
CS 581 Algorithmic Computational Genomicsweb.engr.illinois.edu/~warnow/CS581-Jan16-2018.pdf ·...
67
CS 581 Algorithmic Computational Genomics Tandy Warnow University of Illinois at Urbana-Champaign
Transcript of CS 581 Algorithmic Computational Genomicsweb.engr.illinois.edu/~warnow/CS581-Jan16-2018.pdf ·...

CS581AlgorithmicComputationalGenomics
TandyWarnowUniversityofIllinoisatUrbana-Champaign

Today
• Explainthecourse• Introducesomeoftheresearchinthisarea• Describesomeopenproblems• Talkaboutcourseprojects!

CS581
• Algorithmdesignandanalysis(largelyinthecontextofstatisticalmodels)for– Multiplesequencealignment– PhylogenyEstimation– Genomeassembly
• Iassumemathematicalmaturityandfamiliaritywithgraphs,discretealgorithms,andbasicprobabilitytheory,equivalenttoCS374andCS361.Nobiologybackgroundisrequired!

Textbook
• ComputationalPhylogenetics:AnintroductiontoDesigningMethodsforPhylogenyEstimation
• ThismaybeavailableintheUniversityBookstore.Ifnot,youcangetitfromAmazon.
• Thetextbookprovidesallthebackgroundyou’llneedfortheclass,andhomeworkassignmentswillbetakenfromthebook.

Grading
• Homeworks:25%(worstgradedropped)• Takehomemidterm:40%• Finalproject:25%• Courseparticipationandpaperpresentation:10%

CourseProject
• Courseprojectisduethelastdayofclass,andcanbearesearchprojectorasurveypaper.
• Ifyoudoaresearchproject,youcandoitwithsomeoneelse(includingsomeoneinmyresearchgroup);thegoalwillbetoproduceapublishablepaper.
• Ifyoudoasurveypaper,youwilldoitbyyourself.

Orangutan Gorilla Chimpanzee Human
From the Tree of the Life Website, University of Arizona
Phylogeny (evolutionary tree)
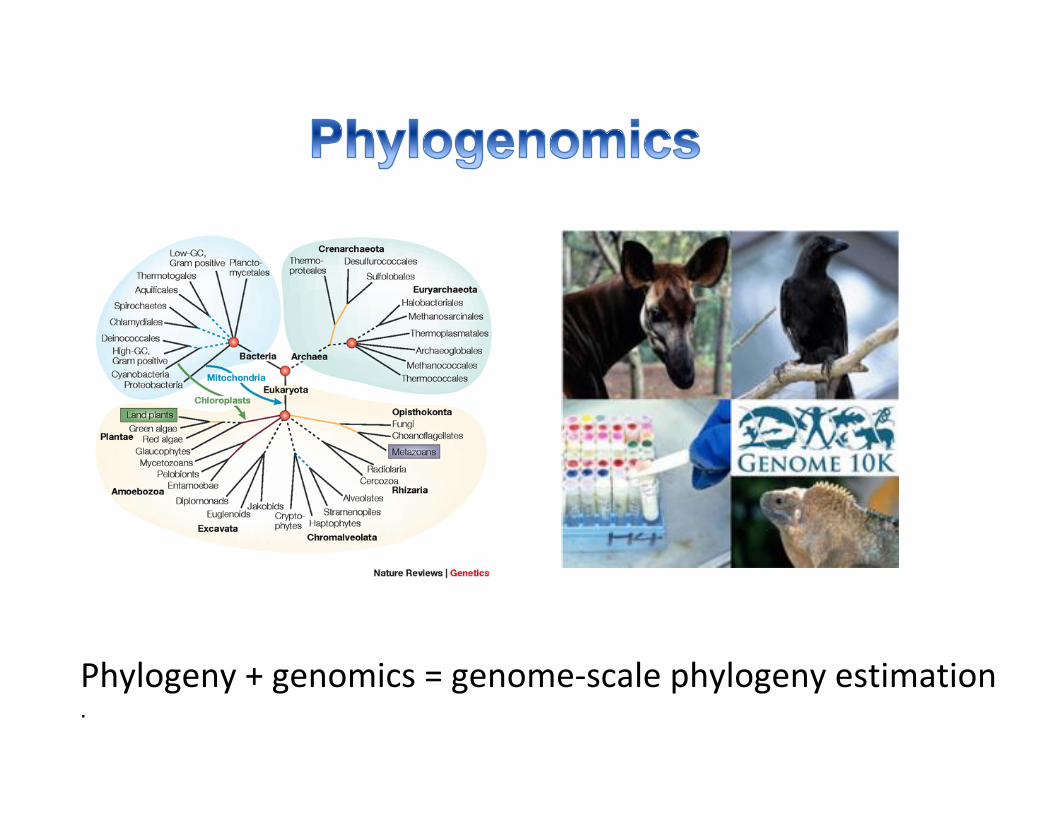
Phylogeny+genomics=genome-scalephylogenyestimation.
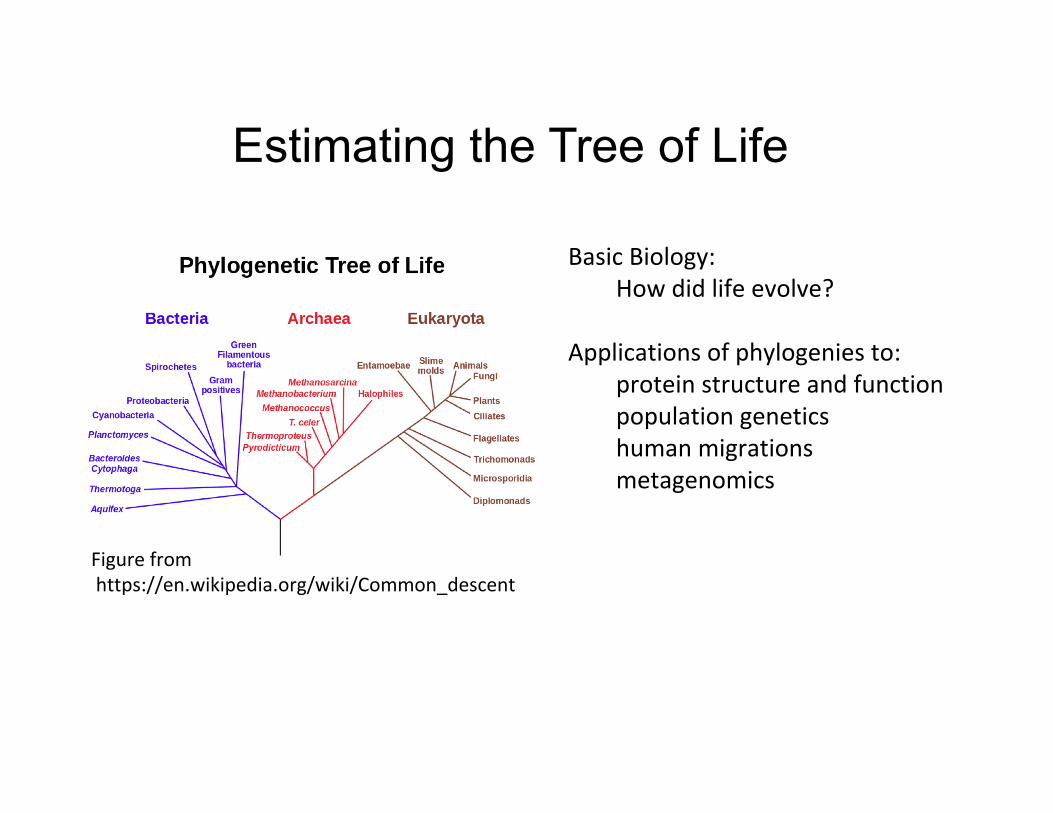
Estimating the Tree of Life
BasicBiology:Howdidlifeevolve?
Applicationsofphylogeniesto:proteinstructureandfunctionpopulationgeneticshumanmigrationsmetagenomics
Figurefromhttps://en.wikipedia.org/wiki/Common_descent
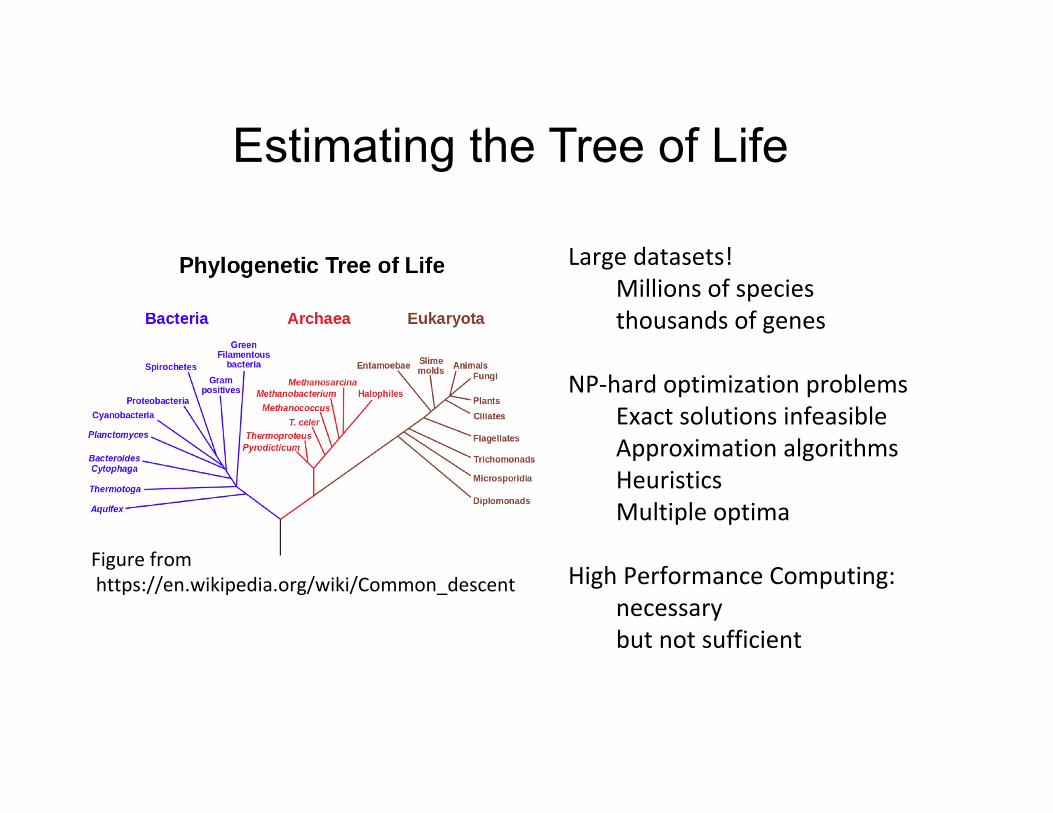
Estimating the Tree of Life
Largedatasets!Millionsofspeciesthousandsofgenes
NP-hardoptimizationproblemsExactsolutionsinfeasibleApproximationalgorithmsHeuristicsMultipleoptima
HighPerformanceComputing:
necessarybutnotsufficient
Figurefromhttps://en.wikipedia.org/wiki/Common_descent

Background Results Conclusions References
Genomic data: challenges and opportunities
Muir et al. [2016]
Muir,2016

ComputerScienceSolvingProblemsinBiologyandLinguistics
• Algorithmdesignusing– Divide-and-conquer– Iteration– Heuristicsearch– Graphtheory
• Algorithmanalysisusing– ProbabilityTheory– GraphTheory
• Simulationsandmodelling• Collaborationswithbiologistsandlinguistsanddataanalysis• Discoveriesabouthowlifeevolvedonearth(andhowlanguages
evolved,too)

ComputationalPhylogenetics(2005)
CourtesyoftheTreeofLifewebproject,tolweb.org
Current methods can use months to
estimate trees on 1000 DNA sequences
Our objective:
More accurate trees and alignments
on 500,000 sequences in under a week

ComputationalPhylogenetics(2018)
CourtesyoftheTreeofLifewebproject,tolweb.org
1997-2001: Distance-based phylogenetic tree estimation from polynomial length sequences
2012: Computing accurate trees (almost) without multiple sequence alignments
2009-2015: Co-estimation of multiple sequence alignments and gene trees, now on 1,000,000 sequences in under two weeks
2014-2015: Species tree estimation from whole genomes in the presence of massive gene tree heterogeneity
2016-2017: Scaling methods to very large heterogeneous datasets using novel machine learning and supertree methods.
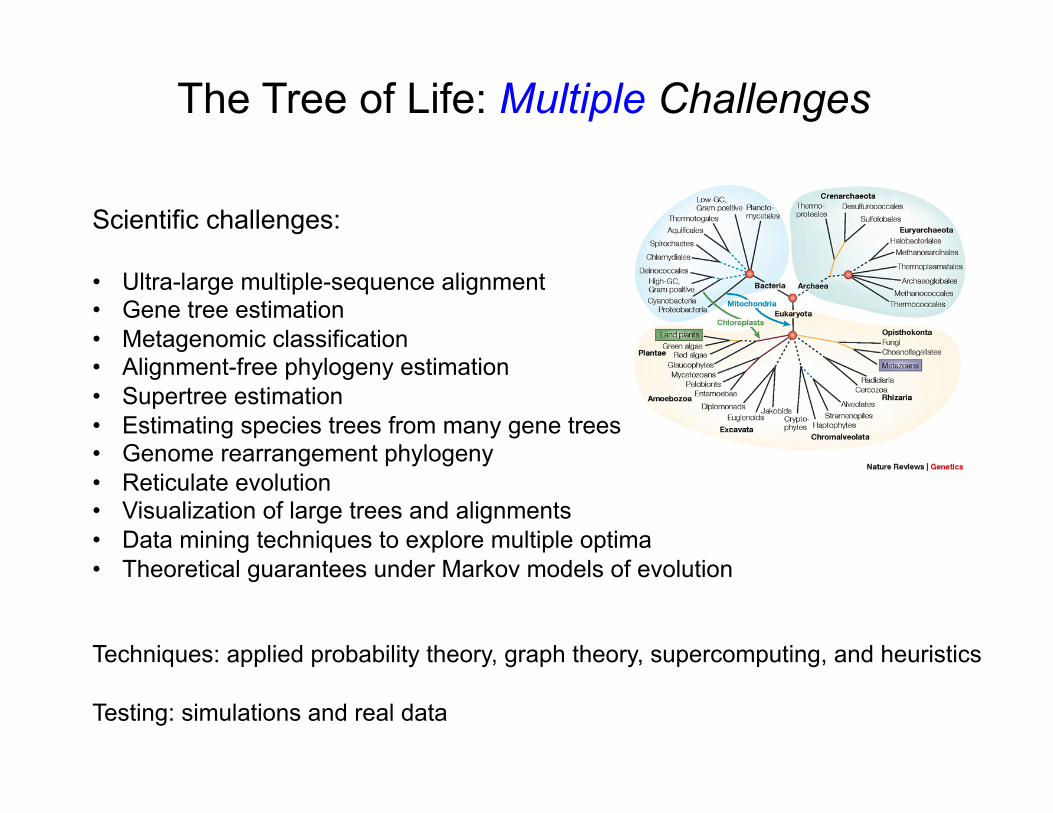
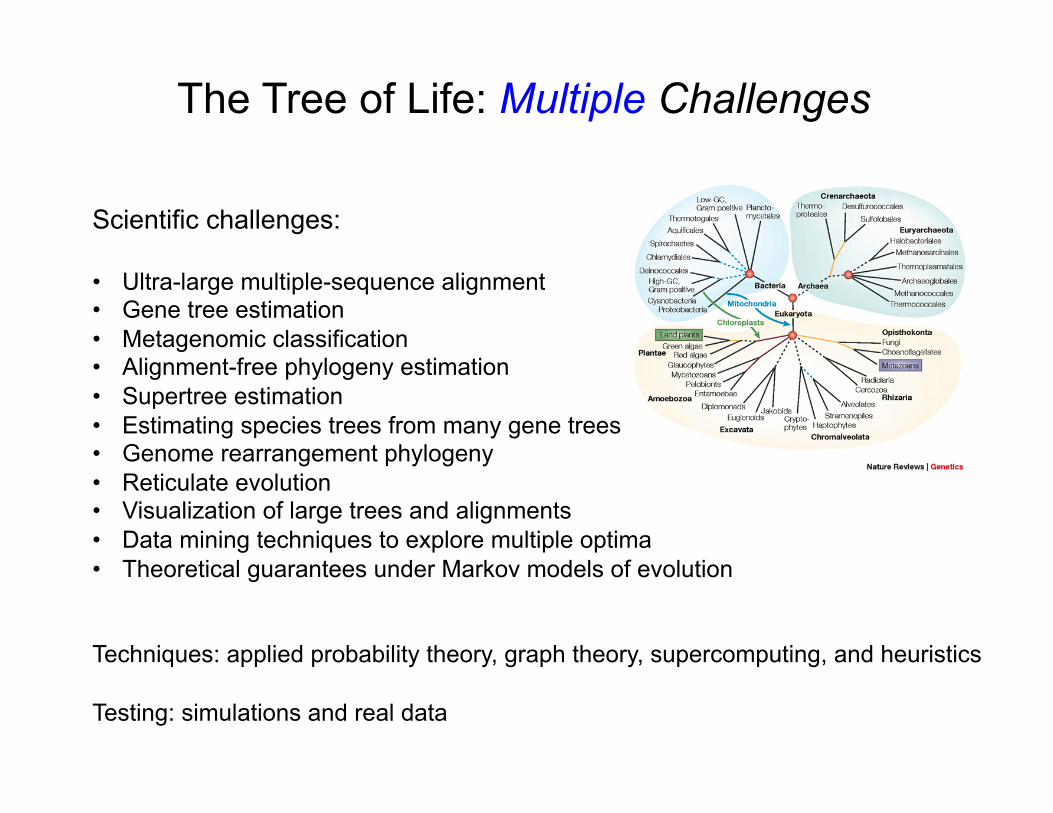
Scientific challenges: • Ultra-large multiple-sequence alignment • Gene tree estimation • Metagenomic classification • Alignment-free phylogeny estimation • Supertree estimation • Estimating species trees from many gene trees • Genome rearrangement phylogeny • Reticulate evolution • Visualization of large trees and alignments • Data mining techniques to explore multiple optima • Theoretical guarantees under Markov models of evolution
Techniques: applied probability theory, graph theory, supercomputing, and heuristics Testing: simulations and real data
The Tree of Life: Multiple Challenges

Thistalk
• Divide-and-conquer:Basicalgorithmdesigntechnique
• Applicationofdivide-and-conquerto:– Phylogenyestimation(andproofof“absolutefastconvergence”)
– Multiplesequencealignment– Almostalignment-freetreeestimation
• Openproblems

Divide-and-Conquer
• Divide-and-conquerisabasicalgorithmictrickforsolvingproblems!
• Threesteps:– divideadatasetintotwoormoresets,– solvetheproblemoneachset,and– combinesolutions.

Sorting
10 3 54 23 75 5 1 25
Objective:sortthislistofintegersfromsmallesttolargest.10,3,54,23,75,5,1,25shouldbecome1,3,5,10,23,25,54,75

MergeSort
10 3 54 23 75 5 1 25
Step1:DivideintotwosublistsStep2:RecursivelysorteachsublistStep3:Mergethetwosortedsublists

Keytechnique:Divide-and-conquer!
• Canbeusedtoproduceprovablycorrectalgorithms(asinMergeSort)
• Butalso–inpractice,divide-and-conquerisusefulforscalingmethodstolargedatasets,becausesmalldatasetswithnottoomuch“heterogeneity”areeasytoanalyzewithgoodaccuracy.

Threedivide-and-conqueralgorithms
• DCM1(2001):Improvingdistance-basedphylogenyestimation
• SATé(2009,2012)andPASTA(2015):Co-estimationofmultiplesequencealignmentsandgenetrees(upto1,000,000sequences)
• DACTAL(2012):Almostalignment-freetreeestimation

Threedivide-and-conqueralgorithms
• DCM1(2001):Improvingdistance-basedphylogenyestimation
• SATé(2009,2012)andPASTA(2015):Co-estimationofmultiplesequencealignmentsandgenetrees(upto1,000,000sequences)
• DACTAL(2012):Almostalignment-freetreeestimation
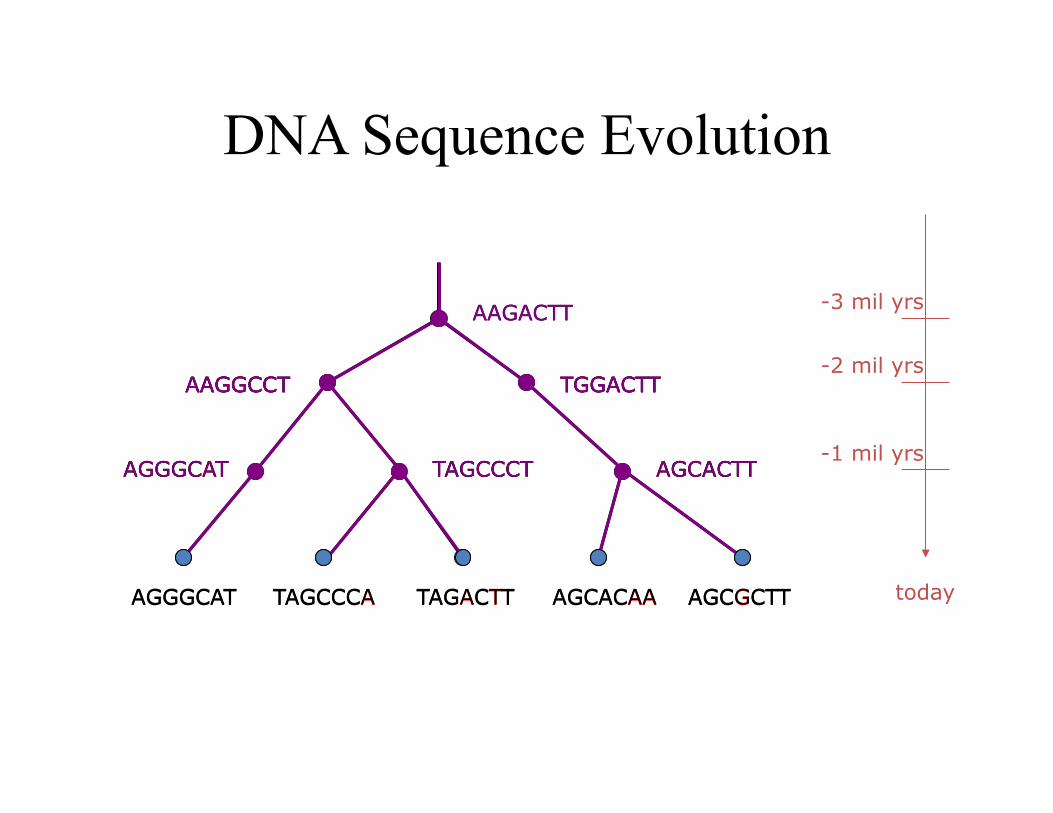
DNA Sequence Evolution
AAGACTT
TGGACTT AAGGCCT
-3 mil yrs
-2 mil yrs
-1 mil yrs
today
AGGGCAT TAGCCCT AGCACTT
AAGGCCT TGGACTT
TAGCCCA TAGACTT AGCGCTT AGCACAA AGGGCAT
AGGGCAT TAGCCCT AGCACTT
AAGACTT
TGGACTT AAGGCCT
AGGGCAT TAGCCCT AGCACTT
AAGGCCT TGGACTT
AGCGCTT AGCACAA TAGACTT TAGCCCA AGGGCAT
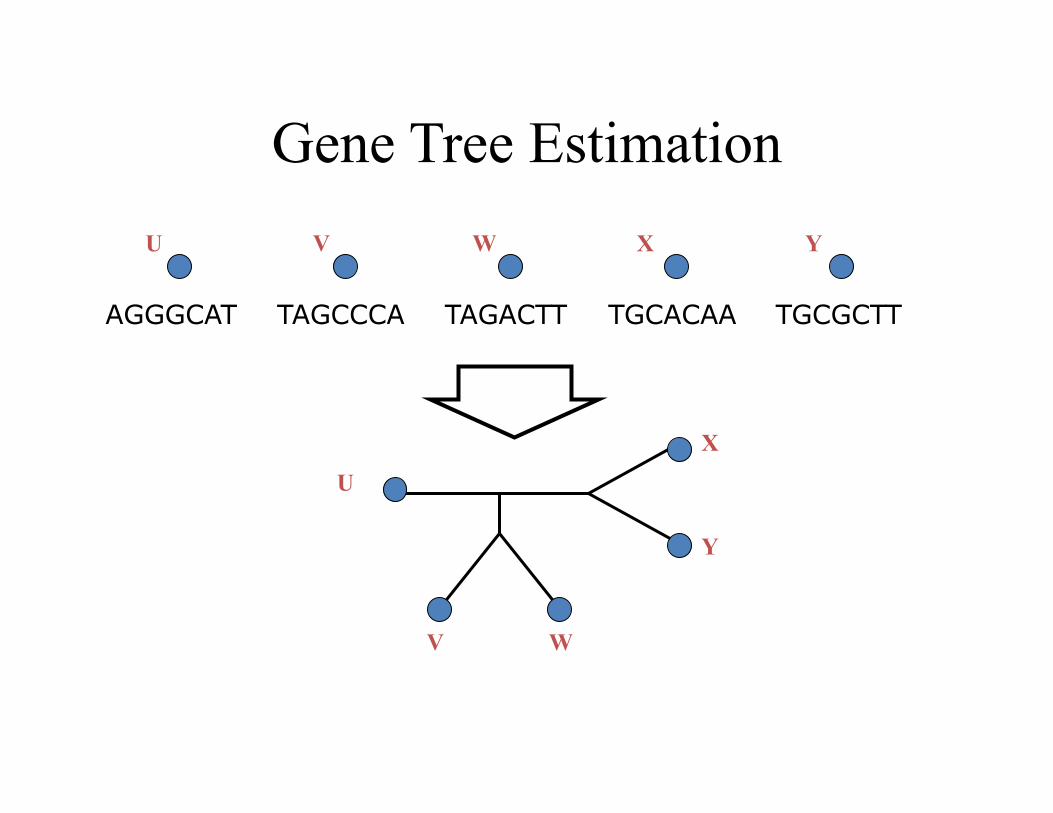
Gene Tree Estimation
TAGCCCA TAGACTT TGCACAA TGCGCTT AGGGCAT
U V W X Y
U
V W
X
Y
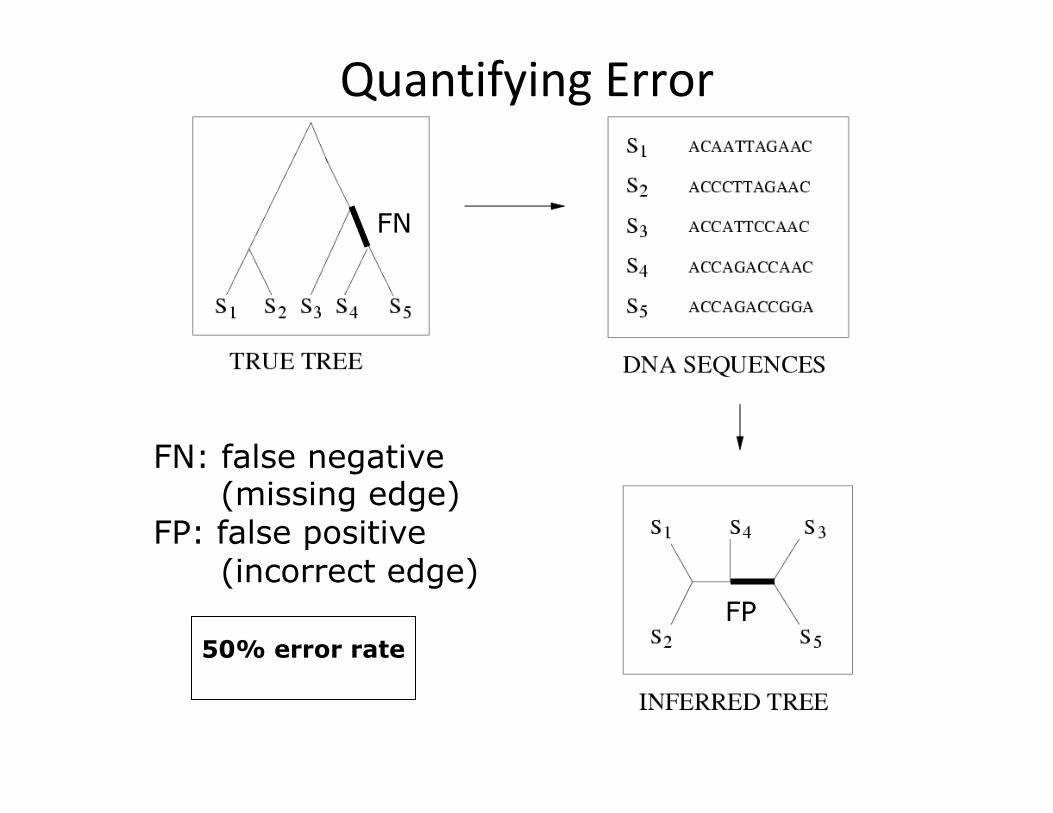
QuantifyingError
FN: false negative (missing edge) FP: false positive (incorrect edge)
FN
FP 50% error rate

MarkovModelofSiteEvolutionSimplest(Jukes-Cantor,1969):• ThemodeltreeTisbinaryandhassubstitutionprobabilitiesp(e)on
eachedgee.• Thestateattherootisrandomlydrawnfrom{A,C,T,G}(nucleotides)• Ifasite(position)changesonanedge,itchangeswithequalprobability
toeachoftheremainingstates.• TheevolutionaryprocessisMarkovian.Thedifferentsitesareassumedtoevolveindependentlyandidenticallydownthetree(withratesthataredrawnfromagammadistribution).
Morecomplexmodels(suchastheGeneralMarkovmodel)arealsoconsidered,oftenwithlittlechangetothetheory.

Sequencelengthrequirements
Thesequencelength(numberofsites)thataphylogenyreconstructionmethodMneedstoreconstructthetruetreewithprobabilityatleast1-εdependson
• M(themethod)• ε• f=minp(e),• g=maxp(e),and• n,thenumberofleaves
Wefixeverythingbutn.
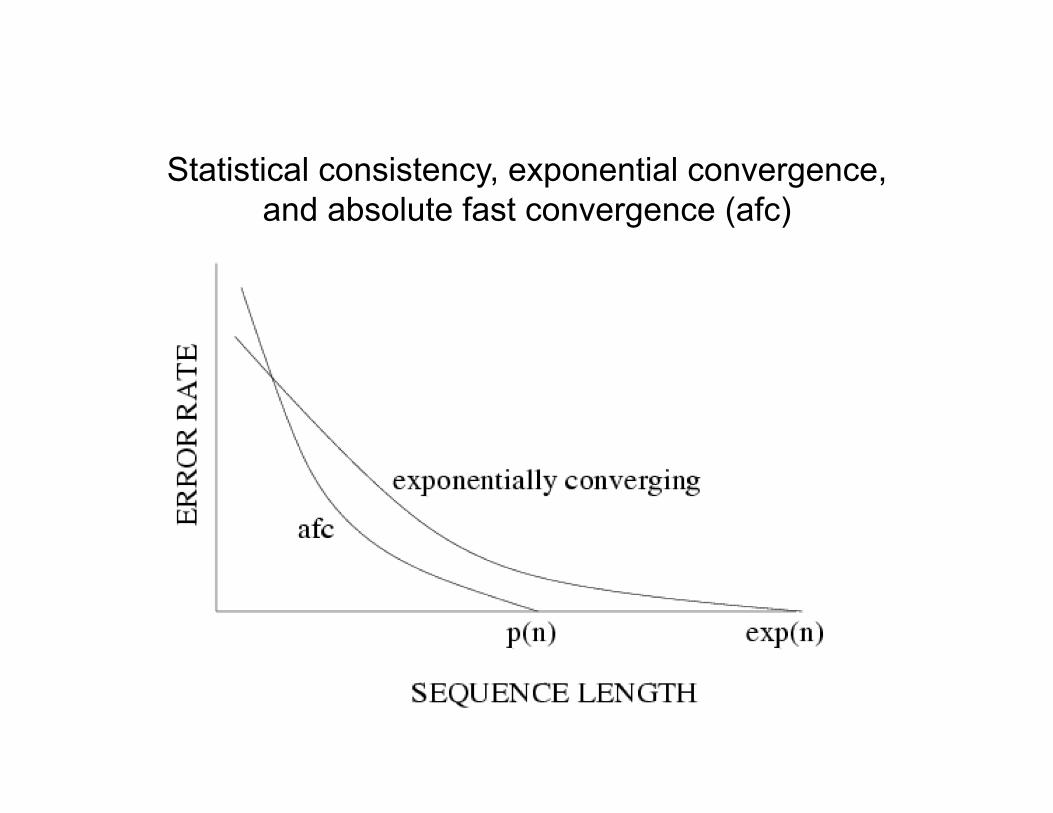
Statistical consistency, exponential convergence, and absolute fast convergence (afc)
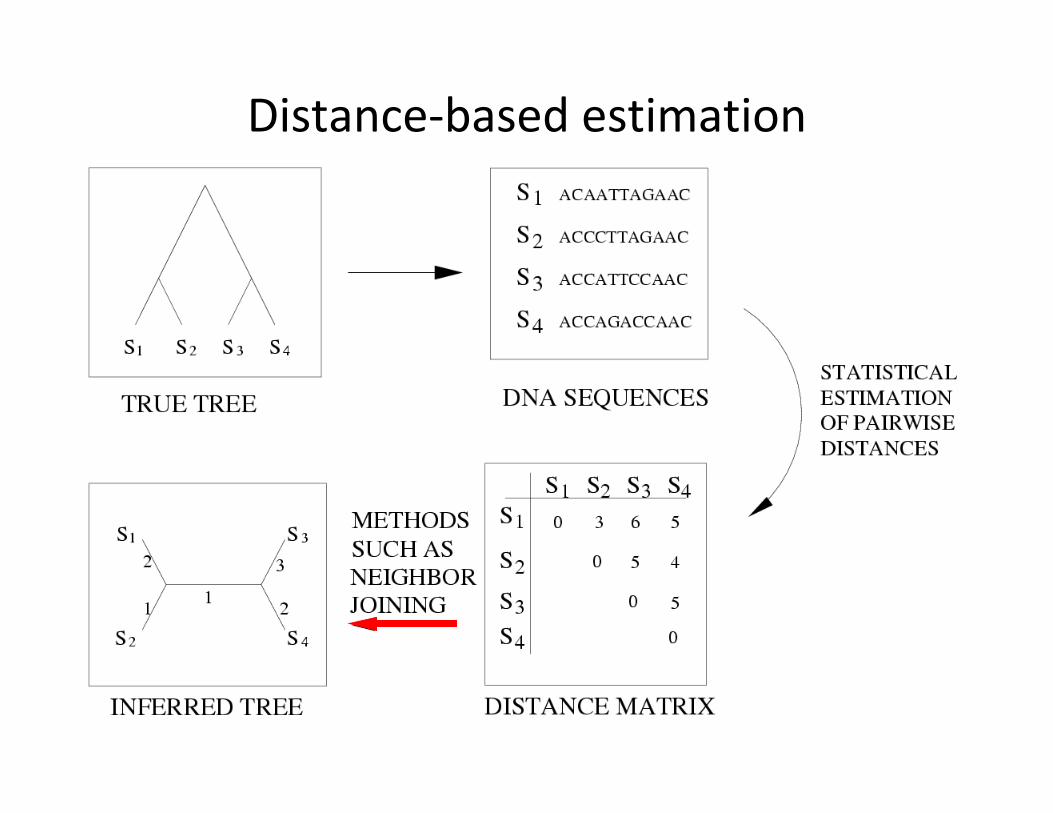
Distance-basedestimation
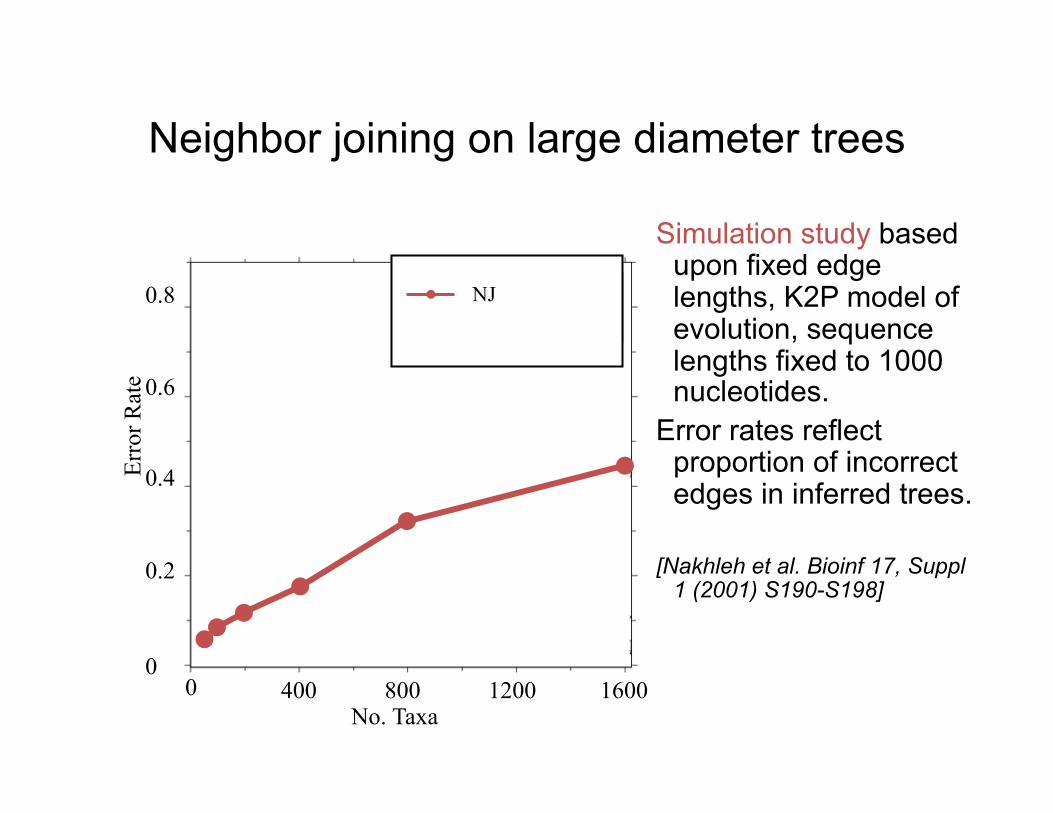
Neighbor joining on large diameter trees
Simulation study based
upon fixed edge lengths, K2P model of evolution, sequence lengths fixed to 1000 nucleotides.
Error rates reflect proportion of incorrect edges in inferred trees.
[Nakhleh et al. Bioinf 17, Suppl
1 (2001) S190-S198]
NJ
0 400 800 1600 1200 No. Taxa
0
0.2
0.4
0.6
0.8
Erro
r Rat
e

DCMs: Divide-and-conquer for improving phylogeny reconstruction

DCM1Decompositions
DCM1 decomposition : Compute maximal cliques
Input: Set S of sequences, distance matrix d, threshold value
1. Compute threshold graph
}),(:),{(,),,( qjidjiESVEVGq ≤===2. Perform minimum weight triangulation (note: if d is an additive matrix, then the threshold graph is provably triangulated).
}{ ijdq∈
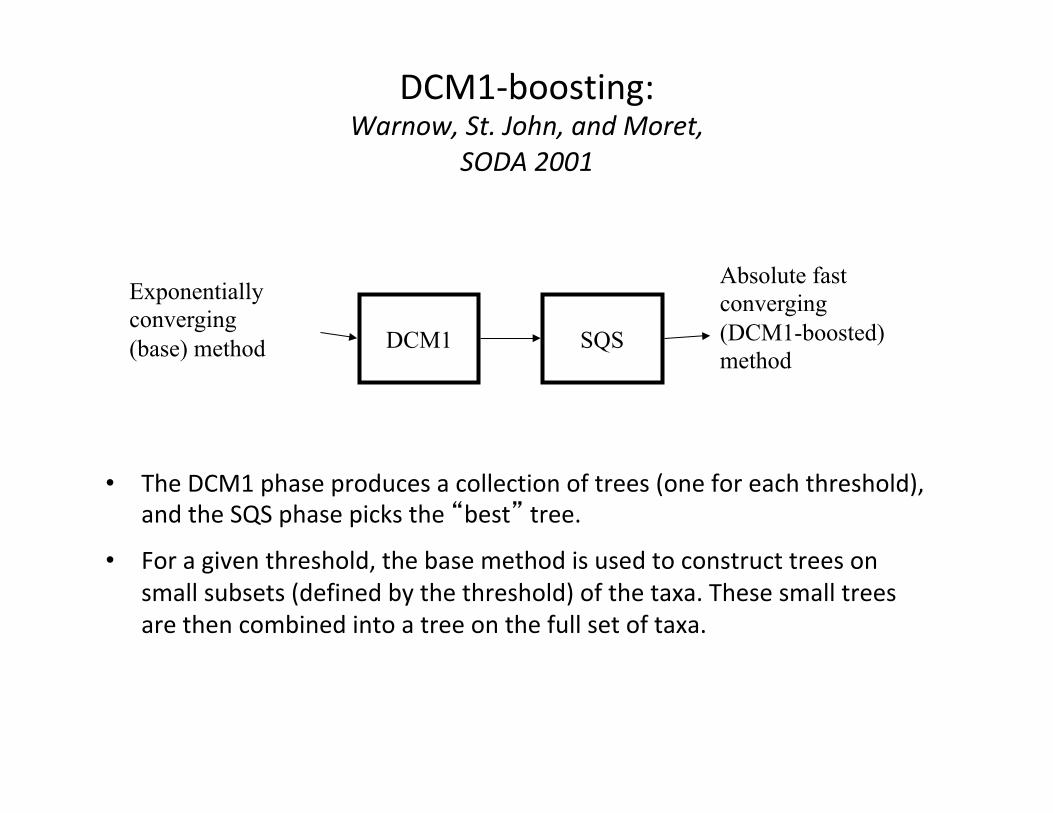
DCM1-boosting:Warnow,St.John,andMoret,
SODA2001
• TheDCM1phaseproducesacollectionoftrees(oneforeachthreshold),andtheSQSphasepicksthe“best”tree.
• Foragiventhreshold,thebasemethodisusedtoconstructtreesonsmallsubsets(definedbythethreshold)ofthetaxa.Thesesmalltreesarethencombinedintoatreeonthefullsetoftaxa.
DCM1 SQS
Exponentially converging (base) method
Absolute fast converging (DCM1-boosted) method
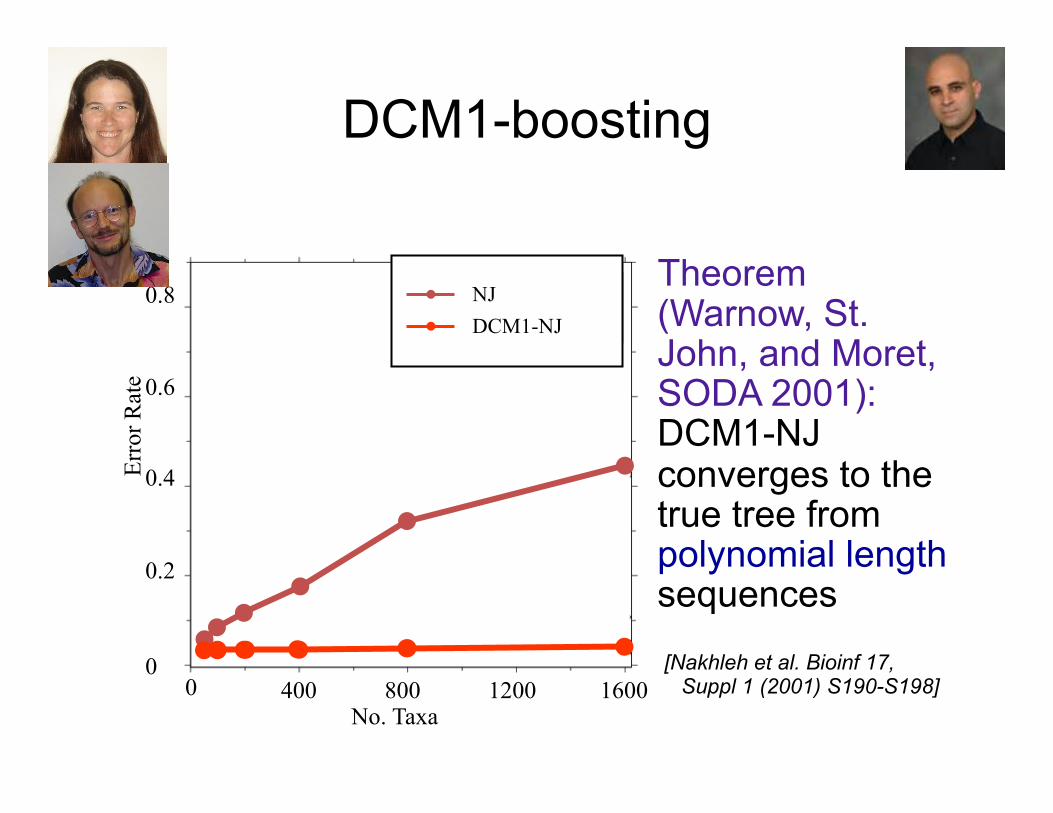
DCM1-boosting
• Theorem (Warnow, St. John, and Moret, SODA 2001): DCM1-NJ converges to the true tree from polynomial length sequences
NJ DCM1-NJ
0 400 800 1600 1200 No. Taxa
0
0.2
0.4
0.6
0.8
Erro
r Rat
e
[Nakhleh et al. Bioinf 17,
Suppl 1 (2001) S190-S198]

Threedivide-and-conqueralgorithms
• DCM1(2001):Improvingdistance-basedphylogenyestimation
• SATé(2009,2012)andPASTA(2015):Co-estimationofmultiplesequencealignmentsandgenetrees(upto1,000,000sequences)
• DACTAL(2012):Almostalignment-freetreeestimation
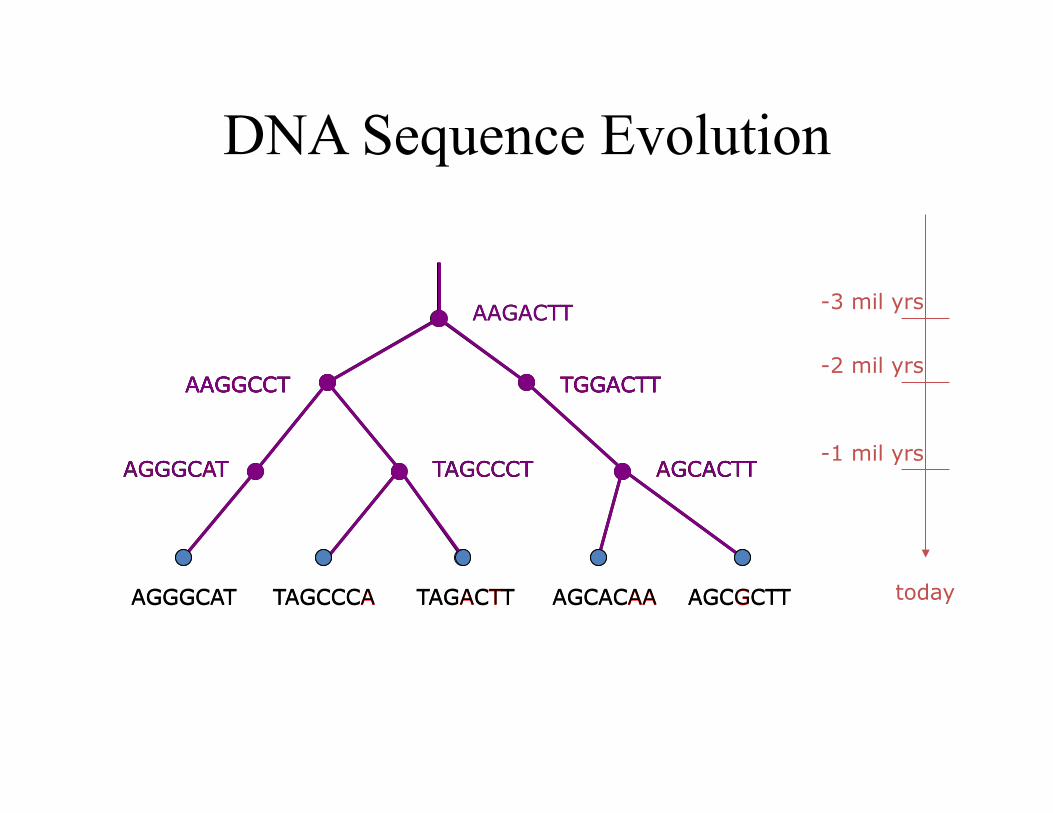
DNA Sequence Evolution
AAGACTT
TGGACTT AAGGCCT
-3 mil yrs
-2 mil yrs
-1 mil yrs
today
AGGGCAT TAGCCCT AGCACTT
AAGGCCT TGGACTT
TAGCCCA TAGACTT AGCGCTT AGCACAA AGGGCAT
AGGGCAT TAGCCCT AGCACTT
AAGACTT
TGGACTT AAGGCCT
AGGGCAT TAGCCCT AGCACTT
AAGGCCT TGGACTT
AGCGCTT AGCACAA TAGACTT TAGCCCA AGGGCAT

…ACGGTGCAGTTACCA…
Mutation Deletion
…ACCAGTCACCA…
Indels (insertions and deletions)
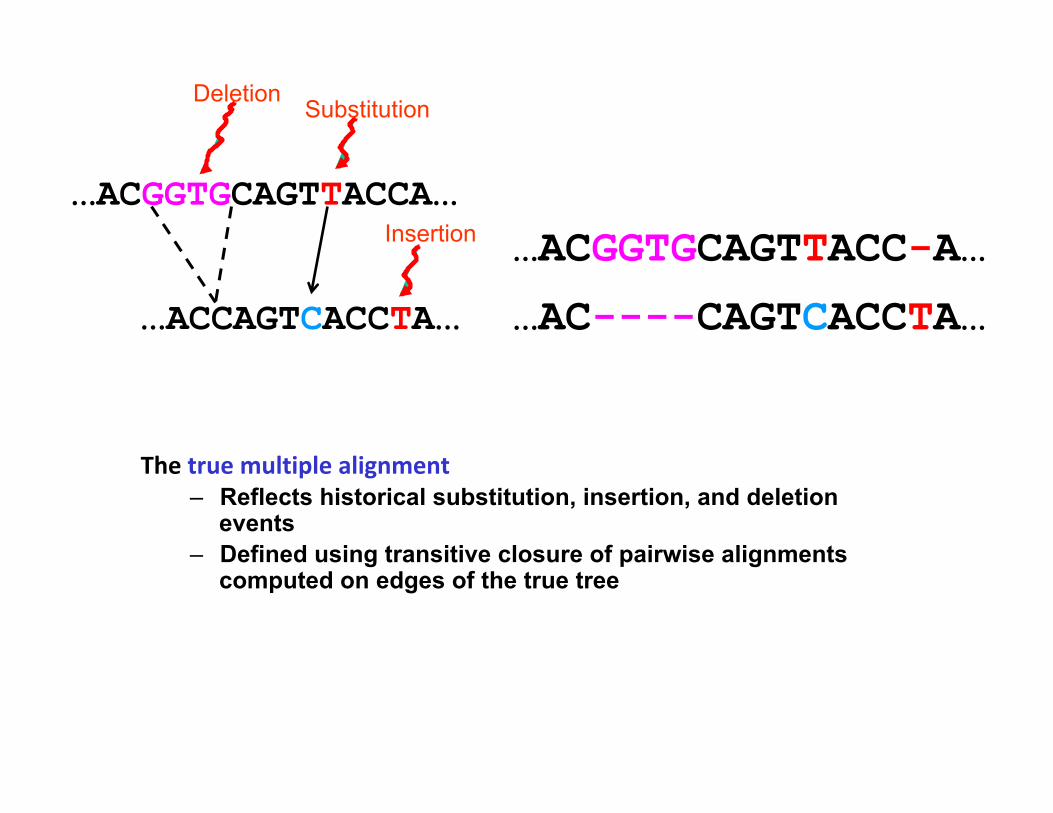
…ACGGTGCAGTTACC-A…
…AC----CAGTCACCTA…
Thetruemultiplealignment– Reflects historical substitution, insertion, and deletion
events – Defined using transitive closure of pairwise alignments
computed on edges of the true tree
…ACGGTGCAGTTACCA…
Substitution Deletion
…ACCAGTCACCTA…
Insertion

Gene Tree Estimation
S1 = AGGCTATCACCTGACCTCCA S2 = TAGCTATCACGACCGC S3 = TAGCTGACCGC S4 = TCACGACCGACA

Input: unaligned sequences
S1 = AGGCTATCACCTGACCTCCA S2 = TAGCTATCACGACCGC S3 = TAGCTGACCGC S4 = TCACGACCGACA
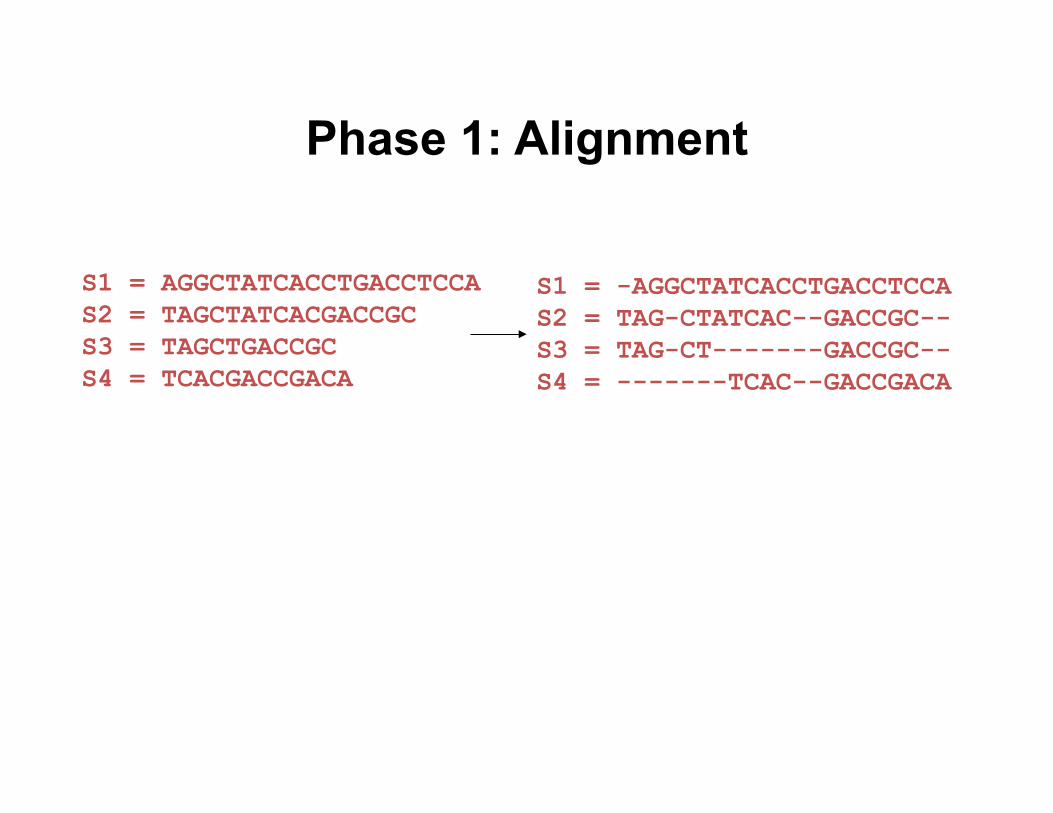
Phase 1: Alignment
S1 = -AGGCTATCACCTGACCTCCA S2 = TAG-CTATCAC--GACCGC-- S3 = TAG-CT-------GACCGC-- S4 = -------TCAC--GACCGACA
S1 = AGGCTATCACCTGACCTCCA S2 = TAGCTATCACGACCGC S3 = TAGCTGACCGC S4 = TCACGACCGACA
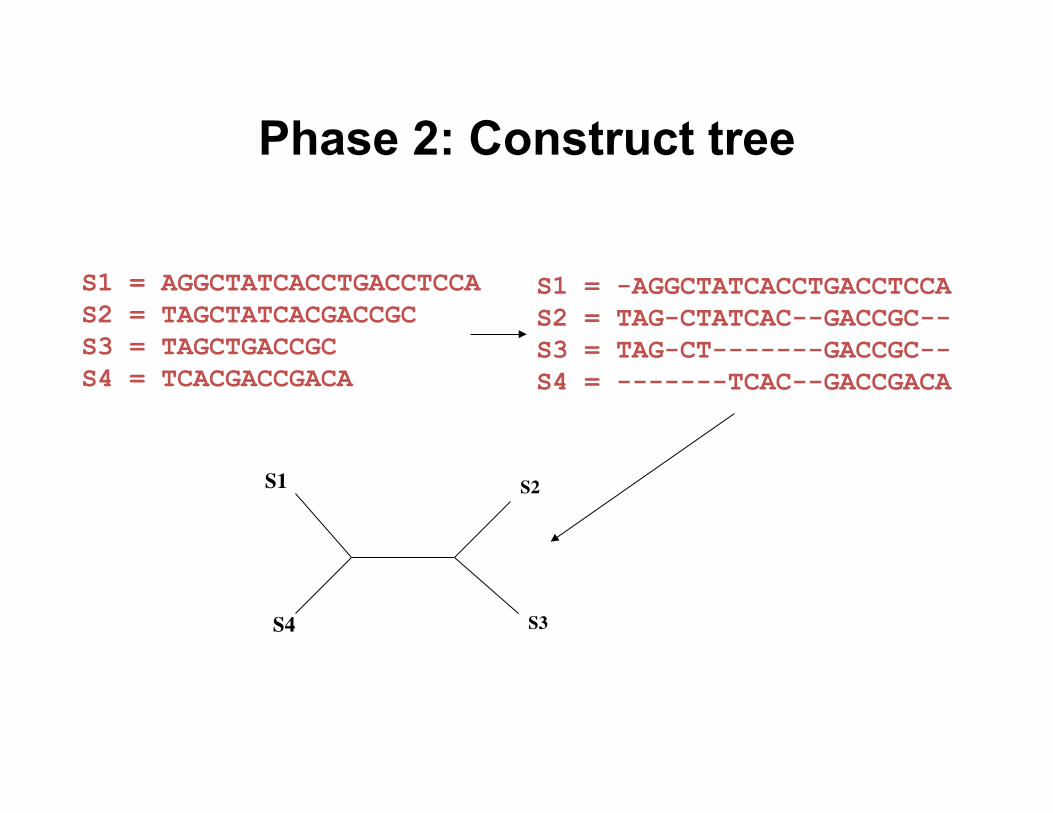
Phase 2: Construct tree
S1 = -AGGCTATCACCTGACCTCCA S2 = TAG-CTATCAC--GACCGC-- S3 = TAG-CT-------GACCGC-- S4 = -------TCAC--GACCGACA
S1 = AGGCTATCACCTGACCTCCA S2 = TAGCTATCACGACCGC S3 = TAGCTGACCGC S4 = TCACGACCGACA
S1
S4
S2
S3

Two-phaseestimationAlignmentmethods• Clustal• POY(andPOY*)• Probcons(andProbtree)• Probalign• MAFFT• Muscle• Di-align• T-Coffee• Prank(PNAS2005,Science2008)• Opal(ISMBandBioinf.2007)• FSA(PLoSComp.Bio.2009)• Infernal(Bioinf.2009)• Etc.
Phylogenymethods• BayesianMCMC• Maximumparsimony• Maximumlikelihood• Neighborjoining• FastME• UPGMA• Quartetpuzzling• Etc.

Two-phaseestimationAlignmentmethods• Clustal• POY(andPOY*)• Probcons(andProbtree)• Probalign• MAFFT• Muscle• Di-align• T-Coffee• Prank(PNAS2005,Science2008)• Opal(ISMBandBioinf.2007)• FSA(PLoSComp.Bio.2009)• Infernal(Bioinf.2009)• Etc.
Phylogenymethods• BayesianMCMC• Maximumparsimony• Maximumlikelihood• Neighborjoining• FastME• UPGMA• Quartetpuzzling• Etc.

Two-phaseestimationAlignmentmethods• Clustal• POY(andPOY*)• Probcons(andProbtree)• Probalign• MAFFT• Muscle• Di-align• T-Coffee• Prank(PNAS2005,Science2008)• Opal(ISMBandBioinf.2007)• FSA(PLoSComp.Bio.2009)• Infernal(Bioinf.2009)• Etc.
Phylogenymethods• BayesianMCMC• Maximumparsimony• Maximumlikelihood• Neighborjoining• FastME• UPGMA• Quartetpuzzling• Etc.
RAxML:heuristicforlarge-scaleMLoptimization
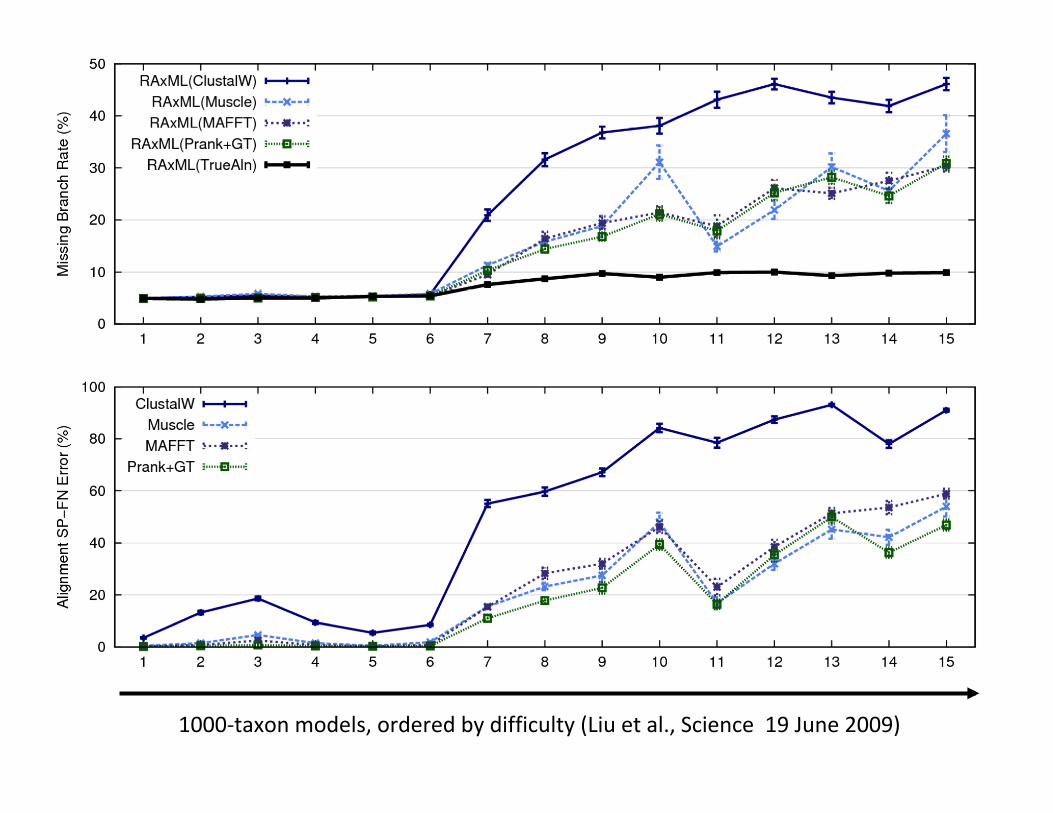
1000-taxonmodels,orderedbydifficulty(Liuetal.,Science19June2009)

Large-scaleCo-estimationofalignmentsandtrees
• SATé:Liuetal.,Science2009(upto10,000sequences)andSystematicBiology2012(upto50,000sequences)
• PASTA:Mirarabetal.,J.ComputationalBiology2015(upto1,000,000sequences)
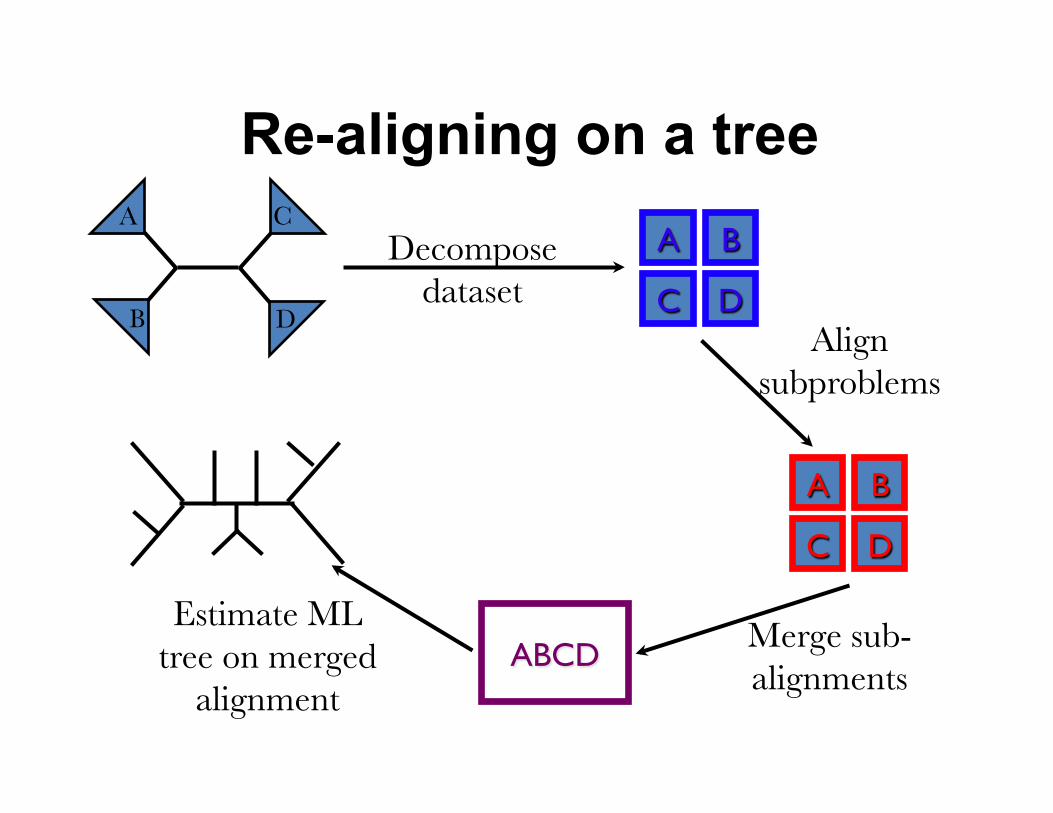
Re-aligning on a tree A
B D
C
Merge sub-alignments
Estimate ML tree on merged
alignment
Decompose dataset
A B
C DAlign
subproblems
A B
C D
ABCD

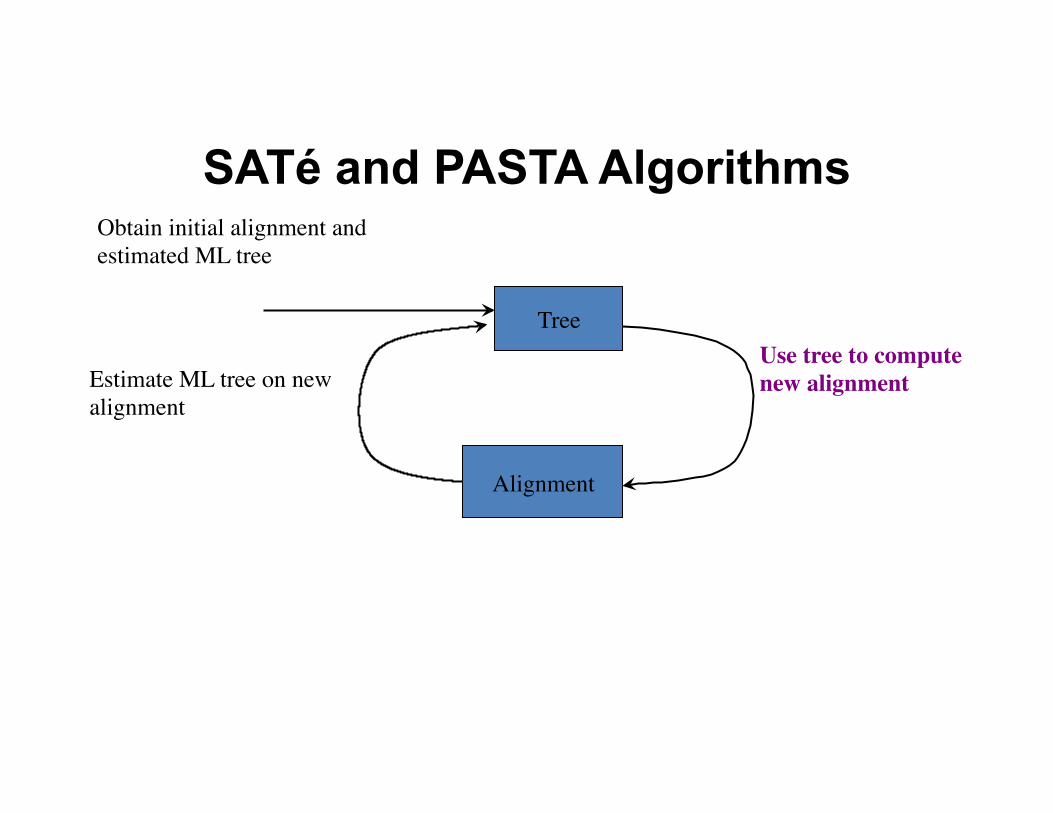
SATé and PASTA Algorithms
Tree
Obtain initial alignment and estimated ML tree

SATé and PASTA Algorithms
Tree
Obtain initial alignment and estimated ML tree
Use tree to compute new alignment
Alignment
SATé and PASTA Algorithms
Estimate ML tree on new alignment
Tree
Obtain initial alignment and estimated ML tree
Use tree to compute new alignment
Alignment
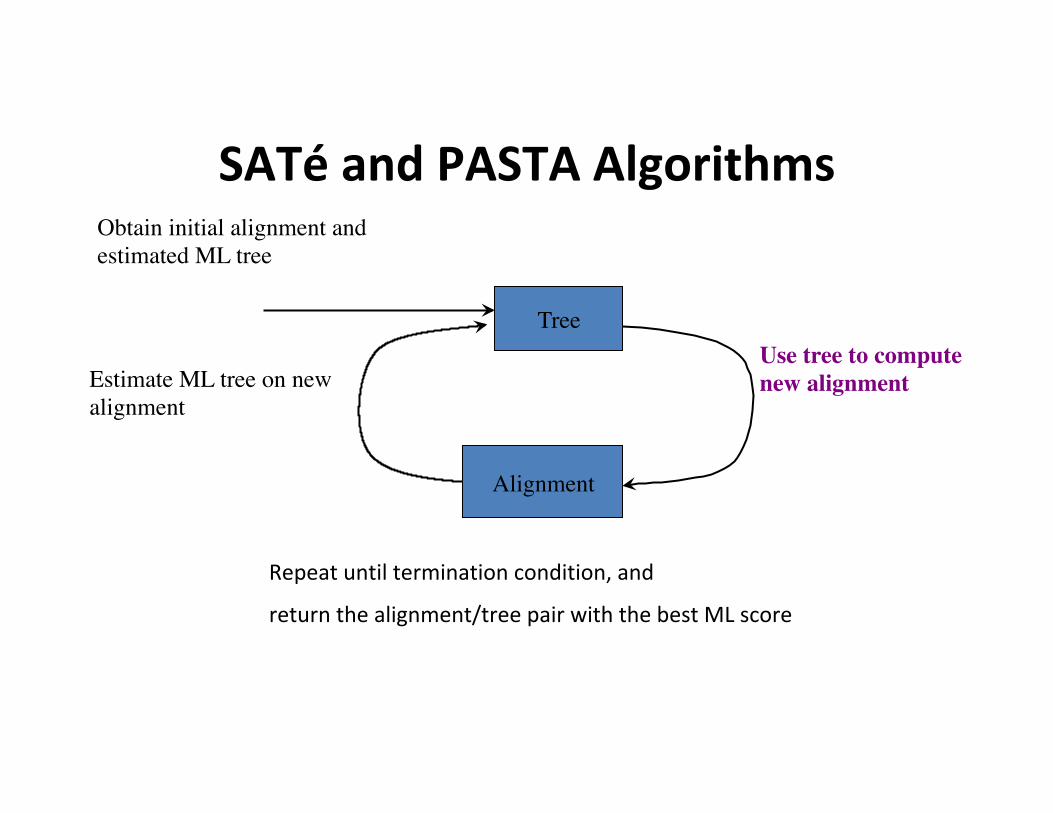
SATéandPASTAAlgorithms
Estimate ML tree on new alignment
Tree
Obtain initial alignment and estimated ML tree
Use tree to compute new alignment
Alignment
Repeatuntilterminationcondition,and
returnthealignment/treepairwiththebestMLscore

1000-taxonmodels,orderedbydifficulty(Liuetal.,Science19June2009)
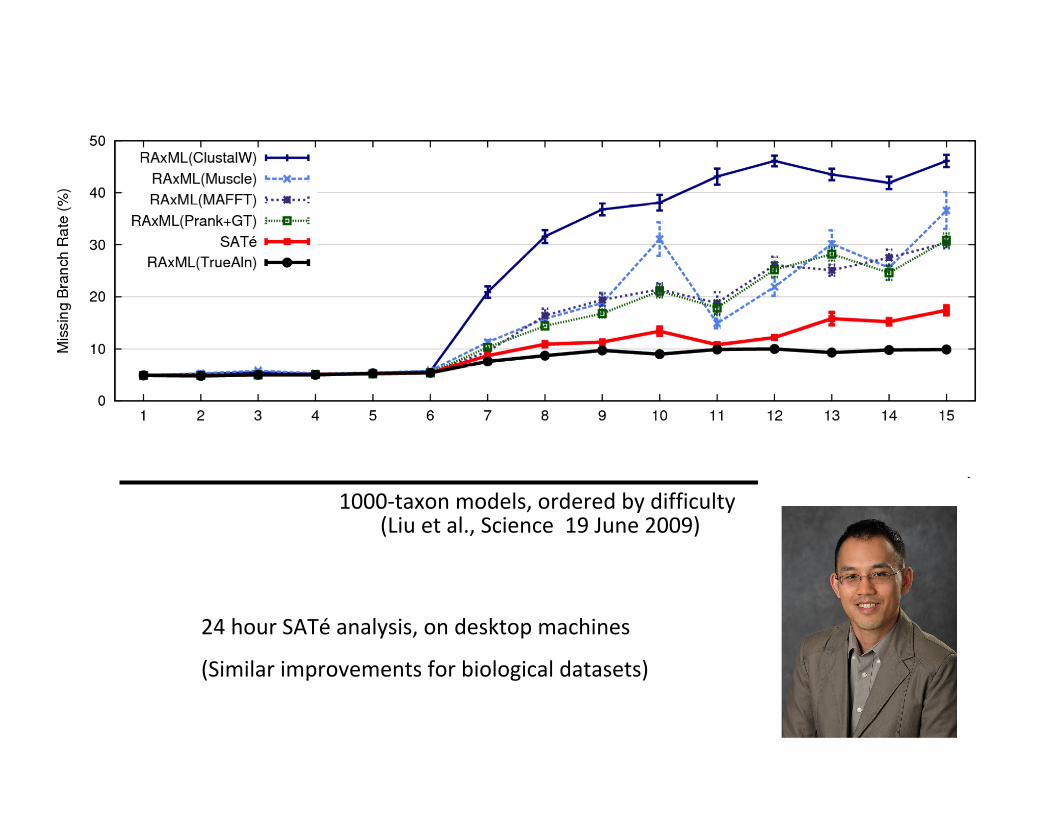
1000-taxonmodels,orderedbydifficulty(Liuetal.,Science19June2009)
24hourSATéanalysis,ondesktopmachines
(Similarimprovementsforbiologicaldatasets)
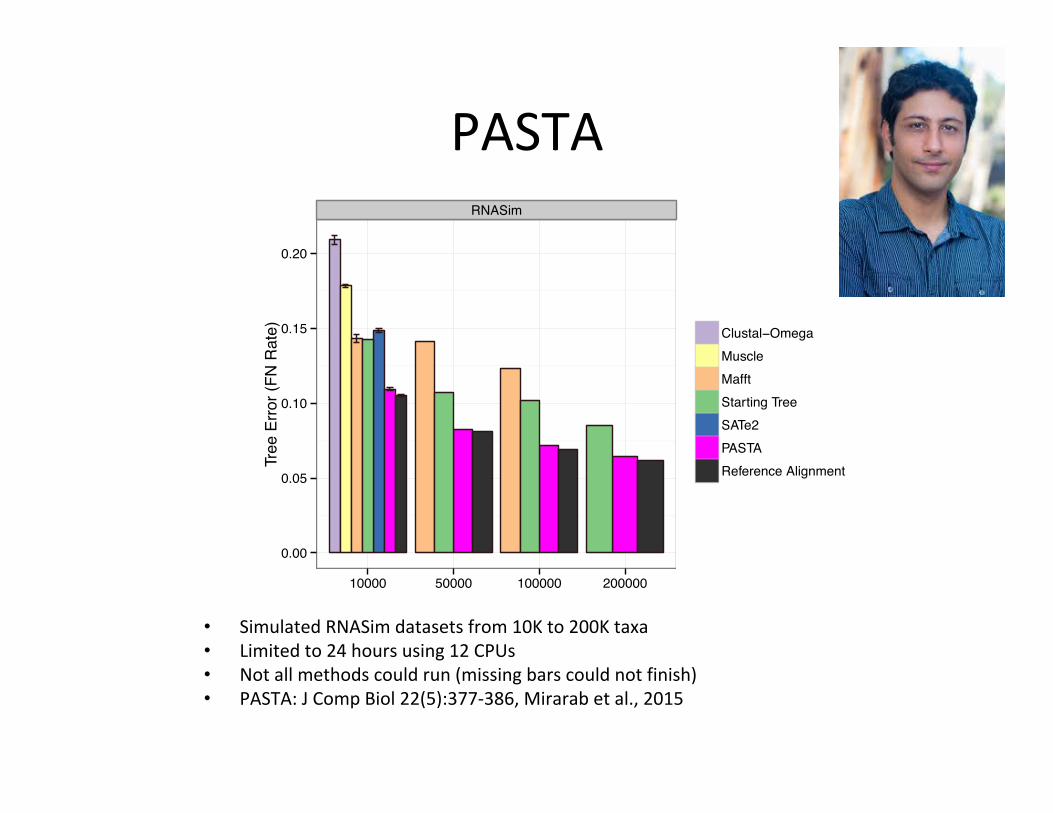
RNASim
0.00
0.05
0.10
0.15
0.20
10000 50000 100000 200000
Tree
Erro
r (FN
Rat
e) Clustal−OmegaMuscleMafftStarting TreeSATe2PASTAReference Alignment
• SimulatedRNASimdatasetsfrom10Kto200Ktaxa• Limitedto24hoursusing12CPUs• Notallmethodscouldrun(missingbarscouldnotfinish)• PASTA:JCompBiol22(5):377-386,Mirarabetal.,2015
PASTA

Multiple Sequence Alignment (MSA): a scientific grand challenge1
S1 = -AGGCTATCACCTGACCTCCA S2 = TAG-CTATCAC--GACCGC-- S3 = TAG-CT-------GACCGC-- … Sn = -------TCAC--GACCGACA
S1 = AGGCTATCACCTGACCTCCA S2 = TAGCTATCACGACCGC S3 = TAGCTGACCGC … Sn = TCACGACCGACA
Novel techniques needed for scalability and accuracy NP-hard problems and large datasets Current methods do not provide good accuracy Few methods can analyze even moderately large datasets Many important applications besides phylogenetic estimation
1 Frontiers in Massive Data Analysis, National Academies Press, 2013

Threedivide-and-conqueralgorithms
• DCM1(2001):Improvingdistance-basedphylogenyestimation
• SATé(2009,2012)andPASTA(2015):Co-estimationofmultiplesequencealignmentsandgenetrees(upto1,000,000sequences)
• DACTAL(2012):Almostalignment-freetreeestimation

DACTAL
• Divide-And-ConquerTrees(Almost)withoutalignments
• Nelesenetal.,ISMB2012andBioinformatics2012
• Input:unalignedsequences• Output:Tree(butnoalignment)
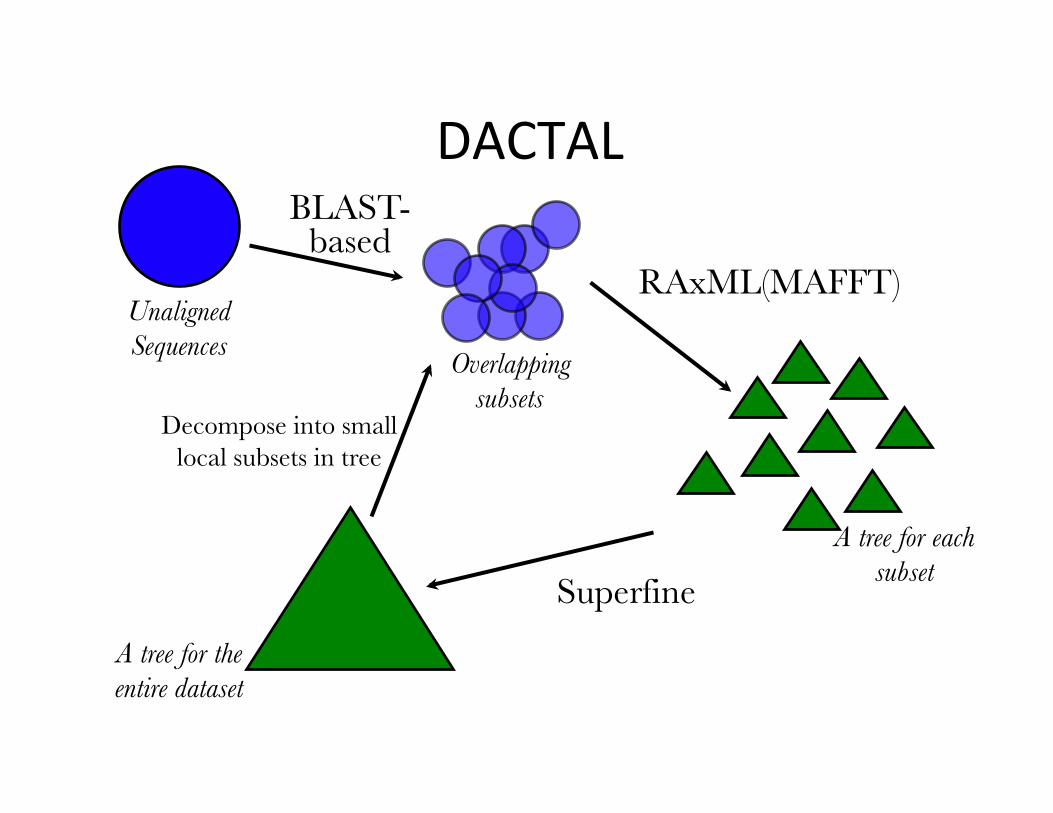
DACTAL
Superfine
RAxML(MAFFT)
Decompose into small local subsets in tree
BLAST-based
Overlapping subsets
A tree for each subset
Unaligned Sequences
A tree for the entire dataset

Resultsonthreebiologicaldatasets–6000to28,000sequences.Weshowresultswith5DACTALiterations
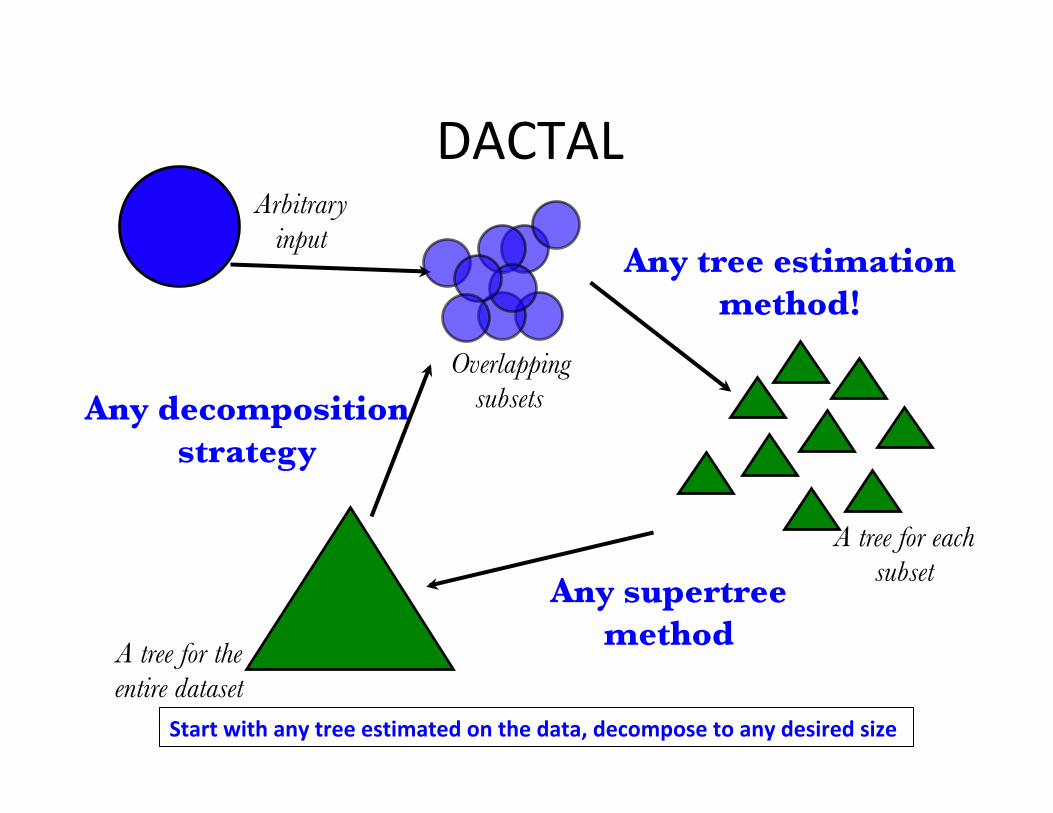
DACTAL
Any supertree method
Any tree estimation method!
Any decomposition strategy
Overlapping subsets
A tree for each subset
Arbitrary input
A tree for the entire dataset
Startwithanytreeestimatedonthedata,decomposetoanydesiredsize

“Boosters”, or “Meta-Methods”
• Meta-methods use divide-and-conquer and iteration (or other techniques) to “boost” the performance of base methods (phylogeny reconstruction, alignment estimation, etc)
Meta-methodBase method M M*

Summary
• Weshowedthreetechniquestoimproveaccuracyandscalability,thatuseddivide-and-conquer(andsometimesiteration):– DCM1(improvesaccuracyandscalabilityfordistance-basedmethods)
– SATeandPASTA(improvesaccuracyandscalabilityformultiplesequencealignmentmethods)
– DACTAL(generaltechniqueforimprovingtreeestimation)
• Innovativealgorithmdesigncanimproveaccuracyaswellasreducerunningtime.

Openproblems(andpossiblecourseprojects)
• MultipleSequenceAlignment:– Mergingtwoalignments– Consensusalignments
• Supertreeestimation– Needtoscaleto100,000+specieswithhighaccuracy– Approximationalgorithms
• Speciestreeandphylogeneticnetworkestimation– Addressgenetreeheterogeneityduetomultiplecauses– Theoreticalguaranteesunderstochasticmodels– Scalabilitytolargenumbersofspecieswithwholegenomes
• StatisticalModelsofevolution– Allmodelsareseriouslyunrealistic– Weneedbetterstatisticalmodels– Newtheory
• Applicationstohistoricallinguistics,microbiomeanalysis,proteinstructureandfunctionprediction,tumorevolution,etc.

Somepriorcourseprojectsthatwerepublished
• ASTRAL:genome-scalecoalescent-basedspeciestreeestimation,Mirarabetal.2014(ECCB2014andBioinformatics2014)
• Phylogenomicspeciestreeestimationinthepresenceofincompletelineagesortingandhorizontalgenetransfer,Davidsonetal.2014(RECOMB-CG2014andBMCGenomics2014)
• AcomparativestudyofSVDquartetsandothercoalescent-basedspeciestreeestimationmethods,Chouetal.(RECOMB-CGandBMCGenomics2014)
• ASTRID:Accuratespeciestreesfrominternodedistances,VachaspatiandWarnow(RECOMB-CG2015andBMCGenomics2015)
• Scalingstatisticalmultiplesequencealignmenttolargedatasets,NuteandWarnow(RECOMB-CG2016andBMCGenomics2016)
Scientific challenges: • Ultra-large multiple-sequence alignment • Gene tree estimation • Metagenomic classification • Alignment-free phylogeny estimation • Supertree estimation • Estimating species trees from many gene trees • Genome rearrangement phylogeny • Reticulate evolution • Visualization of large trees and alignments • Data mining techniques to explore multiple optima • Theoretical guarantees under Markov models of evolution
Techniques: applied probability theory, graph theory, supercomputing, and heuristics Testing: simulations and real data
The Tree of Life: Multiple Challenges

OtherInformation
• Tandy’sofficehours:Tuesdays3:15-4:15(andbyappointment).
• Tandy’semail:[email protected]• TA:SarahChristensen([email protected])• HomeworkwillbesubmittedthroughMoodle.• Coursewebpage:http://tandy.cs.illinois.edu/581-2018.html

![cs581-lec1 - Tandy Warnowtandy.cs.illinois.edu/cs581-lec1.pdf · Additive Matrices •A square matrix D=[d ij] is additive if and only if there is a tree T and edge-weighting w such](https://static.fdocuments.in/doc/165x107/5f184af3ba4adb33030b4ce2/cs581-lec1-tandy-additive-matrices-aa-square-matrix-dd-ij-is-additive-if.jpg)
















