
Constitutive CTGF expression in scleroderma fibroblasts is ...

2053Research Article
IntroductionMetastasis, the major cause of mortality for cancer patients, isa complex and multi-stage process in which secondary tumorsare formed in distant sites (Van’t Veer and Weigelt, 2003).Typically, the development of metastasis involves several stepsthat comprise cellular transformation and tumor growth,angiogenesis and lymphangiogenesis, entry of cancer cells intothe circulation by intravasation, anchorage and/or attachmentin the target organ, invasion of the target organ byextravasation, and proliferation within the organ parenchyma(Hanahan and Weinberg, 2000). The migratory ability of acancer cell is important for many of these steps, and thereforeis correlated with tumor metastasis. In particular, severalsecreted proteins, including vascular endothelial growth factor(VEGF), PC-cell-derived growth factor (PCDGF/GP88,progranulin), epidermal growth factor (EGF), and stromal-cell-
derived factor 1 (CXCL12) confer ameliorated migratoryability to breast cancer cells and are correlated with moreadvanced-stages of breast cancer (Darash-Yahana et al., 2004;Miralem et al., 2001; Price et al., 1999; Tangkeangsirisin andSerrero, 2004). These molecules have been proposed astherapeutic targets (Barrett-Lee, 2005). Identification andmolecular characterization of new molecules that are involvedin tumor progression, therefore, have important clinicalimplications.
In order to invade, a tumor cell must undergo major changesin shape. Cellular motility depends on localized actinpolymerization at the leading edge of the cells (Kirfel et al.,2004), and the polymerization and depolymerization of actinfilaments must be under dynamic control. Simultaneously,paxillin and vinculin interact at focal contacts of the actin stressfibers, providing a link to the extracellular matrix (e.g.
Connective tissue growth factor (CTGF) expression iselevated in advanced stages of breast cancer, but theregulatory role of CTGF in invasive breast cancer cellphenotypes is unclear. Presently, overexpression of CTGFin MCF-7 cells (MCF-7/CTGF cells) enhanced cellularmigratory ability and spindle-like morphologicalalterations, as evidenced by actin polymerization and focal-adhesion-complex aggregation. Reducing the CTGF levelin MDA-MB-231 (MDA231) cells by antisense CTGF cDNA(MDA231/AS cells) impaired cellular migration andpromoted a change to an epithelial-like morphology. Aneutralizing antibody against integrin �v�3 significantlyattenuated CTGF-mediated ERK1/2 activation andcellular migration, indicating that the integrin-�v�3–ERK1/2 signaling pathway is crucial in mediatingCTGF function. Moreover, the cDNA microarray analysisrevealed CTGF-mediated regulation of the prometastaticgene S100A4. Transfection of MCF-7/CTGF cells with AS-
S100A4 reversed the CTGF-induced cellular migratoryability, whereas overexpression of S100A4 in MDA231/AScells restored their high migratory ability. Genetic andpharmacological manipulations suggested that the CTGF-mediated S100A4 upregulation was dependent on ERK1/2activation, with expression levels of CTGF and S100A4being closely correlated with human breast tumors. Weconclude that CTGF plays a crucial role inmigratory/invasive processes in human breast cancer by amechanism involving activation of the integrin-�v�3–ERK1/2–S100A4 pathway.
Supplementary material available online athttp://jcs.biologists.org/cgi/content/full/120/12/2053/DC1
Key words: Connective tissue growth factor, Breast cancer, ERK1/2,Integrin, S100A4, Cell migration, F-actin
Summary
CTGF enhances the motility of breast cancer cells viaan integrin-�v�3–ERK1/2-dependent S100A4-upregulated pathwayPai-Sheng Chen1,2,*, Ming-Yang Wang1,2,3,*, Shin-Ni Wu1, Jen-Liang Su4,5,6, Chih-Chen Hong7,Shuang-En Chuang7, Min-Wei Chen1, Kuo-Tai Hua1, Yu-Ling Wu1, Shih-Ting Cha1, Munisamy Suresh Babu1,Chiung-Nien Chen2,3, Po-Huang Lee2,3, King-Jen Chang2,3,‡,§ and Min-Liang Kuo1,2,‡,§
1Laboratory of Molecular and Cellular Toxicology, Institute of Toxicology, College of Medicine and 2Angiogenesis Research Center, National TaiwanUniversity, Taipei, Taiwan3Department of Surgery, National Taiwan University Hospital, Taipei 100, Taiwan4Graduate Institute of Cancer Biology, College of Medicine, China Medical University, Taichung 404, Taiwan5Center for Molecular Medical, China Medical University Hospital, Taichung 404, Taiwan6Department of Biotechnology and Bioinformatics, Asia University, Taichung 41354, Taiwan7Division of Cancer Research, National Health Research Institutes, Taipei 10016, Taiwan*These authors contributed equally to this work‡Authors for correspondence (e-mails: [email protected]; [email protected])§These authors contributed equally to this work
Accepted 11 April 2007Journal of Cell Science 120, 2053-2065 Published by The Company of Biologists 2007doi:10.1242/jcs.03460
Jour
nal o
f Cel
l Sci
ence

2054
fibronectin and vitronectin). These cytoskeletal changes enablethe invading cell to pass through the stromal cells, extracellularmatrix and endothelial cell layer. Integrins, paxillin, selectins,transmembrane receptor tyrosine kinases, phospholipids, focaladhesion kinases (FAKs), GTPases and the S100 calciumbinding protein A4 (S100A4) calcium binding protein havebeen described as being involved in regulating the organizationof the actin cytoskeleton (Kim and Helfman, 2003; Turner,2000; Brunton et al., 2004). Some of these molecules have alsobeen implicated in the malignant phenotype of certaincarcinomas, such as ERBB-2, in mammary carcinomas(Mariani et al., 2005). ERBB2 increases the potential formetastasis by upregulating the expression of the prometastaticS100A4 in medulloblastoma (Hernan et al., 2003).
Connective tissue growth factor (CTGF, also known asCCN2) belongs to the CCN family (Bork, 1993). This familyconsists of six members, CTGF, NOVH, CYR61, WISP1,WISP2 and WISP3 (Perbal, 2004) that all possess an N-terminal signal peptide identifying them as secreted proteins.CCN proteins probably carry out their biological activitythrough binding and activating of the cell surface integrins,accompanied by activation of Akt and/or MAPK signalcascades (Perbal, 2004). The biological properties of CCNproteins involve the stimulation of cellular proliferation,migration, adhesion, extracellular matrix formation, and alsothe regulation of angiogenesis and tumorigenesis (Lau andLam, 1999). Overexpression of CTGF, WISP1, and CYR61 inbreast tumor cells have been linked to tumor size and lymphnode metastasis (Xie et al., 2001), suggesting that these CCNproteins are involved in the progression of breast cancer.Recently, we reported that CYR61 influences the resistance tochemotherapeutic-agent-induced apoptosis in human breastcancer MCF-7 cells (Lin et al., 2004). However, the biologicalactivities of CTGF in breast cancer have not yet been explored.CTGF serves as an angiogenic factor in collaboration withmatrix metalloproteinases (Kondo et al., 2002). Use of in vitroand in vivo selection models and large-scale microarrayanalysis has revealed that CTGF crucial for the formation ofosteolytic bone metastasis in breast cancer (Kang et al., 2003;Minn et al., 2005). Thus, CTGF plays an important role inbreast cancer progression. However, the precise role of CTGFin breast cancer metastasis is still unknown.
In this study, we demonstrate that CTGF can modulate thecytoskeletal reorganization and in vitro migratory behavior ofbreast cancer cells. We show that activation of ERK1/2 throughintegrin �v�3 confers the enhanced cellular motility.Microarray and reverse transcription (RT)-PCR analysisrevealed that the crucial prometastatic S100A4 is significantlyupregulated in cells and breast tumors that overexpress CTGF.Our results support a new mechanism, in which integrin-�v�3–ERK1/2-dependent upregulation of S100A4 contributesto CTGF-enhanced migratory ability.
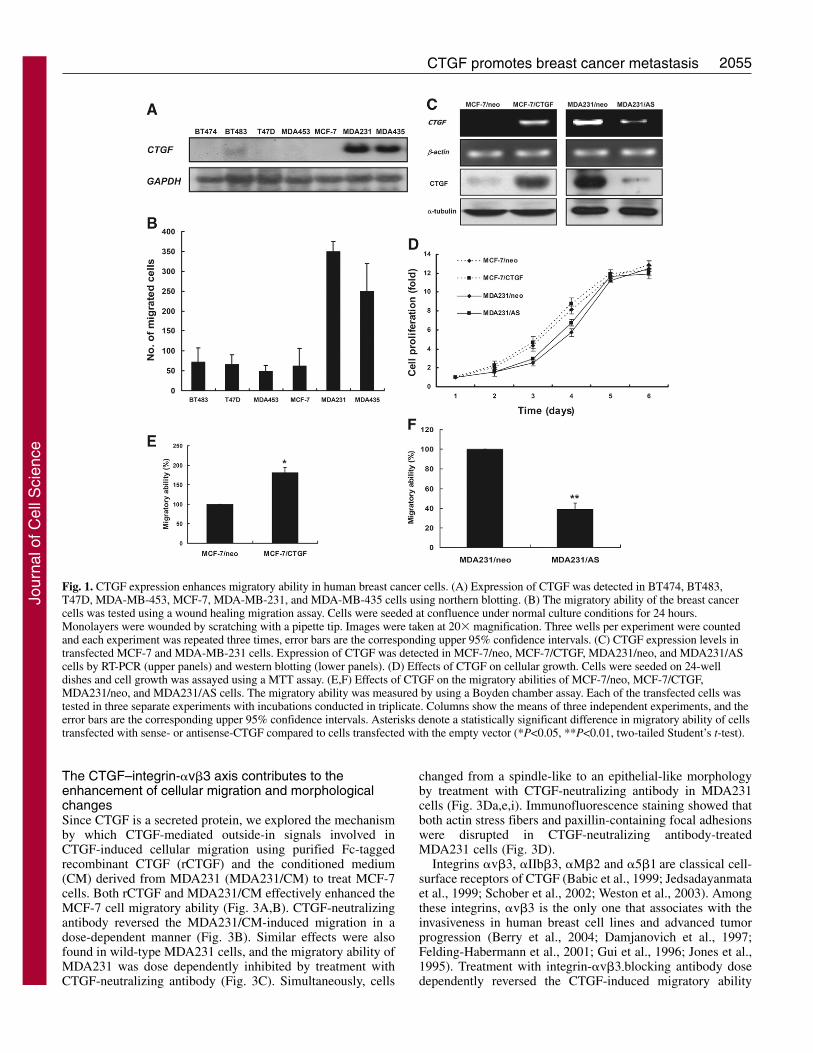
ResultsCTGF enhances the migratory ability of human breastcancer cellsCTGF levels are correlated with most of the advanced stagesof breast cancers (Xie et al., 2001). Hence, we investigatedexpression levels of CTGF by northern blot analysis in severaladeno/ductal carcinoma of human breast cancer cell linesincluding BT474, BT483, T47D, MDA-MB-453, MCF-7,
MDA231 and MDA-MB-435. The breast cancer cell linesBT474, BT483, T47D, MDA-MB-453 and MCF-7 expressedextremely low or undetectable levels of CTGF mRNA,whereas expression was high in MDA231 and MDA-MB-435(Fig. 1A). No correlation was found between CTGFexpression and their histological types. Since cell migration iscrucial for cancer cell invasiveness, we next tested the cellularmigratory ability by a wound healing migration assay.MDA231 and MDA-MB-435 cells that exuberantly expressedCTGF mRNA exhibited a higher level of migratory abilitythan the cell lines with a lower level of CTGF mRNA (Fig.1B). To assess whether CTGF was directly involved in theregulation of breast cancer cell motility, both non-migratingMCF-7 and migrating MDA231 cells were employed togenerate the CTGF-overexpression (MCF-7/CTGF) andantisense-CTGF expression (MDA231/AS) cells, respectively.The CTGF expression levels in vector control cells and stabletransfectants was compared using RT-PCR and westernblotting. Both mRNA and protein levels of CTGF weresignificantly higher in MCF-7/CTGF than in MCF-7/neo cells(Fig. 1C). However, expression of CTGF was dramaticallyinhibited by antisense-CTGF orientation in MDA231/AScells. Since, CTGF has already been reported to act as amitogen in endothelial cells (Shimo et al., 1999), we soughtto characterize the cellular growth rate of control cells andtransfectants, by performing the MTT assay 1-6 days after cellseeding (Fig. 1D). No appreciable difference in cell growthability was evident among these cells (Fig. 1D), suggestingthat CTGF does not have any mitogenic effect in human breastcancer cells. Furthermore, the migratory ability of thesetransfectants was analyzed using a Boyden-chamber migrationassay. As shown in Fig. 1E, the overexpression of CTGFincreased the migratory ability by approximately 1.7-fold inMCF-7 cells. Knockdown of CTGF expression inhibited themigratory ability by approximately 60% in MDA231 cells(Fig. 1F).
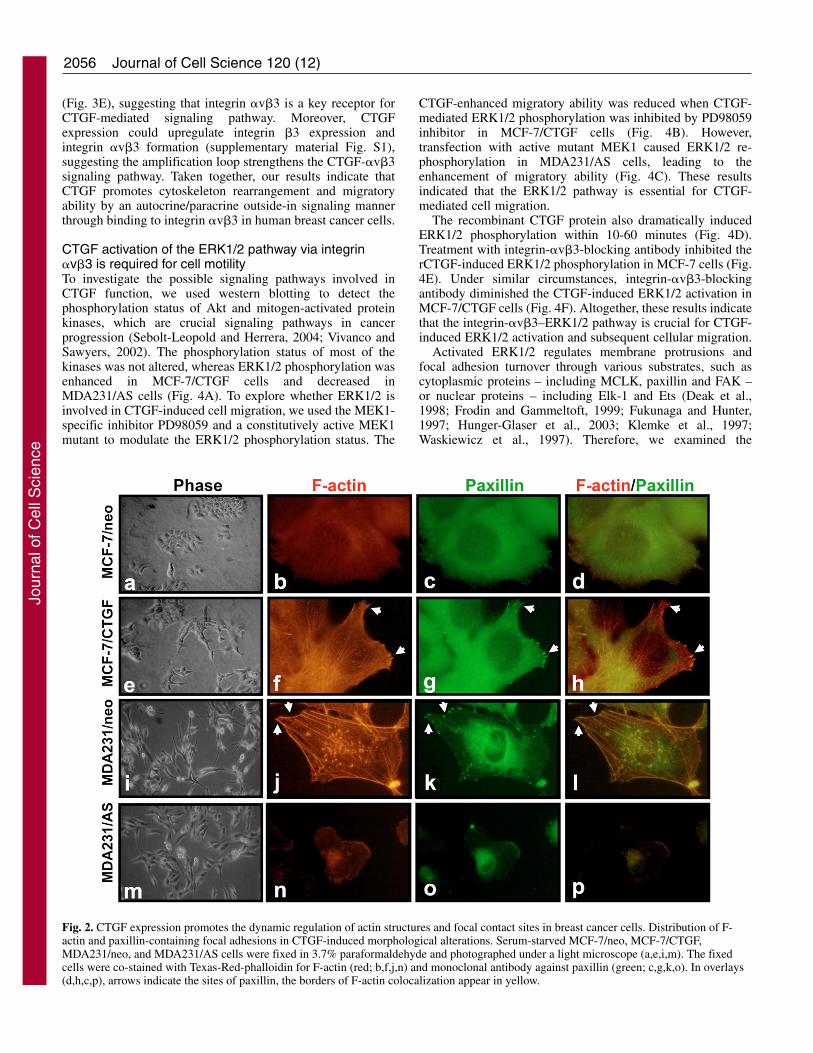
CTGF promotes the dynamic regulation of actinstructures and the formation of focal contact sites inbreast cancer cellsTransfection of MCF-7 cells with CTGF led to a change incellular morphology (Fig. 2a,e). MCF-7/CTGF cells becamemore spindle-like with long, protruding filamentous processes.Double staining of the MCF-7/neo cells using antibodiesconjugated to Texas-Red-conjugated phalloidin andmonoclonal anti-paxillin (the latter as a marker for focaladhesions) revealed few, if any, actin stress fibers, whereaspaxillin was diffusely distributed in the cells (Fig. 2b,c,d). TheMCF-7/CTGF cells exhibited numerous stress fibers (Fig. 2f)with patches of paxillin staining appearing at the leading edgesof actin stress fibers (Fig. 2f,g,h). By contrast, overexpressionof CTGF changed the MDA231 cell morphology from spindle-like to epithelial-like (Fig. 2i,m). In MDA231/neo cells, aplethora of both actin stress fibers and paxillin were evident,and paxillin was macroaggregated at focal adhesions connectedto actin stress fibers (Fig. 2i,j,k). Reducing the level ofendogenous CTGF by transfection with CTGF/ASsignificantly decreased the amount of actin stress fibers andpaxillin (Fig. 2n,o,p). These results support the hypothesis thatCTGF promotes actin contractility events and the dynamicregulation of focal contact structures in breast cancer cells.
Journal of Cell Science 120 (12)
Jour
nal o
f Cel
l Sci
ence
2055CTGF promotes breast cancer metastasis
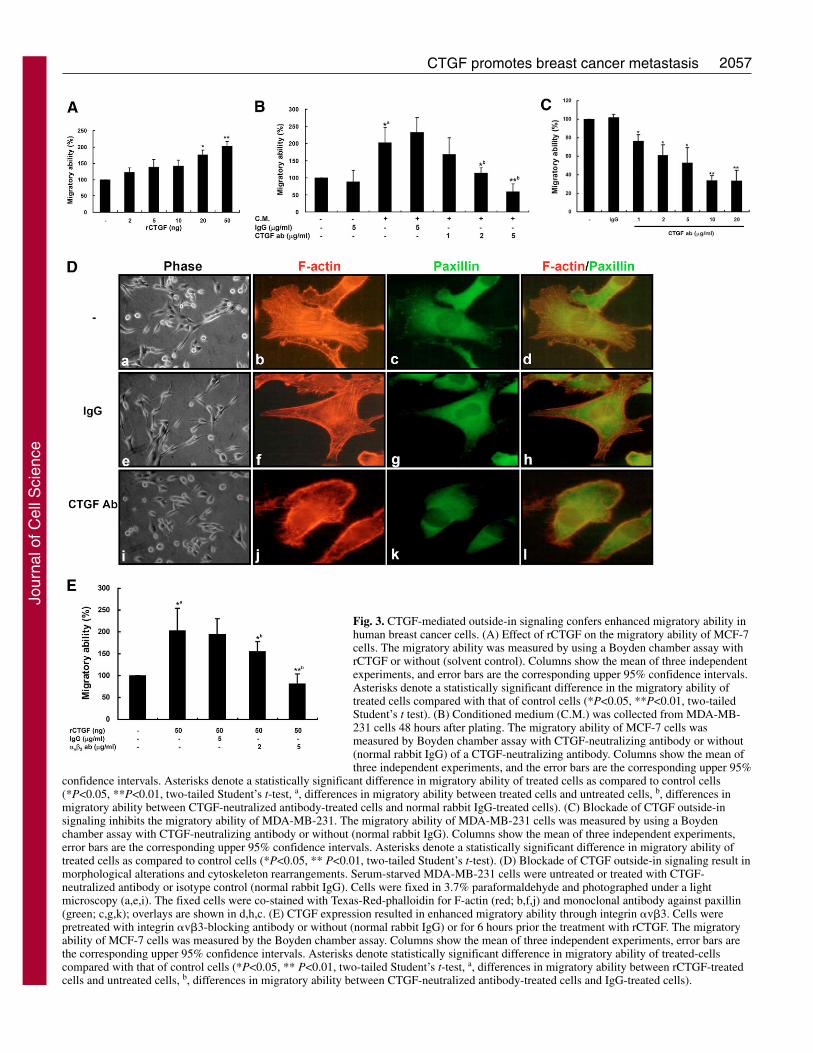
The CTGF–integrin-�v�3 axis contributes to theenhancement of cellular migration and morphologicalchangesSince CTGF is a secreted protein, we explored the mechanismby which CTGF-mediated outside-in signals involved inCTGF-induced cellular migration using purified Fc-taggedrecombinant CTGF (rCTGF) and the conditioned medium(CM) derived from MDA231 (MDA231/CM) to treat MCF-7cells. Both rCTGF and MDA231/CM effectively enhanced theMCF-7 cell migratory ability (Fig. 3A,B). CTGF-neutralizingantibody reversed the MDA231/CM-induced migration in adose-dependent manner (Fig. 3B). Similar effects were alsofound in wild-type MDA231 cells, and the migratory ability ofMDA231 was dose dependently inhibited by treatment withCTGF-neutralizing antibody (Fig. 3C). Simultaneously, cells
changed from a spindle-like to an epithelial-like morphologyby treatment with CTGF-neutralizing antibody in MDA231cells (Fig. 3Da,e,i). Immunofluorescence staining showed thatboth actin stress fibers and paxillin-containing focal adhesionswere disrupted in CTGF-neutralizing antibody-treatedMDA231 cells (Fig. 3D).
Integrins �v�3, �IIb�3, �M�2 and �5�1 are classical cell-surface receptors of CTGF (Babic et al., 1999; Jedsadayanmataet al., 1999; Schober et al., 2002; Weston et al., 2003). Amongthese integrins, �v�3 is the only one that associates with theinvasiveness in human breast cell lines and advanced tumorprogression (Berry et al., 2004; Damjanovich et al., 1997;Felding-Habermann et al., 2001; Gui et al., 1996; Jones et al.,1995). Treatment with integrin-�v�3-blocking antibody dosedependently reversed the CTGF-induced migratory ability
Fig. 1. CTGF expression enhances migratory ability in human breast cancer cells. (A) Expression of CTGF was detected in BT474, BT483,T47D, MDA-MB-453, MCF-7, MDA-MB-231, and MDA-MB-435 cells using northern blotting. (B) The migratory ability of the breast cancercells was tested using a wound healing migration assay. Cells were seeded at confluence under normal culture conditions for 24 hours.Monolayers were wounded by scratching with a pipette tip. Images were taken at 20� magnification. Three wells per experiment were countedand each experiment was repeated three times, error bars are the corresponding upper 95% confidence intervals. (C) CTGF expression levels intransfected MCF-7 and MDA-MB-231 cells. Expression of CTGF was detected in MCF-7/neo, MCF-7/CTGF, MDA231/neo, and MDA231/AScells by RT-PCR (upper panels) and western blotting (lower panels). (D) Effects of CTGF on cellular growth. Cells were seeded on 24-welldishes and cell growth was assayed using a MTT assay. (E,F) Effects of CTGF on the migratory abilities of MCF-7/neo, MCF-7/CTGF,MDA231/neo, and MDA231/AS cells. The migratory ability was measured by using a Boyden chamber assay. Each of the transfected cells wastested in three separate experiments with incubations conducted in triplicate. Columns show the means of three independent experiments, and theerror bars are the corresponding upper 95% confidence intervals. Asterisks denote a statistically significant difference in migratory ability of cellstransfected with sense- or antisense-CTGF compared to cells transfected with the empty vector (*P<0.05, **P<0.01, two-tailed Student’s t-test).
Jour
nal o
f Cel
l Sci
ence
2056
(Fig. 3E), suggesting that integrin �v�3 is a key receptor forCTGF-mediated signaling pathway. Moreover, CTGFexpression could upregulate integrin �3 expression andintegrin �v�3 formation (supplementary material Fig. S1),suggesting the amplification loop strengthens the CTGF-�v�3signaling pathway. Taken together, our results indicate thatCTGF promotes cytoskeleton rearrangement and migratoryability by an autocrine/paracrine outside-in signaling mannerthrough binding to integrin �v�3 in human breast cancer cells.
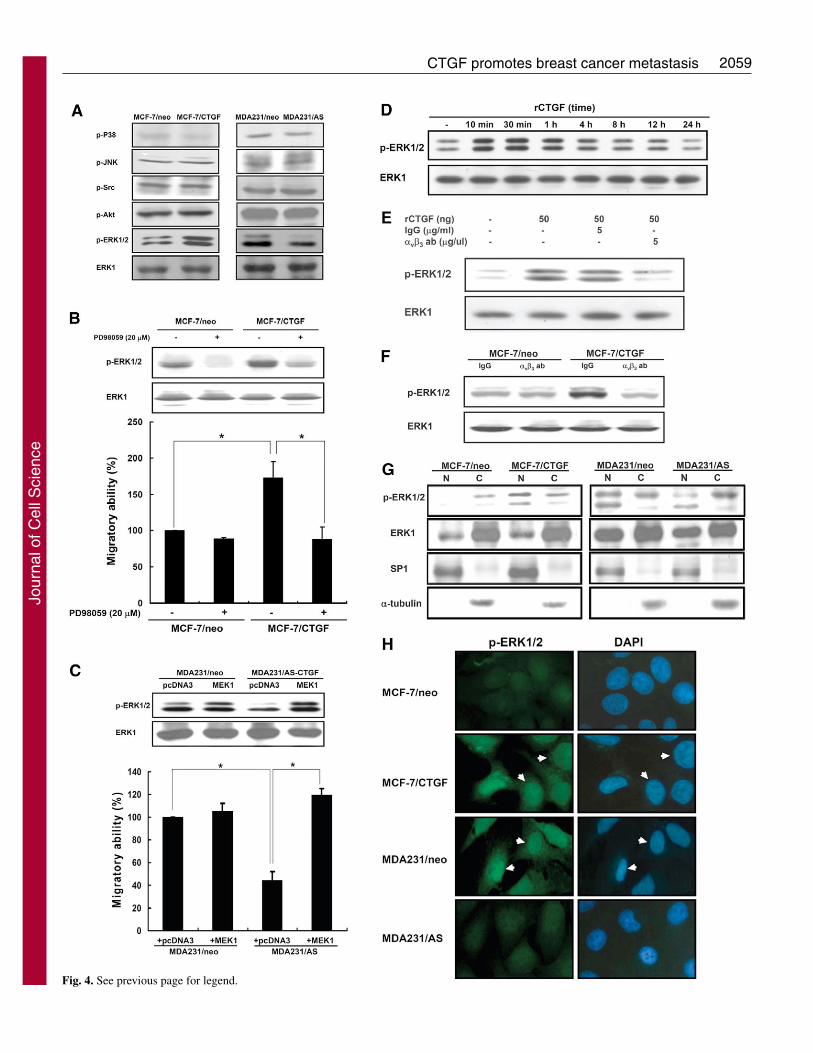
CTGF activation of the ERK1/2 pathway via integrin�v�3 is required for cell motilityTo investigate the possible signaling pathways involved inCTGF function, we used western blotting to detect thephosphorylation status of Akt and mitogen-activated proteinkinases, which are crucial signaling pathways in cancerprogression (Sebolt-Leopold and Herrera, 2004; Vivanco andSawyers, 2002). The phosphorylation status of most of thekinases was not altered, whereas ERK1/2 phosphorylation wasenhanced in MCF-7/CTGF cells and decreased inMDA231/AS cells (Fig. 4A). To explore whether ERK1/2 isinvolved in CTGF-induced cell migration, we used the MEK1-specific inhibitor PD98059 and a constitutively active MEK1mutant to modulate the ERK1/2 phosphorylation status. The
CTGF-enhanced migratory ability was reduced when CTGF-mediated ERK1/2 phosphorylation was inhibited by PD98059inhibitor in MCF-7/CTGF cells (Fig. 4B). However,transfection with active mutant MEK1 caused ERK1/2 re-phosphorylation in MDA231/AS cells, leading to theenhancement of migratory ability (Fig. 4C). These resultsindicated that the ERK1/2 pathway is essential for CTGF-mediated cell migration.
The recombinant CTGF protein also dramatically inducedERK1/2 phosphorylation within 10-60 minutes (Fig. 4D).Treatment with integrin-�v�3-blocking antibody inhibited therCTGF-induced ERK1/2 phosphorylation in MCF-7 cells (Fig.4E). Under similar circumstances, integrin-�v�3-blockingantibody diminished the CTGF-induced ERK1/2 activation inMCF-7/CTGF cells (Fig. 4F). Altogether, these results indicatethat the integrin-�v�3–ERK1/2 pathway is crucial for CTGF-induced ERK1/2 activation and subsequent cellular migration.
Activated ERK1/2 regulates membrane protrusions andfocal adhesion turnover through various substrates, such ascytoplasmic proteins – including MCLK, paxillin and FAK –or nuclear proteins – including Elk-1 and Ets (Deak et al.,1998; Frodin and Gammeltoft, 1999; Fukunaga and Hunter,1997; Hunger-Glaser et al., 2003; Klemke et al., 1997;Waskiewicz et al., 1997). Therefore, we examined the
Journal of Cell Science 120 (12)
Fig. 2. CTGF expression promotes the dynamic regulation of actin structures and focal contact sites in breast cancer cells. Distribution of F-actin and paxillin-containing focal adhesions in CTGF-induced morphological alterations. Serum-starved MCF-7/neo, MCF-7/CTGF,MDA231/neo, and MDA231/AS cells were fixed in 3.7% paraformaldehyde and photographed under a light microscope (a,e,i,m). The fixedcells were co-stained with Texas-Red-phalloidin for F-actin (red; b,f,j,n) and monoclonal antibody against paxillin (green; c,g,k,o). In overlays(d,h,c,p), arrows indicate the sites of paxillin, the borders of F-actin colocalization appear in yellow.
Jour
nal o
f Cel
l Sci
ence
2057CTGF promotes breast cancer metastasis
Fig. 3. CTGF-mediated outside-in signaling confers enhanced migratory ability inhuman breast cancer cells. (A) Effect of rCTGF on the migratory ability of MCF-7cells. The migratory ability was measured by using a Boyden chamber assay withrCTGF or without (solvent control). Columns show the mean of three independentexperiments, and error bars are the corresponding upper 95% confidence intervals.Asterisks denote a statistically significant difference in the migratory ability oftreated cells compared with that of control cells (*P<0.05, **P<0.01, two-tailedStudent’s t test). (B) Conditioned medium (C.M.) was collected from MDA-MB-231 cells 48 hours after plating. The migratory ability of MCF-7 cells wasmeasured by Boyden chamber assay with CTGF-neutralizing antibody or without(normal rabbit IgG) of a CTGF-neutralizing antibody. Columns show the mean ofthree independent experiments, and the error bars are the corresponding upper 95%
confidence intervals. Asterisks denote a statistically significant difference in migratory ability of treated cells as compared to control cells(*P<0.05, **P<0.01, two-tailed Student’s t-test, a, differences in migratory ability between treated cells and untreated cells, b, differences inmigratory ability between CTGF-neutralized antibody-treated cells and normal rabbit IgG-treated cells). (C) Blockade of CTGF outside-insignaling inhibits the migratory ability of MDA-MB-231. The migratory ability of MDA-MB-231 cells was measured by using a Boydenchamber assay with CTGF-neutralizing antibody or without (normal rabbit IgG). Columns show the mean of three independent experiments,error bars are the corresponding upper 95% confidence intervals. Asterisks denote a statistically significant difference in migratory ability oftreated cells as compared to control cells (*P<0.05, ** P<0.01, two-tailed Student’s t-test). (D) Blockade of CTGF outside-in signaling result inmorphological alterations and cytoskeleton rearrangements. Serum-starved MDA-MB-231 cells were untreated or treated with CTGF-neutralized antibody or isotype control (normal rabbit IgG). Cells were fixed in 3.7% paraformaldehyde and photographed under a lightmicroscopy (a,e,i). The fixed cells were co-stained with Texas-Red-phalloidin for F-actin (red; b,f,j) and monoclonal antibody against paxillin(green; c,g,k); overlays are shown in d,h,c. (E) CTGF expression resulted in enhanced migratory ability through integrin �v�3. Cells werepretreated with integrin �v�3-blocking antibody or without (normal rabbit IgG) or for 6 hours prior the treatment with rCTGF. The migratoryability of MCF-7 cells was measured by the Boyden chamber assay. Columns show the mean of three independent experiments, error bars arethe corresponding upper 95% confidence intervals. Asterisks denote statistically significant difference in migratory ability of treated-cellscompared with that of control cells (*P<0.05, ** P<0.01, two-tailed Student’s t-test, a, differences in migratory ability between rCTGF-treatedcells and untreated cells, b, differences in migratory ability between CTGF-neutralized antibody-treated cells and IgG-treated cells).
Jour
nal o
f Cel
l Sci
ence

2058
subcellular localization of phosphorylated (P)-ERK1/2 usingwestern blotting and immunofluorescence staining. CTGFoverexpression promoted their activation and nucleartranslocation in MCF-7 cells (Fig. 4G,H), while consistentlydecreasing the levels of P-ERK1/2 in the nucleus ofMDA231/AS cells (Fig. 4G,H). Thus, CTGF can promoteERK1/2 phosphorylation and nuclear translocation, which thenmay activate gene expression.
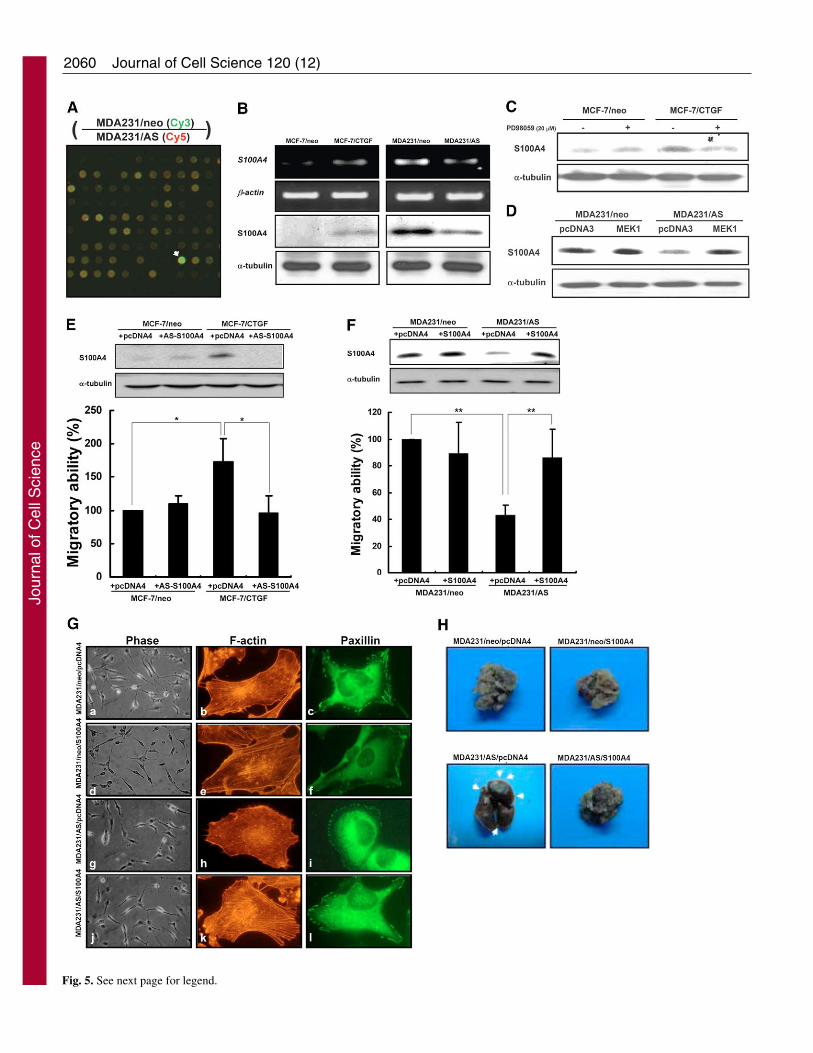
S100A4 as a downstream effecter of CTGFThe question remained as to which gene was the possibledownstream effecter that contributed to CTGF-mediatedERK1/2-dependent cell migratory effect. Since CTGF inducedP-ERK1/2 nuclear translocation, transcriptional regulationmight occur. We used a cDNA microarray to identify thegenetic expression profile (Tables 1 and 2). S100A4 (Fig. 5A)and E-cadherin are of particular interest because they functionas regulators of metastasis of human tumors, and have beenimplicated in cytoskeleton-membrane interaction, migratorybehaviors and malignancy in cancer cells (Cho et al., 2003; Cuiet al., 2004; Davies et al., 1996; Ebralidze et al., 1989; Glenney,Jr et al., 1989; Hernan et al., 2003; Jiang, 1996; Kim andHelfman, 2003; Masiakowski and Shooter, 1988;Mazzucchelli, 2002; Missiaglia et al., 2004; Nikitenko et al.,2000; Platt-Higgins et al., 2000; Rudland et al., 2000). Toconfirm the expression levels of S100A4 and E-cadherin in thestable transfactants and their control cells, we assayed S100A4
and E-cadherin levels by RT-PCR and western blotting. Theexpression of E-cadherin mRNA (data not shown) and proteinwas undetectable both in MDA231/neo and MDA231/AS cells(supplementary material Fig. S2); moreover, E-cadherin levelwas not altered under ectopic expression of CTGF in MCF-7cells (supplementary material Fig. S2), indicating that thederegulation of E-cadherin mRNA in microarray data (Table1) is a false-positive and without biological significance.However, both the mRNA and protein expression levels ofS100A4 were upregulated in MCF-7/CTGF cells butdownregulated in MDA231/AS cells (Fig. 5B). We also foundthat S100A4 was overexpressed in MDA231 and MDA435cells, suggesting a strong association with CTGF expressionand cellular aggressiveness (supplementary material Fig. S3).Subsequently, treatment with the MEK1 inhibitor PD98059effectively abolished the CTGF-induced S100A4 upregulation(Fig. 5C), whereas transfection with the constitutively activeMEK1 mutant significantly increased S100A4 expression inMDA231/AS cell (Fig. 5D). These results strongly suggest thatCTGF-mediated S100A4 upregulation depend on the ERK1/2signaling pathway.
To ascertain the role of S100A4 in CTGF-mediated cellmigration, we generated the pcDNA4-S100A4 and pcDNA4-AS-S100A4 constructs to establish the doubly transfectedstable transfectants of MDA231/AS and MCF-7/CTGF cells,respectively. S100A4 expression levels were reduced by AS-
Journal of Cell Science 120 (12)
Fig. 4. ERK1/2 is activated by CTGF via integrin �v�3 and confersenhanced migratory ability. (A) Effect of CTGF on the activation ofAkt and MAPKs. Cells were serum-starved for 24 hours; activatedlevel of Akt and MAPKs were measured by western blotting withphosphorylation-specific antibodies. (B) The role of P-ERK1/2 inCTGF-mediated cellular migration. Migration assays wereperformed as described above, MCF-7/neo and MCF-7/CTGF cellswere treated with dimethylsulfoxide or PD98059 (20 �M), whereascells were seeded on the upper chambers (lower figure). Cell lysateswere simultaneously analyzed by western blotting with antibodiesagainst P-ERK1/2 and ERK1 (upper panel). (C) MDA231/neo andMDA231/AS cells were transiently transfected active MEK1, 48hours after transfection, cells were trypsinized and assayed using aBoyden chamber (lower figure). Simultaneously, cell lysates wereanalyzed by western blotting with antibodies against P-ERK1/2 andERK1/2 (upper panel). (D) Effect of rCTGF on ERK1/2 activation inMCF-7 cells. Wild-type MCF-7 cells (5�105) were seeded in 6-cmdishes, serum-starved for 24 hours and then treated with 50 ng/mlrCTGF for 20 minutes to 24 hours. P-ERK1/2 and ERK1 levels weredetected by western blotting. (E) Wild-type MCF-7 cells (5�105)were seeded in 6-cm dishes. Cells were serum-starved, pretreatedwith 5 ug/ml IgG or integrin-�v�3-blocking antibodies for 24 hoursand then treated with 50 ng/ml rCTGF for 10 minutes. P-ERK1/2and ERK1 were detected by western blotting. (F) p-ERK1/2 andERK1 expression in MCF-7/neo and MCF-7/CTGF cells treated withintegrin-�v�3-blocking antibody. Cells were treated with 5 ug/mlIgG or integrin-�v�3-blocking antibodies for 24 hours and P-ERK1/2 and ERK1 levels was measured by Western blotting.(G) Subcellular localization of CTGF-activated ERK1/2 analyzed bywestern blotting. Cells were serum-starved for 24 hours, and thenuclear and cytosolic fractions were isolated (see Materials andMethods); P-ERK1/2 and ERK1 were detected by western blotting.(H) Subcellular localization of CTGF-induced P-ERK1/2 wasanalyzed by immunofluorescence staining. Cells were fixed in 3.7%paraformaldehyde and co-stained with anti-P-ERK1/2 antibody andDAPI. Arrows indicate increased expression of CTGF-induced P-ERK1/2 in nuclei of breast cancer cells.
Table 1. Metastasis-associated genes downregulated(>fourfold reduction) by AS-CTGF in MDA231 cells
Gene name GenBank Acc. No.
CDH1 NM_004360ANXA8 NM_001630ITGB3 NM_000212AREG NM_001657TFF2 NM_005423CDH11 NM_033664S100A4 NM_002961MMP15 NM_002428ITGB4 NM_000213NRG1 NM_013958IL8RB NM_001557KIT NM_000222PDGFRA NM_006206PTGES NM_004878CSF1 NM_000757TGFA NM_003236
Table 2. Metastasis-associated genes upregulated(>fourfold induction) by AS-CTGF in MDA231 cells
Gene name GenBank Acc. No.
MYO1D NM_015194FGF13 NM_004114F3 NM_001993PCDH7 NM_002589LDB2 NM_001290NGFR NM_002507MGMT NM_002412IL8 NM_000584CCNG2 NM_004354IL1RN NM_173841TPD52L1 NM_001003395RASSF2 NM_014737IL11RA NM_147162
Jour
nal o
f Cel
l Sci
ence
2059CTGF promotes breast cancer metastasis
Fig. 4. See previous page for legend.
Jour
nal o
f Cel
l Sci
ence
2060 Journal of Cell Science 120 (12)
Fig. 5. See next page for legend.
Jour
nal o
f Cel
l Sci
ence

2061CTGF promotes breast cancer metastasis
S100A4 in MCF-7/CTGF cells, which also decreased themigratory ability (Fig. 5E). Restoring levels of S100A4 inMDA231/AS cells significantly enhanced the migratory ability(Fig. 5F). Immunofluorescence staining further showed thattransfection with S100A4 significantly promoted re-polymerization of actin stress fibers and aggregation of focaladhesion complexes in MDA231/AS cells (Fig. 5G). Theseresults indicate that S100A4 is a crucial downstream effecterinvolved in CTGF-mediated cytoskeleton rearrangement andcellular migration.
To investigate whether the CTGF-induced S100A4expression plays a causal role in tumor metastasis, we injectedMDA231/neo/pcDNA4, MDA231/neo/S100A4, MDA231/ AS/pcDNA4, and MDA231/AS/S100A4 cells intravenously intothe tail vein of nude mice and measured the metastaticcolonization. Whereas tumors expressing the AS-CTGFformed very few macroscopically visible metastases in thelungs, tumors that co-expressed AS-CTGF and S100A4formed a large number of metastases (Fig. 5H). In theMDA231/neo/pcDNA4 group, the mean lung nodule numberwas 39; in the MDA231/AS/pcDNA4 group, the mean lungnodule number was 9. The numbers of visible metastaticnodules in mice injected with MDA231/neo/pcDNA4 cells
were higher than those in MDA231/AS/pcDNA4-bearing mice(Table 3). In the MDA231/AS/S100A4 group, the mean lungnodule number was 24 (Table 3), indicating that transfectionwith S100A4 dramatically increased the mean number ofmetastatic nodules and the occurrence of lung metastasis inMDA231/AS tumor-bearing mice. Together, these datademonstrate that CTGF-mediated tumor metastasis requiresthe expression of S100A4
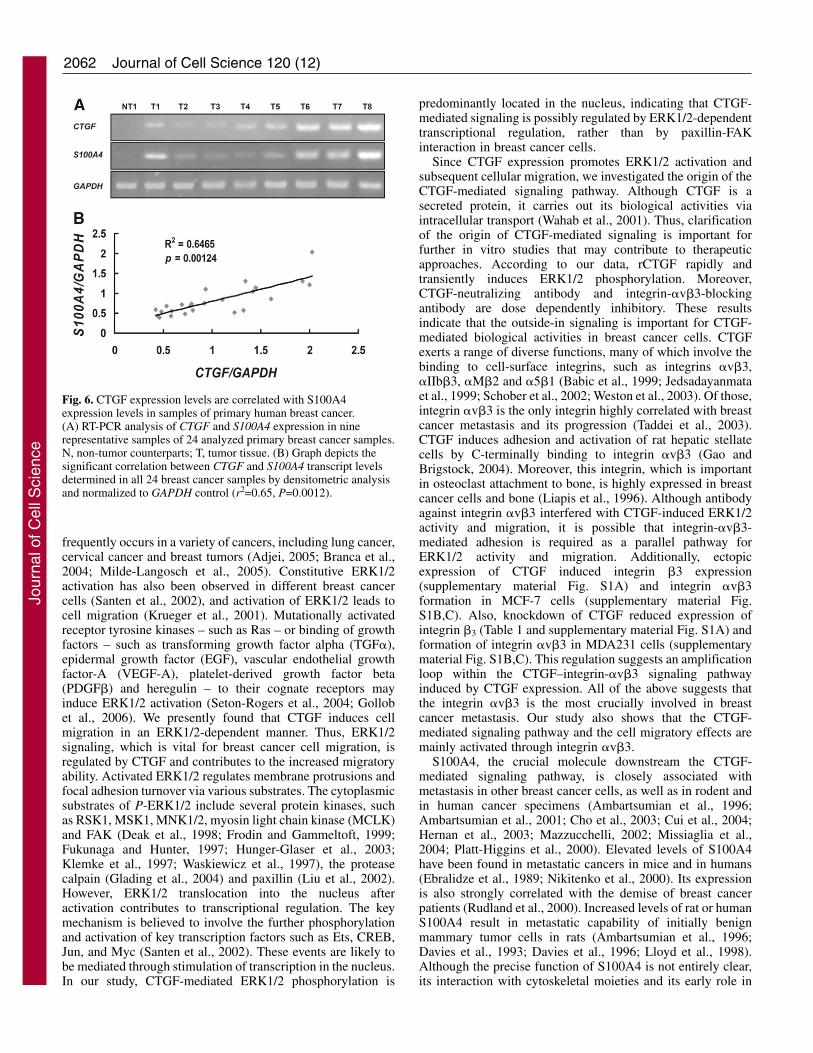
CTGF expression levels are correlated with S100A4expression levels in primary human breast cancerTo establish whether expression levels of CTGF and S100A4are correlated in clinical breast cancer, 24 primary humanbreast cancer specimens and their non-tumor counterparts werecollected, and analyzed semi-quantitatively by RT-PCR andquantitatively by densitometry (Fig. 6). Non-tumor cellsdisplayed extremely low or undetectable levels of CTGF andS100A4 expression. Conversely, all tumor tissues expressedboth CTGF and S100A4 (Fig. 6A). Based on our observationsin breast cancer cells, the expression levels of CTGF andS100A4 were found to closely correlate in-patient samples(Fig. 6B, r2=0.65, P=0.0012). These data strongly support thehypothesis that CTGF signaling upregulates the expression ofS100A4 in primary human breast cancer.
DiscussionIn this study, we demonstrate that the molecular mechanism bywhich CTGF-confers cellular metastatic ability is mediated byS100A4 upregulation by integrin �v�3 and/or ERK1/2. Thisis based on the following evidence. First, overexpression ofCTGF appreciably increases the migratory ability of MCF-7cells. Conversely, knockdown of CTGF abolishes themigratory ability of MDA231 cells. Second, CTGF expressionleads to morphological alterations and formation of F-actin andfocal adhesions. Third, ERK1/2 activation is essential for theCTGF-mediated migratory effects. Fourth, blockade of theCTGF–integrin-�v�3 axis attenuates CTGF-induced ERK1/2activation and subsequent cellular migration. Fifth, theprometastatic S100A4 gene is regulated by the signalingcascades of CTGF–integrin-�v�3–ERK1/2 and contributes tothe metastatic ability. Finally, CTGF expression levels arecorrelated with S100A4 expression levels in primary humanbreast tumors.
When CTGF-overexpressing cells were treated with theMEK1 inhibitor PD98059 their migratory ability was inhibited.Moreover, constitutively active MEK1 also prevented themigratory ability in MDA231/AS cells. Thus, our data suggestthat the ERK1/2 signaling pathway is crucial for CTGF-induced cell migration. In support of our observations, othershave demonstrated that constitutive activation of ERK1/2
Fig. 5. S100A4 acts as a crucial downstream effecter of CTGF.(A) cDNA microarray analysis (Cy3-Cy5 merged image) showingthe expression of S100A4, arrow indicates human S100A4.(B) Expression of S100A4 mRNA and protein by RT-PCR andwestern blotting, respectively. (C) Effects of ERK1/2 activation onCTGF-regulated S100A4 expression. MCF-7/neo and MCF-7/CTGFcells were treated with DMSO or PD98059 (20 �M) for 24 hours,and the levels of S100A4 were evaluated by western blotting.(D) MDA231/neo and MDA231/AS cells were transientlytransfected by constitutively activated MEK1; 48 hours aftertransfection, expression levels of S100A4 were evaluated by westernblotting. (E) Effects of S100A4 on CTGF-mediated cellular motility.MCF-7/neo and MCF-7/CTGF cells were stably transfected withpcDNA4 or pcDNA4-AS-S100A4, respectively (see Materials andMethods). Expression levels of S100A4 were detected by westernblotting (upper panel). Cellular motility was measured by migrationassay (lower panel). (F) MDA231/neo and MDA231/AS cells werestably transfected with pcDNA4 or pcDNA4-S100A4, respectively.Expression levels of S100A4 were detected by western blotting(upper panel). Cellular motility was measured by migration assay(lower panel). (G) S100A4 is involved in CTGF-mediatedcytoskeletal changes. Cells were fixed in 3.7% paraformaldehyde andphotographed under a light microscope (a,d,g,i). The fixed cells wereco-stained with Texas-Red-phalloidin for F-actin (red; b,e,h,k) andmonoclonal antibody against paxillin (green; c,f,i,l). (H) S100A4expression is required for CTGF-mediated metastatic colonization.Lungs of female BALE/cAnN-Foxn1nu/CrlNarl nude mice wereexcised and photographed after the experimental metastasis assay.
Table 3. Regulation of lung metastasis in nude mice by the CTGF-S100A4 pathwayLung weight (mg) Number of mice with lung Mean lung
Cell lines Mean ± s.d. metastasis vs total number of mice nodules (range)
MDA231/neo/pcDNA4 468±69 6/10 39 (0-58)MDA231/neo/S100A4 472±60 7/10 33 (0-61)MDA231/AS/pcDNA4 424±41 4/10 9 (0-26)a,**MDA231/AS/S100A4 461±56 6/10 28 (0-55)b,**
aMDA231/AS/pcDNA4 versus MDA231/neo/pcDNA4 by the Student’s t-test; bMDA231/AS/S100A4 versus MDA231/neo/pcDNA4 by the Student’s t-test,**P<0.01.
Jour
nal o
f Cel
l Sci
ence
2062
frequently occurs in a variety of cancers, including lung cancer,cervical cancer and breast tumors (Adjei, 2005; Branca et al.,2004; Milde-Langosch et al., 2005). Constitutive ERK1/2activation has also been observed in different breast cancercells (Santen et al., 2002), and activation of ERK1/2 leads tocell migration (Krueger et al., 2001). Mutationally activatedreceptor tyrosine kinases – such as Ras – or binding of growthfactors – such as transforming growth factor alpha (TGF�),epidermal growth factor (EGF), vascular endothelial growthfactor-A (VEGF-A), platelet-derived growth factor beta(PDGF�) and heregulin – to their cognate receptors mayinduce ERK1/2 activation (Seton-Rogers et al., 2004; Gollobet al., 2006). We presently found that CTGF induces cellmigration in an ERK1/2-dependent manner. Thus, ERK1/2signaling, which is vital for breast cancer cell migration, isregulated by CTGF and contributes to the increased migratoryability. Activated ERK1/2 regulates membrane protrusions andfocal adhesion turnover via various substrates. The cytoplasmicsubstrates of P-ERK1/2 include several protein kinases, suchas RSK1, MSK1, MNK1/2, myosin light chain kinase (MCLK)and FAK (Deak et al., 1998; Frodin and Gammeltoft, 1999;Fukunaga and Hunter, 1997; Hunger-Glaser et al., 2003;Klemke et al., 1997; Waskiewicz et al., 1997), the proteasecalpain (Glading et al., 2004) and paxillin (Liu et al., 2002).However, ERK1/2 translocation into the nucleus afteractivation contributes to transcriptional regulation. The keymechanism is believed to involve the further phosphorylationand activation of key transcription factors such as Ets, CREB,Jun, and Myc (Santen et al., 2002). These events are likely tobe mediated through stimulation of transcription in the nucleus.In our study, CTGF-mediated ERK1/2 phosphorylation is
predominantly located in the nucleus, indicating that CTGF-mediated signaling is possibly regulated by ERK1/2-dependenttranscriptional regulation, rather than by paxillin-FAKinteraction in breast cancer cells.
Since CTGF expression promotes ERK1/2 activation andsubsequent cellular migration, we investigated the origin of theCTGF-mediated signaling pathway. Although CTGF is asecreted protein, it carries out its biological activities viaintracellular transport (Wahab et al., 2001). Thus, clarificationof the origin of CTGF-mediated signaling is important forfurther in vitro studies that may contribute to therapeuticapproaches. According to our data, rCTGF rapidly andtransiently induces ERK1/2 phosphorylation. Moreover,CTGF-neutralizing antibody and integrin-�v�3-blockingantibody are dose dependently inhibitory. These resultsindicate that the outside-in signaling is important for CTGF-mediated biological activities in breast cancer cells. CTGFexerts a range of diverse functions, many of which involve thebinding to cell-surface integrins, such as integrins �v�3,�IIb�3, �M�2 and �5�1 (Babic et al., 1999; Jedsadayanmataet al., 1999; Schober et al., 2002; Weston et al., 2003). Of those,integrin �v�3 is the only integrin highly correlated with breastcancer metastasis and its progression (Taddei et al., 2003).CTGF induces adhesion and activation of rat hepatic stellatecells by C-terminally binding to integrin �v�3 (Gao andBrigstock, 2004). Moreover, this integrin, which is importantin osteoclast attachment to bone, is highly expressed in breastcancer cells and bone (Liapis et al., 1996). Although antibodyagainst integrin �v�3 interfered with CTGF-induced ERK1/2activity and migration, it is possible that integrin-�v�3-mediated adhesion is required as a parallel pathway forERK1/2 activity and migration. Additionally, ectopicexpression of CTGF induced integrin �3 expression(supplementary material Fig. S1A) and integrin �v�3formation in MCF-7 cells (supplementary material Fig.S1B,C). Also, knockdown of CTGF reduced expression ofintegrin �3 (Table 1 and supplementary material Fig. S1A) andformation of integrin �v�3 in MDA231 cells (supplementarymaterial Fig. S1B,C). This regulation suggests an amplificationloop within the CTGF–integrin-�v�3 signaling pathwayinduced by CTGF expression. All of the above suggests thatthe integrin �v�3 is the most crucially involved in breastcancer metastasis. Our study also shows that the CTGF-mediated signaling pathway and the cell migratory effects aremainly activated through integrin �v�3.
S100A4, the crucial molecule downstream the CTGF-mediated signaling pathway, is closely associated withmetastasis in other breast cancer cells, as well as in rodent andin human cancer specimens (Ambartsumian et al., 1996;Ambartsumian et al., 2001; Cho et al., 2003; Cui et al., 2004;Hernan et al., 2003; Mazzucchelli, 2002; Missiaglia et al.,2004; Platt-Higgins et al., 2000). Elevated levels of S100A4have been found in metastatic cancers in mice and in humans(Ebralidze et al., 1989; Nikitenko et al., 2000). Its expressionis also strongly correlated with the demise of breast cancerpatients (Rudland et al., 2000). Increased levels of rat or humanS100A4 result in metastatic capability of initially benignmammary tumor cells in rats (Ambartsumian et al., 1996;Davies et al., 1993; Davies et al., 1996; Lloyd et al., 1998).Although the precise function of S100A4 is not entirely clear,its interaction with cytoskeletal moieties and its early role in
Journal of Cell Science 120 (12)
Fig. 6. CTGF expression levels are correlated with S100A4expression levels in samples of primary human breast cancer.(A) RT-PCR analysis of CTGF and S100A4 expression in ninerepresentative samples of 24 analyzed primary breast cancer samples.N, non-tumor counterparts; T, tumor tissue. (B) Graph depicts thesignificant correlation between CTGF and S100A4 transcript levelsdetermined in all 24 breast cancer samples by densitometric analysisand normalized to GAPDH control (r2=0.65, P=0.0012).
Jour
nal o
f Cel
l Sci
ence

2063CTGF promotes breast cancer metastasis
EMT indicates that S100A4 expression is involved in themigratory process (Okada et al., 1997). This may result fromits role in the reorganization of cytoskeletal components, suchas nonmuscle myosin II (Ford et al., 1995; Kriajevska et al.,1994), actin (Watanabe et al., 1993) and tropomycin (Takenagaet al., 1994), which indicates that S100A4 is involved inmotility by interacting with various components of thecytoskeleton (Okada et al., 1997). In addition, S100A4 can actas an angiogenic factor (Ambartsumian et al., 2001), and cannegatively regulate bone mineralization and osteoblastdifferentiation (Duarte et al., 2003). In endothelial cells,S100A4 activates transcription of matrix metallopeptidasesMMP11, MMP13 and MMP14 (Schmidt-Hansen et al., 2004a;Schmidt-Hansen et al., 2004b). Accordingly, we hypothesizethat CTGF-regulated S100A4 expression not only confersincreased metastasis, but also regulates angiogenesis, bonehomeostasis and the transcription of MMPs. Together, ourstudy and studies by others provide the evidence that S100A4is a crucial factor in modulating motility in human breastcancer cells, and that its expression can be regulated by CTGF.
In conclusion, we provide a detailed mechanism describingthe potential role of CTGF in migratory behavior via anintegrin-�v�3–P-ERK1/2-dependent upregulation of S100A4.This is the first identification of a new molecular mechanismregulating the CTGF-driven prometastatic pathway, possiblyproviding a new target for therapeutic intervention inmetastatic breast cancer.
Materials and MethodsCell culture, antibodies and reagentsThe human breast cancer cell lines MCF-7 and MDA231 were obtained from theAmerican Type Culture Collection. BT474, BT483, MDA-MB-453, MDA231 andMDA-MB-435 cells were provided generously by the National Health ResearchInstitute (Taipei, Taiwan). These cell lines were maintained in minimum essentialmedium (MEM) supplemented with 10% fetal bovine serum (FBS; Gibco BRL,Gaithersburg, MD), 2 mM L-glutamine (Life Technologies, Carlsbad, CA),100 �g/ml streptomycin and 100 U/ml penicillin in a humidified 5% CO2
atmosphere. Monoclonal mouse anti-human CTGF antibody was purchased fromR&D Systems (Minneapolis, MN). Antibodies against integrin �v�3, �-tubulin, P-ERK1/2 and ERK1/2 were purchased from Santa Cruz Biotechnology (Santa Cruz,CA). Polyclonal rabbit anti-human S100A4 antibody was purchased from AbnovaBiotechnology (Taipei, Taiwan). Anti-paxillin antibody was from BD Biosciences(Franklin Lakes, NJ). Goat anti-rabbit or anti-mouse IgG conjugated with eitherfluorescein isothiocynate (FITC) or tetramethylrhodamine isothiocyanate (TRITC)were purchased from Jackson Immunoresearch (West Grove, PA). Diamidinophenylindole dimethylsulfoxide (DAPI) was purchased from Molecular Probes(Eugene, OR). The constitutively active MEK1 mutant construct (substitution of theregulatory phosphorylation sites S218D and S222D) as described previously(Brunet et al., 1994) was gift from Ruey-Hwa Chen.
Boyden chamber migration assaysMigration of human breast cancer cells through polycarbonate filters was examinedin 24-well modified Boyden chambers (pore size 8 �m). The lower wells of thechamber were loaded with 1 ml of MEM. Breast cancer cells (50,000 cells per 100�l) were placed in the upper wells. After 6 hours of incubation, cells on the lowersurface of the filter were fixed and stained with Crystal Violet and counted using amicroscope (type 090-135.001; Leica Microsystems, Wetzlar, Germany).Incubations were performed in triplicate and each experiment was repeated at leastthree times.
Establishment of stably transfected cellsA 1.2-kb pair sense- or antisense-orientation cDNA fragment of human CTGF waseach cloned into the vector pcDNA3 to produce sense- and antisense-oriented CTGFexpression vectors. Furthermore, 0.4 kb sense- or antisense cDNA fragments ofhuman S100A4 were each cloned into the vector pcDNA4 to produce sense-oriented(pcDNA4-S100A4) and antisense-oriented (pcDNA4-AS-S100A4) S100A4expression vectors. Cells were transfected with plasmids using the lipofectintransfection reagent (Invitrogen, Carlsbad, CA). Transfected cells were grown in anatmosphere of 5% CO2 at 37°C in MEM supplemented with 10% FBS. After 48hours of transfection with pcDNA3-based plasmids, cells were trypsinized and
replated in MEM containing 10% FBS 800 �g/ml G418. G418-resistant clones(MCF-7/neo, MCF-7/CTGF, MDA231/neo, and MDA231/AS) were selected andpooled for further studies. Doubly transfected cells harboring another pcDNA4-based plasmid were trypsinized after 48 hours of transfection, and replated in MEMsupplemented with 10% FBS, 50 �g/ml G418 and 500 ug/ml zeocin. Zeocin-resistant clones were selected, expanded and pooled for further studies.
Western blottingProteins in the total cell lysate (40 �g of protein) were separated on 10% SDS-PAGE and electrotransferred to a polyvinylidene difluoride membrane (Immobilon-P membrane; Millipore, Bedford, MA). After the blot was blocked in a solution of5% skimmed milk, 0.1% Tween 20 and PBS, membrane-bound proteins wereprobed with primary antibodies against CTGF, S100A4, P-ERK1/2, ERK1/2, SP1(Santa Cruz Biotechnology, Santa Cruz, CA). The membrane was washed and thenincubated with horseradish peroxidase-conjugated secondary antibodies for 30minutes. Antibody-bound protein bands were detected with enhancedchemiluminescence reagents (Amersham Pharmacia Biotech, Piscataway, NJ) andphotographed with Kodak X-Omat Blue autoradiography film (Perkin Elmer LifeSciences, Boston, MA).
RNA isolation and RT-PCRTotal RNA was isolated using Trizol (Invitrogen, Carlsbad, CA) according to themanufacturer’s instructions, and 1 �g was reverse transcribed into single-strandedcDNA with M-MLV reverse transcriptase and random hexamers (Promega,Madison, WI). Amplification of cDNAs was performed by PCR with specific primerpairs. Primers were, for CTGF 5�-ACTGTCCCGGAGACAATGAC-3� (forward)and 5�-TGCTCCTAAAGCCACACCTT-3� (reverse), for S100A4 5�-TCTCTCC -TCAGCGCTTCTTC-3� (forward) and 5�-CTTCCTGGGCTGCTTA TCTG-3�(reverse), for GAPDH 5�-ACCCAGAAGACTGTGGATGG-3� (forward) and 5�-GTCCACCACCCTGTTGCTGT-3� (reverse). PCR conditions were: denaturingonce at 95°C (10 minutes), 95°C (1 minute), 52°C (1 minute) and 72°C (1 minute)for 30 cycles, once at 72°C (10 minutes). PCR products were analysed using agarosegel electrophoresis.
Immunofluorescence stainingFor immunofluorescence staining, all fixation and staining procedures wereconducted at room temperature. Cultures were grown in a Lab-Tek four-chamberslide apparatus (Nalge NUNC, Rochester, NY), fixed with cold 4%paraformaldehyde for 15 minutes, washed three times with phosphate-bufferedsaline (PBS) and permeabilized with 0.1% Triton X-100 in PBS for 5 minutes. Afterwashing three times with 0.05% Tween 20 in PBS (PBST), cultures were blockedin 5% nonfat milk (Carnation) in PBST for 30 minutes. Cells were incubated withprimary antibodies against paxillin or P-ERK1/2) for 1 hour and incubated at roomtemperature with Texas-Red-conjugated phalloidin (Molecular Probes) as specifiedin each experiment. After washing, the cells were incubated with secondaryantibody (1:100) and DAPI (1:5000) for 60 minutes. The control samples includedthose treated with either secondary antibody alone or with pre-immune mouseserum. The slides were mounted with VectorShield (Vector Laboratories,Burlingame, CA) and examined under a Nikon Eclipse E600 upright microscopeequipped with fluorescent devices.
Northern blot analysisTotal RNA was isolated using Trizol (Invitrogen, Carlsbad, CA) according to themanufacturer’s instructions and 10 �g were separated by electrophoresis on 1.2%agarose. RNA was transferred to Hybond-XL membranes (Amersham PharmaciaBiotech, Piscataway, NJ), then hybridized with cDNA probe and randomly labeledwith �[32P]dATP (3000 Ci/mmol/l; DuPont/NEN, Boston, MA). Probes werelabeled using the Prime-It II Random Primer kit (Stratagene, La Jolla, CA).Membranes were stripped and reprobed with cDNA for GAPDH mRNA andexposed on Kodak X-OMAT LS film (Eastman Kodak, Rochester, NY) was exposedfor various periods of time to the blots.
Flow cytometryCells were washed once with PBS and harvested in 0.05% trypsin/0.025% EDTA.Detached cells were washed with PBS containing 1% FCS and 1% penicillin-streptomycin (wash buffer), and resuspended in wash buffer (106 cells per 100 �l).Cells were then incubated with anti-integrin �v�3 monoclonal antibodies (SantaCruz Biotechnology) or respective isotype controls and FITC-conjugated secondaryantibodies (Sigma-Aldrich) for 30 minutes on ice. The labeled cells were washedin wash buffer, fixed in PBS containing 1% paraformaldehyde and then analyzedon a FACSVantage (BD Biosciences).
Microarray analysisThe Agilent human 1 cDNA microarray (Agilent Technology) containing 18,564spots of 13,574 different genes was used in this study. Fifteen micrograms of purifiedtotal RNA were converted to cDNA using a 3DNATM Array 50 Expression ArrayDetection Kit (Genisphere). RNA was labeled with Cy3, and RNA from UniversalHuman Reference RNA was labeled with Cy5. Correspondingly synthesized cDNA
Jour
nal o
f Cel
l Sci
ence

2064
products were combined and concentrated by ethanol precipitation and suspended inhybridization buffer. Labeled cDNA was hybridized to Agilent human 1 cDNAmicroarray (Agilent Technologies) at 65°C for 17 hours. After hybridization, slideswere washed in 5�SSC with 0.01% SDS at room temperature for 5 minutes and0.06�SSC at room temperature for 2 minutes. Washed microarrays were thenhybridized with Cy3 and Cy5 dendrimers in formamide-based buffer at 53°C for 3hours. After hybridization with dendrimers, slides were washed in 2�SSC with0.01% SDS at 42°C for 15 minutes, 2�SSC at room temperature for 10 minutes,and 0.2�SSC at room temperature for 10 minutes. Washed microarrays were scannedwith a Virtek fluorescence reader (Virtek, CA) at 532 nm and 635 nm for Cy3 andCy5, respectively. Scanned images were analyzed by Array-Pro image acquisitionsoftware (Media Cybernetics); an image analysis algorithm was used to quantifysignal and background intensity for each target spot (gene). The mean intensity ofthe spot area was computed as the microarray raw data. The data were furtheranalyzed with software package S-Plus 6.1 (Venables, 2002).
Experimental metastasisCells were washed and resuspended in PBS. Subsequently, a single-cell suspensioncontaining 106 cells in 0.1 ml of PBS was injected into the lateral tail vein of 7-week-old female nude mice (supplied by the animal center in the College ofMedicine, National Taiwan University, Taipei, Taiwan). Mice were killed after 18weeks. (Our preliminary study in this animal model indicated that MDA231 cellsdeveloped numerous lung metastasis nodules by 18 weeks.) All organs wereexamined for metastasis formation. Lungs were removed, weighed and fixed in 10%formalin. Lung tumor colonies were counted under a dissecting microscope. Allanimal work was performed usinmg protocols approved by the Institutional AnimalCare and Use Committee of the College of Medicine, National Taiwan University.
Wound healing assayCell monolayers were wounded 24 hours after plating by scratching with a pipettetip. Debris was removed by washing. Images were taken at 20� magnification(Nikon Ph1 DL; NA 0.4) with a Nikon TMS microscope equipped with a Nikon F-601 camera. Distances between wound edges were measured at five sites/image(n=3). Alternatively, wounded monolayers were subjected to time-lapse studies for3 hours at 10� magnification using a Zeiss Axiovert 200 microscope equipped withthe AxioCam digital system.
Purification of the rCTGF proteinTo purify the recombinant CTGF protein (rCTGF) protein, 293T cells were used toexpress rCTGF. Protein A Sepharose 4 Fast Flow was used with an Atka-fast proteinliquid chromoatography system (Amersham Pharmacia, Freiburg, Germany).Columns were equilibrated with PBS (pH 7.0), the supernatant (15-45 ml) wasapplied at a flow rate of 2 ml/minute. Columns were washed with ten columnvolumes of PBS and the protein was eluted with elution buffer (0.1 M glycine pH2.5). Directly thereafter, eluted fractions were neutralized using 3 M Tris-HCl pH8. The rCTGF protein was desalted using a PD-10 column (Amersham Pharmacia)and the purity of the protein checked by silver staining and western blotting.
Preparation of CTGF-neutralizing antibodyAnti-CTGF antibody was raised in rabbits by immunization with a synthetic CTGFpeptide composed of 20 amino acids (aa 243 to aa 263; EADLEENIKKGKK -CIRTPKIS). This sequence is shared with CTGF (also known as Fisp12 in mouse)(the mouse homologue of), but not CYR61, NOV, WISP1 (also known as Elm1 inmouse), WISP2 (also known as rCop1 in mouse) or WISP3. Anti-CTGF antibodywas purified from the serum as previously described (Shimo et al., 1999).
We thank the anonymous reviewers for helpful comments. Thiswork was supported by grants from the Department of IndustrialTechnology, Ministry of Economic Affairs, Taipei, Taiwan (95-EC-17-A-19S1-016), the National Science Council, Taiwan (NSC95-2314-B-002-318-MY3, NSC94-2320-B-002-012, and NSC94-2323-B-002-010), and the National Taiwan University Hospital (NTUH-94M04). We thank Tung-Tien Sun (Department of Urology, New YorkUniversity School of Medicine, NY) for revising this paper. Ruey-Hwa Chen is thanked for providing the active MEK1 plasmid.
ReferencesAdjei, A. A. (2005). The role of mitogen-activated ERK-kinase inhibitors in lung cancer
therapy. Clin. Lung Cancer 7, 221-223.Ambartsumian, N. S., Grigorian, M. S., Larsen, I. F., Karlstrom, O., Sidenius, N.,
Rygaard, J., Georgiev, G. and Lukanidin, E. (1996). Metastasis of mammarycarcinomas in GRS/A hybrid mice transgenic for the mts1 gene. Oncogene 13, 1621-1630.
Ambartsumian, N., Klingelhofer, J., Grigorian, M., Christensen, C., Kriajevska, M.,Tulchinsky, E., Georgiev, G., Berezin, V., Bock, E., Rygaard, J. et al. (2001). The
metastasis-associated Mts1 (S100A4) protein could act as an angiogenic factor.Oncogene 20, 4685-4695.
Babic, A. M., Chen, C. C. and Lau, L. F. (1999). Fisp12/mouse connective tissue growthfactor mediates endothelial cell adhesion and migration through integrin alphavbeta3,promotes endothelial cell survival, and induces angiogenesis in vivo. Mol. Cell. Biol.19, 2958-2966.
Barrett-Lee, P. J. (2005). Growth factor signalling in clinical breast cancer and its impacton response to conventional therapies: a review of chemotherapy. Endocr. Relat.Cancer. 12 Suppl. 1, S125-S133.
Berry, M. G., Gui, G. P., Wells, C. A. and Carpenter, R. (2004). Integrin expressionand survival in human breast cancer. Eur. J. Surg. Oncol. 30, 484-489.
Bork, P. (1993). The modular architecture of a new family of growth regulators relatedto connective tissue growth factor. FEBS Lett. 327, 125-130.
Branca, M., Ciotti, M., Santini, D., Bonito, L. D., Benedetto, A., Giorgi, C., Paba, P.,Favalli, C., Costa, S., Agarossi, A. et al. (2004). Activation of the ERK/MAP kinasepathway in cervical intraepithelial neoplasia is related to grade of the lesion but not tohigh-risk human papillomavirus, virus clearance, or prognosis in cervical cancer. Am.J. Clin. Pathol. 122, 902-911.
Brunet, A., Pages, G. and Pouyssegur, J. (1994). Constitutively active mutants of MAPkinase kinase (MEK1) induce growth factor-relaxation and oncogenicity whenexpressed in fibroblasts. Oncogene 9, 3379-3387.
Brunton, V. G., MacPherson, I. R. and Frame, M. C. (2004). Cell adhesion receptors,tyrosine kinases and actin modulators: a complex three-way circuitry. Biochim.Biophys. Acta 1692, 121-144.
Cho, Y. G., Nam, S. W., Kim, T. Y., Kim, Y. S., Kim, C. J., Park, J. Y., Lee, J. H.,Kim, H. S., Lee, J. W., Park, C. H. et al. (2003). Overexpression of S100A4 is closelyrelated to the aggressiveness of gastric cancer. APMIS 111, 539-545.
Cui, J. F., Liu, Y. K., Pan, B. S., Song, H. Y., Zhang, Y., Sun, R. X., Chen, J., Feng,J. T., Tang, Z. Y., Yu, Y. L. et al. (2004). Differential proteomic analysis of humanhepatocellular carcinoma cell line metastasis-associated proteins. J. Cancer Res. Clin.Oncol. 130, 615-622.
Damjanovich, L., Fulop, B., Adany, R. and Nemes, Z. (1997). Integrin expression onnormal and neoplastic human breast epithelium. Acta Chir. Hung. 36, 69-71.
Darash-Yahana, M., Pikarsky, E., Abramovitch, R., Zeira, E., Pal, B., Karplus, R.,Beider, K., Avniel, S., Kasem, S., Galun, E. et al. (2004). Role of high expressionlevels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 18, 1240-1242.
Davies, B. R., Davies, M. P., Gibbs, F. E., Barraclough, R. and Rudland, P. S. (1993).Induction of the metastatic phenotype by transfection of a benign rat mammaryepithelial cell line with the gene for p9Ka, a rat calcium-binding protein, but not withthe oncogene EJ-ras-1. Oncogene 8, 999-1008.
Davies, M. P., Rudland, P. S., Robertson, L., Parry, E. W., Jolicoeur, P. andBarraclough, R. (1996). Expression of the calcium-binding protein S100A4 (p9Ka)in MMTV-neu transgenic mice induces metastasis of mammary tumours. Oncogene13, 1631-1637.
Deak, M., Clifton, A. D., Lucocq, J. M. and Alessi, D. R. (1998). Mitogenand stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38,and may mediate activation of CREB. EMBO J. 17, 4426-4441.
Duarte, W. R., Shibata, T., Takenaga, K., Takahashi, E., Kubota, K., Ohya, K.,Ishikawa, I., Yamauchi, M. and Kasugai, S. (2003). S100A4: a novel negativeregulator of mineralization and osteoblast differentiation. J. Bone Miner. Res. 18, 493-501.
Ebralidze, A., Tulchinsky, E., Grigorian, M., Afanasyeva, A., Senin, V., Revazova, E.and Lukanidin, E. (1989). Isolation and characterization of a gene specificallyexpressed in different metastatic cells and whose deduced gene product has a highdegree of homology to a Ca2+-binding protein family. Genes Dev. 3, 1086-1093.
Felding-Habermann, B., O’Toole, T. E., Smith, J. W., Fransvea, E., Ruggeri, Z. M.,Ginsberg, M. H., Hughes, P. E, Pampori, N., Shattil, S. J., Saven, A. et al. (2001).Integrin activation controls metastasis in human breast cancer. Proc. Natl. Acad. Sci.USA 98, 1853-1858.
Ford, H. L., Salim, M. M., Chakravarty, R., Aluiddin, V. and Zain, S. B. (1995).Expression of Mts1, a metastasis-associated gene, increases motility but not invasionof a nonmetastatic mouse mammary adenocarcinoma cell line. Oncogene 11, 2067-2075.
Frodin, M. and Gammeltoft, S. (1999). Role and regulation of 90 kDa ribosomal S6kinase (RSK) in signal transduction. Mol. Cell. Endocrinol. 151, 65-77.
Fukunaga, R. and Hunter, T. (1997). MNK1, a new MAP kinase-activated proteinkinase, isolated by a novel expression screening method for identifying protein kinasesubstrates. EMBO J. 16, 1921-1933.
Gao, R. and Brigstock, D. R. (2004). Connective tissue growth factor (CCN2) inducesadhesion of rat activated hepatic stellate cells by binding of its C-terminal domain tointegrin alpha(v)beta(3) and heparan sulfate proteoglycan. J. Biol. Chem. 279, 8848-8855.
Glading, A., Bodnar, R. J., Reynolds, I. J., Shiraha, H., Satish, L., Potter, D. A., Blair,H. C. and Wells, A. (2004). Epidermal growth factor activates m-calpain (calpain II),at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol.Cell. Biol. 24, 2499-2512.
Glenney, J. R., Jr, Kindy, M. S. and Zokas, L. (1989). Isolation of a new member ofthe S100 protein family: amino acid sequence, tissue, and subcellular distribution. J.Cell Biol. 108, 569-578.
Gollob, J. A., Wilhelm, S., Carter, C. and Kelley, S. L. (2006). Role of Raf kinase incancer: therapeutic potential of targeting the Raf/MEK/ERK signal transductionpathway. Semin. Oncol. 33, 392-406.
Gui, G. P., Wells, C. A., Yeomans, P., Jordan, S. E., Vinson, G. P. and Carpenter, R.
Journal of Cell Science 120 (12)
Jour
nal o
f Cel
l Sci
ence

2065CTGF promotes breast cancer metastasis
(1996). Integrin expression in breast cancer cytology: a novel predictor of axillarymetastasis. Eur. J. Surg. Oncol. 22, 254-258.
Hanahan, D. and Weinberg, R. A. (2000). The hallmarks of cancer. Cell 100, 57-70.Hernan, R., Fasheh, R., Calabrese, C., Frank, A. J., Maclean, K. H., Allard, D.,
Barraclough, R. and Gilbertson, R. J. (2003). ERBB2 up-regulates S100A4 andseveral other prometastatic genes in medulloblastoma. Cancer Res. 63, 140-148.
Hunger-Glaser, I., Salazar, E. P., Sinnett-Smith, J. and Rozengurt, E. (2003).Bombesin, lysophosphatidic acid, and epidermal growth factor rapidly stimulate focaladhesion kinase phosphorylation at Ser-910: requirement for ERK activation. J. Biol.Chem. 278, 22631-22643.
Jedsadayanmata, A., Chen, C. C., Kireeva, M. L., Lau, L. F. and Lam, S. C. (1999).Activation-dependent adhesion of human platelets to Cyr61 and Fisp12/mouseconnective tissue growth factor is mediated through integrin alpha(IIb)beta(3). J. Biol.Chem. 274, 24321-24327.
Jiang, W. G. (1996). E-cadherin and its associated protein catenins, cancer invasion andmetastasis. Br. J. Surg. 83, 437-446.
Jones, J. I., Doerr, M. E. and Clemmons, D. R. (1995). Cell migration: interactionsamong integrins, IGFs and IGFBPs. Prog. Growth Factor Res. 6, 319-327.
Kang, Y., Siegel, P. M., Shu, W., Drobnjak, M., Kakonen, S. M., Cordon-Cardo, C.,Guise, T. A. and Massague, J. (2003). A multigenic program mediating breast cancermetastasis to bone. Cancer Cell 3, 537-549.
Kim, E. J. and Helfman, D. M. (2003). Characterization of the metastasis-associatedprotein, S100A4. Roles of calcium binding and dimerization in cellular localizationand interaction with myosin. J. Biol. Chem. 278, 30063-30073.
Kirfel, G., Rigort, A., Borm, B. and Herzog, V. (2004). Cell migration: mechanisms ofrear detachment and the formation of migration tracks. Eur. J. Cell Biol. 83, 717-724.
Klemke, R. L., Cai, S., Giannini, A. L., Gallagher, P. J., deLanerolle, P. and Cheresh,D. A. (1997). Regulation of cell motility by mitogen-activated protein kinase. J. CellBiol. 137, 481-492.
Kondo, S., Kubota, S., Shimo, T., Nishida, T., Yosimichi, G., Eguchi, T., Sugahara,T. and Takigawa, M. (2002). Connective tissue growth factor increased by hypoxiamay initiate angiogenesis in collaboration with matrix metalloproteinases.Carcinogenesis 23, 769-776.
Kriajevska, M. V., Cardenas, M. N., Grigorian, M. S., Ambartsumian, N. S.,Georgiev, G. P. and Lukanidin, E. M. (1994). Non-muscle myosin heavy chain as apossible target for protein encoded by metastasis-related mts-1 gene. J. Biol. Chem.269, 19679-19682.
Krueger, J. S., Keshamouni, V. G., Atanaskova, N. and Reddy, K. B. (2001). Temporaland quantitative regulation of mitogen-activated protein kinase (MAPK) modulates cellmotility and invasion. Oncogene 20, 4209-4218.
Lau, L. F. and Lam, S. C. (1999). The CCN family of angiogenic regulators: the integrinconnection. Exp. Cell Res. 248, 44-57.
Liapis, H., Flath, A. and Kitazawa, S. (1996). Integrin alpha V beta 3 expression bybone-residing breast cancer metastases. Diagn. Mol. Pathol. 5, 127-135.
Lin, M. T., Chang, C. C., Chen, S. T., Chang, H. L., Su, J. L., Chau, Y. P. and Kuo,M. L. (2004). Cyr61 expression confers resistance to apoptosis in breast cancer MCF-7 cells by a mechanism of NF-kappaB-dependent XIAP up-regulation. J. Biol. Chem.279, 24015-24023.
Liu, Z. X., Yu, C. F., Nickel, C., Thomas, S. and Cantley, L. G. (2002). Hepatocytegrowth factor induces ERK-dependent paxillin phosphorylation and regulates paxillin-focal adhesion kinase association. J. Biol. Chem. 277, 10452-10458.
Lloyd, B. H., Platt-Higgins, A., Rudland, P. S. and Barraclough, R. (1998). HumanS100A4 (p9Ka) induces the metastatic phenotype upon benign tumour cells. Oncogene17, 465-473.
Mariani, A., Sebo, T. J., Katzmann, J. A., Riehle, D. L., Dowdy, S. C., Keeney, G. L.,Lesnick, T. G. and Podratz, K. C. (2005). HER-2/neu overexpression and hormonedependency in endometrial cancer: analysis of cohort and review of literature.Anticancer Res. 25, 2921-2927.
Masiakowski, P. and Shooter, E. M. (1988). Nerve growth factor induces the genes fortwo proteins related to a family of calcium-binding proteins in PC12 cells. Proc. Natl.Acad. Sci. USA 85, 1277-1281.
Mazzucchelli, L. (2002). Protein S100A4: too long overlooked by pathologists? Am. J.Pathol. 160, 7-13.
Milde-Langosch, K., Bamberger, A. M., Rieck, G., Grund, D., Hemminger, G.,Muller, V. and Loning, T. (2005). Expression and prognostic relevance of activatedextracellular-regulated kinases (ERK1/2) in breast cancer. Br. J. Cancer 92, 2206-2215.
Minn, A. J., Kang, Y., Serganova, I., Gupta, G. P., Giri, D. D., Doubrovin, M.,Ponomarev, V., Gerald, W. L., Blasberg, R. and Massague, J. (2005). Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J.Clin. Invest. 115, 44-55.
Miralem, T., Steinberg, R., Price, D. and Avraham, H. (2001). VEGF(165) requiresextracellular matrix components to induce mitogenic effects and migratory response inbreast cancer cells. Oncogene 20, 5511-5524.
Missiaglia, E., Blaveri, E., Terris, B., Wang, Y. H., Costello, E., Neoptolemos, J. P.,Crnogorac-Jurcevic, T. and Lemoine, N. R. (2004). Analysis of gene expression in
cancer cell lines identifies candidate markers for pancreatic tumorigenesis andmetastasis. Int. J. Cancer 112, 100-112.
Nikitenko, L. L., Lloyd, B. H., Rudland, P. S., Fear, S. and Barraclough, R. (2000).Localisation by in situ hybridisation of S100A4 (p9Ka) mRNA in primary humanbreast tumour specimens. Int. J. Cancer 86, 219-228.
Okada, H., Danoff, T. M., Kalluri, R. and Neilson, E. G. (1997). The early role of FSP1in epithelial-mesenchymal transformation. Am. J. Physiol. 273, F563-F574.
Perbal, B. (2004). CCN proteins: multifunctional signalling regulators. Lancet 363, 62-64.
Platt-Higgins, A. M., Renshaw, C. A., West, C. R., Winstanley, J. H., De SilvaRudland, S., Barraclough, R. and Rudland, P. S. (2000). Comparison of themetastasis-inducing protein S100A4 (p9ka) with other prognostic markers in humanbreast cancer. Int. J. Cancer 89, 198-208.
Price, J. T., Tiganis, T., Agarwal, A., Djakiew, D. and Thompson, E. W. (1999).Epidermal growth factor promotes MDA-MB-231 breast cancer cell migration througha phosphatidylinositol 3�-kinase and phospholipase C-dependent mechanism. CancerRes. 59, 5475-5478.
Rudland, P. S., Platt-Higgins, A., Renshaw, C., West, C. R., Winstanley, J. H.,Robertson, L. and Barraclough, R. (2000). Prognostic significance of the metastasis-inducing protein S100A4 (p9Ka) in human breast cancer. Cancer Res. 60, 1595-1603.
Santen, R. J., Song, R. X., McPherson, R., Kumar, R., Adam, L., Jeng, M. H. andYue, W. (2002). The role of mitogen-activated protein (MAP) kinase in breast cancer.J. Steroid Biochem. Mol. Biol. 80, 239-256.
Schmidt-Hansen, B., Klingelhofer, J., Grum-Schwensen, B., Christensen, A.,Andresen, S., Kruse, C., Hansen, T., Ambartsumian, N., Lukanidin, E. andGrigorian, M. (2004a). Functional significance of metastasis-inducing S100A4(Mts1)in tumor-stroma interplay. J. Biol. Chem. 279, 24498-24504.
Schmidt-Hansen, B., Ornas, D., Grigorian, M., Klingelhofer, J., Tulchinsky, E.,Lukanidin, E. and Ambartsumian, N. (2004b). Extracellular S100A4(mts1)stimulates invasive growth of mouse endothelial cells and modulates MMP-13 matrixmetalloproteinase activity. Oncogene 23, 5487-5495.
Schober, J. M., Chen, N., Grzeszkiewicz, T. M., Jovanovic, I., Emeson, E. E.,Ugarova, T. P., Ye, R. D., Lau, L. F. and Lam, S. C. (2002). Identification of integrinalpha(M)beta(2) as an adhesion receptor on peripheral blood monocytes for Cyr61(CCN1) and connective tissue growth factor (CCN2): immediate-early gene productsexpressed in atherosclerotic lesions. Blood 99, 4457-4465.
Sebolt-Leopold, J. S. and Herrera, R. (2004). Targeting the mitogen-activated proteinkinase cascade to treat cancer. Nat. Rev. Cancer 4, 937-947.
Seton-Rogers, S. E., Lu, Y., Hines, L. M., Koundinya, M., LaBaer, J., Muthuswamy,S. K. and Brugge, J. S. (2004). Cooperation of the ErbB2 receptor and transforminggrowth factor beta in induction of migration and invasion in mammary epithelial cells.Proc. Natl. Acad. Sci. USA 101, 1257-1262.
Shimo, T., Nakanishi, T., Nishida, T., Asano, M., Kanyama, M., Kuboki, T.,Tamatani, T., Tezuka, K., Takemura, M., Matsumura, T. et al. (1999). Connectivetissue growth factor induces the proliferation, migration, and tube formation of vascularendothelial cells in vitro, and angiogenesis in vivo. J. Biochem. 126, 137-145.
Taddei, I., Faraldo, M. M., Teuliere, J., Deugnier, M. A., Thiery, J. P. amd Glukhova,M. A. (2003). Integrins in mammary gland development and differentiation ofmammary epithelium. J. Mammary Gland Biol. Neoplasia 8, 383-394.
Takenaga, K., Nakamura, Y., Endo, H. and Sakiyama, S. (1994). Involvement of S100-related calcium-binding protein pEL98 (or mts1) in cell motility and tumor cellinvasion. Jpn. J. Cancer Res. 85, 831-839.
Tangkeangsirisin, W. and Serrero, G. (2004). PC cell-derived growth factor(PCDGF/GP88, progranulin) stimulates migration, invasiveness and VEGF expressionin breast cancer cells. Carcinogenesis 25, 1587-1592.
Turner, C. E. (2000). Paxillin interactions. J. Cell Sci. 23, 4139-4140.Van’t Veer, L. J. and Weigelt, B. (2003). Road map to metastasis. Nat. Med. 9, 999-
1000.Vivanco, I. and Sawyers, C. L. (2002). The phosphatidylinositol 3-Kinase AKT pathway
in human cancer. Nat. Rev. Cancer 2, 489-501.Wahab, N. A, Brinkman, H. and Mason, R. M. (2001). Uptake and intracellular
transport of the connective tissue growth factor: a potential mode of action. Biochem.J. 359, 89-97.
Waskiewicz, A. J., Flynn, A., Proud, C. G. and Cooper, J. A. (1997). Mitogen-activatedprotein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 16,1909-1920.
Watanabe, Y., Usada, N., Minami, H., Morita, T., Tsugane, S., Ishikawa, R., Kohama,K., Tomida, Y. and Hidaka, H. (1993). Calvasculin, as a factor affecting themicrofilament assemblies in rat fibroblasts transfected by src gene. FEBS Lett. 324,51-55.
Weston, B. S., Wahab, N. A. and Mason, R. M. (2003). CTGF mediates TGF-beta-induced fibronectin matrix deposition by upregulating active alpha5beta1 integrin inhuman mesangial cells. J. Am. Soc. Nephrol. 14, 601-610.
Xie, D., Nakachi, K., Wang, H., Elashoff, R. and Koeffler, H. P. (2001). Elevated levelsof connective tissue growth factor, WISP-1, and CYR61 in primary breast cancersassociated with more advanced features. Cancer Res. 61, 8917-8923.
Jour
nal o
f Cel
l Sci
ence


















