
Editing function of Escherichia coli cysteinyl-tRNA
6
Nucleic Acids Research, 1994, Vol. 22, No. 7 1155-1160 Editing function of Escherichia coli cysteinyl-tRNA synthetase: cyclization of cysteine to cysteine thiolactone Hieronim Jakubowski Department of Microbiology and Molecular Genetics, UMDNJ- New Jersey Medical School, 185 South Orange Avenue, Newark, NJ 07103, USA Received January 12, 1994; Revised and Accepted March 4, 1994 ABSTRACT A cyclic sulfur compound, identified as cysteine thiolactone by several chemical and enzymatic tests, is formed from cysteine during in vitro tRNACYs aminoacylation catalyzed by Escherichia coli cysteinyl- tRNA synthetase. The mechanism of cysteine thiolactone formation involves enzymatic deacylation of Cys-tRNACYs (k = 0.017 s-1) in which nucleophilic sulfur of the side chain of cysteine in Cys-tRNACYs attacks its carboxyl carbon to yield cysteine thiolactone. Nonenzymatic deacylation of Cys-tRNACYS (k = 0.0006 s-1) yields cysteine, as expected. Inhibition of enzymatic deacylation of Cys-tRNACyS by cysteine and Cys-AMP, but not by ATP, indicates that both synthesis of Cys-tRNACYS and cyclization of cysteine to the thiolactone occur in a single active site of the enzyme. The cyclization of cysteine is mechanistically similar to the editing reactions of methionyl-tRNA synthetase. However, in contrast to methionyl-tRNA synthetase which needs the editing function to reject misactivated homocysteine, cysteinyl-tRNA synthetase is highly selective and is not faced with a problem in rejecting noncognate amino acids. Despite this, the present day cysteinyl-tRNA synthetase, like methionyl-tRNA synthetase, still retains an editing activity toward the cognate product, the charged tRNA. This function may be a remnant of a chemistry used by an ancestral cysteinyl-tRNA synthetase. INTRODUCTION All living cells must conduct protein synthesis with a high degree of accuracy maintained in the transmission and flow of information from gene to finished protein product. One crucial 'quality control' point in maintaining a high level of accuracy is the selectivity by which aminoacyl-tRNA synthetases furnish correctly activated amino acids, attached to tRNA species, as the building blocks for growing protein chains. In many cases, the intrinsic binding energies of amino acids to aminoacyl-tRNA synthetases are inadequate to give the required accuracy of translation. This has necessitated the evolution of a second determinant of specificity, proofreading or editing mechanisms that involve the expenditure of energy to remove errors in amino acid selection (reviewed in refs. 1-3). Editing mechanism of methionyl-tRNA synthetase is directed against the non protein amino acid homocysteine, a metabolic precursor of methionine. Homocysteine is misactivated by methionyl-tRNA synthetase at unacceptably high level (4-6) but then cyclized to homocysteine thiolactone (7) to prevent transfer to tRNAMet both in vitro (5,7) and in vivo (8-10). Because of limited selectivity of the editing site of methionyl-tRNA synthetase, some of the cognate substrate methionine is also cyclized to a corresponding thiolactone, S-methyl homocysteine thiolactone (1 1). Editing of the cognate amino acid is minimized in vivo so that only 0.03% of activated methionine is cyclized by methionyl-tRNA synthetase in bacterial cells (12). An important question which needs to be answered is whether cyclization of an activated amino acid is a general editing mechanism. So far the cyclization has been established as an editing mechanism for methionyl-tRNA synthetase. The cyclization reaction has two important features. First, it utilizes a nucleophilic function of the side chain of an amino acid to accomplish chemistry of editing. Second, cyclization, in which both a side chain and a carboxyl group of an amino acid are involved in a chemical reaction, assures that an incorrect amino acid is not simply regenerated but destroyed. This prevents the edited amino acid from re-entering the active site of an aminoacyl- tRNA synthetase and eliminates futile recycling of an incorrect amino acid between synthetic and editing pathways, thereby minimizing dissipation of energy. An editing reaction regenerating intact amino acid would be more costly energetically because an incorrect amino acid will be recycled through synthetic and editing pathways at the expense of ATP. An amino acid that is cyclized during editing passes through synthetic and editing pathways only once. Like methionyl-tRNA synthetase, other aminoacyl-tRNA synthetases could have evolved to employ the reactivity of the side chain of an amino acid to accomplish editing. In addition to homocysteine and methionine, majority of other cellular amino acids have nucleophilic functions on their side chains that would be capable of reacting with their activated carboxyl carbon. To determine if cyclization is a general editing mechanism, we have systematically analyzed products of reactions of aminoacyl-tRNA synthetases with amino acids. As shown in this paper, cysteinyl-tRNA synthetase catalyzes cyclization of cysteine to a thioester, cysteine thiolactone. The mechanism of cysteine thiolactone formation involves enzymatic deacylation of Cys- tRNACYS by cysteinyl-tRNA synthetase, a reaction mecha- 6.-D 1994 Oxford University Press
Transcript of Editing function of Escherichia coli cysteinyl-tRNA

Nucleic Acids Research, 1994, Vol. 22, No. 7 1155-1160
Editing function of Escherichia coli cysteinyl-tRNAsynthetase: cyclization of cysteine to cysteine thiolactone
Hieronim JakubowskiDepartment of Microbiology and Molecular Genetics, UMDNJ- New Jersey Medical School,185 South Orange Avenue, Newark, NJ 07103, USA
Received January 12, 1994; Revised and Accepted March 4, 1994
ABSTRACT
A cyclic sulfur compound, identified as cysteinethiolactone by several chemical and enzymatic tests,is formed from cysteine during in vitro tRNACYsaminoacylation catalyzed by Escherichia coli cysteinyl-tRNA synthetase. The mechanism of cysteinethiolactone formation involves enzymatic deacylationof Cys-tRNACYs (k = 0.017 s-1) in which nucleophilicsulfur of the side chain of cysteine in Cys-tRNACYsattacks its carboxyl carbon to yield cysteinethiolactone. Nonenzymatic deacylation of Cys-tRNACYS(k = 0.0006 s-1) yields cysteine, as expected.Inhibition of enzymatic deacylation of Cys-tRNACyS bycysteine and Cys-AMP, but not by ATP, indicates thatboth synthesis of Cys-tRNACYS and cyclization ofcysteine to the thiolactone occur in a single active siteof the enzyme. The cyclization of cysteine ismechanistically similar to the editing reactions ofmethionyl-tRNA synthetase. However, in contrast tomethionyl-tRNA synthetase which needs the editingfunction to reject misactivated homocysteine,cysteinyl-tRNA synthetase is highly selective and is notfaced with a problem in rejecting noncognate aminoacids. Despite this, the present day cysteinyl-tRNAsynthetase, like methionyl-tRNA synthetase, still retainsan editing activity toward the cognate product, thecharged tRNA. This function may be a remnant of achemistry used by an ancestral cysteinyl-tRNAsynthetase.
INTRODUCTIONAll living cells must conduct protein synthesis with a high degreeof accuracy maintained in the transmission and flow ofinformation from gene to finished protein product. One crucial'quality control' point in maintaining a high level of accuracyis the selectivity by which aminoacyl-tRNA synthetases furnishcorrectly activated amino acids, attached to tRNA species, as thebuilding blocks for growing protein chains. In many cases, theintrinsic binding energies of amino acids to aminoacyl-tRNAsynthetases are inadequate to give the required accuracy oftranslation. This has necessitated the evolution of a seconddeterminant of specificity, proofreading or editing mechanismsthat involve the expenditure of energy to remove errors in aminoacid selection (reviewed in refs. 1-3).
Editing mechanism of methionyl-tRNA synthetase is directedagainst the non protein amino acid homocysteine, a metabolicprecursor of methionine. Homocysteine is misactivated bymethionyl-tRNA synthetase at unacceptably high level (4-6) butthen cyclized to homocysteine thiolactone (7) to prevent transferto tRNAMet both in vitro (5,7) and in vivo (8-10). Because oflimited selectivity of the editing site of methionyl-tRNAsynthetase, some of the cognate substrate methionine is alsocyclized to a corresponding thiolactone, S-methyl homocysteinethiolactone (1 1). Editing of the cognate amino acid is minimizedin vivo so that only 0.03% of activated methionine is cyclizedby methionyl-tRNA synthetase in bacterial cells (12).An important question which needs to be answered is whether
cyclization of an activated amino acid is a general editingmechanism. So far the cyclization has been established as anediting mechanism for methionyl-tRNA synthetase. Thecyclization reaction has two important features. First, it utilizesa nucleophilic function of the side chain of an amino acid toaccomplish chemistry of editing. Second, cyclization, in whichboth a side chain and a carboxyl group of an amino acid areinvolved in a chemical reaction, assures that an incorrect aminoacid is not simply regenerated but destroyed. This prevents theedited amino acid from re-entering the active site of an aminoacyl-tRNA synthetase and eliminates futile recycling of an incorrectamino acid between synthetic and editing pathways, therebyminimizing dissipation of energy. An editing reactionregenerating intact amino acid would be more costly energeticallybecause an incorrect amino acid will be recycled through syntheticand editing pathways at the expense of ATP. An amino acid thatis cyclized during editing passes through synthetic and editingpathways only once. Like methionyl-tRNA synthetase, otheraminoacyl-tRNA synthetases could have evolved to employ thereactivity of the side chain of an amino acid to accomplish editing.In addition to homocysteine and methionine, majority of othercellular amino acids have nucleophilic functions on their sidechains that would be capable of reacting with their activatedcarboxyl carbon.To determine if cyclization is a general editing mechanism,
we have systematically analyzed products of reactions ofaminoacyl-tRNA synthetases with amino acids. As shown in thispaper, cysteinyl-tRNA synthetase catalyzes cyclization of cysteineto a thioester, cysteine thiolactone. The mechanism of cysteinethiolactone formation involves enzymatic deacylation of Cys-tRNACYS by cysteinyl-tRNA synthetase, a reaction mecha-
6.-D 1994 Oxford University Press

1156 Nucleic Acids Research, 1994, Vol. 22, No. 7
nistically similar to cyclization of methionine by methionyl-tRNAsynthetase. Thus, cysteinyl-tRNA synthetase possesses an editingactivity toward the cognate product Cys-tRNA.
MATERIALS AND METHODSCysteinyl-tRNA synthetasePlasmid pCysS2 (13) containing the cysteinyl-tRNA synthetasegene cloned into pUC18 was obtained from Gilbert Eriani. E. colistrain JM1O1 harboring the plasmid pCys2 was used as a sourceof cysteinyl-tRNA synthetase. Cells (1.6 g) harvested from a 300ml overnight culture grown in LB medium containing 50 /tg/mlampicillin were disrupted by sonication in 2 x 5 ml 10 mMpotassium phosphate buffer, pH 6.8, containing 1 mM2-mercaptoethanol and 10% glycerol (buffer A). Nucleic acidswere precipitated from the crude extract (10 ml) with 3 ml 6%streptomycin sulfate and discarded. Protein was precipitated withammonium sulfate (75% saturation), collected by centrifugation,dissolved in 3 ml of buffer A, and loaded onto 2.5 x70 cmSephacryl S-200 (Pharmacia) gel filtration column equilibratedwith buffer A. The enzyme co-eluted with a third major proteinpeak. Fractions containing the highest cysteinyl-tRNA synthetaseactivity (27 ml) were further purified on a 1.5 x5 cm Mono Q(Pharmacia) column with a gradient from 0-0.4 M KCI in bufferA. The enzyme activity eluted as a single peak correspondingto a major protein peak at 0.2 M KCI. Cysteinyl-tRNA synthetasewas concentrated for storage in 20 mM Tris-HCl, pH 8.0, 1mM dithiotreitol, 50% glycerol. The enzyme was >95% pureas judged by SDS-polyacrylamide gel electrophoresis. Theconcentration of the cysteinyl-tRNA synthetase was calculatedfrom A280 using a mol wt of 59000 and a value of 2 cm- mg- 1(14). Active site titration established that at least 93% of theprotein was functional. Measurements of rates of tRNAcYsaminoacylation at pH 8.0, 37°C as a function of [35S]cysteineconcentration (20-320 14M) yielded kcat = 3.2 s' and Km =60 ytM.
Preparation of [35S]Cys-tRNAAminoacylation mixtures contained 50 mM HEPES, pH 8.0, 10mM MgCl2, 0.1 mM EDTA, 2 mM ATP, 20,uM tRNACYs(1400 pmoles/A260; Subriden RNA), 25 ,tM [35S]cysteine 0.5mCi/ml (1 Ci = 37 GBq) (Amersham), and 2.5 ,aM cysteinyl-tRNA synthetase. After 5 min at 37°C the charged tRNA waspurified by phenol extraction and recovered by precipitation withethanol. Phenol extraction was necessary to remove cysteinyl-tRNA synthetase which was not denatured by ethanolprecipitation.
Enzymatic deacylation of [35S]Cys-tRNAThe reactions were carried out in 50 mM HEPES, pH 8.0,10 mM MgCl2, 0.1 mM EDTA, 10 mM 2-mercaptoethanol at37°C. In one set of experiments the disappearance of radiolabeledCys-tRNA was monitored by trichloroacetic acid precipitation.In another set of experiments in which all forms of radioactivecysteine were followed, the aliquots were quenched with 1 Mformic acid at 0°C and analyzed by TLC.
TLC analysisThe TLC system (cellulose plates from Sigma developed withbutanol:acetic acid:water, 4:1:1, v/v) is the same as the one usedbefore for determination of aminoacyl adenylates (15) andhomocysteine thiolactone (7). An authentic cysteine (Sigma)
standard was co-chromatographed with samples and visualizedby staining with ninhydrin (7). In experiments with [35S]cysteineor [35S]Cys-tRNACYs, the TLC plates were autoradiographedusing XAR-5 Kodak X-ray film.For unknown reasons, unlabeled cysteine as well as 35S-
labeled cysteine and Spot X (see the Results section) migratedon TLC plates used in these experiments as double spots. Thisis not immediately obvious in experiments with the commercialpreparations of [35S]cysteine illustrated in Fig. 1, because of asignificant levels of impurities present in these preparations.However, it is clear in experiments with purified [35S]Cys-tRNACYs because only [35S]cysteine is attached to tRNACYs bycysteinyl-tRNA synthetase. [35S]Cysteine released nonenzy-matically from [35S]Cys-tRNACYs at pH 8 or by treatment withNaOH migrated on TLC plates as a double spot (Fig. 3). Adouble spot of Spot X yielded a double spot of cysteine uponsimilar treatments (Fig. 2). Another batch ofTLC plates obtainedafter completion of these experiments did not produce doublespots.
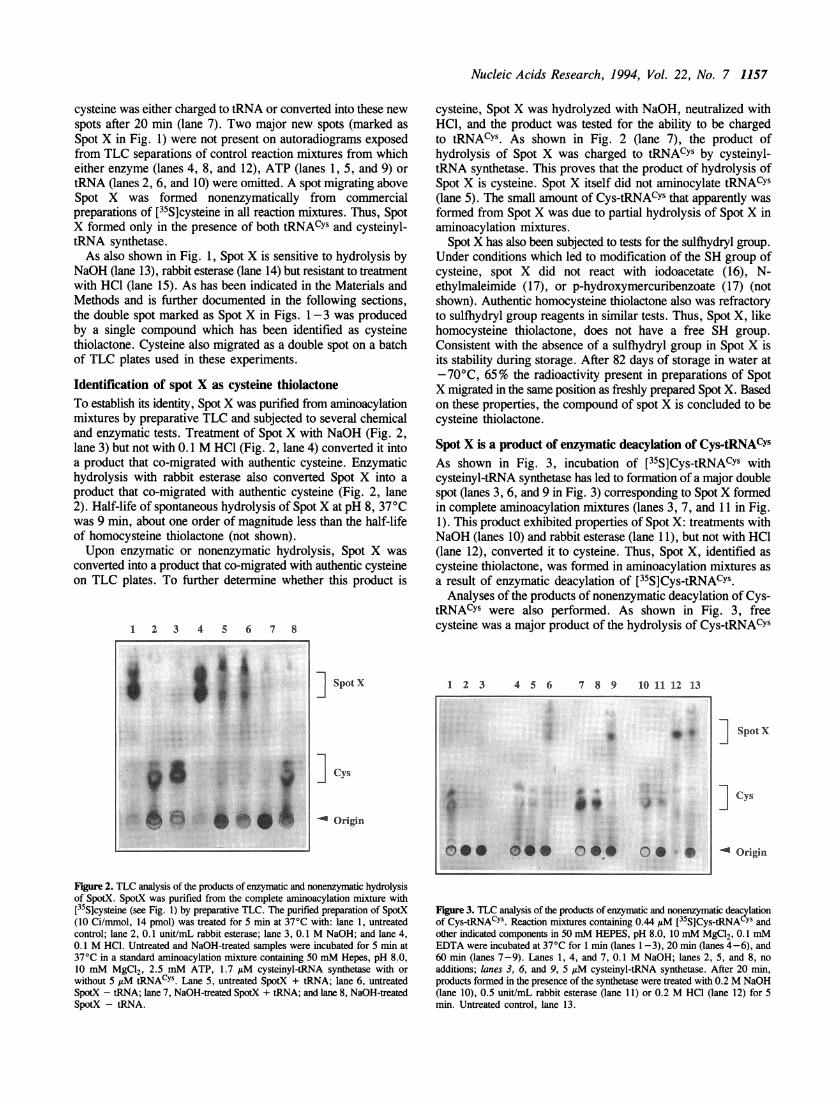
RESULTSA cysteine-derived compound is formed during amino-acylation of tRNACYSReaction mixtures containing [35S]cysteine, ATP, tRNACYs, andcysteinyl-tRNA synthetase were incubated at 37°C. Products ofthe reactions were separated by TLC. An autoradiogram exposedfrom these chromatograms is presented in Fig. 1. In additionto the spot of [35S]Cys-tRNACYs at the origin of the TLC plate(lanes 3, 7, and 11 in Fig. 1), other major new spots appearedon the autoradiogram exposed from the TLC separation of thecomplete reaction mixture after 20 min incubation and were stillpresent after 60 min incubation (lanes 7 and 11). Almost all
2i2 .i4I5( S l 1 4 15 6
'I* .*.. 0 *
.*. .i. *
...: _:
.6F ws0 .. .r e
* ~*S
-Ji
Sp(..i X__j
(--. V.i
'O Origli
Figure 1. TLC analysis of products formed in tRNACYS aminoacylation mixtures.Reaction mixtures containing 5 AM cysteinyl-tRNA synthetase, 4.4 IAM tRNACYS,2 mM ATP, 2 jsM [35S]cysteine (5 Ci/mmol, 0.1 mCi/mL), 50 mM HEPES,pH 8.0, 10 mM MgC12, 0.1 mM EDTA, and 10 mM 2-mercaptoethanol wereincubated at 37°C for 1 min (lanes 1-4), 20 min (lanes 5-8) and 60 min (lanes9-12). Lanes 1, 5, and 9,-ATP; lanes 2, 6, and 10,-tRNA; lanes 3, 7, and11, complete reaction mixture; lanes 4, 8, and 12,-cysteinyl-tRNA synthetase.After 20 min, the products formed in the complete reaction mixture were treatedwith either 0.2 M NaOH (lane 13), 0.5 unit/mL rabbit esterase (lane 14), or0.2 M HCl, 80°C (lane 15) for another 20 min. Untreated control, lane 16. Spotsmigrating just below Cys and between Cys and Spot X are contaminants presentin commercial preparations of [35S]cysteine. [35S]Cys-tRNACYs present in somereaction mixtures stays at the origin.
Nucleic Acids Research, 1994, Vol. 22, No. 7 1157
cysteine was either charged to tRNA or converted into these newspots after 20 min (lane 7). Two major new spots (marked asSpot X in Fig. 1) were not present on autoradiograms exposedfrom TLC separations of control reaction mixtures from whicheither enzyme (lanes 4, 8, and 12), ATP (lanes 1, 5, and 9) or
tRNA (lanes 2, 6, and 10) were omitted. A spot migrating aboveSpot X was formed nonenzymatically from commercialpreparations of [35S]cysteine in all reaction mixtures. Thus, SpotX formed only in the presence of both tRNAcYs and cysteinyl-tRNA synthetase.As also shown in Fig. 1, Spot X is sensitive to hydrolysis by
NaOH (lane 13), rabbit esterase (lane 14) but resistant to treatmentwith HCl (lane 15). As has been indicated in the Materials andMethods and is further documented in the following sections,the double spot marked as Spot X in Figs. 1-3 was producedby a single compound which has been identified as cysteinethiolactone. Cysteine also migrated as a double spot on a batchof TLC plates used in these experiments.
Identification of spot X as cysteine thiolactoneTo establish its identity, Spot X was purified from aminoacylationmixtures by preparative TLC and subjected to several chemicaland enzymatic tests. Treatment of Spot X with NaOH (Fig. 2,lane 3) but not with 0.1 M HCI (Fig. 2, lane 4) converted it intoa product that co-migrated with authentic cysteine. Enzymatichydrolysis with rabbit esterase also converted Spot X into a
product that co-migrated with authentic cysteine (Fig. 2, lane2). Half-life of spontaneous hydrolysis of Spot X at pH 8, 37°Cwas 9 min, about one order of magnitude less than the half-lifeof homocysteine thiolactone (not shown).Upon enzymatic or nonenzymatic hydrolysis, Spot X was
converted into a product that co-migrated with authentic cysteineon TLC plates. To further determine whether this product is
1 2 3 4 5 6 7 8
Spot X
] Cys
` Origin
Figure 2. TLC analysis of the products of enzymatic and nonenzymatic hydrolysisof SpotX. SpotX was purified from the complete aminoacylation mixture with[35S]cysteine (see Fig. 1) by preparative TLC. The purified preparation of SpotX(10 Ci/mmol, 14 pmol) was treated for 5 min at 37°C with: lane 1, untreatedcontrol; lane 2, 0.1 unit/mL rabbit esterase; lane 3, 0.1 M NaOH; and lane 4,0.1 M HC1. Untreated and NaOH-treated samples were incubated for 5 min at37°C in a standard aminoacylation mixture containing 50 mM Hepes, pH 8.0,10 mM MgCl2, 2.5 mM ATP, 1.7 4tM cysteinyl-tRNA synthetase with or
without 5 itM tRNAcYs. Lane 5, untreated SpotX + tRNA; lane 6, untreatedSpotX - tRNA; lane 7, NaOH-treated SpotX + tRNA; and lane 8, NaOH-treatedSpotX - tRNA.
cysteine, Spot X was hydrolyzed with NaOH, neutralized withHCl, and the product was tested for the ability to be chargedto tRNAcys. As shown in Fig. 2 (lane 7), the product ofhydrolysis of Spot X was charged to tRNAcYs by cysteinyl-tRNA synthetase. This proves that the product of hydrolysis ofSpot X is cysteine. Spot X itself did not aminocylate tRNACYs(lane 5). The small amount of Cys-tRNACYs that apparently wasformed from Spot X was due to partial hydrolysis of Spot X inaminoacylation mixtures.
Spot X has also been subjected to tests for the sulthydryl group.Under conditions which led to modification of the SH group ofcysteine, spot X did not react with iodoacetate (16), N-ethylmaleimide (17), or p-hydroxymercuribenzoate (17) (notshown). Authentic homocysteine thiolactone also was refractoryto sulfhydryl group reagents in similar tests. Thus, Spot X, likehomocysteine thiolactone, does not have a free SH group.Consistent with the absence of a sulfhydryl group in Spot X isits stability during storage. After 82 days of storage in water at-70°C, 65% the radioactivity present in preparations of SpotX migrated in the same position as freshly prepared Spot X. Basedon these properties, the compound of spot X is concluded to becysteine thiolactone.
Spot X is a product of enzymatic deacylation of Cys-tRNACYsAs shown in Fig. 3, incubation of [35S]Cys-tRNAcYs withcysteinyl-tRNA synthetase has led to formation of a major doublespot (lanes 3, 6, and 9 in Fig. 3) corresponding to Spot X formedin complete aminoacylation mixtures (lanes 3, 7, and 11 in Fig.1). This product exhibited properties of Spot X: treatments withNaOH (lanes 10) and rabbit esterase (lane 11), but not with HCI(lane 12), converted it to cysteine. Thus, Spot X, identified ascysteine thiolactone, was formed in aminoacylation mixtures asa result of enzymatic deacylation of [35S]Cys-tRNAcYs.Analyses of the products of nonenzymatic deacylation of Cys-
tRNAcys were also performed. As shown in Fig. 3, freecysteine was a major product of the hydrolysis of Cys-tRNACYs
1 2 3 4 5 6 7 8 9 10 11 12 13
] Spot X
] Cys
origin
Figure 3. TLC analysis of the products of enzymatic and nonenzymatic deacylationof Cys-tRNACYs. Reaction mixtures containing 0.44 1tM [3SS]Cys-tRNACYs andother indicated components in 50 mM HEPES, pH 8.0, 10 mM MgCl2, 0.1 mMEDTA were incubated at 37°C for 1 min (lanes 1-3), 20 min (lanes 4-6), and60 min (lanes 7-9). Lanes 1, 4, and 7, 0.1 M NaOH; lanes 2, 5, and 8, no
additions; lanes 3, 6, and 9, 5 JLM cysteinyl-tRNA synthetase. After 20 min,products formed in the presence of the synthetase were treated with 0.2 M NaOH(lane 10), 0.5 unit/mL rabbit esterase (lane 11) or 0.2 M HCI (lane 12) for 5min. Untreated control, lane 13.
.......
.:.o.: . * *..... :w* .. t w. ...tZ ... W.:ti: *:* ::@ X . :...:.X.} . . : .. : : ::: ::::o* e o* o > *

1158 Nucleic Acids Research, 1994, Vol. 22, No. 7
j80
z
cc 60
40
20 -
0 2 4 6 8 1 0
Time (min)
Figure 4. Time course of enzymatic deacylation of Cys-tRNACYS. Reactions were
carried out at 370C in mixtures containing 1 tM [3SS]Cys-tRNACYs (5 Ci/mmol),2 /M cysteinyl-tRNA synthetase, 10 mM MgCl2, 10 mM 2-mercaptoethanol,50 mM HEPES buffer, pH 8.0, and following additions: (O), control, no
additions; (*), 25 mM cysteine, (U), 2.5 mM ATP; (o) 25 mM cysteine +2.5 mM ATP. % remaining [3"S]Cys-tRNACYs was determined by trichloroaceticacid precipitation at indicated time intervals.
with either NaOH (lanes 1, 4, and 7) or the pH 8.0 buffer (lanes2, 5, and 8) at 370C.
Inhibition of enzymatic deacylation of Cys-tRNACYs bycysteineTime course of enzymatic deacylation of [35S]Cys-tRNACYs inthe presence of an excess cysteinyl-tRNA synthetase at pH 8.0,37°C was monitored by trichloroacetic acid precipitation. Fig.4 illustrates typical time courses of the reaction. It is unclear whythe reaction stopped after about 80% of the substrate was
consumed and did not go to the completion (Fig. 4). Similarincomplete kinetics of enzymatic deacylation of Cys-tRNACYs(14) and Ile-tRNAPhe (18) have been observed by others. Thekinetics were first order until reactions were about 70%completed. Rate constants calculated from half-lives of enzymaticdeacylation of Cys-tRNACYs at pH 7.4 and pH 8.0 in thepresence and absence of various tested compounds are
summarized in Table 1. As shown in Fig. 4, cysteine inhibitedenzymatic deacylation of [35S]Cys-tRNAcYs. The inhibition was95%, both at pH 8.0 and 7.4 (Table 1). Slightly greater inhibition,99%, was observed in the presence of Cys-AMP, formed in situ.ATP did not affect the reaction (Fig. 4 and Table 1). Controlexperiments showed that the inhibition was specific for L-cysteine. Noncognate amino acids (homocysteine, methionine,serine, and D-cysteine were tested) did not inhibit enzymaticdeacylation of Cys-tRNA (Table 1), indicating that these aminoacids did not bind to the active site of the enzyme. These resultssuggest that enzymatic deacylation of Cys-tRNACYs occurs in a
single active site that also carries out the synthesis of Cys-AMPand Cys-tRNACYs.
Increasing concentration of cysteinyl-tRNA synthetase from2 AM (Fig. 4) to 8 itM (Table 1) did not accelerate enzymaticdeacylation of 1 AtM Cys-tRNA. The rate of enzymaticdeacylation became dependent on the enzyme concentration when[enzyme] < [Cys-tRNA] (not shown), as expected. Theseobservations are consistent with cysteinyl-tRNA synthetasecatalyzing deacylation of Cys-tRNA and make it unlikely that
Table 1. Inhibition of enzymatic deacylation of Cys-tRNACYs by cysteine
Additions kpH 8.0 pH 7.4
103 x s-J-Enzyme 0.6 0.3None 17 6L-Cys 1.3 0.6D-Cys 6ATP 17L-Cys, ATP 0.8D,L-Homocysteine, L-Met or L-Ser 17 6
Conditions were: 1 AM [3"S]Cys-tRNACYS (5 Ci/mmol), 8 jiM cysteinyl-tRNAsynthetase, 10 mM MgCl2, 10 mM 2-mercaptoethanol, 37°C, 50 mM HEPESbuffer adjusted to pH 7.4 or 8.0 with KOH. 25 mM amino acid and 3.3 mMATP were used where indicated. First order rate constants of disappearance of[31S]Cys-tRNACYs , kcat, were calculated from experimentally determined half-lives, t1/2 (see Fig. 4), according to kcat = ln2/tl/2.
the reaction is due to another enzyme that might be present asa contamination in cysteinyl-tRNA synthetase preparation.
ATP pyrophosphatase activity of cysteinyl-tRNA synthetaseThe ATP pyrophosphatase reaction measures the totalconsumption of ATP as an amino acid is first activated in thesynthetic reaction and the product (an enzyme-bound aminoacyladenylate and/or aminoacyl-tRNA) is then destroyed in the editingreaction that yields AMP. Here, the ATP pyrophosphataseactivity of the enzyme (1 ,uM) has been measured in the presenceof 1 uM tRNACYs, 0.2 mM [3H]ATP, 1 unit/ml inorganicpyrophosphatase, and 25 mM amino acid. Out of 9 amino acidstested (Cys, homocysteine, Ser, homoserine, Thr, Asp, Met, Val,a-aminobutyrate) only Cys gave significant ATP pyrophosphataseactivity. No ATP pyrophosphatase has been detected in thepresence of Ala, the only amino acid tested before (14). In thepresence of cysteine, ATP was hydrolyzed at 0.023 (at pH 8.0)or 0.007 (at pH 7.4) mol per mol of enzyme per s. These waluesare in good agreement with k, values for enzymatic deacylationof Cys-tRNACYs, indicating that most of ATP hydrolysis is dueto recharging of deacylated tRNACYs. The ATP pyrophosphataseactivity in the presence of the other amino acids was 10 timesor more lower, which was not significantly different from thecontrols without an amino acid. The lack ofATP pyrophosphataseactivity in the presence of noncognate amino acids is due to theirinability to bind to the active site of the enzyme as demonstratedpreviously for Ala, Ser, and cx-aminobutyrate (14) and here forhomocysteine, Met, and Ser (Table 1). No ATP hydrolysis hasbeen observed in the absence of tRNACYS. The requirement fortRNACYs in cysteine-dependent ATP hydrolysis indicates that theenzyme-bound Cys-AMP is stable whereas the enzyme-boundCys-tRNACYs is not.
DISCUSSIONThis work demonstrates an unexpected chemistry of enzymaticdeacylation of Cys-tRNACYs by cysteinyl-tRNA synthetase.During deacylation the enzyme makes use of a nucleophilicfunction of the side chain of cysteine in Cys-tRNACYs (I) toaccomplish intramolecular cyclization of the amino acid tocysteine thiolactone (II) (equation 1) . In contrast to the enzyme-bound Cys-tRNACYs, the enzyme-bound Cys-AMP is stable and

Nucleic Acids Research, 1994, Vol. 22, No. 7 1159
does not undergo cyclization. Spontaneous deacylation of Cys-tRNACYs in solution yields free cysteine.
NH+3 0
\ 4@tRNA -a
SH
I
3 0
K-is
II
The conclusion that a product (Spot X) of enzymaticdeacylation of Cys-tRNACYs by cysteinyl-tRNA synthetase iscysteine thiolactone is supported by chemical and enzymatic tests.For example, Spot X was converted to cysteine chemically bytreatment with NaOH or enzymatically by treatment with rabbitesterase. The product of hydrolysis of spot X has also been shownto be esterified into tRNACYs in the presence of ATP andcysteinyl-tRNA synthetase. Spot X itself did not appear to esterifytRNAcYs under standard aminoacylation conditions. Finally,Spot X has been found to be refractive to several sulfhydryl groupreagents, a property expected of cysteine thiolactone.Enzymatic deacylation of Cys-tRNACYs (kcat = 0.017 s') is
about 200-times slower than tRNACYs aminoacylation withcysteine catalyzed by cysteinyl-tRNA synthetase (kcat = 3.2 s'-)(this work). Similar ratios of deacylating and synthetic activitiesinvolving cognate substrates have been observed with othertRNAs and synthetases (10,18,19). Because of a relative slownessof enzymatic deacylation, in each case other explanations for theorigin of the observed deacyaltion of aminoacyl-tRNA, inparticular a possibility of a contaminating enzyme, must be ruledout. In the case of cysteinyl-tRNA synthetase described in thiswork, the possibility that enzymatic deacylation of Cys-tRNAaffording cysteine thiolactone is catalyzed by a contaminatingenzyme is unlikely. First, cysteinyl-tRNA synthetase used in theseexperiments was at least 95% pure as judged by SDSpolyacrylamide gel electrophoresis. Maximum concentration ofany single contaminating protein in cysteinyl-tRNA synthetasepreparation can be estimeted to be at most 1 %. Second, the rateof enzymatic deacylation of Cys-tRNA was independent of theenzyme concentration when [enzyme] > [Cys-tRNA], whichexcludes that a contaminating enzyme is responsible for thereaction.The enzymatic deacylation of Cys-tRNACYs by cysteinyl-
tRNA synthetase is mechanistically similar to the editing reactionsof methionyl-tRNA synthetase. Both cysteinyl-tRNA synthetaseand methionyl-tRNA synthetase deacylate their cognate chargedtRNAs affording cyclic forms of their cognate amino acidsubstrates, cysteine thiolactone (H, equation 1; this work) andS-methyl homocysteine thiolactone (IV, equation 2; ref. 10),respectively.
NH3
\tRNAS\
CH3
(III)
+-NHI
-o=+
CH3
(IV
+ tRNA
The presence of the cyclization function also in cysteinyl-tRNAsynthetase further supports close evolutionary relatedness betweencysteinyl- and methionyl-tRNA synthetases (13,21,22).Moreover, cysteinyl-tRNA synthetase (this work), like methionyl-tRNA synthetase (11, 23), carries out the synthetic anddeacylating functions in one active site. With both enzymes,enzymatic deacylation of charged tRNA and formation ofrespective cyclized amino acids is inhibited by a cognate aminoacid. It is therefore concluded that cyclization of cysteine tocysteine thiolactone is a manifestation of the editing function ofcysteinyl-tRNA synthetase.
Cyclization of methionine to S-methyl homocysteine thiolactoneby methionyl-tRNA synthetase occurs as a result of the limitedability of the enzyme to discriminate against the cognatemethionine at the editing site designed for the noncognatehomocysteine. The discovery of a similar ability of cysteinyl-tRNA synthetase to cyclize cysteine to cysteine thiolactonesuggests that this enzyme also has an editing function. However,the editing function of cysteinyl-tRNA synthetase is directed onlyagainst its cognate product Cys-tRNACYs and there is no knownnoncognate amino acid that would need to be edited by theenzyme. As originally demonstrated by others (14) and confirmedhere, any noncognate amino acid that might compete with cysteinefor the active site of cysteinyl-tRNA synthetase is effectivelyexcluded from binding to the active site and therefore there isno need for its editing. For example, serine, a-aminobutyrate,and alanine are activated by the enzyme 106- 108 times lessefficiently than cysteine (14). The inability of homocysteine,methionine, and serine to inhibit enzymatic deacylation of Cys-tRNA Table 1) indicates that these amino acids are also excludedfrom binding to the active site of cysteinyl-tRNA synthetase.
It is not clear, why, in the absence of a need to edit noncognateamino acids, the editing function is preserved in the present daycysteinyl-tRNA synthetase. A possible reason may come fromthe consideration of the energy costs of editing and evolution.It has been demonstrated that most of the energy costs of editingin vivo are associated with the destruction of an incorrect product.Only about 3% of total energy expended on editing is due to thedestruction of a correct product in bacterial cells (12). Thus, onecan expect that there will be a much greater evolutionary pressureon minimizing editing of a noncognate product (major energy-consuming process) than on editing of a cognate product (minorenergy-consuming process). Early in biotic evolution whenselection of amino acids by synthetases may have been lessaccurate than today, an ancestral cysteinyl-tRNA synthetase mayhave utilized the editing function to correct errors in amino acidselection. Because energy costs associated with proofreading canbe substantial, the principle of parsimony would require thatediting be minimized and intrinsic binding energy optimized toprovide adequate selectivity. Thus, as the active site of cysteinyl-tRNA synthetase evolved to ultimately prevent any significantmisactivation in the first place, and therefore minimize the needto dissipate energy on editing, the editing function becamesuperfluous, but apparently not harmful. As a result, the presentday cysteinyl-tRNA synthetase relies exclusively on intrinsicbinding energies to assure adequate accuracy of protein synthesis(14).Another possible explanation for the ability to cyclize cysteine
by the present day cysteinyl-tRNA synthetase is suggested bythe chemistry of the thioester bond. The thioester bond is a highenergy bond and thiolactones, including cysteine thiolactone, areexpected to acylate any nucleophilic group. Thus, cysteine

1160 Nucleic Acids Research, 1994, Vol. 22, No. 7
thiolactone may have been a source of activated cysteine forprotein synthesis in early biotic systems. Although we were notable to accomplish this, the deacylation of Cys-tRNA to cysteinethiolactone should in principle be reversible and allow synthesisof Cys-tRNA from cysteine thiolactone under appropriateconditions. Homocysteine thiolactone forms spontaneously fromhomocysteine at acidic pH. The equilibrium of formation ofcysteine thiolactone from cysteine is expected to be shiftedtowards cysteine because of the unfavorable strain induced inthe four member ring of the thiolactone. The formation of cysteinethiolactone may be enhanced by its removal from the solution,for example by binding to a solid surface. Thus, a source ofactivated amino acids may have existed in early prebioticconditions. S-Methyl homocysteine thiolactone, which canpossibly be formed by methylation of homocysteine thiolactone,may have provided a source of activated methionine.
ACKNOWLEDGEMENTSThis research was supported by a grant from the National ScienceFoundation (MCB-9218358).
REFERENCES1. Fersht, A. R. (1986) in Accuracy in Molecular Processes, eds. Kirkwood,
T. B. L., Rosenberger, R. F., Galas, D. J. (Chapman & Hall, N. Y.), pp.67-82.
2. Jakubowski, H. and Goldman, E. (1992) Microbiol. Rev. 56, 412-429.3. Jakubowski, H. (1994) Ann. N. Y Acad. Sci., in press.4. Old, J.M. and Jones, D.S. (1976) Biochem. J. 159, 503-511.5. Fersht, A.R. and Dingwall, C. (1979) Biochemistry 18, 1250-1256.6. Smith, L.T. and Cohn, M. (1981) Biochemistry 20, 385-391.7. Jakubowski, H. and Fersht, A. R. (1981) NucleicAcidsRes. 9,3105-3117.8. Jakubowski, H. (1990) Proc. Natl. Acad. Sci. USA 87, 4504-4508.9. Jakubowski, H. (1991) EMBO J. 10, 593 -598.
10. Jakubowski, H. and Goldman, E. (1993) FEBS Lent. 317, 237-240.11. Jakubowski, H. (1993) J. Biol. Chem. 268, 6549-6553.12. Jakubowski, H. (1993) FASEB J. 7, 168-172.13. Eriani, G., Dirheimer, G. and Gangloff, J. (1991) Nucleic Acids Res. 19:
265-269.14. Fersht, A.R. and Dingwall, C. (1979) Biochemistry 18: 1245-1249.15. Jakubowski, H., Pastuszyn, A. and Loftfield, R.B. (1977) Anal. Biochem.
82, 29-37.16. Gurd, F.R.N. (1967) Meth. Enzymol. 11: 532-541.17. Riordan, J.F. and Vallee, B.L. (1967) Meth. Enzymol. 11: 541-548.18. Yarus, M. (1972) Proc. Natl. Acad. Sci. USA 69: 1915-1919.19. Schreier, A.A. and Schimmel, P. (1971) Biochemistry 11: 1582-1589.20. Fersht, A.R. and Dingwall, C. (1979) Biochemistry 18: 1238-1245.21. Hou, Y.M., Shiba, K., Mottes, C. and Schimmel, P. (1991) Proc. Natl.
Acad. Sci. USA 88: 976-980.22. Avalos, J., Corrochano, L.M. and Brenner, S. (1991) FEBS Lett. 286:
176-180.23. Kim, H.Y., Ghosh, G., Schulman, L.H., Brunie, S. and Jakubowski, H.
(1993) Proc. Natl. Acad. Sci. USA 90: 11553-11557.






![An Aminoacyl-tRNA Synthetase Complex Escherichia coli(iii) Freezepress. Cellpellets weesuspendedin 2volumes ofbuffer Aandaddedto afreeze press (Eatonmodification oftheHughespress[7]),](https://static.fdocuments.in/doc/165x107/5e4dfce8f29b5d54b52a0e06/an-aminoacyl-trna-synthetase-complex-escherichia-coli-iii-freezepress-cellpellets.jpg)







![RESEARCH ARTICLE Open Access Fragmentation of ... - SLU.SE · 18–46 nt pieces derived from mature tRNA or the 3 ′ end of precursor-tRNA (pre-tRNA) [14-16]. tRNA fragmenta-tion](https://static.fdocuments.in/doc/165x107/60474a078cb48655a57c0958/research-article-open-access-fragmentation-of-sluse-18a46-nt-pieces-derived.jpg)


