
Dipartimento di Medicina Sperimentale e Scienze ... s - neural... · *Dipartimento di Medicina...
13
*Dipartimento di Medicina Sperimentale e Scienze Biochimiche, Sezione di Biochimica e Biologia Molecolare, Universita ` di Perugia, Perugia, Italy San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET), Milan, Italy àStem Cell Research Institute, San Raffaele Scientific Institute, Milano, Italy §Department of Medical Chemistry, Biochemistry and Biotechnology, University of Milan, Milan, Italy ¶Kekule-Institut fu ¨r Organische Chemie und Biochemie der Universita ¨t Bonn, Gerhard-Domagk-Strasse, Bonn, Germany **Department of Biotechnology and Biosciences, University of Milano Bicocca, Piazza della Scienza, Milano, Italy Tay-Sachs (TS) and Sandhoff disease (SD) (GM2 ganglios- idosis) are lysosomal storage disorders with severe CNS involvement. Inherited defects in the a-subunit gene (HEXA, chromosome 15) or in the b-subunit gene (HEXB, chromo- some 5) lead to the absence of hexosaminidase (Hex, E.C.3.2.1.52) isoenzymes: HexA (ab), HexS (aa), and HexB (ab) respectively (Sandhoff 2001). This results in accumu- lation of GM2 ganglioside and related lipids within the CNS (Gravel et al. 1991; Martino et al. 2002a,b; Martino et al. 2005a), culminating with death in the second or third year of life. No therapy is currently available and treatment is restricted to supportive care. An effective therapy for GM2 gangliosidosis requires first and foremost widespread distribution of the functional therapeutic protein to the brain and repair of the damaged tissue. Different therapeutic approaches based on pharmaco- logical treatment, gene, and/or cell therapy have been proposed (Jeyakumar et al. 2005; Platt et al. 2005; Lee et al. 2007). The injection of the herpes simplex viral-1 Received December 3, 2008; revised manuscript received January 9, 2009; accepted January 14, 2009. Address correspondence and reprint requests to Aldo Orlacchio or Angela Gritti, Dipartimento di Medicina Sperimentale e Scienze Biochimiche, Sezione di Biochimica e Biologia Molecolare, Universita ` di Perugia, Via del Giochetto, Perugia 06100, Italy; San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET), Via Olgettina 02100 Milan. E-mail: [email protected]; [email protected] Abbreviations used: ARSA, Arylsulfatase A; DEAE, Diethylaminoethyl Cellulose; Hex, hexosaminidase; Hex-I, Hex intermediate; MS, mass spectrometry; MUG, 4-methylumbelliferyl-N-acetyl-b-D-glucosaminide; MUGS, 4-methylumbelliferone-6-sulfo-2-acetamido-2-deoxy-b-D-gluco- pyranoside; NPC, neural precursor cell; P10, postnatal day 10; SD, Sand- hoff disease; SVZ, subventricular zone; TS, Tay-Sachs; WT, wild-type. Abstract In this work we showed that genotype-related patterns of hexosaminidase activity, isoenzyme composition, gene expression and ganglioside metabolism observed during embryonic and postnatal brain development are recapitulated during the progressive stages of neural precursor cell (NPC) differentiation to mature glia and neurons in vitro. Further, by comparing NPCs and their differentiated progeny established from Tay-Sachs (TS) and Sandhoff (SD) animal models with the wild-type counterparts, we studied the events linking the accumulation of undegraded substrates to hexosaminidase activity. We showed that similarly to what observed in brain tissues in TS NPCs and progeny, the stored GM2 was partially converted by sialidase to GA2, which can be then degraded in the lysosomes to its components. The latter can be used in a salvage pathway for the formation of GM3. Interestingly, results obtained from ganglioside feeding assays and from measurement of lysosomal sialidase activity suggest that a similar pathway might work also in the SD model. Keywords: brain development, lysosomal enzymes, neural precursor cells, Sandhoff, Tay-Sachs. J. Neurochem. (2009) 109, 135–147. JOURNAL OF NEUROCHEMISTRY | 2009 | 109 | 135–147 doi: 10.1111/j.1471-4159.2009.05919.x ȑ 2009 The Authors Journal Compilation ȑ 2009 International Society for Neurochemistry, J. Neurochem. (2009) 109, 135–147 135
Transcript of Dipartimento di Medicina Sperimentale e Scienze ... s - neural... · *Dipartimento di Medicina...

*Dipartimento di Medicina Sperimentale e Scienze Biochimiche, Sezione di Biochimica e Biologia Molecolare,
Universita di Perugia, Perugia, Italy
�San Raffaele Telethon Institute for Gene Therapy (HSR-TIGET), Milan, Italy
�Stem Cell Research Institute, San Raffaele Scientific Institute, Milano, Italy
§Department of Medical Chemistry, Biochemistry and Biotechnology, University of Milan, Milan, Italy
¶Kekule-Institut fur Organische Chemie und Biochemie der Universitat Bonn, Gerhard-Domagk-Strasse, Bonn, Germany
**Department of Biotechnology and Biosciences, University of Milano Bicocca, Piazza della Scienza, Milano, Italy
Tay-Sachs (TS) and Sandhoff disease (SD) (GM2 ganglios-idosis) are lysosomal storage disorders with severe CNSinvolvement. Inherited defects in the a-subunit gene (HEXA,chromosome 15) or in the b-subunit gene (HEXB, chromo-some 5) lead to the absence of hexosaminidase (Hex,E.C.3.2.1.52) isoenzymes: HexA (ab), HexS (aa), and HexB(ab) respectively (Sandhoff 2001). This results in accumu-lation of GM2 ganglioside and related lipids within the CNS(Gravel et al. 1991; Martino et al. 2002a,b; Martino et al.2005a), culminating with death in the second or third year oflife. No therapy is currently available and treatment isrestricted to supportive care.
An effective therapy for GM2 gangliosidosis requires firstand foremost widespread distribution of the functionaltherapeutic protein to the brain and repair of the damagedtissue. Different therapeutic approaches based on pharmaco-
logical treatment, gene, and/or cell therapy have beenproposed (Jeyakumar et al. 2005; Platt et al. 2005; Leeet al. 2007). The injection of the herpes simplex viral-1
Received December 3, 2008; revised manuscript received January 9,2009; accepted January 14, 2009.Address correspondence and reprint requests to Aldo Orlacchio or
Angela Gritti, Dipartimento di Medicina Sperimentale e ScienzeBiochimiche, Sezione di Biochimica e Biologia Molecolare, Universita diPerugia, Via del Giochetto, Perugia 06100, Italy; San Raffaele TelethonInstitute for Gene Therapy (HSR-TIGET), Via Olgettina 02100 Milan.E-mail: [email protected]; [email protected] used: ARSA,ArylsulfataseA;DEAE,Diethylaminoethyl
Cellulose; Hex, hexosaminidase; Hex-I, Hex intermediate; MS, massspectrometry; MUG, 4-methylumbelliferyl-N-acetyl-b-D-glucosaminide;MUGS, 4-methylumbelliferone-6-sulfo-2-acetamido-2-deoxy-b-D-gluco-pyranoside; NPC, neural precursor cell; P10, postnatal day 10; SD, Sand-hoff disease; SVZ, subventricular zone; TS, Tay-Sachs;WT, wild-type.
Abstract
In this work we showed that genotype-related patterns of
hexosaminidase activity, isoenzyme composition, gene
expression and ganglioside metabolism observed during
embryonic and postnatal brain development are recapitulated
during the progressive stages of neural precursor cell (NPC)
differentiation to mature glia and neurons in vitro. Further, by
comparing NPCs and their differentiated progeny established
from Tay-Sachs (TS) and Sandhoff (SD) animal models with
the wild-type counterparts, we studied the events linking the
accumulation of undegraded substrates to hexosaminidase
activity. We showed that similarly to what observed in brain
tissues in TS NPCs and progeny, the stored GM2 was partially
converted by sialidase to GA2, which can be then degraded in
the lysosomes to its components. The latter can be used in a
salvage pathway for the formation of GM3. Interestingly,
results obtained from ganglioside feeding assays and from
measurement of lysosomal sialidase activity suggest that a
similar pathway might work also in the SD model.
Keywords: brain development, lysosomal enzymes, neural
precursor cells, Sandhoff, Tay-Sachs.
J. Neurochem. (2009) 109, 135–147.
JOURNAL OF NEUROCHEMISTRY | 2009 | 109 | 135–147 doi: 10.1111/j.1471-4159.2009.05919.x
� 2009 The AuthorsJournal Compilation � 2009 International Society for Neurochemistry, J. Neurochem. (2009) 109, 135–147 135

vector encoding for HEXA gene in the brain internal capsuleof adult TS murine models led to reconstitution of HexAactivity and to almost complete clearance of storage in thebrain and spinal cord of treated mice (Martino et al. 2005a).Similarly, the efficacy of adeno-associated viral vectors inimproving pathology and restoring behavior in the SDmurine model has been documented (Cachon-Gonzalez et al.2006). However, the potential of these and other approachesto prevent/arrest the disease progression and to contrast theextensive neuronal loss remains to be determined, suggestingthat an effective treatment might not rely exclusively onclearance of lipid storage. Moreover, the early onset ofsymptoms and the rapid disease progression impose a narrowwindow for intervention which has to be clearly defined inorder to achieve the best therapeutic efficacy.
To address these issues we investigated the pathophysiol-ogy of GM2 gangliosidosis during the disease progression inSD (hexb)/)) and TS (hexa)/)) animal models which showa severe and mild disease phenotype, respectively. Weperformed a comparative study in which lipid storage, Hexactivity, isoenzyme composition, and gene expression wereinvestigated in brain tissues isolated from mutant mice andfrom age-matched wild-type (WT) controls and from theembryonic to the adult age. A similar analysis was performedon mutant and WT subventricular zone (SVZ)-derived neuralprecursor cells (NPCs) (Gritti et al. 1999, 2002) at differentstages of differentiation/maturation in vitro, in view of theirpotential use as a reliable experimental model to study themechanisms of the disease and as potential donors in cell-based therapeutic approaches. Further, by comparing NPCcultures established from SD and TS mice with the WTcounterparts, we investigated the events linking the accumu-lation of undegraded substrates to Hex deficiency.
Material and methods
Animal modelSD (hexb)/); kindly provided by Dr. Platt F. M.) and TS mice
(hexa)/)) were generated as previously described (Yamanaka et al.1994; Sango et al. 1995) and were bred in the HSR SPF animal
house. C57/bL6 (Charles-River, Calco, LC, Italy) and hexb+/+
littermates were used as WT controls for hexa)/) and hexb)/)mice, respectively. Human fibroblasts were obtained from the ‘Cell
line and DNA bank from patients affected by Genetic diseases –
Telethon Genetic Biobank’.
ChemicalsSee Supporting Information.
Brain tissue preparationBrains were isolated from SD, TS, and WT mice and the two
hemispheres were separated and used for biochemical and molecular
assays. Tissues were immediately frozen in liquid nitrogen or treated
to obtain brain extracts for biochemical analysis or for RNA
extraction. We collected tissues at different ages: E13 and E18
(embryonic stage); P0, P3, P7, P10, P13, P17, P21 (postnatal stage);
P30, P120 (SD), and P150 (WT, TS) (adult).
Isolation and culture propagation of NPCsNeonatal (P3) and adult mice (P90) were anaesthetised in crushed
ice and by i.p. injection of Avertin (2-2-2 Tribromoethanol;
0.25 mg/g body weight), respectively, before being decapitated.
Brains were removed and tissue containing the subventricular zone
(SVZ) of the forebrain lateral ventricles was dissected. We
established independent cell lines from the SVZ of SD, TS, and
WT mice (n = 5 for each genotype). NPCs cultures were established
and expanded as previously described (Gritti et al. 1999, 2002). Forall the experiments we used serially passaged NPCs (passage 5–15).
Cultures of NPCs at different stages of maturationCultures were prepared as previously described (Cavazzin et al.2006). Briefly, serially passaged 5-day-old neurospheres were
dissociated and grown for 24 h in serum-free medium (Dulbecco’s
modified Eagle’s medium/F12, 1 : 1, vol : vol containing insulin,
apo-transferrin, putrescine, and progesterone) containing Fibroblast
growth factor 2 (FGF2) and epidermal growth factor (EGF) in order
to obtain a population enriched in proliferating undifferentiated cells
(precursor cells). The population of committed progenitors con-
sisted of cells grown for 3 days in the presence of an adhesion
substrate and in serum-free medium containing Fibroblast growth
factor 2 (FGF2). These cultures which contained glial and neuronal
progenitors at different stages of commitment were exposed to
serum-free medium containing 2% fetal calf serum (FCS) and grown
for additional 4–7 days in order to achieve terminal differentiation
of neural progenitors into neurons, astrocytes, and oligodendrocytes
(differentiated cells). The extent of neuronal and glial differentia-
tion/maturation in the different cell populations was assessed by
using antibodies against lineage- and stage-specific markers, as
described below.
Immunostaining of NPC culturesDouble-labeling immunofluorescence was performed as previously
described (Gritti et al. 1999). Details can be found in Supporting
Information.
Brain and cell extractsTissues were homogenized with an Elveheim type homogenizer in
buffer A (10 mM sodium phosphate buffer, pH 6.0, containing 0.1%
(v/v) Nonidet NP40 detergent (Sigma-Aldrich, St Louis, MO, USA).
Brain lysates were subjected to three round of sonication and
centrifuged at 13 400 g for 10 min in a Eppendorf microfuge.
Supernatants were used as tissue extracts. Cells were harvested,
washed in phosphate-buffered saline, and resuspended in buffer A.
Cell extracts were prepared by employing three rounds of
sonication. All procedures were carried out at 4�C. Proteins were
measured by the Bradford method using bovine serum albumin as
standard.
Enzyme activity assaysHexosaminidase activity was determined using 3 mM 4-methylum-
belliferyl-N-acetyl-b-D-glucosaminide (MUG) and 3 mM 4-methyl-
umbelliferone-6-sulfo-2-acetamido-2-deoxy-b-D-glucopyranoside
Journal Compilation � 2009 International Society for Neurochemistry, J. Neurochem. (2009) 109, 135–147� 2009 The Authors
136 | S. Martino et al.

(MUGS) substrates dissolved in 0.1 M citrate/0.2 M disodium
phosphate buffer at pH 4.5. b-Galactosidase and b-glucuronidaseactivities were determined using 1.5 mM 4-methylumbelliferyl-b-D-galactoside and 3 mM 4-methylumbelliferyl-b-D-glucuronide sub-
strate, respectively. Both substrates were dissolved in 0.1 M citric
acid/0.2 M disodium phosphate buffer, pH 4.5. a-Mannosidase
activity was determined using 3 mM 4-methylumbelliferyl-a-D-mannopyranoside substrate dissolved in 50 mM sodium acetate/
acetic acid buffer, pH 4.0. Arylsulfatase A (ARSA) and arylsulfatase
B (ARSB) were determined using 5 mM 4-methylumbelliferylsul-
phate, dissolved in 50 mM sodium acetate/acetic acid buffer, pH
5.5, in the presence and absence of 0.75 mM AgNO3 (a specific
ARSA inhibitor). ARSA activity was calculated by subtracting the
value obtained in the presence of AgNO3 in the substrate solution
(ARSB activity) from that measured in the absence of the inhibitor
(ARSA + ARSB activity) (Martino et al. 2005b). Sialidase activity
was measured using the fluorogenic substrate, 0.6 mM 4-methyl-
umbelliferyl-N-acetyl-a-D-neuraminic acid, dissolved in 50 mM
sodium acetate/acetic acid buffer, pH 4.7 (Kato et al. 2001).
Detailed methods can be found in Supporting Information.
b-Hexosaminidase isoenzyme analysisTissue and cell extracts were analyzed by ion-exchange chroma-
tography on Diethylaminoethyl Cellulose (DEAE). Chromatography
was performed using 0.5 mL column equilibrated with 10 mM
sodium phosphate buffer, pH 6.0 (buffer B). The flow rate was
0.5 mL/min. Enzyme activity retained by the column was eluted by
a linear gradient of NaCl (0.0–0.5 M in 20 mL of buffer B starting
from fraction no. 20). Finally, the column was eluted with 1.0 M
NaCl in buffer B. Fractions (0.5 mL) were collected and assayed for
the Hex activity. Detailed methods for b-Hex isoenzyme character-
ization can be found in Supporting Information.
Real time RT-PCRTotal RNA was isolated using an Rneasy Mini Kit (Qiagen, Hilden,
Germany). Reverse transcription was carried out using 1 lg of total
RNA in the presence of 200 U of Super Script IITM Reverse
Transcriptase (Invitrogen, Carlsbad, CA, USA) and 10 ng/mL of
random hexamers as reverse primers (Invitrogen, Carlsbad, CA,
USA). Details on RT-PCR assay can be found in Supporting
Information.
Other analytical methodsDetails of methods for HPLC and electrospray ionization-mass
spectrometry (MS), lipid analysis, and feeding experiments can be
found in Supporting Information.
Results
Hexosaminidase activity increases during braindevelopmentWe analyzed Hex activity in SD, TS, and WT mice brainfrom embryonic day (E) 13 to the adult age (3 or 5 months ofage). Enzyme activity was measured using two artificialsubstrates: MUG, hydrolyzed by HexA (ab), HexB (bb), andHexS (aa), and MUGS hydrolyzed by HexA and HexS.
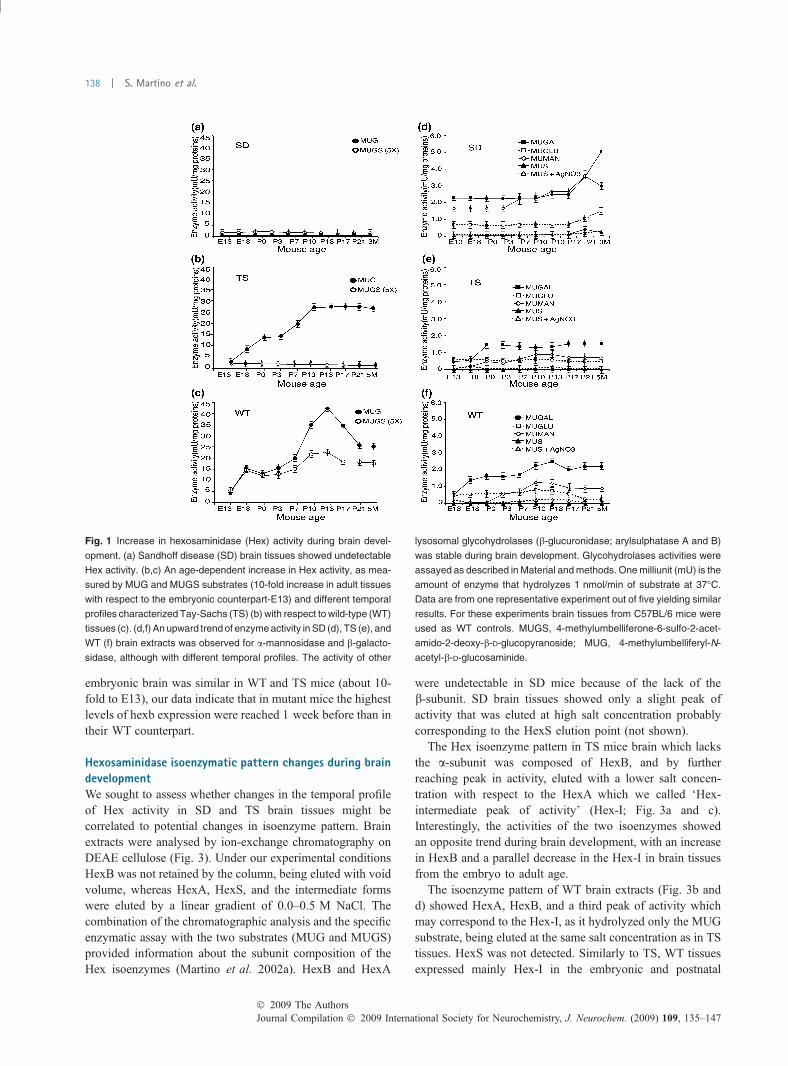
Sandhoff diseased brain extracts showed only a residualenzyme activity toward both substrates (Fig. 1a). Enzymeactivity measured in TS brain extracts with MUG substrateshowed a 9.5-fold increase from E13 to postnatal day (P) 10,with an intermediate plateau between P0 and P3 (Fig. 1b).From P10, enzyme activity remained stable up to adulthood.As expected, we did not detect MUGS activity. The timecourse of Hex activity up-regulation observed in TS brainextracts was strikingly different from what observed in theWT counterparts (Fig. 1c). In the latter, a threefold increasewas observed with MUG substrate at E18 with respect toE13. After a plateau between E18 and P3, enzyme activitypeaked until P13 (9.5-fold increase with respect to E13), thendecreased until P21, reaching levels comparable with thoseobserved in P21 TS tissues. Activity measured using MUGSshowed similar results, with peaks at E18 and P10–P13 (2-and 4-fold increase with respect to E13, respectively)(Fig. 1c).
As an internal control, we measured enzyme activity of thelysosomal glycohydrolases b-galactosidase, b-glucuronidase,a-mannosidase, and arylsulfatase A and B (Fig. 1d–f).Activity of b-glucuronidase and arylsulfatase A and B didnot change at increasing ages in SD, TS, and WT brainextracts (Fig. 1d–f). In TS and WT mice, a-mannosidase andb-galactosidase activity increased around E18–P0 up to P7–P13, then reached levels that after a slight decrease remainedstable throughout adulthood. These variations were lessevident in TS brain extracts with respect to WT (Fig. 1eand f), while in SD tissues the increase was delayed, startingaround P13–P17 (Fig. 1d).
hexa and hexb genes are differentially regulated duringbrain developmentIn order to assess if Hex activity might be correlated tochanges in gene expression, we evaluated the expression ofhex genes in SD, TS, and WT brain tissues by real-time RTPCR (Fig. 2). Hexa expression in SD tissues moderatelyincreased from the postnatal to adult age (0.4 and 0.8 fold atP13 and P21) with respect to E13 (our reference value)(Fig. 2a). As hexa expression in WT tissues was stable fromthe embryonic to the adult age (Fig. 2c), we suggest that thehexb null mutation might alter the expression of hexa. In TSbrains we observed three substantial increments at increasingdevelopmental ages: the first one between E13 and E18 (3.5-fold increase), the second between P0 and P3 (1.5-foldincrease), and the third between P6 and P13 (1.5-foldincrease). Hexb levels were then relatively stable up to adultage (Fig. 2b). In WT brains the hexb up-regulation showed adifferent trend with respect to age-matched TS counterparts(Fig. 2c). The first consistent increase was observed betweenP0 and P3 (about 2.5-fold increase) and the second betweenP13 and P21 (1.5-fold increase). Thus, although the extent ofhexb up-regulation found in the adult with respect to the
� 2009 The AuthorsJournal Compilation � 2009 International Society for Neurochemistry, J. Neurochem. (2009) 109, 135–147
Hexosaminidase activity in brain development and neural precursor cell differentiation | 137
embryonic brain was similar in WT and TS mice (about 10-fold to E13), our data indicate that in mutant mice the highestlevels of hexb expression were reached 1 week before than intheir WT counterpart.
Hexosaminidase isoenzymatic pattern changes during braindevelopmentWe sought to assess whether changes in the temporal profileof Hex activity in SD and TS brain tissues might becorrelated to potential changes in isoenzyme pattern. Brainextracts were analysed by ion-exchange chromatography onDEAE cellulose (Fig. 3). Under our experimental conditionsHexB was not retained by the column, being eluted with voidvolume, whereas HexA, HexS, and the intermediate formswere eluted by a linear gradient of 0.0–0.5 M NaCl. Thecombination of the chromatographic analysis and the specificenzymatic assay with the two substrates (MUG and MUGS)provided information about the subunit composition of theHex isoenzymes (Martino et al. 2002a). HexB and HexA
were undetectable in SD mice because of the lack of theb-subunit. SD brain tissues showed only a slight peak ofactivity that was eluted at high salt concentration probablycorresponding to the HexS elution point (not shown).
The Hex isoenzyme pattern in TS mice brain which lacksthe a-subunit was composed of HexB, and by furtherreaching peak in activity, eluted with a lower salt concen-tration with respect to the HexA which we called ‘Hex-intermediate peak of activity’ (Hex-I; Fig. 3a and c).Interestingly, the activities of the two isoenzymes showedan opposite trend during brain development, with an increasein HexB and a parallel decrease in the Hex-I in brain tissuesfrom the embryo to adult age.
The isoenzyme pattern of WT brain extracts (Fig. 3b andd) showed HexA, HexB, and a third peak of activity whichmay correspond to the Hex-I, as it hydrolyzed only the MUGsubstrate, being eluted at the same salt concentration as in TStissues. HexS was not detected. Similarly to TS, WT tissuesexpressed mainly Hex-I in the embryonic and postnatal
Fig. 1 Increase in hexosaminidase (Hex) activity during brain devel-
opment. (a) Sandhoff disease (SD) brain tissues showed undetectable
Hex activity. (b,c) An age-dependent increase in Hex activity, as mea-
sured by MUG and MUGS substrates (10-fold increase in adult tissues
with respect to the embryonic counterpart-E13) and different temporal
profiles characterized Tay-Sachs (TS) (b) with respect to wild-type (WT)
tissues (c). (d,f) An upward trend of enzyme activity in SD (d), TS (e), and
WT (f) brain extracts was observed for a-mannosidase and b-galacto-
sidase, although with different temporal profiles. The activity of other
lysosomal glycohydrolases (b-glucuronidase; arylsulphatase A and B)
was stable during brain development. Glycohydrolases activities were
assayed as described in Material and methods. One milliunit (mU) is the
amount of enzyme that hydrolyzes 1 nmol/min of substrate at 37�C.
Data are from one representative experiment out of five yielding similar
results. For these experiments brain tissues from C57BL/6 mice were
used as WT controls. MUGS, 4-methylumbelliferone-6-sulfo-2-acet-
amido-2-deoxy-b-D-glucopyranoside; MUG, 4-methylumbelliferyl-N-
acetyl-b-D-glucosaminide.
Journal Compilation � 2009 International Society for Neurochemistry, J. Neurochem. (2009) 109, 135–147� 2009 The Authors
138 | S. Martino et al.

specimens (maximum around P13–P21) (Fig. 3b and d). Onthe contrary, the relative amount of HexB was low in theembryonic and postnatal period and increased in the adult
age. HexA levels were stable at all ages, except for a markeddecrease between P13 and P21 (second postnatal period) thatwas paralleled by an increase in Hex-I, being HexB stable(Fig. 3b and d). Characterization of Hex isoenzymes from TSand WT brain tissues for thermal stability, N-acetyl-gluco-samine inhibition, optimum pH, and subunit compositionafter DEAE chromatography separation indicated that theyhave similar biochemical properties and that the Hex-I,although it is composed of b subunit, actually expressesintermediate biochemical properties between HexA andHexB isoenzymes (see Methods and Results in SupportingInformation; Fig. S1).
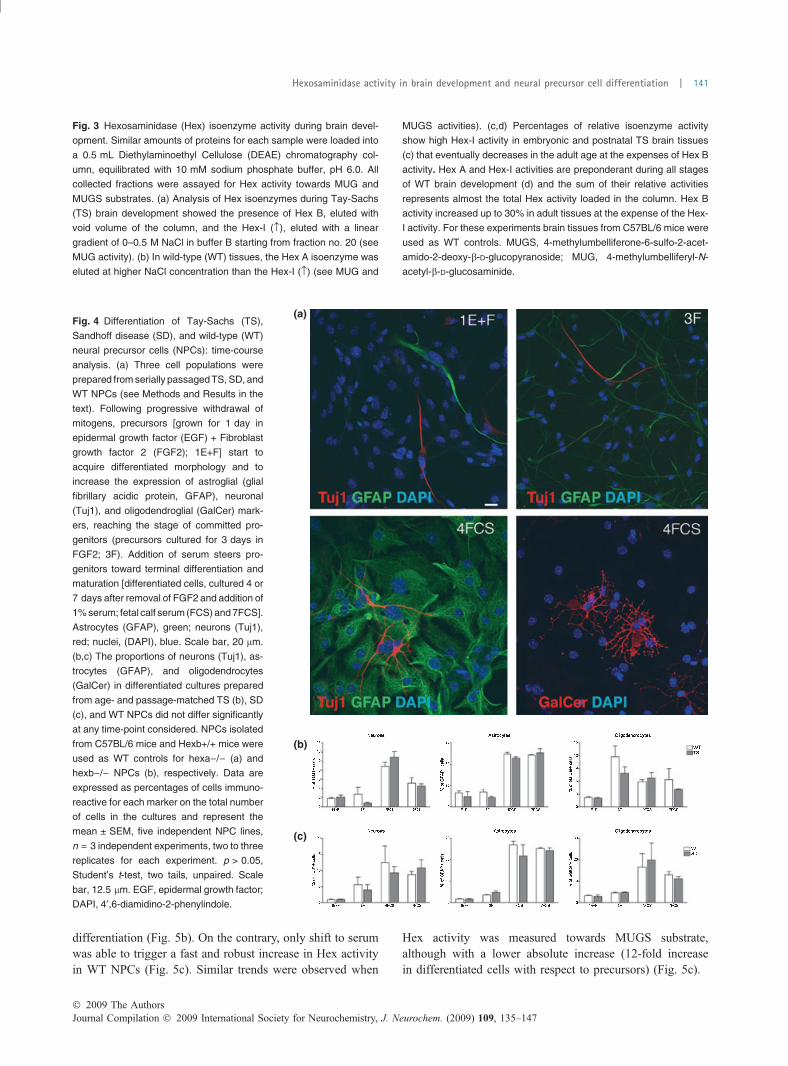
Up-regulation of Hex activity and hex gene expressionduring NPC differentiationWe applied the neurosphere assay (Galli et al. 2002; Grittiet al. 2002; Reynolds and Rietze 2005) to isolate, expand,and differentiate NPCs from the SVZ of SD, TS, and WTmice. All NPC lines displayed stable proliferation and self-renewal over time in culture (not shown). In order to evaluateHex activity in NPCs and to test whether this activity mightbe regulated during the process of NPC differentiation wegenerated from neurospheres, three types of cultures: (i) acell population enriched in proliferating and undifferentiatedcells (precursor cells); (ii) a cell population of neuronal andglial progenitors that started expressing markers of differen-tiation [i.e., b-tubulin III, GalCer, and glial fibrillary acidicprotein (GFAP) for neurons, oligodendrocytes, and astro-cytes, respectively] (committed progenitors), and (iii) themature progeny of progenitor cells which express high levelsof either neuronal or glial markers (differentiated cells). Theextent of cell differentiation/maturation in the different cellpopulations was assessed and quantified at different time-points. The proportions of cells expressing the proliferationmarker Ki67 and the neuroepithelial marker, nestin,decreased at increasing stages of cell differentiation, being85 and 90%, 45 and 63%, and 1.33 and 0.95% in precursors,committed progenitors, and differentiated cells, respectively.On the contrary, increasing numbers of cells expressingneuronal, astroglial, and oligodendroglial markers wereprogressively observed as cell differentiation proceeded fromprecursors (Figs. 4a, 1e and f) to progenitors (Figs. 4a and3f) to differentiated cells [Fig. 4a; fetal calf serum (FCS)].We did not observe significant differences in the ability of TSand SD NPCs to give rise to neurons, astrocytes, andoligodendrocytes with respect to the WT counterpart (Fig. 4band c), irrespectively of the time in culture (up to 15subculturing passages, 3 months in culture).
On these NPC populations we studied Hex activity andhex gene expression using the same assays described forbrain tissue extracts. Hex activity increased in SD-, TS-, andWT-derived NPC cultures at progressive stages of differen-tiation, although with different temporal profiles (Fig. 5a–c).In SD-derived cells the residual Hex activity increased after
(a)
(b)
(c)
Fig. 2 Hexosaminidase gene expression was up-regulated during
brain development. We studied the expression of hex genes in
Sandhoff disease (SD), Tay-Sachs (TS), and wild-type (WT) mice
brain at different developmental ages by real-time RT-PCR. (a) Hexa
gene was up-regulated during SD brain development. (b,c) Age-
dependent up-regulation of Hexb gene was present in TS (b) and in
WT brain tissues (c), although with different temporal profiles. The
highest levels of hexb expression were reached in WT mice only at
P21, while in mutant animals the maximum up-regulation was ob-
served starting from P13. On the contrary, hexa gene expression was
stable during brain development. The fold increase is calculated using
the DDCt method. DCt corresponds to the difference between Ct of hex
gene and the 18S RNA gene taken as control gene for normalization.
DDCt corresponds to the difference between DCt of hex gene at dif-
ferent period of development and DCt of hex gene at the E13. The
expression fold level is calculated as 2)DDCt. For these experiments
brain tissues from C57BL/6 mice are used as WT controls.
� 2009 The AuthorsJournal Compilation � 2009 International Society for Neurochemistry, J. Neurochem. (2009) 109, 135–147
Hexosaminidase activity in brain development and neural precursor cell differentiation | 139

adding serum to the culture media (Fig. 5a). Remarkably, inboth TS and WT differentiated cells, the activity of Hextowards MUG activity was 20-fold higher with respect to theprecursor counterpart. Also, the increase in Hex activity as afunction of the differentiation stage resembled that observed
in brain tissues during development. In fact, a robust increasein Hex activity was present in TS-derived early committedprogenitors as early as 1 day after removal of epidermalgrowth factor. Hex activity increased following shiftto serum, a process that triggers terminal NPC cell
Journal Compilation � 2009 International Society for Neurochemistry, J. Neurochem. (2009) 109, 135–147� 2009 The Authors
140 | S. Martino et al.
differentiation (Fig. 5b). On the contrary, only shift to serumwas able to trigger a fast and robust increase in Hex activityin WT NPCs (Fig. 5c). Similar trends were observed when
Hex activity was measured towards MUGS substrate,although with a lower absolute increase (12-fold increasein differentiated cells with respect to precursors) (Fig. 5c).
Fig. 3 Hexosaminidase (Hex) isoenzyme activity during brain devel-
opment. Similar amounts of proteins for each sample were loaded into
a 0.5 mL Diethylaminoethyl Cellulose (DEAE) chromatography col-
umn, equilibrated with 10 mM sodium phosphate buffer, pH 6.0. All
collected fractions were assayed for Hex activity towards MUG and
MUGS substrates. (a) Analysis of Hex isoenzymes during Tay-Sachs
(TS) brain development showed the presence of Hex B, eluted with
void volume of the column, and the Hex-I (›), eluted with a linear
gradient of 0–0.5 M NaCl in buffer B starting from fraction no. 20 (see
MUG activity). (b) In wild-type (WT) tissues, the Hex A isoenzyme was
eluted at higher NaCl concentration than the Hex-I (›) (see MUG and
MUGS activities). (c,d) Percentages of relative isoenzyme activity
show high Hex-I activity in embryonic and postnatal TS brain tissues
(c) that eventually decreases in the adult age at the expenses of Hex B
activity. Hex A and Hex-I activities are preponderant during all stages
of WT brain development (d) and the sum of their relative activities
represents almost the total Hex activity loaded in the column. Hex B
activity increased up to 30% in adult tissues at the expense of the Hex-
I activity. For these experiments brain tissues from C57BL/6 mice were
used as WT controls. MUGS, 4-methylumbelliferone-6-sulfo-2-acet-
amido-2-deoxy-b-D-glucopyranoside; MUG, 4-methylumbelliferyl-N-
acetyl-b-D-glucosaminide.
(a)
(b)
(c)
Fig. 4 Differentiation of Tay-Sachs (TS),
Sandhoff disease (SD), and wild-type (WT)
neural precursor cells (NPCs): time-course
analysis. (a) Three cell populations were
prepared from serially passaged TS, SD, and
WT NPCs (see Methods and Results in the
text). Following progressive withdrawal of
mitogens, precursors [grown for 1 day in
epidermal growth factor (EGF) + Fibroblast
growth factor 2 (FGF2); 1E+F] start to
acquire differentiated morphology and to
increase the expression of astroglial (glial
fibrillary acidic protein, GFAP), neuronal
(Tuj1), and oligodendroglial (GalCer) mark-
ers, reaching the stage of committed pro-
genitors (precursors cultured for 3 days in
FGF2; 3F). Addition of serum steers pro-
genitors toward terminal differentiation and
maturation [differentiated cells, cultured 4 or
7 days after removal of FGF2 and addition of
1% serum; fetal calf serum (FCS) and 7FCS].
Astrocytes (GFAP), green; neurons (Tuj1),
red; nuclei, (DAPI), blue. Scale bar, 20 lm.
(b,c) The proportions of neurons (Tuj1), as-
trocytes (GFAP), and oligodendrocytes
(GalCer) in differentiated cultures prepared
from age- and passage-matched TS (b), SD
(c), and WT NPCs did not differ significantly
at any time-point considered. NPCs isolated
from C57BL/6 mice and Hexb+/+ mice were
used as WT controls for hexa)/) (a) and
hexb)/) NPCs (b), respectively. Data are
expressed as percentages of cells immuno-
reactive for each marker on the total number
of cells in the cultures and represent the
mean ± SEM, five independent NPC lines,
n = 3 independent experiments, two to three
replicates for each experiment. p > 0.05,
Student’s t-test, two tails, unpaired. Scale
bar, 12.5 lm. EGF, epidermal growth factor;
DAPI, 4¢,6-diamidino-2-phenylindole.
� 2009 The AuthorsJournal Compilation � 2009 International Society for Neurochemistry, J. Neurochem. (2009) 109, 135–147
Hexosaminidase activity in brain development and neural precursor cell differentiation | 141

Gene expression analysis performed on TS and WT NPCsand in their progeny showed that hexb up-regulationoccurring during the transition from precursor to progenitors(2-fold increase) was maintained in differentiated cells(Fig. 5e and f). Hexa gene was up-regulated in SD (2.5-fold increase with respect to the precursors) but not in WT-differentiated cultures (Fig. 5d and f).
We also analysed the Hex isoenzymes pattern in precur-sors, progenitors, and differentiated cells (Fig. 5g and h). SD
cells showed only a small peak of activity corresponding toHexS (not shown). TS cells were characterized by absence ofHexA, while they maintain HexB and Hex-I isoenzymes(Fig. 5g). Interestingly, higher levels of Hex-I were ex-pressed in WT precursors and progenitors with respect todifferentiated cells. We did not detect Hex S (Fig. 5h). Allthe results described above are from adult (P90)-derivedNPC cultures. Overlapping results were obtained usingneonatal (P3)-derived NPC cultures (not shown).
(a)
(b)
(c) (f)
(e)
(d)
(g) (h)
Fig. 5 Hexosaminidase (Hex) activity
expression increase during neural precur-
sor cell (NPC) differentiation. Hex activity
was tested in Sandhoff disease (SD), Tay-
Sachs (TS), and wild-type (WT) NPC cul-
tures at different stages of differentiation
(precursors, progenitors, and differentiated
cells). Time-points of cell differentiation are
the same as those indicated in Fig. 4. The
time-course analysis shows residual Hex
activity in Sandhoff disease NPCs (a) and
a consistent (up to 20-fold) increase in Hex
activity in TS (b) and WT (c) NPC-derived
differentiated cells as compared with pre-
cursors. Increase in Hex activity was rap-
idly induced soon after epidermal growth
factor (EGF) removal in TS-derived NPCs
(b), while only serum addition triggered a
comparable increase in WT NPCs (c). (d)
hexa gene was up-regulated (2.5-fold)
during differentiation of SD NPCs. In both
TS (e) and WT (f) NPCs, the hexb gene
was up-regulated in progenitors and the
2-fold increase in gene expression level
remained stable in differentiated cells.
Expression of hexa gene did not change
following NPC differentiation. (g) Diethyl-
aminoethyl Cellulose (DEAE) chromato-
graphy of TS NPCs during differentiation
displayed the complete absence of HexA,
the presence of the HexB isoform, and
the Hex-I (›). (h) Hex-I activity (›) was
present in WT precursors and progeni-
tors, being barely detectable in differenti-
ated WT cells. We did not detect the
presence of Hex S in both TS- and
WT-differentiated cells. NPCs isolated
from C57BL/6 mice and hexb+/+ mice
were used as WT controls for hexa)/)and hexb)/) NPCs, respectively. MUGS,
4-methylumbelliferone-6-sulfo-2-acetamido-
2-deoxy-b-D-glucopyranoside; MUG, 4-methyl-
umbelliferyl-N-acetyl-b-D-glucosaminide.
Journal Compilation � 2009 International Society for Neurochemistry, J. Neurochem. (2009) 109, 135–147� 2009 The Authors
142 | S. Martino et al.

Lysosomal sialidase activity during SD, TS, and WT braindevelopment and NPC differentiationLysosomal sialidase (E.C.3.2.1.18) converts GM2 ganglio-side into the asialo-derivative GA2 ganglioside that is thenhydrolyzed by HexB. This pathway accounts for reducedGM2 storage in TS brain tissues. We evaluated the activity ofthis enzyme in brain tissues during development and in NPCsduring in vitro differentiation (Fig. 6). We showed thatsimilarly to what was described for HexA and HexB,sialidase activity in TS and SD brain tissues increased, earlypostnatally (from P7 to P10), then reached a plateau(Fig. 6a), while it was stable in WT tissues during the wholepostnatal ages until P21, and then reached the levelsobserved in mutants adult age (Fig. 6a). Notably, sialidaseactivity increased up to threefold during SD, TS, and WTNPC differentiation, with no significant genotype-relateddifferences (Fig. 6b).
Ganglioside pattern during NPC differentiationIn order to establish a potential correlation between Hexactivity and ganglioside storage we evaluated the gangliosidepattern in SD, TS, and WT NPCs and in their differentiated
progeny (Fig. 7a). The main gangliosides were separated byHPLC and characterized with negative electrospray ioniza-tion-mass spectrometry by means of identification of ionsobtained by MS1 and MS2 (see Methods in SupportingInformation for details).
In WT cells GM2 ganglioside was a substrate for theproduction of GM3 ( fi GD3 fi GD2 fi GD1b fi GT1b),GA2, and GM1 ( fi GD1a). The parallel increase in thelevels of GM2 (eightfold to NPC) and GD1a (3.5-fold toNPC) in NPC-differentiated progeny cells suggests aprevalent activity of the GM2 fi GM1 fi GD1a pathwayin these cells (Fig. 7a). In mutant NPCs, levels of GM2 werein the range or lower to WT while GD1a was undetectable.However, while GM2 accumulated in mutant-differentiatedcells (TS > SD; 70- and 22-fold to precursors, respectively),GD1a was still low. Increased levels of GD3 and GD2(TS > SD) were observed in mutant NPCs and in theirprogeny with respect to WT cells (Fig. 7a).
We further investigated this ganglioside pathway byincubating WT, TS, SD NPCs, and their differentiatedprogeny with [3-3H(sphingosine)]GM1 or [3-3H(sphingo-sine)]GM2 (Fig. 7b). In the presence of labeled GM1, weobserved production of GD1a and GM2, which wasconverted to GM3 in WT NPCs (Fig. 7b; precursors).Production of GM3 and increased levels of GA2 gangliosidewere detected in mutant NPCs (Fig. 7b; precursors). Thesedata were confirmed when the same cells were incubatedwith labeled GM2 (Fig. 7b; precursors).
A comparable pathway of the labeled gangliosides wasobserved in SD-, TS-, and WT-differentiated cells. Notably,we detected a strong increase in GA2 ganglioside in SD cellswhile GM3 was barely detectable in both mutant and WTcells. Control feeding experiments, performed in TS bonemarrow-derived stem cells, demonstrated storage of GM2ganglioside, absence of GM3 production, and absence ofinternalized HexA from the culture medium (not shown;Martino et al. 2002b). All together these data suggest that theGM2 ganglioside, indirectly through the GA2 pathway, mightbe partially converted to GM3 ( fi GD3 fi GD2) also in SDNPCs and in their differentiated progeny (Figs. 7 and 8).
Discussion
In this work we showed that NPC cultures represent areliable experimental paradigm in which the expression andregulation of lysosomal enzymes may be studied in normaland mutant NPCs and in their differentiated progeny. Wedemonstrated that the temporal profile of Hex activity, geneexpression, and ganglioside pattern observed in brain tissuesduring postnatal development were recapitulated during thein vitro differentiation of NPCs to mature neurons and glia.Importantly, by comparing NPCs and their progeny estab-lished from SD and TS animal models with the WTcounterparts, we clarified the events linking the accumulation
(a)
(b)
Fig. 6 Lysosomal sialidase activity increases during brain develop-
ment and neural precursor cell (NPC) differentiation. Lysosomal
sialidase activity in Tay-Sachs (TS) and Sandhoff disease (SD) brain
tissues increased from P7 to P10, then reaching a plateau (a), while it
was stable in wild-type (WT) tissues during the whole postnatal ages
until P21, then reaching the levels observed in adult mutants (a).
Sialidase activity increased during SD, TS, and WT NPC differentia-
tion, with no significant genotype-related differences (b).
� 2009 The AuthorsJournal Compilation � 2009 International Society for Neurochemistry, J. Neurochem. (2009) 109, 135–147
Hexosaminidase activity in brain development and neural precursor cell differentiation | 143

of undegraded substrates to Hex activity, highlighting apotential alternate GM2 ganglioside pathway that might beactive in the SD model.
We found an age-dependent increase in Hex activityduring postnatal brain development with distinct temporalprofiles and peculiar isoenzyme composition in brain tissuesof the murine models of disease compared with those ofunaffected mice. Despite an overall 10-fold up-regulation ofbrain Hex activity in adulthood with respect to the embry-onic stage, temporal profiles were consistently different inhexa)/) mice compared with WT littermates. Interestingly,the sharp increase in Hex activity observed between P7 andP13 in WT brain tissue was absent in TS counterparts inwhich steady Hex activity was maintained from P10throughout adulthood. Neurodevelopment at P7–P13 agesin mice can be comparable with late-stage fetal and earlyneonatal human brain development (Clancy et al. 2001,2007). This period was characterized by active cerebellardevelopment, dendrite arborisation, axon outgrowth, syna-ptogenesis, and myelinogenesis processes in which the roleof gangliosides had been extensively studied in normal andpathological conditions (Svennerholm et al. 1989, 1991;Kroll et al. 1995; Shen et al. 1998; Folkerth et al. 2000;Walkley et al. 2000; Buccoliero et al. 2002; Alkan et al.
2003; Buccoliero and Futerman 2003; van der Voorn et al.2004; Di Rocco et al. 2005; Sohn et al. 2006; Ngamukoteet al. 2007). A large body of literature indicated that in theseearly stages of human and murine CNS development, thelevels of GM3 and GM2 gangliosides were barely detectablewhereas the levels of ganglioside GD1a were elevated,suggesting a high rate of GM1 fi GD1a conversion (Iwa-mori and Nagai 1979; Rosengren et al. 1987; Ishii et al.2007; Ngamukote et al. 2007; Baek et al. 2008). SD and TSbrain tissues were characterized by GM2 ganglioside storage(Yamanaka et al. 1994; Sango et al. 1995) by levels of GM1and GD1a comparable with those present in WT tissues(Rosengren et al. 1987; Platt et al. 1997; Ngamukote et al.2007; Baek et al. 2008) and by increased GA2 levels(Phaneuf et al. 1996; Platt et al. 1997). This correlates withhigh levels of lysosomal sialidase activity which we foundstrongly increased in pathological compared with WT brains.The GA2 was formed in the lysosomal compartment andcannot be converted directly to GM3, GD3 or GD2, etc.However, it can be degraded to its components which mayleave the lysosome and can be re-used for biosynthesis ofcomplex gangliosides (Gillard et al. 1998a,b) in the Golgiand trans-golgi network compartment (Schwarzmann andSandhoff 1990) (Sandhoff 2001).
(a)
(b)
Fig. 7 Ganglioside pattern in wild-type
(WT), Sandhoff disease (SD) and Tay-
Sachs (TS) neural precursor cell (NPCs)
and in their differentiated progeny. (a)
Ganglioside content in WT, SD, and TS
NPCs and in their differentiated progeny.
Note the high levels of GD3 and GD2 in
mutant cells with respect to WT cells.
Ganglioside extraction and measurement
are described in methods in Supporting
Information. (b) Feeding experiments were
assessed with gangliosides containing tri-
tium al position 3 of sphingosine (see Sup-
porting Information for details). TLC showed
metabolic GM1 and GM2 pathways. Note
the presence of GM3 ganglioside in
SD > TS NPCs, and to a lower extent, in
their differentiated progeny, and the high
production of GA2 in mutant NPCs and
differentiated cells. The labeled GM1 and
GM2 used for the feeding experiments were
used as standards.
Journal Compilation � 2009 International Society for Neurochemistry, J. Neurochem. (2009) 109, 135–147� 2009 The Authors
144 | S. Martino et al.

Our data suggest that sialidase might be active in convertingGM2 into GA2 also in SDmice. GA2 can be rapidly convertedto other compounds (Fig. 8), probably increasing GD3 andGD2production inmutant brain indirectly (Iwamori andNagai1979; Takamiya et al. 1996). A similar glycolipid compositionwas observed in adult human and murine mutant brains(Rosengren et al. 1987; Platt et al. 1997; Ngamukote et al.2007; Baek et al. 2008).
Temporal differences in Hex expression observed in SD,TS, and WT brain tissues might be the consequence of adifferential expression of hexa and hexb genes. In fact, whilehexb was up-regulated during brain development, hexa wasstable, even in the time-window (P13–P21) in which therelative amount of HexA activity increased. In turn, hexagene was up-regulated in SD tissue, in which hexb wasdisrupted. This suggests that regulation of hexb is acheckpoint for enhancing Hex activity and determining theparticular isoenzyme pattern observed, including the peak of
activity that we called Hex-I. Our data showed that Hex-Iwas composed of b subunit precursors, with intermediatecharacteristics between HexB and HexA isoenzymes. Itsrelative preponderance in a restricted postnatal periodsuggests its potential role in physiologic CNS development.
The biological significance of this Hex paradigm wasfurther confirmed by the demonstration that age- andgenotype-related patterns of lipid storage, Hex activity,isoenzyme composition, and gene expression were recapit-ulated during the progressive stages of differentiation thatSVZ-derived NPCs undergo to generate mature glia andneurons in vitro (Gritti et al. 2002; Reynolds and Rietze2005). The high increase in enzyme activity and the peculiarHex isoenzymes composition observed in NPC-deriveddifferentiated progeny compared with their precursors (likelya consequence of hexb and/or hexa up-regulation) indicate astrong correlation between Hex activity and NPC commit-ment/differentiation, regardless of the age of the animalsfrom which NPCs were isolated (neonatal or adult). Thissituation closely mimics that observed in isolated braintissues during the steps of neurodevelopment. Data onenzyme activity were further confirmed by data on gangli-oside levels. In fact, the high level of GA2 and the relevantincrease in GD3 and GD2 ganglioside levels in mutant cells(NPCs and differentiated progeny) compared with the WTcounterpart resembled that previously described in braintissues. In TS NPCs and progeny the stored GM2 waspartially converted by sialidase to GA2 (Sandhoff andJatkewitz 1967), which can be then degraded to its compo-nents in lysosomes. The latter can be used in a salvagepathway (Gillard et al. 1998a,b) for the formation of GM3ganglioside, which in turn, might be rapidly converted toGD3 and then to GD2 (Fig. 8). Here, we show for the firsttime that a similar alternative metabolic route could be activealso in hexb)/) NPCs and in their progeny.
Overall our data indicate that NPC cultures recapitulate thebiochemical and molecular hallmarks of GM2 gangliosidosisand represent a valuable model to study the gangliosidemetabolism in these diseases.Our findings also suggest that thesharp increase in brain Hex activity occurring in physiologicalconditions during a narrow postnatal period in mice might becritical to ensure proper ganglioside balance required forphysiologicalCNSdevelopment (see also the temporal profilesof other glycohydrolases activity, and particularly of b-galac-tosidase, the hydrolytic enzyme for GM1). This implies thatthe efficacy of any therapeutic approach will largely rely on itseffectiveness in providing appropriate enzyme levels withinthis time-window, which roughly corresponds to the late fetal/early postnatal period in humans.
Acknowledgments
We thank T. Neri and C. Maderna for technical advice, Dr. J. L.
Stirling (Kings College of London) for helpful comments and Hilary
(a)
(b)
Fig. 8 Pathways of ganglioside metabolism in physiological condi-
tions and in GM2 gangliosidosis. (a) In wild-type (WT) neural precursor
cell (NPCs) (and in brain tissues) GM2 can be converted to GM3,
GA2, and GM1 ( fi GD1a). GA2 was then converted by HexB and
GM2 activator protein to LacCer that could implement the GM3 syn-
thesis and GD3 fi GD2 metabolism. (b) In Tay-Sachs (TS) and
Sandhoff disease (SD) neural cells (NPCs < differentiated progeny)
and in brain tissues (Embryonic/fetal < postnatal) stored GM2 can be
only converted to GA2 and GM1. Conversion of GM1 to GD1a was
highest in brain tissues while it was barely detectable in both NPCs
and their differentiated progeny. In TS neural cells/brain tissues GA2
was converted by HexB and GM2 activator protein to LacCer that
could implement the GM3 synthesis and GD3 fi GD2 metabolism
(blue arrows). In SD cells the high rate of GD3 fi GD2 metabolism
(red arrows) suggests an active GM3 synthesis that could be alter-
natively implemented by GA2 catabolism.
� 2009 The AuthorsJournal Compilation � 2009 International Society for Neurochemistry, J. Neurochem. (2009) 109, 135–147
Hexosaminidase activity in brain development and neural precursor cell differentiation | 145

Giles (MA) for language advice. We thank the ‘Diagnosi PrePost-
natale Malattie Metaboliche’ Laboratory (G. Gaslini Institute) for
providing us with specimens from the Cell line and DNA bank from
patients affected by Genetic diseases – Telethon Genetic Biobank
Network (project no. GTB07001A).
This work was supported by grants from MIUR (FIRB-
RBAU01M5FR_001 to AG; FIRB-RBNE0121W8_001 to AO),
CIB-2004–2007, Fondazione Cassa Risparmio di Perugia (Project
2007.0149.02), and Consorzio INNB to AO, Fondazione Mariani
Onlus (Project R-05-43 2005-2006), National Tay-Sachs and Allied
Diseases (NTSAD; Project 1 R21 NS052750-01), and Italian
Telethon Foundation (Grant no. TGT06B02) to AG. This study is
dedicated to P. Vigezzi.
Supporting Information
Additional Supporting Information may be found in the online
version of this article:
MethodsResultsFigure S1 Biochemical characterization of hexosaminidase
isoenzymes.
Please note: Wiley-Blackwell are not responsible for the content
or functionality of any supporting materials supplied by the authors.
Any queries (other than missing material) should be directed to the
corresponding author for the article.
References
Alkan A., Kutlu R., Yakinci C., Sigirci A., Aslan M. and Sarac K. (2003)Infantile Sandhoff’s disease: multivoxel magnetic resonancespectrosecopy findings. J. Child Neurol. 18, 425–428.
Baek R. C., Kasperzyk J. L., Platt F. M. and Seyfried T. N. (2008) N-butyldeoxygalactonojirimycin reduces brain ganglioside and GM2content in neonatal Sandhoff disease mice. Neurochem. Int. 52,1125–1133.
Buccoliero R. and Futerman A. H. (2003) The roles of ceramide andcomplex sphingolipids in neuronal cell function. Pharmacol. Res.47, 409–419.
Buccoliero R., Bodennec J. and Futerman A. H. (2002) The role ofsphingolipids in neuronal development: lessons from models ofsphingolipid storage diseases. Neurochem. Res. 27, 565–574.
Cachon-Gonzalez M. B., Wang S. Z., Lynch A., Ziegler R., Cheng S. H.and Cox T. M. (2006) Effective gene therapy in an authentic modelof Tay-Sachs-related diseases. Proc. Natl Acad. Sci. USA 103,10373–10378.
Cavazzin C., Ferrari D., Facchetti F., Russignan A., Vescovi A. L., LaPorta C. A. and Gritti A. (2006) Unique expression and localizationof aquaporin-4 and aquaporin-9 in murine and human neural stemcells and in their glial progeny. Glia 2, 167–181.
Clancy B., Darlington R. B. and Finlay B. L. (2001) Translating devel-opmental time across mammalian species. Neuroscience 105, 7–17.
Clancy B., Finlay B. L., Darlington R. B. and Anand K. J. (2007)Extrapolating brain development from experimental species tohumans. Neurotoxicology 28, 931–937.
Di Rocco M., Rossi A., Parenti G., Allegri A. E., Filocamo M., PessagnoA., Tortori-Donati P., Minetti C. and Biancheri R. (2005) Differentmolecular mechanisms leading to white matter hypomyelination ininfantile onset lysosomal disorders. Neuropediatrics 36, 265–269.
Folkerth R. D., Alroy J., Bhan I. and Kaye E. M. (2000) Infantile G(M1)gangliosidosis: complete morphology and histochemistry of two
autopsy cases, with particular reference to delayed central nervoussystem myelination. Pediatr. Dev. Pathol. 3, 73–86.
Galli R., Fiocco R., De Filippis L., Muzio L., Gritti A., Mercurio S.,Broccoli V., Pellegrini M., Mallamaci A. and Vescovi A. L. (2002)Emx2 regulates the proliferation of stem cells of the adult mam-malian central nervous system. Development 129, 1633–1644.
Gillard B. K., Clement R. G. and Marcus D. M. (1998a) Variationsamong cell lines in the synthesis of sphingolipids in de novo andrecycling pathways. Glycobiology 8, 885–890.
Gillard B. K., Clement R., Colucci-Guyon E., Babinet C., SchwarzmannG., Taki T., Kasama T. and Marcus D. M. (1998b) Decreasedsynthesis of glycosphingolipids in cells lacking vimentin inter-mediate filaments. Exp. Cell Res. 242, 561–572.
Gravel R. A., Triggs-Raine B. L. and Mahuran D. J. (1991) Biochem-istry and genetics of Tay-Sachs disease. Can. J. Neurol. Sci. 18,419–423.
Gritti A., Frolichsthal-Schoeller P., Galli R., Parati E. A., Cova L., PaganoS. F., Bjornson C. R. and Vescovi A. L. (1999) Epidermal andfibroblast growth factors behave as mitogenic regulators for a singlemultipotent stem cell-like population from the subventricular regionof the adult mouse forebrain. J. Neurosci. 19, 3287–3297.
Gritti A., Bonfanti L., Doetsch F., Caille I., Alvarez-Buylla A., Lim D.A., Galli R., Verdugo J. M., Herrera D. G. and Vescovi A. L.(2002) Multipotent neural stem cells reside into the rostralextension and olfactory bulb of adult rodents. J. Neurosci. 22, 437–445.
Ishii A., Ikeda T., Hitoshi S., Fujimoto I., Torii T., Sakuma K., NakakitaS., Hase S. and Ikenaka K. (2007) Developmental changes in theexpression of glycogenes and the content of N-glycans in themouse cerebral cortex. Glycobiology 17, 261–276.
Iwamori M. and Nagai Y. (1979) Ganglioside-composition of brain inTay-Sachs disease: increased amounts of GD2 and N-acetyl-beta-D-galactosaminyl GD1a ganglioside. J. Neurochem. 32, 767–777.
Jeyakumar M., Dwek R. A., Butters T. D. and Platt F. M. (2005) Storagesolutions: treating lysosomal disorders of the brain. Nat. Rev.Neurosci. 6, 713–725.
Kato T., Wang Y., Yamaguchi K., Milner C. M., Shineha R., Satomi S.and Miyagi T. (2001) Overexpression of lysosomal-type sialidaseleads to suppression of metastasis associated with reversion ofmalignant phenotype in murine B16 melanoma cells. Int. J. Cancer92, 797–804.
Kroll R. A., Pagel M. A., Roman-Goldstein S., Barkovich A. J.,D’Agostino A. N. and Neuwelt E. A. (1995) White matter changesassociated with feline GM2 gangliosidosis (Sandhoff disease):correlation of MR findings with pathologic and ultrastructuralabnormalities. AJNR Am. J. Neuroradiol. 16, 1219–1226.
Lee J. P., Jeyakumar M., Gonzalez R. et al. (2007) Stem cells actthrough multiple mechanisms to benefit mice with neurodegener-ative metabolic disease. Nat. Med. 13, 439–447.
Martino S., Cavalieri C., Emiliani C. et al. (2002a)Restoration of theGM2ganglioside metabolism in bone marrow-derived stromal cells fromTay-Sachs disease animal model. Neurochem. Res. 27, 793–800.
Martino S., Emiliani C., Tancini B., Severini G. M., Chigorno V.,Bordignon C., Sonnino S. and Orlacchio A. (2002b) Absence ofmetabolic cross-correction in Tay-Sachs cells: implications forgene therapy. J. Biol. Chem. 277, 20177–20184.
Martino S., Marconi P., Tancini B. et al. (2005a) A direct gene transferstrategy via brain internal capsule reverses the biochemical defectin Tay-Sachs disease. Hum. Mol. Genet. 14, 2113–2123.
Martino S., Consiglio A., Cavalieri C., Tiribuzi R., Costanzi E., SeveriniG. M., Emiliani C., Bordignon C. and Orlacchio A. (2005b)Expression and purification of a human, soluble arylsulfatase Afor metachromatic leukodystrophy enzyme replacement therapy.J. Biotechnol. 117, 243–251.
Journal Compilation � 2009 International Society for Neurochemistry, J. Neurochem. (2009) 109, 135–147� 2009 The Authors
146 | S. Martino et al.

Ngamukote S., Yanagisawa M., Ariga T., Ando S. and Yu R. K. (2007)Developmental changes of glycosphingolipids and expression ofglycogenes in mouse brains. J. Neurochem. 103, 2327–2341.
Phaneuf D., Wakamatsu N., Huang J. Q. et al. (1996) Dramaticallydifferent phenotypes in mouse models of human Tay-Sachs andSandhoff diseases. Hum. Mol. Genet. 5, 1–14.
Platt F. M., Neises G. R., Reinkensmeier G., Townsend M. J., Perry V.H., Proia R. L., Winchester B., Dwek R. A. and Butters T. D.(1997) Prevention of lysosomal storage in Tay-Sachs mice treatedwith N-butyldeoxynojirimycin. Science 276, 428–431.
Platt F. M., Jeyakumar M., Andersson U., Dwek R. A. and Butters T. D.(2005) New developments in treating glycosphingolipid storagediseases. Adv. Exp. Med. Biol. 564, 117–126.
Reynolds B. A. and Rietze R. L. (2005) Neural stem cells and neuro-spheres: re-evaluating the relationship. Nat. Methods 2, 333–336.
Rosengren B., Mansson J. E. and Svennerholm L. (1987) Compositionof gangliosides and neutral glycosphingolipids of brain in classicalTay-Sachs and Sandhoff disease: more lyso-GM2 in Sandhoffdisease? J. Neurochem. 49, 834–840.
Sandhoff K. (2001) The GM2-gangliosidoses and the elucidation of thebeta-hexosaminidase system. Adv. Genet. 44, 67–91.
Sandhoff K. and Jatkewitz H. (1967) A particle-bound sialyl lactosido-ceramide splitting mammalian sialidase. Biochim. Biophys. Acta141, 442–444.
Sango K., Yamanaka S., Hoffmann A. et al. (1995) Mouse models ofTay-Sachs and Sandhoff diseases differ in neurologic phenotypeand ganglioside metabolism. Nat. Genet. 11, 170–176.
Schwarzmann G. and Sandhoff K. (1990) Metabolism and intracellulartransport of glycosphingolipids. Biochemistry 29, 10865–10871.
Shen W. C., Tsai F. J. and Tsai C. H. (1998) Myelination arrest dem-onstrated using magnetic resonance imaging in a child with type IGM1 gangliosidosis. J. Formos. Med. Assoc. 97, 296–299.
Sohn H., Kim Y. S., Kim H. T., Kim C. H., Cho E. W., Kang H. Y., KimN. S., Ryu S. E., Lee J. H. and Ko J. H. (2006) Ganglioside GM3 isinvolved in neuronal cell death. FASEB J. 20, 1248–1250.
Svennerholm L., Bostrom K., Fredman P., Mansson J. E., Rosengren B.and Rynmark B. M. (1989) Human brain gangliosides: develop-mental changes from early fetal stage to advanced age. Biochim.Biophys. Acta 1005, 109–117.
Svennerholm L., Rynmark B. M., Vilbergsson G., Fredman P., GottfriesJ., Mansson J. E. and Percy A. (1991) Gangliosides in human fetalbrain. J. Neurochem. 56, 1763–1768.
Takamiya K., Yamamoto A., Furukawa K. et al. (1996) Mice with dis-rupted GM2/GD2 synthase gene lack complex gangliosides butexhibit only subtle defects in their nervous system. Proc. NatlAcad. Sci. USA 93, 10662–10667.
van der Voorn J. P., Kamphorst W., van der KnaapM. S. and Powers J. M.(2004) The leukoencephalopathy of infantile GM1 gangliosidosis:oligodendrocytic loss and axonal dysfunction. Acta Neuropathol.(Berl.) 107, 539–545.
Walkley S. U., Zervas M. and Wiseman S. (2000) Gangliosides asmodulators of dendritogenesis in normal and storage disease-affected pyramidal neurons. Cereb. Cortex 10, 1028–1037.
Yamanaka S., Johnson M. D., Grinberg A., Westphal H., Crawley J. N.,Taniike M., Suzuki K. and Proia R. L. (1994) Targeted disruptionof the Hexa gene results in mice with biochemical and pathologicfeatures of Tay-Sachs disease. Proc. Natl Acad. Sci. USA 91, 9975–9979.
� 2009 The AuthorsJournal Compilation � 2009 International Society for Neurochemistry, J. Neurochem. (2009) 109, 135–147
Hexosaminidase activity in brain development and neural precursor cell differentiation | 147


















