
Determination of selenomethionine by high performance liquid chromatography-direct hydride...
9
Ž . Microchemical Journal 69 2001 179187 Determination of selenomethionine by high performance liquid chromatography-direct hydride generation-atomic absorption spectrometry Amit Chatterjee , Y. Shibata, M. Morita Department of Chemistry and Biochemistry, 600 Forbes A enue, Mellon Hall, Duquesne Uni ersity, Pittsburgh, PA 15282, USA Received 1 December 2000; received in revised form 31 January 2001; accepted 1 February 2001 Abstract Ž . A simple, reliable, trace determination of selenomethionine Semet based on a direct hydride generation atomic Ž . absorption spectrometric method was developed using sodium tetrahydroborate 0.3% in 0.2% NaOH and hydro- Ž . Ž . chloric acid 3 M . The method excluded any chemical pretreatment prior to hydride generation HG . The optimized Ž . HG system was successfully coupled with the HPLC system. The detection limit 3 of blank; n 5 , reproducibility Ž . Ž R.S.D. of three successive analysesday, performed on three different days , and repeatability R.S.D. of three . 1 1 1 successive analyses of the method were 1.08 ng ml , 9.8% for 9.04 ng ml and 2.1 9.5% for 30.0 1.27 ng ml Ž . 1 Semet as Se standards prepared in Milli-Q water . Calibration graph was linear up to 30 ng ml . This Ž . HPLC-HG-AAS method is very promising and successfully determined Semet spiked in human urine. 2001 Elsevier Science B.V. All rights reserved. Keywords: Selenium; Selenomethionine; Flow injection; Hydride generation atomic absorption spectrometry; Hydride generation; AAS 1. Introduction Ž . Selenium Se has been recognized as an essen- Corresponding author. Tel.: 81-298-61-8301; fax: 81- 298-61-8308. Ž . E-mail address: [email protected] A. Chatterjee . tial nutrient for humans based on its presence at the active sites of the enzyme glutathione peroxi- dase that protects membranes from damage 1. During past decades, selenium compounds have gained much popularity as food supplements in order to prevent various diseases 2 4 . However, there is a relatively narrow range of selenium intake above which toxicity symptoms may de- 0026-265X01$ - see front matter 2001 Elsevier Science B.V. All rights reserved. Ž . PII: S 0 0 2 6 - 2 6 5 X 01 00081-9
-
Upload
amit-chatterjee -
Category
Documents
-
view
215 -
download
3
Transcript of Determination of selenomethionine by high performance liquid chromatography-direct hydride...

Ž .Microchemical Journal 69 2001 179�187
Determination of selenomethionine by highperformance liquid chromatography-direct hydride
generation-atomic absorption spectrometry
Amit Chatterjee�, Y. Shibata, M. MoritaDepartment of Chemistry and Biochemistry, 600 Forbes A�enue, Mellon Hall, Duquesne Uni�ersity, Pittsburgh, PA 15282,
USA
Received 1 December 2000; received in revised form 31 January 2001; accepted 1 February 2001
Abstract
Ž .A simple, reliable, trace determination of selenomethionine Semet based on a direct hydride generation atomicŽ .absorption spectrometric method was developed using sodium tetrahydroborate 0.3% in 0.2% NaOH and hydro-
Ž . Ž .chloric acid 3 M . The method excluded any chemical pretreatment prior to hydride generation HG . The optimizedŽ .HG system was successfully coupled with the HPLC system. The detection limit 3� of blank; n�5 , reproducibility
Ž . ŽR.S.D. of three successive analyses�day, performed on three different days , and repeatability R.S.D. of three. �1 �1 �1successive analyses of the method were 1.08 ng ml , 9.8% for 9.04 ng ml and 2.1�9.5% for 30.0�1.27 ng ml
Ž . �1Semet as Se standards prepared in Milli-Q water . Calibration graph was linear up to 30 ng ml . ThisŽ .HPLC-HG-AAS method is very promising and successfully determined Semet spiked in human urine. � 2001
Elsevier Science B.V. All rights reserved.
Keywords: Selenium; Selenomethionine; Flow injection; Hydride generation atomic absorption spectrometry; Hydride generation;AAS
1. Introduction
Ž .Selenium Se has been recognized as an essen-
� Corresponding author. Tel.: �81-298-61-8301; fax: �81-298-61-8308.
Ž .E-mail address: [email protected] A. Chatterjee .
tial nutrient for humans based on its presence atthe active sites of the enzyme glutathione peroxi-
� �dase that protects membranes from damage 1 .During past decades, selenium compounds havegained much popularity as food supplements in
� �order to prevent various diseases 2�4 . However,there is a relatively narrow range of seleniumintake above which toxicity symptoms may de-
0026-265X�01�$ - see front matter � 2001 Elsevier Science B.V. All rights reserved.Ž .PII: S 0 0 2 6 - 2 6 5 X 0 1 0 0 0 8 1 - 9

( )A. Chatterjee et al. � Microchemical Journal 69 2001 179�187180
� �velop 5 . It is well known that the bioavailabilityand the toxicity of selenium compounds are closelyrelated. Selenoamino acids attract much interest,because these compounds take part in the biolog-ical selenium cycle and are incorporated intoproteins. Selenoamino acids such as selenome-
�Žthionine Semet; CH �Se�CH �CH �CH�NH3 2 2 2Ž .�COOH are used as selenium supplements forhumans and animals. Inorganic selenium com-pounds are less absorbed in the human body thanselenomethionine. Moreover, Semet serves for se-lenium storage in proteins and for the incorpora-
� �tion of Se into selenium-specific enzymes 6�8 .Nevertheless, Semet has been reported to be less
� �toxic than inorganic selenium compounds 9 .Thus, free selenomethionine and selenized yeast,active components in the nutritional supplementformulations are commercially available for hu-man consumption. Semet, which is chemicallysimilar to methionine, has been identified in food
� � � �materials 10 , selenium enriched yeast 11 and in� �urine 12 . Semet has pK values of 2.28 and 9.21a
� � Ž13 , carries a positive charge a localized proto-.nated amino group at relatively low pH, a zwitte-
Ž .rionic amino and carboxylate group at moder-Žately high pH and becomes anionic carboxylate
.group at higher pH. Semet is measured in theurine of supplemented individuals. The minimumlethal dose of Semet in rats is 4.25 mg Se kg�1,
� �over 1.3 times higher than for selenite 14 . Withincreased use of Semet for dietary supplementa-tion in animals and humans, sensitive and selec-tive methods are needed for the determination ofSemet in nutritional supplements, urine and otherbiological and environmental samples so that themetabolic fate of selenium is better understood.Availability of analytical techniques for the sepa-ration and determination of Semet at trace levelshas gained considerable importance. Edmonds
� � � �and Morita 15 , Pyrzynska 16 and others� �14,17,18 have reviewed methods for the determi-nation of selenoamino acids including Semet. Thepopular and reliable methods for the determina-tion of Semet are AAS, AES, AFS, MIP-MS,
� �ICP-MS and GC-ICP-MS 14�25 . The hydrideŽ .generation HG system has become one of the
useful techniques for the determination of sele-nium, as it decreases interference problems with
Ž .increasing transport efficiency �100% and ulti-mately increases overall sensitivity. Sensitive tech-niques combined with a HG system include HG-
� �AAS 17 , HG-atomic fluorescence spectrometryŽ . � � � � � �AFS 17,24 , HG-AES 24 , HG-ICP-MS 11
� �and HG-MIP-MS 25 . HG methods are based ona reduction of an element from a higher oxidationstate to the gaseous hydride with NaBH How-4.ever, one potential drawback of HG is that itrequires elements to be in a particular oxidationstate before a hydride may be formed effectively� � Ž .24 , this being the selenite �IV for selenium.Unlike selenite, Semet does not directly formvolatile selenium compounds in a HG system with
� �NaBH and HCl 19 . Thus, for determination of4Semet, a treatment prior to HG is mandatory� �14�16,18,26 . A number of manuscripts contain-ing different procedures for decomposition ofSemet to selenate and further reduction to selen-
� �ite are reported 14�16 . The decomposition ofSemet to selenate using a microwave unit with
� � � �K S O 20 , HBr�KBrO 20 , and KBr�HCl2 2 8 3with UV irradiation, is widely used for the estima-
� �tion of Semet by HPLC-HG-AAS 19 . However,� �we have found 17,25 that organic selenium com-
pounds are HG active and generate volatile sele-nium compounds in the HG system.
This study reports the development of aHPLC-direct HG-AAS methodology using NaBH4and HCl for the determination of Semet. Themethodology has excluded chemical pretreatmentbefore HG, and the developed HG system issuccessfully coupled with HPLC for the on-lineseparation and estimation of selenium com-pounds in human urine.
2. Experimental
ŽA Perkin-Elmer Zeeman 5100 with D back-2.ground corrector flame atomic absorption spec-
trometer fitted with electrically heated quartz T-tube served as the selenium specific detector. Theselenium compounds were reacted with sodiumtetrahydroborate selectively in a Perkin-ElmerFIAS 100 system equipped with a gas�liquid sep-arator. Gaseous products were separated fromthe liquid in the gas�liquid separator and flushed

( )A. Chatterjee et al. � Microchemical Journal 69 2001 179�187 181
into the quartz tube by a stream of argon. APerkin-Elmer selenium electrodeless dischargeŽ . ŽEDL lamp was part no. N305-0672, serial no.
.1209 M676 operated at 210 mA. Fig. 1 shows theHPLC-HG-AAS system. The operation condi-tions for HG-AAS are summarized in Table 1.Background corrected Se signals were read into apersonal computer via the Perkin-Elmer GEMsoftware. A PRP-X100, anion-exchange columnŽHamilton, Reno, NV, USA, 25 cm�4.1 mm i.d.;spherical 10-�m particles of a styrene�divinyl-benzene copolymer with trimethylammonium ex-change sites; stable between pH 1 and 13; ex-
�1 .change capacity 0.19 meq. g was used for theseparation study. The column was equilibrated by
Ž �1 .passing at least 100 ml flow rate 1.0 ml min ofthe mobile phase through the column before in-jection of selenium compounds. Outlet of thecolumn was connected to a HG system via a300-mm 1�16-inch i.d. polyether ether ketoneŽ .PEEK capillary tubing. The outlet of the HGsystem was connected with a 400-mm-long Tygon
Ž .tubing 1.0 mm i.d. to the QT of an AAS. Fromthe chromatograms, peak areas were calculated,and concentrations were determined with exter-nal calibration curves established for each of theSe compounds.
Table 1Operating conditions for FI-HG-AAS
Atomic absorption spectrometer Perkin-Elmer 5100
Ž .Lamp current EDL 210 mAWavelength 196.0 nmElectrically heated quartz T-tube �790�C
temperature�1Ž .NaBH 0.3% in 0.2% NaOH 4.5 ml min4�1Ž .Carrier flow rate 3.0 M HCl 5.6 ml min
Sample loop 500 �lFill time 10 sInject time 15 s
�1Argon gas flow 116 ml min
2.1. Chemicals and reagents
All reagents were of analytical grade and wereused without further purification. Sodium sele-
Ž .nate No. 71948 was purchased from Fluka,Žsodium selenite pentahydrate from Merck No.
. Ž106607 . Seleno DL-methionine S-3875; Lot.18H0561 was purchased from Sigma. Stock solu-
Žtions were prepared in Milli-Q water 18.2 M�cm, Milli-Q SP. TOC. Reagent water system, Ni-
.hon Millipore Ltd. Yonezawa, Japan from sodiumŽselenite pentahydrate 1666 mg to 500 ml; 1000
�1 .�g Se ml , and from Seleno DL-methionine
Fig. 1. Schematic diagram of HPLC-HG-AAS.

( )A. Chatterjee et al. � Microchemical Journal 69 2001 179�187182
Ž �1 .124 mg to 100 ml, 500 �g Se ml . Stocksolutions were stored in a freezer at �20�C be-fore use. Solutions of the selenium compoundswith concentrations in the range 1.00�50.0 ng Seml�1 were prepared by appropriate dilution of
Žthe stock solutions with Milli-Q water standard. Ž .solutions , tap water and urine spike analyses .
ŽSodium tetrahydroborate solutions 0.2�1.5%.m�v were prepared freshly by dissolving NaBH4
Ž .powder Merck, 1063710 of 96% purity in Milli-Qwater and stabilizing the solutions with sodium
Ž .hydroxide 0.2% m�v; Merck, 71690 . Hydrochlo-Ž .ric acid solutions 2.0�6.0 M were prepared by
Ždilution of concentrated HCl Merck P.A., No.100319, further purified in a quartz sub-boiling
.distillation unit . Mobile phase for the anion-ex-change chromatography was prepared by dissolv-
Ž .ing 1.73 g NH H PO Merck, p.a. to 1000 ml4 2 4Ž .0.015 M and adjusting the pH to 7.0 by addition
Ž .of 2.0 M aqueous NH solution Merck, p.a. .3
3. Results and discussion
3.1. Optimization of instrumental and non-instrumental parameters
In order to obtain the maximum sensitivity,repeatability and reproducibility for the determi-nation of Semet, the influence of several instru-mental and non-instrumental parameters on theanalytical performance of the HPLC-HG-AASsystem were optimized in detail. Peaks arerecorded as peak areas, since peak areas insteadof peak heights improve the signal-to-noise ratioby a factor of � 4. Detailed optimizationprocesses are explained in the following sections.
3.2. Sodium tetrahydroborate and hydrochloric acid
The dependence of the signal intensities ofŽ �1 .Semet 20 ng Se ml on the NaBH concentra-4Ž .tions 0.3�1.5% NaBH in 0.2% NaOH in the4
flow mode was investigated. Results indicate thatthe signal intensity increases with increasingNaBH concentration and a maximum is ob-4tained at 0.3% NaBH concentration. Moreover,4signal intensities decrease with further increasing
NaBH concentrations. 0.3% NaBH solution4 4yielded good results and was used as the optimum
Ž .condition maximum signal intensity for the HGŽ .system. It is observed that the quartz cell QT is
contaminated with elevated sample concentra-tions and NaBH when the latter is used in4excessive concentrations. Due to excess use ofNaBH concentrations, the reaction is vagarious4in the gas�liquid separator. So, in the gas�liquidseparator, acidic water droplets containing NaBH4were generated. A part of these NaBH contain-4ing liquid droplets were transported to QT by thecarrier gas, and formed a layer inside the quartzsurface along with the sample components. Since
Ž .the temperature of QT is very high 800�1000�C ,contaminants were burnt into the surface, gener-ating a kind of catalyst film which acceleratesradical recombination and therefore reduces theconcentration of H� radicals within QT. Conse-quently, equilibrium concentration of H� radicals,required for the atomization of gaseous hydrides,is disturbed. Hence, a significant signal depres-sion and lower sensitivity occurred. Thus, in orderto maintain the highest sensitivity, contaminantsfrom QT should be removed by frequently wash-ing either by rinsing with 40% HF for 15 min or
Ž .by heating annealing the QT at approximately1100�C. It was observed that the risk for contami-nation or formation of film is minimized whenlow concentrations of NaBH were used. Hence,4low concentration NaBH increases the lifetime4of the QT.
As signals for hydride forming elements aredependent on the concentration of hydrogen ions,so, efficiency on the generation of volatile sele-
Ž .nium compound in various HCl 2.0�6.0 M solu-tions was investigated. The most sensitive concen-tration is at 3.0 M HCl. More details about theoptimization of HCl and NaBH have been dis-4
� �cussed in a previous publication 17 . TheNaBH �HCl solution flow ratio influences4volatile selenium compound formation fromSemet. An optimum signal is achieved when theflow ratio was 0.80. Further increase of the flowratio decreases signal intensity with enhancingthe baseline noise, and liquid overflowed from thegas�liquid separator.

( )A. Chatterjee et al. � Microchemical Journal 69 2001 179�187 183
3.3. Carrier gas flow
A varying argon flow rate from the gas�liquidŽ .separator to a quartz-tube QT is a significant
parameter to transport volatile selenium com-pounds, which are formed in the gas�liquid sepa-rator to the quartz tube. With increasing Ar flowfrom 89�3 to 312�18 ml min�1, analyte signalis increased up to 116 ml min�1 and soon after-ward descended sharply with further increasingAr flow rate up to 220 ml min�1. Signal intensityis almost steady with increasing beyond 220 mlmin�1. An ideal flow rate of 116�5 ml min�1
was used for the measurements. With enhancingŽ �1 .Ar flow rate up to 316 ml min Semet signal
repeatability was improved, background noise de-creased but an irregular doublet shape peak ap-peared.
3.4. Quartz tube temperature
To adjust correct and optimized temperaturefor the decomposition of gaseous Se-hydride thatis formed from Semet and to obtain the highestsignal intensities, initially, the temperature of
Ž .quartz tube atomizer was varied 600�960�C . Themaximum intensities for a given concentration of
ŽSemet is achieved at a temperature of 790�C Fig..4 . Signal intensities form a plateau up to 800�CŽ .Fig. 2 . A further increase in temperature causesa decrease in signal, with increasing baseline noiseŽ .Fig. 2 . However, peak area and height have a
Ž .better repeatability R.S.D.� 3.3%, n � 5 attemperatures above 940�C; 790�C was used as the
Ž .optimum temperature maximum signal for themeasurement. Moreover, a temperature of 900�Cis recommended by the manufacturers of theatomic absorption spectrometer and was gener-ally used. An increase of approximately 15�20%of signal intensities at the optimized temperatureŽ .790�C compared with the manufacturer recom-
Ž .mended temperature 900�C was noticed. In somecases the QT was also heated using anair�acetylene flame system by placing the QT atthe top of the burner. However, it is very difficultto keep the temperature of the QT steady using aflame heating system. Shifting to different tem-perature of QT with flame heating is also verydifficult, as the temperature of a gaseous flamecannot be regulated properly. Even though, back-
� Ž .�Fig. 2. Dependence of signal intensity of Semet on the temperature of the quartz tube atomizer mean�S.D. n�3 .

( )A. Chatterjee et al. � Microchemical Journal 69 2001 179�187184
ground absorption in the flame heated QT ishigher than the electrically heated QT. Further-more, sensitivity for Semet depends on QT tem-perature, therefore, correct adjustment of the op-timized temperature is very important to acquirethe proper decomposition temperature for Se-hy-dride and to reach maximum sensitivity for Semet.This can be achieved by electrically heatedatomization QT rather than flame heatedatomization QT. Thus, we have used electricallyheated atomization QT for the experiments.
3.5. Performance
Linearity of standard curve, limits of detection,reproducibility and repeatability characterize theperformance of the developed HPLC-HG-AAS
Žsystem. The detection limit of Semet in Milli-Q.water based on the variability of reagent blank
Ž . �13� is 1.08 ng ml . The reproducibility of re-peated measurements for standard solutions was
Žassessed R.S.D., three successive analyses�day,.performed on three different days 9.8% for 9.04
ng of Se ml�1 as Semet. Calibration graphs areobtained from the areas of the HGAAS signalsŽ .three replicates with standard solutions of Semetat concentrations of 1.27, 2.53, 5.05, 10.3, 15.2,20.0 and 30.0 ng Se ml�1. The graph is linear inthis concentration range with a correlation coef-
ficient of 0.998. Relative standard deviations�Ž . �R.S.D. , n�3, repeatability of the peak areasdo not exceed 9.5% even at the lowest concentra-tion of 1.27 ng Se ml�1. The detection limits ofSemet with atomic and other spectroscopic tech-niques are listed in Table 2. The detection limitof Semet obtained with this HGAAS method iscomparable with the other spectroscopic methodsŽ .Table 2 . Measuring the sensitivity of the HGtechnique depends strongly on the atomizationtemperature and the state of QT inside the sur-face, where atomization of the element hydrideoccurs. In order to maintain the quartz surface inan optimum state, it is essential to anneal or washthe QT periodically.
3.6. Application to real samples
To ensure accuracy and precision of the devel-oped analytical procedure, and in order to de-monstrate that the system works for practicalanalysis, the method was applied to the determi-nation of Semet in tap water and urine afterHPLC separation. No Semet is detected in tapwater and urine. Hence, spikes of different Semet
Ž �1 .concentrations 10.0�40.2 ng Se ml are addedto tap water and urine. Recoveries of Semet spikefrom tap water and urine are in the range97�103% and 92�101%. It is well known that by
Table 2Ž .Detection limits DLs of Semet in different spectroscopic techniques
�1Ž .Methods DLs �g l References
� �HPLC-UV-HG-QTAAS 11.8 27� �LC-Microwave digestion- HG-QTAAS 8 19� �103 26� �Solid phase living cell extraction-ETAAS 1.5 28� �Capillary electrophoresis with UV detection 279 29� �Quartz thermochemical HG-AAS 1.4 30� �HPLC-offline-ETAAS 5.0 20
a � �HPLC-FAAS 2.2 31� �HPLC-ICP-MS 0.7 31� �HPLC-ICP-AES 100 20� �HPLC-HG-MIP-MS 8.7 25
Ž . � �GC-ICP-MS TFA derivatization of Semet 0.8 23Ž . � �GC-ICP-MS ECF derivatization of Semet 13 23
HPLC-HG-QTAAS 1.08 This work
a mg l�1 . TFA, trifloroacetic acid anhydride; ECF, ethyl chloroformate.
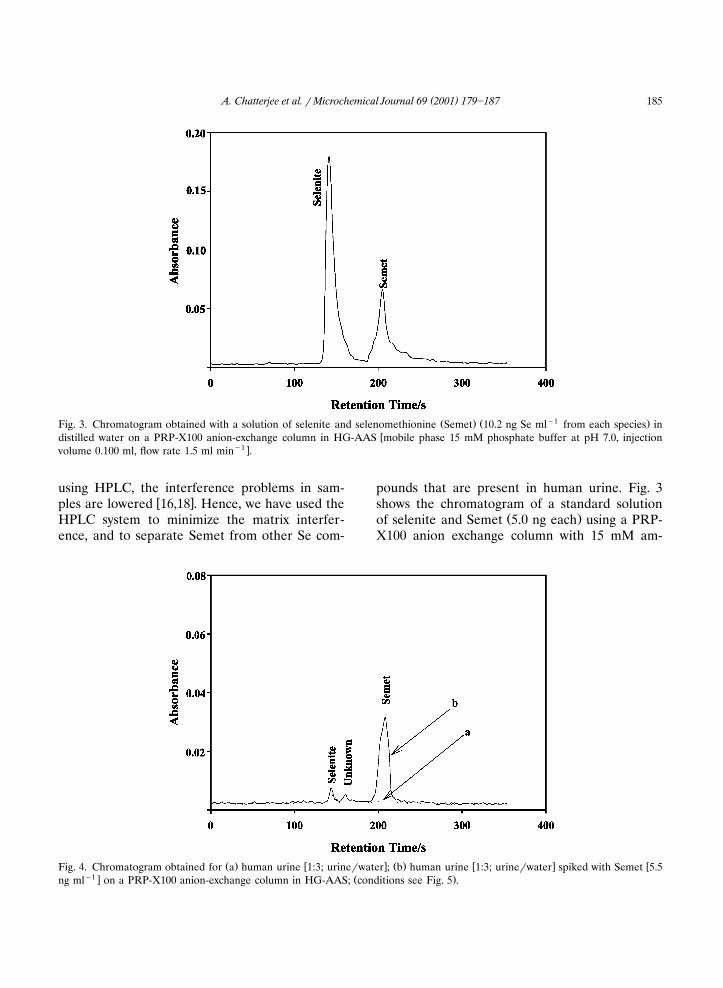
( )A. Chatterjee et al. � Microchemical Journal 69 2001 179�187 185
Ž . Ž �1 .Fig. 3. Chromatogram obtained with a solution of selenite and selenomethionine Semet 10.2 ng Se ml from each species in�distilled water on a PRP-X100 anion-exchange column in HG-AAS mobile phase 15 mM phosphate buffer at pH 7.0, injection
�1 �volume 0.100 ml, flow rate 1.5 ml min .
using HPLC, the interference problems in sam-� �ples are lowered 16,18 . Hence, we have used the
HPLC system to minimize the matrix interfer-ence, and to separate Semet from other Se com-
pounds that are present in human urine. Fig. 3shows the chromatogram of a standard solution
Ž .of selenite and Semet 5.0 ng each using a PRP-X100 anion exchange column with 15 mM am-
Ž . � � Ž . � � �Fig. 4. Chromatogram obtained for a human urine 1:3; urine�water ; b human urine 1:3; urine�water spiked with Semet 5.5�1 � Ž .ng ml on a PRP-X100 anion-exchange column in HG-AAS; conditions see Fig. 5 .

( )A. Chatterjee et al. � Microchemical Journal 69 2001 179�187186
monium dihydrogen phosphate buffer at pH 7.0.Fig. 4 shows selenium species present in the urineunder the above analytical separation conditions.
Ž .Peaks of selenite and Semet spiked are wellŽ .separated in the urine matrix Fig. 4 , and per-
centage recoveries are in the range of 92�101%.Ž .Selenate most probable in the urine does not
interfere, as selenate is HG inactive with the� �present HG conditions 17 . However, Seet
Ž .selenoethionine gives a broad peak, and elutesŽafter Semet. Nevertheless, TmSe retention time
.60 s elutes before selenite at pH 7.0 in thePRP-X100 column. Thus, the present chromato-graphic system is adequate for the separation ofSemet in human urine.
Sodium tetrahydroborate is a versatile reagent,which is widely used for its reducing and hydride
� �transfer properties 32 . It is used for conversionof element species present in aqueous solutioninto volatile hydrides. In this role, tetrahydro-borate reagent can act as a reductant and as ahydride source. In its reaction with selenium oxy-anion, which contains selenium in tetravalent oxi-dation state, the reaction with tetrahydroboratetakes selenium compounds through the corre-sponding selenide:
Ž . � �OSe OH �BH �H �SeH �H BO �H2 4 2 3 3 2
The reaction between NaBH and an ion in4solution is sensitive to pH, and it appears that, forthe rapid reaction, target species must not bepresent in solution as a negatively charged species.As pK for selenous acid is approximately 2.5,1which is fully protonated below pH 2. Hence, thereaction must be carried out at very low pH and
� �we have used 3 M HCl 32 . Under this condition,selenous acid reacts with NaBH and reduces to4selenide. Furthermore, selenic acid is not fully
Žprotonated at 3 M HCl pK �2.0 implying a pK2 1.value of approx. �3 . Hence, selenate is inactive
in the HG system and this agrees well with ourexperimental findings. The HG forming capabili-ties of Semet may be described in a similar way asselenite, because Semet has a pK �2.28 and1pK �9.21. Thus, in 3 M HCl, it is protonated.2Under these conditions Semet reacted withNaBH , decomposed and produced methylhy-4
drogenselenide, which dimerized to volatile di-methyldiselenide. This dimethyldiselenide wastransported to the quartz atomizer by carrier gas.There, it was decomposed to Se atoms and gener-ated a signal intensity. Hence, Semet is HG sys-tem active.
4. Conclusions
This report concerns the determination ofSemet in urine by a direct HG system coupledwith HPLC-AAS. Recent literature has noted that
� �Semet is HG inactive 16,24 . Previous re-searchers established that the decomposition ofSemet to selenate�selenite is necessary before
� �HG 16,24 and has complicated its measurement.The current developed method has its own advan-tage and potential for directly determining Semetin real samples. This HG method eliminates the
Ž .error caused by pretreatment step�s and orindirect methods, because Semet itself is HGactive. Previous literature revealed that only aHG-active oxidation state of selenium is seleniteŽ . � ��IV 18,24 . However, current investigationsalso proved that the �II oxidation of organicselenium compounds is HG active, and is safe as
Žit excludes all pretreatment UV�microwave�.acids prior to HG. It decreases sample and
reagent consumption, waste generation, and riskof contamination. The procedure has been suc-cessfully applied for the on-line determination of
Ž .Semet spiked from human urine by HPLC-HG-AAS.
Acknowledgements
Authors gratefully acknowledge JISTEC andSTA Japan for financial support.
References
� �1 J.P. Rotruck, A.L. Pope, H.E. Ganther, A.B. Swanson,Ž .D.G. Hafeman, W.G. Hoekstra, Science 179 1973 588.
� � Ž .2 A.M. Fan, K.W. Kizer, West J. Med. 153 1990 160.� � Ž .3 A.D. Salbe, O.A. Levander, J. Nutr. 120 1990 207.� �4 L.H. Foster, S. Sumar, Crit. Rev. Food Sci. Nutr. 37
Ž .1997 211.

( )A. Chatterjee et al. � Microchemical Journal 69 2001 179�187 187
� �5 W.N. Choy, P.R. Henika, C.C. Willhite, A.F. Tarantal,Ž .Environ. Mol. Mutagen 21 1993 73.
� � Ž .6 T.C. Stadtman, J. Biol. Chem. 266 1991 16257.� � Ž .7 M.A. Beilstein, P.D. Whanger, J. Nutr. 116 1986 1711.� � Ž .8 F. Dubois, F. Belleville, Pathol. Biol. 36 1988 1017.� �9 W.C. Willett, M.J. Stampfer, Acta Pharmacol. Toxical.
Ž .Copenh. 59 1986 240.� �10 H. Ge, X.J. Cia, J.F. Tyson, P.C. Uden, E.R. Denoyer, E.
Ž .Block, Anal. Commun. 33 1996 279.� �11 S.P. Mendez, E.B. Gonzalez, M.L.F. Sanchez, A.S.
Ž .Medel, J. Anal. At. Spectrom. 13 1998 893.� �12 R.J. Kraus, S.J. Foster, H.E. Ganther, Anal. Biochem.
Ž .147 1985 432.� �13 M. Albert, C. Demesmay, J.L. Rocca, Fresenius J. Anal.
Ž .Chem. 351 1995 426.� � Ž .14 Y. Shibata, M. Morita, K. Fuwa, Adv. Biophys. 28 1992
31.� �15 J. Edmonds, M. Morita, Appl. Organomet. Chem. 14
Ž .2000 133.� � Ž .16 K. Pyrzynska, Anal. Sci. 14 1998 479.� � Ž .17 A. Chatterjee, K.J. Irgolic, Anal. Commun. 35 1998
Ž .337 and references cited therein .� �18 G. Kolbl, K. Kalcher, K.J. Irgolic, R. Magee, Appl.
Ž .Organomet. Chem. 7 1993 443.� �19 M.M. Gomez, T. Gasparic, M.A. Palacios, C. Camara,
Ž .Anal. Chim. Acta. 374 1998 241.� �20 J.M.M. Gayon, J.M. Gonzalez, M.L. Fernandez, E.
Blanco, A. Sanz-Medel, Fresenius J. Anal. Chem. 355Ž .1996 615.
� �21 B. Gammelgaard, P. Jons, J. Anal. At. Spectrom. 15Ž .2000 945.
� �22 K. Tirez, W. Brusten, S.V. Roy, N.D. Brucker, L. Diels,Ž .J. Anal. At. Spectrom. 15 2000 1087.
� �23 M.V. Pelaez, M.M. Bayon, J.I.G. Alonso, A. Sanz-Medel,Ž .J. Anal. At. Spectrom. 15 2000 1217.
� �24 Y. He, J. Moreda-Pineiro, M.L. Cervera, M.J. DelaŽ . ŽGuardia, J. Anal. At. Spectrom. 13 1998 289 and
.references cited therein .� �25 A. Chatterjee, Y. Shibata, M. Morita, J. Anal. At. Spec-
Ž .trom. 15 2000 913.� �26 N. Ellend, C. Rohrer, M. Grasserbauer, J.A.C.
Ž .Broekaert, Fresenius J. Anal. Chem. 356 1996 99.� �27 M. Vilano, A. Padro, R. Rubio, G. Rauret, J. Chro-
Ž .matogr. A 819 1998 211.� � Ž .28 A.J. Aller, L.C. Robles, Analyst 123 1998 919.� � Ž .29 N. Gilon, M. Potin-Gautier, J. Chromatogr. A 732 1996
369.� � Ž .30 T. Lei, W.D. Marshall, Appl. Organomet. Chem. 9 1995
149.� �31 G.A. Pederson, E.H. Larsen, Fresenius J Anal. Chem.
Ž .358 1997 591.� � Ž . Ž32 A.G. Howard, J. Anal. At. Spectrom. 12 1997 267 and
.references cited therein .


















