
Deactivation of STAT6 through Serine 707 Phosphorylation by JNK ...
9
Deactivation of STAT6 through Serine 707 Phosphorylation by JNK * □ S Received for publication, July 26, 2010, and in revised form, November 11, 2010 Published, JBC Papers in Press, December 1, 2010, DOI 10.1074/jbc.M110.168435 Takashi Shirakawa ‡ , Yoshinori Kawazoe § , Tomoko Tsujikawa § , Dongju Jung ‡ , Shin-ichi Sato ‡ , and Motonari Uesugi ‡§1 From the ‡ Institute for Integrated Cell-Material Sciences, Kyoto University, Kyoto 611-0011, Japan and the § Institute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan Signal transducer and activator of transcription 6 (STAT6), which plays a critical role in immune responses, is activated by interleukin-4 (IL-4). Activity of STAT family members is regu- lated primarily by tyrosine phosphorylations and possibly also by serine phosphorylations. Here, we report a previously unde- scribed serine phosphorylation of STAT6, which is activated by cell stress or by the pro-inflammatory cytokine, interleu- kin-1 (IL-1). Our analyses suggest that Ser-707 is phos- phorylated by c-Jun N-terminal kinase (JNK). Phosphorylation decreases the DNA binding ability of IL-4-stimulated STAT6, thereby inhibiting the transcription of STAT6-responsive genes. Inactivation of STAT6 by JNK-dependent Ser-707 phos- phorylation may be one mechanism of controlling the balance between IL-1 and IL-4 signals. STAT proteins are transcription factors that are activated by a variety of cytokines and growth factors. Seven mamma- lian STAT proteins have been identified, which contain a con- served structure composed of SH2, DNA binding, and trans- activation domains. Extracellular binding of cytokines or growth factors to the receptors of these STAT proteins in- duces the activation of intracellular Janus kinases (JAK), which phosphorylate STAT proteins on a specific tyrosine residue. The phosphorylated STAT proteins promote forma- tion of homo- or heterodimers from the phosphorylated tyro- sine residue and its partner SH2 domains. The dimers are transported into the nucleus, where they induce transcription of the target genes (1– 4). One of the seven STAT proteins, STAT6, was originally cloned as an IL-4-activated transcrip- tion factor (5). Studies of STAT6-deficient mice showed that STAT6 plays key roles in the differentiation of TH2 cells, the switching of B-cell immunoglobulin isotype to IgE, and the induction of allergic disease (6 –9). The mitogen-activated protein kinases (MAPK) include three families of serine/threonine-protein kinases: extracellu- lar signal-regulated kinase (ERK), JNK, and p38. JNK and p38 are activated by environmental stress and pro-inflammatory cytokines. JNK, also known as stress-activated protein kinase (SAPK), is activated by IL-1, tumor necrosis factor (TNF), UV radiation, osmotic stress, anisomycin, and other stress factors (10 –14). JNK activation leads to Ser/Thr phosphorylation of several transcription factors and other cellular substrates that are implicated in cell survival, insulin receptor signaling, and mRNA stabilization (15–19). In addition to the tyrosine phosphorylations, those of ser- ine residues are also important for regulation of STAT activi- ties. The serine residue in a conserved Pro-X-Ser-Pro se- quence at the COOH termini of STAT1, STAT3, STAT4, STAT5a, and STAT5b is phosphorylated in response to cyto- kines and growth factors (20 –23). Serine phosphorylation of STAT6 has also been demonstrated. Following IL-4 stimula- tion, Ser-756 in the transactivation domain of STAT6 is con- currently phosphorylated with Tyr-641, which is essential for the activation of STAT6 (24). However, the biological signifi- cance of this phosphorylation remains unclear. Phosphoryla- tion of other multiple serine residues at unspecified locations in the transactivation domain of STAT6 has also been de- tected, following treatment of cells with protein phosphatase 2A (PP2A) 2 inhibitors, such as calyculin A (25, 26). These ser- ine phosphorylations led to reduced transcriptional activity of STAT6. Despite the previous findings, the biological roles of serine phosphorylations in STAT6 and the specific kinases involved are not clear. Results of the present study show that Ser-707 in the transactivation domain of STAT6 is directly phosphorylated by JNK in response to stress treatments or IL-1 stimulation. Ser-707 phosphorylation appears to play a role in a crosstalk between the intracellular signals of IL-4 and IL-1 by negatively regulating IL-4-induced transcriptional activation of STAT6. EXPERIMENTAL PROCEDURES Cell Culture and Transfection—Human HeLa cells and HEK293 cells were maintained in DMEM supplemented with 10% (v/v) fetal bovine serum and penicillin/streptomycin (50 units/ml and 50 g/ml, respectively) at 37 °C, under humidi- fied 5% CO 2 . For transient transfection studies, the cells were transfected with an expression plasmid of STAT6 or its mu- tants, using Lipofectamine 2000 (Invitrogen). All analyses were performed 24 h after transfection. * This work was supported in part by grants from JSPS (21310140, to M. U., and 20611007, to Y. K.), the Uehara Memorial Foundation (to M. U.), and the Naito Foundation (to S. S.). □ S The on-line version of this article (available at http://www.jbc.org) con- tains supplemental Figs. S1–S4. 1 To whom correspondence should be addressed: Institute for Chemical Research, Kyoto University, Uji, Kyoto, 611-0011, Japan. Tel.: 81-774-38- 3225; Fax: 81-774-38-3226; E-mail: [email protected]. 2 The abbreviations used are: PP2A, protein phosphatase 2A; DSS, disuccin- imidyl suberate. THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 286, NO. 5, pp. 4003–4010, February 4, 2011 © 2011 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A. FEBRUARY 4, 2011 • VOLUME 286 • NUMBER 5 JOURNAL OF BIOLOGICAL CHEMISTRY 4003 by guest on April 16, 2018 http://www.jbc.org/ Downloaded from
Transcript of Deactivation of STAT6 through Serine 707 Phosphorylation by JNK ...

Deactivation of STAT6 through Serine 707 Phosphorylationby JNK*□S
Received for publication, July 26, 2010, and in revised form, November 11, 2010 Published, JBC Papers in Press, December 1, 2010, DOI 10.1074/jbc.M110.168435
Takashi Shirakawa‡, Yoshinori Kawazoe§, Tomoko Tsujikawa§, Dongju Jung‡, Shin-ichi Sato‡,and Motonari Uesugi‡§1
From the ‡Institute for Integrated Cell-Material Sciences, Kyoto University, Kyoto 611-0011, Japan and the §Institute forChemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan
Signal transducer and activator of transcription 6 (STAT6),which plays a critical role in immune responses, is activated byinterleukin-4 (IL-4). Activity of STAT family members is regu-lated primarily by tyrosine phosphorylations and possibly alsoby serine phosphorylations. Here, we report a previously unde-scribed serine phosphorylation of STAT6, which is activatedby cell stress or by the pro-inflammatory cytokine, interleu-kin-1� (IL-1�). Our analyses suggest that Ser-707 is phos-phorylated by c-Jun N-terminal kinase (JNK). Phosphorylationdecreases the DNA binding ability of IL-4-stimulated STAT6,thereby inhibiting the transcription of STAT6-responsivegenes. Inactivation of STAT6 by JNK-dependent Ser-707 phos-phorylation may be one mechanism of controlling the balancebetween IL-1� and IL-4 signals.
STAT proteins are transcription factors that are activatedby a variety of cytokines and growth factors. Seven mamma-lian STAT proteins have been identified, which contain a con-served structure composed of SH2, DNA binding, and trans-activation domains. Extracellular binding of cytokines orgrowth factors to the receptors of these STAT proteins in-duces the activation of intracellular Janus kinases (JAK),which phosphorylate STAT proteins on a specific tyrosineresidue. The phosphorylated STAT proteins promote forma-tion of homo- or heterodimers from the phosphorylated tyro-sine residue and its partner SH2 domains. The dimers aretransported into the nucleus, where they induce transcriptionof the target genes (1–4). One of the seven STAT proteins,STAT6, was originally cloned as an IL-4-activated transcrip-tion factor (5). Studies of STAT6-deficient mice showed thatSTAT6 plays key roles in the differentiation of TH2 cells, theswitching of B-cell immunoglobulin isotype to IgE, and theinduction of allergic disease (6–9).The mitogen-activated protein kinases (MAPK) include
three families of serine/threonine-protein kinases: extracellu-lar signal-regulated kinase (ERK), JNK, and p38. JNK and p38are activated by environmental stress and pro-inflammatory
cytokines. JNK, also known as stress-activated protein kinase(SAPK), is activated by IL-1, tumor necrosis factor (TNF), UVradiation, osmotic stress, anisomycin, and other stress factors(10–14). JNK activation leads to Ser/Thr phosphorylation ofseveral transcription factors and other cellular substrates thatare implicated in cell survival, insulin receptor signaling, andmRNA stabilization (15–19).In addition to the tyrosine phosphorylations, those of ser-
ine residues are also important for regulation of STAT activi-ties. The serine residue in a conserved Pro-X-Ser-Pro se-quence at the COOH termini of STAT1, STAT3, STAT4,STAT5a, and STAT5b is phosphorylated in response to cyto-kines and growth factors (20–23). Serine phosphorylation ofSTAT6 has also been demonstrated. Following IL-4 stimula-tion, Ser-756 in the transactivation domain of STAT6 is con-currently phosphorylated with Tyr-641, which is essential forthe activation of STAT6 (24). However, the biological signifi-cance of this phosphorylation remains unclear. Phosphoryla-tion of other multiple serine residues at unspecified locationsin the transactivation domain of STAT6 has also been de-tected, following treatment of cells with protein phosphatase2A (PP2A)2 inhibitors, such as calyculin A (25, 26). These ser-ine phosphorylations led to reduced transcriptional activity ofSTAT6. Despite the previous findings, the biological roles ofserine phosphorylations in STAT6 and the specific kinasesinvolved are not clear. Results of the present study show thatSer-707 in the transactivation domain of STAT6 is directlyphosphorylated by JNK in response to stress treatments orIL-1� stimulation. Ser-707 phosphorylation appears to play arole in a crosstalk between the intracellular signals of IL-4 andIL-1� by negatively regulating IL-4-induced transcriptionalactivation of STAT6.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection—Human HeLa cells andHEK293 cells were maintained in DMEM supplemented with10% (v/v) fetal bovine serum and penicillin/streptomycin (50units/ml and 50 �g/ml, respectively) at 37 °C, under humidi-fied 5% CO2. For transient transfection studies, the cells weretransfected with an expression plasmid of STAT6 or its mu-tants, using Lipofectamine 2000 (Invitrogen). All analyseswere performed 24 h after transfection.
* This work was supported in part by grants from JSPS (21310140, to M. U.,and 20611007, to Y. K.), the Uehara Memorial Foundation (to M. U.), andthe Naito Foundation (to S. S.).
□S The on-line version of this article (available at http://www.jbc.org) con-tains supplemental Figs. S1–S4.
1 To whom correspondence should be addressed: Institute for ChemicalResearch, Kyoto University, Uji, Kyoto, 611-0011, Japan. Tel.: 81-774-38-3225; Fax: 81-774-38-3226; E-mail: [email protected].
2 The abbreviations used are: PP2A, protein phosphatase 2A; DSS, disuccin-imidyl suberate.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 286, NO. 5, pp. 4003–4010, February 4, 2011© 2011 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
FEBRUARY 4, 2011 • VOLUME 286 • NUMBER 5 JOURNAL OF BIOLOGICAL CHEMISTRY 4003
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Antibodies and Reagents—The following primary antibod-ies were used: anti-pY-STAT6, anti-JNK, anti-ERK, and anti-AKT (Cell Signaling Technology); anti-STAT6, anti-actin,and anti-PARP (Santa Cruz Biotechnology); anti-Flag (Sigma-Aldrich); and anti-Myc (Novus Biologicals). Recombinanthuman IL-4, IFN-�, and IL-1� (PeproTech) were used at finalconcentrations of 10 ng/ml. Anisomycin, MG-132, SB202190,and SP600125 were obtained from Sigma-Aldrich. Nocoda-zole, taxol, colchicine, and calyculin A were obtained fromWako Chemicals.Plasmid Constructs—The cDNA of full-length STAT6 was
cloned into pCMV-3Tag-1A (Flag-tagged) and pCMV-3Tag-9(Myc-tagged) (Stratagene). For overexpression experiments,five alanine substitution mutants of STAT6 (T168A, S583A,T658A, S707A, and S756A) were obtained by overlap PCRfrom STAT6 cDNA.In Vitro Phosphatase Assay—Whole cell lysates (50 �g)
from anisomycin-treated HeLa cells were prepared in a phos-phatase reaction buffer (50 mM Tris-HCl, pH 9.0, 1 mM
MgCl2) and incubated with alkaline phosphatase (CIAP: 10units) for 30 min at 30 °C in the presence or absence of phos-phatase inhibitors. The reactions were terminated by addingan SDS sample buffer, separated by SDS-PAGE, and analyzedby Western blotting.In Vitro Kinase Assay—cDNA encoding, human
STAT6621–847 or ATF21–109 was inserted into pET41a expres-sion vector (Novagen). Their GST fusion proteins were in-duced in BL21 cells of Escherichia coli by adding 0.4 mM iso-propyl-�-d-thiogalactopyranoside, and were purified byglutathione-Sepharose 4B (Amersham Biosciences). Five micro-grams of GST-STAT6621–847 or GST-ATF21–109 were incubatedwith 100 ng of JNK1 (Carna Biosciences, Inc.) or p38� (UpstateBiotechnology) for 30min at 30 °C in 30 �l of kinase buffer (20mMTris-HCl, pH 8.0, 10 mMMgCl2, 2 mMDTT, 5mMNaF, 0.2mMNa3VO4, 3 �Ci [�-32P]ATP). The kinase reactions were ter-minated by adding an SDS sample buffer, separated by SDS-PAGE, and visualized by autoradiography.Electrophoretic Gel Mobility-Shift Assay—For electro-
phoretic gel mobility-shift assays, HeLa or HEK293 cells werelysed in buffer C (20 mM HEPES, 25% glycerol, 0.42 M NaCl,1.5 mM MgCl2, 0.2 mM EDTA). Twenty micrograms of the celllysates were incubated with 200 ng of poly-dI-dC (Sigma-Al-drich) and 32P-labeled N6-GAS oligonucleotide (5�-GATCGCTCTTCTTCCCAGGAACTCAATG) (5) for 30 minon ice in 15 �l of a reaction buffer (20 mM Tris-HCl, 1 M
NaCl, 0.1 M EDTA, 0.1 M DTT, 37.6% glycerol, 1.5% NonidetP-40, 5 mg/ml BSA). The samples were separated by 4% (w/v)Tris borate EDTA (TBE)-PAGE and visualized byautoradiography.Cross-linking Experiments—HeLa cells grown on 6-well
plates were washed twice with phosphate-buffered saline (150mM NaCl, 10 mM sodium phosphate, pH 7.4) and collectedinto a lysis buffer (phosphate-buffered saline containing 1%Triton X-100). The cell lysates were incubated with or with-out disuccinimidyl suberate (DSS, 0.5 mM) for 30 min on ice.The reaction was stopped by adding 4 mM glycine. The cross-linked products were separated by SDS-PAGE and analyzedby Western blotting.
Immunoprecipitation of STAT6 Homodimers—HEK293cells were transiently transfected with expression vectors ofFlag-tagged and Myc-tagged STAT6. The transfected cellswere treated with 1% (v/v) DMSO or 500 ng/ml anisomycinfor 1 h, then stimulated with 10 ng/ml IL-4 for 30 min. Thecells were lysed in buffer C and centrifuged at 65,000 rpm for10 min at 4 °C. The supernatant was incubated with anti-c-Myc agarose beads (Sigma) at 4 °C for 2 h. Bound fractionswere eluted with an SDS sample buffer, separated by SDS-PAGE, and analyzed by Western blotting with an anti-Flagantibody.Nuclear and Cytoplasmic Extracts—HeLa cells cultured on
100 mm dishes were transferred into 1 ml of ice-cold phos-phate-buffered saline. The cells were centrifuged at 1500 rpmfor 5 min and lysed in 150 �l of low salt buffer (10 mM
HEPES, 10 mM KCl, 1.5 mM MgCl2, and 0.5 mM DTT) on ice.After a 20-min incubation, the cell suspension was homoge-nized by passage through a 27-gauge needle. The supernatantwas collected as a cytoplasmic extract after centrifugation at4000 rpm for 10 min at 4 °C. The nuclear pellet was resus-pended in buffer C and centrifuged at 14,000 rpm for 10 minat 4 °C. The supernatant was saved as a nuclear extract.Reverse Transcription PCR—Total cellular RNA was ex-
tracted with QIAshredder (Qiagen) and further isolated withan RNeasy Mini Kit (Qiagen). First-strand cDNAs were syn-thesized using Superscript II (Invitrogen) and amplified usingthe following primers: 5�-GGAACTGCCACACGTGGGAG-TGAC and 5�-CTCTGGGAGGAAACACCCTCTCC forEotaxin-3 (CCL26); 5�-CACGCACTTCCGCACATTCC and5�-TCCAGCAGCTCGAAGAGGCA for SOCS-1; 5�-CTCA-AGACCTTCAGCTCCAA and 5�-TTCTCATAGGAGTCC-AGGTG-3� for SOCS-3; 5�-GACCACAGTCCATGCCAT-CACT and 5�-TCCACCACCCTGTTGCTGTAG forGAPDH.
RESULTS
Cell Stress Induces Phosphorylation of STAT6 in HeLa Cells—During the course of our investigation, we discovered a mo-bility shift of STAT6 in Western blot analyses of HeLa cellstreated with a range of bioactive small molecules. Amongthirteen molecules with distinct pharmacological effects, ani-somycin (a protein synthesis inhibitor), nocodazole and chol-chitin (microtubule inhibitors), taxol (a microtubule stabi-lizer), and MG-132 (a proteasome inhibitor) exhibited clearband shifts or doublet formations of STAT6 bands on an SDSgel (Fig. 1A). In particular, 500 ng/ml of anisomycin induced acomplete band shift of STAT6. The pharmacological diversityof these five molecules suggested that the mobility shift mightbe induced by cell stress. Indeed, 0.5 mM NaCl, an osmoticstress inducer, produced a band shift similar to that producedby anisomycin.The band shift implied post-translational modification of
STAT6 upon cell stress. A number of post-translational modi-fications are known to exhibit such band shifts and inducebiologically significant outputs, including phosphorylation,acetylation, glycosylation, and ubiqutination (27–30). We hy-pothesized that the stress-induced post-translational modifi-cation plays a role in the regulation of STAT6. Our further
Serine Phosphorylation of STAT6
4004 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 • NUMBER 5 • FEBRUARY 4, 2011
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from
studies used anisomycin as an inducer of the modification ofSTAT6, because anisomycin induced a most prominent bandshift on an SDS gel (Fig. 1A).Phosphorylation is the most general post-translational
modification that often induces an upper band shift. There-fore, we performed in vitro phosphatase assays, in whichwhole cell lysates from anisomycin-treated HeLa cells weretreated with CIAP, a universal protein phosphatase. Thephosphatase treatment converted the slower-migrating bandback to the faster-migrating band. In contrast, co-treatmentof cells with CIAP and a phosphatase inhibitor (Na2PO4 orNa3VO4) restored the slower-migrating band (Fig. 1B). Thus,the band shift caused by anisomycin did appear to be due toprotein phosphorylation.JNK Directly Phosphorylates Ser-707 of STAT6—Cell stress
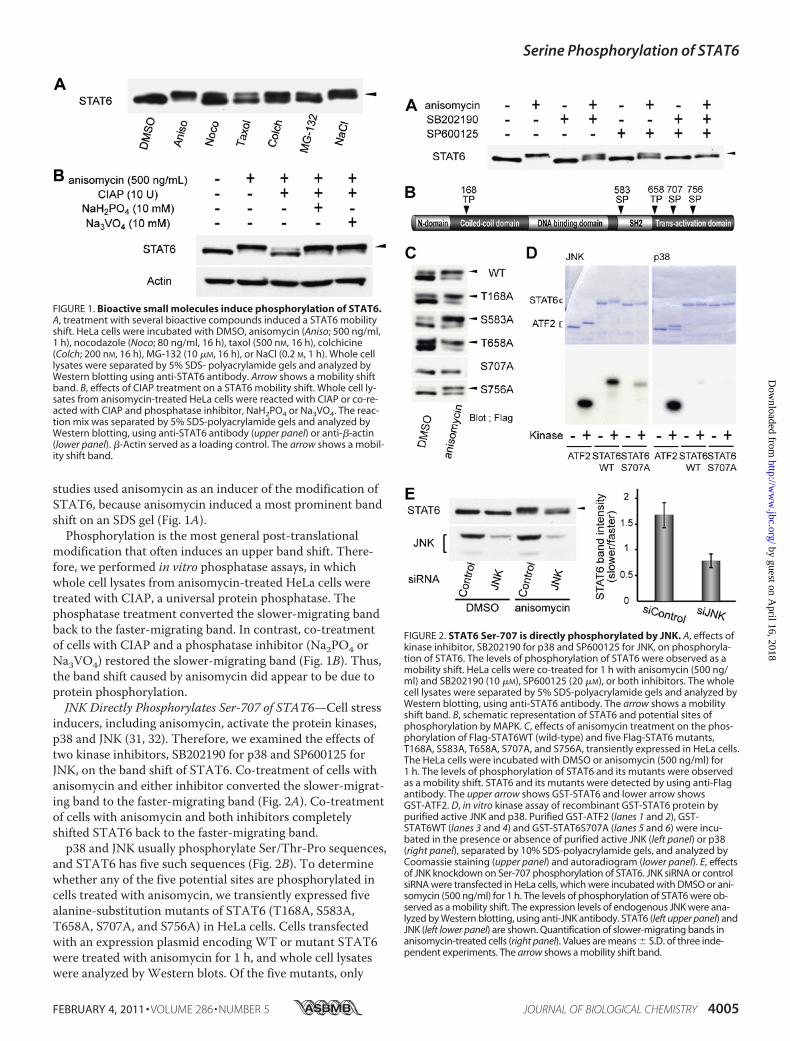
inducers, including anisomycin, activate the protein kinases,p38 and JNK (31, 32). Therefore, we examined the effects oftwo kinase inhibitors, SB202190 for p38 and SP600125 forJNK, on the band shift of STAT6. Co-treatment of cells withanisomycin and either inhibitor converted the slower-migrat-ing band to the faster-migrating band (Fig. 2A). Co-treatmentof cells with anisomycin and both inhibitors completelyshifted STAT6 back to the faster-migrating band.p38 and JNK usually phosphorylate Ser/Thr-Pro sequences,
and STAT6 has five such sequences (Fig. 2B). To determinewhether any of the five potential sites are phosphorylated incells treated with anisomycin, we transiently expressed fivealanine-substitution mutants of STAT6 (T168A, S583A,T658A, S707A, and S756A) in HeLa cells. Cells transfectedwith an expression plasmid encoding WT or mutant STAT6were treated with anisomycin for 1 h, and whole cell lysateswere analyzed by Western blots. Of the five mutants, only
FIGURE 1. Bioactive small molecules induce phosphorylation of STAT6.A, treatment with several bioactive compounds induced a STAT6 mobilityshift. HeLa cells were incubated with DMSO, anisomycin (Aniso; 500 ng/ml,1 h), nocodazole (Noco; 80 ng/ml, 16 h), taxol (500 nM, 16 h), colchicine(Colch; 200 nM, 16 h), MG-132 (10 �M, 16 h), or NaCl (0.2 M, 1 h). Whole celllysates were separated by 5% SDS- polyacrylamide gels and analyzed byWestern blotting using anti-STAT6 antibody. Arrow shows a mobility shiftband. B, effects of CIAP treatment on a STAT6 mobility shift. Whole cell ly-sates from anisomycin-treated HeLa cells were reacted with CIAP or co-re-acted with CIAP and phosphatase inhibitor, NaH2PO4 or Na3VO4. The reac-tion mix was separated by 5% SDS-polyacrylamide gels and analyzed byWestern blotting, using anti-STAT6 antibody (upper panel) or anti-�-actin(lower panel). �-Actin served as a loading control. The arrow shows a mobil-ity shift band.
FIGURE 2. STAT6 Ser-707 is directly phosphorylated by JNK. A, effects ofkinase inhibitor, SB202190 for p38 and SP600125 for JNK, on phosphoryla-tion of STAT6. The levels of phosphorylation of STAT6 were observed as amobility shift. HeLa cells were co-treated for 1 h with anisomycin (500 ng/ml) and SB202190 (10 �M), SP600125 (20 �M), or both inhibitors. The wholecell lysates were separated by 5% SDS-polyacrylamide gels and analyzed byWestern blotting, using anti-STAT6 antibody. The arrow shows a mobilityshift band. B, schematic representation of STAT6 and potential sites ofphosphorylation by MAPK. C, effects of anisomycin treatment on the phos-phorylation of Flag-STAT6WT (wild-type) and five Flag-STAT6 mutants,T168A, S583A, T658A, S707A, and S756A, transiently expressed in HeLa cells.The HeLa cells were incubated with DMSO or anisomycin (500 ng/ml) for1 h. The levels of phosphorylation of STAT6 and its mutants were observedas a mobility shift. STAT6 and its mutants were detected by using anti-Flagantibody. The upper arrow shows GST-STAT6 and lower arrow showsGST-ATF2. D, in vitro kinase assay of recombinant GST-STAT6 protein bypurified active JNK and p38. Purified GST-ATF2 (lanes 1 and 2), GST-STAT6WT (lanes 3 and 4) and GST-STAT6S707A (lanes 5 and 6) were incu-bated in the presence or absence of purified active JNK (left panel) or p38(right panel), separated by 10% SDS-polyacrylamide gels, and analyzed byCoomassie staining (upper panel) and autoradiogram (lower panel). E, effectsof JNK knockdown on Ser-707 phosphorylation of STAT6. JNK siRNA or controlsiRNA were transfected in HeLa cells, which were incubated with DMSO or ani-somycin (500 ng/ml) for 1 h. The levels of phosphorylation of STAT6 were ob-served as a mobility shift. The expression levels of endogenous JNK were ana-lyzed by Western blotting, using anti-JNK antibody. STAT6 (left upper panel) andJNK (left lower panel) are shown. Quantification of slower-migrating bands inanisomycin-treated cells (right panel). Values are means � S.D. of three inde-pendent experiments. The arrow shows a mobility shift band.
Serine Phosphorylation of STAT6
FEBRUARY 4, 2011 • VOLUME 286 • NUMBER 5 JOURNAL OF BIOLOGICAL CHEMISTRY 4005
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

S707A failed to exhibit a mobility shift in the presence of ani-somycin (Fig. 2C). These results suggested that p38 and/orJNK phosphorylate STAT6 at Ser-707.To test this hypothesis, we performed an in vitro kinase
assay using purified recombinant proteins. Kinase activity ofp38 and JNK was verified by phosphorylation of a GST-taggedNH2-terminal fragment of ATF2 (amino acids 1–109), aknown substrate of p38 and JNK (33). Ser-707 of STAT6 islocated in the transactivation domain (amino acids 621–847),so we prepared a GST-tagged transactivation domain ofSTAT6 (GST-STAT6) and determined whether or not thisfusion protein is phosphorylated by p38 or JNK. RecombinantJNK did phosphorylate GST-STAT6 and shifted its band onan SDS gel; recombinant p38 failed to do so (Fig. 2D). We alsoprepared a Ser-707-mutated version of GST-STAT6 and usedit as a substrate. JNK showed little, if any, phosphorylation ofthis Ser-707 mutant and failed to induce a band shift. Theseresults indicated that Ser-707 in STAT6 is directly phos-phorylated by JNK, and that the phosphorylation of Ser-707induces a mobility shift of STAT6 on an SDS gel.We further examined the phosphorylation of STAT6 in-
duced by anisomycin, using siRNA knockdown of JNK. Tran-sient knockdown of JNK converted the slower migrating bandto the faster migrating band, and the percentages of theslower migrating band decreased, while the faster-migratingband increased (Fig. 2E). These results confirmed that STAT6is directly phosphorylated at Ser-707 by JNK in cells treatedwith anisomycin.JNK Specifically Phosphorylates Ser-707 of STAT6—To con-
firm that JNK is a specific kinase for the phosphorylation ofSer-707, we performed two series of experiments. First, ERK,another Ser/Thr MAP kinase, and AKT, a major Ser/Thr pro-tein kinase, were tested for their ability to phosphorylateSTAT6 in vitro, in addition to p38 and JNK. Only JNK exhib-ited strong phosphorylation (Fig. 3A). Second, each of thefour Ser/Thr protein kinases was transiently knocked down bysiRNA. Although all of the siRNAs reduced the expression ofcorresponding kinases at comparable levels, only the siRNA ofJNK blocked the STAT6 band-shift induced by anisomycin(Fig. 3B). These results indicated that Ser-707 of STAT6 isspecifically phosphorylated by JNK.Ser-707 Phosphorylation of STAT6 Negatively Regulates Its
Transcriptional Activity—Small molecule inhibitors of PP2A,a Ser/Thr-protein phosphatase, are known to induce multipleserine phosphorylations in the transactivation domain ofSTAT6, which repress its transcriptional activity (20, 24). Ser-ine phosphorylations in the transactivation domains ofSTAT1 and STAT3 also regulate their transcriptional activi-ties. Therefore, we hypothesized that Ser-707 phosphoryla-tion of STAT6 similarly regulates the transcriptional activityof STAT6. To test this hypothesis, we compared expressionlevels of CCL26, a representative STAT6-responsive gene(34), in anisomycin-treated and untreated HeLa cells. Cellswere exposed to IL-4 after incubation with anisomycin orDMSO control for 1 h. RT-PCR analysis of extracted mRNAsamples revealed a decrease of CCL26 expression in anisomy-cin-treated cells compared with control cells (Fig. 4A). Similarrepression of CCL26 was observed in nocodazole-treated cells
(supplemental Fig. S1). Expression levels of CCL26 did notdecrease in HeLa cells co-treated with anisomycin andSP600125, a well-known JNK inhibitor (Fig. 4B).We next tested the hypothesis that overexpression of the
S707A mutant of STAT6 would maintain transcriptional ac-tivity in anisomycin-treated cells. HEK293 cells, which do notexpress endogenous functional STAT6, were used in this ex-periment. Anisomycin had no detectable effects on the ex-pression level of CCL26 in the S707A-overexpressed cells (Fig.4C), further confirming that Ser-707 phosphorylation by JNKinhibits IL-4-induced gene activation by STAT6.Ser-707 Phosphorylation Decreases the DNA Binding Activ-
ity of STAT6—To determine how phosphorylation of Ser-707down-regulates STAT6, we first examined the effects of ani-somycin on Tyr-641 phosphorylation induced by IL-4. HeLacells were incubated with anisomycin for 1 h, then stimulatedwith IL-4 for 30 min. Whole cell lysates were subsequentlyanalyzed by Western blotting, using an anti-pY STAT6 anti-body. Anisomycin had no effects on the amount of IL-4-in-duced tyrosine-phosphorylated STAT6, and the bands of Tyr-641-phosphorylated STAT6 were shifted by anisomycintreatment (Fig. 5A).To determine whether Ser-707 phosphorylation induced by
anisomycin inhibits IL-4-induced dimerization of STAT6,
FIGURE 3. Specificity of kinases for Ser-707 phosphorylation of STAT6.A, purified GST-STAT6WT was incubated in the presence or absence of puri-fied active JNK (lanes 1 and 2), p38 (lanes 3 and 4), ERK (lanes 5 and 6), or AKT(lanes 7 and 8). Products were separated on a 10% SDS-polyacrylamide gel,and analyzed by Coomassie staining (upper panel) or autoradiogram (lowerpanel). B, effects of JNK, p38, ERK, or AKT knockdown on Ser-707 phosphor-ylation of STAT6. siRNAs were transfected into HeLa cells, which were incu-bated with DMSO (control) or anisomycin (500 ng/ml) for 45 min. The levelsof Ser-707 phosphorylation of STAT6 were observed as a mobility shift inWestern blots. The arrow indicates a shifted band.
Serine Phosphorylation of STAT6
4006 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 • NUMBER 5 • FEBRUARY 4, 2011
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

HeLa cells were treated with anisomycin, and cell lysates werereacted with a cross-linking agent, so that endogenous STAT6dimers were covalently crosslinked. Western blot analysisusing anti-pY STAT6 antibody showed that treatment withanisomycin did not affect the band whose molecular weightmatched that of the STAT6 homodimer (Fig. 5B).We also performed co-immunoprecipitation experiments
for the STAT6 homodimer. HEK293 cells were co-transfectedwith two expression plasmids encoding Flag-tagged STAT6and Myc-tagged STAT6. The transfected cells were treatedwith anisomycin for 1 h, then stimulated with IL-4 for 30 min.Myc-tagged STAT6 in the lysates was immunoprecipitatedwith anti-Myc agarose beads, and the co-immunoprecipitatedFlag-tagged STAT6 was analyzed by Western blotting with ananti-Flag antibody. Anisomycin treatment had no detectableimpact on the amount of Flag-tagged STAT6 co-immunopre-cipitated with Myc-tagged STAT6 (Fig. 5C), consistent withthe cross-linking experiments. Therefore, we concluded thatSer-707 phosphorylation of STAT6 has no significant effecton its IL4-induced dimerization.We next checked the nuclear translocation of STAT6.
HeLa cells were incubated with anisomycin for 1 h, and then
stimulated with IL-4 for 30 min. The cell lysates were frac-tionated into nuclear and cytoplasmic components. Eachcomponent was subsequently analyzed by Western blotting,using anti-STAT6 and anti-pY STAT6 antibodies. Anisomy-cin treatment had no detectable effect on the levels of STAT6and pY-STAT6 in nuclear extracts, indicating that Ser-707phosphorylation does not affect the IL4-induced nucleartranslocation of STAT6 (Fig. 5D).Finally, electrophoretic mobility shift assays (EMSA) were
performed to analyze the DNA-binding activity of STAT6.HeLa cells were incubated with anisomycin for 1 h, then stim-ulated with IL-4 for 30 min, and whole cell lysates were pre-pared. The DNA binding affinity of endogenous STAT6 wasanalyzed using N6-GAS, an oligonucletide containing a highaffinity binding site for STAT6, as a probe (5, 35). The DNAbinding activity of STAT6 decreased in anisomycin-treatedcells compared with control (DMSO-treated) cells (Fig. 5E),and restored by addition of SP600125, a JNK inhibitor (sup-plemental Fig. S2A). The decrease of the DNA binding activ-ity of STAT6 was also observed in nocodazole-treated cells(supplemental Fig. S2B).Similar experiments were conducted with HEK293 cells,
which do not express functional endogenous STAT6. HEK293cells were transfected with an expression vector encodingwild-type STAT6 or its S707A mutant. The DNA binding af-finity of wild-type STAT6 decreased upon anisomycin treat-ment, while the binding affinity of the S707A mutant was notaffected (Fig. 5F). Overall, these results suggest that Ser-707phosphorylation impairs the DNA binding activity of IL4-activated STAT6, resulting in the suppression of STAT6-re-sponsive genes.IL-1� Stimulation Induces Ser-707 Phosphorylation and
Suppresses Transcriptional Activity of STAT6—Ser-707 ofSTAT6 is directly phosphorylated by JNK, which is activatedby a number of endogenous and exogenous factors, includingcytokines, ultraviolet irradiation, heat shock, and osmoticshock. Based on the relevance of STAT6 to cytokine signaling,the effects of JNK-activating cytokines on Ser-707 phosphor-ylation of STAT6 were examined. We first tested the ability ofIL-1�, a typical pro-inflammatory cytokine that leads to acti-vation of JNK (36, 37), to induce the STAT6 band shift.Whole cell lysates from HeLa cells stimulated by IL-1�showed the predicted STAT6 mobility shift (Fig. 6A). How-ever, the S707A alanine-substitution mutant of STAT6 failedto show a mobility shift in response to IL-1� treatment (Fig.6B). These results indicated that IL-1� stimulation inducesSer-707 phosphorylation of STAT6.Because Ser-707 phosphorylation of STAT6 represses its
transcriptional activity, we examined the effects of IL-1� onthe DNA binding affinity of STAT6. Treatment with IL-1�decreased of STAT6-DNA complexes upon IL-4 stimulationby an amount similar to the decrease caused by treatmentwith anisomycin (Fig. 6C). IL-1� stimulation also repressedthe IL-4-induced expression of CCL26, a representativeSTAT6-responsive gene. HeLa cells were stimulated by IL-1�for 1 h, then by IL-4 for 6 h. RT-PCR analyses of extractedmRNA samples showed that the IL-4-induced expression ofCCL26 was reduced by IL-1� to the levels in non-stimulated
FIGURE 4. Repression of IL-4-induced transcriptional activity of STAT6by Ser-707 phosphorylation. A, effects of anisomycin treatment on IL-4-indcued transcriptional activity of STAT6. HeLa cells were incubated withDMSO or anisomycin (Aniso; 500 ng/ml) for 1 h, and stimulated by IL-4 (10ng/ml) for 0 – 8 h. The mRNA levels of CCL26 (upper panel) and GAPDH (lowerpanel) were quantified by RT-PCR. GAPDH served as a loading control.B, effects of JNK inhibitor on IL-4-indcued transcriptional activity of STAT6 inanisomycin-treated cells. HeLa cells were co-incubated with anisomycin(Aniso; 500 ng/ml) and JNK inhibitor, SP600125 (20 �M), for 1 h, then stimu-lated by IL-4 (10 ng/ml) for 6 h. The CCL26 levels were quantified by RT-PCR.C, IL-4-induced transcriptional activity of STAT6 in anisomycin-treated cellswith WT and S707A mutant STAT6. WT or S707A mutant-expressed HEK293cells were incubated with anisomycin for 1 h, then stimulated by IL-4 (10ng/ml) for 6 h. The CCL26 levels were quantified by RT-PCR.
Serine Phosphorylation of STAT6
FEBRUARY 4, 2011 • VOLUME 286 • NUMBER 5 JOURNAL OF BIOLOGICAL CHEMISTRY 4007
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

cells or cells stimulated by interferon-� (IFN-�) (Fig. 7A).IFN-�, a T helper 1 (Th1) cytokine, which plays a critical rolein host immune responses, suppresses IL-4-induced STAT6activation through the induction of expression of suppresserof cytokine signal (SOCS) genes (38, 39). Expression ofSOCS-1 and -3 genes in HeLa cells, measured by RT-PCR,was not induced by IL-1�, but was induced by IFN-� (Fig. 7B).Western blot analysis also showed that IFN-� stimulationfailed to induce Ser-707 phosphorylation of STAT6 in HeLacells (Fig. 7C). The mechanism by which IL-1� deactivatesSTAT6 is clearly distinct from that of IFN-�, and phosphory-lation of Ser-707 is likely to mediate the deactivation ofSTAT6 by IL-1�.
DISCUSSION
Results of the present study demonstrate that JNK phos-phorylates Ser-707 and, thereby, suppresses the IL-4-inducedtranscriptional activation of STAT6. When cells were treatedwith either p38 or JNK inhibitors, the Ser-707 phosphoryla-tion of STAT6 was impaired. However, Ser-707 of STAT6was phosphorylated in vitro only by JNK, and not by p38; and
FIGURE 5. Analysis of repression mechanism for transcriptional activityof Ser-707-phosphorylated STAT6. A, effects of anisomycin treatment ontyrosine phosphorylation levels of STAT6. HeLa cells were incubated withanisomycin (Aniso; 500 ng/ml) for 1 h, then stimulated by IL-4 (10 ng/ml) for30 min. The whole cell lysates were separated by 5% SDS-polyacrylamidegels and analyzed by Western blotting, using anti-pY STAT6 (upper panel),anti-STAT6 (middle panel), or anti-�-actin antibodies (lower panel). �-Actinserved as a loading control. B, effects of anisomycin treatment on dimeriza-tion levels of STAT6. Whole cell lysates prepared as described for A werereacted by cross-linking reagent (DSS, 0.5 mM) for 30 min, then the reactionmixtures were analyzed as described for A using anti-pY STAT6 antibody.The arrow shows a band of STAT6 dimer. C, effects of anisomycin treatmenton dimerization levels of STAT6. HEK293 cells were transiently co-trans-fected with expression vectors of Myc-tagged and Flag-tagged STAT6. After24 h, the cells were incubated with anisomycin (Aniso; 500 ng/ml) for 1 h,and then stimulated by 10 ng/ml IL-4 for 30 min. The cell extracts were im-munoprecipitated with anti-Myc-agarose beads. The immunoprecipitatedsamples and the input sample (1%) were resolved by 5% SDS-PAGE andanalyzed by Western blotting with anti-Flag (upper panel) and anti-Myc
(lower panel) antibodies. D, effects of anisomycin treatment on nucleartranslocation of STAT6. HeLa cells were incubated with anisomycin (Aniso;500 ng/ml) for 1 h, then stimulated by 10 ng/ml IL-4 for 30 min. The cell ly-sates were fractionated into nuclear and cytoplasmic compartments. Eachcompartment was subsequently analyzed by Western blotting, using an-ti-pY STAT6 (upper panel), anti-STAT6 (upper middle panel), anti-PARP (lowermiddle panel), or anti-�-tubulin (lower panel) antibodies. PARP and �-tubulinserved as nuclear and cytoplasmic controls, respectively. E, effects of aniso-mycin treatment on DNA binding affinity of endogenous STAT6. HeLa cellswere incubated with anisomycin (Aniso; 500 ng/ml) for 1 h, then stimulatedby 10 ng/ml IL-4 for 30 min. Twenty micrograms of whole cell lysates, pre-pared using buffer C, were used for EMAS, with N6-GAS as a probe. F, DANbinding activities of STAT6WT and S707A mutant in the presence of aniso-mycin. HEK293 cells expressing WT or S707A mutant of STAT6 were incu-bated with anisomycin (Aniso; 500 ng/ml) for 1 h, and then stimulated by 10ng/ml IL-4 for 30 min. DNA binding activities were analyzed by EMSA asdescribed in E.
FIGURE 6. Ser-707 phosphorylation and down-regulation of STAT6 byIL-1�. A, induction of the STAT6 mobility shift by IL-1� treatment. HeLacells were stimulated by 10 ng/ml IL-1� for 1 h, then the whole cell lysateswere separated by 5% SDS-polyacrylamide gels and analyzed by Westernblotting using anti-STAT6 antibody. B, induction of STAT6 Ser-707 phos-phorylation by IL-1� stimulation. S707A mutant was transiently overex-pressed in HeLa cells, which were stimulated by 10 ng/ml IL-1� for 1 h. Thewhole cell lysates were separated by 5% SDS-polyacrylamide gels and ana-lyzed by Western blotting using anti-Flag antibody. C, effects of IL-1� stimu-lation on DNA binding affinity of STAT6. HeLa cells were stimulated by 10ng/ml IL-1� for 1 h, then stimulated by 10 ng/ml IL-4 for 30 min. Twentymicrograms of whole cell lysates, prepared using buffer C, were used forEMSA, with N6-GAS as a probe.
Serine Phosphorylation of STAT6
4008 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 • NUMBER 5 • FEBRUARY 4, 2011
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

knockdown of p38 had no detectable effects on Ser-707 phos-phorylation. Although the p38 inhibitor, SB202190, has beenreported to have little inhibitory activities against JNK at theconcentration used in the present study, SB202190 did blockthe Ser-707 phosphorylation in cell-based assays. SB202190inhibits cyclin G-associated kinase (GAK) and glycogen syn-thase kinase 3 (GSK3) at levels similar to p38, and inhibitsreceptor-interacting protein 2 (RIP2) even more than p38 (40,41), suggesting that Ser/Thr-protein kinases other than JNKmight be involved in the phosphorylation of Ser-707 ofSTAT6. Indeed, in STAT1 and STAT3, identical serine resi-dues are phosphorylated by a few distinct kinases (21, 22, 42,43). Further investigations are needed to determine if addi-tional kinases are involved in Ser-707 phosphorylation ofSTAT6.Our studies identified a new phosphorylation site, Ser-707,
in STAT6, which controls the transcriptional activity ofSTAT6 upon cell stress or exposure to IL-1�. AlthoughSTAT6 possesses five potential phosphorylation sites thatmatch the consensus substrate sequences of JNK, we detectedphosphorylation only of Ser-707 in the transactivation do-main. Phosphorylation of multiple serine residues at un-known locations in the transactivation domain of STAT6 hasbeen reported in cells treated with calyculin A, a small mole-cule that inhibits PP2A (22). To determine if calyculin Atreatment induces Ser-707 phosphorylation, the S707A mu-tant of STAT6 was transiently expressed in HeLa cells, andthe cells were treated with calyculin A for 45 min. Westernblot analyses revealed that calyculin A treatment induced alarge shift of the S707A mutant band, similar to the band shiftobserved in wild-type STAT6 (supplemental Fig. S3), suggest-ing that the sites of phosphorylation induced by PP2A inhibi-tion are distinct from those induced by Ser-707.The transactivation domain of STAT6 (amino acids 621–
847) is one of a family of proline-rich transactivation domains
in a variety of transcription factors, including B cell-specificactivator protein (44) and erythroid kruppel-like factor (45),which are proposed to recruit various chromatin remodelingproteins and coactivators. The transactivation domain ofSTAT6 has been suggested to interact with a number of coac-tivators, including p100 (46) and NcoA-1 (47). There are nu-merous examples in which phosphorylation of transactivationdomains modulates interactions with coactivators. Therefore,we examined the effects of anisomycin on the protein-proteininteraction between STAT6 and one of its co-activators,NcoA-1. Anisomycin treatment had no detectable impact onthe amount of NcoA-1 co-immunoprecipitated with Flag-tagged STAT6 (supplemental Fig. S4), suggesting that the in-hibition of STAT6 by Ser-707 phosphorylation is not due todecreased interaction with NcoA-1. Indeed, the LXXLL motif,a typical binding site for NcoA-1, is 100 amino acids awayfrom Ser-707.It remains unknown how phosphorylation in the transacti-
vation domain, which is �280 amino acids away from theCOOH terminus of the DNA-binding domain in the primarysequence, reduces the DNA binding activity of STAT6. Re-sults of previous investigations indicated that phosphorylationof various transcription factors modulates their DNA bindingactivities primarily by two mechanisms. First, phosphoryla-tion of residues within or close to the DNA-binding site candirectly affect DNA binding (48–51). Second, phosphoryla-tion can lead to a conformational change, resulting in an indi-rect modulation of DNA-binding activity (52–54). In STAT6,Ser-707 is located away from the DNA-binding domain(amino acids 268–430) in the primary sequence, and there-fore, direct inhibition of DNA binding by the negativelycharged phosphorous group seems unlikely. The secondmechanism, in which phosphorylation of Ser-707 induces aconformational alteration extending to the DNA-binding do-main, appears more likely. The peptide segment adjacent toSer-707 is rich in acidic residues (707Ser-Pro-Glu-Glu-Ser),which upon phosphorylation, might mask the positivelycharged DNA binding interface of STAT6 through intramo-lecular interactions.Inhibition of IL-4 signals by other classes of cytokines, such
as IFN-�, has been well described. This interference is usuallymediated by induction of SOCS genes, which bind to the IL-4receptor or JAK (55). In contrast, our results suggest thatIL-1� interferes with IL-4 signals by phosphorylating Ser-707of STAT6 in HeLa cells without inducing SOCS genes. Ser-707 phosphorylation of STAT6 may be an alternate mecha-nism for deactivating IL-4 signals by other classes ofcytokines.Our results showed that JNK is one of the kinases that
phosphorylate Ser-707 of STAT6. Results of previous studieshave suggested a role of JNK in immune responses. The JNKsignaling pathway has been implicated to play important rolesin the differentiation and activation of T cells (56). Moreover,JNK activation negatively regulates production of Th2 cyto-kines, such as IL-4 (57, 58). Although further studies areneeded for confirmation, the ability of JNK to phosphorylateSer-707 of STAT6 might be involved in its roles in immuneresponses.
FIGURE 7. Ser-707 phosphorylation of STAT6 inhibits overlap betweenIL-1� and IL-4 signals. A, effects of IL-1� and IFN-� signals on IL-4-inducedtranscriptional activity of STAT6. HeLa cells were stimulated with 10 ng/mlIL-1� or IFN-� for 1 h, then stimulated by 10 ng/ml IL-4 for 6 h. The mRNAlevels of CCL26 (upper panel) and GAPDH (lower panel) were quantified byRT-PCR. B, induction of SOCS-1 and -3 gene expression by stimulation withIL-1� or IFN-�. HeLa cells were stimulated with 10 ng/ml IL-1� or IFN-� for6 h, then the mRNA levels of SOCS-1 (upper panel), SOCS-3 (middle panel),and GAPDH (lower panel) were quantified by RT-PCR. GAPDH served as aloading control. C, comparison of Ser-707 phosphorylation levels with IL-1�and IFN-� stimulation. Whole cell lysates were separated by 5% SDS-poly-acrylamide gels and analyzed by Western blotting, using anti-STAT6antibody.
Serine Phosphorylation of STAT6
FEBRUARY 4, 2011 • VOLUME 286 • NUMBER 5 JOURNAL OF BIOLOGICAL CHEMISTRY 4009
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Acknowledgments—We thank H. Osada and T. Sudo (Riken) forproviding DNA constructs and the members of the Uesugi researchgroup for helpful discussion and support. The Kyoto research groupparticipates in the Global COE program “Integrated Material Sci-ences” (B-09).
REFERENCES1. Darnell, J. E., Jr., Kerr, I. M., and Stark, G. R. (1994) Science 264,
1415–14212. Darnell, J. E., Jr. (1997) Science 277, 1630–16353. Ivashkiv, L. B., and Hu, X. (2004) Arthritis Res. Ther. 6, 159–1684. Ihle, J. N. (1996) Cell 84, 331–3345. Hou, J., Schindler, U., Henzel, W. J., Ho, T. C., Brasseur, M., and McK-
night, S. L. (1994) Science 265, 1701–17066. Akimoto, T., Numata, F., Tamura, M., Takata, Y., Higashida, N.,
Takashi, T., Takeda, K., and Akira, S. (1998) J. Exp. Med. 187,1537–1542
7. Kaplan, M. H., Schindler, U., Smiley, S. T., and Grusby, M. J. (1996) Im-munity 4, 313–319
8. Takeda, K., Tanaka, T., Shi, W., Matsumoto, M., Minami, M., Kashi-wamura, S., Nakanishi, K., Yoshida, N., Kishimoto, T., and Akira, S.(1996) Nature 380, 627–630
9. Shimoda, K., van Deursen, J., Sangster, M. Y., Sarawar, S. R., Carson,R. T., Tripp, R. A., Chu, C., Quelle, F. W., Nosaka, T., Vignali, D. A.,Doherty, P. C., Grosveld, G., Paul, W. E., and Ihle, J. N. (1996) Nature380, 630–633
10. Chen, Y. R., Wang, X., Templeton, D., Davis, R. J., and Tan, T. H. (1996)J. Biol. Chem. 271, 31929–31936
11. Derijard, B., Hibi, M., Wu, I. H., Barrett, T., Su, B., Deng, T., Karin, M.,and Davis, R. J. (1994) Cell 76, 1025–1037
12. Marshall, S. W., Kawachi, I., Cryer, P. C., Wright, D., Slappendel, C., andLaird, I. (1994) N ZMed J 107, 434–437
13. Mansour, S. J., Matten, W. T., Hermann, A. S., Candia, J. M., Rong, S.,Fukasawa, K., Vande Woude, G. F., and Ahn, N. G. (1994) Science 265,966–970
14. Sluss, H. K., Barrett, T., Derijard, B., and Davis, R. J. (1994)Mol. Cell.Biol. 14, 8376–8384
15. Wei, Y., Pattingre, S., Sinha, S., Bassik, M., and Levine, B. (2008)MolCell 30, 678–688
16. Gupta, R. K., Bhatia, V., Poptani, H., and Gujral, R. B. (1995) J Pediatr.126, 389–392
17. Galcheva-Gargova, Z., Derijard, B., Wu, I. H., and Davis, R. J. (1994) Sci-ence 265, 806–808
18. Aguirre, V., Uchida, T., Yenush, L., Davis, R., and White, M. F. (2000)J. Biol. Chem. 275, 9047–9054
19. Chen, C. Y., Del Gatto-Konczak, F., Wu, Z., and Karin, M. (1998) Sci-ence 280, 1945–1949
20. Yamashita, H., Xu, J., Erwin, R. A., Farrar, W. L., Kirken, R. A., and Rui,H. (1998) J. Biol. Chem. 273, 30218–30224
21. Wen, Z., Zhong, Z., and Darnell, J. E., Jr. (1995) Cell 82, 241–25022. Lim, C. P., and Cao, X. (1999) J. Biol. Chem. 274, 31055–3106123. Morinobu, A., Gadina, M., Strober, W., Visconti, R., Fornace, A., Mon-
tagna, C., Feldman, G. M., Nishikomori, R., and O’Shea, J. J. (2002) Proc.Natl. Acad. Sci. U.S.A. 99, 12281–12286
24. Wang, Y., Malabarba, M. G., Nagy, Z. S., and Kirken, R. A. (2004) J. Biol.Chem. 279, 25196–25203
25. Maiti, N. R., Sharma, P., Harbor, P. C., and Haque, S. J. (2005) J. Inter-feron Cytokine Res. 25, 553–563
26. Woetmann, A., Brockdorff, J., Lovato, P., Nielsen, M., Leick, V., Rieneck,K., Svejgaard, A., Geisler, C., and Ødum, N. (2003) J. Biol. Chem. 278,2787–2791
27. Wilkinson, K. A., and Henley, J. M. (2010) Biochem. J. 428, 133–14528. Bode, A. M., and Dong, Z. (2004) Nat. Rev. Cancer 4, 793–80529. Golks, A., and Guerini, D. (2008) EMBO Rep. 9, 748–75330. Gu, W., and Roeder, R. G. (1997) Cell 90, 595–60631. Shifrin, V. I., and Anderson, P. (1999) J. Biol. Chem. 274, 13985–1399232. Cano, E., Hazzalin, C. A., and Mahadevan, L. C. (1994)Mol. Cell. Biol.
14, 7352–736233. Yamagishi, S., Yamada, M., Ishikawa, Y., Matsumoto, T., Ikeuchi, T., and
Hatanaka, H. (2001) J. Biol. Chem. 276, 5129–513334. Ahn, H. J., Kim, J. Y., and Nam, H. W. (2009) Korean J Parasitol 47,
117–12435. Schindler, U., Wu, P., Rothe, M., Brasseur, M., and McKnight, S. L.
(1995) Immunity 2, 689–69736. Uciechowski, P., Saklatvala, J., von der Ohe, J., Resch, K., Szamel, M.,
and Kracht, M. (1996) FEBS Lett. 394, 273–27837. Li, X., Commane, M., Jiang, Z., and Stark, G. R. (2001) Proc. Natl. Acad.
Sci. U.S.A. 98, 4461–446538. Dickensheets, H. L., Venkataraman, C., Schindler, U., and Donnelly,
R. P. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 10800–1080539. Losman, J. A., Chen, X. P., Hilton, D., and Rothman, P. (1999) J. Immu-
nol. 162, 3770–377440. Uddin, S., Sassano, A., Deb, D. K., Verma, A., Majchrzak, B., Rahman,
A., Malik, A. B., Fish, E. N., and Platanias, L. C. (2002) J. Biol. Chem. 277,14408–14416
41. Bain, J., Plater, L., Elliott, M., Shpiro, N., Hastie, C. J., McLauchlan, H.,Klevernic, I., Arthur, J. S., Alessi, D. R., and Cohen, P. (2007) Biochem. J.408, 297–315
42. Chung, J., Uchida, E., Grammer, T. C., and Blenis, J. (1997)Mol. Cell.Biol. 17, 6508–6516
43. Becker, S., Groner, B., and Muller, C. W. (1998) Nature 394, 145–15144. Anderson, K. P., Kern, C. B., Crable, S. C., and Lingrel, J. B. (1995)Mol.
Cell. Biol. 15, 5957–596545. Shen, C. H., and Stavnezer, J. (1998)Mol. Cell. Biol. 18, 3395–340446. Valineva, T., Yang, J., Palovuori, R., and Silvennoinen, O. (2005) J. Biol.
Chem. 280, 14989–1499647. Litterst, C. M., and Pfitzner, E. (2001) J. Biol. Chem. 276, 45713–4572148. Boyle, W. J., Smeal, T., Defize, L. H., Angel, P., Woodgett, J. R., Karin,
M., and Hunter, T. (1991) Cell 64, 573–58449. Lin, A., Frost, J., Deng, T., Smeal, T., al-Alawi, N., Kikkawa, U., Hunter,
T., Brenner, D., and Karin, M. (1992) Cell 70, 777–78950. Caelles, C., Hennemann, H., and Karin, M. (1995)Mol. Cell. Biol. 15,
6694–670151. Coqueret, O., Martin, N., Berube, G., Rabbat, M., Litchfield, D. W., and
Nepveu, A. (1998) J. Biol. Chem. 273, 2561–256652. Bullock, B. P., and Habener, J. F. (1998) Biochemistry 37, 3795–380953. Cowley, D. O., and Graves, B. J. (2000) Genes Dev. 14, 366–37654. Yang, S. H., Shore, P., Willingham, N., Lakey, J. H., and Sharrocks, A. D.
(1999) EMBO J. 18, 5666–567455. Yoshimura, A., Naka, T., and Kubo, M. (2007) Nat. Rev. Immunol. 7,
454–46556. Rincon, M., Flavell, R. A., and Davis, R. A. (2000) Free Radic. Biol. Med.
28, 1328–133757. Dong, C., Yang, D. D., Tournier, C., Whitmarsh, A. J., Xu, J., Davis, R. J.,
and Flavell, R. A. (2000) Nature 405, 91–9458. Dong, C., Yang, D. D., Wysk, M., Whitmarsh, A. J., Davis, R. J., and Fla-
vell, R. A. (1998) Science 282, 2092–2095
Serine Phosphorylation of STAT6
4010 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 • NUMBER 5 • FEBRUARY 4, 2011
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Sato and Motonari UesugiTakashi Shirakawa, Yoshinori Kawazoe, Tomoko Tsujikawa, Dongju Jung, Shin-ichi
Deactivation of STAT6 through Serine 707 Phosphorylation by JNK
doi: 10.1074/jbc.M110.168435 originally published online December 1, 20102011, 286:4003-4010.J. Biol. Chem.
10.1074/jbc.M110.168435Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2010/12/01/M110.168435.DC1
http://www.jbc.org/content/286/5/4003.full.html#ref-list-1
This article cites 58 references, 34 of which can be accessed free at
by guest on April 16, 2018
http://ww
w.jbc.org/
Dow
nloaded from


















