
CXC Chemokine Ligand (CXCL) 9 and CXCL10 Are … Costimulation Molecules during the Priming of ......
12
of June 3, 2018. This information is current as T Cell Effectors Molecules during the Priming of Alloreactive CXCL10 Are Antagonistic Costimulation CXC Chemokine Ligand (CXCL) 9 and Joshua M. Farber and Robert L. Fairchild Schenk, Howard Zhang, Danielle D. Kish, Karen Keslar, Joshua M. Rosenblum, Naohiko Shimoda, Austin D. http://www.jimmunol.org/content/184/7/3450 doi: 10.4049/jimmunol.0903831 March 2010; 2010; 184:3450-3460; Prepublished online 1 J Immunol References http://www.jimmunol.org/content/184/7/3450.full#ref-list-1 , 22 of which you can access for free at: cites 54 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2010 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on June 3, 2018 http://www.jimmunol.org/ Downloaded from by guest on June 3, 2018 http://www.jimmunol.org/ Downloaded from
Transcript of CXC Chemokine Ligand (CXCL) 9 and CXCL10 Are … Costimulation Molecules during the Priming of ......

of June 3, 2018.This information is current as
T Cell EffectorsMolecules during the Priming of AlloreactiveCXCL10 Are Antagonistic Costimulation CXC Chemokine Ligand (CXCL) 9 and
Joshua M. Farber and Robert L. FairchildSchenk, Howard Zhang, Danielle D. Kish, Karen Keslar, Joshua M. Rosenblum, Naohiko Shimoda, Austin D.
http://www.jimmunol.org/content/184/7/3450doi: 10.4049/jimmunol.0903831March 2010;
2010; 184:3450-3460; Prepublished online 1J Immunol
Referenceshttp://www.jimmunol.org/content/184/7/3450.full#ref-list-1
, 22 of which you can access for free at: cites 54 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2010 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 3, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 3, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
CXC Chemokine Ligand (CXCL) 9 and CXCL10 AreAntagonistic Costimulation Molecules during the Priming ofAlloreactive T Cell Effectors
Joshua M. Rosenblum,*,† Naohiko Shimoda,‡ Austin D. Schenk,*,† Howard Zhang,x
Danielle D. Kish,† Karen Keslar,† Joshua M. Farber,x and Robert L. Fairchild*,†
Donor Ag-reactive CD4 and CD8 T cell production of IFN-g is a principal effector mechanism promoting tissue injury during
allograft rejection. The CXCR3-binding chemokines CXCL9 and CXCL10 recruit donor-reactive T cells to the allograft, but their
role during the priming of donor-reactive T cells to effector function is unknown. Using a murine model of MHC-mismatched
cardiac transplantation, we investigated the influence of CXCL9 and CXCL10 during donor-reactive T cell priming. In allograft
recipient spleens, CXCL9 and CXCL10 were expressed as early as 24 h posttransplant and increased with similar kinetics,
concurrently with CXCR3 expression on T cells. CXCL9, but not CXCL10, expression required NK cell production of IFN-g.
The absence of CXCL9 in donor allografts, recipients, or both significantly decreased the frequency of donor-reactive CD8 T cells
producing IFN-g and increased the frequency of donor-reactive CD8 T cells producing IL-17A. In contrast, the absence of
CXCL10 increased the frequency of IFN-g–producing CD8 T cells in a CXCL9-dependent manner. These data provide novel
evidence that donor-reactive CD8 T cells use the CXCR3 chemokine axis as a costimulation pathway during priming to allografts
where CXCL9 promotes the development of IFN-g–producing CD8 T cells, and CXCL10 antagonizes this skewing. The Journal
of Immunology, 2010, 184: 3450–3460.
Solid organ transplantation is the sole treatment option forpatients with end-stage organ failure. MHC-mismatchedallografts induce a vigorous antidonor T cell response that
requires aggressive immunosuppression to prevent rejection. Acuterejection of allografts is initiated by the emigration of passengerdendritic cells (DCs) from the transplanted organ to the recipientspleen where they prime donor Ag-specific T cells to express theeffector functions, including cytolytic activities and cytokineproduction, that mediate graft tissue injury. IFN-g is the principalcytokine produced by CD4 and CD8 effector T cells in response toallografts (1–4). The mechanisms that lead to a preferentialskewing of the donor-reactive CD4 and CD8 T cell repertoire topredominantly IFN-g–producing effectors following allografttransplantation is unknown. Although CD4 T cell development toan IFN-g–producing phenotype requires APC production of IL-12(5), CD8 T cell development to IFN-g–producing cells often oc-curs independently of IL-12 (6).Following priming in the spleen, allograft-reactive T cells mi-
grate through the recipient blood stream to the graft, where they are
activated to express the effector functions, including IFN-g pro-duction, that mediate graft injury. Extensive studies have estab-lished that the CXCR3- and CCR5-binding chemokines playprominent roles in the recruitment of effector T cells into allog-rafts (7–10), and correlates to these findings have been found inclinical transplantation (11–13). Studies from this and other lab-oratories have shown a role for CXCL9/monokine induced byIFN-g, CXCL10/IFN-g–inducible protein 10 (IP-10), and CXCR3expression in accelerating acute rejection of MHC-mismatchedallografts (7, 10, 14–16). In cardiac allografts, graft vascular en-dothelial cells and infiltrating neutrophils and macrophages pro-duce these T cell chemoattractants (14, 17). CXCL9 and CXCL10are also produced by DCs, B cells, and macrophages (18) and bindthe G-protein–coupled receptor CXCR3, which is expressed onmultiple cell types but predominantly on memory phenotype cellsand primed effector T cells producing IFN-g (19).In addition to directing leukocyte trafficking to inflammatory
sites, many chemokines are produced at sites of T and B cellactivation in primary and secondary lymphoid tissues (20). A re-cent in vitro study suggested that CXCL9 might influence theproliferation and development of alloantigen-reactive T cells inMLCs (21). Several models of inflammation have also suggestedthat CXCR3-binding chemokines may influence the functionaldevelopment of T cells during Ag priming (22–24). CD4 and CD8T cells express CXCR3 early during priming in response to MHC-disparate allografts, and CXCR3 expression is most pronouncedon effector cells that produce IFN-g. This raises the possibilitythat downstream signaling from CXCR3 early during CD8 T cellpriming may promote preferential polarization of donor-reactiveT cells to an IFN-g–producing phenotype.In this study, we investigated a potential role for CXCL9 during
recipient T cell priming to MHC-mismatched cardiac allografts.We demonstrate that CXCL9 and CXCR3 are coincidentallyexpressed in the graft-draining lymphoid tissue as early as 24 hfollowing transplant and that CXCL9 is induced by NK cell-derived
*Department of Pathology, Case Western Reserve University, Cleveland, OH 44106;†Department of Immunology, Lerner Research Institute, The Cleveland Clinic, Cleve-land, OH 44195; ‡Department of Urology, Hokkaido University School of Medicine,Sapporo, Japan; and xInflammation Biology Section, National Institute of Allergy andInfectious Diseases, National Institutes of Health, Bethesda, MD 20892
Received for publication December 1, 2009. Accepted for publication January 27,2010.
This work was supported by National Institutes of Health Grants RO1 AI51620 andAI40459 (to R.L.F.). J.M.R. was supported in part by the Case Western ReserveUniversity Medical Scientist Training Program (National Institutes of Health T32GM07250) and an individual National Research Service Award (National Institutesof Health F30 HL940052).
Address correspondence and reprint requests to Dr. Joshua M. Rosenblum, ClevelandClinic Lerner Research Institute, Mail Code NB30, 9500 Euclid Avenue, Cleveland,OH 44195. E-mail address: [email protected]
Abbreviations used in this paper: DC, dendritic cell; IP-10, IFN-g–inducible protein;qRT-PCR, quantitative real-time PCR; Treg, regulatory T cell.
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0903831
by guest on June 3, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

IFN-g. The absence of CXCL9 depresses the number of IFN-g–producing, donor-reactive CD8 and CD4 T cells, but this does notprolong graft survival, potentially as the result of increased fre-quencies of donor-specific CD8 T cells producing IL-17A. Finally,we provide evidence that CXCL9 and CXCL10 antagonize eachother as costimulatory molecules during T cell priming to allo-antigen. Taken together, these data implicate an important andunrecognized role for CXCR3-binding chemokines in the acuterejection process. Moreover, we propose that the predominanteffector mechanism in complete MHC-mismatched allograft re-jection, CD8 T cell production of IFN-g, is the consequence ofhigh levels of CXCL9 produced in the allograft recipient spleenduring priming of these T cells, and this is regulated, in part, bythe coincident production of CXCL10 in the priming site.
Materials and MethodsMice
The following mice were used: C57BL/6 (H-2b) and A/J (H-2a) fromCharles River Laboratories (Wilmington, MA); DBA/1 (H-2q), BALB/c(H-2d), and B6.Thy1.1 from The Jackson Laboratory (Bar Harbor, ME);and B6.2C TCR transgenic, B6.CXCL92/2, A/J.CXCL92/2, B6.Rag12/2,B6.CXCL102/2, and BALB/c.CXCL102/2 (bred at our facility). All ex-periments used 8–12-wk-old male mice, and the Cleveland Clinic In-stitutional Animal Care and Use Committee approved all procedures.
Heterotopic cardiac transplantation
Standard methods of murine heterotopic intra-abdominal cardiac trans-plantation were adapted from the method of Corry et al. (25). Total op-erative times averaged 30–35 min, and graft survival was monitored byabdominal palpation, with cessation of beating confirmed by laparotomy.
Ab treatments
For CD8 depletion, 0.2 mg a 1:1 mixture of anti-CD8 mAbs YTS169 andTB-105 (BioExpress, West Lebanon, NH) was administered i.p. on days –3,22, 21, and +4 and every 4 d until rejection. NK cell-depleting anti-NK1.1 mAb (0.25 mg, PK136; BioExpress) was given i.p. on days 23,22, 21, +2, and +4 and every 2 d. In all cases, equal amounts of normalrat IgG (Sigma-Aldrich, St. Louis MO) were administered to controlgroups, and cell depletion was confirmed by flow cytometry analysis ofperipheral blood samples. Mouse anti-mouse CXCL9 mAb was adminis-tered to graft recipients (150 mg, i.p.) on days 1, 3, and 5 posttransplant.
Bone marrow chimera
Anestablishedprotocol for radiosensitivemicewasused (26).A/J.CXCL92/2
mice received a split dose of 2 3 3.5 Gy gamma radiation delivered at 1.21Gy/min with a 4-h interval between treatments. One day after irradiation, 10–15 3 106 wild-type A/J bone marrow cells were given i.p. Recipients weretreated with 0.25 ml gentamicin (Sigma-Aldrich) i.p. on days 0 and +2 afterbone marrow transplantation and were maintained on acid water (pH 2.5) for2 wk post-bone marrow transplantation. After resting for 8 wk, chimerismwas confirmed using standard PCR analysis of DNA purified from PBMCs.
Flow cytometry
Graft-infiltrating leukocytes were analyzed using a modified method ofAfanasyeva et al. (27). Following harvest, graft tissue was incubated for 1 hat 37˚C in RPMI 1640 plus type II collagenase (Sigma-Aldrich). Afterincubation, graft tissue was gently crushed and passed through a 40-mmfilter and washed with RPMI. For flow cytometric analysis of splenocytes,single-cell suspensions were made after RBC lysis, and 5 3 106 cells werestained. In all cases, surface markers were stained using standard methodsand commercially available Abs (eBioscience, San Diego, CA and BDBiosciences, San Jose, CA). Flow cytometry was performed usinga FACSCalibur (BD Biosciences) cytometer and FlowJo analysis software(Tree Star, Ashland, OR).
Intracellular cytokine staining
For intracellular cytokine staining, purified cells were incubated in completeRPMI 1640 with 0.01 mg/ml PMA and 1 nM ionomycin at 37˚C for 2 h.Monensin (2 mM) was added, and the cells were cultured for an additional2 h. Following restimulation, cells were surface stained, fixed with 4%paraformaldehyde, and then permeabilized with 0.1% saponin (permbuffer). Cells were washed and stained for intracellular cytokines usingcommercially available Abs in perm buffer.
In vitro culture assays
Naive 2C transgenic CD44loCD8+ T cells were purified by flow sorting andcultured in 48-well plates with T cell-depleted CXCL92/2 syngeneic stim-ulators pulsed with 10 mM Ld peptide (SIYRYYGL; a generous gift from A.Morelli, University of Pittsburgh) at a 10:1 (responder/stimulator) ratio for 3 d.In some experiments, recombinant murine CXCL9 protein or anti-CXCL10mAb (both from R&D Systems, Minneapolis MN) was added to the cultureson day 0. Following culture, cells were restimulatedwith PMAand ionomycinfor 4 h, with monensin for the final 2 h as above, and were processed andstained for measurement of intracellular IFN-g by flow cytometry.
Quantitative real-time PCR
Snap-frozen graft pieces were crushed, homogenized using Qiashredders,and RNA was isolated using Fibrous Tissue kits (both from Qiagen,Valencia, CA), according to the manufacturer’s instructions. For analysis ofmRNA expression in recipient spleens, spleen pieces were snap-frozen inliquid nitrogen, homogenized as above, and RNA was isolated usingRNeasy Mini kits (Qiagen). For RNA purification from flow-sorted cells,cell pellets were homogenized using Qiashredders, and RNA purificationwas performed using RNeasy Plus Micro kits (Qiagen).
Commercially available reagents and probes were used for reverse tran-scription, and real-time PCR was performed on a 7500 Fast Real-Timethermocycler, all from Applied Biosystems (Foster City, CA). For quantifi-cation of message expression, target gene expression was normalized toMrpl32 gene expression. All samples were plated in triplicate, and the resultsare expressed as mean fold increase6 SEM in gene expression over controls.
ELISPOT
Responder CD8 or CD4 T cells were column-purified (R&D Systems) fromrecipient spleens; self, donor, and third-party stimulator splenocytes weredepleted of T cells using magnetic beads (Invitrogen, Carlsbad, CA).Stimulator and responder cell populations were cocultured in serum-freeHL-1 medium (BioWhittaker, Walkersville, MD) supplemented with 1 mML-glutamine and 1 mM antibiotic for 24 h at 37˚C in 96-well plates coatedwith anti–IFN-g (R4-6A2) or anti–IL-17 (TC11-18H10, BD Biosciences)capture Abs. Cells were washed from the plate, and biotinylated anti–IFN-g(XMG1.2) or biotinylated anti–IL-17 (TC11-8H4.1, BD Biosciences) wasadded, followed by antibiotin alkaline phosphatase. Spots were developedwith 5-bromo-4-chloro-3-indolyl phosphate and NBT (both from Bio-Rad,Hercules, CA), and total spots per well were quantified using an Im-munoSpot Series 2 Analyzer (Cellular Technology, Shaker Heights, OH).
Statistics
All data were analyzed using GraphPad Prism Pro (GraphPad, San Diego,CA). Replicates were used as indicated. Log-rank testing was performed todetermine differences in survival data, and the Student t test was used todetermine significance throughout; p , 0.05 was considered a significantdifference. Error bars throughout indicate SEM.
ResultsCXCL9 is required for maximum generation of donor-reactive,IFN-g–producing T cells
To directly test whether the presence of CXCL9 influences thefunctional development of donor-reactive T cells in vivo, groups ofwild-type or CXCL9-deficient C57BL/6 mice received wild-type orCXCL9-deficient A/J heart allografts. CD8 or CD4 T cells werepurified from recipient spleens on day 7 posttransplant, and thenumbers of donor-reactive, IFN-g–producing cells were enumeratedby ELISPOT. Wild-type cardiac allografts placed in wild-type re-cipients induced more than twice the number of donor-reactive,IFN-g–producing CD8 and CD4 T cells than did CXCL92/2 al-lografts placed in CXCL92/2 recipients; in both cases, there were∼10-fold greater numbers of IFN-g–producing CD8 T cells com-pared with CD4 T cells (Fig. 1A, 1B).In concurrence with the decreased frequency of IFN-g–producing
CD8 andCD4T cells in the spleens of CXCL92/2 allograft recipients,IFN-g mRNA expression in CXCL92/2 allografts retrieved fromCXCL92/2 recipients onday7posttransplantwasone thirdof the levelof expression in wild-type grafts retrieved from wild-type recipients(Fig. 1C). Additionally, when graft-infiltrating cells were purified and
The Journal of Immunology 3451
by guest on June 3, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

restimulated in vitro with PMA and ionomycin, fewer CD8 T cellsproduced IFN-g when recovered from CXCL92/2 allograft combi-nations compared with wild-type allograft combinations (Fig. 1D).
Taken together, these data show that alloimmune responses generatedin the complete absenceofCXCL9 induce significantly fewernumbersof donor-reactive, IFN-g–producing T cells compared with alloim-mune responses in the presence of CXCL9.The data also suggest that there are fewer graft-infiltrating CD8
T cells in the CXCL92/2 allografts retrieved from CXCL92/2
recipients compared with wild-type allografts. At day 5 post-transplant, the number of CD8 T cells was significantly reduced inCXCL9-deficient allografts compared with wild-type allografts;the trend continued at day 7 posttransplant, but it was not statis-tically significant (Fig. 1E). The number of graft-infiltrating CD4T cells in CXCL9-deficient allograft combinations was onlymarginally reduced at days 5 and 7 posttransplant compared withwild-type allograft combinations (data not shown).
Donor- and recipient-derived CXCL9 contribute to CD8 T cellpriming
Because the complete absence of CXCL9 resulted in depressedfrequencies of donor-reactive T cells producing IFN-g, the relativecontribution of donor- and recipient-derived CXCL9 was tested.
FIGURE 1. CXCL9 is required for maximum generation of donor-reactive,
IFN-g–producingCD8Tcells.Wild-type orCXCL92/2B6mice receivedwild-
type orCXCL92/2A/J heart allografts. Recipient spleenswere harvested on day
7 posttransplant, and purifiedCD8 (A) or CD4 (B) T cells producing IFN-gwere
enumerated by ELISPOT assay. Data are representative of at least three in-
dependent experiments.C, mRNAwas purified from total graft homogenates on
day 7 posttransplant. qRT-PCR analysis was performed on five or six samples/
group. Expression of Mrpl32 was used as the endogenous control, and the ex-
pression of IFN-g in each samplewas normalized to the expression in a random
native heart sample. D, Graft-infiltrating cells were purified on day 7 post-
transplant and were restimulated in vitro with PMA/ionomycin for 4 h, with
monensin added for the last 2 h. Intracellular cytokine staining was performed
using standard techniquesand reagents.Numbers ineachplot are thepercentages
of total lymphocytes, and plots represent four or five samples per group. E, Al-
lograftswereharvestedat the times indicated, and thenumberof graft-infiltrating
CD8 T cells was quantitated using flow cytometry. Bars in each panel represent
mean6 SEM (n = 5–6/group). pp, 0.05; ppp, 0.01.
FIGURE 2. Donor- and recipient-derived CXCL9 influence the de-
velopment of donor-reactive IFN-g–producing CD8 T cells. A, Wild-type
or CXCL92/2 B6 mice received wild-type or CXCL92/2 A/J heart al-
lografts. Recipient spleens were harvested on day 7 posttransplant, and
purified CD8 T cells producing IFN-g were enumerated by ELISPOT as-
say. Data are representative of at least three independent experiments (n =
4–5/group). B, Chimerism of donor grafts in B was confirmed by PCR
analysis of DNA from PBMCs collected from mice just prior to transplant.
Data are representative of four mice per group. C, CXCL92/2 A/J mice
were irradiated, and wild-type A/J bone marrow was adoptively trans-
ferred; mice were then rested for 8 wk. These chimeric mice were used as
heart donors for wild-type B6 mice. CD8 T cells were purified from re-
cipient spleens on day 7 posttransplant, and IFN-g–producing cells were
enumerated using ELISPOT. Data are representative of two independent
experiments (n = 3–4/group). pp , 0.05; ppp , 0.01.
3452 COSTIMULATORY CHEMOKINES
by guest on June 3, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
Groups of reciprocal allograft combinations were performed with
wild-type and CXCL92/2 donors and recipients, and the number
of IFN-g–producing, donor-reactive CD8 T cells was enumerated
by ELISPOT on day 7 posttransplant. Although CXCL92/2 recip-
ients of wild-type allografts demonstrated significantly decreased
numbers of IFN-g–producing CD8T cells comparedwith wild-type
recipients (3,1056 951 versus 12,2486 1377 spots/106 responders;
Fig. 2A), CXCL92/2 grafts placed in wild-type recipients induced
a 32% decrease in IFN-g–producing CD8 T cells compared with
wild-type grafts. These data indicate that donor- and recipient-de-
rived CXCL9 influence priming of CD8 T cells, but the primary
contribution comes from recipient-derived CXCL9.CXCL9 can be produced by the cardiac graft endothelium, in-
terstitial/passenger APCs, and infiltrating leukocytes (14, 18). To
determine which population of graft cells (endothelium or pas-
senger APCs) is responsible for producing the CXCL9 that in-
fluences CD8 T cell priming in the recipient spleen, bone marrow
chimeras were generated in which CXCL92/2 A/J mice were le-
thally irradiated and received wild-type A/J bone marrow. After
resting for 8 wk, chimerism was confirmed using PCR to assess
CXCL9 expression in PBMCs (Fig. 2B). B6 recipients of
CXCL92/2 allografts had ∼26% fewer CD8 T cells producing
IFN-g compared with recipients of wild-type grafts, but the use of
a chimeric CXCL92/2 donor graft containing wild-type bone
marrow-derived passenger leukocytes restored the number of IFN-
g–producing CD8 T cells to wild-type levels (Fig. 2C).
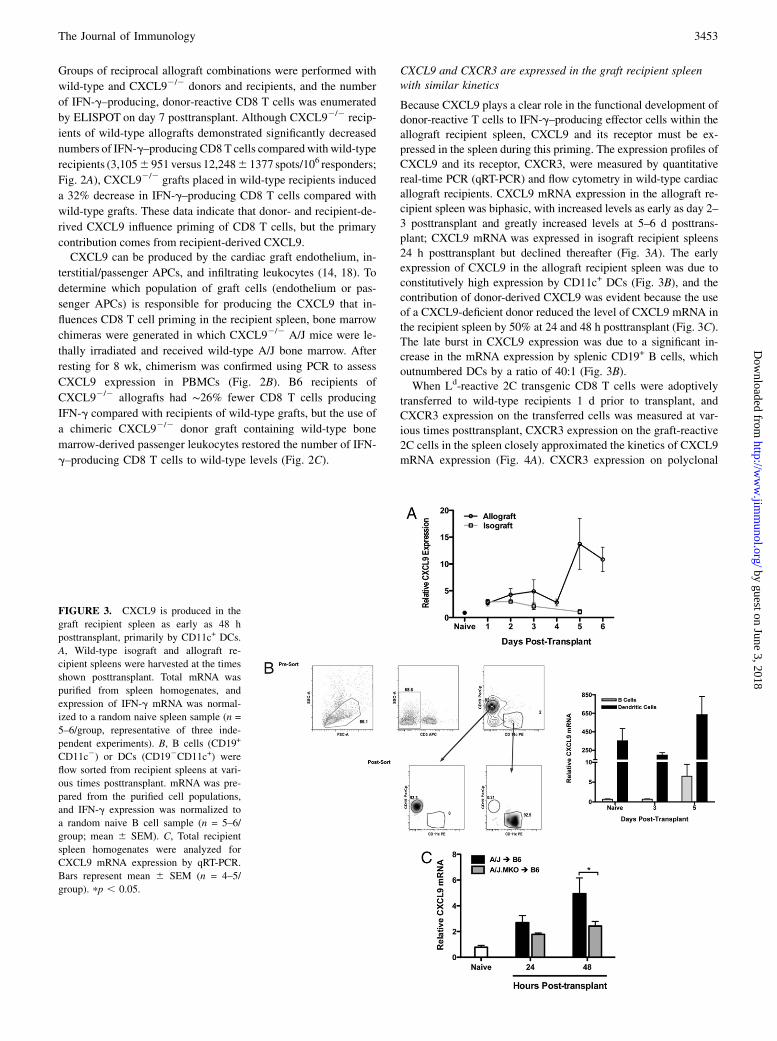
CXCL9 and CXCR3 are expressed in the graft recipient spleenwith similar kinetics
Because CXCL9 plays a clear role in the functional development ofdonor-reactive T cells to IFN-g–producing effector cells within the
allograft recipient spleen, CXCL9 and its receptor must be ex-
pressed in the spleen during this priming. The expression profiles of
CXCL9 and its receptor, CXCR3, were measured by quantitative
real-time PCR (qRT-PCR) and flow cytometry in wild-type cardiac
allograft recipients. CXCL9 mRNA expression in the allograft re-
cipient spleen was biphasic, with increased levels as early as day 2–
3 posttransplant and greatly increased levels at 5–6 d posttrans-
plant; CXCL9 mRNA was expressed in isograft recipient spleens
24 h posttransplant but declined thereafter (Fig. 3A). The early
expression of CXCL9 in the allograft recipient spleen was due to
constitutively high expression by CD11c+ DCs (Fig. 3B), and the
contribution of donor-derived CXCL9 was evident because the use
of a CXCL9-deficient donor reduced the level of CXCL9 mRNA in
the recipient spleen by 50% at 24 and 48 h posttransplant (Fig. 3C).
The late burst in CXCL9 expression was due to a significant in-
crease in the mRNA expression by splenic CD19+ B cells, which
outnumbered DCs by a ratio of 40:1 (Fig. 3B).When Ld-reactive 2C transgenic CD8 T cells were adoptively
transferred to wild-type recipients 1 d prior to transplant, and
CXCR3 expression on the transferred cells was measured at var-
ious times posttransplant, CXCR3 expression on the graft-reactive
2C cells in the spleen closely approximated the kinetics of CXCL9
mRNA expression (Fig. 4A). CXCR3 expression on polyclonal
FIGURE 3. CXCL9 is produced in the
graft recipient spleen as early as 48 h
posttransplant, primarily by CD11c+ DCs.
A, Wild-type isograft and allograft re-
cipient spleens were harvested at the times
shown posttransplant. Total mRNA was
purified from spleen homogenates, and
expression of IFN-g mRNA was normal-
ized to a random naive spleen sample (n =
5–6/group, representative of three inde-
pendent experiments). B, B cells (CD19+
CD11c2) or DCs (CD192CD11c+) were
flow sorted from recipient spleens at vari-
ous times posttransplant. mRNA was pre-
pared from the purified cell populations,
and IFN-g expression was normalized to
a random naive B cell sample (n = 5–6/
group; mean 6 SEM). C, Total recipient
spleen homogenates were analyzed for
CXCL9 mRNA expression by qRT-PCR.
Bars represent mean 6 SEM (n = 4–5/
group). pp , 0.05.
The Journal of Immunology 3453
by guest on June 3, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

CD4 and CD8 T cells in the recipient spleens of wild-type andCXCL9-deficient allograft combinations was measured by flowcytometry. Although the percentage of CD4+ and CD8+ T cellsexpressing CXCR3 in the wild-type allograft recipient spleen wasrelatively constant (20–25% of CD8 T cells and 10–15% of CD4T cells; Fig. 4B) over time posttransplant, the percentage of CD4and CD8 T cells expressing CXCR3 in CXCL9-deficient allograftrecipients continuously increased with time posttransplant (40–45% of CD8 T cells and 20–25% of CD4 T cells on day 5 post-transplant). Because CXCR3 is internalized upon ligand binding,these data suggest that in the wild-type allograft recipient spleen,CXCL9 binds to CXCR3 on CD4 and CD8 T cells, maintaininga constant frequency of CXCR3-expressing T cells in the spleen.
In the allograft recipient spleen, ∼50–60% of divided CD8 andCD4 T cells expressed CXCR3, whereas only 2% of undividedT cells expressed CXCR3 (Fig. 4C). A significant majority ofCD8+CD44hi effector T cells recovered from wild-type allograftrecipient spleens on day 5 posttransplant expressed CXCR3, butvery few naive CD44lo T cells expressed CXCR3 (Fig. 4D).
NK cell IFN-g production induces early CXCL9 production inthe recipient spleen
Inmurineandhumanmodels of inflammation anddisease,CXCL9 isspecifically induced by IFN-g (28–30). Thus, because CXCL9 wasexpressed in the allograft recipient spleen andwehave shown that itspresence influences the functional phenotype of donor-reactive
FIGURE 4. CXCR3 is expressed in the recipient spleen by proliferating, activated T cells with kinetics similar to CXCL9 expression. A, A total of
2.5 3 106 2C CD8 T cells were adoptively transferred to wild-type B6 recipients of A/J heart allografts 1 d prior to transplant. Recipient spleens were
harvested on the days indicated, and CXCR3 expression on 2C CD8 T cells was measured by flow cytometry. Data represent the percentage of total 2C
CD8 T cells expressing CXCR3 (n = 3–4/time point; mean 6 SEM). B, The percentage of endogenous polyclonal CD8 (top panel) or CD4 (bottom panel)
T cells expressing CXCR3 was determined at the times shown posttransplant. Data are representative of two independent experiments (n = 3–4/time
point; mean 6 SEM). C, A total of 2.5 3 106 CD8 or CD4 T cells were labeled with CFSE and transferred to wild-type B6 recipients of A/J heart
allografts 1 d prior to transplant. Recipient spleens were harvested on day 5 posttransplant, and CXCR3 expression on CFSE-labeled proliferating cells
was measured by flow cytometry. Numbers represent the percentage of total transferred cells in each quadrant; plots are representative of three mice per
group. D, Spleens from wild-type B6 recipients of wild-type A/J cardiac allografts were harvested on day 5 posttransplant, and cells were processed using
standard techniques for flow cytometry. Numbers represent the percentage of total CD8 T cells expressing CXCR3; plots are representative of four
individual mice.
3454 COSTIMULATORY CHEMOKINES
by guest on June 3, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

T cells, the IFN-g requirements for induction were investigated. Inisograft and allograft recipient spleens, IFN-gmRNAwas expressedat elevated levels during the first 3 d posttransplant (Fig. 5A). Giventhat splenic IFN-g mRNA levels were elevated in isograft and al-
lograft recipients and that use of an IFN-g2/2 donor did not alter
recipient splenic IFN-g mRNA expression (data not shown), it was
hypothesized that an innate immune cell of recipient origin was
responsible for IFN-g production after being activated by danger
signals or the systemic mediators of ischemia/reperfusion injury.
When isograft and allograft recipients were treated with anti–
NK1.1-depletingmAb, spleen levels of IFN-g andCXCL9 at 24 and
48 h posttransplant were significantly reduced compared with
controls (Fig. 5B), and these levels were also reduced in NK1.1-
depleted RAG12/2 allograft recipients (Fig. 5C), indicating that the
early IFN-g driving early CXCL9 production was produced by NK
cells and not NK T cells.Because the presence of CXCL9 was important for optimum gen-
eration of donor-reactive, IFN-g–producingT cells, andNKcellswere
required to induce early CXCL9 in the recipient spleen, the effect of
NK cell depletion on donor-reactive T cell development was tested.
Whenwild-type cardiac allograft recipientsweredepleted ofNKcells,
the frequency of IFN-g–producing CD8 T cells in the recipient spleen
at day 7 posttransplant was significantly lower (∼50%) than in IgG-
treated controls (Fig. 5D). Taken together, these data indicate that NK
cells produce IFN-g in the allograft recipient spleen within 2 d post-
transplant, and this subsequently induces the CXCL9 production that
influences donor-reactive T cell development in the spleen.
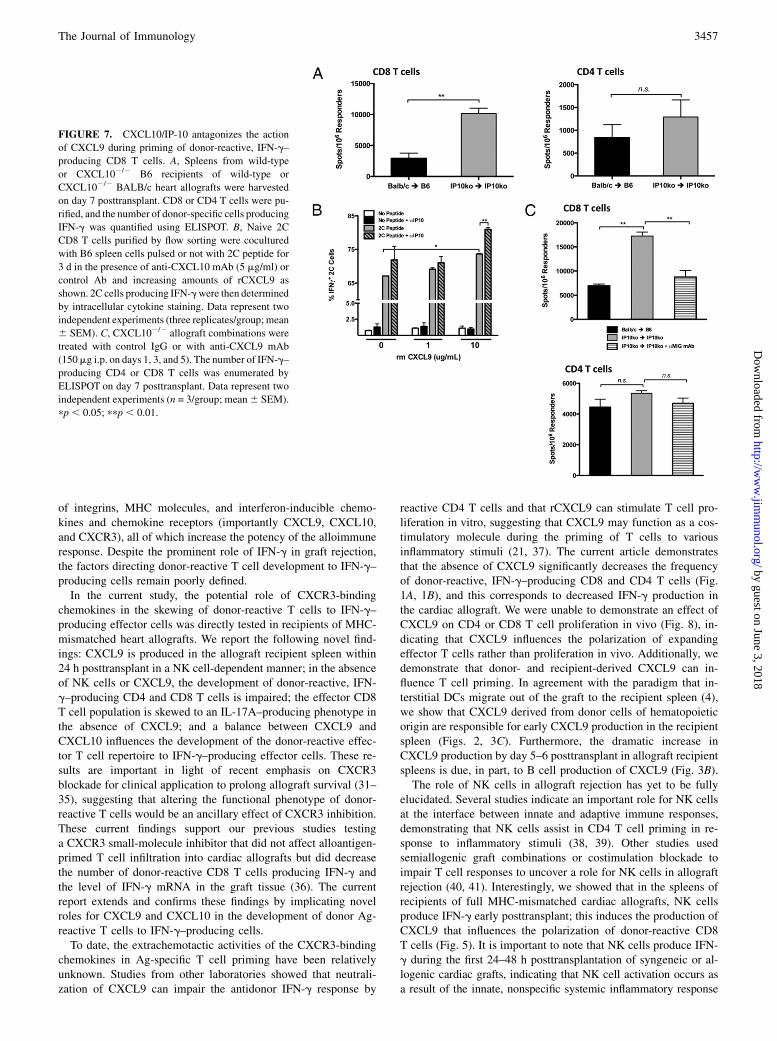
CXCL10 antagonizes CXCL9 during T cell priming
CXCL9,CXCL10, andCXCL11all bind the same receptor,CXCR3.Because the presence of CXCL9 in the allograft recipient spleen
clearly influenced the functionalphenotypeofdonor-reactiveTcells,
the role of CXCL10/IP10 in this process was tested. Similar to
CXCL9 expression in the wild-type allograft recipient spleen,
CXCL10 mRNA is expressed at increasing levels with time post-
transplant, principally by DCs early and by B cells only by day 5
posttransplant (Fig. 6A, 6B). Interestingly, CXCL10 mRNA ex-
pression was relatively insensitive to NK cell depletion with anti-
NK1.1 mAb (Fig. 6C).Because CXCL10 is expressed in the allograft recipient spleen in
a similar manner as CXCL9, and CXCL9 influences the functional
developmentofdonor-reactiveTcells, theeffectofCXCL10deficiency
onTcellprimingwas tested.Unexpectedly,whenCD8andCD4Tcells
were purified from CXCL102/2 recipients of CXCL102/2 complete
MHC-mismatched cardiac allografts on day 7 posttransplant, there
was amarked increase in the number of IFN-g–producingCD8T cells
compared with wild-type allograft recipients (Fig. 7A).To further investigate whether and how CXCL9 and CXCL10
strike a balance as antagonistic costimulatory molecules during
the functional polarization of donor-reactive CD8 T cells, we
FIGURE 5. Recipient NK cells produce IFN-g during the first 48 h posttransplant in the recipient spleen. A, Wild-type isograft or allograft recipient
spleens were harvested at the times shown posttransplant. Total mRNA was purified from spleen homogenates, and IFN-g mRNA expression was de-
termined by qRT-PCR (n = 4–6/group, mean 6 SEM). Wild-type (B) or RAG12/2 (C) isograft or allograft recipients were treated with control IgG or with
NK-depleting mAb (PK136, 250 mg i.p. on days 23, 22, 21, and 1). Total mRNA was purified from spleen homogenates at the times shown. qRT-PCR
analysis was performed as in A (n = 4–5/group, mean6 SEM). pp, 0.05; ppp, 0.01. D, Wild-type B6 allograft recipients (n = 3/group) were treated with
control IgG or PK136 (250 mg i.p. on days23, 22, 21, 1, 3, and 5). Recipient spleens were harvested on day 7 posttransplant, and the numbers of purified
CD8 T cells producing IFN-g were enumerated by ELISPOT assay. Data are representative of two independent experiments (mean 6 SEM). pp , 0.05.
The Journal of Immunology 3455
by guest on June 3, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

attempted to recapitulate our in vivo findings using an in vitroapproach. When increasing amounts of rCXCL9 was added to 2CCD8 T cells stimulated with CXCL92/2 syngeneic splenocytespulsed with Ld peptide (SIYRYYGL), the percentage of IFN-g–producing cells increased concomitantly (Fig. 7B). The magnitudeof increase over baseline was heightened when anti–IP-10 mAbwas added to the cultures (Fig. 7B).Toextendthesefindings invivo, thenumberofdonor-reactiveCD4
and CD8 T cells producing IFN-g was determined at day 7 post-transplant in wild-type allograft recipients, CXCL10-deficient al-lograft recipients, and CXCL10-deficient allograft recipientstreatedwith anti-CXCL9mAb. In agreementwith previous findings,the number of donor-reactive, IFN-g–producing CD8 T cells wassignificantly higher in CXCL10-deficient recipients of CXCL102/2
allografts compared with wild-type recipients; however, the treat-ment of CXCL102/2 recipients with anti-CXCL9 mAb reduced thefrequency of IFN-g–producing CD8T cells towild-type levels (Fig.7C, upper panel). These data suggest that CXCL9 promotes thedevelopment of IFN-g–producing CD8 T cell effectors and thatCXCL10 impedes this process. Of note, CD4 T cells were relatively
insensitive to the presence or absence of CXCL10 (Fig. 7C, lowerpanel). Taken together with the findings in Fig. 1, these data suggestthat CXCL9 and CXCL10 are antagonistic in providing costim-ulation to influence the effector phenotype of donor-reactive CD8T cells to IFN-g–producing cells during priming.
Acute rejection in the absence of CXCL9
Graft-survival studies using CXCL92/2 donors, recipients, or bothindicated no prolongation in graft survival in these combinations(CXCL92/2 to CXCL92/2 allograft: mean survival time = 8.5 d;wild-type control allograft: mean survival time = 8 d; data notshown), despite the marked decrease in T cell priming to IFN-g–producing effector cells. These results raised the possibility that,in the absence of CXCL9, donor-reactive T cell development wasskewed to another functional phenotype that was effective inmediating allograft rejection. In such a case, the absence ofCXCL9 in the T cell priming environment would not be expectedto affect the clonal proliferation of the donor-reactive T cellsduring priming. To determine whether CXCL9 influences pro-liferation in response to alloantigen in vivo, Ld-reactive 2C CD8T cells or polyclonal CD90.1+ CD4 T cells were labeled withCFSE and were adoptively transferred to wild-type or CXCL92/2
CD90.2 B6 recipients of wild-type or CXCL92/2 A/J heart grafts1 d prior to transplant. Grafts and recipient spleens were recoveredon day 5 posttransplant, and proliferation of transferred cells wasquantified by CFSE dilution using flow cytometry. Equivalentpercentages of CD8 and CD4 T cells (Fig. 8A, 8B, respectively)proliferated in recipient spleens and grafts, regardless of thepresence or absence of CXCL9. Moreover, NK cell depletion didnot affect donor-reactive CD8 T cell proliferation (Fig. 8C). Thesedata indicate that the decrease in the number of donor-reactive,IFN-g–producing T cells resulting from CXCL9 deficiency is notdue to a defect in clonal expansion of donor-reactive cells in al-lograft recipients.Taken together, the above findings suggest that graft rejection in
a CXCL9-deficient environment may rely on other effectormechanisms. Initial testing by ELISPOT of CD8 T cells recoveredfrom wild-type and CXCL9-deficient allograft recipients demon-strated no difference in the frequency of donor-reactive cellsproducing granzyme B or IL-4 (data not shown). However, whenCD8 T cells were purified from the spleens of CXCL92/2 recip-ients of CXCL92/2 cardiac allografts, the frequency of IL-17A–producing cells was significantly higher than CD8 T cells re-covered from wild-type allograft recipients, but there was nodifference in the frequency of CD4 T cells producing IL-17A (Fig.9A). Consistent with these results, intragraft cytokine mRNA ex-pression in wild-type or CXCL9-deficient allograft combinationsat day 7 posttransplant revealed that IL-17A mRNA expression(but not IL-4 or -5, perforin, or granzyme B; data not shown) wassubstantially increased in CXCL92/2 allografts placed inCXCL92/2 recipients compared with wild-type allografts (Fig.9B). Thus, donor-specific production of IL-17 by CD8 T cells maybe an alternative mechanism leading to graft rejection in the ab-sence of CXCL9.
DiscussionThe inflammatory environment within allografts during rejection iscomposed of a complex organization of infiltrating cells, cytokines,and other mediators contributing to graft tissue injury. In clinicaland experimental transplantation, graft-infiltrating T cell pro-duction of IFN-g is a principal mechanism mediating tissue injury.IFN-g has direct and indirect effects on the donor graft, includingstimulation of macrophages and neutrophils to produce enzymesthat generate toxic molecular products and upregulated expression
FIGURE 6. CXCL10/IP-10 is produced in the allograft recipient spleen
by DCs and B cells, independent of NK cells. A, mRNA expression of
CXCL10 was determined in allograft and isograft recipient spleens at
various times posttransplant (n = 3–4/group). Data were analyzed as in Fig.
3A. B, B cells and DCs were purified by flow sorting as in Fig. 3B, and
CXCL10 mRNA expression was determined as described. C, Wild-type
allograft recipients were depleted of NK cells as described, and total spleen
homogenates were taken at the times indicated for qRT-PCR analysis of
CXCL10 expression. Data represent two individual experiments (n = 3–4/
group; mean 6 SEM).
3456 COSTIMULATORY CHEMOKINES
by guest on June 3, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
of integrins, MHC molecules, and interferon-inducible chemo-kines and chemokine receptors (importantly CXCL9, CXCL10,and CXCR3), all of which increase the potency of the alloimmuneresponse. Despite the prominent role of IFN-g in graft rejection,the factors directing donor-reactive T cell development to IFN-g–producing cells remain poorly defined.In the current study, the potential role of CXCR3-binding
chemokines in the skewing of donor-reactive T cells to IFN-g–producing effector cells was directly tested in recipients of MHC-mismatched heart allografts. We report the following novel find-ings: CXCL9 is produced in the allograft recipient spleen within24 h posttransplant in a NK cell-dependent manner; in the absenceof NK cells or CXCL9, the development of donor-reactive, IFN-g–producing CD4 and CD8 T cells is impaired; the effector CD8T cell population is skewed to an IL-17A–producing phenotype inthe absence of CXCL9; and a balance between CXCL9 andCXCL10 influences the development of the donor-reactive effec-tor T cell repertoire to IFN-g–producing effector cells. These re-sults are important in light of recent emphasis on CXCR3blockade for clinical application to prolong allograft survival (31–35), suggesting that altering the functional phenotype of donor-reactive T cells would be an ancillary effect of CXCR3 inhibition.These current findings support our previous studies testinga CXCR3 small-molecule inhibitor that did not affect alloantigen-primed T cell infiltration into cardiac allografts but did decreasethe number of donor-reactive CD8 T cells producing IFN-g andthe level of IFN-g mRNA in the graft tissue (36). The currentreport extends and confirms these findings by implicating novelroles for CXCL9 and CXCL10 in the development of donor Ag-reactive T cells to IFN-g–producing cells.To date, the extrachemotactic activities of the CXCR3-binding
chemokines in Ag-specific T cell priming have been relativelyunknown. Studies from other laboratories showed that neutrali-zation of CXCL9 can impair the antidonor IFN-g response by
reactive CD4 T cells and that rCXCL9 can stimulate T cell pro-liferation in vitro, suggesting that CXCL9 may function as a cos-timulatory molecule during the priming of T cells to variousinflammatory stimuli (21, 37). The current article demonstratesthat the absence of CXCL9 significantly decreases the frequencyof donor-reactive, IFN-g–producing CD8 and CD4 T cells (Fig.1A, 1B), and this corresponds to decreased IFN-g production inthe cardiac allograft. We were unable to demonstrate an effect ofCXCL9 on CD4 or CD8 T cell proliferation in vivo (Fig. 8), in-dicating that CXCL9 influences the polarization of expandingeffector T cells rather than proliferation in vivo. Additionally, wedemonstrate that donor- and recipient-derived CXCL9 can in-fluence T cell priming. In agreement with the paradigm that in-terstitial DCs migrate out of the graft to the recipient spleen (4),we show that CXCL9 derived from donor cells of hematopoieticorigin are responsible for early CXCL9 production in the recipientspleen (Figs. 2, 3C). Furthermore, the dramatic increase inCXCL9 production by day 5–6 posttransplant in allograft recipientspleens is due, in part, to B cell production of CXCL9 (Fig. 3B).The role of NK cells in allograft rejection has yet to be fully
elucidated. Several studies indicate an important role for NK cellsat the interface between innate and adaptive immune responses,demonstrating that NK cells assist in CD4 T cell priming in re-sponse to inflammatory stimuli (38, 39). Other studies usedsemiallogenic graft combinations or costimulation blockade toimpair T cell responses to uncover a role for NK cells in allograftrejection (40, 41). Interestingly, we showed that in the spleens ofrecipients of full MHC-mismatched cardiac allografts, NK cellsproduce IFN-g early posttransplant; this induces the production ofCXCL9 that influences the polarization of donor-reactive CD8T cells (Fig. 5). It is important to note that NK cells produce IFN-g during the first 24–48 h posttransplantation of syngeneic or al-logenic cardiac grafts, indicating that NK cell activation occurs asa result of the innate, nonspecific systemic inflammatory response
FIGURE 7. CXCL10/IP-10 antagonizes the action
of CXCL9 during priming of donor-reactive, IFN-g–
producing CD8 T cells. A, Spleens from wild-type
or CXCL102/2 B6 recipients of wild-type or
CXCL102/2 BALB/c heart allografts were harvested
on day 7 posttransplant. CD8 or CD4 T cells were pu-
rified, and the number of donor-specific cells producing
IFN-g was quantified using ELISPOT. B, Naive 2C
CD8 T cells purified by flow sorting were cocultured
with B6 spleen cells pulsed or not with 2C peptide for
3 d in the presence of anti-CXCL10 mAb (5 mg/ml) or
control Ab and increasing amounts of rCXCL9 as
shown. 2C cells producing IFN-gwere then determined
by intracellular cytokine staining. Data represent two
independent experiments (three replicates/group; mean
6 SEM). C, CXCL102/2 allograft combinations were
treated with control IgG or with anti-CXCL9 mAb
(150mg i.p. on days 1, 3, and 5). The number of IFN-g–
producing CD4 or CD8 T cells was enumerated by
ELISPOT on day 7 posttransplant. Data represent two
independent experiments (n = 3/group; mean6 SEM).
pp , 0.05; ppp, 0.01.
The Journal of Immunology 3457
by guest on June 3, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

to cardiac transplantation. Although it is unclear from our studieshow NK cells are activated to produce IFN-g posttransplant, in-creased levels of proinflammatory cytokines like TNF-a are likelyto play a role, because blockade of TNF-a depresses early IFN-g inthe spleen (data not shown). Alternatively, NK cells may be activatedby so-called alarmins or danger signals like high-mobility group boxprotein 1, which are released by activated endothelium following is-chemia-reperfusion injury of syngeneic and allogenic grafts (42–44).
Mounting evidence from several groups has focused on themechanisms underlying allograft rejection in the absence of IFN-g.Although donor-specific production of IFN-g remains a hallmarkof clinical and experimental allograft rejection, uncovering alter-native effector cytokines becomes important when antirejectiontherapy is specifically targeted to limiting IFN-g–induced re-jection. In allograft recipients genetically deficient in IFN-g, CD8-mediated graft rejection is resistant to costimulation blockade andoccurs independently of IL-4 (45). These findings suggest that, inthe absence of donor-reactive T cells capable of producing IFN-g,other effector mechanisms are activated to mediate allograft re-jection. Recent studies using Th1-deficient T-bet2/2 recipientsdemonstrated that cardiac allografts were rejected in an IL-17–dependent manner and that the rejected grafts had histologicalfindings consistent with the development of allograft vasculopathy(46, 47). Because CXCL92/2 allograft combinations were rejectedat the same rate as wild-type grafts, we investigated whether othereffector functions were overexpressed in these groups. IL-17Aproduction by CD8 T cells was significantly increased in CXCL9-deficient allograft recipients, and this correlated with increasedlevels of IL-17A mRNA in the graft tissue. In contrast to recentreports indicating a role for IL-17–mediated accelerated graft lossdue to vasculopathy in an IFN-g–deficient environment, we did notobserve an overt vasculopathy in grafts from CXCL92/2 combi-nations (data not shown).
FIGURE 8. CXCL9 does not influence the proliferation of donor-reactive
CD8 or CD4 T cells in vivo. A, A total of 5 3 106 Ld-reactive naive 2C
transgenic CD8 T cells were labeled with CFSE and transferred to wild-type
or CXCL92/2 B6 mice 1 d prior to transplant with wild-type or CXCL92/2
A/J heart grafts. Grafts and recipient spleens were harvested on day 5 post-
transplant, and CFSE dilution was measured by standard flow cytometry
techniques. Bar graphs are representative of four to six mice per group, and
graphs represent the percentage of 2C CD8 T cells that have divided. B, A
total of 10 3 106 CD90.1 CD4 T cells were labeled with CFSE and adop-
tively transferred toCD90.2mice 1 d prior to transplant. Proliferation ofCD4
T cells was determined at day 5 posttransplant as in A. C, A total of 53 106
CFSE-labeled 2C CD8 T cells were adoptively transferred to wild-type B6
allograft recipients treated with control IgG or PK136 1 d prior to transplant.
Proliferation of 2C cells in the recipient spleens and graftswas determined by
flow cytometry on day 5 posttransplant. Graphs are representative of three
mice per group; numbers representmean percentage6 SEMof total 2C cells
that have divided.
FIGURE 9. Donor-specific production of IL-17 by CD8 T cells is in-
creased in the absence of CXCL9. A, Wild-type or CXCL92/2 B6 mice
received wild-type or CXCL92/2 A/J heart allografts, and CD8 or CD4
T cells were purified from recipient spleens on day 7 posttransplant. The
number of donor-specific cells producing IL-17 was enumerated using
ELISPOT. Data represent three independent experiments (n = 5–6/group,
mean 6 SEM). pp , 0.05. B, Total mRNA was purified from graft ho-
mogenates at day 7 posttransplant. IL-17 mRNA expression was normal-
ized to endogenous Mrpl32 expression using a random native heart sample
as the baseline. Data represent two independent experiments.
3458 COSTIMULATORY CHEMOKINES
by guest on June 3, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

Given that CXCL9 and CXCL10 bind CXCR3, we sought todetermine whether CXCL10 also played a role in the polarizationof donor-reactive CD8 T cells. Surprisingly, the absence ofCXCL10 in allograft combinations resulted in a significant increasein the number of IFN-g–producing CD8 T cells compared withwild-type controls (Fig. 7A). Taken in context with subsequentin vitro experiments (Fig. 7B), these data indicate that CXCL9 andCXCL10 antagonize each other in the allograft recipient spleen topromote effector CD8 T cell polarization. When CXCL9 is neu-tralized, the effect of unopposed CXCL9 in CXCL10-deficientallograft recipients is abrogated (Fig. 7C). We hypothesize thatCD8 T cells use the CXCR3 axis as a costimulatory pathway, inwhich binding of CXCL9 promotes differentiation to an IFN-g–producing phenotype, and binding of CXCL10 may promote anIL-17–producing phenotype or may simply inhibit generation ofan IFN-g–producing phenotype, with development to an IL-17–producing phenotype being the default pathway. Current studiesare aimed at determining the mechanism that provides a differen-tial signaling cascade through CXCR3, depending on which li-gand binds. Reports demonstrating that CXCL9 and CXCL10 bindto CXCR3 in different manners and, at the least, induce differentdownstream signals to mediate chemotaxis suggest that differentsignaling mechanisms may influence the costimulatory nature ofthe receptor (48, 49).Although our data suggest that CXCL9 and CXCL10 act as
antagonistic costimulation molecules, it is possible that an alter-native mechanism underlies our findings. Elegant work from theGermain laboratory (50, 51) demonstrated that in models of in-fection and inflammation, chemokines produced by individualcells within the lymph node create a microchemotactic environ-ment and that CD4 T cell interaction with DCs induces these DCsto produce MIP-1a, which recruits naive CCR5+CD8 T cells tothe DC. The chemokines may promote CD8 T cell migration toa cluster of APC-CD4 T cell complexes (52), or chemokines maybe produced by competent APCs to recruit naive CD8 T cellsfollowing licensing by the CD4 T cells (53). Taken in this context,our results raise the possibility that the CXCR3 axis plays a role inthe priming of naive T cells by acting as a chemotactic factor. Thismay provide a rationale as to why CXCL9 and CXCL10 pro-duction in the allograft recipient spleen have such variant effectson the differentiation of T cells. An APC that has been licensed toprime a CD8 T cell to an IFN-g–producing phenotype may pro-duce high levels of CXCL9. In contrast, CXCL10 production maymark an APC that expresses costimulatory ligands that polarizeT cells to an IL-17–producing phenotype. Reconciling our dis-parate results with CXCL92/2 and CXCL102/2 allograft combi-nations using the microchemotaxis model would require twoseparate populations of DCs migrating from the allograft: oneproducing CXCL9 and one producing CXCL10. Although wehave no direct evidence to support either model at this point, webelieve that the alternate signaling hypothesis outlined above ismore likely, and we have focused our current efforts to test this asa mechanism.Recent evidence has shown that CXCR3 expression on CD4+
regulatory T cells (Tregs) is regulated by T-bet induction fol-lowing exposure to IFN-g during a Th1-type immune response(54). Conceivably, this mechanism may serve to bring regulatorycells to the site of inflammation to dampen aberrant immune re-sponses. Could the expression of CXCR3 on Tregs and the dif-ferential signaling via binding of CXCL9 or CXCL10 contributeto the results observed in this study? First, there is little evidencethat Tregs function effectively in the face of a complete MHC-mismatched alloimmune response, making this possibility un-likely. Second, in the absence of CXCL9, there does not seem to
be any effect on antidonor CD8 T cell proliferation (Fig. 8A),again implicating that Tregs are not effective at dampening thisimmune response. Finally, we demonstrate a measurable differ-ence in CD8 T cell differentiation in the presence and absence ofCXCL9 in vitro, indicating that the effect of CXCL9 and CXCL10on CD8 T cell priming occurs at the level of the CD8 T cell;however, the possibility that Tregs are involved in this process ina limited MHC-mismatch or a minor Ag-mismatched graft com-bination in vivo cannot be ruled out.Collectively, this study provides novel evidence that CXCR3-
binding chemokines function as costimulation molecules for CD8T cells during priming to alloantigen because CXCL9 and CXCL10can skew the balance of effector phenotypes. In addition to thesefindings providing novel insights into T cell development followingalloantigen exposure, they present important implications andtargets for the pharmacological prolongation of graft survival inclinical transplantation.
AcknowledgmentsWe thank the Cleveland Clinic Biological Resources Unit and their staff for
the exceptional care and maintenance of the experimental animals used.
Adrian Morelli, University of Pittsburgh, generously provided the peptide
used in this work.
DisclosuresThe authors have no financial conflicts of interest.
References1. Heeger, P. S. 2003. T-cell allorecognition and transplant rejection: a summary
and update. Am. J. Transplant. 3: 525–533.2. Hall, B. M., and S. E. Dorsch. 1984. Cells mediating allograft rejection. Im-
munol. Rev. 77: 31–59.3. Hidalgo, L. G., and P. F. Halloran. 2002. Role of IFN-gamma in allograft re-
jection. Crit. Rev. Immunol. 22: 317–349.4. Larsen, C. P., P. J. Morris, and J. M. Austyn. 1990. Migration of dendritic leu-
kocytes from cardiac allografts into host spleens. A novel pathway for initiationof rejection. J. Exp. Med. 171: 307–314.
5. Trinchieri, G. 1995. Interleukin-12: a proinflammatory cytokine with immuno-regulatory functions that bridge innate resistance and antigen-specific adaptiveimmunity. Annu. Rev. Immunol. 13: 251–276.
6. Gorbachev, A. V., and R. L. Fairchild. 2004. CD40 engagement enhances antigen-presenting langerhans cell priming of IFN-gamma-producing CD4+ and CD8+T cells independently of IL-12. J. Immunol. 173: 2443–2452.
7. Agostini, C., F. Calabrese, F. Rea, M. Facco, A. Tosoni, M. Loy, G. Binotto,M. Valente, L. Trentin, and G. Semenzato. 2001. Cxcr3 and its ligand CXCL10are expressed by inflammatory cells infiltrating lung allografts and mediatechemotaxis of T cells at sites of rejection. Am. J. Pathol. 158: 1703–1711.
8. Duffner, U., B. Lu, G. C. Hildebrandt, T. Teshima, D. L. Williams, P. Reddy,R. Ordemann, S. G. Clouthier, K. Lowler, C. Liu, et al. 2003. Role of CXCR3-induced donor T-cell migration in acute GVHD. Exp. Hematol. 31: 897–902.
9. el-Sawy, T., N. M. Fahmy, and R. L. Fairchild. 2002. Chemokines: directingleukocyte infiltration into allografts. Curr. Opin. Immunol. 14: 562–568.
10. Hancock, W. W., B. Lu, W. Gao, V. Csizmadia, K. Faia, J. A. King, S. T. Smiley,M. Ling, N. P. Gerard, and C. Gerard. 2000. Requirement of the chemokinereceptor CXCR3 for acute allograft rejection. J. Exp. Med. 192: 1515–1520.
11. Fahmy, N. M., M. H. Yamani, R. C. Starling, N. B. Ratliff, J. B. Young,P. M. McCarthy, J. Feng, A. C. Novick, and R. L. Fairchild. 2003. Chemokineand receptor-gene expression during early and late acute rejection episodes inhuman cardiac allografts. Transplantation 75: 2044–2047.
12. Fahmy, N. M., M. H. Yamani, R. C. Starling, N. B. Ratliff, J. B. Young,P. M. McCarthy, J. Feng, A. C. Novick, and R. L. Fairchild. 2003. Chemokineand chemokine receptor gene expression indicates acute rejection of humancardiac transplants. Transplantation 75: 72–78.
13. Hu, H., B. D. Aizenstein, A. Puchalski, J. A. Burmania, M. M. Hamawy, andS. J. Knechtle. 2004. Elevation of CXCR3-binding chemokines in urine indicatesacute renal-allograft dysfunction. Am. J. Transplant. 4: 432–437.
14. Miura, M., K. Morita, H. Kobayashi, T. A. Hamilton, M. D. Burdick,R. M. Strieter, and R. L. Fairchild. 2001. Monokine induced by IFN-gamma isa dominant factor directing T cells into murine cardiac allografts during acuterejection. J. Immunol. 167: 3494–3504.
15. Yun, J. J., M. P. Fischbein, D. Whiting, Y. Irie, M. C. Fishbein, M. D. Burdick,J. Belperio, R. M. Strieter, H. Laks, J. A. Berliner, and A. Ardehali. 2002. Therole of MIG/CXCL9 in cardiac allograft vasculopathy. Am. J. Pathol. 161: 1307–1313.
The Journal of Immunology 3459
by guest on June 3, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

16. Hancock, W. W., W. Gao, V. Csizmadia, K. L. Faia, N. Shemmeri, andA. D. Luster. 2001. Donor-derived IP-10 initiates development of acute allograftrejection. J. Exp. Med. 193: 975–980.
17. Kobayashi, H., S. Koga, A. C. Novick, H. Toma, and R. L. Fairchild. 2003. T-cellmediated induction of allogeneic endothelial cell chemokine expression.Transplantation 75: 529–536.
18. Park, M. K., D. Amichay, P. Love, E. Wick, F. Liao, A. Grinberg, R. L. Rabin,H. H. Zhang, S. Gebeyehu, T. M. Wright, et al. 2002. The CXC chemokinemurine monokine induced by IFN-gamma (CXC chemokine ligand 9) is madeby APCs, targets lymphocytes including activated B cells, and supports antibodyresponses to a bacterial pathogen in vivo. J. Immunol. 169: 1433–1443.
19. Loetscher, M., B. Gerber, P. Loetscher, S. A. Jones, L. Piali, I. Clark-Lewis,M. Baggiolini, and B. Moser. 1996. Chemokine receptor specific for IP10 andmig: structure, function, and expression in activated T-lymphocytes. J. Exp. Med.184: 963–969.
20. Sallusto, F., C. R. Mackay, and A. Lanzavecchia. 2000. The role of chemokinereceptors in primary, effector, and memory immune responses. Annu. Rev. Im-munol. 18: 593–620.
21. Whiting, D., G. Hsieh, J. J. Yun, A. Banerji, W. Yao, M. C. Fishbein, J. Belperio,R. M. Strieter, B. Bonavida, and A. Ardehali. 2004. Chemokine monokine in-duced by IFN-gamma/CXC chemokine ligand 9 stimulates T lymphocyte pro-liferation and effector cytokine production. J. Immunol. 172: 7417–7424.
22. Campbell, J. D., V. Gangur, F. E. Simons, and K. T. HayGlass. 2004. Allergic hu-mans are hyporesponsive to a CXCR3 ligand-mediated Th1 immunity-promotingloop. FASEB J. 18: 329–331.
23. Gangur, V., F. E. Simons, and K. T. Hayglass. 1998. Human IP-10 selectivelypromotes dominance of polyclonally activated and environmental antigen-drivenIFN-gamma over IL-4 responses. FASEB J. 12: 705–713.
24. Manicone, A. M., K. M. Burkhart, B. Lu, and J. G. Clark. 2008. CXCR3 ligandscontribute to Th1-induced inflammation but not to homing of Th1 cells into thelung. Exp. Lung Res. 34: 391–407.
25. Corry, R. J., H. J. Winn, and P. S. Russell. 1973. Primarily vascularized allograftsof hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection.Transplantation 16: 343–350.
26. Cui, Y. Z., H. Hisha, G. X. Yang, T. X. Fan, T. Jin, Q. Li, Z. Lian, and S. Ikehara.2002. Optimal protocol for total body irradiation for allogeneic bone marrowtransplantation in mice. Bone Marrow Transplant. 30: 843–849.
27. Afanasyeva, M., D. Georgakopoulos, D. F. Belardi, A. C. Ramsundar,J. G. Barin, D. A. Kass, and N. R. Rose. 2004. Quantitative analysis of myo-cardial inflammation by flow cytometry in murine autoimmune myocarditis:correlation with cardiac function. Am. J. Pathol. 164: 807–815.
28. Farber, J. M. 1990. A macrophage mRNA selectively induced by gamma-interferonencodes a member of the platelet factor 4 family of cytokines. Proc. Natl. Acad.Sci. USA 87: 5238–5242.
29. Amichay, D., R. T. Gazzinelli, G. Karupiah, T. R. Moench, A. Sher, andJ. M. Farber. 1996. Genes for chemokines MuMig and Crg-2 are induced inprotozoan and viral infections in response to IFN-gamma with patterns of tissueexpression that suggest nonredundant roles in vivo. J. Immunol. 157: 4511–4520.
30. Ebnet, K., M. M. Simon, and S. Shaw. 1996. Regulation of chemokine geneexpression in human endothelial cells by proinflammatory cytokines and Bor-relia burgdorferi. Ann. N. Y. Acad. Sci. 797: 107–117.
31. Schenk, A. D., J. M. Rosenblum, and R. L. Fairchild. 2008. Chemokine-directedstrategies to attenuate allograft rejection. Clin. Lab. Med. 28: 441–454, vii.
32. Akashi, S., M. Sho, H. Kashizuka, K. Hamada, N. Ikeda, Y. Kuzumoto,Y. Tsurui, T. Nomi, T. Mizuno, H. Kanehiro, et al. 2005. A novel small-moleculecompound targeting CCR5 and CXCR3 prevents acute and chronic allograftrejection. Transplantation 80: 378–384.
33. Schnickel, G. T., S. Bastani, G. R. Hsieh, A. Shefizadeh, R. Bhatia,M. C. Fishbein, J. Belperio, and A. Ardehali. 2008. Combined CXCR3/CCR5blockade attenuates acute and chronic rejection. J. Immunol. 180: 4714–4721.
34. Turner, J. E., O. M. Steinmetz, R. A. Stahl, and U. Panzer. 2007. Targeting ofTh1-associated chemokine receptors CXCR3 and CCR5 as therapeutic strategyfor inflammatory diseases. Mini Rev. Med. Chem. 7: 1089–1096.
35. Wijtmans, M., D. Verzijl, R. Leurs, I. J. de Esch, and M. J. Smit. 2008. Towardssmall-molecule CXCR3 ligandswith clinical potential.ChemMedChem 3: 861–872.
36. Rosenblum, J. M., Q. W. Zhang, G. Siu, T. L. Collins, T. Sullivan, D. J. Dairaghi,J. C. Medina, and R. L. Fairchild. 2009. CXCR3 antagonism impairs the de-velopment of donor-reactive, IFN-gamma-producing effectors and prolongs al-lograft survival. Transplantation 87: 360–369.
37. Dufour, J. H., M. Dziejman, M. T. Liu, J. H. Leung, T. E. Lane, and A. D. Luster.2002. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveala role for IP-10 in effector T cell generation and trafficking. J. Immunol. 168:3195–3204.
38. Martın-Fontecha, A., L. L. Thomsen, S. Brett, C. Gerard, M. Lipp,A. Lanzavecchia, and F. Sallusto. 2004. Induced recruitment of NK cells tolymph nodes provides IFN-gamma for T(H)1 priming. Nat. Immunol. 5: 1260–1265.
39. Bajenoff, M., B. Breart, A. Y. Huang, H. Qi, J. Cazareth, V. M. Braud,R. N. Germain, and N. Glaichenhaus. 2006. Natural killer cell behavior in lymphnodes revealed by static and real-time imaging. J. Exp. Med. 203: 619–631.
40. McNerney, M. E., K. M. Lee, P. Zhou, L. Molinero, M. Mashayekhi, D. Guzior,H. Sattar, S. Kuppireddi, C. R. Wang, V. Kumar, and M. L. Alegre. 2006. Role ofnatural killer cell subsets in cardiac allograft rejection. Am. J. Transplant. 6:505–513.
41. Maier, S., C. Tertilt, N. Chambron, K. Gerauer, N. Huser, C. D. Heidecke, andK. Pfeffer. 2001. Inhibition of natural killer cells results in acceptance of cardiacallografts in CD282/2 mice. Nat. Med. 7: 557–562.
42. Andrassy, M., H. C. Volz, J. C. Igwe, B. Funke, S. N. Eichberger, Z. Kaya,S. Buss, F. Autschbach, S. T. Pleger, I. K. Lukic, et al. 2008. High-mobility groupbox-1 in ischemia-reperfusion injury of the heart. Circulation 117: 3216–3226.
43. Huang, Y., H. Yin, J. Han, B. Huang, J. Xu, F. Zheng, Z. Tan, M. Fang, L. Rui,D. Chen, et al. 2007. Extracellular hmgb1 functions as an innate immune-mediatorimplicated inmurine cardiac allograft acute rejection.Am. J. Transplant. 7: 799–808.
44. Rao, D. A., and J. S. Pober. 2008. Endothelial injury, alarmins, and allograftrejection. Crit. Rev. Immunol. 28: 229–248.
45. Bishop, D. K., S. Chan Wood, E. J. Eichwald, and C. G. Orosz. 2001. Im-munobiology of allograft rejection in the absence of IFN-gamma: CD8+ effectorcells develop independently of CD4+ cells and CD40-CD40 ligand interactions.J. Immunol. 166: 3248–3255.
46. Burrell, B. E., K. Csencsits, G. Lu, S. Grabauskiene, and D. K. Bishop. 2008.CD8+ Th17 mediate costimulation blockade-resistant allograft rejection in T-bet-deficient mice. J. Immunol. 181: 3906–3914.
47. Yuan, X., J. Paez-Cortez, I. Schmitt-Knosalla, F. D’Addio, B. Mfarrej,M. Donnarumma, A. Habicht, M. R. Clarkson, J. Iacomini, L. H. Glimcher, et al.2008. A novel role of CD4 Th17 cells in mediating cardiac allograft rejectionand vasculopathy. J. Exp. Med. 205: 3133–3144.
48. Colvin, R. A., G. S. Campanella, L. A. Manice, and A. D. Luster. 2006. CXCR3requires tyrosine sulfation for ligand binding and a second extracellular looparginine residue for ligand-induced chemotaxis. Mol. Cell. Biol. 26: 5838–5849.
49. Colvin, R. A., G. S. Campanella, J. Sun, and A. D. Luster. 2004. Intracellulardomains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J.Biol. Chem. 279: 30219–30227.
50. Germain, R. N., M. Bajenoff, F. Castellino, M. Chieppa, J. G. Egen,A. Y. Huang, M. Ishii, L. Y. Koo, and H. Qi. 2008. Making friends in out-of-the-way places: how cells of the immune system get together and how they conducttheir business as revealed by intravital imaging. Immunol. Rev. 221: 163–181.
51. Bajenoff, M., J. G. Egen, H. Qi, A. Y. Huang, F. Castellino, and R. N. Germain.2007. Highways, byways and breadcrumbs: directing lymphocyte traffic in thelymph node. Trends Immunol. 28: 346–352.
52. Castellino, F., A. Y. Huang, G. Altan-Bonnet, S. Stoll, C. Scheinecker, andR. N. Germain. 2006. Chemokines enhance immunity by guiding naive CD8+T cells to sites of CD4+ T cell-dendritic cell interaction. Nature 440: 890–895.
53. Beuneu, H., Z. Garcia, and P. Bousso. 2006. Cutting edge: cognate CD4 helppromotes recruitment of antigen-specific CD8 T cells around dendritic cells. J.Immunol. 177: 1406–1410.
54. Koch, M. A., G. Tucker-Heard, N. R. Perdue, J. R. Killebrew, K. B. Urdahl, andD. J. Campbell. 2009. The transcription factor T-bet controls regulatory T cell ho-meostasis and function during type 1 inflammation. Nat. Immunol. 10: 595–602.
3460 COSTIMULATORY CHEMOKINES
by guest on June 3, 2018http://w
ww
.jimm
unol.org/D
ownloaded from


















