
Endocrine tumours
55
Endocrine Tumours khaing zay aung 18.5.2015
-
Upload
khaing-zay-aung -
Category
Health & Medicine
-
view
91 -
download
0
Transcript of Endocrine tumours

Endocrine Tumours
khaing zay aung
18.5.2015

Endocrine tumours of pancreas
Neuroendocrine tumours of the bronchi
Neuroendocrine tumours of the stomach
Neuroendocrine tumours of the small bowel
Multiple endocrine neoplasia

Endocrine tumours of the pancreas
Insulinomas (17%) Gastrinomas (15%) Non functioning tumours Rare functioning tumours (RFTs)
VIPomas (2%) Glucagonomas (1%) Carcinoid (1%) Somastinomas (1%)

Insulinomas
The most frequent cause of primary hyperinsulinism
80% are benign solitary tumours An even distribution of tumour in head, body and
tail of pancreas rarely, they may be located in the duodenum,
splenic hilum, or gastrocolic ligament 90% < 2 cm in size

Multiple tumours in 10%, associated with MEN I syndrome they are encapsulated, firm, yellow-brown nodules that are
typically hypervascular release large amounts of proinsulin (C-peptide and insulin) which
cause hypoglycemia
Carcinoma in 5-10% Characterized by local invasion and metastatic spread to
regional lymph node and liver

Clinical feature and diagnosis
Hypoglycaemia Symptoms reflecting the activation of ANS and release
of epinephrine together with cerebral dysfunction Symptoms of adrenergic overactivity
Weakness Sweating Hunger Palpitation Tremulousness

Clinical feature and diagnosis contd:
Neuroglycopenia Headache Visual disturbances Dizziness Confusion Seizures Coma

Clinical feature and diagnosis contd:
Whipple's Triad low glucose level (<50 mg/dL) symptoms of hypoglycemia symptoms resolve with administration of glucose

Clinical feature and diagnosis contd:
The most important clue to early correct diagnosis is
The relationship of symptoms to periods of food deprivation, exercise
And relieve of symptoms after taking food

Clinical feature and diagnosis contd:
Investigations
Laboratory Studieslow glucose levels (< 50 mg/dL)insulin levels > 7 U/mLinsulin/glucose ratio > 0.3C-peptide to confirm endogenous source of
insulin (>2.5 ng/ml)

Clinical feature and diagnosis contd:
LocalizationCT and MRI for larger tumorsEUS can detect small tumors (<2 cm in size)angiography showing a “blush”

Management:
Preoperative Administration of diazoxide to prevent
hypoglycaemic attacks Other agents such as verapamil, growth
hormone or corticosteroids can be given Perioperative
Glucose infusions

Management:
Operative Tumour enucleation Pancreatico duodenectomy Tumour debulking
Post operative Octeriotide infusion Systemic chemotherapy

Gastrinomas (Zollinger-Ellison syndrome ZES)
About 20% of all PETs Second common after insulinoma Male > female (3:2) Definition
1. Fulminating ulcer diathesis in the stomach, duodenum or atypical sites
2. Recurrent ulceration despite adequate therapy
3. Non-beta islet cell tumour of the pancreas

¼ of ZES pations have gastrinomas as part of MEN-1 syndrome Gastrinoma in MEN-1 are less malignant but frequently multifocal Whereas sporadic types are more malignant and solitary Arises in pancreas in about 75% Extrapancreatic sites
Duodenum – most common Omenta Liver Gastric antrum
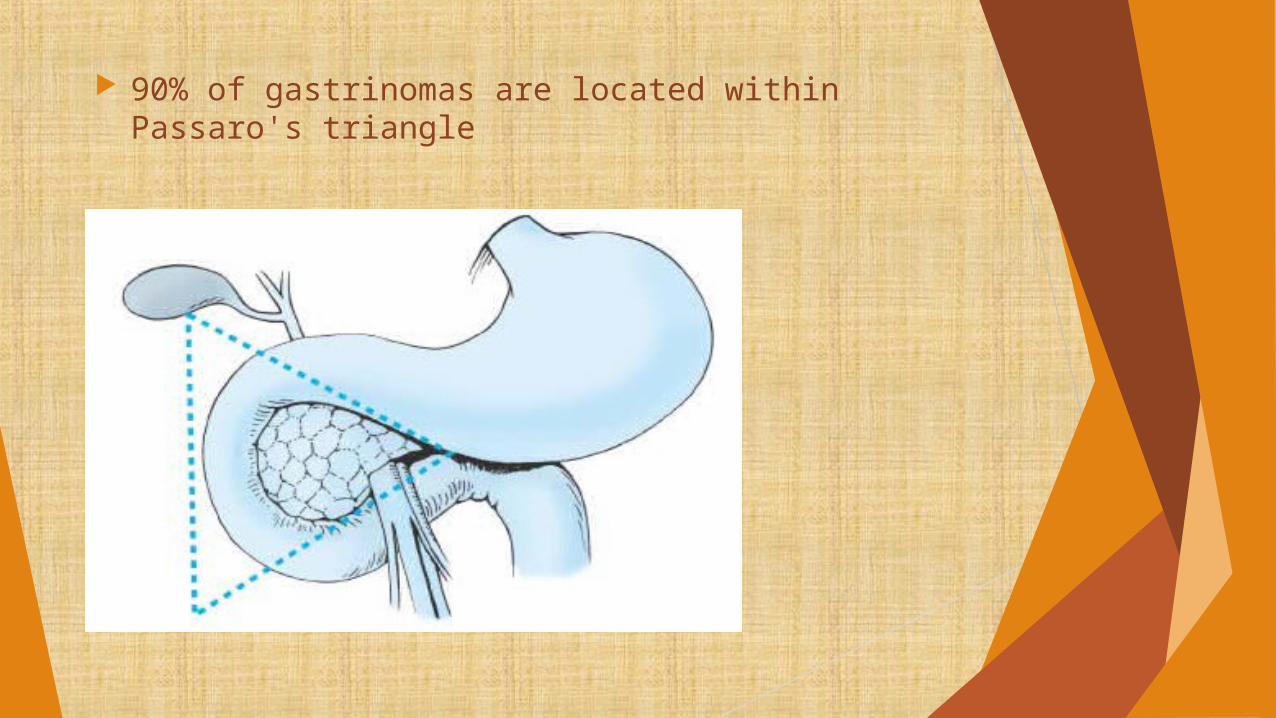
90% of gastrinomas are located within Passaro's triangle

Clinical feature and diagnosis:
Present with severe peptic ulcer disease Typical dyspeptic pain which is more severe and
less responsive to medical treatment May complaint of co-existing diarrhea Or steatorrhoea Peptic ulcers in duodenum or even in jejunum
because of mucosal injury May present with ulcer complications

Investigations
1. Laboratory Gastric pH below 2.5 Serum gastrin concentration >1000 pg/ml Secretin test – positive when serum gastrin of
>200 pg/ml over pretreatment value
2. Imaging Endoscopy
multiple ulcerslarge gastric rugal foldsmucosal edemajejunal hypermotility

LocalizationCT (not for small tumors)MRI (liver metastases)SRS with radiolabeled octreotideEUS

Differential diagnosis
Idiopathic peptic ulcer disease Chronic idiopathic diarrhea GORD Chronic atrophic gastritis Gastric outlet stenosis Retained antrum after gastric resection

Management
Medical treatment Proton pump inhibitors Octreotide – to control acid hypersecretion Systemic chemotherapy – in patients with
diffuse metastatic gastrinomas
(streptozotocin in combination with 5-FU or doxorubicin is the first line of treatment)

Management contd:
Surgical treatment
Indications for surgery
Surgical exploration should be performed in all
patients without diffuse metastases, to remove known malignant gastrinomas or benign ones

Management contd:
Pancreatic gastrinomas Most are solitary
Located in the head of the gland or uncinate process
Enucelation with peripancreatic lymph node dissection is the procedure of choice
Body or tail – enucleation with distal resection
Duodenotomy is indicated in patients with MEN-1 syndrome

Management contd:
Duodenal gastrinomas Smaller tumours <5mm can be enucleated with
the overlying mucosa Larger ones are excised with full thickness of the
duodenal wall

Non-functional endocrine pancreatic tumours NF-PETs – when they do not cause clinical
syndrome Incidence
30-50% of all PETs 5th to 6th decade of life

Pathology Cannot differentiated from functional ones by
immunohistochemistry Usually larger (>5cm) Unifocal May distribute throughout the pancreas (head to
tail ratio of 7:1:1:5)

Clinical features Usually late May present with various non-specific symptoms Jaundice, abdominal pain, weight loss and pancreatitis
Biochemical diagnosis Increased level of chromogranin A (50-80%) Chromogranin combined with PP (sensitivity 84-96%)
Differential diagnosis Important to differentiate from exocrine counterpart Because of their good prognosis

Differences between pancreatic cancer and NF-PETs
Pancreatic cancer NF-PETs
Tumour size <5 cm >5 cm
CT scan Hypodense & no calcifications
Hyperdense & calcifications possible
Chromogranin A in blood Negative Positive
Somatostatin receptor scintigraphy
Negative Positive

Management
Medical treatment When surgical excision is not possible Chemotherapy – streptozotocin, octreotide &
interferon

Surgical treatment Should be considered in malignant NF-PETs,
even with distant metastasis Pre-op tumour localization by USG or CT as they
are usually large Major goal is the potentially curative resection Partial pancreaticoduodenectomy as well as the
synchronous or metachronous resection of liver metastases
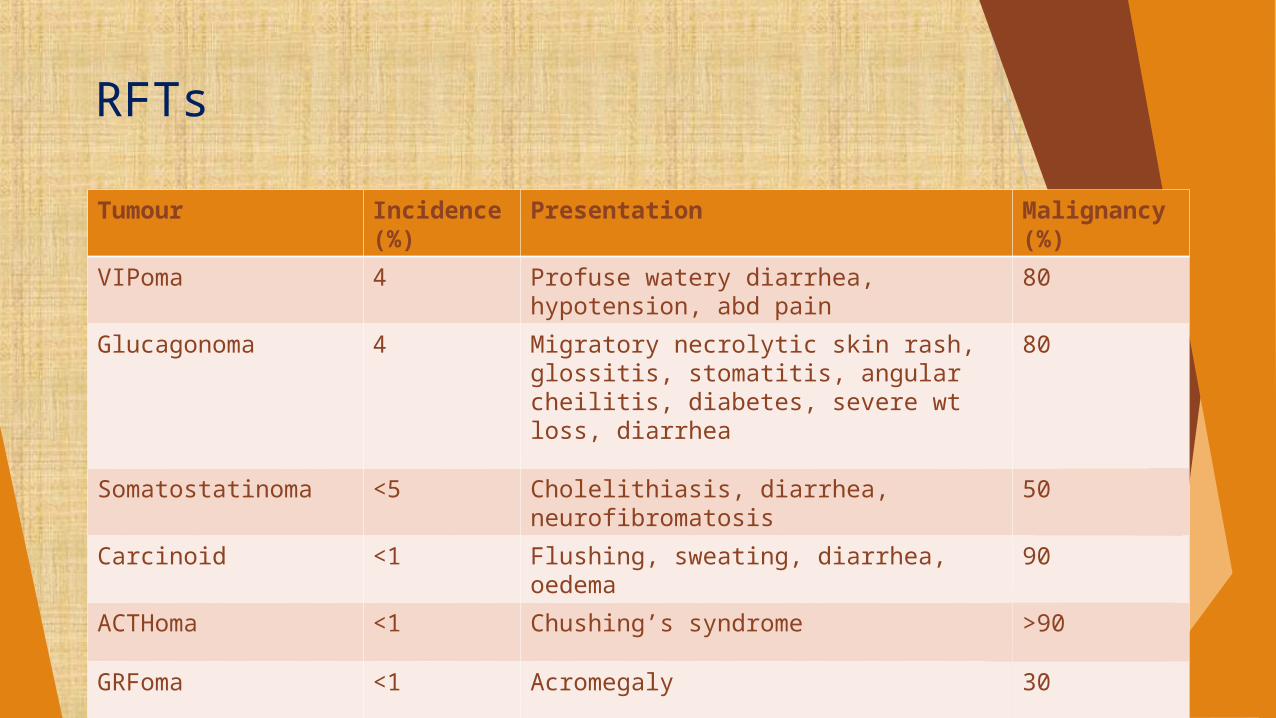
RFTs
Tumour Incidence (%)
Presentation Malignancy (%)
VIPoma 4 Profuse watery diarrhea, hypotension, abd pain
80
Glucagonoma 4 Migratory necrolytic skin rash, glossitis, stomatitis, angular cheilitis, diabetes, severe wt loss, diarrhea
80
Somatostatinoma <5 Cholelithiasis, diarrhea, neurofibromatosis
50
Carcinoid <1 Flushing, sweating, diarrhea, oedema 90
ACTHoma <1 Chushing’s syndrome >90
GRFoma <1 Acromegaly 30

Neuroendocrine tumours of stomach & small bowel
All cells of the system secret neuroendocrine markers such as synaptophysin,
chromogranin A and neurone-specific enolase (NSE)
Peptide hormones that are stored in granules s/a serotonin, somatostatin, PP or gastrin

Neuroendocrine tumours of stomach
5% of all NETs of the GIT ~0.2 cases per 100,000 population 4 different types of gastric NETs Type 1 & 2 are small benign tumours Arise from endochromaffin cells of gastric mucosa

Neuroendocrine tumours of stomach contd: Type 1
Most frequent of all gastric NETs ~ 80% Mostly in elderly women Chronic atrophic gastritis and achlorhydria resulting in chronic
hypergastrinaemia Usually no symptoms Usually detected during gastroscopy for other reasons

Neuroendocrine tumours of stomach contd:
Endoscopic resection is the treatment of choice Antrectomy and resection of ECLomas - only when recurrent
disease and multiple ( at least >1 cm & infiltration into submucosa)
Type 2 Pathogenesis & treatment is similar to that of type 1 Hypergastrinaemia in type 2 is the result of MEN-1 syndrome With multiple gastrinomas in the duodenum or rarely in the
pancreas

Type 3 Rare Sporadic Solitary tumours of unknown origin Usually larger than 2 cm Serum gastrin level is usually normal Gastrectomy and lymph node dissection and
resection of liver metastases is the treatment of choice

Neuroendocrine tumours of stomach contd:
Type 4 Present as large ulcerative malignancies Resemble to adenocarcinomas Should be treated accordingly
Type 3 & type 4 tumours usually carries poor prognosis

Neuroendocrine tumours of the small bowel
Mostly refered to as carcinoid tumours Also called midgut tumours Secrete serotonin and substance P Almost always malignant Metastasize early to regional lymph nodes and to
the liver

They produce serotonin causing carcinoid syndrome But only in the patients with large volume of
liver metastases Or tumour infiltrating into IVC bypassing the
liver

Clinical features
Acute/chronic, recurrent/ persistant abdominal pain Ileus or Rarely lower GI bleeding may occur Sudden painful reddening of the face & chest Diarrhoea Bronchospasm
Carcinoid syndrome

Clinical features cont:
Abdominal symptoms are also caused by obstruction of the lumen of appendix and undergone appendicectomy for symptoms of acute appendicitis
Polypoid NET of terminal ileum may cause intussception Pain is caused by chronic ischaemia of the bowel
Resulting from mesenteric lymph node metastasis Or constrition of the mesenteric arteries and fibrosis
of the mesentery So called desmoplastic reaction

Clinical features cont:
Diagnosis Assessment of 5-HIAA in 24 hr urine sample CT or MRI may show the primary tumour,
mesenteric lymph node and liver metastasis Best method for staging NET is octerotide scan Will show all the tumour deposits if they are
large enough and have a high somatostatin receptor density

Management
Surgical procedure Should be undertaken as soon as the diagnosis is
made The main goal is resection of the primary tumour and
lymph node metastases Liver metastases can be treated by chemotherapy of
tumour embolization Also liver transplation can be performed Symptoms of carcinoid syndrome can be treated by
somatostatin and its analog

Bronchopulmonary carcinoid tumours
80% are found in main bronchi Slow growing and highly vascular Mostly benign but ~ 15% are metastasize Patient may present as recurrent pneumonia or
haemoptysis or rarely with carcinoid syndrome Surgical excision is preferred because of excellent
prognosis after complete excision Small peripheral tumours – segmental or wedge
resection is sufficient Lobectemy or pneumonectomy may be needed in
central tumours

Multiple Endocrine Neoplasia
An inherited syndrome Characterized by a combination of benign and
malignant tumours in different endocrine glands Two main types
MEN-1 MEN-2

MEN-1
Wermer’s syndrome Caused by germline mutations in the menin gene
located on chromosome 11
Tumours in anterior pituitary gland Mostly prolactinoma or non functioning tumours Or microadenomas

MEN-1
Hyperplasia of parathyroidsCausing primary hyperparathyroidism (90-100%)
Pancreaticoduodenal endocrine tumoursMost common syndrome-associated cause of
deathMost common functional tumour is gastrinoma
followed by insulinoma

Adrenal tumours and other organ manifestation Nearly 40-50% of patientsMostly non functioning adenomas are found Rarely adrenocortical carcinoma and
pheochromocytomas are found

Operative therapy for MEN 1
Parathyroids Total parathyroidectomy including cervical
thymectomy or 3 ½ gland resection leaving approximately 50 g of gland
Endocrine pancreas Should be operated to prevent liver metastasis Pylorus preserving pancreaticoduodenectomy is
recommended

Anterior pituitary gland Indicated for non functional tumours or medical
therapy for prolactinoma fails
Adrenal tumours Functional tumours and >4 cm non functional
tumours

MEN-2
Subdivided into FMTC, MEN 2a & MEN 2b MEN 2 is caused by mutations in RET proto-onco
gene located on chromosome 10 MEN 2a
MTC pHPT Mostly bilateral pheochromocytoma MTC + pheochromocytoma alone is called Sipple’s
syndrome

MEN-2
MEN 2b MTC Pheochromocytomas Characteristic facial and oral mucosal neuromas
and intestinal ganglioneuromatosis accompanied by marfanoid habitus

MEN-2
Medullary thyroid carcinoma Multicentricity and often accompanied by C-cell
hyperplasia More aggressive in MEN 2b
Primary hyperparathyroidism Less common in MEN 2b Milder clinical course
Phaeochromocytoma 10-15% Almost always benign Can be bilateral

Operative therapy for MEN 2
Medullary thyroid carcinoma Mutation carriers can be operated on before the
disease develops Pheochromocytoma
Unilateral or bilateral subtotal resection is feasible Primary hyperparathyroidism
More difficult because associated with MTC Localization procedures should be done with
targeted approach



















