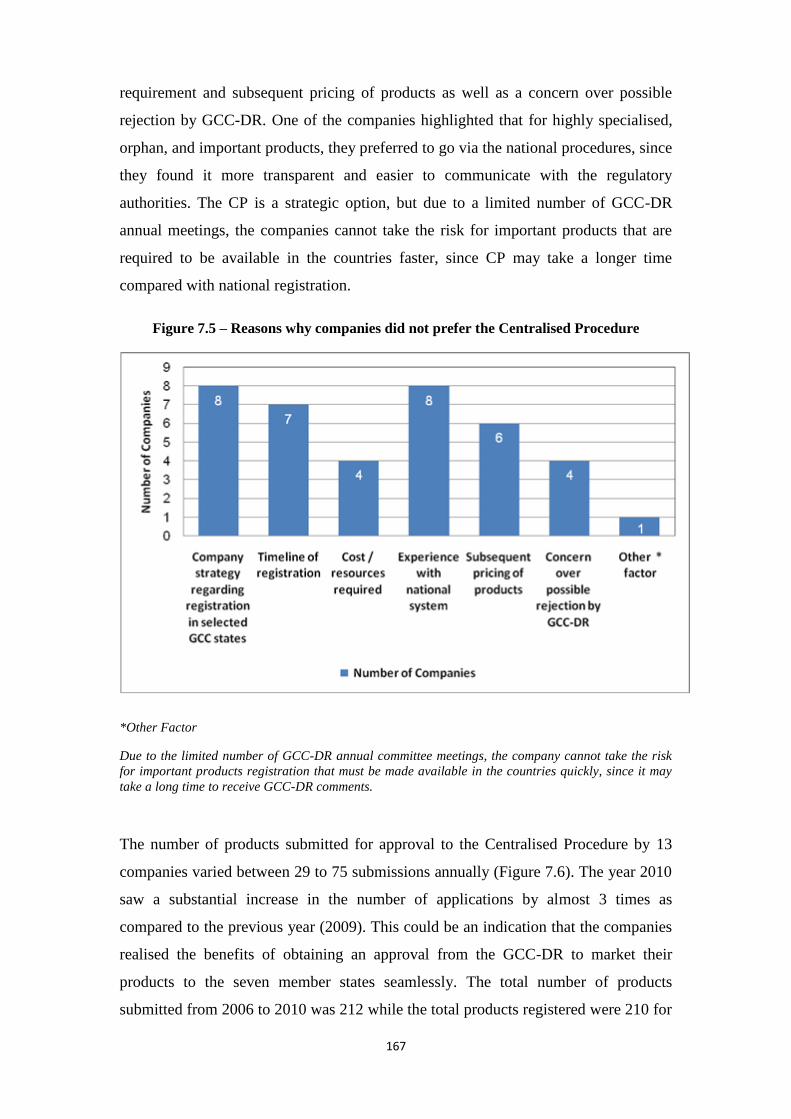
167
requirement and subsequent pricing of products as well as a concern over possible
rejection by GCC-DR. One of the companies highlighted that for highly specialised,
orphan, and important products, they preferred to go via the national procedures, since
they found it more transparent and easier to communicate with the regulatory
authorities. The CP is a strategic option, but due to a limited number of GCC-DR
annual meetings, the companies cannot take the risk for important products that are
required to be available in the countries faster, since CP may take a longer time
compared with national registration.
Figure 7.5 – Reasons why companies did not prefer the Centralised Procedure
*Other Factor
Due to the limited number of GCC-DR annual committee meetings, the company cannot take the risk
for important products registration that must be made available in the countries quickly, since it may
take a long time to receive GCC-DR comments.
The number of products submitted for approval to the Centralised Procedure by 13
companies varied between 29 to 75 submissions annually (Figure 7.6). The year 2010
saw a substantial increase in the number of applications by almost 3 times as
compared to the previous year (2009). This could be an indication that the companies
realised the benefits of obtaining an approval from the GCC-DR to market their
products to the seven member states seamlessly. The total number of products
submitted from 2006 to 2010 was 212 while the total products registered were 210 for

168
the four years (2006 to 2009). An upward trend can be seen in product submissions
and product registrations under the CP system. This clearly indicates that though there
were challenges in processing the products for approval at GCC-DR level, it certainly
is gaining recognition as a viable option. Twenty companies believed the quality of
decision-making of the Centralised Procedure to be good or excellent while seven
companies stated that it was only satisfactory.
This is an indication that although there are issues with the centralised registration
system, the quality of its decision-making is still satisfactory for most companies
(Figure 7.7). Although some companies had no experience of registering their
products through the CP they were still able to provide their opinion about this
system.
Figure 7.6 – Number of products submitted and approved through the Centralised
Procedure
Twenty companies stated that it took them more time to register their products
through the centralised system. The pharmaceutical companies also stated that the
limited number of meetings by the Central Committee was one of the main causes of
the delay in product registration; thus their preference for national registration. The
delay in product registration is also attributed to the fact that the submission of the
documents for review and approval by the Committee has to be done earlier before
the summer hiatus if the companies wish to complete the procedure in good time.

169
Figure 7.7 –The quality of decision-making of the Centralised Procedure
However, unlike the national registration procedure in which both the product and the
pricing registration could be done concurrently, the company has then to wait for the
product approval centrally before proceeding with the pricing approval locally.
Further delays in product approval and registration in the CP were caused by long
technical documents evaluation, distribution of the files for the reference countries for
evaluation and analysis feedback and a long queue for submission and review by the
committee. The issue of sequentially processing for national registration after the
Centralised Procedure added a further delay to the approval process and it could take
between 1.5 to 2.5 years for approval to be attained for each product. Furthermore,
with so many issues to be discussed in the CP Committee meetings coupled with the
limited number of meetings per year meant approval and registration issues would
take a longer time to be resolved, hence lengthening the approval process. More
delays are caused when evaluators waited until after the CP meetings to pass their
comments and requests for additional information instead of asking for this
immediately on reviewing at the first stage.
Twenty-seven companies indicated that the timelines for registration in the
Centralised Procedure could be shortened and twenty of these companies also stated
that by having more GCC-DR meetings this could be achieved. Others believed that
having additional technical expertise and conducting parallel processing of the
laboratory tests and the CP registration could also shorten the process however,

171
Parallel processing was induced in 2011. Figure 7.8 summarises how certain aspects
of the registration procedure could be simplified. Twenty of the companies also
indicated that certain aspects of the registration procedure could be simplified. The
majority of these respondents believed that electronic submissions and automation of
some of the processes could assist in simplifying the Centralised Procedure as a whole
and hence shorten the whole process. Others indicated that the technical evaluation
could be simplified by relying on documents submitted to GCC since these were the
same documents already submitted and approved in the US/UK and other reference
countries. Finally, 16 of the companies indicated that the current registration fee for
the Centralised Procedure is reasonable while 11 of the companies stated that it is too
high. Hence, the cost factor from the registration perspective is not the key reason
why companies preferred the national registration system to the Centralised Procedure
to obtain approval for their products.
Figure 7.8– Companies’ suggestions on how certain aspects of the registration procedure
could be simplified
*Others
1. The pricing procedure in the individual GCC states should be in parallel with the CP
Registration.
2. The communication with the companies could be via email or other means of electronic
transmission.
3. Stop the site inspection if the site was in a reference country and has the appropriate GMP
and manufacturing license (same as the national registration).
4. Have one registration certificate for all GCC states from GCC-DR (once registered, no need
to submit again per each country to gain approval).

171
Section 2 - Centralised Registration Procedure
The companies experiences in submitting and registering products under the CP is
reviewed in this section. Generally the companies were satisfied with the quality of
the guidelines and scientific advice as well as their opinions on the implementation of
the Common Technical documents, pharmacovigilance procedures, and transparency
of the Centralised Procedure. In addition, the majority of the companies also agreed
that the CP led to the harmonisation of national procedures in the GCC legislation.
Half of the companies indicated that the Centralised Procedure guidelines were easy
to understand (eight generic and seven innovative companies), 10 companies (six
innovative and four generic companies) stated that the guidelines were not always
clear and another three companies indicated that they were generally unclear (Figure
7.9). Those who stated that the guidelines were not always clear further provided their
explanation for their choice. They pointed out that there were no guidelines for all
registration requirements (e.g. available guidelines are for Stability Studies and
Bioequivalence study only). Variations to the guidelines were also not available. The
biotechnology products registration guidelines were very short and ambiguous and
many of the requirements, that the committee asked for, were not included. In
addition, they commented that the committee sometimes went outside the guidelines
and asked for additional requirements found in other international guidelines
(oscillating between EU and FDA, especially with stability and Bioequivalence).
They also stated that the patient information leaflets (PIL) requirements were not clear
and many times the committee asked for amendments for the PIL that was
internationally recognized. Hence, they were not provided with clear criteria in this
matter. Finally, they added that the registration certificates for manufacturing sites
were not harmonized with regards to the nomenclature of the production lines which
created confusion when required to register a product from such a site later.
Quality of GCC-DR Scientific Opinions
The majority of the companies indicated that the quality of the GCC-DR scientific
opinions ranged between good to excellent while five companies stated that it was only
satisfactory (Figure 7.10). It can be concluded that the pharmaceutical companies in
general were satisfied with the quality of the GCC-DR scientific opinions despite the
hassle and the issues faced in getting their products registered through the Centralised
Procedure.

172
Figure 7.9 – Companies’ view on the Centralised Procedure guidelines
Figure 7.10 – Companies’ views on the quality of the GCC-DR scientific advice,
efficiency and effectiveness of the centralised pharmacovigilance procedures and the
transparency of the Centralised Procedure
Similarly, the majority of the companies also stated that the current appeal procedure
was sufficient in the case of a negative opinion issued by the GCC-DR, although, 10
companies felt otherwise. They further added that there should be meetings between
the GCC-DR and the companies so that these companies could explain the reasons for
the appeal which may not have been understood by the committee previously.
Therefore, these views should be considered by the member states. Also, the GCC
needs to take in to account the registration certificates issued from the mature

173
regulatory authorities such as FDA and EMA. Other respondents also stated that the
GCC-DR had no experience in dealing with appeal cases and hence there was neither
any clear timelines nor clear answers provided to the companies as justification for the
rejection. This made it very difficult for the companies to convince all the members of
the committee of their positive opinion or challenge the rejection decision. There were
also no response received by the companies in case of a negative opinion by the GCC
and the submission of their appeal document.
Seven companies indicated that the implementation of the common technical
document format would be a barrier to their company‟s submission of product
registrations through the Centralised Procedure. Nevertheless, they also stated that to
work in the same CTD format for all the GCC countries individually in addition to
GCC-CP, would encourage many manufacturing companies in the region to obtain the
approvals from GCC-CP. However, the resources, specialised personnel and hands-on
training are not enough. At present, not all the member states are at the same level for
receiving and processing CTD applications. Most medium size and Arab companies
did not have any idea about such a format and they would need a considerable time
before they could fulfil these requirements. This would be very difficult for these
companies and would be costly to submit the same documentation but in a “different”
ICH format. Moreover, such a requirement has been applied without prior notice or
time allowance. As a result, it may take a much longer time to get the final approval.
Some form of flexibility should be accepted as there are old and rare products which
do not have a CTD dossier. This new requirement would also mean that there would
be additions in the required documents. Some of these documents and tests may
require more time to be prepared, thus, this would delay the process of registration.
The efficiency and effectiveness of the GCC centralised pharmacovigilance
procedures
The majority of the companies who responded were satisfied with the efficiency and
effectiveness of the GCC centralised pharmacovigilance procedures as the GCC-DR
had continued to be vigilant and monitor Adverse Drug Reactions (ADR) of
medicines speedily. In addition, currently the companies believe that the current GCC
guidelines for pharmacovigilance are not clear, even though the Saudi FDA guidelines
are well recognised. However five companies (Figure 7.10) indicated that although
the efficiency and effectiveness of the GCC centralised pharmacovigilance procedures

174
were poor, they were not aware of any pharmacovigilance systems in the region being
implemented. However, the five companies had strict internal systems related to
safety, PV and Risk Management Plans (RMP).
The transparency of Centralised Procedure
Five companies indicated that the transparency of the Centralised Procedure was
excellent because the GCC-DR was open in their communications; it was easy to meet
the central organization team member who was willing to simplify the issues and had
great understanding with a high level of transparency. On the other hand half of the
companies stated that although the CP transparency was good as there were clear
guidelines, refinement was needed in the follow-up of the application process.
Overall, 70% of the companies disagreed that the Centralised Procedure should be
made compulsory for certain range of products. However, six companies agreed that
the Centralised Procedure should be made compulsory for the following range of
products:
Prescription only Medicine
Narcotics and controlled drugs
Medicinal, Herbal and Heath products
Biotechnology products especially coming from non stringent countries.
Vaccines
Life saving medicines and products that are crucially required in these markets
Finally, 18 companies agreed that the Centralised Procedure has contributed to the
harmonisation of the legislation throughout the GCC while eight of the respondents
did not agree.
Section 3 - Interaction with Gulf Central Committee for Drug Registration
(GCC-DR)
Generally, the companies were satisfied with the level of interaction with the GCC-
DR in terms of the quality of the information received and the relationship between
the authorities. The majority of them also agreed that the pharmaceutical industry can
influence the GCC-DR Centralised Procedure through sharing of knowledge and
experiences and promoting closer collaboration.

175
Based on the same data, more than 15 companies were satisfied with the timing of the
registration procedure at GCC-DR while six companies indicated that the timing of
the registration was poor. Nineteen companies indicated that their relationship with
the Registration Department of the GCC-DR was good or excellent and 22 companies
indicated that they were satisfied with the relationship (Figure 7.11).
Figure 7.11 – Companies’ view of the level of interaction on the quality of information
received, timing of the registration procedure and their relationship with the
Registration Department of the GCC-DR
Although many companies refrained from commenting on their relationship with the
GCC-DR because of its sensitivity, however they indicated that the relationship could
be improved. Some of the ways in which this could be improved are summarised in
Figure 7.12. Three companies suggested that the increased sharing of knowledge
through training, seminars and workshops would promote more interactions between
the companies and the GCC-DR especially in discussing changes to regulations and
procedures. This would significantly improve the relationship between companies and
the GCC-DR and could contribute to providing healthcare benefits to GCC states.

176
Figure 7.12 – Companies’ views on some of the ways to improve the relationship
between the companies and the GCC-DR
Ten companies suggested that there should be an increased number of meetings as
well as improved communication between the GCC-DR and the pharmaceutical
companies. Through openness and regular communication with the companies and
having open dialogue and meetings to determine companies needs, the relationship
between the two organizations would be improved. This would also promote
consistency in decision-making and provide transparency and clear justifications
should there be any rejections in the future.
One of the companies indicated that the number of GCC-DR staff is low as compared
to the considerable amount of work they have to carry out; therefore the quality of the
relationship is adversely affected. Hence, increasing the number of GCC-DR staff is
essential to ease the workload and improve their relationship with the companies.
Others suggested that the GCC-DR must work as the centre for all GCC
Pharmaceutical companies. They should promote regular activities such as running
conferences and training courses in different fields of pharmaceutical manufacturing,
case-studies, training on registration guidelines in GCC and in other regulated
markets. Also, it should include distributing statistics on GCC pharmaceutical
companies and medicines, improving knowledge on pharmaceutical technology
among pharmaceutical companies and linking with other international conferences on
medicines and related issues. In addition, they suggested that by providing electronic
online services, the companies would be able to track the progress of their product‟s

177
registration status and comments, anytime, anywhere. This would greatly facilitate
communication with the GCC-DR and expedite information requests. Twenty
companies agreed that the pharmaceutical industry can influence the GCC-DR
procedure and that it could be done in the following manner;
Through sharing of knowledge, experiences and interaction, the
pharmaceutical companies could provide support at any level through their
worldwide experience with authorities and regulators. As an important partner
in the registration process, the companies would be able to provide the GCC-
DR with effective and efficient services and quality products.
The pharmaceutical companies could contribute to drafting and refining the
guidelines and GMP inspection systems to make them more effective and
efficient.
They could assist the GCC-DR in streamlining the procedure in order to
improve and expedite the registration process.
They also added that private accredited laboratories could be engaged to help
facilitate the procedure and by giving priority to the registration of new
products from local industries.
Section 4 - Scientific Opinion
Twenty companies indicated that the scientific opinions from the CP were relevant
with respect to their regulatory expertise. Generally, they were satisfied with the
quality and safety data, bioequivalence studies and vaccines data provided by the
GCC-DR experts for the pharmaceutical products registration and approval process.
Generally, half of the companies indicated that the quality of the assessment provided
by CP experts for safety, efficacy, pharmacovigilance, bioequivalence studies,
biological/biosimilar, vaccines data and GMP inspections were good or excellent
(Table 7.1) with only six companies indicating that the assessment for efficacy data,
pharmacovigilance data, biological/biosimilar and GMP inspections were poor. There
was also a high percentage of companies (36%) who did not provide their responses
for this question which probably indicated that they may not have had the appropriate
experience in these areas.

178
Table 7.1 – Companies’ opinion regarding regulatory expertise within the GCC-DR
Review Expertise Excellent Good Satisfactory Poor Total
Quality data 5 16 3 0 24
Safety data 5 13 5 0 23
Efficacy data 5 14 3 1 23
Pharmacovigilance data 2 6 8 3 19
Bioequivalence studies 6 7 6 0 19
Biological/biosimilar 2 8 1 1 12
Vaccines data 3 7 1 0 11
GMP inspections 9 10 1 1 21
Section 5 - General observations
The majority of the companies believe that the CP has a more efficient registration
system as compared to the national system and indicated the main advantages and
disadvantages of the CP system based on their experiences. Three companies
indicated that the current centralised evaluation system always provides a better
scientific opinion/outcome than the national system, while five companies stated that
this occurs frequently. Nine companies indicated that the GCC-DR sometimes
provided better scientific opinion/outcome than the national system while three
companies stated that this happened rarely. However, more than half of the companies
agreed that the current centralised evaluation system did provide better scientific
opinion/outcome than the national system (Figure 7.13).
Nine companies agreed that the current centralised system provided consistent
evaluations in comparison with the national evaluation while ten companies indicated
that this happened sometimes and only six companies stated that this happened rarely
(Figure 7.13). Clearly, attaining a standard consistency of the evaluation reports
continuously is critical to the quality and efficacy of the centralised GCC-DR
procedure. Half of the companies did not agree with the idea of having a standard
price for specific medicines throughout the GCC states and the reasons for their
disagreement are shown (Figure 7.14). The most critical aspect, as cited by 14
companies, is the difference in economic aspects of the various GCC states as
differences in the cost of living and demographics would impact the volume of the
products, hence the price of the medicines.

179
Figure 7.13 – A comparison of the companies’ views of the national versus the
centralised system with respect to scientific opinion and consistency of evaluations
Figure 7.14 – Companies’ views as to why a standardised pricing is not appropriate
It is therefore a complex task to set a standard price for specific medicines throughout
the GCC states. Finally, 80% of the companies stated that they would support the
continuity of a national registration in parallel to the Centralised Procedure while 20%
of the companies would not.

181
Advantages of the Centralised Procedure compared to the National System
Fourteen companies who responded to the survey clearly indicated that one decision
from the GCC-DR has a considerable advantage as the product could be promoted
faster, easier and is highly recognisable in the seven GCC states. The advantages of
the Centralised Procedure as compared to the national system are as follows;
a. Harmonisation of procedures
One approval procedure, one set of documents for the entire approval process
and one file with the same format for one submission. The procedure is much
simpler and less complicated. This would save time and effort and promote
transparency.
b. One decision for all seven countries
In this way medicinal products can be available to all GCC States at the same
time once they have been approved by GCC-DR. This facilitates the
registration of manufacturing sites in one step. Hence, it would reduce the
burden on internal company resources to prepare “dossiers” for additional
registration.
c. Others
Collective and diversified opinions from the different GCC states add
more value to the CP decision.
Products registered at the GCC level will add value to the company
and it will open new markets for its products,
The implementation of CTD format in the central and national levels.
Proceed with other countries with less documentation.
If there are no major objections from the committee, then the approval
is faster

181
Disadvantages of the Centralised Procedure compared to the National
System
The major disadvantage of the CP as compared to the national system is that it is a time
consuming and costly process, followed by the inconsistency of the process and the
insufficient number of meetings. The companies also stated that the need to re-submit
product files at a national level after approval at the Centralised Procedure is another
disadvantage. Sometimes more details are required in terms of the assessment of products
by the national system compared to the Centralised Procedure. The companies are also
concerned about the possible rejection by GCC-DR that would affect other GCC
countries national registration. They stated that although the products are acknowledged
by national authorities as final approval in one or more of the member states, they still
need to go through the full CP process. By not using communication such as electronic
mail, the GCC-DR further delays any correspondence with the companies which could
facilitate the registration processes. In addition, the companies also indicated that inter-
committee politics may affect certain decision-making outcomes which are not
standardised.
The three major disadvantages of the Centralised Procedure as compared to the national
system are as follows;
Time consuming procedure and costly process - The procedure of registration
involves a very lengthy period both at the quality control laboratory for analysis as
well as at the committee level. Feedback is not clear and there is no direct
communication with principal companies. Coupled with the high registration fee, the
long registration process is the key disadvantage of the Centralised Procedure.
Inconsistency of the process – The absence of guidelines leads to inconsistency in
the opinions and some radical differences occur at national level. Many of the
documents submitted to the GCC are requested again at a national level after GCC
approval. Some states do not accept registration without prior GCC-DR approval.
National authorities do not approve the product for distribution even after attaining
GCC-DR registration through the CP system.
Quarterly meetings - Once a quarter committee meetings is not sufficient to
process the application. Hence, the long delay in the approval process.

182
Practicality and Applicability of the CACP
An evaluation of the companies‟ experiences in participating and completing the
questionnaire for this study was carried out. Twenty-two companies (88%) found the
questionnaire easy to complete while 12% of the companies found it difficult (Figure
7.15). Twenty-four (96%) of the respondents found the questionnaire easy to
understand while only one company found it difficult to understand. Nineteen (76%)
of the respondents found the questionnaire relevant while only 3 companies (12%)
found the questionnaire not relevant. One of the companies stated that the
questionnaire was not always relevant. Twenty-three (95%) of the respondents found
the questionnaire clear while only one company stated that the questionnaire was not
clear. Twenty-one 83% of the companies participating in this study found the length
of the questionnaires about right. Only 17% of the companies indicated that the
questionnaire was too long. Nine (30 %) of the companies took about 30 minutes to
complete the questionnaire while 35% took more than 30 minutes and the remaining
35% took less than 30 minutes. Thirteen percent of the companies took only 10
minutes to complete the questionnaire which was the shortest time while 5 (21%) of
the companies took more than an hour and the longest time taken was 120 minutes.
Figure 7.15 Summary of responses on the questionnaire

183
DISCUSSION
An efficient review allows the regulatory authorities to fulfill their public health
mission to ensure that safe and effective medicines are made available to patients in a
timely manner and allows for the efficient use of resources. Patients benefit from the
timely access to safe and effective therapies while pharmaceutical companies are able
to market their products sooner and generate revenues (Booz Allen Hamilton, 2008).
The regulatory challenge is to allow access to the safest and most effective
pharmaceutical products in the shortest possible time with the highest degree of
certainty (Alder, 2001). In the GCC states, pharmaceutical companies can choose
whether to register new products via each regulatory authority individually or via the
central registration system and the length of the expected review time will influence
this decision. Therefore, evaluating the pharmaceutical industry‟s views about the
advantages and disadvantages of the GCC-DR system and how this has impacted the
speed of the marketing authorisation time of their products in each Gulf State was
essential.
Pharmaceutical companies were able to provide critical information on the strengths
and weaknesses of the CP registration system. Based on this experience, only 13
companies surveyed preferred the CP registration system, of which nine were generic
companies. The main reasons for choosing the centralised registration system over the
national system include:
The ability of the pharmaceutical companies to meet the requirement of GCC
tender specification.
The convenience of the system,
An improvement of patients‟ access to safe and effective medicines in the GCC
region.
The central registration team is more accessible and easier in communication,
The companies‟ strategy and economic-related factors.
The pharmaceutical companies which preferred the national system stated that their
product strategy was aligned to the registration in selected GCC states for the
following reasons and hence their preference:

184
Timeline of registration.
Experience with national system.
Concern over possible rejection by GCC-DR.
More transparent and communication easier with local national regulatory
authorities.
Limited number of GCC-DR annual meetings.
However, despite the majority of the companies‟ preferences for a national
registration, the companies indicated that the CP has a better and efficient registration
system as compared to the national system. The companies agreed that the CP
provides better scientific opinion/outcome than the national system and that the CP
provides consistent evaluations as compared to the national system. The companies
indicated that through harmonisation of the processes, which would entail one
approval procedure, one set of documents for the entire approval process and one file
with the same format for one submission, the procedure is much simpler and less
complicated.
This saves time and effort and promotes transparency. Furthermore, a medicinal
product can be available to all GCC states at the same time once it has been approved
centrally. This also facilitates the registration of manufacturing sites in one step and
hence, reduces the burden on internal company resources. However, the time
consuming procedure and costly process coupled with unclear feedback and lack of
direct communication between the GCC-DR and the companies are some of the major
issues that hampered the CP registration system. The absence of guidelines leads to
inconsistency and therefore some radical differences occur at national levels. Many of
the documents submitted for the GCC-CP are requested again at a national level and
some GCC countries do not approve the product for distribution even after attaining
GCC-DR registration. Moreover, committee meetings held only four times a year is
insufficient to address all the product registration issues which results in further
delays.
Although more than 60% of pharmaceutical companies stated that it took more time to
register their products through the centralised system, they still felt that the GCC-DR
Centralised Procedure is the way forward for several reasons:

185
By registering their products at GCC level they would have gained access to
seven markets in the GCC states.
The CP leads to the harmonisation of the procedures at the national level and
better quality scientific opinions.
The current centralised system provides consistent evaluations in comparison
with national evaluation.
The majority of these respondents indicated that electronic submission and
automation of some of the processes could assist in simplifying the Centralised
Procedure and hence shorten the whole process. Others indicated that the technical
evaluation could be simplified by relying on documents submitted to the GCC since
these were the same documents already submitted and approved in the US/UK and
other reference countries.
Pharmaceutical companies in general were satisfied with the quality of the GCC-DR
scientific opinions despite the hassle and the issues faced in getting their products
registered through the Centralised Procedure. In addition, the pharmaceutical
companies were satisfied with the efficiency and effectiveness of the GCC centralised
pharmacovigilance procedures as the GCC-DR, through this procedure, has monitored
Adverse Drug Reactions (ADR) of medicines rapidly.
All the companies indicated they were satisfied with the level of transparency of the
Centralised Procedure. They cited that the GCC-DR were open in their
communication, and it was easy to meet the central organization team member. They
were willing to simplify the issues, had great understanding and there was a high level
of transparency in the GCC executive office. However, there is still room for
improvement in the follow-up of the application process. Generally, the majority of
the companies indicated that they were satisfied with the relationship that they had
established with the Registration Department of the GCC-DR. They suggested that
increasing the sharing of knowledge through training, seminars and workshops could
promote more interactions between the companies and the GCC-DR authority
especially in discussing changes to regulations and procedures. This will significantly
improve the relationship between companies and GCC-DR authorities and can
contribute more in providing healthcare benefits to GCC States. The companies also

186
suggested that there should be an increase in the number of meetings and communications
between the GCC-DR committee and the pharmaceutical companies. Through openness
and regular communication with the companies and having open meetings to define
companies needs, the relationship between the two organisations would be improved.
This would also promote consistency in decision-making and provide transparency and a
clear justification should there be any rejections in the future. In addition, they suggested
that providing electronic online services would greatly facilitate communications with the
GCC-DR and expedite information requests. Pharmaceutical companies also agreed that
the pharmaceutical industry can influence the GCC-DR procedure through knowledge
sharing, interactions and collaboration as a partnership basis. Through such activities, the
pharmaceutical companies could provide support at any level through their worldwide
experience with authorities and regulators. The pharmaceutical companies could
contribute in drafting and refining guidelines and the GMP inspection system to make
them more effective and efficient. They could also assist the GCC-DR in streamlining the
procedure in order to improve and expedite the registration process.
Finally, the GCC authorities are facing an increasing number of pharmaceutical
companies who are demanding more efficient, effective and transparent regulatory
services. This places a significant pressure on the Gulf States to improve the quality of the
regulatory review process as well as to expedite the marketing authorization of
pharmaceutical products without affecting the quality of the new medicines. The GCC-
DR challenge in the beginning was to convince companies to consider submitting their
dossiers to the centralised procedure. The submission process is voluntary as GCC-DR
cannot implement a compulsory system until the required level of standardization in the
regulatory review systems has been reached. However, pharmaceutical companies still
have mixed feelings about whether they can gain faster marketing authorization through
the national regulatory systems or the regional centralised system (Al Essa, 2011). After
all, the goal of any pharmaceutical company is to complete the registration requirements
and gain access to the national GCC markets in the shortest possible time. In the end, two
approval routes, national and centralised, are permitted to exist side-by-side in the GCC
region. But for the centralised system to dominate, member states should seek ways to
increase their collaborative efforts to bring their systems closer towards standardization
that would facilitate the regional registration process and maintain patients‟ access to safe
and effective medicines within a reasonable time frame.

187
SUMMARY
The pharmaceutical companies who responded to the survey were able to provide
critical information on the strengths and weaknesses of the CP registration
system.
In general pharmaceutical companies were satisfied with the quality of the GCC-
DR scientific opinions despite the difficulties and the issues faced in getting their
products registered through the CP.
The participant companies were satisfied with the efficiency and effectiveness of
the GCC centralised pharmacovigilance procedures as the GCC-DR, through this
procedure, rapidly monitors adverse drug reactions.
All the companies indicated they were satisfied with the level of transparency of
the CP as the GCC-DR were open in their communication, it was easy to meet the
central organisation team member, they were willing to simplify the issues and
had great understanding.
The GCC-DR has a significant advantage as the product could be promoted faster,
easier and is highly recognisable in the seven GCC States.
The major disadvantage of the CP as compared to the national system is the time
and the expense involved in the process, followed by the inconsistency of the
process and the insufficient number of meetings. The companies also stated that
the need to re-submit product files at a national level after approval at the
Centralised Procedure is a further disadvantage.
The study confirmed that although more than half of the 30 pharmaceutical
companies preferred the national registration system as compared to the GCC-DR
Centralised Procedure, they agreed that the CP is the way forward for the future
as it will bring about harmonisation of the registration process, quality scientific
opinions and faster access to the GCC market.

188
CHAPTER 8
The Gulf Centralised Procedure- A
Comparison between the Experiences of
Regulatory Authorities and Pharmaceutical
Companies: a Proposed new model

189
INTRODUCTION
The Centralised Procedure was designed to improve the operation of the „Single
Market‟ for medicinal products, the avoidance of duplication of scientific evaluation
and the reduction of the administrative burden. Apart from these, product safety
enhancement and harmonisation are also important for this region. However, the
situation in the Gulf Centralised Registration procedure is complex because each state
has its own government, constitution, legislation, regulatory requirements and
healthcare systems, as well as differences in history and medical practices.
Today regulatory authorities are thinking in terms of Global and Regional themes, for
example the European Medicines Agency (EMA), the African Regulatory
Harmonization Initiative (AMRH), and the International Conference on
Harmonisation (ICH) for Europe, Japan and USA. However, such efforts are not risk-
free due to the influence of regional groups in pursuing harmonisation of drug
registration. In addition, more stringent registration requirements may displace
substandard medicines from the market.
The Ministers of Health in most of the Gulf States have expressed their doubts about
the ability to secure the expertise required for registration while at the same time there
is the fear of losing their sovereignty to the centralised authority (A. Rahman, 2005).
The successful development and registration of a medicinal product requires effective
dialogue between the pharmaceutical industry and the health authorities. For the
pharmaceutical industry, good communication with the health authorities is essential
during the entire lifecycle of a medicinal product, which include the registration
procedure and the subsequent maintenance phase (Heidenreich, 2004). The main goal
of both the pharmaceutical industry and the health authorities is to establish a positive
benefit/risk ratio and an appropriate benefit/risk management system for medicinal
products. Pharmaceutical companies should contact the health authorities at an early
stage in the development in order to achieve this goal. Early and frequent dialogue
between these two stakeholders can build a good relationship between the future
assessors of the marketing authorisation applications and the responsible managers of
the company. This may rule out problems and issues with the development of the
product and thus reduce the risk of an application been rejected.

191
The result will be more rapid access to essential priority medicines, including
important new treatment options such as generic fixed-dose combinations of assured
quality and affordable prices by the communities and patients in Gulf States. National
drug regulatory authorities need to be better equipped to register medicines in a cost
effective and timely manner and this will lead to increased efficiencies across GCC
Countries. Capacity building will enhance the quality of the registration decision,
whilst streamlined processes and improved information management lead to effective
medicines control. As a result drug regulatory authorities may enjoy substantial
savings and increased patient reach with generic equivalents (in the context of high
volume and high value essential medicines. Beyond this, Governments could enjoy
additional economies of scale via pooled procurement which is only possible when
the same medicine is registered across all participating countries.
OBJECTIVES
The main objectives of this comparison were to:
Identify the similarities and differences between the experiences of regulatory
authorities and pharmaceutical companies with the Gulf Centralised
Procedure.
Develop a new model for the GCC-DR based on evaluation and the outcome
of the centralized procedure.
Propose a business plan which would enable the implementation of the new
model
METHODS
An assessment of the Centralised Procedure was carried out with two groups of
stakeholders in the GCC. The first study Gulf Assessment of Centralised Procedure
(GACP) evaluated the experience and opinions of all seven Gulf regulatory authorities
with the GCC Centralised Registration. The second study Companies Assessment of
Central Procedure (CACP) evaluated the experiences and views of pharmaceutical
companies with respect to the centralised registration review process. In this chapter,
a comparative analysis between the GACP and the CACP is presented to highlight the

191
issues and challenges faced by these stakeholders as a prelude to the
recommendations to be made to the GCC-DR. Information from chapter 5, 6 and 7
was used to develop the new GCC-CP model and proposed business plan which
would help to implement the new model.
RESULTS AND DISCUSSION
The results of this comparison will be presented in three parts:
Part I: The Similarities and Differences between the Experiences of the Drug
Regulatory Authorities and the Pharmaceutical Companies.
Part II: The proposed model for GCC-CP.
Part III: The proposed business plan which would help to implement the new GCC-
CP model.
Part I - The Similarities and Differences between the Experiences of
the Drug Regulatory Authorities and the Pharmaceutical Companies
At the introduction of the GCC-DR system in 1999, government officials were
concerned about losing sovereignty to the centralised authority (Hashan, 2005). The
central registration system has faced criticism for some aspects and approval for
others from both the member states of the centralised authority and the
pharmaceutical companies dealing with the system. An interesting development
which contributed to the improvement of the GCC-CP was the appointment of
“Cameron McKenna and Andersen Consulting” to audit the EMA's centralised and
decentralised drug registration procedures. An EMA audit survey mandated a review
of that agency's activity within six years of its inception.
The report of this survey namely the Evaluation of Community Producers for the
Authorisation of Medicinal Products was published in October 2000. It was based on
interviews with and/or questionnaire responses from trade associations, national drug
registration authorities, professional and patient associations, national ministries with
interests in drug approval and pharmaceutical companies. The audit report contained
the following summary of survey responses:

192
1) The centralised and decentralised procedures are both perceived to have
contributed to the creation of a harmonised community market for
medicinal products.
2) There is some criticism of particular aspects of both systems. However, in
general, the two systems provide a strong foundation for future progress to
a harmonised and efficient regulatory environment. Because of their
different attributes, there is a strong desire on the part of both applicants
for marketing authorisations and the competent regulatory authorities to
maintain the parallel systems.
The survey examined many aspects of both the centralised and mutual recognition
procedures. The auditors did not limit their research to the EMA's activities, but they
investigated member states' response to EMA decisions. This audit survey examined
the level of satisfaction with the Centralised Procedure. Of the 32 companies that had
obtained marketing authorizations by this route, 88% were satisfied with the system
and 3% was very satisfied. These results are identical to the outcomes of this study.
The member states regulatory authorities of EMA were more enthusiastic about the
Centralised Procedure with 73% being satisfied while 20% were very satisfied with its
operation. These results from Europe also mirrored the outcomes of this study in the
Gulf States.
A well coordinated, reliable centralised regulatory framework effectively reduces the
administrative burden and duplication of scientific evaluations by participating
member states. The companies believe that if this procedure was streamlined, was
faster and transparent it could be the system of choice. As the documents for a
medicinal product are submitted as a common single registration, this results in cost
saving and an efficient follow up opportunity in all the seven Gulf States. At the
beginning of 2014, the GCC-DR will initiate the CTD electronic submission (as NeeS
[Non-eCTD electronic Submissions] format), which is currently implemented by
internationally recognised agencies and as from January 2015 only the eCTD format
will be accepted. This approach is useful in that it assists the pharmaceutical
companies in understanding the rules of the submission process and thus helps both
the industry and the authority to make better decisions (TGA, 2009). The findings
from this study highlight the ten years achievement of the Centralised procedure and

193
its future potential as identified by the health authorites as well as the pharmaceutical
companies. The pharmaceutical companies shed some light on the advantages and
disadvantages of the centralised system which were similar to the views of the
national regulatory authorities of the member states, although in some ways they were
different. Both the regulatory authorities and the pharmaceutical companies agreed
that the centralised procedure is an effective system for authorizing the marketing of
medicinal products in all of the GCC countries in one procedure and is the way
forward, but there is room for improvement in the procedure and the follow ups.
With regard to submission preference the majority of the pharmaceutical companies,
which took part in this study, prefered the review at a national level mainly due to
their experience with the national regulatory authorities, therefore making it easier to
gain market authorization. The long duration of the CP process contributes to the
preference for the national submission as well as the fear of rejection by a centralised
authority. The preference of the pharmaceutical companies to apply to national
authorities is confirmed by the responses of the national regulatory authorities which
stated that the number of national applications was greater than those submitted to the
centralised authority adding to the workload of the national regulatory authorities in
addition to their CP workload.
Despite the preference of the pharmaceutical companies for national submissions,
they find the registration at the CP to be better and more efficient. Ease of
communication has been reported for both the national procedure and CP although
there is no direct access to CP by the pharmaceutical companies. Most of the national
regulatory authorities of the member states concur with the fact that the duration of
the process to obtain market authorisaiton through the centralised procedure should be
shorter to make it more appealing to applicants. The duration of the centralised
procedure could be shortened by several means which include increasing the number
of CP committee meetings per year which has been clearly recognized by both the
pharmaceutical companies and the national regulatory authorities. The lack of
resources, mainly human expertise on the side of national regulatory authorities,
contributes as well to the delays even though there is effective collaboration and
sharing of information between the national regulatory authorities. Recruiting external
experts can aid the national regulatory authorities with the shortage of human
expertise but there is a fear that this will impact the quality of the review. The demand

194
for additional requirements by different national regulatory authorities adds to the
delay and length of the process. The most effective way is to optimise the process of
evaluation by the centralised authority by finding a more appropriate model.
According to the regulatory authorities‟ views, standardisation of the system would
also be a means to improve and facilitate the regional registration process and
maintain the supply of safe and effective medicines within a reasonable time frame.
The national regulatory authorities in the Gulf have found the shared experience, ideas
and knowledge at the centralised meetings to have enriched their own experience and
knowledge which is reflected in their performance at the national level. The clear
outcome of this collaboration and joint effort between the national regulatory
authorities produces an improved scientific opinion and a higher quality of decision-
making. Pharmaceutical companies have also indicated their agreement with this
where they found consistent evaluation of submission and a better scientific opinion
received from the centralised authority than from the national regulatory authorities
leading to a more efficient system.
One major aspect of CP approval reported by both the pharmaceutical companies and
the member states is the centralised pharmacovigilance procedures and reporting
system which are efficient and effective. In addition, member states of the centralised
authority find the GCC guidelines to be sufficient and appropriate for their intended
purpose. On the other hand, pharmaceutical companies, submitting to the centralised
authority, struggle due to the absence of appropriate guidelines leading to
inconsistency. Also there are differences when accessing the market at a national level
such as re-requesting documents already submitted for the CP or not even approving
the product for distribution after attaining GCC-DR registration.
Certain aspects of the registration procedure can be modified in order to move
forward and improve the quality of the experience. Both the national regulatory
authorities of the member states and the pharmaceutical companies have agreed that
the use of electronic on-line submissions to the centralised authority, as well as
electronic communication between the member states, is one way to expedite the
process. This is the current method implemented in the recognised established
authorities worldwide such as EMA and FDA. Further, a recognition of the
assessment by established mature regulatory authorities through implementation of
the verification procedure is a means of improving the process and easing the burden

195
of the high workload on the national regulatory authorities and for the pharmaceutical
companies as well. Harmonisation of requests and clarifications sent to the
pharmaceutical companies is yet another way agreed by both the pharmaceutical
companies and the national regulatory authorities. The national regulatory authorities
identified the need for training, seminars and workshops to improve the experience of
their expert staff. Pharmaceutical companies have suggested the same where the
additional interactions between pharmaceutical companies and the national regulatory
authorities would improve the overall knowledge of the national regulatory authorities
especially as the pharmaceutical companies have worldwide experience. Also it would
further enhance the relationship and communication between them. The
pharmaceutical companies even suggested helping to draft, improve and refine
guidelines including GMP. One of the key concerns, as highlighted by the companies,
is the limited number of committee meetings per year. The quarterly meetings to
validate the pharmaceutical products are clearly insufficient to meet the increasing
demands for product registration and approval. The companies recommended that the
frequency of the GCC – DR committee meetings needs to be at least 8 per year or
every 45 days. In addition, sub-committees could be formed with more frequent
meetings to handle specialised matters such the registration of life saving products.
The companies recommended that the comments and opinions of the countries, where
the products have been registered nationally, should be considered when products are
being evaluated by the GCC-DR. In this way, the approval process would be
expedited and this would facilitate faster approvals at a national level after the
products have been approved by the GCC-DR. Over half of the companies supported
the GCC-DR procedure while five companies out of thirty prefered the national
registration system. The main reason companies move toward local registration is
because of the delay and queuing at CP. Nevertheless, the companies agreed that if
the GCC-CP improves it will increase its acceptability of the marketing authorisation
issued centrally in other markets. They also suggested the use of University
laboratories to speed up „product testing‟ against additional fees.
The only way to improve support for the GCC registration is to give some advantages
over the national registration for companies who choose this route. It is very
important to note that most companies are seeking to establish a single registration
system, either national or centralised, for their products. Dealing with two registration

196
systems in the same region is impractical. For all these reasons, companies usually
adopt the most economically viable registration system that meets their corporate
objectives. In conclusion, figure 8.1 summarises the key issues to be consider in the
new proposed GCC model.
Figure 8.1: Key issues to be consider in the new proposed GCC model
Part II - The proposed model for GCC-CP
The time taken to register a pharmaceutical product differs from country to country
and from product to product. However, it is possible to complete the review process
within a reasonable time frame if the data are available and adequate. Many countries
have legislative maximum times allowed for the review of a dossier. For example, the
target time-frame for completing the review process in the EU centralized system is
210 days, although the clock is stopped if it is necessary to ask the applicant for
clarification or further supporting data (EMA, 2009). This compares to the median of 304
Enhance the relationship
and communication between the stakeholders
Standardizatio
n of procedures
Increase in the number of committee meetings
On-line
submission
Improve the outcome of the review
Registration time should
be shortened
Verification procedure is a
means of improving the
process

197
days taken by the US FDA (CIRS, 2013). Several challenges faced the GCC health
authorities to successfully operate the centralised system. Therefore, they encountered the
need for regulatory reforms during the last 12 years, in order to improve their individual
systems and to unify their procedures to expedited improved patients‟ access to high
quality medicines throughout the region. The GCC-DR challenge in the beginning was to
convince companies to consider submitting their dossiers via this procedure. The
pharmaceutical companies still have mixed feelings about whether they can gain faster
marketing authorization through the national regulatory systems or the regional
centralized system. After all, the goal of any pharmaceutical company is to complete the
registration requirements and gain access to the national GCC markets in the shortest
possible time.
It is crucial for the GCC states to identify the most prevalent driving forces for improving
the centralised procedure and to attract the pharmaceutical companies to use this system.
From the results of this study and the information in chapter 5, 6 and 7 a model can be
proposed based on the current GCC-DR centralised procedure (Figure 8.2) and taking
account of the comments in part I of this chapter. The model‟s main features are as
follows:
Maintain the main structure of GCC-DR centralised procedure.
Amend the parts that are time consuming in the current GCC-DR centralised
procedure.
Allow all member states to participate in the review process and the conclusions
reached in a timely manner.
Adopt an electronic system of review for the proposed model to overcome
infrequency of meetings and the possibility of all member states participating in
the review process in a remote manner.
Adopt electronic communication with applicants for deficiencies for improved
review process.
Adopt a clock-stop timing for the review process based on the automation of the
review process.
This model (Figure 8.3) is based on the current GCC review process. It maintains the
main features of the approval process namely receipt & validation, management of the
overall dossier review cycle, registration approval by heads of authorities and upholding
the independence and sovereignty of member states within the approval process.

198
However, the proposed model eliminates the steps in the approval process which are time
consuming and present redundancy in the process. The main step to be eliminated relates
to the questions raised by member states following the review. Clearly this step raises the
question as to why member states would raise questions that are not highlighted by the
two reviewing authorities. This at best produces doubts regarding the review capabilities
of certain member states while at worst raises doubts about the GCC review process in
general. On one hand each member state must be allowed the possibility to state their
views regarding the application, while on the other hand there is a need to achieve review
time efficiency. To realize such a target the model proposed is as follows:
A. Receipt phase: Receipt of the application is carried out in accordance to the
published GCC guidelines for NAS‟s and EAS‟s. The applicant should request a
submission date for the application which would be provided by the GCC office. It is
of importance that the applicant would provide a statement that the application being
submitted has observed all the relevant GCC guidelines. This statement has a
functional effect on the clock-stop as it is quite frequently observed that applicants
submit partially completed dossiers, especially the local industry, making promises to
bring missing or complete data at a later date. The applicant must be held accountable
in such cases and this can only be done according to local laws if such documents are
provided, otherwise the receiving officer will usually be held responsible.
Furthermore, the statement will serve as time zero for the “clock-stop”. The receipt
process should be conducted at the GCC Executive Office and performed by GCC
staff. This phase is actually maintained as the current practice of application receipt at
the GCC office.
B. Validation phase: The validation phase may be critical for agencies like Health
Canada, US FDA or EMA, when many dossiers are submitted by the applicant
whereby the submission is screened to ensure that the data are complete and of
suitable quality for review. In this phase for the GCC-CP the requirements are
checked against the requested formal requirements. This should include the
administrative registration (reference number) and checks on legal requirements, the
status of the company, manufacturer etc. as well as a „checklist‟ validation of the
application content (e.g. technical sections, CPP status).

199
Figure 8.2 The current GCC-CP registration system

211
Figure 8.3 The proposed model for GCC-CP

211
Further checks on patent compliance are necessary and any contractual
arrangements such as contract manufacturing or licensing agreements are also
checked. The allocated clock-stop time for both processes of administrative and
legal formalities should be 30 days. The validation processes are carried out at the
GCC office. In effect the validation process should make sure that the submitted
application is in compliance with the formal requirements as stated in module 1 of
the CTD specifications together with further checks that all the required technical
data is provided. This phase is maintained as the current practice of application
validation by the GCC office.
C. Review phase: The scientific assessment stage is the major part of the review
process and requires considerable amount of evaluation of data relevant to the
safety, efficacy and quality of the pharmaceutical product. Therefore, it is
essential to focus the attention on providing the appropriate skill sets and the
facilities as well as establishing guidelines, SOPs, training and continuing
education programmes and the electronic handling of the review process to
implement the desired good review practices (GRPs). In the new proposed model
the scientific review should be overhauled to allow all member states to
participate in the review process in a one-step approach. This necessarily means
that steps (C), (D) and most of (E) of the current GCC model of practice are
merged into one step in the proposed model. In this phase, two committees are
established from all member states. Therefore each member state is represented in
each of the two committees to ensure the active participation of all states. The two
committees are as follows:
a. Clinical committee: this committee should consist of experts from the
member states and should be responsible for reviewing the clinical
requirements for both NAS‟s and EAS‟s dossiers. In effect this committee
is concerned with safety and efficacy/modules 4 and 5 of the approved
GCC CTD standard.
b. Technical committee: this committee should consist of experts from the
member states and should be responsible for reviewing and approving
API‟s and excipients in the formula as well as the finished product. This
committee is very similar to the EDQM committee in the EU in addition

212
to the technical review committee in respective EU member states. This
committee would focused on module 3 of the GCC CTD standard.
The clinical and technical committees cover all aspects with regard to the safety,
efficacy and quality of the product while any legal aspect that may relate to the
product would have already been checked during the application validation
process. The clinical and technical committees approach to the product‟s review
compared to the two step approach of the current model would lead to
considerable time saving and review process efficiency and transparency. Each
member state would be able to state their approval or concerns during the review
process in that particular committee. A letter of deficiency relating to that
committee would be issued to the applicant with a time limit for the applicant to
respond. The committee‟s meeting time can be organised in a manner to allow for
the review of new applications while keeping about 30% of the meeting time for
replies to issued letters of deficiencies. The committees should meet at least once
a month to keep the process efficient and timely. The allocated clock-stop time
for both committees concurrently is 150 days. The tasks performed in the
committees are done primarily by members of staff from health authorities of the
member states and not from the GCC office staff.
c. Laboratory analysis: the required laboratory tests should be performed
concurrently with the application review process of both clinical and
technical committees. The allocated clock-stop time for the laboratory to
complete the required tasks is 120 days which is well within the 150 days
clock-stop time allocated for both clinical and technical committees. This
should be the default and implies that the work of both clinical and
technical committees and the laboratory is harmonized and synchronized
to complete the work within the 150 days time limit.
D. Approval phase: Once the two committees grant approval for their part as well as
the laboratory decision to approve the product, their respective decisions are
transferred to the head of authorities meeting to give the approval to issue the
registration certificate. If, for any reason, the heads of the authorities are
concerned regarding any aspect of the application they will issue a letter of
concern to the appropriate organisation which might be the GCC management

213
staff, specified committee(s), the laboratory or the applicant. Once the concern is
answered then an instruction to issue the registration certificate is mandated. The
heads of authority‟s approval is to check that the approval process steps and an
adequate review is observed. This may represent a quality assurance task
performance to guarantee good review practices at the GCC office. The allocated
clock-stop time for this phase should be 30 days and the tasks are entrusted to the
heads of health authorities of the GCC member states. The heads of authorities in
this case should be considered as the approval council (head of agencies) and they
should meet, every month. The allocated clock-stop time for this committee is 30
days. This takes into account the meeting frequency of this committee making
sure that once the applications passes all requested formal and substantive
requirements the application‟s final approval by the heads of authorities shall be
compliant with the clock-stop. Once the approval is granted by the approval
council then the GCC office should issue the registration certificate for the
product under application.
In order for the above proposed model to function the following are required:
a. Clear guidelines for the review process are laid down by the council of
heads of authorities. Good review practices should be mandated in such
guidelines. The GCC should elect one authority to host a training academy
for all aspects related to the sciences and skills needed by the GCC and
member states staff involved in the review process.
b. Electronic management (Computerisation) of the whole process of the
review including an electronic review system which allows committees to
hold meetings, while each committee member is in their office, utilising
modern computer software and communication tools and devices. Using
modern meeting techniques will result in cost savings, process efficiency
and significant review time saving. Further electronic process
management will enable external experts to participate in meetings of the
various committees if required without the need to make travel
arrangements giving expert support to the work of the GCC reviewing
process.

214
This new model would allow the GCC to adopt a clock-stop approach to the review
process. The GCC‟s main bottle neck is the “questions to the applicants” raised by
non-reviewing member states. This proposed model eliminates such a bottle neck
through the participation of all member states in the reviewing committees. The time
saving should allow GCC to promise 210 calendar days for NAS‟s and of course
fewer days for EAS‟s is expected, but at worst would be the 210 days.
However, adopting the clock-stop dictates that the applicant‟s expected response time
and date is indicated to the applicant. Failing to meet the response date by the
applicant should allow the GCC to remove the application from the review process
after warning, in order to reduce the review pipeline. Clear guidelines for the clock-
stop concept and the use of such a concept should be clearly documented and
announced.
The laboratory analysis requirement should be performed in a manner that eliminates
delays in the analysis process. This is based on member states laboratories declaring
their readiness to meet the clock-stop requirement and availability of time for
analysis. Even though each of Kuwait, Oman, UAE and KSA Quality Control (QC)
laboratories are well equipped for conducting analysis of pharmaceutical products,
member states should agree on accreditation criteria for QC laboratories in order to
officially identify that these laboratories are able to meet these accreditation criteria.
In addition, each member state with the ability to meet the accreditation criteria
should also be able to accommodate 120 days clock-stop to complete the assigned
task. Analysis tasks are assigned to laboratories that declare their ability to meet the
clock-stop requirements and tasks are given to member states laboratories in order to
give analysis tasks to member states laboratories in a fair manner. This would allow
all participating laboratories equal opportunities to contribute to the process of
product approval in the GCC system. In this way, laboratory tasks, assigned in this
model, would eliminate the long analysis time and undue waiting experienced
currently.

215
Part III - A proposed business plan underpinning the implementation
of the new GCC-CP model
Quality management practices are an integral part of several established drug
regulatory authorities (eg. EMA, FDA) including governance configuration and its
business processes. These practices help to ensure that the agency operates to
consistently high levels of quality, efficiency and cost-effectiveness. In terms of
financial resources, it is worth investigating the impact of different budget structures
on performance across authorities (Anderson, 2007).
The agency charges a fee for processing applications from companies that want to
bring a medicine to the market. It also charges fees for services related to the
marketing of medicines in the EU in areas such as scientific advice, inspections,
renewals and variations and the establishment of maximum residue limits. The total
budget of EMA was 194.4 M€ in 2009 (EMA, 2010) and it is expected to be 239.1M€
in 2013. The budget process allows the EMA to cover its needs, mainly by fee
revenue which is subject to fluctuation.
The pharmaceutical industry considers EMA fees fair and appropriate „to the services
provided‟. On the other hand the US FDA's federal budget request for fiscal year (FY)
2012 totalled $4.36 billion (El Boghdady et al, 2012). About $2 billion of this budget
is generated by user fees and pharmaceutical firms pay the majority of these fees,
which are used to expedite drug reviews. Even though the procedures and
classifications differ considerably between EMA and FDA, the majority of
stakeholders consider both agencies provide high quality evaluations for their
products. It is noteworthy that the European pharmaceutical market size is 2/3 of the
US pharmaceutical sector.
The EMA fee structure (EMA, 2010) appears complex in comparison with the FDA
fee structure; there are specific cases for three different types of variations, such as
type Ia, Ib, and II, beyond extension, additional fees for specific strengths or
presentations, as well as referral fees. Several fee waivers favouring generics,
biosimilars, orphan and paediatric products, small and medium sized enterprises exist
in the EMA structure.

216
On the other hand, the FDA levies establishment fees (US$, 457,200) every year from
the authorised drug manufacturers. In a similar fashion, FDA also gives fee waivers
like EMA, in special cases. Spending on medicines will reach nearly $1,100Bn in
2015 (IMS, 2012). The U.S. share of global spending will decline from 41% in 2005
to 31% in 2015, while the share of spending from the top five European countries
(Germany, France, UK, Italy and Spain) will decline from 20% to 13% over the same
period. In contrast to EMA, the FDA fee requirements include application fees for
previously filed applications or supplements , fees for designated orphan drugs or
indications ( both for prescription products or for a supplement) .
The FDA user fee programmes support the safe and effective review for human and
animal medicines, biological products, medical devices and the review of other FDA-
regulated products. User fees also allow FDA programs to achieve enhanced
premarket review performance. Other FDA user fees support the regulation of tobacco
products, the inspection of mammography facilities, the certification of color
additives and the certification of FDA-regulated products exported from the United
States. New user fees enacted by the FDA Food Safety Modernization Act support
essential food safety activities. The budget includes inflationary increases for FDA
user fee programmes, as authorized by law.
The Prescription Drug User Fee Act (PDUFA), enacted in 1992, authorizes FDA to
collect fees from companies that produce certain human drugs and biological
products. Since the establishment of PDUFA, user fees have played an important role
in expediting the drug approval process. On July 9, 2012, the President sanctioned
the Food and Drug Administration Safety and Innovation Act (FDASIA), which
includes the reauthorization of PDUFA through to September 2017. PDUFA V will
provide for the continued timely review of new medicines and biologic license
applications in the US. The new law ensures that FDA will continue to receive a
source of stable and consistent funding during fiscal years 2013-2017 and allow the
agency to fulfill its mission to protect and promote public health by helping to bring to
market critical new medicines for patients.

217
Government support in the form of a budget is the method of financing employed in
most of the GCC States, however, only Saudi Arabia and Yemen are self-financed by
fees. Governments are committed to support the GCC authorities financially and
prioritise funding of the regulatory review process. In any case, arrangements should
be made so that the financial sustainability is maintained for continuous and effective
implementation of the various drug regulatory functions (Ratanawijitrasin, 2002).
The quality of the review process, the technological and scientific skills
improvements remain a limiting factor without Governmental support (Hashan, 2006).
The range of fees, however, varies significantly from country to country according to
the funding structure and the services provided by each authority. The relatively high
registration fees charged by the Saudi Food and Drug Authority (SFDA), compared to
the other GCC authorities, are related to the autonomous support of the SFDA for its
own practices, facilities and services through the direct access to the fees, rather than
being collected by the central government revenue as in the other GCC States which
may or may not be returned to the authority undertaking the work (Hill and Johnson,
2004).
The Yemen regulatory authority is an autonomous authority as well and therefore the
registration fees are slightly higher than those charged by the rest of the GCC States,
but considerably lower than SFDA probably to attract pharmaceutical companies to
the local market in Yemen. It is agreed by all the GCC authorities that in order to
improve the regulatory review process, the authorities need to increase their resources
such as the number of experts, developing the information technology structure and
establishing training and continuous education programmes.
Without appropriate funding, the authorities will always face difficulties in improving
their regulatory systems. It can clearly be seen (Table 8.1) the parity among member
states' fees ranging from no fees in Qatar to as high as USD 25,341 in Saudi Arabia.
This wide difference in the range of the requested fees at national level from each
member state will cause those with higher national fees to resist the GCC Centralised
Procedure.

218
Table 8.1: Registration fees in the GCC States
*The proposed fees structure calculated according to the ratio of current national fees to the total fees for all member states in addition to 10% as administration fees. (The basic fees in EMA for a single strength= 274,400 Euro) (FDA New Drug
Application (With Clinical Data) fees for 2014 fiscal year (FY) =$2,169,100).
It is recommended that if the proposed model is implanted then each member state
should decline their national fees in favour of GCC-CP fees and that the GCC-CP fees
should be shared amongst member states where each share is calculated according to
the ratio of current national fees to the total fees for all member states in addition to
10% as administration fees. For this model to be feasible in the future, the overall fees
of the GCC-CP must be increased. The increased fees would allow member health
authorities to benefit from these application fees and for tasks they perform during the
application approval process. Further to the above, the fees for analysis should be
awarded to the assigned QC laboratory of the member state performing the analysis
task. Training is a key element for capacity building for the success of the proposed
model. Training should be carried out to achieve higher performance with improved
efficiency for member states for the review of GCC-CP applications. The training fees
should be awarded to the member state hosting the training activity. The comparison
of the views from pharmaceutical companies and regulatory authorities has identified
the most important outcome as the desire for improving the GCC centralised
procedure model.
Bahrain Kuwait Oman Qatar Saudi Arabia UAE Yemen GCC-
CP
Proposed
GCC-CP
fees
Company
Registration
5 BD =
USD 13.3
250 KD =
USD
887.2
100 OR =
USD
259.7
NO
FEE
(ONLY
INSPECTION
FEE)
1000 DH
=
USD 272.3
3000 USD
10000 SR
=
USD 2,667.5
(ONLY INSPEC-
TION
FEE)
Products
NASs
5 BD
= USD
13.3
100 KD
= USD
354.9
75 OR
= USD
194.8
NO
FEE
95,000 SR
= USD
25,340.8
1000
DH =
USD
272.3
1500
USD
10000
SR =
USD
2,667.5
USD 30,4444*
Products
EASs
5 BD
= USD
13.3
100 KD
= USD
354.9
75 OR
= USD
194.8
NO
FEE
40,000 SR
= USD
10,669.8
1000
DH =
USD
272.3
1500
USD
10000
SR =
USD
2,667.5
USD 14,305*

219
SUMMARY
Agencies and companies agreed that the centralised procedure had
contributed to an effective system for authorising medicinal products in the
GCC by providing the best possible scientific opinion for the quality of CP
decision-making as compared to the national system.
Enhancing sufficient resources like qualified and trained experts together with
technical facilities would improve the quality of the GCC regulatory
performance.
It is crucial for GCC states to identify the most prevalent driving forces for
improvement to the GCC centralised procedure such as on-line submissions
and an increase in the number of committee meetings in order attract
pharmaceutical companies to use this system.
Quality management practices are an integral part of several established drug
regulatory authorities and its business processes. These practices help to
ensure that an Agency operates to consistently high levels of quality,
efficiency and cost-effectiveness.
The overall fees of the GCC-CP should be increased in order to allow member
health authorities to benefit from application fees and for tasks they perform
during the application approval process.
The comparison of the views from pharmaceutical companies and regulatory
authorities has identified the most important outcome as the desire for
improving the GCC centralised procedure model.

211
CHAPTER 9
General Discussion

211
A systematic examination of pharmaceutical regulation and its environment across
countries can help shed light on a country situation, provide a new perspective on the
constraints facing it and suggest options for improvement. A comparative approach is
used in this study, on the grounds that countries can benefit from learning from one
another. There are three basic reasons for conducting systematic comparisons between
countries (Booz Allen Hamilton, 2006).
Strategy development: comparing different ways of managing similar
problems can provide both positive and negative lessons, i.e. guidance on what
to do and what not to do. Comparing cross-country experiences is a useful way
of developing policy instruments for problem-solving in a particular country.
Understanding: comparing public policies can help improve understanding of
how government institutions operate within their environment and suggest
possibilities for improvement.
Interdependence: the interdependence of nations as reflected in international
agreements, regional political-economic groupings, bilateral treaties and
collaboration is constantly increasing. Accordingly, problems that occur in one
country can spill over into other countries more easily and rapidly today than
at any other time in history. Similarly, policies adopted in one country often
have important implications for others. In other words, knowledge about what
has occurred in other countries can help a country prepare for new challenges
of its own.
Making a positive decision about a pharmaceutical product requires careful weighing
up of the potential benefits and harms and the scientific complexity of such decision-
making should not be underestimated (Hill, 2004). From the authority‟s perspective,
success is the licensing of quality medicines to enable patients to have access in a
reasonable time frame without compromising safety and/or efficacy (Hashan, 2005).
Therefore, a thorough review is carried out by the assessors with particular emphasis
on the quality, safety and efficacy studies. The use of ineffective, harmful, poor
quality medicines can result in therapeutic failure, deterioration of the disease being
treated, resistance to medicines and sometimes death. It also undermines confidence

212
in the health review system, health professionals, pharmaceutical manufacturers and
distributors (Rago and Santoso, 2008). The complexity and challenges of the drug
registration system should not be overlooked. It is simplistic to believe that speeding
up the registration process will improve patients‟ access to quality, safe and effective
medicines. This is the industry‟s argument but neglects to take into account important
issues such as the quality of the review. Given the pressures that arise from the
legitimate business interests of the multinational pharmaceutical manufacturers, there
needs to be support for regional activities designed to ensure the quality in the
registration systems. This includes ensuring that there is adequate capacity to cover
the required resources and appropriate guidelines at a country level to assess and
control the quality of medicines.
Information sharing and harmonisation reduces workload and improves overall
regulatory performance. Harmonization is the value added that directs expert
knowledge and resources to functions that improve public and personal health and
facilitates access to essential medicines. Formation of effective networks among
national regulatory authorities participating in various harmonization initiatives
facilitates sharing of scarce resources; eliminates duplication of activities; saves
money for all; supports cooperation, collaboration and international understanding;
facilitates in building regulatory capacity and enhances public trust in regulatory
efforts.
The Gulf States faced significant challenges in dealing with their established
regulatory bodies who were reluctant to give up their independence to the newly
established GCC Central Drug Registration (GCC-DR) system (Al-Essa, 2011). The
idea of implementing a central registration system came originally from the
industrialised world, especially from the success stories of the European Medicine
Agency which encouraged gulf regulatory organisations to structure the central
registration system. The centralised process of the European Union drug regulatory
agencies under one roof resulted in the development of EMA which has attained
improved trust, transparency, communication and mutual recognition through the
unification process. The European approach to national and central procedures differs
greatly from that adopted in the GCC. The difference is conceptual and structural
which is reflected in the procedural aspects and furthered the outcome of the

213
European approach. The missing link in the comparison between the European
approach and the GCC approach is the following:
1. The European system was initiated based on the need for mutual recognition
between some member states. This lead to the mutual recognition procedure
between some member states such as UK, France, Germany, etc. The system
was further refined to the centralised procedure covering all member states. In
this evolution the European approach presented a coherent system for review
of medicinal products with standards acceptable to all member states. The
system has inherent flexibilities for applicants to choose the national route for
submission or the centralised procedure. Furthermore, the system allowed
applicants to switch from the centralised procedure to the decentralised
procedure. On the other hand, the GCC system evolved from the need for
communal procurement of medicinal products not from mutual recognition
needs. The foundation of the system is based on the GCC tendering procedure
rather than communal review system. The review standards and consequently
review quality leads to a non-harmonized system between member states. It is
this non-harmonized system that led to the difficulties expressed clearly by
GCC member state staff. In order to overcome this difficulty most member
states are advocating national harmonization with GCC-DR.
2. The European system of review and approval leads a formal grant of
marketing authorization for applicants without the need for follow up
submission by member states. In this regard the centralised procedure in
Europe represents a viable alternative to submission in multi-states in the EU
while the GCC-DR does not offer the same opportunity and applicants need to
go through the national procedure as well as following the central procedure
approval.
3. For a review system to be effective all elements of the review system must be
clearly defined through guidelines and procedure. One of the most important
elements in any review system is the pricing guideline and procedure. The
GCC-DR does not include the pricing element which is of course emanating
from the intended purpose of the GCC-DR being the unified route for
tendering in the GCC procurement of medicinal products.

214
4. The European Medicines Agency (EMA) has established systems of training
its own staff and extends it to the training of scientific experts. Training takes
into account entry level of review staff up to the continuous training of senior
reviewers. Even non-employed personnel have their own training (EMA,
2011). The main purpose of the training is to ensure the review process is
measured from different aspects namely effectiveness, efficiency, long-term
effectiveness on EU citizens, impact on EU internal market and on different
stakeholders (EMA, 2010). Training is the main tool to ensure a quality review
and the absence of a GCC-DR training system directly affects the quality of
the review procedure and consequently impacts all other features of the GCC-
DR system.
From the above it becomes evident that a comparison between EMA and GCC-DR is
not an objective assessment. Certain elements will look common between both central
procedures by virtue of them being central procedures e.g. the participation of
reviewers from all member states. However, for the EMA the criteria for a reviewer
appointment is based on review skills attested by EMA for any national member state,
while in the GCC-DR it is a formal procedure based on member state‟s discretion.
The anomalies between GCC-DR and EMA could render the comparison misleading.
The quality of the review process is a comprehensive and multifaceted concept
(Brown et al., 2002). Previous studies have concluded that it is not sufficient to
measure regulatory performance in terms of the speed of the approval process alone.
Maxwell (2004) indicated that: “Concern about quality of care must be as old as
medicine. But honest concern about quality, however genuine, is not the same as
methodical assessment based on reliable evidence”. Maxwell also identified six key
dimensions of the quality, and these are (1) Accessibility (is the service accessible in
terms of distance, time, and social barriers?) (2) Effectiveness (does the service
produce the desired outcome?) (3) Efficiency (does the service produce the desired
outcome with the least waste/at low cost?) (4) Equity (indicating the quality of access
to services) (5) Relevance to need (is the service appropriate to the needs of its users?)
(6) Acceptability (what is provided and the manner in which it is provided) (Moss,
2004). The quality of the regulatory review process, from the construction of the
dossier to the final regulatory decision must also be examined (McAuslane and Cone,

215
2006). The importance of implementing and maintaining Good Review Practices
(GRPs) are critical measures that need to be considered by the Gulf States to provide
consistency and to improve efficiency, clarity, and transparency of the review
process. It is important to adopt GRPs as standard processes through formal training
of the review staff (US FDA, 2009).
The GCC authorities were found to be carrying out a number of activities to bring
about continuous improvement in their regulatory review process. Various reasons
were stated for introducing quality measures into their activities but the most common
ones were to ensure consistency and efficiency and to minimize errors in the system
(Al-Essa, 2011). Therefore, quality systems should be regularly reviewed to ensure
that they are working effectively (Mallia-Milanes, 2010).
The purpose of this research has been to evaluate the regulatory review process in the
GCC central registration and its impact on patients‟ access to medicines in the GCC
States. The focus of chapter three was on evaluating the regulatory review process in
Oman in order to identify areas of improvements in the system. Chapter four analysed
the approval time over the period 2008-2010 in the GCC States. The evaluation of the
review times for New Active Substances (NASs) and Existing Active Substances
(EASs) in the GCC Centralised Registration system (January 2006 to December 2010)
was the focus of chapter five while chapters six and seven assessed the views and
experiences of the GCC regulatory authorities and pharmaceutical companies in order
to identify the strengths and weakness of the current GCC centralised procedure.
Finally, the similarities and differences between the experiences of regulatory
authorities and pharmaceutical companies with the Gulf Centralised Procedure and a
proposed new model for the GCC-DR was the focus of chapter eight.
The research began by thoroughly assessing the trend in the regulatory approval times
in Oman for the period 2006 to 2010. The findings showed that although there is an
increase in the approval time for all pharmaceutical company products, the median
approval time for the five year period was 117 days. This is within the time limit (4
months) which is fixed by the health authority for the overall registration time. Oman
is facing significant challenges reflecting the rapid advancement of regulatory
services with limited resources possibly influencing patients' timely access to
medicines. These challenges gave the regulatory agency no choice but to review and
update its regulatory practices. The ability to regulate medicines effectively is

216
determined by a number of factors including the availability of guidelines, good
written procedures and the provision of the appropriate resources to fulfill the
regulatory needs. The lack of resources can be compensated to some extent by
effective collaboration among countries and information sharing (WHO, 2008). The
Directorate General of Pharmaceutical Affairs and Drug Control (DGPA&DC) budget
is not separated from the MOH, so development is minimized, due to priority within
the government system. Increase in the registration fee and its use to develop the
authority could help to improve the quality of the review.
Another important aspect that needs to be highlighted in Omani regulatory practices is
the use of the verification model for the assessment of a new registration dossier. This
model is acceptable for the majority of applications if they are registered in countries
with recognized regulatory authorities. If not, it is critical to consider a thorough and
more specialized review for products which are not registered elsewhere and for
biotechnology and biological products. Singapore, for example, conducts a
verification review for all types of medicines which are previously authorized by at
least two reference authorities (e.g FDA, EMA), except for biological and
biotechnology products (Health Science Authority (HAS, 2011).
In a further study a downward trend was demonstrated in the median approval time
for most of the GCC States during 2008 to 2010. However, the approval time for all
approved products in the GCC States during this period varied from about 60 days in
Qatar and Oman (2009 and 2010), and to about 609 days in Saudi Arabia (2008).
There are many factors which can affect and explain the differences in approval time
in the GCC States. The main factor is the difference in the positions of milestones
within the approval process, for example, the analytical step starts with the scientific
assessment in Oman, Saudi Arabia, and Qatar (parallel procedure), but in Kuwait and
Bahrain the analytical step comes after the scientific assessment (sequential
procedure).
Regulatory approval times have also been influenced by the type of assessment
carried out by different authorities. Bahrain and Kuwait uses a verification review for
all types of products registered in countries with competent authorities. Saudi Arabia
performs a full review on all types of application while Oman and Qatar perform an

217
abridged review for all products. The GCC states should seek to increase the level of
funding to bring about the required expertise and resources to conduct a more
extensive review of important medicines, including biological and biotechnology
products. A clock stop is another important approach which is not fully enforced in
the GCC authorities to control the overall approval time. The EMA is obliged by the
regulation to reach a decision within 210 days, although the clock is stopped if it is
necessary to ask the applicant for clarification or further supporting data (EMA,
2009).
Furthermore, this research project investigated the pattern of total regulatory approval
times in the Gulf Centralised Procedure over the period from 2006 to 2010. It also
addressed the various factors which may have a positive or negative effect on total
approval times. During the five year period of the study (2006 – 2010), the median
approval time significantly increased due to several factors which included the limited
number of meetings by the GCC-DR and increased number of applications for
registration. Other factors that delayed the registration were the assessment review
process where two authorities are selected alphabetically to review the registration
dossier. In addition, the lack of a standard evaluation template for product assessment
leads to an increase in the correspondence which is sent to the sponsor in several
batches and at different times. It is recommended to have one standard assessment
template and rather than selecting the reviewing authorities alphabetically, the dossier
should be sent to all countries and all assessments combined into one final report on
which the final decision in the GCC-DR could be based. Other factors that could
speed up the registration process is an increase in the number of the committee
meetings and using information technology and e-mails in the communication
between the Executive Office and the companies.
The findings from this study also highlight the ten years achievement of the
Centralised procedure and its future potential as identified by the health authorites as
well as the pharmaceutical companies. Although, both the regulatory authorities and
the pharmaceutical companies agreed that the centralised procedure is an effective system
for authorizing the marketing of medicinal products, the pharmaceutical companies still
have mixed feelings about whether they can gain faster marketing authorization through
the national regulatory systems or the regional centralized system. After all, the goal of

218
any pharmaceutical company is to complete the registration requirements and gain access
to the national GCC markets in the shortest possible time (AL-Essa, 2011).
In the end, two approval routes, national and centralized, are permitted to exist side-by-
side in the GCC region. But for the centralized system to dominate, member states should
seek ways to increase their collaborative efforts to bring their systems closer towards
standardization that would facilitate the regional registration process and maintain
patients‟ access to safe and effective medicines within a reasonable time frame. It is
recommended that the comments and opinions of the countries, where the products have
been registered nationally, should be considered when products are being evaluated at
GCC-DR. In this way, the approval process would be expedited and this would facilitate
faster approvals at a national level after the products have been approved by the GCC-
DR. The only way to improve support for the GCC registration is to give some
advantages over the national registration for companies who choose this route. It is very
important to note that most companies require a single registration system, either national
or centralised, for their products. Dealing with two registration systems in the same region
is thought by some to be inefficient.
The GCC as a conceptual framework for harmonization among the member states has
been extended to areas like pharmaceutical procurement. In this regard the centralised
procedure was established as a step in the overall harmonization efforts of member states.
The GCC-DR is gaining momentum and importance for the private sector as reflected in
the responses of companies. Furthermore, the GCC-DR is contributing in a positive way,
especially in smaller member states although efficiency needs to be improved. Efficiency
can be clearly linked to the quality of decision-making during the review process and all
member states considered the GCC-DR review to be of the same quality as the national
review or better except for Saudi Arabia who considered the review quality as worse than
national review. Member states sought to find a solution to improve the review process by
utilising a standardised review template. This would avoid efficiency issues due to
requests for additional data. In addition raising the scientific and review skills of
participating staff from member states in the central procedure and by adopting clear
SOPs for the review process would be of benefit. Furthermore, harmonization of national
review guidelines and procedures within the GCC-DR would be an advantage as well.
Needless to say the positive influence of such harmonization will result in efficiency at
both national and central procedure level. For all these reasons, companies usually adopt
the most economically viable registration system that meets their corporate objectives.

219
The GCC-DR system can be better executed by the formulation of policies and guidelines
that are practical, concise and clear. A healthy interaction between regulatory agencies
and pharmaceutical companies at an early planning stage may boost the overall central
drug registration process. A competent registration process which is rooted in
transparency of procedures and unbiased scientific opinions would lead to a smooth drug
registration process among the GCC States.
Limitations of the study
As with any research there were a number of limitations including the following:
For the evaluation of regulatory review times in the Gulf States it was not possible to
obtain information of the review system or timelines for UAE or Yemen. This was
despite several e-mails and follow-up phone calls. Although for the sake of
completion it would have been ideal to have received data from both these States, it
did not have a detrimental effect on the overall conclusions.
The evaluation of the GCC Centalised regulatory review process only focused on
approved products. It would have been an advantage in reviewing the timelines to
have access to those products that were rejected, however, these were not available in
an electronic form and so would have necessitated identifying these manually. Again,
this would not have influenced the evaluation of the efficiency and overall
performance of the GCC-DR.
The evaluation of the pharmaceutical companies experiences with the GCC
Centralised Procedure was limited by the response rate of 30%. This was despite an
extensive follow-up of the companies who did not respond or who had limited
experience with the centralised system. In such circumstances, the problem is not so
much ending up with a low number, but rather the differences between responders
and non- responders.

221
Recommendations
As a result of this research there are a number of recommendations that can be made.
There is defiantly scope for improvement whether at national level or at the GCC-DR.
Although many, if not most, of the desired improvements are a common to all
member states some of them are specific to each member state. Common
recommendations for all member states include the following:
Adoption of harmonized regulations
Member states in the GCC-DR have collaborated to produce regulations that
govern the approval process at the GCC-DR. The regulations are in effect the
result of a common acceptance of the required standard for marketing
authorization of pharmaceutical products. It is logical for each member state to
adopt, at a national level, such regulations which would be the basis for greater
harmonization within the GCC-DR and among member states should mutual
recognition be contemplated between two or more states.
Training programmes
Common training programmes are needed for reviewers in all member states.
Such training programmes should provide the required knowledge and skills
for reviewers. In order for such programmes to be effective, acceptable criteria
for the attainment of training objectives must be agreed as well. This could be
at a national, inter-state or common for all GCC states. Only qualified trainees
should be allowed to progress to be official reviewers. Almost all developed
health authorities have similar programmes for training employees to be
reviewers and this is an essential element for any health authority.
Electronic process management
The adoption of computer based systems for task management is often absent
in most Gulf States despite the fact that they have adopted electronic
governance in their countries. Computerization is seen in almost all aspects of
life in the Gulf States and it is surprising that computer systems are not yet
used in many Gulf States for the regulatory review process. Computerizations

221
of process management remain one of the most challenging tasks for most, if
not all, GCC states. This task is assisted in that all member states and the
GCC-DR have adopted the CTD (Common Technical Document) standard for
dossier submission. Furthermore, the eCTD (electronic Common Technical
Document) standard is already published by ICH and should allow the Gulf
States to manage the computerization of the review process in an enlightened
manner.
Qualifying programs for Quality Control Laboratories (QCL)
In order to harmonize the standards for analytical procedures in each member
state, common standards must be agreed by member states for each state QCL.
This should be followed by appropriate training for all QCL staff and adopting
of common accreditation criteria.
Batch analysis
Like all Arab states, the GCC states perform release analysis for all submitted
products for approval. Further, the QCL performs release analysis for every
batch marketed. This places a burden on QCL in any country and prevents the
QCL from performing their main task of assuring the product‟s quality in the
market place. The GCC states need therefor to agree on the required function
of the QCL in member states which should be similar to the European
approach to QCL in European regulatory authorities.
Further recommendations are also suggested for the GCC-DR.
Adopt a new model for the regulatory review
As already outlined a new model is proposed for the GCC-DR. Irrespective of
whether the proposed model is acceptable or not, a new process model is
needed to improve the performance of the GCC-DR. Any model must take
into account the effectiveness and efficiency needs of the GCC-DR.

222
Proceed to full approval with pricing
For the GCC-DR to operate effectively the mandate foundation of GCC-DR
needs to be revised. The new mandate should state that the GCC-DR is the
way forward for approvals within the GCC states for medicine and health
products and this should include pricing of submitted products for approval.
Validation of authorized products in member states
To minimize the work load, the GCC-DR should adopt and validate approved
products already authorised in member states. In this regard the GCC-DR
should develop guidelines for such validation and seek member states
approval. The validation process should be a quality assurance process and
ensure that this is within an acceptable standard for member state.
There are also a number of recommendations for individual member state,
Harmonize regulations with GCC-DR
GCC-DR regulations should be adopted by each member state and this should
be a formality within the state legal framework.
Adopt mutual recognition
Member states should adopt mutual recognition with other member states with
a similar review system. This would allow the development of the GCC-DR
as an organisation similar to that of the European Medicines Agency.
Mutual recognition of QCL batch certificates
Member states should adopt mutual recognition procedure for QCL batch
certificates in order to reduce the work load and avoid unnecessary repetition
in samples analysis.

223
Exchange programmes for reviewers
Member states should adopt a programme of exchange for reviewers to allow
experience to be shared between member states.
Electronic processes
Member states should adopt the electronic management of processes and
documents. This will relieve the bottle necks in work performance including
the review procedures. Commercially available work flow systems, document
management system and electronic review systems are readily available and in
use by many health authorities worldwide but especially in Europe this has
been an established practice. Most European authorities including the
European Medicines Agency use validated software which is well accepted
and commercially available.
Future work
It would be of value to continue the evaluation of the Oman pharmaceutical
review process and analysis of the timelines for NAS‟s and EAS‟s for the
period 2011-2014. In addition it would be helpful to understand if there have
been any improvements in good review practice.
It would be advantageous to continue the evaluation of regulatory review
times in the Gulf States for the period 2011-2014 and to make sure that
responses are obtained from all states including UAE and Yemen.
As an evaluation of the GCC centralised procedure is fundamental to any
future model that might be implemented, it would be essential to update the
timeline data for products that have been reviewed between 2011-2014.
It would also be of value to repeat the evaluation of the Gulf States views and
experiences, as well as pharmaceutical companies with the GCC centralised
procedure over the past three years.

224
It would be of value both to the member states and the rest of the world to
understand from the seven Gulf States and the pharmaceutical industries what
are the benefits of this new proposed system as well as the challenges.
Conclusion
In this research the Gulf Cooperation Council centralised procedure for the approval
of pharmaceutical products was evaluated. The results demonstrated that the GCC-DR
centralised procedure has contributed to a better review process both at the GCC-DR
and national level. The GCC-DR needs to be revised and developed to an independent
marketing authorization granting authority providing applicants with one license that
covers all seven Gulf States. A new model for the GCC-DR CP has been proposed
which should resolve many of the difficulties facing the GCC-DR currently and
should improve its effectiveness and efficiency.

225
References

226
Alder, S. (2001) Methods for setting parameters for submission quality: How do we
Know id we have got them right? In: Hynes, C., McAuslane, N. and Walker, S. (Eds).
Building quality measures into the regulatory review process: Assessing the needs of
industry and regulators. Surrey: CMR International. Pg 39-42.
Al-Essa R. (2011). An evaluation of the regulatory review processes, the quality of
decision-making and strategic planning in the Gulf Cooperation Council (GCC)
States. The Welsh School of Pharmacy, Cardiff University. Doctor of Philosophy
thesis.
Al-Essa, R., Salek, S. and Walker, S. (2011) A comparison of regulatory review
processes and the factors influencing the quality of decision-making in the Gulf
Cooperation Council (GCC) States. Presented at the 9th
Middle East Regulatory
Conference (MERC), February 1-2, 2011. Amman, Jordan
Al-Essa, R., Salek, S. and Walker, S. (2012) An Appraisal of Good Regulatory
Review Practices in the Gulf Cooperation Council States. Drug Information Journal
46: 57-64
Al-Essa, R., Salek, S. and Walker, S. (2012) Regulatory Review Process in the Gulf
Cooperation Council States: Similarities and Differences. Drug Information Journal
46: 65-72 Anderson, C. (2004). "An evaluation of harmonisation in the Regulatory
environment and its impact on patients' access to new medicines." School of
Pharmacy: Cardiff. University of Wales. Doctor of Philosophy.
Annells, M., Deroche, M., Koch, T., Lewin, G. and Lucke, J. (2003). "District nursing
research priorities according to a panel of district nurses in Australia. Research
report." royal district nursing service of South Australia inc.
http://www.rdns.net.au/research_publications/dn%20priorities_section%203.pdf
Anon (2000). "Guidelines for planning effective surveys" statistical services centre
and the University of reading. 2005.
Anon (2001). "Approaches to the analysis of survey data" Statistical Services Centre
and The University of Reading. 2005.
Barrocliffes.S. (1994). Developing a global regulatory strategy, Drug Information
Journal, 28,525-531.
Booz Allen Hamilton Inc. (2006) Independent evaluation of FDA‟s first cycle review
performance-retrospective analysis final report. Available at:
http://www.fda.gov/ope/pdufa/PDUFA1stCycle/default.htm.

227
Booz Allen Hamilton Inc. (2008) Independent evaluation of FDA‟s first cycle review
performance Final Report. Available at: http://www.fda.gov /For
Industry/UserFees/PrescriptionDrugUserFee/ucm127117.htm.
Bourque, L.B. and Fielder, E.P. (2003a) How to conduct self-administered and mail
surveys. London: Sage Publications, Inc.
Bradburn, and Seymour Sudman, eds. (1982). Asking Questions. San Francisco:
Jossey Bass Inc., Publishers.
Brower, V. (2002). Fast Tracking Drugs to Patients. European Molecular Biology
Organization. EMBO reports 3 (1).
Cameron McKenna and Anderson Consulting “Evaluation of the Community
Procedures for the Authorisation of Medicinal Products” (2001), available on
http://pharmacos.eudra.org.
Canadian drug review process (2006) available from: http://www.hc-sc.gc.ca/hpb-
dgps/therapeut.
CIRS Centre for Innovation in Regulatory Science (2013). New Drug Approvals in
ICH Countries 2003–2012: Focus on 2012 R&D Briefing 52:2013.
CMR International Institute for Regulatory Science (2001). Asia Update: Key Issues
in the Registration of Pharmaceuticals, R&D Briefing No. 32B. [Online] Surrey:
CMR International. Available from: http://www.cmr.org/institute
Cocchetto, D. (2000). "The Food and Drug Administration's fast track designation."
Drug Information Journal. 34: 753-760.
Cole F.L. (1988) Content analysis: process and application. Clinical Nurse Specialist
2 (1), 53–57.
Coplan, PM, Noel RA, Levitan BS, Ferguson J and Mussen, F (2011). Development
of a Framework for Enhancing the Transparency, Reproducibility and
Communication of the Benefit–Risk Balance of Medicines. Clinical Pharmacology &
Therapeutics, 89(2): 312–315.
Crichton, N. (2000a). "Mann-Whitney test." Journal of clinical nursing. 9: 583
http://www.blackwellpublishing.com/specialarticles/jcn _9_583. Pdf.
Crichton, N. (2000b). "Wilcoxon signed rank test." Journal of clinical nursing. (9):
584. Http://www.blackwellpublishing.com/specialarticles/ jcn_9_584.pdf.

228
Crow, Graham and Wiles, Rose (2008) Managing anonymity and confidentiality in
social research: the case of visual data in community research. London,
GB, Economic & Social Research Council (NCRM Working Paper Series, 8/08).
Lumley and Walker (Edit.) (1996) Improving the regulatory review process, CMR
workshop series. Industry and regulatory initiatives edited by, Kluwer Academic
Publishers, Lancaster UK, 1996.1-20.
Diem. K.G. (2002a) Choosing a data collection method for survey research. Fact sheet
FS996. USA: The State University of New Jersey Rutgers Cooperative Extension.
New Jersey Agricultural Experiment Station. Available at:
http://www.rce.rutgers.edu/pubs/publication.asp?pid=FS996
Diem, K.G. (2002b) A step-by-step guide to developing effective questionnaires and
survey procedures for program evaluation and research. Fact sheet FS995. The State
University of New Jersey Rutgers Cooperative Extension. New Jersey Agricultural
Experiment Station. Available at:
http://www.rce.rutgers.edu/pubs/publication.asp?pid=FS9965
Dillman, Don A. 1978. Mail and Telephone Surveys. New York: John Wiley & Sons.
Inc.
Donnelly E, Ritchie J, and French S, (1996), company strategies to ensure a quick and
efficient review: A transnational company. In Lunlay CE and Walker SR (Eds)
Improving the regulatory review process: Industry and regulatory initiatives, Kluwer
Academic publishers, Lancaster, pp 41-52.
Dureja, H. (2010) New Drug Approval Process: Regulatory View. Pharmainfo.net 8
(4). Available at: http://www.pharmainfo.net/reviews/new-drug-approval-proces
Easton,V.McColl,J.(2001)"StepsStatisticsGlossary=v1.1"http://www.cas.lancs.ac.uk/g
lossary_v1.1/.
El Boghdady, Dina (February 13, 2012). "FDA budget stays about the same at $2.5
billion". The Washington Post. Retrieved August 30, 2012.
European Medicines Agency (EMA),(2010):Available at:
http://www.ema.europa.eu/ema/index.jsp?curl=pages/about_us/general/general_
content_000130.jsp= mid=WC0b01ac0580029336
European Medicines Agency (EMA) (2009) Survey 2008 on the performance of
EMEA scientific procedures for medicinal products for human use.
EMEA/MB/30754/2009.London:EMA.Available.at:
http://www.ema.europa.eu/docs/en_GB/document_library/Other/2009/10/WC500006
220.pdf

229
European Medicines Agency (EMA) (2011) Training manual Review of European
Medicines Agency documents addressed to the general public by patients and
consumers. Available.at:
http://www.ema.europa.eu/docs/en_GB/document_library/Other/2010/05/WC500090
127.pdf.
European Medicines Agency (EMA) (2012) Benefit-risk methodology project
Work package 4 report: Benefit-risk tools and process. Available at:
http://www.ema.europa.eu/docs/en_GB/document_library/Report/2012/03/WC50012
3819.pdf.
European Medicines Agency (EMA) (2012) Evaluation of the European Medicine
Agency –final report-January 2010, Ernst & Young associes.
Explanatory note on fees payable to the European Medicines Agency
http://www.ema.europa.eu/docs/en_GB/document_library/Other/2013/07/WC500146
978.pdf
Fink, A., Jacqueline, K., Chassin M. and Brook, R.H. (1984). "Consensus Methods:
Characteristics and Guidelines for Use." American Journal of Public Health 74(9):
979-983.
Foo, Y. T. (2007) Submission management: Singapore‟s model. Drug Information
Journal 41, 415-416
GCC (2003) Procedures manual of central drug registration. Riyadh, Saudi Arabia.
Available online: www.sgh.org.sa.
GCC (2012). The Cooperation Council for the Arab States of Gulf Secretariat
General, Statistical Bulletin, Information Sector - Statistical Department Volume
Twenty, 2012. Available at: http://www.gcc-sg.org
Guyatt, G.H., Feeny, D.H. and Patrick, D.L. (1993) Measuring health-related quality
of life. Annals of Internal Medicine 118: 622-629
Hashan, H. (2005). Evaluation of the review process for marketing pharmaceutical
products in the Gulf States and its impact on patients' access to medicines. . The
Welsh School of Pharmacy, Cardiff University. Unpublished Doctor of Philosophy.
Health Canada (HC) (2006). Access to Therapeutic Products: The Regulatory Process
in Canada. Available at: http://www.hc-sc.gc.ca/ahc-asc/pubs/hpfb-dgpsa/access-
therapeutic_acces-therapeutique-eng.php

231
Health Technology Advisory Committee (OHTAC) (2010). Decision Determinant
Guidance Document. M. A. Secretariat and M. o. H. a. L.-t. Care.
Ontario.Available.at:http://www.health.gov.on.ca/english/providers/program/mas/pub
/guide_decision.pdg
Higginson, I.J. and Carr, A.J. (2001) Measuring quality of life using quality of life
measures in the clinical setting. British Medical Journal 322: 1297-1300.
Hill, S. and K. Johnson (2004). Emerging Challenges and Opportunities in Drug
Registration and Regulation in Developing Countries London, DFID Health Systems
Resources Centre.
Hirako, M., McAuslane, N., Salek, S., Anderson, C. and Walker, S. (2007) A
comparison of the drug review process at five international regulatory agencies. Drug
Information Journal 41: 291-308.
Hynes, C., McAuslane, N. and Walker, S. (Eds) (2001) Building quality measures into
the regulatory review process: Assessing the needs of industry and regulators. Surrey:
CMR International Institute for Regulatory Science.
International Conference on Harmonisation (ICH) Requirements for registration of
pharmaceuticals for human use Available at: http://www.ich.org
International Conference on Harmonisation (ICH) (2011) Global Cooperation Group.
Available at: http://www.ich.org/about/organisation-of-ich/coopgroup.html
Jamel E. Zarrouk. (1998) Arab free trade area: Potentialities and effects, Benefiting
from Globalization Workshop, September 3 - 6, 1998, Marrakech,
Morocco.avalible.at.http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.203.23
29&rep=rep1&type=pdf.
Jefferys, D. (1997). "How do you measure the quality of the scientific assessment and
the review process? The view of the MCA". Improving the regulatory review process:
Assessing performance and setting targets. N. a. W. McAuslane, S, Kluwer Academic
Publishers: 115-123.
Jones, J. and Hunter, D. (1995). "Qualitative research: Consensus methods for
medical and health services research. Education and debate." British Medical Journal.
311: 376-380.
Jones, J. and Hunter, D. (2000). "Using the Delphi and nominal group technique in
health services research". Qualitative research in healthcare. Second edition. C. Pope
and n. Mays, BMJ books.

231
Kaplan, A., Skogstad, A. L. and Girshick, M. A. (1950): The Prediction of Social and
Technological Events, in: The Public Opinion Quarterly, XIV, pp. 93-110.
Karlton, P.C. and Johnson, D. (1997) An integrated approach to the preparation of
global CMC dossiers. Drug Information Journal 31,237-242.
Karin Heidenreich, (2004) Dialogue Between Companies and the EMEA Regarding
the Centralized Procedure from an Industry‟s Point of View, the First DIA Workshop
on Regulatory Affairs in Japan, Tokyo, October 1, 2004.
Kidder, L., and Fine, M. (1987). Qualitative and Quantitative Methods: When Stories
Converge. Multiple Methods in Program Evaluation. New Directions for Program
Evaluation, No. 35. San Francisco, CA: Jossey-Bass.
Krippendorff K. (1980) Content Analysis: An Introduction to its Methodology. Sage
Publications, Newbury Park.
Laila A. Rahman, (2002) Centralized drug registration system in the Gulf Region the
Tenth International Conference of Drug Regulatory Authorities (ICDRA) Hong Kong,
China, 24 - 27 June 2002 ; 166.
Laila A-Rahman, L. (2005). "Drug Registration Activities", Bahrain MOH. Pharmacy
& Drug Control Directorate. Manama, Bahrain.
Lumpkin, M. (2000). "Assessing Quality of the Regulatory Review Function". Drug
Information Association, Annual Meeting, San Diego, California.
Mallia-Milanes, A. (2010). The Development and Application of Scorecards to Assess
the Quality of Regulatory Submissions and their Review. Welsh School of Pharmacy.
Cardiff, Cardiff University. Unpublished Doctor of Philosophy.
Marcelo J. Vernengo, Kees de Joncheere, Enrique Fefer, (1998)Advances in
Pharmaceutical Market Integration in MERCOSUR and other Latin American
Countries, Drug Information Journal Vol. 32(3): 831-839.
McAuslane, N., Anderson, C. and Walker, S. (2004) The changing regulatory
environment: reality and perception? R&D Briefing No. 42. Surry: CMR
International. Available at: http://www.cmr.org/institute
McAuslane N, Cone M, Collins J, Walker S. (2006) Building Quality into Regulatory
Activities: What does it mean? R&D Briefing No. 46. Surrey: CMR International
Institute for Regulatory Science. Available at: http://www.cmr.org/institute.

232
McAuslane, N., Cone and Collins, J. (2006) A cross-regional comparison of the
regulatory environment in emerging markets. R&D Briefing No 50. Surry: CMR
International. Available at: http://www.cmr.org.
McAuslane, N., Cone, M., Collins, J. and Walker, S. (2009) Emerging markets and
emerging agencies: A comparative study of how key regulatory agencies in Asia,
Latin America, the Middle East and Africa are developing processes and review
models for new medicinal products. Drug Information Journal 43, 349-359.
Mead, D. and Mosely, L. (2001). "The use of Delphi as a research approach." Nurse
researcher. 8: 4-37.
Miles, M. B. and M. Huberman, Eds. (1994). Qualitative Data Analysis. London,
Sage Publications.
Molzon, J.A (2010) .The value and benefits of ICH to drug regulatory authorities-
advancing harmonization for better health. ICH 20th
anniversary publication.
Available at: http://www.ich.org/home.html
Molzon JA.,Giaquinto A., Lindstorm L., Tominaga T., Ward M., Doerr P (2011). The
value and benefits of the International conference on harmonization to Drug
Regulatory Authorities: Advancing Harmonization for better Public Health .Clinical
Pharmacology and Therapeutics 2011; Vol.89 (4):503-512.
Morgan SG, McMahon M, Mitton C, Roughead E, Kirk R, Kanavos P, (2006)
Centralized drug review processes in Australia, Canada, New Zealand, and the United
Kingdom. Health Aff. (Millwood). 25(2):337-47
Moss, F. (2004). "Transforming Health: From Policy to Action." 29-30. University of
Cambridge, Judge Institute of Management, Cambridge's business school.
Passmore, C., Dobbie, A.E., Parchman, M. and Tysinger, J. (2002) Guidelines for
constructing a survey. Family Medicine 34 (4), 281-286.
Perez, A (2008) Argentina. In: Walker, S., Cone, M., McAuslane, N. and Collins, J.
(Eds.) Emerging markets: Models of best practice for the regulatory review of new
medicines. CMR International Institute workshop report. Surrey: CMR International
Institute for Regulatory Science. Pg 15-16.
Philip R L , Jessica Herzstein (1986) .International Drug regulation, Ann.Rev.Public
Health 7: 217-35.
Pinar R (2002). A study on the reliability and validity of the Turkish version of the
MQOLS-CA2 in people with cancer. Turkish Journal of Cancer Vol. 32/No.4

233
Polit D.F. & Beck C.T. (2004) Nursing Research. Principles and Methods. Lippincott
Williams & Wilkins, Philadelphia, PA.
Ratanawijitrasin, S. and Wondemagegnehu, E. (2002). "Effective drug regulation - A
multicountry study."[Online] Geneva, Switzerland. WHO. Available from:
http://pdfoo.com/ebook-4146/effective-drug-regulation.html
Rawson, N. S. B. (2000). "Time required for approval of new drugs in Canada,
Australia, Sweden, the United Kingdom and the United States in 1996-1998."
Canadian Medical Association Journal 4(162): 501-504.
Rehnquist, J. (2003). FDA‟s Review Process for New Drug Applications: A
Management Review. Department of Health and Human Services (DHHS), Office of
Inspector General. Available at: http://oig.hhs.gov/oei/reports/oei-01-01-00590.pdf
Salant, P. and D. A. Dilman, Eds. (1994). How to conduct your own survey?
Singapore, John Wiley and Sons, Inc.
Saillot, J and Paxton, M. (2009) Industry efforts on simultaneous global development.
Drug Information Journal 43, 339-347.
Salek, S, Mallia-Millanes, A, McAuslane, N and Walker, S. (2012) Development and
application of scorecards to Assess the Quality of a Regulatory Submission and
Review. Drug Information Journal 46: 73-83
Sandelowski M. (1995) Qualitative analysis: what it is and how to begin? Research in
Nursing & Health 18, 371–375.
Sauwakon R., Eshetu W. (2002) Effective drug regulation: a multicountry study,
World Health Organization.
Saw, S.M. (2001) The design and assessment of questionnaires in clinical research.
Singapore Medical Journal 42 (3), 131-135.
Skulmoski, G. J., F. T. Hartman, et al. (2007). "The Delphi Method for Graduate
Research." Journal of Information Technology Education 6: 1-21.
Stewart AL (1990). “Psychometric considerations in functional status instruments.”
In: WONCA Classification Committee (ed.), Functional Status Measurement in
Primary Care. New York: Springer-Verlag.
Trochim W.M.K (2006) types of surveys. Available at:
http://www.socialresearchmethods.net/kb/survtype.php

234
Trouiller P, Folb P, Weerasuriya K. (2002). Legal and regulatory issues affecting drug
development for neglected diseases: Harmonization of Technical Requirements for
Registration of Pharmaceuticals for Human Use (ICH) and Neglected Diseases
Act, MSF/DND Working Group. The Crisis of Neglected Diseases, Developing
Treatments and Ensuring Access. New York, March 2002.
Trouiller P, Salmen R, Myhr K, Folb P, Weerasuriya K, Gray A.(2002) The
globalization of regulatory requirements, and the development and availability of
medicinal products in developing countries: quality, efficacy and safety
issues. MSF/DND/Working Group: The Crisis of Neglected Diseases, Developing
Treatments and Ensuring Access. New York.
Tsui, S.E. (2009) The role of the Asia-Pacific region in global drug development
strategy. Drug Information Journal 43, 35-40
US FDA (2013), The Prescripation Drug User Fee Act Available online:
http://www.fda.gov/ForIndustry/UserFees/PrescriptionDrugUserFee/ucm272170.htm
Valovich McLeod T C, Alison R S, John T P, R Curtis B, L A, and Eric LS, (2008)
Using Disablement Models and Clinical Outcomes Assessment to Enable Evidence-
Based Athletic Training Practice, Part II: Clinical Outcomes Assessment. J Athl
Train. 43(4): 437–445.
Veronica Franco.(2011)MarketResearch.com
http://www.marketresearch.com/Espicom-Healthcare-Intelligence-v1129/Outlook-
Pharmaceuticals-Middle-East-North 6666035
Walker, S., Cone, M., and Collins, J. (2005a) Assessing the regulatory environment
and its impact on patients‟ access to new medicines- South-East Asia and Western
Pacific. R&D Briefing No.47. Surrey: CMR International.
Walker, S., Cone, M., and Collins, J. (2005b) Assessing the regulatory environment
and its impact on patients‟ access to new medicines- Middle East and Africa. R&D
Briefing No.48. Surrey: CMR International.
Walker, S., Cone, M., and Collins, J. (2005c) Assessing the regulatory environment
and its impact on patients‟ access to new medicines- Latin America. R&D Briefing
No.49. Surrey: CMR International.
Walker, S., Cone, M., and McAuslane, N. (2006) Quality decision-making:
procedures and practices in drug development amd the regulatory review. CMR
International Institute workshop report. Surrey: CMR International.

235
Walker, S., Cone, M., McAuslane, N and Collins, J. (2007) Emerging markets
models of best practice for the regulatory review of new medicines. Report of the
CMR International Institute worshop. Surrey: CMR International.
Walker.S and Cone M, (2004) “Benefit-Risk Assessment: Summary Report on the
Workshop on the Development of a Model for Benefit-Risk Assessment of Medicines
Based on Multi-Criteria Decision Analysis” Surrey, U.K.: CMR International
Ware, J.E., Brook, R.H., Davies-Avery, A., et al. (1980a) Conceptualization and
Measurement of Health for Adults in the Health Insurance Study: Vol. I, Model of
Health and Methodology, The RAND Corporation, R-1987/1-H E.
Ware, J.E., Brook, R.H., Davies, A.R. and Lohr, K.N. (1981) Choosing measures of
health status for individuals in general population. American Journal of Public Health
71 (6): 620-629.
Wei, L. and M. G. Moyer, Eds. (2008). The Blackwell Guide to Research Methods in
Bilinguali sm and Multilingualism, Blackwell Publishing Ltd.
World Health Organisation (WHO) (1999). "Globalization and Assess to
Drugs"7.Health Economics and Drugs. Geneva, WHO, 1211 Geneva 27, Switzerland.
World Health Organisation (WHO) (2003a). "Annual Report 2002, Essential Drugs
and Medicines Policy: Supporting countries to close the access gap." 9. WHO, 1211
Geneva 27, Switzerland.
World Health Organisation (WHO) (2003b). "Improving drug regulation." Essential
drug monitor. (32): 2
World Health Organisation (WHO) (2004). "WHO Medicines Strategy Countries at
the core 2004-2007." 2. WHO, 1211 Geneva 27, Switzerland.
World Health Organization (WHO). 2006. World Health Report 2006: Working
Together for Health. Geneva: WHO. Available at:
www.who.int/whr/2006/en/index.html.
World Health Organization (WHO) (2008) WHO Drug Information, 22 (2). Available
at:http://apps.who.int/medicinedocs/index/assoc/s14886e/s14886e.pdf
World Health Organization (2010).Regulatory harmonization. Updating medicines
regulatory systems in Sub-Saharan African countries. WHO drug information 24(1):
620.
Woudenberg, F. (1991): An Evaluation of Delphi, in: Technological Forecasting and
Social Change, vol. 40, pp. 131 – 150.

236
Y Greg Nixon and Emmanuelle Trombe. (2013) The Bureau of National Affairs, Inc.
(800-372-1033) http://www.bna.com.
Zellerhoff, K. (2001). How the quality of the dossier affects the quality of the review:
a company‟s perspective. In: Hynes, C, McAuslane, N., Walker, S., (Eds). Building
quality measures into the regulatory review process: Assessing the needs of industry
and regulators. Surrey: CMR International Institute for Regulatory Science Pg 33-37.

237
Publications

238
Poster presentations
Al-Rubaie M., Salek, S. and Walker, S. (2011). Comparisons of the Gulf States and
Pharmaceutical Companies Perspective on the Effectiveness of the GCC
Centralized Procedure. Presented at the 47th
DIA annual Meeting, June 19-23, 2011.
Chicago, USA
Al-Rubaie M., Salek, S. and Walker, S. (2012). An Evaluation of the Gulf
Cooperation Council (GCC) Centralized Regulatory Review Process for
Pharmaceutical Products. Presented at the 48th
DIA annual Meeting, June 24-28,
2012. Philadelphia, USA
Oral Presentation
Al-Rubaie M., Salek, S. and Walker, S. (2011). Regulatory collaboration in the Gulf
Cooperation Council states. Presented at the Centre for Innovation in Regulatory
Science workshop, Evolving the regulatory review process: What are the features that
enable a transparent, timely, predictable and good-quality review, 6-7 December
2011, Kuala Lumpur, Malaysia.
Al-Rubaie M, Salek, S. and Walker, S. (2013). Ten years experience with the GCC CP
the challenges and opportunities. Presented at the 10th
Middle East Regulatory
Conference (MERC), 24-25 September 2013, Muscat (Oman).
Al-Rubaie M, Salek, S. and Walker, S. (2013). Oman regulatory review process.
Presented at the 10th
Middle East Regulatory Conference (MERC), 24-25 September
2013, Muscat (Oman).

239
Appendix 1

241
List of pharmaceutical companies respond to CACP
1 Abbott
2 Amriya Pharma
3 Apopharma Inc
4 AstraZeneca
5 B.Braun
6 CSL
7 Eli-Lilly
8 Glaxo Smith Kline
9 Glaxo Smith Kline Consumer Healthcare
10 Hospira
11 Hovid
12 Janssen
13 Jazeera Pharmaceutical Industries
14 Joriver
15 Kilitch
16 KSPICO
17 Leo Pharma
18 National Pharmaceutical Industries
19 Novartis Pharma AG, Basel, Switzerland
20 Oman Pharmaceutical Products
21 Pfizer
22 Pharmascience
23 Qatar Pharma
24 Ranbaxy
25 Reckitt Benckiser
26 Riyadh Pharma
27 Sandoz
28 SEDICO
29 Servier
30 SPIMACO


















