
DNA Repair: From Genome Maintenance to Biomarker and...
13
Review DNA Repair: From Genome Maintenance to Biomarker and Therapeutic Target Shadia Jalal 1 , Jennifer N. Earley 1 , and John J. Turchi 1,2 Abstract A critical link exists between an individual’s ability to repair cellular DNA damage and cancer develop- ment, progression, and response to therapy. Knowledge gained about the proteins involved and types of damage repaired by the individual DNA repair pathways has led to the development of a variety of assays aimed at determining an individual’s DNA repair capacity. These assays and their use in the analysis of clinical samples have yielded useful though somewhat conflicting data. In this review article, we discuss the major DNA repair pathways, the proteins and genes required for each, assays used to analyze activity, and the relevant clinical studies to date. With the recent results from clinical trials targeting specific DNA repair proteins for the treatment of cancer, accurate, reproducible, and relevant analysis of DNA repair takes on an even greater significance. We highlight the strengths and limitations of these DNA repair studies and assays, with respect to the clinical assessment of DNA repair capacity to determine cancer development and response to therapy. Clin Cancer Res; 17(22); 6973–84. Ó2011 AACR. Introduction The human genome is subject to constant damage through a combination of endogenous and exogenous factors. Multiple pathways are required to restore the struc- ture and the sequence of DNA once damage has occurred, and these systems are essential to maintain genomic integ- rity and stability. The response to DNA damage is exqui- sitely regulated, often specific to the type of damage incurred. Five main repair pathways have been described: the nucleotide excision repair (NER) pathway; base excision repair (BER) pathway; mismatch repair (MMR) pathway; and the 2 double-strand break (DSB) repair pathways, nonhomologous end-joining (NHEJ) and homology- directed repair (HDR). The HDR pathway is also involved in the repair of interstrand DNA cross-links (ICL) in con- junction with the Fanconi anemia pathway (1). Collective- ly, these pathways are orchestrated by more than 150 proteins, which enable the response to a wide array of DNA-damaging events. Over the past decade, our knowledge about the roles of DNA repair pathways and how deficiencies or abnormali- ties in them affect the development of numerous disease processes has increased greatly. Mutations in DNA repair genes have been implicated in the development of neuro- logic diseases (2), aging (3), cancer risk (4), cancer therapy outcomes (5), inflammation, and other genetic syndromes with a variety of distinct phenotypes (6). Because of the importance of DNA repair in human disease, interest in the measurement or determination of an individual’s DNA repair capacity is great. Here, we review DNA repair path- ways, the current assays available for analysis of DNA repair capacity, their strengths and limitations, and their clinical applicability. DNA Damage Signaling The wide variety of DNA damage that occurs necessi- tates a flexible and sensitive DNA damage response net- work to signal the presence of an insult and coordinate the cellular response to the damage. DNA damage response is initiated with the recognition of the damage and often results in the activation of cell-cycle check- points to arrest the eukaryotic cell-cycle progression (7). The cellular response to DNA damage is propagated through signal transduction and posttranslational mod- ification of proteins involved in the various DNA repair pathways, as well as other signaling complexes that do not directly participate in repair reactions. As the DNA damage incurred typically disrupts nucleic acid metabo- lism, affecting either DNA replication or transcription, repair is often coupled to these pathways either directly or indirectly (8, 9). It should be noted that some DNA lesions escape repair and are bypassed during replication by error-prone polymerases in a process termed transle- sion synthesis. For a recent discussion of this pathway, see the review from McCulloch and Kunkel (10). Authors' Affiliations: 1 Division of Hematology and Oncology, Depart- ments of Medicine, and 2 Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, Indiana Corresponding Author: John J. Turchi, Indiana University School of Medicine, Joseph E. Walther Hall, R3-C562, 980 West Walnut Street, Indianapolis, IN 46202. Phone: 317-278-1996; Fax: 317-274-0396; E-mail: [email protected] doi: 10.1158/1078-0432.CCR-11-0761 Ó2011 American Association for Cancer Research. Clinical Cancer Research www.aacrjournals.org 6973 Research. on May 18, 2019. © 2011 American Association for Cancer clincancerres.aacrjournals.org Downloaded from Published OnlineFirst September 9, 2011; DOI: 10.1158/1078-0432.CCR-11-0761
Transcript of DNA Repair: From Genome Maintenance to Biomarker and...

Review
DNA Repair: From Genome Maintenance to Biomarkerand Therapeutic Target
Shadia Jalal1, Jennifer N. Earley1, and John J. Turchi1,2
AbstractA critical link exists between an individual’s ability to repair cellular DNA damage and cancer develop-
ment, progression, and response to therapy. Knowledge gained about the proteins involved and types of
damage repaired by the individual DNA repair pathways has led to the development of a variety of assays
aimed at determining an individual’s DNA repair capacity. These assays and their use in the analysis of
clinical samples have yielded useful though somewhat conflicting data. In this review article, we discuss the
majorDNA repair pathways, theproteins and genes required for each, assays used to analyze activity, and the
relevant clinical studies to date. With the recent results from clinical trials targeting specific DNA repair
proteins for the treatment of cancer, accurate, reproducible, and relevant analysis of DNA repair takes on an
even greater significance. We highlight the strengths and limitations of these DNA repair studies and assays,
with respect to the clinical assessment of DNA repair capacity to determine cancer development and
response to therapy. Clin Cancer Res; 17(22); 6973–84. �2011 AACR.
Introduction
The human genome is subject to constant damagethrough a combination of endogenous and exogenousfactors. Multiple pathways are required to restore the struc-ture and the sequence of DNA once damage has occurred,and these systems are essential to maintain genomic integ-rity and stability. The response to DNA damage is exqui-sitely regulated, often specific to the type of damageincurred. Five main repair pathways have been described:the nucleotide excision repair (NER) pathway; base excisionrepair (BER) pathway; mismatch repair (MMR) pathway;and the 2 double-strand break (DSB) repair pathways,nonhomologous end-joining (NHEJ) and homology-directed repair (HDR). The HDR pathway is also involvedin the repair of interstrand DNA cross-links (ICL) in con-junction with the Fanconi anemia pathway (1). Collective-ly, these pathways are orchestrated by more than 150proteins, which enable the response to a wide array ofDNA-damaging events.Over the past decade, our knowledge about the roles of
DNA repair pathways and how deficiencies or abnormali-ties in them affect the development of numerous disease
processes has increased greatly. Mutations in DNA repairgenes have been implicated in the development of neuro-logic diseases (2), aging (3), cancer risk (4), cancer therapyoutcomes (5), inflammation, and other genetic syndromeswith a variety of distinct phenotypes (6). Because of theimportance of DNA repair in human disease, interest in themeasurement or determination of an individual’s DNArepair capacity is great. Here, we review DNA repair path-ways, the current assays available for analysis of DNA repaircapacity, their strengths and limitations, and their clinicalapplicability.
DNA Damage Signaling
The wide variety of DNA damage that occurs necessi-tates a flexible and sensitive DNA damage response net-work to signal the presence of an insult and coordinatethe cellular response to the damage. DNA damageresponse is initiated with the recognition of the damageand often results in the activation of cell-cycle check-points to arrest the eukaryotic cell-cycle progression (7).The cellular response to DNA damage is propagatedthrough signal transduction and posttranslational mod-ification of proteins involved in the various DNA repairpathways, as well as other signaling complexes that donot directly participate in repair reactions. As the DNAdamage incurred typically disrupts nucleic acid metabo-lism, affecting either DNA replication or transcription,repair is often coupled to these pathways either directlyor indirectly (8, 9). It should be noted that some DNAlesions escape repair and are bypassed during replicationby error-prone polymerases in a process termed transle-sion synthesis. For a recent discussion of this pathway,see the review from McCulloch and Kunkel (10).
Authors' Affiliations: 1Division of Hematology and Oncology, Depart-ments of Medicine, and 2Biochemistry and Molecular Biology, IndianaUniversity School of Medicine, Indianapolis, Indiana
Corresponding Author: John J. Turchi, Indiana University School ofMedicine, Joseph E. Walther Hall, R3-C562, 980 West Walnut Street,Indianapolis, IN 46202. Phone: 317-278-1996; Fax: 317-274-0396; E-mail:[email protected]
doi: 10.1158/1078-0432.CCR-11-0761
�2011 American Association for Cancer Research.
ClinicalCancer
Research
www.aacrjournals.org 6973
Research. on May 18, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 9, 2011; DOI: 10.1158/1078-0432.CCR-11-0761

DNA mismatch repairThe MMR pathway is responsible for correcting replica-
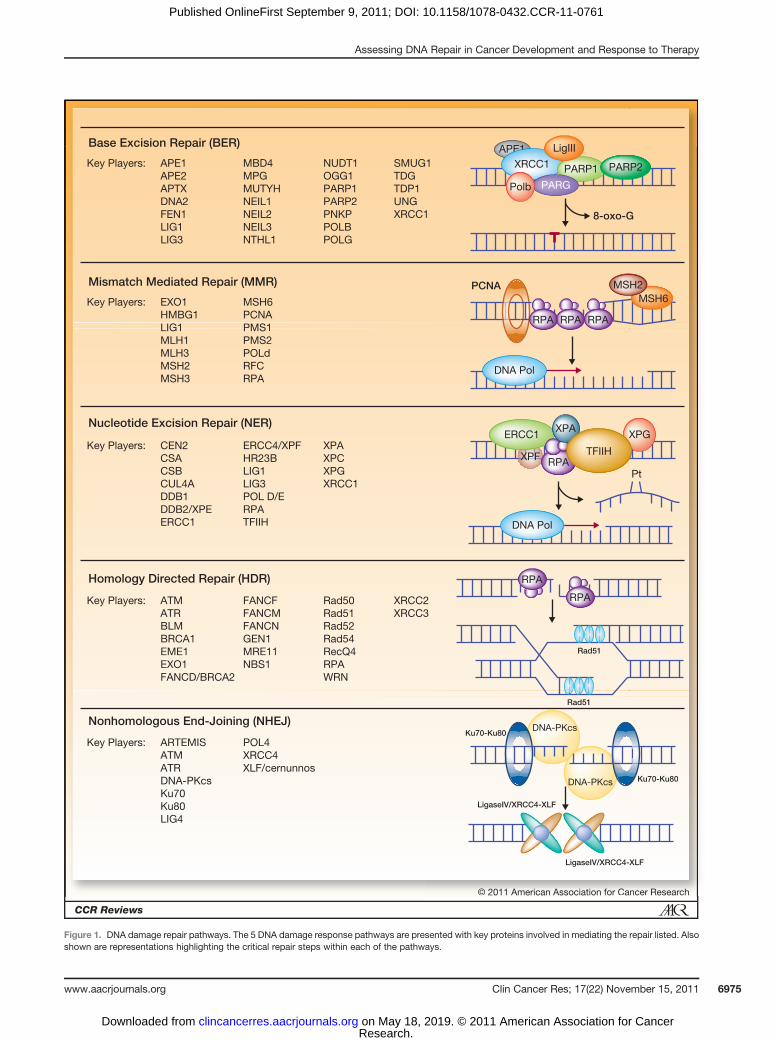
tion errors that escape processing by the 30- to 50-proofread-ing exonuclease activity of replicative DNA polymerases.Mismatches and insertion–deletion loops arise from poly-merase slippage. Defects or mutations that arise in certainMMRproteins have been ascribed to clinicalmanifestations(Fig. 1; refs. 11, 12). The most well described is hereditarynonpolyposis colorectal cancer, also known as Lynch syn-drome, an autosomal dominant disorder characterized, in90% of the cases, by germ-line mutations in one of thealleles of the MSH2 or MLH1 genes (13). The resultingfailure to repair DNA mismatches is associated with theincreased risk of development of colorectal, endometrial,ovarian, upper gastrointestinal, and genitourinary cancers(14, 15). The most common clinical assessment of MMRinvolves analysis of microsatellite instability, which servesas a robust and validated marker for MMR deficiency(16).
Base excision repairThe BER pathway repairs oxidative damage to the bases
of DNA, which can be caused by reactive oxygen species(ROS). ROS can be produced by intracellular or extracel-lular processes, including therapeutic exposures and ion-izing radiation. This pathway is orchestrated by DNAglycosylases, AP-endonuclease (APE) activity, DNAligases, polymerases, XRCC1, PCNA, and other proteins(Fig. 1). PARP1 is also involved in BER, recognizingsingle-strand and double-strand DNA breaks. This pro-tein has become a subject of considerable clinical interestin the past few years with the development of small-molecule inhibitors of this protein and the demonstra-tion of anticancer activity (17, 18). Genetic diseasescaused by mutations in BER genes seem less commonthan those described with other DNA repair pathways;
however, increased levels of APE1 have been described ingerm cell tumors (19). Mutations or overexpression ofDNA polymerase b have also been linked to increased riskof multiple cancers, including colorectal, lung, breast,gastric, and prostate cancers (20). The analysis of BERactivity is relevant in the context of cancer therapy; bothtemozolomide and dacarbazine induce base damagerepaired by BER.
Nucleotide excision repairThe NER pathway recognizes larger, helix-distorting
lesions that occur by chemical modification of DNAbases upon exposure to environmental mutagens suchas UV light, tobacco smoke, ROS, radiation, and chemo-therapeutic agents (21). The 2 subpathways of NER,transcription-coupled NER and global genomic NER,differ only in the initial recognition step and involvethe assembly and coordination of more than 30 proteins(Fig. 1; ref. 22). Hereditary disorders related to defects inthe NER pathway have been described and includexeroderma pigmentosum (XP), which predisposes affect-ed individuals to certain cancers (6). Consistent withthese findings, a subset of NER genes has been shown tohave both prognostic and predictive value in the clinicalassessment of certain cancers. NER is also relevant totherapeutic response as a function of the intrastrandDNA adducts formed by the platinum-based therapeuticscisplatin, carboplatin, and oxaliplatin being repaired viaNER.
DNA double-strand break repairRepair of DNA DSBs is mediated by the HDR and NHEJ
pathways. HDR involves the Rad52 group of proteins,BRCA1/2 and XRCC2/3, in addition to EME1 and NBS1(Fig. 1). Multiple hereditary disorders have been asso-ciated with defects in HDR, including mutations inBRCA1 or BRCA2, which have been associated withhereditary breast and ovarian cancer (23). In contrast,DSB repair via the NHEJ pathway is potentially moremutagenic and requires the coordinated assembly of anumber of proteins at the DNA termini to facilitate endjoining (Fig. 1). These proteins include the Ku hetero-dimer (Ku70/Ku80), DNA protein kinase catalytic sub-unit (DNA-PKcs), ligase IV, XRCC4, XLF, Artemis, andpolymerases m and l (24). Mutations in NHEJ compo-nents have been described and confer radiation sensitivityand defective immune function via reduced V(D)J recom-bination (25), whereas complete abrogation of NHEJcomponents seems to be incompatible with life (26).The role of NHEJ and HDR in the repair of ionizingradiation–induced DNA damage has also made thesepathways popular as targets for the development of radio-sensitizing agents (27). DNA DSBs can also arise via theenzymatic processing of interstrand DNA cross-links.Repair of interstrand DNA cross-links involves the HDRpathway as well as the Fanconi anemia pathway, andmutations in these pathways confer increased suscepti-bility to cross-linking agents (1, 28, 29).
Translational Relevance
An individual’s DNA repair capacity hasmajor clinicalimplications, which include risk of development ofvarious illnesses including cancer, the response toDNA-damaging cancer therapies, and likelihood of ther-apy-induced toxicities. Multiple assays analyzing DNArepair capacity have yielded useful information, but theyhave limitations in applicability, reproducibility, andinterpretation. These assays analyze different aspects ofDNA repair capacity under specific conditions and arenot necessarily amarker of an individual’s absoluteDNArepair capacity. As we aim to personalize cancer therapy,a deeper understanding of DNA repair pathways and thespecific assays used to analyze their activity is criticalwhen determining associations with complexmolecularand cellular processes involved in disease susceptibilityand response to therapeutics.
Jalal et al.
Clin Cancer Res; 17(22) November 15, 2011 Clinical Cancer Research6974
Research. on May 18, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 9, 2011; DOI: 10.1158/1078-0432.CCR-11-0761
© 2011 American Association for Cancer Research© 2011 American Association for Cancer Research
DNA-PKcs
DNA-PKcs
APE1
XRCC1 PARP1
ERCC1
XPF
DNA Pol
PCNA
8-oxo-G
DNA Pol
PARGPolb
LigIII
MSH6
XPG
PtRPA
PARP2
MSH2
RPA RPA RPA
RPA
RPA
TFIIH
XPA
Key Players:
Key Players:
Key Players:
APE1
APE2
APTX
DNA2
FEN1
LIG1
LIG3
MBD4
MPG
MUTYH
NEIL1
NEIL2
NEIL3
NTHL1
SMUG1
TDG
TDP1
UNG
XRCC1
NUDT1
OGG1
PARP1
PARP2
PNKP
POLB
POLG
EXO1
HMBG1
LIG1
MLH1
MLH3
MSH2
MSH3
CEN2
CSA
CSB
CUL4A
DDB1
DDB2/XPE
ERCC1
POL4
XRCC4
XLF/cernunnos
LigaseIV/XRCC4-XLF
LigaseIV/XRCC4-XLF
Ku70-Ku80
Ku70-Ku80
Rad51
Rad51
ARTEMIS
ATM
ATR
DNA-PKcs
Ku70
Ku80
LIG4
XRCC2
XRCC3
Rad50
Rad51
Rad52
Rad54
RecQ4
RPA
WRN
FANCF
FANCM
FANCN
GEN1
MRE11
NBS1
ATM
ATR
BLM
BRCA1
EME1
EXO1
FANCD/BRCA2
Base Excision Repair (BER)
Mismatch Mediated Repair (MMR)
Nucleotide Excision Repair (NER)
Key Players:
Homology Directed Repair (HDR)
Key Players:
Nonhomologous End-Joining (NHEJ)
MSH6
PCNA
PMS1
PMS2
POLd
RFC
RPA
ERCC4/XPF
HR23B
LIG1
LIG3
POL D/E
RPA
TFIIH
XPA
XPC
XPG
XRCC1
Figure 1. DNA damage repair pathways. The 5 DNA damage response pathways are presented with key proteins involved in mediating the repair listed. Alsoshown are representations highlighting the critical repair steps within each of the pathways.
Assessing DNA Repair in Cancer Development and Response to Therapy
www.aacrjournals.org Clin Cancer Res; 17(22) November 15, 2011 6975
Research. on May 18, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 9, 2011; DOI: 10.1158/1078-0432.CCR-11-0761

© 2011 American Association for Cancer Research© 2011 American Association for Cancer Research
C C C C C C C CCA A A A A A AA A AT T T T T T T T T T T T TG G G G G G G G G G
CKTYCVGPF60 70 80 90
L S* WP
Treatment of
cells with
mutagen
of choice
Read-out based
on reporter
gene selection
Electric field is
applied across the
gel; broken DNA
escapes the cell
Negative
response:
DNA damage
persists; migration
out of the cell
Positive
response:
DNA damage
is repaired
(no migration)
A blood or tissue sample
is retrieved from a patient
and submitted for DNA
sequencing
Cells are obtained
then treated
and fixed into
a gel matrix
Positive
response:
gene is activated
upon repair
Negative
response:
gene is not
repaired
Transfection of
damaged (∗) plasmid
into a host cell line
Verify with
cell viability
testing to
determine
results
Mutagen Sensitivity Assay
Host Cell Reactivation Assay
∗
∗
Comet Assay
SNP Analysis
– +
O
OHO
NH
NH
N
S
N
S
S +
HN
O
O
O
O
O
O
O
HO
O
O
H2N
H2N
H2NNH2
HN
OH
OHOH
OH
HO
HO
NH2HN
NH
NH
N
N N O
O
HO
O
OH
H2N
H2N
CI
CI
Pt
Jalal et al.
Clin Cancer Res; 17(22) November 15, 2011 Clinical Cancer Research6976
Research. on May 18, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 9, 2011; DOI: 10.1158/1078-0432.CCR-11-0761

Assays and Methods Analyzing DNA RepairCapacity
Hereditary disorders caused by defects in specific DNArepair genes, as described above, lead to reducedDNA repaircapacity and distinct phenotypes. Although these disordersare exceedingly rare, they highlight the importance of DNArepair in influencing biologic and physiologic processes.The interindividual variations in DNA repair capacities inthe general population are likely to be subtle; however, theimpact of these subtle differences may be significant and acontributing factor for the predisposition to cancer andresponse to cancer therapy. As genomic instability is oneof the hallmarks of cancer development (30), a large num-ber of studies have aimed to compare DNA repair capacitiesbetween patients with cancer and healthy control subjects,with separate studies assessing DNA repair capacity as apredictor of response to chemotherapy or radiation. Each ofthese studies uses assays of human tissue to analyze DNArepair capacity. The most popular assays are discussedbelow, along with the clinical trials highlighting the limita-tions of the assays and how these limitations affect theconclusions. Themost important limitation for the biologicassays is the difficulty in adapting these methodologies tothe clinical setting. Routine clinical use is often limited bythe need for preparation of isolated, viable cells or extractsfrompatient samples, preparation of nonstandard reagents,and specialized, often expensive, instrumentation.
Mutagen sensitivity assaysThe most commonly used assay to indirectly measure
DNA repair capacity is themutagen sensitivity assay (Fig. 2).This cytogenetic assay quantifies chromatid breaks in cul-tured peripheral blood lymphocytes (PBL) after exposure todifferent mutagens (31, 32). Numerous studies have usedmutagen sensitivity assays to evaluate DNA repair capacityin patients with a variety of malignancies (including headand neck cancer, bladder cancer, breast cancer, non–smallcell lung cancer, and basal cell carcinoma), and in general,they showed higher mutagen sensitivity in patients withcancer compared with healthy control subjects (33–37). Astrength of this assay is its reproducibility when conductedby different laboratories (38); however, a weakness is thatthe assay readout does not provide an analysis of the directdamage induced by themutagen or its repair. The detectionof chromatid breaks, which are indicative of cellular muta-genicity and hence risk for cancer development, can beinfluenced by many factors of which DNA damage andDNA repair are only a subset. The vast majority of studies
use a singlemutagen and, thus, limit the assessment ofDNArepair to the pathway responsible for repair of the specificdamage induced, which may or may not be clinicallyrelevant. Thus, assessment of mutagen sensitivity is appro-priate, although both positive and negative correlationsmust be viewed cautiously and in the context of the muta-gen employed.
Host cell reactivationThe host cell reactivation (HCR) assay analyzes DNA
repair capacity using a mammalian expression vector har-boring DNA damage within a reporter gene that is tran-siently transfected into the host cell, typically PBLs fromstudy participants, withDNA repair activity beingmeasuredvia removal or repair of the damage to reactivate reportergene expression (Fig. 2; ref. 39). Various DNA-damagingagents can be used in this assay and, thus, similar to themutagen sensitivity assay, restrict the conclusions to aspecific repair pathway. HCR assays are useful for assess-ment of minimal protein components necessary for repairactivity, but they do not take into account important issuesthat can affect DNA repair, including chromatin effects andDNA damage signaling. Thus, results from these studiesmust be viewed as not the absolute activity but the repairactivity possible under a limited set of conditions. As thedamaged DNA is introduced to the cell, it does not neces-sarily initiate the same series of cellular responses and,therefore, although this assay is useful for assessing themechanics of repair, the true repair capacity is a function ofadditional factors that cannot be measured in this type ofassay. Despite these shortcomings, studies using the HCRassay in cancer patients have shown reduced DNA repaircapacity (40–46).
Single-cell analysisThe comet assay (also known as single-cell micro gel
electrophoresis assay) measures single-stand breaks andDSBs in a semiquantitative manner from whole cells (Fig.2; ref. 47). Similar to HCR assays, comet assays requireobtaining PBLs from patients, followed by treatment with aspecific DNA-damaging agent that leads to strand breakage.The basic principle of interpreting this assay is that themoreDNA damage induced, the farther the DNAmigrates duringelectrophoresis into a "tail" region and the less DNAremains in the "head" region, giving the appearance of acomet (48). This assay has beenused to analyze damage andrate of repair in epidemiologic studies similar to thosementioned above, and it showed reduced DNA repaircapacity in patients with lung cancer comparedwith control
Figure 2. Commonly used assays to analyze DNA damage or repair. Themutagen sensitivity assay involves treatment of cells with a chemical or radiation andobservation of the effect on the cells. In this depiction, cells are sorted using fluorescence-activated cell sorting to determine viable (green box) or apoptotic(red box) cells following treatment. The HCR assay takes advantage of the host cell's DNA repair machinery to repair a pre-damaged plasmid DNA,which is transfected into the host cells. The damage is typically incorporated into a selection gene (i.e., luciferase), which remains inactive if not repaired, oractive when repaired. The comet assay involves obtaining cells, which can be cultured from an animal or from a patient. The cells are then treated withDNA-damaging agents (i.e., radiation), then embedded into a thin layer of agarose. An electric field is applied across the gel, and broken DNA will migrateout of the cell. Single-nucleotude polymorphism (SNP) analysis involves obtaining a sample of cells fromwhich DNA can be extracted and sent for sequenceanalysis. The sequencing information obtained will give the genotype for the marker selected.
Assessing DNA Repair in Cancer Development and Response to Therapy
www.aacrjournals.org Clin Cancer Res; 17(22) November 15, 2011 6977
Research. on May 18, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 9, 2011; DOI: 10.1158/1078-0432.CCR-11-0761

Table 1. Clinical correlations of DNA repair gene single-nucleotide polymorphisms
Pathway Gene SNPs Reference SNPidentifier
Clinical correlations
BER APE1 c.-656T > G (promoter region) rs1760944 Decreased risk of lung cancerc.-141T > G (promoter region) rs1760944 Decreased risk of lung cancer,
Chinese populationsc.2197T > G (p.Asp148Glu) rs1130409 Reduced risk of bladder cancer
FEN1 c.-69G > A (promoter region) rs174538 Increased risk of lung cancerc.4150G > T rs4246215 Increased risk of lung cancer
MBD4 c.1036G > A (p.Glu346Lys) rs140693 Reduced risk of lung cancer,Chinese population
MUTYH c.494A > G (p.Tyr165Cys) rs34612342 Associated with colorectaladenomas and carcinoma
c.36þ11C > T rs2275602 Increase colorectal cancer risk,Japanese population
OGG1 g.748–15C > G rs2072668 Increased risk of gallbladder carcinomain females, northern India
p.Ser326Cys rs1052133 Increased risk of gallbladder carcinomain females, northern India; also notedin several places an increased riskof lung cancer
PARP1 c.2285T > C (p.Val762Ala) rs1136410 Increased risk of multiple malignancies,decreased risk of non-Hodgkinlymphoma in Korean patients
POLB c.725C > G (p.Pro242Arg) rs3136797 Increased risk of breast cancer developmentNEIL2 g.4102972C > G rs804270 Increased risk of squamous cell carcinoma
of the oral cavityXRCC1 c.580C > T (p.Arg194Trp) rs1799782 Platinum sensitivity
MMR EXO1 c.1765C > A (p.Glu589Lys) rs1047840 Associated with increased risk of gastriccancer and breast cancer
MLH1 c.655A > G rs1799977 Increased risk of colorectal cancer butfavorable outcome
MSH2 c.2006–6T > C [gIVS12–6T > C] rs2303428 Poor prognostic factor in NSCLCMSH3 c.693G > A (p.Pro222Pro) rs1805355 Increased risk of prostate cancer
NER DDB2/XPE c.457–314G > C rs830083 Possible association with lung cancer riskERCC1 C8092A > CA rs3212986 Poor prognosis in NSCLC patients
C8092A C/T,TT rs11615 Better survival in ovarian cancerERCC2/XPD c.934G > A (p.Asp312Asn) rs1799793 Increased risk of lung cancer; improved
responses in patients with secondary AMLc.2251A > C (p.Lys751Gln) rs13181 Increased risk of lung cancer; improved
responses in patients with secondary AMLXPA c.-4A > G (50 noncoding region) rs1800975 Reduced risk of lung cancerXPC c.1496C > T (p.Ala499Val) rs2228000 Associated with bladder cancer riskXPG c.138T > C (p.His46His) rs1047768 Affects sensitivity to chemotherapy
HDR ATM c.-111G > A rs189037 Increased risk of NSCLC in never smokersc.496þ448G > A rs228597c.1803–355C > A rs228592c.3078–77C > T rs664677
NBS1 c.553G > C (p.Glu185Gln) rs1805794 Positive association with bladder cancerc.2016A > G (p.Pro672Pro) rs1061302 Decreased association with laryngeal cancer;
positively associated with liver cancerRad51 c.-98G > C (50 UTR) rs1801320 Protective against breast and ovarian cancer
in BRCA1 carriers
(Continued on the following page)
Jalal et al.
Clin Cancer Res; 17(22) November 15, 2011 Clinical Cancer Research6978
Research. on May 18, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 9, 2011; DOI: 10.1158/1078-0432.CCR-11-0761

subjects (46, 49, 50). The comet assay has the advantage ofbeing adaptable to be more specific to the type of lesion inquestion through the use of specific enzymes (51). How-ever, performance of the comet assay requires a substantialamount of preparation and hands-on manipulation. Theseanalyses have been plagued by issues with interlaboratoryreproducibility and variations in quantification, andalthough standardization is being addressed, no unifyingstandard has been established to date (52).
Biomarkers of DNA damage and repairA number of other assays are used occasionally, including
measurement of the expression or activation of certain bio-markers or surrogates of DNA repair. The most commonassays include g-H2AX foci, whichmeasure the expression ofa histone variant (53–55). The analysis typically can generatea robust signal because megabases of DNA include thismodification around a single DNA DSB. However, howgamma-H2AX foci relates to repair and, ultimately, sensitiv-ity to DNA damaging agents remains to be determined.Similarly, the assessment of Rad51, 53BP1, and RPA foci isbeing investigated as they also indicate that recombinationand repair machinery is accumulating at specific sites in thegenome, likely in the vicinity of DNA damage. Althoughnone of these analyses are routinely used in clinical practice,theirpotential asbiomarkers isbeing investigatedand shoulddetermine their effectiveness as prognostic or predictivefactors.
Candidate gene analysesFinally, genetic-based assays have been used as surrogates
to evaluate DNA repair capacity via quantitative real-timePCR of gene expression (56) and single-nucleotide poly-morphism (SNP) analyses of DNA repair genes. The can-didate gene approach for these studies has substantialadvantages over genome-wide association studies and cansupport hypothesis-driven research. Multiple studies havecorrelated a variety of polymorphisms with the risk ofdevelopment of different solid organ malignancies, andnumerous DNA repair gene SNPs have been shown to be
prognostic in patients with cancer (Table 1). SNPs are animportant genetic tool, but the interpretation of studiesevaluating individual SNPs as they relate to variables, suchas therapeutic efficacy, cancer risk, or prognosis, is hinderedby numerous limitations, including linkage disequilibrium,inadequate statistical analyses (e.g., small sample size,multiple testing, and reproducibility), and the publicationbias of positive associations. Thus biologic, pathway-drivenselection of candidate genes and independent corrobora-tion of the effect of candidate SNPs on biologic activityshould be the minimal requirement for studies measuringassociations with therapeutic efficacy or prognosis.
Tissue analysis and the use of surrogatesAnother factor that will likely influence the results of
studies evaluating DNA repair capacity would be the select-ed tissue used for analysis. A wide range of surrogates hasbeen used, including PBLs (freshly isolated), cryopreservedEpstein–Barr virus (EBV)–transformed PBLs [also known aslymphoblastoid cell lines (LCL)], blood cultures (36), pri-mary tumor samples, and, on rare occasions,metastatic sites(57). The effectiveness of LCLs as surrogates of cryopre-served isolated lymphocytes for the analysis of DNA repairgenotype–phenotype correlations is unclear, but they arecontinuously used in these studies. At least one studyshowedhigh variability andpoor reproducibility of analysisof DNA DSBs using LCLs compared with PBLs, possiblybecause of chromosomal instability resulting from EBVtransformation (58). Another study showed significantlyincreased 8-oxoG DNA glycosylase activity and expressionin LCLs compared with PBLs (59). Even unmodified freshlyisolated PBLs are unlikely to reflect DNA repair capacity oftumor tissue, as all cancers (unlike PBLs) will display somedefect in DNA repair. In fact, one study compared the repaircapacity of lymphocytes and colon tumor cells throughmeasuring rates of removal of DNA cross-links induced byoxaliplatin using the comet assay and found significantdifferences (60). With recent data showing genomic differ-ences between primary tumors and their metastases, inaddition to the increased understanding of intratumor
Table 1. Clinical correlations of DNA repair gene single-nucleotide polymorphisms (Cont'd )
Pathway Gene SNPs Reference SNPidentifier
Clinical correlations
BLM c.2603C > T (p.Pro868Leu) rs11852361 Increased risk of rectal cancerXRCC2 c.563G > A (p.Arg188His) rs3218536 Possible protective role against breast
cancer in women that never breast fedXRCC3 c.722C > T (p.Thr241Met) rs861539 Weak association with bladder cancer risk
NHEJ Ku70 T 991C rs5751129 Increased susceptibility to oral cancerKu80 G1041T rs828907 Increased risk of colon cancerXRCC4 c.26C > T (p.Thr9Ile) rs1805388 Prognostic in NSCLC
c.894–1(7)G > A rs1805377 Prognostic in NSCLC
Abbreviations: AML, acute myelogenous leukemia; NSCLC, non–small cell lung cancer; SNP, single-nucleotide polymorphism;UTR, untranslated region.
Assessing DNA Repair in Cancer Development and Response to Therapy
www.aacrjournals.org Clin Cancer Res; 17(22) November 15, 2011 6979
Research. on May 18, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 9, 2011; DOI: 10.1158/1078-0432.CCR-11-0761

heterogeneity, caution should be used when interpretingthe available data evaluating DNA repair capacity.
It is possible that newer circulating tumor cell technologywill prove to be a very useful tool (61). Until then, tumortissue should be used for analysis, as its DNA repair defectsare more likely to be successfully exploited. Use of surro-gates, though more easily obtainable, is of limited clinicalvalue and should be avoided.
Impact of DNA Repair Capacity on Cancer Risk
Many retrospective studies have used the previously dis-cussed assays to measure DNA repair capacity and, ingeneral, have reported decreased DNA repair capacity inpatients with numerous solid organmalignancies (Table 2).These studies had multiple limitations, including reversecausation biases due to their retrospective nature. A limitednumber of prospective studies have examined the possibleassociation between cancer risk and reduced DNA repaircapacity (31, 62, 63). The largest prospective trial was
reported recently as part of the prostate, lung, colorectal,and ovarian (PLCO) cancer-screening trial. It comparedprospective cases with lung cancer with their controls. HCRand comet assays were unrelated to lung cancer risk, but themutagen sensitivity assay, which used bleomycin as theDNA-damaging agent, showed a positive association withan OR of 2.1 in the quartile with the highest chromatidbreaks per cell (64). Cases and controls were well matchedand stratified for pertinent variables, including age, gender,weight, and smoking history. Among the limitations of thistrial was the use of EBV-transformed LCLs for the analysis,which, as stated above, might not be the best surrogate, inaddition to the small sample size. The study againhighlightsthe importance of the choice of DNA repair assay used.
DNA Repair Capacity's Impact on Response toCancer Therapies
In addition to the evaluation of DNA repair capacity inrelation to the risk of cancer development, multiple studies
Table 2. Evaluation of DNA repair capacity in cancer patients
Assay and/or agent Patient population Cases/Controls Outcome Reference
Mutagen sensitivityBleomycin NSCLC 90/119 OR ¼ 3.7 (95% CI, 1.4–9.4) 35BPDE Lung cancer 977/977 Higher mutagen sensitivity in
lung cancer patients36
BPDE Squamous cell carcinomaof the head and neck
895/898 Higher frequency of BPDE-inducedchromatid breaks in patients
33
BPDE Breast cancer 100/105 Higher frequency of chromatid breaksin breast cancer patients;OR ¼ 3.11 (95% CI, 1.72–5.64)
37
UV Melanoma and nonmelanomaskin cancer
329/469 Higher frequency of UVB-inducedchromatid breaks in nonmelanomaskin cancer patients
34
HCRBPDE Squamous cell carcinoma
of the head and neck744/753 Reduced DNA repair capacity; OR ¼ 1.91
(95% CI, 1.52–2.40)41
BPDE NSCLC 467/488 Reduced DNA repair capacity; OR ¼ 1.85(95% CI, 1.42–2.42)
42
BPDE Head and neck cancer 55/61 Reduced DNA repair capacity 43BPDE Lung cancer 51/56 Reduced DNA repair capacity 44BPDE Lung cancer 316/316 Reduced DNA repair capacity in patients;
OR 1.8 (95% CI, 1.1–3.1)45
UV Basal cell carcinoma and/orsquamous cell carcinoma
333/255 16% reduction in DNA repair capacityin patients
40
Comet assayH202 Lung cancer 30/90 Higher level of H2O2-induced DNA damage
in lung cancer patients49
BPDE Patients with multiple versussingle NSCLC
108/99 Higher BPDE-induced damage and repairin cases
50
Bleomycin and BPDE Laryngeal carcinomas 52/56 Higher levels of mutagen-induced damagein patients
46
Abbreviations: BPDE, benzo(a)pyrene diol epoxide;CI, confidence interval; NSCLC, non–small cell lung cancer; UVB, ultraviolet light B.
Jalal et al.
Clin Cancer Res; 17(22) November 15, 2011 Clinical Cancer Research6980
Research. on May 18, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 9, 2011; DOI: 10.1158/1078-0432.CCR-11-0761

have aimed to evaluate the impact of repair capacity in thecontext of cancer patients’ response to different cancertherapies. Although reduced innate DNA repair capacity isundesirable from a cancer risk perspective, reduced tumoralDNA repair capacity is desirable because it can be exploitedtherapeutically with DNA-damaging therapeutics, and viasynthetic lethality (18, 65).Platinum has been the most intensely studied anticancer
agent due to themajor role it plays in cancer treatment for anumber of solid organ malignancies. Platinum efficacy ismediated by the formation of DNA adducts, themajority ofwhich are repaired via the NER pathway (Fig. 1; ref. 66).ERCC1 is probably the most-studied protein as both aprognostic and predictive marker for the survival benefitfrom adjuvant platinum-based chemotherapy (67),although all NER-deficient cells display sensitivity to cis-platin. Despite the plethora of clinical data, skepticismpersists about the usefulness of this marker due to multiplefactors, including the retrospective nature of the trials andthe known limitations of immunohistochemistry, includ-ing controversies around the optimal primary antibody forERCC1 detection (68, 69). The definition of ERCC1 posi-tivity is also arbitrary and varies between studies. DNArepair is a complex process that is unlikely to be measuredby the expression of one protein. The prospective data usingERCC1 as a biomarker for platinum response, althoughstatistically significant, were unimpressive. It is possible thatsome patients who were designated as DNA repair deficient(because of low ERCC1 expression) would, in fact, beconsidered DNA repair proficient if other DNA repair pro-teins had been measured. In addition, the mechanisticexplanation for why ERCC1 would confer platinum sensi-tivity is unclear. The recognition step inNER is thought to berate limiting, and ERCC1-XPF is the last factor recruited tothe preincision complex (70). Does the likely role ofERCC1-XPF in the repair of interstrand cisplatin cross-linksexplain its possible importance (71, 72)?Mutations inHDRgenes confer extreme sensitivity to cisplatin, suggesting thatHDR proteins are involved in some capacity in the cellularresponse to platinum lesions. These findings raise anotherinteresting point that it may not be as simple as targetingone pathway, as the efficacy of platinum treatment may bethe combination of the inter- and intrastrand cross-linksthat arise or cross-talk that may occur between differentDNA damage pathways.DNA repair is also the focus of predicting responses to
other DNA-damaging agents. For example, bothMMR- andBER-related protein levels are thought to correlate withclinical response to alkylating agents (dacarbazine andtemozolomide) in patients with metastatic melanoma(73, 74). Expression of aprataxin (APTX), a DNA repairprotein, was recently found to regulate sensitivity to irino-tecan in colorectal cancer, in which low tumor levels ofAPTX correlated with good response (75). Survival benefitin glioblastoma multiforme patients treated with temozo-lomide was only noted in the subset with methylatedMGMT promoter leading to its silencing (76). In a recentbreast cancer study, complete pathologic response to
neoadjuvant chemotherapy was associated with lowerRad51 foci in tumor biopsies obtained after chemotherapy(77).
Finally, various polymorphisms of DNA repair geneshave been studied in association with sensitivity to plati-num or other DNA-damaging agents (Table 1). However,the significance of the SNPs identified to date must befurther investigated because of conflicting observations anda general lack of follow-up linking specific SNPs to specificpathway deficiencies. The specific defect conferred by eachSNP should be tied to a molecular phenotype and func-tional significance within a biologic pathway.
DNA Repair Proteins as Drug Targets
Interest in targeting DNA repair proteins has increasedgreatly over the past decade. The impact of DNA repair onresistance to cisplatin is well documented in numerouscancers. The holy grail of reversing either innate or acquiredcisplatin resistance is being pursued by targeting specificproteins that are involved directly in the repair of thecisplatin lesions (78–81) or in the pathways responsiblefor signaling DNA damage (82–84). Currently, the mostadvanced inhibitors of DNA repair in the clinical setting areinhibitors of PARP. PARP is activated by DNA breaks and isinvolved in multiple DNA damage responses includingBER, HDR, NHEJ, and replication restart, in addition to itsrole in transcription (85, 86). PARP inhibition was noted tobemost effective, in preclinical studies, in BRCA-deficient ormutant tumor cell lines, possibly because of their greaterdependence on PARP and BER for maintenance of genomicintegrity in the presence of HDR defects, establishing asynthetic lethal interaction (87, 88). Two genes are synthet-ically lethal if a mutation in either alone is compatible withcell survival, whereas amutation in both leads to cell death.The concept of synthetic lethality is very attractive becausenormal cells should be less likely to be affected, reducingtoxicity. Iniparib (BSI-201), a PARP inhibitor, was recentlyevaluated in combination with chemotherapy in triple-negative breast cancer because of the similarities betweentriple-negative breast cancer and BRCA-deficient breastcancers (89). The combination showed improvement inprogression-free and overall survival in a phase II trial (90);however, preliminary reports of a phase III trial evaluatingthe same combinationwere negativewith a lack of improve-ment in overall survival in women with triple-negativemetastatic breast cancer who received iniparib combinedwith chemotherapy (91). Importantly, PARP is not directlyinvolved in the repair of platinum DNA lesions, whichraises many mechanistic questions about the reasons forbenefit initially reported with the addition of the PARPinhibitor in conjunction with cisplatin. Another possiblereason for the lack of survival improvement could be relatedto the fact that iniparib is a much less potent inhibitor ofPARP1 (with approximately 0.1% of the potency) thanmost other agents of this class (92). Both olaparib andiniparib are currently being studied in a number of solidorgan malignancies and seem to be especially promising in
Assessing DNA Repair in Cancer Development and Response to Therapy
www.aacrjournals.org Clin Cancer Res; 17(22) November 15, 2011 6981
Research. on May 18, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 9, 2011; DOI: 10.1158/1078-0432.CCR-11-0761

relapsed ovarian cancer, either in combination with che-motherapy or as maintenance therapy. The negative phaseIII study in triple-negative breast cancer, nonetheless, high-lights the importance of a deeper understanding of DNArepair and the intimate connection between the damageinduced and the pathway responsible for its repair. Onlyarmed with this information can rational combinationsof therapies be developed, tested, and, ultimately, be effec-tive in the treatment of complex diseases such as cancer.The activity of the topoisomerase II inhibitor, etoposide, incancers with inactivation of the retinoblastoma tumorsuppressor protein is another example of synthetic lethality(93).
Conclusions
Genome instability is a hallmark of cancer, and accuratedetermination of one’s capacity for maintaining genomestability holds the potential to assess risk of cancer devel-opment. The complexity and specificity of the pathways thatgovern genomic integrity necessitate accurate analyses ofeach specific pathway to ultimately determine risk. Perhaps
more important than assessing an individual’s risk ofmalig-nancy is the potential to personalize therapy with a betterunderstanding of DNA repair pathways, because DNAdamage continues to be the mainstay of cancer therapy.The development of reproducible, patient- and laboratory-friendly assays for the analysis of DNA repair is critical.Limitations with current methodologies provide the impe-tus for improvement, miniaturization, and automation tobring to fruition the goal of assessing individual DNA repaircapacity.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Grant Support
This work was supported by National Cancer Institute grantsR01CA82741 and R21CA128628 and NIH grants CA82741 and CA128628to J.J. Turchi.
Received March 21, 2011; revised August 5, 2011; accepted August 10,2011; published OnlineFirst September 9, 2011.
References1. Kennedy RD, D'Andrea AD. DNA repair pathways in clinical practice:
lessons from pediatric cancer susceptibility syndromes. J Clin Oncol2006;24:3799–808.
2. Rolig RL,McKinnon PJ. Linking DNA damage and neurodegeneration.Trends Neurosci 2000;23:417–24.
3. Akbari M, Krokan HE. Cytotoxicity and mutagenicity of endogenousDNA base lesions as potential cause of human aging. Mech AgeingDev 2008;129:353–65.
4. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—anevolving hallmark of cancer. Nat Rev Mol Cell Biol 2010;11:220–8.
5. Olaussen KA, Dunant A, Fouret P, Brambilla E, Andr�e F, Haddad V,et al. IALT Bio Investigators. DNA repair by ERCC1 in non-small-celllung cancer and cisplatin-based adjuvant chemotherapy.NEngl JMed2006;355:983–91.
6. de Boer J, Hoeijmakers JH. Nucleotide excision repair and humansyndromes. Carcinogenesis 2000;21:453–60.
7. Warmerdam DO, Kanaar R. Dealing with DNA damage: relationshipsbetween checkpoint and repair pathways. Mutat Res 2010;704:2–11.
8. Cimprich KA, Cortez D. ATR: an essential regulator of genome integ-rity. Nat Rev Mol Cell Biol 2008;9:616–27.
9. Tornaletti S. DNA repair in mammalian cells: Transcription-coupledDNA repair: directing your effort where it's most needed. Cell Mol LifeSci 2009;66:1010–20.
10. McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryoticreplicative and translesion synthesis polymerases. Cell Res2008;18:148–61.
11. Ricciardone MD, Ozcelik T, Cevher B, Ozda�g H, Tuncer M, G€urgey A,et al. Human MLH1 deficiency predisposes to hematological malig-nancy and neurofibromatosis type 1. Cancer Res 1999;59:290–3.
12. Whiteside D, McLeod R, Graham G, Steckley JL, Booth K, SomervilleMJ, et al. A homozygous germ-line mutation in the humanMSH2 genepredisposes to hematological malignancy and multiple caf�e-au-laitspots. Cancer Res 2002;62:359–62.
13. Lynch HT, de la Chapelle A. Genetic susceptibility to non-polyposiscolorectal cancer. J Med Genet 1999;36:801–18.
14. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria forhereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome)proposed by the International Collaborative group on HNPCC. Gas-troenterology 1999;116:1453–6.
15. Barrow E, Alduaij W, Robinson L, Shenton A, Clancy T, Lalloo F, et al.Colorectal cancer in HNPCC: cumulative lifetime incidence, survivaland tumour distribution. A report of 121 familieswith provenmutations.Clin Genet 2008;74:233–42.
16. Laghi L, Bianchi P,Malesci A.Differencesandevolutionof themethodsfor the assessment of microsatellite instability. Oncogene 2008;27:6313–21.
17. AnnunziataCM, Bates SE. PARP inhibitors in BRCA1/BRCA2germlinemutation carriers with ovarian and breast cancer. F1000 Biol Rep2010;2:10.
18. Lord CJ, Ashworth A. Targeted therapy for cancer using PARP inhi-bitors. Curr Opin Pharmacol 2008;8:363–9.
19. Robertson KA, Bullock HA, Xu Y, Tritt R, Zimmerman E, Ulbright TM,et al. Altered expression of Ape1/ref-1 in germ cell tumors and over-expression in NT2 cells confers resistance to bleomycin and radiation.Cancer Res 2001;61:2220–5.
20. AlbertellaMR, Lau A, O'ConnorMJ. The overexpression of specializedDNA polymerases in cancer. DNA Repair (Amst) 2005;4:583–93.
21. Friedberg EC, Aguilera A, Gellert M, Hanawalt PC, Hays JB, LehmannAR, et al. DNA repair: from molecular mechanism to human disease.DNA Repair (Amst) 2006;5:986–96.
22. Shuck SC, Short EA, Turchi JJ. Eukaryotic nucleotide excision repair:from understanding mechanisms to influencing biology. Cell Res2008;18:64–72.
23. KingMC,Marks JH,Mandell JBNewYorkBreastCancer StudyGroup.Breast and ovarian cancer risks due to inherited mutations in BRCA1and BRCA2. Science 2003;302:643–6.
24. Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining.Biochem J 2009;417:639–50.
25. Schwarz K, Ma YM, Pannicke U, Lieber MR. Human severe com-bined immune deficiency and DNA repair. Bioessays 2003;25:1061–70.
26. Wang Y, Ghosh G, Hendrickson EA. Ku86 represses lethal telomeredeletion events in human somatic cells. Proc Natl Acad Sci U S A2009;106:12430–5.
27. O'ConnorMJ,Martin NM, Smith GC. Targeted cancer therapies basedon the inhibition of DNA strand break repair. Oncogene 2007;26:7816–24.
Jalal et al.
Clin Cancer Res; 17(22) November 15, 2011 Clinical Cancer Research6982
Research. on May 18, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 9, 2011; DOI: 10.1158/1078-0432.CCR-11-0761

28. Wang LC, Stone S, Hoatlin ME, Gautier J. Fanconi anemia proteinsstabilize replication forks. DNA Repair (Amst) 2008;7:1973–81.
29. McKinnon PJ, Caldecott KW. DNA strand break repair and humangenetic disease. Annu Rev Genomics Hum Genet 2007;8:37–55.
30. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57–70.
31. Wu X, Gu J, Spitz MR. Mutagen sensitivity: a genetic predispositionfactor for cancer. Cancer Res 2007;67:3493–5.
32. Li C, Wang LE, Wei Q. DNA repair phenotype and cancer suscepti-bility—a mini review. Int J Cancer 2009;124:999–1007.
33. Wang LE, XiongP, ZhaoH, SpitzMR, Sturgis EM,WeiQ.Chromosomeinstability and risk of squamous cell carcinomas of head and neck.Cancer Res 2008;68:4479–85.
34. Wang LE, Xiong P, Strom SS, Goldberg LH, Lee JE, Ross MI, et al. Invitro sensitivity to ultraviolet B light and skin cancer risk: a case-controlanalysis. J Natl Cancer Inst 2005;97:1822–31.
35. Spitz MR, Hsu TC, Wu X, Fueger JJ, Amos CI, Roth JA. Mutagensensitivity as a biological marker of lung cancer risk in African Amer-icans. Cancer Epidemiol Biomarkers Prev 1995;4:99–103.
36. Wu X, Lin J, Etzel CJ, Dong Q, Gorlova OY, Zhang Q, et al. Interplaybetween mutagen sensitivity and epidemiological factors in modula-tinglung cancer risk. Int J Cancer 2007;120:2687–95.
37. Xiong P, Bondy ML, Li D, Shen H, Wang LE, Singletary SE, et al.Sensitivity to benzo(a)pyrene diol-epoxide associated with risk ofbreast cancer in young women and modulation by glutathione S-transferase polymorphisms: a case-control study. Cancer Res 2001;61:8465–9.
38. Erdei E, Lee SJ,Wei Q,Wang LE, Song YS, Bovbjerg D, et al. Reliabilityof mutagen sensitivity assay: an inter-laboratory comparison. Muta-genesis 2006;21:261–4.
39. Athas WF, Hedayati MA, Matanoski GM, Farmer ER, Grossman L.Development and field-test validation of an assay for DNA repair incirculating human lymphocytes. Cancer Res 1991;51:5786–93.
40. WangLE, Li C, StromSS,Goldberg LH,Brewster A,GuoZ, et al. Repaircapacity for UV light induced DNA damage associated with risk ofnonmelanoma skin cancer and tumor progression. Clin Cancer Res2007;13:6532–9.
41. Wang LE, Hu Z, Sturgis EM, Spitz MR, Strom SS, Amos CI, et al.Reduced DNA repair capacity for removing tobacco carcinogen-induced DNA adducts contributes to risk of head and neck cancerbut not tumor characteristics. Clin Cancer Res 2010;16:764–74.
42. ShenH,SpitzMR,QiaoY,GuoZ,WangLE,BoskenCH, et al. Smoking,DNA repair capacity and risk of nonsmall cell lung cancer. Int J Cancer2003;107:84–8.
43. Cheng L, Eicher SA, Guo Z, HongWK, Spitz MR,Wei Q. ReducedDNArepair capacity in head and neck cancer patients. Cancer EpidemiolBiomarkers Prev 1998;7:465–8.
44. WeiQY,ChengL,HongWK,SpitzMR.ReducedDNA repair capacity inlung cancer patients. Cancer Res 1996;56:4103–7.
45. Wei Q, Cheng L, Amos CI, Wang LE, Guo Z, Hong WK, et al. Repair oftobacco carcinogen-induced DNA adducts and lung cancer risk: amolecular epidemiologic study. J Natl Cancer Inst 2000;92:1764–72.
46. Gajecka M, Rydzanicz M, Jaskula-Sztul R, Wierzbicka M, Szyfter W,Szyfter K. ReducedDNA repair capacity in laryngeal cancer subjects. Acomparison of phenotypic and genotypic results. Adv Otorhinolaryn-gol 2005;62:25–37.
47. Schmezer P, Rajaee-Behbahani N, Risch A, Thiel S, Rittgen W, DringsP, et al. Rapid screening assay for mutagen sensitivity and DNA repaircapacity in human peripheral blood lymphocytes. Mutagenesis2001;16:25–30.
48. McArt DG, McKerr G, Saetzler K, Howard CV, Downes CS, WassonGR. Comet sensitivity in assessing DNA damage and repair in differentcell cycle stages. Mutagenesis 2010;25:299–303.
49. El-Zein RA, Monroy CM, Cortes A, Spitz MR, Greisinger A, Etzel CJ.Rapid method for determination of DNA repair capacity in humanperipheral blood lymphocytes amongst smokers. BMC Cancer2010;10:439.
50. Orlow I, Park BJ, Mujumdar U, Patel H, Siu-Lau P, Clas BA, et al. DNAdamage and repair capacity in patients with lung cancer: prediction ofmultiple primary tumors. J Clin Oncol 2008;26:3560–6.
51. Olive PL, Ban�ath JP. The comet assay: a method to measure DNAdamage in individual cells. Nat Protoc 2006;1:23–9.
52. Rosenberger A, Rossler U, Hornhardt S, Sauter W, Bickeboller H,Wichmann HE, et al. Validation of a fully automated COMET assay:1.75 million single cells measured over a 5 year period. DNA Repair(Amst) 2011;10:322–37.
53. Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molec-ular marker of DNA damage and repair. Leukemia 2010;24:679–86.
54. YuanJ,AdamskiR,Chen J. Focusonhistone variantH2AX: tobeor notto be. FEBS Lett 2010;584:3717–24.
55. Podhorecka M, Skladanowski A, Bozko P. H2AX phosphorylation: itsrole in DNA damage response and cancer therapy. J Nucleic Acids2010;2010:920161.
56. Saviozzi S, Ceppi P, Novello S, Ghio P, Lo Iacono M, Borasio P, et al.Non-small cell lung cancer exhibits transcript overexpression of genesassociatedwith homologous recombination andDNA replication path-ways. Cancer Res 2009;69:3390–6.
57. McLaren DB, Pickles T, Thomson T, Olive PL. Impact of nicotinamideon human tumour hypoxic fraction measured using the comet assay.Radiother Oncol 1997;45:175–82.
58. Zijno A, Porcedda P, Saini F, Allione A, Garofalo B, Marcon F, et al.Unsuitability of lymphoblastoid cell lines as surrogate of cryopreservedisolated lymphocytes for the analysis of DNA double-strand breakrepair activity. Mutat Res 2010;684:98–105.
59. Mazzei F, Guarrera S, Allione A, Simonelli V, Narciso L, Barone F, et al.8-Oxoguanine DNA-glycosylase repair activity and expression: acomparison between cryopreserved isolated lymphocytes and EBV-derived lymphoblastoid cell lines. Mutat Res 2011;718:62–7.
60. Herrera M, Dominguez G, Garcia JM, Pe~na C, Jimenez C, Silva J, et al.Differences in repair of DNA cross-links between lymphocytes andepithelial tumor cells from colon cancer patientsmeasured in vitro withthe comet assay. Clin Cancer Res 2009;15:5466–72.
61. Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, et al.Isolation of circulating tumor cells using a microvortex-generatingherringbone-chip. Proc Natl Acad Sci U S A 2010;107:18392–7.
62. Wu X, Gu J, Dong Q, Huang M, Do KA, Hong WK, et al. Joint effect ofmutagen sensitivity and insulin-like growth factors in predicting the riskof developing secondary primary tumors and tumor recurrence inpatients with head and neck cancer. Clin Cancer Res 2006;12:7194–201.
63. ChaoDL,MaleyCC,WuX, FarrowDC,GalipeauPC, SanchezCA, et al.Mutagen sensitivity and neoplastic progression in patients with Bar-rett's esophagus: a prospective analysis. Cancer Epidemiol Biomar-kers Prev 2006;15:1935–40.
64. Sigurdson AJ, Jones IM, Wei Q, Wu X, Spitz MR, Stram DA, et al.Prospective analysis of DNA damage and repair markers of lungcancer risk from the Prostate, Lung, Colorectal and Ovarian (PLCO)Cancer Screening Trial. Carcinogenesis 2011;32:69–73.
65. Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNArepair pathways as targets for cancer therapy. Nat Rev Cancer 2008;8:193–204.
66. Wo�zniak K, Bøasiak J. Recognition and repair of DNA-cisplatinadducts. Acta Biochim Pol 2002;49:583–96.
67. Olaussen KA, Mountzios G, Soria JC. ERCC1 as a risk stratifier inplatinum-based chemotherapy for nonsmall-cell lung cancer. CurrOpin Pulm Med 2007;13:284–9.
68. Bhagwat NR, Roginskaya VY, Acquafondata MB, Dhir R, Wood RD,Niedernhofer LJ. Immunodetection of DNA repair endonucleaseERCC1-XPF in human tissue. Cancer Res 2009;69:6831–8.
69. Niedernhofer LJ, Bhagwat N, Wood RD. ERCC1 and non-small-celllung cancer. N Engl J Med 2007;356:2538–40, author reply 2540–1.
70. Wakasugi M, Sancar A. Assembly, subunit composition, and footprintof human DNA repair excision nuclease. Proc Natl Acad Sci U S A1998;95:6669–74.
71. McHugh PJ, Spanswick VJ, Hartley JA. Repair of DNA interstrandcrosslinks: molecular mechanisms and clinical relevance. LancetOncol 2001;2:483–90.
72. Arora S, Kothandapani A, Tillison K, Kalman-Maltese V, Patrick SM.Downregulation of XPF-ERCC1 enhances cisplatin efficacy in cancercells. DNA Repair (Amst) 2010;9:745–53.
www.aacrjournals.org Clin Cancer Res; 17(22) November 15, 2011 6983
Assessing DNA Repair in Cancer Development and Response to Therapy
Research. on May 18, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 9, 2011; DOI: 10.1158/1078-0432.CCR-11-0761

73. Trivedi RN,AlmeidaKH, Fornsaglio JL, SchamusS,SobolRW.The roleof base excision repair in the sensitivity and resistance to temozolo-mide-mediated cell death. Cancer Res 2005;65:6394–400.
74. Pollack IF, Hamilton RL, Sobol RW, Burnham J, Yates AJ, Holmes EJ,et al. O6-methylguanine-DNA methyltransferase expression stronglycorrelates with outcome in childhood malignant gliomas: results fromthe CCG-945 Cohort. J Clin Oncol 2006;24:3431–7.
75. Dopeso H, Mateo-Lozano S, Elez E, Landolfi S, Ramos Pascual FJ,Hern�andez-Losa J, et al. Aprataxin tumor levels predict response ofcolorectal cancer patients to irinotecan-based treatment. Clin CancerRes 2010;16:2375–82.
76. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M,et al. MGMT gene silencing and benefit from temozolomide in glio-blastoma. N Engl J Med 2005;352:997–1003.
77. Graeser M, McCarthy A, Lord CJ, Savage K, Hills M, Salter J, et al. Amarker of homologous recombination predicts pathologic completeresponse to neoadjuvant chemotherapy in primary breast cancer. ClinCancer Res 2010;16:6159–68.
78. Shuck SC, Turchi JJ. Targeted inhibition of Replication Protein Areveals cytotoxic activity, synergy with chemotherapeutic DNA-dam-aging agents, and insight into cellular function. Cancer Res 2010;70:3189–98.
79. Neher TM, Shuck SC, Liu JY, Zhang JT, Turchi JJ. Identification ofnovel small molecule inhibitors of the XPA protein using in silico basedscreening. ACS Chem Biol 2010;5:953–65.
80. Anciano Granadillo VJ, Earley JN, Shuck SC, Georgiadis MM, FitchRW, Turchi JJ. Targeting the OB-folds of replication protein A withsmall molecules. J Nucleic Acids 2010;2010:304035.
81. Andrews BJ, Turchi JJ. Development of a high-throughput screen forinhibitors of replication protein A and its role in nucleotide excisionrepair. Mol Cancer Ther 2004;3:385–91.
82. Walton MI, Eve PD, Hayes A, Valenti M, De Haven Brandon A, BoxG, et al. The preclinical pharmacology and therapeutic activity of thenovel CHK1 inhibitor SAR-020106. Mol Cancer Ther 2010;9:89–100.
83. Willmore E, Elliott SL,Mainou-Fowler T, SummerfieldGP, JacksonGH,O'Neill F, et al. DNA-dependent protein kinase is a therapeutic targetand an indicator of poor prognosis in B-cell chronic lymphocyticleukemia. Clin Cancer Res 2008;14:3984–92.
84. Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC,et al.Mechanismof radiosensitizationby theChk1/2 inhibitor AZD7762involves abrogation of theG2 checkpoint and inhibition of homologousrecombinational DNA repair. Cancer Res 2010;70:4972–81.
85. KrishnakumarR,KrausWL.PARP-1 regulates chromatin structure andtranscription through a KDM5B-dependent pathway. Mol Cell 2010;39:736–49.
86. Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecularactions, physiological outcomes, and clinical targets. Mol Cell 2010;39:8–24.
87. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB,et al. Targeting the DNA repair defect in BRCA mutant cells as atherapeutic strategy. Nature 2005;434:917–21.
88. Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribosepolymerase inhibitors for the treatment of cancers deficient in DNAdouble-strand break repair. J Clin Oncol 2008;26:3785–90.
89. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al.Inhibition of poly(ADP-ribose) polymerase in tumors from BRCAmuta-tion carriers. N Engl J Med 2009;361:123–34.
90. O'Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha C,et al. Iniparib plus chemotherapy in metastatic triple-negative breastcancer. N Engl J Med 2011;364:205–14.
91. O'Shaughnessy J, Schwartzberg LS, Danso MA, Rugo HS, Miller K,Yardley DA, et al. A randomized phase III study of iniparib (BSI-201) incombination with gemcitabine/carboplatin (G/C) in metastatic triple-negative breast cancer (TNBC). J Clin Oncol 2011;29 (suppl 15): abstr1007.
92. He JX, Yang CH, Miao ZH. Poly(ADP-ribose) polymerase inhibitors aspromising cancer therapeutics. Acta Pharmacol Sin 2010;31:1172–80.
93. Kaelin WG Jr. The concept of synthetic lethality in the context ofanticancer therapy. Nat Rev Cancer 2005;5:689–98.
Clin Cancer Res; 17(22) November 15, 2011 Clinical Cancer Research6984
Jalal et al.
Research. on May 18, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 9, 2011; DOI: 10.1158/1078-0432.CCR-11-0761

2011;17:6973-6984. Published OnlineFirst September 9, 2011.Clin Cancer Res Shadia Jalal, Jennifer N. Earley and John J. Turchi Therapeutic TargetDNA Repair: From Genome Maintenance to Biomarker and
Updated version
10.1158/1078-0432.CCR-11-0761doi:
Access the most recent version of this article at:
Cited articles
http://clincancerres.aacrjournals.org/content/17/22/6973.full#ref-list-1
This article cites 92 articles, 35 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/17/22/6973.full#related-urls
This article has been cited by 6 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://clincancerres.aacrjournals.org/content/17/22/6973To request permission to re-use all or part of this article, use this link
Research. on May 18, 2019. © 2011 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
Published OnlineFirst September 9, 2011; DOI: 10.1158/1078-0432.CCR-11-0761


















